Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Non – CME – dSTRIDE™-HR: A Functional Biomarker for In Situ, ‘real-time’ Detection and Quantification of Homologous Recombination Activity.
Magda Kordon-Kiszala, PhD, CEO and co-founder,intoDNA
12:35-12:55
Epigenetic Plasticity and Tumor Evolution: Mechanisms of Resistance in Precision Oncology
Johnathan R. Whetstine, PhD, Director, Cancer Epigenetics Institute, Director, Geonomics Resource, Fox Chase Cancer Center
Title: Epigenetic plasticity a gatekeeper to generating extrachromosomal DNA amplification and rearrangements
genetic events in cancer are actually controlled not random as he says
Fox Chase Cancer Center Epigenetics Institute; 5th year goal to understand epigenetic mechanisms to understand resistance and biomarker development; bring others and break down silos; they are expanding and hiring and bringing into a network; March 5 2026 5th Annual Symposium Philadelphia Franklin Institute
DNA amplification is also chromosomal: integrated same locus or different regions or chromosomal duplication
KDM4A epigenetic demethylase controls transiet site specific DNA re-replication; can have focal control of DNA regions
you can control regional control of like EGFR amplification
can use Cy3 to find local regions
KDM3B inhibitor promotes transiet copy gains in KMT2A/MLL
EHMT2 is lysine demethylase is a driver of this copy amplification
this demethylase can change expression locally in one hour.. very fast
demethylases are very specific for their gene locus they control and so this demethylase only controls MLL gene
doxorubicin topoisomerase inhibitor can cause LOH in MLL locus and methylase inhibitor can reverse this
over twenty combinatorial regulators so this field is just budding
11:30-12:30
Companion Diagnostics in Hereditary and Chronic Diseases – Development, Regulatory Approval, and Commercialization – Non-CME Discussion
Huw Ricketts PhD, Senior Director, CLIA Business Development, QIAGEN
Tricia Carrigan, PhD, BC Biosolutions
Arushi Agarwal, MS, Partner, Health Advances
Melissa Reuter, MS, MBA, Director, Precision Medicine Program Strategy, GSK
This is a session panel Discussion on the current state of companion diagnostic development, not just in oncology. Regulatory aspects will be discussed
Arushi: There are alot of opportunities in non-oncology areas for companion diagnostics, and time to development may be an obstacle
Huw Rickets: From a development standpoint most people are not looking at the diagnostic side but more on the therapeutic side.
Tricia: There needs to be a shift in oncology drug development world, and pharma sees developing diagnostic is too expensive.
Meliisa: They try to engage early with the agencies to understand the regulatory landscape; GSK is very strong in their oncology platform but there are gaps in diagnostics and non-oncology programs
Arushi: seems in Pharma oncology and non-oncology programs seems siloed
for non-oncology many of the biomarkers may be rare… well under 25% of population
Huw: Qiagen trying to develop diagnostics for Parkinson’s but those rare genetic diseases are easier to develop
Arushi: neurodegenerative, NASH, and immuno diseases are big areas where companies are looking to make companion diagnostics
Huw: kidney disease is a big focus to develop companion diagnostics for
12:30-12:40
Non – CME – dSTRIDE™-HR: A Functional Biomarker for In Situ, ‘real-time’ Detection and Quantification of Homologous Recombination Activity.
Magda Kordon-Kiszala, PhD, CEO and co-founder,intoDNA
Amanda Paulovich, Professor, Aven Foundation Endowed Chair
Fred Hutchinson Cancer Center
Susan Monarezm Deputy Director ARPA-H
Henry Rodriguez, NCI/NIH
Eric Schadt, Pathos
Ezra Cohen, Tempus
Jennifer Leib, Innovation Policy Solutions
Nick Seddon, Optum Genomics
Giselle Sholler, Penn State Hershey Children’s Hospital
Janet Woodcock, formerly FDA
Amanda Paulovich: Frustrated by the variability in cancer therapy results. Decided to help improve cancer diagnostics
We have plateaued on relying on single gene single protein companion diagnostics
She considers that regulatory, economic, and cultural factors are hindering the innovation and resulting in the science way ahead of the clinical aspect of diagnostics
Diagnostic research is not as well funded as drug discovery
Biomarkers, the foundation for the new personalized medicine, should be at forefront Read the Tipping Point by Malcolm Gladwell
FDA is constrained by statutory mandates
Eric Schadt
Pathos
Multiple companies trying to chase different components of precision medicine strategy including all the one involved in AI
He is helping companies creating those mindmaps, knowledge graphs, and create more predictive systems
Population screening into population groups will be using high dimensional genomic data to determine risk in various population groups however 60% of genomic data has no reported ancestry
He founded Sema4 but many of these companies are losing $$ on these genomic diagnostics
So the market is not monetizing properly
Barriers to progress: arbitrary evidence thresholds for payers, big variation across health care system, regulatory framework
Beat Childhood Cancer Consortium Giselle
Consortium of university doctors in pediatrics
They had a molecular tumor board to look at the omics data
Showed example of choroid plexus tumor success with multi precision meds vs std chemo
Challenges: understanding differences in genomics test (WES, NGS, transcriptome etc.
Precision medicine needs to be incorporated in med education.. Fellowships.. Residency
She spends hours with the insurance companies providing more and more evidence to justify reimbursements
She says getting that evidence is a challenged; biomedical information needs to be better CURATED
Dr. Ezra Cohen, Tempest
HPV head and neck cancer, good prognosis, can use cituximab and radiation
$2 billion investment at Templest of AI driven algorithm to integrate all omics; used LLM models too
Dr. Janet Woodcock
Our theoretical problem with precision and personalized medicine is that we are trained to think of the average patient
ISPAT II trial a baysian trial; COVID was a platform trial
She said there should there be NIH sponsored trials on adaptive biomarker platform trials
This event will be covered by the LPBI Group on Twitter. Follow on
Real Time Coverage Morning Session on Precision Oncology: Advancing Precision Medicine Annual Conference, Philadelphia PA November 1 2024
Reporter: Stephen J. Williams, Ph.D.
