Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Live Conference Coverage: International Dialogue in Gynecological Oncology, From Bench to Bedside, Ovarian Cancer
Reporter: Stephen J. Williams, Ph.D.
Join Live on Wednesday May 22, 2024 for an international discussion on the current state of ovarian cancer diagnostics and therapeutics, and potential therapies and biomarkers, and biotargets. Topics including potential new molecular targets for development of ovarian therapeutics, current changes in ovarian cancer clinical treatment protocols, chemo-resistance, and the use of Artificial Intelligence (AI) in the diagnosis and treatment of cancer will be discussed.
10/15.10 We Have Never Been Only Human: a new perspective to defeat ovarian cancer (C. Martinelli)
Molecular Section
20/15.20 DNA Repair mechanisms: understanding their role in cancer development and chemoresistance (L. Alfano)
35/15.35 Progranulins: a new target for oncological treatment (A. Morrione)
50/15.50 Modulation of gene expression and its applications (M. Cuomo)
10.05/16.05 Commanding the cell cycle: the role of CDKs (S.R. Burk
10.20/16.20 Drug development from nature (M. D’Angelo
Clinical Section
05/17.05 Core principles of Radiologic Diagnosis & Staging in Ovarian Cancer(A. Blandino)
20/17.20 Key Indications for Nuclear Medicine in Ovarian Cancer (S. Baldari)
35/17.35 Cutting Edge Decision: Understanding Surgical Indications and Outcomes in Ovarian Cancer (A. Ercoli)
50/17.50 Gold Standard in Oncology for Ovarian Cancer (N. Silvestris)
12.05/18.05 Role of Radiotherapy in Ovarian Cancer (S. Pergolizzi)
Conclusion
12.20/18.20 AI Applied to medical science (V. Carnevale)
Speakers
– Professor Alfredo Blandino: Professor Blandino holds the esteemed positions of Head of school of Radiology and director of the department of radiology at the University of Messina. He has made significant contributions to diagnostic imaging with over hundreds of publications to his name, Professor Blandino’s work exemplifies excellence and innovation in radiology.
– Professor Alfredo Ercoli, serves as the Director of the Department of Gynecology and Obstetrics at the “G. Martino” University Hospital in Messina. He is also head of school of gynecology and obstetrics at Messina University. Starting his research in France with studies on pelvic anatomy that became a cornerstone in medical literature, He is a pioneer in advanced gynecologic surgery, including laparoscopic and robotic procedures, having performed over thousands of surgical interventions. His research focuses on gynecologic oncology, advanced gynecologic surgery, and endometriosis, urogynecology. Professor Ercoli’s dedication to education and his numerous publications have significantly advanced the field of gynecology.
–Professor Sergio Baldari, an eminent figure in nuclear medicine. Professor Baldari is the Director of the department of nuclear medicine and head of school of nuclear medicine at the University of Messina. He has authored or co-authored over 500 publications, with a focus on diagnostic imaging and the use of PET and radiopharmaceuticals in cancer treatment. His leadership and expertise have been recognized through various prestigious positions and awards within the medical community.
– Professor Nicola Silvestris is the Director of UOC Oncologia Medica at the University of Messina. His extensive research in cancer, has led to over 360 peer-reviewed publications. Professor Silvestris has made significant contributions to translational research and the development of guidelines for managing complex oncological conditions. His work continues to shape the future of cancer treatment.
–Professor Stefano Pergolizzi, a leading expert in radiation oncology. Professor Pergolizzi serves as the Director of the department of radiotherapy and head of the school of radiotherapya at the University of Messina. He is also the president of the Italian Association of Radiotherapy and Clinical Oncology (AIRO) His research focuses on advanced radiotherapy techniques for cancer treatment. With a career spanning several decades, Professor Pergolizzi has published numerous papers and has been instrumental in developing innovative therapeutic approaches. His dedication to patient care and education is exemplary.
Margherita D’angelo: Graduated in Molecular Biology with honors from the Federico II University of Naples.
Third year intern in Food Science at the Luigi Vanvitelli University of Naples.
Research intern in Molecular oncology with the project of developing novel drugs starting from food waste at the Sbarro Institute for Cancer Research and Molecular Medicine at Temple University, Philadelphia (USA), directed by Dr A. Giordano.
Dr. Carnevale is an Associate Professor in the Institute for Computational Molecular Science in the College of Science & Technology, Temple University. He holds multiple NIH RO1 and NSF grants. Vincenzo Carnevale received B.Sc. and M.Sc. degrees in Physics from the University of Pisa and a PhD from SISSA – Scuola Internazionale Superiore di Studi Avanzati in Trieste, Italy. The Carnevale research group uses statistical physics and machine learning approaches to investigate sequence-structure-function relations in proteins. A central theme of the group’s research is how interactions give rise to collective phenomena and complex emergent behaviors. At the level of genes, the group is interested in epistasis – the complex entanglement phenomenon that causes amino acids to evolve in a concerted fashion – and how this shapes molecular evolution. At the cellular level, the group investigates how intermolecular interactions drive biomolecules toward self-organization and pattern formation. A long-term goal of the group is understanding the molecular underpinnings of electrical signaling in excitable cells. Toward these goals, the group applies and actively develops an extensive arsenal of theoretical and computational approaches including statistical (mean)field theories, Monte Carlo and molecular dynamics simulations, statistical inference of generative models, and deep learning.
Professor Andrea Morrione, Ph.D: Research Associate Professor, CST Temple University; After his studies in Biochemistry at Universita’ degli Studi Milano, Milan Italy, Dr. Morrione moved to USA in 1993 and has been working in the field of cancer biology since his postdoctoral training at the Kimmel Cancer Institute, Thomas Jefferson University, Philadelphia, PA in the laboratory of Dr. Renato Baserga, one of the leading experts in IGF-IR oncogenic signaling. In 1997 Dr. Morrione joined the Faculty of Thomas Jefferson University in the Department of Microbiology. In 2002 after receiving an NIH/NIDDK Career Development Award Dr. Morrione joined the Department of Urology at Jefferson where from 2008 to 2018 serves as the Director for Urology Basic Science and Associate Professor. Dr. Morrione joined the Department of Biology and the Sbarro Institute for Cancer Research and Molecular Medicine and Center for Biotechnology as Associate Professor of Research, and he is currently professor of Research and Deputy Director of the Sbarro Institute for Cancer Research and Molecular Medicine and Center for Biotechnology. He is a full member of the AACR.
Canio Martinelli, M.D.: Dr. Marinelli received his MD from Catholic University of the Sacred Heart in Rome, Visiting researcher at SHRO Temple University in Philadelphia, PhD candidate in Translational Molecular Medicine and Surgery & GYN-OB resident at UNIME. He has published numerous clinical papers in gynecologic oncology, risk reduction, and therapy and, most recently investigating clinical utilities of generative AI in gynecologic oncology.
Sharon Burk, Sharon Burk is a PhD student with Professor Antonio Giordano at the University of Siena, Italy in the department of Medical Biotechnologies, studying the role of Cyclin Dependent Kinase 10 in Triple Negative Breast Cancer. She received her Bachelor’s of Arts Degree from the University of California, Berkeley with a double major in molecular and cell biology and Italian studies. She is a member of AACR.
Optimization of CRISPR Gene Editing with Gold Nanoparticles
Reporter: Irina Robu, PhD
The CRISPR-Cas9 gene editing system has been welcomed as a hopeful solution to a range of genetic diseases, but the expertise has proven hard to deliver into cells. One plan is to open the cell membrane using an electric shock, but that can accidentally kill the cell. Another is to use viruses as couriers. Problem is, viruses can cause off-target side effects.
CRISPR-Cas9 is a unique technology that enables geneticists and medical researchers to edit parts of the genome by removing, adding or altering sections of DNA sequence. It is faster, cheaper and more accurate than previous techniques of editing DNA and can have a wide range of potential applications.
The CRISPR-Cas9 system consists of two key molecules that introduce a change into the DNA. One is an enzyme called Cas9 which acts as a pair of molecular scissors that can cut the two strands of DNA at a specific location in the genome where bits of DNA can be added or removed. The other one, is a piece of RNA which consists of a small piece of pre-designed RNA sequence located within a longer RNA scaffold. The scaffold part binds to the DNA and pre-designed sequence which contains Cas9. The RNA sequence is designed to find and locate specific sequence in the DNA. The Cas9 trails the guide RNA to the same location in the DNA sequence and makes a cut across both strands of DNA. At this point the cell distinguishes that the DNA is damaged and tries to repair it.
Researchers at Fred Hutchinson Cancer Research Center published new findings in Nature Materials suggested an alternative delivery method such as gold nanoparticles. The gold nanoparticles are packed with all the CRISPR components necessary to make clean gene edits. When the gold nanoparticles were tested in lab models of inherited blood disorders and HIV, between 10% and 20% of the targeted cells were effectively edited, with no toxic side effects.
The researchers use gold nanoparticles to deliver CRISPR to blood stem cells. Each gold nanoparticle contains four CRISPR components, including the enzyme needed to make the DNA cuts. But Fred Hutchinson researchers chose Cas12a, which they believed would lead to more efficient edits. Plus, Cas12a only needs one molecular guide, while Cas9 requires two.
In one experiment, they sought to disturb the gene CCR5 to make cells resistant to HIV. In the second, they created a gene mutation that can protect against blood disorders, including sickle cell disease. They observed the cells encapsulated the nanoparticles within six hours and began the gene-editing process within 48 hours. In mice, gene editing peaked eight weeks after injection, and the edited cells were still in circulation 22 weeks after the treatment.
Researchers at Fred Hutchinson are now working on improving the efficiency of the gold-based CRISPR delivery system so that 50% or more of the targeted cells are edited and are also looking for a commercial partner to bring the technology to clinical phase in the next few years.
Gender affects the prevalence of the cancer type, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
Gender of a person can affect the kinds of cancer-causing mutations they develop, according to a genomic analysis spanning nearly 2,000 tumours and 28 types of cancer. The results show striking differences in the cancer-causing mutations found in people who are biologically male versus those who are biologically female — not only in the number of mutations lurking in their tumours, but also in the kinds of mutations found there.
Liver tumours from women were more likely to carry mutations caused by a faulty system of DNA mending called mismatch repair, for instance. And men with any type of cancer were more likely to exhibit DNA changes thought to be linked to a process that the body uses to repair DNA with two broken strands. These biases could point researchers to key biological differences in how tumours develop and evolve across sexes.
The data add to a growing realization that sex is important in cancer, and not only because of lifestyle differences. Lung and liver cancer, for example, are more common in men than in women — even after researchers control for disparities in smoking or alcohol consumption. The source of that bias, however, has remained unclear.
In 2014, the US National Institutes of Health began encouraging researchers to consider sex differences in preclinical research by, for example, including female animals and cell lines from women in their studies. And some studies have since found sex-linked biases in the frequency of mutations in protein-coding genes in certain cancer types, including some brain cancers and advanced melanoma.
But the present study is the most comprehensive study of sex differences in tumour genomes so far. It looks at mutations not only in genes that code for proteins, but also in the vast expanses of DNA that have other functions, such as controlling when genes are turned on or off. The study also compares male and female genomes across many different cancers, which can allow researchers to pick up on additional patterns of DNA mutations, in part by increasing the sample sizes.
Researchers analysed full genome sequences gathered by the International Cancer Genome Consortium. They looked at differences in the frequency of 174 mutations known to drive cancer, and found that some of these mutations occurred more frequently in men than in women, and vice versa. When they looked more broadly at the loss or duplication of DNA segments in the genome, they found 4,285 sex-biased genes spread across 15 chromosomes.
There were also differences found when some mutations seemed to arise during tumour development, suggesting that some cancers follow different evolutionary paths in men and women. Researchers also looked at particular patterns of DNA changes. Such patterns can, in some cases, reflect the source of the mutation. Tobacco smoke, for example, leaves behind a particular signature in the DNA.
