Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Jennifer A. Lewis: Revolutionizing Materials Science with the 2025 James Prize
Curator: Dr. Sudipta Saha, Ph.D.
Jennifer A. Lewis, the Hansjörg Wyss Professor of Biologically Inspired Engineering at Harvard University, has been awarded the prestigious 2025 James Prize in Science and Technology Integration by the National Academy of Sciences. This recognition highlights her ground breaking research in the programmable assembly of soft functional, structural, and biological materials.
Lewis has pioneered work in integrating various scientific fields, including materials science, soft matter physics, additive manufacturing, bioengineering, and stem cell biology. Her lab focuses on developing advanced materials, such as electrically and ionically conductive inks for micro-scale printed devices like electronics and batteries. Additionally, Lewis’s work on stem cell-derived organoids has enabled the creation of 3D organ-on-chip models and vascularized tissues, which hold promise for drug screening, disease modeling, and therapeutic applications.
The James Prize, awarded by the National Academy of Sciences, recognizes outstanding contributions made by individuals who integrate knowledge across multiple disciplines to address pressing challenges. Lewis’s innovative approach, exemplified in her multidisciplinary work, has transformed the way soft materials and biological systems are designed and utilized. The prize includes a $50,000 award, underscoring her exceptional impact on science and technology.
With numerous accolades to her name, including the NSF Presidential Faculty Fellow Award and election to the National Academy of Sciences and the National Academy of Engineering, Lewis’s work continues to reshape the future of biologically inspired engineering.
Eight Subcellular Pathologies driving Chronic Metabolic Diseases – Methods for Mapping Bioelectronic Adjustable Measurements as potential new Therapeutics: Impact on Pharmaceuticals in Use
In this curation we wish to present two breaking through goals:
Goal 1:
Exposition of a new direction of research leading to a more comprehensive understanding of Metabolic Dysfunctional Diseases that are implicated in effecting the emergence of the two leading causes of human mortality in the World in 2023: (a) Cardiovascular Diseases, and (b) Cancer
Goal 2:
Development of Methods for Mapping Bioelectronic Adjustable Measurements as potential new Therapeutics for these eight subcellular causes of chronic metabolic diseases. It is anticipated that it will have a potential impact on the future of Pharmaceuticals to be used, a change from the present time current treatment protocols for Metabolic Dysfunctional Diseases.
According to Dr. Robert Lustig, M.D, an American pediatric endocrinologist. He is Professor emeritus of Pediatrics in the Division of Endocrinology at the University of California, San Francisco, where he specialized in neuroendocrinology and childhood obesity, there are eight subcellular pathologies that drive chronic metabolic diseases.
These eight subcellular pathologies can’t be measured at present time.
In this curation we will attempt to explore methods of measurement for each of these eight pathologies by harnessing the promise of the emerging field known as Bioelectronics.
Unmeasurable eight subcellular pathologies that drive chronic metabolic diseases
Glycation
Oxidative Stress
Mitochondrial dysfunction [beta-oxidation Ac CoA malonyl fatty acid]
Insulin resistance/sensitive [more important than BMI], known as a driver to cancer development
Membrane instability
Inflammation in the gut [mucin layer and tight junctions]
Epigenetics/Methylation
Autophagy [AMPKbeta1 improvement in health span]
Diseases that are not Diseases: no drugs for them, only diet modification will help
Image source
Robert Lustig, M.D. on the Subcellular Processes That Belie Chronic Disease
These eight Subcellular Pathologies driving Chronic Metabolic Diseases are becoming our focus for exploration of the promise of Bioelectronics for two pursuits:
Will Bioelectronics be deemed helpful in measurement of each of the eight pathological processes that underlie and that drive the chronic metabolic syndrome(s) and disease(s)?