Notes from Precision Medicine for Rare Diseases 9:00AM – 10:50
Precision Medicine and markers Cure models vs disease models Dr Ekker from UT MD Anderson
UT MD Anderson zebrafish disease model program now focusing more on figuring the mechanisms by which a disease model is reverted to normal upon CRISPR screens
Traditional drug development process long and expensive
2nd in class only takes 4 years while 3rd in class drugs take only 1.5 years
Health-in-a-fish: using a CRE system to go from disease to normal
The theory is making a CRE or CURE avatar; taking a diseased zebrafish and reverse engineering the disease genome
He used transposon based CRE mutational mutants with protein trap and 3’ exon trap (transposon based mutagenesis)
He reverted the diseased gene by CRE
He feels that can scale up to using organoids to develop more cure based models
FDA Christine Nguyen MD regulatory perspective of framework of drug approval for rare diseases
1 in 10 Amercians have rare diseases; 70% genetic and half are children
Due to Orphan Drug Act in 2023 half of novel drugs approved for rare diseases
CDER and FDA 550 unique drugs for over 1000 rare diseases
Clinical and surrogate validated endpoints are important for traditional approvals
For accelerated approval need predictive surrogate endpoint of clinical benefit
For accelerated approval needs completion of a confirmatory trials so FDA has new authority under FDORA; FDA can dictate trial milestones
Candidate surrogate endpoints: known to predict (validated) for traditional approval but reasonably likely to predict for accelerated approval
Does surrogate endpoint associated with a causal pathway? Also important to understand the magnitude of benefit so surrogate should be quantitative not just qualitative
RDEA is a series of 3 public workshops at FY2027 to promote innovation and novel endpoints and guidance
Frank Sasinowski FDA regulatory flexibility beyond One Positive Adequate and Well Controlled Trial
As we move to rare diseases we may only have one well controlled study so FDA feels we need new regulatory frameworks and guidelines especially for rare disease clinical trails especially with precision medicine
Accelerated approval does not mean your evidence is any less stringent that traditional approval (only difference is endpoint but quality of evidence the same)
Confirmatory evidence is a primary concern
In 2021 FDA coordinated with the two divisions CBER and CDER
Sometimes a primary endpoint shows positive benefit but secondary endpoints may not; FDA now feels that results from one well designed AWC gives confirmatory evidence
FDA can be flexible by taking in consideration the quantity and quality of confirmatory evidence and the totality of evidence
So pharmacology studies, natural history etc. can be enough
For a drug like Lamzede for mannosidosis there were no positive endpoint studies or for ADA SCID disease there was other compelling evidence
The FDA does have flexibility when it comes to advanced precision medicines and ultr rare diseases
10:50 Do we Really Need Liquid Biopsy? A Panel Discussion on the Issues Hampering the full Adoption of Liquid Biopsy
In Mexico leading cancer is colorectal but only have the FIT test and noone except one organization who issupplying health access
Access to precision medicine is a concern: the communication between the patient, who is pushing this more than healthcare, needs to be coordinated better with all stakeholders in care
We also need to educate many physicians even oncologists (like in Virginia) a better understanding of genetics and omics
FT3 consortium does testing to therapy (multistakeholder group comprised of patient advocacy groups); focus on amplifying global efforts to increase access; they are trying to make a roadmap to help access in other countries; when it comes to precision medicine it is usually the nurses that are aksing for training because they are usually the first responders for the patient’s questions
In rural areas just getting access to liquid biopsy is a concern and maybe satellite sites might be useful because the time to schedule is getting worse (like 3 or more months)
A recent paper showed that liquid biopsy may actually perpetuate health disparities and not ameliorate them
BloodPAC: there are barriers to LB access and adoption so consortium felt that there were many areas that need to be addressed: financial, access, disparities, education
ctDNA to define variants was the past focus; there is growing realization that there are representatives populations in your R&D studies
Submission of data to BloodPac is easier to do for tissue not for liquid biopsy; there is lack of harmonization across many of these databanks
Reimbursement: is a barrier to access for liquid biopsy
Illumina: challenge finding clinical utility for payers; FDA approval is not as hard; show improved outcomes for patients; Medicare is starting to approve some tests but the criteria bar keeps changing with payers;
How do we leverage the on-market data to support performance of your diagnostic test or genomic panel
This event will be covered by the LPBI Group on Twitter. Follow on
Entering the last day of the American College of Cardiology’s annual conference, the Big Pharma is trotting out new phase 2 data of its anti-PCSK9 drug, finding that it reduced particular kinds of cholesterol by up to 61% compared to placebo.
Meanwhile, expanded phase 3 data of sotatercept, added onto background therapy, has exceeded the expectations of Chief Medical Officer Eliav Barr, M.D. “It just hits the right receptor,” he said in an interview with Fierce Biotech.
Sotatercept was the prized jewel in the company’s $11.5 billion purchase of Acceleron Pharma in 2021. The cardio med aimed at treating pulmonary arterial hypertension improved patients’ six-minute walk distance by more than 40 meters after 24 weeks compared to placebo, hitting the primary endpoint of the 323-patient trial.
The therapy also reduced the risk of clinical worsening or death by 84% compared to placebo for a median follow-up of 32.7 weeks, according to the conference presentation.What’s more, sotatercept had a slightly lower discontinuation rate due to treatment-related side effects than placebo patients.
While sotatercept has accrued much of the acclaim for the cardio team, Barr was also riding the high of positive phase 2 data from the company’s oral PCSK9 inhibitor to treat high cholesterol. The trial compared four doses of MK-0616 in patients with high cholesterol compared to placebo; all four were found to significantly reduce LDL cholesterol levels.
The highest dose of the med reduced levels of this cholesterol by more than 60% compared to placebo and the number of side effects across all dose levels was consistent with placebo.
The data is naturally a critical checkpoint as Barr and Merck tout the value of the first oral version of the therapy class currently dominated by Amgen’s Repatha and Regeneron’s Praluent. Next on the clinical docket is a phase 3 trial slated for the second half of the year, but Barr also hopes to launch a cardiovascular outcomes trial before year-end as well.