Taken together, the results highlight the importance of accounting for sex, not only in clinical trials but also in preclinical studies. This could eventually allow researchers to pin down the sources of many of the differences found in this study. Liver cancer is roughly three times as common in men as in women in some populations, and its incidence is increasing in some countries. A better understanding of its aetiology may turn out to be really important for prevention strategies and treatments.
Recent Progress in Gene Editing Error Reduction, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Recent Progress in Gene Editing Error Reduction
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Advances in Genome Editing
Researchers develop a CRISPR-based technique that efficiently corrects point mutations without cleaving DNA.
Illustration of DNA ligase, one of the cell proteins involved in repairing double-strand breaks in DNA WIKIMEDIA; WASHINGTON UNIVERSITY SCHOOL OF MEDICINE IN ST. LOUIS, TOM ELLENBERGER
Most genetic diseases in humans are caused by point mutations—single base errors in the DNA sequence. However, current genome-editing methods cannot efficiently correct these mutations in cells, and often cause random nucleotide insertions or deletions (indels) as a byproduct. Now, researchers at Harvard University have modified CRISPR/Cas9 technology to get around these problems, creating a new “base editor,” described today (April 20) in Nature, which permanently and efficiently converts cytosine (C) to uracil (U) bases with low error in human and mouse cell lines.
“There are a lot of genetic diseases where you would want, in essence, to swap bases in and out,” said Jacob Corn, scientific director of the Innovative Genomics Initiative at the University of California, Berkeley, who was not involved in the research. “Trying to get this to work is one of the big challenges in the field, and I think this is a really exciting approach.”
To date, CRISPR/Cas9 genome-editing approaches have relied on a cellular mechanism called homology-directed repair, which is triggered by double-strand breaks in DNA. Researchers supply cells with a template containing the desired sequence, make a targeted double-strand break with the Cas9 enzyme, and then wait to see whether homology-directed repair incorporates the template to reconnect the strands. Unfortunately, this method is inefficient (incorporation is rare) and often introduces new errors in the form of random indels around the break, making it impractical for therapeutic correction of point mutations.
So researchers at Harvard, led by chemist and chemical biologist David Liu, tried a different approach. First, they inactivated part of Cas9 so that it couldn’t make the double-strand break. They then tethered Cas9 to an enzyme called cytidine deaminase that directly catalyzes conversion of C to U (essentially an equivalent of thymine, T), without DNA cleavage. Sending this machinery into cells creates a mismatched pair at the target, comprising the newly introduced U, and an original guanine base (G) on the opposite strand. “This [mismatch] distorts the DNA,” Liu explained. “It creates a funny little bulge that doesn’t look normal.”
The bulge alerts a different cellular repair mechanism, mismatch repair, which removes one of the mismatched bases and replaces it with the complement to the remaining one. Without any information about which base is incorrect, mismatch repair produces the desired G to A conversion about 50 percent of the time; the rest of the time it converts the U back into a C.
But mismatch repair does incorporate further information when available: it detects tiny breaks in the DNA backbone called nicks. “Cells have evolved mismatch repair machinery to prioritize old DNA over newly synthesized DNA,” said Liu. “Newly synthesized DNA tends to have some nicks in it. So we reasoned that we could manipulate mismatch repair to favor correcting the DNA strand that we don’t want, namely the strand containing the G.”
The team again modified Cas9, this time so that it would create a nick in the nonedited, G-containing strand, while leaving the edited, U-containing strand intact. “Now the cell says, ‘Aha, there’s a mismatch here, and the base at fault must be the G, because that must be a newly synthesized strand because it has a nick in it,’” said Liu. “It will preferentially correct that G, using the other strand as a template.”
Using the technique at six loci in human cells, the team reported a targeted base correction rate of up to 37 percent, with only around 1 percent of the sequences showing indels. By contrast, a normal Cas9 editing technique tested on three of those loci showed less than one percent efficiency, and more than four percent formation of indels. The researchers also demonstrated the technique’s potential to correct disease-associated mutations by converting a variant of APOE, a gene linked with Alzheimer’s, into a lower risk version in mouse cells.
“By engineering this Cas9, they’ve figured out a really nice way to trick the cell into preferring pathways that it would normally not prefer,” said Corn. However, because the method is currently only able to convert C-G to U-A (i.e., T-A) base pairs, and in some cases edits other C bases in the immediate vicinity of the target, “it’s certainly not a panacea,” he cautioned. “It doesn’t mean that you can now cure every genetic disease out there. But there are probably going to be quite a few that fit into this category.”
The University of Oxford’s Tudor Fulga called the technique “an extremely ingenious idea” to get around inefficient homology-directed repair, and to reduce unwanted indel formation. “I think this will set up a paradigm shift in the field,” he told The Scientist. “It is very likely that the impact of Cas9-mediated base editing is going to be massive—both in terms of answering basic research questions and in genome engineering–based therapeutic applications.”
Also appearing in Nature today are two studies addressing a potential alternative to Cas9: the Cpf1 enzyme. CRISPR/Cpf1 creates “sticky ends”—overhangs in cleaved DNA that leave unpaired bases either side of the break—rather than the blunt ends made by Cas9’s double-strand DNA cleavage.
Emmanuelle Charpentier and colleagues at the Max Planck Institute for Infection Biology in Germany have shown that, unlike Cas9, Cpf1 processes RNA in addition to cleaving DNA. Meanwhile, Zhiwei Huang of the Harbin Institute of Technology, China, and colleagues have described the crystal structure of CRISPR/Cpf1.
“Sticky ends are more efficient [than blunt ends] for DNA repair in cells,” Huang told The Scientist. “We believe that [understanding] the structure of Cpf1 will help us not only to know the working mechanism of Cpf1 but also to design more specific and more efficient genome-editing tools.”
D. Dong et al., “The crystal structure of Cpf1 in complex with CRISPR RNA,” Nature, doi:10.1038/nature17944, 2016.
I. Fonfara et al., “The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA,” Nature, doi:10.1038/nature17945, 2016.
A.C. Komor et al., “Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage,” Nature, doi:10.1038/nature17946, 2016.
Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage
Current genome-editing technologies introduce double-stranded (ds) DNA breaks at a target locus as the first step to gene correction1, 2. Although most genetic diseases arise from point mutations, current approaches to point mutation correction are inefficient and typically induce an abundance of random insertions and deletions (indels) at the target locus resulting from the cellular response to dsDNA breaks1, 2. Here we report the development of ‘base editing’, a new approach to genome editing that enables the direct, irreversible conversion of one target DNA base into another in a programmable manner, without requiring dsDNA backbone cleavage or a donor template. We engineered fusions of CRISPR/Cas9 and a cytidine deaminase enzyme that retain the ability to be programmed with a guide RNA, do not induce dsDNA breaks, and mediate the direct conversion of cytidine to uridine, thereby effecting a C→T (or G→A) substitution. The resulting ‘base editors’ convert cytidines within a window of approximately five nucleotides, and can efficiently correct a variety of point mutations relevant to human disease. In four transformed human and murine cell lines, second- and third-generation base editors that fuse uracil glycosylase inhibitor, and that use a Cas9 nickase targeting the non-edited strand, manipulate the cellular DNA repair response to favour desired base-editing outcomes, resulting in permanent correction of ~15–75% of total cellular DNA with minimal (typically ≤1%) indel formation. Base editing expands the scope and efficiency of genome editing of point mutations.
The CRISPR/Cas9 technique is revolutionizing genetic research: scientists have already used it to engineer crops, livestock and even human embryos, and it may one day yield new ways to treat disease.
But now one of the technique’s pioneers thinks that he has found a way to make CRISPR even simpler and more precise. In a paper published in Cell on 25 September, a team led by synthetic biologist Feng Zhang of the Broad Institute in Cambridge, Massachusetts, reports the discovery of a protein1 called Cpf1 that may overcome one of CRISPR/Cas9’s few limitations; although the system works well for disabling genes, it is often difficult to truly edit them by replacing one DNA sequence with another.
The CRISPR/Cas9 system evolved as a way for bacteria and archaea to defend themselves against invading viruses. It is found in a wide range of these organisms, and uses an enzyme called Cas9 to cut DNA at a site specified by ‘guide’ strands of RNA. Researchers have turned CRISPR/Cas9 into a molecular-biology powerhouse that can be used in other organisms. The cuts made by the enzyme are repaired by the cell’s natural DNA-repair processes.
Good, better, best?
CRISPR is much simpler than previous gene-editing methods, but Zhang thought there was still room for improvement.
So he and his colleagues searched the bacterial kingdom to find an alternative to the Cas9 enzyme commonly used in laboratories. In April, they reported that they had discovered a smaller version of Cas9 in the bacterium Staphylococcus aureus2. The small size makes the enzyme easier to shuttle into mature cells — a crucial destination for some potential therapies.
The team was also intrigued by Cpf1, a protein that looks very different from Cas9, but is present in some bacteria with CRISPR. The scientists evaluated Cpf1 enzymes from 16 different bacteria, eventually finding two that could cut human DNA.
They also uncovered some curious differences between how Cpf1 and Cas9 work. Cas9 requires two RNA molecules to cut DNA; Cpf1 needs only one. The proteins also cut DNA at different places, offering researchers more options when selecting a site to edit. “This opens up a lot of possibilities for all the things we could not target before,” says epigeneticist Luca Magnani of Imperial College London.
Cpf1 also cuts DNA in a different way. Cas9 cuts both strands in a DNA molecule at the same position, leaving behind what molecular biologists call ‘blunt’ ends. But Cpf1 leaves one strand longer than the other, creating a ‘sticky’ end. Blunt ends are not as easy to work with: a DNA sequence could be inserted in either end, for example, whereas a sticky end will only pair with a complementary sticky end.
“The sticky ends carry information that can direct the insertion of the DNA,” says Zhang. “It makes the insertion much more controllable.”
Zhang’s team is now working to use these sticky ends to improve the frequency with which researchers can replace a natural DNA sequence. Cuts left by Cas9 tend to be repaired by sticking the two ends back together, in a relatively sloppy repair process that can leave errors. Although it is possible that the cell will instead insert a designated, new sequence at that site, that kind of repair occurs at a much lower frequency. Zhang hopes that the unique properties of how Cpf1 cuts may be harnessed to make such insertions more frequent.
For Bing Yang, a plant biologist at the Iowa State University in Ames, this is the most exciting aspect of Cpf1. “Boosting the efficiency would be a big step for plant science,” he says. “Right now, it is a major challenge.”
Will the new enzyme surpass Cas9 in popularity? “It’s too early to tell,” says Zhang. “It certainly has some distinct advantages.” The CRISPR/Cas9 system is so popular — and potentially lucrative — that it has sparked a fierce patent fight between the University of California, Berkeley, and the Broad Institute and its ally, the Massachusetts Institute of Technology in Cambridge. Zhang says that his lab will make the CRISPR/Cpf1 components available to academic researchers, as it has done with its CRISPR/Cas9 tools.
For now, the results stand as a testament that researchers still have more to learn from the genome-editing systems that bacteria have evolved. “This study powerfully demonstrates that the natural evolutionary diversity of CRISPR systems is rich with potential solutions to the challenges facing the use of genome-editing agents,” says David Liu, a chemical biologist at Harvard University in Cambridge. (Zhang and Liu are both scientific advisers to Editas Medicine, a company in Cambridge that aims to develop CRISPR-based therapies.)
Microbiologist John van der Oost of Wageningen University in the Netherlands, who collaborated on the latest study with Zhang, plans to keep searching for new methods. “You never know whether one of these systems will be suitable for genome editing,” he says. “There are still surprises ahead of us.”