IF we will be able to suggest new measurements to currently unmeasurable health harming processes THEN we will attempt to conceptualize new therapeutic targets and new modalities for therapeutics delivery – WE ARE HOPEFUL
In the Bioelecronics domain we are inspired by the work of the following three research sources:
Michael Levin is an American developmental and synthetic biologist at Tufts University, where he is the Vannevar Bush Distinguished Professor. Levin is a director of the Allen Discovery Center at Tufts University and Tufts Center for Regenerative and Developmental Biology. Wikipedia
THE VOICE of Dr. Justin D. Pearlman, MD, PhD, FACC
PENDING
THE VOICE of Stephen J. Williams, PhD
Ten TakeAway Points of Dr. Lustig’s talk on role of diet on the incidence of Type II Diabetes
25% of US children have fatty liver
Type II diabetes can be manifested from fatty live with 151 million people worldwide affected moving up to 568 million in 7 years
A common myth is diabetes due to overweight condition driving the metabolic disease
There is a trend of ‘lean’ diabetes or diabetes in lean people, therefore body mass index not a reliable biomarker for risk for diabetes
Thirty percent of ‘obese’ people just have high subcutaneous fat. the visceral fat is more problematic
there are people who are ‘fat’ but insulin sensitive while have growth hormone receptor defects. Points to other issues related to metabolic state other than insulin and potentially the insulin like growth factors
At any BMI some patients are insulin sensitive while some resistant
Visceral fat accumulation may be more due to chronic stress condition
Fructose can decrease liver mitochondrial function
A methionine and choline deficient diet can lead to rapid NASH development
Rare earth-doped nanoparticles applications in biological imaging and tumor treatment
Reporter: Irina Robu, PhD
Bioimaging aims to interfere as little as possible with life processes and can be used to gain information on the 3-D structure of the observed specimen from the outside. Bioimaging ranges from the observation of subcellular structures and the entire cells over tissues up to entire multicellular organisms. The technology uses light, fluorescence, ultrasound, X-ray, magnetic resonance as sources of imaging. The more common imaging is fluorescence imaging which is used to monitor the dynamic interaction between the drug molecules and tumor cells and the ability to monitor the real time dynamic process in biological tissues.
Researchers from the Xi’an Institute of Optics and Precision Mechanics (XIOPM) of the Chinese Academy of Sciences (CAS) described the recent progress they made in the rare earth-doped nanoparticles in the field of bio-engineering and tumor treatment. It is well known that producing small nanoparticles with good dispersion and exploitable optical coherence properties is highly challenging. According to them, these rare earth-doped nanoparticles can be vested with additional capabilities such as water solubility, biocompatibility, drug-loading ability and the target ability for different tumors by surface functionalization. The luminescent properties and structure design were also looked at.
According to the Chinese researchers, for applying the RE-doped NPs to the diagnosis and treatment of tumors, their first goal is to improve water solubility and biocompatibility. The second goal would be to give the nanoparticles the ability to target tumors by surface functionalization. Lastly, biocompatible water-soluble tumor-targeting NPs can be used as carriers to load drugs for treatment of tumor cells. All things considered, the recent research progress on the development of fluorescence intensity of NPs, surface modification, and tumor targeted diagnosis and treatment has also been emphasized.
The cardiovascular team at SSM Health Cardinal Glennon Children’s Hospital found a solution for better surgical planning using 3D printing. As a pediatric center, Glennon Children’s Hospital deals with the most complex patients, which requires surgeries within days or weeks of birth. According to the center, one of the pediatric patients was an infant diagnosed in utero via fetal ultrasound with an unusual form of switch of great arteries. Deoxygenated blue blood entered the right atrium which connected to the left ventricle, then to the aorta and the oxygenated red blood entered the left atrium which connects to the right ventricle and then to the pulmonary artery. The pediatric patients had a very large ventricular septal defect connecting both ventricles and severe narrowing between the left ventricle and the aorta.
It is obvious that the patient was fairly blue as deoxygenated blood was directed toward the aorta. The balloon atrial septostomy made in the first few days of life. Yet, the tachycardia persisted. The surgical team from SSM Health Cardinal Glennon Children’s Hospital, led by Charles Huddleston, MD used 3D printing to identify the anatomy of the patient clearly and provided them with the ability to repair the mitral valve. It seems that the neonatal atrial switch appeared to be the best plan, even if the operation proved challenging.
The team knew that they could go into the procedure knowing that the tissue can be safely removed without damage to the mitral valve. The team was able to show that the 3D model was essential in determining the optimal surgical approach and with the help of the 3D printed heart model, the neonatal atrial switch, the VSD closure and the subaortic stenosis resection was performed effectively on a 20-day infant. The surgery allowed the mitral valve function to remain intact. The pediatric patient cardiac function improved gradually and is expected to have an excellent recovery.
3D printing is a fabrication technique used to transform digital objects into physical models, which builds structures of arbitrary geometry by depositing material in successive layers on the basis of specific digital design. Even though, the use of 3D bioprinting in cardiovascular medicine is relatively new development, advancement within this discipline is occurring at such a rapid rate. Most cardiologists believed the costs would be too high for routine use such that the price tag was better for academic applications.
