Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Some Recent Challenging News from Gene Therapy Companies: Sarepta’s Gene Therapy Halted by FDA, Spark Therapeutics Program Gets a Realignment and Review from Roche
Curator: Stephen J.Williams, Ph.D.
Sarepta Therapeutics has received a order from the FDA to halt clinical trials on its Duchenne Muscular Dystrophy gene therapy Elevidys on July 18, 2025 following three deaths.
FDA Requests Sarepta Therapeutics Suspend Distribution of Elevidys and Places Clinical Trials on Hold for Multiple Gene Therapy Products Following 3 Deaths
For Immediate Release:
July 18, 2025
The U.S. Food and Drug Administration today announced it has placed Sarepta Therapeutics investigational gene therapy clinical trials for limb girdle muscular dystrophy on clinical hold following three deaths potentially related to these products and new safety concerns that the study participants are or would be exposed to an unreasonable and significant risk of illness or injury. The FDA has also revoked Sarepta’s platform technology designation.
The FDA leadership also met with Sarepta Therapeutics and requested it voluntarily stop all shipments of Elevidys today. The company refused to do so.
“Today, we’ve shown that this FDA takes swift action when patient safety is at risk.” said FDA Commissioner Marty Makary, M.D., M.P.H. “We believe in access to drugs for unmet medical needs but are not afraid to take immediate action when a serious safety signal emerges.”
The three deaths appear to have been a result of acute liver failure in individuals treated with Elevidys or investigational gene therapy using the same AAVrh74 serotype that is used in Elevidys. One of the fatalities occurred during a clinical trial conducted under an investigational new drug application for the treatment of Limb Girdle Muscular Dystrophy.
“Protecting patient safety is our highest priority, and the FDA will not allow products whose harms are greater than benefits. The FDA will halt any clinical trial of an investigational product if clinical trial participants would be exposed to an unreasonable and significant risk of illness or injury,” said Director of the FDA’s Center for Biologics Evaluation and Research Vinay Prasad, M.D., M.P.H.
Elevidys is an adeno-associated virus vector-based gene therapy using Sarepta Therapeutics, Inc.’s AAVrh74 Platform Technology for the treatment of Duchenne muscular dystrophy (DMD). It is designed to deliver into the body a gene that leads to production of Elevidys micro-dystrophin, a shortened protein (138 kDa, compared to the 427 kDa dystrophin protein of normal muscle cells) that contains selected domains of the dystrophin protein present in normal muscle cells. The product is administered as a single intravenous dose.
Duchenne muscular dystrophy is a rare and serious genetic condition which worsens over time, leading to weakness and wasting away of the body’s muscles. The disease occurs due to a defective gene that results in abnormalities in, or absence of, dystrophin, a protein that helps keep the body’s muscle cells intact.
Further, today, the FDA revoked the platform technology designation for Sarepta’s AAVrh74 Platform Technology because, among other things, given the new safety information, the preliminary evidence is insufficient to demonstrate that AAVrh74 Platform Technology has the potential to be incorporated in, or utilized by, more than one drug without an adverse effect on safety.
Elevidys received traditional approval for use in ambulatory DMD patients 4 years of age and older with a confirmed mutation in the DMD gene on June 20, 2024. It was approved for non-ambulatory patients on June 22, 2023 under the accelerated approval pathway. This pathway can allow earlier approval based on an effect on a surrogate endpoint or intermediate clinical endpoint that is reasonably likely to predict clinical benefit, while the company conducts confirmatory studies to verify the predicted clinical benefit. Continued approval for non-ambulatory patients is contingent upon verification and description of clinical benefit in a confirmatory trial. Given the new safety information, The FDA has notified the company that the indication should be restricted to use in ambulatory patients. The FDA is committed to further investigating the safety of the product in ambulatory patients and will take additional steps to protect patients as needed.
On July 18 Sarepta appeared to be disregarding the FDA release (according to the New York Times)
In a remarkable public dispute between drugmaker and regulator, the biotech company Sarepta Therapeutics is defying the Food and Drug Administration’s request that it halt distribution of its treatment for a deadly muscle-wasting disease.
In a news release on Friday evening, the agency said that it requested that the company voluntarily stop all shipments of the therapy, known as Elevidys, citing the deaths of three patients from liver failure who had taken the product or a similar therapy.
In its own news release later on Friday evening, Sarepta, which is based in Cambridge, Mass., said that it would continue to ship the treatment for patients who do not use wheelchairs. The company said its analysis showed no new safety problems in those patients and that it was committed to patient safety.
Dr. Marty Makary, the F.D.A. commissioner, said in the agency’s statement that its request to Sarepta demonstrated that the F.D.A. “takes swift action when patient safety is at risk.”
“We believe in access to drugs for unmet medical needs but are not afraid to take immediate action when a serious safety signal emerges,” he said.
In the past, the F.D.A. has sometimes asked companies to pause distribution of a drug until a new problem is better understood and mitigated. However, it can also press its case, and begin a process to revoke the drug’s license, which would begin with a formal notification and opportunity to respond and participate in a public hearing.
On July 21, 2025 Sarepta announces on their website in press release
Sarepta Therapeutics Announces Voluntary Pause of ELEVIDYS Shipments in the U.S.
07/21/25 7:40 PM EDT
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Jul. 21, 2025– Sarepta Therapeutics, Inc. (NASDAQ:SRPT), the leader in precision genetic medicine for rare diseases, today issued the following statement:
Today, Sarepta Therapeutics notified the U.S. Food and Drug Administration (FDA) of its decision to voluntarily and temporarily pause all shipments of ELEVIDYS (delandistrogene moxeparvovec) for Duchenne muscular dystrophy in the United States, effective close of business Tuesday, July 22, 2025.
This proactive step will allow Sarepta the necessary time to respond toany requests for information and allow Sarepta and FDA to complete the ELEVIDYS safety labeling supplement process. The Company looks forward to a collaborative, science-driven review process and dialogue with the FDA.
“As a patient-centric organization, the decision to voluntarily and temporarily pause shipments of ELEVIDYS was a painful one, as individuals with Duchenne are losing muscle daily and in need of disease-modifying options,” said Doug Ingram, chief executive officer, Sarepta. “It is important for the patients we serve that Sarepta maintains a productive and positive working relationship with FDA, and it became obvious that maintaining that productive working relationship required this temporary suspension while we address any questions that FDA may have and complete the ELEVIDYS label supplement process.”
Sarepta remains committed to transparency and patient safety and will continue to provide timely updates to patients, families, healthcare providers, and the broader Duchenne community as additional information becomes available.
About ELEVIDYS (delandistrogene moxeparvovec-rokl) ELEVIDYS (delandistrogene moxeparvovec-rokl) is a single-dose, adeno-associated virus (AAV)-based gene transfer therapy for intravenous infusion designed to address the underlying genetic cause of Duchenne muscular dystrophy – mutations or changes in the DMD gene that result in the lack of dystrophin protein – through the delivery of a transgene that codes for the targeted production of ELEVIDYS micro-dystrophin in skeletal muscle.
ELEVIDYS is indicated for the treatment of Duchenne muscular dystrophy (DMD) in individuals at least 4 years of age.
For patients who are ambulatory and have a confirmed mutation in the DMD gene
For patients who are non-ambulatory and have a confirmed mutation in the DMD gene.
However this is not the first time Sarepta has been in the hot seat…
Read this interesting article from Derrick Lowe of Science. I will put it in its entirety as Derick Lowe really writes some great articles in his blog.
I really, really wish that I were not writing about Sarepta again. But here we are. Perhaps a quick review will explain my reluctance.
Back in 2013, the company was trying to get approval for an unusual “exon skipping” molecule (eteplirsen) as a therapy for Duchenne muscular dystropy. Nothing wrong with that – in fact, there’s a lot that’s right with that, since Duchenne is a perfect “unmet medical need” situation, and the exon-skipping idea was an innovative approach ten years ago (and it’s still not exactly a standard-issue therapy). Attacking very hard-to-treat diseases with new mechanisms of action is just what we’re supposed to be doing in this business.
The approval, though, was having trouble for some very good reasons. Sarepta’s trial was very, very small and the FDA later found that their trial design was very, very flawed. But in 2016 eteplirsen was suddenly approved, to the surprise of many observers (including me). A few years later, a follow-up drug (golodirsen) from the company (golodirsen) was also rejected by the FDA (with a Complete Response Letter) but then was later suddenly approved, although no new data had been presented. That was particularly mystifying since the eventually-published CRL detailed a number of real problems with eteplirsen since its approval, problems that looked to be possibly even greater with the follow-up drug. To the best of my knowledge, the confirmatory Phase III trial that was required at the time of golodirsen’s approval is still going on and is expected to read out next year. In 2021, another Sarepta exon-skipping drug (different exon this time) was approved (casimirsen) on the basis of biomarker levels that were expected to show eventual clinical benefit, and I believe that its confirmatory trial is part of the golodirsen one. That one at least did not go through the first-rejected-then-approved pathway.
More recently the company has been working on an outright gene therapy (elevidys) for Duchenne, and the initial results were quite promising. The company got accelerated FDA approval for that one last June for 4- and 5-year-old patients, even though actual clinical benefit had not yet been established. But gene therapy is a winding road, and last October the Phase III results for Elevidys were a complete miss in the primary endpoint. Arguing commenced, with the company saying that the results in the secondary endpoints showed that the drug was “modifying the trajectory” of the disease, and the CEO called the results a “massive win” and said that the company would use them to ask for a much wider label approval from the FDA. Apparently during the conference call, when he was asked about why he was so confident, he said that the FDA’s CBER head Peter Marks was “very supportive”. (It should be noted that since then another Duchenne gene therapy effort, this one from Pfizer, also failed its Phase III, so it’s not like this is a straightforward area).
Boy, was that the truth. The agency has just granted that use expansion, and it turns out that it was all due to Peter Marks, who completely overruled three review teams and two of his highest-level staffers (all of whom said that Sarepta had not proven its case). Honestly, I’m starting to wonder why any of us go to all this trouble. It appears that all you need is a friend high up in the agency and your clinical failures just aren’t an issue any more. Review committees aren’t convinced? Statisticians don’t buy your arguments? Who cares! Peter Marks is here to deliver hot, steaming takeout containers full of Hope.
Back in 2016, when eteplirsen first came up for its advisory committee vote, I wrote that there was a matrix of possible votes and interpretations, which I summed up this way:
(1) A negative vote, which is a rejection of the potential of the drug, the suffering of DMD patients, and their right to try a therapy which apparently does no harm, for a disease that has no other options.
(2) A negative vote, which is the only possible one, considering that the company’s trial data are far too sparse and unconvincing to allow a recommendation to approve the drug. If this gets recommended, what doesn’t? Why do we require new drugs to show efficacy at all?
(3) A positive vote, which is a victory for patient advocates everywhere, and in particular for the extremely ill boys who suffer from this disease, or. . .
(4) A positive vote, which marks an undeserved and potentially hazardous victory of emotional rhetoric and relentless patient advocacy over the scientific and medical evidence.
As I’ve said many times since, including just a few days ago, I believe that the FDA is tilting very, very noticeably towards #4 while proclaiming the wonderful new world of #3. And while I realize that this may make me sound like a heartless SOB, I think this is a huge mistake that we will be paying for for a long time.
Note that there has been reported deaths in 2024.
The following was from some data published in Nature in 2025 from Clinical Trial ClinicalTrials.gov: NCT05096221.
Mendell JR, Muntoni F, McDonald CM, Mercuri EM, Ciafaloni E, Komaki H, Leon-Astudillo C, Nascimento A, Proud C, Schara-Schmidt U, Veerapandiyan A, Zaidman CM, Guridi M, Murphy AP, Reid C, Wandel C, Asher DR, Darton E, Mason S, Potter RA, Singh T, Zhang W, Fontoura P, Elkins JS, Rodino-Klapac LR. AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial.Nat Med. 2025 Jan;31(1):332-341. doi: 10.1038/s41591-024-03304-z
Abstract
Duchenne muscular dystrophy (DMD) is a rare, X-linked neuromuscular disease caused by pathogenic variants in the DMD gene that result in the absence of functional dystrophin, beginning at birth and leading to progressive impaired motor function, loss of ambulation and life-threatening cardiorespiratory complications. Delandistrogene moxeparvovec, an adeno-associated rh74-viral vector-based gene therapy, addresses absent functional dystrophin in DMD. Here the phase 3 EMBARK study aimed to assess the efficacy and safety of delandistrogene moxeparvovec in patients with DMD. Ambulatory males with DMD, ≥4 years to <8 years of age, were randomized and stratified by age group and North Star Ambulatory Assessment (NSAA) score to single-administration intravenous delandistrogene moxeparvovec (1.33 × 1014 vector genomes per kilogram; n = 63) or placebo (n = 62). At week 52, the primary endpoint, change from baseline in NSAA score, was not met (least squares mean 2.57 (delandistrogene moxeparvovec) versus 1.92 (placebo) points; between-group difference, 0.65; 95% confidence interval (CI), -0.45, 1.74; P = 0.2441). Secondary efficacy endpoints included mean micro-dystrophin expression at week 12: 34.29% (treated) versus 0.00% (placebo). Other secondary efficacy endpoints at week 52 (between-group differences (95% CI)) included: Time to Rise (-0.64 (-1.06, -0.23)), 10-meter Walk/Run (-0.42 (-0.71, -0.13)), stride velocity 95th centile (0.10 (0.00, 0.19)), 100-meter Walk/Run (-3.29 (-8.28, 1.70)), time to ascend 4 steps (-0.36 (-0.71, -0.01)), PROMIS Mobility and Upper Extremity (0.05 (-0.08, 0.19); -0.04 (-0.24, 0.17)) and number of NSAA skills gained/improved (0.19 (-0.67, 1.06)). In total, 674 adverse events were recorded with delandistrogene moxeparvovec and 514 with placebo. There were no deaths, discontinuations or clinically significant complement-mediated adverse events; 7 patients (11.1%) experienced 10 treatment-related serious adverse events. Delandistrogene moxeparvovec did not lead to a significant improvement in NSAA score at week 52. Some of the secondary endpoints numerically favored treatment, although no statistical significance can be claimed. Safety was manageable and consistent with previous delandistrogene moxeparvovec trials.
