Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Article SELECTION from Collection of Aviva Lev-Ari, PhD, RN Scientific Articles on PULSE on LinkedIn.com for Training Small Language Models (SLMs) in Domain-aware Content of Medical, Pharmaceutical, Life Sciences and Healthcare by 15 Subjects Matter
Article selection: Aviva Lev-Ari, PhD, RN
#1 – February 20, 2016
Contributions to Personalized and Precision Medicine & Genomic Research
Eight Subcellular Pathologies driving Chronic Metabolic Diseases – Methods for Mapping Bioelectronic Adjustable Measurements as potential new Therapeutics: Impact on Pharmaceuticals in Use
In this curation we wish to present two breaking through goals:
Goal 1:
Exposition of a new direction of research leading to a more comprehensive understanding of Metabolic Dysfunctional Diseases that are implicated in effecting the emergence of the two leading causes of human mortality in the World in 2023: (a) Cardiovascular Diseases, and (b) Cancer
Goal 2:
Development of Methods for Mapping Bioelectronic Adjustable Measurements as potential new Therapeutics for these eight subcellular causes of chronic metabolic diseases. It is anticipated that it will have a potential impact on the future of Pharmaceuticals to be used, a change from the present time current treatment protocols for Metabolic Dysfunctional Diseases.
According to Dr. Robert Lustig, M.D, an American pediatric endocrinologist. He is Professor emeritus of Pediatrics in the Division of Endocrinology at the University of California, San Francisco, where he specialized in neuroendocrinology and childhood obesity, there are eight subcellular pathologies that drive chronic metabolic diseases.
These eight subcellular pathologies can’t be measured at present time.
In this curation we will attempt to explore methods of measurement for each of these eight pathologies by harnessing the promise of the emerging field known as Bioelectronics.
Unmeasurable eight subcellular pathologies that drive chronic metabolic diseases
Glycation
Oxidative Stress
Mitochondrial dysfunction [beta-oxidation Ac CoA malonyl fatty acid]
Insulin resistance/sensitive [more important than BMI], known as a driver to cancer development
Membrane instability
Inflammation in the gut [mucin layer and tight junctions]
Epigenetics/Methylation
Autophagy [AMPKbeta1 improvement in health span]
Diseases that are not Diseases: no drugs for them, only diet modification will help
Image source
Robert Lustig, M.D. on the Subcellular Processes That Belie Chronic Disease
These eight Subcellular Pathologies driving Chronic Metabolic Diseases are becoming our focus for exploration of the promise of Bioelectronics for two pursuits:
Will Bioelectronics be deemed helpful in measurement of each of the eight pathological processes that underlie and that drive the chronic metabolic syndrome(s) and disease(s)?
IF we will be able to suggest new measurements to currently unmeasurable health harming processes THEN we will attempt to conceptualize new therapeutic targets and new modalities for therapeutics delivery – WE ARE HOPEFUL
In the Bioelecronics domain we are inspired by the work of the following three research sources:
Michael Levin is an American developmental and synthetic biologist at Tufts University, where he is the Vannevar Bush Distinguished Professor. Levin is a director of the Allen Discovery Center at Tufts University and Tufts Center for Regenerative and Developmental Biology. Wikipedia
THE VOICE of Dr. Justin D. Pearlman, MD, PhD, FACC
PENDING
THE VOICE of Stephen J. Williams, PhD
Ten TakeAway Points of Dr. Lustig’s talk on role of diet on the incidence of Type II Diabetes
25% of US children have fatty liver
Type II diabetes can be manifested from fatty live with 151 million people worldwide affected moving up to 568 million in 7 years
A common myth is diabetes due to overweight condition driving the metabolic disease
There is a trend of ‘lean’ diabetes or diabetes in lean people, therefore body mass index not a reliable biomarker for risk for diabetes
Thirty percent of ‘obese’ people just have high subcutaneous fat. the visceral fat is more problematic
there are people who are ‘fat’ but insulin sensitive while have growth hormone receptor defects. Points to other issues related to metabolic state other than insulin and potentially the insulin like growth factors
At any BMI some patients are insulin sensitive while some resistant
Visceral fat accumulation may be more due to chronic stress condition
Fructose can decrease liver mitochondrial function
A methionine and choline deficient diet can lead to rapid NASH development
The following paper in Cells describes the discovery of protein interactors of endoglin, which is recruited to membranes at the TGF-β receptor complex upon TGF-β signaling. Interesting a carbohydrate binding protein, galectin-3, and an E3-ligase, TRIM21, were found to be unique interactors within this complex.
Gallardo-Vara E, Ruiz-Llorente L, Casado-Vela J, Ruiz-Rodríguez MJ, López-Andrés N, Pattnaik AK, Quintanilla M, Bernabeu C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells. 2019 Sep 13;8(9):1082. doi: 10.3390/cells8091082. PMID: 31540324; PMCID: PMC6769930.
Abstract
Endoglin is a 180-kDa glycoprotein receptor primarily expressed by the vascular endothelium and involved in cardiovascular disease and cancer. Heterozygous mutations in the endoglin gene (ENG) cause hereditary hemorrhagic telangiectasia type 1, a vascular disease that presents with nasal and gastrointestinal bleeding, skin and mucosa telangiectases, and arteriovenous malformations in internal organs. A circulating form of endoglin (alias soluble endoglin, sEng), proteolytically released from the membrane-bound protein, has been observed in several inflammation-related pathological conditions and appears to contribute to endothelial dysfunction and cancer development through unknown mechanisms. Membrane-bound endoglin is an auxiliary component of the TGF-β receptor complex and the extracellular region of endoglin has been shown to interact with types I and II TGF-β receptors, as well as with BMP9 and BMP10 ligands, both members of the TGF-β family. To search for novel protein interactors, we screened a microarray containing over 9000 unique human proteins using recombinant sEng as bait. We find that sEng binds with high affinity, at least, to 22 new proteins. Among these, we validated the interaction of endoglin with galectin-3, a secreted member of the lectin family with capacity to bind membrane glycoproteins, and with tripartite motif-containing protein 21 (TRIM21), an E3 ubiquitin-protein ligase. Using human endothelial cells and Chinese hamster ovary cells, we showed that endoglin co-immunoprecipitates and co-localizes with galectin-3 or TRIM21. These results open new research avenues on endoglin function and regulation.
