Wnt/β-catenin Signaling
Writer and Curator: Larry H. Bernstein, MD, FCAP
7.10 Wnt/β-catenin signaling
7.10.1 Wnt signaling and hepatocarcinogenesis. The hepatoblastoma model
7.10.2 The Wnt.β-catenin pathway in ovarian cancer : a review.
7.10.3 Wnt Signaling in the Niche Enforces Hematopoietic Stem Cell Quiescence and Is Necessary to Preserve Self-Renewal In Vivo
7.10.4 Wnt.β-Catenin Signaling in Development and Disease
7.10.5 Wnt.β-Catenin Signaling. Components, Mechanisms, and Diseases
7.10.6 Wnt.β-Catenin Signaling. Turning the Switch
7.10.7 Wnt–β-catenin signaling
7.10.8 Extracellular modulators of Wnt signaling
7.10.9 FOXO3a modulates WNT.β-catenin signaling and suppresses epithelial-to-mesenchymal transition in prostate cancer cells
7.10.1 Wnt signalinbg pathway in liver cancer
7.10.1.1 Wnt signaling and hepatocarcinogenesis. The hepatoblastoma model
Armengol C1, Cairo S, Fabre M, Buendia MA.
Int J Biochem Cell Biol. 2011 Feb; 43(2):265-70.
http://dx.doi.org:/10.1016/j.biocel.2009.07.012
The Wnt/β-catenin pathway plays a key role in liver development, regeneration and tumorigenesis. Among human cancers tightly linked to abnormal Wnt/β-catenin signaling, hepatoblastoma (HB) presents with the highest rate (50-90%) of β-catenin mutations. HB is the most common malignant tumor of the liver in childhood. This embryonic tumor differs from hepatocellular carcinoma by the absence of viral etiology and underlying liver disease, and by distinctive morphological patterns evoking hepatoblasts, the bipotent precursors of hepatocytes and cholangiocytes. Recent studies of the molecular pathogenesis of hepatoblastoma have led to identify two major tumor subclasses resembling early and late phases of prenatal liver development and presenting distinctive chromosomal alterations. It has been shown that the molecular signature of Wnt/β-catenin signaling in hepatoblastoma is mainly imposed by liver context, but differs according to developmental stage. Finally, the differentiation stage of tumor cells strongly influences their invasive and metastatic properties, therefore affecting clinical behavior.
7.10.1.2 Targeting the Wnt/β-Catenin Signaling Pathway in Liver Cancer Stem Cells and Hepatocellular Carcinoma Cell Lines with FH535
Roberto Gedaly ,Roberto Galuppo, Michael F. Daily, Malay Shah, Erin Maynard, et al.
PLoS ONE 2014; 9(6): e99272. http://dx.doi.org:/10.1371/journal.pone.0099272
Activation of the Wnt/β-catenin pathway has been observed in at least 1/3 of hepatocellular carcinomas (HCC), and a significant number of these have mutations in the β-catenin gene. Therefore, effective inhibition of this pathway could provide a novel method to treat HCC. The purposed of this study was to determine whether FH535, which was previously shown to block the β-catenin pathway, could inhibit β-catenin activation of target genes and inhibit proliferation of Liver Cancer Stem Cells (LCSC) and HCC cell lines. Using β-catenin responsive reporter genes, our data indicates that FH535 can inhibit target gene activation by endogenous and exogenously expressed β-catenin, including the constitutively active form of β-catenin that contains a Serine37Alanine mutation. Our data also indicate that proliferation of LCSC and HCC lines is inhibited by FH535 in a dose-dependent manner, and that this correlates with a decrease in the percentage of cells in S phase. Finally, we also show that expression of two well-characterized targets of β-catenin, Cyclin D1 and Survivin, is reduced by FH535. Taken together, this data indicates that FH535 has potential therapeutic value in treatment of liver cancer. Importantly, these results suggest that this therapy may be effective at several levels by targeting both HCC and LCSC.
Hepatocellular carcinoma (HCC), the most common liver cancer, is the fifth most common cancer and the third highest cause of cancer-related mortality worldwide [1]–[2]. The alarming rise in HCC incidence in Europe and North America in recent years is related mainly to hepatitis C virus infection, although other factors such as excessive alcohol consumption and obesity also contribute to this increase [3]. The etiology of HCC is complex and involves numerous genetic and epigenetic alterations and the disruption of various signaling pathways including the Wnt/β-catenin, Ras/Raf/MAPK, PI3K/AKT/mTOR, HGF/c-MET, IGF, VEGF and PDGF pathways. Among these, the Wnt/β-catenin pathway is considered among the most difficult to inhibit [4]. Currently, few chemical agents targeting the Wnt/β-catenin pathway are available or under investigation [5].
Activation of the canonical Wnt/β-catenin pathway involves the binding of Wnt proteins to cell surface Frizzled receptors and LRP5/6 co-receptors. In the absence of Wnt proteins, much of the cellular β-catenin is bound to E-cadherin on the cell membrane. Cytosolic β-catenin is constitutively phosphorylated at specific serine residues by an enzymatic complex that includes adenomatous polyposis coli (APC), Axin, and the kinases glycogen synthase kinase-3β (GSK-3β) and casein kinase I, marking it for ubiquitin-mediated proteolysis. Under these conditions, the TCF/LEF transcription factors are bound to their cognate DNA recognition elements along with members of the Groucho family of co-repressors, insuring the transcriptional silencing of β-catenin target genes. Engagement of Wnt proteins with the Frizzled receptor activates the Dishevelled protein, resulting in the dissociation of the cytosolic destructive complex and inhibition of GSK-3β. This leads to the stabilization and accumulation of cytoplasmic β-catenin, which then enters the nucleus, binds TCF/LEF proteins and leads to the subsequent dissociation of groucho co-repressors, recruitment of the coactivator p300 and activation of β-catenin target genes [6]–[9]. Many of the β-catenin targets, including Cyclin D1, c-myc and Survivin, promote cell cycle progression and inhibit apoptosis [10]–[12]. Consistent with this data, activation of the Wnt/β-catenin pathway is seen in a variety of cancers, including HCC. Aberrant activation of the Wnt/β-catenin pathway has been observed in at least 1/3 of HCC, and roughly 20% of HCCs have mutations in the β-catenin gene. More than 50% of HCC tumors display nuclear accumulation of β-catenin indicating that other factors may be involved such as aberrant methylation of the tumor suppressors APC and E-cadherin, inactivation of casein kinase and GSK-3β, or increased secretion of Wnt ligants [4]–[5].
There has been increasing interest in the role of liver cancer stem cells (LCSC) in tumorigenesis, tumor progression, invasion and metastases. The cancer stem cell theory suggests that a tumor is comprised of a heterogeneous population of cells that form a distinct cellular hierarchy. Recent studies have provided convincing evidence that these cells do exist in solid tumors of many types including, brain, breast, colorectal, liver, pancreas and prostate cancers. In 2006, two different groups isolated a CD133+ subpopulation from HCC cell lines and described higher proliferative and tumorigenic potential, consistent with stem cell properties. CD44 was also found as an important marker used in combination with other stem cell markers to better define the surface phenotype of LCSC. It has been demonstrated that CD133+ and CD90+ cells co-expressing CD44+ are more aggressive than those expressing CD133 or CD90 alone [13]–[14].
The chemical agents used to target Wnt-/β-catenin pathway are at the membrane, cytosol and transcription factor levels [5]. The small molecular agent FH535 is a dual inhibitor of peroxisome proliferator-activated receptor (PPAR) and β-catenin/TCF/LEF. FH535 has been shown to inhibit proliferation of HCC and hepatoblastoma cell lines and its specificity on inhibition of β-catenin/TCF/LEF activity was illustrated in hepatoblastoma cell line HepG2 [15].
The aim of this study was to determine if FH535 can inhibit the activation of β-catenin-regulated genes by endogenous and ectopically expressed β-catenin in the HCC cell lines Huh7, Hep3B and PLC and liver cancer stem cells (LCSC). The specificity of FH535 on inhibition of β-catenin via TCF/LEF activation was assayed in dual luciferase reporter transfected in LCSC and in HCC cells. Proliferation, cell cycle, and other targeted genes and proteins were assayed.
FH535 inhibits transcriptional activation mediated by wild-type and constitutively active β-catenin
FH535 has been shown to block signaling through endogenous β-catenin in several cell lines, including the hepatoblastoma cell line HepG2
[15]. To further explore this regulation and to test whether FH535 could block ectopic β-catenin, co-transfections with β-catenin expression vectors and the TCF4-dependent luciferase reporter vector TOPFlash were performed in the human HCC cell lines Huh7 and Hep3B (
Fig. 1). In both cell lines, co-transfected wild-type β-catenin expression vector increased luciferase activity from TOPFlash nearly 15-fold compared to cells co-transfected with the empty vector (E.V.) control. This β-catenin-dependent increase was inhibited by FH535 in a dose-dependent manner. β-catenin is often mutated in various cancers, including HCC. One natural mutation changes the serine at position 37; this altered form of β-catenin is resistant to degradation by the APC complex and thus has higher stability. To test whether this form of activated form of β-catenin could also be blocked by FH535, an expression vector for βCatS37A, in which the serine at position 37 has been changed to an alanine, was co-transfected with TOPFlash. As expected, βCatS37A-mediated transactivation of TOPFlash was significantly higher than transactivation by wild-type β-catenin. However, in both cell lines, βCatS37A-mediated transactivation was significantly inhibited by FH535. As controls, cells were also co-transfected with FOPFlash, which is identical to TOPFlash except that the TCF4 sites have been mutated and therefore no longer responsive to β-catenin; FOPFlash was not activated by wild-type β-catenin or βCatS37A as shown in
Figure 1.
TOPFlash contains three consensus TCF4 binding motifs that confer responsiveness to β-catenin. To test whether FH535 could also block β-catenin-mediated transactivation of a TCF4 motif in the context of a natural regulatory region, co-transfections were performed with E3-pGL3. E3 is a ~340 bp fragment that contains alpha-fetoprotein (AFP) enhancer element E3, one of three enhancers that control hepatic expression of the mouse AFP gene. E3 contains binding sites for multiple factors, including Foxa/HNF6, C/EBP, orphan nuclear receptors, and TCF4 [26]–[27]. We recently showed that this enhancer is regulated by β-catenin in cells and transgenic mice [21]. E3-pGL3 was transactivated by β-catenin and to a greater extent by βCatS37A (Fig. 1). However, this transactivation by both wild-type and S37A forms of β-catenin was blocked by FH535 in a dose-dependent manner.
3.2 FH535 inhibits β-catenin-mediated transcriptional activation in LCSC
Previous studies have shown that β-catenin signaling is elevated in EpCAM positive cells with LCSC properties [28]. We previously described that CD133+, CD44+, CD24+ LCSC aggressively form tumors when small numbers of these cells are injected into nude mice [29]. To test the ability of FH535 to inhibit β-catenin in these LCSCs, transient transfections were performed with TOPFlash. As controls, TOPFlash was also transfected into the HCC cell lines Huh7 and PLC (Fig. 2). In all three populations, untreated cells exhibited low luciferase levels. When treated with the GSK-3β inhibitor LiCl, which leads to endogenous β-catenin activation[30], TOPFlash activity increased dramatically. FH535 effectively blocked LiCl-mediated activation of TOPFlash in a dose-dependent manner. Interestingly, this inhibition was more robust in LCSC than in either HCC cell line. As a control, transfections were also performed with FOPFlash, which is no longer responsive to β-catenin. As expected, luciferase activity in FOPFlash-transfected cells was neither increased by LiCl nor inhibited by FH535.
3.3 FH535 inhibits proliferation of LCSC and HCC cell lines
Numerous studies have demonstrated that β-catenin plays an important role in proliferation during normal development and in cellular transformation in many tissues, including the liver. Liver development is impaired in the absence of β-catenin, and mutations that activate the β-catenin pathway are found in about 1/3 of HCC [4]–[5]. Furthermore, the growth of adult liver progenitor stem cells (oval cells) can be inhibited by blocking the β-catenin pathway. Since our data indicated that FH535 can block β-catenin-mediated transcriptional activation, we also tested whether proliferation of LCSC and HCC cell lines was affected by this compound. LCSC were cultured in the presence of 10% or 1% serum and with between 5 µM and 30 µM FH535 for 72 hours, and cell proliferation was monitored by 3H-thymidine incorporation (Figs. 3A and 3B, respectively). Proliferation decreased with increasing amounts of FH535, with a more dramatic reduction observed in cells grown in the presence of lower serum; the concentration of FH535 to cause a 50% inhibition of cell grown (IC50) was 13.8 µM for cells grown in 10% serum and 5.1 µM for cells grown in 1% serum. This inhibition was more potent than that seen with XAV939 (IC50 = 55 µM), which inhibits tankyrase, thus stabilizing axin and promoting β-catenin degradation (Fig. 3C) [31]. FH535 also blocked proliferation of HCC cells at concentrations that were similar to that seen with LCSC (IC50 of 10.9 µM, 9.25 µM and 6.6 µM for Huh7, PLC and Hep3B, respectively; Fig.3D). To confirm that FH535 indeed inhibited cell proliferation and did not lead to increased cell death, FH535 and 3H-thymidine were added simultaneously to Huh7 cells, which were then cultured for 18 h. In this scenario, we observed a significant inhibition of proliferation at 2.5, 5, 10 and 15 µM of FH535 treatment as compared to control (p<0.05, n = 6), with FH535 at 15 µM causing a 41% inhibition (Figure S3). This data indicates that FH535 is inhibiting cell proliferation rather than increasing cell death.
3.4 FH535 induces cell cycle arrest in the HCC cell line Huh7 and in LCSC
The ability of FH535 to inhibit cell proliferation prompted us to investigate the cell cycle distribution following treatment. Huh7 cells were synchronized by growth in 0.1% FBS for 24 hours and then cultured in the presence of 10% FBS and with no FH535 or FH535 at 7.5 µM and 15 µM. After 24 hours, cells were harvested and DNA content was analyzed by propidium iodide staining. In the presence of FH535, there was a statistically significant increase in the number of cells in G0/G1 and a corresponding decreased in the percentage of cells in S phase compared to cells grown in the absence of FH535 (Fig. 4A). The number of cells in G2 was not significantly altered by FH535. In addition, there was no sub-G1 peak detected by flow cytometry, indicating that FH535 was not promoting apoptosis at the concentrations being use (see Figure S4). We also did cell cycle analysis in LCSC after FH535 treatment and found FH535 at 15 µM significantly caused G1 phase arrest in LCSC (P = 0.012). FH535 also significantly reduced G2/M phase in the LCSC after 24 h of 7.5 µM and 15 µM FH535 treatment (P = 0.038 and P<0.001 respectively), no significant S phase inhibition was observed in LCSC (p = 0.446) (Fig. 4B.). Our data are similar to previously published results and reflects β-catenin regulation of cell cycle is different in different cell types [32]–[33]. Cell cycle regulators (cyclins, CDKs and regulators) can vary in different cell types, which could lead to different responses after FH535 treatment. This may worth exploring in our future study.
3.5 Expression of β-catenin target genes cyclin D1 and Survivin is inhibited by FH535
β-catenin controls cell proliferation and survival by regulating the expression of numerous targets genes. Two well-established targets are the genes encoding Survivin (Birc5) and Cyclin D1 (CcnD1). Survivin is an anti-apoptotic protein that also regulates progression through mitosis [34]; Cyclin D1 controls proliferation by activating the G1 kinases cdk4 and cdk6 [35]. Survivin and Cyclin D1 transcription are regulated through TCF elements in their promoter regions [36]. To test whether FH535 inhibits expression of these two β-catenin target genes, real-time RT-PCR was performed with LCSC and HCC cells that were treated with increasing amounts of FH535. Cyclin D1 and Survivin mRNA levels were reduced by FH535 in all three cell populations in a dose-dependent manner (Fig. 5). To confirm that this reduction in mRNA levels also led to lower protein levels, western analysis was performed using whole cell extracts from Huh7 cells. Both Cyclin D1 and Survivin protein levels were reduced in a dose-dependent manner, with the greatest reduction seen in the presence of 10 µM FH535 (Fig. 6.). Densitometric analysis indicated that FH535 at 5 and 10 µM inhibited Cyclin D1 28% and 64% respectively; FH535 at 5 and 10 µM inhibited surviving 24% and 48% respectively (Fig. 6).
Discussion
In recent years, numerous signaling pathways have been implicated in hepatic carcinogenesis. The β-catenin pathway is essential in stem cells for self-renewal and maintenance of stem cell properties. Disruption of this balance results in both genetic and epigenetic changes, found in many cancers, including colon cancer and HCC [4]. In this study, we used FH535 as an inhibitor of the β-catenin pathway. This compound has been used previously to inhibit β-catenin expression in cells from colon and lung as well as in cells from hepatoblastoma and HCC [15]. In this report, the authors concluded that FH535 was toxic to a number of cell lines, including Huh7. However, their assays could not distinguish between toxicity and reduced cell proliferation. Our data indicates that FH535 does indeed inhibit cell proliferation; we did not directly measure toxicity.
FH535 inhibition of LCSC proliferation is of interest due to its potential therapeutic effect in chemo-resistant HCC. Our group and others have focused on strategies to inhibit the proliferation of LCSC and differences in resistance patterns with non-liver cancer stem cell lines in vitro and in vivo.
Despite numerous efforts, the etiology of HCC tumorigenesis, whether transformed cells originate from mature hepatocytes or stem/progenitor cells remains unclear. Stem cells are defined by their potential for self-renewal and by their ability to proliferate and differentiate into diverse cell types [37]. In recent years, studies have provided convincing evidence that these cells do exist in solid tumors of many types including, brain, breast, colorectal, liver, pancreas and prostate cancers [27]. In this study we have used LCSC that are 64.4%, 83.2%, 96.4% and 96.9% positive, respectively, for CD133, CD44, CD24 and Aldehyde A1 as determined by flow cytometry. These cells have been previously profiled not only by checking the LCSC markers but also by evaluating their tumorigenic potential using low cell numbers (using 2000 LCSCs instead of 100,000 HCC cells to generate tumors) and studying resistance to several drugs. We previously found that these LCSC have intermediate to high resistance to drugs compare to non- liver cancer stem cell lines using different inhibitors.
In this study, we found that FH535, LCSC inhibition of proliferation was affected by FBS concentration in the culture medium, suggesting that the PPAR pathway may be involved in LCSC proliferation as found in the human cancer cell line HCT116 [15]. This could be explained by a variety of fatty acids and their derivatives present in the FBS that are natural agonists to PPAR. It is possible the PPAR agonists suppress the inhibitory effects of FH535 in cell culture. Indeed, in HCT116 cells, FH535 inhibition of β-catein/TCF-dependent luciferase reporter genes was five times stronger in serum-free medium than in media containing 10% FBS. The ability of FH535 to inhibit tumor growth was dramatically increased when 10% FBS was replaced with 10% BSA [15]. Lysophosphatidic acid was found to be an effective PPAR agonist that could reverse FH535 induced inhibition of HCT116 growth [15]. However, the potential function of PPAR in LCSC is beyond the scope of this study and needs further investigation. Recently, FH535 was found to be the most potent drug among several other Wnt/β-catenin inhibitors on human biliary tract cancer cells cultured in serum-free medium [38]. Our study found that FH535 is much more potent than XAV939 in 10%FBS DMEM. This may be related to the PPAR inhibition potential of FH535. Our study found that FH535 inhibited HCC cell lines Huh7, Hep3B and PLC proliferation, indicating that Wnt/β-catenin signaling plays an important role not only in LCSC but also in HCC.
FH535 inhibition of LCSC and HCC proliferation was illustrated by its ability to inhibit β-catenin/TCF/LEF-dependent luciferase reporter activity. To our knowledge, this is the first report on the ability of FH535 to inhibit β-catenin/TCF/LEF activity in LCSC and in HCC cell lines. Previously, Handeli and Simon reported that FH535 inhibits β-catenin/TCF/LEF activity in the HepG2 cell line, which was mistakenly labeled as HCC by these authors [15]. For over thirty years this cell line was considered HCC by numerous investigators. Lopez et al., who initially isolated these cells, recently concluded that HepG2 cells should in fact be considered a hepatoblastoma cell line [39]. Further studies will be needed to investigate how FH535 inhibition of β-catenin influences LCSCs and HCCs. As shown here, cyclin D1 and Survivin expression are inhibited by FH535. Survivin is an anti-apoptotic protein that also regulates progression through mitosis [26], whereas Cyclin D1 controls proliferation by activating the G1 kinases [35]. Real-time RT-PCR and Western analysis confirmed that the expression of these target genes was evident at the mRNA and protein level. Our preliminary data indicate that FH535 treatment does not alter CD133, CD13 and EPCAM expression in LCSC and HCC cell lines (data not shown). Further analysis of these and other stem cell markers are warranted.
In conclusion, our data show that FH535 is a potent inhibitor of the Wnt/β-catenin pathway in LCSCs and HCC cell lines. Whether its ability to inhibit PPAR also affects the growth of LCSCs and HCC cells will require further investigation. Further studies will also be needed to investigate the in vivo efficacy and toxicity of FH535 on HCC xenografts in an animal model. The role of combination therapy using FH535 with other anti-HCC drugs and the possibility of finding cross-talk of Wnt/β-catenin pathway with other signaling pathways should be investigated.
7.10.1.3 Wnt signaling in hepatocellular carcinoma: analysis of mutation and expression of beta-catenin, T-cell factor-4 and glycogen synthase kinase 3-beta genes.
Hepatocellular carcinoma (HCC) is a common killer cancer in the world. Recently, abnormal activation of the Wnt pathway has been found to be involved in the carcinogenesis of several human cancers including HCC. The goal of the present study was to investigate the mechanism of inappropriate activation of the Wnt pathway in hepatocarcinogenesis. We analyzed the alterations of three key components of the Wnt pathway: beta-catenin, glycogen synthase kinase (GSK)-3beta and T-cell factor (Tcf)-4 in 34 HCC and paracancerous normal liver by immunohistochemistry, polymerase chain reaction (PCR)-single-strand conformation polymorphism (SSCP), direct sequencing, and quantitative real-time reverse transcription (RT)-PCR. We found that 61.8% (21/34) of all HCC examined showed an abnormal beta-catenin protein accumulation in the cytoplasm or nuclei. The RT-PCR-SSCP and direct sequencing showed that beta-catenin exon 3 mutations existed in 44.1% (15/34) of the HCC. No mutations of GSK-3beta or Tcf-4 were detected in HCC. Moreover, messenger RNA of beta-catenin and Tcf-4, but not GSK-3beta, was found to be overexpressed in HCC. On analyzing the relationship between alterations of beta-catenin or Tcf-4 and C-myc or Cyclin D1 expression, we found that mutations of beta-catenin, as well as overexpression of beta-catenin or the Tcf-4 gene were independently correlated with C-myc gene overexpression in HCC. Our present findings strongly suggest that mutations of beta-catenin, as well as overexpression of beta-catenin and the Tcf-4 gene, independently activate the Wnt pathway in HCC, with the target gene most likely to be C-myc.
7.10.1.4 Wnt signaling and cancer
The regulation of cell growth and survival can be subverted by a variety of genetic defects that alter transcriptional programs normally responsible for controlling cell number. High throughput analysis of these gene expression patterns should ultimately lead to the identification of minimal expression profiles that will serve as common denominators in assigning a cancer to a given category. In the course of defining the common denominators, though, we should not be too surprised to find that cancers within a single category may nevertheless exhibit seemingly disparate genetic defects. The wnt pathway has already provided an outstanding example of this. We now know of three regulatory genes in this pathway that are mutated in primary human cancers and several others that promote experimental cancers in rodents (Fig. 1). In all of these cases the common denominator is the activation of gene transcription by β-catenin. The resulting gene expression profile should provide us with a signature common to those cancers carrying defects in the wnt pathway. In this review, the wnt pathway will be covered from the perspective of cancer, with emphasis placed on molecular defects known to promote neoplastic transformation in humans and in animal models.
Figure 1.
Oncogenes and tumor suppressors in the wnt signaling pathway. Lines ending with arrows or bars indicate activating or inhibitory effects, respectively. Green and red indicate proto-oncogenic and tumor suppressive activity, respectively, in human cancer or transgenic animals. Definition of the genes and the basis for their activities are described in the text.
The wnt signaling mechanism
The model illustrated in Figure 2 is a proposed mechanism for wnt signaling and is based on the following literature. Signaling is initiated by the secreted wnt proteins, which bind to a class of seven-pass transmembrane receptors encoded by the frizzled genes (Bhanot et al. 1996; Yang-Snyder et al. 1996; He et al. 1997). Activation of the receptor leads to the phosphorylation of the dishevelled protein which, through its association with axin, prevents glycogen synthase kinase 3β (GSK3β) from phosphorylating critical substrates (Itoh et al. 1998; Kishida et al. 1999; Lee et al. 1999; Peters et al. 1999; Smalley et al. 1999). In vertebrates, the inactivation of GSK3β might result from its interaction with Frat-1 (Thomas et al. 1999; Yost et al. 1998; Li et al. 1999a; Salic et al. 2000). The GSK3β substrates include the negative regulators axin and APC, as well as β-catenin itself (Rubinfeld et al. 1996; Yost et al. 1996; Yamamoto et al. 1999). Unphosphorylated β-catenin escapes recognition by β-TRCP, a component of an E3 ubiquitin ligase, and translocates to the nucleus where it engages transcription factors such as TCF and LEF (Behrens et al. 1996; Molenaar et al. 1996;Hart et al. 1999). Additional components in the pathway include casein kinases I and II, both of which have been proposed to phosphorylate dishevelled (Sakanaka et al. 1999; Willert et al. 1997; Peters et al. 1999). The serine/threonine phosphatase PP2A associates with axin and APC, although its functional role in the pathway remains obscure (Hsu et al. 1999; Seeling et al. 1999). Also obscure is the manner by which the wnt receptors communicate with dishevelled.
Figure 2.
Proposed mechanism for the transmission of wnt signals. In the absence of wnt –wnt) GSK3β phosphorylates APC and axin, increasing their binding affinities for β-catenin, which too is phosphorylated by GSK3β, marking it for destruction. In the presence of wnt (+wnt) FRAT prevents GSK3β from phosphorylating its substrates, and β-catenin is stabilized. Casein kinase1ε (CK1ε) binds to and phosphorylates dishevelled (dvl) modulating the FRAT1/GSK3β interaction. RGS, PDZ, and DIX are protein interaction domains.
Receptors, ligands, and related proteins
The proto-oncogenic effects of wnt were discovered over 18 years ago inciting intense investigation into the role of wnt genes in human cancer (Nusse and Varmus 1982). The subsequent discovery of wingless, the fly homolog of wnt-1, paved the way for assembling a signaling pathway subsequently found to contain cancer causing genes (Cabrera et al. 1987; Rijsewijk et al. 1987). Although wnt was the prototypical oncogene in this pathway, no formal proof for its involvement in human cancer has ever been documented. There have been numerous reports on the overexpression, and sometimes underexpression, of wnt genes in human cancers, but mRNA expression levels are merely correlative. More compelling evidence, such as amplification, rearrangement, or mutation of genes encoding wnt ligands or receptors has not been forthcoming. In lieu of these sorts of findings, we are left to speculate on the consequences of epigenetic events implicating these genes in human cancer. In doing so we can use animal and cell culture models to guide our interpretation.
The wnt ligands, of which there are at least 16 members in vertebrates, are secreted glycoproteins that can be loosely categorized according to their ability to promote neoplastic transformation (for review, seeWodarz and Nusse 1998). For example, the activation of wnt-1, wnt-3, or wnt-10b by retroviral insertion in the mammary gland will promote tumor formation in mice (Lee et al. 1995; Nusse and Varmus 1982; Roelink et al. 1990). Oncogenic potential can also be assessed in cultured mammalian cells, such as C57MG and CH310T1/2, where expression of the proto-oncogenic wnts results in morphological transformation (Bradbury et al. 1994; Wong et al. 1994). These cells are transformed by wnt-1, wnt-2, wnt3a but not by wnt-4, wnt-5a, and wnt-6. The transforming wnt genes also promote the accumulation of β-catenin in some cultured mammalian cells (Shimizu et al. 1997). Some aspects of the wnt cancer pathway are also recapitulated inXenopusdevelopment, where injection of transforming wnts into early embryos results in duplication of the dorsal axis (Wodarz and Nusse 1998). A caveat here is that the lack of specific receptors for certain wnts might also explain their inactivity in some of these assays (He et al. 1997). Nevertheless, identifying those wnts capable of neoplastic transformation will aid the interpretation of epigenetic evidence implicating wnts in cancer. For example, expression of thewnt-16 gene is activated by the E2A–Pbx1 fusion product in acute lymphoblastoid leukemia (McWhirter et al. 1999), but the oncogenic potential of wnt-16 is unknown.
