Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Reporter and Original Article Co-Author: Amandeep Kaur, B.Sc. , M.Sc.
Abstract Since its inception in late 2019, SARS-CoV-2 has evolved resulting in emergence of various variants in different countries. These variants have spread worldwide resulting in devastating second wave of COVID-19 pandemic in many countries including India since the beginning of 2021. To control this pandemic continuous mutational surveillance and genomic epidemiology of circulating strains is very important. In this study, we performed mutational analysis of the protein coding genes of SARS-CoV-2 strains (n=2000) collected during January 2021 to March 2021. Our data revealed the emergence of a new variant in West Bengal, India, which is characterized by the presence of 11 co-existing mutations including D614G, P681H and V1230L in S-glycoprotein. This new variant was identified in 70 out of 412 sequences submitted from West Bengal. Interestingly, among these 70 sequences, 16 sequences also harbored E484K in the S glycoprotein. Phylogenetic analysis revealed strains of this new variant emerged from GR clade (B.1.1) and formed a new cluster. We propose to name this variant as GRL or lineage B.1.1/S:V1230L due to the presence of V1230L in S glycoprotein along with GR clade specific mutations. Co-occurrence of P681H, previously observed in UK variant, and E484K, previously observed in South African variant and California variant, demonstrates the convergent evolution of SARS-CoV-2 mutation. V1230L, present within the transmembrane domain of S2 subunit of S glycoprotein, has not yet been reported from any country. Substitution of valine with more hydrophobic amino acid leucine at position 1230 of the transmembrane domain, having role in S protein binding to the viral envelope, could strengthen the interaction of S protein with the viral envelope and also increase the deposition of S protein to the viral envelope, and thus positively regulate virus infection. P618H and E484K mutation have already been demonstrated in favor of increased infectivity and immune invasion respectively. Therefore, the new variant having G614G, P618H, P1230L and E484K is expected to have better infectivity, transmissibility and immune invasion characteristics, which may pose additional threat along with B.1.617 in the ongoing COVID-19 pandemic in India.
Study: Emergence of a new SARS-CoV-2 variant from GR clade with a novel S glycoprotein mutation V1230L in West Bengal, India
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….
Personalized genomics is the next research in cancer. However not all the mutations found in this disease are targeted equally by the researchers.
“Genetics is changing oncology for the good,” says Benjamin Kipp, an expert in clinical genetics at the Mayo Clinic in Rochester, Minn. “But overinterpretation can harm the patient.”
Genetic profile of tumors offers opportunities for both cancer diagnostics and treatment. For example, bowel cancer tumors with mutations in the KRAS gene respond poorly to the drug Cetuximab, while the drug Vemurafenib works only in melanomas that have a particular mutation in BRAF gene. But such genetic testing can be misleading if it isn’t conducted alongside tests of healthy cells from the same person, says oncologist Victor Velculescu of the Johns Hopkins University School of Medicine. He led a vast analysis comparing the genetic profiles of tumors and normal tissue of more than 800 cancer patients and found that nearly two-thirds of mutations in the studied tumors — many of which might be used to guide treatment — also showed up in patients’ healthy tissues . Thus, there are many “false positive” mutations that appear to contribute to cancer but in reality they are showing up elsewhere in an individual’s health tissue. Sampling both tumor and healthy tissues might provide a way to sort out truly cancerous mutations, the scientists report.
A team of researchers in Baltimore tested tumor tissue and healthy tissue from 815 patients who had various cancers. Using only the tumor analysis, the tests spotted an average of 382 mutations per case that appeared associated with cancer. But nearly two-thirds of these variations, on average, also showed up in healthy tissues, suggesting that they weren’t driving the cancer, the authors report in the April 15 Science Translational Medicine.
For those patients, the mutations were probably just benign variants unrelated to the cancer. Analyzing healthy tissue can also reveal whether mutations found in tumors are heritable or not, Velculescu says, which is important for deciding whether a cancer patient’s family should receive genetic counseling.
Even mutation that have been linked to the cancer not always manifest as cancer making this interpretation even worse. A study published in May examining eyelid skin discovered numerous cancer-associated mutations in normal, healthy patches of the skin. Researchers had previously thought that the types of mutations that fuel tumor growth were rare and happened just before a cell becomes cancerous. But a study of the eyelids of four people who don’t have cancer reveals that such mutations “are staggeringly common in normal skin,” says Philip Jones, a clinical scientist at the University of Cambridge. Thus, Jones and his colleagues collected 234 skin samples from four people ages 55 to 73 who had plastic surgery to correct droopy eyelids. DNA sequencing showed that about 20 percent of the skin cells had mutations in the NOTCH1 gene, the team reports in the May 22 Science. When mutated, that gene is a driving force in some cancers, including skin cancers called squamous cell carcinomas.
As genetic testing of tumors becomes more widespread, best practices will emerge, as will a better understanding of the disease. “We are trying to change the way we look at cancer,” says Sameek Roychowdhury, a medical oncologist at the Ohio State University Comprehensive Cancer Center in Columbus. “But we are just seeing the tip of the iceberg.”
Conclusion: We have to be really careful when are making interpretation of mutated genes that may cause cancer and identify those mutations in both healthy and cancer tissues as well as find the expression of those genes that may lead to cancer, being said that only cancer mutations that are expressed may have an importance in cancer appearance.
Bioinformatic Tools for Cancer Mutational Analysis: COSMIC and Beyond, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Bioinformatic Tools for Cancer Mutational Analysis: COSMIC and Beyond
Curator: Stephen J. Williams, Ph.D.
Updated 7/26/2019
Updated 04/27/2019
Signatures of Mutational Processes in Human Cancer (from COSMIC)
The genomic landscape of cancer. The COSMIC database has a fully curated and annotated database of recurrent genetic mutations founds in various cancers (data taken form cancer sequencing projects). For interactive map please go to the COSMIC database here: http://cancer.sanger.ac.uk/cosmic
Somatic mutations are present in all cells of the human body and occur throughout life. They are the consequence of multiple mutational processes, including the intrinsic slight infidelity of the DNA replication machinery, exogenous or endogenous mutagen exposures, enzymatic modification of DNA and defective DNA repair. Different mutational processes generate unique combinations of mutation types, termed “Mutational Signatures”.
The current set of mutational signatures is based on an analysis of 10,952 exomes and 1,048 whole-genomes across 40 distinct types of human cancer. These analyses are based on curated data that were generated by The Cancer Genome Atlas (TCGA), the International Cancer Genome Consortium (ICGC), and a large set of freely available somatic mutations published in peer-reviewed journals. Complete details about the data sources will be provided in future releases of COSMIC.
The profile of each signature is displayed using the six substitution subtypes: C>A, C>G, C>T, T>A, T>C, and T>G (all substitutions are referred to by the pyrimidine of the mutated Watson–Crick base pair). Further, each of the substitutions is examined by incorporating information on the bases immediately 5’ and 3’ to each mutated base generating 96 possible mutation types (6 types of substitution ∗ 4 types of 5’ base ∗ 4 types of 3’ base). Mutational signatures are displayed and reported based on the observed trinucleotide frequency of the human genome, i.e., representing the relative proportions of mutations generated by each signature based on the actual trinucleotide frequencies of the reference human genome version GRCh37. Note that only validated mutational signatures have been included in the curated census of mutational signatures.
Additional information is provided for each signature, including the cancer types in which the signature has been found, proposed aetiology for the mutational processes underlying the signature, other mutational features that are associated with each signature and information that may be relevant for better understanding of a particular mutational signature.
The set of signatures will be updated in the future. This will include incorporating additional mutation types (e.g., indels, structural rearrangements, and localized hypermutation such as kataegis) and cancer samples. With more cancer genome sequences and the additional statistical power this will bring, new signatures may be found, the profiles of current signatures may be further refined, signatures may split into component signatures and signatures
COSMIC v75 includes curations across GRIN2A, fusion pair TCF3-PBX1, and genomic data from 17 systematic screen publications. We are also beginning a reannotation of TCGA exome datasets using Sanger’s Cancer Genome Project analyis pipeline to ensure consistency; four studies are included in this release, to be expanded across the next few releases. The Cancer Gene Census now has a dedicated curator, Dr. Zbyslaw Sondka, who will be focused on expanding the Census, enhancing the evidence underpinning it, and developing improved expert-curated detail describing each gene’s impact in cancer. Finally, as we begin to streamline our ever-growing website, we have combined all information for each gene onto one page and simplified the layout and design to improve navigation
may be found in cancer types in which they are currently not detected.
Signature 1 has been found in all cancer types and in most cancer samples.
Proposed aetiology:
Signature 1 is the result of an endogenous mutational process initiated by spontaneous deamination of 5-methylcytosine.
Additional mutational features:
Signature 1 is associated with small numbers of small insertions and deletions in most tissue types.
Comments:
The number of Signature 1 mutations correlates with age of cancer diagnosis.
Signature 2
Cancer types:
Signature 2 has been found in 22 cancer types, but most commonly in cervical and bladder cancers. In most of these 22 cancer types, Signature 2 is present in at least 10% of samples.
Proposed aetiology:
Signature 2 has been attributed to activity of the AID/APOBEC family of cytidine deaminases. On the basis of similarities in the sequence context of cytosine mutations caused by APOBEC enzymes in experimental systems, a role for APOBEC1, APOBEC3A and/or APOBEC3B in human cancer appears more likely than for other members of the family.
Additional mutational features:
Transcriptional strand bias of mutations has been observed in exons, but is not present or is weaker in introns.
Comments:
Signature 2 is usually found in the same samples as Signature 13. It has been proposed that activation of AID/APOBEC cytidine deaminases is due to viral infection, retrotransposon jumping or to tissue inflammation. Currently, there is limited evidence to support these hypotheses. A germline deletion polymorphism involving APOBEC3A and APOBEC3B is associated with the presence of large numbers of Signature 2 and 13 mutations and with predisposition to breast cancer. Mutations of similar patterns to Signatures 2 and 13 are commonly found in the phenomenon of local hypermutation present in some cancers, known as kataegis, potentially implicating AID/APOBEC enzymes in this process as well.
Signature 3
Cancer types:
Signature 3 has been found in breast, ovarian, and pancreatic cancers.
Proposed aetiology:
Signature 3 is associated with failure of DNA double-strand break-repair by homologous recombination.
Additional mutational features:
Signature 3 associates strongly with elevated numbers of large (longer than 3bp) insertions and deletions with overlapping microhomology at breakpoint junctions.
Comments:
Signature 3 is strongly associated with germline and somatic BRCA1 and BRCA2 mutations in breast, pancreatic, and ovarian cancers. In pancreatic cancer, responders to platinum therapy usually exhibit Signature 3 mutations.
Signature 4
Cancer types:
Signature 4 has been found in head and neck cancer, liver cancer, lung adenocarcinoma, lung squamous carcinoma, small cell lung carcinoma, and oesophageal cancer.
Proposed aetiology:
Signature 4 is associated with smoking and its profile is similar to the mutational pattern observed in experimental systems exposed to tobacco carcinogens (e.g., benzo[a]pyrene). Signature 4 is likely due to tobacco mutagens.
Additional mutational features:
Signature 4 exhibits transcriptional strand bias for C>A mutations, compatible with the notion that damage to guanine is repaired by transcription-coupled nucleotide excision repair. Signature 4 is also associated with CC>AA dinucleotide substitutions.
Comments:
Signature 29 is found in cancers associated with tobacco chewing and appears different from Signature 4.
Signature 5
Cancer types:
Signature 5 has been found in all cancer types and most cancer samples.
Proposed aetiology:
The aetiology of Signature 5 is unknown.
Additional mutational features:
Signature 5 exhibits transcriptional strand bias for T>C substitutions at ApTpN context.
Comments:
Signature 6
Cancer types:
Signature 6 has been found in 17 cancer types and is most common in colorectal and uterine cancers. In most other cancer types, Signature 6 is found in less than 3% of examined samples.
Proposed aetiology:
Signature 6 is associated with defective DNA mismatch repair and is found in microsatellite unstable tumours.
Additional mutational features:
Signature 6 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 6 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 15, 20, and 26.
Signature 7
Cancer types:
Signature 7 has been found predominantly in skin cancers and in cancers of the lip categorized as head and neck or oral squamous cancers.
Proposed aetiology:
Based on its prevalence in ultraviolet exposed areas and the similarity of the mutational pattern to that observed in experimental systems exposed to ultraviolet light Signature 7 is likely due to ultraviolet light exposure.
Additional mutational features:
Signature 7 is associated with large numbers of CC>TT dinucleotide mutations at dipyrimidines. Additionally, Signature 7 exhibits a strong transcriptional strand-bias indicating that mutations occur at pyrimidines (viz., by formation of pyrimidine-pyrimidine photodimers) and these mutations are being repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 8
Cancer types:
Signature 8 has been found in breast cancer and medulloblastoma.
Proposed aetiology:
The aetiology of Signature 8 remains unknown.
Additional mutational features:
Signature 8 exhibits weak strand bias for C>A substitutions and is associated with double nucleotide substitutions, notably CC>AA.
Comments:
Signature 9
Cancer types:
Signature 9 has been found in chronic lymphocytic leukaemias and malignant B-cell lymphomas.
Proposed aetiology:
Signature 9 is characterized by a pattern of mutations that has been attributed to polymerase η, which is implicated with the activity of AID during somatic hypermutation.
