Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
A magnetic wire could replace the lottery of cancer blood tests
Reporter: Irina Robu, PhD
Stanford University scientists developed a magnetic wire which doctors can use to detect cancer before symptoms are detected in patients. The device is threaded into a vein, screens for the disease by attracting scarce and hard to capture tumor cells just like a magnet. The wire would be predominantly valuable to detect ‘silent killers’ such as pancreatic, ovarian and kidney cancer where symptoms only seem in the late stages when it has spread too far to treat. The magnetic wire can save thousands of lives by catching the disease at a time when drugs would be effective. Cells that have broken off a tumor to wander the bloodstream easily can assist as cancer biomarkers signaling the presence of the disease.
Dr. Gambhir’s team published the results in Nature Biomedical Engineering which described how using a wire that has magnetic nano-particles engineered to stick to cancerous cells. The original experiment is on pigs, which are structurally alike to humans and suffer from the same genetic malfunctions that cause cancer. The wire captured 10 to 80 times more tumor cells and was placed in a vein near the pig’s ear which can be removed from and the cells can be used for analysis. In real standings it chosen up 500 to 5,000 more cancerous cells than normal blood samples.
The circulating tumor cells were magnetized with nanoparticles containing an antibody that latch onto them. When attached, the cell carries the tiny magnet around with it and flows past the wire to veer from its regular path in the bloodstream and stick to the wire. Professor Gambhir hopes that this approach will enrich detection capability and give insight how circulating tumor cells are and how early on they exist once the cancer is present. Once the technology is accepted for humans, the goal is to mature it into a multi-pronged tool that will increase detection, diagnosis, treatment and evaluation of cancer therapy.
It can also be used to gather genetic information about tumors located in places from where it’s hard to take biopsies.
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….
Mapping the dreaming brain through neuroimaging and studies of brain damage
By Karen Zusi | March 1, 2016
Prefrontal leucotomies—surgeries to cut a section of white matter in the front of the brain, thus severing the frontal lobe’s connections to other brain regions—were all the rage through the 1950s as treatments for psychoses. The operations drastically altered the mental state of most patients. But along with personality changes, dulled initiative, and reduced imagination came a seemingly innocuous effect of many of these procedures: the patients stopped dreaming.
Mark Solms, a neuropsychologist at the University of Cape Town in South Africa, uncovered the correlation in historical data from around the globe as part of a long-term study to assess the impact, on dreams and dreaming, of damage to different parts of the brain. Between 1985 and 1995, Solms interviewed 332 of his own patients at hospitals in Johannesburg and London who had various types of brain trauma, asking them about their nightly experiences.
Solms identified two brain regions that appeared critical for the experience of dreaming. The first was at the junction of the parietal, temporal, and occipital lobes—a cortical area that supports spatial cognition and mental imagery. The second was the ventromesial quadrant of the frontal lobes, a lump of white matter commonly associated with goal-seeking behavior that links the limbic structures to the frontal cortex. “This lesion site rang a historical bell in my mind—that’s where the prefrontal leucotomy used to be done,” says Solms, adding that the operation controlled the hallucinations and delusions that came with psychosis. “That sort of struck me as, ‘Gosh, that’s what dreaming is.’” Lesions in other areas could intensify or reduce certain aspects of dreams, but damage to either of the regions Solms pinpointed reportedly caused dreaming to cease completely (Psychoanal Q, 64:43-67, 1995).
Advances in neuroimaging have lent more support to Solms’s brain map, and pinned down other areas that researchers now understand play a part in dream development. In 2013, Bill Domhoff, a psychologist from the University of California, Santa Cruz, and colleagues from the University of British Columbia published results that combined neuroimaging scans from separate studies of REM sleep and daydreaming. They discovered that brain regions that light up when there’s a high chance that one is dreaming overlapped with parts of the brain’s default mode network—regions active when the brain is awake but not focused on a specific external task (Front Hum Neurosci, 7:412, 2013). “It very much lines up,” says Domhoff. “It’s just stunning.”
The default mode network allows us to turn our attention inward, and dreaming is the extreme example, explains Jessica Andrews-Hanna, a cognitive scientist at the University of Colorado Boulder. The network takes up a large amount of cortical real estate. Key players are regions on the midline of the brain that support memories and future planning; these brain sections connect to other areas affecting how we process social encounters and imagine other individuals’ thoughts. “When people are sleeping—in particular, when they’re dreaming—the default mode network actually stays very active,” says Andrews-Hanna. With external stimuli largely cut off, the brain operates in a closed loop, and flights of fancy often ensue.
We usually take the bizarre nature of these experiences at face value. “Even in a completely crazy dream, we all think that it’s normal,” says Martin Dresler, a cognitive neuroscientist at Radboud University in the Netherlands. Dresler and many other researchers attribute this blasé acceptance to the deactivation of a brain region called the dorsolateral prefrontal cortex. When we sleep, the dorsolateral prefrontal cortex powers down, and higher executive control—which would normally flag a nonsensical concern, such as running late for a class when you haven’t been in school for a decade, as unimportant—evaporates. “You have this overactive default mode network with no connectivity, with no communication with regions that are important for making sense of the thoughts,” says Andrews-Hanna.
In healthy sleeping subjects, these executive functions can be unlocked in what’s known as lucid dreaming, when the prefrontal cortex reactivates and sleepers gain awareness of and control over their imagined actions. A lucid dreamer can actually “direct” a dream as it unfolds, deciding to fly, for example, or turning a nightmarish monster into a docile pet.
Records of lucid dreaming are limited to REM sleep, the sleep stage where the brain is most active. REM sleep normally induces paralysis to prevent people from acting out their dreams, but the eye muscles are exempt, and this gives skilled lucid dreamers a way to signal their lucidity to researchers.
Dresler’s team is using this phenomenon as a tool to ask specific questions about dreams. Before trained lucid dreamers fall asleep in Dresler’s lab, they agree to flick their eyes from left to right as soon as they realize within a dream that they’re asleep. The dreamed movement causes their actual eyes to move in a similar way under their closed eyelids. Researchers mark this signal as the beginning of a lucid dream, and then track brain patterns associated with specific dreamed actions. Dreaming also occurs in non-REM sleep, but with the brain less active, the eye muscles won’t respond to dream input—so there’s no robust way to tell if lucid dreaming takes place.
When subjects achieved lucidity and consciously dreamed that they performed a predetermined hand movement, Dresler’s research team observed activity in the sensorimotor cortex matching what would occur if the subjects actually moved their hands while awake (Curr Biol, 21:1833-37, 2011). “It’s probably the case that, for most of what we are dreaming about, the very same machinery and the very same brain regions are active compared to wakefulness,” says Dresler. “It’s just that the motor execution is stopped at the spinal level.”
Beyond sleep research, tracking lucid and normal dreaming offers an investigative model to study aspects of psychosis, according to some researchers. “These regions that are activated during lucid dreaming are typically impaired in patients with psychosis,” explains Dresler. “Having insight into your non-normal mental state in dreaming shares neural correlates with having insights into your non-normal state of consciousness in psychosis.” Dresler proposes training patients in early stages of psychosis to dream lucidly, in the hope that it might grant them some therapeutically relevant understanding of their illness.
While executive functions are impaired in many patients suffering from psychosis, their default networks seem to be overactive, says Andrews-Hanna. But how much similarity exists between the brain states of dreaming and psychosis remains controversial. Domhoff emphasizes the unique nature of dreams. “They’re not like schizophrenia, they’re not like meditation, they’re not like any kind of drug trip,” he says. “They’re an enactment of a scenario that is based upon various wishes and concerns.”
Ultimately, says Solms, deciphering dreaming furthers the field’s knowledge of what the brain does, as much as studies conducted during waking hours. “If you’re a clinician, and you understand what the different parts of the brain do in relation to dreaming, then it’s one of the things you can use as a road map for evaluating your patients.”
Dreamed Movement Elicits Activation in the Sensorimotor Cortex
Since the discovery of the close association between rapid eye movement (REM) sleep and dreaming, much effort has been devoted to link physiological signatures of REM sleep to the contents of associated dreams [1, 2, 3 and 4]. Due to the impossibility of experimentally controlling spontaneous dream activity, however, a direct demonstration of dream contents by neuroimaging methods is lacking. By combining brain imaging with polysomnography and exploiting the state of “lucid dreaming,” we show here that a predefined motor task performed during dreaming elicits neuronal activation in the sensorimotor cortex. In lucid dreams, the subject is aware of the dreaming state and capable of performing predefined actions while all standard polysomnographic criteria of REM sleep are fulfilled [5 and 6]. Using eye signals as temporal markers, neural activity measured by functional magnetic resonance imaging (fMRI) and near-infrared spectroscopy (NIRS) was related to dreamed hand movements during lucid REM sleep. Though preliminary, we provide first evidence that specific contents of REM-associated dreaming can be visualized by neuroimaging.
Highlights
► Eye signals can be used to access dream content with concurrent EEG and neuroimaging
► Dreamed hand movements correspond to activity in the contralateral sensorimotor cortex
Lucid dreaming is a rare but robust state of sleep that can be trained [5]. Phenomenologically, it comprises features of both waking and dreaming [7]: in lucid dreams, the sleeping subject becomes aware of his or her dreaming state, has full access to memory, and is able to volitionally control dreamed actions [6]. Although all standard polysomnographic criteria of rapid eye movement (REM) sleep [8] are maintained and REM sleep muscle atonia prevents overt motor behavior, lucid dreamers are able to communicate their state by predefined volitional eye movements [6], clearly discernable in the electrooculogram (EOG) (Figure 1). Combining the techniques of lucid dreaming, polysomnography, and brain imaging via functional magnetic resonance imaging (fMRI) or near-infrared spectroscopy (NIRS), we demonstrate the possibility to investigate the neural underpinnings of specific dream contents—in this case, dreamed hand clenching. Predecided eye movements served as temporal markers for the onset of hand clenching and for hand switching. Previous studies have shown that muscle atonia prevents the overt execution of dreamed hand movements, which are visible as minor muscle twitches at most [3 and 9].
Figure 1.
Exemplary Lucid REM Sleep as Captured by Polysomnography during Simultaneous fMRI
Note high-frequency electroencephalogram (EEG) and minimal electromyogram (EMG) amplitude due to muscle atonia characteristic of rapid eye movement (REM) sleep (left), with wakefulness for comparison (right). Subjects were instructed to communicate the state of lucidity by quick left-right-left-right (LRLR) eye movements. Filter settings are as follows: EEG, bandpass filter 0.5−70 Hz, with additional notch filter at 50 Hz; electrooculogram (EOG), bandpass filter 0.1–30 Hz; EMG, bandpass filter 16–250 Hz.
Figure 2.
Comparison of Sensorimotor Activation during Wakefulness and Sleep
Functional magnetic resonance imaging (fMRI) blood oxygen level-dependent (BOLD)-response increases were contrasted between left and right hand movements (columns) in the three conditions (rows): executed hand movement during wakefulness (WE) (A), imagined hand movement during wakefulness (WI) (B), and dreamed hand movement during lucid REM sleep (LD) (C). Effects of left (right) hand movements were calculated in a fixed-effects analysis as a contrast “left > right” and “right > left,” respectively. Subpanels depict results in an SPM glass-brain view (sagital and coronal orientation) to demonstrate the regional specificity of the associated cortical activation, along with sensorimotor activation overlaid on an axial slice of the subject’s T1-weighted anatomical scan (position indicated on the glass brain for condition A). Clusters of activation in the glass-brain views are marked using the numbering given in Table S1. Red outlines in the glass-brain views mark the extent of activation found in the WE condition. This region of interest (ROI) was derived from the respective activation map during executed hand movement (A), thresholded at whole-brain corrected pFWE < 0.005, cluster extent >50 voxels, and served as a ROI for analysis of the WI and LD conditions in (B) and (C), respectively. T values are color-coded as indicated. The time course of the peak voxel inside the ROI is depicted (black) along with the predicted hemodynamic response based on the external pacing (A and B) or the predefined LRLR-eye signals during (C). The maximal difference in activation of the peak voxel between conditions is indicated as percentage of BOLD signal fluctuations of the predicted time course (gray).
FMRI results were confirmed by an independent imaging method in a second subject: NIRS data showed a typical hemodynamic response pattern of increased contralateral oxygenation over the sensorimotor region during successful task performance in lucid REM sleep (Figure 3; Figure 4). Notably, during dreaming, the hemodynamic responses were smaller in the sensorimotor cortex but of similar amplitude in the supplementary motor area (SMA) when compared to overt motor performance during wakefulness.
Figure 3.
Near-Infrared Spectroscopy Topography
Concentration changes of oxygenated (Δ[HbO], upper panel) and deoxygenated hemoglobin (Δ[HbR], lower panel) during executed (WE) and imagined (WI) hand clenching in the awake state and dreamed hand clenching (LD). The optical probe array covered an area of ∼7.5 × 12.5 cm2 over the right sensorimotor area. The solid box indicates the ROI over the right sensorimotor cortex with near-infrared spectroscopy (NIRS)-channels surrounding the C4-EEG electrode position. NIRS channels located centrally over midline and more anterior compared to sensorimotor ROI were chosen as ROI for the supplementary motor area (SMA, dotted box).
Figure 4.
Condition-Related NIRS Time Courses
Time courses of HbO (red traces) and HbR (blue traces) from the right sensorimotor ROI (left panel) and the supplementary motor ROI SMA (right panel) for executed (WE) and imagined (WI) hand clenching in the awake state and dreamed hand clenching (LD). The time courses represent averaged time courses from NIRS channels within the respective ROI (Figure 3). For each condition, 0 s denotes the onset of hand clenching indicated by LRLR-signals. Note that the temporal dynamics, i.e., an increase in HbO and a decrease in HbR, are in line with the typical hemodynamic response. Overt movement during wakefulness (dark red/blue traces) showed the strongest hemodynamic response, whereas the motor-task during dreaming leads to smaller changes (light red/blue traces). In the SMA, the hemodynamic response was stronger during the dreamed task when compared to imagery movement during wakefulness
Neurophysiological studies suggest that during REM sleep, the brain functions as a closed loop system, in which activation is triggered in pontine regions while sensory input is gated by enhanced thalamic inhibition and motor output is suppressed by atonia generated at the brain stem level [4 and 12].
Efforts have been made to correlate REMs to gaze direction during dreams—the “scanning hypothesis” [1 and 2]—and indeed similar cortical areas are involved in eye movement generation in wake and REM sleep [17]. In a similar vein, small muscle twitches during REM sleep were presumed to signal a change in the dream content [3]. Dream research methodology mostly relies on the evaluation of subjective reports of very diverse dream contents.
During dreaming, activation was much more localized in small clusters representing either generally weaker activation or focal activation of hand areas only, with signal fluctuations only in the order of 50% as compared to the actually executed task during wakefulness. The SMA is involved in timing, preparation, and monitoring of movements [21], and linked to the retrieval of a learned motor sequence especially in the absence of external cues [22]. Our NIRS data speak for an activation of SMA even during simple movements. This is in line with several PET and fMRI studies reporting SMA activations for simple tasks such as hand clenching, single finger-tapping, and alternated finger-tapping.
While You Were Sleeping
Assessing body position in addition to activity may improve monitoring of sleep-wake periods.
By Ruth Williams | March 1, 2016
Polysomnography—the combined assessment of brain waves, heart rate, oxygen saturation, muscle activity, and other parameters—is the most precise way to track a person’s sleeping patterns. However, the equipment required for such analyses is expensive, bulky, and disruptive to natural behavior.
Researchers are thus searching for ways to improve the accuracy of wearable devices while maintaining user-friendliness. Maria Angeles Rol of the University of Murcia in Spain and her colleagues have now discovered that by using a device strapped to the patient’s upper arm that measures both arm activity and position (the degree of tilt), they can more precisely detect periods of sleep.
The researchers studied just 13 people in this pilot study, says Barbara Galland of the University of Otago in New Zealand, but adds that nonetheless it “provide[s] an opening for further investigations to demonstrate the value of this novel technique.” (Chronobiol Int, 32:701-10, 2015)
Validation of an innovative method, based on tilt sensing, for the assessment of activity and body position
Since there is less movement during sleep than during wake, the recording of body movements by actigraphy has been used to indirectly evaluate the sleep–wake cycle. In general, most actigraphic devices are placed on the wrist and their measures are based on acceleration detection. Here, we propose an alternative way of measuring actigraphy at the level of the arm for joint evaluation of activity and body position. This method analyzes the tilt of three axes, scoring activity as the cumulative change of degrees per minute with respect to the previous sampling, and measuring arm tilt for the body position inference. In this study, subjects (N = 13) went about their daily routine for 7 days, kept daily sleep logs, wore three ambulatory monitoring devices and collected sequential saliva samples during evenings for the measurement of dim light melatonin onset (DLMO). These devices measured motor activity (arm activity, AA) and body position (P) using the tilt sensing of the arm, with acceleration (wrist acceleration, WA) and skin temperature at wrist level (WT). Cosinor, Fourier and non-parametric rhythmic analyses were performed for the different variables, and the results were compared by the ANOVA test. Linear correlations were also performed between actimetry methods (AA and WA) and WT. The AA and WA suitability for circadian phase prediction and for evaluating the sleep–wake cycle was assessed by comparison with the DLMO and sleep logs, respectively. All correlations between rhythmic parameters obtained from AA and WA were highly significant. Only parameters related to activity levels, such as mesor, RA (relative amplitude), VL5 and VM10 (value for the 5 and 10 consecutive hours of minimum and maximum activity, respectively) showed significant differences between AA and WA records. However, when a correlation analysis was performed on the phase markers acrophase, mid-time for the 10 consecutive hours of highest (M10) and mid-time for the five consecutive hours of lowest activity (L5) with DLMO, all of them showed a significant correlation for AA (R = 0.607, p = 0.028; R = 0.582, p = 0.037; R = 0.620, p = 0.031, respectively), while for WA, only acrophase did (R = 0.621, p = 0.031). Regarding sleep detection, WA showed higher specificity than AA (0.95 ± 0.01 versus 0.86 ± 0.02), while the agreement rate and sensitivity were higher for AA (0.76 ± 0.02 versus 0.66 ± 0.02 and 0.71 ± 0.03 versus 0.53 ± 0.03, respectively). Cohen’s kappa coefficient also presented the highest values for AA (0.49 ± 0.04) and AP (0.64 ± 0.04), followed by WT (0.45 ± 0.06) and WA (0.37 ± 0.04). The findings demonstrate that this alternative actigraphy method (AA), based on tilt sensing of the arm, can be used to reliably evaluate the activity and sleep–wake rhythm, since it presents a higher agreement rate and sensitivity for detecting sleep, at the same time allows the detection of body position and improves circadian phase assessment compared to the classical actigraphic method based on wrist acceleration.