As might be expected from the plethora of wnt genes, there are also numerous wnt receptors. At least 11 vertebrate frizzled genes have been identified, but how they differ in function and ligand specificity is far from clear. The analysis of mere binding specificity may not be sufficient to sort out the appropriate combinations of functional receptor-ligand interactions. Wnt-3a and wnt-5a both bind to Human frizzled 1 (Hfz1), yet only wnt-3a mediates TCF-dependent transcription (Gazit et al. 1999). This suggests that the activation of TCF/LEF-dependent transcription is a good correlate to neoplastic transformation. Implementation of this assay, along with a second assay involving the translocation of PKC to the cell membrane, resulted in the categorization of murine wnt receptors into two exclusive groups (Sheldahl et al. 1999). Human FzE3 fell into the TCF/LEF activation group, consistent with previous work showing that its overexpression resulted in nuclear localization of β-catenin (Tanaka et al. 1998). This receptor was also expressed in numerous human esophageal cancers, but not in matched normal tissue (Tanaka et al. 1998).
In addition to the frizzled receptors, there exists a family of secreted proteins bearing homology to the extracellular cysteine-rich domain of frizzled. The so-called secreted frizzled-related proteins (sFRP) bind to the wnt ligands, thereby exerting antagonistic activity when overexpressed in wnt signaling assays (Leyns et al. 1997; Wang et al. 1997). The vertebrate sFRPs, like the frizzled proteins, exhibit functional specificity with respect to the various wnts. InXenopus assays, the prototypical frizzled related protein frzb, now known as sFRP-3, inhibited wnt-1 and wnt-8, but not wnt-5a (Leyns et al. 1997; Lin et al. 1997; Wang et al. 1997). Assays in mammalian cells showed that FrzA, now termed sFRP-1, inhibited wnt-1-induced accumulation of β-catenin (Dennis et al. 1999;Melkonyan et al. 1997). Again, binding specificity may not relate to functional specificity, as wnt-5a associated with sFRP-3 but was unable to inhibit its activity (Lin et al. 1997). Even the significance of specific functional interactions might be suspect based on recent titration experiments with purified soluble sFRP-1. At low concentrations sFRP-1 enhanced signaling activity by soluble wingless protein, whereas at higher concentrations it was inhibitory (Uren et al. 2000). The authors proposed high and low states of binding affinity that involved the carboxy-terminal heparin binding domain and the amino-terminal cysteine-rich domain of sFRP-1, respectively. Binding to the cysteine-rich domain might confer inhibition while binding to the carboxy-terminal region could facilitate presentation of active ligand to receptor. The potential for some sFRPs to activate wnt signaling is consistent with a previous study in which sFRP-2, then known as SARP-1, increased the intracellular concentration of β-catenin and conferred anti-apoptotic properties to cultured MCF-7 cells (Melkonyan et al. 1997). Functional studies are further complicated by the binding of a sFRP to the putative human receptor frizzled-6, underscoring additional possible modes of regulation (Bafico et al. 1999). The sFRPs have not been directly linked to cancer, but one could speculate that the anti-apoptotic activity observed with the SARP-1 could contribute to tumor progression. Alternatively, the identification of sFRP-2 as a target of the hedgehog signaling pathway might be relevant to human basal cell cancers (Lee et al. 2000). Additional structurally distinct secreted inhibitors of wnt signaling include the recently discovered dickopft-1 and wif-1 proteins (Fedi et al. 1999; Glinka et al. 1998;Hsieh et al. 1999).
GSK3β
The serine/threonine kinase GSK3β binds to and phosphorylates several proteins in the wnt pathway and is instrumental to the down regulation of β-catenin (Dominguez et al. 1995; He et al. 1995; Hedgepeth et al. 1999b; Ikeda et al. 1998;Itoh et al. 1998;Li et al. 1999a; Nakamura et al. 1998b; Rubinfeld et al. 1996;Yamamoto et al. 1999; Yost et al. 1996). As a negative regulator of wnt signaling, GSK3β would qualify as a potential tumor suppressor. However, mutations or deletions in the gene coding for GSK3β were not been detect ed in a survey of colorectal tumors (Sparks et al. 1998). Perhaps GSK3β can compensate for the loss of GSK3β and the biallelic inactivation of both these genes is unlikely in tumor progression. Alternatively, the utilization of GSK3β by pathways independent of wnt could make its overall ablation incompatible with cell viability. Nevertheless, inactivation of GSK3β can still be achieved by a means other than genetic ablation and can occur in a manner that uniquely affects wnt signaling. This mode of inactivation involves the association of GSK3β with Frat-1. Frat-1 was identified by insertional mutagenesis in a screen for genes that enhanced the progression of transplanted T-cell lymphomas in mice (Jonkers et al. 1997). Subsequent transgenic expression of Frat-1 alone did not induce spontaneous lymphomas, but greatly enhanced lymphomagenesis initiated either by leukemia virus M-MuLV or expression of the Pim1 oncogene (Jonkers et al. 1999). A connection to GSK3β was realized by the discovery of the Frat-1 Xenopushomolog GBP, a GSK3β binding protein inhibitory to wnt signaling when expressed in Xenopus embryos (Yost et al. 1998). Frat-1 is also antagonistic to wnt signaling in mammalian cells, presumably because it competes with axin for binding to GSK3β (Li et al. 1999a; Thomas et al. 1999). GBP also inhibited the phosphorylation and degradation of β-catenin in vitro when added to Xenopusextracts (Salic et al. 2000). Although Frat-1 contributes to cancer progression in a transgenic mouse model, its contribution to human cancer has not been documented.
Dishevelled
The genetic analysis of dishevelled in developmental systems has defined it as a positive mediator of wnt signaling positioned downstream of the receptor and upstream of β-catenin (Noordermeer et al. 1994). Overexpression or constitutive activation of dishevelled would be expected to promote neoplastic transformation, but its involvement in human cancers has not been reported. This might reflect the dual function of dishevelled, one that transduces wnt signals for the stabilization of β-catenin and a second that relays signals for the activation of jun kinases (Li et al. 1999b; Moriguchi et al. 1999). Although these two functions are housed in physically separable regions of the protein, dysregulation of one function, without impacting the other, could place severe constraints on selection for potential oncogenic mutations. A possible connection of dishevelled to cancer is through casein kinase II, which binds to and phosphorylates dishevelled and also promotes the formation of lymphomas when expressed in transgenic mice (Seldin and Leder 1995; Song et al. 2000; Willert et al. 1997).
β-catenin
Mutations in the β-catenin gene (CTNNb1) affecting the amino-terminal region of the protein make it refractory to regulation by APC (Morin et al. 1997; Rubinfeld et al. 1997). These mutations affect specific serine and threonine residues, and amino acids adjacent to them, that are essential for the targeted degradation of β-catenin (for review, see Polakis 1999). The mutations abrogate the phosphorylation dependent interaction of β-catenin with β-TRCP, a component of an E3 ubiquitin ligase that makes direct contact with amino terminal sequence in β-catenin (Hart et al. 1999). This regulatory sequence in β-catenin is mutated in a wide variety of human cancers as well as in chemically and genetically induced animal tumors. Importantly, β-catenin mutations in tumors are exclusive to those that inactivate APC. This is particularly apparent in colorectal cancer where the vast majority of these tumors contain APC mutations and the overall frequency of β-catenin mutations is quite low (Samowitz et al. 1999; Sparks et al. 1998;Kitaeva et al. 1997) (Table 1). When colorectal tumors lacking APC mutations were analyzed separately, the likelihood of finding a CTNNb1 mutation was greatly increased (Iwao et al. 1998; Sparks et al. 1998). The exclusivity of CTNNb1 and APC mutations in colorectal cancer was also evident from the analysis of replication error-positive tumors identified by microsatellite instability. Both the hereditary and sporadic forms of replication error-positive colorectal cancers had a relatively high frequency of β-catenin mutations, whereas APC mutations were relatively rare (Mirabelli-Primdahl et al. 1999; Miyaki et al. 1999) (Table 1). Interestingly, this correlation between microsatellite instability andCTNNb1 mutations was not apparent in endometrial cancers (Mirabelli-Primdahl et al. 1999).
Table 1.
Beta-catenin mutations in human cancers
Aggressive fibromatosis, otherwise known as desmoid tumor, is a locally invasive fibrocytic growth that occurs with increased incidence in patients with familial adenomatous polyposis coli (FAP). FAP individuals carry APC mutations in their germline and present with multiple intestinal adenomas at an early age. Desmoids also occur sporadically and, with the exception of colorectal cancer, represent a rare example of biallelic inactivation of APC in individuals without a pre-existing germline mutation in APC (
Alman et al. 1997). Not surprisingly, mutations in
CTNNb1 have also been detected in sporadic desmoid tumors (
Shitoh et al. 1999;
Tejpar et al. 1999). The β-catenin mutations were found in over half of the 42 desmoids analyzed, while inactivating mutations in APC were detected in nine and, again, there was no overlap between APC and β-catenin mutations (
Tejpar et al. 1999). The β-catenin mutations were all of the missense variety and were confined to codons 41 and 45. Some of the desmoids lacked mutations in either β-catenin or APC, but all displayed increased expression of β-catenin, implying that yet unidentified defects in β-catenin regulation exist in some of these tumors.
There appears to be a low probability of accruing biallelic inactivating mutations in APC in most sporadic cancers, despite increased cancer incidence at numerous extracolonic sites in FAP patients. This suggests that the stabilization of β-catenin can promote cancer in many tissue types, but the biallelic inactivation of APC is an unlikely means to this end. Components in the wnt pathway other than APC, such as β-catenin, might make easier targets for oncogenic mutations. Indeed, several mutations in CTNNb1 were recently identified in gastric cancers, which occur with increased incidence in FAP patients (Park et al. 1999). In this study, 27% of intestinal type gastric cancers harbored mutations in β-catenin. Hepatoblastoma also occurs with increased incidence in FAP individuals (Hughes and Michels 1992;Giardiello et al. 1996; Cetta et al. 1997), but biallelic inactivation of APC is uncommon in the sporadic forms of these tumors. In three separate studies, mutations in β-catenin were identified at high frequency in hepatoblastoma, while no APC mutations were found (Koch et al. 1999; Jeng et al. 2000; Wei et al. 2000). Hepatoblastoma is also associated with Beckwidth–Wiedemann syndrome (BWS), however, a direct link between wnt signaling and the genetic defects underlying BWS are unlikely as a tumor from one of these patients also contained a somatic mutation in β-catenin (Wei et al. 2000). By contrast, a subset of patients with Turcot’s syndrome harbor germline mutations in APC and are at increased risk of medulloblastoma (Hamilton et al. 1995; Lasser et al. 1994). Although inactivating mutations in APC have not been detected in the sporadic forms of medulloblastoma, CTNNb1mutations were found in a small percentage (Zurawel et al. 1998).
Hepatocellular carcinoma (HCC) has become one of the most common tumors harboring mutations in the wnt pathway. Based on five separate studies, the frequency of CTNNb1 mutations in hepatocellular carcinoma (HCC) was ∼20% overall and perhaps higher still for HCCs associated with hepatitis C virus (de La Coste et al. 1998; Miyoshi et al. 1998;Huang et al. 1999; Legoix et al. 1999; Van Nhieu et al. 1999) (Table1). Preliminary data indicated a poorer prognosis associated with nuclear accumulation of β-catenin in HCC and histological data indicated enhanced nuclear staining in the invasive and intravascular compartments of the tumors (Huang et al. 1999; Van Nhieu et al. 1999). In one of these studies an inverse correlation between β-catenin mutations and loss of heterozygosity in the genome was noted (Legoix et al. 1999). This suggests that chromosomal instability and mutations inCTNNb1 represent alternative modes of tumor progression in HCC.
It is noteworthy that c-myc and cyclin D genes are amplified in a subset of HCCs and both these genes are downstream targets of β-catenin (He et al. 1998; Nishida et al. 1994; Peng et al. 1993;Shtutman et al. 1999; Tetsu and McCormick 1999). It would be of interest to determine whether any overlap exists between their amplification and CTNNb1mutations in HCC. Animal models of HCC have provided some clues toward understanding the relationship between these genes in cancer. HCCs induced by transgenic expression of SV40 T antigen in murine liver did not contain mutations in CTNNb1 (Umeda 2000). As T antigen activates cyclin D kinase by sequestration of Rb, the activation of the cyclin D gene by mutant β-catenin may no longer be required. By contrast, activating mutations inCTNNb1 were identified in half of the HCCs generated by transgenic expression of c-myc in murine liver (de La Coste et al. 1998). This animal model suggests that β-catenin mutations occur as a second “hit” in HCC tumor progression in cooperation with a distinct cancer pathway initiated by c-myc. That CTNNb1mutations can occur subsequent to other oncogenic defects is also evident from their occurrence in Wilm’s tumor. Mutations in β-catenin were detected in 15% of these pediatric kidney cancers and in two of these cases they were concomitant with mutations in the Wilm’s tumor gene WT1 (Koesters et al. 1999). One of these cases was associated with Denys-Drash syndrome, a familial disorder attributable to germline mutations in WT1.
It makes sense that extracolonic tumors associated with FAP, such as desmoids, medulloblastoma, and HCC, would contain CTNNb1mutations in their sporadic forms. Thyroid cancers also occur with increased incidence in FAP and, not surprisingly, a high frequency ofCTNNb1 mutations was recently reported for anaplastic thyroid cancers (Cetta et al. 2000; Garcia-Rostan et al. 1999). Although many of these mutations affected amino acids known to influence the regulation of β-catenin, many of them affected residues for which the consequence of their mutation is unknown (Garcia-Rostan et al. 1999). In particular, the substitution K49R was detected nine times. This mutation was frequently detected in the context of independentCTNNb1 mutations in the same thyroid tumor, and up to four independent CTNNb1 mutations were found in some tumors. The occurrence of multiple independent CTNNb1 mutations was also noted in some HCCs and might reflect the multifocal origin of some cancers (Huang et al. 1999; Legoix et al. 1999; Van Nhieu et al. 1999). In one HCC study, examination of different tumor areas from the same patient revealed distinct CTTNb1 mutations in two independent cases (Huang et al. 1999).
Some cancers, such as endometrial ovarian tumors, do not occur with increased incidence in patients with FAP, yet they contain activating mutations in CTNNb1(Palacios and Gamallo 1998; Gamallo et al. 1999; Wright et al. 1999). Perhaps inactivation of the remaining wild-type APC allele in FAP individuals is unlikely in this tissue, or the expression of an alternative APC gene compensates for its loss. The CTNNb1 mutations associated with ovarian cancer appeared to be confined to the endometrioid subtype. In this tissue, cancers with activated β-catenin signaling were reported to be less aggressive than their nonactivated counterparts. In one report, a more favorable prognosis was associated with cancers exhibiting enhanced nuclear staining of β-catenin and another indicated higher frequency ofCTNNb1 mutations in lower grade tumors (Palacios and Gamallo 1998; Wright et al. 1999). A similar inverse correlation between tumor grade and occurrence ofCTNNb1 mutations was also reported for uterine endometrial cancers (Fukuchi et al. 1998). The overlap between mutations in CTNNb1 and other gene defects in ovarian cancers has not been explored in detail, although one study noted coexisting mutations in the PTEN tumor suppressor andCTNNb1 in endometrioid tumors (Wright et al. 1999).
Additional types of cancers with CTNNb1 mutations, albeit at low frequency, include melanoma and prostate. Although only one of sixty-five melanomas contained detectable mutations, nuclear localization of the protein was seen in one-third (Rimm et al. 1999). Thus, additional mechanisms for β-catenin activation likely occur in these tumors. Possibly the highest percentage ofCTNNb1mutations occurs in a common skin tumor known as pilomatricomas (Chan et al. 1999). That these tumors might contain CTNNb1 mutations was surmised from the genesis of similar tumors in transgenic mice expressing mutant β-catenin in the skin (Gat et al. 1998). The tumors appeared to originate from the hair follicle, which is consistent with the lack of hair in mice homozygous for mutations in LEF, a transcription factor responsive to β-catenin (van Genderen et al. 1994).
Axin
Axin was originally identified as an inhibitor of wnt signaling inXenopus embryos and was subsequently shown to bind directly to APC, β-catenin, GSK3β and dishevelled (for review, see Peifer and Polakis 2000). A plethora of in vitro and in vivo studies inXenopus, Drosophila, and cultured mammalian cells has demonstrated that axin is central to the down regulation of β-catenin (Zeng et al. 1997; Behrens et al. 1998; Hart et al. 1998;Ikeda et al. 1998; Nakamura et al. 1998a; Sakanaka et al. 1998; Fagotto et al. 1999; Hedgepeth et al. 1999a; Li et al. 1999a; Willert et al. 1999a; Farr et al. 2000). It is not entirely clear how axin functions, but it has been proposed to facilitate the phosphorylation of β-catenin and APC by GSK3β (Hart et al. 1998; Ikeda et al. 1998). Thus axin would be viewed as a tumor suppressor based on its ability to downregulate signaling, and this has now been verified by documentation of its biallelic inactivation in human hepatocellular cancers and cell lines (Satoh et al. 2000). Importantly, these mutations were identified in those HCCs that lacked activating mutations inCTNNb1. All of the mutations were predicted to truncate the axin protein in a manner that eliminated the β-catenin binding sites. Axin, which should now be regarded as a tumor suppressor, constitutes the third genetic defect in the wnt pathway that contributes to human cancer. There also exists a close homolog of axin termed conductin, which exhibits of all the binding and regulatory functions of axin (Behrens et al. 1998). That this apparent redundancy did not suppress axin mutations in HCC suggests conductin is either not functionally equivalent to axin or not expressed at levels sufficient to compensate for its loss in HCCs.
PP2A
The dependence upon serine/threonine kinases for the regulation of β-catenin implies that phosphatases are also involved. Indeed, the rapid dephosphorylation of the axin protein is a consequence of wnt signaling and has been proposed to both destabilize axin and reduce its affinity for β-catenin (Willert et al. 1999b;Yamamoto et al. 1999). Although axin binds directly to the PP2A catalytic subunit, the phosphatase affecting axin in response to wnt signaling has not been identified (Hsu et al. 1999). If PP2A is this phosphatase, it would be viewed as proto-oncogenic because it downregulates the tumor suppressor axin. On the contrary, expression of the PP2A regulatory subunit B56 in human colon cancer cells results in the downregulation of β-catenin, consistent with a tumor suppressive function in the wnt pathway (Seeling et al. 1999). Moreover, the beta isoform of the PP2A A subunit is deleted in some human colon tumors, again implying tumor suppression (Wang et al. 1998). Also, disruption of twins, aDrosophila gene coding for a PP2A subunit, complemented the overexpression and underexpression of the β-catenin homolog armadillo, in a manner consistent with negative regulation of wnt signaling (Greaves et al. 1999). By all accounts, PP2A plays a role in wnt signaling, but its potential role as proto-oncogene or tumor suppressor might be dependent upon the precise nature of the defect.
APC
Genetic analysis of FAP families led to the identification of theAPC gene, and subsequent studies demonstrating an interaction with β-catenin placed it tentatively in the wnt pathway (Groden et al. 1991; Kinzler et al. 1991; Munemitsu et al. 1995; Rubinfeld et al. 1993; Su et al. 1993). Experiments in Drosophilaultimately revealed that genetic ablation of APC indeed resulted in upregulation of β-catenin signaling (Ahmed et al. 1998). In some systems, such as Xenopus andCaenorhabditis elegans, a positive role for APC in the wnt pathway has been proposed, but the former study suffers from potential overexpression artifacts and the latter from a lack of relatedness to the vertebrate APC protein (Rocheleau et al. 1997; Vleminckx et al. 1997). In any case, APC is a tumor suppressor in human cancers and its mutation relates strongly to the regulation of β-catenin. The spectrum of APC mutations, which typically truncate the protein, suggest selection against β-catenin regulatory domains, albeit not necessarily against β-catenin binding (for review, see Polakis 1999). The selective pressure might be directed against the presence of Axin binding sites, of which there are three, dispersed across the central region of the APC protein (Behrens et al. 1998). The presence of axin binding sites are critical to APC in the regulation of β-catenin levels and signaling in cultured cells (Kawahara et al. 2000). Moreover, mice lacking wild-type APC but expressing a truncated mutant APC retaining a single axin binding site are viable and do not develop intestinal neoplasia (Smits et al. 1999). This has not been the case for mice with more extensive truncations in APC (Oshima et al. 1995a; Su et al. 1992). Also, milder forms of colorectal polyposis, as well as familial infiltrative fibromatosis (desmoid tumors), have been associated with germline mutations in the 3′ region of the APC open reading frame. These mutations predict truncated proteins that retain only one or two of the three axin binding sites in APC (Walon et al. 1997; Kartheuser et al. 1999; Scott et al. 1996;van der Luijt et al. 1996). A recent study has also demonstrated that the expression of just the central region of APC, which contains all of the axin and β-catenin binding sites, was sufficient to elicit cellular growth suppression (Shih et al. 2000). This effect is consistent with previous work showing that a like fragment of APC was sufficient to downregulate β-catenin levels in cancer cells (Munemitsu et al. 1995).
Although both copies of the APC gene are typically inactivated in colorectal cancers, it remains possible that a mutant truncated APC could contribute to cancer progression. This was tested by transgenic expression of two different APC mutants in a wild-type intestinal background (Oshima et al. 1995b). This did not result in cancer-prone mice, despite high levels of expression of mutant proteins and, therefore, argues against a dominant negative effect by these particular mutants. However, it does not rule out an additive contribution to tumor progression by mutant APC protein in a background lacking wildtype APC. In fact, genetic evidence argues in favor of selection for a somewhat specific mutant APC protein. The mutation cluster region (MCR) in APC, roughly defined by codons 1250–1500, is not only consistent with selection against specific sequence, but also retention of an APC molecule that extends into the MCR (Fig.3.). A correlation between the presence of a germline mutation in the MCR and the severity of polyposis has been noted (Ficari et al. 2000; Nagase et al. 1992; Wu et al. 1998). The enhanced severity of polyposis suggests there should also be selective pressure for somatic mutations in the MCR, which indeed appears to be the case (Miyoshi et al. 1992). Selective pressure for an MCR mutant has also been proposed based on the occurrence of somatic mutations in the MCR relative to the position of the germline mutation in FAP (Lamlum et al. 1999). Tumors from FAP patients with a germline MCR mutation exhibited frequent inactivation of the remaining APC allele by LOH, while those without a germline MCR mutation had frequent somatic mutations in the MCR (Fig. 3). Therefore, a germline mutation in the MCR could relieve the constraint for a subsequent somatic MCR mutation, thereby increasing the likelihood of polyposis. This implies that a truncated MCR APC mutant has an interfering or gain of function property that enhances tumor progression beyond simple loss of APC function. An interfering function would probably not involve interaction with wild-type APC, as recently suggested, because the MCR mutant is still selected for in the absence of a wild-type APC gene copy (Dihlmann et al. 1999). Finally, some of the germline mutations in APC do not disrupt the open reading frame yet correlate with increased risk of colorectal cancer (Frayling et al. 1998; Gryfe et al. 1999; Laken et al. 1997). These mutations have been proposed to increase the occurrence of subsequent truncating mutations by enhancing the mutational susceptibility of the affected nucleotide tract.
Figure 3.
Mutations in APC. A compilation of germline and somatic mutations in APC illustrates selection for mutations in the mutation cluster region (MCR). MCR mutations result in truncated proteins retaining β-catenin binding but not regulatory activity. Somatic MCR mutations are more frequently selected for in FAP patients with germline mutations outside of the MCR.
Transcription factors
Prior to discussing the potential role for LEF/TCF transcription factors in cancer, it is important to outline the mechanism by which they have been proposed to operate. Although LEF/TCFs bind directly to DNA through their HMG domains, they are incapable of independently activating gene transcription (Eastman and Grosschedl 1999; Roose and Clevers 1999). This has best been illustrated for LEF, which through its binding to the cofactor ALY, makes indirect contacts with a second transcription factor AML (Bruhn et al. 1997). The TCFs do not contain the ALY binding site, but like LEF they cannot activate test genes comprised of multimerized TCF/LEF binding sites and a minimal promotor sequence. However, these reporter genes are activated on coexpression of TCF with β-catenin, suggesting that β-catenin supplies additional cofactors required for transcriptional activation (Molenaar et al. 1996). This activity was localized to the carboxy-terminal region of the Drosophila β-catenin armadillo, which when fused directly to TCF resulted in β-catenin independent transcriptional activation (van de Wetering et al. 1997).
The simple interpretation is that the TCF/LEF-β-catenin complex comprises a bipartite positive acting transcription factor in the wnt pathway. This interpretation agrees well with developmental studies in which the manipulation of LEF/TCF function results in phenotypes consistent with the genetic manipulation of wnt/β-catenin signaling (Behrens et al. 1996; Brunner et al. 1997; Huber et al. 1996; van de Wetering et al. 1997). For example, a zygotic homozygous null mutation inDrosophila LEF results in a loss of naked cuticle in the larval epidermis, a phenotype typical of loss of function wingless mutations (Brunner et al. 1997). Moreover, the formation of excess naked cuticle by ectopic expression of armadillo in wild-type embryos does not occur in the LEF null mutants. Exactly how β-catenin contributes to transcriptional activation is unclear, but might involve additional proteins that bridge the TCF/β-catenin complex to the basal transcriptional machinery. The bridging function might be fulfilled by Pontin 52, a TATA-binding protein that was reported to bind to β-catenin (Bauer et al. 1998). Also, a mutant form of β-catenin incapable of binding LEF squelched LEF-dependent reporter gene activation, presumably by titration of an essential cofactor (Prieve and Waterman 1999). Finally, the carboxy-terminal region of armadillo binds to the Zinc finger protein teashirt, a homeotic gene essential for a subset of wingless signaling outputs in Drosophila (Gallet et al. 1999).
The simple model of positive transcriptional activation by the TCF-β-catenin complex is not in accord with all experiments. Mutation of the TCF/LEF binding sites in the promotors of the wingless responsive gene ultrabithorax and the Wnt-responsive Xenopus gene Siamois enhanced their activities under conditions where the wingless/β-catenin signal input was weak (Brannon et al. 1999; Riese et al. 1997). The mammalian cyclin D gene was recently identified as a wnt target and, again, mutation of the corresponding TCF binding sites enhanced its basal activity (Tetsu and McCormick 1999). These results suggest TCF represses transcription of its target genes in unstimulated cells and the binding of β-catenin promotes derepression. Derepression cannot fully account for signaling activity, however, as mutations in the TCF binding sites compromise target gene activation under conditions of active wnt signaling (Brannon et al. 1999; Riese et al. 1997). Repression of gene expression by TCF is consistent with its direct physical interaction with at least three different gene products, the Groucho/TLE and CtBP corepressors, and the CREB binding protein CBP (Brannon et al. 1999;Cavallo et al. 1998; Levanon et al. 1998; Roose et al. 1998; Waltzer and Bienz 1998).
The groucho/TLE proteins bind to the central region of TCF/LEF at a site distinct from that of β-catenin binding and inhibit gene activation of TCF target genes (Levanon et al. 1998; Roose et al. 1998). By contrast, CtBP binds to two independent sites in the carboxy-terminal region of Xtcf-3, which when mutated abrogated the repressor function of this region of Xtcf-3 (Brannon et al. 1999). The binding sites for CtBP are not present in LEF, which might explain the ability of LEF, but not Xtcf-3, to induce axis duplication in Xenopus embryos. Finally, the Drosophila CREB binding protein CBP was reported to bind to the HMG domain of dTCF (Waltzer and Bienz 1998). Loss-of-function CBP mutants displayed some features of wingless over expression and also suppressed phenotypes resulting from loss of the β-catenin homolog armadillo. The genetics imply suppression of wingless by CBP, which is somewhat paradoxical when considering the role of CBP acetyltransferase activity in chromatin remodeling and gene activation. However, it was shown that CBP acetylates a lysine proximal to the armadillo binding site in TCF, thereby reducing its affinity for armadillo. Repression of β-catenin/TCF signaling by CBP does not occur in all settings, though, as two recent studies demonstrated activation ofXenopus TCF target genes by CBP (Hecht et al. 2000;Takemaru and Moon 2000). CBP directly associated with carboxy-terminal sequence in β-catenin and overexpression of E1A, which also directly binds CBP, reduced β-catenin dependent transactivation.