Cholesterol Lowering Novel PCSK9 drugs: Praluent [Sanofi and Regeneron] vs Repatha [Amgen] – which drug cuts CV risks enough to make it cost-effective?
2022 FDA Drug Approval List, 2022 Biological Approvals and Approved Cellular and Gene Therapy Products
Reporter: Aviva Lev-Ari, PhD, RN
SOURCE
Tal Bahar’s post on LinkedIn on 1/17/2023
Novel Drug Approvals for 2022
FDA’s Center for Drug Evaluation and Research (CDER)
New Molecular Entities (“NMEs”)
Some of these products have never been used in clinical practice. Below is a listing of new molecular entities and new therapeutic biological products that CDER approved in 2022. This listing does not contain vaccines, allergenic products, blood and blood products, plasma derivatives, cellular and gene therapy products, or other products that the Center for Biologics Evaluation and Research approved in 2022.
Others are the same as, or related to, previously approved products, and they will compete with those products in the marketplace. See Drugs@FDA for information about all of CDER’s approved drugs and biological products.
Certain drugs are classified as new molecular entities (“NMEs”) for purposes of FDA review. Many of these products contain active moieties that FDA had not previously approved, either as a single ingredient drug or as part of a combination product. These products frequently provide important new therapies for patients. Some drugs are characterized as NMEs for administrative purposes, but nonetheless contain active moieties that are closely related to active moieties in products that FDA has previously approved. FDA’s classification of a drug as an “NME” for review purposes is distinct from FDA’s determination of whether a drug product is a “new chemical entity” or “NCE” within the meaning of the Federal Food, Drug, and Cosmetic Act.
To treat adults with HIV whose HIV infections cannot be successfully treated with other available treatments due to resistance, intolerance, or safety considerations Press Release
To decrease the incidence of infection in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with clinically significant incidence of febrile neutropenia
The Center for Biologics Evaluation and Research (CBER) regulates products under a variety of regulatory authorities. See the Development & Approval Process page for a description of what products are approved as Biologics License Applications (BLAs), Premarket Approvals (PMAs), New Drug Applications (NDAs) or 510Ks.
Biologics License Applications and Supplements
New BLAs (except those for blood banking), and BLA supplements that are expected to significantly enhance the public health (e.g., for new/expanded indications, new routes of administration, new dosage formulations and improved safety).
Other Applications Approved or Cleared by the Center for Biologics Evaluation and Research (CBER)
Medical devices involved in the collection, processing, testing, manufacture and administration of licensed blood, blood components and cellular products.
There are five FDA-approved CAR-T treatments for blood cancers and two gene therapies to treat rare diseases now on the market in the U.S. The late-stage pipeline could produce several more cancer CAR-Ts and gene therapies to treat a range of diseases.
One of the biggest races to watch in the cell therapy space will be that between Gilead Sciences’ Yescarta and Bristol Myers Squibb’s Breyanzi, both of which are gunning to move their CAR-Ts into earlier lines of treatment in large B-cell lymphoma (LBCL). At ASH, both companies rolled out impressive data from their trials in the second-line setting, but Gilead could have the upper hand by virtue of its three-year head start in the market, analysts said. Gilead expects to hear from the FDA on a label expansion in the second-line setting in April.
#TUBiol5227: Biomarkers & Biotargets: Genetic Testing and Bioethics
Curator: Stephen J. Williams, Ph.D.
The advent of direct to consumer (DTC) genetic testing and the resultant rapid increase in its popularity as well as companies offering such services has created some urgent and unique bioethical challenges surrounding this niche in the marketplace. At first, most DTC companies like 23andMe and Ancestry.com offered non-clinical or non-FDA approved genetic testing as a way for consumers to draw casual inferences from their DNA sequence and existence of known genes that are linked to disease risk, or to get a glimpse of their familial background. However, many issues arose, including legal, privacy, medical, and bioethical issues. Below are some articles which will explain and discuss many of these problems associated with the DTC genetic testing market as well as some alternatives which may exist.
As you can see,this market segment appears to want to expand into the nutritional consulting business as well as targeted biomarkers for specific diseases.
Rising incidence of genetic disorders across the globe will augment the market growth
Increasing prevalence of genetic disorders will propel the demand for direct-to-consumer genetic testing and will augment industry growth over the projected timeline. Increasing cases of genetic diseases such as breast cancer, achondroplasia, colorectal cancer and other diseases have elevated the need for cost-effective and efficient genetic testing avenues in the healthcare market.
For instance, according to the World Cancer Research Fund (WCRF), in 2018, over 2 million new cases of cancer were diagnosed across the globe. Also, breast cancer is stated as the second most commonly occurring cancer. Availability of superior quality and advanced direct-to-consumer genetic testing has drastically reduced the mortality rates in people suffering from cancer by providing vigilant surveillance data even before the onset of the disease. Hence, the aforementioned factors will propel the direct-to-consumer genetic testing market overt the forecast timeline.
Nutrigenomic Testing will provide robust market growth
The nutrigenomic testing segment was valued over USD 220 million market value in 2019 and its market will witness a tremendous growth over 2020-2028. The growth of the market segment is attributed to increasing research activities related to nutritional aspects. Moreover, obesity is another major factor that will boost the demand for direct-to-consumer genetic testing market.
Nutrigenomics testing enables professionals to recommend nutritional guidance and personalized diet to obese people and help them to keep their weight under control while maintaining a healthy lifestyle. Hence, above mentioned factors are anticipated to augment the demand and adoption rate of direct-to-consumer genetic testing through 2028.
