The Vibrant Philly Biotech Scene: Recent Happenings & Deals
Curator: Stephen J. Williams, Ph.D.
UPDATED: 07/06/2026
This update is a review on the current biotech funding situation as of Q1 of 2026. The situation in Philadelphia biotech has turned a bit dire, as CRE that went on massive construction projects are finding vacancy rates up in certain neighborhoods and hubs in the Philadelphia area. Presented here are some of the observations from CRE, the investment community, and some recent issues in biotech buildouts that have reshaped the Philly landscape.
The following is a post form Technical.ly in Philadelphia on the issues in finding Venture Capital to stay and grow in the Philadelphia Area. As noted, the dearth of venture capitalist firms in the city or commited to the cities’ biotech initiative is a major reason for some of the recent setbacks to Philly Biotech.
Source: https://technical.ly/entrepreneurship/life-sciences-year-in-review-2025/
Philly’s life sciences scene saw wins this year, but there’s still a big problem: Money
Partnerships with giants like Eli Lilly and Thermo Fisher Scientific put biotech in the spotlight again, though founders still struggle to find investments in their hometown.
Written by Sarah Huffman
December 18, 2025
Edited by Katie Malone
HiveBio Accelerator 2025 cohort at its Investor Showcase (Courtesy HiveBio)
2025 pulled into focus the cracks in Philly’s life sciences ecosystem.
Philly has the talent, research institutions and collaboration to build a strong life sciences community. That’s helped the region build a reputation in the sector, but stakeholders say there’s not enough local funding to keep companies growing here.
With exciting partnerships started this year and more opportunities coming in 2026, capital is needed if Philadelphia wants the sector to reach its full potential.
“The fact that we’ve got excellent research capabilities and spin-off capabilities in Philadelphia speaks to the intellectual horsepower,” Ira Spector, CEO of SFA Therapeutics, told Technical.ly, “but Philadelphia does not have a very well-developed venture capital infrastructure.”
This year, Philly attracted major outside interest. Most recently, global pharmaceutical company Eli Lilly announced plans to open a new Lilly Gateway Labs in Center City, an incubator for early-stage biotech companies.
“Access to capital is a critical factor in moving forward, and that’s been a frustrating thing for us.”
Ira Spector, CEO of SFA Therapeutics
Other national orgs have taken steps to increase their regional presence, too. Thermo Fisher Scientific, a global equipment and services company, announced a partnership and opened its second Advanced Therapies Collaboration Center with BioLabs Philadelphia.
New local resources sprouted, catering directly to startups’ VC needs. The HiveBio accelerator, which supports minority founders in the life sciences space, launched this year. After wrapping up its pilot this month, the program reported successful fundraising efforts for most of its cohort companies.
These wins happened against a backdrop of internal company turmoil in 2025. The region’s cell and gene therapy standout Spark Therapeutics took a hit when its parent company Roche designated it as a financial loss, eventually laying off about half of its employees. Other companies like Century Therapeutics and Adaptimmune announced significant layoffs this year, too.
For life sciences — and the entire startup ecosystem — venture capital has also become harder to come by as the market fluctuated throughout 2025.
Developing a drug or therapy and bringing it to market is extremely expensive work. Oftentimes, investors don’t want to give money to companies that are still high risk, but it takes money to support development to derisk the product, Spector said.
Without that cash, Philly could lose its rising stars — like GEMMA Bio, a standout that raised $34 million this year — to nearby life sciences hotspots like Boston. While Philly consistently lags behind Boston, San Diego and San Francisco, it still ranks high for life sciences talent, which continues to attract new companies.
“Philly is finally starting to step on the map,” Rakesh Shah, founder of Newtown-based medical device company DRS.LINQ, said. “We have so much talent in the biomedical engineering and med tech space. It’s just we need to harness that energy, bring it all together.”
A well-connected community, but few investors to maintain it
Founders are already looking for major investments outside of Philly, as it’s often their only option, according to Jean Cho, CEO of Trevarx Biomedical.
Jenkintown-based biotech company SFA Therapeutics is experiencing this firsthand. As it raises its next funding round, the majority of the money is coming from investors outside the region, CEO Spector said.
The company is tied to Philly because of its affiliation with Temple University and investments from Ben Franklin Technology Partners. It’s also integrated with Pennsylvania’s life sciences ecosystem, attending programming hosted by orgs like Life Sciences PA, although there aren’t usually a ton of investors at those events, Spector said.
The lack of local funding has caused Spector to consider what the company’s future would look like in other parts of the country. In Boston, for example, the VC ecosystem is very well developed, he said, and the company has raised money from investors there.
A bright spot: Millions raised by just one accelerator
Despite the lack of a strong local investor network, Philly isn’t alone in having a VC slowdown.
In general, the life sciences sector has been struggling this year, especially for early-stage companies. Since the IPO market for biotech companies has slowed down, investors aren’t making their money back and are focusing instead on existing later-stage firms.
But there are some programs in the city helping founders to secure much-needed investments.
The HiveBio accelerator exceeded its first-year goal, helping eight out of 10 companies raise a total of $3.6 million during the nine-month program, Tia Lyles-Williams, founder and CIO of HiveBio, told Technical.ly. Plus, a lot of that money came from local investors like Ben Franklin Technology Partners and Robin Hood Ventures, she said.
The key, she said, was giving founders as many opportunities as possible to have face time with investors.
“They had the opportunity, behind closed doors, with these mentors to be totally transparent about what they know, what they didn’t know, and what they needed,” she said. “These mentors and these experts in our local investment community delivered on that, they received it, implemented it, and that’s how these checks got written.”
“This is an industry that runs on capital,” Spector said. “It requires capital to prove that a drug or a therapeutic or a diagnostic or device works, and that means that access to capital is a critical factor in moving forward, and that’s been a frustrating thing for us.”
