Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Bacterial multidrug resistance problem solved by a broad-spectrum synthetic antibiotic
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
There is an increasing demand for new antibiotics that effectively treat patients with refractory bacteremia, do not evoke bacterial resistance, and can be readily modified to address current and anticipated patient needs. Recently scientists described a promising compound of COE (conjugated oligo electrolytes) family, COE2-2hexyl, that exhibited broad-spectrum antibacterial activity. COE2-2hexyl effectively-treated mice infected with bacteria derived from sepsis patients with refractory bacteremia, including a CRE K. pneumoniae strain resistant to nearly all clinical antibiotics tested. Notably, this lead compound did not evoke drug resistance in several pathogens tested. COE2-2hexyl has specific effects on multiple membrane-associated functions (e.g., septation, motility, ATP synthesis, respiration, membrane permeability to small molecules) that may act together to abrogate bacterial cell viability and the evolution of drug-resistance. Impeding these bacterial properties may occur through alteration of vital protein–protein or protein-lipid membrane interfaces – a mechanism of action distinct from many membrane disrupting antimicrobials or detergents that destabilize membranes to induce bacterial cell lysis. The diversity and ease of COE design and chemical synthesis have the potential to establish a new standard for drug design and personalized antibiotic treatment.
Recent studies have shown that small molecules can preferentially target bacterial membranes due to significant differences in lipid composition, presence of a cell wall, and the absence of cholesterol. The inner membranes of Gram-negative bacteria are generally more negatively charged at their surface because they contain more anionic lipids such as cardiolipin and phosphatidylglycerol within their outer leaflet compared to mammalian membranes. In contrast, membranes of mammalian cells are largely composed of more-neutral phospholipids, sphingomyelins, as well as cholesterol, which affords membrane rigidity and ability to withstand mechanical stresses; and may stabilize the membrane against structural damage to membrane-disrupting agents such as COEs. Consistent with these studies, COE2-2hexyl was well tolerated in mice, suggesting that COEs are not intrinsically toxic in vivo, which is often a primary concern with membrane-targeting antibiotics. The COE refinement workflow potentially accelerates lead compound optimization by more rapid screening of novel compounds for the iterative directed-design process. It also reduces the time and cost of subsequent biophysical characterization, medicinal chemistry and bioassays, ultimately facilitating the discovery of novel compounds with improved pharmacological properties.
Additionally, COEs provide an approach to gain new insights into microbial physiology, including membrane structure/function and mechanism of drug action/resistance, while also generating a suite of tools that enable the modulation of bacterial and mammalian membranes for scientific or manufacturing uses. Notably, further COE safety and efficacy studies are required to be conducted on a larger scale to ensure adequate understanding of the clinical benefits and risks to assure clinical efficacy and toxicity before COEs can be added to the therapeutic armamentarium. Despite these limitations, the ease of molecular design, synthesis and modular nature of COEs offer many advantages over conventional antimicrobials, making synthesis simple, scalable and affordable. It enables the construction of a spectrum of compounds with the potential for development as a new versatile therapy for the emergence and rapid global spread of pathogens that are resistant to all, or nearly all, existing antimicrobial medicines.
From High-Throughput Assay to Systems Biology: New Tools for Drug Discovery
Curator: Stephen J. Williams, PhD
Marc W. Kirschner*
Department of Systems Biology Harvard Medical School
Boston, Massachusetts 02115
With the new excitement about systems biology, there is understandable interest in a definition. This has proven somewhat difficult. Scientific fields, like species, arise by descent with modification, so in their earliest forms even the founders of great dynasties are only marginally different than their sister fields and species. It is only in retrospect that we can recognize the significant founding events. Before embarking on a definition of systems biology, it may be worth remembering that confusion and controversy surrounded the introduction of the term “molecular biology,” with claims that it hardly differed from biochemistry. Yet in retrospect molecular biology was new and different. It introduced both new subject matter and new technological approaches, in addition to a new style.
As a point of departure for systems biology, consider the quintessential experiment in the founding of molecular biology, the one gene one enzyme hypothesis of Beadle and Tatum. This experiment first connected the genotype directly to the phenotype on a molecular level, although efforts in that direction can certainly be found in the work of Archibald Garrod, Sewell Wright, and others. Here a protein (in this case an enzyme) is seen to be a product of a single gene, and a single function; the completion of a specific step in amino acid biosynthesis is the direct result. It took the next 30 years to fill in the gaps in this process. Yet the one gene one enzyme hypothesis looks very different to us today. What is the function of tubulin, of PI-3 kinase or of rac? Could we accurately predict the phenotype of a nonlethal mutation in these genes in a multicellular organism? Although we can connect structure to the gene, we can no longer infer its larger purpose in the cell or in the organism. There are too many purposes; what the protein does is defined by context. The context also includes a history, either developmental or physiological. Thus the behavior of the Wnt signaling pathway depends on the previous lineage, the “where and when” questions of embryonic development. Similarly the behavior of the immune system depends on previous experience in a variable environment. All of these features stress how inadequate an explanation for function we can achieve solely by trying to identify genes (by annotating them!) and characterizing their transcriptional control circuits.
That we are at a crossroads in how to explore biology is not at all clear to many. Biology is hardly in its dotage; the process of discovery seems to have been perfected, accelerated, and made universally applicable to all fields of biology. With the completion of the human genome and the genomes of other species, we have a glimpse of many more genes than we ever had before to study. We are like naturalists discovering a new continent, enthralled with the diversity itself. But we have also at the same time glimpsed the finiteness of this list of genes, a disturbingly small list. We have seen that the diversity of genes cannot approximate the diversity of functions within an organism. In response, we have argued that combinatorial use of small numbers of components can generate all the diversity that is needed. This has had its recent incarnation in the simplistic view that the rules of cis-regulatory control on DNA can directly lead to an understanding of organisms and their evolution. Yet this assumes that the gene products can be linked together in arbitrary combinations, something that is not assured in chemistry. It also downplays the significant regulatory features that involve interactions between gene products, their localization, binding, posttranslational modification, degradation, etc. The big question to understand in biology is not regulatory linkage but the nature of biological systems that allows them to be linked together in many nonlethal and even useful combinations. More and more we come to realize that understanding the conserved genes and their conserved circuits will require an understanding of their special properties that allow them to function together to generate different phenotypes in different tissues of metazoan organisms. These circuits may have certain robustness, but more important they have adaptability and versatility. The ease of putting conserved processes under regulatory control is an inherent design feature of the processes themselves. Among other things it loads the deck in evolutionary variation and makes it more feasible to generate useful phenotypes upon which selection can act.
Systems biology offers an opportunity to study how the phenotype is generated from the genotype and with it a glimpse of how evolution has crafted the phenotype. One aspect of systems biology is the development of techniques to examine broadly the level of protein, RNA, and DNA on a gene by gene basis and even the posttranslational modification and localization of proteins. In a very short time we have witnessed the development of high-throughput biology, forcing us to consider cellular processes in toto. Even though much of the data is noisy and today partially inconsistent and incomplete, this has been a radical shift in the way we tear apart problems one interaction at a time. When coupled with gene deletions by RNAi and classical methods, and with the use of chemical tools tailored to proteins and protein domains, these high-throughput techniques become still more powerful.