Endoglin is an auxiliary TGF-β co-receptor predominantly expressed in endothelial cells, which is involved in vascular development, repair, homeostasis, and disease [1,2,3,4]. Heterozygous mutations in the human ENDOGLIN gene (ENG) cause hereditary hemorrhagic telangiectasia (HHT) type 1, a vascular disease associated with nasal and gastrointestinal bleeds, telangiectases on skin and mucosa and arteriovenous malformations in the lung, liver, and brain [4,5,6]. The key role of endoglin in the vasculature is also illustrated by the fact that endoglin-KO mice die in utero due to defects in the vascular system [7]. Endoglin expression is markedly upregulated in proliferating endothelial cells involved in active angiogenesis, including the solid tumor neovasculature [8,9]. For this reason, endoglin has become a promising target for the antiangiogenic treatment of cancer [10,11,12]. Endoglin is also expressed in cancer cells where it can behave as both a tumor suppressor in prostate, breast, esophageal, and skin carcinomas [13,14,15,16] and a promoter of malignancy in melanoma and Ewing’s sarcoma [17]. Ectodomain shedding of membrane-bound endoglin may lead to a circulating form of the protein, also known as soluble endoglin (sEng) [18,19,20]. Increased levels of sEng have been found in several vascular-related pathologies, including preeclampsia, a disease of high prevalence in pregnant women which, if left untreated, can lead to serious and even fatal complications for both mother and baby [2,18,19,21]. Interestingly, several lines of evidence support a pathogenic role of sEng in the vascular system, including endothelial dysfunction, antiangiogenic activity, increased vascular permeability, inflammation-associated leukocyte adhesion and transmigration, and hypertension [18,22,23,24,25,26,27]. Because of its key role in vascular pathology, a large number of studies have addressed the structure and function of endoglin at the molecular level, in order to better understand its mechanism of action.
Galectin-3 Interacts with Endoglin in Cells
Galectin-3 is a secreted member of the lectin family with the capacity to bind membrane glycoproteins like endoglin and is involved in the pathogenesis of many human diseases [52]. We confirmed the protein screen data for galectin-3, as evidenced by two-way co-immunoprecipitation of endoglin and galectin-3 upon co-transfection in CHO-K1 cells. As shown in Figure 1A, galectin-3 and endoglin were efficiently transfected, as demonstrated by Western blot analysis in total cell extracts. No background levels of endoglin were observed in control cells transfected with the empty vector (Ø). By contrast, galectin-3 could be detected in all samples but, as expected, showed an increased signal in cells transfected with the galectin-3 expression vector. Co-immunoprecipitation studies of these cell lysates showed that galectin-3 was present in endoglin immunoprecipitates (Figure 1B). Conversely, endoglin was also detected in galectin-3 immunoprecipitates (Figure 1C).
Figure 1. Protein–protein association between galectin-3 and endoglin. (A–C). Co-immunoprecipitation of galectin-3 and endoglin. CHO-K1 cells were transiently transfected with pcEXV-Ø (Ø), pcEXV–HA–EngFL (Eng) and pcDNA3.1–Gal-3 (Gal3) expression vectors. (A) Total cell lysates (TCL) were analyzed by SDS-PAGE under reducing conditions, followed by Western blot (WB) analysis using specific antibodies to endoglin, galectin-3 and β-actin (loading control). Cell lysates were subjected to immunoprecipitation (IP) with anti-endoglin (B) or anti-galectin-3 (C) antibodies, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin or anti-galectin-3 antibodies, as indicated. Negative controls with an IgG2b (B) and IgG1 (C) were included. (D) Protein-protein interactions between galectin-3 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 7.3 µM recombinant human galectin-3/6xHis at the C-terminus (LGALS3), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Figure 2.Galectin-3 and endoglin co-localize in human endothelial cells. Human umbilical vein-derived endothelial cell (HUVEC) monolayers were fixed with paraformaldehyde, permeabilized with Triton X-100, incubated with the mouse mAb P4A4 anti-endoglin, washed, and incubated with a rabbit polyclonal anti-galectin-3 antibody (PA5-34819). Galectin-3 and endoglin were detected by immunofluorescence upon incubation with Alexa 647 goat anti-rabbit IgG (red staining) and Alexa 488 goat anti-mouse IgG (green staining) secondary antibodies, respectively. (A) Single staining of galectin-3 (red) and endoglin (green) at the indicated magnifications. (B) Merge images plus DAPI (nuclear staining in blue) show co-localization of galectin-3 and endoglin (yellow color). Representative images of five different experiments are shown.
Endoglin associates with the cullin-type E3 ligase TRIM21
Figure 3.Protein–protein association between TRIM21 and endoglin. (A–E) Co-immunoprecipitation of TRIM21 and endoglin. A,B. HUVEC monolayers were lysed and total cell lysates (TCL) were subjected to SDS-PAGE under reducing (for TRIM21 detection) or nonreducing (for endoglin detection) conditions, followed by Western blot (WB) analysis using antibodies to endoglin, TRIM21 or β-actin (A). HUVECs lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or negative control antibodies, followed by WB analysis with anti-endoglin (B). C,D. CHO-K1 cells were transiently transfected with pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (E) or pcDNA3.1–HA–hTRIM21 (T) expression vectors, as indicated. Total cell lysates (TCL) were subjected to SDS-PAGE under nonreducing conditions and WB analysis using specific antibodies to endoglin, TRIM21, and β-actin (C). Cell lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or anti-endoglin antibodies, followed by SDS-PAGE under reducing (upper panel) or nonreducing (lower panel) conditions and WB analysis with anti-TRIM21 or anti-endoglin antibodies. Negative controls of appropriate IgG were included (D). E. CHO-K1 cells were transiently transfected with pcDNA3.1–HA–hTRIM21 and pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (FL; full-length), pDisplay–HA–EngEC (EC; cytoplasmic-less) or pDisplay–HA–EngTMEC (TMEC; cytoplasmic-less) expression vectors, as indicated. Cell lysates were subjected to immunoprecipitation with anti-TRIM21, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin antibodies, as indicated. The asterisk indicates the presence of a nonspecific band. Mr, molecular reference; Eng, endoglin; TRIM, TRIM21. (F) Protein–protein interactions between TRIM21 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 5.4 µM recombinant human TRIM21/6xHis at the N-terminus (R052), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Table 1. Human protein-array analysis of endoglin interactors1.