This story is made possible thanks to support from Ben Franklin Technology Partners of Southeastern Pennsylvania, a nonprofit that leads the Philadelphia region’s equitable economic growth by nurturing and investing in innovative, early-stage companies, and through purposeful involvement in regional and national initiatives. All stories are independently reported, with no partner review.
This is an analysis of the risk associated to bond tranches in the Philly Biotech niche
When looking at the riskiest secured bond tranches for the biotechnology sector in the Greater Philadelphia area, the risk is completely split between two totally different financial assets: commercial real estate debt backing empty lab spaces and corporate first-lien loans issued by struggling drug-development firms.
While “secured” bonds are theoretically lower risk because they are backed by physical or intellectual property collateral, specific sub-sectors in Philadelphia are facing unprecedented headwinds. [1, 2, 3, 4]
1. The Real Estate Angle: Speculative Lab Building Bonds
The absolute highest concentration of risk for secured debt sits with senior secured credit facilities, CMBS tranches, and construction bank bonds backing recently completed, unleased life sciences real estate. From 2019 to 2021, developers rushed to build or retrofit lab spaces, creating a massive oversupply just as venture capital funding slowed down. [1, 5, 6]
- University City Speculative Developments: University City is the epicenter of Philly’s biotech boom, but the submarket currently faces a staggering 39.1% to 40% vacancy rate. Secured bonds or construction debt tranches tied directly to newly built, unleased towers in this zone (such as Brandywine’s mostly empty 3151 Market St. lab facility) are highly vulnerable. Without tenant cash flows to service the debt, these properties face imminent refinancing or modification risks. [1, 5, 7, 8, 9]
- Suburban “Mega-Lab” Tranches: Out in the suburbs, massive suburban lab conversions are driving negative absorption. A prime example is the The Discover Labs in King of Prussia, where huge blocks of lab space became vacant. Tranches of commercial mortgage-backed securities (CMBS) or private credit heavily exposed to these massive suburban footprints carry elevated risk as local tenants shrink their physical boundaries. [10, 11]
2. The Corporate Angle: Asset-Backed Pre-Revenue Biotech Debt
For actual biotechnology companies (as opposed to their landlords), public bond issuance is rare. Instead, their secured debt takes the form of First-Lien Term Loans or Senior Secured Notes. [12]
Because biotech venture capital funding in Philadelphia dropped sharply—falling from $1.2 billion in 2021 to roughly $600 million—companies are facing a severe cash crunch. The riskiest secured corporate debt tranches share distinct characteristics: [13, 14, 15]
- Maturity Term Loans: The riskiest tranches are senior secured facility loans facing upcoming refinancing walls. Lenders hold first-lien rights over the company’s patent portfolio, drug pipelines, and lab equipment. If a clinical trial fails, the “secured” collateral (intellectual property) loses almost all market value overnight. [16]
- Sidecar & PIK (Payment-in-Kind) Tranches: To avoid outright defaults, distressed Philly biotechs are increasingly utilizing structured “sidecar facilities” or paying interest using more debt (PIK). These specific tranches are highly speculative because they delay the cash crunch rather than fixing the underlying capital structure. [8, 17]
Summary of Risk Profile
|
Tranche Type [1, 5, 10, 11, 13, 18, 19] |
Location/Focus |
Current Risk Driver |
Collateral Value |
|---|---|---|---|
|
University City Lab CMBS/Construction Bonds |
Downtown Hub |
~40% vacancy; tenants pivoting back to office use. |
High (Prime real estate). |
|
Suburban Life Science Mortgages |
King of Prussia |
Heavy negative absorption and tenant consolidation. |
Medium (Harder to repurpose). |
|
First-Lien Corporate Biotech Loans |
Regional Startups |
50% drop in local VC funding over recent years. |
Low/Binary (Dependent on FDA approval). |
Are you looking to evaluate specific real estate debt instruments (like CMBS or commercial mortgages) tied to Philly’s University City developments, or are you assessing the corporate high-yield loan market for localized clinical-stage drug developers? Let me know how you would like to filter the data. [5, 19, 20]
[1] https://www.bizjournals.com
[2] https://www.investopedia.com
[4] https://www.thefixedincome.com
[7] https://www.bizjournals.com
[9] https://www.bizjournals.com
[10] https://biobuzz.io
[11] https://www.cbre.com
[12] https://www.key.com
[13] https://technical.ly
[14] https://biobuzz.io
[16] https://www.biopharmawatch.com
[17] https://octus.com
[18] https://technical.ly
[20] https://www.fitchratings.com
It has been surprising how fast the landscape in Philadelphia has changed. A few years ago, Philadelphia was being touted as “Cellicon Valley” due to the development of the CAR-T therapies and immuno and cell based therapy startups being spun-out from local universities. I have posted on this tranformation on this journal HERE.
In fact a huge campus in the Philadelphia suburbs including the Discovery Labs site (a former GSK site) was being buildout to be a major cGMP biomanufacturing facility which would handle University of Pennsylvania’s cell and gene therapy manufacturing needs as well as a contract manufacturing facility for pharma and biotech for cell and gene therapies.
A summary of the issues and refinancing problems of Discovery Labs can be seen below:
- Contractor Payment Delays: The site, owned by MLP Ventures (CEO Brian O’Neill), was hit with roughly $100 million in mechanics’ liens for unpaid contracting work. [1]
- Legal Battles with Key Tenants: In late 2025, MLP Ventures filed a civil lawsuit against its largest tenant, the Center for Breakthrough Medicines (CBM), and its majority stakeholder, South Korea’s SK Inc.. This involved a landlord/tenant dispute seeking more than $50,000 in damages. [1]
- Massive Data Center Pivot & Protests: To offset debts, MLP Ventures proposed a sprawling 4.6 million-square-foot data center project across the Swedeland and Conshohocken areas. This has triggered massive pushback from local Upper Merion Township residents, who have cited heavy concerns over noise, pollution, and health risks. [1]
- Financial Restructuring: The owner has sought extensive refinancing to manage the site’s debts and attempt to compensate the unpaid contractors, though progress has been slow. [1, 2]
Original Article
As the office and retail commercial real estate market has been drying up since the COVID pandemic, commercial real estate developers in the Philadelphia area have been turning to the health science industry to suit their lab space needs. This includes refurbishing old office space as well as new construction.
Gattuso secures $290M construction loan for life sciences building on Drexel campus

