Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
There has always been Personalized Medicine if you consider the time a physician spends with a patient, which has dwindled. But the current recognition of personalized medicine refers to breakthrough advances in technological innovation in diagnostics and treatment that differentiates subclasses within diagnoses that are amenable to relapse eluding therapies. There are just a few highlights to consider:
We live in a world with other living beings that are adapting to a changing environmental stresses.
Nutritional resources that have been available and made plentiful over generations are not abundant in some climates.
Despite the huge impact that genomics has had on biological progress over the last century, there is a huge contribution not to be overlooked in epigenetics, metabolomics, and pathways analysis.
A Reconstructed View of Personalized Medicine
There has been much interest in ‘junk DNA’, non-coding areas of our DNA are far from being without function. DNA has two basic categories of nitrogenous bases: the purines (adenine [A] and guanine [G]), and the pyrimidines (cytosine [C], thymine [T], and no uracil [U]), while RNA contains only A, G, C, and U (no T). The Watson-Crick proposal set the path of molecular biology for decades into the 21st century, culminating in the Human Genome Project.
There is no uncertainty about the importance of “Junk DNA”. It is both an evolutionary remnant, and it has a role in cell regulation. Further, the role of histones in their relationship the oligonucleotide sequences is not understood. We now have a large output of research on noncoding RNA, including siRNA, miRNA, and others with roles other than transcription. This requires major revision of our model of cell regulatory processes. The classic model is solely transcriptional.
DNA-> RNA-> Amino Acid in a protein.
Redrawn we have
DNA-> RNA-> DNA and
DNA->RNA-> protein-> DNA.
Neverthess, there were unrelated discoveries that took on huge importance. For example, since the 1920s, the work of Warburg and Meyerhoff, followed by that of Krebs, Kaplan, Chance, and others built a solid foundation in the knowledge of enzymes, coenzymes, adenine and pyridine nucleotides, and metabolic pathways, not to mention the importance of Fe3+, Cu2+, Zn2+, and other metal cofactors. Of huge importance was the work of Jacob, Monod and Changeux, and the effects of cooperativity in allosteric systems and of repulsion in tertiary structure of proteins related to hydrophobic and hydrophilic interactions, which involves the effect of one ligand on the binding or catalysis of another, demonstrated by the end-product inhibition of the enzyme, L-threonine deaminase (Changeux 1961), L-isoleucine, which differs sterically from the reactant, L-threonine whereby the former could inhibit the enzyme without competing with the latter. The current view based on a variety of measurements (e.g., NMR, FRET, and single molecule studies) is a ‘‘dynamic’’ proposal by Cooper and Dryden (1984) that the distribution around the average structure changes in allostery affects the subsequent (binding) affinity at a distant site.
What else do we have to consider? The measurement of free radicals has increased awareness of radical-induced impairment of the oxidative/antioxidative balance, essential for an understanding of disease progression. Metal-mediated formation of free radicals causes various modifications to DNA bases, enhanced lipid peroxidation, and altered calcium and sulfhydryl homeostasis. Lipid peroxides, formed by the attack of radicals on polyunsaturated fatty acid residues of phospholipids, can further react with redox metals finally producing mutagenic and carcinogenic malondialdehyde, 4-hydroxynonenal and other exocyclic DNA adducts (etheno and/or propano adducts). The unifying factor in determining toxicity and carcinogenicity for all these metals is the generation of reactive oxygen and nitrogen species. Various studies have confirmed that metals activate signaling pathways and the carcinogenic effect of metals has been related to activation of mainly redox sensitive transcription factors, involving NF-kappaB, AP-1 and p53.
I have provided mechanisms explanatory for regulation of the cell that go beyond the classic model of metabolic pathways associated with the cytoplasm, mitochondria, endoplasmic reticulum, and lysosome, such as, the cell death pathways, expressed in apoptosis and repair. Nevertheless, there is still a missing part of this discussion that considers the time and space interactions of the cell, cellular cytoskeleton and extracellular and intracellular substrate interactions in the immediate environment.
There is heterogeneity among cancer cells of expected identical type, which would be consistent with differences in phenotypic expression, aligned with epigenetics. There is also heterogeneity in the immediate interstices between cancer cells. Integration with genome-wide profiling data identified losses of specific genes on 4p14 and 5q13 that were enriched in grade 3 tumors with high microenvironmental diversity that also substratified patients into poor prognostic groups. In the case of breast cancer, there is interaction with estrogen , and we refer to an androgen-unresponsive prostate cancer.
Finally, the interaction between enzyme and substrates may be conditionally unidirectional in defining the activity within the cell. The activity of the cell is dynamically interacting and at high rates of activity. In a study of the pyruvate kinase (PK) reaction the catalytic activity of the PK reaction was reversed to the thermodynamically unfavorable direction in a muscle preparation by a specific inhibitor. Experiments found that in there were differences in the active form of pyruvate kinase that were clearly related to the environmental condition of the assay – glycolitic or glyconeogenic. The conformational changes indicated by differential regulatory response were used to present a dynamic conformational model functioning at the active site of the enzyme. In the model, the interaction of the enzyme active site with its substrates is described concluding that induced increase in the vibrational energy levels of the active site decreases the energetic barrier for substrate induced changes at the site. Another example is the inhibition of H4 lactate dehydrogenase, but not the M4, by high concentrations of pyruvate. An investigation of the inhibition revealed that a covalent bond was formed between the nicotinamide ring of the NAD+ and the enol form of pyruvate. The isoenzymes of isocitrate dehydrogenase, IDH1 and IDH2 mutations occur in gliomas and in acute myeloid leukemias with normal karyotype. IDH1 and IDH2 mutations are remarkably specific to codons that encode conserved functionally important arginines in the active site of each enzyme. In this case, there is steric hindrance by Asp279 where the isocitrate substrate normally forms hydrogen bonds with Ser94.
Personalized medicine has been largely viewed from a lens of genomics. But genomics is only the reading frame. The living activities of cell processes are dynamic and occur at rapid rates. We have to keep in mind that personalized in reference to genotype is not complete without reconciliation of phenotype, which is the reference to expressed differences in outcomes.
A new periodic table presents a systematic, ordered view of protein assembly, providing a visual tool for understanding biological function. [EMBL-EBI / Spencer Phillips]
Move over Mendeleev, there’s a new periodic table in science. Unlike the original periodic table, which organized the chemical elements, the new periodic table organizes protein complexes, or more precisely, quaternary structure topologies. Though there are other differences between the old and new periodic tables, they share at least one important feature—predictive power.
When Mendeleev introduced his periodic table, he predicted that when new chemical elements were discovered, they would fill his table’s blank spots. Analogous predictions are being ventured by the scientific team that assembled the new periodic table. This team, consisting of scientists from the Wellcome Genome Campus and the University of Cambridge, asserts that its periodic table reveals the regions of quaternary structure space that remain to be populated.
The periodic table of protein complexes not only offers a new way of looking at the enormous variety of structures that proteins can build in nature, it also indicates which structures might be discovered next. Moreover, it could point protein engineers toward entirely novel structures that never occurred in nature, but could be engineered.
The new table appeared December 11 in the journal Science, in an article entitled, “Principles of assembly reveal a periodic table of protein complexes.” The “principles of assembly” referenced in this title amount to three basic assembly types: dimerization, cyclization, and heteromeric subunit addition. In dimerization, one protein complex subunit doubles, and becomes two; in cyclization, protein complex subunits from a ring of three or more; and in heteromeric subunit addition, two different proteins bind to each other.
These steps, repeated in different combinations, gives rise to enormous number of proteins of different kinds. “Evolution has given rise to a huge variety of protein complexes, and it can seem a bit chaotic,” explained Joe Marsh, Ph.D., formerly of the Wellcome Genome Campus and now of the MRC Human Genetics Unit at the University of Edinburgh. “But if you break down the steps proteins take to become complexes, there are some basic rules that can explain almost all of the assemblies people have observed so far.”
The authors of the Science article noted that many protein complexes assemble spontaneously via ordered pathways in vitro, and these pathways have a strong tendency to be evolutionarily conserved. “[There] are strong similarities,” the authors added, “between protein complex assembly and evolutionary pathways, with assembly pathways often being reflective of evolutionary histories, and vice versa. This suggests that it may be useful to consider the types of protein complexes that have evolved from the perspective of what assembly pathways are possible.”
To explore this rationale, the authors examined the fundamental steps by which protein complexes can assemble, using electrospray mass spectrometry experiments, literature-curated assembly data, and a large-scale analysis of protein complex structures. Ultimately, they derived their approach to explaining the observed distribution of known protein complexes in quaternary structure space. This approach, they insist, provides a framework for understanding their evolution.
“In addition, it can contribute considerably to the prediction and modeling of quaternary structures by specifying which topologies are most likely to be adopted by a complex with a given stoichiometry, potentially providing constraints for multi-subunit docking and hybrid methods,” the authors concluded. “Lastly, it could help in the bioengineering of protein complexes by identifying which topologies are most likely to be stable, and thus which types of essential interfaces need to be engineered.”
The rows and columns of the periodic table of the elements, called periods and groups, were originally determined by each element’s atomic mass and chemical properties, later by atomic number and electron configuration. In contrast, the rows and columns of the periodic table of protein complexes correspond to the number of different subunit types and the number of times these subunits are repeated. The new table is not, it should be noted, periodic in the same sense as the periodic table of the elements. It is in principle open-ended.
Although there are no theoretical limitations to quaternary structure topology space in either dimension, the abridged version of the table presented in the Science article can accommodate the vast majority of known structures. Moreover, when the table’s creators compared the large variety of countenanced topologies to observed structures, they found that about 92% of known protein complex structures were compatible with their model.
“Despite its strong predictive power, the basic periodic table model does not account for about 8% of known protein complex structures,” the authors conceded. “More than half of these exceptions arise as a result of quaternary structure assignment errors.
“A benefit of this approach is that it highlights likely quaternary structure misassignments, particularly by identifying nonbijective complexes with even subunit stoichiometry. However, this still leaves about 4% of known structures that are correct but are not compatible with the periodic table.” The authors added that the exceptions to their model are interesting in their own right, and are the subject of ongoing studies.
The Periodic Table of Protein Complexes, published today in Science, offers a new way of looking at the enormous variety of structures that proteins can build in nature, which ones might be discovered next, and predicting how entirely novel structures could be engineered. Created by an interdisciplinary team led by researchers at the Wellcome Genome Campus and the University of Cambridge, the Table provides a valuable tool for research into evolution and protein engineering.
Different ballroom dances can be seen as an endless combination of a small number of basic steps. Similarly, the ‘dance’ of protein complex assembly can be seen as endless variations on dimerization (one doubles, and becomes two), cyclisation (one forms a ring of three or more) and subunit addition (two different proteins bind to each other). Because these happen in a fairly predictable way, it’s not as hard as you might think to predict how a novel protein would form.
“We’re bringing a lot of order into the messy world of protein complexes,” explains Sebastian Ahnert of the Cavendish Laboratory at the University of Cambridge, a physicist who regularly tangles with biological problems. “Proteins can keep go through several iterations of these simple steps, , adding more and more levels of complexity and resulting in a huge variety of structures. What we’ve made is a classification based on these underlying principles that helps people get a handle on the complexity.”
The exceptions to the rule are interesting in their own right, adds Sebastian, as are the subject of on-going studies.
“By analysing the tens of thousands of protein complexes for which three-dimensional structures have already been experimentally determined, we could see repeating patterns in the assembly transitions that occur – and with new data from mass spectrometry we could start to see the bigger picture,” says Joe.
“The core work for this study is in theoretical physics and computational biology, but it couldn’t have been done without the mass spectrometry work by our colleagues at Oxford University,” adds Sarah Teichmann, Research Group Leader at the European Bioinformatics Institute (EMBL-EBI) and the Wellcome Trust Sanger Institute. “This is yet another excellent example of how extremely valuable interdisciplinary research can be.”
The assembly of proteins into complexes is crucial for most biological processes. The three-dimensional structures of many thousands of homomeric and heteromeric protein complexes have now been determined, and this has had a broad impact on our understanding of biological function and evolution. Despite this, the organizing principles that underlie the great diversity of protein quaternary structures observed in nature remain poorly understood, particularly in comparison with protein folds, which have been extensively classified in terms of their architecture and evolutionary relationships.
RATIONALE
In this work, we sought a comprehensive understanding of the general principles underlying quaternary structure organization. Our approach was to consider protein complexes in terms of their assembly. Many protein complexes assemble spontaneously via ordered pathways in vitro, and these pathways have a strong tendency to be evolutionarily conserved. Furthermore, there are strong similarities between protein complex assembly and evolutionary pathways, with assembly pathways often being reflective of evolutionary histories, and vice versa. This suggests that it may be useful to consider the types of protein complexes that have evolved from the perspective of what assembly pathways are possible.
RESULTS
We first examined the fundamental steps by which protein complexes can assemble, using electrospray mass spectrometry experiments, literature-curated assembly data, and a large-scale analysis of protein complex structures. We found that most assembly steps can be classified into three basic types: dimerization, cyclization, and heteromeric subunit addition. By systematically combining different assembly steps in different ways, we were able to enumerate a large set of possible quaternary structure topologies, or patterns of key interfaces between the proteins within a complex. The vast majority of real protein complex structures lie within these topologies. This enables a natural organization of protein complexes into a “periodic table,” because each heteromer can be related to a simpler symmetric homomer topology. Exceptions are mostly the result of quaternary structure assignment errors, or cases where sequence-identical subunits can have different interactions and thus introduce asymmetry. Many of these asymmetric complexes fit the paradigm of a periodic table when their assembly role is considered. Finally, we implemented a model based on the periodic table, which predicts the expected frequencies of each quaternary structure topology, including those not yet observed. Our model correctly predicts quaternary structure topologies of recent crystal and electron microscopy structures that are not included in our original data set.
CONCLUSION
This work explains much of the observed distribution of known protein complexes in quaternary structure space and provides a framework for understanding their evolution. In addition, it can contribute considerably to the prediction and modeling of quaternary structures by specifying which topologies are most likely to be adopted by a complex with a given stoichiometry, potentially providing constraints for multi-subunit docking and hybrid methods. Lastly, it could help in the bioengineering of protein complexes by identifying which topologies are most likely to be stable, and thus which types of essential interfaces need to be engineered.
Protein assembly steps lead to a periodic table of protein complexes and can predict likely quaternary structure topologies.
Three main assembly steps are possible: cyclization, dimerization, and subunit addition. By combining these in different ways, a large set of possible quaternary structure topologies can be generated. These can be arranged on a periodic table that describes most known complexes and that can predict previously unobserved topologies.
Classification of protein structure has had a broad impact on our understanding of biological function and evolution, yet this work has largely focused on individual protein domains and their pairwise interactions. In contrast, the assembly of individual polypeptides into protein complexes, which are ubiquitous in cells, has received comparatively little attention. The periodic table of protein complexes is a new framework for analysis of complexes based on the principles of self-assembly. This reveals that sequence-identical subunits almost always have identical assembly roles within a complex and allows us to unify the vast majority of complexes of known structure (~32,000) into about 120 topologies. This facilitates the exhaustive enumeration of unobserved protein complex topologies and has significant practical applications for quaternary structure prediction, modelling and engineering.
Chloroplast genomes encode ∼37 proteins that integrate into the thylakoid membrane. The mechanisms that target these proteins to the membrane are largely unexplored. We used ribosome profiling to provide a comprehensive, high-resolution map of ribosome positions on chloroplast mRNAs in separated membrane and soluble fractions in maize seedlings. The results show that translation invariably initiates off the thylakoid membrane and that ribosomes synthesizing a subset of membrane proteins subsequently become attached to the membrane in a nucleaseresistant fashion. The transition from soluble to membraneattached ribosomes occurs shortly after the first transmembrane segment in the nascent peptide has emerged from the ribosome. Membrane proteins whose translation terminates before emergence of a transmembrane segment are translated in the stroma and targeted to the membrane posttranslationally. These results indicate that the first transmembrane segment generally comprises the signal that links ribosomes to thylakoid membranes for cotranslational integration. The sole exception is cytochrome f, whose cleavable N-terminal cpSecA-dependent signal sequence engages the thylakoid membrane cotranslationally. The distinct behavior of ribosomes synthesizing the inner envelope protein CemA indicates that sorting signals for the thylakoid and envelope membranes are distinguished cotranslationally. In addition, the fractionation behavior of ribosomes in polycistronic transcription units encoding both membrane and soluble proteins adds to the evidence that the removal of upstream ORFs by RNA processing is not typically required for the translation of internal genes in polycistronic chloroplast mRNAs.
Significance Proteins in the chloroplast thylakoid membrane system are derived from both the nuclear and plastid genomes. Mechanisms that localize nucleus-encoded proteins to the thylakoid membrane have been studied intensively, but little is known about the analogous issues for plastid-encoded proteins. This genome-wide, high-resolution analysis of the partitioning of chloroplast ribosomes between membrane and soluble fractions revealed that approximately half of the chloroplast encoded thylakoid proteins integrate cotranslationally and half integrate posttranslationally. Features in the nascent peptide that underlie these distinct behaviors were revealed by analysis of the position on each mRNA at which elongating ribosomes first become attached to the membrane.
Structures of the HIN Domain:DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor
Electrostatic attraction underlies innate dsDNA recognition by the HIN domains
Both OB folds and the linker between them engage the dsDNA backbone
An autoinhibited state of AIM2 is activated by DNA that liberates the PYD domain
DNA serves as an oligomerization platform for the inflammasome assembly
Summary
Recognition of DNA by the innate immune system is central to antiviral and antibacterial defenses, as well as an important contributor to autoimmune diseases involving self DNA. AIM2 (absent in melanoma 2) and IFI16 (interferon-inducible protein 16) have been identified as DNA receptors that induce inflammasome formation and interferon production, respectively. Here we present the crystal structures of their HIN domains in complex with double-stranded (ds) DNA. Non-sequence-specific DNA recognition is accomplished through electrostatic attraction between the positively charged HIN domain residues and the dsDNA sugar-phosphate backbone. An intramolecular complex of the AIM2 Pyrin and HIN domains in an autoinhibited state is liberated by DNA binding, which may facilitate the assembly of inflammasomes along the DNA staircase. These findings provide mechanistic insights into dsDNA as the activation trigger and oligomerization platform for the assembly of large innate signaling complexes such as the inflammasomes.