High-throughput biology has opened up another important area of systems biology: it has brought us out into the field again or at least made us aware that there is a world outside our laboratories. Our model systems have been chosen intentionally to be of limited genetic diversity and examined in a highly controlled and reproducible environment. The real world of ecology, evolution, and human disease is a very different place. When genetics separated from the rest of biology in the early part of the 20th century, most geneticists sought to understand heredity and chose to study traits in the organism that could be easily scored and could be used to reveal genetic mechanisms. This was later extended to powerful effect to use genetics to study cell biological and developmental mechanisms. Some geneticists, including a large school in Russia in the early 20th century, continued to study the genetics of natural populations, focusing on traits important for survival. That branch of genetics is coming back strongly with the power of phenotypic assays on the RNA and protein level. As human beings we are most concerned not with using our genetic misfortunes to unravel biology’s complexity (important as that is) but with the role of our genetics in our individual survival. The context for understanding this is still not available, even though the data are now coming in torrents, for many of the genes that will contribute to our survival will have small quantitative effects, partially masked or accentuated by other genetic and environmental conditions. To understand the genetic basis of disease will require not just mapping these genes but an understanding of how the phenotype is created in the first place and the messy interactions between genetic variation and environmental variation.
Extracts and explants are relatively accessible to synthetic manipulation. Next there is the explicit reconstruction of circuits within cells or the deliberate modification of those circuits. This has occurred for a while in biology, but the difference is that now we wish to construct or intervene with the explicit purpose of describing the dynamical features of these synthetic or partially synthetic systems. There are more and more tools to intervene and more and more tools to measure. Although these fall short of total descriptions of cells and organisms, the detailed information will give us a sense of the special life-like processes of circuits, proteins, cells in tissues, and whole organisms in their environment. This meso-scale systems biology will help establish the correspondence between molecules and large-scale physiology.
You are probably running out of patience for some definition of systems biology. In any case, I do not think the explicit definition of systems biology should come from me but should await the words of the first great modern systems biologist. She or he is probably among us now. However, if forced to provide some kind of label for systems biology, I would simply say that systems biology is the study of the behavior of complex biological organization and processes in terms of the molecular constituents. It is built on molecular biology in its special concern for information transfer, on physiology for its special concern with adaptive states of the cell and organism, on developmental biology for the importance of defining a succession of physiological states in that process, and on evolutionary biology and ecology for the appreciation that all aspects of the organism are products of selection, a selection we rarely understand on a molecular level. Systems biology attempts all of this through quantitative measurement, modeling, reconstruction, and theory. Systems biology is not a branch of physics but differs from physics in that the primary task is to understand how biology generates variation. No such imperative to create variation exists in the physical world. It is a new principle that Darwin understood and upon which all of life hinges. That sounds different enough for me to justify a new field and a new name. Furthermore, the success of systems biology is essential if we are to understand life; its success is far from assured—a good field for those seeking risk and adventure.
Biologically active small molecules have a central role in drug development, and as chemical probes and tool compounds to perturb and elucidate biological processes. Small molecules can be rationally designed for a given target, or a library of molecules can be screened against a target or phenotype of interest. Especially in the case of phenotypic screening approaches, a major challenge is to translate the compound-induced phenotype into a well-defined cellular target and mode of action of the hit compound. There is no “one size fits all” approach, and recent years have seen an increase in available target deconvolution strategies, rooted in organic chemistry, proteomics, and genetics. This review provides an overview of advances in target identification and mechanism of action studies, describes the strengths and weaknesses of the different approaches, and illustrates the need for chemical biologists to integrate and expand the existing tools to increase the probability of evolving screen hits to robust chemical probes.
5.1.5. Large-Scale Proteomics
While FITExP is based on protein expression regulation during apoptosis, a study of Ruprecht et al. showed that proteomic changes are induced both by cytotoxic and non-cytotoxic compounds, which can be detected by mass spectrometry to give information on a compound’s mechanism of action. They developed a large-scale proteome-wide mass spectrometry analysis platform for MOA studies, profiling five lung cancer cell lines with over 50 drugs. Aggregation analysis over the different cell lines and the different compounds showed that one-quarter of the drugs changed the abundance of their protein target. This approach allowed target confirmation of molecular degraders such as PROTACs or molecular glues. Finally, this method yielded unexpected off-target mechanisms for the MAP2K1/2 inhibitor PD184352 and the ALK inhibitor ceritinib [97]. While such a mapping approach clearly provides a wealth of information, it might not be easily attainable for groups that are not equipped for high-throughput endeavors.
All-in-all, mass spectrometry methods have gained a lot of traction in recent years and have been successfully applied for target deconvolution and MOA studies of small molecules. As with all high-throughput methods, challenges lie in the accessibility of the instruments (both from a time and cost perspective) and data analysis of complex and extensive data sets.
5.2. Genetic Approaches
Both label-based and mass spectrometry proteomic approaches are based on the physical interaction between a small molecule and a protein target, and focus on the proteome for target deconvolution. It has been long realized that genetics provides an alternative avenue to understand a compound’s action, either through precise modification of protein levels, or by inducing protein mutations. First realized in yeast as a genetically tractable organism over 20 years ago, recent advances in genetic manipulation of mammalian cells have opened up important opportunities for target identification and MOA studies through genetic screening in relevant cell types [98]. Genetic approaches can be roughly divided into two main areas, with the first centering on the identification of mutations that confer compound resistance (Figure 3a), and the second on genome-wide perturbation of gene function and the concomitant changes in sensitivity to the compound (Figure 3b). While both methods can be used to identify or confirm drug targets, the latter category often provides many additional insights in the compound’s mode of action.
Figure 3. Genetic methods for target identification and mode of action studies. Schematic representations of (a) resistance cloning, and (b) chemogenetic interaction screens.
5.2.1. Resistance Cloning
The “gold standard” in drug target confirmation is to identify mutations in the presumed target protein that render it insensitive to drug treatment. Conversely, different groups have sought to use this principle as a target identification method based on the concept that cells grown in the presence of a cytotoxic drug will either die or develop mutations that will make them resistant to the compound. With recent advances in deep sequencing it is now possible to then scan the transcriptome [99] or genome [100] of the cells for resistance-inducing mutations. Genes that are mutated are then hypothesized to encode the protein target. For this approach to be successful, there are two initial requirements: (1) the compound needs to be cytotoxic for resistant clones to arise, and (2) the cell line needs to be genetically unstable for mutations to occur in a reasonable timeframe.
In 2012, the Kapoor group demonstrated in a proof-of-concept study that resistance cloning in mammalian cells, coupled to transcriptome sequencing (RNA-seq), yields the known polo-like kinase 1 (PLK1) target of the small molecule BI 2536. For this, they used the cancer cell line HCT-116, which is deficient in mismatch repair and consequently prone to mutations. They generated and sequenced multiple resistant clones, and clustered the clones based on similarity. PLK1 was the only gene that was mutated in multiple groups. Of note, one of the groups did not contain PLK1 mutations, but rather developed resistance through upregulation of ABCBA1, a drug efflux transporter, which is a general and non-specific resistance mechanism [101]. In a following study, they optimized their pipeline “DrugTargetSeqR”, by counter-screening for these types of multidrug resistance mechanisms so that these clones were excluded from further analysis (Figure 3a). Furthermore, they used CRISPR/Cas9-mediated gene editing to determine which mutations were sufficient to confer drug resistance, and as independent validation of the biochemical relevance of the obtained hits [102].
While HCT-116 cells are a useful model cell line for resistance cloning because of their genomic instability, they may not always be the cell line of choice, depending on the compound and process that is studied. Povedana et al. used CRISPR/Cas9 to engineer mismatch repair deficiencies in Ewing sarcoma cells and small cell lung cancer cells. They found that deletion of MSH2 results in hypermutations in these normally mutationally silent cells, resulting in the formation of resistant clones in the presence of bortezomib, MLN4924, and CD437, which are all cytotoxic compounds [103]. Recently, Neggers et al. reasoned that CRISPR/Cas9-induced non-homologous end-joining repair could be a viable strategy to create a wide variety of functional mutants of essential genes through in-frame mutations. Using a tiled sgRNA library targeting 75 target genes of investigational neoplastic drugs in HAP1 and K562 cells, they generated several KPT-9274 (an anticancer agent with unknown target)-resistant clones, and subsequent deep sequencing showed that the resistant clones were enriched in NAMPT sgRNAs. Direct target engagement was confirmed by co-crystallizing the compound with NAMPT [104]. In addition to these genetic mutation strategies, an alternative method is to grow the cells in the presence of a mutagenic chemical to induce higher mutagenesis rates [105,106].