Additional mutational features:
Comments:
Chronic lymphocytic leukaemias that possess immunoglobulin gene hypermutation (IGHV-mutated) have elevated numbers of mutations attributed to Signature 9 compared to those that do not have immunoglobulin gene hypermutation.
Signature 10
Cancer types:
Signature 10 has been found in six cancer types, notably colorectal and uterine cancer, usually generating huge numbers of mutations in small subsets of samples.
Proposed aetiology:
It has been proposed that the mutational process underlying this signature is altered activity of the error-prone polymerase POLE. The presence of large numbers of Signature 10 mutations is associated with recurrent POLE somatic mutations, viz., Pro286Arg and Val411Leu.
Additional mutational features:
Signature 10 exhibits strand bias for C>A mutations at TpCpT context and T>G mutations at TpTpT context.
Comments:
Signature 10 is associated with some of most mutated cancer samples. Samples exhibiting this mutational signature have been termed ultra-hypermutators.
Signature 11
Cancer types:
Signature 11 has been found in melanoma and glioblastoma.
Proposed aetiology:
Signature 11 exhibits a mutational pattern resembling that of alkylating agents. Patient histories have revealed an association between treatments with the alkylating agent temozolomide and Signature 11 mutations.
Additional mutational features:
Signature 11 exhibits a strong transcriptional strand-bias for C>T substitutions indicating that mutations occur on guanine and that these mutations are effectively repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 12
Cancer types:
Signature 12 has been found in liver cancer.
Proposed aetiology:
The aetiology of Signature 12 remains unknown.
Additional mutational features:
Signature 12 exhibits a strong transcriptional strand-bias for T>C substitutions.
Comments:
Signature 12 usually contributes a small percentage (<20%) of the mutations observed in a liver cancer sample.
Signature 13
Cancer types:
Signature 13 has been found in 22 cancer types and seems to be commonest in cervical and bladder cancers. In most of these 22 cancer types, Signature 13 is present in at least 10% of samples.
Proposed aetiology:
Signature 13 has been attributed to activity of the AID/APOBEC family of cytidine deaminases converting cytosine to uracil. On the basis of similarities in the sequence context of cytosine mutations caused by APOBEC enzymes in experimental systems, a role for APOBEC1, APOBEC3A and/or APOBEC3B in human cancer appears more likely than for other members of the family. Signature 13 causes predominantly C>G mutations. This may be due to generation of abasic sites after removal of uracil by base excision repair and replication over these abasic sites by REV1.
Additional mutational features:
Transcriptional strand bias of mutations has been observed in exons, but is not present or is weaker in introns.
Comments:
Signature 2 is usually found in the same samples as Signature 13. It has been proposed that activation of AID/APOBEC cytidine deaminases is due to viral infection, retrotransposon jumping or to tissue inflammation. Currently, there is limited evidence to support these hypotheses. A germline deletion polymorphism involving APOBEC3A and APOBEC3B is associated with the presence of large numbers of Signature 2 and 13 mutations and with predisposition to breast cancer. Mutations of similar patterns to Signatures 2 and 13 are commonly found in the phenomenon of local hypermutation present in some cancers, known as kataegis, potentially implicating AID/APOBEC enzymes in this process as well.
Signature 14
Cancer types:
Signature 14 has been observed in four uterine cancers and a single adult low-grade glioma sample.
Proposed aetiology:
The aetiology of Signature 14 remains unknown.
Additional mutational features:
Comments:
Signature 14 generates very high numbers of somatic mutations (>200 mutations per MB) in all samples in which it has been observed.
Signature 15
Cancer types:
Signature 15 has been found in several stomach cancers and a single small cell lung carcinoma.
Proposed aetiology:
Signature 15 is associated with defective DNA mismatch repair.
Additional mutational features:
Signature 15 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 15 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 6, 20, and 26.
Signature 16
Cancer types:
Signature 16 has been found in liver cancer.
Proposed aetiology:
The aetiology of Signature 16 remains unknown.
Additional mutational features:
Signature 16 exhibits an extremely strong transcriptional strand bias for T>C mutations at ApTpN context, with T>C mutations occurring almost exclusively on the transcribed strand.
Comments:
Signature 17
Cancer types:
Signature 17 has been found in oesophagus cancer, breast cancer, liver cancer, lung adenocarcinoma, B-cell lymphoma, stomach cancer and melanoma.
Proposed aetiology:
The aetiology of Signature 17 remains unknown.
Additional mutational features:
Comments:
Signature 1Signature 18
Cancer types:
Signature 18 has been found commonly in neuroblastoma. Additionally, Signature 18 has been also observed in breast and stomach carcinomas.
Proposed aetiology:
The aetiology of Signature 18 remains unknown.
Additional mutational features:
Comments:
Signature 19
Cancer types:
Signature 19 has been found only in pilocytic astrocytoma.
Proposed aetiology:
The aetiology of Signature 19 remains unknown.
Additional mutational features:
Comments:
Signature 20
Cancer types:
Signature 20 has been found in stomach and breast cancers.
Proposed aetiology:
Signature 20 is believed to be associated with defective DNA mismatch repair.
Additional mutational features:
Signature 20 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 20 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 6, 15, and 26.
Signature 21
Cancer types:
Signature 21 has been found only in stomach cancer.
Proposed aetiology:
The aetiology of Signature 21 remains unknown.
Additional mutational features:
Comments:
Signature 21 is found only in four samples all generated by the same sequencing centre. The mutational pattern of Signature 21 is somewhat similar to the one of Signature 26. Additionally, Signature 21 is found only in samples that also have Signatures 15 and 20. As such, Signature 21 is probably also related to microsatellite unstable tumours.
Signature 22
Cancer types:
Signature 22 has been found in urothelial (renal pelvis) carcinoma and liver cancers.
Proposed aetiology:
Signature 22 has been found in cancer samples with known exposures to aristolochic acid. Additionally, the pattern of mutations exhibited by the signature is consistent with the one previous observed in experimental systems exposed to aristolochic acid.
Additional mutational features:
Signature 22 exhibits a very strong transcriptional strand bias for T>A mutations indicating adenine damage that is being repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 22 has a very high mutational burden in urothelial carcinoma; however, its mutational burden is much lower in liver cancers.
Signature 23
Cancer types:
Signature 23 has been found only in a single liver cancer sample.
Proposed aetiology:
The aetiology of Signature 23 remains unknown.
Additional mutational features:
Signature 23 exhibits very strong transcriptional strand bias for C>T mutations.
Comments:
Signature 24
Cancer types:
Signature 24 has been observed in a subset of liver cancers.
Proposed aetiology:
Signature 24 has been found in cancer samples with known exposures to aflatoxin. Additionally, the pattern of mutations exhibited by the signature is consistent with that previous observed in experimental systems exposed to aflatoxin.
Additional mutational features:
Signature 24 exhibits a very strong transcriptional strand bias for C>A mutations indicating guanine damage that is being repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 25
Cancer types:
Signature 25 has been observed in Hodgkin lymphomas.
Proposed aetiology:
The aetiology of Signature 25 remains unknown.
Additional mutational features:
Signature 25 exhibits transcriptional strand bias for T>A mutations.
Comments:
This signature has only been identified in Hodgkin’s cell lines. Data is not available from primary Hodgkin lymphomas.
Signature 26
Cancer types:
Signature 26 has been found in breast cancer, cervical cancer, stomach cancer and uterine carcinoma.
Proposed aetiology:
Signature 26 is believed to be associated with defective DNA mismatch repair.
Additional mutational features:
Signature 26 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 26 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 6, 15 and 20.
Signature 27
Cancer types:
Signature 27 has been observed in a subset of kidney clear cell carcinomas.
Proposed aetiology:
The aetiology of Signature 27 remains unknown.
Additional mutational features:
Signature 27 exhibits very strong transcriptional strand bias for T>A mutations. Signature 27 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 28
Cancer types:
Signature 28 has been observed in a subset of stomach cancers.
Proposed aetiology:
The aetiology of Signature 28 remains unknown.
Additional mutational features:
Comments:
Signature 29
Cancer types:
Signature 29 has been observed only in gingivo-buccal oral squamous cell carcinoma.
Proposed aetiology:
Signature 29 has been found in cancer samples from individuals with a tobacco chewing habit.
Additional mutational features:
Signature 29 exhibits transcriptional strand bias for C>A mutations indicating guanine damage that is most likely repaired by transcription-coupled nucleotide excision repair. Signature 29 is also associated with CC>AA dinucleotide substitutions.
Comments:
The Signature 29 pattern of C>A mutations due to tobacco chewing appears different from the pattern of mutations due to tobacco smoking reflected by Signature 4.
Signature 30
Cancer types:
Signature 30 has been observed in a small subset of breast cancers.
Proposed aetiology:
The aetiology of Signature 30 remains unknown.
Examples in the literature of deposits into or analysis from the COSMIC database
“analysis of exons representing 20,857 transcripts from 18,191 genes, we conclude that the genomic landscapes of breast and colorectal cancers are composed of a handful of commonly mutated gene “mountains” and a much larger number of gene “hills” that are mutated at low frequency. “
found cellular pathways with multiple pathways
analyzed a highly curated database (Metacore, GeneGo, Inc.) that includes human protein-protein interactions, signal transduction and metabolic pathways
There were 108 pathways that were found to be preferentially mutated in breast tumors. Many of the pathways involved phosphatidylinositol 3-kinase (PI3K) signaling
the cancer genome landscape consists of relief features (mutated genes) with heterogeneous heights (determined by CaMP scores). There are a few “mountains” representing individual CAN-genes mutated at high frequency. However, the landscapes contain a much larger number of “hills” representing the CAN-genes that are mutated at relatively low frequency. It is notable that this general genomic landscape (few gene mountains and many gene hills) is a common feature of both breast and colorectal tumors.
developed software to analyze multiple mutations and mutation frequencies available from Harvard Bioinformatics at
R Software for Cancer Mutation Analysis (download here)
CancerMutationAnalysis Version 1.0:
R package to reproduce the statistical analyses of the Sjoblom et al article and the associated Technical Comment. This package is build for reproducibility of the original results and not for flexibility. Future version will be more general and define classes for the data types used. Further details are available in Working Paper 126.
CancerMutationAnalysis Version 2.0:
R package to reproduce the statistical analyses of the Wood et al article. Like its predecessor, this package is still build for reproducibility of the original results and not for flexibility. Further details are available in Working Paper 126
Update 04/27/2019
Review 2018. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Z. Sondka et al. Nature Reviews. 2018.
The Catalogue of Somatic Mutations in Cancer (COSMIC) Cancer Gene Census (CGC) reevaluates the cancer genome landscape periodically and curates the findings into a database of genetic changes occurring in various tumor types. The 2018 CGC describes in detail the effect of 719 cancer driving genes. The recent expansion includes functional and mechanistic descriptions of how each gene contributes to disease etiology and in terms of the cancer hallmarks as described by Hanahan and Weinberg. These functional characteristics show the complexity of the cancer mutational landscape and genome and suggest ” multiple cancer-related functions for many genes, which are often highly tissue-dependent or tumour stage-dependent.” The 2018 CGC expands a second tier of genes, expanding the list of cancer related genes.
Criteria for curation of genes into CGC (curation process)
choosing candidate genes are selected from published literature, conference abstracts, large cancer genome screens deposited in databases, and analysis of current COSMIC database
COSMIC data are analyzed to determine presence of patterns of somatic mutations and frequency of such mutations in cancer
literature review to determine the role of the gene in cancer
Minimum evidence
– at least two publications from different groups shows increased mutation frequency in at least one type of cancer (PubMed)
– at least two publications from different groups showing experimental evidence of functional involvement in at least one hallmark of cancer in order to classify the mutant gene as oncogene, tumor suppressor, or fusion partner (like BCR-Abl)
independent assessment by at least two postdoctoral fellows
gene must be classified as either Tier 1 of Tier 2 CGC gene
inclusion in database
continued curation efforts
definitions:
Tier 1 gene: genes which have strong evidence from both mutational and functional analysis as being involved in cancer
Tier 2 gene: genes with mutational patterns typical of cancer drivers but not functionally characterized as well as genes with published mechanistic description of involvement in cancer but without proof of somatic mutations in cancer
Tier 2 genes (719 genes): include 103 oncogenes, 181 tumor suppressors, 134 fusion partners and 31 with unknown function
Updated 7/26/2019
The COSMIC database is undergoing an extensive update and reannotation, in order to ensure standardisation and modernisation across COSMIC data. This will substantially improve the identification of unique variants that may have been described at the genome, transcript and/or protein level. The introduction of a Genomic Identifier, along with complete annotation across multiple, high quality Ensembl transcripts and improved compliance with current HGVS syntax, will enable variant matching both within COSMIC and across other bioinformatic datasets.
As a result of these updates there will be significant changes in the upcoming releases as we work through this process. The first stage of this work was the introduction of improvedHGVS syntax compliance in our May release. The majority of the changes will be reflected in COSMIC v90, which will be released in late August or early September, and the remaining changes will be introduced over the next few releases.
The significant changes in v90 include:
Updated genes, transcripts and proteins from Ensembl release 93 on both the GRCh37 and GRCh38 assemblies.
Full reannotation of COSMIC variants with known genomic coordinates using Ensembl’s Variant Effect Predictor (VEP). This provides accurate and standardised annotation uniformly across all relevant transcripts and genes that include the genomic location of the variant.