Complexity of Protein-Protein Interactions, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Complexity of Protein-Protein Interactions
Curator: Larry H. Bernstein, MD, FCAP
Cracking the Complex
Using mass spec to study protein-protein interactions
Mass spectrometry is a proteomics workhorse. By precisely measuring polypeptide masses, researchers can identify and sequence those molecules, and characterize whether and how they have been chemically modified. To twist a phrase, by their masses you shall know them.
But many proteins do not act in isolation. Critical biological processes such as DNA replication, transcription, translation, cell division, and energy generation rely on the action of massive protein assemblies, many of which comprise dozens of subunits. While these clusters are ripe for study, few traditional mass spectrometric methods can handle them.
Indeed, protein complexes are unwieldy for many types of analysis, says Philip Compton, director of instrumentation at the Proteomics Center of Excellence at Northwestern University in Evanston, Illinois. Most complexes are held together by noncovalent interactions, assemble only transiently, or are located in the cell membrane—all of which complicate sample preparation, he explains. Also, while some complexes are relatively abundant, others are rare, further thwarting detection and analysis.
For mass spectrometry specifically, however, the problem with analyzing protein complexes, which can weigh in at 500 kDa, is size. “In a mass spec, things of that size have traditionally been fairly difficult to handle,” Compton says. Even if you can deliver them into the spectrometer itself, you need a way to figure out which proteins are present, and in what stoichiometry. Plus, normal sample preparation procedures tend to denature proteins, ripping complexes apart.
Still, researchers are increasingly keen to train their mass specs on intact protein assemblies. The Scientistasked four protein-complex experts about the approaches they use in their own labs. This is what they said.
GETTING TOGETHER: Lactate dehydrogenase from human skeletal muscle comprises four identical M subunits, shown here in different colors. FVASCONCELLOS/WIKIMEDIA COMMONS
RESEARCHER:Philip Compton, Director of Instrumentation, Proteomics Center of Excellence, Northwestern University
PROJECT: High-throughput top-down proteomics
SOLUTION: If protein complexes are onions, Compton needs a way to iteratively peel off the layers to see what’s inside. Working with researchers at Thermo Fisher Scientific, Compton is developing an Orbitrap-based mass spectrometer that can do just that, or perform what is called an MS3 study.
Basically, an MS3 experiment involves weighing all the complexes in a sample fraction—there could be as many as 10 or 15 at a time—grabbing one, smashing it into inert-gas molecules to eject a subunit, weighing and sequencing the cast-off piece, and then repeating the process.
That’s the goal, but because that instrument is not yet built, Compton must temporarily content himself with what he calls a “pseudo-MS3” experiment. Basically, instead of one seamless workflow, the instrument shatters the complex, weighs the pieces that come off it, and then repeats the process, only this time capturing and fragmenting those ejected pieces for subsequent analysis (Anal Chem, 85:11163-73, 2013). “We’re kind of splitting it into these two different steps; that accomplishes essentially the same thing,” Compton says.
Compton and his team are still ironing out the kinks, but they have begun applying the approach to protein complexes involved in metabolism. One of these, lactate dehydrogenase (LDH), is a 145-kDa tetramer comprising M (muscle) and H (heart) subunits that can exist in any of five configurations (MMMM, MMMH, MMHH, MHHH, and HHHH). Using the MS3 workflow, Compton says he can differentiate these “multiproteoform assemblies,” as well as any posttranslational modifications those subunits may bear, and determine the abundance of each. Now he hopes to apply the approach to quantify LDH differences between cell and tissue types.
From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry
Native mass spectrometry (MS) is becoming an important integral part of structural proteomics and system biology research. The approach holds great promise for elucidating higher levels of protein structure: from primary to quaternary. This requires the most efficient use of tandem MS, which is the cornerstone of MS-based approaches. In this work, we advance a two-step fragmentation approach, or (pseudo)-MS3, from native protein complexes to a set of constituent fragment ions. Using an efficient desolvation approach and quadrupole selection in the extended mass-to-charge (m/z) range, we have accomplished sequential dissociation of large protein complexes, such as phosporylase B (194 kDa), pyruvate kinase (232 kDa), and GroEL (801 kDa), to highly charged monomers which were then dissociated to a set of multiply charged fragmentation products. Fragment ion signals were acquired with a high resolution, high mass accuracy Orbitrap instrument that enabled highly confident identifications of the precursor monomer subunits. The developed approach is expected to enable characterization of stoichiometry and composition of endogenous native protein complexes at an unprecedented level of detail.
EXTEND YOUR RANGE: Compton’s team uses a souped-up version of Thermo Fisher’s Orbitrap-based Q Exactive HF mass spectrometer, which among other things features a fourfold wider mass range. Other researchers can perform similar work using Thermo’s Exactive Plus EMR Orbitrap system, an off-the-shelf, “extended mass range” instrument. But, because the EMR lacks the “high-mass isolation capabilities” of Compton’s bespoke hardware, the application range is more limited, he says. “You can still do a similar experiment to us, provided that you have one clean [purified] complex.”
Mapping protein-protein interaction interfaces RESEARCHER:Igor Kaltashov, Professor of Chemistry, University of Massachusetts Amherst
PROJECT: Probing the interactions of candidate protein therapeutics with their molecular targets
SOLUTION: Most attempts at studying protein complexes deliver them to the mass spec intact. Kaltashov takes a different approach, using a technique called hydrogen-deuterium exchange (HDX).
It works like this: proteins (like other molecules) pass hydrogen atoms back and forth with the solvent that surrounds them. Normally, one hydrogen is simply swapped for another, and nobody is the wiser. But in deuterated (“heavy”) water, as hydrogens are swapped at the protein surface, the protein gets slightly heavier as deuterium molecules replace some of the hydrogens. This allows researchers to probe how accessible different pieces of the protein are to the solvent, based on how much deuterium they pick up from the buffer, and how quickly they do so.
As Kaltashov explains, HDX can be used to study any event that might alter the accessibility of different protein regions to the solvent that surrounds them. Those events include protein folding and aggregation, but also protein-protein interactions. “Once two proteins bind to each other, solvent would be excluded from the interface, and that would be reflected in the hydrogen-deuterium exchange kinetics,” he says. That change is evident when compared to the proteins in isolation.
In a 2009 review, Kaltashov demonstrated the process with transferrin, an iron transport protein, and its receptor. After undergoing the exchange reaction, the proteins were fragmented to peptides and analyzed piecemeal. Some peptides exhibited no hydrogen-deuterium exchange, he says. That suggests they were never exposed to solvent because they were buried inside the protein core. Other peptides exchanged hydrogens with the solvent at the same rate regardless of receptor binding, indicating they are not part of the protein-receptor interface. A third set of peptides, though, exhibited clear differences in the presence and absence of receptor, marking those as elements of the protein-protein interaction domain (Anal Chem, 81:7892-99, 2009).
“You can actually localize these sites and obtain information both on the strength of the binding [interactions] and the structural characteristics of the interface region,” Kaltashov says.
H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: Is there a need for a top-down approach?
Hydrogen/deuterium exchange (HDX) combined with mass spectrometry (MS) detection has matured in recent years to become a powerful tool in structural biology and biophysics. Several limitations of this technique can and will be addressed by tapping into ever expanding arsenal of methods to manipulate ions in the gas phase offered by mass spectrometry.
Keywords: hydrogen/deuterium exchange (HDX), mass spectrometry (MS), protein ion fragmentation, collision-induced dissociation (CAD), electron-capture dissociation (ECD), electron-transfer dissociation (ETD), protein conformation, protein dynamics
Introduction: HDX MS in the context of structural proteomics
The spectacular successes of proteomics and bioinformatics in the past decade have resulted in an explosive growth of information on the composition of complex networks of proteins interacting at the cellular level and beyond. However, a simple inventory of interacting proteins is insufficient for understanding how the components of sophisticated biological machinery work together. Protein interactions with each other, small ligands and other biopolymers are governed by their higher order structure, whose determination on a genome scale is a focus of structural proteomics. Realization that “the structures of individual macromolecules are often uninformative about function if taken out of context”1 is shifting the focus of the inquiry from comprehensive characterization of individual protein structures to structural analysis of protein complexes.
X-ray crystallography remains the mainstay in this field, and high resolution structures of proteins and protein complexes often provide important clues as to how they carry out their diverse functions in vivo. However, individual proteins are not static objects, and their behavior cannot be adequately described based solely on information derived from static snapshots and without taking into consideration their dynamic character.2Conformation and dynamics of small proteins can be probed at high spatial resolution on a variety of time scales using NMR spectroscopy; however, rather unforgiving molecular weight limitations make this technique less suited for the studies of larger proteins and protein complexes.
Mass spectrometry (MS) is playing an increasingly visible role in this field, as it can provide information on protein dynamics on a variety of levels, ranging from interactions with their physiological partners by forming dynamic assemblies3 to large-scale conformational transitions within individual subunits.4 Perhaps one of the most powerful MS-based tools to characterize protein conformation and dynamics is HDX MS, a technique that combined hydrogen/deuterium exchange in solution5 with MS detection of the progress of exchange reactions.6 This technique is certainly not new,7 and in fact already made lasting impact in diverse fields ranging from structural proteomics8 to analysis of biopharmaceutical products.9 Nevertheless, HDX MS methodology is still in a phase where dramatic progress is made, fed by the continued expansion of the experimental armamentarium offered by MS. In particular, better integration of new methods of manipulating ions in the gas phase into HDX MS routine is likely to result in truly transformative changes. This sea change in HDX MS methodology will transform it to a potent tool rivaling NMR in terms of resolution, but without suffering the limitations of this technique.
What information can be deduced from HDX MS measurements? The classic “bottom-up” approach, its challenges and limitations
While the concept of HDX experiment may appear rather transparent (Figure 1), interpretation of the results is usually not. The backbone protection measured in a typical HDX MS experiment is a combination of several factors, as the exchange reaction of each labile hydrogen atom is a convolution of two processes.5The first is a protein motion that makes a particular hydrogen atom exposed to solvent and therefore available for the exchange. This could be a small-scale event, such as relatively frequent local structural fluctuations transiently exposing hydrogen atoms residing close to the protein surface, or a rare global unfolding event exposing atoms sequestered from the solvent in the protein core. The second process is a chemical reaction of exchanging the unprotected labile hydrogen atom with the solvent. The kinetics of this reaction (intrinsic exchange rate) strongly depends on solution temperature and pH (with a minimum at pH 2.5-3 for backbone amides), parameters that obviously have a great influence on the protein dynamics as well.
Schematic representation of HDX MS experiments: bottom-up (A) and top-down (B) HDX MS.
Since the majority of HDX MS studies target protein dynamics under near-native conditions, the experiments are typically carried out at physiological pH, where the progress of the exchange is followed by monitoring the protein mass change. The direct infusion scheme offers the simplest way to carry out such measurements, either in real time7 or by using on-line rapid mixing.10 However, in many cases these straightforward approaches cannot be used, as they limit the choice of exchange buffer systems to those compatible with electrospray ionization (ESI). To avoid this, HDX can be carried out in any suitable buffer followed by rapid quenching (lowering pH to 2.5-3 and temperature to near 0°C). Dramatic deceleration of the intrinsic exchange rate for backbone amides under these conditions allows the protein solution to be de-salted prior to MS analysis. Additionally, the slow exchange conditions denature most proteins, resulting in facile removal of various binding partners, ranging from small ligands to receptors (their binding to the protein of interest inevitably complicates the HDX MS data interpretation by making accurate mass measurements in the gas phase less straightforward).
An example of such experiments is shown in Figure 2, where HDX is used to probe the higher order structure and conformational dynamics of metal transporter transferrin (Fe2Tf) alone and in the receptor-bound form. Both Tf-metal and Tf-receptor complexes dissociate under the slow exchange conditions prior to MS analysis; therefore, the protein mass evolution in each case reflects solely deuterium uptake in the course of exchange in solution. The extra protection afforded by the receptor binding to Tf persists over an extended period of time, and it may be tempting to assign it to shielding of labile hydrogen atoms at the protein-receptor interface. However, this view is overly simplistic, as the conformational effects of protein binding are frequently felt well beyond the interface region. The difference in the backbone protection levels of receptor-free and receptor-bound forms of Fe2Tf appears to grow during the initial hour of the exchange (Figure 2), reflecting significant stabilization of Fe2Tf higher order structure by the receptor binding. Indeed, while the fast phase of HDX is typically ascribed to frequent local fluctuations (transient perturbations of higher order structure) affecting relatively small protein segments, the slower phases of HDX usually reflect relatively rare, large-scale conformational transitions (transient partial or complete unfolding). This is why global HDX MS measurements similar to those presented in Figure 2 are can be used to obtain quantitative thermodynamic characteristics for protein interaction with a variety of ligands, ranging from metal ions11 and small organic molecules 12 to other proteins13 and oligonucleotides.14
HDX MS of Fe2Tf in the presence (blue) and the absence (red) of the cognate receptor. The exchange was carried out by diluting the protein stock solution 1:10 in exchange solution (100 mM NH4HCO3 in D2O, pH adjusted to 7.4) and incubating for a certain period of time as indicated on each diagram followed by rapid quenching (lowering pH to 2.5 and temperature to near 0°C). The black trace shows unlabeled protein.
While global HDX MS measurements under near-native conditions provide valuable thermodynamic information on proteins and their interaction with binding partners, structural studies (e.g., localizing the changes in Tf that occur as a result of receptor binding) must rely on the knowledge of exchange kinetics at the local level. This is typically accomplished by carrying out proteolysis under the slow exchange conditions following the quench of HDX.6 Here we will refer to this approach as “bottom-up” HDX MS, by drawing analogy to a bottom-up approach to obtain sequence information.15 An example is shown in Figure 3, where Fe2Tf undergoes exchange in solution in the absence and in the presence of the receptor, followed by rapid quenching of HDX reactions, protein reduction and digestion with pepsin and LC/MS analysis of the deuterium content of individual proteolytic peptides.
Localizing the influence of the receptor binding on backbone protection of Fe2Tf using bottom-up HDX MS on the physiologically relevant time scale. The panels show isotopic distributions of representative peptic fragments derived from the protein subjected to HDX in the presence (blue) and the absence (red) of the receptor and followed by rapid quenching. Dotted lines indicate deuterium content of unlabeled and fully exchanged peptides. Colored segments within the Fe2Tf/receptor complex show location of the peptic fragments.
Evolution of deuterium content of various peptic fragments in Figure 3 reveals a wide spectrum of protection, which is clearly distributed very unevenly across the protein sequence. While some peptides exhibit nearly complete protection of backbone amides (e.g., segment [396-408] sequestered in the core of the protein C-lobe), exchange in some other segments is fast (e.g., peptide [612-621] in the solvent-exposed loop of the C-lobe). The influence of the receptor binding on the backbone protection is also highly localized. While many segments appear to be unaffected by the receptor binding, there are a few regions where exchange kinetics noticeably decelerates (e.g., segment [71-81] of the N-lobe, which contains several amino acid residues that form Tf/receptor interface according to the available model of the complex based on low-resolution cryo-EM data16).
Although the increased protection of backbone amides proximal to the protein/receptor binding interface is hardly surprising, HDX MS data also reveal a less trivial trend, acceleration of exchange kinetics in some segments of the protein as a result of receptor binding (such behavior is illustrated in Figure 3 with segment [113-134], a part of the N-lobe that is distal to the receptor). Therefore, in addition to mapping binding interface regions, HDX MS also provides a means to localize the protein segments that are affected by the binding indirectly via allosteric mechanisms. However, this example also highlights one of the limitations of HDX MS, namely inadequate spatial resolution. This peptic fragment spans several distinct regions of the protein (an α-helical segment, a β-strand, and two loops). The moderate level of protection observed in this segment in the absence of the receptor binding (fast exchange of three protons followed by slow exchange of the rest) is likely to be a result of averaging out very uneven protection patterns across this peptide. Even smaller peptides may comprise two or more distinct structural elements, such as segment [71-81] spanning three distinct regions of the protein (an α-helical segment, a β-strand, and a loop connecting them).
In some favorable cases spatial resolution in HDX MS of small proteins (<15 kDa) may be enhanced up to a single residue level by analyzing deuterium content of a set of overlapping proteolytic fragments.17However, single-residue resolution has never been demonstrated in HDX MS studies of proteins falling out of the mass range routinely accessible by NMR, although overlapping peptic fragments frequently provide moderate improvement of spatial resolution.
In addition to limited spatial resolution, the “classic” HDX MS scheme frequently suffers from incomplete sequence coverage, especially when applied to larger and extensively glycosylated proteins. Proteins with multiple disulfide bonds constitute another class of targets for which adequate sequence coverage is difficult to achieve, although certain changes in experimental protocol can alleviate this problem, at least for smaller proteins.18 Typically, an 80% level of sequence coverage is considered good, although significantly lower levels may also be adequate, depending on the context of the study.
Protein processing in HDX MS experiments is carried out under the conditions that minimize the exchange rates for backbone amides. Since these slow exchange conditions are highly denaturing for most proteins, both intact protein and its proteolytic fragments lack any protection and inevitably begin to lose their labile isotopic labels, despite low (but finite) intrinsic exchange rates.19 This phenomenon, known as “back-exchange,” may be accelerated during various stages of protein processing, e.g. during the chromatographic step.20 Although back-exchange was frequently evaluated in early HDX MS studies using unstructured model peptides, the utility of this procedure is questionable, since the intrinsic exchange rates are highly sequence-dependent. In many instances, back-exchange may be estimated using algorithms based on context-specific kinetics data (e.g., http://hx2.med.upenn.edu/download.html); it may also be determined experimentally for each proteolytic fragment by processing a fully labeled protein using a series of steps that precisely reproduce those used in HDX MS measurements.9 Typical back-exchange levels reported in recent literature range from 10% to 50%, although significantly higher numbers have also been reported. Even if back-exchange can be accounted for, it nonetheless has very detrimental influence on the quality of HDX MS measurements by reducing the available dynamic range.