Sleep’s Kernel
Surprisingly small sections of brain, and even neuronal and glial networks in a dish, display many electrical indicators of sleep.
Sleep is usually considered a whole-brain phenomenon in which neuronal regulatory circuits impose sleep on the brain. This paradigm has its origins in the historically important work of Viennese neurologist Constantin von Economo, who found that people who suffered from brain infections that damaged the anterior hypothalamus slept less. The finding was a turning point in sleep research, as it suggested that sleep was a consequence of active processes within the brain. This stood in stark contrast to the ideas of renowned St. Petersburg physiologist Ivan Pavlov, who believed that sleep resulted from the passive withdrawal of sensory input. Although the withdrawal of sensory input remains recognized as playing a role in sleep initiation, there is now much evidence supporting the idea that neuronal and glial activity in the anterior hypothalamus leads to the inhibition of multiple excitatory neuronal networks that project widely throughout the brain.
But we also know from millions of stroke cases that cause brain damage and from experimentally induced brain damage in animal models that, regardless of where a lesion occurs in the brain, including the anterior hypothalamus, all humans or animals that survive the brain damage will continue to sleep. Further, a key question remains inadequately answered: How does the hypothalamus know to initiate sleep? Unless one believes in the separation of mind and brain, then, one must ask: What is telling the hypothalamus to initiate sleep? If an answer is found, it leads to: What is telling the structure that told the hypothalamus? This is what philosophers call an infinite regress, an unacceptable spiral of logic.
For these reasons, 25 years ago the late Ferenc Obál Jr. of A. Szent-Györgyi Medical University in Szeged, Hungary, and I (J.K.) began questioning the prevailing ideas of how sleep is regulated. The field needed answers to fundamental questions. What is the minimum amount of brain tissue required for sleep to manifest? Where is sleep located? What actually sleeps? Without knowing what sleeps or where sleep is, how can one talk with any degree of precision about sleep regulation or sleep function? A new paradigm was needed.
There is no direct measure of sleep, and no single measure is always indicative of sleep. Quiescent behavior and muscle relaxation usually occur simultaneously with sleep but are also found in other circumstances, such as during meditation or watching a boring TV show. Sleep is thus defined in the clinic and in experimental animals using a combination of multiple parameters that typically correlate with sleep.
The primary tool for assessing sleep state in mammals and birds is the electroencephalogram (EEG). High-amplitude delta waves (0.5–4 Hz) are a defining characteristic of the deepest stage of non–rapid eye movement (non-REM) sleep. However, similar waves are evident in adolescents who hyperventilate for a few seconds while wide awake. Other measures used to characterize sleep include synchronization of electrical activity between EEG electrodes and the quantification of EEG delta wave amplitudes. Within specific sensory circuits, the cortical electrical responses induced by sensory stimulation (called evoked response potentials, or ERPs) are higher during sleep than during waking. And individual neurons in the cerebral cortex and thalamus display action potential burst-pause patterns of firing during sleep.
Using such measures, researchers have shown that different parts of the mammalian brain can sleep independently of one another. Well-characterized sleep regulatory substances, or somnogens, such as growth hormone releasing hormone (GHRH) and tumor necrosis factor α (TNF-α), can induce supranormal EEG delta waves during non-REM sleep in the specific half of the rat brain where the molecules were injected. Conversely, if endogenous TNF-α or GHRH production is inhibited, spontaneous EEG delta waves during non-REM sleep are lower on the side receiving the inhibitor. A more natural example of sleep lateralization is found in the normal unihemispheric sleep of some marine mammals. (See “Who Sleeps?”)
Much smaller parts of the brain also exhibit sleep-like cycles. As early as 1949, Kristian Kristiansen and Guy Courtois at McGill University and the Montreal Neurological Institute showed that, when neurons carrying input from the thalamus and surrounding cortical tissue are surgically severed, clusters of neurons called cerebral cortical islands will alternate between periods of high-amplitude slow waves that characterize sleep and low-amplitude fast waves typical of waking, independently of surrounding tissue.1 This suggests that sleep is self-organizing within small brain units.
In 1997, Ivan Pigarev of the Russian Academy of Sciences in Moscow and colleagues provided more-concrete evidence that sleep is a property of local networks. Measuring the firing patterns of neurons in monkeys’ visual cortices as the animals fell asleep while performing a visual task, they found that some of the neurons began to stop firing even while performance persisted. Specifically, the researchers found that, within the visual receptive field being engaged, cells on the outer edges of the field stopped firing first. Then, as the animal progressed deeper into a sleep state, cells in more-central areas stopped firing. This characteristic spatial distribution of the firing failures is likely a consequence of network behavior. The researchers thus concluded that sleep is a property of small networks.2
More recently, David Rector at Washington State University and colleagues provided support for the idea of locally occurring sleep-like states. In a series of experiments, they recorded electrical activity from single cortical columns using a small array of 60 electrodes placed over the rat somatosensory cortex. The sensory input from individual facial whiskers maps onto individual cortical columns. As expected, ERPs in the cortical columns induced by twitching a whisker were higher during sleep than during waking. But looking at the activity of individual columns, the researchers observed that they could behave somewhat independently of each other. When a rat slept, most—but not all—of the columns exhibited the sleep-like high-amplitude ERPs; during waking, most—but not all—of the columns were in a wake-like state. Interestingly, the individual cortical columns also exhibited patterns that resembled a sleep rebound response: the longer a column was in the wake-like state, the higher the probability that it would soon transition into a sleep-like state.3
To test how cortical-column state can affect whole-animal behavior, Rector and his team trained rats to lick a sucrose solution upon the stimulation of a single whisker, then characterized the whisker’s cortical-column state. If the column receiving input from the stimulated whisker was in a wake-like state (low-magnitude ERP), the rats did not make mistakes. But if the column was in the sleep-like state (high-magnitude ERP), the animals would fail to lick the sucrose when stimulated and would sometimes lick it even when their whisker was not flicked.4 Even though the animal was awake, if a cortical column receiving stimulation was asleep, it compromised the animal’s performance. These experiments indicate that even very small neuronal networks sleep and that the performance of learned behavior can depend on the state of such networks.
Given that sleep can manifest in relatively small brain regions, perhaps it should not be too surprising that co-cultures of neurons and glia possess many of the electrophysiological sleep phenotypes that are used to define sleep in intact animal brains. During sleep, cortical and thalamic neurons display bursts of action potentials lasting about 500 ms, followed by periods of hyperpolarization lasting about the same length of time. The synchronization of this firing pattern across many neurons is thought to generate the EEG activity characteristic of delta-wave sleep, and undisturbed co-cultures of glia and neurons display periodic bursts of action potentials, suggesting that the culture default state is sleep-like. In contrast, if neuronal and glia networks are stimulated with excitatory neurotransmitters, the culture’s “burstiness”—the fraction of all action potentials found within bursts—is reduced, indicating a transition to a wake-like state. Treatment of co-cultures with excitatory neurotransmitters also converts their gene expression profile from a spontaneous sleep-like pattern to a wake-like pattern.5
Cell cultures also respond to sleep-inducing agents similarly to whole organisms. If a neuronal and glial culture is treated with TNF-α, the synchronization and amplitudes of slow-wave electrical activity increase, indicating a deeper sleep-like state. Moreover, ERPs are of greater magnitude after cultures are treated with TNF-α than during the sleep-like default state, suggesting that the somnogen induces a deeper sleep-like state in vitro as it does in vivo.6
Researchers have even studied the developmental pattern of such sleep phenotypes, using multielectrode arrays to characterize network activity throughout the culture, and the emergence of network properties follows a similar time course as in intact mouse pups. Spontaneous action potentials occur during the first few days in culture, but network emergent properties are not evident until after about 10 days. Then, synchronization of electrical potentials begins to emerge, and the network’s slow waves begin to increase in amplitude. If the cultures are electrically stimulated, slow-wave synchronization and amplitudes are reduced, suggesting the networks wake up. This is followed by rebound-enhanced slow-wave synchronization and amplitudes the next day, suggesting sleep homeostasis is also a characteristic of cultured networks.6
Clearly, even small neural networks can exhibit sleep-like behavior, in a dish or in the brain. But the question remains: What is driving the oscillations between sleep- and wake-like states?
Sleep emerges
In the intact brain, communication among neurons and between neurons and other cells is ever changing. Bursts of action potentials trigger the release of multiple substances and changes in gene expression, both of which alter the efficacy of signal transmission. For instance, neural or glial activity induces the release of ATP into the local extracellular space. Extracellular ATP, in turn, induces changes in the expression of TNF-α and other somnogens known to induce a sleep-like state. Because these effects take place in the immediate vicinity of the cell activity, they target sleep to local areas that were active during prior wakefulness.
In 1993, Obál and I (J.K.) proposed that sleep is initiated within local networks as a function of prior activity.7 The following year, Derk-Jan Dijk and Alex Borbely of the University of Zurich provided support for this idea when they had volunteers hold hand vibrators in one hand during waking to stimulate one side of the somatosensory cortex. In subsequent sleep, the side of the brain that received input from the stimulated hand exhibited greater sleep intensity, determined from amplitudes of EEG slow waves, than the opposite side of the brain. And in 2006, Reto Huber, then at the University of Wisconsin, showed that if an arm is immobilized during waking, amplitudes of EEG slow waves from the side of the brain receiving input from that arm are lower in subsequent sleep.
These experiments indicate that local sleep depth is a function of the activity of the local network during waking—an idea that has been confirmed by multiple human and animal studies. Moreover, local network state oscillations strongly indicate that sleep is initiated within local networks such as cortical columns. But how do the states of a population of small networks translate into whole-animal sleep?
Small local clusters of neurons and glia are loosely connected with each other via electrophysiological and biochemical signaling, allowing for constant communication between local networks. Steven Strogatz of Cornell University showed that dynamically coupled entities, including small neuronal circuits, will synchronize with each other spontaneously without requiring direction by an external actor. Synchronization of loosely coupled entities occurs at multiple levels of complexity in nature from intact animals to molecules—for example, birds flocking, or the transition from water to ice. The patterns generated by bird flocking, or the hardness of ice, are called emergent properties.
We, Obál, and our colleagues proposed that whole-brain sleep is an emergent property resulting from the synchronization of local neuronal network states.7,8,9 This would explain why sleep continues to occur after brain damage: because the remaining local circuits will spontaneously synchronize with each other. This view also allows one to easily envision variations in the depth or degree of sleep and waking because it allows for some parts of the brain to be in sleep-like states while other areas are in wake-like states, just as Rector observed. These independent states of local networks may account for sleep inertia, the minutes-long period upon awakening of poor cognitive performance and fuzzy-mindedness, and may also play a role in the manifestation of dissociated states such as sleepwalking. Most importantly, this paradigm frees sleep regulation from the dualism trap of mind/brain separation: top-down imposition of state is not required for the initiation of local state oscillations or for subsequent whole-organism sleep to ensue.
Our theory is also consistent with the modulation of sleep and wakefulness by sleep regulatory circuits such as those in the hypothalamus. For example, if interleukin-1, a sleep regulatory substance, is applied locally to the surface of the rat cortex, it induces local high-amplitude EEG slow waves indicative of a greater local depth of sleep.10 The responses induced by interleukin-1 in the cortex enhanced neuronal activity in anterior hypothalamic sleep regulatory areas.11 That hypothalamic neuronal activity likely provides information on local sleep- and wake-like states occurring in the cortex to the hypothalamus, where it can modulate the orchestration of the sleep initiated within the smaller brain units.
Finally, our ideas may inform the study of how sleep influences the formation of memories. A fundamental problem a living brain faces is the incorporation of new memories and behaviors while conserving existing ones. We know that cell activity enhances neuronal connectivity and the efficacy of neurotransmission within active circuits, a phenomenon that has been posited to be a mechanism by which memories are formed and solidified. By themselves, however, these use-dependent mechanisms would lead to unchecked growth of connectivity (in response to activity patterns) and positive feedback (since increased connectivity leads to reuse), ultimately resulting in a rigid, non-plastic network.7 Instead, we suggest that biochemical mechanisms—specifically, the use-dependent expression of genes involved in sleep regulation and memory—induce oscillations, representing local wake- and sleep-like states, which serve to stabilize and preserve brain plasiticity.7
For more than a century, researchers have struggled to understand how sleep works and what it does. Perhaps this lack of answers stems from a fundamental misconception about what sleeps. By thinking about sleep in smaller units, such as individual networks in the brain, hopefully the field will start to understand what exactly is going on during this enigmatic—but very common—phenomenon.
James M. Krueger is a regents professor of neuroscience and Sandip Roy is an associate professor of electrical engineering at Washington State University.
K. Kristiansen, G. Courtois, “Rhythmic electrical activity from isolated cerebral cortex,” Electroen Clin Neuro, 1:265-72, 1949.
I.N. Pigarev et al., “Evidence for asynchronous development of sleep in cortical areas,” Neuroreport, 8:2557-60, 1997.
D.M. Rector et al., “Local functional state differences between rat cortical columns,” Brain Res, 1047:45-55, 2005.
J.M. Krueger et al., “Sleep: A synchrony of cell activity-driven small network states,” Eur J Neurosci, 38:2199-09, 2013.
V. Hinard et al., “Key electrophysiological, molecular, and metabolic signatures of sleep and wakefulness revealed in primary cortical cultures,” J Neurosci, 32:12506-17, 2012.
K.A. Jewett et al., “Tumor necrosis factor enhances the sleep-like state and electrical stimulation induces a wake-like state in co-cultures of neurons and glia,” Eur J Neurosci, 42:2078-90, 2015.
J.M. Krueger, F. Obál, “A neuronal group theory of sleep function,” J Sleep Res, 2:63-69, 1993.
J.M. Krueger et al., “Sleep as a fundamental property of neuronal assemblies,” Nat Rev Neurosci, 9: 910-19, 2008.
S. Roy et al., “A network model for activity-dependent sleep regulation,” J Theor Biol, 253:462-68, 2008.
T. Yasuda et al., “Interleukin-1 beta has a role in cerebral cortical state-dependent electro-encephalographic slow-wave activity,” Sleep, 28:177-84, 2005.
K. Yasuda et al., “Unilateral cortical application of interleukin-1β (IL1β) induces asymmetry in Fos- and IL1β-immunoreactivity: Implications for sleep regulation,” Brain Res, 1131:44-59, 2007.
In Dogged Pursuit of Sleep
Unearthing the root causes of narcolepsy keeps Emmanuel Mignot tackling one of sleep science’s toughest questions.
In November 1986, Emmanuel Mignot arrived at Stanford University’s Center for Sleep Sciences and Medicine for a 16-month stint as a research associate. His goal was to find effective drugs to treat narcolepsy; his study subjects belonged to a colony of canines that suffered from the malady. “[When I got there], the dogs were being maintained, but not much was being done with them other than some chemistry studies on known neurotransmitters,” says Mignot, a professor of psychiatry and behavioral sciences at Stanford University and now director of the center. “As a pharmacologist, I wanted to study potential treatments for narcolepsy and understand the molecular biology to improve treatment in humans.”
The first narcoleptic dog, a French poodle named Monique, was brought to Stanford in 1974 byWilliam Dement, the so-called “father of sleep medicine,” who had founded the center in 1970, the first in the world dedicated to the study of sleep. Dement and other researchers there established a full breeding colony in 1977 when dogs with a genetic form of the neurological disorder were discovered—initially, some puppies from a litter of Dobermans and, later, some Labradors. Narcoleptic dogs and humans both exhibit a combination of symptoms: perpetual sleepiness, cataplexy—muscle paralysis attacks triggered by emotions—and abnormal rapid eye movement (REM) sleep. While the condition in humans and dogs is treatable, there is no cure.
To study which narcolepsy drugs increased wakefulness and decreased cataplexy in the dogs, Mignot and psychiatry professor Seiji Nishino used a food-elicited cataplexy test: administration of the drug followed by release into a room with pieces of food on the floor and careful observation. “The dog would rush into the room and be so happy to eat the treats, and then would have an attack and collapse on the floor.” The researchers counted the number and duration of the attacks after treatment with a drug at various doses. In humans, cataplexy episodes are triggered by a positive emotion such as laughter at a joke or pleasant surprise. “For the dogs, it is food or the joy of playing. That is what is great about dogs as a model for this condition. When you give a treatment to a rat or mouse and they stop having cataplexy, you really don’t know if it is because they don’t feel good or if it is a genuine effect. But the dogs show you emotions like humans. I knew all of these dogs by name. They were my friends. I could see if they were worried or didn’t feel well.”
Mignot worked mostly with the Dobermans and Labs, but there were also dogs donated to the colony that seemed to have a sporadic form of narcolepsy, “There was Vern, a miniature poodle; Wally, a big poodle; Tucker, a mutt; and Beau, my beloved dachshund.” Using the cataplexy test in animals along with in vitro studies of the drugs’ chemical properties, Mignot and Nishino found that antidepressants suppress cataplexy by inhibiting adrenergic reuptake, and that amphetamine-like stimulants promote wakefulness in narcoleptics by increasing the availability of dopamine. “We improved the then-current treatments and started to understand the kinds of chemicals important to regulate narcolepsy symptoms.”
But Mignot wanted to understand the molecular mechanism of narcolepsy, so he turned his focus to the genetic basis of the disorder. A lack of genetics training and no map of the dog genome to guide him did not deter Mignot. He has tirelessly pursued this previously little-studied and, so far, only known neurological disorder that fundamentally perturbs the nature of sleep states.
Here, Mignot talks about pursuing a master’s, PhD, and MD simultaneously, the paper retraction that has been the most difficult episode in his career so far, and his unexpected devotion to a Chihuahua.