As noted in the adobe abstract everything seemed to fine as reported in this trial.
However there was a report of an immunoloically related death in 2023:
For the first time, in June 2023, delandistrogene moxeparvovec (SRP-9001), a gene replacement therapy based on an adeno-associated virus (AAV) vector, was approved in the USA for children aged 4-5 years with DMD. Other promising gene therapies are in preclinical development or clinical trials, including CRISPR/Cas9-mediated strategies to restore dystrophin expression. Two deaths following DMD gene therapy with high-dose AAV vectors were attributed to AAV-mediated immune responses. The pre-existing disease underlying the therapy is most likely involved in the fatal AAV toxicity.
Now this may have been dose related as the patient was given a high dose.
DMD gene therapy death exposes risks of treating older patients
Cure Rare Disease plans to continue its programs with alternative vectors. (iStock / Getty Images Plus)
Cure Rare Disease has shared a deep dive into the death of the only participant in a gene therapy trial. The nonprofit and its collaborators tied the death of a patient with Duchenne muscular dystrophy (DMD) to an immune reaction to the viral vector, raising concerns about dosing older, more advanced people.
Commercial development of DMD gene therapies has focused on younger patients, with Sarepta Therapeutics limiting enrollment in its phase 3 trial to children aged 4 to 8 years old. The restrictive recruitment criteria have stopped many DMD patients from accessing gene therapies in clinical trials run by Sarepta and its rivals. The patient dosed in the Cure Rare Disease clinical trial was 27 years of age, and the therapy had been designed for him.
Last year, the nonprofit reported that the patient, who was the brother of its CEO, died after receiving the therapy. The death led to an investigation into what happened after the patient received the therapy, which was designed to use CRISPR transactivation to upregulate an alternate form of a key DMD protein.
Writing in preprint journal medRxiv (PDF), Cure Rare Disease described the findings of the investigation. A post-mortem showed injuries to the patient’s lungs, likely caused by a strong immune reaction to the high dose of the adeno-associated virus (AAV) vector that was given to try to ensure sufficient expression to achieve a therapeutic effect. There was minimal expression of the transgene in the liver.
At 1×1014 vg/kg, the studied dose was similar to that tested in other clinical trials but resulted in a higher vector genome load, a finding the researchers attributed to the patient’s lower lean muscle mass, 45%. The analysis suggests the patient had “a more severe innate immune reaction than others receiving similar or slightly higher doses of rAAV in microdystrophin gene therapy trials.”
Based on the finding, the researchers identified a need for more data on the characteristics that may predispose people to severe innate immune reactions and concluded “dose determination will remain a challenge for custom-designed AAV-mediated therapies, as by definition the precise therapeutic dose will not have been established.”
As for the application of CRISPR, the researchers said the toxicity and eventual death of the patient meant that an assessment of the safety and efficacy of the treatment was not possible.
AAV related clinical trials have been halted for drug-induced liver injury, predominantly due to severe immune reaction. In many cases it appears when high dose AAV therapy is used.
.10.015. Epub 2023 Oct 10. PMID: 37822079; PMCID: PMC10638066.
Abstract
High-dose systemic gene therapy with adeno-associated virus (AAV) is in clinical trials to treat various inherited diseases. Despite remarkable success in spinal muscular atrophy and promising results in other diseases, fatality has been observed due to liver, kidney, heart, or lung failure. Innate and adaptive immune responses to the vector play a critical role in the toxicity. Host factors also contribute to patient death. This mini-review summarizes clinical findings and calls for concerted efforts from all stakeholders to better understand the mechanisms underlying lethality in AAV gene therapy and to develop effective strategies to prevent/treat high-dose systemic AAV-gene-therapy-induced immunotoxicity.
Table 1.
Fatality cases following high-dose systemic AAV delivery
Roche Decides to Stop backing Sparks Therapeutics Hemophilia A Gene Therapy Program
In 2019, Roche acquired Children’s Hospital of Pennsylvania (CHOP) spinout Spark Therapeutics for $4.8 billion, one of the largest pharma acquisitions up to that time. It was reported on this site here
However as reported by Fierce Biotech (and updated above link) at https://www.fiercepharma.com/pharma/roche-overhauls-spark-gene-therapy-unit-recording-24b-full-impairment Roche will reorganize the company and deal, bringing in Spark into the corporate fold. However this meant massive layoffs and possibly either end of the gene therapy program in order to integrate it with Roche’s current programs. The Spark gene therapy has met with success so it will be interesting to see how Roche continues this program in the future.
However it has been a rough year for many gene therapies.
Other Articles in this Open Access Scientific Journol of Gene Therapy
Use of 3D Bioprinting for Development of Toxicity Prediction Models
Curator: Stephen J. Williams, PhD
SOT FDA Colloquium on 3D Bioprinted Tissue Models: Tuesday, April 9, 2019
The Society of Toxicology (SOT) and the U.S. Food and Drug Administration (FDA) will hold a workshop on “Alternative Methods for Predictive Safety Testing: 3D Bioprinted Tissue Models” on Tuesday, April 9, at the FDA Center for Food Safety and Applied Nutrition in College Park, Maryland. This workshop is the latest in the series, “SOT FDA Colloquia on Emerging Toxicological Science: Challenges in Food and Ingredient Safety.”
Human 3D bioprinted tissues represent a valuable in vitro approach for chemical, personal care product, cosmetic, and preclinical toxicity/safety testing. Bioprinting of skin, liver, and kidney is already appearing in toxicity testing applications for chemical exposures and disease modeling. The use of 3D bioprinted tissues and organs may provide future alternative approaches for testing that may more closely resemble and simulate intact human tissues to more accurately predict human responses to chemical and drug exposures.
A synopsis of the schedule and related works from the speakers is given below:
8:40 AM–9:20 AM
Overview and Challenges of Bioprinting
Sharon Presnell, Amnion Foundation, Winston-Salem, NC
9:20 AM–10:00 AM
Putting 3D Bioprinting to the Use of Tissue Model Fabrication
Y. Shrike Zhang, Brigham and Women’s Hospital, Harvard Medical School and Harvard-MIT Division of Health Sciences and Technology, Boston, MA
10:00 AM–10:20 AM
Break
10:20 AM–11:00 AM
Uses of Bioprinted Liver Tissue in Drug Development
Jean-Louis Klein, GlaxoSmithKline, Collegeville, PA
11:00 AM–11:40 AM
Biofabrication of 3D Tissue Models for Disease Modeling and Chemical Screening
Marc Ferrer, National Center for Advancing Translational Sciences, NIH, Rockville, MD
Dr. Sharon Presnell was most recently the Chief Scientific Officer at Organovo, Inc., and the President of their wholly-owned subsidiary, Samsara Sciences. She received a Ph.D. in Cell & Molecular Pathology from the Medical College of Virginia and completed her undergraduate degree in biology at NC State. In addition to her most recent roles, Presnell has served as the director of cell biology R&D at Becton Dickinson’s corporate research center in RTP, and as the SVP of R&D at Tengion. Her roles have always involved the commercial and clinical translation of basic research and early development in the cell biology space. She serves on the board of the Coulter Foundation at the University of Virginia and is a member of the College of Life Sciences Foundation Board at NC State. In January 2019, Dr. Presnell will begin a new role as President of the Amnion Foundation, a non-profit organization in Winston-Salem.
Integrating Kupffer cells into a 3D bioprinted model of human liver recapitulates fibrotic responses of certain toxicants in a time and context dependent manner. This work establishes that the presence of Kupffer cells or macrophages are important mediators in fibrotic responses to certain hepatotoxins and both should be incorporated into bioprinted human liver models for toxicology testing.
Abstract: Modeling clinically relevant tissue responses using cell models poses a significant challenge for drug development, in particular for drug induced liver injury (DILI). This is mainly because existing liver models lack longevity and tissue-level complexity which limits their utility in predictive toxicology. In this study, we established and characterized novel bioprinted human liver tissue mimetics comprised of patient-derived hepatocytes and non-parenchymal cells in a defined architecture. Scaffold-free assembly of different cell types in an in vivo-relevant architecture allowed for histologic analysis that revealed distinct intercellular hepatocyte junctions, CD31+ endothelial networks, and desmin positive, smooth muscle actin negative quiescent stellates. Unlike what was seen in 2D hepatocyte cultures, the tissues maintained levels of ATP, Albumin as well as expression and drug-induced enzyme activity of Cytochrome P450s over 4 weeks in culture. To assess the ability of the 3D liver cultures to model tissue-level DILI, dose responses of Trovafloxacin, a drug whose hepatotoxic potential could not be assessed by standard pre-clinical models, were compared to the structurally related non-toxic drug Levofloxacin. Trovafloxacin induced significant, dose-dependent toxicity at clinically relevant doses (≤ 4uM). Interestingly, Trovafloxacin toxicity was observed without lipopolysaccharide stimulation and in the absence of resident macrophages in contrast to earlier reports. Together, these results demonstrate that 3D bioprinted liver tissues can both effectively model DILI and distinguish between highly related compounds with differential profile. Thus, the combination of patient-derived primary cells with bioprinting technology here for the first timedemonstrates superior performance in terms of mimicking human drug response in a known target organ at the tissue level.
A great interview with Dr. Presnell and the 3D Models 2017 Symposium is located here:
Please clickhere for Web based and PDF version of interview
Some highlights of the interview include
Exciting advances in field showing we can model complex tissue-level disease-state phenotypes that develop in response to chronic long term injury or exposure
Sees the field developing a means to converge both the biology and physiology of tissues, namely modeling the connectivity between tissues such as fluid flow
Future work will need to be dedicated to develop comprehensive analytics for 3D tissue analysis. As she states “we are very conditioned to get information in a simple way from biochemical readouts in two dimension, monocellular systems” however how we address the complexity of various cellular responses in a 3D multicellular environment will be pertinent.
Additional challenges include the scalability of such systems and making such system accessible in a larger way
Shrike Zhang, Brigham and Women’s Hospital, Harvard Medical School and Harvard-MIT Division of Health Sciences and Technology
Dr. Zhang currently holds an Assistant Professor position at Harvard Medical School and is an Associate Bioengineer at Brigham and Women’s Hospital. His research interests include organ-on-a-chip, 3D bioprinting, biomaterials, regenerative engineering, biomedical imaging, biosensing, nanomedicine, and developmental biology. His scientific contributions have been recognized by >40 international, national, and regional awards. He has been invited to deliver >70 lectures worldwide, and has served as reviewer for >400 manuscripts for >30 journals. He is serving as Editor-in-Chief for Microphysiological Systems, and Associate Editor for Bio-Design and Manufacturing. He is also on Editorial Board of Bioprinting, Heliyon, BMC Materials, and Essays in Biochemistry, and on Advisory Panel of Nanotechnology.
Skardal A, Murphy SV, Devarasetty M, Mead I, Kang HW, Seol YJ, Shrike Zhang Y, Shin SR, Zhao L, Aleman J, Hall AR, Shupe TD, Kleensang A, Dokmeci MR, Jin Lee S, Jackson JD, Yoo JJ, Hartung T, Khademhosseini A, Soker S, Bishop CE, Atala A.
Sci Rep. 2017 Aug 18;7(1):8837. doi: 10.1038/s41598-017-08879-x.
Bhise NS, Manoharan V, Massa S, Tamayol A, Ghaderi M, Miscuglio M, Lang Q, Shrike Zhang Y, Shin SR, Calzone G, Annabi N, Shupe TD, Bishop CE, Atala A, Dokmeci MR, Khademhosseini A.
Biofabrication. 2016 Jan 12;8(1):014101. doi: 10.1088/1758-5090/8/1/014101.
Marc Ferrer, National Center for Advancing Translational Sciences, NIH
Marc Ferrer is a team leader in the NCATS Chemical Genomics Center, which was part of the National Human Genome Research Institute when Ferrer began working there in 2010. He has extensive experience in drug discovery, both in the pharmaceutical industry and academic research. Before joining NIH, he was director of assay development and screening at Merck Research Laboratories. For 10 years at Merck, Ferrer led the development of assays for high-throughput screening of small molecules and small interfering RNA (siRNA) to support programs for lead and target identification across all disease areas.
