Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
A highly effective platforms for the ex utero culture of post-implantation mouse embryos have been developed in the present study by scientists of the Weizmann Institute of Science in Israel. The study was published in the journal Nature. They have grown more than 1,000 embryos in this way. This study enables the appropriate development of embryos from before gastrulation (embryonic day (E) 5.5) until the hindlimb formation stage (E11). Late gastrulating embryos (E7.5) are grown in three-dimensional rotating bottles, whereas extended culture from pre-gastrulation stages (E5.5 or E6.5) requires a combination of static and rotating bottle culture platforms.
At Day 11 of development more than halfway through a mouse pregnancy the researchers compared them to those developing in the uteruses of living mice and were found to be identical. Histological, molecular and single-cell RNA sequencing analyses confirm that the ex utero cultured embryos recapitulate in utero development precisely. The mouse embryos looked perfectly normal. All their organs developed as expected, along with their limbs and circulatory and nervous systems. Their tiny hearts were beating at a normal 170 beats per minute. But, the lab-grown embryos becomes too large to survive without a blood supply. They had a placenta and a yolk sack, but the nutrient solution that fed them through diffusion was no longer sufficient. So, a suitable mechanism for blood supply is required to be developed.
Till date the only way to study the development of tissues and organs is to turn to species like worms, frogs and flies that do not need a uterus, or to remove embryos from the uteruses of experimental animals at varying times, providing glimpses of development more like in snapshots than in live videos. This research will help scientists understand how mammals develop and how gene mutations, nutrients and environmental conditions may affect the fetus. This will allow researchers to mechanistically interrogate post-implantation morphogenesis and artificial embryogenesis in mammals. In the future it may be possible to develop a human embryo from fertilization to birth entirely outside the uterus. But the work may one day raise profound questions about whether other animals, even humans, should or could be cultured outside a living womb.
Anti-Müllerian Hormone (AMH), is secreted by growing follicles that contains the egg or ovum. According to regular practice low AMH and high Follicle Stimulating Hormone (FSH) are generally considered as indicators of diminished egg quantity in a female. But, there are several cases the female conceived absolutely normally without any support even after low AMH was reported.
Therefore, a new research published in the Journal of the American Medical Association declares that AMH doesn’t dictate a woman’s reproductive potential. Although AMH testing is one of the most common ways that doctors assess a woman’s fertility. Present research says that all it takes is one egg each cycle and AMH is not a marker of whether a female can or cannot become pregnant. So, for women who haven’t yet tried to get pregnant and who are wondering whether they are fertile, an AMH value isn’t going to be helpful in that context. In addition, AMH is not necessarily a good marker to predict that whether one has to cryopreserve her eggs. So, practically doctors don’t yet have a way to definitively predict egg quality or a woman’s long-term ability to conceive, but age is obviously one of the most important factors.
The above mentioned study followed 750 women between the ages of 30 and 44 who had been trying to conceive for three months or less. During the 12-month observation period, those with low AMH values of less than 0.7 were not less likely to conceive than those who had normal AMH values. The study had various limitations, however, that are worth noting. The researchers only included women who did not have a history of infertility. Women who sought fertility treatments (about 6 percent) were withdrawn. And only 12 percent of the women were in the 38-to-44 age range. In addition, the number of live births was unavailable.
Among women aged 30 to 44 years without a history of infertility who had been trying to conceive for 3 months or less, biomarkers indicating diminished ovarian reserve compared with normal ovarian reserve were not associated with reduced fertility. These findings do not support the use of urinary or blood FSH tests or AMH levels to assess natural fertility for women with these characteristics. The researchers’ next want to see whether low AMH is associated with a higher risk of miscarriage among the women who conceived.
Although AMH testing isn’t designed to be an overall gauge of a woman’s fertility, it can still provide valuable information, especially for women who are infertile and seeking treatment. It can assist in diagnosing polycystic ovarian syndrome, and identify when a woman is getting closer to menopause. Previous research also showed that AMH is good predictor of a woman’s response to ovarian stimulation for in vitro fertilization and therefore it can predict the probability of conceiving via in vitro fertilization (I.V.F.).
Disease related changes in proteomics, protein folding, protein-protein interaction, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Disease related changes in proteomics, protein folding, protein-protein interaction
Curator: Larry H. Bernstein, MD, FCAP
LPBI
Frankenstein Proteins Stitched Together by Scientists
The Frankenstein monster, stitched together from disparate body parts, proved to be an abomination, but stitched together proteins may fare better. They may, for example, serve specific purposes in medicine, research, and industry. At least, that’s the ambition of scientists based at the University of North Carolina. They have developed a computational protocol called SEWING that builds new proteins from connected or disconnected pieces of existing structures. [Wikipedia]
Unlike Victor Frankenstein, who betrayed Promethean ambition when he sewed together his infamous creature, today’s biochemists are relatively modest. Rather than defy nature, they emulate it. For example, at the University of North Carolina (UNC), researchers have taken inspiration from natural evolutionary mechanisms to develop a technique called SEWING—Structure Extension With Native-substructure Graphs. SEWING is a computational protocol that describes how to stitch together new proteins from connected or disconnected pieces of existing structures.
“We can now begin to think about engineering proteins to do things that nothing else is capable of doing,” said UNC’s Brian Kuhlman, Ph.D. “The structure of a protein determines its function, so if we are going to learn how to design new functions, we have to learn how to design new structures. Our study is a critical step in that direction and provides tools for creating proteins that haven’t been seen before in nature.”
Traditionally, researchers have used computational protein design to recreate in the laboratory what already exists in the natural world. In recent years, their focus has shifted toward inventing novel proteins with new functionality. These design projects all start with a specific structural “blueprint” in mind, and as a result are limited. Dr. Kuhlman and his colleagues, however, believe that by removing the limitations of a predetermined blueprint and taking cues from evolution they can more easily create functional proteins.
Dr. Kuhlman’s UNC team developed a protein design approach that emulates natural mechanisms for shuffling tertiary structures such as pleats, coils, and furrows. Putting the approach into action, the UNC team mapped 50,000 stitched together proteins on the computer, and then it produced 21 promising structures in the laboratory. Details of this work appeared May 6 in the journal Science, in an article entitled, “Design of Structurally Distinct Proteins Using Strategies Inspired by Evolution.”
“Helical proteins designed with SEWING contain structural features absent from other de novo designed proteins and, in some cases, remain folded at more than 100°C,” wrote the authors. “High-resolution structures of the designed proteins CA01 and DA05R1 were solved by x-ray crystallography (2.2 angstrom resolution) and nuclear magnetic resonance, respectively, and there was excellent agreement with the design models.”
Essentially, the UNC scientists confirmed that the proteins they had synthesized contained the unique structural varieties that had been designed on the computer. The UNC scientists also determined that the structures they had created had new surface and pocket features. Such features, they noted, provide potential binding sites for ligands or macromolecules.
“We were excited that some had clefts or grooves on the surface, regions that naturally occurring proteins use for binding other proteins,” said the Science article’s first author, Tim M. Jacobs, Ph.D., a former graduate student in Dr. Kuhlman’s laboratory. “That’s important because if we wanted to create a protein that can act as a biosensor to detect a certain metabolite in the body, either for diagnostic or research purposes, it would need to have these grooves. Likewise, if we wanted to develop novel therapeutics, they would also need to attach to specific proteins.”
Currently, the UNC researchers are using SEWING to create proteins that can bind to several other proteins at a time. Many of the most important proteins are such multitaskers, including the blood protein hemoglobin.
Histone Mutation Deranges DNA Methylation to Cause Cancer
In some cancers, including chondroblastoma and a rare form of childhood sarcoma, a mutation in histone H3 reduces global levels of methylation (dark areas) in tumor cells but not in normal cells (arrowhead). The mutation locks the cells in a proliferative state to promote tumor development. [Laboratory of Chromatin Biology and Epigenetics at The Rockefeller University]
They have been called oncohistones, the mutated histones that are known to accompany certain pediatric cancers. Despite their suggestive moniker, oncohistones have kept their oncogenic secrets. For example, it has been unclear whether oncohistones are able to cause cancer on their own, or whether they need to act in concert with additional DNA mutations, that is, mutations other than those affecting histone structures.
While oncohistone mechanisms remain poorly understood, this particular question—the oncogenicity of lone oncohistones—has been resolved, at least in part. According to researchers based at The Rockefeller University, a change to the structure of a histone can trigger a tumor on its own.
This finding appeared May 13 in the journal Science, in an article entitled, “Histone H3K36 Mutations Promote Sarcomagenesis Through Altered Histone Methylation Landscape.” The article describes the Rockefeller team’s study of a histone protein called H3, which has been found in about 95% of samples of chondoblastoma, a benign tumor that arises in cartilage, typically during adolescence.
The Rockefeller scientists found that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo.
After the scientists inserted the H3 histone mutation into mouse mesenchymal progenitor cells (MPCs)—which generate cartilage, bone, and fat—they watched these cells lose the ability to differentiate in the lab. Next, the scientists injected the mutant cells into living mice, and the animals developed the tumors rich in MPCs, known as an undifferentiated sarcoma. Finally, the researchers tried to understand how the mutation causes the tumors to develop.
The scientists determined that H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases.
“Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation,” the authors of the Science study wrote. “After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation.”
Essentially, when the H3K36M mutation occurs, the cell becomes locked in a proliferative state—meaning it divides constantly, leading to tumors. Specifically, the mutation inhibits enzymes that normally tag the histone with chemical groups known as methyls, allowing genes to be expressed normally.
In response to this lack of modification, another part of the histone becomes overmodified, or tagged with too many methyl groups. “This leads to an overall resetting of the landscape of chromatin, the complex of DNA and its associated factors, including histones,” explained co-author Peter Lewis, Ph.D., a professor at the University of Wisconsin-Madison and a former postdoctoral fellow in laboratory of C. David Allis, Ph.D., a professor at Rockefeller.
The finding—that a “resetting” of the chromatin landscape can lock the cell into a proliferative state—suggests that researchers should be on the hunt for more mutations in histones that might be driving tumors. For their part, the Rockefeller researchers are trying to learn more about how this specific mutation in histone H3 causes tumors to develop.
“We want to know which pathways cause the mesenchymal progenitor cells that carry the mutation to continue to divide, and not differentiate into the bone, fat, and cartilage cells they are destined to become,” said co-author Chao Lu, Ph.D., a postdoctoral fellow in the Allis lab.
Once researchers understand more about these pathways, added Dr. Lewis, they can consider ways of blocking them with drugs, particularly in tumors such as MPC-rich sarcomas—which, unlike chondroblastoma, can be deadly. In fact, drugs that block these pathways may already exist and may even be in use for other types of cancers.
“One long-term goal of our collaborative team is to better understand fundamental mechanisms that drive these processes, with the hope of providing new therapeutic approaches,” concluded Dr. Allis.
Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape
Missense mutations (that change one amino acid for another) in histone H3 can produce a so-called oncohistone and are found in a number of pediatric cancers. For example, the lysine-36–to-methionine (K36M) mutation is seen in almost all chondroblastomas. Lu et al. show that K36M mutant histones are oncogenic, and they inhibit the normal methylation of this same residue in wild-type H3 histones. The mutant histones also interfere with the normal development of bone-related cells and the deposition of inhibitory chromatin marks.
Several types of pediatric cancers reportedly contain high-frequency missense mutations in histone H3, yet the underlying oncogenic mechanism remains poorly characterized. Here we report that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo. H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases. Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation. After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation. Our findings are mirrored in human undifferentiated sarcomas in which novel K36M/I mutations in H3.1 are identified.
Mitochondria? We Don’t Need No Stinking Mitochondria!
Diagram comparing typical eukaryotic cell to the newly discovered mitochondria-free organism. [Karnkowska et al., 2016, Current Biology 26, 1–11]
The organelle that produces a significant portion of energy for eukaryotic cells would seemingly be indispensable, yet over the years, a number of organisms have been discovered that challenge that biological pretense. However, these so-called amitochondrial species may lack a defined organelle, but they still retain some residual functions of their mitochondria-containing brethren. Even the intestinal eukaryotic parasite Giardia intestinalis, which was for many years considered to be mitochondria-free, was proven recently to contain a considerably shriveled version of the organelle.
Now, an international group of scientists has released results from a new study that challenges the notion that mitochondria are essential for eukaryotes—discovering an organism that resides in the gut of chinchillas that contains absolutely no trace of mitochondria at all.
“In low-oxygen environments, eukaryotes often possess a reduced form of the mitochondrion, but it was believed that some of the mitochondrial functions are so essential that these organelles are indispensable for their life,” explained lead study author Anna Karnkowska, Ph.D., visiting scientist at the University of British Columbia in Vancouver. “We have characterized a eukaryotic microbe which indeed possesses no mitochondrion at all.”
Mysterious Eukaryote Missing Mitochondria
Researchers uncover the first example of a eukaryotic organism that lacks the organelles.
Monocercomonoides sp. PA203VLADIMIR HAMPL, CHARLES UNIVERSITY, PRAGUE, CZECH REPUBLIC
Scientists have long thought that mitochondria—organelles responsible for energy generation—are an essential and defining feature of a eukaryotic cell. Now, researchers from Charles University in Prague and their colleagues are challenging this notion with their discovery of a eukaryotic organism,Monocercomonoides species PA203, which lacks mitochondria. The team’s phylogenetic analysis, published today (May 12) in Current Biology,suggests that Monocercomonoides—which belong to the Oxymonadida group of protozoa and live in low-oxygen environments—did have mitochondria at one point, but eventually lost the organelles.
“This is quite a groundbreaking discovery,” said Thijs Ettema, who studies microbial genome evolution at Uppsala University in Sweden and was not involved in the work.
“This study shows that mitochondria are not so central for all lineages of living eukaryotes,” Toni Gabaldonof the Center for Genomic Regulation in Barcelona, Spain, who also was not involved in the work, wrote in an email to The Scientist. “Yet, this mitochondrial-devoid, single-cell eukaryote is as complex as other eukaryotic cells in almost any other aspect of cellular complexity.”
Charles University’s Vladimir Hampl studies the evolution of protists. Along with Anna Karnkowska and colleagues, Hampl decided to sequence the genome of Monocercomonoides, a little-studied protist that lives in the digestive tracts of vertebrates. The 75-megabase genome—the first of an oxymonad—did not contain any conserved genes found on mitochondrial genomes of other eukaryotes, the researchers found. It also did not contain any nuclear genes associated with mitochondrial functions.
“It was surprising and for a long time, we didn’t believe that the [mitochondria-associated genes were really not there]. We thought we were missing something,” Hampl told The Scientist. “But when the data kept accumulating, we switched to the hypothesis that this organism really didn’t have mitochondria.”
Because researchers have previously not found examples of eukaryotes without some form of mitochondria, the current theory of the origin of eukaryotes poses that the appearance of mitochondria was crucial to the identity of these organisms.
“We now view these mitochondria-like organelles as a continuum from full mitochondria to very small . Some anaerobic protists, for example, have only pared down versions of mitochondria, such as hydrogenosomes and mitosomes, which lack a mitochondrial genome. But these mitochondrion-like organelles perform essential functions of the iron-sulfur cluster assembly pathway, which is known to be conserved in virtually all eukaryotic organisms studied to date.
Yet, in their analysis, the researchers found no evidence of the presence of any components of this mitochondrial pathway.
Like the scaling down of mitochondria into mitosomes in some organisms, the ancestors of modernMonocercomonoides once had mitochondria. “Because this organism is phylogenetically nested among relatives that had conventional mitochondria, this is most likely a secondary adaptation,” said Michael Gray, a biochemist who studies mitochondria at Dalhousie University in Nova Scotia and was not involved in the study. According to Gray, the finding of a mitochondria-deficient eukaryote does not mean that the organelles did not play a major role in the evolution of eukaryotic cells.
To be sure they were not missing mitochondrial proteins, Hampl’s team also searched for potential mitochondrial protein homologs of other anaerobic species, and for signature sequences of a range of known mitochondrial proteins. While similar searches with other species uncovered a few mitochondrial proteins, the team’s analysis of Monocercomonoides came up empty.
“The data is very complete,” said Ettema. “It is difficult to prove the absence of something but [these authors] do a convincing job.”
To form the essential iron-sulfur clusters, the team discovered that Monocercomonoides use a sulfur mobilization system found in the cytosol, and that an ancestor of the organism acquired this system by lateral gene transfer from bacteria. This cytosolic, compensating system allowed Monocercomonoides to lose the otherwise essential iron-sulfur cluster-forming pathway in the mitochondrion, the team proposed.
“This work shows the great evolutionary plasticity of the eukaryotic cell,” said Karnkowska, who participated in the study while she was a postdoc at Charles University. Karnkowska, who is now a visiting researcher at the University of British Columbia in Canada, added: “This is a striking example of how far the evolution of a eukaryotic cell can go that was beyond our expectations.”
“The results highlight how many surprises may await us in the poorly studied eukaryotic phyla that live in under-explored environments,” Gabaldon said.
Ettema agreed. “Now that we’ve found one, we need to look at the bigger picture and see if there are other examples of eukaryotes that have lost their mitochondria, to understand how adaptable eukaryotes are.”
Karnkowska et al., “A eukaryote without a mitochondrial organelle,” Current Biology,doi:10.1016/j.cub.2016.03.053, 2016.
•Monocercomonoides sp. is a eukaryotic microorganism with no mitochondria
•The complete absence of mitochondria is a secondary loss, not an ancestral feature
•The essential mitochondrial ISC pathway was replaced by a bacterial SUF system
The presence of mitochondria and related organelles in every studied eukaryote supports the view that mitochondria are essential cellular components. Here, we report the genome sequence of a microbial eukaryote, the oxymonad Monocercomonoides sp., which revealed that this organism lacks all hallmark mitochondrial proteins. Crucially, the mitochondrial iron-sulfur cluster assembly pathway, thought to be conserved in virtually all eukaryotic cells, has been replaced by a cytosolic sulfur mobilization system (SUF) acquired by lateral gene transfer from bacteria. In the context of eukaryotic phylogeny, our data suggest that Monocercomonoides is not primitively amitochondrial but has lost the mitochondrion secondarily. This is the first example of a eukaryote lacking any form of a mitochondrion, demonstrating that this organelle is not absolutely essential for the viability of a eukaryotic cell.
This method catches a bait protein together with its associated protein partners in virus-like particles that are budded from human cells. Like this, cell lysis is not needed and protein complexes are preserved during purification.
With his feet in both a proteomics lab and an interactomics lab, VIB/UGent professor Sven Eyckerman is well aware of the shortcomings of conventional approaches to analyze protein complexes. The lysis conditions required in mass spectrometry–based strategies to break open cell membranes often affect protein-protein interactions. “The first step in a classical study on protein complexes essentially turns the highly organized cellular structure into a big messy soup”, Eyckerman explains.
Inspired by virus biology, Eyckerman came up with a creative solution. “We used the natural process of HIV particle formation to our benefit by hacking a completely safe form of the virus to abduct intact protein machines from the cell.” It is well known that the HIV virus captures a number of host proteins during its particle formation. By fusing a bait protein to the HIV-1 GAG protein, interaction partners become trapped within virus-like particles that bud from mammalian cells. Standard proteomic approaches are used next to reveal the content of these particles. Fittingly, the team named the method ‘Virotrap’.
The Virotrap approach is exceptional as protein networks can be characterized under natural conditions. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. The researchers showed the method was suitable for detection of known binary interactions as well as mass spectrometry-based identification of novel protein partners.
Virotrap is a textbook example of bringing research teams with complementary expertise together. Cross-pollination with the labs of Jan Tavernier (VIB/UGent) and Kris Gevaert (VIB/UGent) enabled the development of this platform.
Jan Tavernier: “Virotrap represents a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. It is complementary to the arsenal of existing interactomics methods, but also holds potential for other fields, like drug target characterization. We also developed a small molecule-variant of Virotrap that could successfully trap protein partners for small molecule baits.”
Kris Gevaert: “Virotrap can also impact our understanding of disease pathways. We were actually surprised to see that this virus-based system could be used to study antiviral pathways, like Toll-like receptor signaling. Understanding these protein machines in their natural environment is essential if we want to modulate their activity in pathology.“
Trapping mammalian protein complexes in viral particles
Cell lysis is an inevitable step in classical mass spectrometry–based strategies to analyse protein complexes. Complementary lysis conditions, in situ cross-linking strategies and proximal labelling techniques are currently used to reduce lysis effects on the protein complex. We have developed Virotrap, a viral particle sorting approach that obviates the need for cell homogenization and preserves the protein complexes during purification. By fusing a bait protein to the HIV-1 GAG protein, we show that interaction partners become trapped within virus-like particles (VLPs) that bud from mammalian cells. Using an efficient VLP enrichment protocol, Virotrap allows the detection of known binary interactions and MS-based identification of novel protein partners as well. In addition, we show the identification of stimulus-dependent interactions and demonstrate trapping of protein partners for small molecules. Virotrap constitutes an elegant complementary approach to the arsenal of methods to study protein complexes.
Proteins mostly exert their function within supramolecular complexes. Strategies for detecting protein–protein interactions (PPIs) can be roughly divided into genetic systems1 and co-purification strategies combined with mass spectrometry (MS) analysis (for example, AP–MS)2. The latter approaches typically require cell or tissue homogenization using detergents, followed by capture of the protein complex using affinity tags3 or specific antibodies4. The protein complexes extracted from this ‘soup’ of constituents are then subjected to several washing steps before actual analysis by trypsin digestion and liquid chromatography–MS/MS analysis. Such lysis and purification protocols are typically empirical and have mostly been optimized using model interactions in single labs. In fact, lysis conditions can profoundly affect the number of both specific and nonspecific proteins that are identified in a typical AP–MS set-up. Indeed, recent studies using the nuclear pore complex as a model protein complex describe optimization of purifications for the different proteins in the complex by examining 96 different conditions5. Nevertheless, for new purifications, it remains hard to correctly estimate the loss of factors in a standard AP–MS experiment due to washing and dilution effects during treatments (that is, false negatives). These considerations have pushed the concept of stabilizing PPIs before the actual homogenization step. A classical approach involves cross-linking with simple reagents (for example, formaldehyde) or with more advanced isotope-labelled cross-linkers (reviewed in ref. 2). However, experimental challenges such as cell permeability and reactivity still preclude the widespread use of cross-linking agents. Moreover, MS-generated spectra of cross-linked peptides are notoriously difficult to identify correctly. A recent lysis-independent solution involves the expression of a bait protein fused to a promiscuous biotin ligase, which results in labelling of proteins proximal to the activity of the enzyme-tagged bait protein6. When compared with AP–MS, this BioID approach delivers a complementary set of candidate proteins, including novel interaction partners7, 8. Such particular studies clearly underscore the need for complementary approaches in the co-complex strategies.
The evolutionary stress on viruses promoted highly condensed coding of information and maximal functionality for small genomes. Accordingly, for HIV-1 it is sufficient to express a single protein, the p55 GAG protein, for efficient production of virus-like particles (VLPs) from cells9, 10. This protein is highly mobile before its accumulation in cholesterol-rich regions of the membrane, where multimerization initiates the budding process11. A total of 4,000–5,000 GAG molecules is required to form a single particle of about 145 nm (ref. 12). Both VLPs and mature viruses contain a number of host proteins that are recruited by binding to viral proteins. These proteins can either contribute to the infectivity (for example, Cyclophilin/FKBPA13) or act as antiviral proteins preventing the spreading of the virus (for example, APOBEC proteins14).
We here describe the development and application of Virotrap, an elegant co-purification strategy based on the trapping of a bait protein together with its associated protein partners in VLPs that are budded from the cell. After enrichment, these particles can be analysed by targeted (for example, western blotting) or unbiased approaches (MS-based proteomics). Virotrap allows detection of known binary PPIs, analysis of protein complexes and their dynamics, and readily detects protein binders for small molecules.
Concept of the Virotrap system
Classical AP–MS approaches rely on cell homogenization to access protein complexes, a step that can vary significantly with the lysis conditions (detergents, salt concentrations, pH conditions and so on)5. To eliminate the homogenization step in AP–MS, we reasoned that incorporation of a protein complex inside a secreted VLP traps the interaction partners under native conditions and protects them during further purification. We thus explored the possibility of protein complex packaging by the expression of GAG-bait protein chimeras (Fig. 1) as expression of GAG results in the release of VLPs from the cells9, 10. As a first PPI pair to evaluate this concept, we selected the HRAS protein as a bait combined with the RAF1 prey protein. We were able to specifically detect the HRAS–RAF1 interaction following enrichment of VLPs via ultracentrifugation (Supplementary Fig. 1a). To prevent tedious ultracentrifugation steps, we designed a novel single-step protocol wherein we co-express the vesicular stomatitis virus glycoprotein (VSV-G) together with a tagged version of this glycoprotein in addition to the GAG bait and prey. Both tagged and untagged VSV-G proteins are probably presented as trimers on the surface of the VLPs, allowing efficient antibody-based recovery from large volumes. The HRAS–RAF1 interaction was confirmed using this single-step protocol (Supplementary Fig. 1b). No associations with unrelated bait or prey proteins were observed for both protocols.
Figure 1: Schematic representation of the Virotrap strategy.
Expression of a GAG-bait fusion protein (1) results in submembrane multimerization (2) and subsequent budding of VLPs from cells (3). Interaction partners of the bait protein are also trapped within these VLPs and can be identified after purification by western blotting or MS analysis (4).
Virotrap for the detection of binary interactions
We next explored the reciprocal detection of a set of PPI pairs, which were selected based on published evidence and cytosolic localization15. After single-step purification and western blot analysis, we could readily detect reciprocal interactions between CDK2 and CKS1B, LCP2 and GRAP2, and S100A1 and S100B (Fig. 2a). Only for the LCP2 prey we observed nonspecific association with an irrelevant bait construct. However, the particle levels of the GRAP2 bait were substantially lower as compared with those of the GAG control construct (GAG protein levels in VLPs; Fig. 2a, second panel of the LCP2 prey). After quantification of the intensities of bait and prey proteins and normalization of prey levels using bait levels, we observed a strong enrichment for the GAG-GRAP2 bait (Supplementary Fig. 2).
…..