•Cpf1 is a CRISPR-associated two-component RNA-programmable DNA nuclease
•Targeted DNA is cleaved as a 5-nt staggered cut distal to a 5′ T-rich PAM
•Two Cpf1 orthologs exhibit robust nuclease activity in human cells
The microbial adaptive immune system CRISPR mediates defense against foreign genetic elements through two classes of RNA-guided nuclease effectors. Class 1 effectors utilize multi-protein complexes, whereas class 2 effectors rely on single-component effector proteins such as the well-characterized Cas9. Here, we report characterization of Cpf1, a putative class 2 CRISPR effector. We demonstrate that Cpf1 mediates robust DNA interference with features distinct from Cas9. Cpf1 is a single RNA-guided endonuclease lacking tracrRNA, and it utilizes a T-rich protospacer-adjacent motif. Moreover, Cpf1 cleaves DNA via a staggered DNA double-stranded break. Out of 16 Cpf1-family proteins, we identified two candidate enzymes from Acidaminococcus and Lachnospiraceae, with efficient genome-editing activity in human cells. Identifying this mechanism of interference broadens our understanding of CRISPR-Cas systems and advances their genome editing applications.
CRISPR–Cas systems that provide defence against mobile genetic elements in bacteria and archaea have evolved a variety of mechanisms to target and cleave RNA or DNA1. The well-studied types I, II and III utilize a set of distinct CRISPR-associated (Cas) proteins for production of mature CRISPR RNAs (crRNAs) and interference with invading nucleic acids. In types I and III, Cas6 or Cas5d cleaves precursor crRNA (pre-crRNA)2, 3, 4, 5 and the mature crRNAs then guide a complex of Cas proteins (Cascade-Cas3, type I; Csm or Cmr, type III) to target and cleave invading DNA or RNA6, 7, 8, 9, 10, 11, 12. In type II systems, RNase III cleaves pre-crRNA base-paired withtrans-activating crRNA (tracrRNA) in the presence of Cas9 (refs 13, 14). The mature tracrRNA–crRNA duplex then guides Cas9 to cleave target DNA15. Here, we demonstrate a novel mechanism in CRISPR–Cas immunity. We show that type V-A Cpf1 from Francisella novicida is a dual-nuclease that is specific to crRNA biogenesis and target DNA interference. Cpf1 cleaves pre-crRNA upstream of a hairpin structure formed within the CRISPR repeats and thereby generates intermediate crRNAs that are processed further, leading to mature crRNAs. After recognition of a 5′-YTN-3′ protospacer adjacent motif on the non-target DNA strand and subsequent probing for an eight-nucleotide seed sequence, Cpf1, guided by the single mature repeat-spacer crRNA, introduces double-stranded breaks in the target DNA to generate a 5′ overhang16. The RNase and DNase activities of Cpf1 require sequence- and structure-specific binding to the hairpin of crRNA repeats. Cpf1 uses distinct active domains for both nuclease reactions and cleaves nucleic acids in the presence of magnesium or calcium. This study uncovers a new family of enzymes with specific dual endoribonuclease and endonuclease activities, and demonstrates that type V-A constitutes the most minimalistic of the CRISPR–Cas systems so far described.
The crystal structure of Cpf1 in complex with CRISPR RNA
The CRISPR–Cas systems, as exemplified by CRISPR–Cas9, are RNA-guided adaptive immune systems used by bacteria and archaea to defend against viral infection1, 2, 3, 4, 5, 6, 7. The CRISPR–Cpf1 system, a new class 2 CRISPR–Cas system, mediates robust DNA interference in human cells1, 8, 9, 10. Although functionally conserved, Cpf1 and Cas9 differ in many aspects including their guide RNAs and substrate specificity. Here we report the 2.38 Å crystal structure of the CRISPR RNA (crRNA)-bound Lachnospiraceae bacterium ND2006 Cpf1 (LbCpf1). LbCpf1 has a triangle-shaped architecture with a large positively charged channel at the centre. Recognized by the oligonucleotide-binding domain of LbCpf1, the crRNA adopts a highly distorted conformation stabilized by extensive intramolecular interactions and the (Mg(H2O)6)2+ ion. The oligonucleotide-binding domain also harbours a looped-out helical domain that is important for LbCpf1 substrate binding. Binding of crRNA or crRNA lacking the guide sequence induces marked conformational changes but no oligomerization of LbCpf1. Our study reveals the crRNA recognition mechanism and provides insight into crRNA-guided substrate binding of LbCpf1, establishing a framework for engineering LbCpf1 to improve its efficiency and specificity for genome editing
AstraZeneca’s WEE1 protein inhibitor AZD1775 Shows Success Against Tumors with a SETD2 mutation
Stephen J. Williams, Ph.D., Curator
There have been multiple trials investigating the utility of cyclin inhibitors as anti-tumoral agents (see post) with the idea of blocking mitotic entry however another potential antitumoral mechanism has been to drive the cell into mitosis in the presence of DNA damage or a defective DNA damage repair capacity. A recent trial investigating an inhibitor or the cell cycle checkpoint inhibitor Wee1 showed positive results in select cohorts of patients with mutations in DNA repair, indicating the therapeutic advantage of hijacking the cell’s own DNA damage response, much like how PARP inhibitor Olaparib works in BRCA1 mutation positive ovarian cancer patients.
Investigators at Oxford University say that one of AstraZeneca’s ($AZN) pipeline drugs proved particularly effective in killing cancer cells with a particular genetic mutation.
The ex-Merck ($MRK) drug is AstraZeneca’s WEE1 protein inhibitor AZD1775, which proved particularly lethal to genes with a SETD2 mutation, which the researchers see as a potential ‘Achilles heel’ often found in kidney cancer and childhood brain tumors.
“When WEE1 was inhibited in cells with a SETD2 mutation, the levels of deoxynucleotides, the components that make DNA, dropped below the critical level needed for replication,” noted Oxford’s Andy Ryan. “Starved of these building blocks, the cells die. Importantly, normal cells in the body do not have SETD2 mutations, so these effects of WEE1 inhibition are potentially very selective to cancer cells.”
AstraZeneca landed rights to the drug back in 2013, when incoming Merck R&D chief Roger Perlmutter opted to spin it out while focusing an immense effort around the development of its PD-1 checkpoint inhibitor KEYTRUDA® (pembrolizumab). Since then, AstraZeneca has made it available to academic investigators through their open innovation program.
Wee1, DNA damage checkpoint and cell cycle regulation
In fission yeast, Wee1 delays entry into mitosis by inhibiting the activity of Cdk1, the cyclin-dependent kinase that promotes entry into mitosis (Cdk1 is encoded by the cdc2+ gene in fission yeast and the CDC28 gene in budding yeast) (Russell and Nurse, 1987a). Wee1 inhibits Cdk1 by phosphorylating a highly conserved tyrosine residue at the N-terminus (Featherstone and Russell, 1991; Gould and Nurse, 1989; Lundgren et al., 1991; Parker et al., 1992; Parker and Piwnica-Worms, 1992). The phosphatase Cdc25 promotes entry into mitosis by removing the inhibitory phosphorylation (Dunphy and Kumagai, 1991; Gautier et al., 1991; Kumagai and Dunphy, 1991; Millar et al., 1991; Russell and Nurse, 1986; Strausfeld et al., 1991). Loss of Wee1 activity causes cells to enter mitosis before sufficient growth has occurred and cytokinesis therefore produces two abnormally small daughter cells (Fig. 1A) (Nurse, 1975). Conversely, increasing the gene dosage of wee1 causes delayed entry into mitosis and an increase in cell size, indicating that the levels of Wee1 activity determine the timing of entry into mitosis and can have strong effects on cell size (Russell and Nurse, 1987a). Similarly, cdc25– mutants undergo delayed entry into mitosis, producing abnormally large cells, and an increase in the gene dosage of cdc25 causes premature entry into mitosis and decreased cell size (Russell and Nurse, 1986). Despite these difficulties, early work in fission yeast suggested that the Wee1 kinase plays an important role in a checkpoint that coordinates cell growth and cell division at the G2/M transition (Fantes and Nurse, 1978; Nurse, 1975; Thuriaux et al., 1978). WEE1 is an evolutionarily conserved nuclear tyrosine kinase (Table 2) that is markedly active during the S/G2 phase of the cell cycle [24, 25]. It was first discovered 25 years ago as a cell division cycle (cdc) mutant-wee1– in the fission yeast, Schizosaccharomyces pombe [26]. Fission yeast lacking WEE1 are characterized by a smaller cell size, and this phenotype has been attributed to the ability of WEE1 to negatively regulate the activity of cyclin dependent kinase, Cdc2 (Cdc28 in budding yeast and CDK1 in human), in the Cdc2/CyclinB complex [27]. Recently, WEE1 was shown to directly phosphorylate the mammalian core histone H2B at tyrosine 37 in a cell cycle dependent manner. Inhibition of WEE1 kinase activity either by a specific inhibitor (MK-1775) or suppression of its expression by RNA interference abrogated H2B Y37-phosphorylation with a concurrent increase in histone transcription [17].
As shown in the Below figure Wee1 is a CDK cyclin kinase which results in an inactivating phosphorylation event on CDK/Cyclin complexes
Figure 1. Schematic representation of the effects of Chk1 and Wee1 inhibition on CDK-CYCLIN complex regulation, that gets more activated being unphosphorylated from Cell cycle, checkpoints and cancer by Laura Carrassa.
Figure 2. Schematic representation of the role of Chk1 and Wee1 in regulation of the CDK-cyclin complexes involved in S phase and M phase entry from Cell cycle, checkpoints and cancer by Laura Carrassa.
The following articles discuss how Wee1 can be a target and synergize with current chemotherapy
p53 mutation Frequency in Ovarian Cancer and contribution to chemo-resistance
The following is from the curated database TCGA and cBioPortal TCGA Data Viewer for mutations found in ovarian cancer sequencing studies in the literature
Confirmed that mutations in gene TP53 are present in more than 96 percent of ovarian cases (>57% mutation frequency) while SETD2 mutations are present in only 1% of cases (1.1% mutation frequency).
In general, ovarian cancers with TP53 are considered to have increased resistance to commonly used cytotoxic agents used for this neoplasm, for example cisplatin and taxol, as TP53 is a major tumor suppressor/transcription factor involved in cell cycle, DNA damage response, and other chemosensitivity mechanisms. One subtype of TP53 mutations, widely termed gain-of-function (GOF) mutations, surprisingly converts this protein from a tumor suppressor to an oncogene. We term the resulting change an oncomorphism. In this review, we discuss particular TP53 mutations, including known oncomorphic properties of the resulting mutant p53 proteins. For example, several different oncomorphic mutations have been reported, but each mutation acts in a distinct manner and has a different effect on tumor progression and chemoresistance.
Figure 1. The spectrum of protection against cancer provided by WT p53. As copies of WT p53 (TP53+/+) are lost, cancer protection decreases. When oncomorphic mutations are acquired, cancer susceptibility is increased.
Oncomorphic p53 proteins were first identified over two decades ago, when different TP53 mutants were introduced into cells devoid of endogenous p53 [38,39]. Among all cancers, the most common oncomorphic mutations are at positions R248, R273, and R175, and in ovarian cancers the most common oncomorphic TP53 mutations are at positions R273, R248, R175, and Y220 at frequencies of 8.13%, 6.02%, 5.53%, and 3.74%, respectively [33,34]. In in vitro studies, cells with oncomorphic p53 demonstrate increased invasion, migration, angiogenesis, survival, and proliferation as well as resistance to chemotherapy [35,37,40,41].
Figure 2. Hotspots for TP53 mutations. Mutations that occur at a frequency greater than 3% are highlighted. Certain p53 mutants have oncomorphic activity (denoted by *), functioning through novel protein interactions as well as novel transcriptional targets to promote cell survival and potentially chemoresistance. Codons in the “other” category include those that produce non-functional p53 or have not been characterized to date.
Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, Neskey DM, Frederick MJ, Woo SH, Caulin C, Hsu TK, McDonald TO, Kimmel M, Meyn RE, Lichtarge O, Myers JN.
Mol Cancer Ther. 2015 Feb;14(2):608-19. doi: 10.1158/1535-7163.MCT-14-0735-T. Epub 2014 Dec 10.
Mol Cancer Ther. 2015 Jan;14(1):90-100. doi: 10.1158/1535-7163.MCT-14-0496. Epub 2014 Nov 5.