At NCATS, Ferrer leads the implementation of probe development programs, discovery of drug combinations and development of innovative assay paradigms for more effective drug discovery. He advises collaborators on strategies for discovering small molecule therapeutics, including assays for screening and lead identification and optimization. Ferrer has experience implementing high-throughput screens for a broad range of disease areas with a wide array of assay technologies. He has led and managed highly productive teams by setting clear research strategies and goals and by establishing effective collaborations between scientists from diverse disciplines within industry, academia and technology providers.
Ferrer has a Ph.D. in biological chemistry from the University of Minnesota, Twin Cities, and completed postdoctoral training at Harvard University’s Department of Molecular and Cellular Biology. He received a B.Sc. degree in organic chemistry from the University of Barcelona in Spain.
Wilson KM, Mathews-Griner LA, Williamson T, Guha R, Chen L, Shinn P, McKnight C, Michael S, Klumpp-Thomas C, Binder ZA, Ferrer M, Gallia GL, Thomas CJ, Riggins GJ.
SLAS Technol. 2019 Feb;24(1):28-40. doi: 10.1177/2472630318803749. Epub 2018 Oct 5.
Ability of gut microbiota to influence the bioavailability of levodopa in Parkinson’s disease – The presence of more bacteria producing the tyrosine decarboxylase (TDC) enzyme means less levodopa in the bloodstream
Reporter: Aviva Lev-Ari, PhD, RN
Decarboxylase enzymes can convert levodopa into dopamine. In contrast to levodopa, dopamine cannot cross the blood-brain barrier, so patients are also given a decarboxylase inhibitor. “But the levels of levodopa that will reach the brain vary strongly among Parkinson’s disease patients.
The bacterial tyrosine decarboxylase enzyme, which normally converts tyrosine into tyramine, but was found to also convert levodopa into dopamine. “We then determined that the source of this decarboxylase was Enterococcus bacteria.” The researchers also showed that the conversion of levodopa was not inhibited by a high concentration of the amino acid tyrosine, the main substrate of the bacterial tyrosine decarboxylase enzyme.
Carbidopa is over 10,000 times more potent in inhibiting the human decarboxylase,
the higher abundance of bacterial enzyme in the small intestines of rats reduced levels of levodopa in the bloodstream,
positive correlation between disease duration and levels of bacterial tyrosine decarboxylase.
Some Parkinson’s disease patients develop an overgrowth of small intestinal bacteria including Enterococci due to frequent uptake of proton pump inhibitors, which they use to treat gastrointestinal symptoms associated with the disease.
Altogether, these factors result in a vicious circle leading to an increased levodopa/decarboxylase inhibitor dosage requirement in a subset of patients.El Aidy concludes that
the presence of the bacterial tyrosine decarboxylase enzyme can explain why some patients need more frequent dosages of levodopa to treat their motor fluctuations. “This is considered to be a problem for Parkinson’s disease patients, because a higher dose will result in dyskinesia, one of the major side effects of levodopa treatment.“
Human gut microbiota senses its environment and responds by releasing metabolites, some of which are key regulators of human health and disease. In this study, we characterize gut-associated bacteria in their ability to decarboxylate levodopa to dopamine via tyrosine decarboxylases. Bacterial tyrosine decarboxylases efficiently convert levodopa to dopamine, even in the presence of tyrosine, a competitive substrate, or inhibitors of human decarboxylase. In situ levels of levodopa are compromised by high abundance of gut bacterial tyrosine decarboxylase in patients with Parkinson’s disease. Finally, the higher relative abundance of bacterial tyrosine decarboxylases at the site of levodopa absorption, proximal small intestine, had a significant impact on levels of levodopa in the plasma of rats. Our results highlight the role of microbial metabolism in drug availability, and specifically, that abundance of bacterial tyrosine decarboxylase in the proximal small intestine can explain the increased dosage regimen of levodopa treatment in Parkinson’s disease patients.
Leadership we provide on curation of scientific findings in the eScientific publishing for Medical Education contents.
In Section 1, the Leadership we provide on curation of scientific findings in the eScientific publishing for Medical Education contents is demonstrated by a subset of several outstanding curations with high electronic Viewer volume. Each article included presents unique content contribution to Medical Clinical Education.
· These articles are extracted from the list of all Journal articles with >1,000 eReaders, 4/28/2012 to 1/29/2018.
Article Title, # of electronic Viewers, Author(s) Name
As BioMed e-Series Editor–in-Chief, I was responsible for the following functions of product design and product launch
· 16 Title creations for e-Books
· Designed 16 Cover Pages for a 16-Volume e-Books e-Series in BioMed
· Designed Series A eTOCs and approved of all 16 electronic Table of Contents (eTOCs), working in tandem with all the Editors of each volume and all the Author contributors of article contents in the Journal.
· Commissioned Articles by Authors/Curators per Author’s expertise on a daily basis
The Immune System, Stress Signaling, Infectious Diseases and Therapeutic Implications
9/4/2017
3747 pages
The VOICES of Patients, Hospitals CEOs, Health Care Providers, Caregivers and Families: Personal Experience with Critical Care and Invasive Medical Procedures
10/16/2017
826 pages
Medical Scientific Discoveries for the 21st Century & Interviews with Scientific Leaders
12/9/2017
2862 pages
Milestones in Physiology: Discoveries in Medicine, Genomics and Therapeutics
12/27/2015
11125 KB
Medical 3D BioPrinting – The Revolution in Medicine, Technologies for Patient-centered Medicine: From R&D in Biologics to New Medical Devices
12/30/2017
1005 pages
Pharmacological Agents in Treatment of Cardiovascular Disease
Work-in-Progress, Expected Publishing date in 2018
???
Interventional Cardiology and Cardiac Surgery for Disease Diagnosis and Guidance of Treatment
Work-in-Progress, Expected Publishing date in 2018
Scientists from Duke-NUS Medical School (Duke-NUS) have derived a structural model of a transporter at the blood-brain barrier called Mfsd2a. This is the first molecular model of this critical transporter, and could prove important for the development of therapeutic agents that need to be delivered to the brain — across the blood-brain barrier. In future, this could help treat neurological disorders such as glioblastoma.
Currently, there are limitations to drug delivery to the brain as it is tightly protected by the blood-brain barrier. The blood-brain barrier is a protective barrier that separates the circulating blood from the central nervous system which can prevent the entry of certain toxins and drugs to the brain. This restricts the treatment of many brain diseases. However, as a transporter at the blood-brain barrier, Mfsd2a is a potential conduit for drug delivery directly to the brain, thus bypassing the barrier.
In this study, recently published in the Journal of Biological Chemistry, first author Duke-NUS MD/PhD student Debra Quek and senior author Professor David Silver used molecular modeling and biochemical analyses of altered Mfsd2a transporters to derive a structural model of human Mfsd2a. Importantly, the work identifies new binding features of the transporter, providing insight into the transport mechanism of Mfsd2a.
“Our study provides the first glimpse into what Mfsd2a looks like and how it might transport essential lipids across the blood-brain barrier,” said Ms Quek. “It also facilitates a structure-guided search and design of scaffolds for drug delivery to the brain via Mfsd2a, or of drugs that can be directly transported by Mfsd2a.”
Currently this information is being used by Duke-NUS researchers to design novel therapeutic agents for direct drug delivery across the blood brain barrier for the treatment of neurological diseases. This initiative by the Centre for Technology and Development (CTeD) at Duke-NUS, is one of many collaborative research efforts aimed at translating Duke-NUS’ research findings into tangible commercial and therapeutic applications for patients.
Ms Quek plans to further validate her findings by purifying the Mfsd2a protein in order to further dissect how it functions as a transporter.
J Biol Chem. 2016 Mar 4. pii: jbc.M116.721035. [Epub ahead of print]
Structural insights into the transport mechanism of the human sodium-dependent lysophosphatidylcholine transporter Mfsd2a.
Major Facilitator Superfamily Domain containing 2A (Mfsd2a) was recently characterized as a sodium-dependent lysophosphatidylcholine (LPC) transporter expressed at the blood-brain barrier endothelium. It is the primary route for importation of docosohexaenoic acid and other long-chain fatty acids into foetal and adult brain, and is essential for mouse and human brain growth and function. Remarkably, Mfsd2a is the first identified MFS family member that uniquely transports lipids, implying that Mfsd2a harbours unique structural features and transport mechanism. Here, we present three 3D structural models of human Mfsd2a derived by homology modelling using MelB- and LacY-based crystal structures, and refined by biochemical analysis. All models revealed 12 transmembrane helices and connecting loops, and represented the partially outward-open, outward-partially occluded, and inward-open states of the transport cycle. In addition to a conserved sodium-binding site, three unique structural features were identified: A phosphate headgroup binding site, a hydrophobic cleft to accommodate a hydrophobic hydrocarbon tail, and three sets of ionic locks that stabilize the outward-open conformation. Ligand docking studies and biochemical assays identified Lys436 as a key residue for transport. It is seen forming a salt bridge with the negative charge on the phosphate headgroup. Importantly, Mfsd2a transported structurally related acylcarnitines but not a lysolipid without a negative charge, demonstrating the necessity of a negative charged headgroup interaction with Lys436 for transport. These findings support a novel transport mechanism by which LPCs are flipped within the transporter cavity by pivoting about Lys436 leading to net transport from the outer to the inner leaflet of the plasma membrane.
Brain and eye contain membrane phospholipids that are enriched in the omega-3 fatty acid docosohexaenoic acid (DHA). It is widely accepted that DHA is important for brain and eye function and brain development (1,2), although mechanisms for DHA function in these tissues are not well defined. The mechanism by which DHA and other conditionally essential and essential fatty acids cross the blood-brain barrier (BBB) has been a long-standing mystery. Recently, we identified Major Facilitator Superfamily Domain containing 2a (Mfsd2a, aka NLS1) as the primary transporter by which the brain obtains DHA. Importantly, Mfsd2a does not transport unesterified DHA, but transports DHA in the chemical form of lysophosphatidylcholine (LPC) that are synthesized by the liver and circulate largely on albumin (3). This is consistent with biochemical evidence that the brain does not transport unesterified fatty acids (4) and that LPC is the preferred carrier of DHA to the brain (5,6). Mfsd2a is a sodium-dependent transporter that is part of the Major Facilitator Superfamily (MFS) of proteins. Members of this family with elucidated structures have 12 transmembrane domains composed of two evolutionarily duplicated 6 transmembrane units (7). Transporting an LPC is a unique feature of Mfsd2a, since most members of this family transport water-soluble and minimally polar substrates such as sugars (GLUT, MelB, LacY), and amino acids (TAT1). Mfsd2a transport is not limited to LPCs containing DHA, as it can transport LPCs containing a variety of fatty acyl chains, with higher specificity for LPCs with unsaturated fatty acyl chains with a minimum chain length of 14 carbons (6,8). Crystal structures have been solved for more than a dozen members of the MFS family, with more than 19 structures, including that of Melibiose permease (MelB) of S. typhimurium (9), Lactose permease (LacY) of Escherichia coli (10), glycerol-3-phosphate transporter of E. coli (11) and the mammalian glucose transporters 1, 3, and 5 (GLUT1, GLUT3, GLUT5) (12-14). A common transport mechanism has emerged from both biochemical and structural analyses of MFSs, in which they transport via a rocker-switch, alternating access mechanism (7,15). In the rocker-switch model, rigid-body relative motion of the N- and C-termini domains renders the substrate-binding site alternatively accessible from either side of the membrane.
Mfsd2a is highly expressed at the bloodbrain barrier in both mouse and human (6,16). Mfsd2a deficient mice (KO) have significantly reduced brain DHA as a result of a 90% reduction in brain uptake of LPC containing DHA as well as other LPCs. The most prominent phenotype of Mfsd2a KO mice is microcephaly, and KO mice additionally exhibit motor dysfunction, and behavioral disorders including anxiety and memory and learning deficits (6). In line with the mouse KO phenotypes, human patients with partially or completely inactivating mutations in Mfsd2a presented with severe microcephaly, intellectual disability, and motor dysfunction (8,16). Plasma LPCs are significantly elevated in both KO mice and human patients with Mfsd2a mutations, consistent with reduced uptake at the blood-brain barrier. Taken together, these findings demonstrate that LPCs are essential for normal brain development and function in mouse and humans.
The fact that Mfsd2a transports a lysolipid, a non-canonical substrate for an MFS protein, might indicate unique structure features and a novel transport mechanism. However, no structural information or mechanism of transport of Mfsd2a is known. Human Mfsd2a is composed of 530 amino acids, with two glycosylation sites at Asn217 and Asn227. Mfsd2a is evolutionarily conserved from teleost fish to humans. Although not a functional ortholog of bacterial MFS transporters, Mfsd2a shares 25% and 26% amino acid sequence identity with S. typhimurium MelB (9,17), and LacY from E. coli (10), respectively. Given the high conservation of the MFS fold, the use of homology modeling to gain insight into the structure of S. typhimurium MelB, for example, has proven to be highly accurate and largely consistent with subsequent X-ray crystal data (9,18). Here, we take advantage of two recently derived high resolution X-ray crystal structures of S. typhimurium MelB (9), and a high resolution X-ray crystal structure of LacY (10) to generate three predictive structural models of human Mfsd2a. These models reveal three unique regions critical for function – an LPC headgroup binding site, a hydrophobic cleft occupied by the LPC fatty acyl tail, and three sets of ionic locks. These structural features indicate a novel mechanism of transport for LPCs.