Does the activation of TCF/LEF target genes by β-catenin cause cancer? Good evidence to this effect was provided by the expression of a chimeric protein consisting of the LEF DNA binding sequence fused to the transcriptional activation domain of either VP16 or the estrogen receptor (Aoki et al. 1999). Expression of these constructs in chicken embryo fibroblasts resulted in their neoplastic transformation. The proliferative potential of TCF was also apparent from the phenotype resulting from homozygous disruption of TCF-4 in the germline of mice. These animals were incapable of maintaining a proliferative stem cell compartment in the small intestine and died shortly after birth (Korinek et al. 1998). Whether the TCF/LEF genes are directly activated by mutations in cancer is unclear, but mutations in TCF-4 have been detected in a subset of colorectal tumors (Duval et al. 1999). The mutations all occur as single base deletions in an (A)9 nucleotide repeat within the 3′ coding region of the gene. These deletions generate frame shifts predicted to effect the proportion of the long and short forms of TCF that normally result from alternative mRNA splicing. The mutations were also found in cancer cell lines, all of which possessed mutations in either APC or β-catenin. This indicates that the TCF mutations do not substitute for APC/β-catenin mutations but could act in an additive manner.
An additional mechanism by which TCFs could contribute to cancer was gleaned from the phenotype of mice homozygous for mutations in TCF-1 (Roose et al. 1999). Fifteen percent of these animals developed adenomatous intestinal polyps by one year of age, implicating TCF-1 as a tumor suppressor. The major isoforms of TCF-1 do not contain a β-catenin binding site and could therefore act in a dominant negative manner in wnt signaling. Crossing TCF-1 null mice with cancer-prone ApcMin/+ mice resulted in offspring with ten times the number of intestinal polyps relative to ApcMin/+ littermates. This experimental model suggests that the genetic ablation of TCF-1 could modify, or even independently contribute to cancer progression in humans. Additional potential mechanisms for cancer would include the inactivation of corepressors such as CtBP and TLE/groucho.
Cross talk
Defects leading to activation of the wnt pathway could also occur in signaling systems that are seemingly unrelated to wnt signaling. One potential mode of cross talk includes the kinase TAK1, which can substitute for MAPK kinase kinase in the yeast pheromone pathway. TAK1 (TGF-β activatedkinase) is activated by TGF-β in mammalian cells and has also been implicated in interleukin-1 activation of NFκB (Ninomiya-Tsuji et al. 1999; Yamaguchi et al. 1995). In c. elegans, the TAK1 homolog MOM-4 negatively regulates the TCF homolog POP-1 by activating another kinase LIT-1, which then phosphorylates POP-1 (Meneghini et al. 1999;Shin et al. 1999). LIT-1 is thought to gain access to POP-1 through its direct binding to the β-catenin homolog WRM-1 (Shin et al. 1999). Parallel interactions have been demonstrated for the mammalian counterparts of these proteins where the phosphorylation of TCF, by the LIT-1 homolog NLK, reduces its DNA binding affinity (Ishitani et al. 1999). Thus a MAPK-like signaling system might downregulate the wnt-1 pathway. A second opportunity for cross talk with wnt signaling was realized by a physical interaction between the β-catenin-TCF complex and SMAD4, a mediator of TGF-β signaling (Nishita et al. 2000). This interaction was proposed to be synergistic with respect to the activation of theXenopus wnt target gene twin. How this relates to cancer is somewhat puzzling when considering that TGF-β signaling is typically compromised by genetic and epigenetic defects during tumor progression.
An additional mode of cross regulation was recently revealed by the discovery that retinoids inhibit β-catenin dependent gene transcription (Easwaran et al. 1999). β-catenin associated with a retinoic acid receptor (RAR) and cooperated with retinoids to enhance activation of a retinoic acid responsive promotor. Moreover, the identification of RAR-γ as a target of wnt signaling inXenopus also points to an interaction between these signaling systems (McGrew et al. 1999). Signaling by β-catenin was also reported to be repressed by expression of sox3 and sox17 transcription factors, which associated directly with β-catenin (Zorn et al. 1999). Although inhibition of β-catenin signaling was clearly demonstrated, it is also possible that β-catenin drives gene activation independent of LEF/TCF, through its association with the sox proteins. Finally, the activation of the WISP genes by β-catenin is highly dependent upon the presence of a CREB binding site in the WISP promotor (Xu et al. 2000). This implies that cAMP-dependent protein kinase A feeds into wnt signaling and might cooperate with the activation of some wnt target genes. The binding of CBP to β-catenin is particularly relevant with respect to this proposal (Hecht et al. 2000; Takemaru and Moon 2000).
Conclusion
It is apparent that wnt signaling causes cancer and that tumor promotion by this pathway can proceed through a number of different genetic defects. Additional mechanisms by which defects in the regulation of wnt signaling contribute to tumor progression probably remain undiscovered. The manifestation of cancer by aberrant wnt signaling most likely results from inappropriate gene activation mediated by stabilized β-catenin. Target genes need not contain TCF/LEF binding sites in their promotors, though, as new potential modes of gene activation by β-catenin are becoming apparent. Several target genes of β-catenin signaling have now been identified and some of their functions are consistent with control of cellular growth, differentiation, and survival. For an excellent summary of wnt target genes, and a wealth of information on wnt signaling in general, I refer the reader to the Wnt Home Page posted by the Nusse lab (http://www.stanford.edu/rnusse/wntwindow.html).
7.10.2 The Wnt.β-catenin pathway in ovarian cancer : a review.
Arend RC1, Londoño-Joshi AI, Straughn JM Jr, Buchsbaum DJ.
Gynecol Oncol. 2013 Dec; 131(3):772-9.
http://dx.doi.org:/10.1016/j.ygyno.2013.09.034.
Objective: Ovarian cancer is the deadliest gynecologic malignancy and the fifth leading cause of death from cancer in women in the U.S. Since overall survival remains poor, there is a need for new therapeutic paradigms. This paper will review the Wnt/β-catenin pathway as it relates to epithelial ovarian cancer, specifically its role in chemoresistance and its potential role as a target for chemosensitization. Methods: A PubMed search was performed for articles published pertaining to Wnt/β-catenin pathway specific to ovarian cancer. Wnt/β-catenin signaling pathways play an active role in cancer stem cells (CSCs) and carcinogenesis of all ovarian cancer subtypes. Studies also have shown that ovarian CSCs are involved in chemoresistance, metastasis, and tumor recurrence. Results: Wnt/β-catenin target genes regulate cell proliferation and apoptosis, thereby mediating cancer initiation and progression. The Wnt/β-catenin pathway is one of the major signaling pathways thought to be involved in epithelial-to-mesenchymal transition (EMT). Alterations affecting Wnt pathway proteins on the cell membrane, in the cytoplasm, and in the nucleus have been shown to play important roles in the tumorigenesis of ovarian cancer. Conclusions: Wnt signaling is activated in epithelial ovarian cancer. Given the role of the Wnt/β-catenin pathway in carcinogenesis, more pre-clinical studies are warranted to further investigate other Wnt inhibitors in ovarian cancer. The Wnt pathway should also be investigated as a potential target in the development of new drugs for ovarian cancer as a single agent and in combination with chemotherapy or other targeted agents.
Introduction
Ovarian cancer is the deadliest gynecologic malignancy and the fifth leading cause of death from cancer in women in the U.S. In 2013, there will be an estimated 22,240 newly diagnosed cases of ovarian cancer and an estimated 14,030 deaths in the United States [1].A major contributor to the high mortality rate is the fact that 70% of women with ovarian cancer initially present with metastases throughout the peritoneal cavity. Over the last two decades, advances in chemotherapy have improved the overall survival in patients with advanced stage ovarian cancer [2]. These advances include the introduction of taxane/platinum-based chemotherapy, intraperitoneal delivery of chemotherapy,dose-dense chemotherapy, and the availability of novel agents such as bevacizumab [3,4].Since overall survival remains poor, there is a need for new therapeutic paradigms. Further research is needed to understand how molecular pathways contribute to the development of metastasis, recurrence, and resistance of ovarian cancer to chemotherapeutic agents. Studies have shown that ovarian cancer stem cells (CSCs) are also involved in chemoresistance, metastasis, and tumor recurrence [5]. CSCs area subpopulation of cancer cells that possess characteristics associated with normal stem cells and are able to generate tumors through the stem cell processes of self-renewal and differentiation.These cells are proposed to persist in tumors as a distinct population that cause recurrence and metastasis by giving rise to new tumors. Recently, chemoresistance has been reported to be associated with acquiring epithelial to mesenchymal transition (EMT) in ovarian cancer cells [6].CancercellsundergoingEMT are unique in that they have stem-like properties that enable cancer cell dissemination and metastasis formation [7]. Major signaling pathways involved in EMT include TGF-β, Wnt/ β-catenin, Notch, Hedgehog, and others [8]. Endometrioid ovarian carcinomas often harbor mutations in the β-catenin gene, but mutations in the Wnt/β-catenin pathway are rare in serous, clear cell, and mucinous ovarian carcinomas [9]. There is emerging data that suggests that despite not having mutations, the Wnt/β-catenin pathway plays a role in carcinogenesis of all ovarian cancer subtypes [10–12]. It has been suggested that the Wnt/β-catenin target genes can be divided into two groups: a “stemness/proliferation group” that is active early in tumor progression and an “EMT/ dissemination group” that is expressed in late stage tumors. The Wnt/ β-catenin pathway has been shown to be a therapeutic molecular target for CSCs[13].Wnt/β-catenin target genes regulate cell proliferation and apoptosis,thereby mediating cancer initiation and progression [14,15]. Given the role of the Wnt/β-catenin pathway in carcinogenesis, we will review the Wnt/β-catenin pathway as it relates to epithelial ovarian cancer, specifically its role in chemoresistance and its potential role as a target for chemosensitization.
Historical perspective of Wnt signaling in the ovary
In the late 1990s, the importance of the Wnt pathways in the embryonic development of the ovary was established. Wnt-4, a Wnt ligand, demonstrated a critical role in embryonic ovarian development [16]. Wnt-7a was shown to affect sex-specific differentiation of the reproductive tract [17]. In 2002, Ricken et al. reported that components of the Wnt signaling pathways are expressed in the immature rat ovary, and that their expression is localized to specific ovarian compartments [18]. This study reported the expression of three different Wnt transcripts (Wnt-2b, Wnt-5a, Wnt-11) that were common to five ovarian cancer cell lines derived from histologically varied human ovarian carcinomas.These results raised the possibility that aberrant Wnt expression may be involved in ovarian tumorigenesis in humans. Prior to this study, alterations in Wnt expression had been described in a variety of female human tumors, including breast and endometrial cancer, but this was the first study to suggest its involvement in ovarian cancer. When β-catenin gene mutations were initially discovered in ovarian cancer, they were thought to be limited to the endometrioid subtype [19]. A study by Wu et al. carried out a comprehensive molecular analysis of 45 tumor specimens of primary ovarian endometrioid adenocarcinomas and two ovarian endometrioid adenocarcinomaderived cell lines. They found Wnt/β-catenin pathway defects in both the cell lines and in nearly half of the primary ovarian endometrioid adenocarcinomas analyzed. β-catenin deregulation was most often attributable to a mutation of the β-catenin gene (CTNNB1) itself, although less frequently β-catenin deregulation may have resulted from inactive mutations in the APC, AXIN1, orAXIN2 genes [20]. Depending on the study, a wide range (3–59%) of serous ovarian cancers have also been reported to stain positive for cytoplasmic and nuclear β-catenin by immunohistochemistry even in the absence of mutations in APC, Axin or β-catenin, which are more common in the endometrioid subtype [21–23]. More recent data have shown that although gene mutations in the Wnt/β-catenin pathway are relatively uncommon in ovarian cancer in general, especially in serous ovarian cancer,components of the pathway are still important in the molecular events that lead to ovarian cancer development [12]. There are three main Wnt signaling pathways: the canonical Wnt/β-catenin pathway, the non-canonical planar cell polarity pathway, and the non-canonical Wnt–Ca2+ pathway. These pathways belong to one of two categories: canonical or non-canonical. The difference between these two categories is the presence or absence of β-catenin. The canonical Wnt/β-catenin pathway involves this protein and the non-canonical pathway operates independently of it.
Components of the Wnt signaling pathway
Non-canonical Wnt signaling pathways
Wnt proteins, which serve as ligands for the Wnt pathway, consist of 19 cysteine-rich glycoproteins. They bind to the Frizzled (Fzd) transmembrane receptor, one of the main receptors of the Wnt pathways [24]. When Wnt binds to Fzd, it can activate one of the three distinct intracellular signaling pathways. While the canonical Wnt/β-catenin signaling pathway leads to the accumulation and stabilization of cytosolic, unphosphorylated (“free”) β-catenin, the non-canonical pathways promote an increase in intracellular calcium or mediate cell polarity. In all three pathways, a Wnt ligand binds to Fzd receptor and promotes recruitment of Dishevelled (Dsh) protein (Figs. 1 and 2). In the planar cell polarity non-canonical pathway, this complex binds to the Dsh-associated activator of morphogenesis (Daam1). This cascade of events leads to the activation of Rac and RhoA GTPases which mediate cell polarity (Fig. 1). In the Wnt-Ca2+ noncanonical pathway, the Wnt/Fzd/Dsh complex binds with a G protein (Ror 1/2) as shown in Fig. 1, which leads to activation of calmodulindependent kinase II, protein kinase C and the phosphatase calcineurin. This binding promotes the increase in intracellular calcium levels which then mediates other signaling pathways. The Wnt pathways are critical to embryonic development of a variety of organs including the ovaries. Activation of Wnt signaling occurs via the canonical Wnt/β-catenin pathway and the non-canonical cell polarity pathway and the Wnt/ Ca2+ pathway; however, as it relates to oncology research the Wnt/β-catenin canonical pathwayis the mostrelevant [25].
Canonical (Wnt/β-catenin) signaling pathway
In the canonicalWnt/β-catenin pathway, the pathway is “off” when either there is no Wnt ligand, no receptor, or the receptor is being blocked (Fig. 2A). Dikkopf family (DKK1–4) binds directly to one of the transmembrane receptors (Fzd, LRP5/6) and blocks Wnt from binding. Wnt-inhibitory factor (WIF-1) and the family of secreted Fzd receptor proteins (SFRP1-5) bind to Wnt itself and prevent them from binding to the receptors. If the Wnt ligand does not bind to the receptors, β-catenin is degraded by a destruction complex that is comprised of Axin, adenomatous polyposis coli (APC), and glycogen synthase kinase 3β (GSK3β). β-Catenin is phosphorylated by the kinases casein kinase 1 (CK1) and GSK3β, followed by ubiquitination and proteasomal degradation by the 26S proteasome. Low cytoplasmic levels of β-catenin allow for the recruitment of the corepressor Groucho to lymphoid enhanced factor–T-cell factor (TCF/LEF) transcription factors,which blocks the target genes from being activated and ensures transcriptional repression (Fig. 2A). Activation of the canonical Wnt pathway involves the stabilization of β-catenin through the binding of Wn tligands to cell surface receptors including Fzd family receptors and low-density lipoprotein receptor (LDLR)-related proteins: LRP5 and LRP6. When the Wnt pathway is “on”, cytosolic β-catenin is stabilized (Fig. 2B). LRP6/LRP5 is phosphorylated by the kinases CK1 and GSK3β. Dsh molecules are recruited to the plasma membrane to interact with Fzd. Interaction of Axin with phosphorylated LRP6/LRP5 and Dsh leads to inactivation of the destruction complex and degradation of β-catenin is inhibited. βCatenin accumulates in the cytoplasm and enters the nucleus and activates Wnt target genes by binding to the transcriptional factors of the TCF/LEF family by displacing Groucho and interacting with coactivators such as B-cell lymphoma 9/Legles (BCL9/LGS) and Pygopus (Pygo) to promote transcription of target genes [26]. TCF/LEF, BCL9/ LGS, and Pygo all bind with β-catenin in the nucleus to form a transcriptional activation complex (Fig. 2B). β-Catenin promotes transcription of genes related to proliferation and survival. Some of the key downstream proteins and genes that are activated with the binding of β-catenin to the transcriptional factors of the canonical pathway include c-MYC (MYC), Cyclin D1 (CCND1), Survivin (BIRC5), Axin2 (AXIN2), and matrix metalloproteinases (MMPs). There have been over 100 target genes identified as regulated by the Wnt pathway and 23 of them have been shown to be overexpressed in ovarian cancer [27].
Regulation of the Wnt pathway
The remainder of the review will focus on the canonical Wnt/ β-catenin pathway, because the Wnt/β-catenin pathway has been the most well described in the literature as it relates to cancer research and specifically ovarian cancer. It is regulated at multiple levels: gene mutations, extracellular inhibitors, and intranuclear transcription cofactors. These all contribute to the diverse mechanisms that are involved in the Wnt pathway.When there is no Wnt ligand, a destruction complex regulates β-catenin levels. Specifically, CK1 and unphosphorylated GSK3β phosphorylate β-catenin and target the protein for ubiquitination and proteasomal degradation. Phosphorylation of GSK3β by protein kinases (A, B, and C), Akt/PI3K, and MAPK inhibits its ability to phosphorylate and target β-catenin for degradation. The majority of ovarian cancers have an activation of PI3K (phosphoinositide 3-kinase) by gene amplification, which can potentially phosphorylate GSK3β, impeding the phosphorylation of β-catenin and resulting in cellular differentiation, division, and survival [28,29].
Alterations of the Wnt pathway in ovarian cancer
Membranous factors
The first event in the activation of the Wnt pathway is the binding of a Wnt ligand to Fzd and LRP6/LRP5. Two subtypes of the Fzd receptor are increased in epithelial ovarian cancer, Fzd1 and Fzd5. A higher number of malignant ovarian specimens stained positive for both receptors than normal ovary and the Fzd5-positive tumors had a worse 6-year survival than those that were Fzd5-negative [30]. During metastatic spread of epithelial ovarian cancer, there is adhesion of cancer cells to submesothelial interstitial collagens. When β1 integrin mediated anchoring to the mesothelium and submesothelial matrix occurs, it facilitates the formation of metastatic tumor sites on other peritoneal organs. The engagement of collagen-binding β1 integrins have been shown to upregulate LRP6, WNT5A, MMP9, PTGS2 (COX2), PLAUR (uPAR), VIM (vimentin), SNAII (Snail) at the mRNA level [31]. This suggests tha tmetastatic spread of ovarian cancer is likely facilitated by the upregulation of LRP6 and targeting LRP6 may be an effective strategy for treating ovarian cancer.
There are several proteins that act as antagonists to the Wnt pathway. These proteins include: the Dikkopf family (DKK1–4), Wnt inhibitory factor (WIF-1) and the family of secreted Fzd receptor proteins (SFRP1-5)(Fig.2A). SFRPs bind directly to the Wnt ligand or Fzd receptor and inhibit Wnt from binding to Fzd and activating the pathway. Loss of SFRP4 expression correlates with a more aggressive ovarian cancer phenotype and the level of SFRP4 is directly related to prognosis [32]. Investigators have studied the re-expression of SFRP4 in epithelial ovarian cancer cell lines, and found that re-expression inhibited the Wnt/ β-catenin signaling pathway, thereby inhibiting cell migration and EMT. These proteins provide important potential therapeutic targets by either re-expression, if their expression is lost,or potentially upregulated.
Cytoplasmic and nuclear factors
Endometrioid ovarian carcinomas often have mutations in the βcatenin gene. Table 1 summarizes the studies that show β-catenin mutations in human ovarian cancer, from 16% to 54% in endometrioid cancers and 14% in mucinous cancers. Despite no reported mutations in the CTNNB1 gene in serous and clear cell cancers, nuclear β-catenin has been observed in serous and clear cell ovarian cancer [21]. Lee et al. showed a statistically significant correlation between nuclear β-catenin expression and high-grade serous ovarian cancer [23]. The protooncogene, frequently rearranged in advanced T-cell lymphomas-1 (FRAT1), which inhibits phosphorylation of β-catenin, was found to be overexpressed in serous ovarian cancer and was strongly correlated with the accumulation of cytoplasmic β-catenin, leading to an increase in nuclear β-catenin [21]. Pygo, oneof the co-activators that binds to β-catenin is a necessary component of tumor cell growth and is widely expressed in ovarian cancer, both in cell lines and in primary tumor tissue [33]. RNA expression of BCL9/LGS, also a co-activator,is common in both epithelial ovarian cancer and normal ovaries. Upregulation of these co-activators is further evidence that the Wnt pathway plays a pivotal role in the tumorigenesis of ovarian cancer.
Intercellular interactions
Cells undergoing EMT are known to lose E-cadherin and gain vimentin expression, resulting in tumor cell invasion and metastasis [34]. Epithelial ovarian cancer cells also undergo a mesenchymal to epithelial transition (MET) because the normal ovarian surface epithelium is mesenchymally derived. This dynamic process has been termed EMP (epithelial to mesenchymal plasticity). It is thought that both transitions are equally important for metastasis formation and that the “metastable” state is actually when the cells transition between the two states [34]. Metastatic epithelial ovarian cancer cells adhere to the interstitial collagen of the peritoneal cavity via integrins. Cell–matrix and cell– cell adhesions are paramount to this process and are mediated by integrins and E-cadherins. Integrin engagement has been linked to increased internalization of E-cadherin [31]. In epithelial cancer, the MET component dominates, unlike other epithelial cell-derived cancers where the EMT component dominates; therefore, E-cadherin expression is increased with malignant transformation in ovarian cancer [31]. E-cadherin-based adherens junctions are stabilized by β-catenin, and the loss of stability in the junctions may cause an increase in cytoplasmic and/or nuclear β-catenin. Integrins have also been suggested to inhibit GSK3β, elevate levels of nuclear β-catenin, and increase β-catenin-regulated promoter activation. Burkhalter et al.
showed that an inhibitor of β-catenin and TCF-4, a member of the TCF/LEF transcription factor family, reduced cellular invasion [31]. Most of the regulation of the Wnt pathway ultimately leads to an accumulation or depletion of β-catenin in the nucleus, or affects the binding of nuclear β-catenin to TCF/LEF, which determines whether apoptosis can occur. It is important to note that the transcriptional regulatory activity of β-catenin is also controlled by factors other than Wnt signaling. One example of Wnt-independent regulation of β-catenin is through E-cadherin expression, which selectively depletes the transcriptionally active pool of β-catenin [35]. This is especially significant as epithelial ovarian cancer cells are known to undergo MET which causes an increase in E-cadherin.
Extracellular factors
Not only have membranous and intercellular components of theWnt pathway been found to be upregulated in epithelial ovarian cancer, but extracellular activators also are upregulated. These factors specifically include Wnt-1,Wnt-2b,Wnt-5a, and Wnt-11 [30]. Ricken et al. reported the possibility that Wnt-5a could be involved in ovarian carcinogenesis [18]. This study used RT-PCR on RNA from five ovarian cancer cell lines and confirmed the expression of transcripts for Wnt-2b, Wnt-5a and Wnt-11. Filho et al. showed that upregulation of Wnt-1 and Wnt-5a, detected by immunohistochemistry in patient samples, portended a significantly lower survival than ovarian cancer patient samples that did not have an upregulation of Wnt-1 and Wnt-5a [30].
Gene expression
Kumar et al. analyzed 1500 miRNAs to identify which ones were potentially different between A2780 (parental ovarian cancer cell line) and A2780.cp70 (cisplatin resistant cell line) and found changes in 11 miRNAs [36]. The microRNA data was validated by quantitative realtime PCR for these 11 miRNAs. Ingenuity Pathway Analysis (IPA) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were performed for the 11 miRNAs and their targets to identify the pathways involved in cisplatin resistance. Not only was Wnt signaling one of the pathways identified, but so were MAPK and mTOR signaling pathways which both cross-talk with the Wnt pathway by causing the phosphorylation of GSK3β, blocking its ability to phosphorylate βcatenin to allow it to be ubiquitinated. Four gene expression datasets: Moffitt Cancer Center (MCC), Total Cancer Care (TCC), the Cancer Genome Atlas(TCGA),andMDAnderson (MDA) were analyzed, and only four pathways were noted to be differentially expressed between normal ovarian surface epithelium and ovarian cancer. One of these pathways is the “Cytoskeleton remodeling/TGF–Wnt pathway” [37]. The“Cytoskeleton remodeling/TGF/WNT” pathway was previously described as a common pathway created by the crosstalk between the TGF-β pathway and the Wnt pathway that is involved in cytoskeleton remodeling: cell–cell adhesion and cell–matrix adhesion [38]. This pathway has been associated with metastasis in various cancer types and is critical for cancer cell migration and invasion. The same group at H. Lee Mof fitt Cancer Center found that six common molecular signaling pathways were associated with chemoresistance and survival in ovarian cancer that included the TGF– Wnt pathway and specifically Wnt pathway activated by Wnt-2, one of the 19 Wnt ligands [39]. In addition, this group also used the same novel computer analysis technique to identify genes and molecular signaling pathways associated with cancer cell proliferation. Genes and pathways associated with cancer cell proliferation and survival were analyzed against the NCI 60 cell line-drug screening database to identify agents predicted to have pathway- and gene-specific activity. They identified 81 existing agents that could potentially be repurposed to target the TGF-Wnt pathway that are currently the focus of in vitro functional analyses [40].
Non-canonical pathways
Fig. 1. Non-canonical Wnt signaling pathways. In the planar cell polarity pathway Wnt–Frizzled complex binds to the Dsh-associated activator of morphogenesis (Daam1). This cascade of events leads to the activation of Rac and RhoA GTPases which mediate cell polarity. In the Wnt–Ca2+ pathway, the Wnt/Fzd/Dsh complex binds with a G protein, which leads to activation of calmodulin-dependent kinase II (CaMKII), protein kinase C (PKC), and the phosphatase calcineurin. This binding promotes the increase in intracellular calcium levels which stimulates other signaling pathways.
Fig.2.The canonical Wnt signaling pathway. (A)In the absence of Wnt ligand, β-catenin is degraded through interactions with Axin, APC and GSK3β “destruction complex”. β-Cateninis phosphorylated by the kinases CK1 (casein kinase 1) and GSK3β (glycogen synthase kinase 3β), followed by ubiquitylation and proteasomal degradation. Low cytoplasmic levels of βcatenin allow for the recruitment of the corepressor Groucho to LEF (lymphoid enhanced factor)–TCF (T-cell factor) transcription factors which ensures transcriptional repression. Dikkopf (DKK) family proteins, the Wnt-inhibitory factor (WIF), and the family of secreted Frizzled receptor proteins (SFRPs) all act as antagonists to the Wnt pathway. SFRP binds directly to the Wnt ligand or th eFrizzled receptor to inhibit Wnt binding to Frizzled. (B) In the presence of Wnt ligands, Wnt proteins bind to Frizzled/LRP6/LRP5 receptor complex at the cell surface. LRP6/LRP5 is phosphorylated by the kinases casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β). Dishevelled (Dsh) molecules are recruited to the plasma membrane to interact with Frizzled. Interaction of Axin with phosphorylated LRP6/LRP5 and Dsh leads to inactivation of the destruction complex. Degradation of β-catenin is inhibited. β-Catenin accumulates inthe cytoplasm and nucleus. β-Catenin forms a transcriptionally active complex with TCF/LEF by displacing Groucho and interacting with co-activators suchasBCL9/LGS (B-cell lymphoma 9/Legless) and Pygo (Pygopus) to promote transcription of target genes (Axin, CyclinD1, Survivin). β-Catenin is also a coactivator of CREB binding protein (CBP) which is the binding protein of the cAMP response element-binding protein (CREB). β-Catenin/CBP binds to Wnt-responsive element (WRE) and activates transcription. This leads to cell proliferation, survival, and self-renewal.