Finally, the classic HDX MS scheme is poorly suited for measurements that are carried out under conditions favoring correlated exchange, when HDX kinetics follows the so-called EX1 regime, leading to appearance of bimodal and convoluted multi-modal isotopic distributions of protein ions.21 Carrying out HDX MS measurements under these conditions provides a unique opportunity to visualize and characterize distinct conformational states, which can be populated either transiently10 or at equilibrium.22 The distinction among such states can be made based on the differences in their deuterium contents. However, proteolysis in solution almost always leads to a loss of correlation between the deuterium content of fragment peptides and specific conformers with distinct levels of backbone protection. Therefore, the classic HDX MS scheme does not allow protein higher order structure and dynamics to be characterized in a conformer-specific fashion.
“Top-down” HDX MS: tandem MS allows protein structure to be probed in the conformer-specific fashion but raises the specter of hydrogen scrambling
The problem of characterizing protein conformation and dynamics in a conformer-specific fashion can be addressed using methods of tandem mass spectrometry (the so-called “top-down” HDX MS). Indeed, replacement of proteolysis in solution with protein ion fragmentation in the gas phase following mass selection of precursor ions provides a means to obtain fragment ions originating from a particular conformer with a specific level of deuterium incorporation. Deuterium content of fragment ions would then provide a measure of local protection patterns, assuming there is no internal re-arrangement of labile hydrogen and deuterium atoms during ion activation (vide infra). Although the idea to use polypeptide ion dissociation in the gas phase as an alternative to proteolysis was originally proposed in early 1990s,23 its implementation for proteins only became possible24 following dramatic improvements in FTMS and hybrid TOF analyzers in the late 1990s.
An example of conformer-specific characterization of protein higher order structure using a top-down HDX MS approach is illustrated in Figure 4. The isotopic profile of a fully deuterated 18 kDa protein wt*-CRABPI is recorded following its brief exposure to the 1H-based exchange buffer. The bimodal appearance of the isotopic distribution of the molecular ion (top trace in Figure 4A) clearly indicates the presence of at least two conformers with different levels of backbone protection. Collisional activation of the entire protein ion population generates a set of fragment ions with convoluted isotopic distributions (top trace in Figure 4B). However, mass selection of precursor ions with a specific level of deuterium content allows the top-down HDX MS measurements to be carried out in a conformation-specific fashion, taking full advantage of the HDX MS ability to detect distinct conformers. For example, selective fragmentation of protein ions representing a highly protected conformation is achieved by mass-selecting a narrow population of intact protein ions with high level of retained deuterium (the blue trace in Figure 4A). Mass-selection and subsequent fragmentation of a narrow population of protein ions with significantly lower deuterium content (the red trace in Figure 4A) generates a set of fragment ions whose isotopic distributions provide information on backbone protection within non-native protein states. For example, the data presented in Figure 4 clearly indicate that the C-terminal segment of the protein represented by the y172+ ions retains significant structure even within the partially unfolded conformers: the amount of retained deuterium atoms reduces by only 30% as a result of switching from the precursor ion from highly protected (blue) to less protected (red). At the same time, selection of the precursor ion has a much more dramatic effect on the protection levels exhibited by the N-terminal segment (represented by the b425+ ion), where more than a two-fold decrease in the amount of retained deuterium atoms is observed. Extending this analysis to other protein fragments may allow detailed backbone protection maps to be created for each protein conformer, provided there is no hydrogen scrambling prior to protein ion fragmentation (vide infra).
Characterization of local dynamics in wt*-CRABP I in a conformer-specific fashion using top-down HDX MS (fully deuterated protein was exposed to 1H2O/CH3CO2N1H4 at pH 3.1 for 10 min; the gray trace at the bottom corresponds to HDX end-point). A: mass selection of precursor ions for subsequent CAD (from top to bottom): broad-band selection of the entire ionic population (not conformer-specific); highly protected conformers; narrow population of less protected conformers; HDX end-point. B: isotopic distributions of two representative fragment ions generated by CAD of precursor ions shown in panel A. Selection of different ion populations as precursor ions for subsequent fragmentation was achieved by varying the width of a mass selection window of a quadrupole filter (Q) in a hybrid quadrupole/time-of-flight mass spectrometer (Qq-TOF MS).
The example shown above illustrates a great promise of top-down HDX MS as a technique uniquely capable of probing structure and dynamics of populations of protein conformers coexisting in solution with high selectivity. Furthermore, this approach often allows one to avoid protein handling under the slow exchange conditions prior to MS analysis, thereby eliminating back-exchange as a factor adversely influencing the quality of measurements. Nonetheless, applications of top-down HDX MS have been limited due to concerns over the possibility of hydrogen scrambling accompanying collision-activated dissociation (CAD) of protein ions. Indeed, several reports pointed out that proton mobility in the gas phase may under certain conditions influence the outcome of top-down HDX MS measurements when CAD is employed to fragment protein ions.25, 26
The occurrence (or the absence) of hydrogen scrambling in the gas phase can be reliably detected by using built-in scrambling indicators. One particularly convenient indicator is a Histag, a 6-30 residues long, histidine-rich segment appended to wild-type sequences to facilitate protein purification on metal affinity columns. Such segments are fully unstructured in solution and, therefore, should lack any backbone protection.27 Alternatively, intrinsic scrambling indicators (e.g., internal flexible loops26), as well as other approaches25 can be used to detect occurrence of scrambling. The available experimental evidence suggests that slow protein ion activation (e.g., SORI CAD) always leads to hydrogen scrambling, while fast activation allows it to be minimized or eliminated in top-down HDX MS experiments.26
Another shortcoming of top-down HDX MS schemes utilizing CAD is the limited extent of protein ion fragmentation, which may lead to sizeable gaps in sequence coverage, particularly for larger proteins,28 and insufficient level of spatial resolution (even for smaller proteins29). Our earlier attempts to solve this problem by employing multi-stage CAD (MSn) were unsuccessful due to massive hydrogen scrambling exhibited by the second generation of fragments.
Electron-induced ion fragmentation in top-down schemes: keeping hydrogen scrambling at bay while enhancing sequence coverage and spatial resolution
Some time ago we suggested that the specter of hydrogen scrambling in top-down HDX MS measurements may be alleviated by using non-ergodic fragmentation processes, where dissociation is induced by ion-electron interaction, rather than collisional activation.30 Indeed, the results of earlier work combining hydrogen exchange of polypeptide ions in the gas phase and electron capture dissociation (ECD) were consistent with the notion of intramolecular rearrangement of hydrogen atoms occurring on a slower time scale compared to ion dissociation.31 A recent study demonstrated that the extent of scrambling was indeed negligible when ECD was used as a means to obtain fragment ions in top-down HDX MS characterization of a small protein ubiquitin.32
Our own recent work suggests that hydrogen scrambling can be avoided when top-down HDX MS employs ECD in characterizing higher order structure of larger proteins (approaching 20 kDa), although experimental conditions must be carefully controlled to minimize proton mobility induced by ion-molecule collisions in the ESI interface. The point in question is illustrated in Figure 5, which shows the results of top-down HDX MS analysis of higher order structure of wt*-CRABP I. The protein retains a significant proportion of labile deuterium label following its complete deuteration and then brief exposure to the 1H-based exchange buffer, as indicated by the isotopic distribution of the surviving molecular ions (red and blue traces in Figure 5A). However, the deuterium content of fragment ions derived from the 21-residue long His-tag region of the protein (e.g., c22 in Figure 5B) is indistinguishable from that of the exchange reaction endpoint, as long as moderate ion desolvation conditions are kept in the ESI interface. This clearly signals that hydrogen scrambling does not affect the outcome of local HDX MS measurements. However, once collision-assisted desolvation of protein ions is attempted in the ESI interface, the appearance of isotopic distributions of larger fragment ions derived from the His-tag region (e.g., c22, red trace in Figure 5B) shifts, indicating apparent deuterium retention and signaling the occurrence of limited hydrogen scrambling. We also demonstrated that deuterium distribution across the protein backbone is preserved when another recently introduced fragmentation technique based on cation-electron interactions, electron transfer dissociation (ETD), is used in top-down HDX MS schemes.33
Top-down HDX MS of wt*-CRABP I using ECD of the entire protein ion population (fully deuterated protein was exposed to1H2O/CH3CO2N1H4 at pH 3.5 for varying time periods); the black trace at the bottom of corresponds to HDX end-point). A: isotopic distributions of surviving intact protein ions. B: two representative c-ions. Minimal collision-and temperature-induced desolvation was used for acquisition of all mass spectra, except the one top (red trace).
In addition to allowing scrambling to be easily eliminated in top-down HDX MS experiments, both ECD and ETD appear to be superior to CAD in terms of sequence coverage, at least for the proteins in the 20 kDa range. Unlike CAD, protein backbone cleavage in ECD and ETD is less specific,34 leading to a higher number of fragment ions. This translates not only to improved sequence coverage, but also enhanced spatial resolution. Indeed, in some cases it becomes possible to generate patterns of deuterium distribution across the protein backbone down to the single residue level.
One example of such work is shown in Figure 6, where ETD was used as a protein ion fragmentation tool in top-down HDX MS characterization of a 16 kDa variant of CRABP I. The bar graph shows the levels of deuterium retention in a series of c-ions derived from the N-terminal segment of the protein. The bar height at position n in this diagram shows mass difference between two cn-1 fragments, one derived from the fully deuterated protein that was exposed to the protiated exchange buffer at pH 7 for 5 min and then placed under the slow exchange conditions for the duration of the data acquisition cycle, and another one representing the HDX endpoint (raw data for bars at n=14 and 35 are shown in Figure 7). Unchanged height between two adjacent bars at residues n and n+1 indicates no difference in deuterium content of cn-1 and cn fragments, signaling no backbone amide deuterium retention at residue n+1, while bar height increase by one unit indicates complete retention of deuterium at the nth amide.
Backbone protection pattern of CRABPI mutant (without N-terminal His-tag) obtained from top-down HDX MS measurements using ETD of the entire protein ion population. HDX was initiated by exposing the fully deuterated protein to 1H2O/CH3CO2N1H4 at pH 3.5 for 5 min followed by rapid quenching.
An example of raw HDX MS data used to generate the protection plot shown in Figure 6. Isotopic distributions of c13 and c34 fragments derived from protein subjected to 5 min HDX exchange in solution (red trace) and protein at the HDX end-point (blue trace) were used to calculate the bar heights at n=12 and 35.
The resulting backbone protection pattern in Figure 6 shows clear correlation with the known higher order structure of the protein (the amino acid sequence and the secondary structure assignment are shown at the top of the graph). Furthermore, the diagram clearly shows uneven distribution of backbone protection even within single structural elements (e.g., lower protection at the fringes vs. the middle of helix α1), as well as unequal protection of similar structural elements participating in the same structural motif (e.g., lower protection of helix α2 vs. helix α1, consistent with the available NMR data). A comparable level of spatial resolution can be achieved with ECD, as shown recently in top-down HDX MS analysis of higher order structure of myoglobin.35
The ability to characterize protein conformation and dynamics at the single residue level is certainly very exciting; however, it comes at a price. Since the protein fragmentation is carried out entirely in the gas phase, no fragment separation can be done prior to mass analysis. A large number of fragment ions with different masses and charges are usually confined to a relatively narrow m/z region, leading to inevitable overlaps of fragment ion isotopic distributions (Figure 7). This places rather stringent requirements on the resolving power of the mass analyzer, effectively narrowing the selection of mass spectrometers suitable for this work to FTMS.
Meeting in the middle: integration of top-down strategies into bottom-up HDX MS schemes
The top-down approach to HDX MS measurements clearly shows a promise to solve many problems that mar the commonly employed bottom-up methodology. The fragmentation efficiency afforded by ECD and ETD provides better spatial resolution, at least for proteins in the 20 kDa range, and this number is likely to grow as there are numerous examples of successful use of these fragmentation techniques to obtain sequence information on significantly larger proteins.36 Unlike the classic bottom-up approach, top-down HDX MS provides an elegant solution to the problem of characterizing higher order structure and dynamics in a conformer-specific fashion (see Figure 4 and discussion in the text). Finally, back-exchange can be eliminated, as outsourcing protein fragmentation to the gas phase often eliminates the need to manipulate the protein in solution under the slow exchange conditions prior to MS analysis.
The top-down/bottom-up dichotomy in HDX MS should not be viewed through the “eitheror” prism. In fact, gas phase fragmentation can enhance the quality of HDX MS data derived from experiments that are built around the bottom-up approach. The suggestion to supplement proteolysis in solution with peptide ion fragmentation in the gas phase to achieve better spatial resolution was made over 10 years ago.37 However, earlier attempts to implement this idea using CAD on a variety of platforms yielded mixed results due to apparent scrambling in some (but not all) fragment ions.37, 38 Later reports showed even more extensive scrambling in small peptide ions subjected to collisional activation,39 an obvious anathema to the proposed marriage of CAD and bottom-up HDX MS. Nonetheless, continued search for a scrambling-free solution to this problem has yielded very encouraging results, with both ECD and ETD showing minimal scrambling when applied to short peptides under carefully controlled conditions40, 41 and feasibility of supplementing proteolytic fragmentation in solution with ETD in the gas phase was recently demonstrated using a small model protein.42 Although these initial steps are relatively modest, they certainly warrant further work in this field.
The two complementary approaches to HDX MS measurements share a set of common challenges that inevitably arise as these techniques gain popularity and the scope of their applications expands. One such challenge is presented by membrane proteins, a notoriously difficult class of biological objects. HDX MS has been shown to have a great potential in this field.43 Interestingly, some initial work in this field was done nearly ten years ago using then-infant top-down HDX MS technique,44 while more recent work in this field utilizes both bottomup18 and top-down45 approaches. Another challenge faced by HDX MS is presented by highly heterogeneous proteins, such as proteins conjugated to other biopolymers and/or synthetic polymers, which constitute a significant fraction of the next generation of biopharmaceuticals. Presently, there are no biophysical techniques capable of characterizing conformation and dynamics of these systems, and there is an urgent need to fill this gap. Finally, nearly all HDX MS work reported to date was carried out in vitro under conditions that some regard as “reductionist.” Although initial HDX work with living objects was carried out over 75 years ago,46 as the years passed only one report on in vivo HDX MS studies was published.47 As mass spectrometry at large is being increasingly used in both in vivo and ex vivo studies, there is a growing pressure on HDX MS to follow the trend, although it remains to be seen how this will be done.
It probably is not an exaggeration to say that we are witnessing a renaissance of HDX MS, with the emergence of the top-down approach not only expanding our experimental arsenal by offering new capabilities, but also serving as a catalyst in enhancing the classic bottom-up methodology. The two techniques are highly complementary, and their synergism will certainly bring about new exciting discoveries and accelerate our progress in solving a variety of problems ranging from very fundamental questions in biophysics to applied problems in drug design.
WATCH OUT FOR DISULFIDES: If you’re going to try bottom-up HDX experiments, be careful of disulfide bonds, Kaltashov says. Pepsin is one of the very few proteinases that can efficiently digest a protein into its composite peptides under HDX experimental conditions, but it struggles when multiple disulfide bonds are present. In 2014, Kaltashov’s lab published two solutions to that problem. The first employs a fragmentation technique called electron capture dissociation (ECD) to break the disulfide linkage in the mass spec (Anal Chem, 86:5225-31, 2014); the second skips the pepsin digestion altogether—a strategy called top-down analysis (Anal Chem, 86:7293-98, 2014).
Enhancing the Quality of H/D Exchange Measurements with Mass Spectrometry Detection in Disulfide-Rich Proteins Using Electron Capture Dissociation
Hydrogen/deuterium exchange (HDX) mass spectrometry (MS) has become a potent technique to probe higher-order structures, dynamics, and interactions of proteins. While the range of proteins amenable to interrogation by HDX MS continues to expand at an accelerating pace, there are still a few classes of proteins whose analysis with this technique remains challenging. Disulfide-rich proteins constitute one of such groups: since the reduction of thiol–thiol bonds must be carried out under suboptimal conditions (to minimize the back-exchange), it frequently results in incomplete dissociation of disulfide bridges prior to MS analysis, leading to a loss of signal, inadequate sequence coverage, and a dramatic increase in the difficulty of data analysis. In this work, the dissociation of disulfide-linked peptide dimers produced by peptic digestion of the 80 kDa glycoprotein transferrin in the course of HDX MS experiments is carried out using electron capture dissociation (ECD). ECD results in efficient cleavage of the thiol–thiol bonds in the gas phase on the fast LC time scale and allows the deuterium content of the monomeric constituents of the peptide dimers to be measured individually. The measurements appear to be unaffected by hydrogen scrambling, even when high collisional energies are utilized. This technique will benefit HDX MS measurements for any protein that contains one or more disulfides and the potential gain in sequence coverage and spatial resolution would increase with disulfide bond number.
———
Hydrogen/deuterium exchange (HDX) with mass spectrometry (MS) detection has evolved in the past two decades into a powerful tool that is now used to decipher intimate details of processes as diverse as protein folding, recognition and binding, and enzyme catalysis.1,2 While initially being a tool that was used exclusively in fundamental studies, HDX MS is now becoming an indispensable part of the analytical arsenal in the biopharmaceutical sector, where it is utilized increasingly in all stages of protein drug development from discovery to quality control.3−5 Despite this progress, several areas remain where the application of HDX MS has met with only limited success. Disulfide-rich proteins constitute one such group, where characterization of higher-order structure and dynamics is particularly difficult, because of the suboptimal conditions used for reduction of thiol–thiol bonds following a quench of the exchange reactions. Proteins containing disulfide bonds are encountered very rarely in the protein folding studies where the most popular targets are small proteins lacking cysteine residues (with a notable exception of the oxidative folding studies), as well as in many other fundamental studies focusing on proteins of prokaryotic origin. However, disulfide-rich proteins are encountered very frequently in eukaryotic proteomes6 and constitute a large segment of the biopharmaceutical products,7 where the thiol–thiol bonds are critical elements defining conformation of protein drugs, and also play an important role in stabilizing proteins by endowing them with protease resistance.