When there is already a hypothesis on the pathway involved in compound action, the resistance cloning methodology can be extended to non-cytotoxic compounds. Sekine et al. developed a fluorescent reporter model for the integrated stress response, and used this cell line for target deconvolution of a small molecule inhibitor towards this pathway (ISRIB). Reporter cells were chemically mutagenized, and ISRIB-resistant clones were isolated by flow cytometry, yielding clones with various mutations in the delta subunit of guanine nucleotide exchange factor eIF2B [107].
While there are certainly successful examples of resistance cloning yielding a compound’s direct target as discussed above, resistance could also be caused by mutations or copy number alterations in downstream components of a signaling pathway. This is illustrated by clinical examples of acquired resistance to small molecules, nature’s way of “resistance cloning”. For example, resistance mechanisms in Hedgehog pathway-driven cancers towards the Smoothened inhibitor vismodegib include compound-resistant mutations in Smoothened, but also copy number changes in downstream activators SUFU and GLI2 [108]. It is, therefore, essential to conduct follow-up studies to confirm a direct interaction between a compound and the hit protein, as well as a lack of interaction with the mutated protein.
5.2.3. “Chemogenomics”: Examples of Gene-Drug Interaction Screens
When genetic perturbations are combined with small molecule drugs in a chemogenetic interaction screen, the effect of a gene’s perturbation on compound action is studied. Gene perturbation can render the cells resistant to the compound (suppressor interaction), or conversely, result in hypersensitivity and enhanced compound potency (synergistic interaction) [5,117,121]. Typically, cells are treated with the compound at a sublethal dose, to ascertain that both types of interactions can be found in the final dataset, and often it is necessary to use a variety of compound doses (i.e., LD20, LD30, LD50) and timepoints to obtain reliable insights (Figure 3b).
An early example of successful coupling of a phenotypic screen and downstream genetic screening for target identification is the study of Matheny et al. They identified STF-118804 as a compound with antileukemic properties. Treatment of MV411 cells, stably transduced with a high complexity, genome-wide shRNA library, with STF-118804 (4 rounds of increasing concentration) or DMSO control resulted in a marked depletion of cells containing shRNAs against nicotinamide phosphoribosyl transferase (NAMPT) [122].
The Bassik lab subsequently directly compared the performance of shRNA-mediated knockdown versus CRISPR/Cas9-knockout screens for the target elucidation of the antiviral drug GSK983. The data coming out of both screens were complementary, with the shRNA screen resulting in hits leading to the direct compound target and the CRISPR screen giving information on cellular mechanisms of action of the compound. A reason for this is likely the level of protein depletion that is reached by these methods: shRNAs lead to decreased protein levels, which is advantageous when studying essential genes. However, knockdown may not result in a phenotype for non-essential genes, in which case a full CRISPR-mediated knockout is necessary to observe effects [123].
Another NAMPT inhibitor was identified in a CRISPR/Cas9 “haplo-insufficiency (HIP)”-like approach [124]. Haploinsuffiency profiling is a well-established system in yeast which is performed in a ~50% protein background by heterozygous deletions [125]. As there is no control over CRISPR-mediated loss of alleles, compound treatment was performed at several timepoints after addition of the sgRNA library to HCT116 cells stably expressing Cas9, in the hope that editing would be incomplete at early timepoints, resulting in residual protein levels. Indeed, NAMPT was found to be the target of phenotypic hit LB-60-OF61, especially at earlier timepoints, confirming the hypothesis that some level of protein needs to be present to identify a compound’s direct target [124]. This approach was confirmed in another study, thereby showing that direct target identification through CRISPR-knockout screens is indeed possible [126].
An alternative strategy was employed by the Weissman lab, where they combined genome-wide CRISPR-interference and -activation screens to identify the target of the phase 3 drug rigosertib. They focused on hits that had opposite action in both screens, as in sensitizing in one but protective in the other, which were related to microtubule stability. In a next step, they created chemical-genetic profiles of a variety of microtubule destabilizing agents, rationalizing that compounds with the same target will have similar drug-gene interactions. For this, they made a focused library of sgRNAs, based on the most high-ranking hits in the rigosertib genome-wide CRISPRi screen, and compared the focused screen results of the different compounds. The profile for rigosertib clustered well with that of ABT-571, and rigorous target validation studies confirmed rigosertib binding to the colchicine binding site of tubulin—the same site as occupied by ABT-571 [127].
From the above examples, it is clear that genetic screens hold a lot of promise for target identification and MOA studies for small molecules. The CRISPR screening field is rapidly evolving, sgRNA libraries are continuously improving and increasingly commercially available, and new tools for data analysis are being developed [128]. The challenge lies in applying these screens to study compounds that are not cytotoxic, where finding the right dosage regimen will not be trivial.
SYSTEMS BIOLOGY AND CANCER RESEARCH & DRUG DISCOVERY
Integrative Analysis of Next-Generation Sequencing for Next-Generation Cancer Research toward Artificial Intelligence
The rapid improvement of next-generation sequencing (NGS) technologies and their application in large-scale cohorts in cancer research led to common challenges of big data. It opened a new research area incorporating systems biology and machine learning. As large-scale NGS data accumulated, sophisticated data analysis methods became indispensable. In addition, NGS data have been integrated with systems biology to build better predictive models to determine the characteristics of tumors and tumor subtypes. Therefore, various machine learning algorithms were introduced to identify underlying biological mechanisms. In this work, we review novel technologies developed for NGS data analysis, and we describe how these computational methodologies integrate systems biology and omics data. Subsequently, we discuss how deep neural networks outperform other approaches, the potential of graph neural networks (GNN) in systems biology, and the limitations in NGS biomedical research. To reflect on the various challenges and corresponding computational solutions, we will discuss the following three topics: (i) molecular characteristics, (ii) tumor heterogeneity, and (iii) drug discovery. We conclude that machine learning and network-based approaches can add valuable insights and build highly accurate models. However, a well-informed choice of learning algorithm and biological network information is crucial for the success of each specific research question
1. Introduction
The development and widespread use of high-throughput technologies founded the era of big data in biology and medicine. In particular, it led to an accumulation of large-scale data sets that opened a vast amount of possible applications for data-driven methodologies. In cancer, these applications range from fundamental research to clinical applications: molecular characteristics of tumors, tumor heterogeneity, drug discovery and potential treatments strategy. Therefore, data-driven bioinformatics research areas have tailored data mining technologies such as systems biology, machine learning, and deep learning, elaborated in this review paper (see Figure 1 and Figure 2). For example, in systems biology, data-driven approaches are applied to identify vital signaling pathways [1]. This pathway-centric analysis is particularly crucial in cancer research to understand the characteristics and heterogeneity of the tumor and tumor subtypes. Consequently, this high-throughput data-based analysis enables us to explore characteristics of cancers with a systems biology and a systems medicine point of view [2].Combining high-throughput techniques, especially next-generation sequencing (NGS), with appropriate analytical tools has allowed researchers to gain a deeper systematic understanding of cancer at various biological levels, most importantly genomics, transcriptomics, and epigenetics [3,4]. Furthermore, more sophisticated analysis tools based on computational modeling are introduced to decipher underlying molecular mechanisms in various cancer types. The increasing size and complexity of the data required the adaptation of bioinformatics processing pipelines for higher efficiency and sophisticated data mining methodologies, particularly for large-scale, NGS datasets [5]. Nowadays, more and more NGS studies integrate a systems biology approach and combine sequencing data with other types of information, for instance, protein family information, pathway, or protein–protein interaction (PPI) networks, in an integrative analysis. Experimentally validated knowledge in systems biology may enhance analysis models and guides them to uncover novel findings. Such integrated analyses have been useful to extract essential information from high-dimensional NGS data [6,7]. In order to deal with the increasing size and complexity, the application of machine learning, and specifically deep learning methodologies, have become state-of-the-art in NGS data analysis.