1 Microarrays containing over 9000 unique human proteins were screened using recombinant sEng as a probe. Protein interactors showing the highest scores (Z-score ≥2.0) are listed. GeneBank (https://www.ncbi.nlm.nih.gov/genbank/) and UniProtKB (https://www.uniprot.org/help/uniprotkb) accession numbers are indicated with a yellow or green background, respectively. The cellular compartment of each protein was obtained from the UniProtKB webpage. Proteins selected for further studies (TRIM21 and galectin-3) are indicated in bold type with blue background.
Note: the following are from NCBI Genbank and Genecards on TRIM21
Official Symbol TRIM21provided by HGNC Official Full Name tripartite motif containing 21provided by HGNC Primary source HGNC:HGNC:11312 See related Ensembl:ENSG00000132109MIM:109092;AllianceGenome:HGNC:11312 Gene type protein coding RefSeq status REVIEWED Organism Homo sapiens Lineage Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini; Catarrhini; Hominidae; Homo Also known as SSA; RO52; SSA1; RNF81; Ro/SSA Summary This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008] Expression Ubiquitous expression in spleen (RPKM 15.5), appendix (RPKM 13.2) and 24 other tissues See more Orthologs mouseall NEW Try the new Gene table Try the new Transcript table
This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008]
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression. Enhances the decapping activity of DCP2. Exists as a ribonucleoprotein particle present in all mammalian cells studied and composed of a single polypeptide and one of four small RNA molecules. At least two isoforms are present in nucleated and red blood cells, and tissue specific differences in RO/SSA proteins have been identified. The common feature of these proteins is their ability to bind HY RNAs.2. Involved in the regulation of innate immunity and the inflammatory response in response to IFNG/IFN-gamma. Organizes autophagic machinery by serving as a platform for the assembly of ULK1, Beclin 1/BECN1 and ATG8 family members and recognizes specific autophagy targets, thus coordinating target recognition with assembly of the autophagic apparatus and initiation of autophagy. Acts as an autophagy receptor for the degradation of IRF3, hence attenuating type I interferon (IFN)-dependent immune responses (PubMed:26347139, 16297862, 16316627, 16472766, 16880511, 18022694, 18361920, 18641315, 18845142, 19675099). Represses the innate antiviral response by facilitating the formation of the NMI-IFI35 complex through ‘Lys-63’-linked ubiquitination of NMI (PubMed:26342464). ( RO52_HUMAN,P19474 )
Molecular function for TRIM21 Gene according to UniProtKB/Swiss-Prot
Function:
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression.
Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners
Gallardo-Vara E, Ruiz-Llorente L, Casado-Vela J, Ruiz-Rodríguez MJ, López-Andrés N, Pattnaik AK, Quintanilla M, Bernabeu C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells. 2019 Sep 13;8(9):1082. doi: 10.3390/cells8091082. PMID: 31540324; PMCID: PMC6769930.
Abstract
Endoglin is a 180-kDa glycoprotein receptor primarily expressed by the vascular endothelium and involved in cardiovascular disease and cancer. Heterozygous mutations in the endoglin gene (ENG) cause hereditary hemorrhagic telangiectasia type 1, a vascular disease that presents with nasal and gastrointestinal bleeding, skin and mucosa telangiectases, and arteriovenous malformations in internal organs. A circulating form of endoglin (alias soluble endoglin, sEng), proteolytically released from the membrane-bound protein, has been observed in several inflammation-related pathological conditions and appears to contribute to endothelial dysfunction and cancer development through unknown mechanisms. Membrane-bound endoglin is an auxiliary component of the TGF-β receptor complex and the extracellular region of endoglin has been shown to interact with types I and II TGF-β receptors, as well as with BMP9 and BMP10 ligands, both members of the TGF-β family. To search for novel protein interactors, we screened a microarray containing over 9000 unique human proteins using recombinant sEng as bait. We find that sEng binds with high affinity, at least, to 22 new proteins. Among these, we validated the interaction of endoglin with galectin-3, a secreted member of the lectin family with capacity to bind membrane glycoproteins, and with tripartite motif-containing protein 21 (TRIM21), an E3 ubiquitin-protein ligase. Using human endothelial cells and Chinese hamster ovary cells, we showed that endoglin co-immunoprecipitates and co-localizes with galectin-3 or TRIM21. These results open new research avenues on endoglin function and regulation.
Endoglin is an auxiliary TGF-β co-receptor predominantly expressed in endothelial cells, which is involved in vascular development, repair, homeostasis, and disease [1,2,3,4]. Heterozygous mutations in the human ENDOGLIN gene (ENG) cause hereditary hemorrhagic telangiectasia (HHT) type 1, a vascular disease associated with nasal and gastrointestinal bleeds, telangiectases on skin and mucosa and arteriovenous malformations in the lung, liver, and brain [4,5,6]. The key role of endoglin in the vasculature is also illustrated by the fact that endoglin-KO mice die in utero due to defects in the vascular system [7]. Endoglin expression is markedly upregulated in proliferating endothelial cells involved in active angiogenesis, including the solid tumor neovasculature [8,9]. For this reason, endoglin has become a promising target for the antiangiogenic treatment of cancer [10,11,12]. Endoglin is also expressed in cancer cells where it can behave as both a tumor suppressor in prostate, breast, esophageal, and skin carcinomas [13,14,15,16] and a promoter of malignancy in melanoma and Ewing’s sarcoma [17]. Ectodomain shedding of membrane-bound endoglin may lead to a circulating form of the protein, also known as soluble endoglin (sEng) [18,19,20]. Increased levels of sEng have been found in several vascular-related pathologies, including preeclampsia, a disease of high prevalence in pregnant women which, if left untreated, can lead to serious and even fatal complications for both mother and baby [2,18,19,21]. Interestingly, several lines of evidence support a pathogenic role of sEng in the vascular system, including endothelial dysfunction, antiangiogenic activity, increased vascular permeability, inflammation-associated leukocyte adhesion and transmigration, and hypertension [18,22,23,24,25,26,27]. Because of its key role in vascular pathology, a large number of studies have addressed the structure and function of endoglin at the molecular level, in order to better understand its mechanism of action.