Potential therapeutic targets of the Wnt pathway in ovarian cancer
Identification of the specific membranous, intracellular, and extracellular components of the Wnt pathway gives insight to potential targets for therapy. There currently are several small molecules that have recently entered into phase I clinical trials that target the Wnt pathway (Table 2). In order for the Wnt protein to be secreted by the cell to act as a ligand it must first undergo fatty acyl modification. Once it undergoes palmiteolyation it is shepherded through the secretory pathway by Wntless chaperone protein. PORCN is the founding member of a 16-gene family with acyltransferase activity and Porcupine (Porcn) is the acyltransferase enzyme that adds the fatty acid to Wnt which is a crucial step in the secretion of the Wnt ligand. Without Porcn to catalyze this modification, the Wnt protein remains trapped inside the cell. Currently being studied in a phase 1 trial is the small molecule, LGK974 (Novartis Pharmaceuticals) that inhibits Porcn(NCT01352203) [41]. Drugs that specifically target the Wnt signaling pathway in the nucleus include the small molecule inhibitor, PRI-724, which specifically blocks the recruitment of β-catenin with its coactivator CBP which is the binding protein of the cAMP response element-binding protein CREB. βCatenin/CBP binds to Wnt-responsive element (WRE) and activates transcription; therefore, PRI-724 prevents activated transcription by aberrant Wnt signaling. This drug is being studied in solid tumors and myeloid malignancies (NCT01606579) [41]. Other pathways may cross-talk with the Wnt pathway. In Wnt signaling, Axin is a key scaffolding protein of the destruction complex of β-catenin, and Poly (ADP ribose) polymerases (PARPs) promote the ribosylation of Axin, thereby causing it to become degraded and no longer facilitate β-catenin destruction. If PARP is inhibited, Axin is stabilized, which allows it to degrade β-catenin [42]. There are several PARP inhibitors that are currently being used in clinical trials for ovarian cancer. In addition, preclinical studies have been carried out with XAV939, which is a small-molecule PARP inhibitor that targets tankyrases, a specific type of PARP. Huang et al. used a chemical genetic screen to identify the small molecule, XAV939, which selectively inhibits β-catenin mediated transcription. XAV939 was shown to stimulate β-catenin degradation by stabilizing Axin. They used a quantitative chemical proteomic approach to show that XAV939 stabilizes Axin by inhibiting tankyrase1 and tankyrase2.They showed that both tankyrase isoforms 1 and 2 stimulate Axin degradation through the ubiquitin–proteasome pathway [43]. JW55 (Tocris Bioscience) is a selective tankyrase 1 and 2 inhibitor which has been shown to inhibit the growth of cancer. JW55 inhibits the canonical Wnt signaling pathway in colon carcinoma cells that contained mutations either in the APC locus or in anallele of β-catenin [44]. Frizzled, oneof themembrane receptors that activates thepathway upon Wnt ligand binding, has been reported to be overexpressed in ovarian cancer. There are two drugs that specifically target the Fzd receptorthatarebeingevaluatedinclinicaltrials.OMP-18R5(OncoMed Pharmaceuticals/Bayer) is one of the Wnt-targeted compounds that is in clinical development (NCT01345201) [41]. It is a monoclonal antibody that targets Fzd receptors and blocks their association with Wnt ligands. This drug is being used in combination with the standard chemotherapy for breast, lung, pancreas, and colon cancer. Another drug, OMP-54F28, binds to and sequesters the Wnt ligand and is a fusion protein of the Fzd8 ligand-binding domain with the Fc region of a human immunoglobulin (OncoMed Pharmaceuticals/Bayer) (NCT01352203) [41]. There has been a growing trend in oncology to evaluate“repurposed” drugs which are drugs that have been used in the past for other purposes and are now being screened for their function as anticancer drugs. Several drugs have been shown to work through the Wnt pathway including the FDA-approved anti-helminth compound, niclosamide, non-steroidalanti-inflammatory drugs(NSAIDS), and two antipsychotic drugs: lithium and valproic acid. NSAIDS have been shown to cause degradation of TCF and inhibit Wnt target genes such as COX2. Although they do not target the Wnt pathway directly, they could be a potential anti-Wnt agent. Niclosamide inhibitsWnt/β-catenin pathway activation. In colorectal cancer, it was shown to downregulate Dvl2, a member of the Dsh protein family, which in turn decreased downstreamβ-catenin signaling [45]. Recently, niclosamide has been reported to target not only Wnt/β-catenin but also other signaling pathways involved in CSC maintenance such as NF-κB, Notch, ROS, mTORC1, and Stat3 [46,47]. Niclosamide has also been reported to inhibit Wnt/β-catenin signaling by inducing degradation of the Wnt surface receptor, LRP6 [48]. Our laboratory has seen an increase expression of LRP6 in ovarian cancer patients. Yo et al. identified a subset of chemoresistant ovarian tumor cells that fulfilled the definition of CSCs and subjected these cells to high-throughput drug screening using more than 1200 clinically approved drugs. Sixty-one potential compounds were identified on preliminary screening and after more stringent screening, niclosamide was found to be the best drug to selectively target ovarian CSCs both in vitro and in vivo [49].
Wnt/β-catenin pathway and CSC
TheWnt/β-catenin pathway is an important pathway in cell survival and has been implicated in the mechanism of chemoresistance of ovarian CSCs. CSCs are a subpopulation of tumor cells that possess characteristics associated with normal stem cells and have the ability to self-renew and differentiate. Wnt/β-catenin signaling plays an important role in the transcription of multidrug resistance genes such as ABCB1/MDR-1 [50]. Chemoresistance, which can be a result of the inhibition of apoptosis, has been reported to be associated with acquiring EMT in ovarian cancer cells [51,52]. Ovarian cancer cells undergoing EMT have stem-like properties that enable cancer cell dissemination and metastasis formation. A recent study done at Georgia Institute of Technology confirmed that metastasizing ovarian cancer cells taken from patients have a different molecular structure from primary tumor cells and display genetic signatures consistent with EMT [53]. The Wnt/ β-catenin pathway is one of the major signaling pathways thought to be involved in EMT and thus has been shown to play an integral role in metastasis.
Conclusions
Alterations affectingWnt pathway proteins on the cel lmembrane, in the cytoplasm, and in the nucleus have been shown to play important roles in the tumorigenesis of ovarian cancer. Pre-clinical studies have shown an upregulation of 5 of the 19 known Wnt ligands in ovarian cancer, which leads to increased activity of the Wnt pathway. Fzd is one of the membrane receptors that activates the pathway upon Wnt ligand binding. It has been reported to be overexpressed in ovarian cancer. Our laboratory has also seen an upregulation of LRP6 detected by immunohistochemistry (unpublished data). In ovarian cancer, an increase in nuclear β-catenin has been shown to be the result of an upregulation in the β-catenin gene itself and also mutations in the proteins necessary to degrade cytoplasmic β-catenin such as Axin2 and APC. The β-catenin destruction complex consists of Axin2, APC, and GSK3β, which must not be phosphorylated in order to cause βcatenin degradation. GSK3β is frequently phosphorylated in ovarian cancer through other pathways, such as PI3K, inhibiting its ability to degrade β-catenin. Upregulation of co-activators of β-catenin also contributes to the increase in transcription of the target genes. As many as 23 different target genes that lead to cell proliferation and survival, which is a result of nuclear β-catenin build-up, have been shown to be overexpressed in ovariancancer. Wntsignalingis activated in epithelial ovarian cancer, both directly through ligand activated upregulation of the pathway and through a ligand independent increase in nuclear β-catenin through cross-talk with other pathways. Recently, Yo et al. reported that niclosamide, which has been shown to have anti-Wnt activity inhibits growth in ovariantumor-initiatingcells[49].Morepre-clinicalstudies,specifically animal studies and mechanistic studies, are warranted to further investigate other Wnt inhibitors in ovarian cancer. The Wnt pathway is very complex, and further studies with targeted agents need to be done to see if inhibition of a single component of the pathway will be clinically useful. This paper supports the fact that the Wnt pathway shows promise as an effective target for anti-cancer therapy in ovarian cancer. As more efficacy data is collected from the phase 1 studies with Wnt inhibitors LGK974, OMP-54F28, OMP-18R5, and PRI724: NCT01352203, NCT01608867, NCT01345201, and NCT01606579 (www.clinicaltrials.gov), they should be considered as potential agents in the treatment of ovarian cancer. Given the fact that the Wnt pathway is involved in so many biological pathways, results from these studies will be important to determine if effective Wnt pathway inhibition will be excessively toxic to patients. Future directions for investigating the Wnt pathway in ovarian cancer should include genetic sequencing of ovarian cancer patients with the aim of targeting those patients who specifically have upregulation of Wnt pathway target genes. More quantitative data is needed to specifically look at the mechanisms of these drugs in patients by performing qPCR on tissue obtained before and after treatment. The Wnt pathway should be investigated as a potential target in the development of new drugs for ovarian cancer as a single agent and in combination with chemotherapy or other targeted agents.
References
[1] Morton BA, Beatty BG, Mison AP, Wanek PM, Beatty JD. Role of bone marrow transplantation in 90Y antibody therapy of colon cancer xenografts in nude mice. Cancer Res Suppl 1990;50:1008s–10s. [2] OzolsRF,BundyBN,GreerBE,FowlerJM,Clarke-PearsonD,BurgerRA,etal.PhaseIII trialofcarboplatinand paclitaxelcompared with cisplatinand paclitaxel inpatients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 2003;21:3194–200. [3] Garcia AA, Hirte H,Fleming G,YangD, Tsao-WeiDD, RomanL,etal. Phase IIclinical trialofbevacizumabandlow-dosemetronomicoralcyclophosphamideinrecurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J Clin Oncol 2008;26:76–82. [4] Penson RT, Dizon DS, Cannistra SA, Roche MR, Krasner CN, Berlin ST, et al. Phase II study of carboplatin,paclitaxel, andbevacizumab with maintenance bevacizumab as first-line chemotherapy for advanced mulleriantumors.J ClinOncol2010;28:154–9. [5] Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res 2012;18:869–81. [6] RicciF,BernasconiS,PeregoP,GanzinelliM,RussoG,BonoF,etal.Ovariancarcinoma tumor-initiatingcellshaveamesenchymalphenotype.CellCycle2012;11:1966–76. [7] Raimondi C, Gianni W, Cortesi E, Gazzaniga P. Cancer stem cells and epithelial– mesenchymal transition: revisiting minimal residual disease. Curr Cancer Drug Targets 2010;10:496–508.
[8] Talbot LJ, Bhattacharya SD, Kuo PC. Epithelial–mesenchymal transition, the tumor microenvironment,andmetastaticbehaviorofepithelialmalignancies.IntJBiochem Mol Biol 2012;3:117–36. [9] DubeauL.Thecelloforiginofovarianepithelialtumours.LancetOncol2008;9:1191–7. [10] Boyer A, Goff AK, Boerboom D. WNT signaling in ovarian follicle biology and tumorigenesis. Trends Endocrinol Metab 2010;21:25–32. [11] Rask K, Nilsson A, Brannstrom M, Carlsson P, Hellberg P, Janson PO, et al. Wntsignalling pathway in ovarian epithelial tumours: increased expression of betacatenin and GSK3beta. Br J Cancer 2003;89:1298–304. [12] Gatcliffe TA, Monk BJ, Planutis K, Holcombe RF. Wnt signaling in ovarian tumorigenesis. Int J Gynecol Cancer 2008;18:954–62. [13] Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011;8:97–106. [14] Verras M, Sun Z. Roles and regulation ofWnt signaling and beta-catenin inprostate cancer. Cancer Lett 2006;237:22–32. [15] Wend P, Holland JD, Ziebold U, Birchmeier W. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol 2010;21:855–63. [16] Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP. Female development in mammals is regulated by Wnt-4 signalling. Nature 1999;397:405–9. [17] Parr BA, McMahon AP. Sexually dimorphic development of the mammalian reproductive tract requires Wnt-7a. Nature 1998;395:707–10. [18] Ricken A, Lochhead P, Kontogiannea M, Farookhi R. Wnt signaling in the ovary: identification and compartmentalized expression of wnt-2, wnt-2b, and frizzled-4 mRNAs. Endocrinology 2002;143:2741–9. [19] Saegusa M, Okayasu I. Frequent nuclear beta-catenin accumulation and associated mutations in endometrioid-type endometrial and ovarian carcinomas with squamous differentiation. J Pathol 2001;194:59–67. [20] Wu R, Zhai Y, Fearon ER, Cho KR. Diverse mechanisms of beta-catenin deregulation in ovarian endometrioid adenocarcinomas. Cancer Res 2001;61:8247–55. [21] WangY,HewittSM,LiuS,ZhouX,ZhuH,ZhouC,etal.Tissuemicroarrayanalysisof human FRAT1 expression and its correlation with the subcellular localisation of beta-catenin in ovarian tumours. Br J Cancer 2006;94:686–91. [22] Karbova E, Davidson B, Metodiev K, Trope CG, Nesland JM. Adenomatous polyposis coli (APC) protein expression in primary and metastatic serous ovarian carcinoma. Int J Surg Pathol 2002;10:175–80. [23] LeeCM,ShvartsmanH,DeaversMT,WangSC,XiaW,SchmandtR,etal.Beta-catenin nuclear localization is associated with grade in ovarian serous carcinoma. Gynecol Oncol 2003;88:363–8. [24] Gonzalez-SanchoJM,BrennanKR,Castelo-SoccioLA,BrownAM.Wntproteinsinduce dishevelled phosphorylation via an LRP5/6-independent mechanism, irrespective of theirabilityto stabilize beta-catenin. MolCellBiol 2004;24:4757–68. [25] HarrisPJ,SperanzaG,DanskyUllmannC.Targetingembryonicsignalingpathwaysin cancer therapy. Expert Opin Ther Targets 2012;16:131–45. [26] Kikuchi A, Yamamoto H, Kishida S. Multiplicity of the interactions of Wnt proteins and their receptors. Cell Signal 2007;19:659–71. [27] Barbolina MV, Burkhalter RJ, Stack MS. Diverse mechanisms for activation of Wnt signalling in the ovarian tumour microenvironment. Biochem J 2011;437:1–12. [28] Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 2005;5:921–9. [29] Bast Jr RC, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer 2009;9:415–28. [30] Badiglian Filho L, Oshima CT, De Oliveira Lima F, De Oliveira Costa H, De Sousa DamiaoR,GomesTS,etal.CanonicalandnoncanonicalWntpathway:acomparison among normal ovary, benign ovarian tumor and ovarian cancer. Oncol Rep 2009;21:313–20. [31] Burkhalter RJ, Symowicz J, Hudson LG, Gottardi CJ, Stack MS. Integrin regulation of beta-catenin signaling in ovarian carcinoma. J Biol Chem 2011;286:23467–75. [32] Jacob F, Ukegjini K, Nixdorf S, Ford CE, Olivier J, Caduff R, et al. Loss of secreted frizzled-related protein 4 correlates with an aggressive phenotype and predicts poor outcome in ovarian cancer patients. PLoS One 2012;7:e31885. [33] Popadiuk CM, Xiong J, Wells MG, Andrews PG, Dankwa K, Hirasawa K, et al. Antisense suppression of pygopus2 results in growth arrest of epithelial ovarian cancer. Clin Cancer Res 2006;12:2216–23. [34] Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 2006;66:8319–26. [35] Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol 2001;153:1049–60. [36] Kumar S, Kumar A, Shah PP, Rai SN, Panguluri SK, Kakar SS. MicroRNA signature of cis-platin resistant vs. cis-platin sensitive ovarian cancer cell lines. J Ovarian Res 2011;4:17. [37] Al Sawah DM E, Xiong Y, Ramirez-Diaz I, Abbasi F, Bou Zgheib N, Stickles X, et al. A novel strategy to identify ovarian cancer molecular signaling pathways and drug repurposing candidates. Abstract supplement from the Society of Gynecologic Oncology 44th Annual Meeting on Women’s Cancer; 2013. p. S7. [38] FloresRJ,LiY,YuA,ShenJ,RaoPH,LauSS,etal.Asystemsbiologyapproachreveals common metastatic pathways in osteosarcoma. BMC Syst Biol 2012;6:50. [39] Marchion IR-D D, Xiong Y, Al Sawah E, Abbasi F, Bou Zgheib N, Stickles X, et al. An innovativeinsilicomethodtoidentifyagentsthattargetpathwaysofhumancancer chemoresistance. Abstract supplement from the Society of Gynecologic Oncology 44th Annual Meeting on Women’s Cancer; 2013. p. S151. [40] RamirezDMI,XiongY,AlSawahE,BouZgheibN,BoacB,SticklesX,etal.Identifying drug repurposing opportunities to target genes and molecular pathways associated with cancer cell proliferation. Abstract supplement from the Society of Gynecologic Oncology 44th Annual Meeting on Women’s Cancer; 2013. p. S152.
7.10.3 Wnt Signaling in the Niche Enforces Hematopoietic Stem Cell Quiescence and Is Necessary to Preserve Self-Renewal In Vivo
Fleming HE1, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM, Scadden DT.
Cell Stem Cell. 2008 Mar 6; 2(3):274-83
http://dx.doi.org/10.1016%2Fj.stem.2008.01.003
Wingless (Wnt) is a potent morphogen demonstrated in multiple cell lineages to promote the expansion and maintenance of stem and progenitor cell populations. Pharmacologic modification of Wnt signaling has been shown to increase hematopoietic stem cells (HSC). We explored the impact of Wnt signaling in vivo, specifically within the context of the HSC niche. Using an osteoblast-specific promoter to drive the expression of a pan-inhibitor of canonical Wnt signaling, Dickkopf1 (Dkk1), we noted changes in trabecular bone and in HSC. Wnt signaling was inhibited in HSC and the cells exhibited reduced p21Cip1 expression, increased cell cycling and a progressive decline in regenerative function after transplantation. This effect was microenvironment-determined, but irreversible if the cells were transferred to a normal host. Wnt pathway activation in the niche is required to preserve the reconstituting function of endogenous hematopoietic stem cells.
The regulation of hematopoietic stem cell function is a complex and balanced process that requires coordinated input from inherent HSC programs and moderating signals provided by the surrounding microenvironment. Together, these signals permit the maintenance of the stem cell pool for the life of the organism, while also allowing for sufficient steady-state and injury-responsive blood cell production. These somewhat dichotomous aspects of HSC function require mechanisms that both preserve a quiescent population of stem cells and also promote their activation, expansion, differentiation and circulation under appropriate conditions (Akala and Clarke, 2006; Scadden, 2006). The morphogen family of signaling molecules has been identified as a prominent player in the function of numerous stem cell types, including the hematopoietic lineage. The wingless (Wnt) pathway has been studied extensively in the context of hematopoiesis, and the combined impact of multiple family members binding to a range of receptors leads to activation of canonical and non-canonical signaling pathways (Nemeth and Bodine, 2007). Canonical signals are mediated by TCF/LEF transcription factor activity (Daniels and Weis, 2005), and are considered to be largely dependent on the accumulation of nuclear β- (and/or γ-) catenin (Nemeth and Bodine, 2007).Wnt signals have been implicated in mammalian hematopoiesis by studies not intended to assess normal physiology in which Wnt activation had a strong expansive effect on reconstituting HSCs and multipotent progenitors (Baba et al, 2006; Murdoch et al, 2003; Reya et al, 2003; Trowbridge et al, 2006). With enforced, persistent Wnt activation, however, engineered mice developed hematopoietic failure with impaired differentiation of HSC (Kirstetter et al, 2006; Scheller et al, 2006). In contrast, deletion of members of the Wnt / β-catenin cascade under homeostatic conditions had little to no effect on blood cell production by HSCs (Cobas et al, 2004; Jeannet et al, 2007; Koch et al, 2007), raising the question of what physiological role, if any, Wnt signaling has on this cell type. Some of the variation observed may reflect differing influences exerted by canonical versus non-canonical Wnt signals, particularly given a recent report indicating that Wnt5a can modulate canonical signals mediated by Wnt3a (Nemeth et al, 2007). Wnt signals are also regulated by a host of soluble inhibitors that may interact directly with Wnt ligands, such as the frizzled-related proteins (sFRP) or by preventing Wnt binding to its receptors (Kawano and Kypta, 2003). The Dickkopf (Dkk) family of Wnt inhibitors falls into this latter category, by binding the Wnt coreceptor LRP5/6 in combination with a Kremen receptor, and leading to internalization of the complex (Mao et al, 2001; Mao et al, 2002). In order to specifically examine the impact of Wnt activation in an in vivomicroenvironment that has been shown to regulate HSC number and function, we utilized mice engineered to overexpress the Wnt inhibitor, Dkk1, under control of the osteoblast specific 2.3kb fraction of the collagen1α promoter. This promoter has been previously shown to direct transgene expression to osteoblastic cells, resulting in changes in the number and function of HSCs (Calvi et al, 2001; Calvi et al, 2003)
We noted very little overt phenotype in the hematopoietic compartment of the Dkk1 tg mice at steady-state, and confirmed that transgene expression did not extend to the primitive hematopoietic fraction itself. Clear alterations of bone morphology were observed, however, including a 20% decrease in trabecular bone (manuscript in preparation). Despite the absence of a steady-state hematopoetic phenotype, TCF/LEF activity was specifically reduced within the HSC-containing fraction of Dkk1 transgenic mice, and stem cell function was altered under specific conditions. For example, a highly significant defect in the maintenance of reconstitution potential of HSC was observed, either in settings of serial transplant, or following secondary transplantation of wildtype donor cells previously used to reconstitute Dkk1 tg hosts. In agreement with the functional data, HSC populations had a marked reduction of cells within the G0 fraction of the cell cycle, and displayed enhanced sensitivity to 5-fluorouracil treatment. Wnt signals therefore appear to participate in mediating HSC quiescence in vivo, a result that was largely unpredicted from previous studies, although recent analysis of Hmgb3 mutant mice also supports this conclusion (Nemeth et al., 2006). Our results highlight the importance of studying the impact of a signaling pathway over long-term experiments, and in a physiologic context when seeking to resolve the effects of manipulations on HSC function. In that context, Wnt signaling plays an unanticipated role in maintaining HSC quiescence, which may underlie its requirement in preserving the self-renewing capability of HSC.
Osteoblast expression of Dkk1 does not affect blood or marrow primitive hematopoietic cell populations at steady state
The Wnt inhibitor, Dkk1, has been shown to play an important role in bone formation during development (Niehrs, 2006), and is normally expressed by osteoblasts (Grotewold et al., 1999; MacDonald et al., 2004), hence may have regulatory roles as part of the endosteal HSC niche. To examine the impact of Wnt inhibition on hematopoietic stem cells localized to the periendosteal region, Dkk1 was overexpressed within osteoblastic lineage cells under the control of the truncated 2.3kb collagen 1α promoter (manuscript in preparation). Resulting Col1α2.3-Dkk1 transgenic (Dkk1 tg) mice were backcrossed for at least 5 generations to the C57Bl/6 background and examined for bone and blood phenotypic alterations. No significant differences in peripheral white or red blood cell counts were observed (figure S1a). Bone marrow (BM) and spleen cellularity were also unchanged when Dkk1 tg mice and their littermates were compared, although a slight but not significant trend towards reduced body weight and BM cellularity was apparent in transgenic mice (figure S1b and data not shown). In contrast, significant alterations in bone morphology were observed, as is reported elsewhere (manuscript in preparation, and (Li et al, 2006)) Of note, trabecular bone volume was reduced by approximately 20%, whereas cortical bone was unaffected in Dkk1 tg mice (data not shown). Trabecular bone has been shown by us and others to affect HSC number and function (Adams et al, 2007; Calvi et al, 2003; Jung et al, 2007; Zhang et al, 2003). A panel of antibodies using 7 different flurochromes was used for multiparametric analysis of primitive precursors within the BM of Dkk1 tg mice and their littermates, including populations of LT-HSC, ST-HSC, CMP, GMP, MEP and CLP (figure 1a,c). Subpopulations containing primitive HSCs were not significantly altered at steady-state (figure 1b). However, additional cell surface markers revealed a slight but significant increase in the population containing phenotypically-defined common lymphoid progenitors (figure 1d). The calculated absolute cell numbers based on these frequencies indicated a similar pattern of results (figure S2). Despite the elevation of early lymphoid progenitors in the BM of Dkk1 tg mice, no significant changes were observed in the relative proportion of early B lineage progenitor subsets in the BM (data not shown).

Seven color FACS analysis of primitive populations in wt and Dkk1-tg BM nihms-240191-f0001
Seven color FACS analysis of primitive populations in wt and Dkk1-tg BM
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991120/bin/nihms-240191-f0001.jpg
Figure 1 Seven color FACS analysis of primitive populations in wt and Dkk1-tg BM
BM from Dkk1 tg and littermates was assayed by multiparameter FACS for relative proportion of primitive HSC populations. BM was stained with antibodies against Lineage markers, cKit, Sca-1, CD34, Flk2, CD16/32 and CD127 and gated as shown in panels (A) and (C). At least 10 mice per genotype were compared, over at least three separate experiments. The proportion of BM corresponding to the HSC-containing LK+S+ fraction (A, blue gate) is shown in (B, left axis), and is sub-sectioned according to CD34 and Flk2 expression to yield phenotypic assessments of LT-HSC and ST-HSC fractions (B, right axis). More differentiated progenitors gated in the LK+S− population (A, left, green gate) were sub-sectioned based on CD16/32 and CD34 expression to compare CMP, GMP and MEP progenitors as shown in (C, left panel). Frequencies of each population, from the same samples quantified for HSC frequency in (B) are shown in (D, left axis). The CLP fraction, gated on LKloSlo in (A, red gate), and gated further on CD127+ cells in (C, right panel) are quantified in (D, right axis). Significance was determined by a Student’s 2-tailed T-test. Error bars indicate the SE of the mean.
Dkk1-tg HSCs exhibit impaired Wnt signaling in a non-cell autonomous manner
To confirm that the transgenic expression of Dkk1 leads to the inhibition of Wnt/βcatenin signaling in the Dkk1 tg mice, HSC-containing populations were isolated from Dkk1 transgenic mice that had been intercrossed with the Topgal reporter strain. In these Topgal mice (DasGupta and Fuchs, 1999), multiple TCF/LEF binding sites have been inserted to control the expression of the reporter gene, β-galactosidase. Reporter activity using this construct has been shown to correlate with canonical Wnt signaling. Of note, TCF/LEF transcription has recently been shown to proceed even with the combined loss of β-catenin and γ-catenin, suggesting that canonical Wnt signals can be transduced by alternate intermediates (Jeannet et al, 2007). Reporter activity was examined within the LK+S+ (Lineage-cKit+Sca1+), HSC-containing population, and the LK+S− population which is devoid of LT-HSC potential. When the Wnt reporter activity detected in each of these populations was compared, a dramatic reduction (>100 fold reduction) in β-catenin activation was observed in the HSC-containing LK+S+ population isolated from Dkk1 tg mice (figure 2a). A more modest reduction (<5 fold reduction) was observed in the less-actively signaling LK+S− fraction. This finding indicates that despite the unchanged frequency of phenotypically-defined HSC-containing populations in unmanipulated Dkk1 tg animals, there is evidence that these cells are molecularly altered by osteoblast expression of the Wnt inhibitor. These data provide evidence for direct inhibition of Wnt signaling in the HSC population in addition to any effects that might be mediated by decreased trabecular bone mass. Wnt signaling is regulated, in part, via a negative feedback loop by TCF/LEF-dependent transcription of endogenous Dkk1 (Niida et al, 2004). Consistent with the decrease in Topgal reporter activity, expression of endogenous Dkk1 was also inhibited in the LK+S+ population of Dkk1 tg mice (figure 2b). Using primers specific for the Dkk1 tg, and in comparison its expression in wt and Dkk1 tg tibea, sorted LK+S+ cells do not express the Dkk1 transgene (figure 2c). Together, these results confirm that Dkk1 tg mice inhibit Wnt signaling specifically within the HSC compartment in a non-cell autonomous manner.

Assessment of canonical Wnt signal activity in HSC-containing populations of Dkk1-tg mice nihms-240191-f0002
Assessment of canonical Wnt signal activity in HSC-containing populations of Dkk1-tg mice
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991120/bin/nihms-240191-f0002.gif
Figure 2 Assessment of canonical Wnt signal activity in HSC-containing populations of Dkk1-tg mice
Functional impact of the Dkk1 transgene on BM reconstitution
Analysis of stem/progenitor activity cannot rely exclusively on the quantitation of precursors according to phenotypically-defined parameters. Using functional measures, we detected a consistent defect in multilineage and myeloid colony formation on a per cell basis in BM isolated from Dkk1 transgenic mice (figure 3a). This result was despite the absence of significant alteration of myeloid and more primitive progenitors by immunophenotype, possibly reflecting the elevated lymphoid fraction, whose progeny are not read out under these culture conditions. In vitro methods such as the CFU assay offer an entry-level analysis of hematopoietic activity, however functional reconstitution in vivo more accurately examines true HSC function (Purton and Scadden, 2007). Therefore, in order to better assess the functional capacity of HSCs isolated from the Dkk1 transgenic environment, BM was transplanted from wt or Dkk1 tg littermates with an equivalent dose of competing marrow from congenic donor mice into lethally irradiated recipients. Donor marrow was isolated from a single wt or transgenic mouse to assess any individual-to-individual variation. Following six months of engraftment, no significant changes in reconstitution were observed across the groups of recipients receiving BM isolated from individual wt or Dkk1 tg environments, although a range of reconstitution capacity was apparent in both groups (figure S3a). Using a limiting dilution assay to determine the frequency of repopulating cells present in BM isolated from individual Dkk1-expressing animals revealed a two-fold elevation in the number of functional reconstituting HSCs (Figure 3b). These transplant results indicate that cells isolated from the Dkk1-epressing niche are capable of reconstituting irradiated recipients, and appear to be present at a higher frequency when Wnt has been inhibited in this location. An important additional parameter to test when investigating HSC function is their longevity, or ability to respond to repeated rounds of expansion stress. To assay the longevity of HSCs isolated from Dkk1 tg mice, noncompetitive serial transplants were performed. As expected from the previous transplant experiments, Dkk1 tg BM was able to completely reconstitute wt irradiated recipients (data not shown).