While disulfide bond reduction is a relatively trivial task that can be readily accomplished at neutral pH using a variety of reagents, the acidic, low-temperature environment where proteins are placed to quench HDX narrows down the choice to a single reducing agent, TCEP.8 However, the alkaline pH for optimal disulfide reduction by TCEP is substantially higher, compared to the acidic environment of typical “slow exchange conditions” commonly employed to minimize back exchange within proteins and their peptic fragments prior to MS analysis.9 Furthermore, disulfide reduction in HDX MS measurements is usually carried out within a relatively short period of time (a few minutes) and at low temperature (0–4 °C) to limit the extent of the back-exchange, which in many situations does not allow the complete dissociation of thiol–thiol linkages of individual peptic fragments to be achieved in solution prior to LC separation and MS analysis of their deuterium content. Incomplete reduction of disulfide bonds dramatically increases the pool of candidate peptides that should be considered when analyzing proteolytic fragments in HDX MS measurements and frequently reduces sequence coverage and/or spatial resolution. While the former problem can be solved by employing more powerful and robust search engines for peptide identification, the latter one is more difficult to circumvent and can be very detrimental for the quality of HDX MS data and may require significant changes in experimental protocols. Indeed, a complete failure to reduce a certain disulfide bond in a protein will give rise to a thiol–thiol linked peptide dimer, whose constituent monomers do not necessarily represent a contiguous segment of the protein and may have vastly different conformational and dynamic properties. The total deuterium content of the entire dimer (measured by HDX MS) would not provide any meaningful information under these conditions, thereby effectively reducing the sequence coverage in the corresponding segments of the protein.
———-
Disulfide-rich proteins have traditionally been challenging targets for HDX MS studies, because of incomplete reduction of thiol–thiol linkages, which is a consequence of the quench conditions used to minimize amide back-exchange in peptides prior to MS analysis of their deuterium content: limited time, low temperature, and low pH. Traditionally, the principal strategy to address difficult-to-reduce or high-density disulfides in the HDX MS workflow is a brute force approach utilizing high concentrations of reductant and denaturant prior to (or even in combination with) digestion. The effectiveness of this approach is protein-dependent and extended incubation times frequently employed to enhance exposure to reductant invariably result in an undesirable increase in H/D back exchange. More recently, a novel electrochemical approach to reduce disulfides in solution under quench conditions prior to LC-MS has been reported for insulin.32 While electrochemical reduction shows promise, several limitations were identified, an apparent requirement for low-salt conditions, a higher-than-optimal temperature (10 °C), and a current cell pressure limit of 50 bar. In this work, electron capture dissociation (ECD) was used to circumvent the disulfide problem, since it effectively cleaves external disulfide bonds. Dissociation of the disulfide-linked peptide dimers can be accomplished on the fast LC time scale and produces abundant signals for monomeric subunits without interchain hydrogen scrambling, even when collisional activation of ions is applied prior to ion selection and ECD fragmentation. Inclusion of ECD in the HDX MS workflow results in increased sequence coverage and spatial resolution and provides an attractive alternative to extensive chemical reduction of disulfide-rich proteins.
Approach to Characterization of the Higher Order Structure of Disulfide-Containing Proteins Using Hydrogen/Deuterium Exchange and Top-Down Mass Spectrometry
Top-down hydrogen/deuterium exchange (HDX) with mass spectrometric (MS) detection has recently matured to become a potent biophysical tool capable of providing valuable information on higher order structure and conformational dynamics of proteins at an unprecedented level of structural detail. However, the scope of the proteins amenable to the analysis by top-down HDX MS still remains limited, with the protein size and the presence of disulfide bonds being the two most important limiting factors. While the limitations imposed by the physical size of the proteins gradually become more relaxed as the sensitivity, resolution and dynamic range of modern MS instrumentation continue to improve at an ever accelerating pace, the presence of the disulfide linkages remains a much less forgiving limitation even for the proteins of relatively modest size. To circumvent this problem, we introduce an online chemical reduction step following completion and quenching of the HDX reactions and prior to the top-down MS measurements of deuterium occupancy of individual backbone amides. Application of the new methodology to the top-down HDX MS characterization of a small (99 residue long) disulfide-containing protein β2- microglobulin allowed the backbone amide protection to be probed with nearly a single-residue resolution across the entire sequence. The high-resolution backbone protection pattern deduced from the top-down HDX MS measurements carried out under native conditions is in excellent agreement with the crystal structure of the protein and high-resolution NMR data, suggesting that introduction of the chemical reduction step to the top-down routine does not trigger hydrogen scrambling either during the electrospray ionization process or in the gas phase prior to the protein ion dissociation.
Since its initial introduction in the late 1990s,1−3 top-down hydrogen/deuterium exchange (HDX) with mass spectrometric (MS) detection evolved to become a potent biophysical tool capable of providing valuable information on higher order structure and conformational dynamics of proteins at an unprecedented level of structural detail. Among the many advantages offered by top-down HDX MS compared to conventional (bottom-up) measurements are significant reduction or indeed complete elimination of the back exchange,4 high spatial resolution,5,6 and the ability to study conformational dynamics in the conformer-specific fashion.7,8 However, despite the spectacular recent advances and the broader acceptance of this technique, the scope of the proteins amenable to the analysis by top-down HDX MS remains limited, with the protein size and the presence of disulfide bonds being the two most important limiting factors. The limitations imposed by the physical size of the proteins gradually become more relaxed as the sensitivity, resolution, and dynamic range of modern MS instrumentation continue to improve at an ever accelerating pace. However, the presence of disulfides remains a much less forgiving limitation even for the proteins of relatively modest size.
In this work we demonstrated feasibility of applying top-down HDX MS measurements to characterize higher order structure and conformational dynamics of disulfide-containing proteins, which have been out of the reach of this technique so far. Use of a moderate amount of a reducing agent TCEP is compatible with the ESI process, while allowing a fraction of the protein molecules to be reduced in solution thereby enabling nearcomplete sequence coverage at high resolution. The agreement between the top-down HDX MS and NMR data sets demonstrate that the new experimental approach is capable of capturing the dynamic picture of protein conformation at high spatial resolution without compromising the quality of the data by triggering hydrogen scrambling in the gas phase. Despite its modest size, β2m is known to be able to populate a non-native state,35 which might be a key player in a variety of processes, including amyloidosis. However, the structure of this non-native state of β2m remains elusive since this conformer exists in dynamic equilibrium with the native state of the protein.36,37 Recently we demonstrated that top-down HDX MS provides an elegant way to selectively probe structure of protein states coexisting in solution at equilibrium;8 however, β2m remained out of reach of this technique until recently due to the presence of a disulfide bond. The ability to expand the scope of top-down HDX MS to disulfide-containing proteins opens up a host of exciting possibilities to explore the structure of β2m, interferon, lysozyme, and a variety of other disulfidecontaining proteins in a conformer-specific fashion, where physiologically important non-native states may play important roles in processes as diverse as folding, recognition, signaling, and amyloidosis. ■ ASSOCIATED CONTENT *S Supporting Information Representative examples of isotopic distributions of fragment ions that have (Supplementary Figure 1) and have not (Supplementary Figure 2) been used to calculate the deuterium occupancy at individual backbone amides of β2m in top-down HDX MS measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
SUSSING OUT THE SURFACE: Protein topology can be probed by firing low-energy electrons (white circles) at intact protein complexes within a high-resolution mass spectrometer. That reaction, called electron capture dissociation, causes the protein complex to fracture on its surface, revealing the exposed amino acid residues. COURTESY OF PIRIYA WONGKONGKATHEP AND HUILIN LI, UCLA
RESEARCHER:Joseph Loo, Professor of Biological Chemistry, David Geffen School of Medicine, University of California, Los Angeles)
PROJECT: Studying protein-ligand and protein-protein interactions
SOLUTION: Loo is less interested in complex identification than in how the protein subunits assemble. Specifically, he wants to know which amino acid residues lie on the complex’s surface and which are buried inside or interacting with ligands.
It’s a question of structural biology, he explains: “How is this thing folded in a way that these residues are on the outside?”
To work that out, Loo combines high-resolution Fourier transform ion cyclotron resonance mass spectrometry (FTICR) with electron-capture dissociation (ECD), a mass spec fragmentation method in which an ion in the mass spectrometer interacts with free electrons, causing the protein to fracture along its peptide backbone. By measuring the mass of those fragments with high precision, researchers can determine the protein’s amino acid sequence.
In Loo’s case, though, that fragmentation is not uniform along the length of the protein. Proteins usually are denatured for mass spectrometry analysis, but the protein complexes in his studies are intact—a process called native mass spectrometry. Fragmentation thus occurs preferentially on the surface of the complex, like the cracks in the shell of a hard-boiled egg. “You get limited sequence information, but that sequence information comes from regions that are specific to its 3-D structure,” he says (Anal Chem, 86:317-20, 2014).
Native Top-Down ESI-MS of 158 kDa Protein Complex by High Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry
Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) delivers high resolving power, mass measurement accuracy, and the capabilities for unambiguously sequencing by a top-down MS approach. Here, we report isotopic resolution of a 158 kDa protein complex – tetrameric aldolase with an average absolute deviation of 0.36 ppm and an average resolving power of ~520,000 at m/z 6033 for the 26+ charge state in magnitude mode. Phase correction further improves the resolving power and average absolute deviation by 1.3 fold. Furthermore, native top-down electron capture dissociation (ECD) enables the sequencing of 149 C-terminal amino acid (AA) residues out of 463 total AAs. Combining the data from top-down MS of native and denatured aldolase complexes, a total of 58% of the backbone cleavages efficiency is achieved. The observation of complementary product ion pairs confirms the correctness of the sequence and also the accuracy of the mass fitting of the isotopic distribution of the aldolase tetramer. Top-down MS of the native protein provides complementary sequence information to top-down ECD and CAD MS of the denatured protein. Moreover, native top-down ECD of aldolase tetramer reveals that ECD fragmentation is not limited only to the flexible regions of protein complexes and that regions located on the surface topology are prone to ECD cleavage.
“Native” mass spectrometry (MS) is an emerging technique that has been successfully used to characterize intact, noncovalently-bound protein complexes, providing stoichiometry and structural information that is complementary to data supplied by conventional structural biology techniques.1–3 To confidently characterize protein complexes, electrospray ionization (ESI)-MS measurements acquired with isotopic resolving power (RP) and high mass accuracy and capabilities for deriving primary structure, i.e., sequence, information would be ideal. Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) is prominent for its superior resolving power and mass accuracy and its utility for tandem MS (MS/MS) with a variety of fragmentation techniques; FT-ICR MS is noted for characterizating posttranslational modifications (PTMs) and protein-ligand and protein-protein interactions.4–9 However, it remains challenging to isotopically resolving large biomolecules over 100 kDa due to sample heterogeneity, cation/solvent/buffer addition, space charge effects, and electric and magnetic field inhomogeneity (for FT-ICR).10–13 Unit mass resolution has been achieved for a few denatured proteins, including a 112 kDa protein with 3 Da mass error using a 9.4 T FT-ICR MS,14 a 115 kDa protein by a 7 T instrument with a mass error of 5 ppm,4 and a 148 kDa protein with a mass error of 1 Da by a 9.4 T FTMS.10
Compared to denatured proteins, it is more difficult to achieve isotopic resolution for inherently lower charged (and thus, higher m/z) native protein complexes because (1) the peak height is proportional to its charge state, (2) the resolving power is inversely proportional to mass-to-charge ratio for FT-ICR MS, and (3) the broader isotope distribution of large biomolecules reduces overall signal-to-noise ratio.15 However, the introduction of a new FT-ICR analyzer cell – the ParaCell, by Nikolaev and coworkers has significantly increased the resolving power of FT-ICR MS.16, 17 By dynamically harmonizing the electric field potential at any radius of cyclotron motion in the entire cell volume, a resolving power of 39 M has been achieved for the alkaloid, resperine (m/z 609), using a 7 T system.18 In addition, a few native protein complexes, including enolase dimer (93 kDa, RP ~ 800,000 at m/z 4250), alcohol dehydrogenase tetramer (147 kDa, RP ~ 500,000 at m/z 5465), and enolase tetramer (186 kDa), have been isotopically resolved with a 12 T FT-ICR system with the new ICR cell.18 Although Mitchell and Smith reported that cyclotron phase locking due to Coulombic interactions limits the highest mass that unit mass resolution can be achieved by FT-ICR MS (Mmax ≈ 1×104B, where B is magnetic field strength),19 the ParaCell has made it significantly easier and promising to measure high resolution mass spectra for large native protein complexes.
……
Native top-down CAD and ISD were performed for the aldolase tetramer; dissociation of the tetramer to yield monomer was observed in both approaches and no sequence information was obtained. The cleavage sites from ECD (colored in red) and CAD (colored in green) of the denatured aldolase monomer (26+) are overlaid with the native ECD results for aldolase tetramer (Figure 2B). As shown in Figure 2B, in contrast to the limited number of c-ion fragments observed in the ECD of aldolase tetramer, ECD of denatured aldolase monomer induces extensive c-ion fragments in the N-terminal region and enables the assignment of first 156 N-terminal AA residues. Surprisingly, the number of z•-ions observed from ECD of the denatured aldolase monomer is much less compared to the ECD of the native aldolase tetramer. Although it may be possible that the z•-ions may undergo secondary fragmentation due to excess available energy, electrons, or long ion-electron reaction times during the ECD experiment, ECD experiments with reduced reaction time and bias voltages were performed and the results argue against this assumption. Overall, 58% of the total number of backbone bonds are cleaved from combining top-down MS of native aldolase complex and denatured aldolase monomer (20% for native ECD of aldolase tetramer, 37% for ECD of denatured aldolase, and 5% for CAD of denatured aldolase).
The three dimensional structure of the aldolase tetramer is shown in Figure 3. To compare the flexibility of the structure to the data from ECD of the aldolase tetramer, one of the subunits (B-chain) is presented as B-factor putty and the D-chain is shown with its native ECD backbone cleavage regions colored in red. The remaining A- and C-chains are shown in grey. Although the C-terminal region (AA 340–363) of each subunit is highly flexible based on the crystallography B-factor (see B-chain in Figure 3A), only 4 out of 75 backbone cleavage sites are from the AA 340–363 region. Instead, the native ECD fragments largely originate from surface regions of the protein structure (see D-chain in Figure 3A). The N-terminal regions are not directly involved in the interfaces between subunits, but they are located in regions that are partially buried, which is consistent with the limited c-ions observed. To better show the native ECD backbone cleavage regions, the D-chain is rotated 90 degrees clockwise (Figure 3B). It is clear that, although protein structure flexibility might play a role in the native top-down ECD fragmentation pattern, for aldolase the ECD cleavage sites are not limited to the flexible region. In addition, backbone cleavage regions from CAD (yellow) and ECD (cyan) of denatured aldolase are complementary with the native ECD results.
A) Structure of tetrameric aldolase (1ZAH)29. A- and C-chains are shown as grey ribbons, the B-chain is shown in B-factor putty, and the D-chain is in cartoon with native ECD cleavage sites colored in red, CAD cleavage sites of denatured aldolase in yellow, and ECD cleavage sites of the N-terminal region from ECD of denatured aldolase in cyan. B) The D-chain is rotated 90 degrees clockwise to show the outer surface region of the subunit structure.
Also evident in such data sets are protein–small molecule interactions. As the proteins break apart, Loo explains, ligands often remain attached to the polypeptide shards that are produced. In one recent publication, for instance, his team mapped zinc binding sites in eukaryotic alcohol dehydrogenase, a 147-kDa tetrameric complex (J Am Soc Mass Spectrom, 25:2060-8, 2014).
Revealing Ligand Binding Sites and Quantifying Subunit Variants of Non-Covalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment
“Native” mass spectrometry (MS) has been proven increasingly useful for structural biology studies of macromolecular assemblies. Using horse liver alcohol dehydrogenase (hADH) and yeast alcohol dehydrogenase (yADH) as examples, we demonstrate that rich information can be obtained in a single native top-down MS experiment using Fourier transform ion cyclotron mass spectrometry (FTICR MS). Beyond measuring the molecular weights of the protein complexes, isotopic mass resolution was achieved for yeast ADH tetramer (147 kDa) with an average resolving power of 412,700 at m/z 5466 in absorption mode and the mass reflects that each subunit binds to two zinc atoms. The N-terminal 89 amino acid residues were sequenced in a top-down electron capture dissociation (ECD) experiment, along with the identifications of the zinc binding site at Cys46 and a point mutation (V58T). With the combination of various activation/dissociation techniques, including ECD, in-source dissociation (ISD), collisionally activated dissociation (CAD), and infrared multiphoton dissociation (IRMPD), 40% of the yADH sequence was derived directly from the native tetramer complex. For hADH, native top-down ECD-MS shows that both E and S subunits are present in the hADH sample, with a relative ratio of 4:1. Native top-down ISD MS hADH dimer shows that each subunit (E and S chain) binds not only to two zinc atoms, but also the NAD+/NADH ligand, with a higher NAD+/NADH binding preference for the S chain relative to the E chain. In total, 32% sequence coverage was achieved for both E and S chains.
Studying how proteins interact with one another and assemble on a structural basis is key to understanding biological processes and their function. As a complementary technique to conventional technologies used in structural biology, such as nuclear magnetic resonance (NMR) spectroscopy, X-ray crystallography, and electron microscopy, “native” mass spectrometry (MS) has established its crucial role in the characterization of intact noncovalently-bound protein complexes, revealing the composition, stoichiometry, dynamics, stability, and also spatial information of subunit arrangements in protein assemblies [1–11]. To date, most native MS studies of protein complexes have been performed using quadrupole time-of-flight (Q-TOF) MS instruments with electrospray ionization (ESI). Because of the efficient transmission of high mass and highm/z ions using TOF analyzers, large proteins with molecular weights up to 18 MDa have been studied [12,13]. The coupling of ion mobility spectrometry (IMS) with mass spectrometry provides a new dimension to the analysis of biomolecules [14]. With IMS, ions are separated based on size and shape, and the IMS-derived collision cross-section information can be used to understand the topological properties of gas phase protein complexes. Surface induced dissociation (SID) has been recently added for the purposes of disassembling protein complexes into sub-complexes that appear to better reflect the structure of the solution phase complexes [15–17]. The capability of Orbitrap MS has been extended significantly for the analysis of macromolecules, with greatly improved mass (and m/z) range and resolving power to measure the binding of ADP and ATP to the 800 kDa GroEL complex [18].