Galectin-3 Interacts with Endoglin in Cells
Galectin-3 is a secreted member of the lectin family with the capacity to bind membrane glycoproteins like endoglin and is involved in the pathogenesis of many human diseases [52]. We confirmed the protein screen data for galectin-3, as evidenced by two-way co-immunoprecipitation of endoglin and galectin-3 upon co-transfection in CHO-K1 cells. As shown in Figure 1A, galectin-3 and endoglin were efficiently transfected, as demonstrated by Western blot analysis in total cell extracts. No background levels of endoglin were observed in control cells transfected with the empty vector (Ø). By contrast, galectin-3 could be detected in all samples but, as expected, showed an increased signal in cells transfected with the galectin-3 expression vector. Co-immunoprecipitation studies of these cell lysates showed that galectin-3 was present in endoglin immunoprecipitates (Figure 1B). Conversely, endoglin was also detected in galectin-3 immunoprecipitates (Figure 1C).
Figure 1. Protein–protein association between galectin-3 and endoglin. (A–C). Co-immunoprecipitation of galectin-3 and endoglin. CHO-K1 cells were transiently transfected with pcEXV-Ø (Ø), pcEXV–HA–EngFL (Eng) and pcDNA3.1–Gal-3 (Gal3) expression vectors. (A) Total cell lysates (TCL) were analyzed by SDS-PAGE under reducing conditions, followed by Western blot (WB) analysis using specific antibodies to endoglin, galectin-3 and β-actin (loading control). Cell lysates were subjected to immunoprecipitation (IP) with anti-endoglin (B) or anti-galectin-3 (C) antibodies, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin or anti-galectin-3 antibodies, as indicated. Negative controls with an IgG2b (B) and IgG1 (C) were included. (D) Protein-protein interactions between galectin-3 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 7.3 µM recombinant human galectin-3/6xHis at the C-terminus (LGALS3), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Figure 2.Galectin-3 and endoglin co-localize in human endothelial cells. Human umbilical vein-derived endothelial cell (HUVEC) monolayers were fixed with paraformaldehyde, permeabilized with Triton X-100, incubated with the mouse mAb P4A4 anti-endoglin, washed, and incubated with a rabbit polyclonal anti-galectin-3 antibody (PA5-34819). Galectin-3 and endoglin were detected by immunofluorescence upon incubation with Alexa 647 goat anti-rabbit IgG (red staining) and Alexa 488 goat anti-mouse IgG (green staining) secondary antibodies, respectively. (A) Single staining of galectin-3 (red) and endoglin (green) at the indicated magnifications. (B) Merge images plus DAPI (nuclear staining in blue) show co-localization of galectin-3 and endoglin (yellow color). Representative images of five different experiments are shown.
Endoglin associates with the cullin-type E3 ligase TRIM21
Figure 3.Protein–protein association between TRIM21 and endoglin. (A–E) Co-immunoprecipitation of TRIM21 and endoglin. A,B. HUVEC monolayers were lysed and total cell lysates (TCL) were subjected to SDS-PAGE under reducing (for TRIM21 detection) or nonreducing (for endoglin detection) conditions, followed by Western blot (WB) analysis using antibodies to endoglin, TRIM21 or β-actin (A). HUVECs lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or negative control antibodies, followed by WB analysis with anti-endoglin (B). C,D. CHO-K1 cells were transiently transfected with pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (E) or pcDNA3.1–HA–hTRIM21 (T) expression vectors, as indicated. Total cell lysates (TCL) were subjected to SDS-PAGE under nonreducing conditions and WB analysis using specific antibodies to endoglin, TRIM21, and β-actin (C). Cell lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or anti-endoglin antibodies, followed by SDS-PAGE under reducing (upper panel) or nonreducing (lower panel) conditions and WB analysis with anti-TRIM21 or anti-endoglin antibodies. Negative controls of appropriate IgG were included (D). E. CHO-K1 cells were transiently transfected with pcDNA3.1–HA–hTRIM21 and pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (FL; full-length), pDisplay–HA–EngEC (EC; cytoplasmic-less) or pDisplay–HA–EngTMEC (TMEC; cytoplasmic-less) expression vectors, as indicated. Cell lysates were subjected to immunoprecipitation with anti-TRIM21, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin antibodies, as indicated. The asterisk indicates the presence of a nonspecific band. Mr, molecular reference; Eng, endoglin; TRIM, TRIM21. (F) Protein–protein interactions between TRIM21 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 5.4 µM recombinant human TRIM21/6xHis at the N-terminus (R052), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Table 1. Human protein-array analysis of endoglin interactors1.
1 Microarrays containing over 9000 unique human proteins were screened using recombinant sEng as a probe. Protein interactors showing the highest scores (Z-score ≥2.0) are listed. GeneBank (https://www.ncbi.nlm.nih.gov/genbank/) and UniProtKB (https://www.uniprot.org/help/uniprotkb) accession numbers are indicated with a yellow or green background, respectively. The cellular compartment of each protein was obtained from the UniProtKB webpage. Proteins selected for further studies (TRIM21 and galectin-3) are indicated in bold type with blue background.
Note: the following are from NCBI Genbank and Genecards on TRIM21
This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008]
Expression
Ubiquitous expression in spleen (RPKM 15.5), appendix (RPKM 13.2) and 24 other tissues See more
This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008]
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression. Enhances the decapping activity of DCP2. Exists as a ribonucleoprotein particle present in all mammalian cells studied and composed of a single polypeptide and one of four small RNA molecules. At least two isoforms are present in nucleated and red blood cells, and tissue specific differences in RO/SSA proteins have been identified. The common feature of these proteins is their ability to bind HY RNAs.2. Involved in the regulation of innate immunity and the inflammatory response in response to IFNG/IFN-gamma. Organizes autophagic machinery by serving as a platform for the assembly of ULK1, Beclin 1/BECN1 and ATG8 family members and recognizes specific autophagy targets, thus coordinating target recognition with assembly of the autophagic apparatus and initiation of autophagy. Acts as an autophagy receptor for the degradation of IRF3, hence attenuating type I interferon (IFN)-dependent immune responses (PubMed:26347139, 16297862, 16316627, 16472766, 16880511, 18022694, 18361920, 18641315, 18845142, 19675099). Represses the innate antiviral response by facilitating the formation of the NMI-IFI35 complex through ‘Lys-63’-linked ubiquitination of NMI (PubMed:26342464). ( RO52_HUMAN,P19474 )
Molecular function for TRIM21 Gene according to UniProtKB/Swiss-Prot
Function:
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression.
