Vaccines, Small Peptides, aptamers and Immunotherapy [9]
Writer and Curator: Larry H. Bernstein, MD, FCAP
This contribution has the following structure:
9.1.1 Viruses in carcinogenesis
9.1.2 Simultaneous Humoral and Cellular Immune Response against Cancer–Testis Antigen NY-ESO-1: Definition of Human Histocompatibility Leukocyte Antigen (HLA)-A2–binding Peptide Epitopes
9.1.3 Monoclonal Antibodies in Cancer Therapy
9.1.4 Aptamers
9.1.5 Tumor Suppressors
9.1 Vaccines
9.1.1 Viruses in carcinogenesis
- HPV-associated cervical cancer
- HPV-associated head and neck cancer: a virus-related cancer epidemic
The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide
Risk of pancreatic cancer among individuals with hepatitis C or hepatitis B virus infection: a nationwide study in Sweden.
HIV Infection and Cancer Risk
HIV and cancer of the cervix
Anal cancer: an HIV-associated cancer
The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis
Types of Cancer: AIDS/HIV related malignancies
Cytokines in cancer pathogenesis and cancer therapy
Dendritic Cells as Therapeutic Vaccines against Cancer
9.1.2 Simultaneous Humoral and Cellular Immune Response against Cancer–Testis Antigen NY-ESO-1: Definition of Human Histocompatibility Leukocyte Antigen (HLA)-A2–binding Peptide Epitopes
9.1.3 Monoclonal antibodies
Monoclonal antibodies in cancer therapy
Monoclonal Antibodies in Cancer Therapy: 25 Years of Progress
9.1.4 Aptamers
Nanocarriers as an emerging platform for cancer therapy
Quantum Dot−Aptamer Conjugates for Synchronous Cancer Imaging, Therapy, and Sensing of Drug Delivery Based on Bi-Fluorescence Resonance Energy Transfer
Oligonucleotide Aptamers: New Tools for Targeted Cancer Therapy
9.1 Vaccines
9.1.1 Viruses in carcinogenesis
HPV-associated cervical cancer
http://www.cancer.net/navigating-cancer-care/prevention-and-healthy-living/hpv-and-cancer
Human papillomavirus (HPV) is a virus that is usually passed on during direct skin-to-skin contact, most commonly sex. In fact, HPV is the most common sexually transmitted disease in the United States. Most men and women are not aware they have an HPV infection because they do not develop any symptoms or health problems. Certain HPV types can cause precancerous lesions (areas of abnormal tissue) or cancer.
More than 40 of the viruses are called “genital type” HPVs. These viruses are spread from person to person when their genitals come into contact, usually during vaginal or anal sex. They can also be passed on through oral sex.
Genital HPV types can infect the genital area of women, including the vulva (outer portion of the vagina), the vagina, and the cervix (the lower, narrow part of a woman’s uterus), as well as the genital area of men, including the penis. In both men and women, genital HPV can infect the anus and some areas of the head and neck.
Nearly all cervical cancers are caused by HPV infection. Strong scientific evidence shows that a lasting HPV infection is required for cervical cancer to begin developing. Whether a woman who is infected with HPV will develop cervical cancer depends on a number of factors, including the type of HPV infection she has. Of the cervical cancers related to HPV, about 70% are caused by two strains, HPV-16 or HPV-18. In women who have HPV, smoking may increase the risk of cervical cancer.
Warts and precancerous lesions can be removed through cryotherapy (freezing); loop electrosurgical excision procedure (LEEP), which uses electric current to remove abnormal tissue; or surgery.
Receiving an HPV vaccine reduces your risk of infection. The U.S. Food and Drug Administration (FDA) approved two vaccines that help prevent HPV infection: Gardasil and Cervarix. It is important to note that the vaccines cannot cure an existing HPV infection.
Purpose of the vaccines. The goal of HPV vaccination is to prevent a lasting HPV infection after a person is exposed to the virus. Gardasil, introduced in 2006, helps prevent infection from the two HPVs known to cause most cervical cancers and precancerous lesions in the cervix. The vaccine also prevents against the two low-risk HPVs known to cause 90% of genital warts. Gardasil is approved for the prevention of cervical, vaginal, and vulvar cancers in girls and women ages nine to 26. It is also approved to prevent anal cancer in women and men and genital warts in men and boys in the same age range. Cervarix, introduced in 2009, is approved for the prevention of cervical cancer in girls and women ages 10 to 25.
Effectiveness and safety of the vaccines. Data show the HPV vaccinations are safe and highly effective in preventing a lasting infection of the HPV types they target. Because it takes many years before a precancerous lesion develops into an invasive cancer, it will likely take several more years before there is evidence that the number of cancer cases in vaccinated individuals has been reduced.
HPV-associated head and neck cancer: a virus-related cancer epidemic
Shanthi Marur, Gypsyamber D’Souza, William H Westra, Arlene A Forastiere
Lancet Oncol 2010; 11: 781–89
http://dx.doi.org:/10.1016/S1470-2045(10)70017-6
A rise in incidence of oropharyngeal squamous cell cancer—specifically of the lingual and palatine tonsils—in white men younger than age 50 years who have no history of alcohol or tobacco use has been recorded over the past decade. This malignant disease is associated with human papillomavirus (HPV) 16 infection. The biology of HPV-positive oropharyngeal cancer is distinct with P53 degradation, retinoblastoma RB pathway inactivation, and P16 upregulation. By contrast, tobacco-related oropharyngeal cancer is characterized by TP53 mutation and downregulation of CDKN2A (encoding P16). The best method to detect virus in tumor is controversial, and both in-situ hybridization and PCR are commonly used; P16 immunohistochemistry could serve as a potential surrogate marker. HPV-positive oropharyngeal cancer seems to be more responsive to chemotherapy and radiation than HPV-negative disease. HPV 16 is a prognostic marker for enhanced overall and disease-free survival, but its use as a predictive marker has not yet been proven. Many questions about the natural history of oral HPV infection remain under investigation. For example, why does the increase in HPV-related oropharyngeal cancer dominate in men? What is the potential of HPV vaccines for primary prevention? Could an accurate method to detect HPV in tumor be developed? Which treatment strategies reduce toxic effects without compromising survival? Our aim with this review is to highlight current understanding of the epidemiology, biology, detection, and management of HPV-related oropharyngeal head and neck squamous cell carcinoma, and to describe unresolved issues.
Cancers of the head and neck arise from mucosa lining the oral cavity, oropharynx, hypopharynx, larynx, sinonasal tract, and nasophaynx. By far the most common histological type is squamous cell carcinoma, and grade can vary from well-differentiated keratinizing to undifferentiated non-keratinizing. An increase in incidence of oropharyngeal squamous cell carcinoma—specifically in the tonsil and tongue base—has been seen in the USA, most notably in individuals aged 40–55 years. Patients with oropharyngeal cancer are mainly white men. Unlike most tobacco-related head and neck tumors, patients with oropharyngeal carcinoma usually do not have a history of tobacco or alcohol use. Instead, their tumors are positive for oncogenic forms of the human papillomavirus (HPV), particularly 16 type. About 60% of oropharyngeal squamous cell cancers in the USA are positive for HPV 16. HPV-associated head and neck squamous cell carcinoma seems to be a distinct clinical entity, and this malignant disease has a better prognosis than HPV-negative tumors, due in part to increased sensitivity of cancers to chemotherapy and radiation therapy. Although HPV is now recognized as a causative agent for a subset of oropharyngeal squamous cell carcinomas, the biology and natural history of oropharyngeal HPV infection and the best clinical management of patients with HPV-related head and neck squamous cell tumors is not well understood.
Head and neck cancer is the sixth most common cancer worldwide, with an estimated annual burden of 563 826 incident cases (including 274 850 oral cavity cancers, 159 363 laryngeal cancers, and 52 100 oropharyngeal cancers) and 301 408 deaths.1 Although HPV has been long known to be an important cause of anogenital cancer, only in recent times has it been recognized as a cause of a subset of head and neck squamous cell carcinomas.2 More than 100 different types of HPV exist,3 and at least 15 types are thought to have oncogenic potential.4 However, most (>90%) HPV-associated head and neck squamous cell cancers are caused by one virus type, HPV 16, the same type that leads to HPV-associated anogenital cancers. The proportion of head and neck squamous cell carcinomas caused by HPV varies widely (figure 1),5–16 largely because of the burden of tobacco-associated disease in this population of tumors. Tobacco, alcohol, poor oral hygiene, and genetics remain important risk factors for head and neck tumors overall, but HPV is now recognized as one of the primary causes of oropharyngeal squamous cell cancers. In the USA, about 40–80% of oropharyngeal cancers are caused by HPV, whereas in Europe the proportion varies from around 90% in Sweden to less than 20% in communities with the highest rates of tobacco use (figure 1).
The incidence of head and neck cancers overall in the USA has fallen in recent years, consistent with the decrease in tobacco use in this region. By contrast, incidence of HPV-associated oropharyngeal cancer seems to be rising, highlighting the increasing importance of this causal association.17–19 In a US study in which data of the Surveillance, Epidemiology, and End Results (SEER) program were used, incidence of oropharyngeal tumors (which are most likely to be HPV-associated) rose by 1·3% for base of tongue cancers and by 0·6% for tonsillar cancers every year between 1973 and 2004. By contrast, incidence of oral cavity cancers (not associated with HPV) declined by 1·9% every year during the same period.17 The age-adjusted incidence of tonsillar cancer increased 3·5-fold in women and 2·6-fold in men between 1970 and 2002.24 Augmented incidence of HPV-associated oropharyngeal cancers represents an emerging viral epidemic of cancer.
Why is increased incidence of HPV-associated oropharyngeal cancer most pronounced in young individuals? This effect could be attributable to changes in sexual norms (i.e., more oral sex partners or oral sex at an earlier age in recent than past generations) combined with fewer tobacco-associated cancers in young cohorts, making the outcomes of HPV-positive cancers more visible. Can the higher rates of HPV-associated oropharyngeal cancers in men compared with women be accounted for solely by differences in sexual behavior, or are biological differences in viral clearance present that could contribute to the higher burden of these cancers in men? HPV prevalence in cervical rather than penile tissue might boost the chances of HPV infection when performing oral sex on a woman, contributing to the higher rate of HPV-associated oropharyngeal cancer in men.
Tobacco use has fallen in past decades, and the corresponding rise in proportion of head and neck cancers that are oropharyngeal in origin has been striking, both in the USA and internationally. SEER data suggest that about 18% of all head and neck carcinomas in the USA were located in the oropharynx in 1973, compared with 31% of such squamous cell tumors in 2004.19 Similarly, in Sweden, the proportion of oropharyngeal cancers caused by HPV has steadily increased, from 23% in the 1970s to 57% in the 1990s, and as high as 93% in 2007.13,25 These data indicate that HPV is now the primary cause of tonsillar malignant disease in North America and Europe.
Findings of initial studies suggest that oral HPV frequency increases with age. Prevalent oral HPV infection is detected in 3–5% of adolescents26–28 and 5–10% of adults.14,29 We do not yet know whether the natural history of oral HPV or risk factors for persistent HPV infection in the oropharynx differ from those known for anogenital HPV infection (table 1). Data suggest oral HPV prevalence is amplified with number of sexual partners and is more typical in men, in HIV-infected individuals, and in current tobacco users.26–28,30,31
In view of the importance of tobacco use in head and neck squamous cell carcinoma, most cases of this malignant disease seen in non-smokers are unsurprisingly HPV-related. However, oral HPV infection is common in smokers and non-smokers and is an important cause of oropharyngeal cancer in both groups. For example, in case series, only 13–16% of individuals with HPV-positive head and neck squamous cell cancer did not smoke or drink alcohol.32,33 Although a higher proportion of individuals with HPV-positive compared with HPV-negative tumors are non-smokers or neither smoke nor drink alcohol, many with HPV-positive disease have a history of alcohol and tobacco use. In fact, 10–30% of HPV-positive head and neck squamous cell carcinomas were recorded in heavy tobacco and alcohol users.32,33 This finding underscores that HPV-associated malignant disease not only arises in people who do not smoke or drink alcohol but also occurs in people with the traditional risk factors of tobacco and alcohol use.
HPV detection may ultimately serve a more comprehensive role than mere prognostication. Detection of HPV is emerging as a valid biomarker for discerning the presence and progress of disease encompassing all aspects of patients’ care, from early cancer detection,41 to more accurate tumor staging (e.g., localization of tumor origin),42,43 to selection of patients most likely to benefit from specific treatments,44 to post-treatment tumor surveillance.45,46 Consequently, there is a pressing need for a method of HPV detection that is highly accurate, reproducible from one diagnostic laboratory to the next, and practical for universal application in the clinical arena. Despite growing calls for routine HPV testing of all oropharyngeal carcinomas, the best method for HPV detection is not established. Various techniques are currently in use, ranging from consensus and type-specific PCR methods, real-time PCR assays to quantify viral load, type-specific DNA in-situ hybridization, detection of serum antibodies directed against HPV epitopes, and immunohistochemical detection of surrogate biomarkers (e.g., P16 protein). Although PCR-based detection of HPV E6 oncogene expression in frozen tissue samples is generally regarded as the gold standard for establishing the presence of HPV, selection of assays for clinical use will ultimately be influenced by concerns relating to sensitivity, specificity, reproducibility, cost, and feasibility. Development of non-fluorescent chromogens has enabled visualization of DNA hybridization by conventional light microscope; furthermore, adaptation of in-situ hybridization to formalin-fixed and paraffin-embedded tissues has made this technique compatible with standard tissue-processing procedures and amenable to retrospective analysis of archival tissue blocks. Most PCR-based methods, on the other hand, need a high level of technical skill and are best used with fresh-frozen samples.
Limitations of any one detection assay can be offset by algorithms that combine the strengths of complementary assays.50 A highly feasible strategy incorporates P16 immunohistochemistry and HPV in-situ hybridization. In view of sensitivity that approaches 100%, P16 immunostaining is a good first-line assay for elimination of HPV-negative cases from any additional analysis. Since specificity is almost 100%, a finding positive for HPV 16 on in-situ hybridization reduces the number of false-positive cases by P16 staining alone. A P16-positive, HPV 16-negative result singles out a subset of tumors that qualifies for rigorous analysis for other (i.e., non-HPV 16) oncogenic virus types.
HPV in-situ hybridisation and P16 immunostaining as a practical diagnostic approach to discernment of HPV status can be applied readily to cytological preparations, including fi ne-needle aspirates from patients with cervical lymph-node metastases.41,52 Further expansion of HPV testing to blood and other body fl uids would advance the role of HPV as a clinically relevant biomarker, but these specimens would need other detection platforms. PCR-based detection of HPV DNA in blood (53) and saliva (54) of patients after treatment of their HPV-positive cancers suggests a future role in tumour surveillance. Detection of serum antibodies to HPV-related epitopes can predict the HPV status of head and neck cancers, and this method has been advocated as a way to project clinical outcomes and guide treatment without the constraints of tissue acquisition.53,55
The increasing prevalence of oropharyngeal cancer in young populations and substantially amplified survival rates with current treatment approaches stands in contrast to survival achieved in older individuals with comorbid disorders associated with tobacco and alcohol history. Several characteristics of patients with head and neck cancer have been linked with favorable prognosis, including non-smoker, minimum exposure to alcohol, good performance status, and no comorbid disorders, all of which are related to HPV-positive tumor status. Findings of retrospective analyses suggest that individuals with HPV-positive oropharyngeal cancer have higher response rates to chemotherapy and radiation and increased survival62–65 compared with those with HPV-negative tumors. Augmented sensitivity to chemotherapy and radiotherapy has been attributed to absence of exposure to tobacco and presence of functional unmutated TP53.63,64,66 Increased survival of patients with HPV-positive cancer is also possibly attributable in part to absence of field cancerization related to tobacco and alcohol exposure.67
Survival outcomes for individuals with HPV 16-positive and P16-positive oropharyngeal tumors were similar. Failure data indicated significantly diminished rates of locoregional failure and second primary tumour in patients with HPV-positive oropharyngeal cancer compared with those with HPV-negative tumors; distant metastases did not differ between the two groups. When survival was assessed after adjustment for tobacco exposure, in individuals who smoked, those with HPV-positive oropharyngeal tumors and fewer than 20 pack-years had 2-year overall survival of 95%, compared with 80% in those with HPV-positive cancers and 20 pack-years or more, and 63% in HPV-negative cancers and 20 pack-years or more. By comparison with people with HPV-positive oropharyngeal tumors who smoked and had fewer than 20 pack-years, the hazard of death was raised for those with HPV-negative tumors and 20 pack-years or more (hazard ratio 4·33) and those with HPV-positive cancers and 20 pack-years or more (1·79). These data indicate clearly that tobacco exposure alters the biology of HPV-positive oropharyngeal tumors and is an important prognostic factor.
An association between HPV-positive, P16-positive oropharyngeal tumors and survival outcomes was reported in another retrospective analysis of a large phase 3 trial of chemoradiation, which included more than 800 patients enrolled from international sites.72 This substudy analysis looked at 195 available tumor samples in patients with an oropharyngeal primary cancer, of which 28% were HPV-positive and 58% were P16-positive. Individuals with HPV-positive cancers had 2-year overall survival of 94% and 2-year failure-free survival of 86% compared with 77% (p=0·007) and 75% (p=0·035), respectively, in those with HPV-negative tumors. When co-expression of HPV and P16 was correlated with survival outcomes, individuals with HPV-positive, P16-positive tumors had 2-year overall survival of 95% compared with 88% in those with HPV-negative, P16-positive cancers and 71% (p=0·003) in those with HPV-negative, P16-negative tumors. Similar results were noted for 2-year failure-free survival (89%, 86%, and 69%, respectively; p=0·002) and time to locoregional failure (93%, 95%, and 84%, respectively; p=0·051). By multivariable analysis, HPV 16 and P16 were identified as independent prognostic factors.
ECOG proposes induction chemotherapy with a triple drug regimen to reduce tumor burden to subclinical disease (clinical complete response at primary site) followed by lower dose radiation (total dose 54 Gy) and concurrent cetuximab. Overall survival and progression-free survival outcomes will be assessed and compared with results of the 2008 ECOG study.70 The main aim of this planned study is to assess potential for a lower dose of radiation to control disease and to investigate toxic effects and quality-of-life variables.
In summary, tumor HPV status is a prognostic factor for overall survival and progression-free survival and might also be a predictive marker of response to treatment. The method of in-situ hybridization provides a feasible approach for implementation in most diagnostic pathology laboratories, and immunohistochemical staining for P16 could be useful as a surrogate marker for HPV status. Seemingly, locoregional recurrence—but not the rate of distant disease—is diminished in patients with HPV-positive tumors. Smoking and tobacco exposure might modify survival and recurrence of HPV-positive tumors and should be considered in future trials for risk stratification of patients with HPV-positive malignant disease.
HCV and cancer
The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide
Joseph F. Perz, Armstrong GL, Farrington LA, Hutin YJF, Bell BP
J Hepatol 2006; 45:529-538
http://dx.doi.org:/10.1016/j.jhep.2006.05.013
End-stage liver disease accounts for one in forty deaths worldwide. Chronic infections with hepatitis B virus (HBV) and hepatitis C virus (HCV) are well-recognized risk factors for cirrhosis and liver cancer, but estimates of their contributions to worldwide disease burden have been lacking. Methods: The prevalence of serologic markers of HBV and HCV infections among patients diagnosed with cirrhosis or hepatocellular carcinoma (HCC) was obtained from representative samples of published reports. Attributable fractions of cirrhosis and HCC due to these infections were estimated for 11 WHO-based regions. Results: Globally, 57% of cirrhosis was attributable to either HBV (30%) or HCV (27%) and 78% of HCC was attributable to HBV (53%) or HCV (25%). Regionally, these infections usually accounted for >50% of HCC and cirrhosis. Applied to 2002 worldwide mortality estimates, these fractions represent 929,000 deaths due to chronic HBV and HCV infections, including 446,000 cirrhosis deaths (HBV: n = 235,000; HCV: n = 211,000) and 483,000 liver cancer deaths (HBV: n = 328,000; HCV: n = 155,000). Conclusions: HBV and HCV infections account for the majority of cirrhosis and primary liver cancer throughout most of the world, highlighting the need for programs to prevent new infections and provide medical management and treatment for those already infected.