Mfsd2a is a sodium-dependent lysophosphatidylcholine transporter essential for human brain growth and function (40). Mfsd2a is the only known MFS member or secondary transporter that transports a lipid. In line with its unique function, the current study has identified three unique structural features based on a combination of homology structural modeling and biochemical analysis – (1) a unique headgroup binding site and (2) a hydrophobic cleft for acyl chain binding, and (4) 3 sets of ionic locks that stabilize the outward open conformation. Drawing together these findings with studies of the mechanism of transport of other MFS family members, we propose the following alternatingaccess mechanism for LPC transport (Fig. 6). In the first steps, LPC inserts itself into the outer leaflet of the membrane and diffuses laterally into the transporter’s hydrophobic cleft. As Mfsd2a undergoes conformational changes from the outward open to the inward open conformation, the zwitterionic headgroup is inverted from the outer membrane leaflet to the inner membrane leaflet along a translocation pathway within the transporter, interacting with specific polar and charged residues lining the path. Since LPCs are hydrophobic phospholipids, it is unlikely that they will partition out of the transporter into the aqueous environment of the cytoplasm. We propose that the “flipped” LPC exits the transporter laterally into the membrane environment of the inner leaflet. This model of LPC flipping requires further biochemical proof. Of particular interest is the visualization of the interaction of the negatively charged phosphate headgroup of LPC with Lys436 that is maintained in both outward and inward open conformations. The sidechain of Lys436 is seen to be pointing in the upward direction in the outward open conformation, but pointing downward into the translocation cleft in the inward open conformation. These findings suggest that the Lys436 acts as a tether to push or pivot the headgroup down into the translocation cavity while the N- and C-termini of Mfsd2a rock and switch from outward to inward open.
Interestingly, Lys436 is orthologous to the residue Lys377 in the melibiose transporter of S. typhimurium. Based on the S. typhimurium MelB crystal structure, Lys377 has been predicted to be involved in binding melibiose, and in forming a hydrogen bond with Tyr120, likely separating the sodium binding site from the central hydrophilic cavity (9). In a recent molecular dynamic simulation of E. coli MelB, Lys377 was noted to interact differently with residues involved in the sodium binding site (Asp55, Asp59, and Asp124) in the presence or absence of a sodium ion, and thought to be critical for the spatial organization of the sodium binding site (41). Similarly, in our refined models of Mfsd2a, Lys436 is localized in close proximity to the sodium-binding site residue, Asp93, and the central translocation pathway where it has been identified by docking studies to interact with the charged headgroup of LPC. We hypothesize that Lys436 may shuttle between the two binding sites, communicating and coordinating the occupancy status of the two sites. Interestingly, there is a distinct mobility shift in Mfsd2a bands on SDS-PAGE between wild-type Mfsd2a and the L-3 mutant (R498E, R499E, R500E, K503E, K504E) (Fig. 5I) that is not seen when each of the residues are mutated individually (Fig. S1). These findings are consistent with a conformational change in the L-3 mutant. Given that the L-3 ionic lock is visualized in the outward partially occluded model, we hypothesize that the loss of the L-3 ionic lock results in Mfsd2a being trapped in an energetically more favorable inward open conformation, resulting in the loss of transport function (Fig. 5H).
Patients with the partially inactivating mutation p.(S399L) exhibited significant increases specifically in plasma LPCs having monounsaturated (18:1 – 92%, p=0.004) and polyunsaturated LPCs (18:2, 20:4, 20:3 – 254%, p=0.002; 117%, p=0.007, and 238%, p=0.002), but not in the most abundant LPCs – saturated LPCs (C16:0, C18:0) (8). This is consistent with a greater specificity of Mfsd2a for LPCs with unsaturated fatty acyl chains (6)…A possible explanation for this acyl chain specificity is related to the mobility of the acyl tail in the membrane. It is known that phospholipids with unsaturated fatty acyl chains disrupt the packing of the bilayer, resulting in greater lateral membrane fluidity (42). Therefore, one possible mechanism for LPC specificity is that LPCs with unsaturated fatty acyl chains have greater lateral mobility in the membrane, increasing the Ka for interacting with the transport cleft of Mfsd2a.
Another important structural feature of the physiological ligand, LPC, is a minimum acyl chain length of 14 carbons is required for transport by Mfsd2a. A possible explanation for this requirement is that the hydrocarbon chain must extend beyond the cleft, protruding into the hydrophobic milieu of the phospholipid bilayer core. This interaction of the fatty acyl tail with the acyl chains of the membrane bilayer may provide a hydrophobic force strong enough to pull the molecule through and out of the transporter as the LPC headgroup partitions into the inner leaflet of the membrane. A similar scenario is seen in the Sec translocon where a hydrophobic transmembrane domain of a protein partitions laterally from the Sec61p complex channel into the lipid bilayer (43,44). This proposal that the omega carbon of the fatty acyl chain sticks out of the Mfsd2a pocket is consistent with the observation that Mfsd2a can transport nitrobenzoxadiazole (NBD) or Topfluor when these moieties are attached to the omega carbon of the LPC fatty acyl tail [1].
Other known transmembrane phospholipid transporters include flippases, floppases, and scramblases. Flippases and floppases utilize ATP to drive the uphill transport of aminophospholipids from the outer to the inner leaflet, and specific substrates from the inner to the outer leaflet, respectively (45-47). Scramblases are less well understood, facilitating transport of substrates in either direction down concentration gradients upon activation. While the substrates are similar, several differences make comparisons between Mfsd2a and phospholipid transporters of limited relevance. First, the shapes of the substrates differ in shape and size – lysophospholipids are smaller and conical while phospholipids are cylindrical. Second, unlike flippases and floppases, Mfsd2a is a secondary transporter, utilizing a sodium electrochemical gradient to drive the transport of lysophospholipids from one leaflet to the other. Third, the overall structure of MFS members is different from P4- ATPases and ABC transporters. Consequently, the mechanism of action between Mfsd2a and flippases such as P4-ATPases and ABC transporters, or floppases is expected to differ.
Being expressed at the blood-brain barrier, Mfsd2a is a potential conduit for drug delivery to the brain. The blood-brain barrier is highly impermeable, protecting the brain from bloodderived molecules, pathogens, and toxins. However, its impermeability poses a challenge for pharmacological treatment of brain diseases. It has been predicted that 98% of small molecule drugs are excluded from the brain by the blood-brain barrier (48). Currently, most drugs used to treat brain diseases are lipid soluble small molecules with a molecular weight of less than 400 Da (49). A small number of drugs traverse the blood-brain barrier by carrier-mediated transport. An example of this is Levodopa, a treatment for Parkinson’s Disease, which is a precursor of the neurotransmitter dopamine. Levodopa is transported across the blood-brain barrier by the large neutral amino acid transporter, LAT1 (50). Our findings here provide a further refinement of understanding of the structure-activity relationship of LPCs to their transport, and educates the search and design of drugs that can be transported by Mfsd2a. Candidates for transport, whether as a drug itself or as a LPC scaffold, must have a zwitterionic headgroup, but not necessarily a phosphate, and a minimal threshold of hydrophobic character. As the binding pocket is several times larger than LPC, it is sterically feasible to attach a small molecule drug onto LPC or LPC-like scaffolds for delivery across the blood-brain barrier.
In summary, these studies represent a first structural model of human Mfsd2a based on homology modeling and biochemical interrogation. We expect that this model will serve as a foundation for the future development of X-ray crystal structures of the protein, which would provide further insight into the structure and function of this physiologically important transporter required for human brain growth and function.
REFERENCES
1. Salem, N., Jr., Litman, B., Kim, H. Y., and Gawrisch, K. (2001) Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 36, 945-959
2. Bazan, N. G. (2009) Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. Journal of lipid research 50 Suppl, S400- 405
3. Baisted, D. J., Robinson, B. S., and Vance, D. E. (1988) Albumin stimulates the release of lysophosphatidylcholine from cultured rat hepatocytes. The Biochemical journal 253, 693-701
4. Edmond, J., Higa, T. A., Korsak, R. A., Bergner, E. A., and Lee, W. N. (1998) Fatty acid transport and utilization for the developing brain. Journal of neurochemistry 70, 1227-1234
5. Lagarde, M., Bernoud, N., Brossard, N., Lemaitre-Delaunay, D., Thies, F., Croset, M., and Lecerf, J. (2001) Lysophosphatidylcholine as a preferred carrier form of docosahexaenoic acid to the brain. Journal of molecular neuroscience : MN 16, 201-204; discussion 215-221
6. Nguyen, L. N., Ma, D., Shui, G., Wong, P., Cazenave-Gassiot, A., Zhang, X., Wenk, M. R., Goh, E. L., and Silver, D. L. (2014) Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503-506
7. Law, C. J., Maloney, P. C., and Wang, D. N. (2008) Ins and outs of major facilitator superfamily antiporters. Annual review of microbiology 62, 289-305
8. Alakbarzade, V., Hameed, A., Quek, D. Q. Y., Chioza, B. A., Baple, E. L., Cazenave-Gassiot, A., Nguyen, L. N., Wenk, M. R., Ahmad, A. Q., Sreekantan-Nair, A., Weedon, M. N., Rich, P., Patton, M. A., Warner, T. T., Silver, D. L., and Crosby, A. H. (2015) A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat Genet 47, 814-817
9. Ethayathulla, A. S., Yousef, M. S., Amin, A., Leblanc, G., Kaback, H. R., and Guan, L. (2014) Structure-based mechanism for Na(+)/melibiose symport by MelB. Nature communications 5, 3009
10. Guan, L., Mirza, O., Verner, G., Iwata, S., and Kaback, H. R. (2007) Structural determination of wild-type lactose permease. Proceedings of the National Academy of Sciences of the United States of America 104, 15294-15298
Chemotherapy Benefit in Early Breast Cancer Patients
Larry H Bernstein, MD, FCAP, Curator
LPBI
Agendia’s MammaPrint® First and Only Genomic Assay to Receive Level 1A Clinical Utility Evidence for Chemotherapy Benefit in Early Breast Cancer Patients
Clinical high-risk patients with a low-risk MammaPrint® result, including 48 percent node-positive, had five-year distant metastasis-free survival rate in excess of 94 percent, whether randomized to receive adjuvant chemotherapy or not
MammaPrint could change clinical practice by substantially de-escalating the use of adjuvant chemotherapy and sparing many patients an aggressive treatment they will not benefit from
Forty-six percent overall reduction in chemotherapy prescription among clinically high-risk patients
April 19, 2016 / B3C newswire / —Agendia, Inc., together with the European Organisation for Research and Treatment of Cancer (EORTC) and Breast International Group (BIG), announced results from the initial analysis of the primary objective of the Microarray In Node-negative (and 1 to 3 positive lymph node) Disease may Avoid ChemoTherapy (MINDACT) study at the American Association for Cancer Research Annual Meeting 2016 in New Orleans, LA.
Using the company’s MammaPrint® assay, patients with early-stage breast cancer who were considered at high risk for disease recurrence based on clinical and biological criteria had a distant metastasis-free survival at five years in excess of 94 percent.The MammaPrint test—the first and only genomic assay with FDA 510(k) clearance for use in risk assessment for women of all ages with early stage breast cancer—identified a large group of patients for whom five-year distant metastasis–free survival was equally good whether or not they received adjuvant chemotherapy (chemotherapy given post-surgery).
“The MINDACT trial design is the optimal way to prove clinical utility of a genomic assay,” said Prof. Laura van ’t Veer, CRO at Agendia, Leader, Breast Oncology Program, and Director, Applied Genomics at UCSF Helen Diller Family Comprehensive Cancer Center. “It gives the level 1A clinical evidence (prospective, randomized and controlled) that empowers physicians to clearly and confidently know when chemotherapy is part of optimal early-stage breast cancer therapy. In this trial, MammaPrint (70-gene assay) was compared to the standard of care physicians use today, to decide what is the best treatment option for an early-stage breast cancer patient.”
The MINDACT trial is the first prospective randomized controlled clinical trial of a breast cancer recurrence genomic assay with level 1A clinical evidence and the first prospective translational research study of this magnitude in breast cancer to report the results of its primary objective.
Among the 3,356 patients enrolled in the MINDACT trial, who were categorized as having a high risk of breast cancer recurrence based on common clinical and pathological criteria (C-high), the MammaPrint assay reduced the chemotherapy treatment prescription by 46 percent.Using the 70-gene assay, MammaPrint, 48 percent of lymph-node positive breast cancer patients considered clinically high-risk (Clinical-high) and genomic low-risk (MammaPrint-low) had an excellent distant metastasis-free survival at five years in excess of 94 percent.