Virotrap for unbiased discovery of novel interactions
For the detection of novel interaction partners, we scaled up VLP production and purification protocols (Supplementary Fig. 5 and Supplementary Note 1 for an overview of the protocol) and investigated protein partners trapped using the following bait proteins: Fas-associated via death domain (FADD), A20 (TNFAIP3), nuclear factor-κB (NF-κB) essential modifier (IKBKG), TRAF family member-associated NF-κB activator (TANK), MYD88 and ring finger protein 41 (RNF41). To obtain specific interactors from the lists of identified proteins, we challenged the data with a combined protein list of 19 unrelated Virotrap experiments (Supplementary Table 1 for an overview). Figure 3 shows the design and the list of candidate interactors obtained after removal of all proteins that were found in the 19 control samples (including removal of proteins from the control list identified with a single peptide). The remaining list of confident protein identifications (identified with at least two peptides in at least two biological repeats) reveals both known and novel candidate interaction partners. All candidate interactors including single peptide protein identifications are given in Supplementary Data 2 and also include recurrent protein identifications of known interactors based on a single peptide; for example, CASP8 for FADD and TANK for NEMO. Using alternative methods, we confirmed the interaction between A20 and FADD, and the associations with transmembrane proteins (insulin receptor and insulin-like growth factor receptor 1) that were captured using RNF41 as a bait (Supplementary Fig. 6). To address the use of Virotrap for the detection of dynamic interactions, we activated the NF-κB pathway via the tumour necrosis factor (TNF) receptor (TNFRSF1A) using TNFα (TNF) and performed Virotrap analysis using A20 as bait (Fig. 3). This resulted in the additional enrichment of receptor-interacting kinase (RIPK1), TNFR1-associated via death domain (TRADD), TNFRSF1A and TNF itself, confirming the expected activated complex20.
Figure 3: Use of Virotrap for unbiased interactome analysis
Lysis conditions used in AP–MS strategies are critical for the preservation of protein complexes. A multitude of lysis conditions have been described, culminating in a recent report where protein complex stability was assessed under 96 lysis/purification protocols5. Moreover, the authors suggest to optimize the conditions for every complex, implying an important workload for researchers embarking on protein complex analysis using classical AP–MS. As lysis results in a profound change of the subcellular context and significantly alters the concentration of proteins, loss of complex integrity during a classical AP–MS protocol can be expected. A clear evolution towards ‘lysis-independent’ approaches in the co-complex analysis field is evident with the introduction of BioID6 and APEX25 where proximal proteins, including proteins residing in the complex, are labelled with biotin by an enzymatic activity fused to a bait protein. A side-by-side comparison between classical AP–MS and BioID showed overlapping and unique candidate binding proteins for both approaches7, 8, supporting the notion that complementary methods are needed to provide a comprehensive view on protein complexes. This has also been clearly demonstrated for binary approaches15 and is a logical consequence of the heterogenic nature underlying PPIs (binding mechanism, requirement for posttranslational modifications, location, affinity and so on).
In this report, we explore an alternative, yet complementary method to isolate protein complexes without interfering with cellular integrity. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. This constitutes a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. A comparison of our Virotrap approach with AP–MS shows complementary data, with specific false positives and false negatives for both methods (Supplementary Fig. 7).
The current implementation of the Virotrap platform implies the use of a GAG-bait construct resulting in considerable expression of the bait protein. Different strategies are currently pursued to reduce bait expression including co-expression of a native GAG protein together with the GAG-bait protein, not only reducing bait expression but also creating more ‘space’ in the particles potentially accommodating larger bait protein complexes. Nevertheless, the presence of the bait on the forming GAG scaffold creates an intracellular affinity matrix (comparable to the early in vitro affinity columns for purification of interaction partners from lysates26) that has the potential to compete with endogenous complexes by avidity effects. This avidity effect is a powerful mechanism that aids in the recruitment of cyclophilin to GAG27, a well-known weak interaction (Kd=16 μM (ref. 28)) detectable as a background association in the Virotrap system. Although background binding may be increased by elevated bait expression, weaker associations are readily detectable (for example, MAL—MYD88-binding study; Fig. 2c).
The size of Virotrap particles (around 145 nm) suggests limitations in the size of the protein complex that can be accommodated in the particles. Further experimentation is required to define the maximum size of proteins or the number of protein complexes that can be trapped inside the particles.
….
In conclusion, Virotrap captures significant parts of known interactomes and reveals new interactions. This cell lysis-free approach purifies protein complexes under native conditions and thus provides a powerful method to complement AP–MS or other PPI data. Future improvements of the system include strategies to reduce bait expression to more physiological levels and application of advanced data analysis options to filter out background. These developments can further aid in the deployment of Virotrap as a powerful extension of the current co-complex technology arsenal.
New Autism Blood Biomarker Identified
Researchers at UT Southwestern Medical Center have identified a blood biomarker that may aid in earlier diagnosis of children with autism spectrum disorder, or ASD
In a recent edition of Scientific Reports, UT Southwestern researchers reported on the identification of a blood biomarker that could distinguish the majority of ASD study participants versus a control group of similar age range. In addition, the biomarker was significantly correlated with the level of communication impairment, suggesting that the blood test may give insight into ASD severity.
“Numerous investigators have long sought a biomarker for ASD,” said Dr. Dwight German, study senior author and Professor of Psychiatry at UT Southwestern. “The blood biomarker reported here along with others we are testing can represent a useful test with over 80 percent accuracy in identifying ASD.”
ASD1 – was 66 percent accurate in diagnosing ASD. When combined with thyroid stimulating hormone level measurements, the ASD1-binding biomarker was 73 percent accurate at diagnosis
A Search for Blood Biomarkers for Autism: Peptoids
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impairments in social interaction and communication, and restricted, repetitive patterns of behavior. In order to identify individuals with ASD and initiate interventions at the earliest possible age, biomarkers for the disorder are desirable. Research findings have identified widespread changes in the immune system in children with autism, at both systemic and cellular levels. In an attempt to find candidate antibody biomarkers for ASD, highly complex libraries of peptoids (oligo-N-substituted glycines) were screened for compounds that preferentially bind IgG from boys with ASD over typically developing (TD) boys. Unexpectedly, many peptoids were identified that preferentially bound IgG from TD boys. One of these peptoids was studied further and found to bind significantly higher levels (>2-fold) of the IgG1 subtype in serum from TD boys (n = 60) compared to ASD boys (n = 74), as well as compared to older adult males (n = 53). Together these data suggest that ASD boys have reduced levels (>50%) of an IgG1 antibody, which resembles the level found normally with advanced age. In this discovery study, the ASD1 peptoid was 66% accurate in predicting ASD.
….
Peptoid libraries have been used previously to search for autoantibodies for neurodegenerative diseases19 and for systemic lupus erythematosus (SLE)21. In the case of SLE, peptoids were identified that could identify subjects with the disease and related syndromes with moderate sensitivity (70%) and excellent specificity (97.5%). Peptoids were used to measure IgG levels from both healthy subjects and SLE patients. Binding to the SLE-peptoid was significantly higher in SLE patients vs. healthy controls. The IgG bound to the SLE-peptoid was found to react with several autoantigens, suggesting that the peptoids are capable of interacting with multiple, structurally similar molecules. These data indicate that IgG binding to peptoids can identify subjects with high levels of pathogenic autoantibodies vs. a single antibody.
In the present study, the ASD1 peptoid binds significantly lower levels of IgG1 in ASD males vs. TD males. This finding suggests that the ASD1 peptoid recognizes antibody(-ies) of an IgG1 subtype that is (are) significantly lower in abundance in the ASD males vs. TD males. Although a previous study14 has demonstrated lower levels of plasma IgG in ASD vs. TD children, here, we additionally quantified serum IgG levels in our individuals and found no difference in IgG between the two groups (data not shown). Furthermore, our IgG levels did not correlate with ASD1 binding levels, indicating that ASD1 does not bind IgG generically, and that the peptoid’s ability to differentiate between ASD and TD males is related to a specific antibody(-ies).
ASD subjects underwent a diagnostic evaluation using the ADOS and ADI-R, and application of the DSM-IV criteria prior to study inclusion. Only those subjects with a diagnosis of Autistic Disorder were included in the study. The ADOS is a semi-structured observation of a child’s behavior that allows examiners to observe the three core domains of ASD symptoms: reciprocal social interaction, communication, and restricted and repetitive behaviors1. When ADOS subdomain scores were compared with peptoid binding, the only significant relationship was with Social Interaction. However, the positive correlation would suggest that lower peptoid binding is associated with better social interaction, not poorer social interaction as anticipated.
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
It is interesting that in serum samples from older men, the ASD1 binding is similar to that in the ASD boys. This is consistent with the observation that with aging there is a reduction in the strength of the immune system, and the changes are gender-specific25. Recent studies using parabiosis26, in which blood from young mice reverse age-related impairments in cognitive function and synaptic plasticity in old mice, reveal that blood constituents from young subjects may contain important substances for maintaining neuronal functions. Work is in progress to identify the antibody/antibodies that are differentially binding to the ASD1 peptoid, which appear as a single band on the electrophoresis gel (Fig. 4).
……..
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
Titration of IgG binding to ASD1 using serum pooled from 10 TD males and 10 ASD males demonstrates ASD1’s ability to differentiate between the two groups. (B)Detecting IgG1 subclass instead of total IgG amplifies this differentiation. (C) IgG1 binding of individual ASD (n=74) and TD (n=60) male serum samples (1:100 dilution) to ASD1 significantly differs with TD>ASD. In addition, IgG1 binding of older adult male (AM) serum samples (n=53) to ASD1 is significantly lower than TD males, and not different from ASD males. The three groups were compared with a Kruskal-Wallis ANOVA, H = 10.1781, p<0.006. **p<0.005. Error bars show SEM. (D) Receiver-operating characteristic curve for ASD1’s ability to discriminate between ASD and TD males.
Association between peptoid binding and ADOS and ADI-R subdomains
Higher scores in any domain on the ADOS and ADI-R are indicative of more abnormal behaviors and/or symptoms. Among ADOS subdomains, there was no significant relationship between Communication and peptoid binding (z = 0.04, p = 0.966), Communication + Social interaction (z = 1.53, p = 0.127), or Stereotyped Behaviors and Restrictive Interests (SBRI) (z = 0.46, p = 0.647). Higher scores on the Social Interaction domain were significantly associated with higher peptoid binding (z = 2.04, p = 0.041).
Among ADI-R subdomains, higher scores on the Communication domain were associated with lower levels of peptoid binding (z = −2.28, p = 0.023). There was not a significant relationship between Social Interaction (z = 0.07, p = 0.941) or Restrictive/Repetitive Stereotyped Behaviors (z = −1.40, p = 0.162) and peptoid binding.
Computational Model Finds New Protein-Protein Interactions
Researchers at University of Pittsburgh have discovered 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia.
Using a computational model they developed, researchers at the University of Pittsburgh School of Medicine have discovered more than 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia. The findings, published online in npj Schizophrenia, a Nature Publishing Group journal, could lead to greater understanding of the biological underpinnings of this mental illness, as well as point the way to treatments.
There have been many genome-wide association studies (GWAS) that have identified gene variants associated with an increased risk for schizophrenia, but in most cases there is little known about the proteins that these genes make, what they do and how they interact, said senior investigator Madhavi Ganapathiraju, Ph.D., assistant professor of biomedical informatics, Pitt School of Medicine.
“GWAS studies and other research efforts have shown us what genes might be relevant in schizophrenia,” she said. “What we have done is the next step. We are trying to understand how these genes relate to each other, which could show us the biological pathways that are important in the disease.”
Each gene makes proteins and proteins typically interact with each other in a biological process. Information about interacting partners can shed light on the role of a gene that has not been studied, revealing pathways and biological processes associated with the disease and also its relation to other complex diseases.
Dr. Ganapathiraju’s team developed a computational model called High-Precision Protein Interaction Prediction (HiPPIP) and applied it to discover PPIs of schizophrenia-linked genes identified through GWAS, as well as historically known risk genes. They found 504 never-before known PPIs, and noted also that while schizophrenia-linked genes identified historically and through GWAS had little overlap, the model showed they shared more than 100 common interactors.
“We can infer what the protein might do by checking out the company it keeps,” Dr. Ganapathiraju explained. “For example, if I know you have many friends who play hockey, it could mean that you are involved in hockey, too. Similarly, if we see that an unknown protein interacts with multiple proteins involved in neural signaling, for example, there is a high likelihood that the unknown entity also is involved in the same.”
Dr. Ganapathiraju and colleagues have drawn such inferences on protein function based on the PPIs of proteins, and made their findings available on a website Schizo-Pi. This information can be used by biologists to explore the schizophrenia interactome with the aim of understanding more about the disease or developing new treatment drugs.
Schizophrenia interactome with 504 novel protein–protein interactions
(GWAS) have revealed the role of rare and common genetic variants, but the functional effects of the risk variants remain to be understood. Protein interactome-based studies can facilitate the study of molecular mechanisms by which the risk genes relate to schizophrenia (SZ) genesis, but protein–protein interactions (PPIs) are unknown for many of the liability genes. We developed a computational model to discover PPIs, which is found to be highly accurate according to computational evaluations and experimental validations of selected PPIs. We present here, 365 novel PPIs of liability genes identified by the SZ Working Group of the Psychiatric Genomics Consortium (PGC). Seventeen genes that had no previously known interactions have 57 novel interactions by our method. Among the new interactors are 19 drug targets that are targeted by 130 drugs. In addition, we computed 147 novel PPIs of 25 candidate genes investigated in the pre-GWAS era. While there is little overlap between the GWAS genes and the pre-GWAS genes, the interactomes reveal that they largely belong to the same pathways, thus reconciling the apparent disparities between the GWAS and prior gene association studies. The interactome including 504 novel PPIs overall, could motivate other systems biology studies and trials with repurposed drugs. The PPIs are made available on a webserver, called Schizo-Pi at http://severus.dbmi.pitt.edu/schizo-pi with advanced search capabilities.
Schizophrenia (SZ) is a common, potentially severe psychiatric disorder that afflicts all populations.1 Gene mapping studies suggest that SZ is a complex disorder, with a cumulative impact of variable genetic effects coupled with environmental factors.2 As many as 38 genome-wide association studies (GWAS) have been reported on SZ out of a total of 1,750 GWAS publications on 1,087 traits or diseases reported in the GWAS catalog maintained by the National Human Genome Research Institute of USA3 (as of April 2015), revealing the common variants associated with SZ.4 The SZ Working Group of the Psychiatric Genomics Consortium (PGC) identified 108 genetic loci that likely confer risk for SZ.5 While the role of genetics has been clearly validated by this study, the functional impact of the risk variants is not well-understood.6,7 Several of the genes implicated by the GWAS have unknown functions and could participate in possibly hitherto unknown pathways.8 Further, there is little or no overlap between the genes identified through GWAS and ‘candidate genes’ proposed in the pre-GWAS era.9
Interactome-based studies can be useful in discovering the functional associations of genes. For example,disrupted in schizophrenia 1 (DISC1), an SZ related candidate gene originally had no known homolog in humans. Although it had well-characterized protein domains such as coiled-coil domains and leucine-zipper domains, its function was unknown.10,11 Once its protein–protein interactions (PPIs) were determined using yeast 2-hybrid technology,12 investigators successfully linked DISC1 to cAMP signaling, axon elongation, and neuronal migration, and accelerated the research pertaining to SZ in general, and DISC1 in particular.13 Typically such studies are carried out on known protein–protein interaction (PPI) networks, or as in the case of DISC1, when there is a specific gene of interest, its PPIs are determined by methods such as yeast 2-hybrid technology.
Knowledge of human PPI networks is thus valuable for accelerating discovery of protein function, and indeed, biomedical research in general. However, of the hundreds of thousands of biophysical PPIs thought to exist in the human interactome,14,15 <100,000 are known today (Human Protein Reference Database, HPRD16 and BioGRID17 databases). Gold standard experimental methods for the determination of all the PPIs in human interactome are time-consuming, expensive and may not even be feasible, as about 250 million pairs of proteins would need to be tested overall; high-throughput methods such as yeast 2-hybrid have important limitations for whole interactome determination as they have a low recall of 23% (i.e., remaining 77% of true interactions need to be determined by other means), and a low precision (i.e., the screens have to be repeated multiple times to achieve high selectivity).18,19Computational methods are therefore necessary to complete the interactome expeditiously. Algorithms have begun emerging to predict PPIs using statistical machine learning on the characteristics of the proteins, but these algorithms are employed predominantly to study yeast. Two significant computational predictions have been reported for human interactome; although they have had high false positive rates, these methods have laid the foundation for computational prediction of human PPIs.20,21
We have created a new PPI prediction model called High-Confidence Protein–Protein Interaction Prediction (HiPPIP) model. Novel interactions predicted with this model are making translational impact. For example, we discovered a PPI between OASL and DDX58, which on validation showed that an increased expression of OASL could boost innate immunity to combat influenza by activating the RIG-I pathway.22 Also, the interactome of the genes associated with congenital heart disease showed that the disease morphogenesis has a close connection with the structure and function of cilia.23Here, we describe the HiPPIP model and its application to SZ genes to construct the SZ interactome. After computational evaluations and experimental validations of selected novel PPIs, we present here 504 highly confident novel PPIs in the SZ interactome, shedding new light onto several uncharacterized genes that are associated with SZ.
We developed a computational model called HiPPIP to predict PPIs (see Methods and Supplementary File 1). The model has been evaluated by computational methods and experimental validations and is found to be highly accurate. Evaluations on a held-out test data showed a precision of 97.5% and a recall of 5%. 5% recall out of 150,000 to 600,000 estimated number of interactions in the human interactome corresponds to 7,500–30,000 novel PPIs in the whole interactome. Note that, it is likely that the real precision would be higher than 97.5% because in this test data, randomly paired proteins are treated as non-interacting protein pairs, whereas some of them may actually be interacting pairs with a small probability; thus, some of the pairs that are treated as false positives in test set are likely to be true but hitherto unknown interactions. In Figure 1a, we show the precision versus recall of our method on ‘hub proteins’ where we considered all pairs that received a score >0.5 by HiPPIP to be novel interactions. In Figure 1b, we show the number of true positives versus false positives observed in hub proteins. Both these figures also show our method to be superior in comparison to the prediction of membrane-receptor interactome by Qi et al’s.24 True positives versus false positives are also shown for individual hub proteins by our method in Figure 1cand by Qi et al’s.23 in Figure 1d. These evaluations showed that our predictions contain mostly true positives. Unlike in other domains where ranked lists are commonly used such as information retrieval, in PPI prediction the ‘false positives’ may actually be unlabeled instances that are indeed true interactions that are not yet discovered. In fact, such unlabeled pairs predicted as interactors of the hub gene HMGB1 (namely, the pairs HMGB1-KL and HMGB1-FLT1) were validated by experimental methods and found to be true PPIs (See the Figures e–g inSupplementary File 3). Thus, we concluded that the protein pairs that received a score of ⩾0.5 are highly confident to be true interactions. The pairs that receive a score less than but close to 0.5 (i.e., in the range of 0.4–0.5) may also contain several true PPIs; however, we cannot confidently say that all in this range are true PPIs. Only the PPIs predicted with a score >0.5 are included in the interactome.
Computational evaluation of predicted protein–protein interactions on hub proteins: (a) precision recall curve. (b) True positive versus false positives in ranked lists of hub type membrane receptors for our method and that by Qi et al. True positives versus false positives are shown for individual membrane receptors by our method in (c) and by Qi et al. in (d). Thick line is the average, which is also the same as shown in (b). Note:x-axis is recall in (a), whereas it is number of false positives in (b–d). The range of y-axis is observed by varying the threshold from 1.0–0 in (a), and to 0.5 in (b–d).
SZ interactome
By applying HiPPIP to the GWAS genes and Historic (pre-GWAS) genes, we predicted over 500 high confidence new PPIs adding to about 1400 previously known PPIs.
Schizophrenia interactome: network view of the schizophrenia interactome is shown as a graph, where genes are shown as nodes and PPIs as edges connecting the nodes. Schizophrenia-associated genes are shown as dark blue nodes, novel interactors as red color nodes and known interactors as blue color nodes. The source of the schizophrenia genes is indicated by its label font, where Historic genes are shown italicized, GWAS genes are shown in bold, and the one gene that is common to both is shown in italicized and bold. For clarity, the source is also indicated by the shape of the node (triangular for GWAS and square for Historic and hexagonal for both). Symbols are shown only for the schizophrenia-associated genes; actual interactions may be accessed on the web. Red edges are the novel interactions, whereas blue edges are known interactions. GWAS, genome-wide association studies of schizophrenia; PPI, protein–protein interaction.
We have made the known and novel interactions of all SZ-associated genes available on a webserver called Schizo-Pi, at the addresshttp://severus.dbmi.pitt.edu/schizo-pi. This webserver is similar to Wiki-Pi33 which presents comprehensive annotations of both participating proteins of a PPI side-by-side. The difference between Wiki-Pi which we developed earlier, and Schizo-Pi, is the inclusion of novel predicted interactions of the SZ genes into the latter.
Despite the many advances in biomedical research, identifying the molecular mechanisms underlying the disease is still challenging. Studies based on protein interactions were proven to be valuable in identifying novel gene associations that could shed new light on disease pathology.35 The interactome including more than 500 novel PPIs will help to identify pathways and biological processes associated with the disease and also its relation to other complex diseases. It also helps identify potential drugs that could be repurposed to use for SZ treatment.
Functional and pathway enrichment in SZ interactome
When a gene of interest has little known information, functions of its interacting partners serve as a starting point to hypothesize its own function. We computed statistically significant enrichment of GO biological process terms among the interacting partners of each of the genes using BinGO36 (see online at http://severus.dbmi.pitt.edu/schizo-pi).
Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution
Massimo Stefani · Christopher M. Dobson
Abstract The deposition of proteins in the form of amyloid fibrils and plaques is the characteristic feature of more than 20 degenerative conditions affecting either the central nervous system or a variety of peripheral tissues. As these conditions include Alzheimer’s, Parkinson’s and the prion diseases, several forms of fatal systemic amyloidosis, and at least one condition associated with medical intervention (haemodialysis), they are of enormous importance in the context of present-day human health and welfare. Much remains to be learned about the mechanism by which the proteins associated with these diseases aggregate and form amyloid structures, and how the latter affect the functions of the organs with which they are associated. A great deal of information concerning these diseases has emerged, however, during the past 5 years, much of it causing a number of fundamental assumptions about the amyloid diseases to be reexamined. For example, it is now apparent that the ability to form amyloid structures is not an unusual feature of the small number of proteins associated with these diseases but is instead a general property of polypeptide chains. It has also been found recently that aggregates of proteins not associated with amyloid diseases can impair the ability of cells to function to a similar extent as aggregates of proteins linked with specific neurodegenerative conditions. Moreover, the mature amyloid fibrils or plaques appear to be substantially less toxic than the prefibrillar aggregates that are their precursors. The toxicity of these early aggregates appears to result from an intrinsic ability to impair fundamental cellular processes by interacting with cellular membranes, causing oxidative stress and increases in free Ca2+ that eventually lead to apoptotic or necrotic cell death. The ‘new view’ of these diseases also suggests that other degenerative conditions could have similar underlying origins to those of the amyloidoses. In addition, cellular protection mechanisms, such as molecular chaperones and the protein degradation machinery, appear to be crucial in the prevention of disease in normally functioning living organisms. It also suggests some intriguing new factors that could be of great significance in the evolution of biological molecules and the mechanisms that regulate their behaviour.
The genetic information within a cell encodes not only the specific structures and functions of proteins but also the way these structures are attained through the process known as protein folding. In recent years many of the underlying features of the fundamental mechanism of this complex process and the manner in which it is regulated in living systems have emerged from a combination of experimental and theoretical studies [1]. The knowledge gained from these studies has also raised a host of interesting issues. It has become apparent, for example, that the folding and unfolding of proteins is associated with a whole range of cellular processes from the trafficking of molecules to specific organelles to the regulation of the cell cycle and the immune response. Such observations led to the inevitable conclusion that the failure to fold correctly, or to remain correctly folded, gives rise to many different types of biological malfunctions and hence to many different forms of disease [2]. In addition, it has been recognised recently that a large number of eukaryotic genes code for proteins that appear to be ‘natively unfolded’, and that proteins can adopt, under certain circumstances, highly organised multi-molecular assemblies whose structures are not specifically encoded in the amino acid sequence. Both these observations have raised challenging questions about one of the most fundamental principles of biology: the close relationship between the sequence, structure and function of proteins, as we discuss below [3].
It is well established that proteins that are ‘misfolded’, i.e. that are not in their functionally relevant conformation, are devoid of normal biological activity. In addition, they often aggregate and/or interact inappropriately with other cellular components leading to impairment of cell viability and eventually to cell death. Many diseases, often known as misfolding or conformational diseases, ultimately result from the presence in a living system of protein molecules with structures that are ‘incorrect’, i.e. that differ from those in normally functioning organisms [4]. Such diseases include conditions in which a specific protein, or protein complex, fails to fold correctly (e.g. cystic fibrosis, Marfan syndrome, amyotonic lateral sclerosis) or is not sufficiently stable to perform its normal function (e.g. many forms of cancer). They also include conditions in which aberrant folding behaviour results in the failure of a protein to be correctly trafficked (e.g. familial hypercholesterolaemia, α1-antitrypsin deficiency, and some forms of retinitis pigmentosa) [4]. The tendency of proteins to aggregate, often to give species extremely intractable to dissolution and refolding, is of course also well known in other circumstances. Examples include the formation of inclusion bodies during overexpression of heterologous proteins in bacteria and the precipitation of proteins during laboratory purification procedures. Indeed, protein aggregation is well established as one of the major difficulties associated with the production and handling of proteins in the biotechnology and pharmaceutical industries [5].