Other Articles in this Open Access Scientific Journal on Galectins and Proteosome Include
Placenta lacks molecules required for COVID-19 infection
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
The pandemic of coronavirus disease 2019 (COVID-19) caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has affected more than 10 million people, including pregnant women. To date, no consistent evidence for the vertical transmission of SARS-CoV-2 has been found. The placenta serves as the lungs, gut, kidneys, and liver of the fetus. This fetal organ also has major endocrine actions that modulate maternal physiology and, importantly, together with the extraplacental chorioamniotic membranes shield the fetus against microbes from hematogenous dissemination and from invading the amniotic cavity.
Most pathogens that cause hematogenous infections in the mother are not able to reach the fetus, which is largely due to the potent protective mechanisms provided by placental cells (i.e. trophoblast cells: syncytiotrophoblasts and cytotrophoblasts). Yet, some of these pathogens such as Toxoplasma gondii, Rubella virus, herpesvirus (HSV), cytomegalovirus (CMV), and Zika virus (ZIKV), among others, are capable of crossing the placenta and infecting the fetus, causing congenital disease.
The placental membranes that contain the fetus and amniotic fluid lack the messenger RNA (mRNA) molecule required to manufacture the ACE2 receptor, the main cell surface receptor used by the SARS-CoV-2 virus to cause infection. These placental tissues also lack mRNA needed to make an enzyme, called TMPRSS2, that SARS-CoV-2 uses to enter a cell. Both the receptor and enzyme are present in only miniscule amounts in the placenta, suggesting a possible explanation for why SARS-CoV-2 has only rarely been found in fetuses or newborns of women infected with the virus, according to the study authors.
The single-cell transcriptomic analysis presented by the researchers provides evidence that SARS-CoV-2 is unlikely to infect the placenta and fetus since its canonical receptor and protease, ACE2 and TRMPSS2, are only minimally expressed by the human placenta throughout pregnancy. In addition, it was shown that the SARS-CoV-2 receptors are not expressed by the chorioamniotic membranes in the third trimester. However, viral receptors utilized by CMV, ZIKV, and others are highly expressed by the human placental tissues.
Transcript levels do not always correlate with protein expression, but the data of the present study indicates a low likelihood of placental infection and vertical transmission of SARS-CoV-2. However, it is still possible that the expression of these proteins is much higher in individuals with pregnancy complications related with the renin-angiotensin-aldosterone system, which can alter the expression of ACE2. The cellular receptors and mechanisms that could be exploited by SARS-CoV-2 are still under investigation.
Pancreatic cancer survival is determined by ratio of two enzymes, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
Protein kinase C (PKC) isozymes function as tumor suppressors in increasing contexts. These enzymes are crucial for a number of cellular activities, including cell survival, proliferation and migration — functions that must be carefully controlled if cells get out of control and form a tumor. In contrast to oncogenic kinases, whose function is acutely regulated by transient phosphorylation, PKC is constitutively phosphorylated following biosynthesis to yield a stable, autoinhibited enzyme that is reversibly activated by second messengers. Researchers at University of California San Diego School of Medicine found that another enzyme, called PHLPP1, acts as a “proofreader” to keep careful tabs on PKC.
The researchers discovered that in pancreatic cancer high PHLPP1 levels lead to low PKC levels, which is associated with poor patient survival. They reported that the phosphatase PHLPP1 opposes PKC phosphorylation during maturation, leading to the degradation of aberrantly active species that do not become autoinhibited. They discovered that any time an over-active PKC is inadvertently produced, the PHLPP1 “proofreader” tags it for destruction. That means the amount of PHLPP1 in patient’s cells determines his amount of PKC and it turns out those enzyme levels are especially important in pancreatic cancer.
This team of researchers reversed a 30-year paradigm when they reported evidence that PKC actually suppresses, rather than promotes, tumors. For decades before this revelation, many researchers had attempted to develop drugs that inhibit PKC as a means to treat cancer. Their study implied that anti-cancer drugs would actually need to do the opposite — boost PKC activity. This study sets the stage for clinicians to one day use a pancreatic cancer patient’s PHLPP1/PKC levels as a predictor for prognosis, and for researchers to develop new therapeutic drugs that inhibit PHLPP1 and boost PKC as a means to treat the disease.
The ratio — high PHLPP1/low PKC — correlated with poor prognoses: no pancreatic patient with low PKC in the database survived longer than five-and-a-half years. On the flip side, 50 percent of the patients with low PHLPP1/high PKC survived longer than that. While still in the earliest stages, the researchers hope that this information might one day aid pancreatic diagnostics and treatment. The researchers are next planning to screen chemical compounds to find those that inhibit PHLPP1 and restore PKC levels in low-PKC-pancreatic cancer cells in the lab. These might form the basis of a new therapeutic drug for pancreatic cancer.
Lesson 4 Cell Signaling And Motility: G Proteins, Signal Transduction: Curations and Articles of reference as supplemental information: #TUBiol3373
Curator: Stephen J. Williams, Ph.D.
Updated 7/15/2019
Below please find the link to the Powerpoint presentation for lesson #4 for #TUBiol3373. The lesson first competes the discussion on G Protein Coupled Receptors, including how cells terminate cell signals. Included are mechanisms of receptor desensitization. Please NOTE that desensitization mechanisms like B arrestin decoupling of G proteins and receptor endocytosis occur after REPEATED and HIGH exposures to agonist. Hydrolysis of GTP of the alpha subunit of G proteins, removal of agonist, and the action of phosphodiesterase on the second messenger (cAMP or cGMP) is what results in the downslope of the effect curve, the termination of the signal after agonist-receptor interaction.
Ability of gut microbiota to influence the bioavailability of levodopa in Parkinson’s disease – The presence of more bacteria producing the tyrosine decarboxylase (TDC) enzyme means less levodopa in the bloodstream
Reporter: Aviva Lev-Ari, PhD, RN
Decarboxylase enzymes can convert levodopa into dopamine. In contrast to levodopa, dopamine cannot cross the blood-brain barrier, so patients are also given a decarboxylase inhibitor. “But the levels of levodopa that will reach the brain vary strongly among Parkinson’s disease patients.
The bacterial tyrosine decarboxylase enzyme, which normally converts tyrosine into tyramine, but was found to also convert levodopa into dopamine. “We then determined that the source of this decarboxylase was Enterococcus bacteria.” The researchers also showed that the conversion of levodopa was not inhibited by a high concentration of the amino acid tyrosine, the main substrate of the bacterial tyrosine decarboxylase enzyme.