By Ryan Mulligan – Reporter, Philadelphia Business Journal
Dec 19, 2022
Gattuso Development Partners and Vigilant Holdings of New York have secured a $290 million construction loan for a major life sciences building set to be developed on Drexel University’s campus.
The funding comes from Houston-based Corebridge Financial, with an additional equity commitment from Boston-based Baupost Group, which is also a partner on the project. JLL’s Capital Markets group arranged the loan.
Plans for the University City project at 3201 Cuthbert St. carry a price tag of $400 million. The 11-story building will total some 520,000 square feet, making it the largest life sciences research and lab space in the city when it comes online.
The building at 3201 Cuthbert will rise on what had served as a recreation field used by Drexel and is located next to the Armory. Gattuso Development, which will lease the parcel from Drexel, expects to to complete the project by fall 2024. Robert A.M. Stern Architects designed the building.

A rendering of a $400 million lab and research facility Drexel University and Gattuso Development Partners plan to build at 3201 Cuthbert St. in Philadelphia.
The building is 45% leased by Drexel and SmartLabs, an operator of life sciences labs. Drexel plans to occupy about 60,000 square feet, while SmartLabs will lease two floors totaling 117,000 square feet.
“We believe the project validates Philadelphia’s emergence as a global hub for life sciences research, and we are excited to begin construction,” said John Gattuso, the co-founder and president of Philadelphia-based Gattuso Development.
Ryan Ade, Brett Segal and Christopher Peck of JLL arranged the financing.
The project is another play in what amounts to an arms race for life sciences space and tenants in University City. Spark Therapeutics plans to build a $575 million, 500,000-square-foot gene therapy manufacturing plant on Drexel’s campus. One uCity Square, a $280 million, 400,000-square-foot life sciences building, was recently completed at 38th and Market streets. At 3151 Market St., a $307 million, 417,000-square-foot life sciences building is proposed as part of the Schuylkill Yards development.
Tmunity CEO Usman Azam departing to lead ‘stealth’ NYC biotech firm