Mol Cancer Ther. 2013 Aug;12(8):1442-52. doi: 10.1158/1535-7163.MCT-13-0025. Epub 2013 May 22.
1Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, Maryland.
2Laboratory of Cancer Biology and Genetics, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, Maryland.
3Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, NIH, Bethesda, Maryland.
4Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, Maryland. mgottesman@nih.gov.
Abstract
Despite early positive response to platinum-based chemotherapy, the majority of ovarian carcinomas develop resistance and progress to fatal disease. Protein phosphatase 2A (PP2A) is a ubiquitous phosphatase involved in the regulation of DNA-damage response (DDR) and cell-cycle checkpoint pathways. Recent studies have shown that LB100, a small-molecule inhibitor of PP2A, sensitizes cancer cells to radiation-mediated DNA damage. We hypothesized that LB100 could sensitize ovarian cancer cells to cisplatin treatment. We performed in vitro studies in SKOV-3, OVCAR-8, and PEO1, -4, and -6 ovarian cancer lines to assess cytotoxicity potentiation, cell-death mechanism(s), cell-cycle regulation, and DDR signaling. In vivo studies were conducted in an intraperitoneal metastatic mouse model using SKOV-3/f-Luc cells. LB100 sensitized ovarian carcinoma lines to cisplatin-mediated cell death. Sensitization via LB100 was mediated by abrogation of cell-cycle arrest induced by cisplatin. Loss of the cisplatin-induced checkpoint correlated with decreased Wee1 expression, increased cdc2 activation, and increased mitotic entry (p-histone H3). LB100 also induced constitutive hyperphosphorylation of DDR proteins (BRCA1, Chk2, and γH2AX), altered the chronology and persistence of JNK activation, and modulated the expression of 14-3-3 binding sites. In vivo, cisplatin sensitization via LB100 significantly enhanced tumor growth inhibition and prevented disease progression after treatment cessation. Our results suggest that LB100 sensitizes ovarian cancer cells to cisplatin in vitro and in vivo by modulation of the DDR pathway and cell-cycle checkpoint abrogation.
So Why SETD2 Mutations?
SETD2 is a histone methyltransferase that is specific for lysine-36 of histone H3, and methylation of this residue is associated with active chromatin and chromatin remodeling.
Kanu N, Grönroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, Maya-Mendoza A, Mistrík M, Rowan AJ, Patel H, Rabinowitz A, East P, Wilson G, Santos CR, McGranahan N, Gulati S, Gerlinger M, Birkbak NJ, Joshi T, Alexandrov LB, Stratton MR, Powles T, Matthews N, Bates PA, Stewart A, Szallasi Z, Larkin J, Bartek J, Swanton C.
Oncogene. 2015 Mar 2. doi: 10.1038/onc.2015.24. [Epub ahead of print]
Ahn JW, Kim HS, Yoon JK, Jang H, Han SM, Eun S, Shim HS, Kim HJ, Kim DJ, Lee JG, Lee CY, Bae MK, Chung KY, Jung JY, Kim EY, Kim SK, Chang J, Kim HR, Kim JH, Lee MG, Cho BC, Lee JH, Bang D.
Genome Med. 2014 Feb 27;6(2):18. doi: 10.1186/gm535. eCollection 2014.
#2. Gemcitabine Hydrochloride With or Without WEE1 Inhibitor MK-1775 in Treating Patients With Recurrent Ovarian, Primary Peritoneal, or Fallopian Tube Cancer
This randomized phase II clinical trial studies how well gemcitabine hydrochloride and WEE1 inhibitor MK-1775 work compared to gemcitabine hydrochloride alone in treating patients with ovarian, primary peritoneal, or fallopian tube cancer that has come back after a period of time. Gemcitabine hydrochloride may prevent tumor cells from multiplying by damaging their deoxyribonucleic acid (DNA, molecules that contain instructions for the proper development and functioning of cells), which in turn stops the tumor from growing. The protein WEE1 may help to repair the damaged tumor cells, so the tumor continues to grow. WEE1 inhibitor MK-1775 may block the WEE1 protein activity and may increase the effectiveness of gemcitabine hydrochloride by preventing the WEE1 protein from repairing damaged tumor cells without causing harm to normal cells. It is not yet known whether gemcitabine hydrochloride with or without WEE1 inhibitor MK-1775 may be an effective treatment for recurrent ovarian, primary peritoneal, or fallopian tube cancer.
Primary Outcome Measures:
PFS evaluated using RECIST version 1.1 [ Time Frame: Time from start of treatment to time to progression or death, whichever occurs first, assessed up to 1 year ] [ Designated as safety issue: No ]
Secondary Outcome Measures:
GCIG CA125 response rate [ Time Frame: Up to 1 year ] [ Designated as safety issue: No ]
Incidence of grade 3 or 4 serious adverse events, graded according to the National Cancer Institute CTCAE version 4.0 [ Time Frame: Up to 1 year ] [ Designated as safety issue: Yes ]
Objective response by RECIST version 1.1 [ Time Frame: Up to 1 year ] [ Designated as safety issue: No ]
Overall survival [ Time Frame: Up to 1 year ] [ Designated as safety issue: No ]
Survival estimates will be computed using the Kaplan-Meier method.
p53 protein expression in archival tumor tissue by immunohistochemistry (IHC) [ Time Frame: Baseline ] [ Designated as safety issue: No ]
TP53 mutations (presence and type of mutation) by Sanger sequencing [ Time Frame: Baseline ] [ Designated as safety issue: No ]
These Trials Are Not Investigating TP53 Status of Patient Cohorts
To establish the safety and tolerability of single-agent MK-1775 in patients with refractory solid tumors
To determine the pharmacokinetics of MK-1775 in patients with refractory solid tumors
SECONDARY OBJECTIVES:
To determine the effect of MK-1775 on markers of DNA damage and apoptosis in tumor tissue and circulating tumor cells
To evaluate the antitumor activity of MK-1775 in patients with refractory solid tumors
Note: A further expansion cohort of 6 additional patients with documented tumors harboring BRCA-1 or -2 mutations will lso be enrolled at the MTD to further explore the safety of the agent and obtain preliminary evidence of activity in this patient population
To estimate the maximum tolerated dose (MTD) and/or recommended Phase 2 dose of MK-1775 (WEE1 inhibitor MK-1775) administered on days 1 through 5 every 21 days, in combination with oral irinotecan (irinotecan hydrochloride), to children with recurrent or refractory solid tumors.
To define and describe the toxicities of MK-1775 in combination with oral irinotecan administered on this schedule.
III. To characterize the pharmacokinetics of MK-1775 in children with refractory cancer.
SECONDARY OBJECTIVES:
To preliminarily define the antitumor activity of MK-1775 and irinotecan within the confines of a Phase 1 study.
To obtain initial Phase 2 efficacy data on the anti-tumor activity of MK-1775 in combination with irinotecan administered to children with relapsed or refractory neuroblastoma and in children with relapsed or refractory medulloblastoma/CNS PNET (central nervous system primitive neuroectodermal tumor).
III. To investigate checkpoint over-ride by MK-1775 via the mechanism-based pharmacodynamic (PD) biomarker of decreased cyclin-dependent kinase 1 (CDK1) phosphorylation in correlative and exploratory studies.
To evaluate potential predictive biomarkers of MK-1775 sensitivity, including v-myc avian myelocytomatosis viral oncogene homolog (MYC), v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog (MYCN), phosphorylated-WEE1 G2 checkpoint kinase (p-Wee1), enhancer of zeste homolog 2 (Drosophila) (EZH2) and gamma-H2A histone family, member X (H2AX) in tumor tissues in correlative and exploratory studies.
A series of electron micrographs show the barrel-shaped helicase, which is the enzyme that separates the two DNA strands, along with other components of the replisome, including polymerase-epsilon (green).[Brookhaven National Laboratory]
It may be time to update biology texts to reflect newly published data from a collaborative team of scientists at Rockefeller University, Stony Brook University, and the U.S. Department of Energy’s Brookhaven National Laboratory. Using cutting-edge electron microscopy (EM) techniques, the investigators gathered the first ever images of the fully assembled replisome, providing new insight into the molecular mechanisms of replication.
“Our finding goes against decades of textbook drawings of what people thought the replisome should look like,” remarked co-senior author Michael O’Donnell, Ph.D., professor and head of Rockefeller’s Laboratory of DNA Replication. “However, it’s a recurring theme in science that nature does not always turn out to work the way you thought it did.”
“Our finding goes against decades of textbook drawings of what people thought the replisome should look like,” remarked co-senior author Michael O’Donnell, Ph.D., professor and head of Rockefeller’s Laboratory of DNA Replication. “However, it’s a recurring theme in science that nature does not always turn out to work the way you thought it did.”
Previously (left), the replisome’s two polymerases (green) were assumed to be below the helicase (tan), the enzyme that splits the DNA strands. The new images reveal one polymerase is located at the front of the helicase, causing one strand to loop backward as it is copied (right). [Brookhaven National Laboratory]
The researcher’s findings focused on the replisome found in eukaryotic organisms, a category that includes a broad swath of living things, including humans and other multicellular organisms. Over the past several decades, there has been an array of data describing the individual components comprising the complex nature of replisome. Yet, until now no pictures existed to show just how everything fit together.
“This work is a continuation of our long-standing research using electron microscopy to understand the mechanism of DNA replication, an essential function for every living cell,” explained co-senior author Huilin Li, Ph.D., biologist with joint appointments at Brookhaven Lab and Stony Brook University. “These new images show the fully assembled and fully activated ‘helicase’ protein complex—which encircles and separates the two strands of the DNA double helix as it passes through a central pore in the structure—and how the helicase coordinates with the two ‘polymerase’ enzymes that duplicate each strand to copy the genome.”
The image and implications from this study were described in a paper entitled “The architecture of a eukaryotic replisome,” published recently through Nature Structural & Molecular Biology.
Traditional models of DNA replication show the helicase enzyme moving along the DNA, separating the two strands of the double helix, with two polymerases located at the back where the DNA strand is split. In this configuration, the polymerases would add nucleotides to the side-by-side split ends as they move out of the helicase to form two new complete double helix DNA strands. However, the images that the researchers collected of intact replisomes revealed that only one of the polymerases is located at the back of the helicase. The other is on the front side of the helicase, where the helicase first encounters the double-stranded helix. This means that while one of the two split DNA strands is acted on by the polymerase at the back end, the other has to thread itself back through or around the helicase to reach the front-side polymerase before having its new complementary strand assembled.
“DNA replication is one of the most fundamental processes of life, so it is every biochemist’s dream to see what a replisome looks like,” stated lead author Jingchuan Sun, EM biologist in Dr. Li’s laboratory. “Our lab has expertise and a decade of experience using electron microscopy to study DNA replication, which has prepared us well to tackle the highly mobile therefore very challenging replisome structure. Working together with the O’Donnell lab, which has done beautiful, functional studies on the yeast replisome, our two groups brought perfectly complementary expertise to this project.”
The positioning of one polymerase at the front of the helicase suggests that it may have an unforeseen function—the possibilities of which the collaborative group of scientists is continuing to study. Whatever the function the offset polymerase ends up having, Drs. Li and O’Donnell hope that it will not only provide them better insight into the replication machinery but that they may uncover useful information that can be exploited for disease intervention.
“Clearly, further studies will be required to understand the functional implications of the unexpected replisome architecture reported here,” the scientists concluded.
Scientists at the University of Copenhagen say they have located a previously unknown function for histones, which allows for an improved understanding of how cells protect and repair DNA damages. This new discovery may be of great importance to the treatment of diseases caused by cellular changes such as cancer and immune deficiency syndrome.
The study (“Histone H1 couples initiation and amplification of ubiquitin signaling after DNA damage”) is published in Nature.
“I believe that there’s a lot of work ahead. It’s like opening a door onto a previously undiscovered territory filled with lots of exciting knowledge. The histones are incredibly important to many of the cells’ processes as well as their overall wellbeing,” said Niels Mailand, Ph.D., from the Novo Nordisk Foundation Center for Protein Research at the Faculty of Health and Medical Science.