Figure 1. Next-generation sequencing data can originate from various experimental and technological conditions. Depending on the purpose of the experiment, one or more of the depicted omics types (Genomics, Transcriptomics, Epigenomics, or Single-Cell Omics) are analyzed. These approaches led to an accumulation of large-scale NGS datasets to solve various challenges of cancer research, molecular characterization, tumor heterogeneity, and drug target discovery. For instance, The Cancer Genome Atlas (TCGA) dataset contains multi-omics data from ten-thousands of patients. This dataset facilitates a variety of cancer researches for decades. Additionally, there are also independent tumor datasets, and, frequently, they are analyzed and compared with the TCGA dataset. As the large scale of omics data accumulated, various machine learning techniques are applied, e.g., graph algorithms and deep neural networks, for dimensionality reduction, clustering, or classification. (Created with BioRender.com.)
Figure 2. (a) A multitude of different types of data is produced by next-generation sequencing, for instance, in the fields of genomics, transcriptomics, and epigenomics. (b) Biological networks for biomarker validation: The in vivo or in vitro experiment results are considered ground truth. Statistical analysis on next-generation sequencing data produces candidate genes. Biological networks can validate these candidate genes and highlight the underlying biological mechanisms (Section 2.1). (c) De novo construction of Biological Networks: Machine learning models that aim to reconstruct biological networks can incorporate prior knowledge from different omics data. Subsequently, the model will predict new unknown interactions based on new omics information (Section 2.2). (d) Network-based machine learning: Machine learning models integrating biological networks as prior knowledge to improve predictive performance when applied to different NGS data (Section 2.3). (Created with BioRender.com).
Therefore, a large number of studies integrate NGS data with machine learning and propose a novel data-driven methodology in systems biology [8]. In particular, many network-based machine learning models have been developed to analyze cancer data and help to understand novel mechanisms in cancer development [9,10]. Moreover, deep neural networks (DNN) applied for large-scale data analysis improved the accuracy of computational models for mutation prediction [11,12], molecular subtyping [13,14], and drug repurposing [15,16].
2. Systems Biology in Cancer Research
Genes and their functions have been classified into gene sets based on experimental data. Our understandings of cancer concentrated into cancer hallmarks that define the characteristics of a tumor. This collective knowledge is used for the functional analysis of unseen data.. Furthermore, the regulatory relationships among genes were investigated, and, based on that, a pathway can be composed. In this manner, the accumulation of public high-throughput sequencing data raised many big-data challenges and opened new opportunities and areas of application for computer science. Two of the most vibrantly evolving areas are systems biology and machine learning which tackle different tasks such as understanding the cancer pathways [9], finding crucial genes in pathways [22,53], or predicting functions of unidentified or understudied genes [54]. Essentially, those models include prior knowledge to develop an analysis and enhance interpretability for high-dimensional data [2]. In addition to understanding cancer pathways with in silico analysis, pathway activity analysis incorporating two different types of data, pathways and omics data, is developed to understand heterogeneous characteristics of the tumor and cancer molecular subtyping. Due to its advantage in interpretability, various pathway-oriented methods are introduced and become a useful tool to understand a complex diseases such as cancer [55,56,57].
In this section, we will discuss how two related research fields, namely, systems biology and machine learning, can be integrated with three different approaches (see Figure 2), namely, biological network analysis for biomarker validation, the use of machine learning with systems biology, and network-based models.
2.1. Biological Network Analysis for Biomarker Validation
The detection of potential biomarkers indicative of specific cancer types or subtypes is a frequent goal of NGS data analysis in cancer research. For instance, a variety of bioinformatics tools and machine learning models aim at identify lists of genes that are significantly altered on a genomic, transcriptomic, or epigenomic level in cancer cells. Typically, statistical and machine learning methods are employed to find an optimal set of biomarkers, such as single nucleotide polymorphisms (SNPs), mutations, or differentially expressed genes crucial in cancer progression. Traditionally, resource-intensive in vitro analysis was required to discover or validate those markers. Therefore, systems biology offers in silico solutions to validate such findings using biological pathways or gene ontology information (Figure 2b) [58]. Subsequently, gene set enrichment analysis (GSEA) [50] or gene set analysis (GSA) [59] can be used to evaluate whether these lists of genes are significantly associated with cancer types and their specific characteristics. GSA, for instance, is available via web services like DAVID [60] and g:Profiler [61]. Moreover, other applications use gene ontology directly [62,63]. In addition to gene-set-based analysis, there are other methods that focuse on the topology of biological networks. These approaches evaluate various network structure parameters and analyze the connectivity of two genes or the size and interconnection of their neighbors [64,65]. According to the underlying idea, the mutated gene will show dysfunction and can affect its neighboring genes. Thus, the goal is to find abnormalities in a specific set of genes linked with an edge in a biological network. For instance, KeyPathwayMiner can extract informative network modules in various omics data [66]. In summary, these approaches aim at predicting the effect of dysfunctional genes among neighbors according to their connectivity or distances from specific genes such as hubs [67,68]. During the past few decades, the focus of cancer systems biology extended towards the analysis of cancer-related pathways since those pathways tend to carry more information than a gene set. Such analysis is called Pathway Enrichment Analysis (PEA) [69,70]. The use of PEA incorporates the topology of biological networks. However, simultaneously, the lack of coverage issue in pathway data needs to be considered. Because pathway data does not cover all known genes yet, an integration analysis on omics data can significantly drop in genes when incorporated with pathways. Genes that can not be mapped to any pathway are called ‘pathway orphan.’ In this manner, Rahmati et al. introduced a possible solution to overcome the ‘pathway orphan’ issue [71]. At the bottom line, regardless of whether researchers consider gene-set or pathway-based enrichment analysis, the performance and accuracy of both methods are highly dependent on the quality of the external gene-set and pathway data [72].
2.2. De Novo Construction of Biological Networks
While the known fraction of existing biological networks barely scratches the surface of the whole system of mechanisms occurring in each organism, machine learning models can improve on known network structures and can guide potential new findings [73,74]. This area of research is called de novo network construction (Figure 2c), and its predictive models can accelerate experimental validation by lowering time costs [75,76]. This interplay between in silico biological networks building and mining contributes to expanding our knowledge in a biological system. For instance, a gene co-expression network helps discover gene modules having similar functions [77]. Because gene co-expression networks are based on expressional changes under specific conditions, commonly, inferring a co-expression network requires many samples. The WGCNA package implements a representative model using weighted correlation for network construction that leads the development of the network biology field [78]. Due to NGS developments, the analysis of gene co-expression networks subsequently moved from microarray-based to RNA-seq based experimental data [79]. However, integration of these two types of data remains tricky. Ballouz et al. compared microarray and NGS-based co-expression networks and found the existence of a bias originating from batch effects between the two technologies [80]. Nevertheless, such approaches are suited to find disease-specific co-expressional gene modules. Thus, various studies based on the TCGA cancer co-expression network discovered characteristics of prognostic genes in the network [81]. Accordingly, a gene co-expression network is a condition-specific network rather than a general network for an organism. Gene regulatory networks can be inferred from the gene co-expression network when various data from different conditions in the same organism are available. Additionally, with various NGS applications, we can obtain multi-modal datasets about regulatory elements and their effects, such as epigenomic mechanisms on transcription and chromatin structure. Consequently, a gene regulatory network can consist of solely protein-coding genes or different regulatory node types such as transcription factors, inhibitors, promoter interactions, DNA methylations, and histone modifications affecting the gene expression system [82,83]. More recently, researchers were able to build networks based on a particular experimental setup. For instance, functional genomics or CRISPR technology enables the high-resolution regulatory networks in an organism [84]. Other than gene co-expression or regulatory networks, drug target, and drug repurposing studies are active research areas focusing on the de novo construction of drug-to-target networks to allow the potential repurposing of drugs [76,85].