By John George – Senior Reporter, Philadelphia Business Journal
Feb 7, 2022
The CEO of one of Philadelphia’s oldest cell therapy companies is departing to take a new job in the New York City area.
Usman “Oz” Azam, who has been CEO of Tmunity Therapeutics since 2016, will lead an unnamed biotechnology company currently operating in stealth mode.
In a posting on his LinkedIn page, Azam said, “After a decade immersed in cell therapies and immuno-oncology, I am now turning my attention to a new opportunity, and will be going back to where I started my life sciences career in neurosciences.”
Tmunity, a University of Pennsylvania spinout, is looking to apply CAR T-cell therapy, which has proved to be successful in treating liquid cancers, for the treatment of solid tumors.
Last summer, Tmunity suspended clinical testing of its lead cell therapy candidate targeting prostate cancer after two patients in the study died. Azam, in an interview with the Business Journal in June, said the company, which had grown to about 50 employees since its launch in 2015, laid off an undisclosed number of employees as a result of the setback.
Azam said on LinkedIn he is still a big believer in CAR T-cell therapy, noting Tmunity co-founder Dr. Carl June and his colleagues at Penn just published in Nature the 10-year landmark clinical outcomes study with the first CD19 CAR-T patients and programs.
“It’s just the beginning,” he stated. “I’m excited about the prospect of so many new cell- and gene-based therapies emerging in the next five to 10 years to tackle many solid and liquid tumors, and I hope we all continue to see the remarkable impact this makes on patients and families around the world.”
Azam could not be reached for comment Monday. Tmunity has engaged a search firm to identify his successor.
Tmunity, which is based in Philadelphia, has its own manufacturing operations in East Norriton. Tmunity’s founders include June and fellow Penn cell therapy pioneer Bruce Levine, who led the development of a CAR T-cell therapy now marketed by Novartis as Kymriah, a treatment for certain types of blood cancers.
In therapy using CAR-T cells, a patient’s T cells — part of their immune system — are removed and genetically modified in the laboratory. After they are re-injected into a patient, the T cells are better able to attack and destroy tumors. CAR is an acronym for chimeric antigen receptor. Chimeric antigen receptors are receptor proteins that have been engineered to give T cells their improved ability to target tumors.
PIDC names U.S. Department of Treasury veteran, Philadelphia native as next president

Jodie Harris is a Philadelphia native who has spent the last 15 years in the U.S. Department of Treasury.