Fourier transform ion cyclotron resonance mass spectrometry (FTICR MS) is known for its superior resolving power and mass accuracy and its capabilities for tandem MS (MS/MS) with a variety of fragmentation techniques. Particularly, after the introduction of electron capture dissociation (ECD) [19], FTICR MS quickly established its utility for protein top-down protein sequencing, post-translational modification characterization, and protein gas phase studies [20–34]. Polypeptide backbone bonds are cleaved by ECD, but non-covalent interactions are preserved, which therefore makes the native top-down MS study of the non-covalent interaction sites of protein-ligands complexes more feasible. Our group and others have successfully applied top-down ECD-MS to pinpoint the interaction sites of several protein-ligand system [35–38], and this can be enhanced by “supercharging” [35]. An early attempt of applying ECD-MS to the study of large protein complexes was made by Heeren and Heck, but little topology and sequence information was derived [39]. However, the Gross group starting in 2010 made the first breakthrough for the study of large protein complexes using native top-down ECD with FTICR MS. Besides obtaining molecular weight, sequence, and metal-binding site information in a single MS experiment, they correlated the origins of ECD product ions to the flexible regions of proteins as determined by the “B-factor” from the X-ray crystal structures of protein complexes [40, 41]. Therefore, native top-down ECD has been proposed as a tool to probe the flexible regions of protein complexes. Our group recently also demonstrated the capability of obtaining sequence information and isotopic mass resolution of a noncovalently-bound protein complex of 158 kDa using native top-down FTICR MS, and most importantly, we found that the origin of ECD fragments is not limited only to the flexible region of the protein complex (e.g., tetrameric aldolase), but also largely from the surface of the complex [42].
The application of FTICR MS for native top-down interrogation of large non-covalent bound protein complexes is still in its infancy. Here, for the purpose of further exploring the capability of FTICR MS in the analysis of large protein complexes, various fragmentation techniques including in-source dissociation (ISD), collisionally activated dissociation (CAD), ECD, and infrared multiphoton dissociation (IRMPD) were applied in the native top-down MS studies of a 80 kDa dimeric protein complex and a 147 kDa tetrameric protein complex. The results demonstrate that with the superior resolving power, mass accuracy, and versatile fragmentation techniques of FTICR MS, rich information, including isotopic mass resolution, amino acid sequence, point mutations, metal/ligand binding sites, and identification and quantification of subunit variants can be accomplished in a single native top-down FTICR MS experiment.
Still, Loo admits, the technique “is not really ready for prime time.” His team is collecting ECD data on a bank of proteins of known structure to ensure the data they collect really do reflect protein topology. In the meantime, they are working to extend the size of the complexes they can analyze. The technique’s current limit is 800 kDa.
GO NATIONAL: FTICR mass spectrometers offer top-of-the-line accuracy and resolution, with price tags to match. Few researchers have direct access to them, Loo says, but they can always try the national laboratories. Both the National High Magnetic Field Laboratory at Florida State University and the Environmental Molecular Sciences Laboratory at the Pacific Northwest National Laboratory have user facilities open to worthy projects.
Determining the architecture of protein complexes
RESEARCHER: Vicki Wysocki, Ohio Eminent Scholar and Professor of Chemistry and Biochemistry, Ohio State University
PROJECT: Instrumentation development for whole-complex analysis
SOLUTION: An analytical chemist by training, Wysocki focuses on instrumentation development for protein-complex analysis. Among the discoveries in her lab is a method called surface-induced dissociation (SID).
HIT THE WALL, JACK: When it comes to molecular collision in a mass spectrometer, size matters. Collide a complex with small gas molecules, and proteins in the complex will simply unravel (top). By smacking them into a “wall”—a process called surface-induced dissociation—the complex dissociates to reveal its underlying architecture. COURTESY OF VICKI WYSOCKI
Like many other fragmentation approaches, SID works by forcing an ion in the mass spectrometer to collide with another object. Usually that object is a small gas molecule, with the energy of collision sufficient to crack the peptide backbone. But for large protein complexes, bigger is better, and the collision partner in SID is as big as it can get: the method slams protein ions of interest into a nonreactive surface inside the instrument—essentially, a wall—causing complexes to fracture into subcomplexes that reveal the assembly’s inner architecture.
Wysocki combined this approach with ion-mobility separation—a kind of gas-phase electrophoresis that resolves molecules by their size and shape—to dissect an enzyme involved in antibiotic production. The enzyme, they found, has two copies each of three subunits, alpha, beta, and gamma, arranged as a pair of triads sitting on top of one another, with the alpha and beta subunits of one triad linked more tightly to each other than either is to gamma (Anal Chem, 83:2862-65, 2011).
Such information can be valuable to protein engineers, Wysocki says, especially as this particular complex otherwise falls into a structural biology knowledge gap: “It doesn’t crystallize, and it’s too small for the cryoEM and a little bit large for NMR,” she says. “And so, mass spec turned out to be a great tool.”
Revealing the Quaternary Structure of a Heterogeneous Noncovalent Protein Complex through Surface-Induced Dissociation
As scientists begin to appreciate the extent to which quaternary structure facilitates protein function, determination of the subunit arrangement within noncovalent protein complexes is increasingly important. While native mass spectrometry shows promise for the study of noncovalent complexes, few developments have been made toward the determination of subunit architecture, and no mass spectrometry activation method yields complete topology information. Here, we illustrate the surface-induced dissociation of a heterohexamer, toyocamycin nitrile hydratase, directly into its constituent trimers. We propose that the single-step nature of this activation in combination with high energy deposition allows for dissociation prior to significant unfolding or other large-scale rearrangement. This method can potentially allow for dissociation of a protein complex into subcomplexes, facilitating the mapping of subunit contacts and thus determination of quaternary structure of protein complexes.
The majority of proteins exist and perform their functions as multimers of varing stoichiometries and architecture.1 However, very few methods are available that can provide insights into subunit interactions. Native mass spectrometry (MS) is increasingly being used to study noncovalent protein complexes, as many structural features found in solution may be maintained in the gas phase.2,3 While subunit stoichiometries are readily obtainable by mass measurement alone, the determination of subunit arrangement within protein complexes remains a significant challenge. This is particularly true for heterogeneous complexes with multiple types of subunits. Considerable progress has been made using solution-phase disruption to divide the original protein complex into smaller subcomplexes, which may be readily measured by MS.4,5 The composition of the stable subcomplexes provides insight on the topology of the protein complex. However, MS activation methods used to date have fallen short of providing subunit topology. Here, we present the first evidence for subunit arrangement obtained directly from gas-phase experiments on a heterogeneous complex via surfaceinduced dissociation (SID). We have demonstrated previously the ability of SID to yield unique dissociation pathways for protein complexes, resulting in complementary information to collision-induced dissociation (CID).68 While the SID process is not yet well understood for macromolecules, there is a large body of work concerning SID of small molecules; influential factors such as collision energy, surface composition, and translational-to-vibrational energy conversion have been well-studied.911 The higher effective mass of a surface relative to that of neutral gas atoms used in CID (typically argon) results in significantly higher energy deposited through a single surface collision.9 As SID is a single-collision activation process, rather than activation via thousands of less energetic collisions as in CID, dissociation pathways other than those of the lowest energies become accessible
……
This is the only study to date demonstrating an ion activation method capable of yielding extensive dissociation, as well as the release of intact subcomplexes, thus providing relevant substructure information on a noncovalent, hetero-oligomeric protein complex. The capacity to produce intact, charge-symmetric subcomplexes suggests that dissociation occurs faster than subunit unfolding and that a significant degree of secondary and tertiary structure is maintained up to the point of dissociation and for some period of time afterward. Identification of trimeric substructure in TNH provides insight into a protein with little previous structural characterization and indicates a promising advancement of MS as a tool for structural biology.
Such information can be valuable to protein engineers, Wysocki says, especially as this particular complex otherwise falls into a structural biology knowledge gap: “It doesn’t crystallize, and it’s too small for the cryoEM and a little bit large for NMR,” she says. “And so, mass spec turned out to be a great tool.”
CHOOSE MASS: Mass spec may not be the only method for quickly working out protein structure, but it surely is the fastest, Wysocki says. She recalls one instance when a colleague sent over a complex that his group couldn’t crack. “In one afternoon, my student gave them a prediction of the structure: this one’s a heptamer, with a large subunit sitting atop a hexameric ring.” Even if the experiment doesn’t work, she adds, that fast turnaround time can be a boon, as collaborators can get rapid feedback for tweaking their experimental conditions. “Mass is a great thing.”
Pentaalkylcyclopentadienyl (Cp*R) iridium (Ir) and cobalt (Co) 1,2-diamine complexes were synthesized. Susceptibility of Staphylococcus aureus and recent patient methicillin-resistant S. aureus (MRSA) isolates to the transition metal–diamine complexes were measured by broth microdilution and reported as the MIC and MBC. Hemolytic activities of the transition metal-complexes as well as toxicity toward Vero cells were also measured. The transition metal complex of Cp*RIr with cis-1,2-diaminocyclohexane, had strong antibiotic activity against S. aureus and MRSA (MIC = 4 μg mL−1, MBC = 8 μg mL−1) strains and killed 99% of S. aureus cells in 6 hours. Stronger antibiotic activity was associated with the presence of octyl linked to the cyclopentadienyl group and cyclohexane as the diamine backbone. Activity was greatly diminished by tri- or tetramethylation of the nitrogen of the diamine. A cyclopentadienylcobalt complex of cis-1,2-diaminocyclohexane also showed significant anti-microbial activity against both S. aureus and MRSA strains. The absence of hemolytic activity, Vero cell cytotoxicity and the significant anti-microbial activity of several members of the family of compounds reported suggest this is an area worth further development.
Daniel Sykes, Ahmet J. Cankut, Noorshida Mohd Ali, Andrew Stephenson, Steven J. P. Spall, Simon C. Parker, Julia A. Weinstein and Michael D. Ward
Dalton Trans., 2014,43, 6414-6428
Abstract
In Ir(III)/Eu(III) and Ir(III)/Tb(III) dyads, sensitization of lanthanide luminescence occurs via both energy-transfer and electron-transfer pathways on similar timescales.
Junpei Kuwabara, Tomomi Namekawa, Masa-aki Haga and Takaki Kanbara
Dalton Trans., 2012,41, 44-46
Abstract
Ir(III) complexes that contain benzothiazole-based tridentate ligands were synthesized and their crystal structures and luminescent properties were examined. A neutral complex had a high quantum yield (89%) and performed well as an emissive material for organic light-emitting diodes.
Multivalent glycoconjugates as vaccines and potential drug candidates
Pathogens adhere to the host cells during the first steps of infection through multivalent interactions which involve protein–glycan recognition. Multivalent interactions are also involved at different stages of immune response. Insights into these multivalent interactions generate a way to use suitable carbohydrate ligands that are attached to a basic scaffold consisting of e.g., dendrimer, polymer, nanoparticle, etc., with a suitable linker. Thus a multivalent architecture can be obtained with controllable spatial and topology parameters which can interfere with pathogen adhesion. Multivalent glycoconjugates bearing natural or unnatural carbohydrate antigen epitopes have also been used as carbohydrate based vaccines to stimulate an innate and adaptive immune response. Designing and synthesizing an efficient multivalent architecture with optimal ligand density and a suitable linker is a challenging task. This review presents a concise report on the endeavors to potentially use multi- and polyvalent glycoconjugates as vaccines as well as anti-infectious and anti-inflammatory drug candidates.
Proteochemometric (PCM) modelling is a computational method to model the bioactivity of multiple ligands against multiple related protein targets simultaneously. Hence it has been found to be particularly useful when exploring the selectivity and promiscuity of ligands on different proteins. In this review, we will firstly provide a brief introduction to the main concepts of PCM for readers new to the field. The next part focuses on recent technical advances, including the application of support vector machines (SVMs) using different kernel functions, random forests, Gaussian processes and collaborative filtering. The subsequent section will then describe some novel practical applications of PCM in the medicinal chemistry field, including studies on GPCRs, kinases, viral proteins (e.g. from HIV) and epigenetic targets such as histone deacetylases. Finally, we will conclude by summarizing novel developments in PCM, which we expect to gain further importance in the future. These developments include adding three-dimensional protein target information, application of PCM to the prediction of binding energies, and application of the concept in the fields of pharmacogenomics and toxicogenomics. This review is an update to a related publication in 2011 and it mainly focuses on developments in the field since then.
Bum Jun Park and Daeyeon Lee Soft Matter, 2012,8, 7690-7698
Abstract
The equilibrium configuration of amphiphilic ellipsoids and dumbbells at the oil–water interface strongly depends on the particle characteristics, such as their shape and wettability.
Towards understanding cell penetration by stapled peptides
Hydrocarbon-stapled α-helical peptides are a new class of targeting molecules capable of penetrating cells and engaging intracellular targets formerly considered intractable. This technology has been applied to the development of cell-permeable ligands targeting key intracellular protein–protein interactions. However, the properties governing cell penetration of hydrocarbon-stapled peptides have not yet been rigorously investigated. Herein we report our studies to systematically probe cellular uptake of stapled peptides. We developed a high-throughput epifluorescence microscopy assay to quantitatively measure stapled peptide intracellular accumulation and demonstrated that this assay yielded highly reproducible results. Using this assay, we analyzed more than 200 peptides with various sequences, staple positions and types, and found that cell penetration ability is strongly related to staple type and formal charge, whereas other physicochemical parameters do not appear to have a significant effect. We next investigated the mechanism(s) involved in stapled peptide internalization and have demonstrated that stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that involves, in part, sulfated cell surface proteoglycans, but that also seems to exploit a novel, uncharacterized pathway. Taken together, staple type and charge are the key physical properties in determining the cell penetration ability of stapled peptides, and anionic cell surface proteoglycans might serve as receptors to mediate stapled peptide internalization. These findings improve our understanding of stapled peptides as chemical probes and potential targeted therapeutics, and provide useful guidelines for the design of next-generation stapled peptides with enhanced cell permeability.
Introduction Hydrocarbon stapled a-helical peptides are an exciting new class of investigational agents capable of targeting and interfering with intracellular protein–protein interactions.1,2 (For reviews on hydrocarbon stapled peptides, see ref. 3 and 4, and for reviews on synthetic a-helix stabilization in general, see ref. 5 and 6.) These peptides contain a synthetic brace, referred to as a staple, introduced across one face of an a-helix (Fig. 1), that in favorable cases can increase a-helical content and protease resistance, enhance target binding affinity, promote cell membrane penetration, and suppress clearance in vivo. 7–10
Fig. 1 All-hydrocarbon stapled peptide technology. (a) Schematic illustration of peptide stapling. Two alpha-methylated, alkenyl-bearing non-natural amino acids are incorporated at two or more positions in the peptide chain and then cross-linked by ruthenium-catalyzed ringclosing olefin metathesis. (b) Different types of alkenyl-containing non-natural amino acids with distinct stereochemistry at the a-carbon and varied lengths of alkenyl side chains. (c) Three types of stapled peptides used in this study with optimized combinations of nonnatural amino acids.
Stapled peptides are synthesized via incorporation of two amethyl, a-alkenyl amino acids at defined positions in a synthetic peptide, followed by ring-closing olefin metathesis to close the helix-spanning hydrocarbon bridge (Fig. 1a).11,12 The two components of the staple, namely the hydrocarbon bridge and terminal methyl groups, are both important to obtain maximal effectiveness of the conformationally constrained peptide products. This technology has been successfully utilized to target several classes of proteins formerly considered intractable, including multi-component transcription factor complexes and protein–protein interactions having extended interfaces, such as the NOTCH transcription factor complex,13 the b-catenin–TCF interaction in the oncogenic Wnt signaling pathway,14 and the epigenetic modulator PRC2 complex.15 Given the difficulties of developing traditional small molecule drugs that can successfully target intracellular protein–protein interactions, hydrocarbon stapling technology is widely considered to represent a promising avenue of research for the development of chemical probes and potential targeted therapeutics.
Multiple types of hydrocarbon staples have been obtained by varying the relative placement of the cross-linking a,a-disubstituted amino acids, as well as the stereochemistry at the acarbon and the lengths of the alkenyl substituents (Fig. 1b).16,17 These staple types were optimized to provide robust a-helical stabilization and confer the potential for in vitro and in vivo activity. As a result of the combinatorial search process used to identify helix-stabilizing hydrocarbon staples, the diversity of the resulting macrocyclic bridges has revealed stapled peptides with different physicochemical properties. Recently, a new hyperstable version of stapled peptide with tandem crosslinks, referred to as a stitched peptide, was generated by introduction of S5 at the i position, B5 at the i + 4 position, and S8 at the i + 11 position (Fig. 1c) (Y.-W. Kim and G. L. Verdine, to be published).
Of the physicochemical properties demonstrated by peptide bearing hydrocarbon staples, the capacity to promote cellular membrane penetration is perhaps the most signicant and yet remains the most poorly understood. Independent of hydrocarbon-stapled peptides, several classes of cell penetrating peptides (CPPs) have been discovered, including naturally occuring transcription factor domains such as pennetratin18 and HIV-Tat19 and synthetic cationic peptides such as polyArginine peptides.20 Notably, despite extensive exploration during the past two decades, the mechanism(s) by which CPPs enter cells remain unclear.21–23 In contrast to CPPs, in which cell penetration appears to be sequence-dependent, numerous cell permeable stapled peptides have been discovered for peptide scaffolds with little sequence homology. These divergent observations regarding cell penetration is proposed to result from several features of stapled peptides that differentiate them from typical CPPs. For example, the introduction of an allhydrocarbon cross-link results in a constrained a-helical conformation, which embeds the hydrophilic amide backbone in the core of the folded structure. Furthermore, the hydrocarbon brace itself introduces a significantly hydrophobic patch to one face of the peptide. The exposure of the hydrophobic moiety as well as the masking of the hydrophilic peptide backbone may facilitate the interaction of stapled peptides with the hydrophobic interior of the cell membrane and thereby enhance the cellular uptake. As cell penetration is a critical property of stapled peptides, we sought to develop quantitative methods to correlate a battery of stapled peptide properties with the capacity for cellular uptake. A direct comparison with several well-known CPPs has revealed that stapled peptides, including some stapled versions of the CPPs, exhibit more robust cell penetration. Lastly, we have demonstrated that stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that involves, in part, sulfated cell surface proteoglycans. These findings significantly expand our current understanding of cell penetration by stapled peptides and provide useful information for the future rational design of cell penetrating stapled peptides with novel applications.