Considerable attention is presently focused on a group of protein folding diseases known as amyloidoses. In these diseases specific peptides or proteins fail to fold or to remain correctly folded and then aggregate (often with other components) so as to give rise to ‘amyloid’ deposits in tissue. Amyloid structures can be recognised because they possess a series of specific tinctorial and biophysical characteristics that reflect a common core structure based on the presence of highly organised βsheets [6]. The deposits in strictly defined amyloidoses are extracellular and can often be observed as thread-like fibrillar structures, sometimes assembled further into larger aggregates or plaques. These diseases include a range of sporadic, familial or transmissible degenerative diseases, some of which affect the brain and the central nervous system (e.g. Alzheimer’s and Creutzfeldt-Jakob diseases), while others involve peripheral tissues and organs such as the liver, heart and spleen (e.g. systemic amyloidoses and type II diabetes) [7, 8]. In other forms of amyloidosis, such as primary or secondary systemic amyloidoses, proteinaceous deposits are found in skeletal tissue and joints (e.g. haemodialysis-related amyloidosis) as well as in several organs (e.g. heart and kidney). Yet other components such as collagen, glycosaminoglycans and proteins (e.g. serum amyloid protein) are often present in the deposits protecting them against degradation [9, 10, 11]. Similar deposits to those in the amyloidoses are, however, found intracellularly in other diseases; these can be localised either in the cytoplasm, in the form of specialised aggregates known as aggresomes or as Lewy or Russell bodies or in the nucleus (see below).
The presence in tissue of proteinaceous deposits is a hallmark of all these diseases, suggesting a causative link between aggregate formation and pathological symptoms (often known as the amyloid hypothesis) [7, 8, 12]. At the present time the link between amyloid formation and disease is widely accepted on the basis of a large number of biochemical and genetic studies. The specific nature of the pathogenic species, and the molecular basis of their ability to damage cells, are however, the subject of intense debate [13, 14, 15, 16, 17, 18, 19, 20]. In neurodegenerative disorders it is very likely that the impairment of cellular function follows directly from the interactions of the aggregated proteins with cellular components [21, 22]. In the systemic non-neurological diseases, however, it is widely believed that the accumulation in vital organs of large amounts of amyloid deposits can by itself cause at least some of the clinical symptoms [23]. It is quite possible, however, that there are other more specific effects of aggregates on biochemical processes even in these diseases. The presence of extracellular or intracellular aggregates of a specific polypeptide molecule is a characteristic of all the 20 or so recognised amyloid diseases. The polypeptides involved include full length proteins (e.g. lysozyme or immunoglobulin light chains), biological peptides (amylin, atrial natriuretic factor) and fragments of larger proteins produced as a result of specific processing (e.g. the Alzheimer βpeptide) or of more general degradation [e.g. poly(Q) stretches cleaved from proteins with poly(Q) extensions such as huntingtin, ataxins and the androgen receptor]. The peptides and proteins associated with known amyloid diseases are listed in Table 1. In some cases the proteins involved have wild type sequences, as in sporadic forms of the diseases, but in other cases these are variants resulting from genetic mutations associated with familial forms of the diseases. In some cases both sporadic and familial diseases are associated with a given protein; in this case the mutational variants are usually associated with early-onset forms of the disease. In the case of the neurodegenerative diseases associated with the prion protein some forms of the diseases are transmissible. The existence of familial forms of a number of amyloid diseases has provided significant clues to the origins of the pathologies. For example, there are increasingly strong links between the age at onset of familial forms of disease and the effects of the mutations involved on the propensity of the affected proteins to aggregate in vitro. Such findings also support the link between the process of aggregation and the clinical manifestations of disease [24, 25].
The presence in cells of misfolded or aggregated proteins triggers a complex biological response. In the cytosol, this is referred to as the ‘heat shock response’ and in the endoplasmic reticulum (ER) it is known as the ‘unfolded protein response’. These responses lead to the expression, among others, of the genes for heat shock proteins (Hsp, or molecular chaperone proteins) and proteins involved in the ubiquitin-proteasome pathway [26]. The evolution of such complex biochemical machinery testifies to the fact that it is necessary for cells to isolate and clear rapidly and efficiently any unfolded or incorrectly folded protein as soon as it appears. In itself this fact suggests that these species could have a generally adverse effect on cellular components and cell viability. Indeed, it was a major step forward in understanding many aspects of cell biology when it was recognised that proteins previously associated only with stress, such as heat shock, are in fact crucial in the normal functioning of living systems. This advance, for example, led to the discovery of the role of molecular chaperones in protein folding and in the normal ‘housekeeping’ processes that are inherent in healthy cells [27, 28]. More recently a number of degenerative diseases, both neurological and systemic, have been linked to, or shown to be affected by, impairment of the ubiquitin-proteasome pathway (Table 2). The diseases are primarily associated with a reduction in either the expression or the biological activity of Hsps, ubiquitin, ubiquitinating or deubiquitinating enzymes and the proteasome itself, as we show below [29, 30, 31, 32], or even to the failure of the quality control mechanisms that ensure proper maturation of proteins in the ER. The latter normally leads to degradation of a significant proportion of polypeptide chains before they have attained their native conformations through retrograde translocation to the cytosol [33, 34].
….
It is now well established that the molecular basis of protein aggregation into amyloid structures involves the existence of ‘misfolded’ forms of proteins, i.e. proteins that are not in the structures in which they normally function in vivo or of fragments of proteins resulting from degradation processes that are inherently unable to fold [4, 7, 8, 36]. Aggregation is one of the common consequences of a polypeptide chain failing to reach or maintain its functional three-dimensional structure. Such events can be associated with specific mutations, misprocessing phenomena, aberrant interactions with metal ions, changes in environmental conditions, such as pH or temperature, or chemical modification (oxidation, proteolysis). Perturbations in the conformational properties of the polypeptide chain resulting from such phenomena may affect equilibrium 1 in Fig. 1 increasing the population of partially unfolded, or misfolded, species that are much more aggregation-prone than the native state.
Fig. 1 Overview of the possible fates of a newly synthesised polypeptide chain. The equilibrium ① between the partially folded molecules and the natively folded ones is usually strongly in favour of the latter except as a result of specific mutations, chemical modifications or partially destabilising solution conditions. The increased equilibrium populations of molecules in the partially or completely unfolded ensemble of structures are usually degraded by the proteasome; when this clearance mechanism is impaired, such species often form disordered aggregates or shift equilibrium ② towards the nucleation of pre-fibrillar assemblies that eventually grow into mature fibrils (equilibrium ③). DANGER! indicates that pre-fibrillar aggregates in most cases display much higher toxicity than mature fibrils. Heat shock proteins (Hsp) can suppress the appearance of pre-fibrillar assemblies by minimising the population of the partially folded molecules by assisting in the correct folding of the nascent chain and the unfolded protein response target incorrectly folded proteins for degradation.
……
Little is known at present about the detailed arrangement of the polypeptide chains themselves within amyloid fibrils, either those parts involved in the core βstrands or in regions that connect the various β-strands. Recent data suggest that the sheets are relatively untwisted and may in some cases at least exist in quite specific supersecondary structure motifs such as β-helices [6, 40] or the recently proposed µ-helix [41]. It seems possible that there may be significant differences in the way the strands are assembled depending on characteristics of the polypeptide chain involved [6, 42]. Factors including length, sequence (and in some cases the presence of disulphide bonds or post-translational modifications such as glycosylation) may be important in determining details of the structures. Several recent papers report structural models for amyloid fibrils containing different polypeptide chains, including the Aβ40 peptide, insulin and fragments of the prion protein, based on data from such techniques as cryo-electron microscopy and solid-state magnetic resonance spectroscopy [43, 44]. These models have much in common and do indeed appear to reflect the fact that the structures of different fibrils are likely to be variations on a common theme [40]. It is also emerging that there may be some common and highly organised assemblies of amyloid protofilaments that are not simply extended threads or ribbons. It is clear, for example, that in some cases large closed loops can be formed [45, 46, 47], and there may be specific types of relatively small spherical or ‘doughnut’ shaped structures that can result in at least some circumstances (see below).
…..
The similarity of some early amyloid aggregates with the pores resulting from oligomerisation of bacterial toxins and pore-forming eukaryotic proteins (see below) also suggest that the basic mechanism of protein aggregation into amyloid structures may not only be associated with diseases but in some cases could result in species with functional significance. Recent evidence indicates that a variety of micro-organisms may exploit the controlled aggregation of specific proteins (or their precursors) to generate functional structures. Examples include bacterial curli [52] and proteins of the interior fibre cells of mammalian ocular lenses, whose β-sheet arrays seem to be organised in an amyloid-like supramolecular order [53]. In this case the inherent stability of amyloid-like protein structure may contribute to the long-term structural integrity and transparency of the lens. Recently it has been hypothesised that amyloid-like aggregates of serum amyloid A found in secondary amyloidoses following chronic inflammatory diseases protect the host against bacterial infections by inducing lysis of bacterial cells [54]. One particularly interesting example is a ‘misfolded’ form of the milk protein α-lactalbumin that is formed at low pH and trapped by the presence of specific lipid molecules [55]. This form of the protein has been reported to trigger apoptosis selectively in tumour cells providing evidence for its importance in protecting infants from certain types of cancer [55]. ….
Amyloid formation is a generic property of polypeptide chains ….
It is clear that the presence of different side chains can influence the details of amyloid structures, particularly the assembly of protofibrils, and that they give rise to the variations on the common structural theme discussed above. More fundamentally, the composition and sequence of a peptide or protein affects profoundly its propensity to form amyloid structures under given conditions (see below).
Because the formation of stable protein aggregates of amyloid type does not normally occur in vivo under physiological conditions, it is likely that the proteins encoded in the genomes of living organisms are endowed with structural adaptations that mitigate against aggregation under these conditions. A recent survey involving a large number of structures of β-proteins highlights several strategies through which natural proteins avoid intermolecular association of β-strands in their native states [65]. Other surveys of protein databases indicate that nature disfavours sequences of alternating polar and nonpolar residues, as well as clusters of several consecutive hydrophobic residues, both of which enhance the tendency of a protein to aggregate prior to becoming completely folded [66, 67].
……
Precursors of amyloid fibrils can be toxic to cells
It was generally assumed until recently that the proteinaceous aggregates most toxic to cells are likely to be mature amyloid fibrils, the form of aggregates that have been commonly detected in pathological deposits. It therefore appeared probable that the pathogenic features underlying amyloid diseases are a consequence of the interaction with cells of extracellular deposits of aggregated material. As well as forming the basis for understanding the fundamental causes of these diseases, this scenario stimulated the exploration of therapeutic approaches to amyloidoses that focused mainly on the search for molecules able to impair the growth and deposition of fibrillar forms of aggregated proteins. ….
Structural basis and molecular features of amyloid toxicity
The presence of toxic aggregates inside or outside cells can impair a number of cell functions that ultimately lead to cell death by an apoptotic mechanism [95, 96]. Recent research suggests, however, that in most cases initial perturbations to fundamental cellular processes underlie the impairment of cell function induced by aggregates of disease-associated polypeptides. Many pieces of data point to a central role of modifications to the intracellular redox status and free Ca2+ levels in cells exposed to toxic aggregates [45, 89, 97, 98, 99, 100, 101]. A modification of the intracellular redox status in such cells is associated with a sharp increase in the quantity of reactive oxygen species (ROS) that is reminiscent of the oxidative burst by which leukocytes destroy invading foreign cells after phagocytosis. In addition, changes have been observed in reactive nitrogen species, lipid peroxidation, deregulation of NO metabolism [97], protein nitrosylation [102] and upregulation of heme oxygenase-1, a specific marker of oxidative stress [103]. ….
Results have recently been reported concerning the toxicity towards cultured cells of aggregates of poly(Q) peptides which argues against a disease mechanism based on specific toxic features of the aggregates. These results indicate that there is a close relationship between the toxicity of proteins with poly(Q) extensions and their nuclear localisation. In addition they support the hypotheses that the toxicity of poly(Q) aggregates can be a consequence of altered interactions with nuclear coactivator or corepressor molecules including p53, CBP, Sp1 and TAF130 or of the interaction with transcription factors and nuclear coactivators, such as CBP, endowed with short poly(Q) stretches ([95] and references therein)…..
Concluding remarks
The data reported in the past few years strongly suggest that the conversion of normally soluble proteins into amyloid fibrils and the toxicity of small aggregates appearing during the early stages of the formation of the latter are common or generic features of polypeptide chains. Moreover, the molecular basis of this toxicity also appears to display common features between the different systems that have so far been studied. The ability of many, perhaps all, natural polypeptides to ‘misfold’ and convert into toxic aggregates under suitable conditions suggests that one of the most important driving forces in the evolution of proteins must have been the negative selection against sequence changes that increase the tendency of a polypeptide chain to aggregate. Nevertheless, as protein folding is a stochastic process, and no such process can be completely infallible, misfolded proteins or protein folding intermediates in equilibrium with the natively folded molecules must continuously form within cells. Thus mechanisms to deal with such species must have co-evolved with proteins. Indeed, it is clear that misfolding, and the associated tendency to aggregate, is kept under control by molecular chaperones, which render the resulting species harmless assisting in their refolding, or triggering their degradation by the cellular clearance machinery [166, 167, 168, 169, 170, 171, 172, 173, 175, 177, 178].
Misfolded and aggregated species are likely to owe their toxicity to the exposure on their surfaces of regions of proteins that are buried in the interior of the structures of the correctly folded native states. The exposure of large patches of hydrophobic groups is likely to be particularly significant as such patches favour the interaction of the misfolded species with cell membranes [44, 83, 89, 90, 91, 93]. Interactions of this type are likely to lead to the impairment of the function and integrity of the membranes involved, giving rise to a loss of regulation of the intracellular ion balance and redox status and eventually to cell death. In addition, misfolded proteins undoubtedly interact inappropriately with other cellular components, potentially giving rise to the impairment of a range of other biological processes. Under some conditions the intracellular content of aggregated species may increase directly, due to an enhanced propensity of incompletely folded or misfolded species to aggregate within the cell itself. This could occur as the result of the expression of mutational variants of proteins with decreased stability or cooperativity or with an intrinsically higher propensity to aggregate. It could also occur as a result of the overproduction of some types of protein, for example, because of other genetic factors or other disease conditions, or because of perturbations to the cellular environment that generate conditions favouring aggregation, such as heat shock or oxidative stress. Finally, the accumulation of misfolded or aggregated proteins could arise from the chaperone and clearance mechanisms becoming overwhelmed as a result of specific mutant phenotypes or of the general effects of ageing [173, 174].
The topics discussed in this review not only provide a great deal of evidence for the ‘new view’ that proteins have an intrinsic capability of misfolding and forming structures such as amyloid fibrils but also suggest that the role of molecular chaperones is even more important than was thought in the past. The role of these ubiquitous proteins in enhancing the efficiency of protein folding is well established [185]. It could well be that they are at least as important in controlling the harmful effects of misfolded or aggregated proteins as in enhancing the yield of functional molecules.
Nutritional Status is Associated with Faster Cognitive Decline and Worse Functional Impairment in the Progression of Dementia: The Cache County Dementia Progression Study1
Nutritional status may be a modifiable factor in the progression of dementia. We examined the association of nutritional status and rate of cognitive and functional decline in a U.S. population-based sample. Study design was an observational longitudinal study with annual follow-ups up to 6 years of 292 persons with dementia (72% Alzheimer’s disease, 56% female) in Cache County, UT using the Mini-Mental State Exam (MMSE), Clinical Dementia Rating Sum of Boxes (CDR-sb), and modified Mini Nutritional Assessment (mMNA). mMNA scores declined by approximately 0.50 points/year, suggesting increasing risk for malnutrition. Lower mMNA score predicted faster rate of decline on the MMSE at earlier follow-up times, but slower decline at later follow-up times, whereas higher mMNA scores had the opposite pattern (mMNA by time β= 0.22, p = 0.017; mMNA by time2 β= –0.04, p = 0.04). Lower mMNA score was associated with greater impairment on the CDR-sb over the course of dementia (β= 0.35, p < 0.001). Assessment of malnutrition may be useful in predicting rates of progression in dementia and may provide a target for clinical intervention.
Shared Genetic Risk Factors for Late-Life Depression and Alzheimer’s Disease
Background: Considerable evidence has been reported for the comorbidity between late-life depression (LLD) and Alzheimer’s disease (AD), both of which are very common in the general elderly population and represent a large burden on the health of the elderly. The pathophysiological mechanisms underlying the link between LLD and AD are poorly understood. Because both LLD and AD can be heritable and are influenced by multiple risk genes, shared genetic risk factors between LLD and AD may exist. Objective: The objective is to review the existing evidence for genetic risk factors that are common to LLD and AD and to outline the biological substrates proposed to mediate this association. Methods: A literature review was performed. Results: Genetic polymorphisms of brain-derived neurotrophic factor, apolipoprotein E, interleukin 1-beta, and methylenetetrahydrofolate reductase have been demonstrated to confer increased risk to both LLD and AD by studies examining either LLD or AD patients. These results contribute to the understanding of pathophysiological mechanisms that are common to both of these disorders, including deficits in nerve growth factors, inflammatory changes, and dysregulation mechanisms involving lipoprotein and folate. Other conflicting results have also been reviewed, and few studies have investigated the effects of the described polymorphisms on both LLD and AD. Conclusion: The findings suggest that common genetic pathways may underlie LLD and AD comorbidity. Studies to evaluate the genetic relationship between LLD and AD may provide insights into the molecular mechanisms that trigger disease progression as the population ages.
Association of Vitamin B12, Folate, and Sulfur Amino Acids With Brain Magnetic Resonance Imaging Measures in Older Adults: A Longitudinal Population-Based Study
Importance Vitamin B12, folate, and sulfur amino acids may be modifiable risk factors for structural brain changes that precede clinical dementia.
Objective To investigate the association of circulating levels of vitamin B12, red blood cell folate, and sulfur amino acids with the rate of total brain volume loss and the change in white matter hyperintensity volume as measured by fluid-attenuated inversion recovery in older adults.
Design, Setting, and Participants The magnetic resonance imaging subsample of the Swedish National Study on Aging and Care in Kungsholmen, a population-based longitudinal study in Stockholm, Sweden, was conducted in 501 participants aged 60 years or older who were free of dementia at baseline. A total of 299 participants underwent repeated structural brain magnetic resonance imaging scans from September 17, 2001, to December 17, 2009.
Main Outcomes and Measures The rate of brain tissue volume loss and the progression of total white matter hyperintensity volume.
Results In the multi-adjusted linear mixed models, among 501 participants (300 women [59.9%]; mean [SD] age, 70.9 [9.1] years), higher baseline vitamin B12 and holotranscobalamin levels were associated with a decreased rate of total brain volume loss during the study period: for each increase of 1 SD, β (SE) was 0.048 (0.013) for vitamin B12 (P < .001) and 0.040 (0.013) for holotranscobalamin (P = .002). Increased total homocysteine levels were associated with faster rates of total brain volume loss in the whole sample (β [SE] per 1-SD increase, –0.035 [0.015]; P = .02) and with the progression of white matter hyperintensity among participants with systolic blood pressure greater than 140 mm Hg (β [SE] per 1-SD increase, 0.000019 [0.00001]; P = .047). No longitudinal associations were found for red blood cell folate and other sulfur amino acids.
Conclusions and Relevance This study suggests that both vitamin B12 and total homocysteine concentrations may be related to accelerated aging of the brain. Randomized clinical trials are needed to determine the importance of vitamin B12supplementation on slowing brain aging in older adults.
Notes from Kurzweill
This vitamin stops the aging process in organs, say Swiss researchers
A potential breakthrough for regenerative medicine, pending further studies
Improved muscle stem cell numbers and muscle function in NR-treated aged mice: Newly regenerated muscle fibers 7 days after muscle damage in aged mice (left: control group; right: fed NR). (Scale bar = 50 μm). (credit: Hongbo Zhang et al./Science) http://www.kurzweilai.net/images/improved-muscle-fibers.png
EPFL researchers have restored the ability of mice organs to regenerate and extend life by simply administering nicotinamide riboside (NR) to them.
NR has been shown in previous studies to be effective in boosting metabolism and treating a number of degenerative diseases. Now, an article by PhD student Hongbo Zhang published in Science also describes the restorative effects of NR on the functioning of stem cells for regenerating organs.
As in all mammals, as mice age, the regenerative capacity of certain organs (such as the liver and kidneys) and muscles (including the heart) diminishes. Their ability to repair them following an injury is also affected. This leads to many of the disorders typical of aging.
Mitochondria —> stem cells —> organs
To understand how the regeneration process deteriorates with age, Zhang teamed up with colleagues from ETH Zurich, the University of Zurich, and universities in Canada and Brazil. By using several biomarkers, they were able to identify the molecular chain that regulates how mitochondria — the “powerhouse” of the cell — function and how they change with age. “We were able to show for the first time that their ability to function properly was important for stem cells,” said Auwerx.
Under normal conditions, these stem cells, reacting to signals sent by the body, regenerate damaged organs by producing new specific cells. At least in young bodies. “We demonstrated that fatigue in stem cells was one of the main causes of poor regeneration or even degeneration in certain tissues or organs,” said Zhang.
How to revitalize stem cells
Which is why the researchers wanted to “revitalize” stem cells in the muscles of elderly mice. And they did so by precisely targeting the molecules that help the mitochondria to function properly. “We gave nicotinamide riboside to 2-year-old mice, which is an advanced age for them,” said Zhang.
“This substance, which is close to vitamin B3, is a precursor of NAD+, a molecule that plays a key role in mitochondrial activity. And our results are extremely promising: muscular regeneration is much better in mice that received NR, and they lived longer than the mice that didn’t get it.”
Parallel studies have revealed a comparable effect on stem cells of the brain and skin. “This work could have very important implications in the field of regenerative medicine,” said Auwerx. This work on the aging process also has potential for treating diseases that can affect — and be fatal — in young people, like muscular dystrophy (myopathy).
So far, no negative side effects have been observed following the use of NR, even at high doses. But while it appears to boost the functioning of all cells, it could include pathological ones, so further in-depth studies are required.
Abstract of NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice
Adult stem cells (SCs) are essential for tissue maintenance and regeneration yet are susceptible to senescence during aging. We demonstrate the importance of the amount of the oxidized form of cellular nicotinamide adenine dinucleotide (NAD+) and its impact on mitochondrial activity as a pivotal switch to modulate muscle SC (MuSC) senescence. Treatment with the NAD+ precursor nicotinamide riboside (NR) induced the mitochondrial unfolded protein response (UPRmt) and synthesis of prohibitin proteins, and this rejuvenated MuSCs in aged mice. NR also prevented MuSC senescence in the Mdx mouse model of muscular dystrophy. We furthermore demonstrate that NR delays senescence of neural SCs (NSCs) and melanocyte SCs (McSCs), and increased mouse lifespan. Strategies that conserve cellular NAD+ may reprogram dysfunctional SCs and improve lifespan in mammals.
Discriminating the gene target of a distal regulatory element from other nearby transcribed genes is a challenging problem with the potential to illuminate the causal underpinnings of complex diseases. We present TargetFinder, a computational method that reconstructs regulatory landscapes from diverse features along the genome. The resulting models accurately predict individual enhancer–promoter interactions across multiple cell lines with a false discovery rate up to 15 times smaller than that obtained using the closest gene. By evaluating the genomic features driving this accuracy, we uncover interactions between structural proteins, transcription factors, epigenetic modifications, and transcription that together distinguish interacting from non-interacting enhancer–promoter pairs. Most of this signature is not proximal to the enhancers and promoters but instead decorates the looping DNA. We conclude that complex but consistent combinations of marks on the one-dimensional genome encode the three-dimensional structure of fine-scale regulatory interactions.
Images of the 3D-printed parts of the biomimetic liver tissue: liver cells derived from human induced pluripotent stem cells (left), endothelial and mesenchymal supporing cells (center), and the resulting organized combination of multiple cell types (right). (credit: Chen Laboratory, UC San Diego)
University of California, San Diego researchers have 3D-printed a tissue that closely mimics the human liver’s sophisticated structure and function. The new model could be used for patient-specific drug screening and disease modeling and could help pharmaceutical companies save time and money when developing new drugs, according to the researchers.
The liver plays a critical role in how the body metabolizes drugs and produces key proteins, so liver models are increasingly being developed in the lab as platforms for drug screening. However, so far, the models lack both the complex micro-architecture and diverse cell makeup of a real liver. For example, the liver receives a dual blood supply with different pressures and chemical constituents.
So the team employed a novel bioprinting technology that can rapidly produce complex 3D microstructures that mimic the sophisticated features found in biological tissues.
The liver tissue was printed in two steps.
The team printed a honeycomb pattern of 900-micrometer-sized hexagons, each containing liver cells derived from human induced pluripotent stem cells. An advantage of human induced pluripotent stem cells is that they are patient-specific, which makes them ideal materials for building patient-specific drug screening platforms. And since these cells are derived from a patient’s own skin cells, researchers don’t need to extract any cells from the liver to build liver tissue.
Then, endothelial and mesenchymal supporting cells were printed in the spaces between the stem-cell-containing hexagons.
The entire structure — a 3 × 3 millimeter square, 200 micrometers thick — takes just seconds to print. The researchers say this is a vast improvement over other methods to print liver models, which typically take hours. Their printed model was able to maintain essential functions over a longer time period than other liver models. It also expressed a relatively higher level of a key enzyme that’s considered to be involved in metabolizing many of the drugs administered to patients.
“It typically takes about 12 years and $1.8 billion to produce one FDA-approved drug,” said Shaochen Chen, NanoEngineering professor at the UC San Diego Jacobs School of Engineering. “That’s because over 90 percent of drugs don’t pass animal tests or human clinical trials. We’ve made a tool that pharmaceutical companies could use to do pilot studies on their new drugs, and they won’t have to wait until animal or human trials to test a drug’s safety and efficacy on patients. This would let them focus on the most promising drug candidates earlier on in the process.”
The work was published the week of Feb. 8 in the online early edition of Proceedings of the National Academy of Sciences.