Browse key industry insights spread across 161 pages with 126 market data tables & 10 figures & charts from the report, “Direct-To-Consumer Genetic Testing Market Size By Test Type (Carrier Testing, Predictive Testing, Ancestry & Relationship Testing, Nutrigenomics Testing), By Distribution Channel (Online Platforms, Over-the-Counter), By Technology (Targeted Analysis, Single Nucleotide Polymorphism (SNP) Chips, Whole Genome Sequencing (WGS)), Industry Analysis Report, Regional Outlook, Application Potential, Price Trends, Competitive Market Share & Forecast, 2020 – 2028” in detail along with the table of contents: https://www.gminsights.com/industry-analysis/direct-to-consumer-dtc-genetic-testing-market
Targeted analysis techniques will drive the market growth over the foreseeable future
Based on technology, the DTC genetic testing market is segmented into whole genome sequencing (WGS), targeted analysis, and single nucleotide polymorphism (SNP) chips. The targeted analysis market segment is projected to witness around 12% CAGR over the forecast period. The segmental growth is attributed to the recent advancements in genetic testing methods that has revolutionized the detection and characterization of genetic codes.
Targeted analysis is mainly utilized to determine any defects in genes that are responsible for a disorder or a disease. Also, growing demand for personalized medicine amongst the population suffering from genetic diseases will boost the demand for targeted analysis technology. As the technology is relatively cheaper, it is highly preferred method used in direct-to-consumer genetic testing procedures. These advantages of targeted analysis are expected to enhance the market growth over the foreseeable future.
Over-the-counter segment will experience a notable growth over the forecast period
The over-the-counter distribution channel is projected to witness around 11% CAGR through 2028. The segmental growth is attributed to the ease in purchasing a test kit for the consumers living in rural areas of developing countries. Consumers prefer over-the-counter distribution channel as they are directly examined by regulatory agencies making it safer to use, thereby driving the market growth over the forecast timeline.
Favorable regulations provide lucrative growth opportunities for direct-to-consumer genetic testing
Europe direct-to-consumer genetic testing market held around 26% share in 2019 and was valued at around USD 290 million. The regional growth is due to elevated government spending on healthcare to provide easy access to genetic testing avenues. Furthermore, European regulatory bodies are working on improving the regulations set on the direct-to-consumer genetic testing methods. Hence, the above-mentioned factors will play significant role in the market growth.
Focus of market players on introducing innovative direct-to-consumer genetic testing devices will offer several growth opportunities
Few of the eminent players operating in direct-to-consumer genetic testing market share include Ancestry, Color Genomics, Living DNA, Mapmygenome, Easy DNA, FamilytreeDNA (Gene By Gene), Full Genome Corporation, Helix OpCo LLC, Identigene, Karmagenes, MyHeritage, Pathway genomics, Genesis Healthcare, and 23andMe. These market players have undertaken various business strategies to enhance their financial stability and help them evolve as leading companies in the direct-to-consumer genetic testing industry.
For example, in November 2018, Helix launched a new genetic testing product, DNA discovery kit, that allows customer to delve into their ancestry. This development expanded the firm’s product portfolio, thereby propelling industry growth in the market.
The following posts discuss bioethical issues related to genetic testing and personalized medicine from a clinicians and scientisit’s perspective
Question:Each of these articles discusses certain bioethical issues although focuses on personalized medicine and treatment. Given your understanding of the robust process involved in validating clinical biomarkers and the current state of the DTC market, how could DTC testing results misinform patients and create mistrust in the physician-patient relationship?
Question: If you are developing a targeted treatment with a companion diagnostic, what bioethical concerns would you address during the drug development process to ensure fair, equitable and ethical treatment of all patients, in trials as well as post market?
Articles on Genetic Testing, Companion Diagnostics and Regulatory Mechanisms
Question: What type of regulatory concerns should one have during the drug development process in regards to use of biomarker testing?From the last article on Protecting Your IP how important is it, as a drug developer, to involve all payers during the drug development process?
Science Policy Forum: Should we trust healthcare explanations from AI predictive systems?
Some in industry voice their concerns
Curator: Stephen J. Williams, PhD
Post on AI healthcare and explainable AI
In a Policy Forum article in Science “Beware explanations from AI in health care”, Boris Babic, Sara Gerke, Theodoros Evgeniou, and Glenn Cohen discuss the caveats on relying on explainable versus interpretable artificial intelligence (AI) and Machine Learning (ML) algorithms to make complex health decisions. The FDA has already approved some AI/ML algorithms for analysis of medical images for diagnostic purposes. These have been discussed in prior posts on this site, as well as issues arising from multi-center trials. The authors of this perspective article argue that choice of type of algorithm (explainable versus interpretable) algorithms may have far reaching consequences in health care.
Summary
Artificial intelligence and machine learning (AI/ML) algorithms are increasingly developed in health care for diagnosis and treatment of a variety of medical conditions (1). However, despite the technical prowess of such systems, their adoption has been challenging, and whether and how much they will actually improve health care remains to be seen. A central reason for this is that the effectiveness of AI/ML-based medical devices depends largely on the behavioral characteristics of its users, who, for example, are often vulnerable to well-documented biases or algorithmic aversion (2). Many stakeholders increasingly identify the so-called black-box nature of predictive algorithms as the core source of users’ skepticism, lack of trust, and slow uptake (3, 4). As a result, lawmakers have been moving in the direction of requiring the availability of explanations for black-box algorithmic decisions (5). Indeed, a near-consensus is emerging in favor of explainable AI/ML among academics, governments, and civil society groups. Many are drawn to this approach to harness the accuracy benefits of noninterpretable AI/ML such as deep learning or neural nets while also supporting transparency, trust, and adoption. We argue that this consensus, at least as applied to health care, both overstates the benefits and undercounts the drawbacks of requiring black-box algorithms to be explainable.
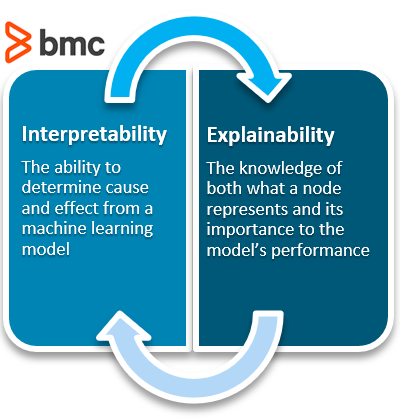
Types of AI/ML Algorithms: Explainable and Interpretable algorithms
Interpretable AI: A typical AI/ML task requires constructing algorithms from vector inputs and generating an output related to an outcome (like diagnosing a cardiac event from an image). Generally the algorithm has to be trained on past data with known parameters. When an algorithm is called interpretable, this means that the algorithm uses a transparent or “white box” function which is easily understandable. Such example might be a linear function to determine relationships where parameters are simple and not complex. Although they may not be as accurate as the more complex explainable AI/ML algorithms, they are open, transparent, and easily understood by the operators.