New stable genomic identifiers (COSV) that indicate the definitive position of the variant on the genome. These unique identifiers allow variants to be mapped between GRCh37 and GRCh38 assemblies and displayed on a selection of transcripts.
Updated cross-reference links between COSMIC genes and other widely-used databases such as HGNC, RefSeq, Uniprot and CCDS.
Complete standardised representation of COSMIC variants, following the most recent HGVS recommendations, where possible.
Remapping of gene fusions on the updated transcripts on both the GRCh37 and GRCh38 assemblies, along with the genomic coordinates for the breakpoint positions.
Reduced redundancy of mutations. Duplicate variants have been merged into one representative variant.
Key points for you
COSMIC variants have been annotated on all relevant Ensembl transcripts across both the GRCh37 and GRCh38 assemblies from Ensembl release 93. New genomic identifiers (e.g. COSV56056643) are used, which refers to the variant change at the genomic level rather than gene, transcript or protein level and can thus be used universally. Existing COSM IDs will continue to be supported and will now be referred to as legacy identifiers e.g. COSM476. The legacy identifiers (COSM) are still searchable. In the case of mutations without genomic coordinates, hence without a COSV identifier, COSM identifiers will continue to be used.
All relevant Ensembl transcripts in COSMIC (which have been selected based on Ensembl canonical classification and on the quality of the dataset to include only GENCODE basic transcripts) will now have both accession and version numbers, so that the exact transcript is known, ensuring reproducibility. This also provides transparency and clarity as the data are updated.
How these changes will be reflected in the download files
As we are now mapping all variants on all relevant Ensembl transcripts, the number of rows in the majority of variant download files has increased significantly. In the download files, additional columns are provided including the legacy identifier (COSM) and the new genomic identifier (COSV). An internal mutation identifier is also provided to uniquely represent each mutation, on a specific transcript, on a given assembly build. The accession and version number for each transcript are included. File descriptions for each of the download files will be available from the downloads page for clarity. We have included an example of the new columns below.
For example: COSMIC Complete Mutation Data (Targeted screens)
[17:Q] Mutation Id – An internal mutation identifier to uniquely represent each mutation on a specific transcript on a given assembly build.
[18:R] Genomic Mutation Id – Genomic mutation identifier (COSV) to indicate the definitive position of the variant on the genome. This identifier is trackable and stable between different versions of the release.
[19:S] Legacy Mutation Id – Legacy mutation identifier (COSM) that will represent existing COSM mutation identifiers.
We will shortly have some sample data that can be downloaded in the new table structure, to give you real data to manipulate and integrate, this will be available on the variant updates page.
How this affects you
We are aware that many of the changes we are making will affect integration into your pipelines and analytical platforms. By giving you advance notice of the changes, we hope much of this can be mitigated, and the end result of having clean, standardised data will be well worth any disruption. The variant updates page on the COSMIC website will provide a central point for this information and further technical details of the changes that we are making to COSMIC.
Evolution can get stuck. When no mutations are available that can improve the fitness of an organism, evolution cannot proceed. However, using environmental fluctuations that are ubiquitous in nature, evolution can proceed. In the preprint that we posted earlier on the bioRxiv and which has recently been published in PNAS we describe how a transcription factor and its binding site, constraint in the different constant environments, can evolve in fluctuating environments.
Stalled evolution
Concept of fitness landscape. From de Visser & Krug, Nature Reviews Genetics 15, 480–490 (2014)
The accessibility of phenotypes in different environments depends on the wiring of the genotype-phenotype map and the translation of this map into fitness. When sub-optimal genotypes are surrounded by valleys in the adaptive landscape, neighboring genotypes are inaccessible and evolution is unable to proceed by single mutational step-by-step positive Darwinian evolution (Figure 1).
To investigate the connectedness of the fitness increasing genotypes in sequence space we used the lac regulatory system in Escherichia coli. Decades of work have elucidated the physiology of the lac regulatory system. And it is known that a few base pairs in the operator DNA, and a few amino acid residues in the transcription factor are responsible for specificity of binding of these operator-repressor pairs. We here experimentally constructed mutants with mutations in these specific residues (6 residues, yielding 26=64 mutants in total) that together constitute 6!=720 direct trajectories.
Assessing the ability to repress the lac operon in one environment (without a lactose-sugar-derivative), and the ability to express the lac operon in the other environment (with a lactose-sugar-derivative), we find that none of the mutational trajectories allows continuous improvements along the mutational trajectories in one of the constant environments. Interestingly, we find that alternating between these environments doesallow for constant improvements along the mutational trajectories. We find that the shortest route towards the final genotype is of Hamming distance 6, there is thus a direct mutational trajectory from the starting sequence to the final combination of transcription-factor and binding site. With a computational method that describes the mutational and environmental transitions as a Markov process, we can further calculate the crossing rates from the initial to the final genotype for all trajectories in the landscape, allowing mutational detours. We find that the crossing rate is maximal when the environmental switches are on the order of the mutation rate (or the rate at which a sweep can be completed).
Tradeoffs are crucial for crossing the adaptive landscape in fluctuating environments
Cross-environmental tradeoffs are responsible for these continuous improvements. In the most extreme case, suboptimal peaks are translated into valleys, by which constraints are resolved. In the more subtle case, descending slopes are turned into ascending slopes upon environmental change, allowing adaptive trajectories to surf over the slopes with positive selective coefficients. Evolutionary constraints can thus be overcome by the environment-dependent ‘ratcheting’ that allows the crossing of otherwise inaccessible regions in sequence space (Figure 2).
We think that this research not only aids the fundamental understanding of ecological and evolutionary transitions in fluctuating environments, but that it can also help rethinking the evolutionary optimization of certain biotechnological processes (for instance the production of antibodies). And in addition, it cautions against the use of cyclic multi-drug protocols in clinical treatments, as these might potentially increase the speed of adaptation to the drugs, instead of halting it.
Breaking evolutionary constraint with a tradeoff ratchetM.G.J. de Vos, A. Dawid, V. Sunderlikova, S.J. Tans
PNAS, (2015) | http://dx.doi.org/10.1073/pnas.1510282112
Notes from Opening Plenary Session – The Genome and Beyond from the 2015 AACR Meeting in Philadelphia PA; Sunday April 19, 2015
Reporter: Stephen J. Williams, Ph.D.
The following contain notes from the Sunday April 19, 2015 AACR Meeting (Pennsylvania Convention Center, Philadelphia PA) 9:30 AM Opening Plenary Session
A) Insights From Cancer Genomes: Michael R. Stratton, Ph.D.; Director of the Wellcome Trust Sanger Institute
How do we correlate mutations with causative factors of carcinogenesis and exposure?
Cancer was thought as a disease of somatic mutations
UV skin exposure – see C>T transversion in TP53 while tobacco exposure and lung cancer see more C>A transversion; Is it possible to determine EXPOSURE SIGNATURES?
Use a method called non negative matrix factorization (like face pattern recognition but a mutation pattern recognition)
Performed sequence analysis producing 12,000 mutation catalogs with 8,000 somatic mutation signatures
Found more mutations than expected; some mutation signatures found in all cancers, while some signatures in half of cancers, and some signatures not found in cancer
For example found 3 mutation signatures in ovarian cancer but 13 for breast cancers (80,000 mutations); his signatures are actually spectrums of mutations
kataegis: defined as localized hypermutation; an example is a signature he found related to AID/APOBEC family (involved in IgG variability); kataegis is more prone in hematologic cancers than solid cancers
recurrent tumors show a difference in mutation signatures than primary tumor before drug treatment
B) Engineering Cancer Genomes: Tyler Jacks, Ph.D.; Director, Koch Institute for Integrative Cancer Research
Cancer GEM’s (genetically engineered mouse models of cancer) had moved from transgenics to defined oncogenes
Observation that p53 -/- mice develop spontaneous tumors (lymphomas)
then GEMs moved to Cre/Lox systems to generate mice with deletions however these tumor models require lots of animals, much time to create, expensive to keep;
figured can use CRSPR/Cas9 as rapid, inexpensive way to generate engineered mice and tumor models
he used CRSPR/Cas9 vectors targeting PTEN to introduce PTEN mutations in-vivo to hepatocytes; when they also introduced p53 mutations produced hemangiosarcomas; took ONLY THREE months to produce detectable tumors
also produced liver tumors by using CRSPR/Cas9 to introduce gain of function mutation in β-catenin
The three “E’s” of cancer immunoediting: Elimination, Equilibrium, and Escape
First evidence for immunoediting came from mice that were immunocompetent resistant to 3 methylcholanthrene (3mca)-induced tumorigenesis but RAG2 -/- form 3mca-induced tumors
RAG2-/- unedited (retain immunogenicity); tumors rejected by wild type mice
Edited tumors (aren’t immunogenic) led to tolerization of tumors
Can use genomic studies to identify mutant proteins which could be cancer specific immunoepitopes
MHC (major histocompatibility complex) tetramers: can develop vaccines against epitope and personalize therapy but only good as checkpoint block (anti-PD1 and anti CTLA4) but personalized vaccines can increase therapeutic window so don’t need to start PD1 therapy right away
E) Report on the Melanoma Keynote 006 Trial comparing pembrolizumab and ipilimumab (PD1 inhibitors)
Results of this trial were published the morning of the meeting in the New England Journal of Medicine and can be found here.
A few notes:
From the paper: The anti–PD-1 antibody pembrolizumab prolonged progression-free survival and overall survival and had less high-grade toxicity than did ipilimumab in patients with advanced melanoma. (Funded by Merck Sharp & Dohme; KEYNOTE-006 ClinicalTrials.gov number, NCT01866319.)
Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG et al: CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514(7522):380-384.
Other related articles on Cancer Genomics and Social Media Coverage were published in this Open Access Online Scientific Journal, include the following:
Reporter and Curator: Larry H. Bernstein, MD, FCAP
This is the FIRST discussion of a several part series leading from the genome, to protein synthesis (1), posttranslational modification of proteins (2), examples of protein effects on metabolism and signaling pathways (3), and leading to disruption of signaling pathways in disease (4), and effects leading to mutagenesis.
DNA carries the information for making all of the cell’s proteins. These proteins implement all of the functions of a living organism and determine the organism’s characteristics. When the cell reproduces, it has to pass all of this information on to the daughter cells.
Before a cell can reproduce, it must first replicate, or make a copy of, its DNA. Where DNA replication occurs depends upon whether the cells is a prokaryote or a eukaryote (see the RNA sidebar on the previous page for more about the types of cells). DNA replication occurs in the cytoplasm of prokaryotes and in the nucleus of eukaryotes. Regardless of where DNA replication occurs, the basic process is the same.
The structure of DNA lends itself easily to DNA replication. Each side of the double helix runs in opposite (anti-parallel) directions. The beauty of this structure is that it can unzip down the middle and each side can serve as a pattern or template for the other side (called semi-conservative replication). However, DNA does not unzip entirely. It unzips in a small area called a replication fork, which then moves down the entire length of the molecule.
Eukaryotic DNA replication (Wikipedia), is a conserved mechanism that restricts DNA replication to only once per cell cycle. Eukaryotic DNA replication of chromosomalDNA is central for the duplication of a cell and is necessary for the maintenance of the eukaryotic genome.
DNA replication is the action of DNA polymerases synthesizing a DNA strand complementary to the original template strand. To synthesize DNA, the double-stranded DNA is unwound by DNA helicases ahead of polymerases, forming a replication fork containing two single-stranded templates.
Replication processes permit the copying of a single DNA double helix into two DNA helices, which are divided into the daughter cells at mitosis. The major enzymatic functions carried out at the replication fork are well conserved from prokaryotes to eukaryotes, but the replication machinery in eukaryotic DNA replication is a much larger complex, coordinating many proteins at the site of replication, forming the replisome.[1]
The replisome is responsible for copying the entirety of genomic DNA in each proliferative cell. This process allows for the high-fidelity passage of hereditary/genetic information from parental cell to daughter cell and is thus essential to all organisms. Much of the cell cycle is built around ensuring that DNA replication occurs without errors.[1]
In G1 phase of the cell cycle, many of the DNA replication regulatory processes are initiated. In eukaryotes, the vast majority of DNA synthesis occurs during S phase of the cell cycle, and the entire genome must be unwound and duplicated to form two daughter copies. During G2, any damaged DNA or replication errors are corrected. Finally, one copy of the genomes is segregated to each daughter cell at mitosis or M phase.[2] These daughter copies each contain one strand from the parental duplex DNA and one nascent antiparallel strand.
This mechanism is conserved from prokaryotes to eukaryotes and is known as semiconservative DNA replication. The process of semiconservative replication for the site of DNA replication is a fork-like DNA structure, the replication fork, where the DNA helix is open, or unwound, exposing unpaired DNA nucleotides for recognition and base pairing for the incorporation of free nucleotides into double-stranded DNA.[3]
Let’s look at the details:
An enzyme called DNA gyrase makes a nick in the double helix and each side separates
An enzyme called helicase unwinds the double-stranded DNA
Several small proteins called single strand binding proteins(SSB) temporarily bind to each side and keep them separated
An enzyme complex called DNA polymerase“walks” down the DNA strands and adds new nucleotides to each strand. The nucleotides pair with the complementary nucleotides on the existing stand (A with T, G with C).