Abstract of Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting
The functional maturation and preservation of hepatic cells derived from human induced pluripotent stem cells (hiPSCs) are essential to personalized in vitro drug screening and disease study. Major liver functions are tightly linked to the 3D assembly of hepatocytes, with the supporting cell types from both endodermal and mesodermal origins in a hexagonal lobule unit. Although there are many reports on functional 2D cell differentiation, few studies have demonstrated the in vitro maturation of hiPSC-derived hepatic progenitor cells (hiPSC-HPCs) in a 3D environment that depicts the physiologically relevant cell combination and microarchitecture. The application of rapid, digital 3D bioprinting to tissue engineering has allowed 3D patterning of multiple cell types in a predefined biomimetic manner. Here we present a 3D hydrogel-based triculture model that embeds hiPSC-HPCs with human umbilical vein endothelial cells and adipose-derived stem cells in a microscale hexagonal architecture. In comparison with 2D monolayer culture and a 3D HPC-only model, our 3D triculture model shows both phenotypic and functional enhancements in the hiPSC-HPCs over weeks of in vitro culture. Specifically, we find improved morphological organization, higher liver-specific gene expression levels, increased metabolic product secretion, and enhanced cytochrome P450 induction. The application of bioprinting technology in tissue engineering enables the development of a 3D biomimetic liver model that recapitulates the native liver module architecture and could be used for various applications such as early drug screening and disease modeling.
I wonder how equivalent are these hepatic cells derived from human induced pluripotent stem cells (hiPSCs) compared with the real hepatic cell populations.
All cells in our organism share the same DNA info, but every tissue is special for what genes are expressed and also because of the specific localization in our body (which would mean different surrounding environment for each tissue). I am not sure about how much of a step forward this is. Induced hepatic cells are known, but this 3-D print does not have liver shape or the different cell sub-types you would find in the liver.
I agree with your observation that having the same DNA information doesn’t account for variability of cell function within an organ. The regulation of expression is in RNA translation, and that is subject to regulatory factors related to noncoding RNAs and to structural factors in protein folding. The result is that chronic diseases that are affected by the synthetic capabilities of the liver are still problematic – toxicology, diabetes, and the inflammatory response, and amino acid metabolism as well. Nevertheless, this is a very significant step for the testing of pharmaceuticals. When we look at the double circulation of the liver, hypoxia is less of an issue than for heart or skeletal muscle, or mesothelial tissues. I call your attention to the outstanding work by Nathan O. Kaplan on the transhydrogenases, and his stipulation that there are significant differences between organs that are anabolic and those that are catabolic in TPNH/DPNH, that has been ignored for over 40 years. Nothing is quite as simple as we would like.
3-D printed liver Larry H. Bernstein, MD, FCAP, Curator LPBI 3D-printing a new lifelike liver tissue for drug …
I wonder how equivalent are these hepatic cells derived from human induced pluripotent stem cells (hiPSCs) compared with the real hepatic cell populations.
All cells in our organism share the same DNA info, but every tissue is special for what genes are expressed and also because of the specific localization in our body (which would mean different surrounding environment for each tissue). I am not sure about how much of a step forward this is. Induced hepatic cells are known, but this 3-D print does not have liver shape or the different cell sub-types you would find in the liver.
There is a growing reproducibility problem across the life sciences. The retraction rate of published papers has increased tenfold over the past decade, and researchers have reported only being able to replicate published results in 11 percent1 or 25 percent2 of attempts. It’s become known as the “reproducibility crisis,” and science is in place to fix it.
One major factor contributing to this problem is the use of poorly validated research antibodies. Lot to lot, antibodies can vary wildly. Some may not bind specifically to their target, or they may bind a different cellular protein altogether. According to one estimate, researchers around the world spend $800 million each year on poorly performing antibodies.3 (See “Exercises for Your Abs” here.)
While many researchers debate the best way to weed out the good antibodies from the bad, others are developing alternatives. Nucleic acid aptamers and protein scaffolds are increasingly being used to detect proteins of interest. Although they currently constitute only a fraction of affinity reagents, with the lion’s share of the market still going to traditional antibodies, these newer options offer an opportunity to rectify the problems stemming from using poorly validated antibodies in research. Researchers can engineer RNA or DNA aptamers and protein scaffolds to a specific target and function, the molecules are consistent from batch to batch, and they can be produced at a fraction of the cost of antibodies. These new reagents can target proteins that remain inaccessible to antibodies. And researchers have designed them to be functional in a wider range of conditions, including intracellular environments that degrade the antibody structure, opening up applications such as super-resolution microscopy and intracellular live-cell imaging to investigate the molecular dynamics of diverse cellular processes.
So, rather than complain about the poor performance of antibodies, perhaps the scientific community should embrace the new antibody alternatives designed to overcome this problem—and, by doing so, begin to resolve the ongoing reproducibility crisis.
Antibodies are large protein molecules composed of two heavy and two light chains linked by disulfide bonds. They play a crucial part in the immune system’s ongoing battle to keep our bodies from falling prey to deadly diseases. Through the diversification of gene segments in the antibody sequence, the mammalian immune system produces different combinations of heavy and light chains to bind a wide variety of foreign proteins. When an invader is detected, those B cells that produce the most specific antibodies undergo hypersomatic mutation to fine-tune the antibody’s affinity to a particular antigen, then differentiate into plasma cells that generate the targeted antibody molecules by the million to mark the disease-causing target for destruction. It has been estimated that the human body can create enough different antibodies to recognize 1012 distinct pathogens.4
For decades, life-science researchers have taken advantage of this natural process to develop tags and assays for a wide array of proteins. In the early 1900s, researchers began to cultivate protein-specific antibodies by immunizing rabbits, chickens, goats, donkeys, and other animals with a desired target protein. B cells within the animal host generate antibodies to different antigenic areas (epitopes) on the protein of interest. The antibodies targeting the desired protein can then be isolated and purified for use in biochemical and cell-based assays to document protein expression under different conditions or to identify potential disease biomarkers. But the reliance on an animal host system for production meant lot-to-lot heterogeneity for such polyclonal antibodies. (See illustration adjacent.)
In 1975, Argentine biochemist César Milstein and German biologist Georges Köhler discovered how to generate batches of individual antibodies, produced by a single B cell to target a specific antigen. Once an animal host produces antibodies to a target, the antibody-producing B cells are isolated from the spleen or lymph nodes and fused with tumor cells to generate immortal hybridoma lines. These lines are then screened to identify clones producing antibodies that bind with a high affinity to a specific epitope on the target protein. These cells are then cultured in large-scale bioreactors.
While heterogeneity can arise from drift in the cell line’s antibody expression and downstream production processes, monoclonal antibodies exhibit far less lot-to-lot variation than polyclonal antibodies, and have become the affinity tool of choice in modern research laboratories. Monoclonal antibodies are now routinely employed to localize proteins within tissues, determine protein network interactions, and analyze protein function. They are now being pushed to the limits of their performance in applications such as nanoimmunoassays and in vivo cell imaging. In medicine, antibody therapeutics represents the fastest growing sector of pharmaceutical sales, with 47 monoclonal antibodies currently on the market and a further 300 in clinical trials.5
But there are many examples where the use of antibodies has actually hindered scientific progress, by providing misleading or inaccurate results. Antibodies have evolved to execute their biological function perfectly, but this does not make them foolproof as investigative tools or therapeutic agents. In fact, many of the very characteristics that aid in antibodies’ function as part of the immune system limit their use in research and medicine.
In the context of an immune reaction, for example, not all B cells produce antibodies that are exquisitely specific. So long as the antibodies exceed a certain threshold of binding affinity for the target, they remain part of the immune system’s defense. In the body, this is a good thing: these less-specific antibodies cross-react with a variety of related antigens, making the antibody defense force more versatile.6 If an invading pathogen mutates or a similar pathogen invades, potentially effective antibodies may already be in circulation. As part of an assay to specifically identify a particular protein, however, such cross-reactivity can be the downfall of the experiment or therapy.
Examined in this light, it is easy to see why taking a molecule that is derived for one purpose and applying it to another may not yield the best results. A clear example of the shortcomings of antibody use in life-science research comes from the Human Protein Atlas project. Mathias Uhlén of the Royal Institute of Technology in Stockholm, Sweden, and colleagues set out to catalog protein expression and localization data across 44 normal human tissue types, 20 different cancers, and 46 cell lines. The team sourced antibodies from 51 different commercial vendors for validation. Of the 5,436 antibodies received, about half failed to detect their target in a Western blot or standard immunohistochemistry assay.7
The development of novel antibodies that bind new protein targets continues to face several challenges. For example, using the conventional route of immunizing lab animals to produce an antibody against a toxic target molecule will often kill the host animal prior to the generation of sufficiently specific antibodies. Conversely, if a protein target is highly homologous to a host protein, the immune system may not recognize the target as foreign in order to generate antibodies against it.
Given the rapid pace at which molecular biology proceeds in the modern era, new protein-binding reagents are desperately needed. A review of 20 million published research articles from 1950 to 2009 showed that three-quarters of the research focused on just 10 percent of the proteins that were known prior to the mapping of the human genome.8 Rational design of antibody alternatives will allow us to target a broader swath of proteins and function across more platforms to better investigate the scientific questions at hand.
Nucleic acid aptamers and protein scaffolds can target proteins that remain inaccessible to antibodies, and researchers have designed them to be functional in a wider range of conditions.
These novel affinity reagents also offer other benefits over antibodies. Both nucleic acid aptamers and protein scaffolds are much smaller than natural antibodies, which typically weigh about 150 kDa. Aptamers and scaffolds are as little as one-tenth that size. This means that their distribution is not restricted in the same manner as that of antibodies, opening up new targets that were previously inaccessible, such as epitopes hidden inside molecular grooves and pockets where antibodies simply can’t fit. Labeling target proteins with these smaller tags in cytochemistry experiments reduces the chance of the target protein being dragged around the cell according to the tag’s biochemistry, and increases the chances of identifying the correct protein localization. Additionally, their smaller size increases these affinity reagents’ tissue penetration, enhancing access to epitopes within tissue sections and decreasing false negative immunohistochemistry results. Smaller molecules are also cleared more rapidly from the body, especially when their size is below the renal cut-off of 45 kDa, making these molecules ideal as imaging agents in the clinic.
Researchers first developed nucleic acid aptamers in 1990 as RNA-based molecules, though DNA variants quickly followed to deal with the low stability of the RNA backbone. Aptamers offer simple chemistry that can be easily functionalized, but they lack stability across a range of temperatures and pH, and in the presence of common buffer components or DNases and RNases found in many media and cell environments. Researchers have used various chemical modifications to increase aptamers’ resistance to nuclease activity, to improve aptamer binding, and to increase their structural diversity, but others have turned to yet another option: protein scaffolds.
Developed around the same time as aptamers, protein scaffold affinity reagents were originally designed to identify potential therapeutic targets. Researchers soon began to apply this technology to screening for binders to completely novel proteins, by presenting a random sequence as the binding surface. Because protein scaffolds lack the disulfide bonds of antibodies, they retain their structure in a greater variety of cell culture and assay environments, without being attacked by other proteins that break these bonds and cause antibodies to fall apart. Scaffolds maintain function and target affinity at temperatures up to 80 °C and in solutions with a pH as low as 2 and as high as 13.
Because protein scaffolds can be delivered to the inside of the cell, researchers can use them in live-cell imaging, ultimately allowing use of the same reagent in both biochemical and cell biology assays. Additionally, protein scaffolds could help to deliver drugs directly into cells, improving targeting of pharmaceutical payloads and reducing side effects. And as new protein scaffolds are often engineered to lack cysteine residues, aberrant folding during their production within the cell factory is unlikely, increasing reproducibility within the reagents.
Importantly, both nucleic acid aptamers and protein scaffolds are far easier to consistently produce than antibodies. In addition to requiring animal hosts to provide an antibody-producing B cell, functional antibodies can only be expressed in higher eukaryotic cell systems. Antibodies are extensively glycosylated with a complex range of sugars that are critical to their function. Lower eukaryotic organisms, such as insects and yeast, and prokaryotic cells are not capable of the full range of complex glycosylation. As a result of these complexities, production times for monoclonal antibodies are six months on average, often making the generation of new antibodies the rate-limiting step in the advance of new research. This production process is also extremely expensive, and the use of such intractable biological systems breeds batch-to-batch inconsistencies.
Aptamers and protein scaffolds can be made without a host immune system. Now that robust and scalable methodologies for creating custom DNA and RNA molecules exist, effective aptamer binders can be chemically synthesized at a fraction of the cost of producing protein-based affinity molecules. And because protein scaffolds do not contain any posttranslational modifications, they can be expressed in bacterial cells, which are cheaper and easier to control than the eukaryotic systems used for antibody production. Both aptamers and scaffolds can often be available to researchers in a matter of weeks. (See illustration above)
So far, the majority of the industry attention for antibody alternatives has largely focused on the therapeutic development of antibody alternatives. Many companies now have initiated Phase 2 and 3 clinical trials of candidate molecules to treat conditions from vision problems to cancer, and two such molecules have already been approved for therapeutic use. In 2004, the RNA aptamer–based therapeutic pegaptanib (Macugen), originally developed by NeXstar Pharmaceuticals, became the first antibody alternative to gain US Food and Drug Administration (FDA) approval for the treatment of neovascular age-related macular degeneration. Pegaptanib is a 28-base-long RNA oligonucleotide with modifications to protect the aptamer from endogenous nucleases and extend its half-life in vivo to 10 days.9 Administered directly into the eye, this aptamer selectively binds the most common isoform of vascular endothelial growth factor (VEGF), preventing angiogenesis and the increased permeability of the blood vessels within the eye associated with neovascular age-related macular degeneration.10 Five years later, in 2009, the FDA approved a protein scaffold called ecallantide (Kalbitor) for the treatment of sudden hereditary angioedema attacks.
In a therapeutic context, the most important characteristic of aptamers and scaffolds is that they lack immunogenicity, thus avoiding harmful immune responses in patients. One way to ensure that protein scaffolds do not trigger host immunity is to model one’s scaffolds after proteins found in the human body. For example, the FDA-approved Kalbitor is based on the common Kunitz domain of protease inhibitors, and clinical trial participants have not suffered immunogenic responses. When brought to market, Kalbitor was one of only two approved therapies to treat cardiovascular attacks of this sort, which can cause rapid and serious swelling of the face or other parts of the body that may result in permanent disfigurement, disability, or death; the other is a protein therapeutic derived from human blood.
A major factor holding back the field of antibody alternatives as therapeutics is their small size. While this improves their intracellular function and use in research applications, their low molecular weight means that they are rapidly cleared from the body via the kidneys, reducing their potential therapeutic impact. Various strategies have been employed by the industry to overcome this rapid renal clearance, such as adding an antibody domain or an albumin-binding domain to the scaffold, or increasing the molecular weight of the protein scaffolds (though they still remain significantly smaller than a corresponding antibody). The fusion of an antibody domain to a protein scaffold can also help engage the immune system for improved therapeutic benefit.
While their small size can be a hurdle in developing antibody alternatives in a clinical setting, it is a big advantage in their use as laboratory tools, allowing them to penetrate bodily tissues that are inaccessible to antibodies and offering more-precise molecular labeling. In 2012, for example, Silvio Rizzoli of the European Neuroscience Institute and Center for Molecular Physiology of the Brain in Göttingen, Germany, and colleagues used 15 kDa aptamers to capture the dynamics of endosomal trafficking in live cells using super-resolution imaging.11 Doubling the molecular weight of the aptamer resulted in a substantial reduction in image quality, showing the importance of the small size of intracellular labels in accurate imaging of the intracellular space.
The increased intracellular stability of protein scaffolds as compared with antibodies is also critical to their function as research tools and offers potential therapeutic benefit for intracellular targets. For instance, protein scaffolds have been used to investigate the function of the small intracellular domain of a matrix metalloproteinase, which was shown to determine protein turnover to help regulate protein function in cell movement.12 Using antibodies in the reducing environment of the cell interior in such a study would be impossible.
When developing antibody alternatives for research, scientists are purposefully mimicking natural proteins to avoid interference by the immune system. For example, Janssen produces protein scaffolds called Centyrins that are based on the fibronectin glycoprotein of the extracellular matrix. Our own company, Avacta Life Sciences, recently introduced Affimer scaffolds, which are based on the cystatin protein family of common protease inhibitors. The use of consensus sequences from a number of species may allow these reagents to be used across a variety of different model systems.
Nucleic acid aptamers and protein scaffolds may also help fight emerging outbreaks of acute infectious disease. Examples of recent outbreaks that have caused considerable social, economic, and political stress are not hard to come by—SARS in 2003, the H1N1 flu pandemic in 2009, the Ebola crisis of recent years, and the continued emerging threat of MERS. It is impossible to predict such episodes, and alternative affinity reagents could be crucial tools in quickly stemming the spread of pandemic diseases. Screening libraries of 10 billion sequences can take as little as 7 weeks. And while the processes required for optimization, scale-up, and subsequent culture and validation of substantial quantities of the required affinity reagent remain to be explored, this all may take only a matter of months.
Over the past few decades, the rate of advancement of genomic technologies has outpaced proteomics. Yet it is the expressed protein within a cell, not the underlying genetic blueprint, that executes correct or aberrant function. In order to unify and make sense of the numerous data sets being produced, scientists need tools that enable the unraveling of proteomics. This requires affinity reagents that can specifically target individual protein isoforms and glycoforms, and that can tag all the proteins within a cell or organism. While the concerns over antibody irreproducibility are increasing, the solution may already be available.
Jane McLeod is a science writer at Avacta Life Sciences, where Paul Ko Ferrigno is the chief scientific officer. Avacta Life Sciences sells peptide aptamers, one of the main forms of antibody alternative.
C.G. Begley, L.M. Ellis, “Drug development: Raise standards for preclinical cancer research,” Nature, 483:531-33, 2012.
F. Prinz et al., “Believe it or not: How much can we rely on published data on potential drug targets?” Nat Rev Drug Discov, 10:712, 2011.
A. Bradbury, A. Plückthun, “Reproducibility: Standardize antibodies used in research,” Nature, 518:27-29, 2015.
B. Alberts et al., Molecular Biology of the Cell, 4th edition (New York: Garland Science, 2002).
D.M. Ecker et al., “The therapeutic monoclonal antibody market,” mAbs, 7:9-14, 2015.
N. Baumgarth, “How specific is too specific? B-cell responses to viral infection reveal the importance of breadth over depth,” Immunol Rev, 255:82-94, 2013.
L. Berglund et al., “A genecentric Human Protein Atlas for expression profiles based on antibodies,” Mol Cell Proteomics, 7:2019-27, 2008.
A.M. Edwards et al., “Too many roads not taken,” Nature, 470:163-65, 2011.
J. Ruckman et al., “2’-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165): Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain,” J Biol Chem, 273:20556-67, 1998.
P. Sundaram et al., “Therapeutic RNA aptamers in clinical trials,” Eur J Pharm Sci, 48:259-71, 2013.
F. Opazo et al., “Aptamers as potential tools for super-resolution microscopy,” Nat Methods, 9:938-39, 2012.
R.D. Wickramasinghe et al., “Peptide aptamers as new tools to modulate clathrin-mediated internalisation—inhibition of MT1-MMP internalisation,” BMC Cell Biol, 11:58, 2010.
Nice article. Your statement in the end identifies correctly a potential conflict of interest. It should probably be a bit more clear and obvious than that though. Perhaps at the top of the page, near the title of the article. My 2 cents.
Recently a German inventor, Clemens Bimek, developed a novel, reversible, hormone free, uncomplicated and lifelong contraceptive device for controlling male fertility. His invention is named as Bimek SLV, which is basically a valve that stops the flow of sperm through the vas deferens with the literal flip of a mechanical switch inside the scortum, rendering its user temporarily sterile. Toggled through the skin of the scrotum, the device stays closed for three months to prevent accidental switching. Moreover, the switch can’t open on its own. The tiny valves are less than an inch long and weigh is less than a tenth of an ounce. They are surgically implanted on the vas deferens, the ducts which carry sperm from the testicles, through a simple half-hour operation.
The valves are made of PEEK OPTIMA, a medical-grade polymer that has long been employed as a material for implants. The device is patented back in 2000 and is scheduled to undergo clinical trials at the beginning of this year. The inventor claims that Bimek SLV’s efficacy is similar to that of vasectomy, it does not impact the ability to gain and maintain an erection and ejaculation will be normal devoid of the sperm cells. The valve’s design enables sperm to exit the side of the vas deferens when it’s closed without any semen blockage. Leaked sperm cells will be broken down by the immune system. The switch to stop sperm flow can be kept working for three months or 30 ejaculations. After switching on the sperm flow the inventor suggested consulting urologist to ensure that all the blocked sperms are cleared off the device. The recovery time after switching on the sperm flow is only one day, according to Bimek SLV. However, men are encouraged to wait one week before resuming sexual activities.
Before the patented technology can be brought to market, it must undergo a rigorous series of clinical trials. Bimek and his business partners are currently looking for men interested in testing the device. If the clinical trials are successful then this will be the first invention of its kind that gives men the ability to control their fertility and obviously this method will be preferred over vasectomy.
Body fluids from individuals with possible Creutzfeldt-Jakob disease (CJD) present distinctive safety challenges for clinical laboratories. Sporadic, iatrogenic, and familial CJD (known collectively as classic CJD), along with variant CJD, kuru, Gerstmann-Sträussler-Scheinker, and fatal familial insomnia, are prion diseases, also known as transmissible spongiform encephalopathies. Prion diseases affect the central nervous system, and from the onset of symptoms follow a typically rapid progressive neurological decline. While prion diseases are rare, it is not uncommon for the most prevalent form—sporadic CJD—to be included in the differential diagnosis of individuals presenting with rapid cognitive decline. Thus, laboratories may deal with a significant number of possible CJD cases, and should have protocols in place to process specimens, even if a confirmatory diagnosis of CJD is made in only a fraction of these cases.
The Lab’s Role in Diagnosis
Laboratory protocols for handling specimens from individuals with possible, probable, and definitive cases of CJD are important to ensure timely and appropriate patient management. When the differential includes CJD, an attempt should be made to rule-in or out other causes of rapid neurological decline. Laboratories should be prepared to process blood and cerebrospinal fluid (CSF) specimens in such cases for routine analyses.
Definitive diagnosis requires identification of prion aggregates in brain tissue, which can be achieved by immunohistochemistry, a Western blot for proteinase K-resistant prions, and/or by the presence of prion fibrils. Thus, confirmatory diagnosis is typically achieved at autopsy. A probable diagnosis of CJD is supported by elevated concentration of 14-3-3 protein in CSF (a non-specific marker of neurodegeneration), EEG, and MRI findings. Thus, the laboratory may be required to process and send CSF samples to a prion surveillance center for 14-3-3 testing, as well as blood samples for sequencing of the PRNP gene (in inherited cases).
Processing Biofluids
Laboratories should follow standard protective measures when working with biofluids potentially containing abnormally folded prions, such as donning standard personal protective equipment (PPE); avoiding or minimizing the use of sharps; using single-use disposable items; and processing specimens to minimize formation of aerosols and droplets. An additional safety consideration is the use of single-use disposal PPE; otherwise, re-usable items must be either cleaned using prion-specific decontamination methods, or destroyed.
Blood. In experimental models, infectivity has been detected in the blood; however, there have been no cases of secondary transmission of classical CJD via blood product transfusions in humans. As such, blood has been classified, on epidemiological evidence by the World Health Organization (WHO), as containing “no detectible infectivity,” which means it can be processed by routine methods. Similarly, except for CSF, all other body fluids contain no infectivity and can be processed following standard procedures.
In contrast to classic CJD, there have been four cases of suspected secondary transmission of variant CJD via transfused blood products in the United Kingdom. Variant CJD, the prion disease associated with mad cow disease, is unique in its distribution of prion aggregates outside of the central nervous system, including the lymph nodes, spleen, and tonsils. For regions where variant CJD is a concern, laboratories should consult their regulatory agencies for further guidance.
CSF. Relative to highly infectious tissues of the brain, spinal cord, and eye, infectivity has been identified less often in CSF and is considered to have “low infectivity,” along with kidney, liver, and lung tissue. Since CSF can contain infectious material, WHO has recommended that analyses not be performed on automated equipment due to challenges associated with decontamination. Laboratories should perform a risk assessment of their CSF processes, and, if deemed necessary, consider using manual methods as an alternative to automated systems.
Decontamination
The infectious agent in prion disease is unlike any other infectious pathogen encountered in the laboratory; it is formed of misfolded and aggregated prion proteins. This aggregated proteinacious material forms the infectious unit, which is incredibly resilient to degradation. Moreover, in vitro studies have demonstrated that disrupting large aggregates into smaller aggregates increases cytotoxicity. Thus, if the aim is to abolish infectivity, all aggregates must be destroyed. Disinfectant procedures used for viral, bacterial, and fungal pathogens such as alcohol, boiling, formalin, dry heat (<300°C), autoclaving at 121°C for 15 minutes, and ionizing, ultraviolet, or microwave radiation, are either ineffective or variably effective against aggregated prions.
The only means to ensure no risk of residual infectious prions is to use disposable materials. This is not always practical, as, for instance, a biosafety cabinet cannot be discarded if there is a CSF spill in the hood. Fortunately, there are several protocols considered sufficient for decontamination. For surfaces and heat-sensitive instruments, such as a biosafety cabinet, WHO recommends flooding the surface with 2N NaOH or undiluted NaClO, letting stand for 1 hour, mopping up, and rinsing with water. If the surface cannot tolerate NaOH or NaClO, thorough cleaning will remove most infectivity by dilution. Laboratories may derive some additional benefit by using one of the partially effective methods discussed previously. Non-disposable heat-resistant items preferably should be immersed in 1N NaOH, heated in a gravity displacement autoclave at 121°C for 30 min, cleaned and rinsed in water, then sterilized by routine methods. WHO has outlined several alternate decontamination methods. Using disposable cover sheets is one simple solution to avoid contaminating work surfaces and associated lengthy decontamination procedures.
With standard PPE—augmented by a few additional safety measures and prion-specific decontamination procedures—laboratories can safely manage biofluid testing in cases of prion disease.
The Microscopic World Inside Us
Emerging Research Points to Microbiome’s Role in Health and Disease
Thousands of species of microbes—bacteria, viruses, fungi, and protozoa—inhabit every internal and external surface of the human body. Collectively, these microbes, known as the microbiome, outnumber the body’s human cells by about 10 to 1 and include more than 1,000 species of microorganisms and several million genes residing in the skin, respiratory system, urogenital, and gastrointestinal tracts. The microbiome’s complicated relationship with its human host is increasingly considered so crucial to health that researchers sometimes call it “the forgotten organ.”