Explainable AI/ML: This type of algorithm depends upon multiple complex parameters and takes a first round of predictions from a “black box” model then uses a second algorithm from an interpretable function to better approximate outputs of the first model. The first algorithm is trained not with original data but based on predictions resembling multiple iterations of computing. Therefore this method is more accurate or deemed more reliable in prediction however is very complex and is not easily understandable. Many medical devices that use an AI/ML algorithm use this type. An example is deep learning and neural networks.
The purpose of both these methodologies is to deal with problems of opacity, or that AI predictions based from a black box undermines trust in the AI.
For a deeper understanding of these two types of algorithms see here:
How interpretability is different from explainability
Why a model might need to be interpretable and/or explainable
Who is working to solve the black box problem—and how
What is interpretability?
Does Chipotle make your stomach hurt? Does loud noise accelerate hearing loss? Are women less aggressive than men? If a machine learning model can create a definition around these relationships, it is interpretable.
All models must start with a hypothesis. Human curiosity propels a being to intuit that one thing relates to another. “Hmm…multiple black people shot by policemen…seemingly out of proportion to other races…something might be systemic?” Explore.
People create internal models to interpret their surroundings. In the field of machine learning, these models can be tested and verified as either accurate or inaccurate representations of the world.
Interpretability means that the cause and effect can be determined.
What is explainability?
ML models are often called black-box models because they allow a pre-set number of empty parameters, or nodes, to be assigned values by the machine learning algorithm. Specifically, the back-propagation step is responsible for updating the weights based on its error function.
To predict when a person might die—the fun gamble one might play when calculating a life insurance premium, and the strange bet a person makes against their own life when purchasing a life insurance package—a model will take in its inputs, and output a percent chance the given person has at living to age 80.
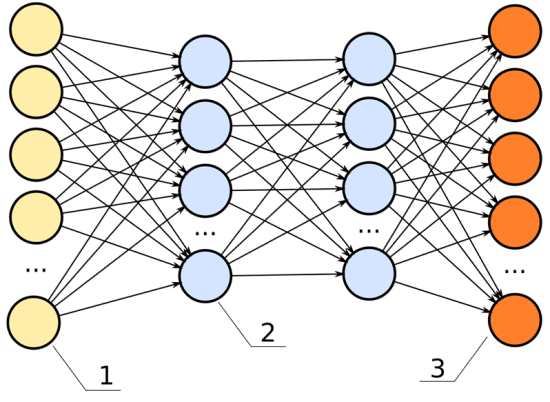
Below is an image of a neural network. The inputs are the yellow; the outputs are the orange. Like a rubric to an overall grade, explainability shows how significant each of the parameters, all the blue nodes, contribute to the final decision.
In this neural network, the hidden layers (the two columns of blue dots) would be the black box.
For example, we have these data inputs:
Age
BMI score
Number of years spent smoking
Career category
If this model had high explainability, we’d be able to say, for instance:
The career category is about 40% important
The number of years spent smoking weighs in at 35% important
The age is 15% important
The BMI score is 10% important
Explainability: important, not always necessary
Explainability becomes significant in the field of machine learning because, often, it is not apparent. Explainability is often unnecessary. A machine learning engineer can build a model without ever having considered the model’s explainability. It is an extra step in the building process—like wearing a seat belt while driving a car. It is unnecessary for the car to perform, but offers insurance when things crash.
The benefit a deep neural net offers to engineers is it creates a black box of parameters, like fake additional data points, that allow a model to base its decisions against. These fake data points go unknown to the engineer. The black box, or hidden layers, allow a model to make associations among the given data points to predict better results. For example, if we are deciding how long someone might have to live, and we use career data as an input, it is possible the model sorts the careers into high- and low-risk career options all on its own.
Perhaps we inspect a node and see it relates oil rig workers, underwater welders, and boat cooks to each other. It is possible the neural net makes connections between the lifespan of these individuals and puts a placeholder in the deep net to associate these. If we were to examine the individual nodes in the black box, we could note this clustering interprets water careers to be a high-risk job.
In the previous chart, each one of the lines connecting from the yellow dot to the blue dot can represent a signal, weighing the importance of that node in determining the overall score of the output.
If that signal is high, that node is significant to the model’s overall performance.
If that signal is low, the node is insignificant.
With this understanding, we can define explainability as:
Knowledge of what one node represents and how important it is to the model’s performance.
So how does choice of these two different algorithms make a difference with respect to health care and medical decision making?
The authors argue:
“Regulators like the FDA should focus on those aspects of the AI/ML system that directly bear on its safety and effectiveness – in particular, how does it perform in the hands of its intended users?”
A suggestion for
Enhanced more involved clinical trials
Provide individuals added flexibility when interacting with a model, for example inputting their own test data
More interaction between user and model generators
Determining in which situations call for interpretable AI versus explainable (for instance predicting which patients will require dialysis after kidney damage)
Other articles on AI/ML in medicine and healthcare on this Open Access Journal include
19 of the 49 New Therapeutic Molecular Entities FDA approved in 2020 — as well as a new Cell-based therapy — are Personalized Medicines
Reporter: Aviva Lev-Ari, PhD, RN
2020 DRUG APPROVALS
19 of the 49 new therapeutic molecular entities FDA approved in 2020 — as well as a new cell-based therapy — are personalized medicines.
Newly Approved Therapeutic Molecular Entities
1. Ayvakit (avapritinib) — for the treatment of metastatic gastrointestinal stromal tumor (GIST). The decision to use this product is informed by the PDGFRA exon 18 biomarker status in the tumors of patients.