Carbidopa is over 10,000 times more potent in inhibiting the human decarboxylase,
the higher abundance of bacterial enzyme in the small intestines of rats reduced levels of levodopa in the bloodstream,
positive correlation between disease duration and levels of bacterial tyrosine decarboxylase.
Some Parkinson’s disease patients develop an overgrowth of small intestinal bacteria including Enterococci due to frequent uptake of proton pump inhibitors, which they use to treat gastrointestinal symptoms associated with the disease.
Altogether, these factors result in a vicious circle leading to an increased levodopa/decarboxylase inhibitor dosage requirement in a subset of patients.El Aidy concludes that
the presence of the bacterial tyrosine decarboxylase enzyme can explain why some patients need more frequent dosages of levodopa to treat their motor fluctuations. “This is considered to be a problem for Parkinson’s disease patients, because a higher dose will result in dyskinesia, one of the major side effects of levodopa treatment.“
Human gut microbiota senses its environment and responds by releasing metabolites, some of which are key regulators of human health and disease. In this study, we characterize gut-associated bacteria in their ability to decarboxylate levodopa to dopamine via tyrosine decarboxylases. Bacterial tyrosine decarboxylases efficiently convert levodopa to dopamine, even in the presence of tyrosine, a competitive substrate, or inhibitors of human decarboxylase. In situ levels of levodopa are compromised by high abundance of gut bacterial tyrosine decarboxylase in patients with Parkinson’s disease. Finally, the higher relative abundance of bacterial tyrosine decarboxylases at the site of levodopa absorption, proximal small intestine, had a significant impact on levels of levodopa in the plasma of rats. Our results highlight the role of microbial metabolism in drug availability, and specifically, that abundance of bacterial tyrosine decarboxylase in the proximal small intestine can explain the increased dosage regimen of levodopa treatment in Parkinson’s disease patients.
↵* Present address: Department of Biological Sciences, New York City College of Technology, City University of New York, Brooklyn, NY 11201, USA, and Arthritis and Tissue Degeneration Program and David Z. Rosensweig Genomics Research Center, Hospital for Special Surgery, New York, NY 10021, USA.
A few years ago, Jihye Yun, then a graduate student at Johns Hopkins University in Baltimore, Maryland, found that colon cancer cells whose growth is driven by mutations in the gene KRAS or a less commonly mutated gene,BRAF, make unusually large amounts of a protein that transports glucose across the cell membrane. The transporter, GLUT1, supplies the cells with the high levels of glucose they need to survive. GLUT1 also transports the oxidized form of vitamin C, dehydroascorbic acid (DHA), into the cell, bad news for cancer cells, because Yun found that DHA can deplete a cell’s supply of a chemical that sops up free radicals. Because free radicals can harm a cell in various ways, the finding suggested “a vulnerability” if the cells were flooded with DHA, says Lewis Cantley at Weill Cornell Medicine in New York City, where Yun is now a postdoc.
Cantley’s lab and collaborators found that large doses of vitamin C did indeed kill cultured colon cancer cells with BRAF or KRAS mutations by raising free radical levels, which in turn inactivate an enzyme needed to metabolize glucose, depriving the cells of energy. Then they gave daily high dose injections—equivalent to a person eating 300 oranges—to mice engineered to develop KRAS-driven colon tumors. The mice developed fewer and smaller colon tumors compared with control mice.
New Studies toward Understanding Alzheimer Disease
Curators: Larry H. Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
There is no unifying concept of Alzheimer Disease beyond the Tau and beta amyloid roles. Recently, Ingenbleek and Bernstein (journal AD) made the connection between the age related decline of liver synthesis of plasma transthyretin and the more dramatic decline of transthyretin at the blood brain barrier, and the relationship to inability to transfer vitamin A via retinol binding protein to the brain. Related metabolic events are reported by several groups.
They show that Aβ oligomerization, a behavior traditionally viewed as intrinsically pathological, may be necessary for the antimicrobial activities of the peptide. Collectively, our data are consistent with a model in which soluble Aβ oligomers first bind to microbial cell wall carbohydrates via a heparin-binding domain. Developing protofibrils inhibited pathogen adhesion to host cells. Propagating β-amyloid fibrils mediate agglutination and eventual entrapment of unatttached microbes….Salmonella Typhimurium bacterial infection of the brains of transgenic 5XFAD mice resulted in rapid seeding and accelerated β-amyloid deposition, which closely colocalized with the invading bacteria.
This is quite interesting in that infection drives the production of acute phase reactants resulting in decreased production of transthyretin. Whether this also has ties to chronic disease in the elderly and risk of AD is not known.
Through whole-genome sequencing of 1345 individuals from 410 families with late-onset AD (LOAD), they identified three highly penetrant variants in PRKCA, the gene that encodes protein kinase Cα (PKCα), in five of the families. All three variants linked with LOAD displayed increased catalytic activity relative to wild-type PKCα as assessed in live-cell imaging experiments using a genetically encoded PKC activity reporter. Deleting PRKCA in mice or adding PKC antagonists to mouse hippocampal slices infected with a virus expressing the Aβ precursor CT100 revealed that PKCα was required for the reduced synaptic activity caused by Aβ. In PRKCA(-/-) neurons expressing CT100, introduction of PKCα, but not PKCα lacking a PDZ interaction moiety, rescued synaptic depression, suggesting that a scaffolding interaction bringing PKCα to the synapse is required for its mediation of the effects of Aβ. Thus, enhanced PKCα activity may contribute to AD, possibly by mediating the actions of Aβ on synapses.
Kim BM, You MH, Chen CH, Suh J, Tanzi RE, Ho Lee T.
Hum Mol Genet. 2016 Apr 19. pii: ddw114.
Extracellular deposition of amyloid-beta (Aβ) peptide, a metabolite of sequential cleavage of amyloid precursor protein (APP), is a critical step in the pathogenesis of Alzheimer’s disease (AD). While death-associated protein kinase 1 (DAPK1) is highly expressed in AD brains and its genetic variants are linked to AD risk, little is known about the impact of DAPK1 on APP metabolism and Aβ generation. This study demonstrated a novel effect of DAPK1 in the regulation of APP processing using cell culture and mouse models. DAPK1, but not its kinase deficient mutant (K42A), significantly increased human Aβ secretion in neuronal cell culture models. Moreover, knockdown of DAPK1 expression or inhibition of DAPK1 catalytic activity significantly decreased Aβ secretion. Furthermore, DAPK1, but not K42A, triggered Thr668 phosphorylation of APP, which may initiate and facilitate amyloidogenic APP processing leading to the generation of Aβ. In Tg2576 APPswe-overexpressing mice, knockout of DAPK1 shifted APP processing toward non-amyloidogenic pathway and decreased Aβ generation. Finally, in AD brains, elevated DAPK1 levels showed co-relation with the increase of APP phosphorylation. Combined together, these results suggest that DAPK1 promotes the phosphorylation and amyloidogenic processing of APP, and that may serve a potential therapeutic target for AD.