Mignot Motivated
Sir Mix-a-Lot. The youngest of six siblings, Mignot had a penchant for collecting fossils and for conducting chemistry experiments in the bathroom of his family’s home in Paris. “I bought chemicals sold by a Chinese shopkeeper on Rue Saint-Dominique to do all kinds of experiments, mixed them, and occasionally made mistakes. There were burn marks and projections on the walls of my bathroom.” In high school, the self-proclaimed “nerd with glasses” became interested in biology, and, after graduation in 1977, went to study for a medical degree at the René Descartes University Faculty of Medicine in Paris.
Collecting degrees. “In the second year of medical school, I got bored from all of the memorization.” He took the entrance exam for the prestigious École Normale Supérieure (ENS), which gives students freedom to pursue their academic interests at other institutions while providing a stipend, housing, and the support of professor mentors. He passed, and entered the ENS in 1979. Mignot worked towards a master’s in biochemistry, and then a PhD in molecular pharmacology while still continuing his medical studies. “Nothing was set up for MD-PhD programs at the time. It was all in parallel, which was crazy. I had an exam every few weeks,” says Mignot. In 1984, he received both his medical degree and, later, a PhD from Pierre and Marie Curie University.
New to narcolepsy. Mignot became interested in the effects of drugs on the brains of psychiatric patients, studying how different compounds affected the metabolism of neurotransmitters in the brains of rats, and pursued a residency in psychiatry to complement his laboratory research. In 1986, he was offered a professorship in pharmacology at the Paris V University School of Medicine. But first, Mignot needed to complete the mandatory military service that he had deferred. “Instead of going to a former French colony to practice medicine, I convinced the French government to send me to Stanford to study modafinil, a wakefulness-promoting drug created by a French pharmaceutical company called Lafon Laboratories for the treatment of narcolepsy. I had never heard about [narcolepsy] during medical school—it must have been a single line in my textbooks. I discovered that Stanford was doing work on sleep and that Dement had started a colony of narcoleptic dogs there. I thought I could study these animals and figure out how modafinil worked.”
So Mignot came to Stanford for 16 months as part of his military service with financial support from Lafon Laboratories. “The company had claimed modafinil worked by a novel mechanism, unrelated to how stimulants work,” says Mignot. But Mignot found that modafinil bound the dopamine transporter, inhibiting the reuptake of the neurotransmitter, boosting wakefulness. “This is a similar mode of action as Ritalin, but the company was claiming otherwise. It took 10 years for my results to be validated, finally, by Nora D. Volkow, now director of the National Institute on Drug Abuse, who showed . . . that indeed the drug displaces the dopamine transporter at doses that increase wakefulness in humans.”
Mignot Moves
Going to the dogs. At Stanford, Mignot immersed himself in his work with the dog colony. “I worked all the time and came home just to sleep. I was definitely not very successful with girls then, because I smelled like dog all the time. I spent all day with the dogs, going to the facility, hugging, playing, and working with them. When we bred them, sometimes the mothers rejected their puppies so we had to come in every few hours, even in the middle of the night, to bottle-feed the puppies. Even after I took a shower, you could still smell the dogs. It was a strange part of my life.”
From pharmacology to genetics. Mignot kept extending his stay at Stanford. “After a few years I realized our pharmacology studies were never going to lead to narcolepsy’s cause. We needed to find the genetic cause in the dog.” In 1988, he resigned a faculty position in Paris—which was being held for him even as he continued to extend his time at Stanford—deciding to search for the mutated gene responsible for narcolepsy in dogs. In 1993, Mignot became the head of the Center for Narcolepsy at Stanford. A connection between an immune gene, the human leukocyte antigen (HLA) allele HLA-DR2, and narcolepsy in humans had already been identified by Yutaka Honda at the University of Tokyo, so Mignot’s lab tried to ascertain whether the same connection was true in the dogs or if the immune gene was simply a genetic linkage marker. These were the days before the dog or human genome had been sequenced, so the work took Mignot’s lab 10 years, and almost 200 narcoleptic Dobermans and Labradors: years of painstaking chromosome walking experiments, DNA fingerprinting, and the construction of a bacterial artificial chromosome library of dog genomic pieces. “What helped us a lot was that we knew the Dobermans and Labs had the same genetic defect because we interbred and got narcoleptic puppies—what’s called a complementation test.” In 1999, Mignot’s team identified the mutated gene as hypocretin receptor 2, whose protein binds hypocretin (also called orexin), a neuropeptide that regulates arousal and wakefulness. Several weeks later, after seeing these findings, Masashi Yanagisawa’s lab independently published a confirmation, showing that hypocretin knockout mice also have narcolepsy.
In parallel narcolepsy studies across ethnic groups, Mignot’s lab found that it was not the initial HLA-DR2allele that predisposed humans to narcolepsy, but another, nearby HLA gene, DQB1*0602.
Humans are not like dogs. “After we found the gene, the research went fast. We decided to look at hypocretin itself and see if it’s abnormal in humans.” Mignot’s lab sequenced the genes for the hypocretin receptor and its ligand in narcoleptic patients, expecting mutations in either to be rare because of the known HLA-narcolepsy linkage and the fact that most cases in humans, unlike in dogs, are not familial. Only one documented case, a child who had narcolepsy onset at six months of age, has been found to harbor a hypocretin gene mutation. “I think you need to knock out both receptor 1 and 2 in humans to get the full narcoleptic phenotype,” says Mignot. “Those with just one mutation may be more prone to tiredness but not full narcolepsy.”
In 2000, Mignot’s and Nishino’s groups reported that hypocretin was not present in narcoleptic patients’ cerebrospinal fluid—a test still used diagnostically today. The same year, independent studies from Mignot’s laboratory and that of Jerome Siegel at the University of California, Los Angeles, found that the lack of hypocretin was not due to gene mutations but to the fact hypocretin cells were missing in the brains of narcoleptic patients. HLA genes were well known to be associated with many autoimmune diseases, and Mignot hypothesized that hypocretin was missing due to an autoimmune attack against hypocretin-secreting neurons. What the abnormality is in those narcolepsy patients with normal hypocretin levels remains a mystery.
Mignot Moves Forward
Still a missing link. “I have been working on this [autoimmunity] hypothesis for 10 years, and we see that this hypothesis is more and more likely, but we cannot find any direct proof. It’s frustrating, but that kind of struggle is the story of my life.” All known autoimmune diseases result in the generation of antibodies in patients, but antibodies against hypocretin or the hypocretin cells have never been detected. So Mignot’s lab tested whether T-cells were the immune component attacking hypocretin. In 2013, his lab published a study identifying the T-cell culprits. But the study was retracted by Mignot himself one year later, when Mignot’s group couldn’t reproduce the results after the scientist who did most of the experiments had left the lab. “It was really painful and the worst time in my career.”
A new lead. “In 2010, a lot of people suddenly started to develop narcolepsy after receiving the Pandemrix vaccine against swine flu. It’s very odd. We still don’t understand why this particular vaccine increased the risk of narcolepsy.” Mignot thinks that a component of the vaccine or the virus itself triggers the immune system to attack hypocretin-producing neurons. “So now I am doing a lot of studies comparing the different vaccines and the wild-type virus to try to understand what could be common to produce this response. I think the vaccine will give us a final clue to isolate the immune T-cells involved in narcolepsy.”
Genetics of sleep. Mignot’s lab is working on a genome-wide association study, which shows that the genetic variants linked to narcolepsy are mostly immune-related, similar to Type 1 diabetes, celiac disease and other autoimmune diseases, further supporting the autoimmune hypothesis. Mignot is also getting a large human study off the ground. “I want to study the genetics of 40,000 people with sleep issues to see if there are genetic traits that cause people to sleep well or not sleep well, to need more sleep or less sleep. This hasn’t been done yet. I think this will help us crack open the mysteries of sleep.”
A new companion. “The dog colony was officially dismantled in 2000 after we found the canine narcolepsy gene. The dogs were adopted and we got Bear, a narcoleptic Schipperke. He passed away over a year ago. I loved that dog and miss him a lot. He was an unusually kind soul. Three months later, a breeder from Vermont called and said he had a narcoleptic Chihuahua. I flew to Vermont and adopted Watson and he’s been with us ever since. I never would have thought to adopt a Chihuahua, but now I can’t think of life without Watson. He is faithful and cuddly. I really think you can bond with any dog.”
The journey continues. “This story of narcolepsy, it’s a difficult story. Finding the gene was very difficult, and finding the autoimmune connection should have been trivial, but it has been an ordeal because there is absolutely no collateral damage. As [Stanford neurologist] Larry Steinman said to me, it’s like a ‘hit and run’—it looks like it was cleaned up and the players disappear. It’s hard, but by learning about this disease, we may discover other diseases where a similar autoimmune destruction happens in the brain but we have never realized it. I wouldn’t be surprised if some forms of depression and schizophrenia have an autoimmune basis in the brain. By experience, the more difficult it is, the most interesting the answer will be.”
Greatest Hits
Identified the gene for hypocretin receptor 2, which, when mutated, causes an inherited form of narcolepsy in Dobermans and Labradors
Identified how antidepressant and stimulant drugs work as treatments for narcolepsy
Identified DQB1*0602 as the main human gene associated with narcolepsy
By genome-wide association, found immune polymorphisms, such as one in the T-cell receptor alpha, that also predispose people to the disease, further suggesting the disease is autoimmune
Found that human narcolepsy, unlike canine narcolepsy, is not caused by mutations in the hypocretin receptor 2 gene but is due to an immune-mediated destruction of hypocretin-producing neurons in the brain
DQB1*0602 and DQA1*0102 (DQ1) are better markers than DR2 for narcolepsy in Caucasian and black Americans.
In the present study, we tested 19 Caucasian and 28 Black American narcoleptics for the presence of the human leucocyte antigen (HLA) DQB1*0602 and DQA1*0102 (DQ1) genes using a specific polymerase chain reaction (PCR)-oligotyping technique. A similar technique was also used to identify DRB1*1501 and DRB1*1503 (DR2). Results indicate that all but one Caucasian patient (previously identified) were DRB1*1501 (DR2) and DQB1*0602/DQA1*102 (DQ1) positive. In Black Americans, however, DRB1*1501 (DR2) was a poor marker for narcolepsy. Only 75% of patients were DR2 positive, most of them being DRB1*1503, but not DRB1*1501 positive. DQB1*0602 was found in all but one Black narcoleptic patient. The clinical and polygraphic results for this patient were typical, thus confirming the existence of a rare, but genuine form of DQB1*0602 negative narcolepsy. These results demonstrate that DQB1*0602/DQA1*0102 is the best marker for narcolepsy across all ethnic groups.
Narcolepsy is a chronic neurologic disorder characterized by excessive daytime sleepiness and abnormal manifestations of REM sleep including cataplexy, sleep paralysis, and hypnagogic hallucinations. Narcolepsy is both a significant medical problem and a unique disease model for the study of sleep. Research in human narcolepsy has led to the identification of specific HLA alleles (DQB1*0602 and DQA1*0102) that predispose to the disorder. This has suggested the possibility that narcolepsy may be an autoimmune disorder, a hypothesis that has not been confirmed to date. Genetic factors other than HLA are also likely to be involved. In a canine model of narcolepsy, the disorder is transmitted as a non-MHC single autosomal recessive trait with full penetrance (canarc-1). A tightly linked marker for canarc-1 has been identified, and positional cloning studies are under way to isolate canarc-1 from a newly developed canine genomic BAC library. The molecular cloning of this gene may lead to a better understanding of sleep mechanisms, as has been the case for circadian rhythms following the cloning of frq, per, and Clock.
Sleep consumes almost one-third of any human lifetime, yet its biological function remains unknown. Electrophysiological studies have shown that sleep is physiologically heterogeneous. Sleep onset is first characterized by light nonrapid eye movement (NREM) sleep (stage I and II), followed by deep NREM sleep or slow-wave sleep (stage III and IV) and finally rapid eye movement (REM) sleep. This sleep cycle is ∼90 min long and is repeated multiple times during nocturnal sleep. REM sleep, also called paradoxical sleep, is characterized by low-voltage fast electroencephalogram activity, increased brain metabolism, skeletal muscle atonia, rapid eye movements, and dreaming. Total sleep deprivation and/or REM sleep deprivation are both lethal in animals.
NREM and REM sleep are mainly regulated by circadian and homeostatic processes. Recent studies have suggested that across the animal kingdom, circadian rhythms are regulated by similar negative feedback loops involving the rhythmic expression of RNAs encoding proteins that act to shut off the genes encoding them (Hall 1995; Dunlap 1996;Rosbash et al. 1996; Young et al. 1996). From a genetic perspective, much less progress has been made in the noncircadian aspects of sleep regulation. This review demonstrates that a genetic approach to narcolepsy will in time provide a novel insight into the molecular basis of sleep control.
Narcolepsy, a Disorder of REM Sleep Regulation
Narcolepsy most often begins in the second decade of life but may be observed at the age of 5 or younger (Honda 1988). The cardinal symptom in narcolepsy is a persistent and disabling excessive daytime sleepiness. Sleep attacks are unpredictable, irresistible, and may lead to continuing activities in a semiconscious manner, a phenomenon referred to as automatic behavior. Naps are usually refreshing, but the restorative effect vanishes quickly.
Sleepiness is not sufficient to diagnose the disorder. Narcoleptic patients also experience symptoms that are secondary to abnormal transitions to REM sleep (Aldrich 1992; Bassetti and Aldrich 1996). The most important of these symptoms is cataplexy, a pathognomonic symptom for the disorder. In cataplexy, humor, laughter, or anger triggers sudden episodes of muscle weakness ranging from sagging of the jaw, slurred speech, buckling of the knees or transient head dropping, to total collapse to the floor (Aldrich 1992; Bassetti and Aldrich 1996). Patients typically remain conscious during the attack, which may last a few seconds or a few minutes. Reflexes are abolished during the attack, as they are during natural REM sleep atonia. Sleep paralysis, another manifestation of REM sleep atonia, is characterized by an inability to move and speak while falling asleep or upon awakening. Episodes last a few seconds to several minutes and can be very frightening. Hypnagogic hallucinations are vivid perceptual dream-like experiences (generally visual) occurring at sleep onset. Sleep paralysis and hypnagogic hallucinations occasionally occur in normal individuals under extreme circumstances of sleep deprivation or after a change in sleep schedule (Aldrich 1992; Bassetti and Aldrich 1996) and thus have little diagnostic value in isolation.
Nocturnal sleep polysomnography is conducted to exclude other possible causes of daytime sleepiness such as sleep apnea or periodic limb movements (Aldrich 1992). The Multiple Sleep Latency Test (MSLT) is also carried out to demonstrate daytime sleepiness objectively. In this test, patients are requested to take four or five naps at 2-hr intervals, during which time to sleep onset (sleep latency) is measured. Short sleep latencies under 5 min are usually observed in narcoleptic patients, together with abnormal REM sleep episodes, referred to as sleep-onset REM periods (SOREMPs). The combination of a history of cataplexy, short sleep latencies, and two or more SOREMPs during MSLT is diagnostic for narcolepsy (Bassetti and Aldrich 1996;Mignot 1996). Note that many naps consist only of NREM sleep suggesting that there is also a broader problem of impaired sleep–wake regulation, with indistinct boundaries between sleep and wakefulness in narcolepsy (Broughton et al. 1986; Bassetti and Aldrich 1996).
The disorder has a large psychosocial impact. Two-thirds of patients have fallen asleep while driving, and 78% suffer from reduced performance at work (Broughton et al. 1981). Depression occurs in up to 23% of cases (Roth 1980). Treatment is purely symptomatic and generally involves amphetamine-like stimulants for excessive daytime sleepiness and antidepressive treatment for cataplexy and other symptoms of abnormal REM sleep (Bassetti and Aldrich 1996; Nishino and Mignot 1997).
Familial and Genetic Aspects of Human Narcolepsy
Narcolepsy–cataplexy affects 0.02%–0.18% of the general population in various ethnic groups (Mignot 1998). A familial tendency for narcolepsy has long been recognized (Roth 1980). The familial risk of a first-degree relative is 0.9%–2.3% for narcolepsy–cataplexy, which is 10–40 times higher than the prevalence in the general population (Mignot 1998).
In a Finnish twin cohort study consisting of 13,888 monozygotic (MZ) and same-sexed dizygotic (DZ) twin pairs, three narcoleptic individuals were found and each of them was discordant DZ with a negative family history (Hublin et al. 1994). In the literature, 16 MZ pairs with at least one affected twin have been reported and five of these pairs were concordant for narcolepsy (Mignot 1998). Although narcolepsy is likely to have a genetic predisposition, the low rate of concordance in narcoleptic MZ twins indicates that environmental factors play an important role in the development of the disease.
HLA DQA1*0102 andDQB1*0602 Are Primary Susceptibility Factors for Narcolepsy
Narcolepsy was shown to be associated with the human leukocyte antigen (HLA) DR2 in the Japanese population (Honda et al. 1984;Juji et al. 1984). DR2 is observed in all Japanese patients versus 33% of Japanese controls (Juji et al. 1984; Matsuki et al. 1988a). A similar association is observed in Caucasians, with >85% versus 22% DR2 positivity (Langdon et al. 1984; Billiard et al. 1986;Rogers et al. 1997). Strikingly however, the DR2association is much lower in African–Americans (65%–67% in narcoleptic patients vs. 27%–38% in controls) (Neely et al. 1987;Matsuki et al. 1992;Rogers et al. 1997). Further studies have shown that HLA DQalleles, located ∼80 kb from the DRregion, are more tightly associated with narcolepsy than HLADR subtypes. More than 90% of narcolepsy–cataplexy patients across all ethnic groups carry a specific allele of HLA DQB1, DQB1*0602 (Matsuki et al. 1992;Mignot et al. 1994); this allele is present in 12%–38% of the general population across many ethnic groups (Matsuki et al. 1992; Mignot et al. 1994; Lin et al. 1997).DQB1*0602 is associated almost exclusively with DR2in Japanese (Lin et al. 1997) and Caucasians (Begovich et al. 1992), whereas it is observed frequently in association with DR2, DR5, or other DRsubtypes in African–Americans (Mignot et al. 1994, 1997a). The increased DR–DQ haplotypic diversity in African–Americans explains the low DR2 association observed in this population.