2.3. Network Based Machine Learning
A network-based machine learning model directly integrates the insights of biological networks within the algorithm (Figure 2d) to ultimately improve predictive performance concerning cancer subtyping or susceptibility to therapy. Following the establishment of high-quality biological networks based on NGS technologies, these biological networks were suited to be integrated into advanced predictive models. In this manner, Zhang et al., categorized network-based machine learning approaches upon their usage into three groups: (i) model-based integration, (ii) pre-processing integration, and (iii) post-analysis integration [7]. Network-based models map the omics data onto a biological network, and proper algorithms travel the network while considering both values of nodes and edges and network topology. In the pre-processing integration, pathway or other network information is commonly processed based on its topological importance. Meanwhile, in the post-analysis integration, omics data is processed solely before integration with a network. Subsequently, omics data and networks are merged and interpreted. The network-based model has advantages in multi-omics integrative analysis. Due to the different sensitivity and coverage of various omics data types, a multi-omics integrative analysis is challenging. However, focusing on gene-level or protein-level information enables a straightforward integration [86,87]. Consequently, when different machine learning approaches tried to integrate two or more different data types to find novel biological insights, one of the solutions is reducing the search space to gene or protein level and integrated heterogeneous datatypes [25,88].
In summary, using network information opens new possibilities for interpretation. However, as mentioned earlier, several challenges remain, such as the coverage issue. Current databases for biological networks do not cover the entire set of genes, transcripts, and interactions. Therefore, the use of networks can lead to loss of information for gene or transcript orphans. The following section will focus on network-based machine learning models and their application in cancer genomics. We will put network-based machine learning into the perspective of the three main areas of application, namely, molecular characterization, tumor heterogeneity analysis, and cancer drug discovery.
3. Network-Based Learning in Cancer Research
As introduced previously, the integration of machine learning with the insights of biological networks (Figure 2d) ultimately aims at improving predictive performance and interpretability concerning cancer subtyping or treatment susceptibility.
3.1. Molecular Characterization with Network Information
Various network-based algorithms are used in genomics and focus on quantifying the impact of genomic alteration. By employing prior knowledge in biological network algorithms, performance compared to non-network models can be improved. A prominent example is HotNet. The algorithm uses a thermodynamics model on a biological network and identifies driver genes, or prognostic genes, in pan-cancer data [89]. Another study introduced a network-based stratification method to integrate somatic alterations and expression signatures with network information [90]. These approaches use network topology and network-propagation-like algorithms. Network propagation presumes that genomic alterations can affect the function of neighboring genes. Two genes will show an exclusive pattern if two genes complement each other, and the function carried by those two genes is essential to an organism [91]. This unique exclusive pattern among genomic alteration is further investigated in cancer-related pathways. Recently, Ku et al. developed network-centric approaches and tackled robustness issues while studying synthetic lethality [92]. Although synthetic lethality was initially discovered in model organisms of genetics, it helps us to understand cancer-specific mutations and their functions in tumor characteristics [91].
Furthermore, in transcriptome research, network information is used to measure pathway activity and its application in cancer subtyping. For instance, when comparing the data of two or more conditions such as cancer types, GSEA as introduced in Section 2 is a useful approach to get an overview of systematic changes [50]. It is typically used at the beginning of a data evaluation [93]. An experimentally validated gene set can provide information about how different conditions affect molecular systems in an organism. In addition to the gene sets, different approaches integrate complex interaction information into GSEA and build network-based models [70]. In contrast to GSEA, pathway activity analysis considers transcriptome data and other omics data and structural information of a biological network. For example, PARADIGM uses pathway topology and integrates various omics in the analysis to infer a patient-specific status of pathways [94]. A benchmark study with pan-cancer data recently reveals that using network structure can show better performance [57]. In conclusion, while the loss of data is due to the incompleteness of biological networks, their integration improved performance and increased interpretability in many cases.
3.2. Tumor Heterogeneity Study with Network Information
The tumor heterogeneity can originate from two directions, clonal heterogeneity and tumor impurity. Clonal heterogeneity covers genomic alterations within the tumor [95]. While de novo mutations accumulate, the tumor obtains genomic alterations with an exclusive pattern. When these genomic alterations are projected on the pathway, it is possible to observe exclusive relationships among disease-related genes. For instance, the CoMEt and MEMo algorithms examine mutual exclusivity on protein–protein interaction networks [96,97]. Moreover, the relationship between genes can be essential for an organism. Therefore, models analyzing such alterations integrate network-based analysis [98].
In contrast, tumor purity is dependent on the tumor microenvironment, including immune-cell infiltration and stromal cells [99]. In tumor microenvironment studies, network-based models are applied, for instance, to find immune-related gene modules. Although the importance of the interaction between tumors and immune cells is well known, detailed mechanisms are still unclear. Thus, many recent NGS studies employ network-based models to investigate the underlying mechanism in tumor and immune reactions. For example, McGrail et al. identified a relationship between the DNA damage response protein and immune cell infiltration in cancer. The analysis is based on curated interaction pairs in a protein–protein interaction network [100]. Most recently, Darzi et al. discovered a prognostic gene module related to immune cell infiltration by using network-centric approaches [101]. Tu et al. presented a network-centric model for mining subnetworks of genes other than immune cell infiltration by considering tumor purity [102].
3.3. Drug Target Identification with Network Information
In drug target studies, network biology is integrated into pharmacology [103]. For instance, Yamanishi et al. developed novel computational methods to investigate the pharmacological space by integrating a drug-target protein network with genomics and chemical information. The proposed approaches investigated such drug-target network information to identify potential novel drug targets [104]. Since then, the field has continued to develop methods to study drug target and drug response integrating networks with chemical and multi-omic datasets. In a recent survey study by Chen et al., the authors compared 13 computational methods for drug response prediction. It turned out that gene expression profiles are crucial information for drug response prediction [105].
Moreover, drug-target studies are often extended to drug-repurposing studies. In cancer research, drug-repurposing studies aim to find novel interactions between non-cancer drugs and molecular features in cancer. Drug-repurposing (or repositioning) studies apply computational approaches and pathway-based models and aim at discovering potential new cancer drugs with a higher probability than de novo drug design [16,106]. Specifically, drug-repurposing studies can consider various areas of cancer research, such as tumor heterogeneity and synthetic lethality. As an example, Lee et al. found clinically relevant synthetic lethality interactions by integrating multiple screening NGS datasets [107]. This synthetic lethality and related-drug datasets can be integrated for an effective combination of anticancer therapeutic strategy with non-cancer drug repurposing.