Disturbances to the microbiome can arise from nutritional deficiencies, antibiotic use, and antiseptic modern life. Imbalances in the microbiome’s diverse microbial communities, which interact constantly with cells in the human body, may contribute to chronic health conditions, including diabetes, asthma and allergies, obesity and the metabolic syndrome, digestive disorders including irritable bowel syndrome (IBS), and autoimmune disorders like multiple sclerosis and rheumatoid arthritis, research shows.
While study of the microbiome is a growing research enterprise that has attracted enthusiastic media attention and venture capital, its findings are largely preliminary. But some laboratorians are already developing a greater appreciation for the microbiome’s contributions to human biochemistry and are considering a future in which they expect to measure changes in the microbiome to monitor disease and inform clinical practice.
Pivot Toward the Microbiome
Following the National Institutes of Health (NIH) Human Genome Project, many scientists noted the considerable genetic signal from microbes in the body and the existence of technology to analyze these microorganisms. That realization led NIH to establish the Human Microbiome Project in 2007, said Lita Proctor, PhD, its program director. In the project’s first phase, researchers studied healthy adults to produce a reference set of microbiomes and a resource of metagenomic sequences of bacteria in the airways, skin, oral cavities, and the gastrointestinal and vaginal tracts, plus a catalog of microbial genome sequences of reference strains. Researchers also evaluated specific diseases associated with disturbances in the microbiome, including gastrointestinal diseases such as Crohn’s disease, ulcerative colitis, IBS, and obesity, as well as urogenital conditions, those that involve the reproductive system, and skin diseases like eczema, psoriasis, and acne.
Phase 1 studies determined the composition of many parts of the microbiome, but did not define how that composition affects health or specific disease. The project’s second phase aims to “answer the question of what microbes actually do,” explained Proctor. Researchers are now examining properties of the microbiome including gene expression, protein, and human and microbial metabolite profiles in studies of pregnant women at risk for preterm birth, the gut hormones of patients at risk for IBS, and nasal microbiomes of patients at risk for type 2 diabetes.
Promising Lines of Research
Cystic fibrosis and microbiology investigator Michael Surette, PhD, sees promising microbiome research not just in terms of evidence of its effects on specific diseases, but also in what drives changes in the microbiome. Surette is Canada research chair in interdisciplinary microbiome research in the Farncombe Family Digestive Health Research Institute at McMaster University
in Hamilton, Ontario.
One type of study on factors driving microbiome change examines how alterations in composition and imbalances in individual patients relate to improving or worsening disease. “IBS, cystic fibrosis, and chronic obstructive pulmonary disease all have periods of instability or exacerbation,” he noted. Surette hopes that one day, tests will provide clinicians the ability to monitor changes in microbial composition over time and even predict when a patient’s condition is about to deteriorate. Monitoring perturbations to the gut microbiome might also help minimize collateral damage to the microbiome during aggressive antibiotic therapy for hospitalized patients, he added.
Monitoring changes to the microbiome also might be helpful for “culture negative” patients, who now may receive multiple, unsuccessful courses of different antibiotics that drive antibiotic resistance. Frustration with standard clinical biology diagnosis of lung infections in cystic fibrosis patients first sparked Surette’s investigations into the microbiome. He hopes that future tests involving the microbiome might also help asthma patients with neutrophilia, community-acquired pneumonia patients who harbor complex microbial lung communities lacking obvious pathogens, and hospitalized patients with pneumonia or sepsis. He envisions microbiome testing that would look for short-term changes indicating whether or not a drug is effective.
Companion Diagnostics
Daniel Peterson, MD, PhD, an assistant professor of pathology at Johns Hopkins University School of Medicine in Baltimore, believes the future of clinical testing involving the microbiome lies in companion diagnostics for novel treatments, and points to companies that are already developing and marketing tests that will require such assays.
Examples of microbiome-focused enterprises abound, including Genetic Analysis, based in Oslo, Norway, with its high-throughput test that uses 54 probes targeted to specific bacteria to measure intestinal gut flora imbalances in inflammatory bowel disease and irritable bowel syndrome patients. Paris, France-based Enterome is developing both novel drugs and companion diagnostics for microbiome-related diseases such as IBS and some metabolic diseases. Second Genome, based in South San Francisco, has developed an experimental drug, SGM-1019, that the company says blocks damaging activity of the microbiome in the intestine. Cambridge, Massachusetts-based Seres Therapeutics has received Food and Drug Administration orphan drug designation for SER-109, an oral therapeutic intended to correct microbial imbalances to prevent recurrent Clostridium difficile infection in adults.
One promising clinical use of the microbiome is fecal transplantation, which both prospective and retrospective studies have shown to be effective in patients with C. difficile infections who do not respond to front-line therapies, said James Versalovic, MD, PhD, director of Texas Children’s Hospital Microbiome Center and professor of pathology at Baylor College of Medicine in Houston. “Fecal transplants and other microbiome replacement strategies can radically change the composition of the microbiome in hours to days,” he explained.
But NIH’s Proctor discourages too much enthusiasm about fecal transplant. “Natural products like stool can have [side] effects,” she pointed out. “The [microbiome research] field needs to mature and we need to verify outcomes before anything becomes routine.”
Hurdles for Lab Testing
While he is hopeful that labs someday will use the microbiome to produce clinically useful information, Surette pointed to several problems that must be solved beforehand. First, molecular methods commonly used right now should be more quantitative and accurate. Additionally, research on the microbiome encompasses a wide variety of protocols, some of which are better at extracting particular types of bacteria and therefore can give biased views of communities living in the body. Also, tests may need to distinguish between dead and live microbes. Another hurdle is that labs using varied bioinfomatic methods may produce different results from the same sample, a problem that Surette sees as ripe for a solution from clinical laboratorians, who have expertise in standardizing robust protocols and in automating tests.
One way laboratorians can prepare for future, routine microbiome testing is to expand their notion of clinical chemistry to include both microbial and human biochemistry. “The line between microbiome science and clinical science is blurring,” said Versalovic. “When developing future assays to detect biochemical changes in disease states, we must consider the contributions of microbial metabolites and proteins and how to tailor tests to detect them.” In the future, clinical labs may test for uniquely microbial metabolites in various disease states, he predicted.
Automated Review of Mass Spectrometry Results
Can We Achieve Autoverification?
Author: Katherine Alexander and Andrea R. Terrell, PhD // Date: NOV.1.2015 // Source:Clinical Laboratory News
Paralleling the upswing in prescription drug misuse, clinical laboratories are receiving more requests for mass spectrometry (MS) testing as physicians rely on its specificity to monitor patient compliance with prescription regimens. However, as volume has increased, reimbursement has declined, forcing toxicology laboratories both to increase capacity and lower their operational costs—without sacrificing quality or turnaround time. Now, new solutions are available enabling laboratories to bring automation to MS testing and helping them with the growing demand for toxicology and other testing.
What is the typical MS workflow?
A typical workflow includes a long list of manual steps. By the time a sample is loaded onto the mass spectrometer, it has been collected, logged into the lab information management system (LIMS), and prepared for analysis using a variety of wet chemistry techniques.
Most commercial clinical laboratories receive enough samples for MS analysis to batch analyze those samples. A batch consists of a calibrator(s), quality control (QC) samples, and patient/donor samples. Historically, the method would be selected (i.e. “analysis of opiates”), sample identification information would be entered manually into the MS software, and the instrument would begin analyzing each sample. Upon successful completion of the batch, the MS operator would view all of the analytical data, ensure the QC results were acceptable, and review each patient/donor specimen, looking at characteristics such as peak shape, ion ratios, retention time, and calculated concentration.
The operator would then post acceptable results into the LIMS manually or through an interface, and unacceptable results would be rescheduled or dealt with according to lab-specific protocols. In our laboratory we perform a final certification step for quality assurance by reviewing all information about the batch again, prior to releasing results for final reporting through the LIMS.
What problems are associated with this workflow?
The workflow described above results in too many highly trained chemists performing manual data entry and reviewing perfectly acceptable analytical results. Lab managers would prefer that MS operators and certifying scientists focus on troubleshooting problem samples rather than reviewing mounds of good data. Not only is the current process inefficient, it is mundane work prone to user errors. This risks fatigue, disengagement, and complacency by our highly skilled scientists.
Importantly, manual processes also take time. In most clinical lab environments, turnaround time is critical for patient care and industry competitiveness. Lab directors and managers are looking for solutions to automate mundane, error-prone tasks to save time and costs, reduce staff burnout, and maintain high levels of quality.
How can software automate data transfer from MS systems to LIMS?
Automation is not a new concept in the clinical lab. Labs have automated processes in shipping and receiving, sample preparation, liquid handling, and data delivery to the end user. As more labs implement MS, companies have begun to develop special software to automate data analysis and review workflows.
In July 2011, AIT Labs incorporated ASCENT into our workflow, eliminating the initial manual peak review step. ASCENT is an algorithm-based peak picking and data review system designed specifically for chromatographic data. The software employs robust statistical and modeling approaches to the raw instrument data to present the true signal, which often can be obscured by noise or matrix components.
The system also uses an exponentially modified Gaussian (EMG) equation to apply a best-fit model to integrated peaks through what is often a noisy signal. In our experience, applying the EMG results in cleaner data from what might appear to be poor chromatography ultimately allows us to reduce the number of samples we might otherwise rerun.
How do you validate the quality of results?
We’ve developed a robust validation protocol to ensure that results are, at minimum, equivalent to results from our manual review. We begin by building the assay in ASCENT, entering assay-specific information from our internal standard operating procedure (SOP). Once the assay is configured, validation proceeds with parallel batch processing to compare results between software-reviewed data and staff-reviewed data. For new implementations we run eight to nine batches of 30–40 samples each; when we are modifying or upgrading an existing implementation we run a smaller number of batches. The parallel batches should contain multiple positive and negative results for all analytes in the method, preferably spanning the analytical measurement range of the assay.
The next step is to compare the results and calculate the percent difference between the data review methods. We require that two-thirds of the automated results fall within 20% of the manually reviewed result. In addition to validating patient sample correlation, we also test numerous quality assurance rules that should initiate a flag for further review.
What are the biggest challenges during implementation and continual improvement initiatives?
On the technological side, our largest hurdle was loading the sequence files into ASCENT. We had created an in-house mechanism for our chemists to upload the 96-well plate map for their batch into the MS software. We had some difficulty transferring this information to ASCENT, but once we resolved this issue, the technical workflow proceeded fairly smoothly.
The greater challenge was changing our employees’ mindset from one of fear that automation would displace them, to a realization that learning this new technology would actually make them more valuable. Automating a non-mechanical process can be a difficult concept for hands-on scientists, so managers must be patient and help their employees understand that this kind of technology leverages the best attributes of software and people to create a powerful partnership.
We recommend that labs considering automated data analysis engage staff in the validation and implementation to spread the workload and the knowledge. As is true with most technology, it is best not to rely on just one or two super users. We also found it critical to add supervisor level controls on data file manipulation, such as removing a sample that wasn’t run from the sequence table. This can prevent inadvertent deletion of a file, requiring reinjection of the entire batch!
What is the relationship of FGF-23 to heart failure?
A Heart failure (HF) is an increasingly common syndrome associated with high morbidity, elevated hospital readmission rates, and high mortality. Improving diagnosis, prognosis, and treatment of HF requires a better understanding of its different sub-phenotypes. As researchers gained a comprehensive understanding of neurohormonal activation—one of the hallmarks of HF—they discovered several biomarkers, including natriuretic peptides, which now are playing an important role in sub-phenotyping HF and in driving more personalized management of this chronic condition.
Like the natriuretic peptides, fibroblast growth factor 23 (FGF-23) could become important in risk-stratifying and managing HF patients. Produced by osteocytes, FGF-23 is a key regulator of phosphorus homeostasis. It binds to renal and parathyroid FGF-Klotho receptor heterodimers, resulting in phosphate excretion, decreased 1-α-hydroxylation of 25-hydroxyvitamin D, and decreased parathyroid hormone (PTH) secretion. The relationship to PTH is important because impaired homeostasis of cations and decreased glomerular filtration rate might contribute to the rise of FGF-23. The amino-terminal portion of FGF-23 (amino acids 1-24) serves as a signal peptide allowing secretion into the blood, and the carboxyl-terminal portion (aa 180-251) participates in its biological action.
How might FGF-23 improve HF risk assessment?
Studies have shown that FGF-23 is related to the risk of cardiovascular diseases and mortality. It was first demonstrated that FGF-23 levels were independently associated with left ventricular mass index and hypertrophy as well as mortality in patients with chronic kidney disease (CKD). FGF-23 also has been associated with left ventricular dysfunction and atrial fibrillation in coronary artery disease subjects, even in the absence of impaired renal function.
FGF-23 and FGF receptors are both expressed in the myocardium. It is possible that FGF-23 has direct effects on the heart and participates in the physiopathology of cardiovascular diseases and HF. Experiments have shown that for in vitro cultured rat cardiomyocytes, FGF-23 stimulates pathological hypertrophy by activating the calcineurin-NFAT pathway—and in wild-type mice—the intra-myocardial or intravenous injection of FGF-23 resulted in left ventricular hypertrophy. As such, FGF-23 appears to be a potential stimulus of myocardial hypertrophy, and increased levels may contribute to the worsening of heart failure and long-term cardiovascular death.
Researchers have documented that HF patients have elevated FGF-23 circulating levels. They have also found a significant correlation between plasma levels of FGF-23 and B-type natriuretic peptide, a biomarker related to ventricular stretch and cardiac hypertrophy, in patients with left ventricular hypertrophy. As such, measuring FGF-23 levels might be a useful tool to predict long-term adverse cardiovascular events in HF patients.
Interestingly, researchers have documented a significant relationship between FGF-23 and PTH in both CKD and HF patients. As PTH stimulates FGF-23 expression, it could be that in HF patients, increased PTH levels increase the bone expression of FGF-23, which enhances its effects on the heart.
The Past, Present, and Future of Western Blotting in the Clinical Laboratory
Much of the discussion about Western blotting centers around its performance as a biological research tool. This isn’t surprising. Since its introduction in the late 1970s, the Western blot has been adopted by biology labs of virtually every stripe, and become one of the most widely used techniques in the research armamentarium. However, Western blotting has also been employed in clinical laboratories to aid in the diagnosis of various diseases and disorders—an equally important and valuable application. Yet there has been relatively little discussion of its use in this context, or of how advances in Western blotting might affect its future clinical use.
Highlighting the clinical value of Western blotting, Stanley Naides, MD, medical director of Immunology at Quest Diagnostics observed that, “Western blotting has been a very powerful tool in the laboratory and for clinical diagnosis. It’s one of many various methods that the laboratorian brings to aid the clinician in the diagnosis of disease, and the selection and monitoring of therapy.” Indeed, Western blotting has been used at one time or the other to aid in the diagnosis of infectious diseases including hepatitis C (HCV), HIV, Lyme disease, and syphilis, as well as autoimmune disorders such as paraneoplastic disease and myositis conditions.
However, Naides was quick to point out that the choice of assays to use clinically is based on their demonstrated sensitivity and performance, and that the search for something better is never-ending. “We’re constantly looking for methods that improve detection of our target [protein],” Naides said. “There have been a number of instances where we’ve moved away from Western blotting because another method proves to be more sensitive.” But this search can also lead back to Western blotting. “We’ve gone away from other methods because there’s been a Western blot that’s been developed that’s more sensitive and specific. There’s that constant movement between methods as new tests are developed.”
In recent years, this quest has been leading clinical laboratories away from Western blotting toward more sensitive and specific diagnostic assays, at least for some diseases. Using confirmatory diagnosis of HCV infection as an example, Sai Patibandla, PhD, director of the immunoassay group at Siemens Healthcare Diagnostics, explained that movement away from Western blotting for confirmatory diagnosis of HCV infection began with a technical modification called Recombinant Immunoblotting Assay (RIBA). RIBA streamlines the conventional Western blot protocol by spotting recombinant antigen onto strips which are used to screen patient samples for antibodies against HCV. This approach eliminates the need to separate proteins and transfer them onto a membrane.
The RIBA HCV assay was initially manufactured by Chiron Corporation (acquired by Novartics Vaccines and Diagnostics in 2006). It received Food and Drug Administration (FDA) approval in 1999, and was marketed as Chiron RIBA HCV 3.0 Strip Immunoblot Assay. Patibandla explained that, at the time, the Chiron assay “…was the only FDA-approved confirmatory testing for HCV.” In 2013 the assay was discontinued and withdrawn from the market due to reports that it was producing false-positive results.
Since then, clinical laboratories have continued to move away from Western blot-based assays for confirmation of HCV in favor of the more sensitive technique of nucleic acid testing (NAT). “The migration is toward NAT for confirmation of HCV [diagnosis]. We don’t use immunoblots anymore. We don’t even have a blot now to confirm HCV,” Patibandla said.
Confirming HIV infection has followed a similar path. Indeed, in 2014 the Centers for Disease Control and Prevention issued updated recommendations for HIV testing that, in part, replaced Western blotting with NAT. This change was in response to the recognition that the HIV-1 Western blot assay was producing false-negative or indeterminable results early in the course of HIV infection.
At this juncture it is difficult to predict if this trend away from Western blotting in clinical laboratories will continue. One thing that is certain, however, is that clinicians and laboratorians are infinitely pragmatic, and will eagerly replace current techniques with ones shown to be more sensitive, specific, and effective. This raises the question of whether any of the many efforts currently underway to improve Western blotting will produce an assay that exceeds the sensitivity of currently employed techniques such as NAT.
Some of the most exciting and groundbreaking work in this area is being done by Amy Herr, PhD, a professor of bioengineering at University of California, Berkeley. Herr’s group has taken on some of the most challenging limitations of Western blotting, and is developing techniques that could revolutionize the assay. For example, the Western blot is semi-quantitative at best. This weakness dramatically limits the types of answers it can provide about changes in protein concentrations under various conditions.
To make Western blotting more quantitative, Herr’s group is, among other things, identifying losses of protein sample mass during the assay protocol. About this, Herr explains that the conventional Western blot is an “open system” that involves lots of handling of assay materials, buffers, and reagents that makes it difficult to account for protein losses. Or, as Kevin Lowitz, a senior product manager at Thermo Fisher Scientific, described it, “Western blot is a [simple] technique, but a really laborious one, and there are just so many steps and so many opportunities to mess it up.”
Herr’s approach is to reduce the open aspects of Western blot. “We’ve been developing these more closed systems that allow us at each stage of the assay to account for [protein mass] losses. We can’t do this exactly for every target of interest, but it gives us a really good handle [on protein mass losses],” she said. One of the major mechanisms Herr’s lab is using to accomplish this is to secure proteins to the blot matrix with covalent bonding rather than with the much weaker hydrophobic interactions that typically keep the proteins in place on the membrane.
Herr’s group also has been developing microfluidic platforms that allow Western blotting to be done on single cells, “In our system we’re doing thousands of independent Westerns on single cells in four hours. And, hopefully, we’ll cut that down to one hour over the next couple years.”
Other exciting modifications that stand to dramatically increase the sensitivity, quantitation, and through-put of Western blotting also are being developed and explored. For example, the use of capillary electrophoresis—in which proteins are conveyed through a small electrolyte-filled tube and separated according to size and charge before being dropped onto a blotting membrane—dramatically reduces the amount of protein required for Western blot analysis, and thereby allows Westerns to be run on proteins from rare cells or for which quantities of sample are extremely limited.
Jillian Silva, PhD, an associate specialist at the University of California, San Francisco Helen Diller Family Comprehensive Cancer Center, explained that advances in detection are also extending the capabilities of Western blotting. “With the advent of fluorescence detection we have a way to quantitate Westerns, and it is now more quantitative than it’s ever been,” said Silva.
Whether or not these advances produce an assay that is adopted by clinical laboratories remains to be seen. The emphasis on Western blotting as a research rather than a clinical tool may bias advances in favor of the needs and priorities of researchers rather than clinicians, and as Patibandla pointed out, “In the research world Western blotting has a certain purpose. [Researchers] are always coming up with new things, and are trying to nail down new proteins, so you cannot take Western blotting away.” In contrast, she suggested that for now, clinical uses of Western blotting remain “limited.”
Adapting Next Generation Technologies to Clinical Molecular Oncology Service
Next generation technologies (NGT) deliver huge improvements in cost efficiency, accuracy, robustness, and in the amount of information they provide. Microarrays, high-throughput sequencing platforms, digital droplet PCR, and other technologies all offer unique combinations of desirable performance.
As stronger evidence of genetic testing’s clinical utility influences patterns of patient care, demand for NGT testing is increasing. This presents several challenges to clinical laboratories, including increased urgency, clinical importance, and breadth of application in molecular oncology, as well as more integration of genetic tests into synoptic reporting. Laboratories need to add NGT-based protocols while still providing old tests, and the pace of change is increasing.What follows is one viewpoint on the major challenges in adopting NGTs into diagnostic molecular oncology service.
Choosing a Platform
Instrument selection is a critical decision that has to align with intended test applications, sequencing chemistries, and analytical software. Although multiple platforms are available, a mainstream standard has not emerged. Depending on their goals, laboratories might set up NGTs for improved accuracy of mutation detection, massively higher sequencing capacity per test, massively more targets combined in one test (multiplexing), greater range in sequencing read length, much lower cost per base pair assessed, and economy of specimen volume.
When high-throughput instruments first made their appearance, laboratories paid more attention to the accuracy of base-reading: Less accurate sequencing meant more data cleaning and resequencing (1). Now, new instrument designs have narrowed the differences, and test chemistry can have a comparatively large impact on analytical accuracy (Figure 1). The robustness of technical performance can also vary significantly depending upon specimen type. For example, LifeTechnologies’ sequencing platforms appear to be comparatively more tolerant of low DNA quality and concentration, which is an important consideration for fixed and processed tissues.
Sequence pile-ups of the same target sequence (2 large genes), all performed on the same analytical instrument. Results from 4 different chemistries, as designed and supplied by reagent manufacturers prior to optimization in the laboratory. Red lines represent limits of exons. Height of blue columns proportional to depth of coverage. In this case, the intent of the test design was to provide high depth of coverage so that reflex Sanger sequencing would not be necessary. Courtesy B. Sadikovic, U. of Western Ontario.
In addition, batching, robotics, workload volume patterns, maintenance contracts, software licenses, and platform lifetime affect the cost per analyte and per specimen considerably. Royalties and reagent contracts also factor into the cost of operating NGT: In some applications, fees for intellectual property can represent more than 50% of the bench cost of performing a given test, and increase substantially without warning.
Laboratories must also deal with the problem of obsolescence. Investing in a new platform brings the angst of knowing that better machines and chemistries are just around the corner. Laboratories are buying bigger pieces of equipment with shorter service lives. Before NGTs, major instruments could confidently be expected to remain current for at least 6 to 8 years. Now, a major instrument is obsolete much sooner, often within 2 to 3 years. This means that keeping it in service might cost more than investing in a new platform. Lease-purchase arrangements help mitigate year-to-year fluctuations in capital equipment costs, and maximize the value of old equipment at resale.
One Size Still Does Not Fit All
Laboratories face numerous technical considerations to optimize sequencing protocols, but the test has to be matched to the performance criteria needed for the clinical indication (2). For example, measuring response to treatment depends first upon the diagnostic recognition of mutation(s) in the tumor clone; the marker(s) then have to be quantifiable and indicative of tumor volume throughout the course of disease (Table 1).
As a result, diagnostic tests need to cover many different potential mutations, yet accurately identify any clinically relevant mutations actually present. On the other hand, tests for residual disease need to provide standardized, sensitive, and accurate quantification of a selected marker mutation against the normal background. A diagnostic panel might need 1% to 3% sensitivity across many different mutations. But quantifying early response to induction—and later assessment of minimal residual disease—needs a test that is reliably accurate to the 10-4 or 10-5 range for a specific analyte.
Covering all types of mutations in one diagnostic test is not yet possible. For example, subtyping of acute myeloid leukemia is both old school (karyotype, fluorescent in situ hybridization, and/or PCR-based or array-based testing for fusion rearrangements, deletions, and segmental gains) and new school (NGT-based panel testing for molecular mutations).
Chemistries that cover both structural variants and copy number variants are not yet in general use, but the advantages of NGTs compared to traditional methods are becoming clearer, such as in colorectal cancer (3). Researchers are also using cell-free DNA (cfDNA) to quantify residual disease and detect resistance mutations (4). Once a clinically significant clone is identified, enrichment techniques help enable extremely sensitive quantification of residual disease (5).
Validation and Quality Assurance
Beyond choosing a platform, two distinct challenges arise in bringing NGTs into the lab. The first is assembling the resources for validation and quality assurance. The second is keeping tests up-to-date as new analytes are needed. Even if a given test chemistry has the flexibility to add analytes without revalidating the entire panel, keeping up with clinical advances is a constant priority.
Due to their throughput and multiplexing capacities, NGT platforms typically require considerable upfront investment to adopt, and training staff to perform testing takes even more time. Proper validation is harder to document: Assembling positive controls, documenting test performance criteria, developing quality assurance protocols, and conducting proficiency testing are all demanding. Labs meet these challenges in different ways. Laboratory-developed tests (LDTs) allow self-determined choice in design, innovation, and control of the test protocol, but can be very expensive to set up.
Food and Drug Administration (FDA)-approved methods are attractive but not always an option. More FDA-approved methods will be marketed, but FDA approval itself brings other trade-offs. There is a cost premium compared to LDTs, and the test methodologies are locked down and not modifiable. This is particularly frustrating for NGTs, which have the specific attraction of extensive multiplexing capacity and accommodating new analytes.
IT and the Evolution of Molecular Oncology Reporting Standards
The options for information technology (IT) pipelines for NGTs are improving rapidly. At the same time, recent studies still show significant inconsistencies and lack of reproducibility when it comes to interpreting variants in array comparative genomic hybridization, panel testing, tumor expression profiling, and tumor genome sequencing. It can be difficult to duplicate published performances in clinical studies because of a lack of sufficient information about the protocol (chemistry) and software. Building bioinformatics capacity is a key requirement, yet skilled people are in short supply and the qualifications needed to work as a bioinformatician in a clinical service are not yet clearly defined.