Among primary liver cancers occurring worldwide, hepatocellular carcinoma (HCC) represents the major histologic type and likely accounts for 70% to 85% of cases [2]. Cirrhosis precedes most cases of HCC, and may exert a promotional effect via hepatocyte regeneration [3,4]. Compared with other causes of cirrhosis, chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) is associated with a higher risk of developing HCC [3,5]. Alcohol abuse represents a leading cause of cirrhosis and is also a major contributor. dietary aflatoxin exposure in parts of Africa and Asia has been associated with primary liver cancer, especially in hosts with chronic HBV infection [8].
An understanding of the relative contribution of various etiologies to disease burden is important for setting public health priorities and guiding prevention programs [10,11]. The World Health Organization’s Global Burden of Disease (GBD) 2000 project aims to quantify the burden of premature morbidity and mortality from over 130 major causes [1,12]. Liver cancer and cirrhosis are included in the analysis, but with the exception of alcohol, the etiologies underlying these diseases have not been well accounted for [1,11,13]. In particular, HBV and HCV infections have been poorly characterized in previous WHO estimates since these were based primarily on the acute effects of infection and omitted the associated burdens of chronic liver disease [10,11].
The attributable fraction represents the proportion of disease occurrence that potentially would be prevented by eliminating a given risk factor. For cirrhosis, a systematic analysis of attributable fractions has been lacking altogether. For HCC, previous estimates of the attributable fractions due to HBV and HCV are available but are not comprehensive and do not correspond to the regional designations and related conventions of the current GBD project [14].
The prevalence of HBV and HCV infection among cirrhosis and HCC patients varied considerably within and between regions (Tables 2 and 3). These variations tended to reflect known patterns of HBV and HCV infection endemicity [99,100]. For example, in countries where HCV infection has long been endemic, such as Japan and Egypt, there were high prevalences of HCV infection among cirrhosis and HCC patients. The same held true for China and most of the African nations in our sample regarding HBV infection. Areas such as these, where HBV infection predominated, appeared to have a younger population of HCC cases, which is thought to reflect the preponderance of infections acquired early in life (e.g., perinatal HBV transmission) [8]. Patterns of HBV and HCV co-infection were also notable.
When we applied the HBV- and HCV-attributable fractions we derived to 2002 worldwide mortality estimates [1], we found that approximately 929,000 deaths from cirrhosis (n = 446,000) and primary liver cancer (n = 483,000) were likely due to chronic viral hepatitis infections. HBV infection accounted for 563,000 deaths (235,000 from cirrhosis and 328,000 from liver cancer) and HCV infection accounted for 366,000 deaths (211,000 from cirrhosis and 155,000 from liver cancer).
We showed that chronic viral hepatitis infections likely account for the majority of both cirrhosis and HCC globally and in nearly all regions of the world. One of the strengths of our analysis was that it employed simple and transparent methods. Our estimates of attributable fractions were derived from reviews of published studies reporting the prevalence of HBV and HCV infections in patients with cirrhosis or HCC in all regions of the world. Alternate approaches rely on estimates of the prevalence of risk factors and corresponding relative risks in the source populations. However, errors associated with extrapolating exposure or hazard from one population to another are a major source of uncertainty in efforts to characterize international health risks [12]. Given the lack of representative data regarding HBV and HCV infection prevalences worldwide along with uncertainties in deriving region specific risk estimates, we believe ours is the preferred approach.
Our findings help illustrate the great need for programs aimed at preventing HBV or HCV transmission. In 1992, WHO recommended that all countries include hepatitis B vaccine in their routine infant immunization programs. As of 2003, WHO/UNICEF estimated 42% hepatitis B vaccination coverage among the global birth cohort [106]. Therefore, implementation of this strategy, which represents the most effective way of preventing chronic HBV infection and related end stage liver disease, is far from complete [107,108]. Other key primary prevention strategies include screening blood donors and maintaining infection control practices to prevent the transmission of healthcare-related HBV and HCV infections [105,109,110]. In countries where these activities have not been fully implemented, they should be given a high priority. In most developed countries, injection drug use and high-risk sexual behaviors represent the major risk factors for HCV infection and HBV infection, respectively, indicating the importance of related prevention efforts (e.g., reducing the numbers of new initiates to injection drug use).
The role of programs to identify, counsel, and provide medical management for the many persons already infected with HBV or HCV requires careful consideration [105,110]. Counseling that includes advice regarding avoidance of alcohol and education regarding modes of transmission can help reduce the risks for developing chronic disease or spreading infection to susceptible persons. The widespread application of therapeutic interventions also has the potential to accelerate the declines in end-stage liver disease that will eventually follow from hepatitis B vaccination and other primary prevention efforts [104,107]. Recent advances have occurred in the therapeutic management of chronic hepatitis B and chronic hepatitis C, but treatments are long and involve substantial costs and side effects [111–113]. Countries will need to consider the potential benefits of treatment while insuring that scarce healthcare resources are allocated in a manner that does not undermine primary prevention efforts [114].
Risk of pancreatic cancer among individuals with hepatitis C or hepatitis B virus infection: a nationwide study in Sweden.
Huang J1, Magnusson M, Törner A, Ye W, Duberg AS.
Br J Cancer. 2013 Nov 26; 109(11):2917-23.
http://dx.doi.org:/10.1038/bjc.2013.689
A few studies indicated that hepatitis C and hepatitis B virus (HCV/HBV) might be associated with pancreatic cancer risk. The aim of this nationwide cohort study was to examine this possible association. Methods: Hepatitis C virus-
and hepatitis B virus-infected individuals were identified from the national surveillance database from 1990 to 2006, and followed to the end of 2008. The pancreatic cancer risk in the study population was compared with the general population by calculation of Standardized Incidence Ratios (SIRs), and with a matched reference population using a Cox proportional hazards regression model to calculate hazard ratios (HRs). Results: In total 340 819 person-years in the HCV cohort and 102 295 in the HBV cohort were accumulated, with 34 and 5 pancreatic cancers identified, respectively. The SIRHCV was 2.1 (95% confidence interval, CI: 1.4, 2.9) and the SIRHBV was 1.4 (0.5, 3.3). In the Cox model analysis, the HR for HCV infection was 1.9 (95% CI: 1.3, 2.7), diminishing to 1.6 (1.04, 2.4) after adjustment for potential confounders.
Conclusion: Our results indicated that HCV infection might be associated with an increased risk of pancreatic cancer but further studies are needed to verify such association. The results in the HBV cohort indicated an excess risk, however, without statistical significance due to lack of power.
Pancreatic cancer is one of the most rapidly fatal malignancies with a 5-year survival rate below 5%. The long-term survival is poor also for early diagnosed patients treated with resection surgery (Jemal et al, 2010). In Europe, it was estimated in a prediction model that in the year 2012 there would be 75 000–80 000 deaths from pancreatic cancer, which is the fourth most common cause of cancer-associated death for both men and women (Malvezzi et al, 2012). The incidence of pancreatic cancer is higher in the Nordic countries and Central Europe than in other parts of the world (Bosetti et al, 2012).
Tobacco smoking is a well-established risk factor for pancreatic cancer (Iodice et al, 2008), and a similar magnitude of excess risk as smoking was found among the users of Scandinavian snus (moist snuff) (Boffettaet al, 2005; Luo et al, 2007). Besides, accumulating evidence consistently shows that old age, male sex, diabetes mellitus, hereditary pancreatitis, chronic pancreatitis and family history are positively associated with this carcinoma (Pandol et al, 2012). Albeit the biological mechanism is unclear, recent epidemiological studies indicated that some infections, such as exposure to Helicobacter pylori (Trikudanathan et al, 2011), poor oral health (Michaud et al, 2007), hepatitis C virus (HCV) (Hassan et al, 2008; El-Serag et al, 2009) or hepatitis B virus (HBV) (Hassan et al, 2008; Iloeje et al, 2010; Wang et al, 2012a, 2012b) might be associated with pancreatic cancer risk.
Globally, ∼170 million people are chronically infected with HCV (World Health Organization, 1997) and an estimated 350 million with HBV (Custer et al, 2004). The prevalence rates of HCV and HBV infection vary widely in the world, and Sweden is a low endemic country with an estimated 0.5% of the population infected with HCV (Duberg et al, 2008a) and even lower rate for HBV infection. Both chronic HCV and HBV infections are main causes of hepatocellular carcinoma (HCC). Previous findings demonstrated that HBV may replicate within the pancreas (Shimoda et al, 1981; Yoshimura et al, 1981) and that HCV could be associated with pancreatitis (Alvares-Da-Silva et al, 2000; Torbenson et al, 2007). Some studies support that HCV and HBV may have a role in the development of pancreatic cancer, but the evidence is far from conclusive (Hassan et al, 2008; El-Serag et al, 2009; Iloeje et al, 2010; Wang et al, 2012a, 2012b), and more studies are needed. Towards this end, we utilised Swedish population-based nationwide registers, with documentation of all diagnosed HCV- and HBV-infected individuals in Sweden, to explore the association of HCV or HBV infection and the risk of pancreatic cancer.
Baseline characteristics of the HCV and HBV cohorts are presented in Table 1. In the HCV and chronic HBV cohorts the mean follow-up time were 9.1 and 9.4 years, with a total of 360 154 and 107 986 person-years at risk, respectively. There was a clear male dominance in the HCV cohort, and median age at entry into the HCV or HBV cohorts (notification date) was 38 and 31 years, respectively. A marked difference between cohorts was observed regarding the aspect of country of origin; HCV-infected individuals were more likely from Nordic countries, but persons with chronic HBV infection were often immigrants from non-Nordic countries.
Hepatitis C virus cohort
In the HCV cohort, there were 34 pancreatic cancer cases observed during 340 819 person-years of follow-up (first 6 months of follow-up excluded), whereas 16.5 were expected, yielding a statistically significant increased risk of pancreatic cancer (SIR: 2.1; 95% CI: 1.4, 2.9). The SIR did not alter substantially across sex or estimated duration of HCV infection (Table 2). The majority of cases were among the patients who were born before 1960.
From the Cox regression model, an ∼90% excessive risk for pancreatic cancer (HR 1.9; 95% CI: 1.3, 2.7) was observed after adjustment for age, sex and county of residence, which is similar to the result from the SIR analysis. This excess risk diminished somewhat but remained statistically significant after further adjustment for potential confounders (HR 1.6; 95% CI: 1.04, 2.4). The results did not vary markedly when stratified by sex (Table 3). In the additional analyses, excluding all individuals ever hospitalized with acute and/or chronic pancreatitis, the results did not alter notably (data not shown).
In the HCV cohort, the Standardized Incidence Ratio (SIR) for lung cancer was 2.3 (95% CI: 1.9, 2.7) and the Hazard Ratio (HR) for lung cancer was 2.2 (95% CI: 1.8, 2.7), decreasing to 1.6 (95% CI: 1.3, 2.1) after adjustment for the potential confounders used in the pancreatic cancer analyses.
Chronic HBV cohort
A total of five pancreatic cancer cases were found during 102 295 person-years of follow-up (first 6 months excluded), whereas 3.5 were expected. Compared with the age- and sex-matched Swedish general population, a 40% excess risk of pancreatic cancer was found in the chronic HBV cohort (SIR: 1.4; 95% CI: 0.5, 3.3), but without statistical significance. Because of the small number of pancreatic cancer cases, there was not enough power for additional stratified analyses (Table 4).
The Cox regression model revealed similar results as the SIR analysis. The point estimates were somewhat higher (HR=2.0 from the model adjusted for only matching factors and HR=1.8 from the fully adjusted model), but still statistically non-significant (Table 5). The SIR for lung cancer in the chronic HBV infection cohort was 1.7 (95% CI: 1.1, 2.5).
This population-based large cohort study revealed a doubled risk of pancreatic cancer among HCV-infected patients compared with the Swedish general population. The excess risk was persistent across strata by sex or duration of infection. Although further adjustment for potential confounders, i.e., chronic obstructive pulmonary disease (related to smoking), diabetes mellitus, chronic pancreatitis and alcohol-related disease, resulted in an attenuated relative risk, this finding still supports the hypothesis that HCV infection might be associated with an increased risk of pancreatic cancer. Besides, the result indicated a moderate excessive risk of pancreatic cancer among HBV-infected patients according to different statistical approaches, but the size of the study cohort and the observed number of cancers were too small to draw a sound conclusion. Pancreatic cancer is more common in older age groups, and the small number of pancreatic cancers among the HBV cohort was probably an effect of the relatively young cohort, concordant with the epidemiology of chronic hepatitis B in Sweden.
The strengths of this register-based study include population-based cohort design, relatively large sample size, independently collected data on documentation of HCV/HBV notifications and pancreatic cancer occurrence and high completeness of follow-up.
The parallel (laboratory and clinician) notification system of HCV/HBV infections in Sweden has a high coverage of those with a diagnosed infection; it is estimated that about 75–80% of HCV infections are diagnosed, but there still remain unknown infections, not yet diagnosed or documented. In addition, a small portion of the reported patients could have a resolved infection, spontaneously or by treatment, this could (probably insignificant) lower the risk in the HCV and HBV cohort.
The number of unidentified HCV/HBV-coinfected individuals is probably low in the studied cohorts. However, in the HCV cohort there could be some patients who were never diagnosed with hepatitis B but have serologic markers of a past HBV infection. In these patients we cannot exclude the possibility of occult hepatitis B.
The biological mechanism of the association between HCV and pancreatic cancer is unclear. However, virtually, the pancreas and liver share the common blood vessels and ducts, and prior evidence demonstrated that the pancreas is a remote location for hepatitis virus inhabitation and replication (Hassan et al, 2008). HCV infection is associated with type 2 diabetes, which is both a risk factor and might be a consequence of pancreatic cancer (Mehta et al, 2000; Sangiorgio et al, 2000). Besides, previous studies reported that subclinical/acute pancreatitis (Katakura et al, 2005) and hyperlipasemia (Yoffe et al, 2003) may be extrahepatic manifestations of HCV infection. In addition, pancreatic involvement was observed among patients who suffered from chronic hepatitis infection, resulting in mild pancreatic damage accompanied with increased serum levels of pancreatic enzyme (Taranto et al, 1989; Katakura et al, 2005). Immune response may lead to chronic inflammation in the targeted organs after long time persistent infection with HCV. Therefore, hepatitis C virus conceivably serves as a biological agent that may indirectly have a role in inflammation-associated pancreatic carcinogenesis. Although still unclear to what extent chronic inflammation contributes to pancreatic cancer development, it is postulated that HCV can induce inflammatory microenvironment with high concentration of growth factors and cytokines. This may exert effects by accumulating alterations in driver genes and promoting cancer cell growth and proliferation.
HIV AIDS and Cancer
http://www.cancer.gov/cancertopics/causes-prevention/risk/infectious-agents/hiv-fact-sheet
Key Points
- People infected with human immunodeficiency virus (HIV) have a higher risk of some types of cancer than uninfected people.
- A weakened immune system caused by infection with HIV, infection with other viruses, and traditional risk factors such as smoking all contribute to this higher cancer risk.
- Highly active antiretroviral therapy and lifestyle changes may reduce the risk of some types of cancer in people infected with HIV.
- The National Cancer Institute (NCI) conducts and supports a number of research programs aimed at understanding, preventing, and treating HIV infection, acquired immunodeficiency syndrome-related cancers, and cancer-associated viral diseases.
- Do people infected with human immunodeficiency virus (HIV) have an increased risk of cancer?
Yes. People infected with HIV have a substantially higher risk of some types of cancer compared with uninfected people of the same age (1). Three of these cancers are known as “acquired immunodeficiency syndrome (AIDS)-defining cancers” or “AIDS-defining malignancies”: Kaposi sarcoma, non-Hodgkin lymphoma, and cervical cancer. A diagnosis of any one of these cancers marks the point at which HIV infection has progressed to AIDS.
People infected with HIV are several thousand times more likely than uninfected people to be diagnosed with Kaposi sarcoma, at least 70 times more likely to be diagnosed with non-Hodgkin lymphoma, and, among women, at least 5 times more likely to be diagnosed with cervical cancer (1).
In addition, people infected with HIV are at higher risk of several other types of cancer (1). These other malignancies include anal, liver, and lung cancer, and Hodgkin lymphoma.
People infected with HIV are at least 25 times more likely to be diagnosed with anal cancer than uninfected people, 5 times as likely to be diagnosed with liver cancer, 3 times as likely to be diagnosed with lung cancer, and at least 10 times more likely to be diagnosed with Hodgkin lymphoma (1).
People infected with HIV do not have increased risks of breast, colorectal, prostate, or many other common types of cancer (1). Screening for these cancers in HIV-infected people should follow current guidelines for the general population
HIV and cancer of the cervix
Z.M. Chirenje
bestpracticeobgyn April 2005; 19(2):269–276
http://dx.doi.org/10.1016/j.bpobgyn.2004.10.002
Cancer of the cervix is the second most common cause of cancer-related death in women worldwide, and in some low resource countries accounts for the highest cancer mortality in women. The highest burden of the HIV/AIDS epidemic is currently in sub-Saharan Africa, where more than half of the people infected are women who have no access to cervical cancer screening. The association between HIV and invasive cervical cancer is complex, with several studies now clearly demonstrating an increased risk of pre-invasive cervical lesions among HIV-infected women. However, there have not been significantly higher incidence rates of invasive cervical cancer associated with the HIV epidemic. The highest numbers of HIV-infected women are in poorly-resourced countries, where the natural progression of HIV disease in the absence of highly active antiretroviral treatment sometimes results in deaths from opportunistic infections before the onset of invasive cervical cancer. This chapter will discuss the association of HIV and cervical intraepithelial neoplasia, the treatment of pre-invasive lesions, and invasive cervical cancer in HIV-infected women. The role of screening and the impact of antiretroviral treatment on the progression of pre-invasive and invasive cancer will also be discussed.
Anal cancer: an HIV-associated cancer
Klencke BJ, Palefsky JM
Hematology/oncology Clinics of North America [2003, 17(3):859-872]
http://dx.doi.org:/1016/S0889-8588(03)00039-X
Although not yet included in the Centers for Disease Control definition of AIDS, anal cancer clearly occurs more commonly in HIV-infected patients. An effective screening program for those groups who are at highest risk might be expected to impact rates of anal cancer just as significantly as did cervical Pap screening programs for the incidence of cervical cancer. Despite a relatively low rate of progression from AIN to invasive cancer, the scope of the problem is enormous based on the prevalence of anal HPV infection and the size of the HIV-infected, at-risk population. Thus, the potential benefits of screening, detection, and the development of more effective therapy also are enormous. Currently, therapeutic HPV vaccines for AIN represent an exciting avenue of research in HPV-related anogenital disease. Invasive anal cancer and HSIL (which is believed to be the precursor lesion) are expected to become increasingly important health problems for both HIV-infected men and women as their life expectancy lengthens. Although HAART may have improved the ability of many to tolerate CMT, it appears that toxicity of this therapy continues to be a problem for a proportion of HIV-infected subjects. The acute side effects present specific challenges to the clinician and patient, have an immediate impact on the patient’s plan of care and dose intensity of the treatment, and ultimately may impact the outcome of the planned treatment. Late toxicity may influence the long-term quality of life. Small patient numbers, variable radiation therapy doses, limited information about viral load, and a potential confounding effect of higher CD4+ levels make it difficult to draw any conclusions about the effect of HAART on anal cancer outcome. Large, prospective studies will be required before solid conclusions about the impact of various factors on anal cancer prognosis and outcome can be drawn.
The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis
Hirokazu Tamamura, and Nobutaka Fujii
Exp Opin on Ther Targets Dec 2005; 9(6): 1267-1282 http://dx.doi.org:/10.1517/14728222.9.6.1267
CXCR4 is the receptor of the chemokine CXCL12, which is involved in progression and metastasis of several types of cancer cells, HIV infection and rheumatoid arthritis. The authors developed selective CXCR4 antagonists, T22 and T140, initially as anti-HIV agents, which inhibit T cell line-tropic (X4-) HIV-1 infection through their specific binding to CXCR4. Recently, T140 analogues have also been shown to inhibit CXCL12-induced migration of breast cancer cells, leukaemia T cells, pancreatic cancer cells, small cell lung cancer cells, chronic lymphocytic leukaemia B cells, pre-B acute lymphoblastic leukaemia cells and so on in vitro. Biostable T140 analogues significantly suppressed pulmonary metastasis of breast cancer cells and melanoma cells in mice. Furthermore, these compounds significantly suppressed the delayed-type hypersensitivity response induced by sheep red blood cells and collagen-induced arthritis, which represent in vivo mouse models of arthritis. Thus, T140 analogues proved to be attractive lead compounds for chemotherapy of these problematic diseases. This article reviews recent research on T140 analogues, referring to several other CXCR4 antagonists.