2. Nexletol (bempedoic acid) — for the treatment of adults with familial hypercholesterolemia who require additional lowering of LDL-C. The use of this product can be informed by the FH biomarker (LOLR, APOB, PCSK9) status in patients.
3. Tukysa (tucatinib) — for the treatment of metastatic breast cancer. The decision to use this product is informed by the HER2 biomarker status in the tumors of patients.
4. Pemazyre (pemigatinib) — for the treatment of cholangiocarcinoma. The decision to use this product is informed by the FGFR2 biomarker status in the tumors of patients.
5. Trodelvy (sacituzumab govitecan-hziy) — for the treatment of metastatic triple-negative breast cancer. The decision to use this product is informed by the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) biomarker statuses in the tumors of patients. Personalized Medicine at FDA 7
6. Tabrecta (capmatinib) — for the treatment of non-small cell lung cancer (NSCLC). The decision to use this product is informed by the MET exon 14 biomarker status in the tumors of patients.
7. Retevmo (selpercatinib) — for the treatment of lung and thyroid cancers. The decision to use this product is informed by the RET fusion biomarker status in the tumors of patients.
8. Uplizna (inebilizumab-cdon) — for the treatment of neuromyelitis optica spectrum disorder. The decision to use this product is informed by the AQP4 biomarker status in patients.
9. Rukobia (fostemsavir) — for the treatment of human immunodeficiency virus (HIV) infection in adults with multidrug-resistant HIV-1 infection. The use of this product can be informed by the HIV-1 expression levels in patients.
10. Evrysdi (risdiplam) — for the treatment of spinal muscular atrophy. This product selectively targets the SMN2 biomarker in patients.
11. Olinvyk (oliceridine) — for the management of acute pain. The use of this product can be informed by the CYP2D6 biomarker status in patients.
12. Viltepso (viltolarsen) — for the treatment of Duchenne muscular dystrophy. This product selectively targets, and its use is informed by, the DMD gene exon 53 biomarker in patients.
13. Enspryng (satralizumab-mwge) — for the treatment of neuromyelitis optica spectrum disorder. The decision to use this product is informed by the AQP4 biomarker status in patients.
14. Gavreto (pralsetinib) — for the treatment of non-small cell lung cancer (NSCLC). The decision to use this product is informed by the RET fusion biomarker status in the tumors of patients.
15. Zokinvy (lonafarnib) — for the treatment of progeroid laminopathies. The decision to use this product is informed by the LMN4 and/or ZMPSTE24 biomarker statuses in patients. 8 Personalized Medicine at FDA Methodology: When evaluating new molecular entities, PMC defined personalized medicines as those therapeutic products for which the label includes reference to specific biological markers, often identified by diagnostic tools, that help guide decisions and/or procedures for their use in individual patients.
16. Oxlumo (lumasiran) — for the treatment of hyperoxaluria type 1. This product selectively targets the hydroxy acid oxidase 1 (HAO1) biomarker in patients.
17. Imcivree (setmelanotide) — for the treatment of obesity due to pro-opiomelanocortin (POMC) deficiency. The decision to use this product is informed by the POMC, PCSK1, or LEPR biomarker statuses in patients.
18. Orladeyo (berotralstat) — for the treatment of hereditary angioedema types I and II. The use of this product can be informed by the C1-INH biomarker status in patients.
19. Margenza (margetuximab-cmkb) — for the treatment of breast cancer. The decision to use this product is informed by the human epidermal growth factor receptor 2 (HER2) biomarker status in the tumors of patients. Newly Approved Cell-Based Therapy
20. Tecartus (brexucabtagene autoleucel) — for the treatment of mantle cell lymphoma (MCL). The treatment is a fully integrated CD19-directed genetically modified autologous T-cell immunotherapy indicated for the treatment of adult patients with refractory MCL.
As part of the all-of-America approach to fighting the COVID-19 pandemic, the U.S. Food and Drug Administration has been working with partners across the U.S. government, academia and industry to expedite the development and availability of critical medical products to treat this novel virus. Today, we are providing an update on one potential treatment called convalescent plasma and encouraging those who have recovered from COVID-19 to donate plasma to help others fight this disease.
Convalescent plasma is an antibody-rich product made from blood donated by people who have recovered from the disease caused by the virus. Prior experience with respiratory viruses and limited data that have emerged from China suggest that convalescent plasma has the potential to lessen the severity or shorten the length of illness caused by COVID-19. It is important that we evaluate this potential therapy in the context of clinical trials, through expanded access, as well as facilitate emergency access for individual patients, as appropriate.
The response to the agency’s recently announced national efforts to facilitate the development of and access to convalescent plasma has been tremendous. More than 1,040 sites and 950 physician investigators nationwide have signed on to participate in the Mayo Clinic-ledExternal Link Disclaimer expanded access protocol. A number of clinical trials are also taking place to evaluate the safety and efficacy of convalescent plasma and the FDA has granted numerous single patient emergency investigational new drug (eIND) applications as well.
FDA issues guidelines on clinical trials and obtaining emergency enrollment concerning convalescent plasma
FDA has issued guidance to provide recommendations to health care providers and investigators on the administration and study of investigational convalescent plasma collected from individuals who have recovered from COVID-19 (COVID-19 convalescent plasma) during the public health emergency.
The guidance provides recommendations on the following:
Because COVID-19 convalescent plasma has not yet been approved for use by FDA, it is regulated as an investigational product. A health care provider must participate in one of the pathways described below. FDA does not collect COVID-19 convalescent plasma or provide COVID-19 convalescent plasma. Health care providers or acute care facilities should instead obtain COVID-19 convalescent plasma from an FDA-registered blood establishment.
Excerpts from the guidance document are provided below.
Background
The Food and Drug Administration (FDA or Agency) plays a critical role in protecting the United States (U.S.) from threats including emerging infectious diseases, such as the Coronavirus Disease 2019 (COVID-19) pandemic. FDA is committed to providing timely guidance to support response efforts to this pandemic.