The Philadelphia Industrial Development Corp. has tapped U.S. Department of Treasury veteran Jodie Harris to be its next president.
Harris succeeds Anne Bovaird Nevins, who spent 15 years in the organization and took over as president in January 2020 before stepping down at the end of last year. Executive Vice President Sam Rhoads has been interim president.
Harris, a Philadelphia native who currently serves as director of the Community Development Financial Institutions Fund for the Department of Treasury, was picked after a regional and national search and will begin her tenure as president on June 1. She becomes the 12th head of PIDC and the first African-American woman to lead the organization.
PIDC is a public-private economic development corporation founded by the city and the Chamber of Commerce for Greater Philadelphia in 1958. It mainly uses industrial and commercial real estate projects to attract jobs, foster business opportunities and spur overall community growth. The organization has spurred over $18.5 billion in financing across its 65 years.
PIDC has its hand in development projects spanning the city, including master planning roles in expansive campuses like the Philadelphia Navy Yard and the Lower Schuylkill Biotech Campus in Southwest Philadelphia.
In a statement, Harris said that it is “a critical time for Philadelphia’s economy.”
“I’m especially excited for the opportunity to lead such an important and impactful organization in my hometown of Philadelphia,” Harris said. “As head of the CDFI Fund, I know first-hand what it takes to drive meaningful, sustainable, and equitable economic growth, especially in historically underserved communities.”
Harris is a graduate of the University of Maryland and received an MBA and master of public administration from New York University. In the Treasury Department, Harris’ most recent work aligns with PIDC’s economic development mission. At the Community Development Financial Institutions Fund, she oversaw a $331 million budget, mainly comprised of grant and administrative funding for various economic programs. Under Harris’ watch, the fund distributed over $3 billion in pandemic recovery funding, its highest level of appropriated grants ever.
Harris has been a part of the Treasury Department for 15 years, including as director of community and economic development policy.
In addition to government work, Harris has previously spent time in the private, academia and nonprofit sectors. In the beginning of her career, Harris worked at Meridian Bank and Accenture before turning to become a social and education policy researcher at New York University. She also spent two years as president of the Urban Business Assistance Corporation in New York.
Mayor Jim Kenney said that Philadelphia is “poised for long-term growth” and Harris will help drive it.
Source: https://www.bizjournals.com/philadelphia/news/2023/02/23/pidc-names-next-president-treasury.html
$250M life sciences conversion planned for Philadelphia’s historic Quartermaster site

A rendering shows the future Quartermaster Science + Technology Park at sunset.

Listen to this article 3 min
Real estate company SkyREM plans to spend $250 million converting the historic Quartermaster site in South Philadelphia to a life sciences campus with restaurants and a hotel.
The redevelopment would feature wet and dry lab space for research, development and bio-manufacturing.
The renamed Quartermaster Science + Technology Park is near the southwest corner of Oregon Avenue and South 20th Street in the city’s Girard Estates neighborhood. It’s east of the Quartermaster Plaza retail center, which sold last year for $100 million.
The 24-acre campus is planned to have six acres of green space, an Aldi grocery store opening by March and already is the headquarters for Indego, the bicycle share program in Philadelphia.
Six buildings totaling 1 million square feet of space would be used for research and development labs. There’s 500,000 square feet of vacant space available for life sciences and high technology companies with availabilities as small as 1,000 square feet up to 250,000 square feet contiguous. There’s also 150,000 square feet of retail space available.
The office park has 200,000 square feet already occupied by tenants. The Philadelphia Job Corps Center and Delaware Valley Intelligence Center are tenants at the site.

A rendering shows part of the Quartermaster Science + Technology Park as a redeveloped mixed-use life science campus.
The campus was previously used by the military as a place to produce clothing, footwear and personal equipment during World War I and II. The clothing factory closed in 1994. The Philadelphia Quartermaster Depot was listed on the National Register of Historic Places in 2010.
“We had a vision to preserve the legacy of this built-to-last historic Philadelphia landmark and transform it to create a vibrant space where the best and brightest want to innovate, collaborate, and work,” SkyREM CEO and Founder Alex Dembitzer said in a statement.
SkyREM, a real estate investor and developer, has corporate offices in New York and Philadelphia. The company acquired the site in 2001.
Vered Nohi, SkyREM’s regional executive director of new business development, called the redevelopment “transformational” for Philadelphia.