Now as the prices are starting to lower, the idea of using 3D printed models of organs vessels and tissue manufactured based on CT, MRI and echocardiography might be beneficial according to Dr. Fadi Matar, professor at University of South Florida. He and his cardiology colleagues use 3D printed models to allow them to view patient’s complex anatomies before deciding what treatments to pursue. The models allow them to calculate the size and exact placement of devices which has led to shorter procedure time and better outcome.
In a study published in Academic Radiology, David Ballard, professor at University School of Medicine appraised the costs of setting up a 3D printing lab including the commercial printer plus software, lab space, materials and staffing. According to Ballard’s team, the commercial printers start at $12,000 but can be as high as high as $500,000.
According to American Medical Association-approved Category III Current Procedural Terminology (CPT) codes allows cardiology relief from setting up a new 3D printing lab such as Codes 0559T and 0560T, for individually prepared 3D-printed anatomical models with one or more components (including arteries and veins) and Codes 0561T and 0562T, which are for the production of personalized 3D-printed cutting or drilling tools that use patient imaging data and often are used to guide or facilitate surgery.
These codes have been met with enthusiasm by teams eyeing 3D printing, but there are noteworthy limitations to Category III codes—which are temporary codes describing emerging technologies, services and procedures that are used for tracking effectiveness data. It is important to note that Category III codes are not reimbursed but often are a step toward reimbursement.
New and improved materials also might lead to a sharper focus on 3D printing in cardiology. Dr. Fadi Matar says companies are working on materials that better mimic elements of the heart. Such “mimicry” ought to enhance the value of 3D-printed models since they will give cardiologists more realistic insights into how specific devices will interact with an individual patient’s heart. Even with the complex modalities of using 3D bioprinting, in time there would be less obstacles to being able to set up a 3D bioprinter lab.
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
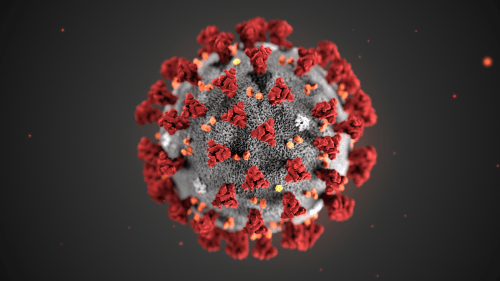
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
Image of newly mapped coronavirus protein, called Nsp15, which helps the virus replicate.
How UC is responding to the coronavirus (COVID-19)
The University of California is vigilantly monitoring and responding to new information about the coronavirus (COVID-19) outbreak, which has been declared a global health emergency.
The 3-D structure of a potential drug target in a newly mapped protein of COVID-19, or coronavirus, has been solved by a team of researchers from the University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University.
The scientists said their findings suggest drugs previously developed to treat the earlier SARS outbreak could now be developed as effective drugs against COVID-19.
The initial genome analysis and design of constructs for protein synthesis were performed by the bioinformatic group of Adam Godzik, a professor of biomedical sciences at the UC Riverside School of Medicine.
The protein Nsp15 from Severe Acute Respiratory Syndrome Coronavirus 2, or SARS-CoV-2, is 89% identical to the protein from the earlier outbreak of SARS-CoV. SARS-CoV-2 is responsible for the current outbreak of COVID-19. Studies published in 2010 on SARS-CoV revealed inhibition of Nsp15 can slow viral replication.This suggests drugs designed to target Nsp15 could be developed as effective drugs against COVID-19.
Adam Godzik, UC Riverside professor of biomedical sciences Credit: Sanford Burnham Prebys Medical Discovery Institute
“While the SARS-CoV-19 virus is very similar to the SARS virus that caused epidemics in 2003, new structures shed light on the small, but potentially important differences between the two viruses that contribute to the different patterns in the spread and severity of the diseases they cause,” Godzik said.
The structure of Nsp15, which will be released to the scientific community on March 4, was solved by the group of Andrzej Joachimiak, a distinguished fellow at the Argonne National Laboratory, University of Chicago Professor, and Director of the Structural Biology Center at Argonne’s Advanced Photon Source, a Department of Energy Office of Science user facility.
“Nsp15 is conserved among coronaviruses and is essential in their lifecycle and virulence,” Joachimiak said. “Initially, Nsp15 was thought to directly participate in viral replication, but more recently, it was proposed to help the virus replicate possibly by interfering with the host’s immune response.”
Mapping a 3D protein structure of the virus, also called solving the structure, allows scientists to figure out how to interfere in the pathogen’s replication in human cells.