A subunit of the DNA polymerase proofreads the new DNA
An enzyme called DNA ligaseseals up the fragments into one long continuous strand
The new copies automatically wind up again
Different types of cells replicated their DNA at different rates. Some cells constantly divide, like those in your hair and fingernails and bone marrow cells. Other cells go through several rounds of cell division and stop (including specialized cells, like those in your brain, muscle and heart). Finally, some cells stop dividing, but can be induced to divide to repair injury (such as skin cells and liver cells). In cells that do not constantly divide, the cues for DNA replication/cell division come in the form of chemicals. These chemicals can come from other parts of the body (hormones) or from the environment.
Pre-replicative_complex
Diagram of the formation of the pre-replicative complex transforming into an active replisome. Mcm 2-7 complex loads onto DNA at replication origins during G1 and unwinds DNA ahead of replicative polymerases.Cdc6 and Cdt1 bring Mcm complexes to replication origins. CDK/DDK-dependent phosphorylation of pre-replicative proteins leads toreplisome assembly and origin firing. Cdc6 and Cdt1 are no longer required and are removed from the nucleus or degraded. Mcms and associated proteins, GINS and Cdc45, unwind DNA to expose template DNA. At this point replisome assembly is completed and replication is initiated. “P” represents phosphorylation.
The assembly of the minichromosome maintenance (Mcm) proteins function together as a complex in the cell. The assembly of the Mcm proteins onto chromatin requires the coordinated function of the Origin Recognition Complex (ORC), Cdc6, and Cdt1.[18] Once the Mcm proteins have been loaded onto the chromatin, ORC and Cdc6 can be removed from the chromatin without preventing subsequent DNA replication. This suggests that the primary role of the pre-replication complex is to correctly load the Mcm proteins.[19]
The Mcm proteins support roles both in the initiation and elongation steps of DNA synthesis.[20] Each Mcm protein is highly related to all others, but unique sequences distinguishing each of the subunit types are conserved across eukaryotes. All eukaryotes have exactly six Mcm protein analogs that each fall into one of the existing classes (Mcm2-7), which suggests that each Mcm protein has a unique and important function.[21]
Minichromosome maintenance proteins have been found to be required for DNA helicase activity and inactivation of any of the six Mcm proteins prevents further progression of the replication fork. This is consistent with the requirement of ORC, Cdc6, and Cdt1 function to assemble the Mcm proteins at the origin of replication.[22] The complex containing all six Mcm proteins creates a hexameric, doughnut like structure with a central cavity.[23] The helicase activity of the Mcm protein complex raises the question of how the ring-like complex is loaded onto the single-stranded DNA. One possibility is that the helicase activity of the Mcm protein complex can oscillate between an open and a closed ring formation to allow single-stranded DNA loading.[6]
Along with the minichromosome maintenance protein complex helicase activity, the complex also has associated ATPase activity.[24] A mutation in any one of the six Mcm proteins reduces the conserved ATP binding sites, which indicates that ATP hydrolysis is a coordinated event involving all six subunits of the Mcm complex.[25] Studies have shown that within the Mcm protein complex are specific catalytic pairs of Mcm proteins that function together to coordinate ATP hydrolysis. For example, Mcm3 but not Mcm6 can activate Mcm6 activity. These studies suggest that the structure for the Mcm complex is a hexamer with Mcm3 next to Mcm7, Mcm2 next to Mcm6, and Mcm4 next to Mcm5. Both members of the catalytic pair contribute to the conformation that allows ATP binding and hydrolysis and the mixture of active and inactive subunits create a coordinated ATPase activity that allows the Mcm protein complex to complete ATP binding and hydrolysis as a whole.[26]
The nuclear localization of the minichromosome maintenance proteins is regulated in budding yeast cells. The Mcm proteins are present in the nucleus in G1 stage and S phase of the cell cycle, but are exported to the cytoplasm during the G2 stage and M phase. A complete and intact six subunit Mcm complex is required to enter into the cell nucleus.[27] InS. cerevisiae, nuclear export is promoted by cyclin-dependent kinase (CDK) activity. Mcm proteins that are associated with chromatin are protected from CDK export machinery due to the lack of accessibility to CDK.[28]
During the G1 stage of the cell cycle, the replication initiation factors, origin recognition complex (ORC), Cdc6, Cdt1, and minichromosome maintenance (Mcm) protein complex, bind sequentially to DNA to form the pre-replication complex (pre-RC). At the transition of the G1 stage to the S phase of the cell cycle, S phase–specific cyclin-dependent protein kinase (CDK) and Cdc7/Dbf4 kinase (DDK) transform the pre-RC into an active replication fork. During this transformation, the pre-RC is disassembled with the loss of Cdc6, creating the initiation complex. In addition to the binding of the Mcm proteins, cell division cycle 45 (Cdc45) protein is also essential for initiating DNA replication.[29][30] Studies have shown that Mcm is critical for the loading of Cdc45 onto chromatin and this complex containing both Mcm and Cdc45 is formed at the onset of the S phase of the cell cycle.[31][32] Cdc45 targets the Mcm protein complex, which has been loaded onto the chromatin, as a component of the pre-RC at the origin of replication during the G1 stage of the cell cycle.[20]
The six minichromosome maintenance proteins and Cdc45 are essential during initiation and elongation for the movement of replication forks and for unwinding of the DNA. GINS are essential for the interaction of Mcm and Cdc45 at the origins of replication during initiation and then at DNA replication forks as the replisome progresses.[37][38] The GINS complex is composed of four small proteins Sld5 (Cdc105), Psf1 (Cdc101), Psf2 (Cdc102) and Psf3 (Cdc103), GINS represents ‘go, ichi, ni, san’ which means ‘5, 1, 2, 3’ in Japanese.[39]
Mcm10 is essential for chromosome replication and interacts with the minichromosome maintenance 2-7 helicase that is loaded in an inactive form at origins of DNA replication. Mcm10 chaperones the catalytic DNA polymerase α and helps stabilize the polymerase.[40]
At the onset of S phase, the pre-replicative complex must be activated by two S phase-specific kinases in order to form an initiation complex at an origin of replication. One kinase is the Cdc7-Dbf4 kinase called Dbf4-dependent kinase (DDK) and the other is cyclin-dependent kinase (CDK).[41] Chromatin-binding assays of Cdc45 in yeast and Xenopus have shown that a downstream event of CDK action is loading of Cdc45 onto chromatin.[30][31]Cdc6 has been speculated to be a target of CDK action, because of the association between Cdc6 and CDK, and the CDK-dependent phosphorylation of Cdc6. The CDK-dependent phosphorylation of Cdc6 has been considered to be required for entry into the S phase.[42]
Eukaryotic replisome complex and associated proteins.
The formation of the pre-replicative complex (pre-RC) marks the potential sites for the initiation of DNA replication. Consistent with the minichromosome maintenance complex encircling double stranded DNA, formation of the pre-RC does not lead to the immediate unwinding of origin DNA or the recruitment of DNA polymerases. Instead, the pre-RC that is formed during the G1 of the cell cycle is only activated to unwind the DNA and initiate replication after the cells pass from the G1 to the S phase of the cell cycle.[2]
Once the initiation complex is formed and the cells pass into the S phase, the complex then becomes a replisome. The eukaryotic replisome complex is responsible for coordinating DNA replication. Replication on the leading and lagging strands is performed by DNA polymerase ε and DNA polymerase δ. Many replisome factors including Claspin, And1, replication factor C clamp loader and the fork protection complex are responsible for regulating polymerase functions and coordinating DNA synthesis with the unwinding of the template strand by Cdc45-Mcm-GINS complex. As the DNA is unwound the twist number decreases. To compensate for this the writhe number increases, introducing positive supercoils in the DNA. These supercoils would cause DNA replication to halt if they were not removed. Topoisomerases are responsible for removing these supercoils ahead of the replication fork.
The replication fork is the junction the between the newly separated template strands, known as the leading and lagging strands, and the double stranded DNA. Since duplex DNA is antiparallel, DNA replication occurs in opposite directions between the two new strands at the replication fork, but all DNA polymerases synthesize DNA in the 5′ to 3′ direction with respect to the newly synthesized strand. Further coordination is required during DNA replication. Two replicative polymerases synthesize DNA in opposite orientations. Polymerase ε synthesizes DNA on the “leading” DNA strand continuously as it is pointing in the same direction as DNA unwinding by the replisome. In contrast, polymerase δ synthesizes DNA on the “lagging” strand, which is the opposite DNA template strand, in a fragmented or discontinuous manner.
The discontinuous stretches of DNA replication products on the lagging strand are known as Okazaki fragments and are about 100 to 200 bases in length at eukaryotic replication forks. The lagging strand usually contains longer stretches of single-stranded DNA that is coated with single-stranded binding proteins, which help stabilize the single-stranded templates by preventing a secondary structure formation. In eukaryotes, these single-stranded binding proteins are a heterotrimeric complex known as replication protein A(RPA).[56]
Each Okazaki fragment is preceded by an RNA primer, which is displaced by the procession of the next Okazaki fragment during synthesis. RNAse H recognizes the DNA:RNA hybrids that are created by the use of RNA primers and is responsible for removing these from the replicated strand, leaving behind a primer:template junction. DNA polymerase α, recognizes these sites and elongates the breaks left by primer removal. In eukaryotic cells,
Depiction of DNA replication at replication fork. a: template strands, b: leading strand, c: lagging strand, d: replication fork, e: RNA primer, f: Okazaki fragment
Leading Strand
Lagging Strand
Replicative DNA Polymerases
After the replicative helicase has unwound the parental DNA duplex, exposing two single-stranded DNA templates, replicative polymerases are needed to generate two copies of the parental genome. DNA polymerase function is highly specialized and accomplish replication on specific templates and in narrow localizations. At the eukaryotic replication fork, there are three distinct replicative polymerase complexes that contribute to DNA replication: Polymerase α, Polymerase δ, and Polymerase ε. These three polymerases are essential for viability of the cell.[66]
Because DNA polymerases require a primer on which to begin DNA synthesis, polymerase α (Pol α) acts as a replicative primase. Pol α is associated with an RNA primase and this complex accomplishes the priming task by synthesizing a primer that contains a short 10 nucleotide stretch of RNA followed by 10 to 20 DNA bases.[3] Importantly, this priming action occurs at replication initiation at origins to begin leading-strand synthesis and also at the 5′ end of each Okazaki fragment on the lagging strand.
However, Pol α is not able to continue DNA replication and must be replaced with another polymerase to continue DNA synthesis.[67] Polymerase switching requires clamp loaders and it has been proven that normal DNA replication requires the coordinated actions of all three DNA polymerases: Pol α for priming synthesis, Pol ε for leading-strand replication, and the Pol δ, which is constantly loaded, for generating Okazaki fragments during lagging-strand synthesis.[68]
The DNA helicases and polymerases must remain in close contact at the replication fork. If unwinding occurs too far in advance of synthesis, large tracts of single-stranded DNA are exposed. This can activate DNA damage signaling or induce DNA repair processes. To thwart these problems, the eukaryotic replisome contains specialized proteins that are designed to regulate the helicase activity ahead of the replication fork. These proteins also provide docking sites for physical interaction between helicases and polymerases, thereby ensuring that duplex unwinding is coupled with DNA synthesis.[73]
To strengthen the interaction between the polymerase and the template DNA, DNA sliding clamps associate with the polymerase to promote the processivity of the replicative polymerase. In eukaryotes, the sliding clamp is a homotrimer ring structure known as the proliferating cell nuclear antigen (PCNA). The PCNA ring has polarity with surfaces that interact with DNA polymerases and tethers them securely to the DNA template. PCNA-dependent stabilization of DNA polymerases has a significant effect on DNA replication because PCNAs are able to enhance the polymerase processivity up to 1,000-fold.[85][86] PCNA is an essential cofactor and has the distinction of being one of the most common interaction platforms in the replisome to accommodate multiple processes at the replication fork, and so PCNA is also viewed as a regulatory cofactor for DNA polymerases.[87)
PCNA loading is accomplished by the replication factor C (RFC) complex. The RFC complex is composed of five ATPases: Rfc1, Rfc2, Rfc3, Rfc4 and Rfc5.[88] RFC recognizes primer-template junctions and loads PCNA at these sites.[89][90] The PCNA homotrimer is opened by RFC by ATP hydrolysis and is then loaded onto DNA in the proper orientation to facilitate its association with the polymerase.[91][92] Clamp loaders can also unload PNCA from DNA; a mechanism needed when replication must be terminated.[92]
Termination
The end replication problem is handled in eukaryotic cells by telomere regions and telomerase. Telomeres extend the 3′ end of the parental chromosome beyond the 5′ end of the daughter strand. This single-stranded DNA structure can act as an origin of replication that recruits telomerase. Telomerase is a specialized DNA polymerase that consists of multiple protein subunits and an RNA component. The RNA component of telomerase anneals to the single stranded 3′ end of the template DNA and contains 1.5 copies of the telomeric sequence.[60] Telomerase contains a protein subunit that is a reverse transcriptase called telomerase reverse transcriptase or TERT. TERT synthesizes DNA until the end of the template telomerase RNA and then disengages.[60] This process can be repeated as many times as needed with the extension of the 3′ end of the parental DNA molecule. This 3′ addition provides a template for extension of the 5′ end of the daughter strand by lagging strand DNA synthesis. Regulation of telomerase activity is handled by telomere-binding proteins.
A depiction of telomerase progressively elongating telomeric DNA.