Types of Cancer: AIDS/HIV related malignancies
http://cancer.northwestern.edu/cancertypes/cancer_type.cfm?category=1
People with HIV/AIDS are at high risk for developing certain cancers, such as Kaposi’s sarcoma, non-Hodgkin lymphoma, and cervical cancer. For people with HIV, these three cancers are often called “AIDS-defining conditions,” meaning that if a person with HIV has one of these cancers it can signify the development of AIDS. The connection between HIV/AIDS and certain cancers is not completely understood, but the link likely depends on a weakened immune system. Most types of cancer begin when normal cells begin to change and grow uncontrollably, forming a mass called a tumor. A tumor can be benign (noncancerous) or malignant (cancerous, meaning it can spread to other parts of the body). The types of cancer most common for people with HIV/AIDS are described in more detail below.
Kaposi’s sarcoma
Kaposi’s sarcoma is a type of skin cancer, which has traditionally occurred in older men of Jewish or Mediterranean descent, young men in Africa, or people who have received organ transplantation. Today, Kaposi’s sarcoma is found most often in homosexual men with HIV/AIDS and related to an infection with the human herpesvirus 8 (HHV-8). Kaposi’s sarcoma in people with HIV is often called epidemic Kaposi’s sarcoma. HIV/AIDS-related Kaposi’s sarcoma causes lesions to arise in multiple sites in the body, including the skin, lymph nodes, and organs such as the liver, spleen, lungs, and digestive tract.
Non-Hodgkin lymphoma
HIV/AIDS-related NHL is the second most common cancer associated with HIV/AIDS, after Kaposi’s sarcoma. There are many different subtypes of NHL. The most common subtypes of NHL in people with HIV/AIDS are primary central nervous system lymphoma (affecting the brain and spinal fluid), found in 20% of all NHL cases in people with HIV/AIDS, primary effusion lymphoma (causing fluid to accumulate around the lungs or in the abdomen), or intermediate and high-grade lymphoma. More than 80% of lymphomas in people with HIV/AIDS are high-grade B-cell lymphoma, while 10% to 15% of lymphomas among people with cancer who do not have HIV/AIDS are of this type. It is estimated that between 4% and 10% of people with HIV/AIDS develop NHL.
Other types of cancer
Other, less common types of cancer that may develop in people with HIV/AIDS are Hodgkin’s lymphoma, angiosarcoma (a type of cancer that begins in the lining of the blood vessels), anal cancer, liver cancer, mouth cancer, throat cancer, lung cancer, testicular cancer, colorectal cancer, and multiple types of skin cancer including basal cell carcinoma, squamous cell carcinoma, and melanoma.
Treatment options for the most common treatments for HIV/AIDS-related cancers are listed by the specific type of cancer. Treatment options and recommendations depend on several factors, including the type and stage of cancer, possible side effects, and the patient’s preferences and overall health.
Palliative care can help a person at any stage of illness. People often receive treatment for the cancer and treatment to ease side effects at the same time. In fact, patients who receive both often have less severe symptoms, better quality of life, and report they are more satisfied with treatment.
Palliative treatments vary widely and often include medication, nutritional changes, relaxation techniques, and other therapies. You may also receive palliative treatments similar to those meant to eliminate the cancer, such as chemotherapy, surgery, and radiation therapy.
It is extremely important that all patients with HIV/AIDS and an associated cancer receive treatment with highly active antiretroviral treatment (HAART) both during the cancer treatments and afterwards. HAART can effectively control the virus in most patients. Better control of the HIV infection decreases the side effects of many of the treatments, may decrease the chance of a recurrence, and can improve a patient’s chance of recovery from the cancer.
The treatment of HIV/AIDS-related Kaposi sarcoma usually cannot cure the cancer, but it can help relieve pain or other symptoms. This can be followed by palliative care for Kaposi sarcoma. Antiviral treatment for HIV/AIDS helps reduce a person’s chance of getting Kaposi sarcoma and can reduce the severity of Kaposi sarcoma. HAART helps treat the tumor and reduce the symptoms associated with Kaposi sarcoma for people with HIV/AIDS. It is usually used before other treatments, such as chemotherapy.
Curettage and electrodesiccation. In this procedure, the cancer is removed with a curette, a sharp, spoon-shaped instrument. The area can then be treated with electrodesiccation, which uses an electric current to control bleeding and kill any remaining cancer cells. Many patients have a flat, pale scar from this procedure.
Cryosurgery. Cryosurgery, also called cryotherapy or cryoablation, uses liquid nitrogen to freeze and kill cells. The skin will later blister and shed off. This procedure will sometimes leave a pale scar. More than one freezing may be needed.
In photodynamic therapy, a light-sensitive substance is injected into the lesion that stays longer in cancer cells than in normal cells. A laser is directed at the lesion to destroy the cancer cells.
Radiation therapy is the use of high-energy x-rays or other particles to destroy cancer cells. A doctor who specializes in giving radiation therapy to treat cancer is called a radiation oncologist. The most common type of radiation treatment is called external-beam radiation therapy, which is radiation given from a machine outside the body. When radiation therapy is given using implants, it is called internal radiation therapy or brachytherapy. External-beam radiation therapy may be given as a palliative treatment. A radiation therapy regimen (schedule) usually consists of a specific number of treatments given over a set period of time.
Side effects from radiation therapy may include fatigue, mild skin reactions, upset stomach, and loose bowel movements. Most side effects go away soon after treatment is finished. Learn more about radiation therapy.
Chemotherapy may help control advanced disease, although curing HIV/AIDS-related Kaposi sarcoma with chemotherapy is extremely rare. Usually, for HIV/AIDS-related Kaposi sarcoma, chemotherapy is used to help relieve symptoms and to lengthen a patient’s life. Common drugs for Kaposi sarcoma include: liposomal doxorubicin (Doxil), paclitaxel (Taxol, LEP-ETU, Abraxane), and vinorelbine (Navelbine, Alocrest).
The side effects of chemotherapy depend on the individual and the dose used, but they can include fatigue, risk of infection, nausea and vomiting, hair loss, loss of appetite, and diarrhea. These side effects usually go away once treatment is finished.
HIV/AIDS-related Kaposi sarcoma may receive alpha-interferon (Roferon-A, Intron A, Alferon), which appears to work by changing the surface proteins of cancer cells and by slowing their growth. Immunotherapy is generally used for people who are in the good-risk category in the immune system (I) factor of the TIS staging system (see Stages). The most common side effects of alpha-interferon are low levels of white blood cells and flu-like symptoms.
The main treatments for HIV/AIDS-related non-Hodgkin lymphoma are chemotherapy, targeted therapy, and radiation therapy.
Treatments for women with the precancerous condition called CIN (see Overview) are generally not as effective for women with HIV/AIDS because of a weakened immune system. Often, the standard treatment for HIV/AIDS can lower the symptoms of CIN.
Women with invasive cervical cancer and HIV/AIDS that is well-controlled with medication, generally receive the same treatments as women who do not have HIV/AIDS. Common treatment options include surgery, radiation therapy, and chemotherapy.
Cytokines in cancer pathogenesis and cancer therapy
Glenn Dranoff
Nature Reviews Cancer Jan 2004; 4(11-22) http://dx.doi.org:/10.1038/nrc1252
The mixture of cytokines that is produced in the tumor microenvironment has an important role in cancer pathogenesis. Cytokines that are released in response to infection, inflammation and immunity can function to inhibit tumor development and progression. Alternatively, cancer cells can respond to host-derived cytokines that promote growth, attenuate apoptosis and facilitate invasion and metastasis. A more detailed understanding of cytokine–tumor-cell interactions provides new opportunities for improving cancer immunotherapy.
Dendritic Cells as Therapeutic Vaccines against Cancer
Jacques Banchereau and A. Karolina Palucka
Nature Reviews Immunology APR 2005; 5:296-306
http://cnc.cj.uc.pt/BEB/private/pdfs/2007-2008/Immunology/E/Rev_paper_E4.pdf
Mouse studies have shown that the immune system can reject tumours, and the identification of tumor antigens that can be recognized by human T cells has facilitated the development of immunotherapy protocols. Vaccines against cancer aim to induce tumor-specific effector T cells that can reduce the tumor mass, as well as tumor-specific memory T cells that can control tumor relapse. Owing to their capacity to regulate T-cell immunity, dendritic cells are increasingly used as adjuvants for vaccination, and the immunogenicity of antigens delivered by dendritic cells has now been shown in patients with cancer. A better understanding of how dendritic cells regulate immune responses will allow us to better exploit these cells to induce effective anti-tumor immunity.
Vaccines against infectious agents are one success of immunology and have spared countless individuals from diseases such as polio, measles, hepatitis B and tetanus8 . However, progress in the development of vaccines against infectious agents has been largely empirical and not always successful, as many infectious diseases still evade the immune system, particularly chronic infections such as tuberculosis, malaria and HIV infection. Further progress will be made through rational design based on our increased understanding of how the immune system works and how the induction of protective immunity is regulated. The same principle applies to vaccines against cancer, particularly as cancer is a chronic disease, and when it becomes clinically visible, tumor cells and their products have already been interacting with and affecting host cells for a considerable time to ensure the survival of the tumor. Ex vivo-generated, antigen-loaded DCs have now been used as vaccines to improve immunity9 . Numerous studies in mice have shown that DCs loaded with tumor antigens can induce therapeutic and protective antitumor immunity10. The immunogenicity of antigens delivered by DCs has been shown in patients with cancer9 or chronic HIV infection11, thereby providing proof of principle that using DCs as vaccines can work. Despite this, the efficacy of therapeutic vaccination against cancer has recently been questioned12 because of the undeniably limited rate of objective tumor regressions that has been observed in clinical studies so far. However, the question is not whether DC vaccines work but how to orient further studies to refine the immunological and clinical parameters of vaccination with DCs to improve its efficacy.
Vaccines against cancer Early studies in mice showed that the immune system can recognize and reject tumours13 and that immunodeficient mice (lacking interferon-γ (IFN-γ) and recombination-activating gene 2) have an increased incidence of cancer14 (BOX 1). In humans, the incidence of some cancers is increased in immunodeficient patients15 and is increased with age, owing to Immunosenescence16. These observations support the scientific rationale for immunotherapy for cancer. The term immunotherapy refers to any approach that seeks to mobilize or manipulate the immune system of a patient for therapeutic benefit17. In this regard, there are numerous strategies for improving the resistance of a patient to cancer. These include non-specific activation of the immune system with microbial components or cytokines, antigen-specific adoptive immunotherapy with antibodies and/or T cells, and antigen-specific active immunotherapy (that is, vaccination). The main limitation of using antibodies is that target proteins need to be expressed at the cell surface. By contrast, targets for T cells are usually peptides derived from intracellular proteins, which are presented at the cell surface in complexes with MHC molecules18. The identification of defined tumor antigens in humans19,20 prompted the development of adoptive T-cell therapy. Yet, the most attractive strategy is vaccination, which is expected to induce both therapeutic T-cell immunity (in the form of tumor-specific effector T cells) and protective T-cell immunity (in the form of tumor-specific memory T cells that can control tumor relapse)21–23. Numerous approaches for the therapeutic vaccination of individuals who have cancer have been developed, including the use of the following: autologous and allogeneic tumor cells (which are often modified to express various cytokines), peptides, proteins and DNA vaccines9,23–26. The observed results are variable; however, in many cases, a tumour-specific immune response has been induced, and tumor regressions, albeit limited, have occurred. These approaches rely on random encounter of the vaccine with host DCs. A lack of encounter of the vaccine antigen with DCs might result in the absence of an immune response. Alternatively, an inappropriate encounter — for example, with unactivated DCs or with the ‘wrong’ subset of DCs — might lead to silencing of the immune response27. Both of these situations could explain some of the shortcomings of current cancer vaccines. Furthermore, we do not know how tumor antigens need to be delivered to DCs in vivo to elicit an appropriate immune response.
Immature and mature dendritic cells have different functions. A | Immature dendritic cells (DCs) induce tolerance. Tissue DCs constantly sample their environment, capture antigens and migrate in small numbers to draining lymph nodes. In the absence of inflammation, the DCs remain in an immature state, and antigens are presented to T cells in the lymph node without costimulation, leading to either the deletion of T cells or the generation of inducible regulatory T cells. B | Mature DCs induce immunity. Tissue inflammation induces the maturation of DCs and the migration of large numbers of mature DCs to draining lymph nodes. The mature DCs express peptide–MHC complexes at the cell surface, as well as appropriate co-stimulatory molecules. This allows the priming of CD4+ T helper cells and CD8+ cytotoxic T lymphocytes (CTLs), the activation of B cells and the initiation of an adaptive immune response. To control the immune response, CD4+CD25+ regulatory T (TReg)-cell populations are also expanded. [ADCC, antibody-dependent cell-mediated cytotoxicity; NK, natural killer; TCR, T-cell receptor].
Box 1 |
Mice
- The immune system can reject tumors
- Immune-mediated rejection of chemically induced tumours13
- Increased cancer incidence in immunodeficient mice14
Humans
- Increased cancer incidence in immunodeficient patients15
- Increased cancer incidence with age (immunosenescence)16
- Cancer regression in patients with paraneoplastic neurological disorders that are mediated by onconeuronal antibodies and specific CD8+ T cells136
Dendritic cells DC subsets. There are thought to be two main pathways of differentiation into DCs2,31 (FIG. 2). The myeloid pathway generates two subsets: Langerhans cells, which are found in stratified epithelia such as the skin; and interstitial DCs, which are found in all other tissues32. The lymphoid pathway generates plasmacytoid DCs (pDCs), which secrete large amounts of type I IFNs (IFN-α and IFN-β) after viral infection33,34. DCs and their precursors show remarkable functional plasticity. For example, pDCs form one of the first barriers to the expansion of intruding viruses, thereby functioning, through the release of type I IFNs, as part of the innate immune response. Subsequently, these cells differentiate into DCs that can present antigens to T cells, thereby functioning as members of the adaptive immune system35,36. Monocytes can differentiate into either macrophages, which function as scavengers, or DCs that induce specific immune responses37,38. Different cytokines skew the in vitro differentiation of monocytes into DCs with different phenotypes and functions (FIG. 3). So, after activation (for example, by granulocyte/ macrophage colony-stimulating factor, GM-CSF), monocytes that encounter interleukin-4 (IL-4) become DCs known as IL-4-DCs29,30,39. By contrast, after encounter with IFN-α, tumour-necrosis factor (TNF) or IL-15, activated monocytes differentiate into IFN-α-DCs40–43, TNF-DCs44 or IL-15-DCs45, respectively. Whether, in vivo, all interstitial DCs are derived from monocytes remains to be established, but myeloid DCs that are isolated from human peripheral blood also give rise to different DC types after exposure to different cytokines. Each of these DC subsets has both common and unique biological functions, which are determined by a unique combination of cell-surface molecule expression and cytokine secretion. For example, whereas IL-4-DCs are a homologous population of immature cells that is devoid of Langerhans cells, TNFDCs are heterogeneous and include both CD1a+ Langerhans cells and CD14+ interstitial DCs44.In vitro experiments showed that Langerhans cells and interstitial DCs that were generated from cultures of CD34+ hematopoietic progenitors differ in their capacity to activate lymphocytes: interstitial DCs induce the differentiation of naive B cells into immunoglobulin-secreting plasma cells4,32, whereas Langerhans cells seem to be particularly efficient activators of cytotoxic CD8+ T cells. They also differ in their cytokine-secretion pattern (only interstitial DCs produce IL-10) and their enzymatic activity4,32, which might be fundamental for the selection of peptides that are presented to T cells. Indeed, different enzymes are likely to degrade a given antigen into different sets of peptides, as has recently been shown for the HIV protein Nef 46. This then leads to different sets of peptide–MHC complexes being presented and thereby to distinct repertoires of antigen-specific T cells. So, these unique DCs are likely to yield unique immune effectors, thereby allowing the broad immune response that is required to combat permanently evolving microorganisms and tumors.
Distinct DC subsets induce distinct types of immune response. DCs have a crucial role in determining the type of response that is induced. There is evidence that either polarized DCs or distinct DC subsets might provide T cells with different signals that determine the class of immune response31. So, in mice, splenic CD8α+ DCs prime naive CD4+ T cells to produce TH1 cytokines in a process that involves IL-12, whereas splenic CD8α– DCs prime naive CD4+ T cells to produce TH2 cytokines47,48. Furthermore, this polarization into different T-cell subsets also depends on the signal received by a DC, as shown by the induction of IL-12 production and polarization towards TH1 cells when DCs are activated with Escherichia coli lipopolysaccharide (LPS), but the absence of IL-12 production and polarization towards TH2 cells when the same type of DC is exposed to LPS from Porphyromonas gingivalis 49. In humans, CD40 ligand (CD40L)-activated monocyte-derived DCs prime TH1-cell responses through an IL-12-dependent mechanism, whereas pDCs activated with IL-3 and CD40L have been shown to secrete negligible amounts of IL-12 and to prime TH2-cell responses50. So, both the type of DC subset and the activation signals to which DCs are exposed are important for polarization of T cells.
Mouse proof-of-principle in vivo studies
- Ex vivo-generated, antigen-loaded dendritic cells (DCs) induce antigen-specific T-cell immunity137
- Ex vivo gene-loaded DCs can induce humoral immunity138
- Ex vivo-generated, antigen-loaded DCs induce tumor-specific immunity139,140
- Ex vivo-generated DCs are superior to other types of vaccine141
- Ex vivo-generated immature DCs induce tolerance142
- Combination therapy with ex vivo-generated DCs improves vaccine efficacy112,113
This is an important parameter in vaccination against cancer, as type 1 immunity (including IFN-γ secretion) is desirable, whereas type 2 immunity (including IL-4 or IL-10 secretion) is considered deleterious. DCs and immune tolerance. DCs can induce and maintain immune tolerance27, both central and peripheral.
Central Tolerance depends on mature thymic DCs, which are essential for the deletion of newly generated T cells that have a receptor that recognizes self-components51. However, central tolerance might not be effective for all antigens. Furthermore, many self-antigens might not have access to the thymus, and others are only expressed later in life. So, there is a requirement for Peripheral Tolerance, which occurs in lymphoid organs and is mediated by immature DCs (FIG. 1a). Immature DCs present tissue antigens to T cells in the absence of appropriate co-stimulation, leading to T-cell Anergy or deletion27 or to the development of IL-10-secreting Inducible Regulatory T Cells52,53. The research groups of Nussenzweig and Steinman54 have elegantly shown that fusion proteins targeted to immature DCs lead to the induction of antigen-specific tolerance. By contrast, concomitant activation of these DCs with CD40- specific antibody results in a potent immune response, because the DCs are induced to express a large number of co-stimulatory molecules55. However, mature DCs might also contribute to peripheral tolerance by promoting the clonal expansion of naturally occurring CD4+CD25+ REGULATORY T (TReg) CELLS56, as discussed later. Therefore, the biology of DCs offers several targets for the control of cellular immunity. The parameters that need to be considered include DC-related factors, host-related factors and combining DC vaccines with other therapies.
Subsets of human dendritic cells. (Fig not shown). The population of dendritic cells (DCs) in the peripheral blood, which can be mobilized by treatment with FLT3L (fms-related tyrosine kinase 3 ligand), contains both CD11c+ myeloid DCs and CD11c– plasmacytoid DCs. So far, most studies of DCs have been carried out with DCs generated by culturing monocytes with granulocyte/macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4); this simple procedure yields a homogenous population of DCs that resemble interstitial DCs, and the population is devoid of Langerhans cells. These DCs are immature and require exogenous factors for maturation. DCs can also be generated by culturing CD34+ haematopoietic progenitor cells (HPCs) or peripheral-blood monocytes with GM-CSF and tumour-necrosis factor (TNF). In this way, two DC subsets can be obtained: Langerhans cells, which might have improved efficacy for eliciting cytotoxic T lymphocytes; and interstitial DCs, which resemble monocyte-derived DCs. Adding IL-4 to CD34+ HPC cultures in the presence of GM-CSF and TNF inhibits the differentiation of Langerhans cells. [Green boxes indicate cell types that can be induced by culture with GM-CSF and TNF. Yellow boxes indicate cell types that can be induced by culture or mobilization with FLT3L].