Results and discussion
Development of a high-throughput assay to quantitatively measure cellular uptake of peptides
Understanding the internalization process of cell penetrating peptides (CPPs), especially stapled peptides, has been a subject of great interest. The majority of previous studies have been performed by either using high-resolution microscopy to show the existence of fluorophore-labeled CPPs inside cells, or by quantitatively measuring intracellular fluorescence by flow cytometry.24,25 Although these two methods can provide important information regarding cell penetration, their respective limitations prompted us to adopt an assay that combines high-resolution imaging with reliable quantitation of intracellular accumulation to better analyze and understand the cell penetration of stapled peptides. In recent years, highthroughput cell-based imaging platforms have become increasingly popular to screen for small molecule modulators of various biological processes.26,27 Taking advantage of one of these platforms, high-content epifluorescence microscopy, we developed a high-throughput quantitative assay to measure stapled peptide intracellular access.
Proof-of-principle experiments were performed to determine whether epifluorescence microscopy could be used to quantitatively compare stapled peptide intracellular access. Human U2OS osteosarcoma cells were seeded in black, clear-bottom 384-well plates and then incubated in serum-containing media supplemented with fluorescein-labeled peptides or DMSO vehicle for 12 hours. After the treatment, cells were washed thoroughly with PBS to remove excess peptide, fixed with 4% formaldehyde, and stained with Hoechst dye to visualize nuclei. Once prepared, the plates were imaged and quantified by epi- fluorescence microscopy according to a protocol developed and discussed in detail in Experimental methods. An initial z-scan was performed using the Hoechst channel to locate the cells, and the microscope parameters were subsequently adjusted to optimize the cell size and fluorescence intensity. The parameters from this acquisition were then applied to the FITC channel, and the microscope scanned and recorded images of the FITC-labeled peptides within the z-plane of the cell. This assay was performed in a high-throughput manner, resulting in a panel of Hoechst/FITC images from individual wells (Fig. S1†). The raw image data was then analyzed using MetaXpress® software (Fig. 2a). Cells were identified based on the Hoechst stain of nuclei, with the requirement that they were a contiguous fluorescent region having a specific intensity above local background as well as having a diameter between defined minimum and maximum to be designated as “positive” cells. The cytoplasm of each cell was then identied according to the spatial location of FITC signal in relation to the nuclei as well as empiric parameters (details in Experimental methods). The FITC intensities in the cytoplasm and nuclei were then quantified separately, and the sum of these two values yielded the FITC signal for the whole cell, which can be considered the relative intracellular peptide intensity. In addition, FITC negative cells were identified on the basis of a positive Hoechst stain, which was accompanied by an absence of appreciable signal in the FITC channel.
Fig. 2 Quantitative measurement of cellular peptide intensity. (a) Hoechst channel (left) showing the location and size of nuclei, FITC channel (middle) showing the fluorescence intensity of the same cells. Information about cell size and fluorescence intensity was integrated to identify the FITC positive (green mask) and negative (red mask) cell (right). For positive cells, additional parameters allowed determination of the fluorescence intensity in the nucleus (inner intense green) and the cytoplasm (outer dim green). (b) The background fluorescence the DMSO vehicle was almost identical among different experiments. (c) Four stapled peptides from different batches of synthesis generated similar intracellular fluorescence intensity in different tests. Error bars represent the S.D. of two measurements.
We found that this system generated highly reproducible and reliable results from assay-to-assay and with different stocks of the same stapled peptides. As shown in Fig. 2b, there were negligible fluorescence differences among experiments for cells treated with DMSO vehicle, which could be used as a fluorescence background for all subsequent experiments. In addition, the same stapled peptides from different batches of synthesis and stocks featured almost identical intracellular fluorescence signals in different tests (Fig. 2c), indicating that the assay developed in this study produces repeatable and reliable results that could be directly combined and compared from a large set of experiments. Furthermore, to determine how this assay performs as a screening tool, we have calculated Z0 factor of 0.54 by using the most penetrant A6 peptide as a positive control and DMSO background as negative control, which also indicates a statistically good assay quality.
Analysis of cell penetration by stapled peptides The development of this quantitative high-throughput assay enabled a broad investigation of the physicochemical properties governing the cell uptake of a diverse set of hydrocarbonstapled peptides synthesized in our laboratory. We postulated that any correlation between cellular uptake and physicochemical properties would illuminate characteristics associated with productive cellular uptake and inform the future design of stapled peptides with improved cell penetration.28 To this end, we screened and analyzed more than 200 discrete FITC-labeled peptides belonging to three different classes: wild-type (unmodified), stapled and stitched peptides. All peptides were converted to two-dimensional structures and analyzed for theoretical physicochemical properties with the publicly available Marvin View software package from ChemAxon. Properties including the molecular weight, theoretical pI, calculated 2D polar surface area (PSA), theoretical log P and formal charge at pH 7.5 were calculated for each peptide (Table S1†). In general, the unmodified, stapled and stitched peptide libraries present in this screen had relatively similar physicochemical characteristics (Fig. S2†). The mean molecular weight and calculated PSA values were nearly identical among the three peptide classes. A notable difference was observed among theoretical log P values, which were significantly higher for the stapled and stitched peptides relative to the unmodified peptides, which is not surprising as these modified peptides contain a solvent exposed hydrocarbon crosslink. Additionally, the stapled peptide class had a mean formal charge of approximately zero while the stitched and unmodified peptide classes exhibited a positive mean charge. Overall, the calculated physicochemical properties indicated that the peptide classes were quite similar in terms of their mean properties, which is useful when making comparisons among their cell penetration properties.
We next performed an intracellular access screen by treating U2OS cells with 1 mM of FITC-labeled peptide for 12 hours in duplicate. All assays contained control DMSO wells and positive control peptides, which were compared among assays to ensure plate-to-plate reproducibility (Fig. 2b and c). The primary readout of the screen was mean cellular fluorescence intensity. As the DMSO background was highly consistent between wells and experiments, a mean background value was subtracted from all data. The results of the screen were used to generate plots comparing cell penetration with peptide physicochemical parameters. Interestingly, as a class, stapled and stitched peptides exhibited significantly higher cell penetration compared with wild-type unmodified peptides, which contained several established cell penetrating peptides (CPPs; Fig. 3a). Given that all three peptide classes have similar physicochemical properties in general, the benefit in cell penetration can be largely attributed to the synthetic stabilization of the a-helical peptides with all-hydrocarbon peptide stapling technology. Furthermore, we found that peptide charge near physiologic pH exhibited a strong correlation with intracellular access and could be fitted into a Gaussian distribution with a population centroid at a formal charge of +4 (Fig. 3b). In particular, peptides exhibiting a net negative charge (7 to 1) exhibited little cellular uptake, whereas peptides of approximately neutral charge (1 to +1) displayed moderate cell penetration above background. Interestingly, peptides with a net positive charge (+1 to +7) showed significantly higher cell penetration as a group. Cellular uptake did not appear to increase linearly with charge, as the cell penetration decreases dramatically for the peptides in this study with charge greater than +7. The same trend between formal charge and cellular uptake were observed for individual stapled and stitched peptide classes as well (Fig. S3†). This observation is not consistent with previously reported models that indicate that peptides/mini-proteins with more positive charge have better penetration properties due to tighter electrostatic interactions with the negatively charged phospholipid membrane.29,30 The lower penetration for highly charged peptides in this study could result from any one of many factors including, for example, peptide aggregation in solution, the disruption of peptide packing during internalization or difficulty in dissociation from cell membrane. Additional tests with a larger number of peptides could further our understanding of this phenomenon. In addition, there was no discernible correlation between cell penetration and peptide molecular weight, log P, pI value or PSA (Fig. S4†). Taken together, these data demonstrated that the staple type and peptide charge are key physical properties correlated with peptide cell penetration ability, whereas the other parameters do not appear to be significantly associated.
In order to further investigate the cell penetration properties for stapled peptides and to systematically analyze the similarities and differences in cellular uptake between stapled peptides and other wild-type cell penetrating peptides, we compared cell penetration of several stapled peptides to that of three well known wild-type CPPs: Tat (48–60), penetratin (Antennapedia 43–58) and poly-Arg8 (Table S2†). First, we investigated the cellular uptake at varied peptide concentrations. As shown in Fig. 4a and b, both wild-type CPPs and stapled peptides showed dose-dependent increases in cell penetration. Strong intracellular fluorescence was detected in the low micromolar range, and although the levels of accumulation were different for distinct peptides, stapled peptides featured more robust dosedependent cell penetration at lower concentrations relative to wild-type CPPs, in general. It is also interesting to note that while significant increases in intracellular fluorescence were mostly evident in the 1–10 mM range for stapled peptides, distinct profiles were observed for specific peptides. For example, TNG147 showed little cell penetration at 1 mM but showed a dramatic increase at 5 mM, which might suggest that concentration-dependent peptide packing or a receptor-mediated mechanism may facilitate the cell penetration process, and these processes may be triggered at different concentrations for distinct peptides. Furthermore, it is worth noting that the stapled peptides studied here were more cell permeable than wild-type CPPs at most concentrations tested, exhibiting nearly an order of magnitude higher intracellular fluorescence at the same treatment concentrations.
Fig. 4 Effects of peptide concentration and incubation time on cellular uptake of stapled and wild-type peptides. (a) Wild-type and (b) stapled peptides showed a dose-dependent increase in cell internalization. Cellular uptake for (c) penetratin and (d) SAHM1 peptides over time at concentrations of 5 and 10 mM. (e) A pulse-chase penetration assay for SAHM1 peptide in which fresh medium containing either a new batch of peptide or DMSO vehicle were exchanged at 12 hours after initial treatment. Error bars represent the S.D. of triplicate samples.
We next performed a time-course penetration assay to better understand the kinetics of peptide internalization using a representative CPP and stapled peptide. Penetratin and SAHM1 showed distinct kinetics of uptake and stabilization throughout a 24 hour time course. 5 mM and 10 mM penetratin peptide exhibited similar intracellular cellular fluorescence after 2 hours, which then decreased until approximately 8 hours and finally stabilized at different intracellular levels until 24 hours (Fig. 4c). On the other hand, the stapled peptide SAHM1 showed time- and dose-dependent cellular uptake, which stabilized after approximately 8 hours (Fig. 4d). Compared to the wild-type penetratin, the SAHM1 profile was unique in that dose-dependent accumulation was evident at all time points and no loss of signal was observed. One explanation for the loss of signal observed with penetratin could be attributed to an equilibrium between cell penetration and subsequent intracellular proteolysis followed by export of the fluorophore. The presence of the all-hydrocarbon crosslink in its peptide sequence and lower net charge of SAHM1 relative to penetratin, could contribute to enhanced cellular uptake and reduced intracellular proteolysis, leading to continuous accumulation in cells. To further explore the equilibrium observed for stapled peptides, we performed a pulse-chase experiment using SAHM1. After 12 hours of incubation with SAHM1, cell culture medium was aspirated and the cells were extensively washed with PBS to completely remove excess peptide. Then fresh medium containing either a new batch of 1 mM peptide or DMSO vehicle was added to cells and incubated for the indicated time points (Fig. 4e). As expected, the cellular uptake increased for the first 12 hours incubation. After medium exchange, cells incubated with fresh medium containing DMSO vehicle retained the intracellular fluorescence intensity. Interestingly, the signal for cells treated with a new batch of staple peptide continued to increase up to 24 hours (Fig. 4e). This observation indicates that despite incubation over a time course previously shown to reach equilibrium, the mechanism(s) responsible for cellular uptake are not saturated, as evidenced by further uptake upon replacement with fresh stapled peptide. Taken together, these data indicate that the mechanism(s) underlying cellular uptake by both CPPs and stapled peptides exhibit time- and dose-dependency that is not saturable at early time points or low micromolar doses and, importantly, appears to be more robustly utilized by stapled peptides.15,31
Given that stapled peptides exhibit better cell penetration properties in general than parent unmodified peptides, we wondered whether the peptide stapling strategy could be applied generally to improve cellular uptake of parent unmodified peptides. To test this hypothesis, we designed a panel of stapled peptides based on Tat (48–60), penetratin and poly-Arg8 (Fig. 5a). These stapled peptides and their parent unmodified peptides were incubated in U2OS cells for 12 hours with a concentration range from 10 nM to 20 mM, mirroring the dosedependent uptake studies shown in Fig. 4. As expected, all peptides showed dose-dependent cell penetration (Fig. 5b–d). Interestingly, stapled peptides derived from penetratin and poly-Arg8 showed improved cell permeability at concentrations starting from 1 mM for stapled penetratin and 5 mM for stapled poly-Arg8. It is noteworthy that the staple position also affected the cellular uptake as the two stapled penetratin peptides with different crosslink positions exhibited varied cell penetration, though both were superior to wild-type penetratin. In contrast, reduced cellular uptake was observed for both stapled peptide variants derived from the Tat sequence (Fig. 5b). This could result from several possible effects, including disruption of peptide secondary structure, masking of residues essential for surface recognition or altering peptide packing interactions involved in cell penetration. Further focused study of these variants is warranted to elucidate the source of altered cellular uptake, however these data clearly demonstrate that peptide stapling may be a general method to further improve the cell permeability of CPPs, which could serve as more efficient transduction domains for molecular cargoes. In addition, while increasing the helical content of stabilized peptides has been stated to be a guiding principle in the successful design of biologically active stapled peptides, it has not been shown to be generally correlated with cell penetration. To specifically address whether increasing the helical content of a peptide is correlated with augmented cell penetration, we have measured the relative helicity of hydrocarbon stapled variants of Tat, penetratin and poly-Arg8 (Fig. S5†). Notably, we did not observe a general correlation between increased helical character and cell penetration of these peptides. Peptide stapling increased the helical content of both Tat and poly-Arg8 peptide sequences, which were largely unstructured when unmodified. In contrast, the unmodified penetratin peptide had signicant helical content (>50%), and the hydrocarbon stapled variants of this sequence largely retained their helicity, albeit lower overall helicity. Intriguingly, these species demonstrated the differing effect of hydrocarbon stapling and increased helical content on cell penetration since introduction of the hydrocarbon staple increased the cellular uptake of both penetratin and poly-Arg8 sequences, while it decreased uptake for Tat peptides. Therefore, we cannot conclusively state, a priori, that the incorporation of a hydrocarbon staple or increased a-helicity will lead to more productive cellular penetration, although in general stapling can increase the uptake of specific sequences (Fig. 5) and as a class stapled and stitched peptides are more cell penetrant (Fig. 3a). A more comprehensive follow-up study with CD analyses on a larger peptide library is needed to better address this question.
Fig. 5 Effects of all-hydrocarbon staples on cell penetration by wild-type cell penetrating peptides. (a) List of wild-type cell penetrating peptides and their stapled derivatives investigated in this study. (b–d) Dose-dependent cell penetration assays showed that stapling strategy greatly improves the cellular uptake of penetratin and poly-Arg8 peptides. Experiments were performed in triplicate, and error bars represent S.D. of three measurements.
Mechanistic studies of cell penetration by stapled peptides The aforementioned studies indicate that stapled peptides exhibit better cellular uptake properties than wild-type peptides in general, and that internalization correlated primarily with hydrocarbon staple type and formal peptide charge. However, the mechanism(s) utilized by peptides to translocate across the cell membrane are still unclear. Therefore, we sought to investigate the uptake mechanism(s) for stapled peptides. The uptake mechanism(s) of wild-type CPPs have been extensively studied. Some evidence indicates that they enter cells via energy-dependent endocytosis, which is an active transport process, however data suggesting passive diffusion for CPPs have also been reported; hence, the mechanism(s) of cell uptake by CPPs remains ambiguous.32–34 We first sought to determine whether cell penetration by stapled peptides and wild-type CPPs occurs via ATP-dependent endocytosis.2 Cells were pre-treated with NaN3 and 2-deoxyglucose (2-DG) to reduce cellular ATP levels, and then incubated with FITC-labeled peptides (wildtype and stapled) for 4 hours and compared to normal cells for intracellular fluorescence. Cellular ATP levels were confirmed to be decreased by approximately 90% after NaN3 and 2-DG treatment (Fig. S6†), but Tat and poly-Arg8 exhibited almost identical cellular uptake in ATP-depleted and normal cells, supporting the model that they utilize passive diffusion to translocate across the cell membrane. However, penetratin and all stapled peptides showed 20–50% lower accumulation in ATP-depleted cells, indicating an active trans-membrane process requiring cellular ATP (Fig. 6a). These data indicate that there may be more than one uptake mechanism for CPPs and stapled peptides, but that for the most robust cell penetrating peptides (penetratin and stapled peptides studied here), the internalization mechanism(s) involves ATP-dependent endocytosis.
Fig. 6 Mechanistic study of cell penetration by stapled peptides and wild-type cell penetrating peptides. (a) Cellular uptake in normal and ATP-depleted cells indicated that stapled peptides penetrate cells via an ATP-dependent endocytosis. (b) Impaired uptake was observed in NaClO3 treated cells, which inhibit proteoglycan biosynthesis. (c) Cell penetration of wild-type and stapled peptides in wild-type CHO and proteoglycan-deficient CHO cells. Experiments were performed in triplicate, and error bars represent S.D. of three measurements.*P < 0.05, **P < 0.01, ***P < 0.001.