Histones enable the tight packaging of DNA strands within cells. The strands are two meters in length and the cells usually about 100,000 times smaller. Generally speaking, there are five types of histones. Four of them are core histones and they are placed like beads on the DNA strands, which are curled up like a ball of wool within the cells. The role of the histones is already well described in research, and in addition to enabling the packaging of the DNA strands they also play a central part in practically every process related to the DNA-code, including repairing possibly damaged DNA.
The four core histones have tails and, among other things, they signal damage to the DNA and thus attract the proteins that help repair the damage. Between the histone “yarn balls” we find the fifth histone, Histone H1, but up until now its function has not been thoroughly examined.
Using a mass spectrometer, Dr. Mailand and his team have discovered that, surprisingly, the H1 histone also helps summon repair proteins.
“In international research, the primary focus has been on the core histones and their functionality, whereas little attention has been paid to the H1 histone, simply because we weren’t aware that it too influenced the repair process. Having discovered this function in the H1 constitutes an important piece of the puzzle of how cells protect their DNA, and it opens a door onto hitherto unknown and highly interesting territory,” noted Dr. Mailand.
He expects the discovery to lead to increased research into Histone H1 worldwide, which will lead to increased knowledge of cells’ abilities to repair possible damage to their DNA and thus increase our knowledge of the basis for diseases caused by cellular changes. It will also generate more knowledge about the treatment of these diseases.
“By mapping the function of the H1 histone, we will also learn more about the repair of DNA damages on a molecular level. In order to provide the most efficient treatment, we need to know how the cells prevent and repair these damages,” point out Dr. Mailand.
Synthetic oligonucleotides have emerged as promising therapeutic agents for the treatment of a variety of diseases, including viral infections and cancer. Researchers are looking at several classes of nucleic acids, such as antisense oligonucleotides, small interfering RNAs (siRNAs), and aptamers, for therapeutic applications.
However, various impurities – product-related, in the starting materials, and arising from incomplete capping of coupling reactions – must be identified and removed and postsynthesis processing must be monitored. Thus, a key challenge in the development and manufacture of oligonucleotide therapeutics is to establish analytical methods that are capable of separating and identifying impurities.
Exploring Better Options for Oligonucleotide LC Separations
Table 1. Options for oligonucleotide LC separations
Ion-pair, reversed-phase separation of the trityl-on oligos and is relatively simple to perform. This method separates the full-length target oligo, which still has the dMT group attached, from the deprotected failure sequences. The analytical information obtained is limited, so this is generally considered a purification method.
An alternate method, ion-exchange separations of the trityl-off, deprotected oligos uses the negative charge on the backbone of the oligo to facilitate the separation. Resolution is good for the shorter oligos but decreases with increasing chain length. Aqueous eluents are used but oligos are highly charged, and high concentrations of salt are needed to achieve elution from the column, making the technique unsuitable for use with LC/MS.
Finally, ion-pair, reversed-phase separation of the trityl-off, deprotected oligos makes use of organic solvents and mobile phase additives such as TEAA (triethylammonium acetate) or TEA-HFIP (triethylamine and hexafluoroisopropanol) to ion-pair with the negatively charged phosphodiester backbone of the oligonucleotide. High-performance columns deliver excellent resolution. What’s more, methods with volatile mobile phase constituents such as TEA-HFIP are suitable for use with LC/MS, providing useful information to help characterize oligonucleotide structures and sequences.
In Table 1 we summarize some of the options for oligonucleotide analysis by liquid chromatography.
Designed for ion-pair, reversed-phase separation of the trityl-off, deprotected oligos using either TEAA or TEA-HFIP mobile phases –Agilent AdvanceBio Oligonucleotide columns meet these challenges.
Single Nucleotide Repair and Tunable DNA-directed Assembly of Nanomaterials, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
single nucleotide repair and tunable DNA-directed assembly of nanomaterials
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Expanding DNAzyme functionality through enzyme cascades with applications in single nucleotide repair and tunable DNA-directed assembly of nanomaterials
Many biological functions require two or more enzymes working together in cascades. While many examples of protein and RNA enzyme cascades are known, few enzyme cascades containing solely DNAzymes have been reported. Herein we demonstrate the combination of an 8–17 DNAzyme with RNA cleavage activity and an E47 DNAzyme with DNA ligation activity to achieve a new function of single ribonucleotide repair in DNA while maintaining the integrity of the original DNA sequence, which is difficult for a single DNAzyme to achieve. In addition, this method is applied to modify the sequences of DNA strands immobilized on the surface ofnanoparticles to control the DNA-directed assembly selectively and sequentially. Such an approach can be applied to other DNAzymes with different activities to expand the functions of DNAzymes and the scope of their applications.
The discovery of deoxyribozymes (DNAzymes) with enzymatic activity in the 1990s1,2 has demonstrated that DNA molecules are not simply inert biopolymers for genetic storage; they can be active catalysts as well.3–8 Since then, many DNAzymes have been obtained with catalytic functions such as cleavage,2,9–13 ligation,14–16 phosphorylation,17 adenylation18 or depurination19 of nucleic acids, as well as other reactions including porphyrin metallation, C–C bond formation, nucleopeptide linkage formation, oxygen transfer and thymine dimer repair.20–26 Because DNAzymes are facile to synthesize and more stable than protein and RNA enzymes, they have been widely used in applications such as nanomaterial assembly,27,28 biosensing,29–31 logical computing,32 nanomachine engineering,33 antiviral or gene therapy,34 and in vitro RNA manipulation.35 Despite these successes, the application of DNAzymes is limited by the narrower range of catalytic functionality compared to protein enzymes. One possible approach to addressing this issue would be to combine enzymes with different reactivities to form a cascade of successive enzymatic reactions, which together create new functionality. Indeed, many such examples exist in biology, since nearly all important biological functions, such as the pathways involved in DNA repair and protein synthesis, require a cascade of multiple protein enzymes to carry out their full function. In contrast, little has been reported about the use of DNAzyme cascades to realize enhanced functionality. Such a strategy could expand the functionality of DNAzymes to a level more on par with protein and RNA enzymes, which should greatly increase the range of possible applications.
One such application is single nucleotide repair, i.e., excision of a misincorporated ribonucleotide in single-stranded DNA and subsequent insertion of the corresponding deoxyribonucleotide at the excision site. The misincorporation of ribonucleotides into DNA strands can occur from exposure to external oxidizing agents or ionizing radiation,36 or spontaneously during DNA replication.37 Misincorporation of ribonucleotide can distort the structure of DNA,38 reduce its stability,39 and interfere with the normal interaction between DNA and DNA polymerases.40 In fact, the overexpression of DNA polymerases that are prone to ribonucleotide misincorporation has been linked to many cancers, including ovarian, prostate, breast and colon cancers.41 In nature, protein enzymes such as RNase H and FEN-1 can efficiently excise misincorporated ribonucleotides in DNA by cleaving the DNA at the ribonucleotide site and then restoring the correct deoxyribonucleotides by DNA polymerases,42,43 which is an example of an enzyme cascade. It would be interesting to nd out if a similar function could be achieved through DNAzyme cascades.
Another potential application is in tuning the properties of DNA-functionalized nanomaterials. For example, DNA-functionalized gold nanoparticles27 have emerged as an attractive platform for biosensing,32,44–50 nanomedicine,45 and as building blocks for controlled nanoassemblies.51–55 Although much research has been focused on the surface modification of gold nanoparticles with DNA for various applications, there are still limited methods to modify the sequences of DNA already immobilized on gold nanoparticles in order to make the properties of the DNA-modified nanomaterials tunable aer fabrication. The use of DNAzymes is a promising approach for DNA modification on nanomaterials56 due to the excellent stability of DNAzymes and their smaller size compared to protein enzymes, thereby minimizing steric effects between the enzyme and the DNA in order to avoid reduction in reaction efficiency. However, it is still very challenging to modify a specific DNA sequence on multiple-DNA-functionalized nanomaterials to tune their functions in a selective and sequential fashion.
Herein, we demonstrate a cascade of two DNAzymes with RNA cleavage and DNA ligation activities, respectively, in order to carry out single nucleotide repair or selective sequence modification of DNA. In a one-pot reaction, a single misincorporated ribonucleotide in a DNA strand was converted to the corresponding deoxyribonucleotide while maintaining sequence integrity. Furthermore, the sequences of DNA strands immobilized on multiple functional nanoparticles were successfully modified in order to control and alter the DNAdirected assembly of nanoparticles in a stepwise and selective fashion.
Results and discussion To demonstrate that single nucleotide repair in DNA can be achieved by the cascade of two DNAzymes, we used a 26-nt DNA strand (O1) containing a misincorporated cytidine (rC) ribonucleotide as an example. The goal was to convert the rC in O1 into a deoxycytidine (C), as seen in O4 (Fig. 1a), while maintaining the integrity of the DNA sequence. The DNAzymes 17Em1 (Fig. 1a, blue) with RNA cleavage activity2,57–59 and E47 (Fig. 1a, red) with DNA ligation activity14,60 were chosen as the cascade pair in this study. The 17Em1 DNAzyme catalyzes the hydrolysis of the 30 phosphodiester linkage of the internal rC in the DNA strand when metal ion cofactors such as Pb2+ and Zn2+ are present (Fig. S1a in ESI†). On the other hand, the E47 DNAzyme can induce the catalytic ligation of the 50 –OH of the DNA substrate with another 30 -phosphorylated DNA strand (activated by imidazole)14 in the presence of Cu2+ or Zn2+ as the metal cofactor (Fig. S1b in ESI†). Therefore, by sequential cleavage and ligation reactions catalyzed by these two DNAzymes on O1 containing rC, O1 could first be cleaved at the 30 phosphodiester of the rC by 17Em1 and then undergo ligation at the cleavage site with another 30 -phosphorylated DNA strand of an identical sequence (except with deoxyribonucleotide C in place of ribonucleotide rC) by E47. The product O4 has a sequence identical to the starting strand O1, with the rC replaced with C.
Fig. 1 (a) Conversion of a single ribonucleotide (rC) in a DNA strand O1 to a deoxyribonucleotide (C) by the cascade of DNAzymes 17Em1 and E47: O1 is cleaved by 17Em1 to afford products of O2 and Oc; O2 is then ligated with O3 (activated by imidazole) to form O4 by E47. (b) Sequence modification of a DNA strand O5 to O4 through a similar protocol by the cascade of DNAzymes 17Em2 and E47.
Initially, 30 -fluorescein-labeled O1 was treated with 17Em1 to form DNA duplex O1-17Em1 via 18 matched base pairs (9 on each binding arm). In the presence of Pb2+, O1 was efficiently cleaved by 17Em1 into fragments O2 and Oc, resulting in the dehybridization of the duplex because the melting temperature of the duplex between 17Em1 and O2 or between 17Em1 and Oc is below room temperature (Fig. 1a). The fluorescence image after polyacrylamide gel electrophoresis (PAGE) suggested the complete cleavage of O1 and formation of O2 (Fig. 2a, lane 1 and 2 for O1 and O2, respectively), while Oc was not visible on the gel due to the lack of a fluorescein label. The cleavage reaction product O2 was also confirmed by the result from matrixassisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrum (Table 1 and Fig. S2 in ESI†). Control experiments using a DNAzyme of a different sequence (17Em2) or without Pb2+ showed negligible cleavage of the substrate O1 (Fig. S3 in ESI†) due to the specificity of the DNAzyme and the essential role of the metal ion cofactor.2,57–59 Subsequently, without any purification of O2 from the mixture solution after the previous cleavage step, E47 and 30 -phosphorylated O3 (imidazole-activated) were added into the solution to generate another DNA complex O2–O3-E47, which gave O4 as the product after the E47-catalyzed ligation reaction in the presence of Cu2+ (Fig. 1a).14,60 The formation of O4 was confirmed by both fluorescent PAGE (Fig. 2a, the upper band of lane 3) and MALDITOF MS (Table 1 and Fig. S2 in ESI†), while some unreacted O2 was also observed on the gel (Fig. 2a, the lower band of lane 3). Here, O3 was invisible due to the lack of a fluorescein label. Considerably lower levels of ligation between O2 and O3 were observed if either E47 or Cu2+ was absent (Fig. S3 in ESI†). Together these results indicate that the reactions catalyzed by the DNAzyme cascade were achieved through a one-pot reaction without isolation and purification of the intermediate O2.