Plasticity of monocyte-derived dendritic cells. (Fig not shown). Activated monocytes can differentiate into different types of dendritic cell (DC) after encounter with different cytokines. These distinct DCs will influence the differentiation of lymphocytes into immune effectors with different functions, leading to varied immune responses. For example, interleukin-15-DCs (IL-15- DCs) are remarkably more efficient at priming and maturation of rare antigen-specific cytotoxic T lymphocytes (CTLs) than are IL-4-DCs. Thymic stromal lymphopoietin-DCs (TSLP-DCs) induce CD4+ T cells to differentiate into pro-inflammatory T helper 2 (TH2) cells, which secrete large amounts of IL-13 and tumor-necrosis factor (TNF)143, whereas interferon-α-DCs (IFN-α-DCs) induce CD4+ T cells to differentiate into TH1 cells, which secrete IFN-γ and IL-10. The properties and function of TNF-DCs remain to be determined. [FLT3L, fms-related tyrosine kinase 3 ligand; GM-CSF, granulocyte/macrophage colony-stimulating factor].
Antigen loading. Loading MHC class I and class II molecules at the cell surface of DCs with peptides derived from defined antigens is the most commonly used strategy for DC-based vaccination22,87. Although this technique is important for proof-of-principle studies, the use of peptides has limitations: the restriction of a peptide to a given HLA type; the limited number of well-characterized Tumor-Associated Antigens; the relatively rapid turnover of exogenous peptide– MHC complexes, resulting in comparatively low antigen presentation by the time that the DC arrives in the draining lymph node after injection; and the induction of a restricted repertoire of T-cell clones, thereby limiting the ability of the immune system to control tumor-antigen variation. Yet another level of complexity is brought about by the use of MODIFIED HETEROCLITIC PEPTIDES. Some synthetic peptides, even those derived from immune-dominant antigens, do not bind MHC class I molecules with high affinity, possibly explaining their limited immunogenicity in vivo88. Therefore, the generation of peptide analogues with increased affinity for MHC class I molecules (known as heteroclitic peptides) could be used to improve peptide immunogenicity89,90. However, recent elegant studies in patients with malignant melanoma show that T cells elicited in vivo by vaccination with heteroclitic MART1 (melanoma antigen recognized by autologous T cells) or glycoprotein 100 (gp100) peptide show poor recognition of the endogenous melanoma-derived peptide and less efficient tumor-cell lysis compared with T cells specific for the native peptide91.
Immunoregulatory mechanisms
Naturally occurring CD4+CD25+ regulatory T cells
Cell-mediated suppression independent of interleukin-10 (IL-10) and/or transforming growth factor-β (TGF-β);
clonal expansion is regulated by mature dendritic cells (DCs)
Inducible regulatory T cells
Cytokine-mediated suppression through IL-10 and/or TGF-β; induction and clonal expansion is regulated by immature DCs
Natural killer T cells
Cytokine-mediated suppression through IL-13
Vaccine-induced B cells?
Cytokine-mediated regulation through IL-4, IL-6 and IL-10; competition with DCs for antigen uptake
Tumor-specific interferon-γ-secreting T cells?
Immunoediting and selection of escape variants (not discussed in main text)
Immune correlates of efficacy of dendritic-cell-based vaccines
- Induction of broad tumour-specific T-cell immunity: T cells specific for several tumour antigens
- Induction of effector T cells: T cells with immediate capacity to recognize tumour antigens and secrete cytokines such as tumour-necrosis factor and interferon-γ
- Induction of memory T cells: T cells that secrete interleukin-2 and proliferate on re-exposure to tumour antigen
- Induction of T cells that kill tumour cells
- Decreased number of T cells with regulatory function
DCs are an attractive target for therapeutic manipulation of the immune system to increase otherwise insufficient immune responses to tumour antigens. However, the complexity of the DC system requires rational manipulation of DCs to achieve protective or therapeutic immunity. So, further research is needed to analyse the immune responses induced in patients by distinct ex vivo-generated DC subsets that are activated through different pathways. The ultimate ex vivo-generated DC vaccine will be heterogeneous and composed of several subsets, each of which will target a specific immune effector. These ex vivo strategies should help to identify the parameters for in vivo targeting of DCs, which is the next step in the development of DC-based vaccination. Indeed, distinct DC subsets express unique cell-surface molecules, such as different lectins131: Langerhans cells express langerin, which is crucial for the formation of Birbeck granules132,133; interstitial DCs express DCSIGN (dendritic-cell-specific intercellular-adhesionmolecule-3-grabbing non-integrin), which is involved in interactions with T cells and in DC migration but is also used by pathogens (such as HIV) to hijack the immune system; and pDCs express yet another lectin, BDCA2 (blood DC antigen 2)134,135. Such differential expression of cell-surface molecules might allow specific in vivo targeting of DC subsets for induction of the desired type of immune response.
9.1.2 Simultaneous Humoral and Cellular Immune Response against Cancer–Testis Antigen NY-ESO-1: Definition of Human Histocompatibility Leukocyte Antigen (HLA)-A2–binding Peptide Epitopes
Elke Jäger, Yao-Tseng Chen, Jan W. Drijfhout, Julia Karbach, et al.
J Exp Med. 1998 Jan 19; 187(2): 265–270.
A growing number of human tumor antigens have been described that can be recognized by cytotoxic T lymphocytes (CTLs) in a major histocompatibility complex (MHC) class I–restricted fashion. Serological screening of cDNA expression libraries, SEREX, has recently been shown to provide another route for defining immunogenic human tumor antigens. The detection of antibody responses against known CTL-defined tumor antigens, e.g., MAGE-1 and tyrosinase, raised the question whether antibody and CTL responses against a defined tumor antigen can occur simultaneously in a single patient. In this paper, we report on a melanoma patient with a high-titer antibody response against the “cancer–testis” antigen NY-ESO-1. Concurrently, a strong MHC class I–restricted CTL reactivity against the autologous NY-ESO-1–positive tumor cell line was found. A stable CTL line (NW38-IVS-1) was established from this patient that reacted with autologous melanoma cells and with allogeneic human histocompatibility leukocyte antigen (HLA)-A2−, NY-ESO-1–positive, but not NY-ESO-1–negative, melanoma cells. Screening of NY-ESO-1 transfectants with NW38-IVS-1 revealed NY-ESO-1 as the relevant CTL target presented by HLA-A2. Computer calculation identified 26 peptides with HLA-A2–binding motifs encoded by NY-ESO-1. Of these, three peptides were efficiently recognized by NW38-IVS-1. Thus, we show that antigen-specific humoral and cellular immune responses against human tumor antigens may occur simultaneously. In addition, our analysis provides a general strategy for identifying the CTL-recognizing peptides of tumor antigens initially defined by autologous antibody.
There is growing evidence for humoral and cellular immune recognition of cancer by the autologous human host (1–6). Based on CTL-dependent lysis of cultured melanoma cell lines, several categories of autoimmunogenic tumor antigens have been characterized, including differentiation antigens of specific cell lineages (7–9), individual antigens caused by point mutations (10, 11), and tumor antigens, such as MAGE, which are expressed in a variable proportion of different tumor types, but are silent in most normal tissues except the testis (12). CTL responses against melanoma antigens induced by peptide vaccines in vivo have been associated with a favorable development of advanced melanoma in some patients (6, 13). As immunoselection of antigen-negative tumor cell variants has been observed during peptide vaccination (14), the molecular characterization of additional CTL-defined tumor antigens is needed to develop polyvalent vaccines with broader immunotherapeutic effects.
Sahin et al. have recently introduced a powerful new methodology for identifying human tumor antigens eliciting humoral immune response (5). The method has been called SEREX, for serological expression cloning of recombinant cDNA libraries of human tumors. Novel and previously defined tumor antigens have been identified by the SEREX method, including MAGE-1 and tyrosinase, both originally identified by cloning the epitopes recognized by CTLs. Thus, antibody screening of cDNA libraries prepared from human tumors can be used to identify antigens eliciting a cellular immune response, including CTLs, circumventing the need for established cultured autologous cell lines and stable CTL lines.
We have recently identified a novel human tumor antigen by SEREX analysis of a human esophageal cancer (15). The antigen, NY-ESO-1, belongs to a growing number of human tumor antigens we have called “cancer–testis” antigens that include MAGE, GAGE, BAGE (1), and SSX2 (HOM-MEL-40) (5, 16). These antigens have the following characteristics: (a) they are expressed in a variable portion of a wide range of cancers, (b) their normal tissue expression is generally restricted to the testis, and (c) they are generally coded for by genes on the X chromosome. In a recent survey of sera from normal individuals and cancer patients, antibodies against NY-ESO-1 were found in ∼10% of patients with melanoma, ovarian cancer, and other cancers, but not in normal individuals (Stockert, E., manuscript in preparation). One patient with a high NY-ESO-1 antibody response was found to have specific CTL reactivity against cultured autologous melanoma cells. In the present study, we report that NY-ESO-1 encodes the CTL target in this patient and identify the NY-ESO-1 peptides that are recognized.
High-titer Antibody Reactivity against NY-ESO-1.
Melanoma patient NW38 presented with extensive metastases to inguinal lymph nodes having large areas of necrosis. Reverse transcriptase PCR of tumor RNA showed that this tumor expressed NY-ESO-1. Based on the hypothesis that exposure of the immune system to large amounts of intracellular tumor proteins released from the necrotic tumor might elicit a strong humoral immune response, the serum of patient NW38 was tested for specific reactivity against recombinant NY-ESO-1 protein. Fig. Fig.11 shows the reactivity of NW38 serum with the recombinant NY-ESO-1 protein, with a lysate of NY-ESO-1–transfected COS-7 cells, and with a lysate of the autologous NY-ESO-1 messenger RNA–positive tumor cell line NW-MEL-38. A 22-kD protein species was identified in both cell lysates, and comigrated with the purified recombinant NY-ESO-1 protein. The identity of this protein species as NY-ESO-1 was further confirmed by using an anti–NY-ESO-1 mouse monoclonal antibody. Reactivity against recombinant NY-ESO-1 protein was still detectable at a serum dilution of 1:100,000. No reactivity was detected against a lysate of untransfected COS-7 cells.
The correlation between NY-ESO-1 expression and NW38-IVS-1 reactivity suggested NY-ESO-1 as the antigenic target. To prove this, COS-7 cells were transfected with NY-ESO-1 cDNA and different MHC class I molecules and used as targets for NW38-IVS-1. Reactivity was measured in a standard TNF-α release assay. TNF release was found after stimulation of NW38-IVS-1 with COS-7 cells cotransfected with HLA-A2− and NY-ESO-1 cDNA. No reactivity was detected after stimulation with cotransfectants of pcDNA3.1(−)-NY-ESO-1− and pcDNA1Amp-HLA-A1 cDNA, COS-7 cells transfected with pcDNA3.1(−), or untransfected COS-7 cells (Fig. (Fig.3).3).
Peptide-specific CTLs.
26 different peptides encoded by NY-ESO-1 with theoretical binding motifs to the HLA-A2.1 molecule were tested for specific recognition by NW38-IVS-1. The target cells were peptide-pulsed T2 cells. Of these 26 peptides, three were recognized by NW38-IVS-1 as determined by a standard51Cr–release assay (Table (Table1).1). The peptide sequences SLLMWITQCFL, SLLMWITQC, and QLSLLMWIT are located between positions 155 and 167 of the NY-ESO-1 protein (15), and show overlapping sequences. The 11-mer SLLMWITQCFL (2 in Table Table1)1) and the 9-mer SLLMWITQC (12 in Table Table1)1) consist of identical amino acids at positions 1–9.
To provide additional confirmation of the peptide specificity, the 26 synthetic peptides were individually incubated with HLA-A2–transfected COS-7 cells and tested in the TNF release assay. Consistent with the results of 51Cr–release assay, specific TNF-α release was detected in tests with peptides SLLMWITQCFL, SLLMWITQC, and QLSLLMWIT. NY-ESO-1/HLA-A2 transfectants were used as a positive control in these assays (Fig. (Fig.4).4).
The search for tumor antigens that induce specific immune responses in cancer patients is the ongoing challenge in tumor immunology. Evidence for a specific humoral response to human cancer came from serological analysis of cell surface reactivity of sera from cancer patients for autologous cancer cells, an approach called autologous typing (4). However, with only a few exceptions, this approach did not allow for the structural definition of the antigenic target. An autologous typing system also provided the first evidence for the development of CTLs with specificity for human melanoma cells (3, 17, 21–24). Using specific antitumor CTLs as probes, a number of CTL targets have been cloned on the basis of MHC class I–restricted recognition (1, 6). However, this approach involves cultured cancer cell lines and stable CTL lines from the same patient, two requirements that cannot easily be met with many tumor types. With the demonstration that genes coding for CTL-recognized tumor antigens elicit humoral immunity and can be cloned by SEREX methodology, a technically less demanding approach defining immunogenic tumor antigens is now available, one that extends the range of analysis to tumor types that are not easily adaptable to in vitro growth and are not sensitive targets for CTLs. A number of novel tumor antigens have been defined by SEREX, including two new members of the cancer–testis antigenic family, SSX2 (HOM-MEL-40) (5, 16), and NY-ESO-1 (15).
In this study, we identified a melanoma patient, NW38, with high-titered antibody against NY-ESO-1. This patient had a large and highly necrotic tumor, and the sustained release of intracellular antigens that are usually inaccessible to the immune system may account for the high NY-ESO-1 titer. The establishment of an autologous cell line that typed NY-ESO-1 positive provided target cells for assessing CTL reactivity in this patient. A CTL line was established from this patient that lysed the autologous melanoma cell line in an HLA-A2–restricted fashion. Using target cells transfected with NY-ESO-1 and HLA-A2, the specificity of CTL reactivity was found to be coded by NY-ESO-1. Computer analysis of the NY-ESO-1 sequence identified 26 peptides with HLA-A2–binding motifs. Screening of these peptides presented by T2 cells identified three sequences that were confirmed to be specifically recognized by NW38-IVS-1. This is the first conclusive demonstration of simultaneous antibody and CTL responses against a cancer–testis antigen in a single patient.
The strategy used in this study to generate and analyze CTL reactivity to a SEREX-defined antigen can be used as a model for investigating cellular immune responses to the growing list of other SEREX antigens. Identification of clones in SEREX requires high-titered IgG antibody, and the development of such antibodies requires the help of CD4+ T cells. In this sense, SEREX can be thought of as a method to define the CD4+ T cell repertoire to human tumor antigens. Also, the presence of both NY-ESO-1 antibody and CTLs in patient NW38 suggests that screening for an antibody response may be a simple and effective way to identify patients with concomitant CTL reactivity, and this possibility is now being tested in other patients with NY-ESO-1 antibody. In the absence of autologous tumor cell lines, CD8+ T cells can be stimulated with autologous antigen-presenting cells that have been transfected with the coding gene or fed purified protein antigens. A similar strategy can be used to identify peptide targets for CD4+ T cells.
A major objective in defining immunogenic human tumor targets is to explore their use in the development of cancer vaccines, and a number of clinical trials with various vaccine constructs are currently underway. Although tumor regression is the desired goal of a therapeutic vaccine, this end point cannot be expected to be an effective way to develop maximally immunogenic tumor vaccines. For this purpose, reliable immunological assays are needed to monitor the specificity and strength of specific immune reactions generated by the vaccine. With the exception of vaccines aimed at inducing a humoral immune response such as GM2 ganglioside vaccines, most vaccine trials are designed to stimulate cellular immunity, particularly the development of CTLs and CD4+ T cells. These have been difficult to detect in vaccine trials with MAGE peptides (25), and difficult to interpret in trials with vaccines containing melanocyte differentiation antigens, since CTLs against these antigens can be generated in vitro from nonvaccinated melanoma patients as well as normal individuals (26, 27). However, de novo induction and increase of preexisting CTL reactivity have been detected after vaccination with melanocyte differentiation antigens and observed to be associated with cancer regressions in a limited number of patients (13). The demonstration of a simultaneous antibody and CTL response to NY-ESO-1 in the same patient suggests that serological methods may be useful in monitoring vaccine trials with NY-ESO-1 and other tumor antigens eliciting a humoral immune response.
9.1.3 Monoclonal Antibodies in Cancer Therapy
R K Oldham
JCO September 1983; 1(9): 582-590
http://jco.ascopubs.org/content/1/9/582.short
The need for improved specificity in cancer therapy is apparent. With the advent of monoclonal antibodies, the possibility of specifically targeted therapy is being considered. Early trials of monoclonal antibody in experimental animals and humans have indicated its ability to traffic to specific tumor sites and to localize on or around the tumor cells displaying antigens to which the antibody is directed. This evidence of specific targeting, along with preliminary evidence of therapeutic efficacy for monoclonal antibodies and immunoconjugates with drugs, toxins, and isotopes is encouraging. The current status of clinical trials with monoclonal antibodies is reviewed and an example of the experimental approach for the development of immunoconjugates in animal models is presented.
Monoclonal Antibodies in Cancer Therapy: 25 Years of Progress
Robert K. Oldham, Robert O. Dillman
JCO Apr 10, 2008: 26(11): 1774-1777
http://dx.doi.org:/10.1200/JCO.2007.15.7438
In 1983, it was apparent that a major problem with current modalities of cancer treatment was the lack of specificity for the cancer cell.1 It was predicted that a major advancement in treatment of cancer would be the development of a class of agents that would have a greater degree of specificity for the tumor cell. Based on many animal studies and the treatment of fewer than 100 patients, it was evident in 1983 that monoclonal antibodies would be that major advance.
The first patient treated in the United States with monoclonal antibody therapy was a patient with non-Hodgkin’s lymphoma.2 Nadler et al2 described the treatment using a murine monoclonal antibody designated AB 89. Although treatment was not successful in inducing a significant clinical response, it did represent the first proof of principle in humans that a monoclonal antibody could induce transient decreases in the number of circulating tumor cells, induce circulating dead cells, and form complexes with circulating antigen, all with minimal toxicity to the patient. Antibody could be detected on the surface of circulating lymphoma cells, and free antigen in the serum decreased with each infusion of antibody. After two courses of milligram doses of AB 89, a final and third course with 1.5 g of antibody was administered during a 6-hour period. A marked reduction in circulating antigen was noted, but these studies suggested to the authors that the quantity of circulating antigen was too great to effectively deliver AB 89 to the patient’s tumor cells in a therapeutically effective manner.2
In the Journal of Clinical Oncology review article cited earlier,1 evidence was reviewed from animal tumor models that clearly demonstrated both specificity and therapeutic efficacy with little serious toxicity. Whereas passive serotherapy of human cancer had shown little success,3 it was apparent in the earlier review that monoclonal antibodies could be used in the treatment of leukemia and lymphoma.4,5 In 1983, a review of the literature revealed approximately 10 published studies and one in-press article of therapeutic trials of monoclonal antibody therapy in humans. All of these studies used murine monoclonal antibodies and were phase I/II studies. Most were in leukemia or lymphoma, but the earliest solid tumor studies were also underway in melanoma6 and GI cancer.1
By 1983, the pioneers in monoclonal antibody research believed that a new era of cancer therapy had begun, and for the first time, true specific and targeted therapy was underway using hybridoma technology to produce monoclonal antibodies with exquisite specificity. It was also apparent, based on animal model studies, that monoclonal antibodies could be a vehicle to bring immunoconjugate therapy to the clinic by conjugating monoclonal antibodies to drugs, toxins, and radioisotopes using the specificity of the monoclonal antibody to carry enhanced killing capacity directly to the tumor cells. Thus, the era of monoclonal antibody therapy, as well as immunoconjugate therapy, had begun.
Although there was much excitement (and skepticism) about this new treatment modality (the use of a form of biologic therapy with great specificity in patients with advanced cancer) there were also problems and limitations. As presented in Table 1, there were clinical toxicities with murine monoclonal antibodies, most of which were secondary to the interaction with the target antigen.7 However, the major limitation was their immunogenicity. Murine proteins are highly immunogenic, and it was soon found that only a few infusions of these foreign proteins could be given to patients with cancer because of the development of human antimouse antibody.8 Another problem quickly became apparent, in that some of the antigens on cancer cell surfaces modulated off the surface and into the circulation when antibody attached. Modulation could also cause internalization of the complex. It was recognized that this could represent a therapeutic advantage by using the antibody as carrier to internalize the toxic component of an immunoconjugate, potentially making it more therapeutically active.