To further characterize the DQB1 region in narcoleptic subjects, novel polymorphic markers were isolated and characterized (Mignot et al. 1997a). The markers tested included six novel microsatellite markers (DQCAR, DQCARII, G51152, DQRIV, T16CAR, and G411624R). DQA1, a DQ gene whose product is known to pair with DQB1-encoding polypeptides to form the biologically active DQ heterodimer molecule, was also studied. The results obtained are summarized in Figure1. The association with narcolepsy decreases in theT16CAR–DQB2 region (Mignot et al. 1997a) and in the DRB1 region (Mignot et al. 1994, 1997b). The G411624R andT16CAR microsatellites are complex repeats with drastically different sizes, all of which are frequently observed in narcolepsy susceptibility haplotypes, a result suggesting crossovers in the region. In the DRB1 region, association with narcolepsy is still tight with DRB1*1501 (DR2) in Caucasians and Asians but is significantly lower in African–Americans, which suggests crossovers in the region among ethnic groups.
Figure 1.
Schematic summary of the narcolepsy susceptibility region within the HLA complex. Genes and markers are depicted by vertical bars, alleles observed in narcoleptic patients are listed above each marker.DQB2, DQB3, DQB1, DQA1, andDRB1 are HLA genes and pseudogenes. QBP and QAP are the promoter regions ofDQB1 and DQA1, respectively. G411624R, T16CAR, G51152, DQCAR, and DQCARII are microsatellite CA repeats identified in the HLA DQ region (Mignot et al. 1997a).DQRIV is a compound tandem repeat of 4- and 2-bp units located between DQB1 and G51152. TheDQA1*0102allele is subdivided into 01021 and 01022 based on a codon 109 synonymous substitution. Genomic segments in which frequent recombination was detected are indicated by vertical solid lines. Broken lines indicate rare possible ancestral crossovers detected in the area. Crossovers betweenT16CAR and G51152 occur within ethnic groups; crossovers between QAP and DRB1are frequently observed among ethnic groups (Mignot et al. 1997a). Note that the genomic region shared by most narcoleptic patients extends from a region between T16CAR and G51152 to a region between QAP andDRB1. No other genes were found in 86 kb of genomic sequence surrounding the DQB1*0602 gene (Ellis et al. 1997). Additional diversity is also found at the level ofG51152 andDQRIV, this being most likely due to a slippage mechanism rather than crossover (Lin et al. 1997; Mignot et al. 1997a). (+, Δ, *) Frequent alleles found predominantly in Caucasian, Asian, and African–American populations, respectively; (kb) kilobase pairs. Alleles frequently observed in theDQB1*0602/DQA1*0121 haplotype are underlined.DRB1*1501, DRB1*1503, and DRB1*1602 are DR2subtypes.DRB1*1101 and DRB1*12022 are DR5 subtypes.
The DQA1*0102/DQB1*0602 haplotype is common in narcoleptic patients (Mignot et al. 1994). Other haplotypes withDQA1*0102but not DQB1*0602, such as DQA1*0102andDQB1*0604, are frequent in control populations in all ethnic groups and do not predispose to narcolepsy. DQA1*0102 alone is thus not likely to confer susceptibility but may be involved in addition to DQB1*0602 for the development of narcolepsy (Mignot et al. 1994, 1997a).
Microsatellite analysis in the HLA DQ region revealed that only the area surrounding the coding regions of DQB1 andDQA1 is well conserved across all susceptibility haplotypes. Polymorphism can be observed in microsatellite and/or in the promoter regions flanking the DQB1*0602 and DQA1*0102alleles and in the region between these two genes (Mignot et al. 1997a). Mutations by slippage for some loci, and rare ancestral crossovers in a few instances, contribute to this diversity (Mignot et al. 1997a). Sequence analysis of DQ genes from narcoleptic and control individuals has revealed no sequence variation that correlates with the disease (Lock et al. 1988; Uryu et al. 1989; Ellis et al. 1997;Mignot et al. 1997a). No new gene was found in 86 kb of genomic sequence surrounding the HLA DQ gene (Ellis et al. 1997). A study on the dosage effect of DQB1*0602 allele on narcolepsy susceptibility revealed that DQB1*0602 homozygous subjects are at two to four times greater risk than heterozygous subjects for developing narcolepsy (Pelin et al. 1998). Taken together, these results strongly suggest that the DQA1*0102 andDQB1*0602alleles themselves rather than an unknown gene in the region are the actual susceptibility genes for narcolepsy.
HLA DQB1*0602 Is Neither Sufficient nor Necessary for the Development of Narcolepsy
Of the general population, 12%–38% carry HLADQB1*0602, yet narcolepsy affects only 0.02%–0.18% of the general population. No sequence variation that correlates with the disease was detected in sequence analysis of DQ genes. Nevertheless, a few narcoleptic patients with cataplexy do not carry the DQB1*0602 allele (Mignot et al. 1992, 1997a). HLADQB1*0602 is thus neither necessary nor sufficient for development of narcolepsy–cataplexy.
…….
Canine Narcolepsy as a Model for the Human Disorder
….narcolepsy was identified in numerous canine breeds, including Doberman pinschers, Labrador retrievers, miniature poodles, dachshunds, beagles, and Saint Bernards. All animals display similar symptoms, but the age of onset, severity, and the clinical course vary significantly among breeds (Baker et al. 1982).
….Similar to human narcoleptic patients, animals affected with the disorder display emotionally triggered cataplexy, fragmented sleep, and increased daytime sleepiness. Sleep paralysis and hypnagogic hallucinations cannot be documented because of difficulties in assessing the symptoms in canines. The validity of this model of narcolepsy has also been established through neurophysiological and neuropharmacological similarities with the human disorder. Pharmacological and neurochemical studies suggest abnormal monoaminergic and cholinergic mechanisms in narcolepsy both in human and canines (Aldrich 1991; Nishino and Mignot 1997, 1998). Interestingly, it is also possible to induce brief episodes of cataplexy in otherwise asymptomatic canarc-1 heterozygous animals using specific drug combinations (Mignot et al. 1993).
……
Narcolepsy is both a significant medical problem and a unique disease model. Research in humans has led to the identification of specific HLA alleles that predispose to the disorder. This has suggested the possibility that narcolepsy may be an autoimmune disorder, a hypothesis that has not been confirmed to date. Cells of the central and peripheral nervous systems and immune systems are known to interact at multiple levels (Morganti-Kossmann et al. 1992; Wilder 1995). For example, peripheral immunity is modulated by the brain via autonomic or neuroendocrinal interactions, whereas the immune system affects the nervous system through the release of cytokines. Cytokines have been shown to modulate sleep directly and have established effects on neurotransmission and neuronal differentiation (Krueger and Karnovsky 1995; Mehler and Kessler 1997). It is therefore possible that neuroimmune interactions that are not autoimmune in nature might be involved in the pathophysiology of narcolepsy.
NREM and REM sleep are mainly regulated by circadian and homeostatic processes. Single gene circadian mutations have been isolated from species as diverse as Arabidopsis(toc1),Neurospora (frq), Drosophila (perand tim), and mouse (Clock) (Hall 1995). Theper and Clock genes isolated inDrosophiliaand mouse, respectively, have been shown to belong to the same family, the PAS domain family (Hall 1995; Rosbash et al. 1996; Young et al. 1996; King et al. 1997). Analysis of frq, tim,andper demonstrate that circadian rhythms of diverse species are regulated by similar negative feedback loops in which gene products negatively regulate their own transcripts (Hall 1995;Dunlap 1996;Rosbash et al. 1996; Young et al. 1996). Putative homologs of theper gene have also been isolated in mammals (Albrecht et al. 1997; Tei et al. 1997). In mouse, RNAs for two perhomologs are expressed rhythmically within the suprachiasmatic nucleus (SCN), a brain region with an established role in generating mammalian circadian rhythms (Shearman et al. 1997;Shigeyoshi et al. 1997; Tei et al. 1997).
Much less progress has been made in the noncircadian aspect of sleep regulation. Sleep can only be recognized and characterized electrophysiologically in mammals and birds, and single gene mutants for this behavior have not been described in the mouse. Canine narcolepsy is the only known single gene mutation affecting sleep state organization as opposed to circadian control of behavior. The molecular cloning of this gene may lead to a better understanding of the molecular basis and biological role of sleep, as has been the case for circadian rhythms following the cloning of frq, per, and Clock.
New insomnia drugs are coming on the market, but drug-free therapy remains the most durable treatment.
Selectively driving cholinergic fibers optically in the thalamic reticular nucleus promotes sleep
Kun-MingNi,
Xiao-JunHou
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China; Fuzhou Children’s Hospital, Fujian, China
Contribution: Acquisition of data, Contributed unpublished essential data or reagents
No competing interests declared
Contributed equally with: Kun-Ming Ni
</div>”>Xiao-JunHou,
Ci-HangYang
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China
Contribution: Acquisition of data
No competing interests declared
</div>”>Ci-HangYang,
PingDong
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China
Contribution: Acquisition of data, Analysis and interpretation of data
No competing interests declared
</div>”>PingDong,
YueLi
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China
Contribution: Acquisition of data, Drafting or revising the article
No competing interests declared
</div>”>YueLi,
YingZhang
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China
Contribution: Drafting or revising the article
No competing interests declared
</div>”>YingZhang,
PingJiang
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China
Contribution: Acquisition of data
No competing interests declared
</div>”>PingJiang,
Darwin KBerg
Neurobiology Section, Division of Biological Sciences, Center for Neural Circuits and Behavior, University of California, San Diego, La Jolla, United States
Contribution: Drafting or revising the article
No competing interests declared
</div>”>Darwin KBerg,
ShuminDuan
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China
Contribution: Drafting or revising the article
No competing interests declared
</div>”>ShuminDuan,
Xiao-MingLi
Department of Neurobiology, Institute of Neuroscience, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China; Soft Matter Research Center, Zhejiang University, Hangzhou, China
Contribution: Conception and design, Analysis and interpretation of data, Drafting or revising the article, Contributed unpublished essential data or reagents
Zhejiang University School of Medicine, China; Fuzhou Children’s Hospital, China; University of California, San Diego, United States; Zhejiang University, China
Cholinergic projections from the basal forebrain and brainstem are thought to play important roles in rapid eye movement (REM) sleep and arousal. Using transgenic mice in which channelrhdopsin-2 is selectively expressed in cholinergic neurons, we show that optical stimulation of cholinergic inputs to the thalamic reticular nucleus (TRN) activates local GABAergic neurons to promote sleep and protect non-rapid eye movement (NREM) sleep. It does not affect REM sleep. Instead, direct activation of cholinergic input to the TRN shortens the time to sleep onset and generates spindle oscillations that correlate with NREM sleep. It does so by evoking excitatory postsynaptic currents via α7-containing nicotinic acetylcholine receptors and inducing bursts of action potentials in local GABAergic neurons. These findings stand in sharp contrast to previous reports of cholinergic activity driving arousal. Our results provide new insight into the mechanisms controlling sleep.
“Sex reversed” (Sxr) is an inherited form of sex reversal that causes XX and XO mice to develop as phenotypically normal males. Adult XYSxra mice exhibit varying degrees of spermatogenic deficiency but are usually fertile, while XOSxra mice have severe spermatogenic failure and are always sterile. The present quantitative spermatogenic analysis reports when these anomalies first appear during puberty. The results demonstrate that in XYSxra mice there was increased degeneration of pachytene spermatocytes and, to a lesser extent, meiotic metaphase stages. On average, there were only one-half the number of spermatids compared with the XY controls. The defect in XOSxra mice appeared a little later, with an almost complete arrest and degeneration during the meiotic metaphases.
A minority of XYSxra mice are sterile, and these may have testes as small as those from XOSxra mice. Adult XOSxra mice have consistently small testes and are invariably sterile. The reported results document the testicular defects in XYSxra and XOSxra testes as they first arise during puberty. The only other quantitative data on XYSxra and XOSxra spermatogenesis are for adult mice. A previous report described XYSxra testes as being a “mosaic” of normal and defective spermatogenesis. Recently a more extensive analysis was carried out of adult XYSxra and XOSxra testes. Once again there is good agreement with the present results in that the spermatogenic failure in XYSxra testes was predominantly between pachytene and diplotene, while in XOSxra testes the block was predominantly during the meiotic metaphases. To explain the spermatogenic anomalies in XYSxra and XOSxra testes, Burgoyne and Baker (1984) invoked the “meiotic pairing site” hypothesis of Miklos (1974). The other notable feature of the present study was the demonstration that the testicular deficiency is manifested earlier (with respect to age and spermatogenic stage) in XYSxra testes than in XOSxra testes. Krzanowska (1989) recently reported increased levels of X-Y univalence in pubertal XY males. So, it is suggested that this reduced efficiency of X-Y pairing at puberty that leads to the increased incidence of diploid spermatids in pubertal XYSxra males and to the presence of diploid spermatids in pubertal XY males. The other feature of pubertal XYSxra testes that deserves mention is the increase in the number of differentiating spermatogonia.
The conclusion is that most of the spermatogenic deficiencies in XYSxra and XOSxra testes can be explained in terms of the “meiotic pairing site” hypothesis, which links spermatogenic failure with sex chromosome univalence during meiosis. In XYSxra testes a variable proportion of pachytene spermatocytes have the X and Y unpaired, and the elimination of these cells explains the variable reduction in testis size and fertility. In XOSxra testes all spermatocytes have a univalent sex chromosome, accounting for the almost total spermatogenic block in these mice. It is suggested that the affected spermatocytes are eliminated earlier in XYSxra testes than in XOSxra testes, because two univalent sex chromosomes have more unpaired sites than the univalent X alone.
References:
Sutcliffe M. J., Darling S. M., Burgoyne P. S. (1991) Spermatogenesis in XY, XYSxra and XOSxra Mice: A quantitative analysis of spermatogenesis throughout puberty. Molecular Reprod. Dev. 30(2), 81–89.
Burgoyne P. S., Baker T. G. (1984) Meiotic pairing and gametogenic failure. In CW Evans and HG Dickinson (eds): “Controlling Events in Meiosis (38th Symp SOC Exp Biol).” Cambridge Company of Biologists, pp 349-362.
Miklos G. L. G. (1974) Sex-chromosome pairing and male fertility. Cytogen. Cell Genet. 13, 558-577.
Krzanowska H (1989) X-Y chromosome dissociation in mouse strains differing in efficiency of spermatogenesis: Elevated frequency of univalents in pubertal males. Gamete. Res. 23, 357-365.
Agilent was created as a spin off from Hewlett-Packard Company in 1999.
Agilent Technologies Inc. is engaged in the life sciences, diagnostics and applied chemical markets. The Company provides application focused solutions that include instruments, software, services and consumables for the entire laboratory workflow. The Company has three business segments:
the life sciences and applied markets business,
the diagnostics and genomics business, and
the Agilent Cross Lab business
The Company’s life sciences and applied markets business segment brings together the Company’s analytical laboratory instrumentation and informatics.
The Company’s diagnostics and genomics business segment consists of three businesses: the Dako business, the genomics business and the nucleic acid solutions business.
The Company’s Agilent Cross Lab business segment combines its analytical laboratory services and consumables business
CARPINTERIA, Calif.–(BUSINESS WIRE)–Dako, an Agilent Technologies company and a worldwide provider of cancer diagnostics, today announced the U.S. Food and Drug Administration has approved a new test that can identify PD-L1 expression levels on the surface of non-small cell lung cancer tumor cells and provide information on the survival benefit with OPDIVO® (nivolumab) for patients with non-squamous NSCLC.
Argentina | Australia | Austria | Brazil | Canada |Chile | China | Colombia | Czech Republic | Denmark | Ecuador | Finland | Germany |Hong Kong | Israel | Italy | Japan | Korea | Malaysia | Mexico | New Zealand | Norway | Paraguay | Peru| Philippines | Poland | Romania | Singapore | South Africa | Spain | Sweden |Switzerland | Taiwan ROC | Thailand | Turkey | United Kingdom | Uruguay | Vietnam
Gen9 is building on advances in synthetic biology to power a scalable fabrication capability that will significantly increase the world’s capacity to produce DNA content. The privately held company’s next-generation gene synthesis technology allows for the high-throughput, automated production of DNA constructs at lower cost and higher accuracy than previous methods on the market. Founded by world leaders in synthetic biology, Gen9 aims to ensure the constructive application of synthetic biology in industries ranging from enzyme and chemical production to pharmaceuticals and biofuels.
SERVICES
Synthetic Biology
Gene Synthesis Services
Variant Libraries
Gene Sequence Design Services
INVESTORS
Agilent Technologies : Private Equity
CAMBRIDGE, Mass. and SANTA CLARA, Calif. — April 24, 2013 —Gen9 Receives $21 Million Strategic Investment from Agilent Technologies
GenScript is the largest gene synthesis provider in the USA
GenScript Corporation, a biology contract research organization, provides biological research and drug discovery services to pharmaceutical companies, biotech firms, and research institutions in the United States, Europe, and Japan. It offers bio-reagent, custom molecular biology, custom peptide, protein production, custom antibody production, drug candidates testing, assay development and screening, lead optimization, antibody drug development, gene synthesis, and assay-ready cell line production services.
The company also offers molecular biology, peptide, protein, immunoassay, chemicals, and cell biology products. It offers its products through distributors in Tokyo, Japan; and Seoul, Korea. GenScript Corporation has a strategic partnership with Immunologix, Inc. The company was founded in 2002 and is based in Piscataway, New Jersey. It has subsidiaries in France, Japan, and China.
Note: As of October 24, 2011, Immunologix, Inc. was acquired by Intrexon Corporation. Immunologix, Inc. develops and produces antibody-based therapeutics for various biological targets. It produces human monoclonal antibodies against viral, bacterial, and tumor antigens, as well as human auto antigens.
Intrexon Corporation, founded in 1998, is a leader in synthetic biology focused on collaborating with companies in Health, Food, Energy, Environment and Consumer sectors to create biologically based products that improve quality of life and the health of the planet.