Functional assessment of HSCs isolated from Dkk1-tg mice
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991120/bin/nihms-240191-f0003.jpg
Figure 3 Functional assessment of HSCs isolated from Dkk1-tg mice
(A) BM from 8 pairs of wt and Dkk1-tg mice was plated in methylcellulose with growth factors (SCF, IL-3, IL-6, Epo) and scored for CFU-C (combined scoring for BFU-E, CFU-GM and CFU-GEMM colonies) after 12 days. All live colonies of more than 30 cells were counted for each of three wells plated per sample. Data are shown as mean colonies per well for each of 8 mice studied over three individual experiments. Significance was determined using a two-tailed Student’s T test. (B) Limiting dilution experiments were performed using three doses of test marrow (CD45.1) transplanted with 5×105 competing cells (CD45.2) into groups of at least 9 recipients (CD45.2) per dose. Test marrow was isolated from two wt and two Dkk1-tg mice, and the Dkk1-tg donors shown here were transplanted into separate groups of irradiated recipients. Data points are plotted as the percent of recipients per group that did not exhibit at least 1% multi-lineage PB engraftment at 6 months (percent unreconstituted). LT-HSC frequency and significance were determined using Poisson statistics: wt, 1 in 63,00 (circles) vs tg, 1 in 31,500 or 1: 37,000 (squares); p<0.02. Similar results were obtained in an independent assessment of two Dkk1-tg donors. (C) Non-competitive serial transplants were initiated by transplanting 1×106 whole BM pooled from three wt or Dkk1-tg donors (CD45.1) into each of 10 irradiated recipients (CD45.2). Secondary and tertiary transplants were performed after 14 weeks of engraftment by pooling BM from 3-4 reconstituted recipients to transplant 1×106 whole BM into new groups of 10 irradiated CD45.2 recipients. The Kaplan-Meier survival graph depicts the survival of tertiary recipients, mice receiving BM from Dkk1-tg mice (solid line) or wt controls (dashed line). Similar results were obtained in an independent assessment of 2 wt and 2 Dkk1-tg mice. (D) Prior to transplant into tertiary recipients, BM from 5 secondary recipients of both genotypes was assayed by FACS for the frequency of LT-HSCs (LK+S+CD34loFlk2−). Error bars indicated SD of the mean, and significance was determined by a two-tailed T test
Effect of temporary exposure to endosteal Dkk1 on HSC function
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991120/bin/nihms-240191-f0004.jpg
Figure 4 Transplant analysis of HSC function following residence in a Dkk1-tg environment
Wnt-inhibited HSC-containing populations are less quiescent
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991120/bin/nihms-240191-f0005.gif
Figure 5 Examination of cell cycle status of primitive BM in wt and Dkk1-tg mice
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991120/bin/nihms-240191-f0006.gif
Figure 6 Gene expression by quantitative PCR of sorted primitive populations
Understanding the role of specific signals in the varied regulatory functions of HSC activities is crucial for designing and developing therapeutic interventions involving these cells. The impact of the Wnt family on the expansion and regulation of hematopoietic cells has been examined in a variety of studies. However, the physiologic effects of this pathway remain somewhat ill-defined with often contradicting results. Some have demonstrated that Wnt cascade activation promotes the proliferation of HSCs and their progeny while maintaining at least short-term functional activity (Baba et al, 2006; Murdoch et al, 2003; Reya et al, 2003;Trowbridge et al, 2006). Others, employing persistent genetic activation of the pathway, have also demonstrated an increase in proliferation of cells with an HSC immunophenotype, but with marked impairment of HSC differentiation resulting in animal death (Kirstetter et al, 2006; Scheller et al, 2006). However, induced deletion of β-catenin, the primary downstream mediator of the Wnt cascade resulted in no apparent impact on HSC activity, even in a reconstitution assay that required expansion of β-catenin null transplanted HSCs (Cobas et al, 2004). Furthermore, recent combined deletions of both β-catenin and its homologue, γ-catenin, also maintain HSC function under steady-state and primary reconstitution conditions (Jeannet et al, 2007; Koch et al, 2007).All of these studies have either assayed Wnt activity in broad over- or under-stimulation settings, and the manipulations have been performed on the HSCs themselves, or broadly applied to recipient animals. The context in which morphogens are present is highly relevant to their effect and not previously studied for Wnt effects on hematopoiesis (Trowbridge et al., 2006). Indeed, Wnt ligands can modulate signaling initiated by other Wnt family members, underscoring the concept that context, and different signaling intermediates may have a strong impact on functional outcome (Nemeth et al, 2007).
In the present study, we have established a system that permits the analysis of localized Wnt inhibition, offering the opportunity to assay the impact of chronic or temporary exposure to this inhibited environment. In particular, we have directed expression of the Wnt inhibitor, Dkk1, to a cell population that has been previously demonstrated to exert a regulatory function over HSC activity, and which normally express Dkk-1, albeit at lower levels (Grotewold et al,1999; MacDonald et al, 2004). It should be noted that while an increasing number of reports suggest that phenotypically-identified HSCs inhabit additional physical locations within the bone marrow environment (Hooper et al, 2007; Scadden, 2006), the promoter used in our study has proven to functionally impact the number and activity of HSCs when used to direct modifying signal expression to a population of osteoblastic cells. Given that expression of Dkk1 also results in alterations to bone morphology itself, there is likely to be a dual effect of Dkk1: one altering the niche architecture and the other affecting Wnt signaling in stem/progenitor cells. Our studies demonstrated an effect of Dkk1 overexpression by non-HSCs on Wnt signaling in hematopoietic stem/progenitors, suggesting that this is at least a contributing factor to the phenotype observed. This observation that TCF/LEF reporter activity is reduced, as is expression of endogenous Dkk1, itself a Wnt signaling target (Niida et al, 2004) in BM cells of the transgenic mice indicates altered canonical Wnt signaling. It does not rule out that Dkk1 may exert additional Wnt-independent functions. The results presented here also indicate that the reduced longevity of HSCs does not require constant exposure to exogenous Dkk1, given that we were unable to detect Dkk1 tg expression within populations of primitive hematopoietic cells, and therefore the functional impact on transplanted cells is observed in a Dkk tg-free environment. It is important to note that transplantation of whole BM populations is generally not effective at engrafting non-hematopoietic cells (Koc et al, 1999).
Wnt mediates HSC quiescence and maintains reconstitution function in vivo
The results presented here establish a role for Wnt, in the maintenance of a quiescent fraction of functional HSCs in BM. This was associated with evidence of increased stem cells on limit dilution transplant analysis. However the ability of the same cells to function after serial rounds of transplantation was drastically reduced. The ability of stem cells to persist under the stress conditions of transplantation requires self-renewal capability that is compromised after Dkk1 exposure
The studies of inducible deletion of β- and γ-catenin noted that they were dispensable for HSC function, however did not include sequential transplants out to the extent where we observed our most dramatic phenotype (Cobas et al, 2004; Jeannet et al, 2007; Koch et al, 2007) Alternatively, it is possible that Dkk1 interferes with HSC function through a process that does not depend on β- or γ-catenin signaling (Jeannet et al, 2007; Niehrs, 2006).
Our results emphasize the importance of studying pathways within the context of other signals present in the natural microenvironment, and underscore the potential for unanticipated functional roles. It is clear that different combinations of signals may have a range of effects depending on the context in which they are received. Indeed, we observed an impact of Wnt-inhibition on the activation of the Notch target, Hes-1, raising the possibility that Notch and Wnt coordinate in vivo to maintain quiescence of HSCs, rather than participating in expansive and/or self-renewal functions (Duncan et al, 2005). Notably, elevated Hes-1 and p21 expression have recently been shown to correlate with the maintenance of quiescence and repopulating function of primitive HSCs (Yu et al, 2006). We noted a highly specific impact of the Dkk1 tg on the stem cell enriched LK+S+ fraction in Wnt-dependent pathway activation and inhibition and the Notch target, Hes-1, or the cell cycle regulator, p21 expression.
The effects of Dkk1 on cell cycling were unanticipated given previous reports of constitutively active β-catenin inducing increased stem/progenitor cell proliferation (Kirstetter et al, 2006; Scheller et al, 2006). However, others found that with deletion of the chromatin binding protein, Hmgb3, Wnt signaling was increased, yet stem cells more readily returned to quiescence after 5-FU challenge than controls. (Nemeth et al, 2006) Both increased and decreased activation of the pathway may therefore alter HSC cycling kinetics. This may again be due to the context differences observed with a microenvironmentally-provided signal in the current study contrasted with cell autonomous activation of the pathway in the prior reports. Alternatively, it may be an example of the complex effects of morphogens, which have dose-dependent actions (Delaney et al, 2005; Kielman et al, 2002; MacDonald et al, 2004). It may be that there is a bi-phasic response of cell cycling to the Wnt pathway and that proper control of stem cell quiescence requires a fine-tuned modulation of intermediate Wnt signaling intensity. This has implications for the potential use of Wnts as mediators of stem cell expansion ex vivo and for interruption of this pathway as an anti-leukemic intervention.
In sum, niche related expression of Dkk1 reveals a role for Wnt signaling in the physiologic regulation of the hematopoietic compartment, altering stem cell cycling and longevity following repeated expansion, or self-renewal. The phenotype observed was sufficiently distinct from what cell-autonomous modifications of the pathway would have predicted to argue for niche specific modeling of exogenous factors’ effects on stem cells. This may be particularly true for members of the locally acting morphogen group of cell modifiers.
7.10.4 Wnt.β-Catenin Signaling in Development and Disease
Clevers H1.
Cell. 2006 Nov 3; 127(3):469-80.
http://dx.doi.org/10.1016/j.cell.2006.10.018
A remarkable interdisciplinary effort has unraveled the WNT (Wingless and INT-1) signal transduction cascade over the last two decades. Wnt genes encode small secreted proteins that are found in all animal genomes. Wnt signaling is involved in virtually every aspect of embryonic development and also controls homeostatic self-renewal in a number of adult tissues. Germline mutations in the Wnt pathway cause several hereditary diseases, and somatic mutations are associated with cancer of the intestine and a variety of other tissues.
The mouse wnt1 gene, originally named Int-1, was identified in 1982 by Nusse and Varmus as a preferential integration site for the Mouse Mammary Tumor Virus in virally induced breast tumors ( Nusse and Varmus, 1982). When sequenced, the Wnt1 proto-oncogene was seen to encode a secreted protein that is cysteine rich. Subsequently, Drosophila wingless (wg), which controls segment polarity during larval development ( Nüsslein-Volhard and Wieschaus, 1980), was shown to be a fly homolog of Wnt1 ( Rijsewijk et al., 1987). Segmentation of the epidermis of wg mutant fly embryos is severely impaired as evidenced by abnormalities in the overlying ventral cuticle. In contrast to the wild-type cuticle, which exhibits alternating denticle and naked belts, the wg cuticle is completely covered with denticles. Fly embryos carrying mutations in the porcupine, dishevelled, and armadillo genes display similar cuticle abnormalities to wgmutant embryos, whereas mutations in shaggy/zeste-white 3 cause the opposite phenotype, a naked cuticle. Epistatic analysis of cuticle structure in double mutants indicated that these genes constituted the core of a new signal transduction cascade ( Siegfried et al., 1992, Noordermeer et al., 1994 and Peifer et al., 1994).
In 1989, McMahon and Moon (McMahon and Moon, 1989) observed a duplication of the body axis inXenopus following injection of mouse Wnt1 mRNA into ventral blastomeres of embryos at the 4-cell stage. This observation supported the notion that Wnt signaling was shared between vertebrates and invertebrates and, moreover, provided a rapid and convenient assay to study components of the Wnt pathway in vertebrates. Axis duplication was also induced by Dishevelled (Dsh), β-catenin (the vertebrate homolog of armadillo), and a dominant-negative version of glycogen synthase kinase 3 (GSK3), the vertebrate homolog of shaggy/zeste-white 3 ( Dominguez et al., 1995, Guger and Gumbiner, 1995 and He et al., 1995). Although long elusive, the specific Wnt signal that triggers axis induction in Xenopus was identified as Wnt11 by Heasman and colleagues last year ( Tao et al., 2005).
The combined observations made in Drosophila and Xenopus delineated a highly conserved signaling pathway, activated by secreted Wnt proteins. Independent of these studies, the adenomatous polyposis coli (APC) gene was discovered in a hereditary cancer syndrome termed familial adenomatous polyposis (FAP) ( Kinzler et al., 1991 and Nishisho et al., 1991). Soon after, the large cytoplasmic APC protein was found to interact with β-catenin ( Rubinfeld et al., 1993 and Su et al., 1993). This observation provided the first connection between the Wnt pathway and human cancer.
Genome sequencing has since revealed that mammalian species have roughly 20 secreted Wnt proteins, which can be divided into 12 conserved Wnt subfamilies. Of these, only 6 subfamilies have counterparts in ecdysozoan animals such as Drosophila and Caenorhabditis. In contrast, at least 11 of the Wnt subfamilies occur in the genome of a cnidarian (the sea anemone Nematostella vectensis). This finding suggests that some Wnt subfamilies were lost during the evolution of the ecdysozoan lineage but more importantly reveals that a complex inventory of Wnt factors was present in multicellular animals well before the Cambrian explosion (550 million years ago). Thus, comparative genomic analysis underscores the crucial role that Wnt genes play in organismal patterning throughout the animal kingdom ( Kusserow et al., 2005).
Currently, three different pathways are believed to be activated upon Wnt receptor activation: the canonical Wnt/β-catenin cascade, the noncanonical planar cell polarity (PCP) pathway, and the Wnt/Ca2+ pathway. Of these three, the canonical pathway is best understood and is the primary subject of this review. For recent comprehensive overviews on the other Wnt signaling pathways, the reader is referred to Katoh (2005) and Kohn and Moon (2005). This review discusses how Wnt proteins are produced and secreted and how they activate the canonical Wnt signaling pathway in recipient cells. Further, the review examines the roles of the canonical Wnt pathway in development, tissue self-renewal, and cancer.
Wnt Protein Secretion
Wnt proteins are characterized by a high number of conserved cysteine residues. Although Wnt proteins carry an N-terminal signal peptide and are secreted, they are relatively insoluble. This insolubility has been attributed to a particular protein modification, cysteine palmitoylation, which is essential for Wnt function (Willert et al., 2003). Hofmann (2000) reported that a Drosophila gene required in the Wnt-secreting cell, termed porcupine, displays homology to acyl-transferases, enzymes that acylate a variety of substrates in the endoplasmic reticulum. Thus, porcupine and its worm homolog mom-1 are believed to encode the enzyme that is responsible for Wnt palmitoylation ( Zhai et al., 2004).
Recently, Banziger et al. (2006) and Bartscherer et al. (2006) uncovered in Drosophila another conserved gene that is essential for Wnt secretion, named wntless (wls) and evenness interrupted (evi), respectively. The gene encodes a seven-pass transmembrane protein that is conserved from worms (mom-3) to man (hWLS). In the absence of Wls/evi, Wnts are retained inside the cell that produces them. The Wntless protein resides primarily in the Golgi apparatus, where it colocalizes and physically interacts with Wnts. A genetic screen in C. elegans revealed that the retromer, a multiprotein complex involved in intracellular trafficking and conserved from yeast to man, is also essential for Wnt secretion and for the generation of a Wnt gradient ( Coudreuse et al., 2006). An attractive hypothesis is that the retromer complex is involved in recycling a Wnt cargo receptor (such as Wntless) between the default secretory pathway and a compartment dedicated to Wnt secretion (see Figure 1).

wnt-secretion
http://ars.els-cdn.com/content/image/1-s2.0-S0092867406013444-gr1.jpg
Figure 1. Wnt Secretion
To be secreted, Wnt proteins in the endoplasmic reticulum (ER) need to be palmitoylated by the action of Porcupine. Wnt proteins also require Wntless (Wls/Evi) in order to be routed to the outside of the cell. Loading onto lipoprotein particles may occur in a dedicated endo/exocytic compartment. The retromer complex may shuttle Wls between the Golgi and the endo/exocytic compartment.
Wnt is thought to act as a morphogen (that is, a long-range signal whose activity is concentration dependent) (reviewed in Logan and Nusse, 2004). However, it is unclear how these long-range gradients are generated. It is conceivable that the palmitoyl moiety constrains movement away from membranes or lipid particles. Thus, Wnts may be tethered to intercellular transport vesicles or lipoprotein particles (Panakova et al., 2005). Alternatively, Wnts may be transported by cytonemes, which are long, thin filopodial processes. Additionally, studies in Drosophila suggest a role for extracellular heparan sulfate proteoglycans (HSPG) in the transport or stabilization of Wnt proteins. For instance, flies carrying mutations in Dally, a GPI-anchored HSPG, or in genes encoding enzymes that modify HSPGs resemblewingless mutants (reviewed in Lin, 2004).
Receptors, Agonists, and Antagonists for Wnt
Wnts bind Frizzled (Fz) proteins, which are seven-pass transmembrane receptors with an extracellular N-terminal cysteine-rich domain (CRD) (Bhanot et al., 1996). The Wnt-Fz interaction appears promiscuous, in that a single Wnt can bind multiple Frizzled proteins (e.g., Bhanot et al., 1996) and vice versa. In binding Wnt, Fzs cooperate with a single-pass transmembrane molecule of the LRP family known as Arrow inDrosophila ( Wehrli et al., 2000) and LRP5 and -6 in vertebrates ( Pinson et al., 2000 and Tamai et al., 2000). The transport of Arrow/LRP5/6 to the cell surface is dependent on a chaperone called Boca inDrosophila and Mesd in mice ( Culi and Mann, 2003 and Hsieh et al., 2003). And consistent with a role of the Boca/Mesd chaperone in the transport of Arrow/LRP5/6 transport, mutations in Boca and Mesdresemble loss of Arrow/LRP5/6. Although it has not been formally demonstrated that Wnt molecules form trimeric complexes with LRP5/6 and Frizzled, surface expression of both receptors is required to initiate the Wnt signal.
Derailed, a transmembrane tyrosine kinase receptor from the RYK subfamily, is an unusual Wnt receptor.Drosophila Wnt5 controls axon guidance in the central nervous system. Embryos lacking Dwnt-5 resemble those lacking Derailed, that is, they generate aberrant neuronal projections across the midline ( Yoshikawa et al., 2003). Derailed binds DWnt-5 through its extracellular WIF (Wnt inhibitory factor) domain. Signaling events downstream of this alternative Wnt receptor remain unclear. Somewhat unexpectedly, the Derailed kinase domain may be dispensable for signaling. Lu et al. (2004) propose that, unlike the Drosophila Ryk homolog Derailed, mammalian Ryk functions as a coreceptor along with Fz. Mammalian Ryk binds Dishevelled to activate the canonical Wnt/β-catenin signaling pathway. Another tyrosine kinase receptor, Ror2, harbors a Wnt binding CRD motif. Wnt5a can engage Ror2 to inhibit the canonical Wnt signaling pathway, although paradoxically Wnt5a can also activate the canonical pathway by directly engaging Fz4 (Mikels and Nusse, 2006) and Fz5 ( He et al., 1997).
At least two types of proteins that are unrelated to Wnt factors activate the Frizzled/LRP receptors. One of these factors is the cysteine-knot protein Norrin, which is mutated in Norrie disease, a developmental disorder characterized by vascular abnormalities in the eye and blindness. Norrin binds with high affinity to Frizzled-4 and activates the canonical signaling pathway in an LRP5/6-dependent fashion (Xu et al., 2004). Other factors that activate the canonical Wnt signaling pathway are R-spondins, which are thrombospondin domain-containing proteins. In Xenopus, R-spondin-2 is a Wnt agonist that synergizes with Wnts to activate β-catenin ( Kazanskaya et al., 2004). Human R-spondin-1 has been found to strongly promote the proliferation of intestinal crypt cells, a process which involves the stabilization of β-catenin (Kim et al., 2005). Indeed, studies in cultured cells demonstrate that R-spondins can physically interact with the extracellular domains of LRP6 and Fzd8 and activate Wnt reporter genes ( Nam et al., 2006).
The secreted Dickkopf (Dkk) proteins inhibit Wnt signaling by direct binding to LRP5/6 (Glinka et al., 1998). Through this interaction, Dkk1 crosslinks LRP6 to another class of transmembrane molecules, the Kremens (Mao et al., 2002), thus promoting the internalization and inactivation of LRP6. An unrelated secreted Wnt inhibitor, Wise, also acts by binding to LRP (Itasaki et al., 2003), as does the WISE family member SOST (Li et al., 2005 and Semenov et al., 2005).
Soluble Frizzled-Related Proteins (SFRPs) resemble the ligand-binding CRD domain of the Frizzled family of Wnt receptors (Hoang et al., 1996). WIF proteins are secreted molecules with similarity to the extracellular portion of the Derailed/RYK class of transmembrane Wnt receptors (Hsieh et al., 1999). SFRPs and WIFs are believed to function as extracellular Wnt inhibitors (reviewed in Logan and Nusse, 2004) but, depending on context, may also promote signaling by Wnt stabilization or by facilitating Wnt secretion or transport.
Canonical Wnt Signaling
Once bound by their cognate ligands, the Fz/LRP coreceptor complex activates the canonical signaling pathway (Figure 2). Fz can physically interact with Dsh, a cytoplasmic protein that functions upstream of β-catenin and the kinase GSK-3. Wnt signaling controls phosphorylation of Dsh (reviewed in Wallingford and Habas, 2005). However, it remains unclear whether the binding of Wnt to Fz regulates a direct Fz-Dsh interaction, nor is it known how Dsh phosphorylation is controlled or how phosphorylated Dsh functions in Wnt signal transduction.

canonical-wnt-signaling
Figure 2. Canonical Wnt Signaling
(Left panel) When Wnt receptor complexes are not bound by ligand, the serine/threonine kinases, CK1 and GSK3α/β, phosphorylate β-catenin. Phosphorylated β-catenin is recognized by the F box/WD repeat protein β-TrCP, a component of a dedicated E3 ubiquitin ligase complex. Following ubiquitination, β-catenin is targeted for rapid destruction by the proteasome. In the nucleus, the binding of Groucho to TCF (T cell factor) inhibits the transcription of Wnt target genes. (Right panel) Once bound by Wnt, the Frizzled(Fz)/LRP coreceptor complex activates the canonical signaling pathway. Fz interacts with Dsh, a cytoplasmic protein that functions upstream of β-catenin and the kinase GSK3β. Wnt signaling controls phosphorylation of Dishevelled (Dsh). Wnts are thought to induce the phosphorylation of LRP by GSK3β and casein kinase I-γ (CK1γ), thus regulating the docking of Axin. The recruitment of Axin away from the destruction complex leads to the stabilization of β-catenin. In the nucleus, β-catenin displaces Groucho from Tcf/Lef to promote the transcription of Wnt target genes.
Recent studies have indicated that the coreceptor LRP5/6 interacts with Axin through five phosphorylated PPP(S/T)P repeats in the cytoplasmic tail of LRP (Davidson et al., 2005 and Zeng et al., 2005). Wnts are thought to induce the phosphorylation of the cytoplasmic tail of LRP, thus regulating the docking of Axin. GSK3 phosphorylates the PPP(S/T)P motif, whereas caseine kinase I-γ (CK1γ) phosphorylates multiple motifs close to the GSK3 sites. CK1γ is unique within the CK1 family in that it is anchored in the membrane through C-terminal palmitoylation. Both kinases are essential for signal initiation. It remains presently debated whether Wnt controls GSK3-mediated phosphorylation of LRP5/6 (Zeng et al., 2005) or whether CK1γ is the kinase regulated by Wnt (Davidson et al., 2005). When bound to their respective membrane receptors, Dsh and Axin may cooperatively mediate downstream activation events by heterodimerization through their respective DIX (Dishevelled-Axin) domains.
The Cytoplasmic Destruction Complex
The central player in the canonical Wnt cascade is β-catenin, a cytoplasmic protein whose stability is regulated by the destruction complex. The tumor suppressor protein Axin acts as the scaffold of this complex as it directly interacts with all other components—β-catenin, the tumor suppressor protein APC, and the two kinase families (CK1α, -δ, -ɛ and GSK3α and -β [reviewed in Price, 2006]). When WNT receptor complexes are not engaged, CK1 and GSK3α/β sequentially phosphorylate β-catenin at a series of highly conserved Ser/Thr residues near its N terminus (Figure 2). Phosphorylated β-catenin is then recognized by the F box/WD repeat protein β-TrCP, a component of a dedicated E3 ubiquitin ligase complex. As a consequence, β-catenin is ubiquitinated and targeted for rapid destruction by the proteasome (Aberle et al., 1997). Note that the CK1 and GSK3 kinases perform paradoxical roles in the Wnt pathway. At the level of the LRP coreceptor they act as agonists, whereas in the destruction complex they act as antagonists
Although genetic observations imply an essential role for APC in the destruction complex, there is no consensus on its specific molecular activity. APC has a series of 15 and 20 amino acid repeats with which it interacts with β-catenin. Three Axin-binding motifs are interspersed between these β-catenin-binding motifs. Increasing the expression of Axin in cancer cells that lack APC restores the activity of the destruction complex, implying that APC is only essential when Axin levels are limiting. Quantitatively, Axin indeed appears to be the limiting factor (Lee et al., 2003) and may be the key scaffolding molecule that promotes the rapid assembly and disassembly of the destruction complex.
Given that CK1, Dsh, β-TrCP, and GSK3 participate in other signaling pathways, low levels of Axin may insulate the Wnt pathway from changes in the abundance or activity of these signaling components. It has been proposed that APC is required for efficient shuttling and loading/unloading of β-catenin onto the cytoplasmic destruction complex. Both APC and Axin can themselves be phosphorylated by their associated kinases, which changes their affinity for other components of the destruction complex. Our understanding of the relevance of these phosphorylation events in the regulation of Wnt signaling remains incomplete. For a comprehensive discussion of the kinases in the Wnt pathway, the reader is referred to a recent review (Price, 2006)
β-catenin plays a second role in simple epithelia, that is, as a component of adherens junctions. It is an essential binding partner for the cytoplasmic tail of various cadherins, such as E-cadherin (Peifer et al., 1992). Unlike the signaling pool of β-catenin, the pool that is bound to the adherens junction is highly stable. It is currently unclear whether the adhesive and signaling properties of β-catenin are interconnected. In a likely scenario, newly synthesized β-catenin first saturates the pool that is part of the adhesion junction, which never becomes available for signaling. “Excess,” free cytoplasmic β-catenin protein is then efficiently degraded by the APC complex. It is only this second, highly unstable pool that is subject to regulation by Wnt signals. In support of this model, these two functions of β-catenin are separately performed by two different β-catenin homologs in C. elegans ( Korswagen et al., 2000).
Upon receptor activation by WNT ligands, the intrinsic kinase activity of the APC complex for β-catenin is inhibited. It is unclear how this occurs, but it likely involves the Wnt-induced recruitment of Axin to the phosphorylated tail of LRP and/or to Fz-bound Dsh. As a consequence, stable, nonphosphorylated β-catenin accumulates and translocates into the nucleus, where it binds to the N terminus of LEF/TCF (lymphoid enhancer factor/T cell factor) transcription factors (Behrens et al., 1996, Molenaar et al., 1996 and van de Wetering et al., 1997).
It has been suggested that protein phosphatases may regulate β-catenin stability as antagonists of the serine kinases (reviewed in Price, 2006). For example, heterotrimeric PP2A is required for the elevation of β-catenin levels that is dependent on Wnt. Moroever, PP2A can bind Axin and APC, suggesting that it might function to dephosphorylate GSK3 substrates. If and how PP2A activity is regulated by Wnt signals remains to be resolved.
Crystallographic studies are starting to provide insights into the structure of the destruction complex. The central region of β-catenin (to which most partners bind) was the first component of the pathway to be crystallized. It consists of 12 armadillo repeats, which adopt a superhelical shape with a basic groove running along its length. Subsequently, structural interactions of Axin, APC, E-cadherin, and TCF with β-catenin have been visualized (Choi et al., 2006, and references therein). APC, E-cadherin, and TCF bind the central part of the basic groove in a mutually exclusive fashion. Despite very limited conservation of primary sequence in the respective interaction domains, the modes of binding are structurally very similar. Axin utilizes a helix that occupies the groove formed by the third and fourth armadillo repeats of β-catenin. Axin binding precludes the simultaneous interaction with other β-catenin partners in this region. Based on this observation, it is suggested that a key function of APC is to remove phosphorylated β-catenin from the active site of the complex (Xing et al., 2003). In a further study, the structure of Axin bound to APC (Spink et al., 2000) was solved. These studies form stepping stones to a better understanding of the dynamics of the destruction complex. Unfortunately, biochemical studies of the destruction complex in its different activation states are sorely lacking.