The “amyloid β hypothesis” of Alzheimer’s disease (AD) has been the reigning hypothesis explaining pathogenic mechanisms of AD over the last two decades. However, this hypothesis has not been fully validated in animal models, and several major unresolved issues remain. Our 3D human neural cell culture model system provides a premise for a new generation of cellular AD models that can serve as a novel platform for studying pathogenic mechanisms and for high-throughput drug screening in a human brain-like environment.
The two key pathological hallmarks of AD are senile plaques (amyloid plaques) and neurofibrillary tangles (NFTs), which develop in brain regions responsible for memory and cognitive functions (i.e. cerebral cortex and limbic system) 3. Senile plaques are extracellular deposits of amyloid-β (Aβ) peptides, while NFTs are intracellular, filamentous aggregates of hyperphosphorylated tau protein 4.
The identification of Aβ as the main component of senile plaques by Drs. Glenner and Wong in 1984 5 resulted in the original formation of the “amyloid hypothesis.” According to this hypothesis, which was later renamed the “amyloid-β cascade hypothesis” by Drs. Hardy and Higgins 6, the accumulation of Aβ is the initial pathological trigger in the disease, subsequently leading to hyperphosphorylation of tau, causing NFTs, and ultimately, neuronal death and dementia 4,7–10. Although the details have been modified to reflect new findings, the core elements of this hypothesis remain unchanged: excess accumulation of the pathogenic forms of Aβ, by altered Aβ production and/or clearance, triggers the vicious pathogenic cascades that eventually lead to NFTs and neuronal death.
Over the last two decades, the Aβ hypothesis of AD has reigned, providing the foundation for numerous basic studies and clinical trials 4,7,10,11. According to this hypothesis, the accumulation of Aβ, either by altered Aβ production and/or clearance, is the initial pathological trigger in the disease. The excess accumulation of Aβ then elicits a pathogenic cascade including synaptic deficits, altered neuronal activity, inflammation, oxidative stress, neuronal injury, hyperphosphorylation of tau causing NFTs and ultimately, neuronal death and dementia 4,7–10.
One of the major unresolved issues of the Aβ hypothesis is to show a direct causal link between Aβ and NFTs 12–14. Studies have demonstrated that treatments with various forms of soluble Aβ oligomers induced synaptic deficits and neuronal injury, as well as hyperphosphorylation of tau proteins, in mouse and rat neurons, which could lead to NFTs and neurodegeneration in vivo18–21. However, transgenic AD mouse models carrying single or multiple human familial AD (FAD) mutations in amyloid precursor protein (APP) and/or presenilin 1 (PS1) do not develop NFTs or robust neurodegeneration as observed in human patients, despite robust Aβ deposition 13,22,23. Double and triple transgenic mouse models, harboring both FAD and tau mutations linked with frontotemporal dementia (FTD), are the only rodent models to date displaying both amyloid plaques and NFTs. However, the NFT pathology in these models stems mainly from the overexpression of human tau as a result of the FTD, rather than the FAD mutations24,25.
Human neurons carrying FAD mutations are an optimal model to test whether elevated levels of pathogenic Aβ trigger pathogenic cascades including NFTs, since those cells truly share the same genetic background that induces FAD in humans. Indeed, Israel et al., observed elevated tau phosphorylation in neurons with an APP duplication FAD mutation 33. Blocking Aβ generation by β-secretase inhibitors significantly decreased tau phosphorylation in the same model, but γ-secretase inhibitor, another Aβ blocker, did not affect tau phosphorylation 33. Neurons with the APP V717I FAD mutation also showed an increase in levels of phospho tau and total tau levels 28. More importantly, Muratore and colleagues showed that treatments with Aβ-neutralizing antibodies in those cells significantly reduced the elevated total and phospho tau levels at the early stages of differentiation, suggesting that blocking pathogenic Aβ can reverse the abnormal tau accumulation in APP V717I neurons 28.
Recently, Moore et al. also reported that neurons harboring the APP V717I or the APP duplication FAD mutation showed increases in both total and phospho tau levels 27. Interestingly, altered tau levels were not detected in human neurons carrying PS1 FAD mutations, which significantly increased pathogenic Aβ42 species in the same cells 27. These data suggest that elevated tau levels in these models were not due to extracellular Aβ accumulation but may possibly represent a very early stage of tauopathy. It may also be due to developmental alterations induced by the APP FAD mutations.
As summarized, most human FAD neurons showed significant increases in pathogenic Aβ species, while only APP FAD neurons showed altered tau metabolism that may represent very early stages of tauopathy. However, all of these human FAD neurons failed to recapitulate robust extracellular amyloid plaques, NFTs, or any signs of neuronal death, as predicted in the amyloid hypothesis.
In our recent study, we moved one step closer to proving the amyloid hypothesis. By generating human neural stem cell lines carrying multiple mutations in APP together with PS1, we achieved high levels of pathogenic Aβ42 comparable to those in brains of AD patients 44–46.
Platform for AD drug screening in human neural progenitor cells with FAD mutations in a 3D culture system, which successfully reproduce human AD pathogenesis (amyloid plaques-driven tauopathy).
In addition to the impact on toxic Aβ species, our 3D culture model can test if these antibodies can block tau pathologies in 3D human neural cell culture systems 44–46. Human cellular AD models can also be used to determine optimal doses of candidate AD drugs to block Aβ and/or tau pathology without affecting neuronal survival (Fig. 1).
While much progress has been made, many challenges still lie on the path to creating human neural cell culture models that comprehensively recapitulate pathogenic cascades of AD. A major difficulty lies in reconstituting the brain regions most affected in AD: the hippocampus and specific cortical layers. Recent progress in 3D culture technology, such as “cerebral organoids,” may also be helpful in rebuilding the brain structures that are affected by AD in a dish 52,53. These “cerebral organoids” were able to model various discrete brain regions including human cortical areas 52, which enabled them to reproduce microcephaly, a brain developmental disorder. Similarly, pathogenic cascades of AD may be recapitulated in cortex-like structures using this model. Adding neuroinflammatory components, such as microglial cells, which are critical in AD pathogenesis, will illuminate the validity of the amyloid β hypothesis. Reconstitution of robust neuronal death stemming from Aβ and tau pathologies will be the next major step in comprehensively recapitulating AD in a cellular model.