PRODUCTS AND SERVICES
Gene synthesis
Antibody services
Protein Services
Peptide services
INVESTORS
Note: The Balloch Group (‘TBG’) was established in 2001 by Howard Balloch (Canada‘s ambassador to China from 1996 to 2001). TBG has since grown from a market-entry consultancy working with North American clients in China to a leading advisory and merchant banking firm serving both domestic Chinese companies and multinational corporations. TBG was ranked as the number one boutique investment bank in China by ChinaVenture in 2008.
Monica Heger : SAN FRANCISCO (GenomeWeb) – Illumina today announced two new next-generation sequencing platforms, a targeted sequencing system called MiniSeq and a semiconductor sequencer that is still under development.
Illumina disclosed the initiatives during a presentation at the JP Morgan Healthcare conference held here today. During the presentation, Illumina CEO Jay Flatley also announced a new genotyping array called Infinium XT; a partnership with Bio-Rad to develop a single-cell sequencing workflow; preliminary estimates of its fourth-quarter 2015 revenues; and an update on existing products. The presentation followed the company’s announcement on Sunday that it has launched a new company called Grail to develop a next-generation sequencing test for early cancer detection from patient blood samples.
The MiniSeq system, which is based on Illumina’s current sequencing technology, will begin shipping early this quarter and has a list price of $49,500. It can perform a variety of targeted DNA and RNA applications, from single-gene to pathway sequencing, and promises “all-in” prices, including library prep and sequencing, of $200 to $300 per sample, Flatley said during the JP Morgan presentation.
Integrated DNA Technologies, Inc. (IDT), the global leader in nucleic acid synthesis, serving all areas of life sciences research and development, offers products for a broad range of genomics applications. IDT’s primary business is the production of custom, synthetic nucleic acids for molecular biology applications, including qPCR, sequencing, synthetic biology, and functional genomics. The company manufactures and ships an average of 44,000 custom nucleic acids per day to more than 82,000 customers worldwide. For more information, visit idtdna.com.
Dyes GMP for Molecular Diagnostics Large Scale Oligo Synthesis
Note : Skokie, IL – December 1, 2015. Integrated DNA Technologies Inc. (“IDT”), the global leader in custom nucleic acid synthesis, has entered into a definitive agreement to acquire the oligonucleotide synthesis business of AITbiotech Pte. Ltd. in Singapore (“AITbiotech”). With this acquisition, IDT expands its customer base across Southeast Asia making it possible for these additional customers to now have access to its broad range of products for genomic applications. AITbiotech will continue operations in its other core business areas.
With over 20 years of experience in oligonucleotide development and production, and over 1000 sequences manufactured, Avecia has played an integral role in the advancing oligo therapeutic market. Our mission is to continue to build value for our customers, as they progress through drug development into commercialization. And as a member of the Nitto Denko Corporation (nitto.com), Avecia is committed to the future of the oligonucleotide market. We are driven by innovative ideas and flexible solutions, designed to provide our customers with the best in service, quality, and technology.
OriGene Technologies, Inc. develops, manufactures, and sells genome wide research and diagnostic products for pharmaceutical, biotechnology, and academic research applications. The company offers cDNA clones, including TrueORF cDNA, viral ORF, destination vectors, TrueClones (human), TrueClones (mouse), organelle marker plasmids, MicroRNA tools, mutant and variant clones, plasmid purification kits, transfection reagents, and gene synthesis service; and HuSH shRNA, siRNA, miRNA, qPCR reagents, plasmid purification products, transfection reagents, PolyA+ and total RNA products, first-strand cDNA synthesis, and CRISPR/Cas9 genome products. It also provides proteins and lysates, such as purified human proteins, over-expression cell lysates, mass spectrometry standard proteins, and protein purification reagents; UltraMAB IHC antibodies, TrueMAB primary antibodies, anti-tag and fluorescent proteins, ELISA antibodies, luminex antibodies, secondary antibodies, and controls and others; and anatomic pathology products, including IHC antibodies, detection systems, and IHC accessories
The company offers luminex and ELISA antibody pairs, autoantibody profiling arrays, ELISA kits, cell assay kits, assay reagents, custom development, and fluorogenic cell assays; TissueFocus search tools; tissue sections; tissue microarrays, cancer protein lysate arrays, TissueScan cDNA arrays, tissue blocks, and quality control products, as well as tissue RNA, DNA, and protein lysates; and lab essentials. Its research areas include cancer biomarker research, RNAi, pathology IHC, stem cell research, ion channels, and protein kinase products. The company provides gene synthesis and molecular biology services, genome editing, custom cloning, custom shRNA, purified protein, monoclonal antibody development, and assay development. It sells its products through distributors worldwide, as well as online. OriGene Technologies, Inc. was incorporated in 1995 and is based in Rockville, Maryland.
Louis, MO – November 18, 2015 Merck KGaA, Darmstadt, Germany, Completes Sigma-Aldrich Acquisition
Merck KGaA today announced the completion of its $17 billion acquisition of Sigma-Aldrich, creating one of the leaders in the $130 billion global industry to help solve the toughest problems in life science.
Press Release: 18-Nov-2015
Letter to our Life Science Customers from Dr. Udit Batra
The life science business of Merck KGaA, Darmstadt, Germany brings together the world-class products and services, innovative capabilities and exceptional talent of EMD Millipore and Sigma-Aldrich to create a global leader in the life science industry.
“Everything we do starts with our shared purpose – to solve the toughest problems in life science by collaborating with the global scientific community.
This combination is built on complementary strengths, which will enable us to serve you even better as one organization than either company could alone.
This means providing a broader portfolio with a catalog of more than 300,000 products, including many of the most respected brands in the industry, greater geographic reach, and an unmatched combination of industry-leading capabilities.”
Thermo Fisher Scientific Inc. is a provider of analytical instruments, equipment, reagents and consumables, software and services for research, manufacturing, analysis, discovery and diagnostics. The company operates through four segments: Life Sciences Solutions, provides reagents, instruments and consumables used in biological and medical research, discovery and production of new drugs and vaccines as well as diagnosis of disease; Analytical Instruments, provides instruments, consumables, software and services that are used in the laboratory; Specialty Diagnostics, offers diagnostic test kits, reagents, culture media, instruments and associated products, and Laboratory Products and Services, offers self-manufactured and sourced products for the laboratory.
WALTHAM, Mass. & SANTA CLARA, Calif.–(BUSINESS WIRE)–Jan. 8, 2016– Thermo Fisher Scientific Inc. (NYSE:TMO), the world leader in serving science, and Affymetrix Inc. (NASDAQ:AFFX), a leading provider of cellular and genetic analysis products, today announced that their boards of directors have unanimously approved Thermo Fisher’s acquisition of Affymetrix for $14.00 per share in cash. The transaction represents a purchase price of approximately $1.3 billion.
The world’s most innovative intersection, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
The world’s most innovative intersection
Reported by: Irina Robu
The world’s most innovative crossroads in history is the intersection of
Vassar Street and Main Street, in the new world’s Cambridge, Massachusetts, would be a leading candidate.
According to the article published in Wired Magazine in November 2015 “when the Whitehead got too small for genomicist Eric Lander’s ambitions, he launched a flashier and brasher newcomer next door. The Broad Institute’s gargantuan gleaming glass lobby is filled with early gene-sequencing instruments. Its multimedia screens boast that this is one of the world’s largest gene-sequencing and research factories. The Broad’s strategy is different from that of the Whitehead; instead of concentrating a few in an ultra-exclusive bioclub, Broad bridges MIT, Harvard and most of the hospitals in Boston. Its 2,000 members extend outwards, partnering with tens of thousands of others globally. Those working at the Broad are not averse to commerce; its director alone helped to build Foundation Medicine, Verastem, Millennium, Fidelity Biosciences, Courtagen and Aclara among many other leading companies.
The sixth building on this extraordinary corner, Novartis, focuses on private research, and represents a huge migration from Basel in Switzerland towards the MIT campus, becoming Cambridge’s largest employer. Pfizer, Sanofi, Amgen, Biogen-Idec and hundreds of others cluster nearby. “
Attracting the best and the brightest, one can change not just a city but the world.
Nicole Kidman’s new role shines a light on genetics
Nicole Kidman has taken to the London stage to play Rosalind Franklin, one of the most important yet overshadowed scientists of the 20th century. The impact of her work is still revolutionizing genetics work in modern pathology.
Photograph 51 relates Franklin’s contribution to the discovery of the double helix structure of DNA in the 1950s. The play depicts the sometimes confrontational working relationship between the talented Franklin and her laboratory partner, Maurice Wilkins.
The play’s name comes from the X-ray image of DNA that Franklin created. It was this image that led scientists James Watson and Francis Crick to determine the chemical structure of DNA, ushering in the age of modern genetics.
In 1962, the Nobel Prize in Physiology or Medicine was awarded to Watson, Crick and Wilkins, with Franklin notably overlooked. In 1958, Franklin died of cancer, never having been recognised for her work. Photograph 51 attempts to bring Franklin’s role to light.
Dr Melody Caramins is a genetic pathologist working in Sydney. She says modern medicine would look very different without the discovery.
“Genetic testing is widely used, particularly for screening; for example prenatal testing for Down Syndrome and newborn bloodspot testing for life-threatening conditions like Cystic Fibrosis.
Genetic testing can also suggest if a particular cancer drug is likely to be effective for an individual patient. Testing can also indicate an elevated risk of developing a hereditary cancer.”
Dr Caramins says that genetics is an exciting and rapidly developing area to work in as there are so many questions to be answered.
“I encourage anyone willing to work hard to consider pathology and genetics in particular. There is great variety in the work on offer, including lab work and consulting directly with patients.”
This burgeoning profession owes much to genetic pioneers like Rosalind Franklin.
FDA Cellular & Gene Therapy Guidances: Implications for CRSPR/Cas9 Trials, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
FDA Cellular & Gene Therapy Guidances: Implications for CRSPR/Cas9 Trials
Reporter: Stephen J. Williams, PhD
The recent announcement by Editas CEO Katrine Bosley to pursue a CRSPR/Cas9 gene therapy trial to correct defects in an yet to be disclosed gene to treat one form of a rare eye disease called Leber congenital amaurosis (multiple mutant genes have been linked to the disease) have put an interesting emphasis on the need for a regulatory framework to initiate these trials. Indeed at the 2015 EmTechMIT Conference Editas CEO Katrine Bosley had mentioned this particular issue: the need for discourse with FDA and regulatory bodies to establish guidelines for design of clinical trials using the CRSPR gene editing tool.
To this effect, I have listed below, the multiple FDA Guidance Documents surrounding gene therapy to show that, in the past year, the FDA has shown great commitment to devise a regulatory framework for this therapeutic area.
MYBPC3 provided by HGNC Official Full Name – myosin binding protein C, cardiac provided by HGNC Primary source – HGNC:HGNC:7551 ;
See related Ensembl:ENSG00000134571;HPRD:02980;MIM:600958;Vega:OTTHUMG00000166986
Gene type protein coding RefSeq status
REVIEWED Organism Homo sapiens
LineageEukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini; Catarrhini; Hominidae; Homo
Also known asFHC; CMH4; CMD1MM; LVNC10; MYBP-C
SummaryMYBPC3 encodes the cardiac isoform of myosin-binding protein C. Myosin-binding protein C is a myosin-associated protein found in the cross-bridge-bearing zone (C region) of A bands in striated muscle. MYBPC3, the cardiac isoform, is expressed exclussively in heart muscle. Regulatory phosphorylation of the cardiac isoform in vivo by cAMP-dependent protein kinase (PKA) upon adrenergic stimulation may be linked to modulation of cardiac contraction. Mutations in MYBPC3 are one cause of familial hypertrophic cardiomyopathy. [provided by RefSeq, Jul 2008]
What is the official name of the MYBPC3 gene?
The official name of this gene is “myosin binding protein C, cardiac.”
MYBPC3 is the gene’s official symbol. The MYBPC3 gene is also known by other names, listed below.
Read more about gene names and symbols on the About page.
What is the normal function of the MYBPC3 gene?
The MYBPC3 gene provides instructions for making the cardiac myosin binding protein C (cardiac MyBP-C), which is found in heart (cardiac) muscle cells. In these cells, cardiac MyBP-C is associated with a structure called the sarcomere, which is the basic unit of muscle contraction. Sarcomeres are made up of thick and thin filaments. The overlapping thick and thin filaments attach to each other and release, which allows the filaments to move relative to one another so that muscles can contract. Regular contractions of cardiac muscle pump blood to the rest of the body.
In cardiac muscle sarcomeres, cardiac MyBP-C attaches to thick filaments and keeps them from being broken down. Cardiac MyBP-C has chemical groups called phosphate groups attached to it; when the phosphate groups are removed, cardiac MyBP-C is broken down, followed by the breakdown of the proteins of the thick filament. Cardiac MyBP-C also regulates the rate of muscle contraction, although the mechanism is not fully understood.
Does the MYBPC3 gene share characteristics with other genes?
A gene family is a group of genes that share important characteristics. Classifying individual genes into families helps researchers describe how genes are related to each other. For more information, see What are gene families? in the Handbook.
Organization and Sequence of Human Cardiac Myosin Binding Protein C Gene (MYBPC3) and Identification of Mutations Predicted to Produce Truncated Proteins in Familial Hypertrophic Cardiomyopathy
Cardiac myosin binding protein C (MyBP-C) is a sarcomeric protein belonging to the intracellular immunoglobulin superfamily. Its function is uncertain, but for a decade evidence has existed for both structural and regulatory roles. The gene encoding cardiac MyBP-C (MYBPC3) in humans is located on chromosome 11p11.2, and mutations have been identified in this gene in unrelated families with familial hypertrophic cardiomyopathy (FHC). Detailed characterization of the MYBPC3 gene is essential for studies on gene regulation, analysis of the role of MyBP-C in cardiac contraction through the use of recombinant DNA technology, and mutational analyses of FHC. The organization of human MYBPC3 and screening for mutations in a panel of French families with FHC were established using polymerase chain reaction, single-strand conformation polymorphism, and sequencing. The MYBPC3 gene comprises >21 000 base pairs and contains 35 exons. Two exons are unusually small in size, 3 bp each. We found six new mutations associated with FHC in seven unrelated French families. Four of these mutations are predicted to produce truncated cardiac MyBP-C polypeptides. The two others should each produce two aberrant proteins, one truncated and one mutated. The present study provides the first organization and sequence for an MyBP-C gene. The mutations reported here and previously in MYBPC3 result in aberrant transcripts that are predicted to encode significantly truncated cardiac MyBP-C polypeptides. This spectrum of mutations differs from the ones previously observed in other disease genes causing FHC. Our data strengthen the functional importance of MyBP-C in the regulation of cardiac work and provide the basis for further studies.
Cardiac MyBP-C is a member of a family comprising isoforms specific for slow-skeletal, fast-skeletal, and cardiac muscles. The skeletal isoforms were initially described in 1971 [1] and later came to be recognized as proteins with specific myosin- and titin-binding properties located in the A bands of the thick filaments of all vertebrate cross-striated muscle and forming a series of seven to nine transverse stripes, 43 nm apart, in the crossbridgebearing region. [2-5] Subsequent cloning of the three isoforms showed them to belong to the intracellular immunoglobulin superfamily and to share a conserved domain pattern consisting of IgI set domains and fn-3 domains. [6-11]
Comparison of the cardiac and the skeletal MyBP-C isoform sequences reveals three distinct regions that are specific to the cardiac isoform: the N-terminal domain C0 IgI containing 101 residues, the MyBP-C motif (a 105-residue stretch linking the C1 and C2 IgI domains), and a 28-residue loop inserted in the C5 IgI domain. [7,12,13] The MyBP-C motif is not specific to the cardiac isoform, but the alignment of skeletal and cardiac sequences revealed the addition of a nine-residue loop in the cardiac variant, which is the key substrate site for phosphorylation by both protein kinase A and a calmodulin-dependent protein kinase associated with the native protein. [7] As for the 28-residue loop, it is strictly cardiac specific. [14,15] The major myosin-binding site of MyBP-C resides in the C-terminal C10 IgI domain and is mainly restricted to the last 102 amino acids. [16-18] The titin-binding site is also located in the C-terminal region, spanning the C8 to C10 IgI domains of the molecule. [6,13]
The function of MyBP-C is uncertain, but for a decade evidence has existed to indicate both structural and regulatory roles. It should be stressed, however, that most studies were performed on skeletal muscles and that very little functional data exist for cardiac muscle. Several investigators have shown that MyBP-C modulates in vitro the shape and the length of sarcomeric thick filaments [19-21] and that depending on ionic strength and the molar ratio of actin and myosin in solution, the addition of MyBP-C can modulate the actin-activated ATPase activity of skeletal and cardiac myosins. [3,22,23] Partial extraction of the MyBP-C from rat skinned cardiac myocytes and rabbit skeletal muscle fibers alters Ca2+-sensitive tension, supporting the view that contractile function is affected by MyBP-C. [24] This view was very recently strengthened by the elegant studies of Weisberg and Winegrad. [25] These authors showed that phosphorylation of cardiac MyBP-C alters myosin crossbridges in native thick filaments isolated from rat ventricles and suggested that MyBP-C can modify force production in activated cardiac muscles.
The gene encoding the cardiac isoform in humans (MYBPC3) was assigned to the chromosomal location 11p11.2 [7] in a region where we had identified the CMH4 disease locus in FHC. [26] Recently, three mutations in MYBPC3 have been identified in unrelated families with FHC by our group [27] and others. [28] FHC is a genetically and phenotypically heterogeneous disease, transmitted as an autosomal-dominant trait. None of the previous hypotheses of the pathophysiological mechanisms would have predicted that defects in sarcomeric protein genes could be a possible molecular basis for the disease. The results of molecular genetic studies have nevertheless shown that many forms of the disease involve mutations in genes encoding sarcomeric proteins (for reviews, see [29-31]), and the findings that MYBPC3 is one of these disease genes are consistent with the view that cardiac MyBP-C may play a more important role in the regulation of cardiac contraction than was previously thought.