One investigational treatment being explored for COVID-19 is the use of convalescent plasma collected from individuals who have recovered from COVID-19. Convalescent plasma that contains antibodies to severe acute respiratory syndrome coronavirus 2 or SARS-CoV-2 (the virus that causes COVID-19) is being studied for administration to patients with COVID-19. Use of convalescent plasma has been studied in outbreaks of other respiratory infections, including the 2003 SARS-CoV-1 epidemic, the 2009-2010 H1N1 influenza virus pandemic, and the 2012 MERS-CoV epidemic.
Although promising, convalescent plasma has not yet been shown to be safe and effective as a treatment for COVID-19. Therefore, it is important to study the safety and efficacy of COVID-19 convalescent plasma in clinical trials.
Pathways for Use of Investigational COVID-19 Convalescent Plasma
The following pathways are available for administering or studying the use of COVID-19 convalescent plasma:
Clinical Trials
Investigators wishing to study the use of convalescent plasma in a clinical trial should submit requests to FDA for investigational use under the traditional IND regulatory pathway (21 CFR Part 312). CBER’s Office of Blood Research and Review is committed to engaging with sponsors and reviewing such requests expeditiously. During the COVID-19 pandemic, INDs may be submitted via email to CBERDCC_eMailSub@fda.hhs.gov.
Expanded Access
An IND application for expanded access is an alternative for use of COVID-19 convalescent plasma for patients with serious or immediately life-threatening COVID-19 disease who are not eligible or who are unable to participate in randomized clinical trials (21 CFR 312.305). FDA has worked with multiple federal partners and academia to open an expanded access protocol to facilitate access to COVID-19 convalescent plasma across the nation. Access to this investigational product may be available through participation of acute care facilities in an investigational expanded access protocol under an IND that is already in place.
Although participation in clinical trials or an expanded access program are ways for patients to obtain access to convalescent plasma, for various reasons these may not be readily available to all patients in potential need. Therefore, given the public health emergency that the COVID-19 pandemic presents, and while clinical trials are being conducted and a national expanded access protocol is available, FDA also is facilitating access to COVID-19 convalescent plasma for use in patients with serious or immediately life-threatening COVID-19 infections through the process of the patient’s physician requesting a single patient emergency IND (eIND) for the individual patient under 21 CFR 312.310. This process allows the use of an investigational drug for the treatment of an individual patient by a licensed physician upon FDA authorization, if the applicable regulatory criteria are met. Note, in such case, a licensed physician seeking to administer COVID-19 convalescent plasma to an individual patient must request the eIND (see 21 CFR 312.310(b)).
Today, the U.S. Food and Drug Administration issued an emergency use authorization (EUA) for investigational convalescent plasma for the treatment of COVID-19 in hospitalized patients as part of the agency’s ongoing efforts to fight COVID-19. Based on scientific evidence available, the FDA concluded, as outlined in its decision memorandum, this product may be effective in treating COVID-19 and that the known and potential benefits of the product outweigh the known and potential risks of the product.
Today’s action follows the FDA’s extensive review of the science and data generated over the past several months stemming from efforts to facilitate emergency access to convalescent plasma for patients as clinical trials to definitively demonstrate safety and efficacy remain ongoing.
The EUA authorizes the distribution of COVID-19 convalescent plasma in the U.S. and its administration by health care providers, as appropriate, to treat suspected or laboratory-confirmed COVID-19 in hospitalized patients with COVID-19.
Alex Azar, Health and Human Services Secretary:
“The FDA’s emergency authorization for convalescent plasma is a milestone achievement in President Trump’s efforts to save lives from COVID-19,” said Secretary Azar. “The Trump Administration recognized the potential of convalescent plasma early on. Months ago, the FDA, BARDA, and private partners began work on making this product available across the country while continuing to evaluate data through clinical trials. Our work on convalescent plasma has delivered broader access to the product than is available in any other country and reached more than 70,000 American patients so far. We are deeply grateful to Americans who have already donated and encourage individuals who have recovered from COVID-19 to consider donating convalescent plasma.”
Stephen M. Hahn, M.D., FDA Commissioner:
“I am committed to releasing safe and potentially helpful treatments for COVID-19 as quickly as possible in order to save lives. We’re encouraged by the early promising data that we’ve seen about convalescent plasma. The data from studies conducted this year shows that plasma from patients who’ve recovered from COVID-19 has the potential to help treat those who are suffering from the effects of getting this terrible virus,” said Dr. Hahn. “At the same time, we will continue to work with researchers to continue randomized clinical trials to study the safety and effectiveness of convalescent plasma in treating patients infected with the novel coronavirus.”
Scientific Evidence on Convalescent Plasma
Based on an evaluation of the EUA criteria and the totality of the available scientific evidence, the FDA’s Center for Biologics Evaluation and Research determined that the statutory criteria for issuing an EUA criteria were met.
The FDA determined that it is reasonable to believe that COVID-19 convalescent plasma may be effective in lessening the severity or shortening the length of COVID-19 illness in some hospitalized patients. The agency also determined that the known and potential benefits of the product, when used to treat COVID-19, outweigh the known and potential risks of the product and that that there are no adequate, approved, and available alternative treatments.
CLINICAL MEMORANDUM From: , OBRR/DBCD/CRS To: , OBRR Through: , OBRR/DBCD , OBRR/DBCD , OBRR/DBCD/CRS Re: EUA 26382: Emergency Use Authorization (EUA) Request (original request 8/12/20; amended request 8/23/20) Product: COVID-19 Convalescent Plasma Items reviewed: EUA request Fact Sheet for Health Care Providers Fact Sheet for Recipients Sponsor: Robert Kadlec, M.D. Assistant Secretary for Preparedness and Response (ASPR) Office of Assistant Secretary for Preparedness and Response (ASPR) U.S. Department of Health and Human Services (HHS) EXECUTIVE SUMMARY COVID-19 Convalescent Plasma (CCP), an unapproved biological product, is proposed for use under an Emergency Use Authorization (EUA) under section 564 of the Federal Food, Drug, and Cosmetic Act (the Act),(21 USC 360bbb-3) as a passive immune therapy for the treatment of hospitalized patients with COVID-19, a serious or life-threatening disease. There currently is no adequate, approved, and available alternative to CCP for treating COVID-19. The sponsor has pointed to four lines of evidence to support that CCP may be effective in the treatment of hospitalized patients with COVID-19: 1) History of convalescent plasma for respiratory coronaviruses; 2) Evidence of preclinical safety and efficacy in animal models; 3) Published studies of the safety and efficacy of CCP; and 4) Data on safety and efficacy from the National Expanded Access Treatment Protocol (EAP) sponsored by the Mayo Clinic. Considering the totality of the scientific evidence presented in the EUA, I conclude that current data for the use of CCP in adult hospitalized patients with COVID-19 supports the conclusion that CCP meets the “may be effective” criterion for issuance of an EUA from section 564(c)(2)(A) of the Act. It is reasonable to conclude that the known and potential benefits of CCP outweigh the known and potential risks of CCP for the proposed EUA. Current data suggest the largest clinical benefit is associated with high-titer units of CCP administered early course of the disease.