Fig. 2 (a) Fluorescent PAGE (20% denaturing gel) images of the transformation from O1 to O4 by DNAzymes 17Em1 and E47. Lanes in (a): 1, O1; 2, 1 after cleavage by 17Em1 to yield O2 and Oc in the presence of Pb2+; 3, 2 after ligation to yield O4 by E47 in the presence of O3 and Cu2+; 4, 2 after ligation to yield O4 + 8A by E47 in the presence of O3 + 8A and Cu2+; 5, O4 in the presence of Pb2+ and 17Em1. (b) Fluorescent PAGE images of the transformation from O5 to O4 by 17Em2 and E47: Lanes in (b): 1, O5; 2, 1 after cleavage by 17Em2 to yield O2 in the presence of Pb2+; 3, 2 after ligation to yield O4 by E47 in the presence of O3 and Cu2+; 4, 2 after ligation to yield O4 + 8A by E47 in the presence of O3 + 8A and Cu2+; 5, O4 in the presence of Pb2+ and 17Em2
To provide further confirmation of the above successful conversion of rC in O1 to C in O4, while keeping other sequences identical, a longer O3 + 8A (O3 extended by A8 at 50 ) was used in place of O3 (Fig. 1a). Under the same conditions, a longer product O4 + 8A was obtained (Fig. 2a, the upper band of lane 4) with slower gel migration compared to O4 (Fig. 2a, the upper band of lane 3), suggesting that the ligation reaction occurred mostly between O2 and imidazole-activated O3, and not between O2 and un-activated Oc (Oc is the product from the previous cleavage reaction of O1 and 17Em1), in which case a band with the same migration as O4 would have been observed. The presence of C rather than rC in the product O4 was supported by the lower molecular weight of O4 in the MALDI-TOF mass spectrum as compared to that of O1 (Table 1), as well as the increased resistance to hydrolysis of O4 even in the presence of Pb2+ and 17Em1 (Fig. 2a, lane 5), which can catalyze the cleavage of a substrate containing an internal ribonucleotide linkage (O1),2,57 but not a substrate containing entirely deoxyribonucleotides (O4).
Table 1 Measured and calculated molecular weight (m/z) in MALDI-TOF mass spectra of 30 -fluorescein-labeled DNAs (O1, O2, O4 and O5). For full spectra, see Fig. S2 in ESI.†
DNA O1 O2 O4 O5 Measured 8831.6 4824.1 8812.3 8819.1 Calculated 8831.9 4826.3 8815.9 8821.9duct O4 + 8A was obtained (Fig. 2a, the upper band of lane 4) with slower gel migration compared to O4 (Fig. 2a, the upper band of lane 3), suggesting that the ligation reaction occurred mostly between O2 and imidazole-activated O3, and not between O2 and un-activated Oc (Oc is the product from the previous cleavage reaction of O1 and 17Em1), in which case a band with the same migration as O4 would have been observed. The presence of C rather than rC in the product O4 was supported by the lower molecular weight of O4 in the MALDI-TOF mass spectrum as compared to that of O1 (Table 1), as well as the increased resistance to hydrolysis of O4 even in the presence of Pb2+ and 17Em1 (Fig. 2a, lane 5), which can catalyze the cleavage of a substrate containing an internal ribonucleotide linkage (O1),2,57 but not a substrate containing entirely deoxyribonucleotides (O4).
In addition to the single nucleotide repair functionality, it is also possible to use this methodology to edit the sequence of a DNA strand, which was used to convert the DNA strand O5 into O4 using the same cascade and conditions as before (Fig. 1b and S4 in ESI†). The product O4 was confirmed by PAGE (Fig. 2b) and MALDI-TOF MS (Table 1 and Fig. S2 in ESI†) and found to be identical to that obtained from the method in Fig. 1a.
Encouraged by the above results, we applied this method to modify the sequence of DNA immobilized on gold nanoparticles27 (AuNPs) to control the DNA-directed assembly of the AuNPs in a selective manner. DNA-functionalized gold AuNPs have been used in a variety of applications due to both their unique properties and the sequence-dependent hybridization of ssDNA immobilized on the AuNPs for controlled assembly.51–55 As shown in Fig. 3, when AuNPs are functionalized by complementary DNAs, the AuNPs can assemble into an “aggregated” state via DNA hybridization, which shows red-shifted and broadened absorption spectra compared to AuNPs functionalized by non-complementary DNAs.
Fig. 3 Assembly of two types of DNA-functionalized gold nanoparticles. If the sequences of the two DNAs are not complementary, the gold nanoparticles are in a “dispersed” state and exhibit a red color with a sharp absorption band peaked around 532 nm (left). In contrast, the assembly of the gold nanoparticles with complementary DNAs causes the formation of an “aggregated” state with a broad absorption band around 600 nm (right).
By modifying the sequences of DNA on the AuNPs, the assembly of the particles can be effectively controlled. Although methods for fabrication of DNAfunctionalized AuNPs have been developed,27 there are still limited methods to modify the DNA sequences already immobilized on AuNPs in order to tune their functions. It is even more challenging to modify a specific DNA sequence on multiply-functionalized AuNPs with different DNA sequences on each nanoparticle. Selective modification can allow each different function of the AuNP to be controlled in a selective fashion for potential applications.
AuNPs of 13 nm diameter were functionalized with DNA molecules via 30 -end thiols and used for this study. The formation of the AuNP assembly was confirmed by TEM images (Fig. S5 in ESI†) and characterized by large changes in absorption spectra27 (A700/A532 changed from <0.15 to >0.50 as illustrated in Fig. 5 and Table S1 in ESI†). As shown in Fig. 4, O6- functionalized AuNPs (red) were found to be able to form aggregates with O9-functionalized AuNPs (blue) via DNAdirected assembly through 12 complementary base pairs (Fig. 5 and S5 and Table S1†), but not with O10-functionalized AuNPs (purple), because of the 4 mismatched base pairs in the middle of the binding arm (Fig. 5 and S5 and Table S1†). After being treated with 17Em2 and Pb2+, the O6 on the surface of AuNPs was cleaved and converted to O8, which could not hybridize with either O9 or O10 efficiently. Thus no DNA-directed assembly was observed between the resulting O8-functionalized AuNPs and either O9- or O10-functionalized AuNPs (Fig. 5 and S5 and Table S1†). However, after a subsequent ligation reaction catalyzed by E47 in the presence of imidazole-activated O3 and Cu2+, O8 on the surface of AuNPs could be extended to O7, making the AuNPs capable of assembling with O10-, but not O9- functionalized AuNPs (Fig. 5 and S5 and Table S1†).
Fig. 4 Controlling the assembly of DNA-functionalized gold nanoparticles via cascade-mediated modification of the DNA sequences. The solid and dashed lines indicate the successful and unsuccessful formation of aggregates, respectively. The inset shows the assembly of AuNPs modified with the complementary strands O6 and O9 (top) and the lack of assembly between AuNPs modified with noncomplementary strands O9 and O7 (obtained by treating O6-functionalized AuNPs with 17Em2 and E47) (bottom)
Fig. 5 Absorption spectra of O6-, O7- or O8-functionalized AuNPs in the presence of O9- and O10-functionalized AuNPs, respectively. The red-shift of the peak indicates the formation of AuNP aggregations due to the hybridization of complementary DNAs on the AuNPs.27 For the ratios of absorbance (A700/A532), see Table S1 in ESI.†
Interestingly, the product, O7-functionalized AuNPs, showed inverse characteristics in the formation of DNA-directed assembly with O10- and O9-functionalized AuNPs, compared to the original O6-functionalized AuNPs. TEM images of O6- functionalized AuNPs, either with or without treatment with 17Em2/E47, mixed with O10-functionalized AuNPs, are displayed in the inset of Fig. 4. These results clearly demonstrate that the ability to edit and replace DNA on AuNPs allows for exquisite programmable control over the assembly of nanoparticles.
Taking advantage of the specificity of DNAzyme to its nucleic acid substrates by complementary base pairing in the binding arms, selective modification of DNA sequences on surface of multiple functional AuNPs was also achieved in this work. As depicted in Fig. 6, O6 (red) and O11 (blue) bi-functional AuNPs could be modified by 17Em1, 17Em2 and E47 selectively and sequentially. As shown in Fig. 6, AuNPs capable of forming DNA-directed assembly with both (A and E), either (B, C and F), or neither (D) of the O9- and O10-functionalized AuNPs could be obtained by monitoring the significant increase of A700/A532 as indication of assembly formation (Fig. 7 and Table S2 in ESI†). Such a result, which is challenging to achieve by other techniques, can be used for the construction of tunable nanoassemblies for various applications.
Fig. 6 (a) Scheme showing stepwise modification of DNA sequences on multiply-functional gold nanoparticles by the collaboration of 17Em1 or 17Em2 and E47. (b) DNA sequences of O6–O8 and O11.
Fig. 7 Absorption spectra of DNA-functionalized gold nanoparticles (A–F) (Fig. 6a) in the presence of O9- and O10-functionalized AuNPs, respectively. The red-shift of the peak indicates the formation of AuNP aggregations due to the hybridization of complementary DNAs on the AuNPs.27 For the ratios of absorbance (A700/A532), see Table S2 in ESI.†
In summary, by putting together a cascade of two DNAzymes with cleaving and ligating activities, we have generated a new functionality for effective DNA modification. This function was applied in the conversion of a single misincorporated ribonucleotide into the corresponding dexoyribonucleotide in DNA and the modification of DNA sequences on the surface of gold nanoparticles to modify and control their self-assembly through DNA hybridization. The results suggest that combining DNAzymes with different catalytic activities may achieve more interesting functions and thus broaden the applications of DNAzymes.
Notes and references
1 D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467–468.
2 R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229.
3 D. Sen and C. R. Geyer, Curr. Opin. Chem. Biol., 1998, 2, 680– 687.
4 Y. F. Li and R. R. Breaker, Curr. Opin. Struct. Biol., 1999, 9, 315–323.
5 Y. Lu, Chem.–Eur. J., 2002, 8, 4588–4596.
6 R. R. Breaker, Nature, 2004, 432, 838–845.
7 K. Schlosser and Y. F. Li, Chem. Biol., 2009, 16, 311–322.
8 S. K. Silverman, Acc. Chem. Res., 2009, 42, 1521–1531.
9 R. R. Breaker and G. F. Joyce, Chem. Biol., 1995, 2, 655–660.
10 N. Carmi, S. R. Balkhi and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2233–2237.
11 A. R. Feldman and D. Sen, J. Mol. Biol., 2001, 313, 283–294.
12 J. W. Liu, A. K. Brown, X. L. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061.
13 M. Chandra, A. Sachdeva and S. K. Silverman, Nat. Chem. Biol., 2009, 5, 718–720.
14 B. Cuenoud and J. W. Szostak, Nature, 1995, 375, 611–614.
15 A. Sreedhara, Y. F. Li and R. R. Breaker, J. Am. Chem. Soc., 2004, 126, 3454–3460.
16 W. E. Purtha, R. L. Coppins, M. K. Smalley and S. K. Silverman, J. Am. Chem. Soc., 2005, 127, 13124–13125.
17 Y. F. Li and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2746–2751.
18 Y. F. Li, Y. Liu and R. R. Breaker, Biochemistry, 2000, 39, 3106–3114.
19 T. L. Sheppard, P. Ordoukhanian and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7802–7807.
20 Y. F. Li and D. Sen, Nat. Struct. Biol., 1996, 3, 743–747
Lingjie Li, Jing Xu, Jinglei Lei, Jie Zhang, Frank McLarnon, Zidong Wei, Nianbing Li and Fusheng Pan
J. Mater. Chem. A, 2015,3, 1953-1960
Abstract
The Ni(OH)2 hexagonal platelets were in situ fabricated on Ni foam as a binder-free supercapacitor electrode material with high performance and excellent cycling stability by a one-step, cost-effective, green hydrothermal treatment of three-dimensional (3D) Ni foam in a 15 wt% H2O2 aqueous solution.