“The Nsp15 protein has been investigated in SARS as a novel target for new drug development, but that never went very far because the SARS epidemic went away, and all new drug development ended,” said Karla Satchell, a professor of microbiology-immunology at Northwestern, who leads the international team of scientists investigating the structure of the SARS CoV-2 virus to understand how to stop it from replicating. “Some inhibitors were identified but never developed into drugs. The inhibitors that were developed for SARS now could be tested against this protein.”
Rapid upsurge and proliferation of SARS-CoV-2 raised questions about how this virus could become so much more transmissible as compared to the SARS and MERS coronaviruses. The scientists are mapping the proteins to address this issue.
Over the past two months, COVID-19 infected more than 80,000 people and caused at least 2,700 deaths. Although currently mainly concentrated in China, the virus is spreading worldwide and has been found in 46 countries. Millions of people are being quarantined, and the epidemic has impacted the world economy. There is no existing drug for this disease, but various treatment options, such as utilizing medicines effective in other viral ailments, are being attempted.
Godzik, Satchell, and Joachimiak — along with the entire center team — will map the structure of some of the 28 proteins in the virus in order to see where drugs can throw a chemical monkey wrench into its machinery. The proteins are folded globular structures with precisely defined functions and their “active sites” can be targeted with chemical compounds.
The first step is to clone and express the genes of the virus proteins and grow them as protein crystals in miniature ice cube-like trays. The consortium includes nine labs across eight institutions that will participate in this effort.
Above is a modified version of the Northwestern University news release written by Marla Paul.
3D printing is a technique that has gained immense popularity for its ability to create 3D structures in art, jewelry, engineering, medicine. In this case, radiologists use 3D printing to transform a 2D scan into 3D visualization of a patient’s anatomy. Radiologists use their unique skills to visualize the anatomy of the organs of interest which give them a large advantage in communicating with patients as well as surgical teams.
The 3D printed anatomical models have proved valuable in providing a better understanding of complex anatomies and being used as a tangible aid for pre-surgical planning. It gives the patient a clear understanding of what is happening and it provides a great value when it comes to patient specific care. However, 3D modelling is essential at the beginning but it can also be a useful tool for surgeons. The list of medical 3D printing benefits is infinite. Just recently, a scientific team at University of Minnesota constructed their own patient specific 3D organ model based on MRI scans and prostate tissue samples of patients. The organs allow surgeons to plan and rehearse surgery.
In addition to researchers at University of Minnesota, Siemens Health engineers also created a platform to make medical apps that can be accessible throughout hospitals. In addition, Siemens Health partnered up with Materialise to make 3D printing software an integral part of the radiology workflow.
Hence, using 3D bioprinting is a desirable path to follow for radiologist. Not only they get to interpret anatomy, but now they can use 3D bioprinting as a state of the art tool that empower them to provide immense value to an audience that stretches from patient to practitioner.
The impact of Machine Learning (ML) and Artificial Intelligence (AI) during the last decade has been tremendous. With the rise of infobesity, ML/AI is evolving to an essential capability to help mine the sheer volume of patient genomics, omics, sensor/wearables and real-world data, and unravel the knot of healthcare’s most complex questions.
Despite the advancements in technology, organizations struggle to prioritize and implement ML/AI to achieve the anticipated value, whilst managing the disruption that comes with it. In this session, panelists will discuss ML/AI implementation and adoption strategies that work. Panelists will draw upon their experiences as they share their success stories, discuss how to implement digital diagnostics, track disease progression and treatment, and increase commercial value and ROI compared against traditional approaches.
most of trials which are done are still in training AI/ML algorithms with training data sets. The best results however have been about 80% accuracy in training sets. Needs to improve
All data sets can be biased. For example a professor was looking at heartrate using a IR detector on a wearable but it wound up that different types of skin would generate a different signal to the detector so training sets maybe population biases (you are getting data from one group)
clinical grade equipment actually haven’t been trained on a large set like commercial versions of wearables, Commercial grade is tested on a larger study population. This can affect the AI/ML algorithms.
Regulations: The regulatory bodies responsible is up to debate. Whether FDA or FTC is responsible for AI/ML in healtcare and healthcare tech and IT is not fully decided yet. We don’t have the guidances for these new technologies
some rules: never use your own encryption always use industry standards especially when getting personal data from wearables. One hospital corrupted their system because their computer system was not up to date and could not protect against a virus transmitted by a wearable.
pharma companies understand they need to increase value of their products so very interested in how AI/ML can be used.