“Traditionally, physicians have relied on clinical-pathological factors such as age, tumor size, tumor grade, lymph node involvement, and hormone receptor status to make breast cancer treatment decisions,” said Massimo Cristofanilli, MD, Associate Director of Translational Research and Precision Medicine at the Robert H. Lurie Comprehensive Cancer Center, Northwestern University in Chicago. “These findings provide level 1A clinical utility evidence by demonstrating that the detection of low-risk of distant recurrence reported by the MammaPrint test can be safely used in the management of thousands of women by identifying those who can be spared from a toxic and unnecessary treatment.”
MINDACT is a randomized phase III trial that investigates the clinical utility of MammaPrint, when compared (or – “used in conjunction with”) to the standard clinical pathological criteria, for the selection of patients unlikely to benefit from adjuvant chemotherapy. From 2007 to 2011, 6,693 women who had undergone surgery for early-stage breast cancer enrolled in the trial (111 centers in nine countries). Participants were categorized as low or high risk for tumor recurrence in two ways: first, through analysis of tumor tissue using MammaPrint at a central location in Amsterdam; and second, using Adjuvant! Online, a tool that calculates risk of breast cancer recurrence based on common clinical and biological criteria.
Patients characterized in both clinical and genomic assessments as “low- risk” are spared chemotherapy, while patients characterized as “high- risk” are advised chemotherapy. Those with conflicting results are randomized to use either clinical or genomic risk (MammaPrint) evaluation to decide on chemotherapy treatment.
The MINDACT trial is managed and sponsored by the EORTC as part of an extensive and complex partnership in collaboration with Agendia and BIG, and many other academic and commercial partners, as well as patient advocates.
“These MINDACT trial results are a testament that the science of the MammaPrint test is the most robust in the genomic breast recurrence assay market. Agendia will continue to collaborate with pharmaceutical companies, leading cancer centers and academic groups on additional clinical research and in the pursuit of bringing more effective, individualized treatments within reach of cancer patients,” said Mark Straley, Chief Executive Officer at Agendia. “We value the partnership with the EORTC and BIG and it’s a great honor to share this critical milestone.”
Breast cancer is the most frequently diagnosed cancer in women worldwide(1). In 2012, there were nearly 1.7 million new breast cancer cases among women worldwide, accounting for 25 percent of all new cancer cases in women(2).
Imaging of Cancer Cells, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Imaging of Cancer Cells
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Microscope uses nanosecond-speed laser and deep learning to detect cancer cells more efficiently
April 13, 2016
Scientists at the California NanoSystems Institute at UCLA have developed a new technique for identifying cancer cells in blood samples faster and more accurately than the current standard methods.
In one common approach to testing for cancer, doctors add biochemicals to blood samples. Those biochemicals attach biological “labels” to the cancer cells, and those labels enable instruments to detect and identify them. However, the biochemicals can damage the cells and render the samples unusable for future analyses. There are other current techniques that don’t use labeling but can be inaccurate because they identify cancer cells based only on one physical characteristic.
Time-stretch quantitative phase imaging (TS-QPI) and analytics system
The new technique images cells without destroying them and can identify 16 physical characteristics — including size, granularity and biomass — instead of just one.
The new technique combines two components that were invented at UCLA:
A “photonic time stretch” microscope, which is capable of quickly imaging cells in blood samples. Invented by Barham Jalali, professor and Northrop-Grumman Optoelectronics Chair in electrical engineering, it works by taking pictures of flowing blood cells using laser bursts (similar to how a camera uses a flash). Each flash only lasts nanoseconds (billionths of a second) to avoid damage to cells, but that normally means the images are both too weak to be detected and too fast to be digitized by normal instrumentation. The new microscope overcomes those challenges by using specially designed optics that amplify and boost the clarity of the images, and simultaneously slow them down enough to be detected and digitized at a rate of 36 million images per second.
A deep learning computer program, which identifies cancer cells with more than 95 percent accuracy. Deep learning is a form of artificial intelligence that uses complex algorithms to extract patterns and knowledge from rich multidimenstional datasets, with the goal of achieving accurate decision making.
The study was published in the open-access journal Nature Scientific Reports. The researchers write in the paper that the system could lead to data-driven diagnoses by cells’ physical characteristics, which could allow quicker and earlier diagnoses of cancer, for example, and better understanding of the tumor-specific gene expression in cells, which could facilitate new treatments for disease.
The research was supported by NantWorks, LLC.
Abstract of Deep Learning in Label-free Cell Classification
Label-free cell analysis is essential to personalized genomics, cancer diagnostics, and drug development as it avoids adverse effects of staining reagents on cellular viability and cell signaling. However, currently available label-free cell assays mostly rely only on a single feature and lack sufficient differentiation. Also, the sample size analyzed by these assays is limited due to their low throughput. Here, we integrate feature extraction and deep learning with high-throughput quantitative imaging enabled by photonic time stretch, achieving record high accuracy in label-free cell classification. Our system captures quantitative optical phase and intensity images and extracts multiple biophysical features of individual cells. These biophysical measurements form a hyperdimensional feature space in which supervised learning is performed for cell classification. We compare various learning algorithms including artificial neural network, support vector machine, logistic regression, and a novel deep learning pipeline, which adopts global optimization of receiver operating characteristics. As a validation of the enhanced sensitivity and specificity of our system, we show classification of white blood T-cells against colon cancer cells, as well as lipid accumulating algal strains for biofuel production. This system opens up a new path to data-driven phenotypic diagnosis and better understanding of the heterogeneous gene expressions in cells.
references:
Claire Lifan Chen, Ata Mahjoubfar, Li-Chia Tai, Ian K. Blaby, Allen Huang, Kayvan Reza Niazi & Bahram Jalali. Deep Learning in Label-free Cell Classification. Scientific Reports 6, Article number: 21471 (2016); doi:10.1038/srep21471 (open access)
Supplementary Information
Deep Learning in Label-free Cell Classification
Claire Lifan Chen, Ata Mahjoubfar, Li-Chia Tai, Ian K. Blaby, Allen Huang,Kayvan Reza Niazi & Bahram Jalali
Deep learning extracts patterns and knowledge from rich multidimenstional datasets. While it is extensively used for image recognition and speech processing, its application to label-free classification of cells has not been exploited. Flow cytometry is a powerful tool for large-scale cell analysis due to its ability to measure anisotropic elastic light scattering of millions of individual cells as well as emission of fluorescent labels conjugated to cells1,2. However, each cell is represented with single values per detection channels (forward scatter, side scatter, and emission bands) and often requires labeling with specific biomarkers for acceptable classification accuracy1,3. Imaging flow cytometry4,5 on the other hand captures images of cells, revealing significantly more information about the cells. For example, it can distinguish clusters and debris that would otherwise result in false positive identification in a conventional flow cytometer based on light scattering6.
In addition to classification accuracy, the throughput is another critical specification of a flow cytometer. Indeed high throughput, typically 100,000 cells per second, is needed to screen a large enough cell population to find rare abnormal cells that are indicative of early stage diseases. However there is a fundamental trade-off between throughput and accuracy in any measurement system7,8. For example, imaging flow cytometers face a throughput limit imposed by the speed of the CCD or the CMOS cameras, a number that is approximately 2000 cells/s for present systems9. Higher flow rates lead to blurred cell images due to the finite camera shutter speed. Many applications of flow analyzers such as cancer diagnostics, drug discovery, biofuel development, and emulsion characterization require classification of large sample sizes with a high-degree of statistical accuracy10. This has fueled research into alternative optical diagnostic techniques for characterization of cells and particles in flow.
Recently, our group has developed a label-free imaging flow-cytometry technique based on coherent optical implementation of the photonic time stretch concept11. This instrument overcomes the trade-off between sensitivity and speed by using Amplified Time-stretch Dispersive Fourier Transform12,13,14,15. In time stretched imaging16, the object’s spatial information is encoded in the spectrum of laser pulses within a pulse duration of sub-nanoseconds (Fig. 1). Each pulse representing one frame of the camera is then stretched in time so that it can be digitized in real-time by an electronic analog-to-digital converter (ADC). The ultra-fast pulse illumination freezes the motion of high-speed cells or particles in flow to achieve blur-free imaging. Detection sensitivity is challenged by the low number of photons collected during the ultra-short shutter time (optical pulse width) and the drop in the peak optical power resulting from the time stretch. These issues are solved in time stretch imaging by implementing a low noise-figure Raman amplifier within the dispersive device that performs time stretching8,11,16. Moreover, warped stretch transform17,18can be used in time stretch imaging to achieve optical image compression and nonuniform spatial resolution over the field-of-view19. In the coherent version of the instrument, the time stretch imaging is combined with spectral interferometry to measure quantitative phase and intensity images in real-time and at high throughput20. Integrated with a microfluidic channel, coherent time stretch imaging system in this work measures both quantitative optical phase shift and loss of individual cells as a high-speed imaging flow cytometer, capturing 36 million images per second in flow rates as high as 10 meters per second, reaching up to 100,000 cells per second throughput.
Box 1: The pulse train is spatially dispersed into a train of rainbow flashes illuminating the target as line scans. The spatial features of the target are encoded into the spectrum of the broadband optical pulses, each representing a one-dimensional frame. The ultra-short optical pulse illumination freezes the motion of cells during high speed flow to achieve blur-free imaging with a throughput of 100,000 cells/s. The phase shift and intensity loss at each location within the field of view are embedded into the spectral interference patterns using a Michelson interferometer. Box 2: The interferogram pulses were then stretched in time so that spatial information could be mapped into time through time-stretch dispersive Fourier transform (TS-DFT), and then captured by a single pixel photodetector and an analog-to-digital converter (ADC). The loss of sensitivity at high shutter speed is compensated by stimulated Raman amplification during time stretch. Box 3: (a) Pulse synchronization; the time-domain signal carrying serially captured rainbow pulses is transformed into a series of one-dimensional spatial maps, which are used for forming line images. (b) The biomass density of a cell leads to a spatially varying optical phase shift. When a rainbow flash passes through the cells, the changes in refractive index at different locations will cause phase walk-off at interrogation wavelengths. Hilbert transformation and phase unwrapping are used to extract the spatial phase shift. (c) Decoding the phase shift in each pulse at each wavelength and remapping it into a pixel reveals the protein concentration distribution within cells. The optical loss induced by the cells, embedded in the pulse intensity variations, is obtained from the amplitude of the slowly varying envelope of the spectral interferograms. Thus, quantitative optical phase shift and intensity loss images are captured simultaneously. Both images are calibrated based on the regions where the cells are absent. Cell features describing morphology, granularity, biomass, etc are extracted from the images. (d) These biophysical features are used in a machine learning algorithm for high-accuracy label-free classification of the cells.
On another note, surface markers used to label cells, such as EpCAM21, are unavailable in some applications; for example, melanoma or pancreatic circulating tumor cells (CTCs) as well as some cancer stem cells are EpCAM-negative and will escape EpCAM-based detection platforms22. Furthermore, large-population cell sorting opens the doors to downstream operations, where the negative impacts of labels on cellular behavior and viability are often unacceptable23. Cell labels may cause activating/inhibitory signal transduction, altering the behavior of the desired cellular subtypes, potentially leading to errors in downstream analysis, such as DNA sequencing and subpopulation regrowth. In this way, quantitative phase imaging (QPI) methods24,25,26,27 that categorize unlabeled living cells with high accuracy are needed. Coherent time stretch imaging is a method that enables quantitative phase imaging at ultrahigh throughput for non-invasive label-free screening of large number of cells.
In this work, the information of quantitative optical loss and phase images are fused into expert designed features, leading to a record label-free classification accuracy when combined with deep learning. Image mining techniques are applied, for the first time, to time stretch quantitative phase imaging to measure biophysical attributes including protein concentration, optical loss, and morphological features of single cells at an ultrahigh flow rate and in a label-free fashion. These attributes differ widely28,29,30,31 among cells and their variations reflect important information of genotypes and physiological stimuli32. The multiplexed biophysical features thus lead to information-rich hyper-dimensional representation of the cells for label-free classification with high statistical precision.
We further improved the accuracy, repeatability, and the balance between sensitivity and specificity of our label-free cell classification by a novel machine learning pipeline, which harnesses the advantages of multivariate supervised learning, as well as unique training by evolutionary global optimization of receiver operating characteristics (ROC). To demonstrate sensitivity, specificity, and accuracy of multi-feature label-free flow cytometry using our technique, we classified (1) OT-IIhybridoma T-lymphocytes and SW-480 colon cancer epithelial cells, and (2) Chlamydomonas reinhardtii algal cells (herein referred to as Chlamydomonas) based on their lipid content, which is related to the yield in biofuel production. Our preliminary results show that compared to classification by individual biophysical parameters, our label-free hyperdimensional technique improves the detection accuracy from 77.8% to 95.5%, or in other words, reduces the classification inaccuracy by about five times. ……..
Feature Extraction
The decomposed components of sequential line scans form pairs of spatial maps, namely, optical phase and loss images as shown in Fig. 2 (see Section Methods: Image Reconstruction). These images are used to obtain biophysical fingerprints of the cells8,36. With domain expertise, raw images are fused and transformed into a suitable set of biophysical features, listed in Table 1, which the deep learning model further converts into learned features for improved classification.