Nuclear Events
Upon stabilization by Wnt signals, β-catenin enters the nucleus to reprogram the responding cell (Figure 3). There is no consensus on the mechanism by which β-catenin travels between the cytoplasm and the nucleus. In many cases, cells that undergo Wnt signaling may actually display an overall rise in β-catenin protein without a clear nuclear preference. β-catenin’s nuclear import is independent of the Nuclear Localization Signal/importin machinery. β-catenin itself is a close relative of importin/karyopherins and directly interacts with nuclear pore components. Two proteins, Tcf and Pygopus are proposed to anchor β-catenin in the nucleus, although β-catenin can still localize to the nucleus in the absence of either of the two (reviewed in Staedeli et al., 2006). β-catenin can also be actively transported back to the cytoplasm, by either an intrinsic export signal or as cargo of Axin (Cong and Varmus, 2004) or APC (Rosin-Arbesfeld et al., 2000) that shuttle between cytoplasm and nucleus.

transactivation-of-wnt-target-genes
Transactivation of Wnt Target Genes
http://ars.els-cdn.com/content/image/1-s2.0-S0092867406013444-gr3.jpg
Figure 3. Transactivation of Wnt Target Genes
The β-catenin/Tcf complex interacts with a variety of chromatin-remodeling complexes to activate transcription of Wnt target genes. The recruitment of β-catenin to Tcf target genes affects local chromatin in several ways. Bcl9 acts as a bridge between Pygopus and the N terminus of β-catenin. Evidence suggests that this trimeric complex is involved in nuclear import/retention of β-catenin (Townsley et al., 2004), but it may also be involved in the ability of β-catenin to activate transcription (Hoffmans et al., 2005). The C terminus of β-catenin also binds to coactivators such as the histone acetylase CBP, Hyrax, and Brg-1 (a component of the SWI/SNF chromatin-remodeling complex).
Whereas the fly and worm genomes both encode a single Tcf protein, the vertebrate genome harbors fourTcf/Lef genes. Tcf factors bind their cognate motif in an unusual fashion, i.e., in the minor groove of the DNA helix, while inducing a dramatic bend of over 90°. Tcf target sites are highly conserved between the four vertebrate Tcf/Lef proteins and Drosophila Tcf. These sites resemble AGATCAAAGG ( van de Wetering et al., 1997). Wnt/TCF reporter plasmids such as pTOPflash ( Korinek et al., 1997), widely used to measure Wnt pathway activation, consist of concatamers of 3–10 of these binding motifs cloned upstream of a minimal promoter. The four vertebrate TCF/LEF differ dramatically in their embryonic and adult expression domains, yet they are highly similar biochemically, explaining the extensive redundancy unveiled in double knockout experiments (as in Galceran et al., 1999).
In the absence of Wnt signals, Tcf acts as a transcriptional repressor by forming a complex with Groucho/Grg/TLE proteins (Cavallo et al., 1998 and Roose et al., 1998). The interaction of β-catenin with the N terminus of Tcf (Behrens et al., 1996, Molenaar et al., 1996 and van de Wetering et al., 1997) transiently converts it into an activator, translating the Wnt signal into the transient transcription of Tcf target genes. To accomplish this, β-catenin physically displaces Groucho from Tcf/Lef (Daniels and Weis, 2005). The recruitment of β-catenin to Tcf target genes affects local chromatin in several ways. Its C terminus is a potent transcriptional activator in transient reporter gene assays (van de Wetering et al., 1997). It binds coactivators such as the histone acetylase CBP and Brg-1, a component of the SWI/SNF chromatin remodeling complex (reviewed in Staedeli et al., 2006). A recent study implies that the human and fly homologs of yeast Cdc37 (Parafibromin and Hyrax, respectively) also interact with the C-terminal transactivation domain of β-catenin to activate target gene transcription (Mosimann et al., 2006). Cdc37 is a component of the PAF complex. In yeast the PAF complex directly interacts with RNA polymerase II to regulate transcription initiation and elongation.
Two dedicated, nuclear partners of the TCF/β-catenin complex, Legless/Bcl9 and Pygopus, were recently found in genetic screens in Drosophila ( Kramps et al., 2002, Parker et al., 2002 and Thompson et al., 2002). Mutations in these genes result in phenotypes similar to wingless, and overexpression of both genes promotes TCF/β-catenin activity in mammalian cells ( Thompson et al., 2002). Bcl9 bridges Pygopus to the N terminus of β-catenin. The formation of this trimeric complex has been implicated in nuclear import/retention of β-catenin ( Townsley et al., 2004) but may also directly contribute to the ability of β-catenin to transactivate transcription ( Hoffmans et al., 2005). Although most if not all Wnt signaling events in Drosophila appear to be dependent on Bcl9 and Pygopus, it is currently unclear if this holds true in vertebrate development.
Tcf itself can be regulated by phosphorylation. The MAP kinase-related protein kinase NLK/Nemo (Ishitani et al., 1999) phosphorylates Tcf, thereby decreasing the DNA-binding affinity of the β-catenin/Tcf complex and inhibiting transcriptional regulation of Wnt target genes. In C. elegans, LIT-1/NLK-dependent phosphorylation results in PAR-5/14-3-3- and CRM-1-dependent nuclear export of POP-1/Tcf ( Meneghini et al., 1999 and Lo et al., 2004). And lastly, a recent study utilizing chromatin immunoprecipitations suggests that APC, independent of its role in the cytoplasmic destruction complex, acts on chromatin to facilitate CtBP-mediated repression of Wnt target genes in normal, but not in colorectal cancer cells ( Sierra et al., 2006).
Wnt Target Genes
Loss of components of the Wnt pathway can produce dramatic phenotypes that affect a wide variety of organs and tissues. A popular view equates Wnt signaling with maintenance or activation of stem cells (Reya and Clevers, 2005). It should be realized, however, that Wnt signals ultimately activate transcriptional programs and that there is no intrinsic restriction in the type of biological event that may be controlled by these programs. Thus, Wnt signals may promote cell proliferation and tissue expansion but also control fate determination or terminal differentiation of postmitotic cells. Sometimes, these disparate events, proliferation and terminal differentiation, can be activated by Wnt in different cell types within the same structure, such as the hair follicle or the intestinal crypt (Reya and Clevers, 2005).
Numerous Tcf target genes have been identified in diverse biological systems. These studies tend to focus on target genes involved in cancer, as exemplified by the wide interest in the Wnt target genes cMyc and Cyclin D1. For a comprehensive, updated overview of Tcf target genes, the reader is referred to the Wnt homepage (http://www.stanford.edu/∼rnusse/wntwindow.html). The Wnt pathway has distinct transcriptional outputs, which are determined by the developmental identity of the responding cell, rather than by the nature of the signal. In other words, the majority of Wnt target genes appear to be cell type specific. It is not clear whether “universal” Wnt/Tcf target genes exist. The best current candidates in vertebrates are Axin2/conductin (Jho et al., 2002) and SP5 (Weidinger et al., 2005). As noted (Logan and Nusse, 2004), Wnt signaling is autoregulated at many levels. The expression of a variety of positive and negative regulators of the pathway, such as Frizzleds, LRP and HSPG, Axin2, and TCF/Lef are all controlled by the β-catenin/TCF complex.
Wnt Signaling in Self-Renewing Tissues in Adult Mammals
Wnt signaling not only features in many developmental processes; in some self-renewing tissues in mammals it remains essential throughout life. It is this aspect of Wnt signaling that is intricately connected to the development of disease. The examples discussed below illustrate how the Wnt pathway is involved in adult tissue self-renewal. Mutations in the Wnt pathway tip the homoeostatic balance in these tissues to cause pathological conditions such as disturbances in skeletal bone mass or cancer.
Gut
Figure 4. Self-Renewing Tissues in the Adult Mammal
Current evidence indicates that the Wnt cascade is the dominant force in controlling cell fate along the crypt-villus axis. In neonatal mice lacking Tcf4, the differentiated villus epithelium appears unaffected, but the crypt progenitor compartment is entirely absent (Korinek et al., 1998). This implies that physiological Wnt signaling is required for the establishment of this progenitor compartment.
Hair Follicle
Multipotent epidermal stem cells reside in the bulge region of the hair follicle (Figure 4). Bulge stem cells can generate all hair lineages but also the sebocytes and even the stem cells of the interfollicular epidermis (Alonso and Fuchs, 2003). To form a hair, cells migrate downward from the bulge through the outer root sheath. At the base of the hair, the cells enter a transit-amplifying compartment termed the germinative matrix where they undergo terminal differentiation in the precortex compartment of the hair.
Hematopoietic System
Hematopoietic stem cells (HSCs) are the best studied stem cells in mammals. A number of studies have implicated the Wnt signaling pathway as an important regulator of hematopoietic stem and progenitor cells. HSCs themselves as well as the bone marrow microenvironment can produce Wnt proteins. Indeed, Tcf reporters are active in HSCs in their native microenvironment.
Bone
In postnatal and adult life, osteoblasts produce bone matrix, whereas osteoclasts resorb the matrix. Bone density is determined by the relative activities of these two cell types. Gain-of-function mutations in the human LRP5 gene occur in bone diseases, indicating that canonical Wnt signaling may regulate bone mass. This observation has motivated genetic studies in mouse models, which generally confirm the importance of this signaling pathway in bone homeostasis, primarily as a positive regulator of the osteoblast lineage. Similar to humans carrying the gain-of-function LRP5G171V mutation, transgenic mice expressing this allele in osteoblasts display increased bone density and elevated numbers of active osteoblasts (reviewed in Hartmann, 2006).
Wnt Signaling in Cancer
Colon Cancer
The APC gene was among the first tumor suppressors to be cloned. A germline APC mutation is the genetic cause of a hereditary cancer syndrome termed Familiar Adenomatous Polyposis (FAP) (Kinzler et al., 1991 and Nishisho et al., 1991). FAP patients inherit one defective APC allele and as a consequence develop large numbers of colon adenomas, or polyps, in early adulthood. Polyps are benign, clonal outgrowths of epithelial cells in which the second APC allele is inactivated. Inevitably, some of these polyps progress into malignant adenocarcinoma. Loss of both APC alleles occurs in the large majority of sporadic colorectal cancers (Kinzler and Vogelstein, 1996). Mutational inactivation of APC leads to the inappropriate stabilization of β-catenin (Rubinfeld et al., 1996; Figure 4). Indeed, Tcf reporter constructs, normally transcribed only upon Wnt signaling, are inappropriately transcribed in APC mutant cancer cells through the action of constitutive complexes between β-catenin and the intestinal TCF family member Tcf4 (Korinek et al., 1997). In rare cases of colorectal cancer where APC is not mutated, Axin2 is mutant (Liu et al., 2000), or activating (oncogenic) point mutations in β-catenin remove its N-terminal Ser/Thr destruction motif (Morin et al., 1997). Of note, patients with hereditary Axin2 mutations display a predisposition to colon cancer (Lammi et al., 2004).
In intestinal epithelial cells in which APC is mutated, the constitutive β-catenin/Tcf4 complex activates a genetic program in crypt stem/progenitor cells (van de Wetering et al., 2002). In the crypt, the Wnt signaling gradient drives expression of this genetic program to maintain progenitor cell proliferation. The Wnt gradient also controls expression of the EphB/EphrinB sorting receptors and ligands (Battle et al., 2002). The resulting EphB/EphrinB countergradients establish crypt-villus boundaries as well as position the Paneth cells at the bottom of the crypt. Several EphB genes are initially upregulated as Wnt/Tcf4 target genes in early adenomas, but their expression is lost upon cancer progression (Batlle et al., 2005) apparently as the result of a selection process. Activating Wnt pathway mutations are not restricted to cancer of the intestine. Loss-of-function mutations in Axin have also been found in hepatocellular carcinomas, whereas oncogenic β-catenin mutations occur in a wide variety of solid tumors (reviewed inReya and Clevers, 2005).
Several animal models exist for FAP. Dove and colleagues first described the multiple intestinal neoplasia(min) mouse, which carries a stop codon in APC (Apcmin). Unlike FAP patients, Apcmin mice develop adenomas predominantly in the small intestine ( Su et al., 1992). Several additional Apc knockout models have been generated in mice. Invariably, these mice develop neoplastic lesions but they may differ in tumor incidence and tissue type in which tumors first appear. In a recent elegant study, the Wnt cascade was mutationally activated in adult mice by conditional deletion of Apc ( Sansom et al., 2004). Within days, villi were entirely populated by crypt-like cells, demonstrating the direct link between active Wnt signaling and the proliferation of crypt progenitors, which when unrestrained results in cancer. Zebrafish that are mutant in Apc resemble the mouse models in that heterozygous mutants develop adenomas in organs of endodermal origin including the intestine. These fish may prove useful for genetic screens for genes that modify cancer risk ( Haramis et al., 2006).
Hair Follicle Tumors
Leukemia
Drawing from the parallels between self-renewal and cancer in the gut and hair follicle, the effects of Wnt pathway components on hematopoietic progenitors predict that Wnt deregulation may contribute to hematological malignancies. Indeed, a recent report suggests that leukemic growth of both myeloid and lymphoid lineages is dependent on Wnt signaling. Granulocyte-macrophage progenitors from Chronic Myelogenous Leukemia patients and blast crisis cells from patients resistant to therapy display active Wnt signaling as demonstrated by Tcf reporter activity and the accumulation of nuclear β-catenin (Jamieson et al., 2004).
Over the last 20 years, a detailed outline of the canonical Wnt pathway has emerged. Although it is likely that most core components of the pathway have now been identified, much remains to be learned about the biochemical events that connect these components. Many of the gaps in our knowledge are due to the notorious difficulties in the production of purified Wnt proteins. Few good Wnt antibodies exist and, 25 years after the cloning of Wnt1, its structure remains unknown. The routing and the coincident posttranslational modifications of Wnt proteins in the secreting cell are incompletely understood. And the rules that dictate the movement of Wnt proteins between cells remain uncertain. However, a procedure to produce soluble Wnt has recently been developed (Willert et al., 2003), which creates avenues to address many of these issues.
The components of the destruction complex have been long known, yet the biochemistry of its activity has remained elusive. APC is an essential component of the destruction complex, but what is its biochemical activity? How relevant is Dsh for the coupling of Wnt receptors to the destruction complex? And what mechanism inhibits the phosphorylation of β-catenin by the destruction complex when a Wnt signal is being transduced?
In addition, a multitude of proposed pathway components, not discussed here, may activate, modify, or inhibit Wnt signaling or may be involved in crosstalk to other pathways. An updated, comprehensive list of these putative components and interactions appears on http://www.stanford.edu/∼rnusse/wntwindow.html. Often based on single studies, these candidate components remain to be independently confirmed.
Wnt signaling ultimately controls developmental fates through the transcription of cell type-specific programs of Tcf target genes. Recent developments in array-based technology allow detailed analysis of the nuclear transcriptional response to Wnt signals. With these technologies, it is expected that the dissection of the gene programs in various developmental or pathological events will provide a wealth of insight into the biology of these processes.
7.10.5 Wnt.β-Catenin Signaling. Components, Mechanisms, and Diseases
MacDonald BT1, Tamai K, He X.
Dev Cell. 2009 Jul; 17(1):9-26
http://dx.doi.org/10.1016%2Fj.devcel.2009.06.016
Signaling by the Wnt family of secreted glycolipoproteins via the transcription co-activator β-catenin controls embryonic development and adult homeostasis. Here we review recent progresses in this so-called canonical Wnt signaling pathway. We discuss Wnt ligands, agonists and antagonists and their interactions with Wnt receptors. We also dissect critical events that regulate β-catenin stability from Wnt receptors to the cytoplasmic β-catenin destruction complex, and nuclear machinery that mediates β-catenin-dependent transcription. Finally we highlight some key aspects of Wnt/β-catenin signaling in human diseases including congenital malformations, cancer and osteoporosis and potential therapeutic implications.
Signaling by the Wnt family of secreted glycolipoproteins is one of the fundamental mechanisms that direct cell proliferation, cell polarity and cell fate determination during embryonic development and tissue homeostasis (Logan and Nusse, 2004). As a result, mutations in the Wnt pathway are often linked to human birth defects, cancer and other diseases (Clevers, 2006). A critical and most studied Wnt pathway is canonical Wnt signaling, which functions by regulating the amount of the transcriptional co-activator β-catenin that controls key developmental gene expression programs. This review focuses on our current understanding of Wnt/β-catenin signaling, drawing mainly from genetic, developmental and biochemical analyses in Drosophila, Xenopus, mice and humans. For more comprehensive and historic perspective we refer readers to earlier reviews (Clevers, 2006; Logan and Nusse, 2004) and the Wnt homepage (www.stanford.edu/~rnusse/wntwindow.html). The nematode Caenorhabditis elegans exhibits similar but also divergent Wnt/β-catenin pathways, which are covered elsewhere (Mizumoto and Sawa, 2007) and in the accompanying review (Kimble 2009). Wnt also activates a number of non-canonical signaling pathways that are independent of β-catenin and have been recently reviewed (Seifert and Mlodzik, 2007; Wang and Nathans, 2007).
The central logic of Wnt/β-catenin signaling has emerged from two decades of studies (Figure 1). In the absence of Wnt, cytoplasmic β-catenin protein is constantly degraded by the action of the Axin complex, which is composed of the scaffolding protein Axin, the tumor suppressor adenomatous polyposis coli gene product (APC), casein kinase 1 (CK1), and glycogen synthase kinase 3 (GSK3). CK1 and GSK3 sequentially phosphorylate the amino terminal region of β-catenin, resulting in β-catenin recognition by β-Trcp, an E3 ubiquitin ligase subunit, and subsequent β-catenin ubiquitination and proteasomal degradation (He et al., 2004). This continual elimination of β-catenin prevents β-catenin from reaching the nucleus, and Wnt target genes are thereby repressed by the DNA-bound T cell factor/lymphoid enhancer factor (TCF/LEF) family of proteins (Figure 1a). The Wnt/β-catenin pathway is activated when a Wnt ligand binds to a seven-pass transmembrane Frizzled (Fz) receptor and its co-receptor, low-density lipoprotein receptor related protein 6 (LRP6) or its close relative LRP5. The formation of a likely Wnt-Fz-LRP6 complex together with the recruitment of the scaffolding protein Dishevelled (Dvl) results in LRP6 phosphorylation and activation and the recruitment of the Axin complex to the receptors. These events lead to inhibition of Axin-mediated β-catenin phosphorylation and thereby to the stabilization of β-catenin, which accumulates and travels to the nucleus to form complexes with TCF/LEF and activates Wnt target gene expression (Figure 1b).

Overview of Wnt.β-catenin signaling nihms196288f1
Overview of Wnt/β-catenin signaling
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861485/bin/nihms196288f1.gif
Figure 1 Overview of Wnt/β-catenin signaling
Wnt ligands and biogenesis
Wnts are conserved in all metazoan animals. In mammals, complexity and specificity in Wnt signaling are in part achieved through 19 Wnt ligands, which are cysteine rich proteins of approxiamately 350-400 amino acids that contain an N-terminal signal peptide for secretion. Murine Wnt3a represents the first purified and biochemically characterized Wnt protein (Willert et al., 2003) owing to its relatively efficient secretion (in contrast to most other Wnt proteins). In addition to N-linked glycosylation, which is required for Wnt3a secretion (Komekado et al., 2007), Wnt3a undergoes two types of lipid modifications that likely account for the hydrophobicity and poor solubility of Wnt proteins (Hausmann et al., 2007). The first reported lipididation was the addition of palmitate to cysteine 77 (Willert et al., 2003). Its mutation had minimal effect on Wnt3a secretion but diminished the ability of Wnt3a to activate β-catenin signaling (Galli et al., 2007;Komekado et al., 2007; Willert et al., 2003). The second identified lipididation was a palmitoleoyl attached to serine 209, and its mutation resulted in Wnt3a accumulation in the endoplasmic reticulum (ER) and failure in secretion (Takada et al., 2006).
Drosophila Wingless (Wg) is the Wnt molecule most investigated in vivo (Hausmann et al., 2007). These studies plus work in nematodes have identified genes that regulate Wnt biogenesis and secretion. Porcupine (Porc) encodes a multipass transmembrane ER protein that contains an O-acyl transferase domain suggesting a role in Wg lipid modification (Hausmann et al., 2007). Porc deficiency results in Wg and Wnt3a accumulation in the ER and diminished Wnt3a palmitoleoylation at serine 209 (Takada et al., 2006), suggesting that Porc is responsible for this particular lipidation. Whether Porc or a distinct acyltransferase is involved in Wnt3a palmitoylation at cysteine 77 remains unknown.
Two additional proteins/protein complexes were identified for Wg/Wnt secretion: Wntless (Wls), also known as Evenness interrupted (Evi) or Sprinter (Srt), in Drosophila and the retromer complex in nematodes (Hausmann et al., 2007). Wls is a multipass transmembrane protein that localizes to the Golgi, endocytic compartments and the plasma membrane, and is essential for Wg secretion. The retromer complex, which is composed of five subunits, was defined first in yeast. It mediates membrane protein trafficking between endosomes and the Golgi apparatus (Hausmann et al., 2007). Several groups recently reported that the retromer complex is required for retrieval/recycling of Wls from the endosome to the Golgi (Belenkaya et al., 2008; Franch-Marro et al., 2008b; Pan et al., 2008a; Port et al., 2008; Yang et al., 2008), likely mediated by direct interaction between Wls and the retromer Vps35 subunit. Loss of retromer function causes Wls to be degraded in the lysosomes and results in reduction of Wls and thus Wnt secretion. These studies led to an emerging picture of Wnt biogenesis (Figure 2). Wnt is glycosylated and lipid modified by Porc in the ER, and is escorted by Wls from the Golgi to the plasma membrane for secretion. Wls is recycled by endocytosis and trafficked back to Golgi by the retromer. Note that porc, wls and retromer mutants largely phenocopywg/wnt mutants in flies and worms, attesting their dedicated roles in Wnt biogenesis.

Wnt biogenesis and secretion nihms196288f2
Wnt biogenesis and secretion
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861485/bin/nihms196288f2.gif
Figure 2 Wnt biogenesis and secretion
Wnt extracellular distribution and movement
Wnt proteins can function as morphogens that are capable of both short and long range signaling, as best demonstrated for Wg. Wg lipidation raises the issue of its diffusion and distribution through the aqueous extracellular space. Indeed purified Wnt3a exhibits increased activity via artificial liposomal packaging (Morrell et al., 2008). Two distinct Wg secretory pathways for short and long range signaling have been speculated but not fully substantiated. Wg may form multimers to bury lipid modifications inside (Katanaev et al., 2008), or bind to lipoprotein particles, which may be involved in Wg long range signaling (Panakova et al., 2005) (Figure 2). The membrane microdomain protein reggie-1/flotillin-2 specifically promotes Wg long-range secretion (Katanaev et al., 2008). The Wg receptors (see below) and heparan sulfate proteoglycans (HSPGs) such as Dally and Dally-like protein have important roles in the Wg morphogen concentration via regulating Wg degradation, diffusion, endocytosis/transcytosis, and may function in Wg signaling as potential low-affinity co-receptors (Lin, 2004). Note that reggie-1/flotillin-2, lipoprotein particles, Dally and Dally-like protein are important analogously for secreted Hedgehog morphogen, which is also lipid modified (Katanaev et al., 2008; Lin, 2004; Panakova et al., 2005).
Wnt receptors: Frizzled and LRP5/6
Two distinct receptor families are critical for Wnt/β-catenin signaling (Figure 3): the Frizzled (Fz or Fzd) seven-pass transmembrane receptors (Logan and Nusse, 2004) and the LDL receptor-related proteins 5 and 6 (LRP5 and LRP6) (He et al., 2004). The Wnt-receptor relationship is best illustrated for Wg, which binds toDrosophila Fz2 (Dfz2) and Dfz1 with high affinity (1-10 nM) and requires either Fz in a redundant manner (Logan and Nusse, 2004). Wg reception also absolutely depends on Arrow, the LRP5/6 homolog (He et al., 2004). The mammalian genome harbors 10 Fz genes, most of which have variable capacities to activate β-catenin signaling when co-overexpressed with Wnt and LRP5/6 (e.g., Binnerts et al., 2007) and functional redundancy among Fz members is likely prevalent (Logan and Nusse, 2004). Between the two LRPs, LRP6 plays a more dominant role and is essential for embryogenesis whereas LRP5 is dispensable for embryogenesis but critical for adult bone homeostasis. Nonetheless LRP5 and LRP6 are partially redundant as their functions together are required for mouse gastrulation (He et al., 2004). Most data, including Wnt binding to LRP5/6 and Wnt1-Fz8-LRP6 complex formation in vitro and observations that engineered Fz-LRP5/6 proximity is sufficient to activate β-catenin signaling (Cong et al., 2004; Holmen et al., 2005;Tolwinski et al., 2003), support the model that Wnt induces the formation of Fz-LRP5/6 complex (He et al., 2004) (Figure 1). But unambiguous demonstration of this receptor complex in vivo is lacking. It is noteworthy that Wnt3a palmitoylation (at cysteine 77) is important for binding to both Fz and LRP6 (Cong et al., 2004; Komekado et al., 2007), explaining in part the importance of this lipid modification

Secreted Wnt antagonists and agonists nihms196288f3
Secreted Wnt antagonists and agonists
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861485/bin/nihms196288f3.gif
Figure 3 Secreted Wnt antagonists and agonists
A particular Wnt may activate β-catenin and/or non-canonical pathways depending on the receptor complement (van Amerongen et al., 2008). Fz function is involved in β-catenin and non-canonical pathways. The Fz-LRP5/6 co-receptor model stipulates that a Wnt-Fz pair capable of recruiting LRP5/6 activates the β-catenin pathway, consistent with the specific requirement of LRP5/6 in Wnt/β-catenin signaling (He et al., 2004). However some evidence suggests that LRP6 antagonizes non-canonical Wnt signaling in vivo, possibly via competing for Wnt ligands (Bryja et al., 2009) or an unknown mechanism (Tahinci et al., 2007). Other Wnt receptors exist such as Ryk and ROR2, which are not required for, but in some cases may antagonize, Wnt/β-catenin signaling (van Amerongen et al., 2008).
Wnt antagonists and agonists
Several secreted protein families antagonize or modulate Wnt/β-catenin signaling (Figure 3). sFRPs (secreted Frizzled related proteins), and WIF (Wnt inhibitory protein) bind to Wnt, and in the case of sFRPs, also to Fz (Figure 3), and thereby function as Wnt antagonists for both β-catenin and non-canonical signaling (Bovolenta et al., 2008). Loss-of-function studies in mice have revealed significant redundancy for the sFRP genes (Satoh et al., 2008). The Wnt-binding property suggests that sFRPs and WIF may also regulate Wnt stability and diffusion/distribution extracellularly beyond just Wnt inhibitors. Some sFRPs have been shown to have Wnt-independent activity such as regulators of extracellular proteinases (Bovolenta et al., 2008).
Two distinct classes of Wnt inhibitors are the Dickkopf (Dkk) family and the Wise/SOST family (Figure 3). Dkk proteins, exemplified by Dkk1, are LRP5/6 ligands/antagonists and are considered specific inhibitors for Wnt/β-catenin signaling. Although two different models for Dkk1 action have been proposed (Mao et al., 2002; Semenov et al., 2001), recent biochemical and genetic studies (Ellwanger et al., 2008; Semenov et al., 2008; Wang et al., 2008) have argued against the model that Dkk1 inhibits Wnt signaling via inducing LRP6 internalization/degradation through transmembrane Kremen (Krm) proteins (Mao et al., 2002). Dkk1 disruption of Wnt-induced Fz-LRP6 complex remains a more likely mechanism (Semenov et al., 2001), with Krm playing a minor modulatory role in specific tissues (Ellwanger et al., 2008). Wise and SOST constitute another family of LRP5/6 ligands/antagonists (Itasaki et al., 2003; Li et al., 2005; Semenov et al., 2005). Like Dkk1, SOST is able to disrupt Wnt-induced Fz-LRP6 complex in vitro (Semenov et al., 2005). Both Dkk1 and SOST are strongly implicated in human diseases (see below).
Shisa proteins represent a distinct family of Wnt antagonists (Figure 3), which trap Fz proteins in the ER and prevent Fz from reaching the cell surface, thereby inhibiting Wnt signaling cell-autonomously (Yamamoto et al., 2005). Shisa proteins also antagonize FGF (fibroblast growth factor) signaling by trapping FGF receptors in the ER. Other Wnt antagonists with multivalent activities exist. Xenopus Cerberus binds to and inhibits Wnt as well as Nodal and BMP (bone morphogenetic protein) (Piccolo et al., 1999), and IGFBP-4 (Insulin-like growth-factor-binding protein-4) antagonizes Wnt signaling via binding to both Fz and LRP6, in addition to modulating IGF signaling (Zhu et al., 2008).