Next, we sought to identify the specific pathway(s) utilized for cellular uptake, since energy-dependent endocytosis can be accomplished by several different pathways including caveolinand clathrin-mediated endocytosis. We repeated the cell penetration experiments under a variety of conditions that each blocked a different endocytosis pathway (Table S3†).35–37 We found that uptake was partially blocked in cells treated with sodium chlorate (Fig. 6b), which aborts the decoration of cells with sulfated proteoglycans, but was unaffected by inhibitors of other endocytic pathways (Fig. S7†). It thus appears that interaction with sulfated proteoglycans is responsible for some, but not all, endocytic uptake of stapled peptides and wild-type CPPs. It is reasonable to connect this result with the previous discovery that peptide charge is a key factor determining cell penetration. Proteoglycans are negatively charged under physiologic conditions due to the occurrence of sulfate groups, and these might form electrostatic pairs with positively charged peptides to facilitate anchoring on the cell membrane.38–40 To further confirm that sulfated proteoglycans are important to mediate cellular uptake for peptides, we performed a secondary assay using wild-type CHO cells (CHO-K1) and proteoglycan deficient CHO cells (pgsA-745) which harbor a defect in xylosyltransferase, thereby preventing glycosaminoglycan biosynthesis. All peptides showed similar penetration properties in wild-type CHO cells, but uptake was decreased by approximately 50% in proteoglycan-deficient CHO cells, consistent with the experiment using a small molecule inhibitor (Fig. 6c). Taken together, our data suggest that CPPs and stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that is in part mediated by interaction with anionic cell surface proteoglycans. This result is very similar to the previous reports on the mechanism of cellular uptake for supercharged GFP (scGFP), which likewise does not utilize clathrin- or caveolin-mediated endocytosis.41 Notably, scGFP internalization requires actin polymerization, which may not be required for peptide penetration (Fig. S7c†) types, and distinct physicochemical properties. As a result, we found that stapled peptides penetrate cells more efficiently than unmodified peptides, including well-characterized cell penetrating peptides. For the panel of peptides used in this study, only staple type and formal charge were significantly correlated with cell penetration potential, whereas the other physical parameters did not appear to have a signicant effect. We further studied the relationships between cellular uptake and
In conclusion, we sought to investigate the cell penetration properties of stapled peptides, which is one of the most significant yet poorly understood aspects of peptide stapling technology and cellular transduction technologies in general. In order to address this problem, we developed a high-throughput assay to quantitatively measure stapled peptide intracellular accumulation. Using this assay, we analyzed more than 200 discrete peptides with various sequences, staple positions and peptide concentration or incubation time, revealing that stapled peptides accumulate in cells in a dose-dependent fashion and reach steady intracellular levels over a course of a few hours. These studies revealed similar time- and dosedependent behavior for CPPs and stapled peptides, but stapled peptides, including stapled versions of CPPs, were shown to be 10- to 20-fold more penetrant, measured by intracellular fluorescence level at a given dose, than the most potent CPP. We also propose that the specific intracellular accumulation and stabilization kinetics of stapled peptides or unmodified CPPs may be a consequence of equilibria between peptide penetration, cellular proteolysis and/or retrograde transport of the species. Finally, we investigated the mechanism(s) involved in the internalization of stapled peptide and unmodified CPPs and demonstrated that cell penetration occurs through a clathrinand caveolin-independent, energy-dependent endocytosis pathway that utilizes, in part, sulfated cell surface proteoglycans. This dataset provides significant insight into the physicochemical properties correlated with productive cellular penetration as well as a more detailed understanding of the mechanism(s) utilized by stapled peptides to access intracellular compartments, which together should aid in the design of and characterization of novel stapled peptides in the future.
Rational design of protein–protein interaction inhibitors
Protein–protein interactions are at the heart of most physiopathological processes. As such, they have attracted considerable attention for designing drugs of the future. Although initially considered as high-value but difficult to identify, low molecular weight compounds able to selectively and potently modulate protein–protein interactions have recently reached clinical trials. Along with high-throughput screening of compound libraries, combining structural and computational approaches has boosted this formerly minor area of research into a currently tremendously active field. This review highlights the very recent developments in the rational design of protein–protein interaction inhibitors.
Interactions between three hexacationic arene ruthenium metallaprisms and human proteins have been studied using NMR spectroscopy, mass spectrometry and circular dichroism spectroscopy, showing that proteins are potential biological targets for these metallaprisms.
Didier Rognan heads the Laboratory of Structural Chemogenomics at the Faculty of Pharmacy of Strasbourg (France). He studied Pharmacy at the University of Rennes (France) and did a Ph.D. in Medicinal Chemistry in Strasbourg (France) under the supervision of Prof. C.G. Wermuth. Aer a postdoctoral fellowship at the University of Tubingen (Ger- ¨ many), he moved as an Assistant Professor to the Swiss Federal Institute of Technology (ETH) until October 2000. He was then appointed Research Director at the CNRS to build a new group in Strasbourg. He is mainly interested in all aspects (method development and applications) of structurebased drug design, notably on G protein-coupled receptor ligands and protein–protein interaction inhibitors.
Introduction Drug discovery is a long, costly, multi-step endeavour which aims at reducing all possible risks to deliver a novel therapeutic solution to previously unmet clinical needs. To reduce chemical risks, empirical rules are used to filter the chemical space and retain drug-like low molecular weight compounds. Reduction of the biological risk is addressed by considering privileged target families (e.g., G protein-coupled receptors and kinases) whose activation/inhibition by drug-like compounds is likely to correct or reverse pathological states. Until recently, mostly single macromolecules (proteins and nucleic acids) have been considered as potential drug targets. Out of 68 000 proteins currently annotated in UniProt for the human proteome,1 only about 300 targets2 have been addressed by current drugs, and the large majority of single targets is still awaiting first-in class drugs.
Besides single targets, large scale genomics and proteomics3 have identified complex networks of targets and pathways regulating physiopathological processes in a coordinated manner. The current human protein–protein interactome has been estimated between 130 000 (ref. 4) and 650 000 (ref. 5) complexes, out of which only a tiny amount is known, and only a very few6–8 have been the object of a drug discovery initiative. Protein–protein interactions (PPIs) therefore describe a totally new biological space that attracts more and more attention, with 26PPI inhibitors9,10 already under clinical development, notably in the oncology field.11 Despite PPIs may adopt quite different sizes, shapes and electrostatics,12 identifying highaffinity PPI inhibitors is a considerable challenge for many reasons: (i) in contrast to conventional targets, a medicinal chemist cannot start inhibitor design from the structure of endogenous ligands, (i) PPIs often involve flat surfaces delocalized over multiple epitopes, usually lack well-defined buried cavities13 typical of conventional targets, and are significantly larger (ca. 1000–3000 A˚2 ) than enzyme/receptor pockets (300– 1000 A˚2 ), (iii) high-throughput screening of traditional compound libraries often returns no viable hits14 for the main reason that PPI inhibitor chemical space is quite different from that described by traditional drug-like compounds.10 Nonetheless, thanks to bioinformatics and proteomics-guided prioritization of therapeutically relevant protein–protein complexes, more and more PPI inhibitors are currently reported. Several excellent reviews6,7,9,11,15–18 have already been published on experimental methods (high throughput screening, biochemical and cellular assays, and fragment-based approaches) suitable to discover PPI inhibitors. The present report will only cover computer-aided approaches, with a major emphasis on structure-based methods and recent discoveries (2012–2014).
Databases Preliminary access to experimentally validated data is key to launch a drug discovery program on PPI modulators. A multitude of databases storing genomics, proteomics and structural data are currently available to help the medicinal chemist. We will here briefly review these archives, focusing mostly on easily interpretable structural data.
PPI databases Many experimental methods with different throughputs (from low to high) have been developed to characterize binary interactomes in various species, among which the most prominent has been the yeast two-hybrid (Y2H) assays, and mass-spectrometry (MS) coupled with co-immunoprecipitation or coaffinity purification.19 These experimental data are stored in many primary databases (Table 1) that are difficult to mine due to their large heterogeneity. Metadatabases have been derived thereof to facilitate their analysis, among which the most popular are APID and PRIMOS (Table 1). These metadatabases cover a wide range of organisms and notably offer the possibility to mine experimental PPI data according to disease relevance or inter-organism crosstalk, and provide graphic tools to visualize complex networks of interacting proteins and identifying important protein nodes (hubs). It is however very difficult, from this large amount of data, to clearly prioritize PPIs for a drug discovery program. Attempts to classify the PPIs by structural druggability25 (although ligandability26 is probably a better term) are worth mentioning but should be taken with care due to the still insufficient number of existing PPI three-dimensional (3D) structures.
Ligand databases Initially limited to a limited subset of inhibitors able to disrupt few PPIs (e.g. p53/MDM2, Bcl-Xl/Bak, and IL-2/IL-2Ra),7,27 the repertoire of PPI inhibitors rises constantly thanks to exciting developments in biophysical fragment screening.15,28 Three publicly available databases storing information on PPIs and their inhibitors (Table 2) may be used to better describe the structural properties of druggable PPIs and the chemical space associated with their disruptors.
The 2P2Idb database12 is a hand-curated repository of protein–protein complexes of known X-ray structures (X-ray diffraction and nuclear magnetic resonance spectroscopy) for which at least one low molecular weight orthosteric inhibitor has been co-crystallized with one of the two protein partners. It currently describes 71 inhibitors for 14 PPIs, clustered in two groups (Fig. 1) with respect to the nature of the interface (protein–peptide and protein–protein). Companion tools (2P2I inspector,30 2P2I score,30 and 2P2I hunter31) are provided to analyse PPIs at a structural level, predict their structural druggability and design PPI focussed libraries, respectively.
Fig. 1 Prototypical examples of class I (left panel) and class II PPIs (right panel), exemplified by the Bcl-Xl/Bak (PDB id 1BXL) and integrase/LEDGF (PDB id 2B4J) complexes, respectively. Class I PPIs involve the interaction of a globular protein with a peptide or a single secondary structure (a-helix and b-strand) of a second protein partner. Class II PPIs are characterized by the interaction of two globular proteins.
The iPPI-DB10 is another manually curated database from world patents and the medicinal chemistry literature, focussing on low molecular weight orthosteric inhibitors, disease-related protein–protein interfaces and a clear biochemical readout (e.g. fluorescence polarisation and enzyme-linked immunosorbent assay). The database archives 1650 PPI inhibitors targeting 13 families of homologous PPI targets mainly involved in cancer, immune disorders and infectious diseases.
Finally, the TIMBAL database29 reports ca. 7000 inhibitors for 50 known PPIs. In contrast to the two other databases, TIMBAL is maintained through a predefined list of PPIs and automated searches in ChEMBL32 and the Protein Data Bank.33 In contrast to the other databases, TIMBAL also registers short peptides with an upper molecular weight limit of 1200 Da. It should be pointed that most of the 15 000 uncurated biological data present in TIMBAL arise from a single target family (integrins) and should be considered with care.
Analysing the content of these databases enables a first comparison of PPI inhibitors versus drugs, as well as PPIs amenable to disruption versus standard heterodimers. PPI surfaces disrupted by inhibitors tend to be smaller, more hydrophobic and accessible than standard heterodimers.12 As a consequence, low molecular weight PPI inhibitors tend to be larger, more hydrophobic and more aromatic-rich than standard drugs. Interestingly, many of them (ca. 60%) still comply with Lipinski’s rule-of-five, 10 revealing some hopes in the developability of such compounds.
However, it should be stated that the set of empirical rules designed to discriminate druggable from non-druggable PPIs, as well as to distinguish PPI inhibitors from conventional druglike compounds still rely on a very limited set of highly homologous data (PPIs, inhibitors), and should therefore be regarded with caution. Increasing coverage of the PPI repertoire by future experimental screens will undoubtedly lead to a better denition of PPI biological and chemical spaces. We therefore expect in the future the above-mentioned rules to be rened and be more descriptive of the true world of PPI inhibitors, notably with respect to rational design of PPI focussed libraries.
Rational design of PPI modulators
Sequence-based approaches Whatever the nature of the PPI (type I or type II, see the definition above), PPI interfaces are often characterized by the presence of hotspots,34 in other words anchor residues that contribute the most to the binding free energy of the protein– protein complex. The interaction of a single modified amino acid with a single anchor residue might be sufficient to disrupt a PPI as elegantly demonstrated by Lin et al. in a recent study.35 Capitalizing on the presence of a reactive cysteine (C246) at the interface of the complex between caspase-7 (CASP7) and the Xlinked inhibitor of apoptosis protein (XIAP), they designed the N-iodoacetyl-lysine amino acid derivative 1 (Fig. 2) that covalently traps C246 and further disrupts the XIAP–CASP7 complex, therefore triggering CASP7-dependent apoptosis and killing MCF-7 breast cancer cells (EC50 ¼ 0.64 mM) previously resistant to chemotherapy.
The easiest way to inhibit a PPI is to start with the amino acid sequence of one interacting epitope, notably if the latter is part of regular secondary structures (a-helix, b-strand, and b-turn). For example, a-helical peptides mimicking the sequence of protein transmembrane domains may disrupt PPIs quite efficiently.36,37
Fig. 2 Peptidomimetics as PPI disruptors
Due to poor pharmacokinetic profiles, linear peptides are good in vitro tools but usually not efficient clinical candidates. Chemical modifications are required to stabilize their secondary structures in physiological media and prevent early degradation. Among the most exciting developments in this area38,39 is the design of stapled peptides.40,41 Stapled peptides are synthetic analogues of a-helical protein epitopes involved in a PPI, and in which a covalent hydrocarbon linkage connects adjacent turns of the helix. Stapling is known to significantly increase the in vivo half-life of the natural peptide (increasing proteolytic stability), decrease the entropic cost of binding, and even enable cellular uptake.42 Many stapled peptides with potent in vivo activities have already been reported.39 One of these stapled peptides (ATSP-7041, compound 2, Fig. 2) just entered clinical development as a dual nM MDM2/MDMX inhibitor for p53-dependent cancer therapy.43
Heterocyclic scaffolds mimicking secondary structures can also be obtained by solution-phase synthesis to afford peptidomimetic libraries amenable to PPI inhibition. Whitby et al. notably reported the design of 8000 member 4-acetamido-3- alkoxy-benzamide focused library featuring weak p53/MDM2 inhibitors and potent HIV-1/gp41 inhibition (compound 3, Fig. 2).44 When the peptide epitope is not structured, developing macrocyclic analogues is more difficult but still feasible as recently demonstrated by Glas et al.38 who successfully improved 14-3-3 binding of a 11-mer peptide from a bacterial ExoS virulence factor by cross-linking binding amino acids with polymethylene linkers, up to an in vitro 40 nM disruptor of the ExoS/14-3-3 interaction (compound 4, Fig. 2). Interestingly, the cross-linker was not only chosen to rigidify the natural ExoS peptide structure but also to directly provide additional hydrophobic interactions to the 14-3-3 binding site.38 Only in exceptional cases the natural unmodified peptide is directly usable as a PPI inhibitor. One recent example is the 28 amino acid cell-penetrating peptide (p28) from a bacterial azurin redox protein, that binds to the DNA-binding domain of the p53 tumor suppressor and inhibits p53 degradation by interfering with the Cop1-mediated ubiquitination,45 thereby enhancing p53 levels in cancer cells and exhibiting antitumoral efficacy in patients with advanced solid tumors.46
Pharmacophore-based approaches As defined by the IUPAC,47 a pharmacophore is “an ensemble of steric and electronic features that are necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response.” Although pharmacophores are mainly used to align and compare ligands sharing the same target,48 the same concept can be easily transferred to PPIs in which one partner is the “receptor” and the second one the “ligand”. Pharmacophore features (hydrophobic, aromatic, H-bond donor and H-bond acceptor, positively and negatively ionisable) can therefore be manually or automatically mapped to atoms of the ligand in direct interactions with the receptor. The resulting pharmacophore can then be used to identify a compound library for hits fulfilling the defined query. Several tools (e.g. LigandScout,49 Discovery Studio,50 and Pocket Query51) can be directly used to map PPI pharmacophores onto protein–protein X-ray structures (Fig. 3).
Fig. 3 Example of a PPI pharmacophore mapped onto interacting atoms of human LEDGF (yellow ribbons) bound to HIV-1 integrase (red ribbons, PDB ID 2B4J). The PPI pharmacophore is composed of 2 Hbond donors (magenta balls), two H-bond acceptors (green balls), one hydrophobic feature (cyan ball) and 6 exclusion volumes (gray balls).
Using a manual PPI pharmacophore defined from the X-ray structure of the Annexin A2/S100A10 complex, a pro-angiogenic complex, Reddy et al.52 derived a simple pharmacophore (2 hydrophobes, 2 H-bond donors, and 2 H-bond acceptors) using the Unity program,53 and screened a library of 700 000 compounds to select 586 hits which were further docked to the Annexin A2 binding site to retain only 190 candidates with both a good docking and pharmacophore fitness score (Table 3). Out of 190 tested compounds, 7 hits blocked the interaction between S100A10 and the Annexin A2 N-terminus in a competitive fluorescent binding assay, with the most potent PPI inhibitor (compound 5, Fig. 4) exhibiting an IC50 of 24 mM.52 Geppert et al.54 reported the rational discovery of a low molecular weight inhibitor of the complex between interferon-a (IFN-a) and its receptor (IFNAR2). Fortunately, the PPI interface was small enough (ca. 800 A˚2 ) to be targeted by a small heterocyclic compound. After identifying major hotspots at the IFN-a surface, a fuzzy receptor-based pharmacophore was determined using the VirtualLigand approach,55 which assigns pharmacophoric features to Gaussian densities. Screening a collection of 556 000 commercially available compounds retained six virtual hits, out of which two were weak IFN-a inhibitors, but one (compound 6, Fig. 4) was confirmed by NMR and surface plasmon resonance (SPR) to bind to IFN-a with a dissociation constant (Kd) of 4 mM and to inhibit IFN-a responses in various cell assays. The novel inhibitor may be useful to reduce IFN-a titers in autoimmune disorders.
Table 3 Protein–protein pharmacophore searches to identify PPI inhibitors
Target Library size Tested Hits Ref.