Natunen T, Takalo M, Kemppainen S, Leskelä S, Marttinen M, Kurkinen KM, Pursiheimo JP, Sarajärvi T, Viswanathan J, Gabbouj S, Solje E, Tahvanainen E, Pirttimäki T, Kurki M, Paananen J, Rauramaa T, Miettinen P, Mäkinen P, Leinonen V, Soininen H, Airenne K, Tanzi RE, Tanila H, Haapasalo A, Hiltunen M.
Accumulation of β-amyloid (Aβ) and phosphorylated tau in the brain are central events underlying Alzheimer’s disease (AD) pathogenesis. Aβ is generated from amyloid precursor protein (APP) by β-site APP-cleaving enzyme 1 (BACE1) and γ-secretase-mediated cleavages. Ubiquilin-1, a ubiquitin-like protein, genetically associates with AD and affects APP trafficking, processing and degradation. Here, we have investigated ubiquilin-1 expression in human brain in relation to AD-related neurofibrillary pathology and the effects of ubiquilin-1 overexpression on BACE1, tau, neuroinflammation, and neuronal viability in vitro in co-cultures of mouse embryonic primary cortical neurons and microglial cells under acute neuroinflammation as well as neuronal cell lines, and in vivo in the brain of APdE9 transgenic mice at the early phase of the development of Aβ pathology. Ubiquilin-1 expression was decreased in human temporal cortex in relation to the early stages of AD-related neurofibrillary pathology (Braak stages 0-II vs. III-IV). There was a trend towards a positive correlation between ubiquilin-1 and BACE1 protein levels. Consistent with this, ubiquilin-1 overexpression in the neuron-microglia co-cultures with or without the induction of neuroinflammation resulted in a significant increase in endogenously expressed BACE1 levels. Sustained ubiquilin-1 overexpression in the brain of APdE9 mice resulted in a moderate, but insignificant increase in endogenous BACE1 levels and activity, coinciding with increased levels of soluble Aβ40 and Aβ42. BACE1 levels were also significantly increased in neuronal cells co-overexpressing ubiquilin-1 and BACE1. Ubiquilin-1 overexpression led to the stabilization of BACE1 protein levels, potentially through a mechanism involving decreased degradation in the lysosomal compartment. Ubiquilin-1 overexpression did not significantly affect the neuroinflammation response, but decreased neuronal viability in the neuron-microglia co-cultures under neuroinflammation. Taken together, these results suggest that ubiquilin-1 may mechanistically participate in AD molecular pathogenesis by affecting BACE1 and thereby APP processing and Aβ accumulation.
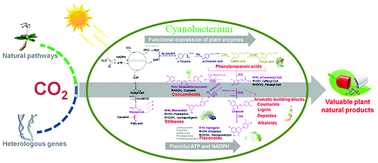
Shared photosynthetic components between plant chloroplasts and cyanobacteria make these microbes ideal hosts for expressing foreign plant enzymes. Ping Xu and colleagues at the Shanghai Jiao Tong University have genetically engineered the cyanobacterium Synechococcus elongatusPC7942 with plant-derived enzymes. In total, the team created 18 bacterial strains expressing different combinations of enzymes. The different strains generate a variety of compounds with a six-carbon, phenyl group and three-carbon propene tail, called phenylpropanoids.
Phenylpropanoids perform diverse functions in plants, ranging from ultraviolet light protection to pathogen defence. One such compound, resveratrol, is made when the bacteria express the plant enzyme stilbene synthase downstream of enzymes tyrosine ammonia lyase and 4-coumarate:coenzyme A-ligase. Found in the skin of grapes and other berries, resveratrol reduces the risk of heart disease and is a valuable pharmaceutical commodity. Different versions of the engineered bacteria can also churn out the phenylpropanoid antioxidants caffeic acid, naringenin and coumaric acid.
The Shanghai Jia Tong University team genetically engineered cyanobacteria to produce compounds like flavonoids, stilbenes and curcuminoids usually only found in plants
What’s more, the team added feedback-inhibition resistant enzymes to the bacteria so that the chemical yields would surpass physiological levels. Photosynthesis within the cyanobacteria generates the chemicals from just water, carbon dioxide and a few mineral nutrients.
The bacterial growth medium houses the products, but isolating them at an industrially relevant yield is currently the biggest challenge. However, by not needing to harvest crops, generating the compounds from bacteria is potentially more sustainable. Xu stresses the potential of this point: ‘For the production of 1 tonne of natural resveratrol, our method may save about 485 hectare of farmland at its current production level.’
‘The approach deftly sidesteps major economic challenges by targeting chemicals with high intrinsic value,’ comments Paul Fowler, executive director of the Wisconsin Institute for Sustainable Technology in the US. A world-scale production plant under these circumstances is not a pre-requisite for commercialising this research.’
Many plant natural products have remarkable pharmacological activities. They are mainly produced directly by extraction from higher plants, which can hardly keep up with the surging global demand. Furthermore, the over-felling of many medicinal plants has undesirable effects on the ecological balance. In this study, we constructed a photoautotrophic platform with the unicellular cyanobacterium Synechococcus elongatus PCC7942 to directly convert the greenhouse gas CO2 into an array of valuable healthcare products, including resveratrol, naringenin, bisdemethoxycurcumin, p-coumaric acid, caffeic acid, and ferulic acid. These six compounds can be further branched to many other precious and useful natural products. Various strategies including introducing a feedback-inhibition-resistant enzyme, creating functional fusion proteins, and increasing malonyl-CoA supply have been systematically investigated to increase the production. The highest titers of these natural products reached 4.1–128.2 mg L−1 from the photoautotrophic system, which are highly comparable with those obtained by many other heterotrophic microorganisms using carbohydrates. Several advantages such as independence from carbohydrate feedstocks, functionally assembling P450s, and availability of plentiful NADPH and ATP support that this photosynthetic platform is uniquely suited for producing plant natural products. This platform also provides a green route for direct conversion of CO2 to many aromatic building blocks, a promising alternative to petrochemical-based production of bulk aromatic compounds.













{kind=link}