The new technique combines two components that were invented at UCLA:
A “photonic time stretch” microscope, which is capable of quickly imaging cells in blood samples. Invented by Barham Jalali, professor and Northrop-Grumman Optoelectronics Chair in electrical engineering, it works by taking pictures of flowing blood cells using laser bursts (similar to how a camera uses a flash). Each flash only lasts nanoseconds (billionths of a second) to avoid damage to cells, but that normally means the images are both too weak to be detected and too fast to be digitized by normal instrumentation. The new microscope overcomes those challenges by using specially designed optics that amplify and boost the clarity of the images, and simultaneously slow them down enough to be detected and digitized at a rate of 36 million images per second.
A deep learning computer program, which identifies cancer cells with more than 95 percent accuracy. Deep learning is a form of artificial intelligence that uses complex algorithms to extract patterns and knowledge from rich multidimenstional datasets, with the goal of achieving accurate decision making.
The study was published in the open-access journal Nature Scientific Reports. The researchers write in the paper that the system could lead to data-driven diagnoses by cells’ physical characteristics, which could allow quicker and earlier diagnoses of cancer, for example, and better understanding of the tumor-specific gene expression in cells, which could facilitate new treatments for disease.
The optical loss images of the cells are affected by the attenuation of multiplexed wavelength components passing through the cells. The attenuation itself is governed by the absorption of the light in cells as well as the scattering from the surface of the cells and from the internal cell organelles. The optical loss image is derived from the low frequency component of the pulse interferograms. The optical phase image is extracted from the analytic form of the high frequency component of the pulse interferograms using Hilbert Transformation, followed by a phase unwrapping algorithm. Details of these derivations can be found in Section Methods. Also, supplementary Videos 1 and 2 show measurements of cell-induced optical path length difference by TS-QPI at four different points along the rainbow for OT-II and SW-480, respectively.
Table 1: List of extracted features.
Feature Name Description Category
Figure 3: Biophysical features formed by image fusion.
(a) Pairwise correlation matrix visualized as a heat map. The map depicts the correlation between all major 16 features extracted from the quantitative images. Diagonal elements of the matrix represent correlation of each parameter with itself, i.e. the autocorrelation. The subsets in box 1, box 2, and box 3 show high correlation because they are mainly related to morphological, optical phase, and optical loss feature categories, respectively. (b) Ranking of biophysical features based on their AUCs in single-feature classification. Blue bars show performance of the morphological parameters, which includes diameter along the interrogation rainbow, diameter along the flow direction, tight cell area, loose cell area, perimeter, circularity, major axis length, orientation, and median radius. As expected, morphology contains most information, but other biophysical features can contribute to improved performance of label-free cell classification. Orange bars show optical phase shift features i.e. optical path length differences and refractive index difference. Green bars show optical loss features representing scattering and absorption by the cell. The best performed feature in these three categories are marked in red.
Figure 4: Machine learning pipeline. Information of quantitative optical phase and loss images are fused to extract multivariate biophysical features of each cell, which are fed into a fully-connected neural network.
The neural network maps input features by a chain of weighted sum and nonlinear activation functions into learned feature space, convenient for classification. This deep neural network is globally trained via area under the curve (AUC) of the receiver operating characteristics (ROC). Each ROC curve corresponds to a set of weights for connections to an output node, generated by scanning the weight of the bias node. The training process maximizes AUC, pushing the ROC curve toward the upper left corner, which means improved sensitivity and specificity in classification.
…. How to cite this article: Chen, C. L. et al. Deep Learning in Label-free Cell Classification.
To better characterize the functional context of genomic variations in cancer, researchers developed a new computer algorithm called REVEALER. [UC San Diego Health]
Scientists at the University of California San Diego School of Medicine and the Broad Institute say they have developed a new computer algorithm—REVEALER—to better characterize the functional context of genomic variations in cancer. The tool, described in a paper (“Characterizing Genomic Alterations in Cancer by Complementary Functional Associations”) published in Nature Biotechnology, is designed to help researchers identify groups of genetic variations that together associate with a particular way cancer cells get activated, or how they respond to certain treatments.
REVEALER is available for free to the global scientific community via the bioinformatics software portal GenePattern.org.
“This computational analysis method effectively uncovers the functional context of genomic alterations, such as gene mutations, amplifications, or deletions, that drive tumor formation,” said senior author Pablo Tamayo, Ph.D., professor and co-director of the UC San Diego Moores Cancer Center Genomics and Computational Biology Shared Resource.
Dr. Tamayo and team tested REVEALER using The Cancer Genome Atlas (TCGA), the NIH’s database of genomic information from more than 500 human tumors representing many cancer types. REVEALER revealed gene alterations associated with the activation of several cellular processes known to play a role in tumor development and response to certain drugs. Some of these gene mutations were already known, but others were new.
For example, the researchers discovered new activating genomic abnormalities for beta-catenin, a cancer-promoting protein, and for the oxidative stress response that some cancers hijack to increase their viability.
REVEALER requires as input high-quality genomic data and a significant number of cancer samples, which can be a challenge, according to Dr. Tamayo. But REVEALER is more sensitive at detecting similarities between different types of genomic features and less dependent on simplifying statistical assumptions, compared to other methods, he adds.
“This study demonstrates the potential of combining functional profiling of cells with the characterizations of cancer genomes via next-generation sequencing,” said co-senior author Jill P. Mesirov, Ph.D., professor and associate vice chancellor for computational health sciences at UC San Diego School of Medicine.
Characterizing genomic alterations in cancer by complementary functional associations
Jong Wook Kim, Olga B Botvinnik, Omar Abudayyeh, Chet Birger, et al.
Systematic efforts to sequence the cancer genome have identified large numbers of mutations and copy number alterations in human cancers. However, elucidating the functional consequences of these variants, and their interactions to drive or maintain oncogenic states, remains a challenge in cancer research. We developed REVEALER, a computational method that identifies combinations of mutually exclusive genomic alterations correlated with functional phenotypes, such as the activation or gene dependency of oncogenic pathways or sensitivity to a drug treatment. We used REVEALER to uncover complementary genomic alterations associated with the transcriptional activation of β-catenin and NRF2, MEK-inhibitor sensitivity, and KRAS dependency. REVEALER successfully identified both known and new associations, demonstrating the power of combining functional profiles with extensive characterization of genomic alterations in cancer genomes
Figure 2: REVEALER results for transcriptional activation of β-catenin in cancer.close
(a) This heatmap illustrates the use of the REVEALER approach to find complementary genomic alterations that match the transcriptional activation of β-catenin in cancer. The target profile is a TCF4 reporter that provides an estimate of…
An imaging-based platform for high-content, quantitative evaluation of therapeutic response in 3D tumour models
Jonathan P. Celli, Imran Rizvi, Adam R. Blanden, Iqbal Massodi, Michael D. Glidden, Brian W. Pogue & Tayyaba Hasan
While it is increasingly recognized that three-dimensional (3D) cell culture models recapitulate drug responses of human cancers with more fidelity than monolayer cultures, a lack of quantitative analysis methods limit their implementation for reliable and routine assessment of emerging therapies. Here, we introduce an approach based on computational analysis of fluorescence image data to provide high-content readouts of dose-dependent cytotoxicity, growth inhibition, treatment-induced architectural changes and size-dependent response in 3D tumour models. We demonstrate this approach in adherent 3D ovarian and pancreatic multiwell extracellular matrix tumour overlays subjected to a panel of clinically relevant cytotoxic modalities and appropriately designed controls for reliable quantification of fluorescence signal. This streamlined methodology reads out the high density of information embedded in 3D culture systems, while maintaining a level of speed and efficiency traditionally achieved with global colorimetric reporters in order to facilitate broader implementation of 3D tumour models in therapeutic screening.
The attrition rates for preclinical development of oncology therapeutics are particularly dismal due to a complex set of factors which includes 1) the failure of pre-clinical models to recapitulate determinants of in vivo treatment response, and 2) the limited ability of available assays to extract treatment-specific data integral to the complexities of therapeutic responses1,2,3. Three-dimensional (3D) tumour models have been shown to restore crucial stromal interactions which are missing in the more commonly used 2D cell culture and that influence tumour organization and architecture4,5,6,7,8, as well as therapeutic response9,10, multicellular resistance (MCR)11,12, drug penetration13,14, hypoxia15,16, and anti-apoptotic signaling17. However, such sophisticated models can only have an impact on therapeutic guidance if they are accompanied by robust quantitative assays, not only for cell viability but also for providing mechanistic insights related to the outcomes. While numerous assays for drug discovery exist18, they are generally not developed for use in 3D systems and are often inherently unsuitable. For example, colorimetric conversion products have been noted to bind to extracellular matrix (ECM)19 and traditional colorimetric cytotoxicity assays reduce treatment response to a single number reflecting a biochemical event that has been equated to cell viability (e.g. tetrazolium salt conversion20). Such approaches fail to provide insight into the spatial patterns of response within colonies, morphological or structural effects of drug response, or how overall culture viability may be obscuring the status of sub-populations that are resistant or partially responsive. Hence, the full benefit of implementing 3D tumour models in therapeutic development has yet to be realized for lack of analytical methods that describe the very aspects of treatment outcome that these systems restore.
Motivated by these factors, we introduce a new platform for quantitative in situ treatment assessment (qVISTA) in 3D tumour models based on computational analysis of information-dense biological image datasets (bioimage-informatics)21,22. This methodology provides software end-users with multiple levels of complexity in output content, from rapidly-interpreted dose response relationships to higher content quantitative insights into treatment-dependent architectural changes, spatial patterns of cytotoxicity within fields of multicellular structures, and statistical analysis of nodule-by-nodule size-dependent viability. The approach introduced here is cognizant of tradeoffs between optical resolution, data sampling (statistics), depth of field, and widespread usability (instrumentation requirement). Specifically, it is optimized for interpretation of fluorescent signals for disease-specific 3D tumour micronodules that are sufficiently small that thousands can be imaged simultaneously with little or no optical bias from widefield integration of signal along the optical axis of each object. At the core of our methodology is the premise that the copious numerical readouts gleaned from segmentation and interpretation of fluorescence signals in these image datasets can be converted into usable information to classify treatment effects comprehensively, without sacrificing the throughput of traditional screening approaches. It is hoped that this comprehensive treatment-assessment methodology will have significant impact in facilitating more sophisticated implementation of 3D cell culture models in preclinical screening by providing a level of content and biological relevance impossible with existing assays in monolayer cell culture in order to focus therapeutic targets and strategies before costly and tedious testing in animal models.
Using two different cell lines and as depicted in Figure 1, we adopt an ECM overlay method pioneered originally for 3D breast cancer models23, and developed in previous studies by us to model micrometastatic ovarian cancer19,24. This system leads to the formation of adherent multicellular 3D acini in approximately the same focal plane atop a laminin-rich ECM bed, implemented here in glass-bottom multiwell imaging plates for automated microscopy. The 3D nodules resultant from restoration of ECM signaling5,8, are heterogeneous in size24, in contrast to other 3D spheroid methods, such as rotary or hanging drop cultures10, in which cells are driven to aggregate into uniformly sized spheroids due to lack of an appropriate substrate to adhere to. Although the latter processes are also biologically relevant, it is the adherent tumour populations characteristic of advanced metastatic disease that are more likely to be managed with medical oncology, which are the focus of therapeutic evaluation herein. The heterogeneity in 3D structures formed via ECM overlay is validated here by endoscopic imaging ofin vivo tumours in orthotopic xenografts derived from the same cells (OVCAR-5).
Figure 1: A simplified schematic flow chart of imaging-based quantitative in situ treatment assessment (qVISTA) in 3D cell culture.
(This figure was prepared in Adobe Illustrator® software by MD Glidden, JP Celli and I Rizvi). A detailed breakdown of the image processing (Step 4) is provided in Supplemental Figure 1.
A critical component of the imaging-based strategy introduced here is the rational tradeoff of image-acquisition parameters for field of view, depth of field and optical resolution, and the development of image processing routines for appropriate removal of background, scaling of fluorescence signals from more than one channel and reliable segmentation of nodules. In order to obtain depth-resolved 3D structures for each nodule at sub-micron lateral resolution using a laser-scanning confocal system, it would require ~ 40 hours (at approximately 100 fields for each well with a 20× objective, times 1 minute/field for a coarse z-stack, times 24 wells) to image a single plate with the same coverage achieved in this study. Even if the resources were available to devote to such time-intensive image acquisition, not to mention the processing, the optical properties of the fluorophores would change during the required time frame for image acquisition, even with environmental controls to maintain culture viability during such extended imaging. The approach developed here, with a mind toward adaptation into high throughput screening, provides a rational balance of speed, requiring less than 30 minutes/plate, and statistical rigour, providing images of thousands of nodules in this time, as required for the high-content analysis developed in this study. These parameters can be further optimized for specific scenarios. For example, we obtain the same number of images in a 96 well plate as for a 24 well plate by acquiring only a single field from each well, rather than 4 stitched fields. This quadruples the number conditions assayed in a single run, at the expense of the number of nodules per condition, and therefore the ability to obtain statistical data sets for size-dependent response, Dfrac and other segmentation-dependent numerical readouts.