Meirong Xia, Ying Liu, Zidong Wei, Siguo Chen, Kun Xiong, Li Li, Wei Ding, Jinsong Hu, Li-Jun Wan, Rong Li and Shahnaz Fatima Alvia
J. Mater. Chem. A, 2013,1, 14443-14448
Abstract
A facile and controllable process for preparing Pd@Pt/CNT core@shell catalysts for the oxygen reduction reaction (ORR) via Pd-induced Pt(IV) reduction on Pd/CNT.
Linfeng Xiong, Hui Zhang, Aiqing Zhong, Zidong He and Kun Huang
Chem. Commun., 2014,50, 14778-14781
Abstract
A novel method that enables the formation of core-confined bottlebrush copolymers (CCBCs) as catalyst supports for one-pot cascade reactions is reported for the first time.
The 2015 Nobel Prize in Chemistry was jointly awarded to Tomas Lindahl, Paul Modrich and Aziz Sancar for their “mechanistic studies of DNA repair”. Their research has not only revolutionised our knowledge of how we function but it also lead to the development of life-saving treatments. In celebration of their landmark achievements, we are delighted to present a special Nobel Prize collection of recent Chemical Communications, Chemical Science and Chemical Society Reviews articles on DNA repair.
Thomas Lindahl’s research pieced together a molecular image of how base excision repairs DNA when a base of a nucleotide is damaged and subsequently managed to recreate the human repair process in vitro. The mechanism known as nucleotide excision repair, which excises damage from UV and carcinogenic substances, was then mapped by Aziz Sancar – the molecular details of this process changed the entire research field. Paul Modrich also studied the human version of the repair system. His work focused on DNA mismatch repair, a natural process which corrects mismatches that occur when DNA is copied during cell division.
The research carried out by the three 2015 Nobel Laureates in Chemistry has not only revolutionised our knowledge of how we function but also lead to the development of life – saving treatments.
EDITING Researchers are learning how to use synthetic RNA sequences to control the cutting of any piece of DNA they choose. The cell will repair the cut, but an imperfect repair may disable the gene. Or a snippet of different DNA can be inserted to fill the gap, effectively editing the DNA sequence.
By:
Wallace Ravven
2.2.20 Principles of Gene Editing, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
The New York Times calls it “a scientific frenzy.” Science magazine dubbed it “red hot” — “The CRISPR Craze.”
It’s been less than two years since Berkeley biochemist Jennifer Doudna reported in Science a startlingly versatile strategy to precisely target and snip out DNA at multiple sites in the cells of microbes, plants and animals.
But since her landmark paper, more than 100 labs have already taken up the new genomic engineering technique to delete, add or suppress genes in fruit flies, mice, zebrafish and other animals widely used to model genetic function in human disease.
Jennifer Doudna in her lab. Photo: Roy Kaltschmidt
Last year, Doudna and her colleagues showed that this “molecular scissors” approach, known as CRISPR/Cas9, can be used with great precision to selectively disable or add several genes at once in human cells, offering a potent new tool to understand and treat complex genetic diseases.
Journal articles now appear almost weekly as researchers around the word apply the technique in basic and clinical research. Patents have been filed and licensed, and companies founded last year in Cambridge, London and Berkeley have begun zeroing in on agricultural, industrial and biomedical applications.
“I’ve never experienced anything like the pace of discovery before in my life,” Doudna says of the flurry of experimentation flowing from her 2012 paper co-authored with Emmanuelle Charpentier, now at the Helmholtz Centre for Infection Research in Germany.
HOW can we create a SNIPPET using 3D Printer? The SNIPPET with Transcription Error will be removed REPAIR the gene by a new Snippet wihtout the Transcription error
Zinc-finger proteins of the Cys2His2 type bind DNA-RNA hybrids with affinities comparable to those for DNA duplexes. Such zinc-finger proteins were converted into site-specific cleaving enzymes by fusing them to the FokI cleavage domain. The fusion proteins are active and under optimal conditions cleave DNA duplexes in a sequence-specific manner. These fusions also exhibit site-specific cleavage of the DNA strand within DNA-RNA hybrids albeit at a lower efficiency (approximately 50-fold) compared to the cleavage of the DNA duplexes. These engineered endonucleases represent the first of their kind in terms of their DNA-RNA cleavage properties, and they may have important biological applications.
Construction of vectors producing ZF–FN.
Chembiochem. 2009 May 25;10(8):1279-88. doi: 10.1002/cbic.200900040.
Artificial restriction DNA cutters as new tools for gene manipulation.
The final cut. Two types of artificial tools (artificial restriction DNA cutter and zinc finger nuclease) that cut double-stranded DNA through hydrolysis of target phosphodiester linkages, have been recently developed. The chemical structures, preparation, properties, and typical applications of these two man-made tools are reviewed.Two types of artificial tools that cut double-stranded DNA through hydrolysis of target phosphodiester linkages have been recently developed. One is the chemistry-based artificial restriction DNA cutter (ARCUT) that is composed of a Ce(IV)-EDTA complex, which catalyses DNA hydrolysis, and a pair of pseudo-complementary peptide nucleic acid fragments for sequence recognition. Another type of DNA cutter, zinc finger nuclease (ZFN), is composed of the nuclease domain of naturally occurring FokI restriction endonuclease and a designed zinc finger DNA-binding domain. For both of these artificial tools, the scission site and specificity can be freely chosen according to our needs, so that even huge genomic DNA sequences can be selectively cut at the target site. In this article, the chemical structures, preparation, properties, and typical applications of these two man-made tools are described.
Figure 1: DNA Binding Overview (original image) (crystal image rendered from PDB: 4UN3 Anders et al. 2014.)
CRISPR/Cas9 systems use a guide RNA with a region complementary to the target DNA to specifically bind their target sequences. However, there is an immediate and inherent issue with this. In order to achieve specificity, longer guide RNAs are beneficial, as each nucleotide in the RNA guide increases the specificity of the nuclease about 4-fold. However, in order for the DNA to melt and accommodate base-pairing to the guide RNA, the longer the RNA guide, the less efficient the nuclease. How can CRISPR/Cas9 systems have such dramatically increased specificity over other nucleases such as TALENS and ZFNS and still maintain roughly the same, if not better, efficiency? (Mali et al. 2013)
The answer is that the CRISPR/Cas9 system uses the Protospacer Adjacent Motif (PAM) binding as a preliminary step in locating the target sequence. As was determined by single molecule fluorescence microscopy, the initial binding of Cas9 to PAM (N-G-G) sequences allows the enzyme to quickly screen for potential target sequences. The enzyme will rapidly detach from DNA that does not have the proper PAM sequence. If the protein finds a potential target with the appropriate PAM, it will to melt the remaining DNA on the target to test whether the remaining target sequence is complementary to its guide sequence. The PAM binding step allows the protein to quickly screen potential targets and avoid melting many non-target sequences in its search for fully complementary sequences to cut. (Sternberg et al. 2014)
In July of 2014, Anders et al. published a crystal structure that led to a model for PAM-dependent target DNA binding, unwinding, and recognition by the Cas9 nuclease. The following images are created based off of figure 4 of the paper, or are images rendered inPymol (distributed by Schrödinger) using the crystal structure from that paper (obtained from the Protein Data Bank).
Proposed model for PAM-dependent target DNA binding, melting, and recognition by Cas9:
1. PAM Binding:
The Protospacer Adjacent Motif (PAM) NGG bases of the target DNA strand are shown in yellow. Arginine residues 1333 and 1335 of the PAM Interacting (PI) domain bind to the major groove of the guanine bases in the PAM. A lysine residue in the Phosphate Lock Loop, also in the PI domain, binds the minor groove.
2. Phosphate Lock Loop:
This positions the PAM and target DNA such that serine 1109 in the phosphate lock loop, and two nitrogens of the phosphate lock loop’s backbone, can form hydrogen bonds to the phosphate at position +1 of the PAM. This stabilizes the target DNA such that the first bases of the target sequence (or the protospacer) can melt and rotate upwards towards the guide RNA.
3. Guide RNA:
If the target DNA is complementary to the guide RNA strand, the two strands will base pair. This will allow the target DNA to unzip, as the bases flip up and bind the guide RNA. Without the initial PAM binding and stabilization of the +1 phosphate, the guide RNA would very rarely be able to bind the target DNA, and Cas9 would be very inefficient. This illustrates a mechanism that explains why Cas9 is able to have both high efficiency and high specificity, thus making it a powerful genome editing tool.
4. Cleavage:
Finally, complete annealing of the guide RNA to the target DNA allows the HNH and RuvC nucleases to cleave their respective strands. These nucleases cleave very specifically between the 3rd and 4th nucleotides from the PAM. Again, this specificity of cleavage, as well as the fact that the individual nucleases may be mutated independently and without affecting the ability of Cas9 to bind specific sequences, make the CRISPR/Cas9 system a simultaneously powerful and flexible genome editing tool.
Seminal studies showed that CRISPR-Cas systems provide adaptive immunity in prokaryotes and promising gene-editing tools from bacteria to humans. Yet, reports diverged on whether some CRISPR systems naturally target DNA or RNA. Here, Samai and colleagues unify the studies, showing that a single type III CRISPR-Cas system cleaves both DNA and RNA targets, independently.
More on Cleavage
Supplementary Figure 11: Base-skipping CRISPR mutants mediated efficient cleavage with Cas9 and D10A Cas9.
HEK293T cells were transfected with the indicated plasmids and the genomic DNA harvested 48 h later was assessed using the Surveyor assay. The mutant name is as described in Fig. 2. wt: wild type; UD, undetectable.
September 18, 2015 | After Chinese scientists announced in April that they had edited the genes in human embryos, many researchers said it shouldn’t be done. Scientists in London say they want to do it for research only. NPR.org
October 29, 2015 | BGI ― formerly the Beijing Genomics Institute, China’s contribution to the Human Genome Project, and now a hybrid state agency and private corporation ― is one of the world’s largest scientific research and industrial powers. From its headquarters in Shenzhen and outposts across Asia, Europe and the United States, BGI performs population-scale genomics studies, runs the world’s largest on-demand DNA sequencing service, and sells a small but growing suite of commercial products. Last week, BGIrevealed the first sequencing instrument to be developed and produced in China, the BGISEQ-500, launched exclusively to Chinese markets.
Like other recent Chinese accomplishments in high-tech fields, the sequencer is as much a point of national pride as it is a commercial venture. “Shenzhen has transformed itself from labor-intensive industry to high tech,” says He Jiankui, a specialist in genomics and biochemistry who teaches at the city’s South University of Science and Technology of China. “The government has ambitions. They’re trying to switch from ‘Made in China’ to ‘Invented in China.’”
October 1, 2015 | This Wednesday, in a surprise announcement, Pacific Biosciences of Menlo Park, Calif., confirmed rumors that it has been working on a smaller, more price-effective version of its RS II gene sequencer. But rather than push out a scaled-down benchtop instrument for simple use cases, as many had anticipated, the company unveiled a machine that improves on the RS II in every particular: less than half the cost, a third the size, and most importantly, almost seven times as powerful.
New and Unusual DNA Repair Activity Identified
Click Image To Enlarge +
The new type of DNA repair enzyme, AlkD on the left, can identify and remove a damaged DNA base without forcing it to physically “flip” to the outside of the DNA backbone, which is how all the other DNA repair enzymes in its family work, as illustrated by the human AAG enzyme on the right. The enzymes are shown in grey, the DNA backbone is orange, normal DNA base pairs are yellow, the damaged base is blue and its pair base is green. [Brandt Eichman, Vanderbilt University]
Hot on the heels of the recent announcement of the Nobel Prize in Chemistry being awarded for seminal discoveries in the area of DNA repair, researchers at Vanderbilt University have published data describing new enzymatic activity for a DNA glycosylase discovered previously in the bacteria Bacillus cereus.