DNA replication is a tightly orchestrated process that is controlled within the context of the cell cycle. Progress through the cell cycle and in turn DNA replication is tightly regulated by the formation and activation of pre-replicative complexs (pre-RCs) which is achieved through the activation and inactivation of cyclin-dependent kinases (Cdks). Specifically it is the interactions of cyclins and cyclin dependent kinases that are responsible for the transition from G1 into S-phase.
Cell_Cycle_
– G-quadruplex
It will be exactly 60 years ago in February that James Watson and Francis Crick famously burst into the pub next to their Cambridge laboratory to announce the discovery of the “secret of life”.
What they had actually done was describe the way in which two long chemical chains wound up around each other to encode the information cells need to build and maintain our bodies.
Today, the pair’s modern counterparts in the university city continue to work on DNA’s complexities.
Balasubramanian’s group has been pursuing a four-stranded version of the molecule that scientists have produced in the test tube now for a number of years.
It is called the G-quadruplex. The “G” refers to guanine, one of the four chemical groups, or “bases”, that hold DNA together and which encode our genetic information (the others being adenine, cytosine, and thymine).
The G-quadruplex seems to form in DNA where guanine exists in substantial quantities.
And although ciliates, relatively simple microscopic organisms, have displayed evidence for the incidence of such DNA, the new research is said to be the first to firmly pinpoint the quadruple helix in human cells.
‘Funny target’
The team, led by Giulia Biffi, a researcher in Balasubramaninan’s lab, produced antibody proteins that were designed specifically to track down and bind to regions of human DNA that were rich in the quadruplex structure. The antibodies were tagged with a fluorescence marker so that the time and place of the structures’ emergence in the cell cycle could be noted and imaged.
This revealed the four-stranded DNA arose most frequently during the so-called “s-phase” when a cell copies its DNA just prior to dividing.
Prof Balasubramaninan said that was of key interest in the study of cancers, which were usually driven by genes, or oncogenes, that had mutated to increase DNA replication.
If the G-quadruplex could be implicated in the development of some cancers, it might be possible, he said, to make synthetic molecules that contained the structure and blocked the runaway cell proliferation at the root of tumours.
If the first and core mission of the genetic code is to faithfully replicate the “genetic material” encoded in the DNA and RNA nucleic acids, then every metabolic process must be functioning in a synchronous 24/7 manner. The only way to do this is to use all the purine and pyrmidine nucleotide, nucleoside and bases (ATUIXGC) =7 necessary and sufficient to make RNA first and then with the assistance of Thioredoxin i.e. ferredoxin purple sulphur bacteria to oxidize rna to dna.
In regards to purine metabolism which is my major area of focus. The two purine nucleotides left out of the current genetic code i.e. IMP and XMP have the following functions through their enzymes.1. Begin purine nucleotide synthesis de novo by IMPDH cyclodehydrogenase the last step in closing the purine ring and the current foundation molecular structure for DNA and RNA; 2. HPRT is the main enzyme is purine salvage for IMP and GMP; APRT provides same service for AMP; 3. Finally the last step in purine metabolism is by xanthine oxidase with the assistance of FES and molybendum. In essence the IMP and XMP families were the first to build the nucleic acid molecular structure; design a process to recycle functional side groups while keeping the purine ring intact and finally developing the biochemical pathway to eliminate toxic ammonia NH3 from the CNS and liver/kidneys.
I believe the 7 nucleotide Novagon DNA triplex genetic code should be called the epigenetic code since it works not only in protein metabolism which is 2% of the genome but noncoding intronic regions ie. rna editing, RNAi, piRNA, snMRN, long noncoding RNA and many other small rnas which operate above the level of the dna and rna base pair i.e. epigenesis suppressing or enhancing whole genes and networks of genes which control protein,lipid,carbohydrate and nucleic acid metabolism.
I am in the process of deveoping a 7 code epigenetic primer to control the gene switches which in turn allows the genetic material to be inherited from generation to generation as the species constantly adapts to external and internal stressors and competitive antagonist.
A Conserved Structural Core in Type II Restriction Enzymes.
ultraviolet rays, especially the UV-C rays (~260 nm) that are absorbed strongly by DNA but also the longer-wavelength UV-B that penetrates the ozone shield [Link].
Highly-reactive oxygen radicals produced during normal cellular respiration as well as by other biochemical pathways. [Link to further discussion.]
Chemicals in the environment
many hydrocarbons, including some found in cigarette smoke
One of the most frequent is the loss of an amino group(“deamination”) — resulting, for example, in a C being converted to a U.
Mismatchesof the normal bases because of a failure of proofreading during DNA replication.
Common example: incorporation of the pyrimidineU (normally found only in RNA) instead of T.
Breaksin the backbone.
Can be limited to one of the two strands (a single-stranded break, SSB) or
on both strands(a double-stranded break (DSB).
Ionizing radiation is a frequent cause, but some chemicals produce breaks as well.
CrosslinksCovalent linkagescan be formed between bases
on the same DNA strand (“intrastrand”) or
on the opposite strand (“interstrand”).
Several chemotherapeutic drugs used against cancers crosslink DNA [Link].
Repairing Damaged Bases
Damaged or inappropriate bases can be repaired by several mechanisms:
Direct chemical reversal of the damage
Excision Repair, in which the damaged base or bases are removed and then replaced with the correct ones in a localized burst of DNA synthesis. There are three modes of excision repair, each of which employs specialized sets of enzymes.
Base Excision Repair (BER)
Nucleotide Excision Repair (NER)
Mismatch Repair (MMR)
Gene expression profiles associated with acute myocardial infarction and risk of cardiovascular death
Background: Genetic risk scores have been developed for coronary artery disease and atherosclerosis, but are not predictive of adverse cardiovascular events. We asked whether peripheral blood expression profiles may be predictive of acute myocardial infarction (AMI) and/or cardiovascular death.
Methods: Peripheral blood samples from 338 subjects aged 62 ± 11 years with coronary artery disease (CAD) were analyzed in two phases (discovery N = 175, and replication N = 163), and followed for a mean 2.4 years for cardiovascular death. Gene expression was measured on Illumina HT-12 microarrays with two different normalization procedures to control technical and biological covariates. Whole genome genotyping was used to support comparative genome-wide association studies of gene expression. Analysis of variance was combined with receiver operating curve and survival analysis to define a transcriptional signature of cardiovascular death.
Results: In both phases, there was significant differential expression between healthy and AMI groups with overall down-regulation of genes involved in T-lymphocyte signaling and up-regulation of inflammatory genes. Expression quantitative trait loci analysis provided evidence for altered local genetic regulation of transcript abundance in AMI samples. On follow-up there were 31 cardiovascular deaths. A principal component (PC1) score capturing covariance of 238 genes that were differentially expressed between deceased and survivors in the discovery phase significantly predicted risk of cardiovascular death in the replication and combined samples (hazard ratio = 8.5, P< 0.0001) and improved the C-statistic (area under the curve 0.82 to 0.91, P= 0.03) after adjustment for traditional covariates.
Conclusions: A specific blood gene expression profile is associated with a significant risk of death in Caucasian subjects with CAD. This comprises a subset of transcripts that are also altered in expression during acute myocardial infarction.
Lecture Contents delivered at Koch Institute for Integrative Cancer Research, Summer Symposium 2014: RNA Biology, Cancer and Therapeutic Implications, June 13, 2014 @MIT
TDP-43 is a transcriptional repressor that binds to chromosomally integrated TAR DNA and represses HIV-1 transcription. In addition, this protein regulates alternate splicing of the CFTR gene. In particular, TDP-43 is a splicing factor binding to the intron8/exon9 junction of the CFTR gene and to the intron2/exon3 region of the apoA-II gene.[2] A similar pseudogene is present on chromosome 20.[3]
TDP-43 has been shown to bind both DNA and RNA and have multiple functions in transcriptional repression, pre-mRNA splicing and translational regulation.
TDP-43 was originally identified as a transcriptional repressor that binds to chromosomally integrated trans-activation response element (TAR) DNA and represses HIV-1 transcription.[1] It was also reported to regulate alternate splicing of theCFTR gene and the apoA-II gene.
In spinal motor neurons TDP-43 has also been shown in humans to be a low molecular weight microfilament (hNFL) mRNA-binding protein.[4] It has also shown to be a neuronal activity response factor in the dendrites of hippocampal neurons suggesting possible roles in regulating mRNA stability, transport and local translation in neurons.[5]
HIV-1, the causative agent of acquired immunodeficiency syndrome (AIDS), contains an RNAgenome that produces a chromosomally integrated DNA during the replicative cycle. Activation of HIV-1 gene expression by the transactivator “Tat” is dependent on an RNA regulatory element (TAR) located “downstream” (i.e. to-be transcribed at a later point in time) of the transcription initiation site.
Mutations in the TARDBP gene are associated with neurodegenerative disorders including frontotemporal lobar degeneration and amyotrophic lateral sclerosis (ALS).[9] In particular, the TDP-43 mutants M337V and Q331K are being studied for their roles in ALS.[10][11] Cytoplasmic TDP-43 pathology is the dominant histopathological feature of multisystem proteinopathy.[12]
General annotation (Comments)
Function
DNA and RNA-binding protein which regulates transcription and
splicing. Involved in the regulation of CFTR splicing. It promotes
CFTR exon 9 skipping by binding to the UG repeated motifs in the
polymorphic region near the 3′-splice site of this exon. The resulting
aberrant splicing is associated with pathological features typical of
cystic fibrosis. May also be involved in microRNA biogenesis,
apoptosis and cell division. Can repress HIV-1 transcription by
binding to the HIV-1 long terminal repeat. Stabilizes the low
molecular weight neurofilament (NFL) mRNA through a direct
interaction with the 3′ UTR. Ref.2Ref.12
Subunit structure
Homodimer. Interacts with BRDT By similarity. Binds specifically to
pyrimidine-rich motifs of TAR DNA and to single stranded TG
repeated sequences. Binds to RNA, specifically to UG repeated
sequences with a minimun of six contiguous repeats. Interacts with
ATNX2; the interaction is RNA-dependent. Ref.16
Subcellular location
Nucleus. Note: In patients with frontotemporal lobar degeneration
and amyotrophic lateral sclerosis, it is absent from the nucleus of
affected neurons but it is the primary component of cytoplasmic
ubiquitin-positive inclusion bodies. Ref.2Ref.11
Tissue specificity
Ubiquitously expressed. In particular, expression is high in pancreas,
placenta, lung, genital tract and spleen.
Domain
The RRM domains can bind to both DNA and RNA By similarity.
Post-translational modification
Hyperphosphorylated in hippocampus, neocortex, and spinal cord
from individuals affected with ALS and FTLDU. Ref.11Ubiquitinated in hippocampus, neocortex, and spinal cord from
individuals affected with ALS and FTLDU. Ref.2Ref.11 Cleaved to
generate C-terminal fragments in hippocampus, neocortex, and
spinal cord from individuals affected with ALS and FTLDU.
Involvement in disease
Amyotrophic lateral sclerosis 10 (ALS10) [MIM:612069]: A
neurodegenerative disorder affecting upper motor neurons in the
brain and lower motor neurons in the brain stem and spinal cord,
resulting in fatal paralysis. Sensory abnormalities are absent. The
pathologic hallmarks of the disease include pallor of the corticospinal
tract due to loss of motor neurons, presence of ubiquitin-positive
inclusions within surviving motor neurons, and deposition of
pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. Note: The disease is caused by mutations affecting the gene represented in this
entry.
Deoxyribonucleic acid (DNA) synthesis is a process by which copies of nucleic acid strands are made. In nature, DNA synthesis takes place in cells by a mechanism known as DNA replication. Using genetic engineering and enzyme chemistry, scientists have developed man-made methods for synthesizing DNA. The most important of these is poly-merase chain reaction (PCR). First developed in the early 1980s, PCR has become a multi-billion dollar industry with the original patent being sold for $300 million dollars.
History
DNA was discovered in 1951 by Francis Crick, James Watson, and Maurice Wilkins. Using x-ray crystallography data generated by Rosalind Franklin, Watson and Crick determined that the structure of DNA was that of a double helix. For this work, Watson, Crick, and Wilkins received the Nobel Prize in Physiology or Medicine in 1962. Over the years, scientists worked with DNA trying to figure out the “code of life.” They found that DNA served as the instruction code for protein sequences. They also found that every organism has a unique DNA sequence and it could be used for screening, diagnostic, and identification purposes. One thing that proved limiting in these studies was the amount of DNA available from a single source.
After the nature of DNA was determined, scientists were able to examine the composition of the cellular genes. A gene is a specific sequence of DNA base pairs that provide the code for the construction of a protein. These proteins determine the traits of an organism, such as eye color or blood type. When a certain gene was isolated, it became desirable to synthesize copies of that molecule. One of the first ways in which a large amount of a specific DNA was synthesized was though genetic engineering.
Genetic engineering begins by combining a gene of interest with a bacterial plasmid. A plasmid is a small stretch of DNA that is found in many bacteria. The resulting hybrid DNA is called recombinant DNA. This new recombinant DNA plasmid is then injected into bacterial cells. The cells are then cloned by allowing it to grow and multiply in a culture. As the cells multiply so do copies of the inserted gene. When the bacteria has multiplied enough, the multiple copies of the inserted gene can then be isolated. This method of DNA synthesis can produce billions of copies of a gene in a couple of weeks.