We envision that the system for high-content interrogation of therapeutic response in 3D cell culture could have widespread impact in multiple arenas from basic research to large scale drug development campaigns. As such, the treatment assessment methodology presented here does not require extraordinary optical instrumentation or computational resources, making it widely accessible to any research laboratory with an inverted fluorescence microscope and modestly equipped personal computer. And although we have focused here on cancer models, the methodology is broadly applicable to quantitative evaluation of other tissue models in regenerative medicine and tissue engineering. While this analysis toolbox could have impact in facilitating the implementation of in vitro 3D models in preclinical treatment evaluation in smaller academic laboratories, it could also be adopted as part of the screening pipeline in large pharma settings. With the implementation of appropriate temperature controls to handle basement membranes in current robotic liquid handling systems, our analyses could be used in ultra high-throughput screening. In addition to removing non-efficacious potential candidate drugs earlier in the pipeline, this approach could also yield the additional economic advantage of minimizing the use of costly time-intensive animal models through better estimates of dose range, sequence and schedule for combination regimens.
Microscope Uses AI to Find Cancer Cells More Efficiently
Scientists at the California NanoSystems Institute at UCLA have developed a new technique for identifying cancer cells in blood samples faster and more accurately than the current standard methods.
In one common approach to testing for cancer, doctors add biochemicals to blood samples. Those biochemicals attach biological “labels” to the cancer cells, and those labels enable instruments to detect and identify them. However, the biochemicals can damage the cells and render the samples unusable for future analyses.
There are other current techniques that don’t use labeling but can be inaccurate because they identify cancer cells based only on one physical characteristic.
The new technique images cells without destroying them and can identify 16 physical characteristics — including size, granularity and biomass — instead of just one. It combines two components that were invented at UCLA: a photonic time stretch microscope, which is capable of quickly imaging cells in blood samples, and a deep learning computer program that identifies cancer cells with over 95 percent accuracy.
Deep learning is a form of artificial intelligence that uses complex algorithms to extract meaning from data with the goal of achieving accurate decision making.
The study, which was published in the journal Nature Scientific Reports, was led by Barham Jalali, professor and Northrop-Grumman Optoelectronics Chair in electrical engineering; Claire Lifan Chen, a UCLA doctoral student; and Ata Mahjoubfar, a UCLA postdoctoral fellow.
Photonic time stretch was invented by Jalali, and he holds a patent for the technology. The new microscope is just one of many possible applications; it works by taking pictures of flowing blood cells using laser bursts in the way that a camera uses a flash. This process happens so quickly — in nanoseconds, or billionths of a second — that the images would be too weak to be detected and too fast to be digitized by normal instrumentation.
The new microscope overcomes those challenges using specially designed optics that boost the clarity of the images and simultaneously slow them enough to be detected and digitized at a rate of 36 million images per second. It then uses deep learning to distinguish cancer cells from healthy white blood cells.
“Each frame is slowed down in time and optically amplified so it can be digitized,” Mahjoubfar said. “This lets us perform fast cell imaging that the artificial intelligence component can distinguish.”
Normally, taking pictures in such minuscule periods of time would require intense illumination, which could destroy live cells. The UCLA approach also eliminates that problem.
“The photonic time stretch technique allows us to identify rogue cells in a short time with low-level illumination,” Chen said.
The researchers write in the paper that the system could lead to data-driven diagnoses by cells’ physical characteristics, which could allow quicker and earlier diagnoses of cancer, for example, and better understanding of the tumor-specific gene expression in cells, which could facilitate new treatments for disease. ….. see also http://www.nature.com/article-assets/npg/srep/2016/160315/srep21471/images_hires/m685/srep21471-f1.jpg
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….
The nanoparticles start out relatively large (100 nm) (large blue circle, upper left) to enable smooth transport into the tumor through leaky blood vessels. Then, in acidic conditions found close to tumors, the particles discharge “bomblets” (right, small blue circles) just 5 nm in size. Once inside tumor cells, a second chemical step activates the platinum-based drug cisplatin (bottom) to attack the cancer directly. (credit: Emory Health Sciences)
Scientists have devised a triple-stage stealth “cluster bomb” system for delivering the anti-cancer chemotherapy drug cisplatin, using nanoparticles designed to break up when they reach a tumor:
The nanoparticles start out relatively large — 100 nanometers wide — so that they can move through the bloodstream and smoothly transport into the tumor through leaky blood vessels.
As they detect acidic conditions close to tumors, the nanoparticles discharge “bomblets” just 5 nanometers in size to penetrate tumor cells.
Once inside tumor cells, the bomblets release the platinum-based cisplatin, which kills by crosslinking and damaging DNA.
Doctors have used cisplatin to fight several types of cancer for decades, but toxic side effects — to the kidneys, nerves and inner ear — have limited its effectiveness. But in research with three different mouse tumor models*, the researchers have now shown that their nanoparticles can enhance cisplatin drug accumulation in tumor tissues for several types of cancer.
Details of the research — by teams led by professor Jun Wang, PhD, at the University of Science and Technology of China and by professor Shuming Nie, PhD, in the Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory — were published this week in the journal PNAS.
* When mice bearing human pancreatic tumors were given the same doses of free cisplatin or cisplatin clothed in pH-sensitive nanoparticles, the level of platinum in tumor tissues was seven times higher with the nanoparticles. This suggests the possibility that nanoparticle delivery of a limited dose of cisplatin could restrain the toxic side effects during cancer treatment.
The researchers also showed that the nanoparticles were effective against a cisplatin-resistant lung cancer model and an invasive metastatic breast cancer model in mice. In the lung cancer model, a dose of free cisplatin yielded just 10 percent growth inhibition, while the same dose clothed in nanoparticles yielded 95 percent growth inhibition, the researchers report. In the metastatic breast cancer model, treating mice with cisplatin clothed in nanoparticles prolonged animal survival by weeks; 50 percent of the mice were surviving at 54 days with nanoparticles compared with 37 days for the same dose of free cisplatin.
Abstract of Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy
A principal goal of cancer nanomedicine is to deliver therapeutics effectively to cancer cells within solid tumors. However, there are a series of biological barriers that impede nanomedicine from reaching target cells. Here, we report a stimuli-responsive clustered nanoparticle to systematically overcome these multiple barriers by sequentially responding to the endogenous attributes of the tumor microenvironment. The smart polymeric clustered nanoparticle (iCluster) has an initial size of ∼100 nm, which is favorable for long blood circulation and high propensity of extravasation through tumor vascular fenestrations. Once iCluster accumulates at tumor sites, the intrinsic tumor extracellular acidity would trigger the discharge of platinum prodrug-conjugated poly(amidoamine) dendrimers (diameter ∼5 nm). Such a structural alteration greatly facilitates tumor penetration and cell internalization of the therapeutics. The internalized dendrimer prodrugs are further reduced intracellularly to release cisplatin to kill cancer cells. The superior in vivo antitumor activities of iCluster are validated in varying intractable tumor models including poorly permeable pancreatic cancer, drug-resistant cancer, and metastatic cancer, demonstrating its versatility and broad applicability.
The facts suggest that big pharma represents only a few companies in most fields of disease. They spend an enormous amount of money in lobbying congress and doctors to get them to do their bidding.They wouldn’t spend the money if they didn’t need to do so.The profit motive is central with patient well being only being practiced if it pays off.Cancer is a superb example, with new drugs being offered usually at astronomical prices in this country. Like wise the FDA is controlled by them and it is in their best interests to make the cost of developing new drugs outrageously expensive.Only big pharma can afford to get new drugs approved.
After the phase 3 trials are completed usually the documentation to ask for approval to market a drug is at least 100,000 pages long. The legal talent needed to compile such documents ( and this is only one of many documents produced in the process) is extremely expensive. The time taken for approval stretches into many years and then the drugs are often not approved.(only a small percentage are approved).
Antibiotics were one example of a group of drugs that really did cure many diseases. Big pharma found it didn’t pay to develop new antibiotics because the treatment was short and so successful that patients used the drugs only for a short time.
Over time, as Alexander Fleming forsaw, the bacteria would develop resistance, especially if they were extensively used indiscriminantly. Now many dangerous bacteria are resistant to many or all antibiotics and there is no treatment available. Since bacteria can pass this resistance to specific antibiotics to almost any species of bacteria, its only a matter of time before we will be back in the pre-antibiotic era.
SINCE IT DOES NOT PAY FOR BIG PHARMA TO DEVELOP NEW ANTIBIOTICS THEY ARE NOW NOT DOING SO AT ALL.
…..
“In the metastatic breast cancer model, treating mice with cisplatin clothed in nanoparticles prolonged animal survival by weeks; 50 percent of the mice were surviving at 54 days with nanoparticles compared with 37 days for the same dose of free cisplatin.”
I’m not so convinced after all. But this is perfectly in line with big pharma goals. Only an idiot would kill its main source of income.
…….
It is almost impossible to set up a conspiracy against big pharma’s abusive practices.Every avenue their high priced lawyers can think of to stop budding conspiracies has been blocked by law where possible. One possible road might be to do research and development in other countries outside US legal juristiction, however most drugs without FDA approval can and are stopped at the border and confiscated even if as in Canada the same drug produced in the US is being manufactured in Canada.Almost certainly Cisplatin is under patent in the US and the patent holder has the right to refuse the use of the drug for any reason they want, including being used in this cluster bomb drug. The manufacturer is almost certainly making huge profits from selling Cisplatin and I doubt they want to see a cheap drug cure many cancers. I guess the only way to go is to try and turn to a country like India.A number of cancer drugs were being sold by US patent holders at wholesale prices that were to high for most Indians. The government of India refused to allow these companies to patent their medicines in India and forced them to license the drugs and much cheaper prices.Most US patents are not operative in India, they can produce US style insulin pumps at a fraction of our cost as they can in China and Vietnam or Mexico. It would be difficult to send these pumps to buyers in the US from India but by shipping them from another country, say Canada or Mexico most would make it past customs. As for Cancer treatment, India and china have some very fine trained biochemist and doctors, who could easily apply many of the immunological treatments against cancer. All arms of the immune system have been used to produce miracle treatments that have cured some patients that were on their death beds.The treatments can be tested carefully in these countries, and improved by any methods including some I have suggested.By advertising in the US to cancer patients that they can inexpensively have these working treatments cheaply as a medical tourist, it is only a matter of time before they will cure the disease wholesale and break the medical industrial complex down. As far as generics that are not being produced here, by setting up a non profit corporation that produces any and all drugs that come off patent as a goal, at the cheapest price less a reasonable markup for cost of manufacture etc. one by one they will end the abuse of not producing or overpricing generics.
………
Significance
Successively overcoming a series of biological barriers that cancer nanotherapeutics would encounter upon intravenous administration is required for achieving positive treatment outcomes. A hurdle to this goal is the inherently unfavorable tumor penetration of nanoparticles due to their relatively large sizes. We developed a stimuli-responsive clustered nanoparticle (iCluster) and justified that its adaptive alterations of physicochemical properties (e.g. size, zeta potential, and drug release rate) in accordance with the endogenous stimuli of the tumor microenvironment made possible the ultimate overcoming of these barriers, especially the bottleneck of tumor penetration. Results in varying intractable tumor models demonstrated significantly improved antitumor efficacy of iCluster than its control groups, demonstrating that overcoming these delivery barriers can be achieved by innovative nanoparticle design.
Researchers use optogenetic light to block tumor development
Uses light-triggered bioelectric current
Tufts University biologists have demonstrated (using a frog model*) for the first time that it is possible to prevent tumors from forming (and to normalize tumors after they have formed) by using optogenetics (light) to control bioelectrical signalling among cells.
Light/bioelectric control of tumors
Virtually all healthy cells maintain a more negative voltage in the cell interior compared with the cell exterior. But the opening and closing of ion channels in the cell membrane can cause the voltage to become more positive (depolarizing the cell) or more negative (polarizing the cell). That makes it possible to detect tumors by their abnormal bioelectrical signature before they are otherwise apparent.
The study was published online in an open-access paper in Oncotarget on March 16.
The use of light to control ion channels has been a ground-breaking tool in research on the nervous system and brain, but optogenetics had not yet been applied to cancer.
The researchers first injected cells in Xenopus laevis (frog) embryos with RNA that encoded a mutant RAS oncogene known to cause cancer-like growths.
The researchers then used blue light to activate positively charged ion channels,which induced an electric current that caused the cells to go from a cancer-like depolarized state to a normal, more negative polarized state. The did the same with a green light-activated proton pump, Archaerhodopsin (Arch). Activation of both agents significantly lowered the incidence of tumor formation and also increased the frequency with which tumors regressed into normal tissue.
“Discovering new ways to specifically control this bioelectrical signaling could be an important path towards new biomedical approaches to cancer. This provides proof of principle for a novel class of therapies which use light to override the action of oncogenic mutations,” said Levin. “Using light to specifically target tumors would avoid subjecting the whole body to toxic chemotherapy or similar reagents.”
This work was supported by the G. Harold and Leila Y. Mathers Charitable Foundation.