SkyREM announced the redevelopment of the Quartermaster campus in South Philadelphia into a life sciences campus with restaurants and a hotel. This rendering looks across Oregon Avenue toward the southwest corner of Oregon and 21st Street.
Quartermaster would join a wave of new life sciences projects being developed in the surrounding area and across the region.
The site is near both interstates 76 and 95 and is about 2 miles north of the Philadelphia Navy Yard, which has undergone a similar transformation from a military hub to a major life sciences and mixed-use redevelopment project. The Philadelphia Industrial Development Corp. is also in the process of selecting a developer to create a massive cell and gene therapy manufacturing complex across two sites totaling about 40 acres on Southwest Philadelphia’s Lower Schuylkill riverfront.
At 34th Street and Grays Ferry Avenue, the University of Pennsylvania is teaming with Longfellow Real Estate Partners on proposed a $365 million, 455,000-square-foot life sciences and biomanufacturing building at Pennovation Works.

A rendering shows part of the future Quartermaster Science and Technology Park in South Philadelphia. The 24-acre campus is planned to have six buildings with 1 million square feet of life science space.
SkyREM is working with Maryland real estate firm Scheer Partners to lease the science and technology space. Philadelphia’s MPN Realty will handle leasing of the retail space. Architecture firm Fifteen is working on the project’s design.
Scheer Partners Senior Vice President Tim Conrey said the Quartermaster conversion will help companies solve for “speed to market” as demand for life science space in the region has been strong.
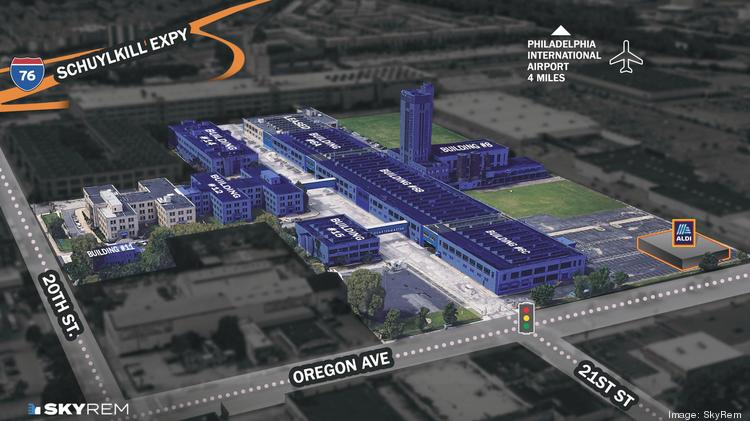
A diagram shows the buildings that are leased (gray) and the buildings that are available (blue) at the Quartermaster site in South Philadelphia.
Brandywine pauses new spec office development, continues to bet big on life sciences

An artist’s rendering of Brandywine Realty Trust’s 168,000-square-foot, four-story life sciences building at 250 King of Prussia Road in Radnor.
Brandywine Realty Trust originally planned to redevelop a Radnor medical office into lab and office space, split 50-50 between the two uses.
After changes in demand for lab and office space, Brandywine (NYSE: BDN) recently completed the 168,000-square-foot, four-story building at 250 King of Prussia Road in Radnor fully for life sciences.
“The pipeline is now 100% life sciences, which, while requiring more capital, is also generating longer term leases at a higher return on cost,” Brandywine CEO Jerry Sweeney of the project said during the company’s fourth-quarter earnings call on Thursday.
At the same time, Brandywine is holding off on developing new office buildings unless it has a tenant lined up in advance.
The shift reflects how Philadelphia-based Brandywine continues to lean into — and bet big — on life sciences.
Brandywine is the city’s largest owner of trophy office buildings and has several major development projects in the works. The company is planning to eventually develop 3 million square feet of life sciences space. For now, 800,000 square feet of life sciences space is under development, including a 12-story, 417,000-square-foot life sciences building at 3151 Market St. and a 29-story building with 200,000 square feet of life sciences space at 3025 John F. Kennedy Blvd. Both are part of the multi-phase Schuylkill Yards project underway near 30th Street Station in University City.
Once its existing projects are completed, Brandywine would have 800,000 square feet of life sciences space, making up 8% of its portfolio.Sweeney said the company wants to grow that figure to 21%.
Brandywine is developing a 145,000-square-foot, build-to-suit office building at 155 King of Prussia Road in Radnor for Arkema, a France-based global supplier of specialty materials. The building will be Arkema’s North American headquarters. Construction began in January and is scheduled to be completed in late 2024.
Brandywine reported that since November it raised over $705 million through fourth-quarter asset sales, an unsecured bond transaction and a secured loan. The company has “complete availability” on its $600 million unsecured line of credit, Sweeney said.
Brandywine sold a 95% leased, 86,000-square-foot office building at 200 Barr Harbor Drive in West Conshohocken for $30.5 million. The company also sold its 50% ownership interest in the 1919 Market joint venture for $83.2 million to an undisclosed buyer. 1919 Market St. is a 29-story building with apartments, office and commercial space. Brandywine co-developed the property with LCOR and the California State Teacher’s Retirement System.
Brandywine declined to comment and LCOR could not be reached.
Brandywine’s core portfolio is 91% leased.
The project at 250 King of Prussia Road cost $103.7 million and was recently completed. The renovation included 12-foot high floor-to-ceiling glass on the second floor, a new roof, lobby, elevator core, common area with a skylight and an added structured parking deck.
Located in the Radnor Life Science Center, a new campus with nearly 1 million square feet of lab, research and office space, Sweeney said it’s a “magnet” for biotech companies. Avantor, a global manufacturer and distributor of life sciences products, is headquartered in the complex.
Sweeney said Brandywine is “very confident” demand will stay strong for life sciences in Radnor. The building at 250 King of Prussia Road is projected to be fully leased by early 2024.
“Larger users we’re talking to, they just tend to take a little bit more time than we would like as they go through technical requirements and space planning requirements,” Sweeney said.