A letter, from Senator Warren, to Commissioner Hahn from Senate Committee asking for documentation for any communication between FDA and White House
August 25, 2020 Dr. Stephen M. Hahn, M.D. Commissioner of Food and Drugs U.S. Food and Drug Administration 10903 New Hampshire Avenue Silver Spring, MD 20993 Dear Commissioner Hahn: We write regarding the U.S. Food and Drug Administration’s (FDA) troubling decision earlier this week to issue an Emergency Use Authorization (EUA) for convalescent plasma as a treatment for coronavirus disease 2019 (COVID-19).1 Reports suggests that the FDA granted the EUA amid intense political pressure from President Trump and other Administration officials, despite limited evidence of convalescent plasma’s effectiveness as a COVID-19 treatment.2 To help us better understand whether the issuance of the blood plasma EUA was motivated by politics, we request copies of any and all communications between FDA and White House officials regarding the blood plasma EUA.
The authorization will allow health-care providers in the U.S. to use the plasma to treat hospitalized patients with Covid-19.
The FDA’s emergency use authorization came a day after President Trump accused the agency of delaying enrollment in clinical trials for vaccines or therapeutics.
The criticism from Trump and action from the FDA led some scientists to believe the authorization, which came on the eve of the GOP national convention, was politically motivated.
FDA Commissioner Dr. Stephen Hahn is walking back comments on the benefits of convalescent plasma, saying he could have done a better job of explaining the data on its effectiveness against the coronavirus after authorizing it for emergency use over the weekend.
In an interview with Bloomberg’s Drew Armstrong, FDA Commissioner Hahn reiterates that his decision was based on hard evidence and scientific fact, not political pressure. The whole interview is at the link below:
Dr. Hahn corrected his initial statement about 35% of people would be cured by convalescent plasma. In the interview he stated:
I was trying to do what I do with patients, because patients often understand things in absolute terms versus relative terms. And I should’ve been more careful, there’s no question about it. What I was trying to get to is that if you look at a hundred patients who receive high titre, and a hundred patients who received low titre, the difference between those two particular subset of patients who had these specific criteria was a 35% reduction in mortality. So I frankly did not do a good job of explaining that.
FDA colleagues had frank discussion after the statement was made. He is not asking for other people in HHS to retract their statements, only is concerned that FDA has correct information for physicians and patients
Hahn is worried that people will not enroll due to chance they may be given placebo
He gave no opinion when asked if FDA should be an independent agency
For more articles on COVID19 please go to our Coronavirus Portal at
National Public Radio interview with Dr. Anthony Fauci on his optimism on a COVID-19 vaccine by early 2021
Reporter: Stephen J. Williams, PhD
Below I am giving a link to an important interview by NPR’s Judy Woodruff with Dr. Anthony Fauci on his thoughts regarding the recent spikes in cases, the potential for a COVID-19 vaccine by next year, and promising therapeutics in the pipeline. The interview link is given below however I will summarize a few of the highlights of the interview.
Some notes on the interview
Judy Woodruff began her report with some up to date news regarding the recent spike and that Miami Florida has just ordered the additional use of facemasks. She asked Dr. Anthony Fauci, head of the National Institute of Allergy and Infectious Diseases (NIAD), about if the measures currently in use are enough to bring this spike down. Dr. Fauci said that we need to reboot our efforts, mainly because people are not doing three things which could have prevented this spike mainly
We have to use the tests we have out there efficiently and effectively And we have to get them out to the right people who can do the proper identification, isolation, and do proper contract tracing and need to test more widely in a surveillance way to get a feel of the extent and penetrance of this community spread. there needs to be support and money for these testing labs
We have a problem and we need to admit and own it but we need to do the things we know are effective to turn this thing around.
On Vaccines
“May be later this year”
His response to Merck’s CEO Ken Frazer who said officials are giving false hop if they say ‘end of year’ but Dr. Fauci disagrees. He says a year end goal is not outlandish.
What we have been doing is putting certain things in line with each other in an unprecedented way.
Dr. Fauci went on to say that, in the past yes, it took a long time, even years to develop a vaccine but now they have been able to go from sequence of virus to a vaccine development program in days, which is unheard of. Sixty two days later we have gone into phase 1 trials. the speed at which this is occurring is so much faster. He says that generally it would take a couple of years to get a neutralizing antibody but we are already there. Another candidate will be undergoing phase 3 trials by end of this month (July 2020).
He is “cautiously optimistic” that we will have one or more vaccines to give to patients by end of year because given the amount of cases it will be able to get a handle on safety and efficacy by late fall.
Now he says the game changer is that the government is working with companies to ramp up the production of doses of the candidate vaccines so when we find which one works we will have ample doses on hand. He is worried about the anti vaccine movement derailing vaccine testing and vaccinations but says if we keep on informing the public we can combat this.
Going back to school
Dr. Fauci is concerned for the safety of the vulnerable in schools, including students and staff. He wants the US to get down to a reasonable baseline of cases but in the US that baseline after the first wave was still significantly higher than in most countries, where the baseline was more like tens of cases not hundreds of cases.
For more information on COVID-19 Please go to our Coronavirus Portal at












")