Norrin and R-spondin (Rspo) proteins are two families of agonists for Wnt/β-catenin signaling (Figure 3). Norrin is a specific ligand for Fz4 and acts through Fz4 and LRP5/6 during retinal vascularization (Xu et al., 2004). Rspo proteins exhibit synergy with Wnt, Fz and LRP6 (Kazanskaya et al., 2004; Kim et al., 2005;Nam et al., 2006; Wei et al., 2007), and show genetic interaction with LRP6 during embryogenesis (Bell et al., 2008), but their mechanism of action is controversial. Results that Rspo binds to both Fz and LRP6 (Nam et al., 2006), to LRP6 primarily (Wei et al., 2007), or to neither (Kazanskaya et al., 2004) have been reported. Another model suggests that Rspo is a ligand for Krm and antagonizes Dkk/Krm-mediated LRP6 internalization (Binnerts et al., 2007), but this seems unlikely given that Krm1 and Krm2 double knockout mice are viable and do not exhibit Rspo mutant phenotypes, and Rspo activates β-catenin signaling in cells lacking both Krm genes (Bell et al., 2008; Ellwanger et al., 2008). Rspo genes are often co-expressed with and depend on Wnt for expression (Kazanskaya et al., 2004), and may represent a means of positive feedback that reinforces Wnt signaling. Mutations in Norrin and Rspo genes cause distinct hereditary diseases (see below).
Wnt signaling
Wnt-off state: β-catenin phosphorylation/degradation by the Axin complex
Cytosolic β-catenin phosphorylation/degradation and its regulation by Wnt are the essence of Wnt signaling (Figure 1). The scaffolding protein Axin uses separate domains to interact with GSK3, CK1α, and β-catenin and coordinates sequential phosphorylation of β-catenin at serine 45 by CK1α and then at threonine 41, serine 37 and serine 33 by GSK3 (Kimelman and Xu, 2006). β-catenin phosphorylation at serine 33 and 37 creates a binding site for the E3 ubiquitin ligase β-Trcp, leading to β-catenin ubiquitination and degradation (Figure 4). Mutations of β-catenin at and surrounding these serine and threonine residues are frequently found in cancers, generating mutant β-catenin that escapes phosphorylation and degradation (Table 1). Axin also contains an RGS (regulator of G protein signaling) domain that interacts with APC, a large multifunctional scaffolding protein that itself binds β-catenin. These core Axin complex components (Kimelman and Xu, 2006) share a common goal of ensuring β-catenin phosphorylation and degradation. Indeed both APC and Axin are tumor suppressor genes, and APC mutations are particularly prevalent in colorectal cancer (Table 1).
Regulation of Axin complex assembly for β-catenin degradation
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861485/bin/nihms196288f4.gif
Figure 4 Regulation of Axin complex assembly for β-catenin degradation
Table 1 Human diseases associated with mutations of the Wnt signaling components
Several aspects of the Axin complex deserve further discussion. (i) In addition to β-catenin, GSK3 and CK1 also phosphorylate Axin and APC, leading to increased association of Axin and APC with β-catenin and thus enhanced β-catenin phosphorylation/degradation (Huang and He, 2008; Kimelman and Xu, 2006) (Figure 4). (ii) Two abundant serine/threonine phosphatases, PP1 and PP2A, both of which associate with Axin and/or APC, counteract the action of GSK3 and/or CK1 in the Axin complex. Thus PP1 dephosphorylates Axin and promotes the disassembly of the Axin complex (Luo et al., 2007), whereas PP2A dephosphorylates β-catenin (Su et al., 2008), each resulting in reduced β-catenin degradation (Figure 4). One should note that PP2A may have multiple and opposing roles in the Wnt pathway depending on the particular associated regulatory subunits and substrates (Kimelman and Xu, 2006). (iii) The assembly of the Axin complex appears to be multivalent and robust. In fly embryos that are null for Axin, expression, at physiological levels, of Axin mutants lacking either the APC-, GSK3-, or β-catenin-binding domain restores a significant degree of normal patterning, implying a quasi-functional Axin complex assembly via multivalent interactions; furthermore, some of these Axin deletion mutants can complement each other and restore fly viability, possibly via Axin dimerization or multimerization (Peterson-Nedry et al., 2008). Indeed Axin has multiple potential dimerization domains (Luo et al., 2005) and the Axin DIX domain may form multimeric polymers (Schwarz-Romond et al., 2007a). (iv) Axin concentration is exceedingly low compared to other components in Xenopus oocytes, indicating that Axin is rate limiting for the complex assembly. This feature may ensure that changes in the Axin protein level will not fluctuate the availability of GSK3 (or other components) for non-Wnt functions, thereby further insulating Wnt and other signaling events (Lee et al., 2003). It is unknown, however, whether the drastic difference between the concentration of Axin versus the other components applies universally, and whether different cells employ quantitative differences in the ratio of Axin and other components to shape their unique Wnt response kinetics (such as the speed and level of β-catenin accumulation). Indeed in Drosophila photoreceptors, APC appears to be present at minimal levels such that a 50% reduction alters the graded Wg response (Benchabane et al., 2008).
Other proteins such as WTX (Wilms tumor gene on the X chromosome) may have roles in β-catenin degradation. Loss of WTX and activating β-catenin mutations seem to have non-overlapping occurrence in Wilms tumor (a pediatric kidney cancer) (Rivera et al., 2007). WTX binds to β-catenin, Axin, APC and β-Trcp to promote β-catenin ubiquitination, although its biochemical role remains unknown (Major et al., 2007). Another Axin-binding protein Diversin can facilitate β-catenin degradation via recruiting CK1ε to phosphorylate β-catenin (Schwarz-Romond et al., 2002).
APC function and APC-Axin cross regulation
The biochemical nature of APC has been enigmatic. A recent study suggested that APC protectsβ-catenin from dephosphorylation by PP2A thereby enhancing β-catenin phosphorylation/degradation (Su et al., 2008) (Figure 4), consistent with the observation that Axin overexpression causes β-catenin degradation even in cells lacking APC function (Behrens et al., 1998). Surprisingly APC (upon phosphorylation by CK1/GSK3) and Axin bind to and compete for the same β-catenin interaction interface, leading to a proposal that APC acts as a “ratchet” to remove phosphorylated β-catenin from Axin for ubiquitination and for making Axin available for a further round of β-catenin phosphorylation (Kimelman and Xu, 2006; Xing et al., 2003). A different model was proposed based on differential β-catenin binding affinity by unphosphorylated versus phosphorylated APC (Ha et al., 2004). APC has also been shown to promote β-catenin nuclear export and to act as a chromatin-associated suppressor for β-catenin target genes, thus functioning in the nucleus (see below).
Another paradoxical observation is that APC has a positive function in physiological and ectopic Wg/Wnt signaling through the promotion of Axin degradation (Lee et al., 2003; Takacs et al., 2008) (Figure 4). One model suggests that this represents a fail-safe mechanism to buffer dramatic β-catenin fluctuations when APC levels vary (Lee et al., 2003). Thus a decrease in the APC level results in higher Axin amounts, compensating for β-catenin degradation. APC-mediated Axin degradation depends on the APC amino terminal domain that is not involved inβ-catenin degradation (Takacs et al., 2008). It is intriguing that colon cancer cells are rarely null for APC but rather retain the amino terminal half, and may have hijacked a part of this fail-safe regulation for tumorigenesis. Conversely Axin can also facilitate APC degradation upon overexpression (Choi et al., 2004), constituting perhaps the other side of the Axin-APC regulation circuit (Figure 4). Mechanisms for Axin and APC degradation, which are proteosome-dependent, have not been characterized.
Wnt-on state
Activation of Wnt receptors
Wnt signaling requires both Fz and LRP6 (or LRP5), likely through a Wnt-induced Fz-LRP6 complex (Figure 1). Wnt-induced LRP6 phosphorylation is a key event in receptor activation (Tamai et al., 2004). LRP6, LRP5 and Arrow each have five reiterated PPPSPxS motifs (P, proline; S, serine or threonine, x, a variable residue), which are essential for LRP6 function and are each transferrable to a heterologous receptor to result in constitutive β-catenin signaling (MacDonald et al., 2008; Tamai et al., 2004; Zeng et al., 2005). These dually phosphorylated PPPSPxS motifs are docking sites for the Axin complex (Davidson et al., 2005;Tamai et al., 2004; Zeng et al., 2005), thereby recruiting Axin to LRP6 upon Wnt stimulation (Mao et al., 2001) (Figure 5).

Models of Wnt receptor activation nihms196288f5
Models of Wnt receptor activation
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861485/bin/nihms196288f5.gif
Figure 5 Models of Wnt receptor activation
The kinases responsible for PPPSPxS phosphorylation have been identified unexpectedly as GSK3 and CK1 (Davidson et al., 2005; Zeng et al., 2005). Although one study argued that only CK1 phosphorylation is Wnt-induced (Davidson et al., 2005), most available data support that Wnt induces PPPSP phosphorylation (Binnerts et al., 2007; Khan et al., 2007; Pan et al., 2008b; Wei et al., 2007), which is carried out by GSK3 and primes xS phosphorylation by CK1, thereby leading to dually induced phosphorylation (Zeng et al., 2005) (Figure 5). Although potential involvement of additional kinases cannot be ruled out, experiments in GSK3α/β null cells indicate that GSK3 accounts for most, if not all, PPPSP phosphorylation (Zeng et al., 2008; Zeng et al., 2005). As in β-catenin phosphorylation, Axin-bound GSK3 appears to mediate LRP6 phosphorylation (Zeng et al., 2008). Thus PPPSPxS phosphorylation exhibits a mirror image of β-catenin phosphorylation in sequential order, in priming requirement, and importantly in functionality, but apparently by the same Axin-GSK3 complex (Huang and He, 2008) (Figure 5). This unusual mechanism, using the same kinase complex for both positive and negative regulation, is reminiscent of another morphogenetic pathway, Hedgehog signaling in Drosophila (Price, 2006), and implies a simple view that Wnt signaling regulates the two opposing activities of the Axin-GSK3 complex. One caveat is that GSK3 is genetically defined as a negative regulator of β-catenin signaling. The positive requirement of GSK3 in LRP6 activation is demonstrated when a membrane-tethered GSK3 inhibitory peptide blocks Wnt signaling (Zeng et al., 2008).
Fz function is required for Wnt-induced LRP6 phosphorylation, and forced Fz-LRP6 association is sufficient to trigger LRP6 phosphorylation (Zeng et al., 2008). Fz function is usually linked to Dsh/Dvl (Wallingford and Habas, 2005), a cytoplasmic scaffolding protein that may directly interact with Fz (Wong et al., 2003). Indeed Fz-Dvl interaction and Dvl function are critical for Wnt-induced LRP6 phosphorylation (Bilic et al., 2007; Zeng et al., 2008). As Dvl interacts with Axin (Wallingford and Habas, 2005), and is required for Axin recruitment to the plasma membrane during Wg signaling (Cliffe et al., 2003) or in Fz overexpression (Zeng et al., 2008), one model stipulates that Fz-Dvl recruitment of the Axin-GSK3 complex initiates LRP6 phosphorylation by GSK3 (Zeng et al., 2008) (Figure 5).
Several features of Wnt receptor activation deserve further discussion. (i) The observation that Axin is required for LRP6 phosphorylation, and phosphorylated LRP6 in turn recruits Axin suggests a positive feed-forward loop, potentially amplifying and ensuring the phosphorylation of all five PPPSPxS motifs (Figure 5). Indeed the phosphorylation of these motifs relies on the presence of one another, and LRP6 activity is particularly sensitive to the PPPSPxS copy number (MacDonald et al., 2008; Wolf et al., 2008). This may explain the distinct roles of Fz and LRP6/Arrow in the “initiation” (which requires both Fz and Arrow) and “amplification” (which requires Arrow only) during Wg signaling (Baig-Lewis et al., 2007) (Figure 5a). (ii) Wnt-induced clustering of Fz-LRP6 receptor has been reported that critically depend on Dvl, Axin and GSK3 for formation (see below) (Bilic et al., 2007; Schwarz-Romond et al., 2007a). Although unambiguous evidence for such aggregation under physiological conditions without overexpression remains to be shown, this “signalsome” model (Figure 5b) and the “initiation-amplification” model (Figure 5a) together provide a spatial and temporal framework for understanding Wnt receptor activation. (iii) Wnt also induces LRP6 phosphorylation by CK1γ outside the PPPSPxS motifs, in particular in a conserved S/T cluster amino-terminal to the first PPPSPxS motif (Davidson et al., 2005). This region upon phosphorylation binds to GSK3 (Piao et al., 2008), potentially accounting for observed LRP6-GSK3 interaction (Mi et al., 2006; Zeng et al., 2005). The significance of this S/T cluster to LRP6 function has not been investigated in the intact receptor, but these results imply multiple interaction interfaces among LRP6, Axin and GSK3. (iv) Wnt may also “activate” Fz, which is structurally related to G-protein coupled receptors (GPCRs). Some genetic and pharmacological evidence suggests that trimeric G proteins, specifically the Gαo and Gαq, are required downstream of Fz and probably upstream of Dvl in Wnt/β-catenin signaling (Katanaev et al., 2005; Liu et al., 2001; Liu et al., 2005). Whether G proteins are involved in Wnt/Fz/Dvl-regulated LRP6 phosphorylation is unknown.
Dvl is involved in Wnt/β-catenin and other Wnt/Fz-dependent pathways and has numerous putative binding partners (Wallingford and Habas, 2005). For example CK1ε (or CK1δ) binds to Dvl and is a potent activator of β-catenin signaling, possibly via phosphorylating Dvl, LRP6 and/or the Axin complex (Price, 2006) (Figure 5). PP2A also associates with Dvl but has a positive or negative influence on Wnt signaling depending on the associated regulatory subunit (Kimelman and Xu, 2006). In addition Dvl is subjected to proteasomal degradation via distinct ubiquitination pathways (Angers et al., 2006; Simons et al., 2005). Some of these Dvl regulation events have been suggested to switch Dvl between β-catenin and non-canonical pathways. Despite these progresses, the mechanism by which Dvl acts in Wnt/β-catenin signaling remains enigmatic. Two recent findings suggest potential new insights. (i) Polymerization/aggregation of Dvl (and Axin). Fz-Dvl and Dvl-Axin interactions are relatively weak (Schwarz-Romond et al., 2007b; Wong et al., 2003). However Dvl and Axin each harbor a homologous DIX domain that exhibit dynamic polymerization (Schwarz-Romond et al., 2007a). This unusual property is proposed to allow Dvl and Axin to form large aggregates that facilitate weak but dynamic protein interactions (Figure 5b). Indeed Wnt-induced receptor clustering requires an intact Dvl DIX domain (Bilic et al., 2007; Schwarz-Romond et al., 2007a). It is unclear whether Wnt regulates DIX-dependent polymerization, and perhaps in a related manner, Fz-Dvl or Dvl-Axin interaction. (ii) Dvl stimulation of phosphatidylinositol 4,5-bisphosphate [PtdIns (4,5)P2 or PIP2] production by sequential actions of phosphatidylinositol 4-kinase type II (PI4KIIα) and phosphatidylinositol-4-phosphate 5-kinase type I (PIP5KI) (Pan et al., 2008b). Wnt induces Dvl, via the DIX domain, to bind to and activate PIP5K, and the resulting PIP2 production is suggested to promote LRP6 clustering and phosphorylation, although the underlying mechanism remains unclear (Figure 5c). Given that PIP2 has pleiotropic functions in cells including receptor endocytosis (see below), other potential mechanisms for PIP2 in LRP6 phosphorylation remain to be explored. Nonetheless Dvl DIX polymerization and stimulation of PIP2 may act in concert to ensure LRP6 clustering/phosphorylation/activation.
Other regulatory events at or proximal to Wnt receptors
A cytoplasmic protein in vertebrates, referred to as Caprin-2, binds to LRP6 and facilitates LRP6 phosphorylation by GSK3 (Ding et al., 2008). Caprin-2 has an oligomerization domain that may enhance LRP6 aggregation, and Caprin-2 additionally may also associate with both GSK3 and Axin and promote LRP6-Axin-GSK3 complex formation (Ding et al., 2008). Besides the requirement of Dvl, recruitment of Axin to the receptor complex may involve a giant protein (600 kD), Macf1 (microtubule actin cross-linking factor 1) (Chen et al., 2006). Macf1 is a member of the spectraplakin family of proteins that link the cytoskeleton to junctional proteins. Defective gastrulation in Macf1−/− mouse embryos phenotypically resembles Lrp5/6−/− double knockout mutants. On Wnt stimulation Macf1 associates with the Axin complex (including APC) in the cytosol and with LRP6 and the Axin complex (but not APC) in the membrane fraction (Chen et al., 2006), and may shuttle Axin to LRP6 (Figure 5). This Macf1 function may be vertebrate-specific as Drosophila Macf1 (shortstop) mutants do not exhibit wg-related phenotypes. …
Inhibition of β-catenin phosphorylation
How receptor activation leads to inhibition of β-catenin phosphorylation remains uncertain, and available data suggest possible parallel mechanisms. In the LRP6-centric view, as constitutively activated forms of LRP6 fully activate β-catenin signaling in an apparently Fz and Dvl-independent manner (He et al., 2004), LRP6 represents the key output whereas Fz and Dvl act upstream to control LRP6 activation. On the other hand, Dsh overexpression in Drosophila or recombinant Dvl in Xenopus egg extracts can activate β-catenin signaling presumably in the absence of Arrow/LRP6 (Salic et al., 2000; Wehrli et al., 2000), and so does a GPCR-Fz chimeric protein in response to the GPCR ligand (Liu et al., 2001). These results argue that Fz/Dvl may activate β-catenin signaling independent of LRP6. The fact that nematodes have a related Wnt/β-catenin pathway (Kimble 2009) but have no LRP6 homolog may be consistent with this notion. Perhaps inDrosophila and vertebrates Wnt signaling components exist under sub-optimal levels and the two parallel branches need to operate together to counteract efficient β-catenin phosphorylation/degradation, whereas over-activation of either branch is sufficient to stabilize β-catenin. …
β-catenin nuclear function
β-catenin nuclear/cytoplasmic shuttling and retention
β-catenin stabilization results in its higher nuclear levels, but how β-catenin is shuttled to and retained in the nucleus is not well understood (Henderson and Fagotto, 2002; Stadeli et al., 2006). Earlier studies suggested that β-catenin enters the nucleus in an NLS (nuclear localization signal)- and importin-independent fashion by interacting directly with nuclear pore proteins (Henderson and Fagotto, 2002). β-catenin also exits the nucleus via export involving APC (Henderson and Fagotto, 2002), Axin (Cong and Varmus, 2004), and RanBP3 (Ran binding protein 3), which binds to β-catenin in a Ran-GTP dependent manner (Hendriksen et al., 2005). Live cell imaging suggests that while Axin and APC can enrich β-catenin in the cytoplasm and TCF and β-catenin co-activators (BCL9 and Pygopus, see below) increase nuclear β-catenin, they do not accelerate the export or import rate of β-catenin, thereby arguing for their roles in β-catenin retention rather than shuttling (Krieghoff et al., 2006). Thus β-catenin nuclear and cytoplasmic partitioning is likely the dynamic sum of both shuttling and retention between the two compartments via multiple mechanisms. ….
TCF/LEF
The TCF/LEF family of DNA-bound transcription factors is the main partner for β-catenin in gene regulation (Arce et al., 2006; Hoppler and Kavanagh, 2007). TCF represses gene expression by interacting with the repressor Groucho (TLE1 in human), which promotes histone deacetylation and chromatin compaction; Wnt-induced β-catenin stabilization and nuclear accumulation leads TCF to complex with β-catenin, which appears to displace Groucho (Daniels and Weis, 2005) and recruits other co-activators for gene activation (Figure 1). While a single TCF gene is found in Drosophila and worm, four TCF genes, TCF1, LEF1, TCF3 and TCF4, exist in mammals. Alternative splicing and promoter usage produce a large number of TCF variants with distinct properties (Arce et al., 2006; Hoppler and Kavanagh, 2007). TCF proteins are HMG (high mobility group) DNA-binding factors, and upon binding to a DNA consensus sequence referred to as the Wnt responsive element (WRE), CCTTTGWW (W represents either T or A), they cause significant DNA bending that may alter local chromatin structure. A genome-wide analysis in colon cancer cells suggests that TCF4/β-catenin target genes are frequently “decorated” with multiple WREs, most of which are located at large distances from transcription start sites (Hatzis et al., 2008). Some TCF1 and TCF4 splicing variants harbor a second DNA-binding domain called C-clamp, which recognizes an additional GC element downstream of the typical WRE, allowing regulation of different sets of target genes (Atcha et al., 2007). These similarities and differences, combined with overlapping and unique expression patterns, underlie in part distinct and sometimes redundant functions of vertebrate/mammalian TCF genes. ….
Three major strategies exist to regulate TCF/β-catenin transcription. (i) Alternative promoter usage in TCF-1 and LEF-1 genes produces dnTCF-1/dnLEF-1, which lack the amino-terminal β-catenin-binding domain and thus act as the endogenous dominant negative TCF/LEF (Arce et al., 2006; Hoppler and Kavanagh, 2007). Indeed the TCF-1 locus acts as an intestinal tumor suppressor primarily due to the production of dnTCF-1, which antagonizes TCF-4 in stem cell renewal. (ii) Nuclear antagonists Chibby and ICAT bind to β-catenin and disrupt β-catenin/TCF and β-catenin/co-activator interactions and promote β-catenin nuclear export (Li et al., 2008; Tago et al., 2000). Besides these devoted inhibitors, many DNA-binding transcription factors interact with β-catenin or TCF and antagonize TCF/β-catenin-dependent transcription (Supplemental Table 1). For example, KLF4 inhibition of β-catenin transcriptional activation is important for intestinal homeostasis and tumor suppression (Zhang et al., 2006). (iii) Post-translational modifications of TCF/LEF exist including phosphorylation, acetylation, sumoylation, and ubiquitination/degradation (Arce et al., 2006; Hoppler and Kavanagh, 2007). For instance, TCF-3 phosphorylation by CK1ε and LEF-1 phosphorylation by CK2 enhances their binding to β-catenin and diminishes LEF-1 binding to Groucho/TLE, whereas LEF-1 and TCF-4 phosphorylation by NLK (Nemo-like kinase) leads to less LEF/TCF/β-catenin complex binding to DNA and to LEF-1/TCF-4 degradation. LEF-1 and TCF-4 sumoylation (by the SUMO ligase PIASy) represses LEF-1 activity by targeting it to nuclear bodies but enhances TCF-4/β-catenin transcription, while CBP-mediated acetylation of TCF results in decreased TCF/β-catenin-binding in Drosophila and increased TCF nuclear retention in nematodes, both leading to transcriptional repression. These diverse modifications are often specific to individual TCF/LEF proteins, conferring differential regulation.
β-catenin associated co-activators
A plethora of β-catenin associated co-activators have been identified. These multi-protein complexes include BCL9 and Pygopus (Pygo), Mediator (for transcription initiation), p300/CBP and TRRAP/TIP60 histone acetyltransferases (HATs), MLL1/2 histone methyltransferases (HMTs), the SWI/SNF family of ATPases for chromatin remodeling, and the PAF1 complex for transcription elongation and histone modifications (Mosimann et al., 2009; Willert and Jones, 2006) (Figure 6). While the central Arm-repeats of β-catenin associate with TCF, and the amino-terminal Arm-repeat binds to BCL9, most of the co-activator complexes interact with the β-catenin carboxyl terminal portion (Figure 6), creating a dazzling interplay between β-catenin and the transcriptional apparatus and the chromatin. Indeed TCF/β-catenin binding to WREs leads to histone acetylation in a CBP-dependent manner over a significant genomic distance (30 kb), suggesting that local TCF/β-catenin recruitment results in widespread chromatin modifications (Parker et al., 2008). …

Nuclear TCF.β-catenin co-activator complexes nihms196288f6
Nuclear TCF/β-catenin co-activator complexes
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861485/bin/nihms196288f6.gif
Figure 6 Nuclear TCF/β-catenin co-activator complexes
…..
Unlike most co-activators that have general roles in transcription, BCL9 and Pygo in Drosophila are specifically required for β-catenin-dependent transcription and their biochemical functions proposed provide a glimpse of the complexity of TCF/β-catenin-coactivator interactions (Mosimann et al., 2009). (i) BCL9 and Pygo function as a “chain of activators” (Hoffmans et al., 2005). β-catenin binding to BCL9 recruits Pygo, which also interacts with Mediator (Carrera et al., 2008) (Figure 6); (ii) Pygo is constitutively nuclear and may have a role in recruiting/retaining BCL9/β-catenin in the nucleus upon Wg/Wnt signaling (Brembeck et al., 2004; Townsley et al., 2004); (iii) Pygo also co-occupies chromatin loci with and via TCF in the absence of Wg signaling (despite a lack of direct TCF-Pygo interaction), and may help capture BCL9/β-catenin for TCF at the onset of Wg signaling (de la Roche and Bienz, 2007); (iv) Pygo has a PHD (plant homology domain) that binds preferentially to dimethylated H3K4 upon interaction with BCL9 (Fiedler et al., 2008). This “histone code” recognition leads to the speculation that Pygo/BCL9 act during the transition from gene silencing to Wnt-induced transcription by participating in histone methylation changes. Alternatively Pygo/BCL9-binding to dimethylated H3K4 may provide a separate β-catenin anchor on chromatin, thereby freeing TCF for interaction with Groucho to pause/terminate transcription (Mosimann et al., 2009); (v) Pygo function is not required when Groucho activity is absent, suggesting that Pygo acts as an anti-repressor (Mieszczanek et al., 2008). Therefore either a single biochemical mechanism of Pygo underlies these diverse observations, or multiple functional properties of Pygo participate in β-catenin signaling. …
Nuclear functions of “cytoplasmic” Wnt signaling components
APC also acts directly on chromatin/WREs to antagonize β-catenin-mediated gene activation via promoting the exchange of co-activators with co-repressors in a stepwise and oscillating manner, as such exchange does not occur in APC mutant cancer cells (Sierra et al., 2006). How APC is recruited to chromatin is a mystery but is unlikely due to β-catenin/TCF, because APC and TCF bind to β-catenin in a mutually exclusive manner. GSK3 and β-Trcp also appear to be associated with the WRE in a cyclic fashion that synchronizes with APC but is opposite to that of β-catenin/co-activators, suggesting that they may have negative roles in TCF/β-catenin-mediated transcription (Sierra et al., 2006). Some studies have also suggested that Dvl is observed in the nucleus (Itoh et al., 2005; Torres and Nelson, 2000) and that nuclear Dvl is a component of the TCF/β-catenin complex and facilitates TCF/β-catenin interaction in conjunction with the c-Jun transcription factor (Gan et al., 2008). …
β-catenin-mediated repression and other transcriptional events
Wnt signaling, via the TCF/β-catenin complex, also represses transcription. Note that this is distinct from TCF-mediated repression in the absence of β-catenin. One mechanism is competitive repression, through which TCF/β-catenin displaces or inhibits other DNA-binding transcription activators (Kahler and Westendorf, 2003; Piepenburg et al., 2000). Another mechanism is direct repression via TCF/β-catenin binding to the canonical WREs by recruiting co-repressors (Jamora et al., 2003; Theisen et al., 2007). A third mechanism is revealed by a novel TCF binding element, AGAWAW, which specifically mediates TCF/β-catenin repression in Drosophila (Blauwkamp et al., 2008). There is evidence that β-catenin is capable of recruiting co-repressors including Groucho/TLE and histone deacetylases (Olson et al., 2006), but the mechanism by which β-catenin recruits co-activators versus co-repressors is unknown. The involvement of co-factors (Theisen et al., 2007) or distinct TCF/β-catenin configurations offers potential explanations. A less understood aspect of β-catenin signaling is that many DNA-binding transcription factors, in addition to TCF/LEF, interact with β-catenin to either activate or repress transcription (Supplemental Table 1b). These β-catenin partners in principle expand significantly the gene expression programs that are regulated by Wnt/β-catenin signaling, but further substantiation of their roles in mediating Wnt signaling is required.