When Watson and Crick first published their now famous double-helix structure of DNA, many scientists imagined the molecule to be extremely chemically stable—acting as the template for passing along inheritable genetic traits. However, over the years investigators have since discovered DNA’s susceptibility to damage and its dynamic nature to repair itself, to maintain genomic stability.
“It’s a double-edged sword,” remarked senior author and project leader Brandt Eichman, Ph.D., associate professor of biological sciences and biochemistry at Vanderbilt. “If DNA were too reactive then it wouldn’t be capable of storing genetic information. But, if it were too stable, then it wouldn’t allow organisms to evolve.”
There are many ways that DNA can become damaged, but they can be classified into two basic groups: environmental sources including ultraviolet light, toxic chemicals, and ionizing radiation and internal sources, which include, reactive oxygen species, a number of chemicals the cell produces during normal metabolism, and even water.
“More than 10,000 DNA damage events occur each day within every cell of the human body, which must be repaired for DNA to function properly,” explained lead author Elwood Mullins, Ph.D., a postdoctoral research associate in Dr. Eichman’s laboratory.
The Vanderbilt team discovered the new repair activity while studying the DNA glycosylase AlkD. Glycosylases are part of a family of enzymes discovered by Tomas Lindahl, Ph.D., who received this year’s Nobel prize for recognizing that these enzymes removed damaged DNA bases through a process called base-excision repair (BER).
Briefly, during BER, a specific glycosylase molecule binds to DNA at the location of a lesion and bends the double-helix in a way that causes the damaged base to flip from the inside of the helix to the outside. The enzyme fits around the flipped out base and holds it in a position that exposes its link to the DNA’s sugar backbone, allowing the enzyme to detach it. After the damaged base has been removed, additional DNA-repair proteins move in to replace it with a new, undamaged base.
Dr. Eichman and his team found that AlkD from B. cereus works in a totally different fashion—as it does not require base flipping to recognize damaged DNA or repair it. Using crystallography techniques, the researchers were able to determine that AlkD forms a series of interactions with the DNA backbone at and around the lesion while the lesion is still stacked in the double helix. Several of these interactions are contributed by three amino acids in the enzyme that catalyze excision of the damaged base.
The findings from this study were published recently in Nature through an article entitled “The DNA glycosylase AlkD uses a non-base-flipping mechanism to excise bulky lesions.”
Additionally, the investigators found that AlkD identifies lesions by interacting with the DNA backbone without contacting the damaged base itself and can repair many different types of lesions as long as they are positively charged. Since the enzyme doesn’t have the same type of binding pocket, it isn’t restricted in the same way as other glycosylases. Lastly, AlkD can excise much bulkier lesions than other glycosylases. Base excision repair is limited to relatively small lesions. A different pathway called nucleotide excision repair typically handles larger lesions like those caused by UV radiation damage. However, Dr. Eichman’s team discovered that AlkD could excise lesions that would normally default to other DNA repair pathways.
“Our discovery shows that we still have a lot to learn about DNA repair and that there may be alternative repair pathways yet to be discovered. It certainly shows us that a much broader range of DNA damage can be removed in ways that we didn’t think were possible,” Dr. Eichman stated. “Bacteria are using this to their advantage to protect themselves against the antibacterial agents they produce. Humans may even have DNA-repair enzymes that operate in similar fashion to remove complex types of DNA damage. This could have clinical relevance because these enzymes if they exist, could be reducing the effectiveness of drugs designed to kill cancer cells by shutting down their ability to replicate.”
New Generation of Platinated Compounds to Circumvent Resistance
Curator/Writer: Stephen J. Williams, Ph.D.
Resistance to chemotherapeutic drugs continues to be a major hurdle in the treatment of neoplastic disorders, irregardless if the drug is a member of the cytotoxic “older” drugs or the cytostatic “newer” personalized therapies like the tyrosine kinase inhibitors. For the platinatum compounds such as cisplatin and carboplatin, which are mainstays in therapeutic regimens for ovarian and certain head and neck cancers, development of resistance is often regarded as the final blow, as new options for these diseases have been limited.
Although there are many mechanisms by which resistance to platinated compounds may develop the purpose of this posting is not to do an in-depth review of this area except to refer the reader to the book Ovarian Cancer and just to summarize the well accepted mechanisms of cisplatin resistance including:
Decreased cellular cisplatin influx
Increased cellular cisplatin efflux
Increased cellular glutathione and subsequent conjugation, inactivation
Increased glutathione-S-transferase activity (GST) and subsequent inactivation, conjugation
Increased γ-GGT
Increased metallothionenes with subsequent conjugation, inactivation
Increased DNA repair: increased excision repair
DNA damage tolerance: loss of mismatch repair (MMR)
altered cell signaling activities and cell cycle protein expression
Williams, S.J., and Hamilton, T.C. Chemotherapeutic resistance in ovarian cancer. In: S.C. Rubin, and G.P. Sutton (eds.), Ovarian Cancer, pp.34-44. Lippincott, Wilkins, and Williams, New York, 2000.
Also for a great review on clinical platinum resistance by Drs. Maritn, Hamilton and Schilder please see the following Clinical Cancer Research link here.
This curation represents the scientific rationale for the development of a new class of platinated compounds which are meant to circumvent mechanisms of resistance, in this case the loss of mismatch repair (MMR) and increased tolerance to DNA damage.
An early step in the production of cytotoxicity by the important anticancer drug cisplatin and its analog carboplatin is the formation of intra- and inter-strand adducts with tumor cell DNA 1-3. This damage triggers a cascade of events, best characterized by activation of damage-sensing kinases (reviewed in 4), p53 stabilization, and induction of p53-related genes involved in apoptosis and cell cycle arrest, such as bax and the cyclin-dependent kinase inhibitor p21waf1/cip1/sdi1 (p21), respectively 5,6. DNA damage significantly induces p21 in various p53 wild-type tumor cell lines, including ovarian carcinoma cells, and this induction is responsible for the cell cycle arrest at G1/S and G2/M borders, allowing time for repair 7,8. DNA lesions have the ability of to result in an opening of chromatin structure, allowing for transcription factors to enter 56-58. Therefore the anti-tumoral ability of cisplatin and other DNA damaging agents is correlated to their ability to bind to DNA and elicit responses, such as DNA breaks or DNA damage responses which ultimately lead to cell cycle arrest and apoptosis. Therefore either repair of such lesions, the lack of recognition of such lesions, or the cellular tolerance of such lesions can lead to resistance of these agents.
Mechanisms of Cisplatin Sensitivity and Resistance. Red arrows show how a DNA lesion results in chemo-sensitivity while the beige arrow show common mechanisms of resistance including increased repair of the lesion, effects on expression patterns, and increased inactivation of the DNA damaging agent by conjugation reactions
Increased DNA Repair Mechanisms of Platinated Lesion Lead to ChemoResistance
Description of Different Types of Cellular DNA Repair Pathways. Nucleotide Excision Repair is commonly up-regulated in highly cisplatin resistant cells
Loss of Mismatch Repair Can Lead to DNA Damage Tolerance
In the following Cancer Research paper Dr. Vaisman in the lab of Dr. Steve Chaney at North Carolina (and in collaboration with Dr. Tom Hamilton) describe how cisplatin resistance may arise from loss of mismatch repair and how oxaliplatin lesions are not recognized by the mismatch repair system.
Defects in mismatch repair are associated with cisplatin resistance, and several mechanisms have been proposed to explain this correlation. It is hypothesized that futile cycles of translesion synthesis past cisplatin-DNA adducts followed by removal of the newly synthesized DNA by an active mismatch repair system may lead to cell death. Thus, resistance to platinum-DNA adducts could arise through loss of the mismatch repair pathway. However, no direct link between mismatch repair status and replicative bypass ability has been reported. In this study, cytotoxicity and steady-state chain elongation assays indicate that hMLH1 or hMSH6 defects result in 1.5-4.8-fold increased cisplatin resistance and 2.5-6-fold increased replicative bypass of cisplatin adducts. Oxaliplatin adducts are not recognized by the mismatch repair complex, and no significant differences in bypass of oxaliplatin adducts in mismatch repair-proficient and -defective cells were found. Defects in hMSH3 did not alter sensitivity to, or replicative bypass of, either cisplatin or oxaliplatin adducts. These observations support the hypothesis that mismatch repair defects in hMutL alpha and hMutS alpha, but not in hMutS beta, contribute to increased net replicative bypass of cisplatin adducts and therefore to drug resistance by preventing futile cycles of translesion synthesis and mismatch correction.
The following are slides I had co-prepared with my mentor Dr. Thomas C. Hamilton, Ph.D. of Fox Chase Cancer Center on DNA Mismatch Repair, Oxaliplatin and Ovarina Cancer.
Multiple Platinum Analogs of Cisplatin (like Oxaliplatin )Had Been Designed to be Sensitive in MMR Deficient Tumors
Please see below video on 2015 Nobel Laureates and their work to elucidate the celluar DNA repair mechanisms.
Clinical genetics expert Kenneth Offit gives an overview of Lynch syndrome, a genetic disorder that can cause colon (HNPCC) and other cancers by defects in the MSH2 DNA mismatch repair gene. (View Video)
References
Johnson, S. W. et al. Relationship between platinum-DNA adduct formation, removal, and cytotoxicity in cisplatin sensitive and resistant human ovarian cancer cells. Cancer Res54, 5911-5916 (1994).
Eastman, A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacology and Therapeutics34, 155-166 (1987).
Zhen, W. et al. Increased gene-specific repair of cisplatin interstrand cross-links in cisplatin-resistant human ovarian cancer cell lines. Molecular and Cellular Biology12, 3689-3698 (1992).
Durocher, D. & Jackson, S. P. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol13, 225-231 (2001).
el-Deiry, W. S. p21/p53, cellular growth control and genomic integrity. Curr Top Microbiol Immunol227, 121-37 (1998).
Ewen, M. E. & Miller, S. J. p53 and translational control. Biochim Biophys Acta1242, 181-4 (1996).
Gartel, A. L., Serfas, M. S. & Tyner, A. L. p21–negative regulator of the cell cycle. Proc Soc Exp Biol Med213, 138-49 (1996).
Chang, B. D. et al. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene19, 2165-70 (2000).
Davies, N. P., Hardman, L. C. & Murray, V. The effect of chromatin structure on cisplatin damage in intact human cells. Nucleic Acids Res28, 2954-2958 (2000).
Vichi, P. et al. Cisplatin- and UV-damaged DNA lure the basal transcription factor TFIID/TBP. Embo J16, 7444-7456 (1997).
Xiao, G. et al. A DNA damage signal is required for p53 to activate gadd45. Cancer Res60, 1711-9 (2000).
Other articles in this Open Access Journal on ChemoResistance Include:













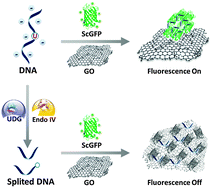
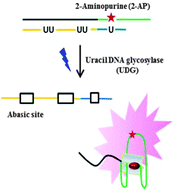
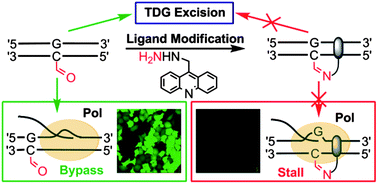
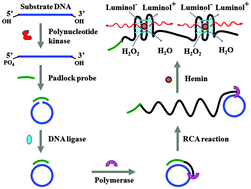
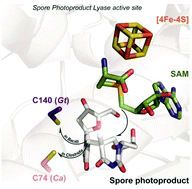























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}