In 1983, the time required to produce copies of DNA was significantly reduced when Kary Mullis developed a process for synthesizing DNA called polymerase chain reaction (PCR). This method is much faster than previous known methods producing billions of copies of a DNA strand in just a few hours. It begins by putting a small section of double stranded DNA in a solution containing DNA polymerase, nucleotides and primers. The solution is heated to separate the DNA strands. When it is cooled, the polymerase creates a copy of each strand. The process is repeated every five minutes until the desired amount of DNA is produced. In 1993, Mullis’s development of PCR earned him the Nobel Prize in Chemistry.
Background
The key to understanding DNA synthesis is understanding its structure. Typically, DNA exists as two chains of chemically linked nucleotides. These links follow specific patterns dictated by the base pairing rules. Each nucleotide is made up of a deoxyribose sugar molecule, a phosphate group, and one of four nitrogen containing bases. The bases include the pyrimidines thymine (T) and cytosine (C)and the purines adenine (A) and guanine (G). In DNA, adenine generally links with thymine and guanine with cytosine. The molecule is arranged in a structure called a double helix which can be imagined by picturing a twisted ladder or spiral staircase. The bases make up the rungs of the ladder while the sugar and phosphate portions make up the ladder sides. The order in which the nucleotides are linked, called the sequence, is determined by a process known as DNA sequencing.
In a eukaryotic cell, DNA synthesis occurs just prior to cell division through a process called replication. When replication begins the two strands of DNA are separated by a variety of enzymes. Thus opened, each strand serves as a template for producing new strands. This whole process is catalyzed by an enzyme called DNA polymerase. This molecule brings corresponding, or complementary, nucleotides in line with each of the DNA strands. The nucleotides are then chemically linked to form new DNA strands which are exact copies of the original strand. These copies, called the daughter strands, contain half of the parent DNA molecule and half of a whole new molecule. Replication by this method is known as semiconservative replication.
Raw Materials
The primary raw materials used for DNA synthesis include DNA starting materials, taq DNA polymerase, primers, nucleotides, and the buffer solution. Each of these play an important role in the production of millions of DNA molecules.
Controlled DNA synthesis begins by identifying a small segment of DNA to copy. This is typically a specific sequence of DNA that contains the code for a desired protein. Called template DNA, this material must be highly purified.
While the process of DNA replication was known before 1980, PCR was not possible because there were no known heat stable DNA polymerases. In the early 1980s, scientists found bacteria living around natural steam vents. It turned out that these organisms, called thermus aquaticus, had a DNA polymerase that was stable and functional at extreme levels of heat. This taq DNA polymerase became the cornerstone for modern DNA synthesis techniques. During a typical PCR process, 2-3 micrograms of taq DNA polymerase is needed.
The polymerase builds the DNA strands by combining corresponding nucleotides on each DNA strand. Chemically speaking, nucleotides are made up of three types of molecular groups including a sugar structure, a phosphate group, and a cyclic base. The sugar portion provides the primary structure for all nucleotides. In general, the sugars are composed of five carbon atoms with a number of hydroxy (-OH) groups attached. For DNA, the sugar is 2-deoxy-D-ribose. The defining part of a nucleotide is the hetero-cyclic base that is covalently bound to the sugar. These bases are either pyrimidine or purine groups, and they form the basis for the nucleic acid code. Two types of purine bases are found including adenine and guanine. In DNA, two types of pyrimidine bases are present, thymine and cytosine. A phosphate group makes up the final portion of a nucleotide. This group is derived from phosphoric acid and is covalently bonded to the sugar structure on the fifth carbon.
The first phase of polymerase chain reaction (PCR) involves the denaturation of DNA. This “opening up” of the DNA molecule provides the template for the next DNA molecule from which to be produced. With the DNA split into separate strands, the temperature is lowered—the primer annealing step. During the next phase, the DNA polymerase interacts with the strands and adds complementary nucleotides along the entire length. The time required at this phase is about one minute for every 1,000 base pairs.
To initiate DNA synthesis, short primer sections of DNA must be used. These primer sections, called oligo fragments, are about 18-25 nucleotides in length and correspond to a section on the template DNA. They typically have a C and G nucleotide concentration of about 60% with even distribution. This provides the maximum efficiency in the synthesis process.
The buffer solution provides the medium in which DNA synthesis can occur. This is an aqueous solution which contains MgCl2, HCI, EDTA, and KCI. The MgCl2 concentration is important because the Mg2+ ions interact with the DNA and the primers creating crucial complexes for DNA synthesis. The pH of this system is critical so it may also be buffered with ammonium sulfate. To energize the reaction, various energy molecules are added such as ATP, GTP, and NTP.
DNA synthesis involves three distinct processes, typically done in separate areas to avoid contamination, including sample preparation, DNA synthesis reaction cycle and DNA isolation. Following these procedures scientists are able to convert a few strands of DNA into millions and millions of exact copies.
Preparation of the samples
1 Typically, all of the starting solutions except the primers, polymerases and the dNTPs are put in an autoclave to kill off any contaminating organism. Two separate solutions are made. One contains the buffer, primers and the polymerase. The other contains the MgCl2 and the template DNA. These solutions are all put into small tubes to begin the reaction.
Kary Banks Mullis.
Kary Banks Mullis was born in Lenoir, North Carolina, in 1944. Upon graduation from Georgia Tech in 1966 with a B.S. in chemistry, Muilis entered the biochemistry doctoral program at the University of California, Berkeley. Earning his Ph.D. in 1973, he accepted a teaching position at the University of Kansas Medical School in Kansas City. In 1977, he assumed a postdoctoral fellowship at the University of California, San Francisco.
Muilis accepted a position as a research scientist in 1979 with a growing biotech firm—Cetus Corporation, in Emeryville, California—that synthesized chemicals used by other scientists in genetic cloning. While there, he designed polymerase chain reaction (PCR), a fast and effective technique for reproducing specific genes or DNA (deoxyribonucleic acid) fragments that can create billions of copies in a few hours. The most effective way to reproduce DNA was by cloning, but it was problematic. It took time to convince Mullis’s colleagues of the importance of this discovery but soon PCR became the focus of intensive research. Scientists at Cetus developed a commercial version of the process and a machine called the Thermal Cycler (with the addition of the chemical building blocks of DNA [nucleotides] and a biochemical catalyst [polymerase], the machine would perform the process automatically on a target piece of DNA).
This is an up-to-date article about the significance of mutations found in 12 major types of cancer.
Word Cloud by Daniel Menzin
UPDATED 4/24/2020 The genomic landscape of pediatric cancers: Curation of WES/WGS studies shows need for more data
Mutational landscape and significance across 12 major cancer types
Cyriac Kandoth1*, Michael D. McLellan1*, Fabio Vandin2, Kai Ye1,3, Beifang Niu1, Charles Lu1, et al.
1The Genome Institute, Washington University in St Louis, Missouri 63108, USA. 2Department of Computer Science, Brown University, Providence, Rhode Island 02912, USA. 3Department of Genetics, Washington University in St Louis, Missouri 63108, USA. 4Department of Medicine, Washington University in St Louis, Missouri 63108, USA. 5Siteman Cancer Center, Washington University in St Louis, Missouri 63108, USA. 6Department of Mathematics, Washington University in St Louis, Missouri 63108, USA.
The Cancer Genome Atlas (TCGA) has used the latest sequencing and analysis methods to identify somatic variants across thousands of tumours. Here we present data and analytical results for point mutations and small insertions/deletions from 3,281 tumours across 12 tumour typesas part of the TCGA Pan-Cancer effort. We illustrate
Using the integrated data sets, we identified 127 significantly mutated genes from well-knownand emerging cellular processes in cancer.
(for example, mitogen-activated protein kinase, phosphatidylinositol-3-OH kinase,Wnt/b-catenin and receptor tyrosine kinase signalling pathways, and cell cycle control)
(for example, histone, histone modification, splicing, metabolism and proteolysis)
The average number of mutations in these significantly mutated genes varies across tumour types;
most tumours have two to six, indicating that the number of driver mutations required during oncogenesis is relatively small.
Mutations in transcriptional factors/regulators show tissue specificity, whereas
histone modifiers are often mutated across several cancer types.
Clinical association analysis identifies genes having a significant effect on survival, and
investigations of mutations with respect to clonal/subclonal architecture delineate their temporal orders during tumorigenesis.
Taken together, these results lay the groundwork for developing new diagnostics and individualizing cancer treatment
Introduction
The advancement of DNA sequencing technologies now enables the processing of thousands of tumours of many types for systematic mutation discovery. This expansion of scope, coupled with appreciable progress in algorithms1–5, has led directly to characterization of significant functional mutations, genes and pathways6–18. Cancer encompasses more than 100 related diseases19, making it crucial to understand the commonalities and differences among various types and subtypes. TCGA was founded to address these needs, and its large data sets are providing unprecedented opportunities for systematic, integrated analysis.
We performed a systematic analysis of 3,281 tumours from 12 cancer types to investigate underlying mechanisms of cancer initiation and progression. We describe variable mutation frequencies and contexts and their associations with environmental factors and defects in DNA repair. We identify 127 significantlymutated genes (SMGs) from diverse signalling and enzymatic processes. The finding of a TP53-driven breast, head and neck, and ovarian cancer cluster with a dearth of other mutations in SMGs suggests common therapeutic strategies might be applied for these tumours. We determined interactions among mutations and correlated mutations in BAP1, FBXW7 and TP53 with detrimental phenotypes across several cancer types. The subclonal structure and transcription status of underlying somatic mutations reveal the trajectory of tumour progression in patients with cancer.
Standardization of mutation data
Stringent filters (Methods) were applied to ensure high quality mutation calls for 12 cancer types: breast adenocarcinoma (BRCA), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), uterine corpus endometrial carcinoma (UCEC), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), colon and rectal carcinoma (COAD, READ),bladder urothelial carcinoma (BLCA), kidney renal clear cell carcinoma (KIRC), ovarian serous carcinoma (OV) and acute myeloid leukaemia (LAML; conventionally called AML) (Supplementary Table 1). A total of 617,354 somatic mutations, consisting of
15,141 frameshift insertions/deletions (indels) and
3,538 inframe indels,
were included for downstream analyses (Supplementary Table 2).
Distinct mutation frequencies and sequence context
Figure 1a shows that AML has the lowest median mutation frequency and LUSC the highest (0.28 and 8.15 mutations per megabase (Mb), respectively). Besides AML, all types average over 1 mutation per Mb, substantially higher than in pediatric tumours20. Clustering21 illustrates that
mutation frequencies for KIRC, BRCA, OV and AML are normally distributed within a single cluster, whereas
other types have several clusters (for example, 5 and 6 clusters in UCEC and COAD/ READ, respectively) (Fig. 1a and Supplementary Table 3a, b).
In UCEC, the largest patient cluster has a frequency of approximately 1.5 mutations per Mb, and
the cluster with the highest frequency is more than 150 times greater.
Multiple clusters suggest that factors other than age contribute to development in these tumours14,16. Indeed,
there is a significant correlation between high mutation frequency and DNA repair pathway genes (for example, PRKDC, TP53 and MSH6) (Supplementary Table 3c). Notably,
PRKDC mutations are associated with high frequency in BLCA, COAD/READ, LUAD and UCEC, whereas
TP53 mutations are related with higher frequencies in AML, BLCA, BRCA, HNSC, LUAD, LUSC and UCEC (all P < 0.05).
Mutations in POLQ and POLE associate with high frequencies in multiple cancer types; POLE association in UCEC is consistent with previous observations14.
Comparison of spectra across the 12 types (Fig. 1b and Supplementary Table 3d) reveals that LUSC and LUAD contain increased C>A transversions, a signature of cigarette smoke exposure10. Sequence context analysis across 12 types revealed
the largest difference being in C>T transitions and C>G transversions (Fig. 1c).
The frequency of thymine 1-bp (base pair)upstream of C>G transversions is markedly higher in BLCA, BRCA and HNSC than in other cancer types (Extended Data Fig. 1). GBM, AML, COAD/READ and UCEC have similar contexts in that
the proportions of guanine 1 base downstreamof C>T transitions are between
59% and 67%, substantially higher than the approximately 40% in other cancer types.
Higher frequencies of transition mutations at CpG in gastrointestinal tumours, including colorectal, were previously reported22. We found three additional cancer types (GBM, AML and UCEC) clustered in the C>T mutation at CpG, consistent with previous findings of
aberrant DNA methylation in endometrial cancer23 and glioblastoma24.
BLCA has a unique signature for C>T transitions compared to the other types (enriched for TC) (Extended Data Fig. 1).
Significantly mutated genes
Genes under positive selection, either in individual or multiple tumour types, tend to display higher mutation frequencies above background. Our statistical analysis3, guided by expression data and curation (Methods), identified 127 such genes (SMGs; Supplementary Table 4). These SMGs are involved in a wide range of cellular processes, broadly classified into 20 categories (Fig. 2), including
The identification of MAPK, PI(3)K and Wnt/ -catenin signaling pathways is consistent with classical cancer studies. Notably, newer categories (for example, splicing, transcription regulators, metabolism, proteolysis and histones) emerge as exciting guides for the development of new therapeutic targets. Genes categorized as histone modifiers (Z = 0.57), PI(3)K signalling (Z = 1.03), and genome integrity (Z = 0.66) all relate to more than one cancer type, whereas
transcription factor/regulator (Z = 0.40), TGF- signalling (Z = 0.66), and Wnt/ -catenin signalling (Z = 0.55) genes tend to associate with single types (Methods).