Jerry Sweeney, CEO of Brandywine Realty Trust.
While Brandywine is aiming to increase its life sciences footprint, the company is being selective about what it builds next. The company may steer away from developments other than life sciences. The Schuylkill Yards project, for example, features a significant life sciences portion in University City.
“Other than fully leased build-to-suit opportunities, our future development starts are on hold,” Sweeney said, “pending more leasing on the existing joint venture pipeline and more clarity on the cost of debt capital and cap rates.”
Brandywine said about 70% to 75%of suburban tenants have returned to offices while that number has been around 50% in Philadelphia. At this point, though, it hasn’t yet affected demand when leasing space. Some tenants, for example, have moved out of the city while others have moved in.
In the fourth quarter, Brandywine had $55.7 million funds from operations, or 32 cents per share. That’s down from $60.4 million, or 35 cents per share, in the fourth quarter of 2021. Brandywine generated $129 million in revenue in the fourth quarter, up slightly from $125.5 in the year-ago period.
Brandywine stock is up 6.4% since the start of the year to $6.70 per share on Monday afternoon.
Many of Brandywine’s properties are in desirable locations, which have seen demand remain strong despite challenges facing offices, on par with industry trends.
Brandywine’s 12-story, 417,000-square-foot building at 3151 Market St. is on budget for $308 million and on schedule to be completed in the second quarter of 2024. Sweeney said Brandywine anticipates entering a construction loan in the second half of 2023, which would help complete the project. The building, being developed along with a global institutional investor,would be used for life sciences, innovation and office space as part of the larger Schuylkill Yards development in University City.
The company’s 29-story building at 3025 John F. Kennedy Blvd. with 200,000 square feet of life sciences space and 326 luxury apartments, is also on budget, costing $287.3 million, and on time, eyeing completion in the third quarter of this year.











 . The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,
. The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,  and
and  . The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……
. The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……














{kind=link}
{kind=link}
{kind=link}
Other posts on the JP Morgan 2019 Healthcare Conference on this Open Access Journal include:
#JPM19 Conference: Dublin HealthBeacon Raises Funding
#JPM19 Conference: Lilly Announces Agreement To Acquire Loxo Oncology
37th Annual J.P. Morgan HEALTHCARE CONFERENCE: #JPM2019 for Jan. 8, 2019; Opening Videos, Novartis expands Cell Therapies, January 7 – 10, 2019, Westin St. Francis Hotel | San Francisco, California
37th Annual J.P. Morgan HEALTHCARE CONFERENCE: News at #JPM2019 for Jan. 8, 2019: Deals and Announcements
37th Annual J.P. Morgan HEALTHCARE CONFERENCE: News at #JPM2019 for Jan. 10, 2019: Deals and Announcements