4. Deep Learning in Cancer Research
DNN models develop rapidly and become more sophisticated. They have been frequently used in all areas of biomedical research. Initially, its development was facilitated by large-scale imaging and video data. While most data sets in the biomedical field would not typically be considered big data, the rapid data accumulation enabled by NGS made it suitable for the application of DNN models requiring a large amount of training data [108]. For instance, in 2019, Samiei et al. used TCGA-based large-scale cancer data as benchmark datasets for bioinformatics machine learning research such as Image-Net in the computer vision field [109]. Subsequently, large-scale public cancer data sets such as TCGA encouraged the wide usage of DNNs in the cancer domain [110]. Over the last decade, these state-of-the-art machine learning methods have been incorporated in many different biological questions [111].
In addition to public cancer databases such as TCGA, the genetic information of normal tissues is stored in well-curated databases such as GTEx [112] and 1000Genomes [113]. These databases are frequently used as control or baseline training data for deep learning [114]. Moreover, other non-curated large-scale data sources such as GEO (https://www.ncbi.nlm.nih.gov/geo/, accessed on 20 May 2021) can be leveraged to tackle critical aspects in cancer research. They store a large-scale of biological data produced under various experimental setups (Figure 1). Therefore, an integration of GEO data and other data requires careful preprocessing. Overall, an increasing amount of datasets facilitate the development of current deep learning in bioinformatics research [115].
4.1. Challenges for Deep Learning in Cancer Research
Many studies in biology and medicine used NGS and produced large amounts of data during the past few decades, moving the field to the big data era. Nevertheless, researchers still face a lack of data in particular when investigating rare diseases or disease states. Researchers have developed a manifold of potential solutions to overcome this lack of data challenges, such as imputation, augmentation, and transfer learning (Figure 3b). Data imputation aims at handling data sets with missing values [116]. It has been studied on various NGS omics data types to recover missing information [117]. It is known that gene expression levels can be altered by different regulatory elements, such as DNA-binding proteins, epigenomic modifications, and post-transcriptional modifications. Therefore, various models integrating such regulatory schemes have been introduced to impute missing omics data [118,119]. Some DNN-based models aim to predict gene expression changes based on genomics or epigenomics alteration. For instance, TDimpute aims at generating missing RNA-seq data by training a DNN on methylation data. They used TCGA and TARGET (https://ocg.cancer.gov/programs/target/data-matrix, accessed on 20 May 2021) data as proof of concept of the applicability of DNN for data imputation in a multi-omics integration study [120]. Because this integrative model can exploit information in different levels of regulatory mechanisms, it can build a more detailed model and achieve better performance than a model build on a single-omics dataset [117,121]. The generative adversarial network (GAN) is a DNN structure for generating simulated data that is different from the original data but shows the same characteristics [122]. GANs can impute missing omics data from other multi-omics sources. Recently, the GAN algorithm is getting more attention in single-cell transcriptomics because it has been recognized as a complementary technique to overcome the limitation of scRNA-seq [123]. In contrast to data imputation and generation, other machine learning approaches aim to cope with a limited dataset in different ways. Transfer learning or few-shot learning, for instance, aims to reduce the search space with similar but unrelated datasets and guide the model to solve a specific set of problems [124]. These approaches train models with data of similar characteristics and types but different data to the problem set. After pre-training the model, it can be fine-tuned with the dataset of interest [125,126]. Thus, researchers are trying to introduce few-shot learning models and meta-learning approaches to omics and translational medicine. For example, Select-ProtoNet applied the ProtoTypical Network [127] model to TCGA transcriptome data and classified patients into two groups according to their clinical status [128]. AffinityNet predicts kidney and uterus cancer subtypes with gene expression profiles [129].
Figure 3. (a) In various studies, NGS data transformed into different forms. The 2-D transformed form is for the convolution layer. Omics data is transformed into pathway level, GO enrichment score, or Functional spectra. (b) DNN application on different ways to handle lack of data. Imputation for missing data in multi-omics datasets. GAN for data imputation and in silico data simulation. Transfer learning pre-trained the model with other datasets and fine-tune. (c) Various types of information in biology. (d) Graph neural network examples. GCN is applied to aggregate neighbor information. (Created with BioRender.com).
4.2. Molecular Charactization with Network and DNN Model
DNNs have been applied in multiple areas of cancer research. For instance, a DNN model trained on TCGA cancer data can aid molecular characterization by identifying cancer driver genes. At the very early stage, Yuan et al. build DeepGene, a cancer-type classifier. They implemented data sparsity reduction methods and trained the DNN model with somatic point mutations [130]. Lyu et al. [131] and DeepGx [132] embedded a 1-D gene expression profile to a 2-D array by chromosome order to implement the convolution layer (Figure 3a). Other algorithms, such as the deepDriver, use k-nearest neighbors for the convolution layer. A predefined number of neighboring gene mutation profiles was the input for the convolution layer. It employed this convolution layer in a DNN by aggregating mutation information of the k-nearest neighboring genes [11]. Instead of embedding to a 2-D image, DeepCC transformed gene expression data into functional spectra. The resulting model was able to capture molecular characteristics by training cancer subtypes [14].
Another DNN model was trained to infer the origin of tissue from single-nucleotide variant (SNV) information of metastatic tumor. The authors built a model by using the TCGA/ICGC data and analyzed SNV patterns and corresponding pathways to predict the origin of cancer. They discovered that metastatic tumors retained their original cancer’s signature mutation pattern. In this context, their DNN model obtained even better accuracy than a random forest model [133] and, even more important, better accuracy than human pathologists [12].
4.3. Tumor Heterogeneity with Network and DNN Model
As described in Section 4.1, there are several issues because of cancer heterogeneity, e.g., tumor microenvironment. Thus, there are only a few applications of DNN in intratumoral heterogeneity research. For instance, Menden et al. developed ’Scaden’ to deconvolve cell types in bulk-cell sequencing data. ’Scaden’ is a DNN model for the investigation of intratumor heterogeneity. To overcome the lack of training datasets, researchers need to generate in silico simulated bulk-cell sequencing data based on single-cell sequencing data [134]. It is presumed that deconvolving cell types can be achieved by knowing all possible expressional profiles of the cell [36]. However, this information is typically not available. Recently, to tackle this problem, single-cell sequencing-based studies were conducted. Because of technical limitations, we need to handle lots of missing data, noises, and batch effects in single-cell sequencing data [135]. Thus, various machine learning methods were developed to process single-cell sequencing data. They aim at mapping single-cell data onto the latent space. For example, scDeepCluster implemented an autoencoder and trained it on gene-expression levels from single-cell sequencing. During the training phase, the encoder and decoder work as denoiser. At the same time, they can embed high-dimensional gene-expression profiles to lower-dimensional vectors [136]. This autoencoder-based method can produce biologically meaningful feature vectors in various contexts, from tissue cell types [137] to different cancer types [138,139].
4.4. Drug Target Identification with Networks and DNN Models
In addition to NGS datasets, large-scale anticancer drug assays enabled the training train of DNNs. Moreover, non-cancer drug response assay datasets can also be incorporated with cancer genomic data. In cancer research, a multidisciplinary approach was widely applied for repurposing non-oncology drugs to cancer treatment. This drug repurposing is faster than de novo drug discovery. Furthermore, combination therapy with a non-oncology drug can be beneficial to overcome the heterogeneous properties of tumors [85]. The deepDR algorithm integrated ten drug-related networks and trained deep autoencoders. It used a random-walk-based algorithm to represent graph information into feature vectors. This approach integrated network analysis with a DNN model validated with an independent drug-disease dataset [15].