In 1983, few specific antigens found only in cancer cells had been identified, and there was much debate about the specificity of these antigens. Many of the antigens to which monoclonal antibodies were made were embryonic antigens or shared antigens found on cancer cells and some normal cells. Therefore, although the specificity of the antibody was exquisite for the antigen, the specificity for the antibody or immunoconjugate for cancer was not absolute. One fairly clear exception occurred early in the 1980s when Levy et al9 developed monoclonal antibodies to the idiotype of B-lymphoma cells. The first patient given this anti-idiotypic antibody had a complete response to therapy, and his lymphoma went into a sustained remission that lasted for years. As a direct result of these early studies with anti-idiotypic antibodies, there is now a series of idiotype vaccines that are in phase III trials in patients with low-grade follicular lymphomas.10 These anti-idiotype vaccines will likely be the first truly custom-tailored, personalized anticancer vaccines to be approved for therapeutic use.
The major limitation of murine monoclonal antibody therapy was the immunogenicity of the mouse protein; a variety of investigators postulated that for monoclonal antibody therapy to be truly successful, human or humanized antibodies would be necessary. It was also known 25 years ago that the half-life of murine antibodies in the circulation was brief, and because of human antimouse antibody, became briefer with each infusion of murine monoclonal antibody. Previous studies of human immunoglobulin in clinical trials had demonstrated a much longer half-life for human immunoglobulin, which predicted that once human or humanized antibodies were available, the therapeutic efficacy of monoclonal antibodies and their immunoconjugates might be considerably enhanced.1
How has the field of monoclonal antibody and immunoconjugate therapy fared since the predictions of the early 1980s? Twenty-five years later, considerable progress has been made in this field.11,12 The US Food and Drug Administration has approved 21 monoclonal antibody products, with six of these biologic drugs approved specifically for cancer (Table 2). It was a landmark date in November 1997 when rituximab became the first monoclonal antibody approved specifically for cancer therapy.13 In addition to these six unconjugated monoclonal antibody therapies, one drug immunoconjugate, gemtuzumab ozogamicin (Mylotarg; Wyeth-Ayerst, Madison, NJ), has been approved. This humanized monoclonal antibody to CD33 is approved for use in acute myelogenous leukemia and uses the antibody conjugated to calicheamicin, a potent enediyene antibiotic originally isolated from aMicromonospora echoinospora.14 Two radioisotope-antibody conjugates, ytrrium-90 ibritumomab tiuxetan (Zevalin; Cell Therapeutics Inc, Seattle, WA) and iodine-131 tositumomab (Bexxar; GlaxoSmithKline, Middlesex, United Kingdom) have been approved.15 The murine form of these antibodies was retained in order to expedite clearance from the circulation. Both radiolabeled antibodies target the CD20 antigen on lymphoma cells.
Unlike the immunoconjugates, which are currently infrequently used, each of the six unconjugated antibodies approved for cancer therapy is currently frequently used in the treatment of humans with cancer. The use of techniques to humanize or chimarize monoclonal antibodies to decrease their murine components has been an important advance in the field. These molecules have a long half-life in the blood stream, and can interact with human complement or effector cells of the patient’s immune system. They behave in a manner similar to naturally occurring immunoglobulin and work along the lines of our normal antibody-based immune response as effective agents in treating patients with cancer.16
Rituximab has become the largest-selling biologic drug in clinical oncology, and is active in a variety of human lymphomas and chronic lymphocytic leukemia.17,18 This is a chimeric monoclonal antibody targeting the CD20 antigen found on both normal B cells and on most low-grade and some higher grade B-cell lymphomas. It is effective as a single agent in induction and maintenance therapy. It is primarily used, however, in combination with standard chemotherapies in the treatment of patients with non-Hodgkin’s B-cell lymphomas and chronic lymphocytic leukemia.19-22
A second monoclonal antibody that has proven highly effective in the clinic is trastuzumab, a humanized antibody that reacts with the second part of the human epidermal growth factor receptor 2.23 Like rituximab, it is effective as a single agent in induction and maintenance therapy, but is used primarily in conjunction with chemotherapy for patients with human epidermal growth factor receptor 2/neu–positive breast cancer.24,25
Alemtuzumab is a humanized monoclonal antibody targeting the CD52 antigen found on B lymphocytes and is used primarily for chronic lymphocytic leukemia.26 Like the two previously cited monoclonal antibody therapies, alemtuzumab is effective as induction and maintenance therapy. Alemtuzumab is also reactive with T lymphocytes, and unlike the other two antibodies, it is typically not combined with chemotherapy because of the increased risk of infection.(26)
Another humanized monoclonal antibody, bevacizumab, has been applied more broadly in human solid tumors because it targets vascular endothelial growth factor, which is the ligand for a receptor found on blood vessels.(27) Because this receptor is on endothelial cells, bevacizumab seems to be effective by reducing the blood supply to tumor nodules, thereby slowing or interrupting growth. Initially approved for advanced colorectal cancer,(28) it is now used in a variety of human solid tumors including cancers of the lung, kidney, and breast.(29-31)
The last two antibodies approved for clinical use were cetuximab (a chimeric antibody), and panitumumab (a completely human antibody). Both target the epidermal growth factor receptors found on a variety of human tumors.(32,33) Cetuximab was originally approved for use in combination with chemotherapy in metastatic colorectal cancer.(34) It also enhances chemotherapy and radiation therapy of squamous cell cancers of the head and neck.(35) Panitumumab was approved based on its single-agent activity in refractory colorectal cancer and is being combined with chemotherapy as well.
At the end of 2007, 25 years of clinical studies have resulted in the approval of six unconjugated, humanized, or chimeric monoclonal antibodies for cancer therapy along with one drug immunoconjugate and two radioisotope immunoconjugates. Although few in number, these monoclonal antibodies are changing the face of cancer therapy, bringing us closer to more specific and more effective biologic therapy of cancer as opposed to nonspecific cytotoxic chemicals.
Modern recombinant techniques have made it possible to rapidly produce both chimeric antibodies and humanized antibodies, and totally human antibodies are also being produced. Identification of surface receptors that are integral to proliferation and apoptosis has provided more targets for monoclonal antibodies beyond those originally identified by the murine immune system. In 2008, there are more than 100 monoclonal antibody–based biologic drugs in hundreds of clinical trials. Many of these are in phase II and phase III and will be coming before the US Food and Drug Administration for approval in the next few months and years. At long last, immunoconjugates are proving efficacious with acceptable toxicity and will extend our diagnostic (36) and therapeutic armamentarium (37) from mainly unconjugated monoclonal antibodies to a broad array of highly active and specific immunoconjugates.
On this silver anniversary for our 1983 review, “Monoclonal Antibodies in Cancer Therapy, ” we can confidently predict that progress toward more specific and less toxic therapy for human cancer is in our near future. The developments during the past 25 years in both biologic drugs and targeted small molecules place us on the verge of more cures with less toxicity for our patients with cancer.
9.1.4 Aptamers
Nanocarriers as an emerging platform for cancer therapy
Dan Peer1,7, Jeffrey M. Karp2,3,7, Seungpyo Hong, et al.
Nature Nanotechnology 2, 751 – 760 (2007)
http://dx.doi.org:/10.1038/nnano.2007.387
Nanotechnology has the potential to revolutionize cancer diagnosis and therapy. Advances in protein engineering and materials science have contributed to novel nanoscale targeting approaches that may bring new hope to cancer patients. Several therapeutic nanocarriers have been approved for clinical use. However, to date, there are only a few clinically approved nanocarriers that incorporate molecules to selectively bind and target cancer cells. This review examines some of the approved formulations and discusses the challenges in translating basic research to the clinic. We detail the arsenal of nanocarriers and molecules available for selective tumor targeting, and emphasize the challenges in cancer treatment.
Quantum Dot−Aptamer Conjugates for Synchronous Cancer Imaging, Therapy, and Sensing of Drug Delivery Based on Bi-Fluorescence Resonance Energy Transfer
Vaishali Bagalkot, L Zhang, E Levy-Nissenbaum, S Jon, PW Kantoff, et al.
Nano Letters 2007; 7(10):3065-3070
http://dx.doi.org:/10.1021/nl071546n
We report a novel quantum dot (QD)−aptamer(Apt)−doxorubicin (Dox) conjugate [QD−Apt(Dox)] as a targeted cancer imaging, therapy, and
sensing system. By functionalizing the surface of fluorescent QD with the A10 RNA aptamer, which recognizes the extracellular domain of the prostate specific membrane antigen (PSMA), we developed a targeted QD imaging system (QD−Apt) that is capable of differential uptake and imaging of prostate cancer cells that express the PSMA protein. The intercalation of Dox, a widely used antineoplastic anthracycline drug with fluorescent properties, in the double-stranded stem of the A10 aptamer results in a targeted QD−Apt(Dox) conjugate with reversible self-quenching properties based on a Bi-FRET mechanism. A donor−acceptor model fluorescence resonance energy transfer (FRET) between QD and Dox and a donor−quencher model FRET between Dox and aptamer result when Dox intercalated within the A10 aptamer. This simple multifunctional nanoparticle system can deliver Dox to the targeted prostate cancer cells and sense the delivery of Dox by activating the fluorescence of QD, which concurrently images the cancer cells. We demonstrate the specificity and sensitivity of this nanoparticle conjugate as a cancer imaging, therapy and sensing system in vitro.
Semiconductor nanocrystals known as quantum dots (QDs)
have been increasingly utilized as biological imaging and labeling probes because of their unique optical properties, including broad absorption with narrow photoluminescence spectra, high quantum yield, low photobleaching, and resistance to chemical degradation. In some cases, these unique properties have conferred advantages over traditional fluorophores such as organic dyes.1-4 The surface modification of QDs with antibodies, aptamers, peptides, or small
molecules that bind to antigens present on the target cells or tissues has resulted in the development of sensitive and specific targeted imaging and diagnostic modalities for in vitro and in vivo applications.5-7 More recently, QDs have been engineered to carry distinct classes of therapeutic agents for simultaneous imaging and therapeutic applications.8,9 While these combined imaging therapy nanoparticles represent an exciting advance in the field of nanomedicine, it would be ideal to engineer “smart” multifunctional nanoparticles that are capable of performing these tasks while sensing the delivery of drugs in a simple and easily detectable manner. One way to achieve this goal is to develop multifunctional nanoparticles capable of sensing the release of the therapeutic modality by a change in the fluorescence of the imaging modality.
Figure 1. (a) Schematic illustration of QD-Apt(Dox) Bi-FRET system. In the first step, the CdSe/ZnS core-shell QD are surface functionalized with the A10 PSMA aptamer. The intercalation of Dox within the A10 PSMA aptamer on the surface of QDs results in the formation of the QD-Apt(Dox) and quenching of both QD and Dox fluorescence through a Bi-FRET mechanism: the fluorescence of the QD is quenched by Dox while simultaneously the fluorescence of Dox is quenched by intercalation within the A10 PSMA aptamer resulting in the “OFF” state. (b)
Schematic illustration of specific uptake of QD-Apt(Dox) conjugates into target cancer cell through PSMA mediate endocytosis. The release of Dox from the QD-Apt(Dox) conjugates induces the recovery of fluorescence from both QD and Dox (“ON” state), thereby sensing the intracellular delivery of Dox and enabling the synchronous fluorescent localization and killing of cancer cells.
Figure 3. Fluorescence spectra. (a) QD-Apt conjugate (1 µM) with increasing molar ratio of Dox (from top to bottom: 0, 0.1, 0.3, 0.6, 1, 1.5, 2.1, 2.8, 3.5, 4.5, 5.5, 7, and 8) at an excitation of 350 nm. (b) Dox (10 µM) with increasing molar ratio of QD-Apt conjugate (from top to bottom: 0.02, 0.04, 0.07, 0.09, 0.12, 0.14, and 0.16) at an excitation of 480 nm.
In conclusion, herein we report to our knowledge the first example of a multifunctional nanoparticle that can detect cancer cells at a single cell level while intracellularly releasing a cytotoxic dose of a therapeutic agent in a reportable manner. We demonstrate the specificity and sensitivity of this cancer imaging, therapy and sensing nanoparticle conjugate system in vitro by using PCa cell lines. By functionalizing the surface of fluorescent QD with the A10 PSMA aptamer, and intercalating Dox into the double-stranded CG sequence of the A10 PSMA aptamer, we developed a targeted QD-Apt(Dox) conjugate with reversible Bi-FRET properties. The incorporation of multiple CG sequences within the stem of the aptamers may further increase the loading efficiency of Dox on these conjugates. The presence of additional Dox may enhance the selfquenching effect of QD-Apt(Dox) conjugates thereby improving their imaging sensitivity, while the higher dose of Dox may enhance the therapeutic efficacy of the conjugates. Furthermore, through the use of other disease-specific aptamers or other targeting molecules, similar multifunctional nanoparticles may potentially be developed for additional important medical applications
Oligonucleotide Aptamers: New Tools for Targeted Cancer Therapy
Hongguang Sun1, Xun Zhu2, Patrick Y Lu3, Roberto R Rosato, et al.
Molecular Therapy Nucleic Acids(2014) 3, e182;
http://dx.doi.org:/10.1038/mtna.2014.32
Aptamers are a class of small nucleic acid ligands that are composed of RNA or single-stranded DNA oligonucleotides and have high specificity and affinity for their targets. Similar to antibodies, aptamers interact with their targets by recognizing a specific three-dimensional structure and are thus termed “chemical antibodies.” In contrast to protein antibodies, aptamers offer unique chemical and biological characteristics based on their oligonucleotide properties. Hence, they are more suitable for the development of novel clinical applications. Aptamer technology has been widely investigated in various biomedical fields for biomarker discovery, in vitro diagnosis, in vivo imaging, and targeted therapy. This review will discuss the potential applications of aptamer technology as a new tool for targeted cancer therapy with emphasis on the development of aptamers that are able to specifically target cell surface biomarkers. Additionally, we will describe several approaches for the use of aptamers in targeted therapeutics, including aptamer-drug conjugation, aptamer-nanoparticle conjugation, aptamer-mediated targeted gene therapy, aptamer-mediated immunotherapy, and aptamer-mediated biotherapy.
The terms “aptamer” and “SELEX” were introduced by two independent groups in 1990.1,2 The term “aptamer” refers to small nucleic acid ligands that exhibit specific therapeutic functions and an unambiguous binding affinity for their targets. Conversely, Systematic Evolution of Ligands by EXponential enrichment (SELEX) technology is the method used for aptamer development. Although using small molecule nucleic acids as therapeutics has been explored for decades, development of SELEX and aptamer technology revolutionized this field.
The most important property of an aptamer, from the Latin aptus (to fit), is its high target selectivity. These short, chemically synthesized, single-stranded (ss) RNA or DNA oligonucleotides fold into specific three-dimensional (3D) structures with dissociation constants usually in the pico- to nano-molar range.3 Moreover, in contrast to other nucleic acid molecular probes, aptamers interact with and bind to their targets through structural recognition (Figure 1), a process similar to that of an antigen-antibody reaction. Thus, aptamers are also referred to as “chemical antibodies.”
Figure 1.
Schematic diagram of aptamer binding to its target.
Full figure (43K)
Due to their small size and oligonucleotide properties, aptamers offer several advantages over protein antibodies in both their extensive clinical applicability and a less challenging industrial synthesis process. Specifically, (i) aptamers can penetrate tissues faster and more efficiently due to their significantly lower molecular weight (8–25 kDa aptamers versus ~150 kDa of antibodies). Therefore, aptamers penetrate tissues barriers and reach their target sites in vivo more efficiently than the larger-sized protein antibodies. (ii) Aptamers are virtually nonimmunogenic in vivo. In principal, as aptamers are oligonucleotides they should not be recognized by the immune system. In practice, a recent clinical study showed that aptamers did not stimulate an immune response in vivo,4,5 as compared to protein antibodies that are highly immunogenic, especially following repeat injections. (iii) Aptamers are thermally stable. Based on the intrinsic property of oligonucleotides, even after a 95 °C denaturation, aptamers can refold into their correct 3D conformations once cooled to room temperature. In comparison, protein-based antibodies permanently lose their activity at high temperatures. More importantly, a well-established synthesis protocol and chemical modification technology lead to (iv) rapid, large-scale aptamer synthesis and modification capacity that includes a variety of functional moieties; (v) low structural variation during chemical synthesis; and (vi) have lower production costs. Moreover, aptamers specifically recognize a wide range of targets, such as ions, drugs, toxins, peptides, proteins, viruses, bacteria, cells, and even tissues.6,7,8,9,10,11,12 In the clinic, aptamer-based therapeutics are gaining momentum. For example, Macugen, a modified RNA aptamer, specifically targets vascular endothelial growth factor. It has been approved by the US Food and Drug Administration (FDA)13 for the treatment of wet age-related macular degeneration and is under evaluation for other conditions.14 In the cancer setting, AS1411 targets nucleolin, a protein over-expressed in a variety of tumors. It is currently being evaluated as a potential treatment option in solid tumors and acute myeloid leukemia.15 An updated list of therapeutic aptamers undergoing clinical trials is included in ref. 16 and Table 1. Taken together, these clinical studies highlight many possible uses that aptamers may have in a variety of biomedical fields, including therapeutics.17
Table 1 – A list of therapeutic aptamers undergoing clinical trials.
Since aptamer technology was first introduced, the RNA-based sequence library has been widely used for SELEX. Based on the existing evidence, it is believed that the presence of a 2′-OH group and non-Watson-Crick base pairing allows RNA aptamer oligonucleotides to fold into more diverse 3D structures than ssDNA molecules. Consequently, using the more flexible RNA sequences simplifies the development of high-affinity and -specificity aptamers. Despite their advantages, RNA sequences are very sensitive to nucleases present in biological environments and can be rapidly degraded.18 To increase nuclease resistance of RNA-based aptamers, several chemical modifications have been investigated. Evidence shows that 2′-OH group and phosphodiester linkages of RNA sequences are the sites of nuclease hydrolysis. Subsequently, substitutions of the 2′-OH functional group by 2′-fluoro, 2′-amino, or 2′-O-methoxy motifs, and/or changes to the phosphodiester backbone with boranophosphate or phosphorothioate are the most common modifications aimed at increasing nuclease resistance.19 More recently, Wu et al. developed a novel chemical modification method to increase siRNA stability, in which phosphorodithioate and 2′-O-Methyl were simultaneously substituted in the same nucleotide.20
This modification method significantly enhanced siRNA stability and represents a potential new direction for utilization of RNA-based therapies in complex biological systems. Other effective modifications recently reported utilize the locked nucleic acid technology16,21 or generate “mirror” RNA sequence structures, termed spiegelmers.22 These modifications result in structural changes to the RNA sequences, which cannot be digested by nucleases.
In addition to RNA aptamers, ssDNA-based aptamers have also been developed. Due to their lack of 2′-OH groups, DNA molecules are naturally resistant to 2′-endonucleases and are stable in biological environments. Recently, our group developed a biostable DNA-based aptamer specific for CD30, a protein biomarker that is over-expressed in Hodgkin and anaplastic large cell lymphomas. Functional analysis demonstrated that this ssDNA-based aptamer exhibited high CD30 binding affinity as low as 2 nmol/l and was stable in human serum for up to 8 hours. Conversely, an RNA-based CD30 aptamer was digested within 10 minutes under similar conditions.23
In summary, unique chemical features and biological functions have made aptamers a very attractive tool in biomedical research over the past two decades. Currently, there are over 4,000 published articles referenced in the PubMed database that include the term “aptamer.” Research areas that include aptamer technology cover bioassays, drug development, cell detection, tissue staining, in vitro and in vivo imaging, nanotechnology, and targeted therapy. As chemical antibodies, aptamers represent an excellent alternative to replace or supplement protein antibodies, which have been extensively used in the clinic.