Notably, 3,053 out of 3,281 total samples (93%) across the Pan-Cancer collection had at least one non-synonymous mutation in at least one SMG. The average number of point mutations and small indels in these genes varies across tumour types, with the highest (,6 mutations per tumour) in UCEC, LUAD and LUSC, and the lowest (,2 mutations per tumour) in AML, BRCA, KIRC and OV. This suggests that the numbers of both cancer-related genes (only 127 identified in this study) and cooperating driver mutations required during oncogenesis are small (most cases only had 2–6) (Fig. 3), although large-scale structural rearrangements were not included in this analysis.
Common mutations
The most frequently mutated gene in the Pan-Cancer cohort is TP53 (42% of samples). Its mutations predominate in serous ovarian (95%) and serous endometrial carcinomas (89%) (Fig. 2). TP53mutations are also associated with basal subtype breast tumours. PIK3CA is the second most commonly mutated gene, occurring frequently (>10%) in most cancer types except OV, KIRC, LUAD and AML. PIK3CA mutations frequented UCEC (52%) and BRCA (33.6%), being specifically enriched in luminal subtype tumours. Tumours lacking PIK3CA mutations often had mutations inPIK3R1, with the highest occurrences in UCEC (31%) and GBM (11%) (Fig. 2).
Many cancer types carried mutations in chromatin re-modelling genes. In particular, histone-lysine N-methyltransferase genes (MLL2 (also known as KMT2D), MLL3 (KMT2C) and MLL4 (KMT2B)) cluster in bladder, lung and endometrial cancers, whereas the lysine (K)-specific demethylase KDM5C is prevalently mutated in KIRC (7%). Mutations in ARID1A are frequent in BLCA, UCEC, LUAD and LUSC, whereas mutations inARID5B predominate in UCEC (10%) (Fig. 2).
Fig. 1. | Distribution of mutation frequencies across 12 cancer types.
Dashed grey and solid white lines denote average across cancer types and median for each type, respectively. b, Mutation spectrum of six transition (Ti) and transversion (Tv) categories for each cancer type. c, Hierarchically clustered mutation context (defined by the proportion of A, T, C and G nucleotides within ±2bp of variant site) for six mutation categories. Cancer types correspond to colours in a. Colour denotes degree of correlation: yellow (r = 0.75) and red (r = 1).
Fig. 2. The 127 SMGs from 20 cellular processes in cancer identified in and Pan-Cancer are shown, with the highest percentage in each gene among 12 (not shown)
Fig. 3. | Distribution of mutations in 127 SMGs across Pan-Cancer cohort.
Box plot displays median numbers of non-synonymous mutations, with outliers shown as dots. In total, 3,210 tumours were used for this analysis (hypermutators excluded).
Figure 4 | Unsupervised clustering based on mutation status of SMGs. Tumours having no mutation or more than 500 mutations were excluded. A mutation status matrix was constructed for 2,611 tumours. Major clusters of mutations detected in UCEC, COAD, GBM, AML, KIRC, OV and BRCA were highlighted.
Complete gene list shown in Extended Data Fig. 3. (not shown)
Figure 5 | Driver initiation and progression mutations and tumour clonal mutation is in the subclone
Survival Analysis
We examined which genes correlate with survival using the Cox proportional hazards model, first analysing individual cancer types using age and gender as covariates; an average of 2 genes (range: 0–4) with mutation frequency 2% were significant (P<_0.05) in each type (Supplementary Table 10a and Extended Data Fig. 6). KDM6A and ARID1A mutations correlate with better survival in BLCA (P = 0.03, hazard ratio (HR) = 0.36, 95% confidence interval (CI): 0.14–0.92) and UCEC (P = 0.03, HR = 0.11, 95% CI: 0.01–0.84), respectively, but mutations in SETBP1, recently identified with worse prognosis in atypical chronic myeloid leukaemia (aCML)31, have a significant detrimental effect in HNSC (P = 0.006, HR = 3.21, 95% CI: 1.39–7.44). BAP1 strongly correlates with poor survival (P = 0.00079, HR = 2.17, 95% CI: 1.38–3.41) in KIRC. Conversely, BRCA2 mutations (P = 0.02, HR = 0.31, 95% CI: 0.12–0.85) associate with better survival in ovarian cancer, consistent with previous reports32,33; BRCA1 mutations showed positive correlation with better survival, but did not reach significance here.
We extended our survival analysis across cancer types, restricting our attention to the subset of 97 SMGs whose mutations appeared in 2% of patients having survival data in 2 tumour types. Taking type, age and gender as covariates, we found 7 significant genes: BAP1, DNMT3A, HGF, KDM5C, FBXW7, BRCA2 and TP53 (Extended Data Table 1). In particular,BAP1 was highly significant (P = 0.00013, HR = 2.20, 95% CI: 1.47–3.29, more than 53 mutated tumours out of 888 total), with mutations associating with detrimental outcome in four tumour typesand notable associations in KIRC(P = 0.00079), consistent with a recent report28, and in UCEC(P = 0.066). Mutations in several other genes are detrimental, including DNMT3A (HR = 1.59), previously identified with poor prognosis in AML34, and KDM5C(HR = 1.63), FBXW7 (HR = 1.57) and TP53(HR = 1.19).TP53 has significant associations with poor outcome in KIRC (P = 0.012), AML(P = 0.0007) and HNSC (P = 0.00007). Conversely, BRCA2(P = 0.05, HR = 0.62, 95% CI: 0.38 to 0.99) correlates with survival benefit in six types, including OV and UCEC (Supplementary Table 10a, b).IDH1 mutations are associated with improved prognosis across the Pan-Cancer set (HR = 0.67, P = 0.16) and also in GBM (HR = 0.42, P = 0.09) (Supplementary Table 10a, b), consistent with previous work.35
Driver mutations and tumour clonal architecture
To understand the temporal order of somatic events, we analysed the variant allele fraction (VAF) distribution of mutations in SMGs across AML, BRCA and UCEC (Fig. 5a and Supplementary Table 11a) and other tumour types (Extended Data Fig. 7). To minimize the effect of copy number alterations, we focused on mutations in copy neutral segments. Mutations in TP53 have higher VAFs on average in all three cancer types, suggesting early appearance during tumorigenesis.
It is worth noting that copy neutral loss of heterozygosity is commonly found in classical tumour suppressors such as TP53, BRCA1, BRCA2 and PTEN, leading to increased VAFs in these genes. In AML, DNMT3A (permutation test P = 0), RUNX1 (P = 0.0003) and SMC3 (P = 0.05) have significantly higher VAFs than average among SMGs (Fig. 5a and Supplementary Table 11b). In breast cancer, AKT1, CBFB, MAP2K4, ARID1A, FOXA1 and PIK3CA have relatively high average VAFs. For endometrial cancer, multiple SMGs (for example, PIK3CA, PIK3R1, PTEN, FOXA2 and ARID1A) have similar median VAFs. Conversely, KRAS and/or NRAS mutations tend to have lower VAFs in all three tumour types (Fig. 5a), suggesting NRAS (for example, P = 0 in AML) and KRAS (for example, P = 0.02 in BRCA) have a progression role in a subset of AML, BRCA and UCEC tumours. For all three cancer types, we clearly observed a shift towards higher expression VAFs in SMGs versus non-SMGs, most apparent in BRCA and UCEC (Extended Data Fig. 8a and Methods).
Previous analysis using whole-genome sequencing (WGS) detected subclones in approximately 50% of AML cases15,36,37; however, analysis is difficult using AML exome owing to its relatively few coding mutations. Using 50 AML WGS cases, sciClone (http://github.com/ genome/sciclone) detected DNMT3A mutations in the founding clone for 100% (8 out of 8) of cases and NRAS mutations in the subclone for 75% (3 out of 4) of cases (Extended Data Fig. 8b). Among 304 and 160 of BRCA and UCEC tumours, respectively, with enough coding mutations for clustering, 35% BRCA and 44% UCEC tumours contained subclones. Our analysis provides the lower bound for tumour heterogeneity, because only coding mutations were used for clustering. In BRCA, 95% (62 out of 65) of cases contained PIK3CA mutations in the founding clone, whereas 33% (3 out of 9) of cases had MLL3 mutations in the subclone. Similar patterns were found in UCEC tumours, with 96% (65 out of 68) and 95% (62 out of 65) of tumours containing PIK3CA and PTEN mutations, respectively, in the founding clone, and 9% (2 out of22) ofKRAS and 14% (1 out of 7) ofNRAS mutations in the subclone (Extended Data Fig. 8b and Supplementary Table 12).
Mutation context (-2 to +2 bp) was calculated for each somatic variant in each mutation category, and hierarchical clustering was then performed using the pairwise mutation context correlation across all cancer types. The mutational significance in cancer (MuSiC)3 package was used to identify significant genes for both individual tumour types and the Pan-Cancer collective. An R function ‘hclust’ was used for complete-linkage hierarchical clustering across mutations and samples, and Dendrix30 was used to identify sets of approximately mutual exclusive mutations. Cross-cancer survival analysis was based on the Cox proportional hazards model, as implemented in the R package ‘survival’ (http://cran.r-project.org/web/ packages/survival/), and the sciClone algorithm (http://github.com/genome/sci-clone) generated mutation clusters using point mutations from copy number neutral segments. A complete description of the materials and methods used to generate this data set and its results is provided in the Methods.
References (20 of 38)
Larson, D. E. et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2012).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Dees, N. D. et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 22, 1589–1598 (2012).
Roth, A. et al. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 28, 907–913 (2012).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnol. 31, 213–219 (2013).
Jones, S. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806 (2008).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008).
Sjo¨blom, T. etal. The consensuscodingsequences of human breast and colorectal cancers. Science 314, 268–274 (2006).
The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008).
Wood, L. D. etal. The genomic landscapesof human breast and colorectal cancers. Science 318, 1108–1113 (2007).
The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013).
The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Ellis, M. J. et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360 (2012).
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 (2013).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Downing, J. R. et al. The Pediatric Cancer Genome Project. Nature Genet. 44, 619–622 (2012).
UPDATED 4/24/2020 The genomic landscape of pediatric cancers: Curation of WES/WGS studies shows need for more data
The past decade has witnessed a major increase in our understanding of the genetic underpinnings of childhood cancer. Genomic sequencing studies have highlighted key differences between pediatric and adult cancers. Whereas many adult cancers are characterized by a high number of somatic mutations, pediatric cancers typically have few somatic mutations but a higher prevalence of germline alterations in cancer predisposition genes. Also noteworthy is the remarkable heterogeneity in the types of genetic alterations that likely drive the growth of pediatric cancers, including copy number alterations, gene fusions, enhancer hijacking events, and chromoplexy. Because most studies have genetically profiled pediatric cancers only at diagnosis, the mechanisms underlying tumor progression, therapy resistance, and metastasis remain poorly understood. We discuss evidence that points to a need for more integrative approaches aimed at identifying driver events in pediatric cancers at both diagnosis and relapse. We also provide an overview of key aspects of germline predisposition for cancer in this age group.
Approximately 300,000 children from infancy to age 14 are diagnosed with cancer worldwide every year (1). Some of the cancer types affecting the pediatric population are also seen in adolescents and young adults (AYA), but it has become increasingly clear that cancers in the latter age group have unique biological characteristics that can affect prognosis and therapy (2). Pediatric and AYA cancer patients present with a heterogeneous set of diseases that can be broadly subclassified as leukemias, brain tumors, and non–central nervous system (CNS) solid tumors. These subgroups contain numerous distinct clinical entities, many of which are still poorly characterized from a molecular standpoint.
Recent large-scale genomic analyses have increased our understanding of the genetic drivers of pediatric cancer and have helped to identify new clinically relevant subtypes. These studies have also underscored the distinct nature of the genetic alterations in pediatric and AYA cancers versus adult cancers. Of particular note, the number of somatic mutations in most pediatric cancers is substantially lower than that in adult cancers (3, 4). Exceptions are tumors in children who carry germline mutations that compromise repair of DNA damage (5). For many pediatric cancers, driver events are conditioned on the developmental stage in which the tumor arises. For example, a mutation occurring in one developmental compartment (e.g., a muscle stem cell) may lead to cancer, whereas the same mutation in another compartment does not (6). Pediatric cancer genomes are also characterized by specific patterns of copy number alterations and structural alterations [chromoplexy (7), chromothripsis (8)] that are prognostic indicators in several cancer subtypes. Gene fusion events have long been recognized as oncogenic drivers in many pediatric cancers; however, advanced sequencing technologies have revealed that the number of fusion partners is greater than previously thought, and that previously undetected gene rearrangements may also function as drivers. Finally, germline mutations in a wide spectrum of genes that predispose to cancer appear to play a greater role in pediatric cancer than previously appreciated (9, 10).
Somatic alterations in pediatric cancers
Genome landscape studies
Early large-scale sequencing studies of pediatric cancers identified novel driver genes while also underscoring the overall low mutational burden (11–14). Whole exome sequencing studies of Wilms tumor, T-cell acute lymphoblastic leukemia (TALL), and acute myeloid leukemia (CML) identified some recurring mutations such as
FLT3-IDT
WT1
NUP98-NST1 gene fusion
however many of the driver genes were subtype specific. Other fusion events were seen (by RNASeq) such as
EWS-FL1
Bcr-Abl
MYB-QK1
as well as multiple epigenetic events such as methylations.