Detailed characterization of the MYBPC3 gene is essential for studies of gene regulation, analysis of the role of cardiac MyBP-C in the sarcomere structure and function through the use of recombinant DNA technology, and, finally, mutational analyses and further studies in FHC. In the present work, we have determined the organization and sequence of the human MYBPC3 gene and shown it to exceed 21 000 bp in size and to contain 35 exons, out of which 34 are coding. We also report that six new mutations in the MYBPC3 gene are associated with FHC in seven unrelated French families. Four of these mutations are predicted to produce truncated cardiac MyBP-C polypeptides in these families. The two others should each produce two aberrant proteins, one truncated and the other mutated or deleted.
Screening the Human MYBPC3 Gene for Mutations
The primers were constructed on the basis of flanking intron sequences and were used to amplify each exon (see Table 1). The touchdown PCR was performed (as described above according to the conditions reported in Table 1) on genomic DNA from unrelated FHC patients. For SSCP, PCR products were denatured for 5 minutes at 96 degrees C in a standard denaturing buffer, kept on ice for 5 minutes, loaded onto 6% to 10% polyacrylamide gels, and then run at 6 mA and at 7 degrees C or 20 degrees C in a Hoeffer apparatus. The bands were visualized after silver staining of the gels (Bio-Rad). Sequencing was performed as described above.
Oligonucleotide Primers and PCR Conditions for Detection of Mutations in Human MYBPC3 Gene
RNA Isolation, cDNA Synthesis, and MYBPC3 cDNA Amplifications
Total cellular RNA was isolated from human lymphoblastoid cell lines using RNA Plus (Bioprobe Systems), and the cDNA synthesis was performed as previously described. [27] The cDNA products were amplified in a 50-micro L PCR reaction using two outer primers (see Table 2). A second round of PCR was performed with a final dilution of 1:100 of the first round products, using nested primers (see Table 2). The primers were determined according to the cDNA sequence (EMBL accession number X84075), and cDNA fragments were amplified using a touchdown PCR protocol between 70 degrees C and 60 degrees C. Sizes of normal and mutated cDNA-PCR fragments were assessed, followed by size-fractionation on agarose gels. After extraction and purification of the normal and the putative mutated cDNAs, they were cloned using pGEM-T System II (Promega) and then sequenced as described above.
Oligonucleotide Primers for MYBPC3 cDNA Amplifications
Genomic Organization and Sequence of Human MYBPC3
The size of introns was first estimated by PCR amplification of DNA segments between exons from control genomic DNA, followed by size-fractionation of the PCR products on agarose gels. The exon/intron boundaries and the entire intronic sequences were then determined by sequencing. The sequences have been deposited with EMBL (accession number Y10129). The schematic organization of the human MYBPC3 gene and the alignment of exons with structural domains in the protein are shown in Figure 1. The gene comprises >21 000 bp and contains 35 exons, out of which 34 are coding. A (GT) repeat was found in intron 20 (data not shown). The 101-residue N-terminal extra IgI domain is encoded by exons 1 to 3; the proline-rich domain (51 residues), by exons 3 and 4; the C1 IgI domain (104 residues), by exons 4 to 6; the MyBP-C motif (105 residues), by exons 6 to 12; the C2 IgI domain (91 residues), by exons 12 to 16; the C3 IgI domain (91 residues), by exons 16 to 18; the C4 IgI domain (90 residues), by exons 18 to 20; the linker (11 residues), by exons 20 and 21; the C5 IgI domain (127 residues), by exons 21 to 24; the C6 fn-3 domain (98 residues), by exons 24 to 26; the C7 fn-3 domain (101 residues), by exons 26 to 28; the C8 IgI domain (95 residues), by exons 28 to 30; the C9 fn-3 domain (115 residues), by exons 30 to 32; and the C-terminal C10 IgI domain (94 residues), by exons 32 to 34.
Schematic organization of the human MYBPC3 gene and alignment of exons with structural domains of the protein. Top, The structural domains of cardiac MyBP-C. The high-affinity myosin heavy chain domain (confined to the C10 IgI repeat), the titin binding site (C8 to C10), and the phosphorylation sites are indicated. Middle, The mRNA with the limits of exons. Bottom, the schematic organization of the gene with locations of exons shown by boxes and introns shown by horizontal lines. The exons are numbered from the 5 prime end of the gene, with exon 1 containing the first codon ATG. The exons coding for structural domains are indicated by interrupted lines.
The sizes of exons and introns are summarized in Table 3. The exon sizes, excluding the 5 prime and 3 prime untranslated regions, vary between 3 and 267 bp. Two of the exons, ie, exons 10 and 14, are unusually small and contain three nucleotides each. The remaining 32 exons vary in size between 18 and 267 bp. Twenty-seven exons finish with a split codon (see Table 3). The intron sizes vary between 85 and [nearly =]2000 bp. The major consensus donor splice site is GTGAG in 53% of the cases, and the major consensus acceptor splice site is CAG in 91% of the cases. Twenty-seven of the 34 introns contain putative branch point sequences located -14 to -51 upstream from each splice acceptor site. Introns 1, 4, 11, 14, 16, 24, and 31 do not contain any known consensus branch point sequence.
Exon-Intron Boundaries in the Human MYBPC3 Gene Identification of Mutations in MYBPC3 Gene Associated With FHC
Because the families were not large enough to assess linkage on the basis of a statistically significant Lod score, we used haplotype analysis to define the disease locus responsible for FHC in each family. Linkage was established on the basis of the transmission of a common haplotype in affected individuals and exclusion on the basis of affected recombinant individuals. Families 717 and 740 presented linkage only to CMH4, and the other five families (families 702, 716, 731, 750, and 754) were less informative but at least potentially linked to CMH4 (data not shown).
All the exon-intron boundaries were analyzed by PCRSSCP according to the conditions described in Table 2. A total of six new mutations were identified in MYBPC3 associated with FHC in seven unrelated French families (Figure 2 andFigure 3, Table 4).
Pedigrees of families with MYBPC3 gene mutations. Clinical affection status is indicated: darkened, affected; clear, unaffected; and clear with a cross, indeterminate. Genetically affected status is indicated by an asterisk. The mutations (M) are as follows: M1, GTGAG[arrow right]GTGAA splice donor site mutation in intron 7; M2, GAA[arrow right]CAA mutation in exon 17; M3, GT[arrow right]AT splice donor site mutation in intron 23; M4, TGAT[arrow right]TGGT transversion in the branch point consensus sequence of intron 23; M5, [-GCGTC] deletion in exon 25; and M6, duplication [+TTCAAGAATGGC]/deletion [-ACCT] in exon 33.
Normal and mutated cardiac MyBP-C polypeptides. N indicates the normal structure of human cardiac MyBP-C; M1 to M6 correspond to the predicted products of the aberrant MyBP-C cDNAs resulting from the different mutations.
M1 is a GTGAG[arrow right]GTGAA transition in the 3 prime splice donor site of intron 7 in family 717. The G residue at position +5 in the intron is a highly conserved nucleotide in the splice donor consensus sequence. [33] The G[arrow right]A mutation inactivates this donor site. Amplification of MYBPC3 cDNA from patients’ lymphocytes identified the skipping of the 49-bp exon 7 that produces a frameshift. No alternative splice donor site was found in intron 7. The aberrant cDNA encodes 258 normal cardiac MyBP-C residues, followed by 25 new amino acids, and a premature termination of translation. This should produce a large truncated protein (-80%) lacking the MyBP-C motif containing the phosphorylation sites and the titin and myosin binding sites.
M2 is a G[arrow right]C transversion at position 1656 in exon 17 in families 702 and 750 that produces a mutated polypeptide in the C3 domain at the position 542 (Glu[arrow right]Gln). Otherwise, this mutation affects the last nucleotide of the exon, which is part of the consensus splicing site. [34] A common feature in human exon-intron boundaries is that 80% of exons finish with a guanine (85% in MYBPC3). This mutation also results in an aberrant transcript in lymphocytes (with the skipping of exon 17) that directly introduces a stop codon. The aberrant cDNA encodes 486 normal cardiac MyBP-C residues, leading to a truncated protein (-62%) that lacks the titin and myosin binding sites.
M3 is a GT[arrow right]AT transition in the 3 prime splice donor site of intron 23 in family 716 that inactivates this splicing site. This mutation produces the skipping of the 160-bp exon 23. No alternative splice donor site was found in lymphocytes. The mutated cDNA identified in lymphocytes encodes 717 normal residues and then 51 novel amino acids, followed by premature termination of the translation in the C5 domain, leading to a potential truncated protein (-44%) that loses the titin and myosin binding domains.
M4 is a TGAT[arrow right]TGGT transition in intron 23 in family 740. This A[arrow right]G mutation inactivates a potential branch point consensus sequence (URAY). Although three potential branch points exist upstream from the mutation, they do not seem to be used, since analysis of the transcripts in lymphocytes indicates the existence of two aberrant cDNAs. One corresponds to the skipping of the 105-bp exon 24 without frameshift and encodes a polypeptide depleted of 35 amino acids in the C6 domain (-50% of C6). The other still contains the 724-bp intron 23. This mutant cDNA is associated with a frameshift: it encodes 770 normal cardiac MyBP-C residues and then 100 novel amino acids, followed by a stop codon, and the corresponding truncated protein (-40%) should not interact with either titin or myosin.
M5 is a 5-bp deletion (-GCGTC) in exon 25 in family 731. This deletion also produces a frameshift: the aberrant cDNA identified in the lymphocytes encodes 845 normal MyBP-C residues and then 35 novel amino acids, followed by a premature stop codon in the C6 domain that should produce a truncated protein (-34%), losing the C-terminal region containing both the titin- and myosin-binding sites.
M6 is a 12-bp duplication (+TTCAAGAATGGC)/4-bp deletion (-ACCT) in exon 33 in family 754. This modification introduces a frameshift at position 3691 that leads to 1220 normal MyBP-C residues and then 19 novel amino acids, followed by a premature stop codon in the last third part of the C10 domain. The predicted truncated protein (-4%) should also lose part of its myosin binding site.
All these six mutations were absent in 200 samples from control unrelated subjects without FHC and also in 42 unrelated probands with FHC (out of which 8 have mutations in MYBPC3, 8 have mutations in the beta-myosin heavy chain gene [MYH7], 1 has a mutation in the cardiac troponin T gene, and 25 have presently undefined mutations).
Discussion
The present work describes the first genomic organization for an MyBP-C. The gene is over 21 000 bp and contains 35 exons. An interesting feature of the organization of this gene is that there is a striking correspondence between the limits of the exons and those of structural domains (Figure 1). The IgI and fn-3 domains are encoded by two or three exons. The linker region between the IgI C4 and IgI C5 domains corresponds to exon 20. Twenty-six of the 28 cardiac-specific amino acids of the IgI C5 domain correspond to exon 22. Finally, the MyBP-C motif is encoded by the most complex exon structure: the nine cardiac-specific amino acids correspond to exon 8, and the four phosphorylation sites described by Gautel et al [7] are encoded by six exons and are located at the end or at the junction of two exons (phosphorylation sites: A, junction of exons 7 and 8; B, end of exon 8; C, end of exon 9, exon 10, and beginning of exon 11; and D, end of exon 12). The correlation between exonic organization and protein structure has also recently been described concerning the titin, [35] suggesting a common feature for the intracellular immunoglobulin superfamily.
We suggest that the new mutations described here cause FHC because they segregate with the disease, are not present in controls, and result in aberrant transcripts that are predicted to encode significantly altered cardiac MyBP-C polypeptide structure and/or function. They are all transcribed into mRNAs in lymphocytes. However, because most, if not all, genes in humans are thought to be transcribed at very low levels in lymphocytes (“illegitimate transcription”), [36] these results do not address the hypothesis that these mutations are expressed in the diseased myocardium. Since cardiac MyBP-C is specifically expressed in heart, ventricular tissue is needed to address this issue, and we had no access to any myocardial specimens. One study documented the expression of a missense mutation in the mRNA for the beta-myosin heavy chain in myocardial tissue from an affected patient with FHC. [37] Because the beta-myosin heavy chain is normally expressed in slow-twitch skeletal fibers, skeletal muscle biopsies can also be used to show that the mutated myosin is produced in the muscle and that the mutation alters the function of the beta-myosin and the contractile properties of the muscle fibers. [38,39] One might thus reasonably assume that the MYBPC3 gene mutations are expressed in the myocardium and that they exert their effect by altering the multimeric complex assembly of the cardiac sarcomere via at least one of these mechanisms: (1) They can act as “poison polypeptides” through a dominant-negative effect. The altered proteins would be incorporated in the sarcomere and would alter the assembly of the sarcomeric filaments, since most truncated MyBP-Cs are unable to cross-link the titin and/or myosin molecules. (2) They can act as “null alleles,” potentially leading to haplo insufficiency; the production of insufficient quantities of normal cardiac MyBP-C would produce an imbalance in stoichiometry of the thick-filament components that would be sufficient to alter the sarcomeric structure and function. (3) Since myosin, titin, and MyBP-C might be translated and assembled cotranslationally, one can also assume that the misfolded, mutated MYBPC3 mRNAs may disturb the translation of the other sarcomeric components that would interfere with the proper assembly of sarcomeric structures.
The full spectrum of mutations of the FHC disease genes is far from known, but it is intriguing to note that most mutations found so far in MYH7 are missense ones, whereas most of those in MYBPC3 disrupt the reading frame and produce premature stop codons. Both genes are large ones, composed of [nearly =]40 exons, and there are no reasons for different types of mutations in the two genes. Thus, one might hypothesize that mutations leading to truncated proteins exist also for MYH7 in humans but have no deleterious effect. In support of this are the reports of two deletions in the C-terminal part of the beta-myosin heavy chain molecule with almost no phenotype. One is a 2.4-kbp deletion including part of intron 39 and exon 40 containing the 3 prime untranslated region and the polyadenylation signal, which was reported in a small pedigree. [40] Only the proband had developed clinically diagnosed hypertrophic cardiomyopathy at a very late onset (age, 59 years), and the other genotypically affected family members had not developed the disease at 10, 32, and 33 years. The other one is a large deletion leaving only a short variant of the beta-myosin heavy chain constituting only the first 53 residues of the molecule (out of 1935). This deletion was found by chance in an unaffected individual. [41] For MYBPC3, in contrast, the majority of the mutations described so far produce the C-terminal truncation of the cardiac MyBP-C polypeptides and are associated with an FHC phenotype. However, no definitive conclusion can be drawn at this stage concerning the pathogenic mechanisms of mutations in these two genes. The present work provides the molecular basis for the production of transgenic animals for cardiac MyBP-C that will help to resolve some of these issues.
Footnotes
Received December 2, 1996; accepted January 10, 1997.
This manuscript was sent to Laurence Kedes, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
. Mammalian skeletal muscle C-protein: purification from bovine muscle, binding to titin and the characterization of a full-length cDNA. J Cell Sci. 1992;102:769–778
. Reinach FC, Fischman DA. Complete sequence of human fast-type and slow type muscle myosin-binding-protein C (MyBP-C): differential expression, conserved domain structure and chromosome assignment.Eur J Biochem. 1993;216:661–669
. Many of the immunoglobulin superfamily domains in cell adhesion molecules and surface receptors belong to a new structural set which is close to that containing variable domains. J Mol Biol. 1994;238:528–539
. Molecular cloning of chicken myosin-binding protein (MyBP) H (86-kDa protein) reveals extensive homology with MyBP-C (C-protein) with conserved immunoglobulin C2 and fibronectin type III motifs. J Biol Chem. 1993;268:3670–3676
. A molecular map of the interactions between titin and myosin-binding protein C: implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem. 1996;235:317–326
. The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J Cell Biol. 1993;123:619–626
. The carboxyl terminus of myosin binding protein C (MyBP-C, C protein) specifies incorporation into A-band of striated muscle. J Cell Sci. 1996;109:101–111
. The aggregation characteristics of column-purified rabbit skeletal myosin in the presence and absence of C-protein at pH 7.0. Biophys J. 1982;37:433–440
http://www.cincinnatichildrens.org/workarea/downloadasset.aspx?id=90111
Hypertrophic Cardiomyopathy (HCM) is relatively common, with a prevalence of 1 in 500 adults (1). HCM is a primary disorder of heart muscle characterized by left ventricular hypertrophy. The most classic finding in HCM is asymmetric septal hypertrophy, with or without left ventricular outflow tract obstruction. The disease demonstrates extensive clinical variability with regard to age of onset, severity and progression of disease. HCM can affect infants and children although it is more typically identified in adolescence or adulthood (2,3).
The MYBPC3 gene codes for cardiac myosin binding protein C. Phosphorylation of this protein modulates contraction and is an important component of the sarcomere (4). The MYBPC3 gene contains 35 exons and is located at chromosome 11p11.2. Up to 40% of individuals with a clinical diagnosis of HCM have MYBPC3 mutations (2). MYBPC3 mutations are inherited in an autosomal dominant manner. The majority of individuals inherit the MYBPC3 from a parent, although de novo mutations do occur. Mutations in MYBPC3 and MYH7 genes are the most common causes of HCM. However, the disease is genetically heterogeneous and sequencing additional genes should be considered if familial HCM is suspected or the underlying etiology remains unknown. Approximately 50-65% of individuals with a known or suspected diagnosis of familial HCM have a mutation in one of a number of genes encoding components of the sarcomere and cytoskeleton (3). Compound heterozygous mutations have been reported in MYBPC3 and other genes associated with HCM (5). Mutations in the MYBPC3 gene have been primarily associated with HCM, but can also be associated with other types of heart muscle disease including dilated cardiomyopathy, restrictive cardiomyopathy and left-ventricular non-compaction (6). Indication MYBPC3 testing is utilized to confirm a diagnosis of HCM in patients with clinically evident disease. Genetic testing also allows for early identification and diagnosis of individuals at greatest risk prior to the expression of typical clinical manifestations. If a mutation is identified in an asymptomatic individual, regular and routine outpatient follow up is indicated. If clinically unaffected members of a family with an identified mutation for HCM are found not to carry that mutation, they can be definitely diagnosed as unaffected and reassured that neither they nor their children will be at higher risk compared to the general population to develop symptoms related to HCM. A negative test result in an individual with a known familial mutation also eliminates the need for routine follow up. Methodology:
All 35 exons of the MYBPC3 gene, as well as the exon/intron boundaries and a portion of untranslated regions of the gene are amplified by PCR. Genomic DNA sequences from both forward and reverse directions are obtained by automatic fluorescent detection using an ABI PRISM® 3730 DNA Analyzer. Sequence variants different from National Center for Biotechnology Information GenBank references are further evaluated for genetic significance. If a mutation is identified, a known familial mutation analysis will be available for additional family members. Sensitivity & Accuracy:
Greater than 98.5% of the mutations in exon 1-35 of MYBPC3 are detectable by sequence based methods. Sequencing does not detect deletions or duplications. Mutations in MYBPC3 account for up to 40% of cases of idiopathic hypertrophic cardiomyopathy. References:
1. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the cardia study. Coronary artery risk development in (young) adults. Circulation. 1995;92:785-789.
2. Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, Elliott PM. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circulation Cardiovascular Genetics. 2009;2:436441.
3. Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, Seidman CE. Shared genetic causes of cardiac hypertrophy in children and adults. The New England Journal of Medicine. 2008;358:1899-1908.
4. van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein c mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473-1483.
5. Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein c mutations and compound heterozygosity in hypertrophic cardiomyopathy. Journal of the American College of Cardiology. 2004;44:1903-1910.
6. Hershberger RE, Norton N, Morales A, Li DX, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. CirculationCardiovascular Genetics. 2010;3:155-161.
The cancer stem-cell hypothesis proposes the existence of a subset of cells within a heterogeneous tumor cell population that have stem-cell like properties [1], and may be essential for the progression and metastases of epithelial malignancies, by providing a reservoir of cells that self-renew and differentiate into the bulk of the tumor [2]. The stem-cell hypothesis implies that similar genetic regulatory pathways might define critical stem-cell like functions, such as self-renewal and pluripotency, in both normal and cancer stem-cells. Indeed, cancer stem-cells have been identified in many tumor types, such as breast [3], pancreas [4] and ovarian [5], based on screening with cellular markers typically found in normal stem-cells such as CD44, ALDH1, and CD133 (reviewed in [2]). A number of studies have suggested that the expression of these stem-cell markers is correlated with poor prognosis [6-9]. The ability to identify and isolate these populations may have a significant impact on design of individualized therapies.
Great general posts and good review on this site about Cancer Stem Cells, their markers, and ability to target them with chemotherapy can be seen here.
However, there has been growing acknowledgement of the ability of cancer stem cell populations to resist the cytotoxic effects of most chemotherapeutic agents, including cisplatin, topoisomerase inhibitors, DNA damaging agents, and even tyrosine kinase inhibitors (TKI). Indeed, some feel that intrinsic resistance to cytotoxic drugs may be a biological feature of cancer stem cells.
Definitions:
Acquired resistance: a resistance to a particular drug which results following continued exposure to said drug. Can take days (in cases of some TKIs) or months to develop. Acquired resistant cells lines are developed by exposure to increasing drug concentration over a time period (either intermittent exposure or continuous exposure)
Intrinsic resistance: a pre-existing resistance usually termed refractory where cancer cells THAT HAVE NOT BEEN EXPOSED to drug, do not respond to initial drug exposure. Can be seen experimentally in panels of unrelated cancer cells lines isolated from untreated patients which show no cytotoxicity to drug exposure in vitro.
Below is one of the first reports which described the drug resistant phenotype of cancer stem cells in an in vivo (mouse) model of breast cancer with videos.
Cancer Res. 2008 May 1;68(9):3243-50. doi: 10.1158/0008-5472.CAN-07-5480.
The majority of BRCA1-associated breast cancers are basal cell-like, which is associated with a poor outcome. Using a spontaneous mouse mammary tumor model, we show that platinum compounds, which generate DNA breaks during the repair process, are more effective than doxorubicin in Brca1/p53-mutated tumors. At 0.5 mg/kg of daily cisplatin treatment, 80% primary tumors (n = 8) show complete pathologic response. At greater dosages, 100% show complete response (n = 19). However, after 2 to 3 months of complete remission following platinum treatment, tumors relapse and become refractory to successive rounds of treatment. Approximately 3.8% to 8.0% (mean, 5.9%) of tumor cells express the normal mammary stem cell markers, CD29(hi)24(med), and these cells are tumorigenic, whereas CD29(med)24(-/lo) and CD29(med)24(hi) cells have diminished tumorigenicity or are nontumorigenic, respectively. In partially platinum-responsive primary transplants, 6.6% to 11.0% (mean, 8.8%) tumor cells are CD29(hi)24(med); these populations significantly increase to 16.5% to 29.2% (mean, 22.8%; P < 0.05) in platinum-refractory secondary tumor transplants. Further, refractory tumor cells have greater colony-forming ability than the primary transplant-derived cells in the presence of cisplatin. Expression of a normal stem cell marker, Nanog, is decreased in the CD29(hi)24(med) populations in the secondary transplants. Top2A expression is also down-regulated in secondary drug-resistant tumor populations and, in one case, was accompanied by genomic deletion of Top2A. These studies identify distinct cancer cell populations for therapeutic targeting in breast cancer and implicate clonal evolution and expansion of cancer stem-like cells as a potential cause of chemoresistance.
Please Watch Videos
Below is a curation of talks and abstracts from the 2015 Annual AACR Meeting in Philadelphia, PA.
The Talk by Dr. Cheresh is an example of this school of thought; that inducing cancer cell stemness can result in development of drug resistance, in this case to a TKI. (For a press release on this finding see here.)
Induction of cancer stemness and drug resistance by EGFR blockade
Presentation Time:
Tuesday, Apr 21, 2015, 12:00 PM -12:15 PM
Abstract Body:
Tumor drug resistance is often accompanied by genetic and biological changes in the tumor cell population reflecting the acquisition of a stem-like state. However, it is not clear whether cancer therapies select for the growth of drug resistance cancer stem cells and/or directly induce the reprograming of tumor cells to a cancer stem-like, drug resistance state. We provide evidence that breast, pancreas and lung carcinomas in the presence of prolonged exposure to EGFR inhibitors undergo an epigenetic reprogramming resulting in a drug resistant stem-like tumor population expressing the cell surface marker CD61 (b3 integrin). In fact, CD61 in the context of KRAS, is necessary and sufficient to account for drug resistance, tumor initiation, self-renewal and expression of the pluripotent genes Oct 4 and Nanog. Once expressed, CD61 in the unligated state recruits KRAS to the plasma membrane leading to the activation of RalB, TBK1 and c-Rel driving both stemness and EGFR inhibitor resistance. Pharmacological targeting this pathway with drugs such as bortezomib or revlimid not only reverses stemness but resensitizes these epithelial tumors to EGFR inhibition. This epigenetic pathway can also be initiated by range of cellular stresses found within the tumor microenvironment such as hypoxia, nutrient deprivation, low pH, and oxidative stress. In normal tissues CD61 is induced during tissue remodeling and repair. For example, CD61 was found to be critical for mammary gland remodeling during pregnancy and as a mediator of pathological neovascularization. Together these findings reveal a stress-induced epigenetic pathway characterized by the upregulation of CD61 that promotes the remodeling of normal tissues but in tumors contributes to EGFR inhibitor resistance and tumor progression.
Molecular and Cellular Biology – Poster Presentations – Proffered Abstracts – Poster Presentations – Cell Death Mechanisms: Abstract 4: ABT-263 is effective in a subset of non-small cell lung cancer cell lines
Aoi Kuroda,
Keiko Ohgino,
Hiroyuki Yasuda,
Junko Hamamoto,
Daisuke Arai,
Kota Ishioka,
Tetsuo Tani,
Shigenari Nukaga,
Ichiro Kawada,
Katsuhiko Naoki,
Kenzo Soejima,
and Tomoko Betsuyaku
Cancer Res August 1, 2015 75:4; doi:10.1158/1538-7445.AM2015-4
Molecular and Cellular Biology – Poster Presentations – Proffered Abstracts – Poster Presentations – Cell Death Mechanisms: Abstract 6: Quantitative assessment of BCL-2:BIM complexes as a pharmacodynamic marker for venetoclax (ABT-199)
Sha Jin,
Paul Tapang,
Donald J. Osterling,
Wenqing Gao,
Daniel H. Albert,
Andrew J. Souers,
Joel D. Leverson,
Darren C. Phillips,
and Jun Chen
Cancer Res August 1, 2015 75:6; doi:10.1158/1538-7445.AM2015-6
Molecular and Cellular Biology – Poster Presentations – Proffered Abstracts – Poster Presentations – Cell Death Mechanisms: Abstract 24: The phosphorylation of p53 at serine 46 is essential to induce cell death through palmdelphin in response to DNA damage
Nurmaa Khund Dashzeveg and
Kiyotsugu Yoshida
Cancer Res August 1, 2015 75:24; doi:10.1158/1538-7445.AM2015-24
Molecular and Cellular Biology – Poster Presentations – Proffered Abstracts – Poster Presentations – Cell Signaling in Cancer 1: Abstract 48: Identification of a novel binding protein playing a critical role in HER2 activation in lung cancer cells
Tomoaki Ohtsuka,
Masakiyo Sakaguchi,
Katsuyoshi Takata,
Shinsuke Hashida,
Mototsugu Watanabe,
Ken Suzawa,
Yuho Maki,
Hiromasa Yamamoto,
Junichi Soh,
Hiroaki Asano,
Kazunori Tsukuda,
Shinichiro Miyoshi,
and Shinichi Toyooka
Cancer Res August 1, 2015 75:48; doi:10.1158/1538-7445.AM2015-48
Abstract 1 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Cell Death Mechanisms
Abstract 4: ABT-263 is effective in a subset of non-small cell lung cancer cell lines
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
Rationale:
ABT-263 (Navitoclax) is one of the BH3 mimetics targeting anti-apoptotic B-cell lymphoma-2 (Bcl-2) family proteins such as Bcl-2, Bcl-XL, and Bcl-w, thereby inducing apoptosis. It has been reported that the response to ABT-263 is associated with expressions of myeloid cell leukemia-1 (Mcl-1), an anti-apoptotic protein. Given its effectiveness as a single agent in preclinical studies, ABT-263 is currently being evaluated in clinical trials for small cell lung cancer (SCLC) and leukemia. However, the efficacy of ABT-263 in non-small cell lung cancer (NSCLC) has not been fully evaluated. We examined the effect of ABT-263 on cell proliferation of NSCLC cell lines and investigated the underlying mechanisms.
Methods:
The following 9 NSCLC cell lines were examined: SK-LU-1, A549, H358, Calu3, H3122, H1975, H460, H441, and BID007. The effects of ABT-263 in NSCLC cell lines were evaluated by MTS assay. Apoptosis was examined by flowcytometry using staining for annexin V and propidium iodide (PI), and also western blotting for cleaved PARP. Quantitative RT-PCR was carried out to assess the mRNA expression levels of anti-apoptotic genes and pro-apoptotic genes. Immunoprecipitation and western blotting were performed to compare the levels of anti-apoptotic and pro-apoptotic proteins between the sensitive and resistant cell lines. In addition, knockdown of Mcl-1 was performed by siRNA.
Results:
By screening 9 NSCLC cell lines using MTS assay, we found Calu3 and BID007were sensitive to ABT-263. We also confirmed that apoptosis was induced only in the ABT-263 sensitive lines but not in the ABT-263 resistant cell lines after ABT-263 treatment. However, the expression levels of Bcl-2 family proteins, including Mcl-1, did not differ significantly among the ABT-263 sensitive and resistant cell lines. Unlike the results in previous reports regarding SCLC, Mcl-1 was not decreased in the sensitive cell lines. The ABT-263 resistant cell lines became sensitive to ABT-263 after knockdown of Mcl-1 by siRNA, while the ABT-263 sensitive cell lines maintained the same sensitivity.
Conclusion:
We found that Calu3 and BID007 were sensitive to ABT-263. In the sensitive NSCLC cell lines, ABT-263 induces apoptosis irrespective of Mcl-1 expression levels.
Citation Format: Aoi Kuroda, Keiko Ohgino, Hiroyuki Yasuda, Junko Hamamoto, Daisuke Arai, Kota Ishioka, Tetsuo Tani, Shigenari Nukaga, Ichiro Kawada, Katsuhiko Naoki, Kenzo Soejima, Tomoko Betsuyaku. ABT-263 is effective in a subset of non-small cell lung cancer cell lines. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 4. doi:10.1158/1538-7445.AM2015-4
Abstract 2 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Cell Death Mechanisms
Abstract 6: Quantitative assessment of BCL-2:BIM complexes as a pharmacodynamic marker for venetoclax (ABT-199)
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
The BCL-2-selective inhibitor venetoclax (ABT-199) binds with high affinity to the BH3-binding groove of BCL-2, thereby competing for binding with the BH3-only protein BIM (Souers et al., 2013). Venetoclax is currently being evaluated in clinical trials for CLL, AML, multiple myeloma and NHL. To facilitate these studies, we developed and validated a 384-well electrochemiluminescent ELISA (MSD, Gaithersburg, MD,USA) that quantifies expression of BCL-2, BCL-XL, and MCL-1protein alone or in complex with BIM. We subsequently quantified expression of BCL-2 and BCL-2:BIM complexes in 16 hematologic tumor cell lines. We found the EC50 of venetoclax in these tumor cell lines to correlate strongly with baseline BCL-2:BIM complex levels. This correlation was superior to the correlation between venetoclax EC50 and absolute BCL-2 expression. We also applied the assay to measure disruption of BCL-2:BIM complexes in vivo. Treatment of the Non-Hodgkin’s Lymphoma (NHL) xenograft model SU-DHL-4 with a BCL-2-selective inhibitor resulted in disruption of tumor BCL-2:BIM complexes that aligned with serum and tumor concentrations of inhibitor. Collectively, these data demonstrate that quantifying BCL-2:BIM complexes offers an accurate means of assessing target engagement by venetoclax and, potentially, predicting its efficacy. The utility of this assay is currently being assessed in clinical trials.
Citation Format: Sha Jin, Paul Tapang, Donald J. Osterling, Wenqing Gao, Daniel H. Albert, Andrew J. Souers, Joel D. Leverson, Darren C. Phillips, Jun Chen. Quantitative assessment of BCL-2:BIM complexes as a pharmacodynamic marker for venetoclax (ABT-199). [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 6. doi:10.1158/1538-7445.AM2015-6
Abstract 3 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Cell Death Mechanisms
Abstract 19: Antitumor activity of selective inhibitors of XPO1/CRM1-mediated nuclear export in diffuse malignant peritoneal mesothelioma: the role of survivin
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
Survivin, which is highly expressed and promotes cell survival in diffuse malignant peritoneal mesothelioma (DMPM), exclusively relies on the nuclear exportin 1 (XPO1/CRM1) to be released in the cytoplasm and perform its anti-apoptotic function. Here, we explored the efficacy of selective inhibitors of nuclear export (SINEs) in patient-derived DMPM preclinical models. Exposure to individual SINE (KPT-251, KPT-276, KPT-330) was able to induce a time- and dose-dependent inhibition of the growth of two DMPM cell lines without affecting normal cell proliferation. Such a cell growth inhibition was preceded by a decline in the nuclear XPO1/CRM1 levels and an increase in the nuclear accumulation of its cargo proteins p53 and p21, which led to a cell cycle arrest at G1-phase. Our results also indicated that survivin is an essential component of the downstream signaling pathway of XPO1/CRM1 inhibition in DMPM cells. In fact, in both cell lines, exposure to SINEs led to a time-dependent reduction of cytoplasmic survivin levels and, after an initial survivin nuclear accumulation, also to a progressive decrease in the nuclear protein abundance, through the ubiquitin-proteasomal degradation pathway, leading to the complete depletion of total survivin levels. In both DMPM cell models, according to survivin anti-apoptotic activity, drug-induced reduction of cytoplasmic survivin levels correlated with the onset of caspase-dependent apoptosis. We further observed that SINEs can be combined with other survivin inhibitors, such as the survivin suppressant YM155 to achieve enhanced growth inhibition in DMPM cells. Initial in vivo experiments with orally administered KPT-251, KPT-276 and the orally available, clinical stage KPT-330 (selinexor) indicated that each compound was able to significantly reduce the growth of early-stage subcutaneous DMPM xenografts. Interestingly, additional experiments carry out with selinexor demonstrated that the compound was also able to inhibit the growth of late-stage subcutaneous DMPM xenografts in nude mice. Most importantly, oral administration of selinexor to SCID mice reduced the growth of orthotopic DMPM xenografts, which properly recapitulate the dissemination pattern in the peritoneal cavity of human DMPM and, for this reason, represent a valuable model for investigating novel therapeutic approaches for the disease. Consistent with an important role of survivin as a determinant of anti-cancer activity of SINE compounds, a reduction of the protein expression was observed in tumor specimens obtained from selinexor treated mice. Overall, our results (i) demonstrate a marked efficacy of SINEs in DMPM preclinical models, which is, at least in part, dependent on the interference with survivin intracellular distribution and function, and (ii) suggest SINE-mediated XPO1/CRM1 inhibition as a novel therapeutic option for the disease.
Citation Format: Nadia Zaffaroni, Michelandrea De Cesare, Denis Cominetti, Valentina Doldi, Alessia Lopergolo, Marcello Deraco, Paolo Gandellini, Yosef Landesman, Sharon Friedlander, Michael G. Kauffman, Sharon Shacham, Marzia Pennati. Antitumor activity of selective inhibitors of XPO1/CRM1-mediated nuclear export in diffuse malignant peritoneal mesothelioma: the role of survivin. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 19. doi:10.1158/1538-7445.AM2015-19
Abstract 4 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Cell Death Mechanisms
Abstract 24: The phosphorylation of p53 at serine 46 is essential to induce cell death through palmdelphin in response to DNA damage
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
Tumor suppressor p53 plays a pivotal role in cell cycle arrest, DNA repair, and apoptosis in response to DNA damage. Promoter selectivity of p53 depends mainly on post-translational modification. Notably, the apoptotic function of p53 is related to its phosphorylation at serine-46 (ser46) to promote pro-apoptotic genes. However, little is known about the pro-apoptotic genes induced by Ser46 phosphorylation. Our research achieved to investigate the pro-apoptotic genes induced by p53 in a phospho-ser46-specific manner using microarray and ChIP sequencing in human cancer cell lines. As a result, palmdelphin (PALMD), an isoform of paralemmin protein, was strongly elicited from the phosphorylation of ser46. The mRNA and protein expression of PALMD increased only in wild type p53 transfected cells, but not in ser46-mutated cells. Importantly, PALMD moved to the nucleus in response to DNA damage and the apoptotic function of PALMD was tightly exerted with localization into nucleus. Interestingly, down-regulation of PALMD by siRNA resulted in necroptosis-like cell death through ATP depletion. Moreover, we found vimentin as a PALMD interacting protein and the depletion of vimentin increased PALMD level to accelerate apoptosis. These results demonstrate that p53 regulates cell death fate (apoptosis or necroptosis-like cell death) through promoting PALMD expression in a phospho-ser46-specific manner in response to DNA damage.