Tumor biology brings another level of complexity. Bioinformatic analysis must distinguish tumor-specific variants from genomic variants. Sequencing of paired normal tissue is often performed as a control, but virtual normal controls may have intriguing advantages (6). One of the biggest challenges is to reproducibly interpret the clinical significance of interactions between different mutations, even with commonly known, well-defined mutations (7). For multiple analyte panels, such as predictive testing for breast cancer, only the performance of the whole panel in a population of patients can be compared; individual patients may be scored into different risk categories by different tests, all for the same test indication.
In large scale sequencing of tumor genomes, which types of mutations are most informative in detecting, quantifying, and predicting the behavior of the tumor over time? The amount and complexity of mutation varies considerably across different tumor types, and while some mutations are more common, stable, and clinically informative than others, the utility of a given tumor marker varies in different clinical situations. And, for a given tumor, treatment effect and metastasis leads to retesting for changes in drug sensitivities.
These complexities mean that IT must be designed into the process from the beginning. Like robotics, IT represents a major ancillary decision. One approach many labs choose is licensed technologies with shared databases that are updated in real time. These are attractive, despite their cost and licensing fees. New tests that incorporate proprietary IT with NGT platforms link the genetic signatures of tumors to clinically significant considerations like tumor classification, recommended methodologies for monitoring response, predicted drug sensitivities, eligible clinical trials, and prognostic classifications. In-house development of such solutions will be difficult, so licensing platforms from commercial partners is more likely to be the norm.
The Commercial Value of Health Records and Test Data
The future of cancer management likely rests on large-scale databases that link hereditary and somatic tumor testing with clinical outcomes. Multiple centers have such large studies underway, and data extraction and analysis is providing increasingly refined interpretations of clinical significance.
Extracting health outcomes to correlate with molecular test results is commercially valuable, as the pharmaceutical, insurance, and healthcare sectors focus on companion diagnostics, precision medicine, and evidence-based health technology assessment. Laboratories that can develop tests based on large-scale integration of test results to clinical utility will have an advantage.
NGTs do offer opportunities for net reductions in the cost of healthcare. But the lag between availability of a test and peer-evaluated demonstration of clinical utility can be considerable. Technical developments arise faster than evidence of clinical utility. For example, immunohistochemistry, estrogen receptor/progesterone receptor status, HER2/neu, and histology are still the major pathological criteria for prognostic evaluation of breast cancer at diagnosis, even though multiple analyte tumor profiling has been described for more than 15 years. Healthcare systems need a more concerted assessment of clinical utility if they are to take advantage of the promises of NGTs in cancer care.
Disruptive Advances
Without a doubt, “disruptive” is an appropriate buzzword in molecular oncology, and new technical advances are about to change how, where, and for whom testing is performed.
• Predictive Testing
Besides cost per analyte, one of the drivers for taking up new technologies is that they enable multiplexing many more analytes with less biopsy material. Single-analyte sequential testing for epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase, and other targets on small biopsies is not sustainable when many more analytes are needed, and even now, a significant proportion of test requests cannot be completed due to lack of suitable biopsy material. Large panels incorporating all the mutations needed to cover multiple tumor types are replacing individual tests in companion diagnostics.
• Cell-Free Tumor DNA
Challenges of cfDNA include standardizing the collection and processing methodologies, timing sampling to minimize the effect of therapeutic toxicity on analytical accuracy, and identifying the most informative sample (DNA, RNA, or protein). But for more and more tumor types, it will be possible to differentiate benign versus malignant lesions, perform molecular subtyping, predict response, monitor treatment, or screen for early detection—all without a surgical biopsy.
cfDNA technologies can also be integrated into core laboratory instrumentation. For example, blood-based EGFR analysis for lung cancer is being developed on the Roche cobas 4800 platform, which will be a significant change from the current standard of testing based upon single tests of DNA extracted from formalin-fixed, paraffin-embedded sections selected by a pathologist (8).
• Whole Genome and Whole Exome Sequencing
Whole genome and whole exome tumor sequencing approaches provide a wealth of biologically important information, and will replace individual or multiple gene test panels as the technical cost of sequencing declines and interpretive accuracy improves (9). Laboratories can apply informatics selectively or broadly to extract much more information at relatively little increase in cost, and the interpretation of individual analytes will be improved by the context of the whole sequence.
• Minimal Residual Disease Testing
Massive resequencing and enrichment techniques can be used to detect minimal residual disease, and will provide an alternative to flow cytometry as costs decline. The challenge is to develop robust analytical platforms that can reliably produce results in a high proportion of patients with a given tumor type, despite using post-treatment specimens with therapy-induced degradation, and a very low proportion of target (tumor) sequence to benign background sequence.
The tumor markers should remain informative for the burden of disease despite clonal evolution over the course of multiple samples taken during progression of the clinical course and treatment. Quantification needs to be accurate and sensitive down to the 10-5 range, and cost competitive with flow cytometry.
• Point-of-Care Test Methodologies
Small, rapid, cheap, and single use point-of-care (POC) sequencing devices are coming. Some can multiplex with analytical times as short as 20 minutes. Accurate and timely testing will be possible in places like pharmacies, oncology clinics, patient service centers, and outreach programs. Whether physicians will trust and act on POC results alone, or will require confirmation by traditional laboratory-based testing, remains to be seen. However, in the simplest type of application, such as a patient known to have a particular mutation, the advantages of POC-based testing to quantify residual tumor burden are clear.
Conclusion
Molecular oncology is moving rapidly from an esoteric niche of diagnostics to a mainstream, required component of integrated clinical laboratory services. While NGTs are markedly reducing the cost per analyte and per specimen, and will certainly broaden the scope and volume of testing performed, the resources required to choose, install, and validate these new technologies are daunting for smaller labs. More rapid obsolescence and increased regulatory scrutiny for LDTs also present significant challenges. Aligning test capacity with approved clinical indications will require careful and constant attention to ensure competitiveness.
References
1. Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012; doi:10.1155/2012/251364.
2. Brownstein CA, Beggs AH, Homer N, et al. An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol 2014;15:R53.
3. Haley L, Tseng LH, Zheng G, et al. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. [Epub ahead of print] Modern Pathol July 31, 2015 as doi:10.1038/modpathol.2015.86.
4. Butler TM, Johnson-Camacho K, Peto M, et al. Exome sequencing of cell-free DNA from metastatic cancer patients identifies clinically actionable mutations distinct from primary disease. PLoS One 2015;10:e0136407.
5. Castellanos-Rizaldos E, Milbury CA, Guha M, et al. COLD-PCR enriches low-level variant DNA sequences and increases the sensitivity of genetic testing. Methods Mol Biol 2014;1102:623–39.
6. Hiltemann S, Jenster G, Trapman J, et al. Discriminating somatic and germline mutations in tumor DNA samples without matching normals. Genome Res 2015;25:1382–90.
7. Lammers PE, Lovly CM, Horn L. A patient with metastatic lung adenocarcinoma harboring concurrent EGFR L858R, EGFR germline T790M, and PIK3CA mutations: The challenge of interpreting results of comprehensive mutational testing in lung cancer. J Natl Compr Canc Netw 2015;12:6–11.
8. Weber B, Meldgaard P, Hager H, et al. Detection of EGFR mutations in plasma and biopsies from non-small cell lung cancer patients by allele-specific PCR assays. BMC Cancer 2014;14:294.
9. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546–58.
10. Heitzer E, Auer M, Gasch C, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res 2013;73:2965–75.
11. Healy B. BRCA genes — Bookmaking, fortunetelling, and medical care. N Engl J Med 1997;336:1448–9.
Complexity of Protein-Protein Interactions, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Complexity of Protein-Protein Interactions
Curator: Larry H. Bernstein, MD, FCAP
Cracking the Complex
Using mass spec to study protein-protein interactions
Mass spectrometry is a proteomics workhorse. By precisely measuring polypeptide masses, researchers can identify and sequence those molecules, and characterize whether and how they have been chemically modified. To twist a phrase, by their masses you shall know them.
But many proteins do not act in isolation. Critical biological processes such as DNA replication, transcription, translation, cell division, and energy generation rely on the action of massive protein assemblies, many of which comprise dozens of subunits. While these clusters are ripe for study, few traditional mass spectrometric methods can handle them.
Indeed, protein complexes are unwieldy for many types of analysis, says Philip Compton, director of instrumentation at the Proteomics Center of Excellence at Northwestern University in Evanston, Illinois. Most complexes are held together by noncovalent interactions, assemble only transiently, or are located in the cell membrane—all of which complicate sample preparation, he explains. Also, while some complexes are relatively abundant, others are rare, further thwarting detection and analysis.
For mass spectrometry specifically, however, the problem with analyzing protein complexes, which can weigh in at 500 kDa, is size. “In a mass spec, things of that size have traditionally been fairly difficult to handle,” Compton says. Even if you can deliver them into the spectrometer itself, you need a way to figure out which proteins are present, and in what stoichiometry. Plus, normal sample preparation procedures tend to denature proteins, ripping complexes apart.
Still, researchers are increasingly keen to train their mass specs on intact protein assemblies. The Scientistasked four protein-complex experts about the approaches they use in their own labs. This is what they said.
GETTING TOGETHER: Lactate dehydrogenase from human skeletal muscle comprises four identical M subunits, shown here in different colors. FVASCONCELLOS/WIKIMEDIA COMMONS
RESEARCHER:Philip Compton, Director of Instrumentation, Proteomics Center of Excellence, Northwestern University
PROJECT: High-throughput top-down proteomics
SOLUTION: If protein complexes are onions, Compton needs a way to iteratively peel off the layers to see what’s inside. Working with researchers at Thermo Fisher Scientific, Compton is developing an Orbitrap-based mass spectrometer that can do just that, or perform what is called an MS3 study.
Basically, an MS3 experiment involves weighing all the complexes in a sample fraction—there could be as many as 10 or 15 at a time—grabbing one, smashing it into inert-gas molecules to eject a subunit, weighing and sequencing the cast-off piece, and then repeating the process.
That’s the goal, but because that instrument is not yet built, Compton must temporarily content himself with what he calls a “pseudo-MS3” experiment. Basically, instead of one seamless workflow, the instrument shatters the complex, weighs the pieces that come off it, and then repeats the process, only this time capturing and fragmenting those ejected pieces for subsequent analysis (Anal Chem, 85:11163-73, 2013). “We’re kind of splitting it into these two different steps; that accomplishes essentially the same thing,” Compton says.
Compton and his team are still ironing out the kinks, but they have begun applying the approach to protein complexes involved in metabolism. One of these, lactate dehydrogenase (LDH), is a 145-kDa tetramer comprising M (muscle) and H (heart) subunits that can exist in any of five configurations (MMMM, MMMH, MMHH, MHHH, and HHHH). Using the MS3 workflow, Compton says he can differentiate these “multiproteoform assemblies,” as well as any posttranslational modifications those subunits may bear, and determine the abundance of each. Now he hopes to apply the approach to quantify LDH differences between cell and tissue types.
From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry
Native mass spectrometry (MS) is becoming an important integral part of structural proteomics and system biology research. The approach holds great promise for elucidating higher levels of protein structure: from primary to quaternary. This requires the most efficient use of tandem MS, which is the cornerstone of MS-based approaches. In this work, we advance a two-step fragmentation approach, or (pseudo)-MS3, from native protein complexes to a set of constituent fragment ions. Using an efficient desolvation approach and quadrupole selection in the extended mass-to-charge (m/z) range, we have accomplished sequential dissociation of large protein complexes, such as phosporylase B (194 kDa), pyruvate kinase (232 kDa), and GroEL (801 kDa), to highly charged monomers which were then dissociated to a set of multiply charged fragmentation products. Fragment ion signals were acquired with a high resolution, high mass accuracy Orbitrap instrument that enabled highly confident identifications of the precursor monomer subunits. The developed approach is expected to enable characterization of stoichiometry and composition of endogenous native protein complexes at an unprecedented level of detail.
EXTEND YOUR RANGE: Compton’s team uses a souped-up version of Thermo Fisher’s Orbitrap-based Q Exactive HF mass spectrometer, which among other things features a fourfold wider mass range. Other researchers can perform similar work using Thermo’s Exactive Plus EMR Orbitrap system, an off-the-shelf, “extended mass range” instrument. But, because the EMR lacks the “high-mass isolation capabilities” of Compton’s bespoke hardware, the application range is more limited, he says. “You can still do a similar experiment to us, provided that you have one clean [purified] complex.”
Mapping protein-protein interaction interfaces RESEARCHER:Igor Kaltashov, Professor of Chemistry, University of Massachusetts Amherst
PROJECT: Probing the interactions of candidate protein therapeutics with their molecular targets
SOLUTION: Most attempts at studying protein complexes deliver them to the mass spec intact. Kaltashov takes a different approach, using a technique called hydrogen-deuterium exchange (HDX).
It works like this: proteins (like other molecules) pass hydrogen atoms back and forth with the solvent that surrounds them. Normally, one hydrogen is simply swapped for another, and nobody is the wiser. But in deuterated (“heavy”) water, as hydrogens are swapped at the protein surface, the protein gets slightly heavier as deuterium molecules replace some of the hydrogens. This allows researchers to probe how accessible different pieces of the protein are to the solvent, based on how much deuterium they pick up from the buffer, and how quickly they do so.
As Kaltashov explains, HDX can be used to study any event that might alter the accessibility of different protein regions to the solvent that surrounds them. Those events include protein folding and aggregation, but also protein-protein interactions. “Once two proteins bind to each other, solvent would be excluded from the interface, and that would be reflected in the hydrogen-deuterium exchange kinetics,” he says. That change is evident when compared to the proteins in isolation.
In a 2009 review, Kaltashov demonstrated the process with transferrin, an iron transport protein, and its receptor. After undergoing the exchange reaction, the proteins were fragmented to peptides and analyzed piecemeal. Some peptides exhibited no hydrogen-deuterium exchange, he says. That suggests they were never exposed to solvent because they were buried inside the protein core. Other peptides exchanged hydrogens with the solvent at the same rate regardless of receptor binding, indicating they are not part of the protein-receptor interface. A third set of peptides, though, exhibited clear differences in the presence and absence of receptor, marking those as elements of the protein-protein interaction domain (Anal Chem, 81:7892-99, 2009).
“You can actually localize these sites and obtain information both on the strength of the binding [interactions] and the structural characteristics of the interface region,” Kaltashov says.
H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: Is there a need for a top-down approach?
Hydrogen/deuterium exchange (HDX) combined with mass spectrometry (MS) detection has matured in recent years to become a powerful tool in structural biology and biophysics. Several limitations of this technique can and will be addressed by tapping into ever expanding arsenal of methods to manipulate ions in the gas phase offered by mass spectrometry.
Keywords: hydrogen/deuterium exchange (HDX), mass spectrometry (MS), protein ion fragmentation, collision-induced dissociation (CAD), electron-capture dissociation (ECD), electron-transfer dissociation (ETD), protein conformation, protein dynamics
Introduction: HDX MS in the context of structural proteomics
The spectacular successes of proteomics and bioinformatics in the past decade have resulted in an explosive growth of information on the composition of complex networks of proteins interacting at the cellular level and beyond. However, a simple inventory of interacting proteins is insufficient for understanding how the components of sophisticated biological machinery work together. Protein interactions with each other, small ligands and other biopolymers are governed by their higher order structure, whose determination on a genome scale is a focus of structural proteomics. Realization that “the structures of individual macromolecules are often uninformative about function if taken out of context”1 is shifting the focus of the inquiry from comprehensive characterization of individual protein structures to structural analysis of protein complexes.
X-ray crystallography remains the mainstay in this field, and high resolution structures of proteins and protein complexes often provide important clues as to how they carry out their diverse functions in vivo. However, individual proteins are not static objects, and their behavior cannot be adequately described based solely on information derived from static snapshots and without taking into consideration their dynamic character.2Conformation and dynamics of small proteins can be probed at high spatial resolution on a variety of time scales using NMR spectroscopy; however, rather unforgiving molecular weight limitations make this technique less suited for the studies of larger proteins and protein complexes.
Mass spectrometry (MS) is playing an increasingly visible role in this field, as it can provide information on protein dynamics on a variety of levels, ranging from interactions with their physiological partners by forming dynamic assemblies3 to large-scale conformational transitions within individual subunits.4 Perhaps one of the most powerful MS-based tools to characterize protein conformation and dynamics is HDX MS, a technique that combined hydrogen/deuterium exchange in solution5 with MS detection of the progress of exchange reactions.6 This technique is certainly not new,7 and in fact already made lasting impact in diverse fields ranging from structural proteomics8 to analysis of biopharmaceutical products.9 Nevertheless, HDX MS methodology is still in a phase where dramatic progress is made, fed by the continued expansion of the experimental armamentarium offered by MS. In particular, better integration of new methods of manipulating ions in the gas phase into HDX MS routine is likely to result in truly transformative changes. This sea change in HDX MS methodology will transform it to a potent tool rivaling NMR in terms of resolution, but without suffering the limitations of this technique.
What information can be deduced from HDX MS measurements? The classic “bottom-up” approach, its challenges and limitations
While the concept of HDX experiment may appear rather transparent (Figure 1), interpretation of the results is usually not. The backbone protection measured in a typical HDX MS experiment is a combination of several factors, as the exchange reaction of each labile hydrogen atom is a convolution of two processes.5The first is a protein motion that makes a particular hydrogen atom exposed to solvent and therefore available for the exchange. This could be a small-scale event, such as relatively frequent local structural fluctuations transiently exposing hydrogen atoms residing close to the protein surface, or a rare global unfolding event exposing atoms sequestered from the solvent in the protein core. The second process is a chemical reaction of exchanging the unprotected labile hydrogen atom with the solvent. The kinetics of this reaction (intrinsic exchange rate) strongly depends on solution temperature and pH (with a minimum at pH 2.5-3 for backbone amides), parameters that obviously have a great influence on the protein dynamics as well.
Schematic representation of HDX MS experiments: bottom-up (A) and top-down (B) HDX MS.
Since the majority of HDX MS studies target protein dynamics under near-native conditions, the experiments are typically carried out at physiological pH, where the progress of the exchange is followed by monitoring the protein mass change. The direct infusion scheme offers the simplest way to carry out such measurements, either in real time7 or by using on-line rapid mixing.10 However, in many cases these straightforward approaches cannot be used, as they limit the choice of exchange buffer systems to those compatible with electrospray ionization (ESI). To avoid this, HDX can be carried out in any suitable buffer followed by rapid quenching (lowering pH to 2.5-3 and temperature to near 0°C). Dramatic deceleration of the intrinsic exchange rate for backbone amides under these conditions allows the protein solution to be de-salted prior to MS analysis. Additionally, the slow exchange conditions denature most proteins, resulting in facile removal of various binding partners, ranging from small ligands to receptors (their binding to the protein of interest inevitably complicates the HDX MS data interpretation by making accurate mass measurements in the gas phase less straightforward).
An example of such experiments is shown in Figure 2, where HDX is used to probe the higher order structure and conformational dynamics of metal transporter transferrin (Fe2Tf) alone and in the receptor-bound form. Both Tf-metal and Tf-receptor complexes dissociate under the slow exchange conditions prior to MS analysis; therefore, the protein mass evolution in each case reflects solely deuterium uptake in the course of exchange in solution. The extra protection afforded by the receptor binding to Tf persists over an extended period of time, and it may be tempting to assign it to shielding of labile hydrogen atoms at the protein-receptor interface. However, this view is overly simplistic, as the conformational effects of protein binding are frequently felt well beyond the interface region. The difference in the backbone protection levels of receptor-free and receptor-bound forms of Fe2Tf appears to grow during the initial hour of the exchange (Figure 2), reflecting significant stabilization of Fe2Tf higher order structure by the receptor binding. Indeed, while the fast phase of HDX is typically ascribed to frequent local fluctuations (transient perturbations of higher order structure) affecting relatively small protein segments, the slower phases of HDX usually reflect relatively rare, large-scale conformational transitions (transient partial or complete unfolding). This is why global HDX MS measurements similar to those presented in Figure 2 are can be used to obtain quantitative thermodynamic characteristics for protein interaction with a variety of ligands, ranging from metal ions11 and small organic molecules 12 to other proteins13 and oligonucleotides.14
HDX MS of Fe2Tf in the presence (blue) and the absence (red) of the cognate receptor. The exchange was carried out by diluting the protein stock solution 1:10 in exchange solution (100 mM NH4HCO3 in D2O, pH adjusted to 7.4) and incubating for a certain period of time as indicated on each diagram followed by rapid quenching (lowering pH to 2.5 and temperature to near 0°C). The black trace shows unlabeled protein.
While global HDX MS measurements under near-native conditions provide valuable thermodynamic information on proteins and their interaction with binding partners, structural studies (e.g., localizing the changes in Tf that occur as a result of receptor binding) must rely on the knowledge of exchange kinetics at the local level. This is typically accomplished by carrying out proteolysis under the slow exchange conditions following the quench of HDX.6 Here we will refer to this approach as “bottom-up” HDX MS, by drawing analogy to a bottom-up approach to obtain sequence information.15 An example is shown in Figure 3, where Fe2Tf undergoes exchange in solution in the absence and in the presence of the receptor, followed by rapid quenching of HDX reactions, protein reduction and digestion with pepsin and LC/MS analysis of the deuterium content of individual proteolytic peptides.
Localizing the influence of the receptor binding on backbone protection of Fe2Tf using bottom-up HDX MS on the physiologically relevant time scale. The panels show isotopic distributions of representative peptic fragments derived from the protein subjected to HDX in the presence (blue) and the absence (red) of the receptor and followed by rapid quenching. Dotted lines indicate deuterium content of unlabeled and fully exchanged peptides. Colored segments within the Fe2Tf/receptor complex show location of the peptic fragments.
Evolution of deuterium content of various peptic fragments in Figure 3 reveals a wide spectrum of protection, which is clearly distributed very unevenly across the protein sequence. While some peptides exhibit nearly complete protection of backbone amides (e.g., segment [396-408] sequestered in the core of the protein C-lobe), exchange in some other segments is fast (e.g., peptide [612-621] in the solvent-exposed loop of the C-lobe). The influence of the receptor binding on the backbone protection is also highly localized. While many segments appear to be unaffected by the receptor binding, there are a few regions where exchange kinetics noticeably decelerates (e.g., segment [71-81] of the N-lobe, which contains several amino acid residues that form Tf/receptor interface according to the available model of the complex based on low-resolution cryo-EM data16).
Although the increased protection of backbone amides proximal to the protein/receptor binding interface is hardly surprising, HDX MS data also reveal a less trivial trend, acceleration of exchange kinetics in some segments of the protein as a result of receptor binding (such behavior is illustrated in Figure 3 with segment [113-134], a part of the N-lobe that is distal to the receptor). Therefore, in addition to mapping binding interface regions, HDX MS also provides a means to localize the protein segments that are affected by the binding indirectly via allosteric mechanisms. However, this example also highlights one of the limitations of HDX MS, namely inadequate spatial resolution. This peptic fragment spans several distinct regions of the protein (an α-helical segment, a β-strand, and two loops). The moderate level of protection observed in this segment in the absence of the receptor binding (fast exchange of three protons followed by slow exchange of the rest) is likely to be a result of averaging out very uneven protection patterns across this peptide. Even smaller peptides may comprise two or more distinct structural elements, such as segment [71-81] spanning three distinct regions of the protein (an α-helical segment, a β-strand, and a loop connecting them).
In some favorable cases spatial resolution in HDX MS of small proteins (<15 kDa) may be enhanced up to a single residue level by analyzing deuterium content of a set of overlapping proteolytic fragments.17However, single-residue resolution has never been demonstrated in HDX MS studies of proteins falling out of the mass range routinely accessible by NMR, although overlapping peptic fragments frequently provide moderate improvement of spatial resolution.
In addition to limited spatial resolution, the “classic” HDX MS scheme frequently suffers from incomplete sequence coverage, especially when applied to larger and extensively glycosylated proteins. Proteins with multiple disulfide bonds constitute another class of targets for which adequate sequence coverage is difficult to achieve, although certain changes in experimental protocol can alleviate this problem, at least for smaller proteins.18 Typically, an 80% level of sequence coverage is considered good, although significantly lower levels may also be adequate, depending on the context of the study.
Protein processing in HDX MS experiments is carried out under the conditions that minimize the exchange rates for backbone amides. Since these slow exchange conditions are highly denaturing for most proteins, both intact protein and its proteolytic fragments lack any protection and inevitably begin to lose their labile isotopic labels, despite low (but finite) intrinsic exchange rates.19 This phenomenon, known as “back-exchange,” may be accelerated during various stages of protein processing, e.g. during the chromatographic step.20 Although back-exchange was frequently evaluated in early HDX MS studies using unstructured model peptides, the utility of this procedure is questionable, since the intrinsic exchange rates are highly sequence-dependent. In many instances, back-exchange may be estimated using algorithms based on context-specific kinetics data (e.g., http://hx2.med.upenn.edu/download.html); it may also be determined experimentally for each proteolytic fragment by processing a fully labeled protein using a series of steps that precisely reproduce those used in HDX MS measurements.9 Typical back-exchange levels reported in recent literature range from 10% to 50%, although significantly higher numbers have also been reported. Even if back-exchange can be accounted for, it nonetheless has very detrimental influence on the quality of HDX MS measurements by reducing the available dynamic range.
Finally, the classic HDX MS scheme is poorly suited for measurements that are carried out under conditions favoring correlated exchange, when HDX kinetics follows the so-called EX1 regime, leading to appearance of bimodal and convoluted multi-modal isotopic distributions of protein ions.21 Carrying out HDX MS measurements under these conditions provides a unique opportunity to visualize and characterize distinct conformational states, which can be populated either transiently10 or at equilibrium.22 The distinction among such states can be made based on the differences in their deuterium contents. However, proteolysis in solution almost always leads to a loss of correlation between the deuterium content of fragment peptides and specific conformers with distinct levels of backbone protection. Therefore, the classic HDX MS scheme does not allow protein higher order structure and dynamics to be characterized in a conformer-specific fashion.