Aptamers Specifically Targeting Cell Surface Biomarkers
Using SELEX technology to develop aptamers for cell surface biomarkers
SELEX, the methodology used to develop aptamers specific for a target of interest, is based on a repetitive amplification and enrichment process. The SELEX process follows several steps: first, a random ssDNA oligonucleotide library is chemically synthesized to contain between 1014–1015 unique random sequences flanked by conserved primer binding sites. This step utilizes the following universal scheme: 5′-sense primer sequence-(random sequence)-antisense primer sequence-3′, where the primer sequence ranges from 18 to 22 bases and the random sequence contains 20–40 nucleic acids. The general procedure consists of labeling the 5′-sense primer with a fluorochrome reporter for monitoring aptamer selection, while the 3′-antisense primer is labeled with an affinity molecule, such as biotin, that is used to separate single-stranded oligonucleotides generated in each amplification round. This random ssDNA library can be used directly to select an initial pool of DNA aptamers. Conversely, generation of RNA aptamers requires two extra steps. Specifically, a pool of random ssDNA oligonucleotides is generated, T7 RNA polymerase promoter sequence is added to the 5′-sense primer, and the DNA is then used as a template for T7 RNA polymerase-based transcription in the 5′ to 3′ direction. During the second SELEX step, the oligonucleotide library is heated and rapidly cooled to promote the formation of 3D structures. The library is then mixed with the target of interest for specific binding enrichment. In the third step, the unbound sequences are discarded through the use of membranes, columns, magnetic beads, and capillary electrophoresis.6,24,25 In the fourth step, the enriched sequences are amplified in vitro by either PCR (DNA aptamers) or RT-PCR (RNA aptamers) to generate a new sequence library for the next round of SELEX. The amplified sequence library may go through further negative-target selection, which eliminates the nonspecific sequences generated by binding of nontarget moieties. Lastly, aptamer selection goes through 4–20 rounds of amplification and enrichment. The exact number of required amplification and selection steps depends on the aptamer target being a purified protein or a living cell, and on the evolution of the aptamer sequence library, as that established by gel electrophoresis, flow cytometry (for target binding), classical cloning or sequencing methods, or by high throughput Next-Generation Sequencing (NGS). In recent years, the traditional SELEX method had also been modified to include the capillary electrophoresis (CE) SELEX, toggle selection, photo-SELEX, bead-based selection, X-Aptamers, and Slow Off-rate Modified Aptamers (SOMAmers) in order to maximize affinity and specificity, to improve the speed of selection and success rate, and to provide additional properties to the selected aptamers.26,27,28,29,30,31
Similar to protein antibody development, purified recombinant proteins or peptides expressed in prokaryotic or eukaryotic systems can be used as targets for aptamers selected by the SELEX method. However, because of the posttranslational modifications, especially in the case of highly glycosylated proteins, purified proteins or peptides often cannot fold into the correct 3D structure that is formed under physiologic conditions.32 Consequently, the newly synthesized aptamers may not be able to selectively recognize and interact with their corresponding targets, which would result in failure of the biomedical application. As this is a common problem, it is very important to choose biomarkers in their native conformation for aptamers selection. Taking this issue into an account, a modified SELEX technology that uses whole living cells, Cell-based SELEX (or Cell-SELEX), was recently established.33 To develop cell-specific aptamers, the Cell-SELEX method uses whole living cells that express surface biomarkers of interest. However, the presence of many different cell surface molecules in addition to the target biomarker(s) results in the synthesis of many unrelated/unwanted aptamers. Therefore, in addition to all the SELEX steps described above, Cell-SELEX technology also utilizes control cells that do not express the target biomarker(s) during the counter-selection step.33
Well-characterized biomarkers that are endogenously expressed at high levels, such as the ErbB superfamily, MUC1, EpCAM, and CD30, offer the best potential for cell-based aptamer development. Subsequently, cell lines that have high endogenous expression of cell-specific or cancer type-specific biomarker(s) are commonly used for Cell-SELEX. However, if such cell lines are unavailable, a biomarker of interest could be over-expressed in a particular cell line via gene transfection and the parental cells used for counter-selection. Using this approach, aptamers targeting the cancer stem cell (CSC) biomarker CD133 have been recently developed.34 In this study, CD133 cDNA was transfected into HEK293T cells that were then used for aptamer enrichment, with the parental HEK293T cells serving as a negative control. Similarly, an aptamer specific for the human receptor tyrosine kinase was recently developed.35
Figure 2.
Schematic diagram of our hybrid-SELEX method for selection of CD30-specific ssDNA aptamer. In our experiment, the hybrid-SELEX process is divided into (a) the cell-based SELEX selection and (b) CD30 protein-based SELEX enrichment. First, CD30-expressing lymphoma cells are used for positive selection and CD30-negative Jurkat cells are used in negative counter-selection. After 20 rounds of selection, the enriched aptamer pool is incubated with CD30 protein immobilized on magnetic beads for five additional rounds of enrichment. SELEX, Systematic Evolution of Ligands by EXponential enrichment.
Full figure and legend (183K)
Aptamers specific for cell surface biomarkers
Cell surface biomarkers are functionally important molecules involved in many biological processes, such as signal transduction, cell adhesion and migration, cell–cell interactions, and communication between the intra- and extra-cellular environments. An abnormal expression of cell surface biomarkers is often related to tumorigenesis.50 Clinically, it is estimated that about 60% of cancer-targeting drugs, including therapeutic antibodies and small molecule inhibitors, target cell surface biomarkers,51 making them attractive for disease treatment. In the last decade, many aptamers targeting cell surface biomarkers have been developed through the advancement of both the protein- and/or cell-based SELEX technologies (see Table 2 for detailed list). These aptamers have been extensively studied for diagnosis and/or treatment of hematological malignancies,7,23,49 lung,52,53,54 liver,55 breast,56,57 ovarian,58 brain,59,60colorectal,61 and pancreatic cancers,46 as well as for identification and characterization of CSCs.34,62
Aptamer-Mediated Targeted Therapies
Traditional cancer treatment approaches, such as chemotherapy, radiotherapy, photodynamic therapy, and photothermal therapy can cause serious side effects in patients due to their associated nonspecific toxicity. To minimize these side effects, a concept of personalized, targeted therapy has been gaining momentum. One of the main clinical approaches for targeted cancer therapy employs antibody-based drugs. Although antibody-mediated therapy is highly specific and results in fewer side effects, potential immunogenicity and high cost of production may limit its clinical applications. To overcome these obstacles, oligonucleotide aptamer-based targeted therapeutics and specific drug delivery systems have recently been explored. These studies revealed numerous advantages offered by the aptamer technology over protein-based antibody therapies, with some of these described in the section below.
Aptamer-drug conjugates
Aptamer-drug conjugation (ApDC) is a very simple yet effective model of noncovalently or covalently conjugating aptamer sequences directly with therapeutic agents (Figure 3). For example, aptamer-conjugated Doxorubicin (Dox), a chemotherapeutic agent extensively used in the treatment of various cancers, has recently been shown to have enhanced therapeutic efficacy over Dox alone. Mechanistically, Dox cytotoxicity is caused by its intercalation into the nucleic acid structure at the preferred paired CG or GC sites with subsequent inhibition of cancer cell proliferation. Taking advantage of its propensity for intercalation, Dox can be noncovalently conjugated to oligonucleotide aptamers containing CG/GC sequences through a simple incubation step. A recent report by Subramanian et al. describes the effectiveness of aptamer-Dox conjugates in the treatment of retinoblastoma.63 In their study, a 2′-fluoro modified RNA aptamer EpDT3 (specific for EpCAM, a CSC marker), was noncovalently conjugated with Dox. After binding to EpCAM molecules expressed at the cancer cell surface, the EpDT3-Dox conjugates were preferentially internalized by the cancer and not by the healthy cells, greatly enhancing therapeutic efficacy and reducing treatment-associated side effects. Several other studies also utilized aptamer-Dox conjugates for cancer therapy, such as HER2 aptamer-Dox conjugates targeting breast cancer,64 MUC1 aptamer-Dox conjugates targeting lung cancer,65 and PSMA aptamer-Dox conjugates targeting prostate cancer.66 Despite their obvious advantages, several concerns related to the use of aptamer-Dox conjugate have been raised. These include (i) instability of the aptamer-drug conjugate due to the reversible nature of noncovalent conjugation process; (ii) short circulating half-life of aptamer-drug conjugates in vivo due to their low molecular weight; and (iii) poor drug payload capacity due to a very simple structure of aptamers. These three disadvantages and technological approaches to improve them are described in greater detail below.
Figure 3.
Schematic diagram of noncovalent or covalent aptamer-drug conjugation.
Full figure (40K)
To enhance the stability of drug loading, Dox can be covalently conjugated to aptamer sequences via a functional linker moiety. For example, the DNA aptamer sgc8 possesses a strong affinity for PTK7 kinase that is abundantly expressed on the surface of CCRF-CEM T-cell acute lymphoblastic leukemia cells. To enhance its stability, this aptamer was covalently conjugated with Dox through an acid-labile linker.67 Once the sgc8 aptamer-Dox conjugate was preferentially bound and internalized by the target cells, the acid-labile linker was easily cleaved in the acidic lysosomal environment, releasing Dox and effectively killing target cells.67 On the other side of the spectrum, covalent conjugation is the most commonly used method of aptamer-drug conjugation, especially for agents that cannot intercalate into the nucleic acid structure or whose intercalation would disrupt aptamer structure.68 Evidence suggests that these covalently conjugated aptamer-drug compounds are significantly more stable than the corresponding noncovalently conjugated intercalations.69
Conjugation of aptamers with high molecular weight polymers, such as polyethylene glycol (PEG), has been examined in order to increase aptamer molecular weight. Specifically, PEG has been widely used in drug modifications, including synthesis of Macugen aptamers. This modification, resulting in PEGylated aptamers, not only increased the aptamer molecular weight and prolonged its circulating half-life, but also enhanced its stability and decreased its toxic accumulation in nontarget tissues.70,71
Finally, in order to increase aptamer-drug payload capacity, an innovative model named aptamer-tethered DNA nanotrains (aptNTrs) was recently introduced by Zhu et al. to deliver Dox to cancer cells.72 In this study, structure of the sgc8 aptamer that targets PTK7 was modified by adding a DNA trigger probe on the 5′-end. Consequently, the modified aptamer acted as a locomotive for targeting, while two hairpin monomers containing Dox intercalation sites acted as boxcars to deliver the drug. After self-assembly, the newly synthesized sgc8 aptamer-NTrs displayed high drug payload capacity, with the drug/sgc8 aptamer-NTr molar ratio of 50:1. Importantly, sgc8 aptamer-NTrs-Dox conjugates were preferentially internalized by the target cells, thereby inhibiting tumor cell growth in vitro and in vivo.72
Another strategy for increasing the aptamer payload capacity involves the construction of polyvalent aptamers. Polyvalent aptamers exhibit an increased target affinity and are more rapidly internalized by their target cells. To demonstrate this, Boyacioglu et al. developed a new DNA aptamer they termed SZTI01 against PSMA.69 First, a dimeric aptamer complex (DAC) was created for specific delivery of Dox to PSMA-expressing cancer cells. Then, the SZTI01aptamer was modified on the 3′-terminus with either a dA16 or dT16 single-stranded tail that contained CpG sites for loading Dox, and the two monomers were annealed in a 1:1 ratio to form the DAC structure. The results of the study showed that DACs have a high Dox payload capacity with the Dox/DAC molar ratio of about 4:1, and the DACs-Dox conjugates were stable under physiological conditions for up to 8 hours.69 In another study, a DNA aptamer targeting MUC1 was truncated and an aptamer containing three repeats of the active targeting region, termed L3, was synthesized. Although the Dox payload capacity was not specifically modified in the L3 aptamer, the L3-Dox conjugates showed a stronger affinity to target cells and lower cytotoxicity to off-target cells than the parental MUC1 aptamer.73 Finally, polyvalent aptamers can also be constructed through the rolling circle amplification (RCA) technology. Using the RCA method and the sgc8 aptamer sequence as a circular template, a polyvalent sgc8 aptamer, termed Poly-Aptamer-Drug, was synthesized.74 It was determined that the Dox payload capacity of the polyvalent sgc8 aptamer increased tenfold, as compared to the monovalent sgc8 aptamer. Moreover, because of their 40-fold greater binding affinity, the Poly-Aptamer-Drug conjugates were more effective than their monovalent counterparts in targeting and killing leukemia cells.74
Although Dox presents itself as a very attractive chemotherapeutic agent for use in aptamer conjugation, other drugs, such as Gemcitabine (Gem) and photosensitizers, can also be targeted to cancer cells through the aptamer technology. Gem is an FDA-approved deoxycytidine analog (dFdC) used for anticancer therapy. To deliver Gem specifically to pancreatic cancer cells, Ray et al. developed a novel aptamer-Gem polymer model. In this model, a single-stranded RNA polymer contained Gem that was enzymatically synthesized through a mutant T7 RNA polymerase-mediated transcription reaction and fused with a nuclease-resistant 2′-fluoro-modified RNA aptamer (E07) that selectively binds to EGFR on pancreatic cancer cells. The E07 aptamer structure was modified by introducing a 24-nucleotide sequence at the 3′ end and using it as an adaptor for Gem polymer binding. Following an annealing step, the Gem polymer complementary bound with the E07 aptamer and preferentially targeted the EGFR-expressing pancreatic cancer cells, inhibiting cell proliferation.75
Compared with the traditional chemotherapeutic agents, controlled conditional prodrug photosensitizers have also been extensively used for aptamer-mediated drug delivery. In this therapeutic approach, termed photodynamic therapy, or photodynamic therapy, photosensitizers are activated by light irradiation and induce production of intracellular reactive oxygen species, resulting in cytotoxicity. A study by Ferreira et al. describes the development of a DNA aptamer specific for MUC1 and covalently conjugated at the 5′ end with the photosensitizer chlorin e6.76 Upon light irradiation, MUC1-expressing epithelial cancer cells were preferentially killed with cytotoxicity about 500-fold higher than that of the control cells. Similar studies have reported using a necleolin aptamer (AS1411)-TMPyP4 for targeting breast cancer77 and the EGFR aptamer (R13)-TF70 for treatment of lung cancer.78
Finally, approaches to extend the scope of aptamer application have also been developed. Similar to bi-specific antibodies, bi-specific or even tri-specific aptamers can be constructed. A bi-specific aptamer for targeting different cells was recently described by Zhu et al. In their study, specific DNA aptamers sgc8 and sgd5a were conjugated through a dsDNA linker. Compared to each mono-aptamer, this bi-specific aptamer (named SD) could recognize its target cell simultaneously with equal specificity and affinity, while Dox intercalation into the dsDNA induced target cell cytotoxicity.79 In the same study, a Y-shape dsDNA linker was used to construct a tri-specific aptamer that also recognized its target cells with high specificity and affinity.79 Clinically, Min et al. proposed using a bi-specific aptamer for prostate cancer therapy. It is well established that prostate tumors may contain both PSMA-positive and -negative cell types. Thus, this study utilized two aptamers, a 2′-fluoro modified RNA aptamer targeting PSMA-expressing cells and a DUP-1 peptide aptamer specific to PSMA-negative cells, conjugated through streptavidin. Moreover, intercalating Dox into the PSMA aptamer of this bi-specific aptamer model could serve as a tool to target all prostate cancer cell types.80
Aptamer-nanoparticle therapeutics
Nanoparticles (NPs) are attractive vehicles to increase both the half-life and the drug payload capacity of aptamer-mediated drug delivery. In addition to their common features, such as biocompatibility for clinical applications, large surface for enhanced aptamer and drug loading, and uniform size and shape for excellent biodistribution, NPs have other individual physical and chemical properties defined by their materials. For example, copolymers and liposomes are biodegradable, while metal materials offer exceptional photothermal and magnetic performance.
Conclusion
Antibody-based targeted therapeutics provide high target specificity and affinity. However, their potential for immunogenicity is of a great concern, as is their high production cost, both of which have limited their clinical applicability. As discussed in this review, when compared to protein antibodies, oligonucleotide aptamers offer many advantages, including simple chemical synthesis, virtual nonimmunogenicity, smaller size, faster tissue penetration, ease of modification with different functional moieties, low cost of production, and high biological stability. Therefore, aptamers have become a promising new class of molecular ligands that could replace or supplement protein antibodies. In summary, aptamer technology has a strong market value and may be applied in various biomedical fields, including in vitro cancer cell detection, in vivo tumor imaging, and targeted cancer therapy (Figure 7).
Figure 7.
Summary of various aptamer applications.
Full figure (58K)
Although aptamer technology has a great potential in the biomedical field, several technical challenges remain and must be addressed. These include: (i) how can aptamers be rapidly adapted for specific targets by decreasing false-positive/-negative selection? Primarily dependent on the natural properties of targets of interest, such as proteins versus cells or tissues, the process of aptamer selection is usually time-consuming, and the success rate is sometimes low. To improve the speed and success rate, novel methods for aptamer selection have been recently described. They include bead-based selection, that can select aptamers as rapidly as a single round of selection,27,28 and the SOMAmer, which improves the aptamer production success rate from less than 30% to over 50%.29,30 More recently, a study by Cho et al. devised a Quantitative Parallel Aptamer Selection System (QPASS) method, which integrates microfluidic selection, NGS, and in situ-synthesized aptamer arrays. This approach allows for the simultaneous measurement of affinity and specificity for thousands of candidate aptamers in parallel.116 In addition to QPASS, evolving modifications to the Cell-SELEX approach are beginning to address difficulties with successful removal of the influence stemming from the presence of dead cells, slow enrichment aptamers recognizing targets of interest, and contamination with unwanted aptamer sequences. As described above, utilization of the above-mentioned FACS-mediated SELEX44,45 and hybrid-SELEX23 offers novel approaches that address these technical challenges.
(ii) How can we select cancer-relevant targets for aptamer development and clinical applications? Tumorigenesis is a dynamic process that includes multiple constantly changing factors. Therefore, a one-size-fits-all cancer-specific biomarker is unlikely to ever be identified. Yet, it has been established that certain biomarkers present in healthy tissues are highly expressed in cancer cells. Moreover, certain biomarkers are associated with particular cancer cell types making them to be considered as useful targets for development of targeted cancer therapy. However, while use of cancer cells to identify biomarkers and to develop therapeutic agents is a reasonable approach, cultured cells, especially immortalized cell lines, greatly differ from tumor tissues in vivo. To overcome these limitations and to select more reliable cancer-relevant biomarkers for aptamer development, several innovative SELEX methods have been recently described. Of particular interest are the tissue-based SELEX117 and the in vivo-SELEX,118 which offer target selection under more relevant pathologic conditions. This cell/tissue-specific biomarker selection can also be utilized for development of noncancer related therapies, as shown for aptamers targeting the adipose tissue in obesity119 and for aptamers designed to penetrate the blood-brain barrier in order to combat brain diseases.120 Hence, we believe that the careful selection of cancer-associated biomarkers and cell/tissue type-specific biomarkers will expand the scopes of aptamer applicability and improve the feasibility of clinical applications.
(iii) What methods could improve aptamer biostability in vivo? Unmodified RNA-based aptamers are very susceptible to the nuclease-mediated degradation in vivo. Although many chemical modifications aimed at increasing biostability of the RNA aptamers have been developed, including 2′-modifications, 3′-modifications, phosphodiester backbone modifications,19,20 and utilizations of novel nucleic acids (locked nucleic acid and Spiegelmers),16,21,22 their effectiveness is still limited. When it was first described, PEGylation was a very attractive strategy for prolonging aptamer circulation half-life and enhancing their biostability. However, a recent report showed that the in vivo use of PEGylated aptamers induced production of anti-PEG antibodies,121 emphasizing the need for the development of alternative approaches.
(iv) How can aptamer technology be modified to achieve a more effective drug delivery? Many drug delivery systems described in this review are tested in vitroor in animal models. Yet, as with any compound that is translated from the bench to the bedside, aptamer-drug conjugates may behave differently in a human patient than they do in laboratory animals. Therefore, aptamer-drug conjugation remains an important challenge that must be considered. Specifically, various coupling approaches lead to different pharmacokinetics, biodistribution, and tolerability in vivo, which in turn greatly affect treatment effectiveness. In the same vein, we must consider the effectiveness of aptamer-mediated target gene therapy. Gene therapy, including siRNA and miRNA aimed at silencing specific genes, is considered the next generation therapeutic approach. However, silencing a single pathogenic gene may not be a viable therapeutic option because tumorigenesis is a process regulated by multiple genes and signaling pathways. Therefore, combining targeted therapeutics with gene therapy may represent the most effective strategy. Such combinational therapy approaches can greatly improve the therapeutic efficacy while reducing the required dosages of both drugs and small molecule RNAs,122 and, more importantly, may offer new alternatives to combat chemotherapy-resistant cancers.110
(v) The last important point to consider is whether aptamer-mediated biotherapies can become effective, FDA-approved medications. Following Macugen approval by the FDA, many aptamer-mediated biotherapies have been evaluated in clinical trials. Of particular interest is AS1411, an antitumor aptamer that has completed several Phase I clinical trials.15 Trial results are promising and offer useful insights into further modifications that could be applied to therapeutic aptamer development.