Annexin A2/S100A 10 700 000 190 7 52
INFAR2/IFN-a 556 000 6 3 54
p53/MDM2 21 287 15 6 56
Nrf2/Keap1 21 199 17 1 57
PKC3/RACK2 330 000 19 1 58
Due to the inherent complexity of PPI pharmacophores (many features covering a large surface), combining several pharmacophores into a consensus model may help to retrieve essential features and simplify pharmacophore queries. Xue et al. applied this approach to the identification of p53–MDM2 inhibitors.56 The p53–MDM2 complex has become a prototypical PPI for its biological background (this interaction plays an important role in regulating the transcriptional activity of tumour cells) and many high affinity low molecular-weight inhibitors of this PPI identified by various screening approaches.59 Starting from a set of 15 MDM2-peptide X-ray structures, a common feature structure-based pharmacophore (2 H-bond donors, one H-bond acceptor, 2 aromatic rings, and one hydrophobe) was first identified. In addition, a receptorligand pharmacophore (five hydrophobes, one aromatic, and one H-bond donor) was generated from a separate set of 10 MDM2-non peptide complexes. Merging both pharmacophores and retaining the most common features led to an ensemble pharmacophore definition (two aromatic rings, two hydrophobes, and one H-bond donor) taking into account both peptide and non-peptide binding. This pharmacophore was used to screen a collection of 21 287 commercially available compounds, and led to a hit list of 15 compounds out of which 6 were confirmed as p53–MDM2 inhibitors using an in vitro uorescence polarization assay.56 The most potent inhibitor (compound 7, Fig. 4) is a 180 nM MDM2 inhibitor. Despite a good selectivity in a MTT tumour cell proliferation assay (p53+/+ vs. p53/ cells), compound 7 was a weak inhibitor (IC50 ¼ 85 mM) of tumour cell growth, because of poor pharmacokinetic properties.
Fig. 4 PPI inhibitors identified by pharmacophore-based virtual screening.
Along the same lines, two X-ray structures were used to derive inhibitors of the PPI between Keap1 and Nrf2, a complex involved in the response to oxidative stress.57 The two PPI pharmacophores were merged into a single query consisting of one H-bond donor, two H-bond acceptors and three negative ionisable centers. To afford some fuzziness in the search, up to two features were allowed to be missed by virtual hits. Since the Keap1-binding epitope of Nrf2 is composed of several acidic residues, only compounds bearing a negative charge were searched among a full commercial library of 251 774 compounds. The remaining 21 199 hit list was matched to the pharmacophore, and led after confirmation with docking and MM-PBSA scoring, to a list of 17 potential hits which were tested for Keap1–Nrf2 inhibition using an in vitro fluorescence polarization assay. A single compound (compound 9, Fig. 3) was confirmed in vitro as a moderately potent Keap1–Nrf2 inhibitor with an EC50 of 9.8 mM.57 Interestingly, the inhibitor activated the Nrf2 transcriptional activity .
When both protein partners involved in the PPI have not been co-crystallized, it is still possible to rationally discover PPI inhibitors, starting from the sole X-ray structure of one of the two proteins. This approach was followed by Rechfeld et al. in the discovery of PKC3–RACK2 inhibitors.58 Starting from the Xray structure of the PKC3 octameric epitope binding to RACK2 (a receptor for activated protein kinase C), a simple peptide-based pharmacophore model (3 H-bond donor/acceptor, one hydrophobe) was defined and used to screen a collection of 330 000 compounds. Out of 19 virtual hits, a thienoquinoline was found to disrupt the PPI in vitro and served as a query for a secondary screen for chemically similar analogues, which led to compound 8 (Fig. 4) as a micromolar potent PKC3-RACK2 inhibitor (IC50 ¼ 5.9 mM) which also inhibited PKC3 downstream signalling, HeLa cancer cell migration and invasion.58
Finally, pharmacophore searches may be used to prioritize privileged scaffolds for synthesizing PPI-focused libraries. For example, Fry et al. reported a rational approach to PPI library design targeting a-helical binding epitopes.60 Starting from the known X-ray structure of an a-helical p53 epitope binding to MDM2, a three point pharmacophore, featuring the three important hydrophobic side chains (Phe19, Trp23, and Leu26) of the p53 peptide, was designed and used to find heterocyclic scaffolds among the CSD database61 of small molecule X-ray structures. Several small-sized libraries (ca. 100 members) were synthesized from each hit and tested for general inhibition of PPIs involving an a-helical epitope (e.g. MDM2, BCL2, BCL-XL, and MCL1). Although no potent hit could be discovered, the average hit rate was far superior (4%) to what should be expected from a random screen. Moreover, many starting hits exhibited good ligand efficiencies,60 and are therefore interesting starting points for hit leading optimization.
Despite its apparent simplicity, PPI-based pharmacophore search is a fast, cost-effective and simple in silico approach to discover the very first inhibitors of a particular PPI. Of course, all successful examples mentioned above imply that the PPI is of manageable size and does not involve a too large and complex binding epitope. Beside the existence of a X-ray or NMR structure of the protein–protein (peptide) complex, it is therefore equally important to properly select PPIs amenable to pharmacophore-based searches, notably with respect to the complexity of the query (5–6 features) and its hydrophobic/ hydrophilic balance.
Docking-based approaches At the first sight, protein–ligand docking should be considered as the most intuitive and logical computational tool to predict likely ligands of any target of known 3D structures.62 Unfortunately, severe drawbacks associated with the scoring of protein– ligand interactions render that tool usually suitable for positioning a ligand into a binding site, but rarely to predict binding free energies or to precisely rank ligands by decreasing affinity.63 Moreover, the ability of docking algorithms to anchor ligands to flat PPI surfaces has long remained elusive. In a benchmark study, Kruger ¨ et al. used two popular docking tools (AutoDock and Glide) to reproduce the known X-ray structure of PPI inhibitors to their target.64 Surprisingly, the performance of these standard docking programs with respect to the positioning of the ligand (rmsd to the X-ray structure) was only moderately affected by switching from conventional targets to PPIs. Although PPI inhibitors with more than 10 rotatable bonds were found more difficult to properly dock, a good pose was generated in ca. 54% of the 80 PPI inhibitors considered. Docking to PPIs providing at least one charge residue was favoured over those purely hydrophobic.64 There are therefore no particular reasons to discard docking-based approaches from rational PPI inhibitor discovery scenarios. Many of the following success stories support this assumption.
We will not here review the many recent reports describing docking as a mean to predict the binding mode of a PPI inhibitor discovered by an experimental screening method.59,65–68 The next section will only focus on inhibitors discovered by a docking-based virtual screening campaign (Table 4).
Table 4 Protein–protein inhibitors discovered by docking-based screening
Target Library size Tested Hits Ref.
TLR4/MD- 2 50 000 14 3 69
uPA–uPAR 5 000 000 50 3 70
IL-6/gp130 9 2 2 71
Keap1–Nrf2 153 611 65 9 72
CRYAB/VEGF 139 735 40 4 73
NRP-1/VEGF- 429 623 1317 56 74
PPxY/Nedd4 4 800 000 20 1 75
p53/MDM2 87 430 295 1 76
Despite an apparent unsuitable large and concave cavity, the MD-2-binding site at the surface of the toll-like receptor 4 (TLR4) was selected for pharmacophore-constrained FlexX77 docking of a library of 49 600 compounds pre-filtered for 3D shape similarity to an existing TLR4 antagonist.69 40 virtual hits were selected for in vitro TLR4 binding and functional antagonism, and 3 of them could be confirmed experimentally. The most potent antagonist (compound 10, Fig. 5) blocked TLR4 in a gene receptor assay with an IC50 of 16.6 mM and inhibited proinflammatory cytokine release (e.g. TNF-a) from human peripheral blood mononuclear cells upon LPS activation. Due to unfavourable aqueous solubility, the compound could not be tested in vivo but represent a good starting hit for developing small molecule TLR4 antagonists for the treatment of neuropathic pain and sepsis.
Fig. 5 PPI inhibitors identified by docking-based virtual screening
To account for the conformational flexibility of proteins, Khanna et al. reported a cascade docking-based virtual screening for discovering inhibitors of the interaction between the urokinase-type plasminogen activator (uPA) and the urokinase receptor (uPAR).70 Two X-ray structures of the uPAR were first used for docking a collection of 5 million commercially available compounds using AutoDock4.78 10 000 top-ranked virtual hits were further docked, still with AutoDock, to 50 molecular dynamics snapshots of the uPAR structure, leading to 500 top-ranked compounds which, in a third step, were docked using a different program (Glide) on the 50 receptor conformers. After clustering the top 250 compounds by chemical similarity, the highest scoring compounds from each of the top 50 clusters were finally selected, purchased and evaluated in vitro in a fluorescence polarization assay. Among the three validated hits, the most potent inhibitor (compound 11, Fig. 5) binds to uPRA with a submicromolar affinity (Kd ¼ 310 nM) and inhibits the uPA–uPAR interaction with an IC50 of 10 mM.70 The hit blocked invasion of breast cancer cells but not their migration or adhesion. A close analogue of compound 11 was recently shown to be efficient in an in vivo breast cancer metastasis assay.79
Docking is not limited to the study of single protein–ligand interactions. In an elegant study, Li et al. reports a computational method enabling the simultaneous docking of multiple fragments to a single binding site, by analogy to experimental fragment screening.71 When applied to the PPI between IL-6 and gp130, simultaneous docking of two fragment pools (6 and 3 fragments, respectively) targeting two different hotspots at the PPI, two theoretical ligands could be reconstructed after tethering the best fragments at each hotspot. Searching for known drugs80 which are chemically similar to the two virtual hits suggested than two estrogen receptor modulators (raloxifene and bazedoxifene) may bind to the gp130/IL-6 PPI. Effective binding of both drugs to gp130 was confirmed experimentally, as well as inhibition of IL-6 induced STAT3 phosphorylation in various cancer cell lines defective in estrogen receptor expression. Bazedoxifene (compound 12, Fig. 5) was the most efficient (IC50 ¼ 25 mM) in inhibiting the ER-independent IL6-induced breast cancer cell proliferation, thereby offering some repositioning potential in the treatment of IL-6/gp130/STAT3 dependent tumours.71
The Nrf2–Keap1 complex, previously investigated using a pharmacophore-based approach (see the previous section), was also used for docking 300 000 commercially available compounds with the program Glide. Among the chemically diverse 65 top-ranking hits, 9 compounds were confirmed to be PPI inhibitors, the most potent disruptor (compound 13, Fig. 5) exhibiting a Kd of 2.9 mM in a fluorescence anisotropy-based assay.
A major hurdle in PPI inhibitor development is the frequently objected high molecular weight and unfavourable pharmacokinetic properties. Chen et al. strikingly contradicted this dogma by reporting a very low molecular weight inhibitor of the aB-crystallin (CRYAB)/VEGF-A interaction.73 CRYAB is a protein overexpressed in triple-negative breast cancer cells that acts as a chaperone to several proteins including the proangiogenic vascular endothelial growth factor (VEGF). Disrupting the interaction between CRYAB and VEGF-A is therefore a potential approach to cancer cell proliferation and invasion. The VEGF-binding site on the surface of the CRYAB X-ray structure was therefore targeted by docking 140 000 compounds from the NCI database using the Dock6.5 program (UCSF). Despite a very modest molecular weight (161.16 Da), one compound (compound 14, Fig. 5) was identified as an in vitro disruptor of the CRYAB/VEGF-A interface with an IC50 of ca. 20 mM. Intraperitoneal injection of compound 14 (200 mg kg1 ) remarkably suppresses tumour growth in vivo in human breast cancer xenograph models. VEGF-A is an important angiogenic factor that interacts with many other partners, notably the family of neuropilin receptors (NRP-1, NRP-2) whose inhibition leads to cancer cell apoptosis. The PPI between the C-terminal end of VEGF-A165 and the tandem b1 and b2 domains of NRP-1 was targeted for docking 430 000 molecules with a consensus docking approach relying on two docking programs (SurflexDock81 and ICM82). A consensus list of 1317 top-scoring compounds was retained for their in vitro anti-proliferative activity and binding to NRP-1 using a chemiluminescent assay.74 56 molecules (hit rate of 4.2%) antagonized the NRP-1/ VEGF-A interaction by at least 30% at the concentration of 10 mM. The best hit (compound 15, Fig. 5) is the first non-peptide NRP-1/VEGF-A antagonist (IC50 ¼ 34 mM) and displays remarkable anti-proliferative effects (IC50 ¼ 0.2 mM) on breast cancer cells. Administered at the dose of 50 mg kg1 in NOG xenographed mice, compound 15 strongly inhibits tumour growth inhibition by inducing cell apoptosis, without any effect on pro-angiogenic kinases.
Although most of the above reported therapeutical indications remain in the oncology field, PPI inhibitors have clear potential in other areas, notably infectious diseases as recently demonstrated by Han et al.75 who reported the structure-based discovery of antiviral compounds inhibiting viral–host interactions. The PPI target is the complex between the conserved Ldomain PPxY sequence of several viral matrix proteins (e.g. Ebola, Marburg, Lassa fever, and VSV) and the ubiquitin ligase Nedd4 protein. Docking ca. 5 million compounds (ZINC database)83 on the Nedd4 X-ray structure with the AutoDock4 program, yielded to the evaluation of 20 compounds, out of which one molecule was confirmed as a true inhibitor of the PPI in a cellular assay. Acquiring close analogs of the initial hit led to two more potent inhibitors (compounds 16 and 17, Fig. 5) as submicromolar inhibitors of the PPxY–Nedd4 interaction in vitro. 75 Both compounds exhibit antibudding activity against Ebola, Lassa fever, Marburg and VSV viruses, thereby decreasing viral titers, without apparent cytotoxicity on HEK293T cells.
Natural compounds are also a major source of potentially interesting PPI inhibitors. By docking a library of commercially available compounds to the p53 binding site, Vogel et al. recently reported lithocholic acid (compound 18, Fig. 5), a secondary bile acid, as a weak binder (Kd of 15 mM) to MDM4 and MDM2 proteins with a slight preference for MDM4.76 The natural compound was further shown to inhibit p53–MDM4 interactions and promote apoptosis in a p53-dependent manner by inducting caspase3/7.
Conclusions
We should acknowledge that peptides usually remain a good starting point to derive PPI inhibitors. Given the increasing number of high resolution X-ray structures of biologically relevant protein–protein complexes, the number of potentially increasing PPIs is likely to significantly rise in the next years. Provided that molecular rules exist to prioritize the most interesting anchoring residues at the interface, continuous protein epitopes can be easily converted into linear peptides for quick experimental validation. Recent progress in peptide stabilisation by chemical stapling next opens an immense eld for deriving either pharmacological tools or drug candidates. Numerous successes in identifying non-peptide PPI inhibitors also exist. The present review has only considered inhibitors mostly discovered by a rational structure-based virtual screening approach. Despite the few cases described herein (15 in total), examples are pretty much indicative of results than can be reasonably achieved. Comparing the properties of PPIs (Fig. 6A and B) and their inhibitors (Fig. 6C) with previously reported larger PPI data,64 some trends could be verified. Considering success as the availability of low micromolar nonpeptide inhibitors, successfully targeted PPIs present a higher proportion of charged residues with respect to conventional targets (sc-PDB data).84 Unsurprisingly, PPI inhibitors bind to smaller cavities (200–350 A˚3 ) than that presented by conventional targets (450–800 A˚3 range). Consequently, PPI inhibitors present a high proportion of aromatic rings, amide moieties and charged groups (Fig. 4 and 5) that hamper their druggability potential, as estimated here by the QED metric85 (Fig. 6C). We notice a significant proportion of negatively charged compounds, suggesting that a strong electrostatic interaction with the target is often mandatory to reach detectable affinity to PPI-participating cavities.
Fig. 6 Properties of PPIs and their inhibitors: (A) cavity properties expressed in percentage according to the cavity detection VolSite program86 (Hydro, hydrophobic; Aro, aromatic; H-bond, H-bond accepting/donating properties; Neg: negatively charged; Pos, positively charged, Du: fully accessible); (B) cavity volumes targeted by PPI inhibitors (this review) and conventional ligands (sc-PDB data84). The box delimits the 25th and 75th percentiles, and the whiskers delimit the 5th and 95th percentiles. The median and mean values are indicated by a horizontal line and an empty square in the box; (C) quantitative estimate of druggability (QED)85 of the inhibitors. QED values for true drug-like compounds should be over 0.5 (red broken line).
However, the current survey also indicates that there is no absolute dogma with respect to PPI inhibitor identification. Very low molecular weight compounds (compounds 1, 6 and 14) have been successfully identified as PPI disruptors.
Beside interfacial inhibitors, there exist promising alternative ways of inhibiting PPIs. For example, PPI stabilizers87,88 (e.g. paclitaxel, rapmycine, and forskolin) bind to rim exposed pockets at or very close to the interface, and also lead to the functional inactivation of the protein–protein complex. Such stabilizers are frequent in the nature, and this area still has not been fully exploited until now. Likewise, the allosteric inhibition of PPIs, at pockets remote from the interface, clearly deserves some consideration. Such pockets have been shown to be frequent at the close vicinity of two protein chains in close interaction,89 and represent, at least for some of them, more ligandable pockets than those presented by PPIs.
Although dominated by a continent of flat and featureless interfaces, the PPI world is also populated by very different islands in terms of shape and electrostatics that should not been discarded. Many factors are likely to increase our knowledge of PPIs and their inhibitors among which: (i) the increasing number of biologically relevant and crystallized protein–protein complexes, (ii) the development of label-free experimental screening techniques, and (iii) the significant contribution of molecular simulations to detect transient interfaces. Medicinal chemistry will be a key factor to transform moderately potent PPI inhibitor hits into clinical candidates with desired pharmacokinetic properties.
2 J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996.
3 P. Legrain and J. C. Rain, J. Proteomics, 2014, 107, 93–97.
4 K. Venkatesan, J. F. Rual, A. Vazquez, U. Stelzl, I. Lemmens, T. Hirozane-Kishikawa, T. Hao, M. Zenkner, X. Xin, K. I. Goh, M. A. Yildirim, N. Simonis, K. Heinzmann, ….A. L. Barabasi and M. Vidal, Nat. Methods, 2009, 6, 83–90.
5 M. P. Stumpf, T. Thorne, E. de Silva, R. Stewart, H. J. An, M. Lappe and C. Wiuf, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6959–6964


 Thursday 15 October 2015, 12:00–13:00
Thursday 15 October 2015, 12:00–13:00

















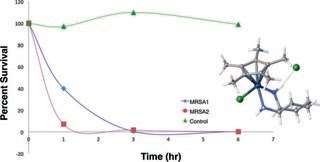




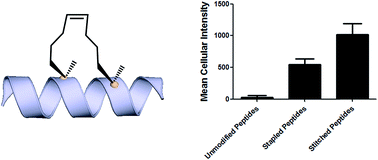
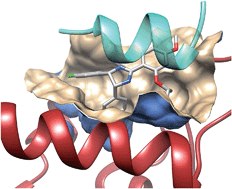
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}