“Top-down” HDX MS: tandem MS allows protein structure to be probed in the conformer-specific fashion but raises the specter of hydrogen scrambling
The problem of characterizing protein conformation and dynamics in a conformer-specific fashion can be addressed using methods of tandem mass spectrometry (the so-called “top-down” HDX MS). Indeed, replacement of proteolysis in solution with protein ion fragmentation in the gas phase following mass selection of precursor ions provides a means to obtain fragment ions originating from a particular conformer with a specific level of deuterium incorporation. Deuterium content of fragment ions would then provide a measure of local protection patterns, assuming there is no internal re-arrangement of labile hydrogen and deuterium atoms during ion activation (vide infra). Although the idea to use polypeptide ion dissociation in the gas phase as an alternative to proteolysis was originally proposed in early 1990s,23 its implementation for proteins only became possible24 following dramatic improvements in FTMS and hybrid TOF analyzers in the late 1990s.
An example of conformer-specific characterization of protein higher order structure using a top-down HDX MS approach is illustrated in Figure 4. The isotopic profile of a fully deuterated 18 kDa protein wt*-CRABPI is recorded following its brief exposure to the 1H-based exchange buffer. The bimodal appearance of the isotopic distribution of the molecular ion (top trace in Figure 4A) clearly indicates the presence of at least two conformers with different levels of backbone protection. Collisional activation of the entire protein ion population generates a set of fragment ions with convoluted isotopic distributions (top trace in Figure 4B). However, mass selection of precursor ions with a specific level of deuterium content allows the top-down HDX MS measurements to be carried out in a conformation-specific fashion, taking full advantage of the HDX MS ability to detect distinct conformers. For example, selective fragmentation of protein ions representing a highly protected conformation is achieved by mass-selecting a narrow population of intact protein ions with high level of retained deuterium (the blue trace in Figure 4A). Mass-selection and subsequent fragmentation of a narrow population of protein ions with significantly lower deuterium content (the red trace in Figure 4A) generates a set of fragment ions whose isotopic distributions provide information on backbone protection within non-native protein states. For example, the data presented in Figure 4 clearly indicate that the C-terminal segment of the protein represented by the y172+ ions retains significant structure even within the partially unfolded conformers: the amount of retained deuterium atoms reduces by only 30% as a result of switching from the precursor ion from highly protected (blue) to less protected (red). At the same time, selection of the precursor ion has a much more dramatic effect on the protection levels exhibited by the N-terminal segment (represented by the b425+ ion), where more than a two-fold decrease in the amount of retained deuterium atoms is observed. Extending this analysis to other protein fragments may allow detailed backbone protection maps to be created for each protein conformer, provided there is no hydrogen scrambling prior to protein ion fragmentation (vide infra).
Characterization of local dynamics in wt*-CRABP I in a conformer-specific fashion using top-down HDX MS (fully deuterated protein was exposed to 1H2O/CH3CO2N1H4 at pH 3.1 for 10 min; the gray trace at the bottom corresponds to HDX end-point). A: mass selection of precursor ions for subsequent CAD (from top to bottom): broad-band selection of the entire ionic population (not conformer-specific); highly protected conformers; narrow population of less protected conformers; HDX end-point. B: isotopic distributions of two representative fragment ions generated by CAD of precursor ions shown in panel A. Selection of different ion populations as precursor ions for subsequent fragmentation was achieved by varying the width of a mass selection window of a quadrupole filter (Q) in a hybrid quadrupole/time-of-flight mass spectrometer (Qq-TOF MS).
The example shown above illustrates a great promise of top-down HDX MS as a technique uniquely capable of probing structure and dynamics of populations of protein conformers coexisting in solution with high selectivity. Furthermore, this approach often allows one to avoid protein handling under the slow exchange conditions prior to MS analysis, thereby eliminating back-exchange as a factor adversely influencing the quality of measurements. Nonetheless, applications of top-down HDX MS have been limited due to concerns over the possibility of hydrogen scrambling accompanying collision-activated dissociation (CAD) of protein ions. Indeed, several reports pointed out that proton mobility in the gas phase may under certain conditions influence the outcome of top-down HDX MS measurements when CAD is employed to fragment protein ions.25, 26
The occurrence (or the absence) of hydrogen scrambling in the gas phase can be reliably detected by using built-in scrambling indicators. One particularly convenient indicator is a Histag, a 6-30 residues long, histidine-rich segment appended to wild-type sequences to facilitate protein purification on metal affinity columns. Such segments are fully unstructured in solution and, therefore, should lack any backbone protection.27 Alternatively, intrinsic scrambling indicators (e.g., internal flexible loops26), as well as other approaches25 can be used to detect occurrence of scrambling. The available experimental evidence suggests that slow protein ion activation (e.g., SORI CAD) always leads to hydrogen scrambling, while fast activation allows it to be minimized or eliminated in top-down HDX MS experiments.26
Another shortcoming of top-down HDX MS schemes utilizing CAD is the limited extent of protein ion fragmentation, which may lead to sizeable gaps in sequence coverage, particularly for larger proteins,28 and insufficient level of spatial resolution (even for smaller proteins29). Our earlier attempts to solve this problem by employing multi-stage CAD (MSn) were unsuccessful due to massive hydrogen scrambling exhibited by the second generation of fragments.
Electron-induced ion fragmentation in top-down schemes: keeping hydrogen scrambling at bay while enhancing sequence coverage and spatial resolution
Some time ago we suggested that the specter of hydrogen scrambling in top-down HDX MS measurements may be alleviated by using non-ergodic fragmentation processes, where dissociation is induced by ion-electron interaction, rather than collisional activation.30 Indeed, the results of earlier work combining hydrogen exchange of polypeptide ions in the gas phase and electron capture dissociation (ECD) were consistent with the notion of intramolecular rearrangement of hydrogen atoms occurring on a slower time scale compared to ion dissociation.31 A recent study demonstrated that the extent of scrambling was indeed negligible when ECD was used as a means to obtain fragment ions in top-down HDX MS characterization of a small protein ubiquitin.32
Our own recent work suggests that hydrogen scrambling can be avoided when top-down HDX MS employs ECD in characterizing higher order structure of larger proteins (approaching 20 kDa), although experimental conditions must be carefully controlled to minimize proton mobility induced by ion-molecule collisions in the ESI interface. The point in question is illustrated in Figure 5, which shows the results of top-down HDX MS analysis of higher order structure of wt*-CRABP I. The protein retains a significant proportion of labile deuterium label following its complete deuteration and then brief exposure to the 1H-based exchange buffer, as indicated by the isotopic distribution of the surviving molecular ions (red and blue traces in Figure 5A). However, the deuterium content of fragment ions derived from the 21-residue long His-tag region of the protein (e.g., c22 in Figure 5B) is indistinguishable from that of the exchange reaction endpoint, as long as moderate ion desolvation conditions are kept in the ESI interface. This clearly signals that hydrogen scrambling does not affect the outcome of local HDX MS measurements. However, once collision-assisted desolvation of protein ions is attempted in the ESI interface, the appearance of isotopic distributions of larger fragment ions derived from the His-tag region (e.g., c22, red trace in Figure 5B) shifts, indicating apparent deuterium retention and signaling the occurrence of limited hydrogen scrambling. We also demonstrated that deuterium distribution across the protein backbone is preserved when another recently introduced fragmentation technique based on cation-electron interactions, electron transfer dissociation (ETD), is used in top-down HDX MS schemes.33
Top-down HDX MS of wt*-CRABP I using ECD of the entire protein ion population (fully deuterated protein was exposed to1H2O/CH3CO2N1H4 at pH 3.5 for varying time periods); the black trace at the bottom of corresponds to HDX end-point). A: isotopic distributions of surviving intact protein ions. B: two representative c-ions. Minimal collision-and temperature-induced desolvation was used for acquisition of all mass spectra, except the one top (red trace).
In addition to allowing scrambling to be easily eliminated in top-down HDX MS experiments, both ECD and ETD appear to be superior to CAD in terms of sequence coverage, at least for the proteins in the 20 kDa range. Unlike CAD, protein backbone cleavage in ECD and ETD is less specific,34 leading to a higher number of fragment ions. This translates not only to improved sequence coverage, but also enhanced spatial resolution. Indeed, in some cases it becomes possible to generate patterns of deuterium distribution across the protein backbone down to the single residue level.
One example of such work is shown in Figure 6, where ETD was used as a protein ion fragmentation tool in top-down HDX MS characterization of a 16 kDa variant of CRABP I. The bar graph shows the levels of deuterium retention in a series of c-ions derived from the N-terminal segment of the protein. The bar height at position n in this diagram shows mass difference between two cn-1 fragments, one derived from the fully deuterated protein that was exposed to the protiated exchange buffer at pH 7 for 5 min and then placed under the slow exchange conditions for the duration of the data acquisition cycle, and another one representing the HDX endpoint (raw data for bars at n=14 and 35 are shown in Figure 7). Unchanged height between two adjacent bars at residues n and n+1 indicates no difference in deuterium content of cn-1 and cn fragments, signaling no backbone amide deuterium retention at residue n+1, while bar height increase by one unit indicates complete retention of deuterium at the nth amide.
Backbone protection pattern of CRABPI mutant (without N-terminal His-tag) obtained from top-down HDX MS measurements using ETD of the entire protein ion population. HDX was initiated by exposing the fully deuterated protein to 1H2O/CH3CO2N1H4 at pH 3.5 for 5 min followed by rapid quenching.
An example of raw HDX MS data used to generate the protection plot shown in Figure 6. Isotopic distributions of c13 and c34 fragments derived from protein subjected to 5 min HDX exchange in solution (red trace) and protein at the HDX end-point (blue trace) were used to calculate the bar heights at n=12 and 35.
The resulting backbone protection pattern in Figure 6 shows clear correlation with the known higher order structure of the protein (the amino acid sequence and the secondary structure assignment are shown at the top of the graph). Furthermore, the diagram clearly shows uneven distribution of backbone protection even within single structural elements (e.g., lower protection at the fringes vs. the middle of helix α1), as well as unequal protection of similar structural elements participating in the same structural motif (e.g., lower protection of helix α2 vs. helix α1, consistent with the available NMR data). A comparable level of spatial resolution can be achieved with ECD, as shown recently in top-down HDX MS analysis of higher order structure of myoglobin.35
The ability to characterize protein conformation and dynamics at the single residue level is certainly very exciting; however, it comes at a price. Since the protein fragmentation is carried out entirely in the gas phase, no fragment separation can be done prior to mass analysis. A large number of fragment ions with different masses and charges are usually confined to a relatively narrow m/z region, leading to inevitable overlaps of fragment ion isotopic distributions (Figure 7). This places rather stringent requirements on the resolving power of the mass analyzer, effectively narrowing the selection of mass spectrometers suitable for this work to FTMS.
Meeting in the middle: integration of top-down strategies into bottom-up HDX MS schemes
The top-down approach to HDX MS measurements clearly shows a promise to solve many problems that mar the commonly employed bottom-up methodology. The fragmentation efficiency afforded by ECD and ETD provides better spatial resolution, at least for proteins in the 20 kDa range, and this number is likely to grow as there are numerous examples of successful use of these fragmentation techniques to obtain sequence information on significantly larger proteins.36 Unlike the classic bottom-up approach, top-down HDX MS provides an elegant solution to the problem of characterizing higher order structure and dynamics in a conformer-specific fashion (see Figure 4 and discussion in the text). Finally, back-exchange can be eliminated, as outsourcing protein fragmentation to the gas phase often eliminates the need to manipulate the protein in solution under the slow exchange conditions prior to MS analysis.
The top-down/bottom-up dichotomy in HDX MS should not be viewed through the “eitheror” prism. In fact, gas phase fragmentation can enhance the quality of HDX MS data derived from experiments that are built around the bottom-up approach. The suggestion to supplement proteolysis in solution with peptide ion fragmentation in the gas phase to achieve better spatial resolution was made over 10 years ago.37 However, earlier attempts to implement this idea using CAD on a variety of platforms yielded mixed results due to apparent scrambling in some (but not all) fragment ions.37, 38 Later reports showed even more extensive scrambling in small peptide ions subjected to collisional activation,39 an obvious anathema to the proposed marriage of CAD and bottom-up HDX MS. Nonetheless, continued search for a scrambling-free solution to this problem has yielded very encouraging results, with both ECD and ETD showing minimal scrambling when applied to short peptides under carefully controlled conditions40, 41 and feasibility of supplementing proteolytic fragmentation in solution with ETD in the gas phase was recently demonstrated using a small model protein.42 Although these initial steps are relatively modest, they certainly warrant further work in this field.
The two complementary approaches to HDX MS measurements share a set of common challenges that inevitably arise as these techniques gain popularity and the scope of their applications expands. One such challenge is presented by membrane proteins, a notoriously difficult class of biological objects. HDX MS has been shown to have a great potential in this field.43 Interestingly, some initial work in this field was done nearly ten years ago using then-infant top-down HDX MS technique,44 while more recent work in this field utilizes both bottomup18 and top-down45 approaches. Another challenge faced by HDX MS is presented by highly heterogeneous proteins, such as proteins conjugated to other biopolymers and/or synthetic polymers, which constitute a significant fraction of the next generation of biopharmaceuticals. Presently, there are no biophysical techniques capable of characterizing conformation and dynamics of these systems, and there is an urgent need to fill this gap. Finally, nearly all HDX MS work reported to date was carried out in vitro under conditions that some regard as “reductionist.” Although initial HDX work with living objects was carried out over 75 years ago,46 as the years passed only one report on in vivo HDX MS studies was published.47 As mass spectrometry at large is being increasingly used in both in vivo and ex vivo studies, there is a growing pressure on HDX MS to follow the trend, although it remains to be seen how this will be done.
It probably is not an exaggeration to say that we are witnessing a renaissance of HDX MS, with the emergence of the top-down approach not only expanding our experimental arsenal by offering new capabilities, but also serving as a catalyst in enhancing the classic bottom-up methodology. The two techniques are highly complementary, and their synergism will certainly bring about new exciting discoveries and accelerate our progress in solving a variety of problems ranging from very fundamental questions in biophysics to applied problems in drug design.
WATCH OUT FOR DISULFIDES: If you’re going to try bottom-up HDX experiments, be careful of disulfide bonds, Kaltashov says. Pepsin is one of the very few proteinases that can efficiently digest a protein into its composite peptides under HDX experimental conditions, but it struggles when multiple disulfide bonds are present. In 2014, Kaltashov’s lab published two solutions to that problem. The first employs a fragmentation technique called electron capture dissociation (ECD) to break the disulfide linkage in the mass spec (Anal Chem, 86:5225-31, 2014); the second skips the pepsin digestion altogether—a strategy called top-down analysis (Anal Chem, 86:7293-98, 2014).
Enhancing the Quality of H/D Exchange Measurements with Mass Spectrometry Detection in Disulfide-Rich Proteins Using Electron Capture Dissociation
Hydrogen/deuterium exchange (HDX) mass spectrometry (MS) has become a potent technique to probe higher-order structures, dynamics, and interactions of proteins. While the range of proteins amenable to interrogation by HDX MS continues to expand at an accelerating pace, there are still a few classes of proteins whose analysis with this technique remains challenging. Disulfide-rich proteins constitute one of such groups: since the reduction of thiol–thiol bonds must be carried out under suboptimal conditions (to minimize the back-exchange), it frequently results in incomplete dissociation of disulfide bridges prior to MS analysis, leading to a loss of signal, inadequate sequence coverage, and a dramatic increase in the difficulty of data analysis. In this work, the dissociation of disulfide-linked peptide dimers produced by peptic digestion of the 80 kDa glycoprotein transferrin in the course of HDX MS experiments is carried out using electron capture dissociation (ECD). ECD results in efficient cleavage of the thiol–thiol bonds in the gas phase on the fast LC time scale and allows the deuterium content of the monomeric constituents of the peptide dimers to be measured individually. The measurements appear to be unaffected by hydrogen scrambling, even when high collisional energies are utilized. This technique will benefit HDX MS measurements for any protein that contains one or more disulfides and the potential gain in sequence coverage and spatial resolution would increase with disulfide bond number.
———
Hydrogen/deuterium exchange (HDX) with mass spectrometry (MS) detection has evolved in the past two decades into a powerful tool that is now used to decipher intimate details of processes as diverse as protein folding, recognition and binding, and enzyme catalysis.1,2 While initially being a tool that was used exclusively in fundamental studies, HDX MS is now becoming an indispensable part of the analytical arsenal in the biopharmaceutical sector, where it is utilized increasingly in all stages of protein drug development from discovery to quality control.3−5 Despite this progress, several areas remain where the application of HDX MS has met with only limited success. Disulfide-rich proteins constitute one such group, where characterization of higher-order structure and dynamics is particularly difficult, because of the suboptimal conditions used for reduction of thiol–thiol bonds following a quench of the exchange reactions. Proteins containing disulfide bonds are encountered very rarely in the protein folding studies where the most popular targets are small proteins lacking cysteine residues (with a notable exception of the oxidative folding studies), as well as in many other fundamental studies focusing on proteins of prokaryotic origin. However, disulfide-rich proteins are encountered very frequently in eukaryotic proteomes6 and constitute a large segment of the biopharmaceutical products,7 where the thiol–thiol bonds are critical elements defining conformation of protein drugs, and also play an important role in stabilizing proteins by endowing them with protease resistance.
While disulfide bond reduction is a relatively trivial task that can be readily accomplished at neutral pH using a variety of reagents, the acidic, low-temperature environment where proteins are placed to quench HDX narrows down the choice to a single reducing agent, TCEP.8 However, the alkaline pH for optimal disulfide reduction by TCEP is substantially higher, compared to the acidic environment of typical “slow exchange conditions” commonly employed to minimize back exchange within proteins and their peptic fragments prior to MS analysis.9 Furthermore, disulfide reduction in HDX MS measurements is usually carried out within a relatively short period of time (a few minutes) and at low temperature (0–4 °C) to limit the extent of the back-exchange, which in many situations does not allow the complete dissociation of thiol–thiol linkages of individual peptic fragments to be achieved in solution prior to LC separation and MS analysis of their deuterium content. Incomplete reduction of disulfide bonds dramatically increases the pool of candidate peptides that should be considered when analyzing proteolytic fragments in HDX MS measurements and frequently reduces sequence coverage and/or spatial resolution. While the former problem can be solved by employing more powerful and robust search engines for peptide identification, the latter one is more difficult to circumvent and can be very detrimental for the quality of HDX MS data and may require significant changes in experimental protocols. Indeed, a complete failure to reduce a certain disulfide bond in a protein will give rise to a thiol–thiol linked peptide dimer, whose constituent monomers do not necessarily represent a contiguous segment of the protein and may have vastly different conformational and dynamic properties. The total deuterium content of the entire dimer (measured by HDX MS) would not provide any meaningful information under these conditions, thereby effectively reducing the sequence coverage in the corresponding segments of the protein.
———-
Disulfide-rich proteins have traditionally been challenging targets for HDX MS studies, because of incomplete reduction of thiol–thiol linkages, which is a consequence of the quench conditions used to minimize amide back-exchange in peptides prior to MS analysis of their deuterium content: limited time, low temperature, and low pH. Traditionally, the principal strategy to address difficult-to-reduce or high-density disulfides in the HDX MS workflow is a brute force approach utilizing high concentrations of reductant and denaturant prior to (or even in combination with) digestion. The effectiveness of this approach is protein-dependent and extended incubation times frequently employed to enhance exposure to reductant invariably result in an undesirable increase in H/D back exchange. More recently, a novel electrochemical approach to reduce disulfides in solution under quench conditions prior to LC-MS has been reported for insulin.32 While electrochemical reduction shows promise, several limitations were identified, an apparent requirement for low-salt conditions, a higher-than-optimal temperature (10 °C), and a current cell pressure limit of 50 bar. In this work, electron capture dissociation (ECD) was used to circumvent the disulfide problem, since it effectively cleaves external disulfide bonds. Dissociation of the disulfide-linked peptide dimers can be accomplished on the fast LC time scale and produces abundant signals for monomeric subunits without interchain hydrogen scrambling, even when collisional activation of ions is applied prior to ion selection and ECD fragmentation. Inclusion of ECD in the HDX MS workflow results in increased sequence coverage and spatial resolution and provides an attractive alternative to extensive chemical reduction of disulfide-rich proteins.
Approach to Characterization of the Higher Order Structure of Disulfide-Containing Proteins Using Hydrogen/Deuterium Exchange and Top-Down Mass Spectrometry
Top-down hydrogen/deuterium exchange (HDX) with mass spectrometric (MS) detection has recently matured to become a potent biophysical tool capable of providing valuable information on higher order structure and conformational dynamics of proteins at an unprecedented level of structural detail. However, the scope of the proteins amenable to the analysis by top-down HDX MS still remains limited, with the protein size and the presence of disulfide bonds being the two most important limiting factors. While the limitations imposed by the physical size of the proteins gradually become more relaxed as the sensitivity, resolution and dynamic range of modern MS instrumentation continue to improve at an ever accelerating pace, the presence of the disulfide linkages remains a much less forgiving limitation even for the proteins of relatively modest size. To circumvent this problem, we introduce an online chemical reduction step following completion and quenching of the HDX reactions and prior to the top-down MS measurements of deuterium occupancy of individual backbone amides. Application of the new methodology to the top-down HDX MS characterization of a small (99 residue long) disulfide-containing protein β2- microglobulin allowed the backbone amide protection to be probed with nearly a single-residue resolution across the entire sequence. The high-resolution backbone protection pattern deduced from the top-down HDX MS measurements carried out under native conditions is in excellent agreement with the crystal structure of the protein and high-resolution NMR data, suggesting that introduction of the chemical reduction step to the top-down routine does not trigger hydrogen scrambling either during the electrospray ionization process or in the gas phase prior to the protein ion dissociation.
Since its initial introduction in the late 1990s,1−3 top-down hydrogen/deuterium exchange (HDX) with mass spectrometric (MS) detection evolved to become a potent biophysical tool capable of providing valuable information on higher order structure and conformational dynamics of proteins at an unprecedented level of structural detail. Among the many advantages offered by top-down HDX MS compared to conventional (bottom-up) measurements are significant reduction or indeed complete elimination of the back exchange,4 high spatial resolution,5,6 and the ability to study conformational dynamics in the conformer-specific fashion.7,8 However, despite the spectacular recent advances and the broader acceptance of this technique, the scope of the proteins amenable to the analysis by top-down HDX MS remains limited, with the protein size and the presence of disulfide bonds being the two most important limiting factors. The limitations imposed by the physical size of the proteins gradually become more relaxed as the sensitivity, resolution, and dynamic range of modern MS instrumentation continue to improve at an ever accelerating pace. However, the presence of disulfides remains a much less forgiving limitation even for the proteins of relatively modest size.
In this work we demonstrated feasibility of applying top-down HDX MS measurements to characterize higher order structure and conformational dynamics of disulfide-containing proteins, which have been out of the reach of this technique so far. Use of a moderate amount of a reducing agent TCEP is compatible with the ESI process, while allowing a fraction of the protein molecules to be reduced in solution thereby enabling nearcomplete sequence coverage at high resolution. The agreement between the top-down HDX MS and NMR data sets demonstrate that the new experimental approach is capable of capturing the dynamic picture of protein conformation at high spatial resolution without compromising the quality of the data by triggering hydrogen scrambling in the gas phase. Despite its modest size, β2m is known to be able to populate a non-native state,35 which might be a key player in a variety of processes, including amyloidosis. However, the structure of this non-native state of β2m remains elusive since this conformer exists in dynamic equilibrium with the native state of the protein.36,37 Recently we demonstrated that top-down HDX MS provides an elegant way to selectively probe structure of protein states coexisting in solution at equilibrium;8 however, β2m remained out of reach of this technique until recently due to the presence of a disulfide bond. The ability to expand the scope of top-down HDX MS to disulfide-containing proteins opens up a host of exciting possibilities to explore the structure of β2m, interferon, lysozyme, and a variety of other disulfidecontaining proteins in a conformer-specific fashion, where physiologically important non-native states may play important roles in processes as diverse as folding, recognition, signaling, and amyloidosis. ■ ASSOCIATED CONTENT *S Supporting Information Representative examples of isotopic distributions of fragment ions that have (Supplementary Figure 1) and have not (Supplementary Figure 2) been used to calculate the deuterium occupancy at individual backbone amides of β2m in top-down HDX MS measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
SUSSING OUT THE SURFACE: Protein topology can be probed by firing low-energy electrons (white circles) at intact protein complexes within a high-resolution mass spectrometer. That reaction, called electron capture dissociation, causes the protein complex to fracture on its surface, revealing the exposed amino acid residues. COURTESY OF PIRIYA WONGKONGKATHEP AND HUILIN LI, UCLA
RESEARCHER:Joseph Loo, Professor of Biological Chemistry, David Geffen School of Medicine, University of California, Los Angeles)
PROJECT: Studying protein-ligand and protein-protein interactions
SOLUTION: Loo is less interested in complex identification than in how the protein subunits assemble. Specifically, he wants to know which amino acid residues lie on the complex’s surface and which are buried inside or interacting with ligands.
It’s a question of structural biology, he explains: “How is this thing folded in a way that these residues are on the outside?”
To work that out, Loo combines high-resolution Fourier transform ion cyclotron resonance mass spectrometry (FTICR) with electron-capture dissociation (ECD), a mass spec fragmentation method in which an ion in the mass spectrometer interacts with free electrons, causing the protein to fracture along its peptide backbone. By measuring the mass of those fragments with high precision, researchers can determine the protein’s amino acid sequence.
In Loo’s case, though, that fragmentation is not uniform along the length of the protein. Proteins usually are denatured for mass spectrometry analysis, but the protein complexes in his studies are intact—a process called native mass spectrometry. Fragmentation thus occurs preferentially on the surface of the complex, like the cracks in the shell of a hard-boiled egg. “You get limited sequence information, but that sequence information comes from regions that are specific to its 3-D structure,” he says (Anal Chem, 86:317-20, 2014).
Native Top-Down ESI-MS of 158 kDa Protein Complex by High Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry
Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) delivers high resolving power, mass measurement accuracy, and the capabilities for unambiguously sequencing by a top-down MS approach. Here, we report isotopic resolution of a 158 kDa protein complex – tetrameric aldolase with an average absolute deviation of 0.36 ppm and an average resolving power of ~520,000 at m/z 6033 for the 26+ charge state in magnitude mode. Phase correction further improves the resolving power and average absolute deviation by 1.3 fold. Furthermore, native top-down electron capture dissociation (ECD) enables the sequencing of 149 C-terminal amino acid (AA) residues out of 463 total AAs. Combining the data from top-down MS of native and denatured aldolase complexes, a total of 58% of the backbone cleavages efficiency is achieved. The observation of complementary product ion pairs confirms the correctness of the sequence and also the accuracy of the mass fitting of the isotopic distribution of the aldolase tetramer. Top-down MS of the native protein provides complementary sequence information to top-down ECD and CAD MS of the denatured protein. Moreover, native top-down ECD of aldolase tetramer reveals that ECD fragmentation is not limited only to the flexible regions of protein complexes and that regions located on the surface topology are prone to ECD cleavage.