Taken together, although some technical challenges remain to be addressed, oligonucleotide aptamers have become an attractive and promising tool for targeted cancer therapy. As more clinical data are accumulated, we and others will be better equipped to optimize aptamer formulations, leading to the expansion of aptamer use in the clinic.
9.1.5 Tumor Suppressors
Intrinsic Disorder in PTEN and its Interactome Confers Structural Plasticity and Functional Versatility
Prerna Malaney, Ravi R Pathak, Bin Xue, VN Uversky & Vrushank Davé
Scientific Reports 20 June 2013; 3(2035)
http://dx.doi.org:/10.1038/srep02035
IDPs, while structurally poor, are functionally rich by virtue of their flexibility and modularity. However, how mutations in IDPs elicit diseases, remain elusive. Herein, we have identified tumor suppressor PTEN as an intrinsically disordered protein (IDP) and elucidated the molecular principles by which its intrinsically disordered region (IDR) at the carboxyl-terminus (C-tail) executes its functions. Post-translational modifications, conserved eukaryotic linear motifs and molecular recognition features present in the C-tail IDR enhance PTEN’s protein-protein interactions that are required for its myriad cellular functions. PTEN primary and secondary interactomes are also enriched in IDPs, most being cancer related, revealing that PTEN functions emanate from and are nucleated by the C-tail IDR, which form pliable network-hubs. Together, PTEN higher order functional networks operate via multiple IDP-IDP interactions facilitated by its C-tail IDR. Targeting PTEN IDR and its interaction hubs emerges as a new paradigm for treatment of PTEN related pathologies.
The concept of “Intrinsic Disorder” in proteins has rapidly gained attention as the preponderance and functional roles of IDPs are increasingly being identified in eukaryotic proteomes1, 2. Structured proteins adopt energetically stable three-dimensional conformations with minimum free energy. In contrast, IDPs, due to their unique amino acid sequence arrangements, cannot adopt energetically favorable conformations and, thus, lack stable tertiary structure in vitro3. This structural plasticity allows IDPs to operate within numerous functional pathways, conferring multiple regulatory functions4, 5, 6. Indeed, mutations in and dysregulation of IDPs are associated with many diseases including cancer1, 6, 7, signifying that IDPs play vital roles in functional pathways. Evidence suggests that ~80% of proteins participating in processes driving cancer contain IDRs6. For example, tumor suppressor p53 as an IDP, functions via its C-terminal IDR, which simultaneously exists in different conformations, each of which function differently1. Since PTEN is the second most frequently mutated tumor suppressor with versatile functions8, we hypothesized that PTEN may contain IDR(s) that can be exploited for therapeutic targeting in cancers and diseases associated with pathogenic PI3K/Akt/mTOR (Phosphoinositide 3-Kinase/Akt/ mammalian Target of Rapamycin) signaling9, 10, 11.
PTEN (phosphatase and tensin homolog), a 403 amino acid dual protein/lipid phosphatase converts phosphatidylinositol(3,4,5)-triphosphate (PIP3) to phosphatidylinositol(4,5)-bisphosphate (PIP2), thereby regulating the PI3K/Akt/mTOR pathway involved in oncogenic signaling, cell proliferation, survival and apoptosis12. PTEN, as a protein phosphatase, autodephosphorylates itself13. Deficiency or dysregulation of PTEN drives endometrial, prostate, brain and lung cancers, and causes neurological defects14, 15. PTEN is activated after membrane association16, providing conformational accessibility to the catalytic phosphatase domain (PD) that converts PIP3 to PIP216(Figure 1a). Because PTEN reduces PIP3 levels and inhibits pathogenic PI3K signaling, therapeutically targeting PTEN to the membrane to enhance its activity is of significance in treating several pathologies including cancer.
Figure 1: PTEN: A newly identified IDP.

PTEN – A newly identified IDP. srep02035-f1
http://www.nature.com/srep/2013/130620/srep02035/images_article/srep02035-f1.jpg
(a) Diagrammatic representation of PTEN structure. PTEN, a 403 amino acid protein, comprises of PBM: PIP2 Binding Module (AA 1–13; in green), a phosphatase Domain (AA 14–185; in pink), C2 Domain (AA 190–350; in blue), C-terminal region or Tail (AA 351–400; in orange) and a PDZ binding domain (AA 401–403; in dark blue). The PDZ-binding motif is considered as a part of the C-terminal region. *Figure not to scale. (b) Crystal structure of PTEN. Only the phosphatase (in pink) and C2 domain (in blue) are amenable to crystallization. The first seven residues and the last 50 residues represent unstructured/loosely-folded regions that are yet to be crystallized. These regions represent the N- and C-termini of PTEN, respectively. (Source: RCSB Protein Data Bank). (c) Disorder analysis of PTEN. PONDR-VLXT and PONDR-FIT prediction tools were used to determine the disorder score of PTEN. Any value above 0.5 indicates intrinsic disorder. There are several disordered stretches within the PTEN protein, however, the most prominent of these disordered regions is a 50 amino-acid stretch located at the C-terminus of the PTEN protein. (d) IDPs are enriched in polar (R, Q, S, T, E, K, D, H) and structure breaking (G, P) amino acids and are depleted in hydrophobic (I, L, V, M, A), aromatic (Y, W, F) and cysteine (C) and asparagine (N) residues. The amino acid sequence of PTEN highlights these classes of residues with their relative distribution. (e) Composition profiling for full-length PTEN (in green), its ordered domain (in yellow) and its IDR (in red). The tool used is Composition Profiler (Vacic et al, 2007). As shown in the graph, the disordered region in PTEN is enriched in polar residues (specifically H, T, D, S and E), structure breaking residues (specifically P) and is depleted in all hydrophobic residues, cysteine and all aromatic residues. (f) Histogram representing the percentage of hydrophobic, polar, aromatic, structure breaking, cysteine and asparagines residues in ordered vs. disordered regions. The disordered region has an amino acid composition in line with the definition of IDPs.
PTEN crystal structure revealed that the PD and membrane-binding C2 domains are ordered (Figure 1b); however, the structures of the N-terminus, the CBR3 loop and the 50 amino-acid C-tail remain undetermined17. The C-tail is of particular significance due to its ability to regulate PTEN membrane association, activity, function, stability18, 19, 20, 21. Herein, we identify PTEN as an IDP with its C-tail being intrinsically disordered. The PTEN C-tail IDR is heavily phosphorylated by a number of kinases and regulates the majority of PTEN functions, including a large number of PPIs that forms the PTEN primary and secondary interactomes, comprising critical functional protein hubs, most of which are related to cancer. Our analysis provides a mechanistic insight into the functioning of the PTEN C-tail IDR at the systems level, including inter- and intra-molecular interactions that will aid in designing drugs to enhance the lipid phosphatase activity of PTEN for the pharmacotherapy of cancers and pathological conditions driven by hyperactive PI3K-signaling.
PTEN is an IDP
Utilizing two disorder prediction software programs, PONDR-VLXT and PONDR-FIT22, 23, we have identified PTEN as a bona fide IDP. PTEN has a highly disordered, functionally versatile, C-tail encompassing amino acids 351–403 (Figure 1a and 1c). A PDZ-binding motif (amino acids 401–403) is part of the disordered region. Thus, the PTEN C-tail IDR facilitates interactions with a vast repertoire of PDZ domain-containing proteins (Figs. 1a and 2d). The unique amino acid composition of IDRs dictates their structural plasticity3, 23, 24. IDRs are enriched in polar and structure-breaking amino acid residues, depleted in hydrophobic and aromatic residues and, rarely, contain Cys and Asn residues1, 23, 24. The ordered region of PTEN (AA 1–350) has 25% hydrophobic, 43% polar, 9% structure breaking, 13% aromatic and 9% Cys and Asn residues. In contrast, the PTEN C-tail (AA 351–403) is enriched in polar (66%) and structure breaking (11%) residues and is depleted in hydrophobic (11%), aromatic (6%) and Cys and Asn residues (6%), indicating an ideal profile for the IDR (Figs. 1d and 1f ). Further, compositional analysis of PTEN using the Composition Profiler24 reveals that the disordered region in PTEN is enriched in polar residues (specifically H, T, D, S and E) and structure breaking residues (specifically P) but is depleted in all aromatic and hydrophobic residues in addition to cysteine. (Figure 1e), again exhibiting universal characteristics of IDPs. Taken together, we establish the PTEN C-tail as a functional IDR and classify PTEN as a new IDP.
Figure 2: The functional relevance of the PTEN IDR.

The functional relevance of the PTEN IDR. srep02035-f2
http://www.nature.com/srep/2013/130620/srep02035/images_article/srep02035-f2.jpg
(a) The number of mutations observed in PTEN over its 403 amino-acid stretch is plotted. Fewer mutations are observed in the tail region (in red) possibly indicating the deleterious nature of mutations in the functionally critical C-terminal region. [Source: Sanger Institute Catalogue of Somatic Mutations in Cancer (COSMIC), Human Gene Mutation Database (HGMD)]. (b) Number of mutations in every successive 50 amino-acid stretch of the PTEN protein. The last 50 amino-acid stretch, representing the tail region has at least one-eighth the number of mutations seen in any other 50 amino-acid stretch along PTEN, pointing to its critical function in cell homeostasis. (c) Correlation of mutations with the amino acid composition of PTEN. The ratio of mutations in specific residues in the disordered vs. ordered region are represented in this graph. The residues considered here are those used to define IDRs: hydrophobic, polar, aromatic, structure-breaking, cysteine and asparagine residues. Compared to the other classes of residues, mutations in aromatic residues are much higher in the disordered region when compared to the ordered region. (d) The PTEN primary interactome. Forty proteins interact with known regions of PTEN. There are approximately 340 more proteins that interact with PTEN at sites that are yet to be determined (see Supplementary Table S2). Proteins shown in pink interact with the phosphatase domain, those in blue interact with the C2 domain and those in orange interact with the disordered tail. (Visualization tool: Cytoscape). (e) The PTEN C-tail has a higher propensity for PPIs. Of the 40 mapped proteins, 60% interact with the disordered indicating a strong correlation between degree of disorder and the number of protein interactions. (f) Most proteins within the PTEN interactome are highly disordered. Approximately 80% of PTEN-interacting proteins within the primary interactome are disordered, as indicated in red. The proteins within the interactome that are ordered are indicated in blue.
Low mutability of PTEN IDR suggests critical biological functions
Mutations in PTEN are associated with several types of cancers14. To correlate PTEN mutations to its structure, we analyzed all human PTEN mutations deposited in the COSMIC Database (http://www.sanger.ac.uk/genetics/CGP/cosmic/). The disordered PTEN C-tail IDR shows unusually low mutability (~8-fold less) compared to any other 50 amino-acid stretch of PTEN (Figure 2a and 2b). To confirm our finding of the low mutability of the C-tail region, we also analyzed all human PTEN mutations deposited in the Human Gene Mutation Database (HGMD,http://www.hgmd.cf.ac.uk/ac/index.php)25 (Figure 2a), cBioPortal for Cancer Genomics26, 27(Supplementary Figure S1) and the Roche Cancer Genome Database28 (Supplementary Figure S1) which was consistent with the COSMIC database mutational data. It is likely that evolutionary pressure maintains a survival advantage and ipso facto abrogates progeny with mutations in highly functional protein sequences29, 30, 31. Thus, the functionally versatile PTEN C-tail IDR cannot afford mutations, hence showing least number of mutations. It is equally likely that mutations in individual residues within the IDR are well tolerated, as the evolutionary pressure may have shifted to maintaining global biophysical properties and structural malleability of the IDR to safeguard the critical protein function29. In either case, on a global scale, the versatile structural pliability of the PTEN IDR dictates functional diversity and biological activities29. Thus, the slightest functional perturbation in the PTEN IDR due to mutations, either within the IDR or in domains interacting with it, could disrupt cellular homeostasis as seen in cancers and neurodegenerative disorders associated with PTEN mutations. This is supported by our data indicating that PTEN, as an IDP when mutated, causes several cancers14.
Moreover, the PTEN C-tail IDR exhibits preferential mutations in aromatic residues compared to the ordered region (Figure 2c). The ratio of mutations in aromatic residues in the disordered to ordered region is much higher than any other class of residues (structure breaking, hydrophobic, polar, Cys and Asn), likely attributed to the structure-imparting property of aromatic residue32. Specifically, aromatic residues within IDRs engage in stacking interactions, enhancing nucleation between distinct residues at functional protein-protein interaction interfaces32. Thus loss of this critical structural and functional property imparted by aromatic residues is associated with a disease phenotype. In summary, the disordered PTEN C-tail IDR has functionally evolved to contain a combination of peptides that cannot tolerate mutations.
Disorderliness in PTEN primary interactome drives functional networks
Protein-Protein Interactions (PPIs) typically occur between conserved, structurally rigid regions of two or more proteins, particularly ordered proteins that display energetically favorable, highly-folded conformations. Intriguingly, IDPs lack tertiary structure, yet engage in PPIs, albeit with lower affinities but high specificity1. The lack of structure within IDPs enhances their biophysical landscape, conferring them with the ability to attain structural complementarities required for PPIs. Since IDPs do not conform to a stable structure, they are less compact, providing a larger physical interface and energetic adaptability to interact with multiple proteins1, 7. Thus, conditional folding within IDPs is effectively utilized for interaction with a multitude of binding partners, enabling them to shuttle between several signaling cascades as efficient “cogs”, mediating and regulating PPIs4,7, 33, 34, 35, 36. Indeed, we discovered that PTEN, being an IDP, interacted with more than 400 proteins (Supplementary Table S1) when a combination of online software, literature search and database mining tools were used. Proteins with known PTEN interaction domains were classified as “mapped” (Figure 2d and Supplementary Table S1), whereas those with uncharacterized/predicted interactions were designated as “unmapped” proteins (Supplementary Table S1). Derivation of PTEN primary interactome from the mapped proteins using Cytoscape (http://www.cytoscape.org/) indicated that PTEN disorderliness is efficiently used for interaction with 40 proteins, most existing in distinct functional pathways (Figure 2d, 2e and Supplementary Table S2).
Interestingly, within the PTEN primary interactome, 60% of interactions occurred within the disordered C-tail region. Furthermore, disorder analysis on the primary interactome revealed that 33 proteins (>82%) were IDPs, of which two-thirds interacted with the C-tail IDR (Figure 2e, 2f andSupplementary Table S3), indicating a high propensity for disorder-disorder (D-D)-type interactions.
In order to study evolutionary conservation of the PTEN C-tail and its interactions across species, several sequence alignments were performed (Figure 3a). Sequence alignment of the entire PTEN protein from different animal species shows a good conservation of the catalytic phosphatase domain between vertebrates and invertebrates with 100% sequence conservation for the dual specificity phosphatase catalytic motif HCKAGKGR8 (Supplementary Figure S2). The C-tail shows good conservation in the vertebrate species, likely indicating the recent emergence of the function of PTEN C-tail region in regulating PTEN activity and enriching its PPI potential, translating to its versatile functions. In order to examine the conservation across species for the PTEN C-tail interacting proteins, a literature search was conducted to identify experimentally verified domains/motifs involved in interaction with the C-tail. The domains involved in these interactions with the C-tail for 13 proteins with relevant literature sources for these interactions are part of Supplementary Figure S3. Subsequent sequence alignments for these thirteen proteins (Supplementary Figure S3) shows good sequence homology for the domains/motifs involved in interaction with the PTEN C-tail. These findings support the concept that the PTEN C-tail has evolved in vertebrates to incorporate features that allow it to interact with these proteins.
Figure 3: Sequence conservation in PTEN and its interacting partners reflects functionality.

Sequence conservation in PTEN and its interacting partners reflects functionality. srep02035-f3
http://www.nature.com/srep/2013/130620/srep02035/images_article/srep02035-f3.jpg
(a) Sequence alignment of the PTEN protein for vertebrate and invertebrate animals. Green color indicates sequence similarity while red indicates sequence dissimilar amino acid residues. All comparisons are made with respect to the human PTEN protein. (b) Network analysis for PTEN was performed to assess its potential as a network hub. The network shows multiple secondary interactions within the 40 mapped proteins, indicating their role in multiple signaling cascades mediated via PTEN. The proteins SMAD2/3, AR, PCAF, ANAPC7, B-arrestin 1 and p53 appear to be critical within these signaling cascades and also happen to be intrinsically disordered (Supplementary Table S3), reinforcing the concept of preferential interactions between disordered proteins. (Analysis Tool: Metacore by GeneGo).
Further, to assess whether PTEN acts as a functional hub protein and regulates pathways through its protein-binding partners, we performed functional network analysis using the Analyze Network option from MetaCore (GeneGo Inc, Thomson Reuters, 2011) (Figure 3b). The PTEN primary interactome was used as input with PTEN as the central node. We identified multiple interactions not only between PTEN (node) and SMAD2/3, AR, PCAF, ANAPC3, ANAPC4, Caveolin, β-arrestin 1 and p53 (edges), but also amongst the edge proteins themselves (Figure 3b). Interestingly, all the edge proteins are themselves highly disordered (Supplementary Table S3). Further supporting this finding, our functional enrichment revealed that 13 proteins (one-third) of the PTEN primary interactome were cancer-related and highly disordered (Figure 4a, Supplementary Table S3 and S4).
Figure 4: Derivation and disorder analysis of the PTEN cancer interactome.

Derivation and disorder analysis of the PTEN cancer interactome. srep02035-f4
http://www.nature.com/srep/2013/130620/srep02035/images_article/srep02035-f4.jpg
- Derivation of the PTEN Cancer Interactome. Functional enrichment of the PTEN primary interactome identified 13 cancer-related proteins which are also intrinsically disordered. Subsequently, the PTEN secondary interactome was derived from the primary PTEN interacting proteins. A subset of the secondary interactome was designated as the PTEN Cancer Interactome and it represents the proteins that interact with the 13 cancer-related proteins of the primary interactome. (b) PTEN Cancer Interactome. PTEN is the primary node that interacts with the 13 cancer-related proteins representing the partial primary interactome. Proteins that interact with each of the 13 cancer-related proteins comprise the secondary interactome. Disordered proteins are represented in red while ordered proteins are shown in blue. Cancer-related proteins in the PTEN primary interactome were identified using IPA (Ingenuity® Systems, ingenuity.com). (c) We identified 40 proteins that are part of the PTEN primary interactome of which 13 are highly disordered (IDP) and identified as potential cancer network hubs based on functional network analysis. We further identify 299 IDPS from the secondary PTEN interactome. A filter for cancer-related proteins revealed that approximately two-thirds of the IDPs that form the secondary interactome (193 out of 299) are involved in oncogenesis, suggesting a high degree of functional enrichment. (Functional network analysis was performed using IPA (Ingenuity® Systems,www.ingenuity.com).Full size image (805 KB)
Pliant PTEN secondary interactome relays function of the primary network
The disorderliness of the PTEN primary interactome prompted us to investigate the possibility that PTEN radiates its function via a malleable network of IDPs that extends beyond the primary interactome. Therefore, we derived the PTEN secondary interactome (Supplementary Table S5) and ascertained the interaction of 13 cancer-related proteins identified in the primary interactome (Figure 4a). The entire PTEN secondary interactome consisted of 299 IDPs, of which 193 IDPs (two-thirds) were associated with the 13 cancer-related proteins, generating a “PTEN-Cancer Interactome” (Figure 4, Supplementary Table S5 and S6). Thus, two-third of the IDPs within the PTEN secondary interactome associates with one-third of the cancer related IDPs within the PTEN primary interactome, indicating that cancer-related functions are driven by IDPs in the PTEN interactome and that the flexibility of IDP-IDP interactions modulates diverse functions; dysregulation of which causes cancers.