* Frogs are a good model for basic science research into cancer because tumors in frogs and mammals share many of the same characteristics. These include rapid cell division, tissue disorganization, increased vascular growth, invasiveness and cells that have an abnormally positive internal electric voltage.
Abstract of Use of genetically encoded, light-gated ion translocators to control tumorigenesis
It has long been known that the resting potential of tumor cells is depolarized relative to their normal counterparts. More recent work has provided evidence that resting potential is not just a readout of cell state: it regulates cell behavior as well. Thus, the ability to control resting potential in vivo would provide a powerful new tool for the study and treatment of tumors, a tool capable of revealing living-state physiological information impossible to obtain using molecular tools applied to isolated cell components. Here we describe the first use of optogenetics to manipulate ion-flux mediated regulation of membrane potential specifically to prevent and cause regression of oncogene-induced tumors. Injection of mutant-KRAS mRNA induces tumor-like structures with many documented similarities to tumors, in Xenopus tadpoles. We show that expression and activation of either ChR2D156A, a blue-light activated cation channel, or Arch, a green-light activated proton pump, both of which hyperpolarize cells, significantly lowers the incidence of KRAS tumor formation. Excitingly, we also demonstrate that activation of co-expressed light-activated ion translocators after tumor formation significantly increases the frequency with which the tumors regress in a process called normalization. These data demonstrate an optogenetic approach to dissect the biophysics of cancer. Moreover, they provide proof-of-principle for a novel class of interventions, directed at regulating cell state by targeting physiological regulators that can over-ride the presence of mutations.
A schematic representation of the miniaturized gold-aluminum oxide hyperbolic metamaterial (HMM) sensor device with a fluid flow channel, showing a scanning electron microscope (SEM) image [gray inset] of the 2D subwavelength gold diffraction grating on top of the hyperbolic metamaterials layers (scale bar, 2 µm) (credit: Kandammathe Valiyaveedu Sreekanth et al./Nature Materials
An optical sensor that’s 1 million times more sensitive than the current best available has been developed by Case Western Reserve University researchers. Based on nanostructured metamaterials, it can identify a single lightweight molecule in a highly dilute solution.The research goal is to provide oncologists a way to detect a single molecule of an enzyme produced by circulating cancer cells. That could allow doctors to diagnose and monitor patients with certain cancers far earlier than possible today.
“The prognosis of many cancers depends on the stage of the cancer at diagnosis,” said Giuseppe “Pino” Strangi, professor of physics at Case Western Reserve and research leader. “Very early, most circulating tumor cells express proteins of a very low molecular weight, less than 500 Daltons,” Strangi explained. “These proteins are usually too small and in too low a concentration to detect with current test methods, yielding false negative results.
“With this platform, we’ve detected proteins of 244 Daltons, which should enable doctors to detect cancers earlier — we don’t know how much earlier yet,” he said. “This biosensing platform may help to unlock the next era of initial cancer detection.”
The researchers believe the sensing technology will also be useful in diagnosing and monitoring other diseases.
A biological sieve
The nanosensor, which fits in the palm of a hand, acts like a biological sieve, capable of isolating a small protein molecule weighing less than 800 quadrillionths of a nanogram from an extremely dilute solution.
To make the device so sensitive, Strangi’s team faced two long-standing barriers: Light waves cannot detect objects smaller than their own physical dimensions (about 500 nanometers, depending on wavelength). And molecules in dilute solutions float in Brownian (random) motion and are unlikely to land on the sensor’s surface.
The solution was to use a microfluidic channel to restrict the molecules’ ability to float around and a plasmon-based metamaterial made of 16 nanostructured layers of reflective and conductive gold and transparent aluminum oxide, a dielectric, each 10s of atoms thick. Light directed onto and through the layers is concentrated into a very small volume much smaller than the wavelength of light.*
“It’s extremely sensitive,” Strangi said. “When a small molecule lands on the surface, it results in a large local modification, causing the light to shift.” Depending on the size of the molecule, the reflecting light shifts different amounts. The researchers hope to learn to identify specific biomarker and other molecules for different cancers by their light shifts.
To add specificity to the sensor, the team added a layer of trap molecules — molecules that bind specifically with the molecules they hunt. In tests, the researchers used two trap molecules to catch two different biomolecules: bovine serum albumin, with a molecular weight of 66,430 Daltons, and biotin, with a molecular weight of 244 Daltons. Each produced a signature light shift.
Other researchers have reported using plasmon-based biosensors to detect biotin in solutions at concentrations ranging from more than 100 micromoles per liter to 10 micromoles per liter. This device proved 1 million times more sensitive, finding and identifying biotin at a concentration of 10 picomoles per liter.
Testing and clinical use in process
Strangi’s lab is working with other oncologists worldwide to test the device and begin moving the sensor toward clinical use.
In Cleveland, Strangi and Nima Sharifi, MD, co-leader of the Genitourinary Cancer Program for the Case Comprehensive Cancer Center, have begun testing the sensor with proteins related to prostate cancers.
“For some cancers, such as colorectal and pancreatic cancer, early detection is essential,” said Sharifi, who is also the Kendrick Family Chair for Prostate Cancer Research at Cleveland Clinic. “High sensitivity detection of cancer-specific proteins in blood should enable detection of tumors when they are at an earlier disease stage.
“This new sensing technology may help us not only detect cancers, but what subset of cancer, what’s driving its growth and spread, and what it’s sensitive to,” he said. “The sensor, for example, may help us determine markers of aggressive prostate cancers, which require treatments, or indolent forms that don’t.”
The research is published online in the journal Nature Materials.
* The top gold layer is perforated with holes, creating a grating that diffuses light shone on the surface into two dimensions. The incoming light, which is several hundreds of nanometers in wavelength, appears to be confined and concentrated in a few nanometers at the interface between the gold and the dielectric layer. As the light strikes the sensing area, it excites free electrons causing them to oscillate and generate a highly confined propagating surface wave, called a surface plasmon polariton. This propagating surface wave will in turn excite a bulk wave propagating across the sensing platform. The presence of the waves cause deep sharp dips in the spectrum of reflecting light. The combination and the interplay of surface plasmon and bulk plasmon waves are what make the sensor so sensitive. Strangi said. By exciting these waves through the eight bilayers of the metamaterial, they create remarkably sharp resonant modes. Extremely sharp and sensitive resonances can be used to detect smaller objects.
Abstract of Extreme sensitivity biosensing platform based on hyperbolic metamaterials
Optical sensor technology offers significant opportunities in the field of medical research and clinical diagnostics, particularly for the detection of small numbers of molecules in highly diluted solutions. Several methods have been developed for this purpose, including label-free plasmonic biosensors based on metamaterials. However, the detection of lower-molecular-weight (<500 Da) biomolecules in highly diluted solutions is still a challenging issue owing to their lower polarizability. In this context, we have developed a miniaturized plasmonic biosensor platform based on a hyperbolic metamaterial that can support highly confined bulk plasmon guided modes over a broad wavelength range from visible to near infrared. By exciting these modes using a grating-coupling technique, we achieved different extreme sensitivity modes with a maximum of 30,000 nm per refractive index unit (RIU) and a record figure of merit (FOM) of 590. We report the ability of the metamaterial platform to detect ultralow-molecular-weight (244 Da) biomolecules at picomolar concentrations using a standard affinity model streptavidin–biotin.
The late Cambridge Mayor Alfred Vellucci welcomed Life Sciences Labs to Cambridge, MA – June 1976
Reporter: Aviva Lev-Ari, PhD, RN
How Cambridge became the Life Sciences Capital
Worth watching is the video below, which captures the initial Cambridge City Council hearing on recombinant DNA research from June 1976. The first speaker is the late Cambridge mayor Alfred Vellucci.
Vellucci hoped to pass a two-year moratorium on gene splicing in Cambridge. Instead, the council passed a three-month moratorium, and created a board of nine Cambridge citizens — including a nun and a nurse — to explore whether the work should be allowed, and if so, what safeguards would be necessary. A few days after the board was created, the pro and con tables showed up at the Kendall Square marketplace.
At the time, says Phillip Sharp, an MIT professor, Cambridge felt like a manufacturing town that had seen better days. He recalls being surrounded by candy, textile, and leather factories. Sharp hosted the citizens review committee at MIT, explaining what the research scientists there planned to do. “I think we built a relationship,” he says.
By early 1977, the citizens committee had proposed a framework to ensure that any DNA-related experiments were done under fairly stringent safety controls, and Cambridge became the first city in the world to regulate research using genetic material.


















{kind=link}
{kind=link}
{kind=link}
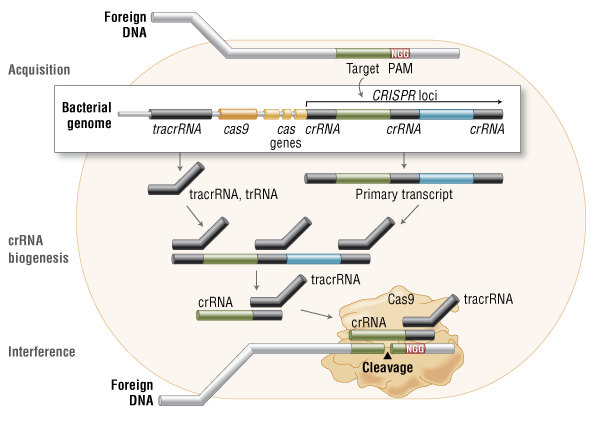{kind=link}
{kind=link}
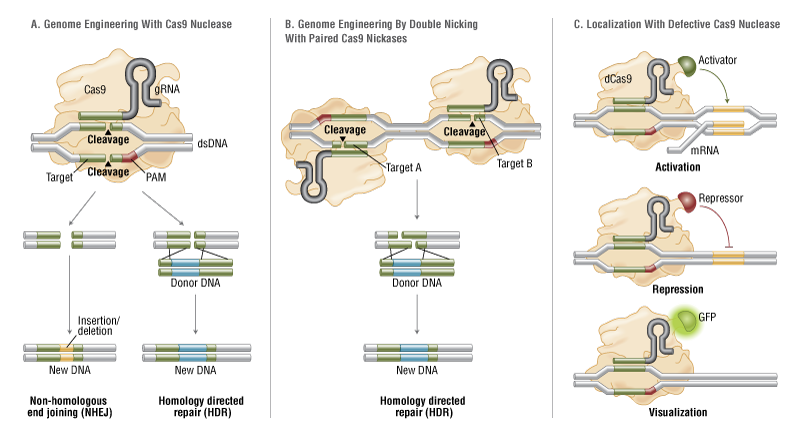{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
It is almost impossible to set up a conspiracy against big pharma’s abusive practices.Every avenue their high priced lawyers can think of to stop budding conspiracies has been blocked by law where possible. One possible road might be to do research and development in other countries outside US legal juristiction, however most drugs without FDA approval can and are stopped at the border and confiscated even if as in Canada the same drug produced in the US is being manufactured in Canada.Almost certainly Cisplatin is under patent in the US and the patent holder has the right to refuse the use of the drug for any reason they want, including being used in this cluster bomb drug. The manufacturer is almost certainly making huge profits from selling Cisplatin and I doubt they want to see a cheap drug cure many cancers. I guess the only way to go is to try and turn to a country like India.A number of cancer drugs were being sold by US patent holders at wholesale prices that were to high for most Indians. The government of India refused to allow these companies to patent their medicines in India and forced them to license the drugs and much cheaper prices.Most US patents are not operative in India, they can produce US style insulin pumps at a fraction of our cost as they can in China and Vietnam or Mexico. It would be difficult to send these pumps to buyers in the US from India but by shipping them from another country, say Canada or Mexico most would make it past customs. As for Cancer treatment, India and china have some very fine trained biochemist and doctors, who could easily apply many of the immunological treatments against cancer. All arms of the immune system have been used to produce miracle treatments that have cured some patients that were on their death beds.The treatments can be tested carefully in these countries, and improved by any methods including some I have suggested.By advertising in the US to cancer patients that they can inexpensively have these working treatments cheaply as a medical tourist, it is only a matter of time before they will cure the disease wholesale and break the medical industrial complex down. As far as generics that are not being produced here, by setting up a non profit corporation that produces any and all drugs that come off patent as a goal, at the cheapest price less a reasonable markup for cost of manufacture etc. one by one they will end the abuse of not producing or overpricing generics.
………
Significance
Successively overcoming a series of biological barriers that cancer nanotherapeutics would encounter upon intravenous administration is required for achieving positive treatment outcomes. A hurdle to this goal is the inherently unfavorable tumor penetration of nanoparticles due to their relatively large sizes. We developed a stimuli-responsive clustered nanoparticle (iCluster) and justified that its adaptive alterations of physicochemical properties (e.g. size, zeta potential, and drug release rate) in accordance with the endogenous stimuli of the tumor microenvironment made possible the ultimate overcoming of these barriers, especially the bottleneck of tumor penetration. Results in varying intractable tumor models demonstrated significantly improved antitumor efficacy of iCluster than its control groups, demonstrating that overcoming these delivery barriers can be achieved by innovative nanoparticle design.
http://www.pnas.org/content/early/2016/03/23/1522080113.full