“Native” mass spectrometry (MS) is an emerging technique that has been successfully used to characterize intact, noncovalently-bound protein complexes, providing stoichiometry and structural information that is complementary to data supplied by conventional structural biology techniques.1–3 To confidently characterize protein complexes, electrospray ionization (ESI)-MS measurements acquired with isotopic resolving power (RP) and high mass accuracy and capabilities for deriving primary structure, i.e., sequence, information would be ideal. Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) is prominent for its superior resolving power and mass accuracy and its utility for tandem MS (MS/MS) with a variety of fragmentation techniques; FT-ICR MS is noted for characterizating posttranslational modifications (PTMs) and protein-ligand and protein-protein interactions.4–9 However, it remains challenging to isotopically resolving large biomolecules over 100 kDa due to sample heterogeneity, cation/solvent/buffer addition, space charge effects, and electric and magnetic field inhomogeneity (for FT-ICR).10–13 Unit mass resolution has been achieved for a few denatured proteins, including a 112 kDa protein with 3 Da mass error using a 9.4 T FT-ICR MS,14 a 115 kDa protein by a 7 T instrument with a mass error of 5 ppm,4 and a 148 kDa protein with a mass error of 1 Da by a 9.4 T FTMS.10
Compared to denatured proteins, it is more difficult to achieve isotopic resolution for inherently lower charged (and thus, higher m/z) native protein complexes because (1) the peak height is proportional to its charge state, (2) the resolving power is inversely proportional to mass-to-charge ratio for FT-ICR MS, and (3) the broader isotope distribution of large biomolecules reduces overall signal-to-noise ratio.15 However, the introduction of a new FT-ICR analyzer cell – the ParaCell, by Nikolaev and coworkers has significantly increased the resolving power of FT-ICR MS.16, 17 By dynamically harmonizing the electric field potential at any radius of cyclotron motion in the entire cell volume, a resolving power of 39 M has been achieved for the alkaloid, resperine (m/z 609), using a 7 T system.18 In addition, a few native protein complexes, including enolase dimer (93 kDa, RP ~ 800,000 at m/z 4250), alcohol dehydrogenase tetramer (147 kDa, RP ~ 500,000 at m/z 5465), and enolase tetramer (186 kDa), have been isotopically resolved with a 12 T FT-ICR system with the new ICR cell.18 Although Mitchell and Smith reported that cyclotron phase locking due to Coulombic interactions limits the highest mass that unit mass resolution can be achieved by FT-ICR MS (Mmax ≈ 1×104B, where B is magnetic field strength),19 the ParaCell has made it significantly easier and promising to measure high resolution mass spectra for large native protein complexes.
……
Native top-down CAD and ISD were performed for the aldolase tetramer; dissociation of the tetramer to yield monomer was observed in both approaches and no sequence information was obtained. The cleavage sites from ECD (colored in red) and CAD (colored in green) of the denatured aldolase monomer (26+) are overlaid with the native ECD results for aldolase tetramer (Figure 2B). As shown in Figure 2B, in contrast to the limited number of c-ion fragments observed in the ECD of aldolase tetramer, ECD of denatured aldolase monomer induces extensive c-ion fragments in the N-terminal region and enables the assignment of first 156 N-terminal AA residues. Surprisingly, the number of z•-ions observed from ECD of the denatured aldolase monomer is much less compared to the ECD of the native aldolase tetramer. Although it may be possible that the z•-ions may undergo secondary fragmentation due to excess available energy, electrons, or long ion-electron reaction times during the ECD experiment, ECD experiments with reduced reaction time and bias voltages were performed and the results argue against this assumption. Overall, 58% of the total number of backbone bonds are cleaved from combining top-down MS of native aldolase complex and denatured aldolase monomer (20% for native ECD of aldolase tetramer, 37% for ECD of denatured aldolase, and 5% for CAD of denatured aldolase).
The three dimensional structure of the aldolase tetramer is shown in Figure 3. To compare the flexibility of the structure to the data from ECD of the aldolase tetramer, one of the subunits (B-chain) is presented as B-factor putty and the D-chain is shown with its native ECD backbone cleavage regions colored in red. The remaining A- and C-chains are shown in grey. Although the C-terminal region (AA 340–363) of each subunit is highly flexible based on the crystallography B-factor (see B-chain in Figure 3A), only 4 out of 75 backbone cleavage sites are from the AA 340–363 region. Instead, the native ECD fragments largely originate from surface regions of the protein structure (see D-chain in Figure 3A). The N-terminal regions are not directly involved in the interfaces between subunits, but they are located in regions that are partially buried, which is consistent with the limited c-ions observed. To better show the native ECD backbone cleavage regions, the D-chain is rotated 90 degrees clockwise (Figure 3B). It is clear that, although protein structure flexibility might play a role in the native top-down ECD fragmentation pattern, for aldolase the ECD cleavage sites are not limited to the flexible region. In addition, backbone cleavage regions from CAD (yellow) and ECD (cyan) of denatured aldolase are complementary with the native ECD results.
A) Structure of tetrameric aldolase (1ZAH)29. A- and C-chains are shown as grey ribbons, the B-chain is shown in B-factor putty, and the D-chain is in cartoon with native ECD cleavage sites colored in red, CAD cleavage sites of denatured aldolase in yellow, and ECD cleavage sites of the N-terminal region from ECD of denatured aldolase in cyan. B) The D-chain is rotated 90 degrees clockwise to show the outer surface region of the subunit structure.
Also evident in such data sets are protein–small molecule interactions. As the proteins break apart, Loo explains, ligands often remain attached to the polypeptide shards that are produced. In one recent publication, for instance, his team mapped zinc binding sites in eukaryotic alcohol dehydrogenase, a 147-kDa tetrameric complex (J Am Soc Mass Spectrom, 25:2060-8, 2014).
Revealing Ligand Binding Sites and Quantifying Subunit Variants of Non-Covalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment
“Native” mass spectrometry (MS) has been proven increasingly useful for structural biology studies of macromolecular assemblies. Using horse liver alcohol dehydrogenase (hADH) and yeast alcohol dehydrogenase (yADH) as examples, we demonstrate that rich information can be obtained in a single native top-down MS experiment using Fourier transform ion cyclotron mass spectrometry (FTICR MS). Beyond measuring the molecular weights of the protein complexes, isotopic mass resolution was achieved for yeast ADH tetramer (147 kDa) with an average resolving power of 412,700 at m/z 5466 in absorption mode and the mass reflects that each subunit binds to two zinc atoms. The N-terminal 89 amino acid residues were sequenced in a top-down electron capture dissociation (ECD) experiment, along with the identifications of the zinc binding site at Cys46 and a point mutation (V58T). With the combination of various activation/dissociation techniques, including ECD, in-source dissociation (ISD), collisionally activated dissociation (CAD), and infrared multiphoton dissociation (IRMPD), 40% of the yADH sequence was derived directly from the native tetramer complex. For hADH, native top-down ECD-MS shows that both E and S subunits are present in the hADH sample, with a relative ratio of 4:1. Native top-down ISD MS hADH dimer shows that each subunit (E and S chain) binds not only to two zinc atoms, but also the NAD+/NADH ligand, with a higher NAD+/NADH binding preference for the S chain relative to the E chain. In total, 32% sequence coverage was achieved for both E and S chains.
Studying how proteins interact with one another and assemble on a structural basis is key to understanding biological processes and their function. As a complementary technique to conventional technologies used in structural biology, such as nuclear magnetic resonance (NMR) spectroscopy, X-ray crystallography, and electron microscopy, “native” mass spectrometry (MS) has established its crucial role in the characterization of intact noncovalently-bound protein complexes, revealing the composition, stoichiometry, dynamics, stability, and also spatial information of subunit arrangements in protein assemblies [1–11]. To date, most native MS studies of protein complexes have been performed using quadrupole time-of-flight (Q-TOF) MS instruments with electrospray ionization (ESI). Because of the efficient transmission of high mass and highm/z ions using TOF analyzers, large proteins with molecular weights up to 18 MDa have been studied [12,13]. The coupling of ion mobility spectrometry (IMS) with mass spectrometry provides a new dimension to the analysis of biomolecules [14]. With IMS, ions are separated based on size and shape, and the IMS-derived collision cross-section information can be used to understand the topological properties of gas phase protein complexes. Surface induced dissociation (SID) has been recently added for the purposes of disassembling protein complexes into sub-complexes that appear to better reflect the structure of the solution phase complexes [15–17]. The capability of Orbitrap MS has been extended significantly for the analysis of macromolecules, with greatly improved mass (and m/z) range and resolving power to measure the binding of ADP and ATP to the 800 kDa GroEL complex [18].
Fourier transform ion cyclotron resonance mass spectrometry (FTICR MS) is known for its superior resolving power and mass accuracy and its capabilities for tandem MS (MS/MS) with a variety of fragmentation techniques. Particularly, after the introduction of electron capture dissociation (ECD) [19], FTICR MS quickly established its utility for protein top-down protein sequencing, post-translational modification characterization, and protein gas phase studies [20–34]. Polypeptide backbone bonds are cleaved by ECD, but non-covalent interactions are preserved, which therefore makes the native top-down MS study of the non-covalent interaction sites of protein-ligands complexes more feasible. Our group and others have successfully applied top-down ECD-MS to pinpoint the interaction sites of several protein-ligand system [35–38], and this can be enhanced by “supercharging” [35]. An early attempt of applying ECD-MS to the study of large protein complexes was made by Heeren and Heck, but little topology and sequence information was derived [39]. However, the Gross group starting in 2010 made the first breakthrough for the study of large protein complexes using native top-down ECD with FTICR MS. Besides obtaining molecular weight, sequence, and metal-binding site information in a single MS experiment, they correlated the origins of ECD product ions to the flexible regions of proteins as determined by the “B-factor” from the X-ray crystal structures of protein complexes [40, 41]. Therefore, native top-down ECD has been proposed as a tool to probe the flexible regions of protein complexes. Our group recently also demonstrated the capability of obtaining sequence information and isotopic mass resolution of a noncovalently-bound protein complex of 158 kDa using native top-down FTICR MS, and most importantly, we found that the origin of ECD fragments is not limited only to the flexible region of the protein complex (e.g., tetrameric aldolase), but also largely from the surface of the complex [42].
The application of FTICR MS for native top-down interrogation of large non-covalent bound protein complexes is still in its infancy. Here, for the purpose of further exploring the capability of FTICR MS in the analysis of large protein complexes, various fragmentation techniques including in-source dissociation (ISD), collisionally activated dissociation (CAD), ECD, and infrared multiphoton dissociation (IRMPD) were applied in the native top-down MS studies of a 80 kDa dimeric protein complex and a 147 kDa tetrameric protein complex. The results demonstrate that with the superior resolving power, mass accuracy, and versatile fragmentation techniques of FTICR MS, rich information, including isotopic mass resolution, amino acid sequence, point mutations, metal/ligand binding sites, and identification and quantification of subunit variants can be accomplished in a single native top-down FTICR MS experiment.
Still, Loo admits, the technique “is not really ready for prime time.” His team is collecting ECD data on a bank of proteins of known structure to ensure the data they collect really do reflect protein topology. In the meantime, they are working to extend the size of the complexes they can analyze. The technique’s current limit is 800 kDa.
GO NATIONAL: FTICR mass spectrometers offer top-of-the-line accuracy and resolution, with price tags to match. Few researchers have direct access to them, Loo says, but they can always try the national laboratories. Both the National High Magnetic Field Laboratory at Florida State University and the Environmental Molecular Sciences Laboratory at the Pacific Northwest National Laboratory have user facilities open to worthy projects.
Determining the architecture of protein complexes
RESEARCHER: Vicki Wysocki, Ohio Eminent Scholar and Professor of Chemistry and Biochemistry, Ohio State University
PROJECT: Instrumentation development for whole-complex analysis
SOLUTION: An analytical chemist by training, Wysocki focuses on instrumentation development for protein-complex analysis. Among the discoveries in her lab is a method called surface-induced dissociation (SID).
HIT THE WALL, JACK: When it comes to molecular collision in a mass spectrometer, size matters. Collide a complex with small gas molecules, and proteins in the complex will simply unravel (top). By smacking them into a “wall”—a process called surface-induced dissociation—the complex dissociates to reveal its underlying architecture. COURTESY OF VICKI WYSOCKI
Like many other fragmentation approaches, SID works by forcing an ion in the mass spectrometer to collide with another object. Usually that object is a small gas molecule, with the energy of collision sufficient to crack the peptide backbone. But for large protein complexes, bigger is better, and the collision partner in SID is as big as it can get: the method slams protein ions of interest into a nonreactive surface inside the instrument—essentially, a wall—causing complexes to fracture into subcomplexes that reveal the assembly’s inner architecture.
Wysocki combined this approach with ion-mobility separation—a kind of gas-phase electrophoresis that resolves molecules by their size and shape—to dissect an enzyme involved in antibiotic production. The enzyme, they found, has two copies each of three subunits, alpha, beta, and gamma, arranged as a pair of triads sitting on top of one another, with the alpha and beta subunits of one triad linked more tightly to each other than either is to gamma (Anal Chem, 83:2862-65, 2011).
Such information can be valuable to protein engineers, Wysocki says, especially as this particular complex otherwise falls into a structural biology knowledge gap: “It doesn’t crystallize, and it’s too small for the cryoEM and a little bit large for NMR,” she says. “And so, mass spec turned out to be a great tool.”
Revealing the Quaternary Structure of a Heterogeneous Noncovalent Protein Complex through Surface-Induced Dissociation
As scientists begin to appreciate the extent to which quaternary structure facilitates protein function, determination of the subunit arrangement within noncovalent protein complexes is increasingly important. While native mass spectrometry shows promise for the study of noncovalent complexes, few developments have been made toward the determination of subunit architecture, and no mass spectrometry activation method yields complete topology information. Here, we illustrate the surface-induced dissociation of a heterohexamer, toyocamycin nitrile hydratase, directly into its constituent trimers. We propose that the single-step nature of this activation in combination with high energy deposition allows for dissociation prior to significant unfolding or other large-scale rearrangement. This method can potentially allow for dissociation of a protein complex into subcomplexes, facilitating the mapping of subunit contacts and thus determination of quaternary structure of protein complexes.
The majority of proteins exist and perform their functions as multimers of varing stoichiometries and architecture.1 However, very few methods are available that can provide insights into subunit interactions. Native mass spectrometry (MS) is increasingly being used to study noncovalent protein complexes, as many structural features found in solution may be maintained in the gas phase.2,3 While subunit stoichiometries are readily obtainable by mass measurement alone, the determination of subunit arrangement within protein complexes remains a significant challenge. This is particularly true for heterogeneous complexes with multiple types of subunits. Considerable progress has been made using solution-phase disruption to divide the original protein complex into smaller subcomplexes, which may be readily measured by MS.4,5 The composition of the stable subcomplexes provides insight on the topology of the protein complex. However, MS activation methods used to date have fallen short of providing subunit topology. Here, we present the first evidence for subunit arrangement obtained directly from gas-phase experiments on a heterogeneous complex via surfaceinduced dissociation (SID). We have demonstrated previously the ability of SID to yield unique dissociation pathways for protein complexes, resulting in complementary information to collision-induced dissociation (CID).68 While the SID process is not yet well understood for macromolecules, there is a large body of work concerning SID of small molecules; influential factors such as collision energy, surface composition, and translational-to-vibrational energy conversion have been well-studied.911 The higher effective mass of a surface relative to that of neutral gas atoms used in CID (typically argon) results in significantly higher energy deposited through a single surface collision.9 As SID is a single-collision activation process, rather than activation via thousands of less energetic collisions as in CID, dissociation pathways other than those of the lowest energies become accessible
……
This is the only study to date demonstrating an ion activation method capable of yielding extensive dissociation, as well as the release of intact subcomplexes, thus providing relevant substructure information on a noncovalent, hetero-oligomeric protein complex. The capacity to produce intact, charge-symmetric subcomplexes suggests that dissociation occurs faster than subunit unfolding and that a significant degree of secondary and tertiary structure is maintained up to the point of dissociation and for some period of time afterward. Identification of trimeric substructure in TNH provides insight into a protein with little previous structural characterization and indicates a promising advancement of MS as a tool for structural biology.
Such information can be valuable to protein engineers, Wysocki says, especially as this particular complex otherwise falls into a structural biology knowledge gap: “It doesn’t crystallize, and it’s too small for the cryoEM and a little bit large for NMR,” she says. “And so, mass spec turned out to be a great tool.”
CHOOSE MASS: Mass spec may not be the only method for quickly working out protein structure, but it surely is the fastest, Wysocki says. She recalls one instance when a colleague sent over a complex that his group couldn’t crack. “In one afternoon, my student gave them a prediction of the structure: this one’s a heptamer, with a large subunit sitting atop a hexameric ring.” Even if the experiment doesn’t work, she adds, that fast turnaround time can be a boon, as collaborators can get rapid feedback for tweaking their experimental conditions. “Mass is a great thing.”
In a unique collaboration, University of New South Wales (UNSW), Sydney, and Australian photographer Tamara Dean set out to “show our knowledge seekers in a different light, in their environment. Not in a way the public normally sees them and their work.” They had the ingenious idea to “help take our research out into the world” to showcase scientists working in the elements to address problems like climate change, endangered species, toxic industrial sites and marine pollution.
The result is a compelling photography series that presents a powerful challenge to the stereotype of nerdy researchers working in a lab coats. From studying heat stress on beach sand dunes in Sydney to combining seafaring with mathematical modeling, and from mathematical analysis of colossal fire tornadoes to subterranean sleuthing, the Wild Researchers exhibition transports viewers outside into the landscapes where researchers work. It gives a glimpse of scientists as they really are, in the real-world environments where they acquire data, collect samples and ponder scientific mysteries and discoveries.
“Imagination: it’s not the first word usually associated with research — with science itself — but it’s a vital one,” writes journalist and science writer Ashley Hay in an essay included in the exhibition catalog.
The exhibition’s 12 images showcase university researchers in the field. Most of the 17 researchers featured are working in scientific areas — from mathematics and astronomy to climate change and biotechnology — along with a landscape architect and a philosopher.
Dean’s body of work “powerfully explores the relationship humans have with the natural world,” and she discusses her inspiration for Wild Researchers and how the project brings together her two loves — nature and art making — in a four-minute video. The exhibition was developed and commissioned in 2014 as part of Dean’s UNSW Artist-in-Residency. Her multi-sensory art installation, Here and Now, premiered to critical acclaim at Studio One in February 2015, assisted by the University’s Creative Practice Lab.
The Australian Museum in Sydney is hosting Wild Researchers from November 4 through December 13, 2015. The exhibition also can be viewed on the University of New South Wales (UNSW) Web site at www.wildresearchers.unsw.edu.au.
“I am thrilled that journalist and author Ashley Hay took up our commission of an essay for the exhibition. Just as Tamara has so evocatively conjured up these images, Ashley has worked a different kind of magic. Her essay is a gorgeous meditation on photography, place and science and I commend it to you,” said Denise Knight, Director of Media at UNSW Australia.
Hay observes: “If I could have any piece of knowledge I would ask what these subjects thought about as they sat still — pinned, pressed — as Tamara Dean fixed these richly made images.” Her companion essay is titled “The Fieldwork of Looking and Seeing.”
Hallie Leighton had dense breasts — a fact she discovered only in her late 30s, via a mammogram. She grew up in an Ashkenazi family in New York, pursued a career in writing and worked with organizations promoting peace between Israelis and Arabs. By 2013 she was making a documentary on her father Jan Leighton, an actor who set the record as an actor for appearing in the most roles (2,407 according to the 1985 Guinness Book of World Records). She was never able to complete it. She died that year, at the age of 42.
Every woman in Leighton’s family had breast cancer, so she began getting annual mammograms at 35 — five years earlier than the recommended age. In 2009 the results of Leighton’s mammogram came in as “negative” or “normal”; by 2013 she was bedridden, undergoing her final days of chemotherapy.
When Leighton was first diagnosed in 2010, her doctor told her, “You have breast cancer, and it was there in 2009.” The tumor in Leighton’s breast went undiscovered until it was palpable — and at that point, the cancer was already in stage 4.
“Happygram,” a documentary which exposes some of the shortcomings in mammography, chronicles Leighton’s struggle with cancer and the implications of having dense breasts.
“Most women simply aren’t informed that they have dense breast tissue,” said Leighton’s best friend Julie Marron. She wrote and directed the documentary, which is currently screening at film festivals around the country.
Breast density is defined by the relative amount of fat in relation to the amount of connective and epithelial tissue (tissue that lines blood vessels and cavities). When more than 50% of breast tissue is connective and epithelial tissue, instead of fatty tissue, the breasts are considered dense. Mammography is the only way to determine breast density.
“If you have dense breasts, what looks dense on a mammogram looks the same as a cancer would look. It tends to confuse or confound the physician, and reduces the sensitivity of the mammogram,” said Gerald Kolb, founder and president of The Breast Group, which counsels clients on different technologies in breast care. “Hallie Leighton’s breasts looked like snowballs; there was no chance they were going to find anything with the mammogram.”
Forty percent of women who are screened for breast cancer have dense breast tissue. These women also account for more than 70% of all invasive cancers. “Mammograms are not very effective screening tools for these women, as they miss between 50% and 75% of all invasive cancers in dense breast tissue,” Marron said. “This is obviously a very critical issue when you are dealing with a population that is more likely to develop cancer.”
Ashkenazi women are even more at risk. They are 1.6 times more likely than the general population to have dense breast tissue, according to Kolb. Moreover, one in 40 Ashkenazi women will test positive for one or both of BRCA gene mutations responsible for breast cancer. For the general population, that number is between one in 350 and one in 800.The BRCA 1 or 2 genes don’t cause cancer, they fight cancer, Kolb says. But if the gene is mutated, the body is not as well equipped to fight the cancer.
“A woman with a BRCA mutation has a lifetime risk of around 33% to 87%, depending on the gene and mutation,” Marron said. “Compare this to a lifetime risk of 12% for developing breast cancer for the overall population.” BRCA gene mutations can be inherited from either or both parents, and therefore they can be present in men as well as in women.
Breast density and BRCA gene mutations are not directly related, but both independently present an increased susceptibility to breast cancer.
“The biggest risk is that a doctor is not going to find the cancer when it’s really small,” Kolb said. When a tumor is detected at a centimeter or smaller, there’s a 95% cure rate. But if the cancer is the size of a golf ball by the time it’s detected, Kolb says, the woman has a 60% chance of living for five years, and then her mortality increases dramatically.
The good news is that mammography isn’t the only method of detecting breast cancer; the bad news is that very few people know this. “What we’re trying to do in the density movement is give women enough information so they can ask appropriate questions of a doctor,” Kolb said.
Kolb advises high-risk women to get a genetic risk analysis, which can be performed by a genetic counselor or a radiologist. He advises getting the risk analysis as early as age 25, but doing so is a personal decision. Not every woman is emotionally prepared to know the results.
“Mammography is a starting point,” said Dr. Dennis McDonald, a California-based women’s imager. Additionally, doctors recommend that women with dense breasts get an MRI, which McDonald says is reserved for high-risk women. It’s an expensive, invasive and time-consuming procedure that requires the injection of fluid in order to read the MRI. As of yet, doctors do not know the side effects of getting an annual MRI.
“A doctor should have started [Leighton] on an MRI right away. She was high risk and they chose to just monitor with a mammogram,” Kolb said. “That’s insufficient.”
Breast ultrasound is another alternative for women with dense breast tissue. “Most of the time, breast density doesn’t present a problem [with ultrasounds],” McDonald said. Though the ultrasound is effective in detecting cancer, he says the downside is that radiologists are often not that comfortable with the technology, simply because they have little experience with it. There are also a lot of false positives, he adds, which result in unnecessary exams or biopsies.
As “Happygram” documents, informing women of their breast density and of alternatives to mammography is a highly charged political issue.
“The whole breast cancer industry has grown up around mammograms,” Marron said. “Physicians weren’t educated on [breast density], deliberately so to a certain extent, and refused to inform patients on this issue, which is really outrageous if you think about it.” Marron says that doctors are required by law and ethical guidelines to inform patients of “material” medical information. “There is no legitimate reason that women have not been informed of this information,” she noted.
After Leighton’s diagnosis, she wanted to ensure that other women didn’t suffer the same misfortune of all-too-late tumor discovery on account of dense breast tissue. She gave media interviews, lobbied in Albany and starred in “Happygram,” all the while undergoing chemotherapy. She died four months after the Breast Density Information Bill passed in New York.
Similar legislation has been passed in more than 20 states throughout the country, but not without objection. Many well-intentioned radiologists, poorly informed about alternative screening options, feared that telling women the limitations of mammography would cause them to lose faith in it altogether and not get tested. Others argued that the information would make women anxious, and that it wouldn’t be fair for those who couldn’t afford additional testing. And still further arguments against informing women were possibly impacted by financial considerations, Marron added.
“Women aren’t getting the benefit of full notification across the board yet,” Marron said. “I think that has to change through education. That’s the primary reason we made this movie. There’s been so much resistance within the medical community to telling women. Change isn’t going to come from the medical community, it has to come from the patients.”
Ashkenazi women shouldn’t panic, Kolb says, but they need to carefully examine their breast density and alternative screening options: “Anytime you have a preventative tragedy like that, you have to do everything in your power to stop it from happening.”
Madison Margolin is a freelance writer based in New York. She writes frequently for the Village Voice.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
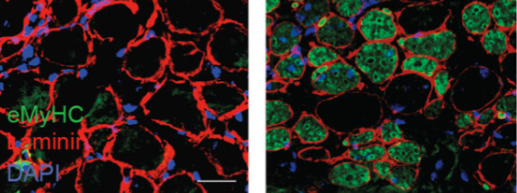{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}