REFERENCES
E. Steliarova-Foucher, M. Colombet, L. A. G. Ries, F. Moreno, A. Dolya, F. Bray, P. Hesseling, H. Y. Shin, C. A. Stiller, IICC-3 contributors, International incidence of childhood cancer, 2001-10: A population-based registry study. Lancet Oncol. 18, 719–731 (2017). 10.1016/S1470-2045(17)30186-9pmid:28410997
2. V. Tricoli, D. G. Blair, C. K. Anders, W. A. Bleyer, L. A. Boardman, J. Khan, S. Kummar, B. Hayes-Lattin, S. P. Hunger, M. Merchant, N. L. Seibel, M. Thurin, C. L. Willman, Biologic and clinical characteristics of adolescent and young adult cancers: Acute lymphoblastic leukemia, colorectal cancer, breast cancer, melanoma, and sarcoma. Cancer 122, 1017–1028 (2016). 10.1002/cncr.29871pmid:26849082
3. S. Lawrence, P. Stojanov, P. Polak, G. V. Kryukov, K. Cibulskis, A. Sivachenko, S. L. Carter, C. Stewart, C. H. Mermel, S. A. Roberts, A. Kiezun, P. S. Hammerman, A. McKenna, Y. Drier, L. Zou, A. H. Ramos, T. J. Pugh, N. Stransky, E. Helman, J. Kim, C. Sougnez, L. Ambrogio, E. Nickerson, E. Shefler, M. L. Cortés, D. Auclair, G. Saksena, D. Voet, M. Noble, D. DiCara, P. Lin, L. Lichtenstein, D. I. Heiman, T. Fennell, M. Imielinski, B. Hernandez, E. Hodis, S. Baca, A. M. Dulak, J. Lohr, D.-A. Landau, C. J. Wu, J. Melendez-Zajgla, A. Hidalgo-Miranda, A. Koren, S. A. McCarroll, J. Mora, B. Crompton, R. Onofrio, M. Parkin, W. Winckler, K. Ardlie, S. B. Gabriel, C. W. M. Roberts, J. A. Biegel, K. Stegmaier, A. J. Bass, L. A. Garraway, M. Meyerson, T. R. Golub, D. A. Gordenin, S. Sunyaev, E. S. Lander, G. Getz, G. Getz, Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). 10.1038/nature12213pmid:23770567
B. Vogelstein, N. Papadopoulos, V. E. Velculescu, S. Zhou, L. A. Diaz Jr.., K. W. Kinzler, Cancer genome landscapes. Science 339, 1546–1558 (2013). 10.1126/science.1235122pmid:23539594
5. B. Campbell, N. Light, D. Fabrizio, M. Zatzman, F. Fuligni, R. de Borja, S. Davidson, M. Edwards, J. A. Elvin, K. P. Hodel, W. J. Zahurancik, Z. Suo, T. Lipman, K. Wimmer, C. P. Kratz, D. C. Bowers, T. W. Laetsch, G. P. Dunn, T. M. Johanns, M. R. Grimmer, I. V. Smirnov, V. Larouche, D. Samuel, A. Bronsema, M. Osborn, D. Stearns, P. Raman, K. A. Cole, P. B. Storm, M. Yalon, E. Opocher, G. Mason, G. A. Thomas, M. Sabel, B. George, D. S. Ziegler, S. Lindhorst, V. M. Issai, S. Constantini, H. Toledano, R. Elhasid, R. Farah, R. Dvir, P. Dirks, A. Huang, M. A. Galati, J. Chung, V. Ramaswamy, M. S. Irwin, M. Aronson, C. Durno, M. D. Taylor, G. Rechavi, J. M. Maris, E. Bouffet, C. Hawkins, J. F. Costello, M. S. Meyn, Z. F. Pursell, D. Malkin, U. Tabori, A. Shlien, Comprehensive Analysis of Hypermutation in Human Cancer. Cell 171, 1042–1056.e10 (2017). 10.1016/j.cell.2017.09.048pmid:29056344
6. Chen, A. Pappo, M. A. Dyer, Pediatric solid tumor genomics and developmental pliancy. Oncogene 34, 5207–5215 (2015). 10.1038/onc.2014.474pmid:25639868
S. C. Baca, D. Prandi, M. S. Lawrence, J. M. Mosquera, A. Romanel, Y. Drier, K. Park, N. Kitabayashi, T. Y. MacDonald, M. Ghandi, E. Van Allen, G. V. Kryukov, A. Sboner, J.-P. Theurillat, T. D. Soong, E. Nickerson, D. Auclair, A. Tewari, H. Beltran, R. C. Onofrio, G. Boysen, C. Guiducci, C. E. Barbieri, K. Cibulskis, A. Sivachenko, S. L. Carter, G. Saksena, D. Voet, A. H. Ramos, W. Winckler, M. Cipicchio, K. Ardlie, P. W. Kantoff, M. F. Berger, S. B. Gabriel, T. R. Golub, M. Meyerson, E. S. Lander, O. Elemento, G. Getz, F. Demichelis, M. A. Rubin, L. A. Garraway, Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013). 10.1016/j.cell.2013.03.021pmid:23622249
P. J. Stephens, C. D. Greenman, B. Fu, F. Yang, G. R. Bignell, L. J. Mudie, E. D. Pleasance, K. W. Lau, D. Beare, L. A. Stebbings, S. McLaren, M.-L. Lin, D. J. McBride, I. Varela, S. Nik-Zainal, C. Leroy, M. Jia, A. Menzies, A. P. Butler, J. W. Teague, M. A. Quail, J. Burton, H. Swerdlow, N. P. Carter, L. A. Morsberger, C. Iacobuzio-Donahue, G. A. Follows, A. R. Green, A. M. Flanagan, M. R. Stratton, P. A. Futreal, P. J. Campbell, Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011). 10.1016/j.cell.2010.11.055pmid:21215367
D. W. Parsons, A. Roy, Y. Yang, T. Wang, S. Scollon, K. Bergstrom, R. A. Kerstein, S. Gutierrez, A. K. Petersen, A. Bavle, F. Y. Lin, D. H. López-Terrada, F. A. Monzon, M. J. Hicks, K. W. Eldin, N. M. Quintanilla, A. M. Adesina, C. A. Mohila, W. Whitehead, A. Jea, S. A. Vasudevan, J. G. Nuchtern, U. Ramamurthy, A. L. McGuire, S. G. Hilsenbeck, J. G. Reid, D. M. Muzny, D. A. Wheeler, S. L. Berg, M. M. Chintagumpala, C. M. Eng, R. A. Gibbs, S. E. Plon, Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. 2, 616 (2016). 10.1001/jamaoncol.2015.5699pmid:26822237
J. Zhang, M. F. Walsh, G. Wu, M. N. Edmonson, T. A. Gruber, J. Easton, D. Hedges, X. Ma, X. Zhou, D. A. Yergeau, M. R. Wilkinson, B. Vadodaria, X. Chen, R. B. McGee, S. Hines-Dowell, R. Nuccio, E. Quinn, S. A. Shurtleff, M. Rusch, A. Patel, J. B. Becksfort, S. Wang, M. S. Weaver, L. Ding, E. R. Mardis, R. K. Wilson, A. Gajjar, D. W. Ellison, A. S. Pappo, C.-H. Pui, K. E. Nichols, J. R. Downing, Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 373, 2336–2346 (2015). 10.1056/NEJMoa1508054pmid:26580448
T. J. Pugh, O. Morozova, E. F. Attiyeh, S. Asgharzadeh, J. S. Wei, D. Auclair, S. L. Carter, K. Cibulskis, M. Hanna, A. Kiezun, J. Kim, M. S. Lawrence, L. Lichenstein, A. McKenna, C. S. Pedamallu, A. H. Ramos, E. Shefler, A. Sivachenko, C. Sougnez, C. Stewart, A. Ally, I. Birol, R. Chiu, R. D. Corbett, M. Hirst, S. D. Jackman, B. Kamoh, A. H. Khodabakshi, M. Krzywinski, A. Lo, R. A. Moore, K. L. Mungall, J. Qian, A. Tam, N. Thiessen, Y. Zhao, K. A. Cole, M. Diamond, S. J. Diskin, Y. P. Mosse, A. C. Wood, L. Ji, R. Sposto, T. Badgett, W. B. London, Y. Moyer, J. M. Gastier-Foster, M. A. Smith, J. M. Guidry Auvil, D. S. Gerhard, M. D. Hogarty, S. J. M. Jones, E. S. Lander, S. B. Gabriel, G. Getz, R. C. Seeger, J. Khan, M. A. Marra, M. Meyerson, J. M. Maris, The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284 (2013). 10.1038/ng.2529pmid:23334666
J. R. Downing, R. K. Wilson, J. Zhang, E. R. Mardis, C.-H. Pui, L. Ding, T. J. Ley, W. E. Evans, The Pediatric Cancer Genome Project. Nat. Genet. 44, 619–622 (2012). 10.1038/ng.2287pmid:22641210
St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project, Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012). 10.1038/ng.1102pmid:22286216
J. Zhang, L. Ding, L. Holmfeldt, G. Wu, S. L. Heatley, D. Payne-Turner, J. Easton, X. Chen, J. Wang, M. Rusch, C. Lu, S.-C. Chen, L. Wei, J. R. Collins-Underwood, J. Ma, K. G. Roberts, S. B. Pounds, A. Ulyanov, J. Becksfort, P. Gupta, R. Huether, R. W. Kriwacki, M. Parker, D. J. McGoldrick, D. Zhao, D. Alford, S. Espy, K. C. Bobba, G. Song, D. Pei, C. Cheng, S. Roberts, M. I. Barbato, D. Campana, E. Coustan-Smith, S. A. Shurtleff, S. C. Raimondi, M. Kleppe, J. Cools, K. A. Shimano, M. L. Hermiston, S. Doulatov, K. Eppert, E. Laurenti, F. Notta, J. E. Dick, G. Basso, S. P. Hunger, M. L. Loh, M. Devidas, B. Wood, S. Winter, K. P. Dunsmore, R. S. Fulton, L. L. Fulton, X. Hong, C. C. Harris, D. J. Dooling, K. Ochoa, K. J. Johnson, J. C. Obenauer, W. E. Evans, C.-H. Pui, C. W. Naeve, T. J. Ley, E. R. Mardis, R. K. Wilson, J. R. Downing, C. G. Mullighan, The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012). 10.1038/nature10725pmid:22237106

































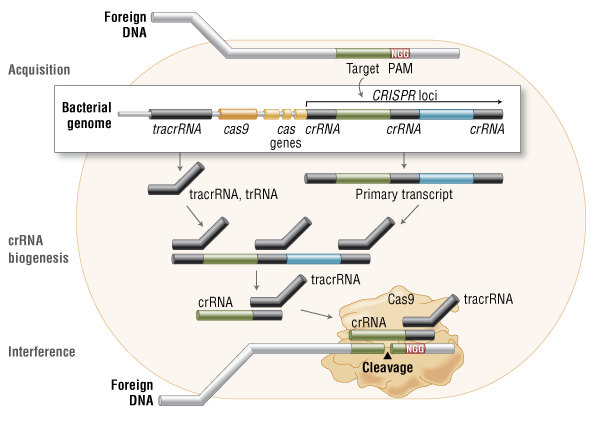{kind=link}
{kind=link}
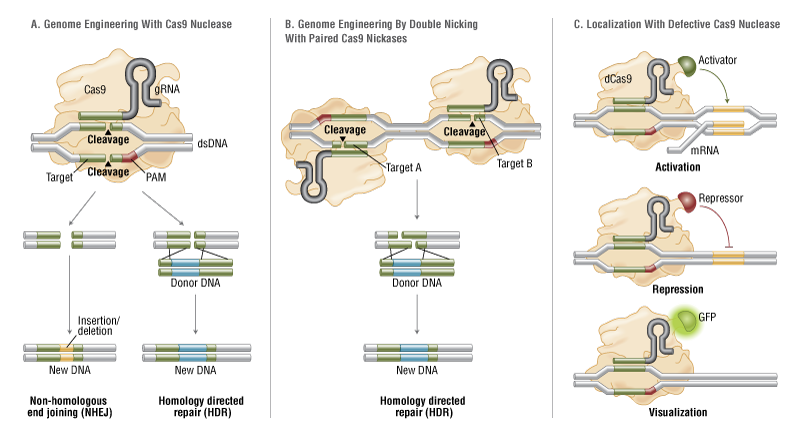{kind=link}
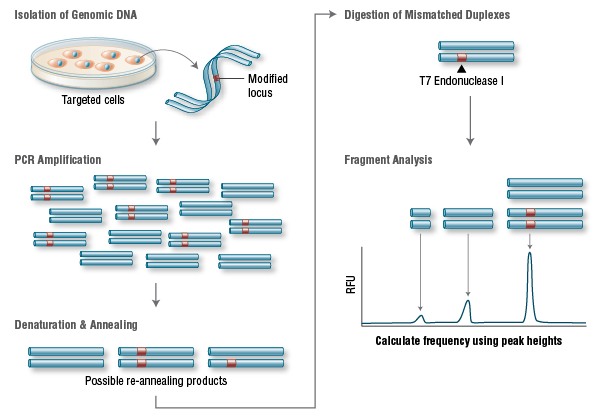{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}