Please follow LIVE on TWITTER using the following @ handles and # hashtags:
Diabetes is a life-long condition where your body does not produce enough insulin (Type 1) or your body cannot use the insulin it has effectively. Since there is no cure for diabetes, the artificial pancreas system comes as a relief for patients that are suffering with this disease.
The artificial pancreas, MiniMed 670G hybrid closed loop system designed by Medtronic is the first FDA-approved device that measures glucose levels and delivers the appropriate dose of basal insulin. The system comprises Medtronic’s MiniMed 670G insulin pump that is strapped to the body, an infusion patch that delivers insulin via catheter from the pump and a sensor which measures glucose levels under the skin and can be worn for 7 days at a time. While the device regulates basal, or background, insulin, patients must still manually request bolus insulin at mealtimes.
The device is intended for people age 14 or older with Type 1 diabetes and is intended to regulate insulin levels with “little to no input” from the patient. The artificial pancreas measures blood sugar levels using a constant glucose monitor (CGM) and communicates the information to an insulin pump which calculates and releases the required amount of insulin into the body, just as the pancreas does in people without diabetes.
The 2016 FDA approval was done in just three months which is a record for any medical device. The agency evaluated data from a clinical trial in which 123 patients with Type 1 diabetes used the system’s hybrid closed-loop feature as repeatedly during a three-month period. The trial presented the device to be safe for use in those 14 and older, showing no serious adverse events. The system is on sale since spring 2017.
While further clinical research is needed to ensure that the strength of the device in different settings is consistent, several researchers support the view that “artificial pancreas systems are a safe and effective treatment approach for people with type 1 diabetes. Medtronic counts this device as a step toward a fully automated, closed-loop system.
Extracellular RNA and their carriers in disease diagnosis and therapy, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
RNA plays various roles in determining how the information in our genes drives cell behavior. One of its roles is to carry information encoded by our genes from the cell nucleus to the rest of the cell where it can be acted on by other cell components. Rresearchers have now defined how RNA also participates in transmitting information outside cells, known as extracellular RNA or exRNA. This new role of RNA in cell-to-cell communication has led to new discoveries of potential disease biomarkers and therapeutic targets. Cells using RNA to talk to each other is a significant shift in the general thought process about RNA biology.
Researchers explored basic exRNA biology, including how exRNA molecules and their transport packages (or carriers) were made, how they were expelled by producer cells and taken up by target cells, and what the exRNA molecules did when they got to their destination. They encountered surprising complexity both in the types of carriers that transport exRNA molecules between cells and in the different types of exRNA molecules associated with the carriers. The researchers had to be exceptionally creative in developing molecular and data-centric tools to begin making sense of the complexity, and found that the type of carrier affected how exRNA messages were sent and received.
As couriers of information between cells, exRNA molecules and their carriers give researchers an opportunity to intercept exRNA messages to see if they are associated with disease. If scientists could change or engineer designer exRNA messages, it may be a new way to treat disease. The researchers identified potential exRNA biomarkers for nearly 30 diseases including cardiovascular disease, diseases of the brain and central nervous system, pregnancy complications, glaucoma, diabetes, autoimmune diseases and multiple types of cancer.
As for example some researchers found that exRNA in urine showed promise as a biomarker of muscular dystrophy where current studies rely on markers obtained through painful muscle biopsies. Some other researchers laid the groundwork for exRNA as therapeutics with preliminary studies demonstrating how researchers might load exRNA molecules into suitable carriers and target carriers to intended recipient cells, and determining whether engineered carriers could have adverse side effects. Scientists engineered carriers with designer RNA messages to target lab-grown breast cancer cells displaying a certain protein on their surface. In an animal model of breast cancer with the cell surface protein, the researchers showed a reduction in tumor growth after engineered carriers deposited their RNA cargo.
Other than the above research work the scientists also created a catalog of exRNA molecules found in human biofluids like plasma, saliva and urine. They analyzed over 50,000 samples from over 2000 donors, generating exRNA profiles for 13 biofluids. This included over 1000 exRNA profiles from healthy volunteers. The researchers found that exRNA profiles varied greatly among healthy individuals depending on characteristics like age and environmental factors like exercise. This means that exRNA profiles can give important and detailed information about health and disease, but careful comparisons need to be made with exRNA data generated from people with similar characteristics.
Next the researchers will develop tools to efficiently and reproducibly isolate, identify and analyze different carrier types and their exRNA cargos and allow analysis of one carrier and its cargo at a time. These tools will be shared with the research community to fill gaps in knowledge generated till now and to continue to move this field forward.