Wnt/β-catenin target genes and Wnt pathway self-regulation
As Wnt/β-catenin signaling regulates proliferation, fate specification and differentiation in numerous developmental stages and adult tissue homeostasis, Wnt target genes are diverse (Vlad et al., 2008) and cell- and context-specific (Logan and Nusse, 2004). An emerging feature is that Wnt signaling components including Fz, LRP6, Axin2, TCF/LEF, Naked (a Dvl antagonist), Dkk1, and Rspo, are often regulated positively or negatively by TCF/β-catenin (Chamorro et al., 2005; Kazanskaya et al., 2004; Khan et al., 2007; Logan and Nusse, 2004). Wnt induction of Axin2, Dkk1 and Naked and suppression of Fz and LRP6 constitute negative feedback loops that dampen Wnt signaling, and the suppression of Fz and LRP6 also enhances Wg/Wnt gradient formation over longer distances (Logan and Nusse, 2004). On the contrary, Wnt induction of Rspo and TCF/LEF genes constitute positive feed-forward circuits that reinforce Wnt signaling, a feature that has been exploited during colon carcinogenesis (Arce et al., 2006; Hoppler and Kavanagh, 2007). These various Wnt pathway self-regulatory loops are mostly utilized in a cell-specific manner, affording additional complexity in the control of amplitude and duration of Wnt responses. …
Wnt/β-catenin signaling in diseases and potential therapeutics
Give the critical roles of Wnt/β-catenin signaling in development and homeostasis it is no surprise that mutations of the Wnt pathway components are associated with many hereditary disorders, cancer and other diseases (Table 1). …
LRP5 activity correlates with bone mass likely via regulation of osteoblast (bone forming cell) proliferation, whereas SOST and DKK1, which are specifically expressed in osteocytes, negatively regulates bone mass by antagonizing LRP5. …
Association of deregulated Wnt/β-catenin signaling with cancer has been well documented, particularly with colorectal cancer (Polakis, 2007) (Table 1). Constitutively activated β-catenin signaling, due to APC deficiency or β-catenin mutations that prevent its degradation, leads to excessive stem cell renewal/proliferation that predisposes cells to tumorigenesis. Indeed APC deletion or β-catenin activation in stem cells is essential for intestinal neoplasia (Fuchs, 2009). Blocking β-catenin signaling for cancer treatment has thus generated significant interests. Indeed the beneficial effect of non-steroidal anti-inflammatory drugs (NSAIDS) in colorectal cancer prevention and therapy has been attributed partially to the perturbation of TCF/β-catenin signaling through the ability of NSAIDS to inhibit Prostaglandin E2 production, which enhances TCF/β-catenin-dependent transcription (Castellone et al., 2005; Shao et al., 2005). Small molecules that disrupt TCF/β-catenin (Lepourcelet et al., 2004) or β-catenin/co-activator (CBP) interaction (Emami et al., 2004) and thereby block TCF/β-catenin signaling have been described. The task of disrupting TCF/β-catenin interaction specifically, however, is a difficult one since β-catenin interacts with TCF and other binding partners such as APC, Axin and E-cadherin via the same or overlapping interface (Barker and Clevers, 2006). Another potential therapeutic target is the kinase CDK8, which, as a Mediator subunit, is often amplified in and is required for β-catenin-dependent transcription and proliferation of colon cancer cells (Firestein et al., 2008; Morris et al., 2008). A new class of small molecules that inhibits β-catenin signaling has recently be identified (Chen et al., 2009), which via an unknown mechanism stabilizes the Axin protein, thereby promoting β-catenin degradation even in cancer cells that lack APC function. As discussed above, since Axin protein levels are the rate-limiting step for β-catenin degradation, manipulation of Axin stabilization represents a promising therapeutic strategy.
Many cancers that do not harbor mutations in the Wnt pathway nonetheless rely on autocrine Wnt signaling for proliferation or survival (Barker and Clevers, 2006). In fact APC mutant colon cancer cells maintain their dependence on Wnt and epigenetically silence the expression of secreted Wnt antagonists (He et al., 2005;Suzuki et al., 2004). Therefore targeting Wnt signaling upstream of TCF/β-catenin is also an important therapeutic option. Reagents against Wnt proteins such as antibodies (He et al., 2005) or a secreted Fz extracellular domain (DeAlmeida et al., 2007), which act outside the cancer cells to block Wnt-receptor interaction, show promise in certain experimental settings, as do small molecule and peptide inhibitors that antagonize Fz-Dvl interaction (Shan et al., 2005; Zhang et al., 2009). Small molecules have also been identified that inhibit Porcupine and thus prevent Wnt lipidation and secretion (Chen et al., 2009). We will likely see additional molecular and chemical agents that can interfere with different steps of Wnt/β-catenin signaling, whose complexity presents many potential therapeutic targets. The challenge will be ensuring that these agents target cancer cells without damaging normal tissue homeostasis.
Since the discovery of the Wnt-1 gene 27 years ago (Nusse and Varmus, 1982), Wnt/β-catenin signaling has cemented its role as a key regulatory system in biology. Studies of different animal models and human diseases have established a complex Wnt signaling network far beyond a linear pathway, with many components having multiple distinct roles and acting in different cellular compartments, and many modulators feeding into and cross-regulating within this network. The patterns of dynamic and kinetic protein phosphorylation/modification and complex assembly/disassembly are beginning to emerge. Challenges and excitement both lie ahead. (i) Novel regulators will likely continue to be identified using classical genetic, molecular, modern genomic and proteomic approaches. (ii) New analytical and imaging technologies should enable us to dissect and visualize the dynamic signaling events in vivo and to shed light on the cell biological aspects of Wnt signaling, including where, when and how signaling occurs inside the cell. (iii) Although we have obtained significant structural information on individual domains and protein interaction interfaces, atomic structures of protein complexes such as the Axin complex and ligand-receptor complexes remain daunting challenges. (iv) Additional specific small molecular inhibitors or activators with defined targets and mechanisms would provide not only leads for therapeutics but also research tools to manipulate the Wnt pathway in precise temporal and spatial manners. (v) Integration of vast amounts of information into quantitative models will allow us to predict the behavior and to study the robustness and evolvability of Wnt signaling in various biological contexts. (vi) The Wnt responsive transcriptome remains a gold mine for digging into Wnt-regulated biology. Unfolding examples include Wnt regulation of intestinal and hair follicle development/homeostasis, which has provided significant insights into stem cell biology and cancer pathogenesis (Clevers, 2006; Fuchs, 2009). As β-catenin is a co-activator for other transcription factors in addition to TCF/LEF, comparative analyses of Wnt responsive transcription programs that depend on TCF/LEF versus others will likely uncover further complexity of Wnt-regulated gene expression. (vii) β-catenin and APC are also key components in the E-cadherin cell adhesion complex and the microtubule network, but how Wnt/β-catenin signaling interacts with these cellular structures remains poorly understood. In addition, the involvement of the primary cilium, a centrosome- and microtubule-based protrusive organelle in vertebrate cells, in Wnt/β-catenin versus non-canonical Wnt signaling remains an intriguing but debated topic (Gerdes et al., 2009).
Since the discovery of the Wnt-1 gene 27 years ago (Nusse and Varmus, 1982), Wnt/β-catenin signaling has cemented its role as a key regulatory system in biology. Studies of different animal models and human diseases have established a complex Wnt signaling network far beyond a linear pathway, with many components having multiple distinct roles and acting in different cellular compartments, and many modulators feeding into and cross-regulating within this network. The patterns of dynamic and kinetic protein phosphorylation/modification and complex assembly/disassembly are beginning to emerge. Challenges and excitement both lie ahead. (i) Novel regulators will likely continue to be identified using classical genetic, molecular, modern genomic and proteomic approaches. (ii) New analytical and imaging technologies should enable us to dissect and visualize the dynamic signaling events in vivo and to shed light on the cell biological aspects of Wnt signaling, including where, when and how signaling occurs inside the cell. (iii) Although we have obtained significant structural information on individual domains and protein interaction interfaces, atomic structures of protein complexes such as the Axin complex and ligand-receptor complexes remain daunting challenges. (iv) Additional specific small molecular inhibitors or activators with defined targets and mechanisms would provide not only leads for therapeutics but also research tools to manipulate the Wnt pathway in precise temporal and spatial manners. (v) Integration of vast amounts of information into quantitative models will allow us to predict the behavior and to study the robustness and evolvability of Wnt signaling in various biological contexts. (vi) The Wnt responsive transcriptome remains a gold mine for digging into Wnt-regulated biology. Unfolding examples include Wnt regulation of intestinal and hair follicle development/homeostasis, which has provided significant insights into stem cell biology and cancer pathogenesis (Clevers, 2006; Fuchs, 2009). As β-catenin is a co-activator for other transcription factors in addition to TCF/LEF, comparative analyses of Wnt responsive transcription programs that depend on TCF/LEF versus others will likely uncover further complexity of Wnt-regulated gene expression. (vii) β-catenin and APC are also key components in the E-cadherin cell adhesion complex and the microtubule network, but how Wnt/β-catenin signaling interacts with these cellular structures remains poorly understood. In addition, the involvement of the primary cilium, a centrosome- and microtubule-based protrusive organelle in vertebrate cells, in Wnt/β-catenin versus non-canonical Wnt signaling remains an intriguing but debated topic (Gerdes et al., 2009).
Finally the study of Wnt signaling in human diseases, and in stem cell biology and regeneration holds promises for translational medicine. In addition to cancer and osteoporosis, both of which will likely see Wnt signaling-based therapeutics moving into clinical trials or even clinics in the near future, potential links between neurological diseases (De Ferrari and Moon, 2006) and a Schizophrenia susceptibility gene product (Mao et al., 2009) to Wnt/β-catenin signaling offer new hopes for the treatment of neurological and psychiatric disorders. Manipulation of Wnt signaling for stem cell regulation also offers exciting opportunities for regenerative medicine (Clevers, 2006; Fuchs, 2009; Goessling et al., 2009; Willert et al., 2003). A better understanding of Wnt/β-catenin signaling will have broad impact on biology and medicine.
7.10.6 Wnt.β-Catenin Signaling. Turning the Switch
Cadigan KM1.
Dev Cell. 2008 Mar; 14(3):322-3
http://dx.doi.org/10.1016/j.devcel.2008.02.006
The regulation of many targets of the Wnt/β-catenin signaling pathway is thought to occur through a transcriptional switch that is achieved by β-catenin binding to TCF transcription factors. Recent work indicates that β-catenin’s intrinsic affinity for TCF is not sufficient for the switch to occur.
The Wnt/β-catenin signaling pathway plays many crucial roles in specifying cell fates during animal development and in regenerating adult tissues. In addition, this pathway is linked to several pathological states, most notably colorectal cancer. Many of the transcriptional responses to Wnt/β-catenin signaling are mediated by the TCF/LEF-1 (TCF) family of transcription factors. Several TCFs are known to repress Wnt targets in the absence of signaling, but upon pathway activation, β-catenin enters the nucleus and binds to TCF on the target chromatin, creating a transcriptional activation complex. Is β-catenin’s intrinsic affinity for TCF sufficient to switch TCF from the repression to the activation state? Two recent papers shed some light on this question. One reports that two previously characterized co-repressor subunits bind to β-catenin and are required to stabilize the β-catenin-TCF interaction. The other suggests that this interaction may be regulated by ubiquitination of APC, a well-known negative regulator of the Wnt/β-catenin pathway.
The first report from Li and Wang (2008) concerns Transducin β-like protein 1 (TBL1) and TBL1-related protein (TBLR1). These proteins are components of the SMRT-nuclear receptor corepressor (N-CoR) complex, where they have been shown to recruit E3 ubiquitin ligases to facilitate the replacement of corepressors with coactivators (Perissi et al., 2004). Similarly, the Drosophila homolog of TBL1, known as Ebi, facilitates proteosomal degradation of the fly N-CoR homolog SMRTER ( Tsuda et al., 2002). In addition, TBL1 binds to the E3 ubiquitin ligase components Siah-1 and Skp1 to promote β-catenin degradation ( Matsuzawa and Reed, 2001). Despite the extensive connections between TBL1, TBLR1, and proteosomal degradation, Li and Wang (2008) found no evidence for these proteins influencing β-catenin turnover in their system. In addition, the proteosome does not appear to contribute to the function of TBL1 and TBLR1 in promoting Wnt/β-catenin signaling.
Using siRNA, Li and Wang found that TBL1 and TBLR1 are required for activation of several targets by Wnt signaling in cell culture. Both proteins interact with β-catenin in coimmunoprecipitation assays. When TBL1 or TBLR1 are depleted, the pathway still promotes nuclear accumulation of β-catenin, but its recruitment to Wnt response element (WRE) chromatin is dramatically reduced. Conversely, TBL1 and TBLR1 are recruited to several WREs in a Wnt- and β-catenin-dependent manner. Thus, binding of β-catenin, TBL1, and TBLR1 to WREs is mutally dependent. Interestingly, TBL1 (but not TBLR1) can be immunoprecipitated by TCF4, and TBL1 is present at some WREs even in the absence of Wnt stimulation. This suggests a model where interactions between TBL1, TCFs, and β-catenin reinforce the complex on WREs, which is required for subsequent recruitment of transcriptional coactivators necessary to activate target gene expression (see Figure 1).

role-of-tbl1-tblr1-and-trabid-in-tcf-ceb2-catenin-gene-regulation
Role of TBL1-TBLR1 and Trabid in TCF-β-Catenin Gene Regulation
http://ars.els-cdn.com/content/image/1-s2.0-S1534580708000762-gr1.jpg
Figure 1. Speculative Model on the Role of TBL1-TBLR1 and Trabid in TCF-β-Catenin Gene Regulation
In the absence of signaling (top panel), a TCF-corepressor complex silences target gene expression. When Wnt signaling causes nuclear accumulation of β-catenin (bottom panel), TBL1 and TBLR1 help recruit β-catenin to TCF at target loci, which nucleates a complex of transcriptional coactivators. APC can inhibit the TCF-β-catenin complex, and Trabid’s positive role in the pathway can be explained by its ability to regulate the ubiquitination state of APC.
This report extends the importance of TBL1 and TBLR1 in Wnt/β-catenin gene regulation in two important ways. First, the key findings were reproduced in Drosophila cell culture. Second, the authors demonstrate that depletion of TBL1 or TBLR1 greatly reduced activation of Wnt targets in a well-characterized colorectal cell line lacking functional APC. This decrease in target gene activation had a striking effect on the ability of these cells to grow on soft agar. In addition, the invasive nature of head and squamous cell carcinoma cells transfected with β-catenin was greatly curtailed by TBL1 or TBLR1 knockdown, as was the growth of these cells into tumors in nude mice. These results clearly demonstrate both the evolutionary conservation of these factors in the pathway and suggest that strategies to interfere with their function might have great therapeutic value.
While most reports (and reviews) focus on the TCF transcriptional switch from the “OFF” to the “ON” state, it is also interesting to consider how the switch works in reverse. For example, a colorectal cell line lacking functional APC can be stably transfected with an inducible full-length APC gene. Without induction, β-catenin is bound to WREs and Wnt target expression is high. Upon induction of APC, Jones and coworkers found that β-catenin and coactivators are rapidly replaced by corepressors at the WRE (Sierra et al., 2006). Interestingly, APC transiently occupies the WRE during this switch. A recent report from Bienz and coworkers (Tran et al., 2008) suggests that ubiquitination of APC may influence its ability to regulate the TCF-β-catenin complex.
This group identified an APC-interacting protein they call Trabid, which contains three tandem Npl4 zinc (NZF) fingers and an ovarian tumor (OTU) domain. They demonstrated that the OTU domain contains a deubiquitylating (DUB) activity that shows marked preference for K63-linked ubiquitin chains. When Trabid is depleted from cells by siRNA, activation of several Wnt targets is reduced, and rescue experiments indicate that both the NZF and OTU domains are required for Trabid’s positive role in Wnt signaling. Epitasis analysis indicates that Trabid is required downstream of β-catenin stabilization but is dispensible for TCF fusion proteins containing transactivation domains. This suggests that Trabid may influence the formation or dynamics of TCF-β-catenin complexes.
7.10.7 Wnt–β-catenin signaling
Tetsu Akiyama
Cyokine & GF Rev Dec 1, 2000; 11(4):273–282
http://dx.doi.org/10.1016/S1359-6101(00)00011-3
The Wnt/Wingless signaling transduction pathway plays an important role in both embryonic development and tumorigenesis. β-Catenin, a key component of the Wnt signaling pathway, interacts with the TCF/LEF family of transcription factors and activates transcription of Wnt target genes. Recent studies have revealed that a number of proteins such as, the tumor suppressor APC and Axin are involved in the regulation of the Wnt signaling pathway. Furthermore, mutations in APC or β-catenin have been found to be responsible for the genesis of human cancers.
7.10.8 Extracellular modulators of Wnt signaling
Boudin E1, Fijalkowski I, Piters E, Van Hul W.
Semin Arthritis Rheum. 2013 Oct; 43(2):220-40
http://dx.doi.org:/10.1016/j.semarthrit.2013.01.004
Objectives: The Wnt signaling pathway is a key pathway in various processes, including bone metabolism. In this review, current knowledge of all extracellular modulators of the canonical Wnt signaling in bone metabolism is summarized and discussed. Methods: The PubMed database was searched using the following keywords: canonical Wnt signaling, β-catenin bone metabolism, BMD, osteoblast, osteoporosis, Wnt, LRPs, Frizzleds, sFRPs, sclerostin or SOST, dickkopfs, Wif1, R-spondins, glypicans, SOST-dc1 and kremen, all separately as well as in different combinations.
Results: Canonical Wnt signaling is considered to be one of the major pathways regulating bone formation. Consequently, a large number of studies were performed to elucidate the role of numerous proteins in canonical Wnt signaling and bone metabolism. These studies led to the identification of novel modulators of the pathway like the R-spondin and glypican protein families. Furthermore novel insights are gained in the regulatory role of the different Wnt proteins. Finally, due to its function in bone formation, the pathway is an interesting target for the development of therapeutics for osteoporosis and other bone diseases. In this review, we discuss the promising results of the Wnt modulators sclerostin, Dkk1 and sFRP1 as targets for osteoporosis treatment.
Conclusion: The increasing number of studies into the exact function of all proteins in the canonical Wnt pathway in general and in bone metabolism already led to novel insights in the regulation of the canonical Wnt pathway. In this review we covered the current knowledge of all extracellular modulators of canonical Wnt signaling.
Fig 1. Activators and inhibitors of the Wnt/b-catenin signaling pathway.
(a) Lipid-modified Wnt protein (green; palmitoleoyl group is shown in red) binds to Frizzled CRD, LRP6 b-propellers 1–2 and/or 3–4, and triggers downstream signaling. CRD of Wnt receptor Frizzled8
is shown in light blue, four b propellers of co-receptor LRP6 are shown in dark blue. Hinge region between b-propellers 1–2 and 3–4 is shown as a blue dot. Dimeric signaling activator Norrin (monomers
shown in magenta and grey) binds specifically to Frizzled4 (grey) and LRP6 b-propellers 1–2. Dotted lines represent interactions between molecules where crystal structures of the complexes are absent.
(b) Extracellular inhibitors bind to Wnt co-receptor LRP6 or Wnt and prevent them from triggering signalling. Both Sclerostin and Dickkopf (Dkk) contain an Asn-X-Ile motif (peptide shown as
connected yellow spheres) recognized by LRP6 b-propeller 1. The C-terminal domain of Dkk1 (red) binds to LRP6 b-propeller 3. WIF1 (pink; WIF1-bound DPPC, light blue) and secreted Frizzled
related protein 3 (sFRP3 CRD, teal) prevent signaling by binding to Wnts. WIF1 binds to HS chains of HSPGs (grey). Sclerostin (as well as other activators and inhibitors) bind to HS-mimic, heparin.
Signaling inhibitor 5T4/Wnt-activated inhibitory factor 1 (WAIF1, purple) acts via unknown binding partners.
Fig 2. Atomic details of Wnt recognition and signaling inhibition.
(a) Zoom-in view of the palmitoleoyl binding site in the CRD of Frizzled8. Molecules are colored as in Figure 1. The Ser187-linked palmitoleoyl group is shown as connected red spheres. Frizzled8 CRD
residues forming the hydrophobic groove are shown as sticks (carbon, blue; oxygen, red) and numbered. Boundaries of the lipid-binding groove are marked with grey lines.
(b) WIF domain of WIF-1 forms a hydrophobic pocket which accommodates DPPC (carbon, grey; oxygen, red; phosphorus, orange; nitrogen, blue). The head group of DPPC is exposed to the solvent
and located proximal to the putative Wnt3a binding site.
(c) The first b propeller of LRP6 recognizes an evolutionarily conserved tripeptide motif Asn-X-Ile (X, variable amino acid) present in two inhibitors of Wnt signaling, Dickkopf1 and Sclerostin. A peptide
derived from human Sclerostin (residues Leu115–Arg121) is shown as sticks (carbon, yellow; oxygen, red; nitrogen, blue).
Regulation of Wnt signaling by R-spondin and its receptors.
(a) Transmembrane ubiquitin (Ub, shown in grey) E3 ligases ZNRF3 (brown) and RNF43 (red) ubiquitinylate Frizzled thus promoting its endocytosis and inhibition of
Wnt signalling. Cytoplasmic regions of both ligases contain RING domains required for ubiquitinylation. The extracellular domains of ZNRF3 form weak dimers in
solution (protomers are shown in brown and grey, respectively; [36]).
(b) R-spondin 1 (RSPO1, green) forms a ternary complex with RNF43 and LGR5 (blue) [35]. Endocytosis of RNF43 and ZNRF3 in complex with RSPOs and LGRs
4–6 prevents ubiquitinylation of Frizzled and promotes Wnt signaling. Dotted lines represent interactions between molecules where crystal structures of complexes
are not determined.
Conclusions and future perspectives
Tremendous progress has been made in structural studies of Wnt signaling receptors and modulators during the past five years. A series of structures of the Wnt co-receptor LRP6, agonists and
antagonists, and, remarkably, the first crystal structure of a Wnt family member, Wnt8, in complex with the Frizzled8 CRD, provide invaluable insights into the basic mechanisms of Wnt
signaling activation and regulation. In 2012, a novel mechanism of Wnt signaling regulation was discovered which centers on the interactions of the ZNRF3/RNF43 E3 ubiquitin ligases,
the R-spondins and LGR4/5/6.
7.10.9 FOXO3a modulates WNT.β-catenin signaling and suppresses epithelial-to-mesenchymal transition in prostate cancer cells
Liu H1, Yin J1, Wang H2, Jiang G3, Deng M1, Zhang G2, Bu X2, Cai S4, Du J5, He Z6.
Cell Signal. 2015 Mar; 27(3):510-8
http://dx.doi.org:/10.1016/j.cellsig.2015.01.001
Highlights
- FOXO3a inhibits β-catenin expression through transactivating miR-34b/c.
- FOXO3a direct binds to β-catenin.
- FOXO3a inhibits β-catenin/TCF transcriptional activity.
- FOXO3a inhibit EMT in prostate cancer cells.
- β-catenin as a regulator of FOXO3a-mediated suppression of EMT.
Emerging evidence has revealed a negative correlation between Forkhead box-O (FOXO) expression and prostate cancer grade and spread, indicating its role as a suppressor of prostate cancer metastasis. However, there is still incomplete understanding about the role of FOXO transcription factors in prostate cancer progression. In this investigation, we demonstrate that FOXO3a significantly inhibits the expression β-catenin in prostate cancer cells. The mechanism of inhibiting β-catenin expression involves the FOXO3a-mediated transactivated microRNA-34b/c, which consequently suppressed β-catenin mRNA expression by targeting the untranslated regions (UTRs) of β-catenin. Additionally, FOXO3a can directly bind to β-catenin, and competes with TCF for interaction with β-catenin, thereby inhibiting β-catenin/TCF transcriptional activity and reducing the expression of β-catenin target genes. Furthermore, prostate cancer cells expressing FOXO3a shRNAs display mesenchymal characteristics, including enhanced cell migration and differential regulation of the EMT markers, whereas knockdown of β-catenin results in reversal of shFOXO3a-mediated EMT phenotypic changes. Collectively, these observations demonstrated that FOXO3a inhibits malignant phenotypes that are dependent on β-catenin-dependent modulation of EMT-related genes, and provided fresh insight into the mechanisms by which a FOXO3a-miR-34b/c axis restrains canonical β-catenin signaling cascades in prostate cancer cell.
Fig.1. FOXO3a activation correlates with downregulation of β-catenin expression in prostate cancer cells. (A) PC3 and DU145 cells were treated with LY294002 for 48h, and Western blot was performed to assess p-FOXO3a, total FOXO3a, and β-catenin expression compared with that of the control cells.(B,C) The PC3 and DU145 cells were transfected with FOXO3a overexpressing and si-FOXO3a knockdown vectors; the mRNA expression (B) and protein expression (C) of β-catenin were assessed by real-time RT-PCR and Western blot, respectively. (D) PC3 cells were transfected with FOXO3a overexpressing vector, immunofluorescence images from PC3 cells stained for FOXO3aand β-catenin. DNA is stained with 4,6-diamidino2-phenylindole (DAPI, blue).Data were presentedasmeans± SDof three independent experiments. *Significant difference from control values with P b 0.05.
Fig.2.FOXO3a inhibits β-catenin expression by modulating miR-34 expression. (A) The miR-34b/c promoter contains consensus FOXO binding sites. miR-34b and miR-34c are encoded by one primary transcript (BC021736). Putative FOXO binding sites were identified at positions−1518,−1512,−1223,and−185 relative to the transcription start site.(B)FOXO3abinds to the BC021736 promoter in vivo. PC3 cells were infected with pCMV-FOXO3a. DNA-bound proteins were crosslinked to chromatin, and FOXO3a was immunoprecipitated with an antibody directed against FOXO3a. Rabbit IgG immune serum was used as IP control. Immunoprecipitated DNA-fragments were amplified by PCR with primers specific for theputative FOXO3 a consensus binding sites(−1518/12,−1223,−185) or a control region.Data are plotted aspercentage ofinput DNA ± SD. (C, D)The PC3 cells were transfectedwith FOXO3a overexpressing(C)andsi-FOXO3aknockdownvectors(D),themRNAexpressionofmiR-34wereassessedbyreal-timeRT-PCR.(E)RealtimeRT-PCRanalysesofβ-cateninmRNAexpression levelswereperformedinPC3 cells 48h after transfectionwith control,miR-34b, ormiR-34cmimics. (F)ThePC3 cells were transfected with pCMV-FOXO3a, anti-miR-34c, pCMVFOXO3a and anti-miR-34c, respectively; or shFOXO3a, miR-34c mimics, shFOXO3a and miR-34c mimics, respectively, the protein expression of FOXO3a and β-catenin were analyzed by Western blot. Data were presented as means± SD of three independent experiments. *Significant difference from control valueswith P < 0.05.
Fig.3. FOXO3a binds to β-catenin, reduces binding of β-Catenin to TCF, and inhibits β-Catenin/TCF-dependent transcription. (A) Total protein extracts of PC3 and DU145 cells were subjected to IP using FOXO3a antibody or control IgG, followed by IB with β-cateninantibody (upper panels). Reciprocal IP was done using β-catenin antibody or control IgG, followed by IB with the FOXO3a antibody (lower panels). (B) Lysates of PC3 cells that stably express FOXO3a or a control vector were subjected to IPusing FOXO3a antibodies, followed by IB with β-catenin antibody.(C) Lysates of PC3 cells that stably express FOXO3a or a control vector were subjected to IP using TCF4 antibodies, followed by IB with β-catenin antibody. Reciprocal IP was done using β-catenin antibody or control IgG, followed by IB with the TCF4 antibody. (D) TOP flash and FOP flash firefly luciferase expression vectors were co-transfected with control, pCMV-FOXO3a, and pCMV-β-catenin plasmid in PC3 cells, and TOP flash activity was measured. (E) PC3 cells were transfected with pCMV-FOXO3a plasmid, or FOXO3 as hRNA, the differential expression of potential β-catenin target genes are shown in the heat map.Data were presented as means±SD of three independent experiments.**Significant difference from control values with P<b 0.01
http://ars.els-cdn.com/content/image/1-s2.0-S0898656815000030-fx1.sml
Read Full Post »








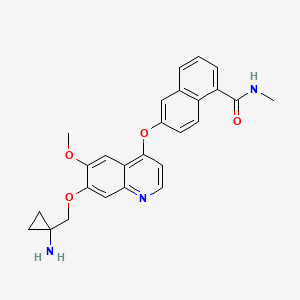


















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Wouldn’t then the preferred target be mTORC instead of Sirtuins if mTORC represses Sirtuin activity?
The answer may not be so simple, perhaps a conundrum.
In conflicting studies, SIRT3 has been shown to be anti-apoptotic. For example, in the cellular response to DNA damage when mitochondrial NAD+ levels fall below critical levels, SIRT3 and SIRT4 display anti-apoptotic activity, protecting cells from death [64].
For anti-cancer activity, apoptosis is a desired effect. This reminds me of the problem 15 years ago with the drug that would be effective against sepsis, the best paper of the year in NEJM. It failed.
We tend to not appeciate the intricacies of biological interactions and fail to see bypass reactions. Pleotropy comes up again and again. The seminal work from Britton Chances lab on the NAD+/NADH ratio have been overlooked.