Functional network analysis of the 193 cancer-related IDPs identified 31 proteins that shared multiple nodes (Figure 5a and Supplementary Table S6). We overlaid this network with the cancer-related IDPs of the primary interactome to predict functionally critical protein hubs (indicated in yellow circles in Figure 5a and b). Our analysis revealed 16 proteins as highly populated hubs, most enriched in disordered regions, again demonstrating that a high degree of structural and functional association between the hubs required IDP-IDP interactions (Figure 5b). The involvement of these hubs in multiple, critical oncogenic signaling pathways make them attractive drug targets in the field of clinical oncology. Our bioinformatic analysis resonates well with observed biological phenomena as seen in the case of MDM2 protein, which is a major PPI hub regulating p53. Interaction of the human androgen receptor (AR) protein and MDM2 influences prostate cell growth and apoptosis37. Mdm2-Daxx interaction activates p53 following DNA damage38, and Daxx binds and inhibits AR function39. Conversely, the breast cancer susceptibility gene 1 (BRCA1) interacts directly with AR and enhances AR target genes, such as p21(WAF1/CIP1), that may result in the increase of androgen-induced cell death in prostate cancer cells40. Further, BRCA1 complexes with Smad3 and is inactivated, leading to early-onset familial breast and ovarian cancer41. Within the same network, MDM2 inhibits the transcriptional activity of SMAD proteins including SMAD342, thereby, emerging as a major player in prostrate, breast and ovarian cancer. Loss of PTEN, on the other hand, results in resistance to apoptosis by activating the MDM2-mediated antiapoptotic mechanism. We also identified proteins like NCL, DAXX and SUMO that play critical roles in mediating cancers as being a part of the PTEN centric cancer interactome (Figure 5b). Interestingly, all of the 16 predicted hubs can be traced back to PTEN (either directly or through other signaling adaptors) reinforcing our analysis (Figure 5c). These findings support the prevailing concept of preferential interaction between disordered regions of two distinct proteins; with PTEN being the common disordered interacting hub, giving functional centrality to PTEN in many critical cellular pathways.
Figure 5: Predicting functionally relevant network hubs in the PTEN cancer interactome.

Predicting functionally relevant network hubs in the PTEN cancer interactome. srep02035-f5
http://www.nature.com/srep/2013/130620/srep02035/images/srep02035-f5.jpg
(a) Methodology to identify functional hubs within the PTEN Cancer Interactome. The PTEN Cancer Interactome contains 193 IDPs that are potential hubs. Over-represented IDPs (or IDPs with multiple occurrences) in the PTEN Cancer Interactome would have a greater propensity to function as hubs. Upon sorting for over-represented IDPs the list of 193 proteins is brought down to 31 proteins. In order to assess the possibility of these 31 proteins as functional hubs a network analysis is warranted. (b) We identified 31 potential hubs based on multiple associations from within the 193 cancer-associated IDPs of the PTEN secondary interactome. Regulatory networks derived from these 31 proteins were overlaid with a similar network from the 13 cancer-related proteins. Based on the number of associations within the network, we identify 16 potential functional hubs in the PTEN cancer interactome (indicated in yellow). Regulatory interactions were generated using the Transcriptome Browser tool (Lopez et al, 2008). (c) Functional network analysis of the 16 predicted hubs. In order to assess the functional association of the 16 predicted hubs with PTEN – a network analysis with PTEN as a central node was done. The analysis identifies MDM2 protein, a major regulator of p53, as one of the major PPI hubs in the PTEN cancer interactome. A number of other critical cancer-related proteins, such as AR, SMAD2/3 and PDGFRB that are part of the PTEN primary interactome, feature prominently in the PTEN cancer interactome. We also identified proteins like NCL, DAXX and SUMO that play critical roles in mediating cancers as being a part of the PTEN centric cancer interactome. Interestingly, all of the 16 predicted hubs can be traced back to PTEN (either directly or through other signaling adaptors) reinforcing our analysis. (Functional network analysis was performed using IPA (Ingenuity® Systems, www.ingenuity.com).
To further validate our methodology in using intrinsic disorder and cancer as filters to identify key signaling hubs, we compared our data sets with a previously published cancer signaling data set. We derived 7 common hubs (Supplementary Table S7), which were extended using the expansive human signaling network described previously43, 44, 45, 46 to obtain the PTEN associated cancer interactome (Figure 6a). An extensive disease associated network analysis using IPA validated our predictions as all the seven predicted hubs had an extensive cross-talk across multiple cancer disease types (Figure 6b).
Figure 7: Biochemical features modulating PTEN PPIs.

Biochemical features modulating PTEN PPIs. srep02035-f7
http://www.nature.com/srep/2013/130620/srep02035/images_article/srep02035-f6.jpg
(a) A PTEN linked cancer network was derived using seven of the 16 predicted cancer hubs that were common with the human cancer associated gene set. The associated partners of the seven hubs were extracted from the human signaling network (Cui et al, 2007, Awan et al, 2007, Li et al, 2012 and Newman et al, 2013). Red color denotes the potential cancer hubs and blue color are their associated partners. Topological analysis identifies p53 as the most significant network hub in the PTEN linked cancer network (Supplementary Table S7). (b) Disease associated network of PTEN cancer hubs. A functional network was constructed with the seven topologically relevant hubs identified previously using the Core Analysis function from the IPA suite to derive the primary network (denoted as MP). A disease network was constructed using the Path Designer option and disease associated biological functions were overlaid on the primary network. Fx denotes the different functions associated with the members of the networks.
Modulation of PTEN PPIs by linear binding motifs
Recent evidence has shown that IDPs mediate PPIs via short linear amino acid sequences (~20 residues) called Molecular Recognition Elements (MoREs) or Molecular Recognition Features (MoRFs)35, 47. MoRFs undergo disorder-to-order transitions upon binding and adopt thermodynamically stable well-defined structures47, increasing the propensity of IDPs to interact with a vast repertoire of proteins. MoRFs also display molecular recognition elements that capture the binding partner proteins with high specificity. These partner-dependent conformational differences are critical to imparting versatile binding properties to IDRs35.
Since the PTEN IDR engages in multiple PPIs, we tested the possibility for the existence of MoRFs. The MORFP red algorithm48 revealed that PTEN contains major MoRF sites at amino acids 273–279 (part of the disordered CBR3 loop of the C2 domain), amino acids 339–347 (in close vicinity of the disordered C-tail) and amino acids 395–403 (part of the disordered C-tail) (Figure 7a and Supplementary Figure S4). The primary restriction of MoRFs to the PTEN C-tail IDR or adjacent regions indicates that these MoRFs directly participate in modulating PPI functions (Figure 7a). However, mutational analysis within MoRFs is required to establish their active role in functional PPIs.
Figure 7: Biochemical features modulating PTEN PPIs.

Biochemical features modulating PTEN PPIs. srep02035-f7
http://www.nature.com/srep/2013/130620/srep02035/images_article/srep02035-f7.jpg
(a) MoRFs in the PTEN C-tail IDR. MoRFpred (Disfani et al, 2012), a computational tool, was used to identify MoRF regions within the PTEN protein (Supplementary Figure S4). The MoRFs in the vicinity of the C-tail IDR are highlighted in red. Interestingly, all of the major MoRFs (with a length greater than 5 residues) are observed in the vicinity of disordered regions (either part of the disordered CBR3 loop of the C2 domain or the C-tail IDR) indicating a positive correlation between intrinsic disorder and PPIs. (b) ELMs in PTEN C-tail IDR. Eukaryotic Linear Motifs (or ELMs) are 3–11 amino acid long sequences that mediate PPIs. IDRs are particularly enriched in ELMs (Dinkel et al, 2012). The linear motifs occurring in the disordered segment of PTEN (tail + PDZ domain) have been highlighted. The motifs with a high conservation score (>0.75) are indicated in red. Interestingly, all of the motifs with a high conservation score are restricted to the C-tail IDR. (c) Phosphorylation sites in the C-tail IDR. Phosphorylation of PTEN, particularly on serine and threonine residues in the disordered region, regulates the function and stability of PTEN. Phosphorylation occurs at Ser 362, Thr 366, Ser 370, Ser 380, Thr 382, Thr 383, Ser 385 by various enzymes such as Casein Kinase II, Glycogen synthase kinase 3-B and Polo-like kinase 3. Each of these phosphorylation events helps regulate the availability and stability of the PTEN molecule within the cell.
Protein-protein interactions are also facilitated by very short motifs (3–10 amino acids) called Short Linear Motifs (SLiMs) or Eukaryotic Linear Motifs (ELMs)49, 50. Because of their short sequences, ELMs arise/disappear by simple point mutations, providing the evolutionary plasticity that the ordered protein domains lack. Thus, ELMs easily adapt to novel interactions in signaling pathways, where rapid assembly/disassembly of multi-protein complexes is a prerequisite. The frequent occurrence of ELMs in a typical proteome indicates their critical cellular functions. Consistent with this notion, a higher density of ELMs are observed in hub proteins and IDPs50. Since ELMs have short sequences, they interact with low-affinity, however, they engage in highly cooperative binding in protein complexes, triggering productive signaling50. Therefore, at increased intracellular local concentrations they competitively bind to mutually overlapping physiological targets of each other as seen with PDZ, SH2 and PTB interaction domains found in cancer-associated proteins and in IDRs49, 50. As PTEN contains a PDZ-binding motif within the IDR (Figure 1a and c), we probed for the existence and features of ELMs in PTEN using The Eukaryotic Linear Motif Resource (http://elm.eu.org). We identified 34 different classes of ELMs in PTEN that mediate PPIs (Supplementary Figure S5). Interestingly, the four ELMs that are most conserved (conservation score>0.75) occurred within the PTEN C-tail IDR, indicating its high level of functional/biological significance (Figure 7b). ELM functions are further modulated by post-translational modifications, mainly by phosphorylation50. Indeed, the PTEN IDR possesses nine phosphorylation sites51, 52(Figure 7c).
PTEN phosphorylation modulates intramolecular association and PPI function
Post-translational Modifications (PTMs) in IDPs facilitate PPIs5. Modifying enzymes readily dock on structurally flexible IDRs, making them a hot spot for PTMs4, 7, 53, 54. Consistent with this notion, regulatory cancer-associated proteins have twice as much disorder and undergo more frequent phosphorylation/dephosphorylation than other cellular proteins as predicted by DISPHOS (a DISorder-enhanced PHOSphorylation prediction software)54, implicating a tight interconnection between protein phosphorylation and disorder. Consistent with the function of PTM in IDRs, clustering of Ser and Thr phosphorylation sites (Figure 7c) in the C-tail IDR regulates PTEN stability, membrane association and activity19, 20. Phosphorylation in the PEST [proline (P), glutamic acid (E), serine (S) and threonine (T)] domain within the C-tail IDR (amino acids 352 to 399) inhibits degradation of PTEN51. Casein kinase II (CK II), Glycogen synthase kinase 3-beta (GSK3-β) and PLK3 (Polo-like kinase 3) phosphorylate Ser and Thr residues within the IDR, each providing a distinct function51 (Figure 7c). The microtubule-associated serine/threonine (MAST), serine/threonine kinase 11(STK11) or LKB1 and casein kinase I (CKI) kinases have also been implicated in PTEN phosphorylation. STK11/LKB1 modifies T383, while CKI modifies T366, S370 and S38552. Indeed, our DISPHOS prediction for C-tail IDRs supports these experimental observations (Supplementary Figure S6).
Substrate-kinase interactions are typically of the disordered-ordered (D-O) type and are stabilized by hydrogen bonding (Figure 7c), a hallmark of IDRs54. Indeed, computational analysis revealed that large ordered regions comprising the catalytic domains of CKII, GSK3B, PLK3, Rak, and Src kinases interact with the C-tail IDR (Supplementary Table S8), indicating that PTEN engages in D-O type intermolecular interactions with the modifying kinases.
At the intramolecular level, phosphorylation at C-tail residues triggers a conformational change in PTEN, inhibiting its membrane association and, therefore, its lipid phosphatase activity18, 19, 21, 55. The phosphorylated C-tail IDR folds onto the PD and C2 domains giving rise to the “closed-closed” conformation of PTEN (Figure 8a) that is incapable of interaction with the membrane18, 20. The “closed- closed” form of PTEN is enzymatically inactive and cannot convert PIP3 to PIP2. The identification of the exact resides involved in this intramolecular interaction remains an active area of research18, 20, 56.
Figure 8: Targeting PTEN C-tail IDR.
http://www.nature.com/srep/2013/130620/srep02035/images_article/srep02035-f8.jpg
Most PTEN functions emanate from the C-tail IDR, including aberrant PPIs that hyper-activate oncogenic pathways. (a) Phosphorylation mediates an intramolecular interaction in the PTEN molecule. Phosphorylation causes a conformational change in PTEN converting it to the enzymatically inactive “closed closed ” form wherein the flexible tail folds onto residues in the C2 and phosphatase domain, thereby making it incapable of interacting with the membrane. Dephosphorylation (by an unknown phosphatase or via auto-dephosphorylation) converts PTEN to the “open-closed” form. Electrostatic interactions, mediated by the PBM, further convert PTEN to the “open-open” form wherein it binds to the membrane and acts as a lipid phosphatase converting PIP3 to PIP2, thereby, abrogating signaling via the PI3K/Akt/mTOR pathways. Subsequent to membrane binding, several E3 ubiquitin ligases polyubiquitinate PTEN marking it for proteasomal degradation. Phosphorylation, by inducing the intramolecular interaction, masks the ubiquitination sites thereby increasing the half-life of the PTEN protein within the cell. Therefore, phosphorylation negatively regulates PTEN function but positively regulates its stability. (b) PTEN IDR engages in PPIs of the disorder:order type (D-O type). As revealed in the present study, this occurs via the use of a MoRF or SLiM region. Therefore, designing a peptidomimetic drug molecule that competes with the PTEN MoRF/SLiM binding to the ordered protein will abrogate PTEN binding, therefore PTEN function. PTEN IDR is highly accessible to multiple kinases that phosphorylate and modulate PTEN function, mainly its inhibition via intra-molecular interactions. PTEN inhibition hyper-activates the PI3K/AKT/mTOR pathway, which increase the oncogenic potential of the cell and drives cancer growth. Therefore, targeting the PTEN C-tail IDR with small molecules that bind and sterically hinder PTEN phosphorylation and/or intra-molecular interactions will be an ideally adjunctive therapy to multiple inhibitor therapy targeting of the PI3/AKT/mTOR pathway.
It was recently shown that the phosphorylation events of PTEN occur in two independent cascades of ordered events, with the S380–S385 cluster being modified prior to the S361–S70 cluster52. Even within the two clusters, the phosphorylation events follow a specific pattern with a distributive kinetic mechanism. Not surprisingly, distributive kinetics is energetically favorable on protein domains that are highly disordered with multiple ensembles of flexible structures52. Thus the dynamic nature of these phosphorylation events is contingent to the inherent flexibility in the PTEN structure driven by intrinsically disordered C-tail crucial for PTEN stability and localization within the cell (Figure 8a).
Targeting intrinsic disorder in PTEN and its interactome
Drug targeting to critical protein regions can mitigate aberrant cellular processes driving oncogenesis57. However, despite numerous clinical trials with molecularly targeted therapies, failure rates for cancer treatments remain high. Conventional therapies targeting pathway-specific kinases suffer from “off-target effects” and often fail due to the emergence of compensatory and alternative pathways58. As a novel approach, facile drug targeting to IDRs within critical signaling hub proteins is highly plausible59, 60, 61. Moreover, as IDRs undergo extensive PTMs53 and engage in PPIs4, 34, 36, the multitude of resulting protein interactions (normal and aberrant) can be targeted concomitantly with a cocktail of distinct inhibitors, which dampens oncogenic signaling60.
Indeed, targeting PPIs is a more selective treatment strategy over conventional enzyme inhibitors60. However, disruption of multiple ordered interfaces within PPIs by small molecule inhibitors remains challenging62. The advantage of targeting IDPs engaged in PPIs is that, unlike ordered proteins, they engage in PPIs via MoRFs or ELMs, which are small peptide regions that bind with low affinity and thus are susceptible to disruption by small molecule inhibitors59. Consistent with this notion, small molecules disrupted highly disordered complexes of p53-Mdm2 and c-Myc-Max interactions by inducing order upon binding60, 63. Likewise, targeting the PTEN C-tail IDR may reduce its intra- and inter-molecular interactions and limit accessibility to enzymes mediating PTMs (Figure 8b), providing a means to increase PTEN activity. Our analysis shows that since the C-tail IDR is rich in conserved MoRFs/SLiMS, targeting these regions will prove to be a rational therapeutic modality for a large number of cancers that show compromised PTEN activity or hyperactivation of the oncogenic PI3K/AKT/mTOR pathway9, 10, 11. Since reductions in the levels and activity of PTEN are sufficient to drive oncogenesis11, 14, 15, increasing PTEN activity is an ideal therapy for cancers associated with hyperactive PI3K-signaling.
Discussion
Recent studies on genome- and proteome-wide molecular alterations in diseases indicate that pathological conditions are caused by perturbations in complex, highly interconnected biological networks64. Thus, current reductionist approach of studying structure-function relationship in diseases has limited our abilities to discover effective targeted therapeutics. In an attempt to overcome these limitations, in the current study, we have undertaken a novel approach to drug discovery that exploits systems and network biology at the structural, topological and functional level. Using PTEN, a tumor suppressor, we have applied computational and systems biology approaches and integrated extensive data-mining and biochemical properties of IDP interactions to reach a finer understanding of PTEN function. These results have identified PTEN C-tail IDR and several hub proteins in PTEN-driven molecular network implicated in human diseases as therapeutic targets, enhancing the repertoire of clinically relevant biological targets for pharmacotherapy.
Our derivation and analysis of PTEN primary and secondary interactome indicates that altered levels or interactions of IDPs perturb myriad cellular signaling pathways, leading to pathological conditions including cancer. IDPs have the propensity to aggregate and cause cellular toxicity65. Therefore, PTEN as an IDP has evolved a mechanism, wherein, the level of active PTEN, its cellular localization and PTEN-PPIs are regulated via phosphorylation of the C-tail IDR. Furthermore, evolutionarily conserved ELMs and MoRFs that we have identified within the C-tail IDR may play a critical role in orchestrating the formation and function of the PTEN interactome.
Increase in complexity of PPIs is either directed by the number and type of proteins or by increasing the number of interactions required to execute cellular functions66. To delineate how PTEN executes myriad functions, we first derived the PTEN primary interactome. We found 40 proteins to directly interact on the PTEN molecule, out of which 25 were associated with the C-tail IDR, consistent with the concept that disorderliness within PTEN executes its myriad functions. To enhance our understanding of PTEN functions in the context of multiple distinct pathways at the systems-level, we delineated functional networks operating within the primary interactome. Our findings showed a high degree of cross-talk between edges, implying that shared regulatory modules, comprised of multiple signaling cascades, operate via PTEN-mediated interaction networks. When these networks are altered, diseases ensue with extreme functional penalties. We also found that the edge proteins were themselves highly disordered indicating that disorderliness within the PTEN primary interactome confers functional versatility. Supporting this notion, 13 proteins that were functionally classified as cancer-related were also highly disordered forming a pliable “PTEN-Cancer Interactome”. Thus, PTEN lesions influence the flexibility of IDP-IDP interactions modulating diverse functions, likely causing cancer.
Owing to the inherent ability of PPIs to be flexible while being complex, specific cellular functions are readily fine-tuned as per the biological demands. Emerging evidence suggests that certain features on the IDRs are recognized as a way of conferring plasticity to protein interaction networks. Consistent with this concept, our data suggest that PTEN, a hub protein containing an IDR, likely utilizes MoRFs and ELMs, gets differentially modified via PTMs, acquiring complementary structures to engage and modulate PPI activity by facilitating adaptive binding to multiple protein partners in many cellular pathways. Thus, our present work provide a novel entrée in targeting intrinsic disorder in PTEN and its interactome to dampen the aberrant PI3K-signaling that drives many cancers. First, imparting order to the PTEN structure may help dampen multiple oncogenic signaling pathways mediated via the 16 hub proteins identified in the present study, by limiting their affinity for PPIs. Second, targeting intrinsic disorder in PTEN and its interactome can become an adjunctive or alternative approach to the use of various kinase inhibitors, which are toxic and have many off-target effects when used to mitigate the aberrant hyperactivation of PI3K/AKT/mTOR oncogenic signaling pathway. Taken together, the present findings provide a novel entrée to design strategies for drug discovery and may become a logical intervention in the pharmacotherapy of cancer and other PTEN-associated disease treatment modalities.
Read Full Post »

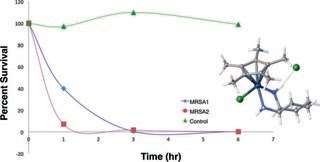




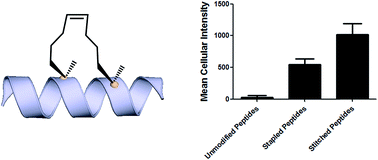
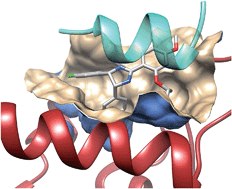










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}