Citation Format: Nurmaa Khund Dashzeveg, Kiyotsugu Yoshida. The phosphorylation of p53 at serine 46 is essential to induce cell death through palmdelphin in response to DNA damage. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 24. doi:10.1158/1538-7445.AM2015-24
Abstract 5 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Cell Signaling in Cancer 1
Abstract 48: Identification of a novel binding protein playing a critical role in HER2 activation in lung cancer cells
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
Human epidermal growth factor receptor 2 (HER2) is a member of epidermal growth factor receptor (EGFR) family. Previous studies have revealed that many kinds of malignant tumors have genetic mutations or amplification of HER2, indicating that HER2 alterations are oncogenic. Many kinds of HER2 targeted therapies are effective to HER2 positive tumors, but those treated tumors often get resistance to drugs. Thus, to elucidate HER2 related pathway in cancer biology is important to develop new therapeutic strategy for cancers.
Recently, we newly identified a protein X (a temporary name) as a novel binding protein to HER2 with immunoprecipitation and following LC-Ms/Ms analysis. The protein generally expressed in lung and breast cancers at remarkable level.
We constructed plasmid vectors carrying wild type HER2 and gene X. These vectors were simultaneously introduced to HEK293T cells to examine the binding ability of protein X and HER2 as well as the effect of gene X on HER2-mediated signal-transduction pathway. The approach clearly showed that the expression of gene X, resulted in phosphorylation of HER2 and subsequent activation of oncogenic effector molecules.
We next constructed several kinds of gene X-truncated variants and subjected to the binding assay to look for the binding domain of gene X to HER2. The analysis showed that N-terminal head domain of gene X was essential for the HER2 binding. This domain has an ability to induce HER2 phosphorylation and subsequent activation of the effector kinase, ERK.
In conclusion, we found that gene X is a novel binding protein to HER2 and has a role in HER2 activation.
Citation Format: Tomoaki Ohtsuka, Masakiyo Sakaguchi, Katsuyoshi Takata, Shinsuke Hashida, Mototsugu Watanabe, Ken Suzawa, Yuho Maki, Hiromasa Yamamoto, Junichi Soh, Hiroaki Asano, Kazunori Tsukuda, Shinichiro Miyoshi, Shinichi Toyooka. Identification of a novel binding protein playing a critical role in HER2 activation in lung cancer cells. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 48. doi:10.1158/1538-7445.AM2015-48
Abstract 6 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Cell Signaling in Cancer 1
Abstract 54: Ezrin enhances signaling and nuclear translocation of the epidermal growth factor receptor in non-small cell lung cancer cells
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
The cytoskeletal cross linker protein ezrin is a member of the ezrin-radixin-moesin (ERM) family and plays important roles not only in cell motility, cell adhesion, and apoptosis, but also in various cell-signaling pathways. Ezrin interacts with EGFR in the cell membrane and involves in cell motility events, but little is known about the effects of this interaction on the EGFR signaling pathway. We investigated the role of Ezrin in EGFR signaling and nuclear trafficking in non-small cell lung cancer (NSCLC) cell lines. The ligand induced interaction between Ezrin and EGFR was evaluated by immunoprecipitation (IP) and immunofluorescence (IF) in H292 and A549 cells. Ezrin levels were reduced using siRNA in these two cell lines. Downstream signaling protein phosphorylation and nuclear localization of EGFR were detected after EGF treatment. Expressions of nuclear EGFR target genes were evaluated by qPCR. Endogenous Ezrin was found in a complex with EGFR in IP and IF. When Ezrin protein expression was inhibited, phosphorylation levels of EGFR at Y1068, Y1101 and Y845 were reduced as well as phosphorylation levels of downstream signaling pathway proteins ERK and STAT3. Cell fractionation revealed that EGFR nuclear translocation after EGF treatment significantly reduced in Ezrin-knockdown cells. Further, mRNA levels of EGFR target genes AuroraK-A, COX2, Cyclin D1 and iNOS were decreased in Ezrin-knockdown A549 cells. Small molecule ezrin inhibitors showed strong synergy with EGFR inhibitors in cytotoxicity assays. These results suggest that Ezrin has a role as an enhancer in the EGFR pathway and targeting ezrin may potentiate anti-EGFR based therapies in NSCLC.
Citation Format: Yasemin Saygideger Kont, Haydar Celik, Hayriye V. Erkizan, Tsion Minas, Jenny Han, Jeffrey Toretsky, Aykut Uren. Ezrin enhances signaling and nuclear translocation of the epidermal growth factor receptor in non-small cell lung cancer cells. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 54. doi:10.1158/1538-7445.AM2015-54
Abstract 7 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Cell Signaling in Cancer 1
Abstract 57: Substrates of protein kinase C drive cell rac1-dependent motility
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
This laboratory has identified and/or characterized substrates of PKC that upon phosphorylation give rise to motility, an aspect of metastasis. By use of the traceable kinase method, we discovered that alpha-tubulin and Cdc42 effector protein-4 (CEP4) are PKC substrates. Phosphorylation of alpha-tubulin stimulates its incorporation into microtubules (MTs), consequently increasing the stability and prolonged growth of MTs and leading to the activation of the small GTPase Rac1. CEP4 undergoes phosphorylation by PKC that results in its release from Cdc42, whereupon CEP4 binds a guanine nucleotide exchange factor (GEF) that in turn activates Rac1 GTPase. These results imply that Rac1 acts as a node in pathways driven by phosphorylated PKC substrates. Since translocation of IQGAP to the membrane is known to be promoted by Rac1, a role is explored in non-transformed human MCF-10A cells that express a specific phospho-mimetic mutant substrate. In addition, the phospho-mimetic mutant for each substrate expressed in human metastatic MDA-MB-231 cells produces different morphologies in 3-D growth assays. This research is being supported by NIH CA125632.
Citation Format: Susan A. Rotenberg, Xin Zhao, Shatarupa De. Substrates of protein kinase C drive cell rac1-dependent motility. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 57. doi:10.1158/1538-7445.AM2015-57
Abstract 8 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Deregulation of Gene Expression in Prostate Cancer and Sarcoma
Abstract 88: The Nkx3.1 homeobox gene maintains prostatic identity while its loss leads to prostate cancer initiation
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
Background
Maintenance of epithelial cell identity is tightly coordinated by tissue-specific gene expression programs, which are often deregulated during tumorigenesis. The homeodomain-containing transcription factor, Nkx3.1, is a key regulator of normal prostatic development and is frequently lost at early stages of prostate cancer initiation. In this study, we aim to elucidate detailed mechanisms governing Nkx3.1-driven maintenance of prostate identity and how deregulation of such can lead to prostate tumorigenesis.
Models and Methods
We evaluated the consequences of Nkx3.1 loss or gain of function in vivo using genetically-engineered mouse models and cell-recombination assays. RNA sequencing was performed to generate gene expression profiles, which were analyzed using Gene Set Enrichment analysis (GSEA), and validated by quantitative real-time PCR. In parallel, protein expression was assessed by immunofluorescence and western blot. Immunoprecipitation (IP) and chromatin-immunoprecipitation (ChIP) assays were performed using RWPE1 prostate epithelial cells.
Results
Here, we show that loss of function of Nkx3.1 leads to the progressive down-regulation of a prostate-specific gene expression program and to aberrant expression of genes that are not typically expressed in the prostate epithelium. Conversely, gain of function of Nkx3.1 in non-prostatic epithelium leads to the acquisition of a prostate-like morphology and expression of prostate-related genes. Our findings indicate that the underlying mechanism by which Nkx3.1 promotes prostatic identity is via epigenetic regulation of gene expression. In particular, we show that Nkx3.1 interacts with the histone methyl-transferase complex G9a/Glp. Finally, we demonstrate that this interaction is necessary for maintenance of prostate identity in vivo and that Nkx3.1 and G9a cooperate to control expression of genes that coordinate prostatic epithelial integrity.
Conclusions
Our results suggest that Nkx3.1 promotes prostatic identity by interacting with histone modifying enzymes to coordinate the expression of prostate-specific genes and that the loss of this function results in a failure to maintain prostate identity associated with early stages of prostate tumorigenesis.
Citation Format: Clémentine Le Magnen, Aditya Dutta, Cory Abate-Shen. The Nkx3.1 homeobox gene maintains prostatic identity while its loss leads to prostate cancer initiation. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 88. doi:10.1158/1538-7445.AM2015-88
Abstract 9 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Deregulation of Gene Expression in Prostate Cancer and Sarcoma
Abstract 90: K63-linked JARID1B ubiquitination by TRAF6 contributes to aberrant elevation of JARID1B in prostate cancer
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
Aberrant elevation of JARID1B and histone H3 Lys4 trimethylations (H3K4me3) is frequently observed in many diseases including prostate cancer (PCa), yet the mechanisms on the regulations of JARID1B and H3K4me3 through epigenetic modifications still remain poorly understood. In this study we performed immunohistochemistry staining, immunofluorescence imaging, immunoprecipitation, shRNA and Western blotting analysis in mouse embryonic fibroblasts (MEFs), mouse models, and cultured human prostate cancer cells. As a result, we discovered that SKP2 modulates JARID1B and H3K4me3 levels in vitro in PTEN null prostate cancer cells and in vivo in Pten/Trp53 mouse models. We demonstrated that levels of SKP2, JARID1B and H3K4me3 are strikingly elevated in vitro and in vivo when both PTEN and P53 are inactivated. Importantly, SKP2 inactivation resulted in a reduction of cell growth, cell migration and malignant transformation of Pten/Trp53 double null MEFs, and further restrained prostate tumorigenesis of Pten/Trp53 mutant mice. Mechanistically, JARID1B is ubiquitinated by E3 ligase TRAF6 through the K63-linkage in prostate cancer cells. Interestingly, SKP2 contributes to JARID1B ubiquitination machinery as a non-E3 ligase regulator by decreasing TRAF6-mediated ubiquitination of JARID1B. SKP2 deficiency resulted in an increase of JARID1B ubiquitination and in turn a reduction of H3K4me3, and induced senescence through JARID1B accumulation in nucleoli of PCa cells and prostate tumors of mice. Furthermore, we showed that the aberrant levels of SKP2, JARID1B, and H3K4me3 are associated with malignant features of castration-resistant prostate cancer (CRPC) in mice. Overall, our findings reveal a novel network of SKP2- JARID1B, and targeting SKP2 and JARID1B may be a potential strategy for PCa control.
Citation Format: Wenfu Lu, Shenji Liu, Bo Li, Yingqiu Xie, Christine Adhiambo, Qing Yang, Billy R. Ballard, Keiichi I. Nakayama, Robert J. Matusik, Zhenbang Chen. K63-linked JARID1B ubiquitination by TRAF6 contributes to aberrant elevation of JARID1B in prostate cancer. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 90. doi:10.1158/1538-7445.AM2015-90
Abstract 10 of 10Molecular and Cellular Biology / Poster Presentations – Proffered Abstracts / Poster Presentations – Histone Methylation and Acetylation
Abstract 97: CARM1 preferentially methylates H3R17 over H3R26 through a random kinetic mechanism
Proceedings: AACR 106th Annual Meeting 2015; April 18-22, 2015; Philadelphia, PA
CARM1 (PRMT4) is a type I arginine methyltransferase involved in the regulation of transcription, pre-mRNA splicing, cell cycle progression and the DNA damage response. Overexpression of CARM1 has been implicated in breast, prostate, and colorectal cancers. Since CARM1 appears to be a good target for the development of therapies against these cancers, we studied the substrate specificity and kinetic mechanism of the full-length human enzyme. CARM1 has been shown to methylate both residues R17 and R26 of histone H3. Substrate specificity was examined by testing CARM1 activity with several H3-based peptide substrates using a radiometric assay. Comparison of kcat/KM values reveal that methylation of H3R17 is preferred over H3R26. An R17/R26K peptide produced 8-fold greater kcat/KM value compared to the corresponding R17K/R26 peptide. These effects are KM-driven as kcat values remain relatively constant for the peptides tested. Shortening the peptide at the C-terminus by 5 amino acid residues greatly reduced the specificity (16-24-fold), demonstrating the contribution of distal residues to substrate binding. In contrast, adding residues to the N-terminus of the shortened peptide had a negative effect on activity. CARM1 displays little preference for monomethylated over unmethylated H3R17 (2-5-fold by kcat/KM) suggesting that it operates through a distributive mechanism. Previous crystallographic studies with mouse CARM1 showed that part of the substrate binding groove was formed by cofactor binding, thereby suggesting an ordered kinetic mechanism (Yue et al., EMBO J., 2007). Our results from dead-end and product inhibition studies performed with human CARM1, however, are consistent with a random kinetic mechanism. SAH and sinefungin demonstrate competitive inhibition with respect to SAM and produced noncompetitive inhibition patterns with respect to peptide. Both dimethylated R17 product peptide and dead-end R17K peptide exhibited noncompetitive inhibition patterns with respect to SAM. Furthermore, binding of SAM and peptide substrates were shown to be independent of each other in initial velocity experiments where both substrates were varied. Together, these results elucidate the kinetic mechanism of CARM1 and highlight elements important for binding affinity.
Citation Format: Suzanne L. Jacques, Katrina P. Aquino, Jodi Gureasko, P Ann Boriack-Sjodin, Robert A. Copeland, Thomas V. Riera. CARM1 preferentially methylates H3R17 over H3R26 through a random kinetic mechanism. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 97. doi:10.1158/1538-7445.AM2015-97
Bonnet D, Dick JE: Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997, 3(7):730-737.
Al-Hajj M, Clarke MF: Self-renewal and solid tumor stem cells. Oncogene 2004, 23(43):7274-7282.
Hughes L, Malone C, Chumsri S, Burger AM, McDonnell S: Characterisation of breast cancer cell lines and establishment of a novel isogenic subclone to study migration, invasion and tumourigenicity. Clin Exp Metastasis 2008, 25(5):549-557.
Li C, Lee CJ, Simeone DM: Identification of human pancreatic cancer stem cells. Methods Mol Biol 2009, 568:161-173.
Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP: Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68(11):4311-4320.
Kakarala M, Wicha MS: Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol 2008, 26(17):2813-2820.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S et al: ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1(5):555-567.
Dontu G: Breast cancer stem cell markers – the rocky road to clinical applications. Breast Cancer Res 2008, 10(5):110.
Ferrandina G, Bonanno G, Pierelli L, Perillo A, Procoli A, Mariotti A, Corallo M, Martinelli E, Rutella S, Paglia A et al: Expression of CD133-1 and CD133-2 in ovarian cancer. Int J Gynecol Cancer 2008, 18(3):506-514.
Additional Articles on this Open Access Journal on Cancer Stem Cells Include















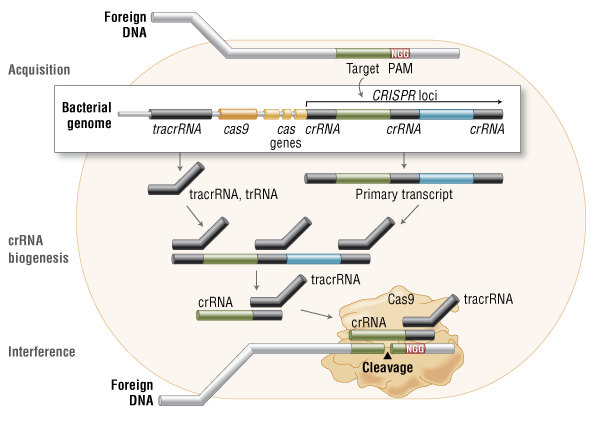{kind=link}
{kind=link}
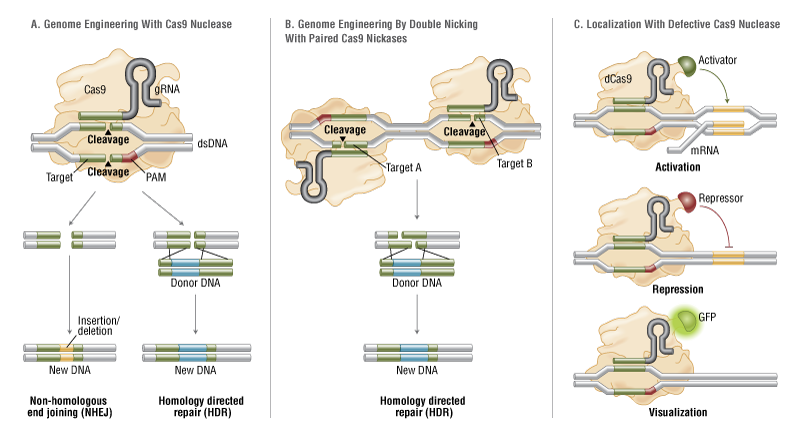{kind=link}
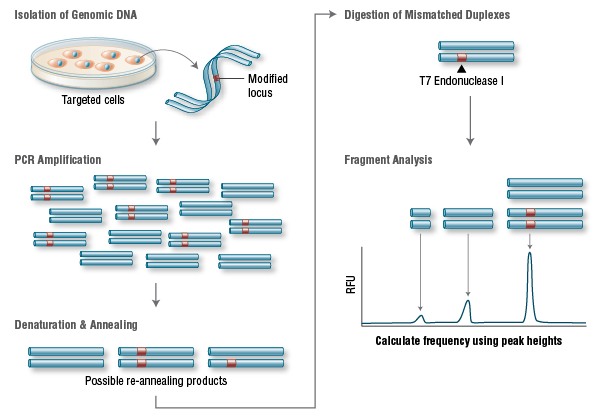{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}