The authors of CDRscan did an integrative analysis of cell-line-based assay datasets and other drug and genomics datasets. It shows that DNN models can enhance the computational model for improved drug sensitivity predictions [140]. Additionally, similar to previous network-based models, the multi-omics application of drug-targeted DNN studies can show higher prediction accuracy than the single-omics method. MOLI integrated genomic data and transcriptomic data to predict the drug responses of TCGA patients [141].
4.5. Graph Neural Network Model
In general, the advantage of using a biological network is that it can produce more comprehensive and interpretable results from high-dimensional omics data. Furthermore, in an integrative multi-omics data analysis, network-based integration can improve interpretability over traditional approaches. Instead of pre-/post-integration of a network, recently developed graph neural networks use biological networks as the base structure for the learning network itself. For instance, various pathways or interactome information can be integrated as a learning structure of a DNN and can be aggregated as heterogeneous information. In a GNN study, a convolution process can be done on the provided network structure of data. Therefore, the convolution on a biological network made it possible for the GNN to focus on the relationship among neighbor genes. In the graph convolution layer, the convolution process integrates information of neighbor genes and learns topological information (Figure 3d). Consequently, this model can aggregate information from far-distant neighbors, and thus can outperform other machine learning models [142].
In the context of the inference problem of gene expression, the main question is whether the gene expression level can be explained by aggregating the neighboring genes. A single gene inference study by Dutil et al. showed that the GNN model outperformed other DNN models [143]. Moreover, in cancer research, such GNN models can identify cancer-related genes with better performance than other network-based models, such as HotNet2 and MutSigCV [144]. A recent GNN study with a multi-omics integrative analysis identified 165 new cancer genes as an interactive partner for known cancer genes [145]. Additionally, in the synthetic lethality area, dual-dropout GNN outperformed previous bioinformatics tools for predicting synthetic lethality in tumors [146]. GNNs were also able to classify cancer subtypes based on pathway activity measures with RNA-seq data. Lee et al. implemented a GNN for cancer subtyping and tested five cancer types. Thus, the informative pathway was selected and used for subtype classification [147]. Furthermore, GNNs are also getting more attention in drug repositioning studies. As described in Section 3.3, drug discovery requires integrating various networks in both chemical and genomic spaces (Figure 3d). Chemical structures, protein structures, pathways, and other multi-omics data were used in drug-target identification and repurposing studies (Figure 3c). Each of the proposed applications has a specialty in the different purposes of drug-related tasks. Sun et al. summarized GNN-based drug discovery studies and categorized them into four classes: molecular property and activity prediction, interaction prediction, synthesis prediction, and de novo drug design. The authors also point out four challenges in the GNN-mediated drug discovery. At first, as we described before, there is a lack of drug-related datasets. Secondly, the current GNN models can not fully represent 3-D structures of chemical molecules and protein structures. The third challenge is integrating heterogeneous network information. Drug discovery usually requires a multi-modal integrative analysis with various networks, and GNNs can improve this integrative analysis. Lastly, although GNNs use graphs, stacked layers still make it hard to interpret the model [148].
4.6. Shortcomings in AI and Revisiting Validity of Biological Networks as Prior Knowledge
The previous sections reviewed a variety of DNN-based approaches that present a good performance on numerous applications. However, it is hardly a panacea for all research questions. In the following, we will discuss potential limitations of the DNN models. In general, DNN models with NGS data have two significant issues: (i) data requirements and (ii) interpretability. Usually, deep learning needs a large proportion of training data for reasonable performance which is more difficult to achieve in biomedical omics data compared to, for instance, image data. Today, there are not many NGS datasets that are well-curated and -annotated for deep learning. This can be an answer to the question of why most DNN studies are in cancer research [110,149]. Moreover, the deep learning models are hard to interpret and are typically considered as black-boxes. Highly stacked layers in the deep learning model make it hard to interpret its decision-making rationale. Although the methodology to understand and interpret deep learning models has been improved, the ambiguity in the DNN models’ decision-making hindered the transition between the deep learning model and translational medicine [149,150].
As described before, biological networks are employed in various computational analyses for cancer research. The studies applying DNNs demonstrated many different approaches to use prior knowledge for systematic analyses. Before discussing GNN application, the validity of biological networks in a DNN model needs to be shown. The LINCS program analyzed data of ’The Connectivity Map (CMap) project’ to understand the regulatory mechanism in gene expression by inferring the whole gene expression profiles from a small set of genes (https://lincsproject.org/, accessed on 20 May 2021) [151,152]. This LINCS program found that the gene expression level is inferrable with only nearly 1000 genes. They called this gene list ’landmark genes’. Subsequently, Chen et al. started with these 978 landmark genes and tried to predict other gene expression levels with DNN models. Integrating public large-scale NGS data showed better performance than the linear regression model. The authors conclude that the performance advantage originates from the DNN’s ability to model non-linear relationships between genes [153].
Following this study, Beltin et al. extensively investigated various biological networks in the same context of the inference of gene expression level. They set up a simplified representation of gene expression status and tried to solve a binary classification task. To show the relevance of a biological network, they compared various gene expression levels inferred from a different set of genes, neighboring genes in PPI, random genes, and all genes. However, in the study incorporating TCGA and GTEx datasets, the random network model outperformed the model build on a known biological network, such as StringDB [154]. While network-based approaches can add valuable insights to analysis, this study shows that it cannot be seen as the panacea, and a careful evaluation is required for each data set and task. In particular, this result may not represent biological complexity because of the oversimplified problem setup, which did not consider the relative gene-expressional changes. Additionally, the incorporated biological networks may not be suitable for inferring gene expression profiles because they consist of expression-regulating interactions, non-expression-regulating interactions, and various in vivo and in vitro interactions.
“ However, although recently sophisticated applications of deep learning showed improved accuracy, it does not reflect a general advancement. Depending on the type of NGS data, the experimental design, and the question to be answered, a proper approach and specific deep learning algorithms need to be considered. Deep learning is not a panacea. In general, to employ machine learning and systems biology methodology for a specific type of NGS data, a certain experimental design, a particular research question, the technology, and network data have to be chosen carefully.”
Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
Hutter, C.; Zenklusen, J.C. The cancer genome atlas: Creating lasting value beyond its data. Cell2018, 173, 283–285. [Google Scholar] [CrossRef]
Chuang, H.Y.; Lee, E.; Liu, Y.T.; Lee, D.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol.2007, 3, 140. [Google Scholar] [CrossRef]
Zhang, W.; Chien, J.; Yong, J.; Kuang, R. Network-based machine learning and graph theory algorithms for precision oncology. NPJ Precis. Oncol.2017, 1, 25. [Google Scholar] [CrossRef] [PubMed]
Ngiam, K.Y.; Khor, W. Big data and machine learning algorithms for health-care delivery. Lancet Oncol.2019, 20, e262–e273. [Google Scholar] [CrossRef]
Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; Sander, C.; et al. Pathway and network analysis of cancer genomes. Nat. Methods2015, 12, 615. [Google Scholar]
Reyna, M.A.; Haan, D.; Paczkowska, M.; Verbeke, L.P.; Vazquez, M.; Kahraman, A.; Pulido-Tamayo, S.; Barenboim, J.; Wadi, L.; Dhingra, P.; et al. Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun.2020, 11, 729. [Google Scholar] [CrossRef]
Luo, P.; Ding, Y.; Lei, X.; Wu, F.X. deepDriver: Predicting cancer driver genes based on somatic mutations using deep convolutional neural networks. Front. Genet.2019, 10, 13. [Google Scholar] [CrossRef]
Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; Danyi, A.; De Ridder, J.; van Herpen, C.; Lolkema, M.P.; Steeghs, N.; et al. A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns. Nat. Commun.2020, 11, 728. [Google Scholar] [CrossRef]
Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res.2018, 24, 1248–1259. [Google Scholar] [CrossRef]
Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis2019, 8, 44. [Google Scholar] [CrossRef]
Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics2019, 35, 5191–5198. [Google Scholar] [CrossRef]
Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature2020, 578, 82. [Google Scholar] [CrossRef] [PubMed]
King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol.2010, 28, 1075. [Google Scholar] [CrossRef] [PubMed]
Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol.2009, 27, 1160. [Google Scholar] [CrossRef]
Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol.2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
Zhao, L.; Lee, V.H.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Brief. Bioinform.2019, 20, 572–584. [Google Scholar] [CrossRef] [PubMed]
Jones, P.A.; Issa, J.P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet.2016, 17, 630. [Google Scholar] [CrossRef] [PubMed]
Huang, S.; Chaudhary, K.; Garmire, L.X. More is better: Recent progress in multi-omics data integration methods. Front. Genet.2017, 8, 84. [Google Scholar] [CrossRef]
Chin, L.; Andersen, J.N.; Futreal, P.A. Cancer genomics: From discovery science to personalized medicine. Nat. Med.2011, 17, 297. [Google Scholar] [CrossRef] [PubMed]
Use of Systems Biology in Anti-Microbial Drug Development
Genomics, Computational Biology and Drug Discovery for Mycobacterial Infections: Fighting the Emergence of Resistance. Asma Munir, Sundeep Chaitanya Vedithi, Amanda K. Chaplin and Tom L. Blundell. Front. Genet., 04 September 2020 | https://doi.org/10.3389/fgene.2020.00965
In an earlier review article (Waman et al., 2019), we discussed various computational approaches and experimental strategies for drug target identification and structure-guided drug discovery. In this review we discuss the impact of the era of precision medicine, where the genome sequences of pathogens can give clues about the choice of existing drugs, and repurposing of others. Our focus is directed toward combatting antimicrobial drug resistance with emphasis on tuberculosis and leprosy. We describe structure-guided approaches to understanding the impacts of mutations that give rise to antimycobacterial resistance and the use of this information in the design of new medicines.
Genome Sequences and Proteomic Structural Databases
In recent years, there have been many focused efforts to define the amino-acid sequences of the M. tuberculosis pan-genome and then to define the three-dimensional structures and functional interactions of these gene products. This work has led to essential genes of the bacteria being revealed and to a better understanding of the genetic diversity in different strains that might lead to a selective advantage (Coll et al., 2018). This will help with our understanding of the mode of antibiotic resistance within these strains and aid structure-guided drug discovery. However, only ∼10% of the ∼4128 proteins have structures determined experimentally.
Several databases have been developed to integrate the genomic and/or structural information linked to drug resistance in Mycobacteria (Table 1). These invaluable resources can contribute to better understanding of molecular mechanisms involved in drug resistance and improvement in the selection of potential drug targets.
There is a dearth of information related to structural aspects of proteins from M. leprae and their oligomeric and hetero-oligomeric organization, which has limited the understanding of physiological processes of the bacillus. The structures of only 12 proteins have been solved and deposited in the protein data bank (PDB). However, the high sequence similarity in protein coding genes between M. leprae and M. tuberculosis allows computational methods to be used for comparative modeling of the proteins of M. leprae. Mainly monomeric models using single template modeling have been defined and deposited in the Swiss Model repository (Bienert et al., 2017), in Modbase (Pieper et al., 2014), and in a collection with other infectious disease agents (Sosa et al., 2018). There is a need for multi-template modeling and building homo- and hetero-oligomeric complexes to better understand the interfaces, druggability and impacts of mutations.
We are now exploiting Vivace, a multi-template modeling pipeline developed in our lab for modeling the proteomes of M. tuberculosis (CHOPIN, see above) and M. abscessus [Mabellini Database (Skwark et al., 2019)], to model the proteome of M. leprae. We emphasize the need for understanding the protein interfaces that are critical to function. An example of this is that of the RNA-polymerase holoenzyme complex from M. leprae. We first modeled the structure of this hetero-hexamer complex and later deciphered the binding patterns of rifampin (Vedithi et al., 2018; Figures 1A,B). Rifampin is a known drug to treat tuberculosis and leprosy. Owing to high rifampin resistance in tuberculosis and emerging resistance in leprosy, we used an approach known as “Computational Saturation Mutagenesis”, to identify sites on the protein that are less impacted by mutations. In this study, we were able to understand the association between predicted impacts of mutations on the structure and phenotypic rifampin-resistance outcomes in leprosy.
FIGURE 2
Figure 2.(A) Stability changes predicted by mCSM for systematic mutations in the ß-subunit of RNA polymerase in M. leprae. The maximum destabilizing effect from among all 19 possible mutations at each residue position is considered as a weighting factor for the color map that gradients from red (high destabilizing effects) to white (neutral to stabilizing effects) (Vedithi et al., 2020). (B) One of the known mutations in the ß-subunit of RNA polymerase, the S437H substitution which resulted in a maximum destabilizing effect [-1.701 kcal/mol (mCSM)] among all 19 possibilities this position. In the mutant, histidine (residue in green) forms hydrogen bonds with S434 and Q438, aromatic interactions with F431, and other ring-ring and π interactions with the surrounding residues which can impact the shape of the rifampin binding pocket and rifampin affinity to the ß-subunit [-0.826 log(affinity fold change) (mCSM-lig)]. Orange dotted lines represent weak hydrogen bond interactions. Ring-ring and intergroup interactions are depicted in cyan. Aromatic interactions are represented in sky-blue and carbonyl interactions in pink dotted lines. Green dotted lines represent hydrophobic interactions (Vedithi et al., 2020).
Examples of Understanding and Combatting Resistance
The availability of whole genome sequences in the present era has greatly enhanced the understanding of emergence of drug resistance in infectious diseases like tuberculosis. The data generated by the whole genome sequencing of clinical isolates can be screened for the presence of drug-resistant mutations. A preliminary in silico analysis of mutations can then be used to prioritize experimental work to identify the nature of these mutations.
FIGURE 3
Figure 3.(A) Mechanism of isoniazid activation and INH-NAD adduct formation. (B) Mutations mapped (Munir et al., 2019) on the structure of KatG (PDB ID:1SJ2; Bertrand et al., 2004).
Other articles related to Computational Biology, Systems Biology, and Bioinformatics on this online journal include:
3.3.11 Unlocking the Microbiome, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Machine-learning technique uncovers unknown features of multi-drug-resistant pathogen
Relatively simple “unsupervised” learning system reveals important new information to microbiologists
According to the CDC, Pseudomonas aeruginosa is a common cause of healthcare-associated infections, including pneumonia, bloodstream infections, urinary tract infections, and surgical site infections. Some strains of P. aeruginosa have been found to be resistant to nearly all or all antibiotics. (illustration credit: CDC)
A new machine-learning technique can uncover previously unknown features of organisms and their genes in large datasets, according to researchers from the Perelman School of Medicine at the University of Pennsylvania and the Geisel School of Medicine at Dartmouth University.
For example, the technique learned to identify the characteristic gene-expression patterns that appear when a bacterium is exposed in different conditions, such as low oxygen and the presence of antibiotics.
The technique, called “ADAGE” (Analysis using Denoising Autoencoders of Gene Expression), uses a “denoising autoencoder” algorithm, which learns to identify recurring features or patterns in large datasets — without being told what specific features to look for (that is, “unsupervised.”)*
Last year, Casey Greene, PhD, an assistant professor of Systems Pharmacology and Translational Therapeutics at Penn, and his team published, in an open-access paper in the American Society for Microbiology’s mSystems, the first demonstration of ADAGE in a biological context: an analysis of two gene-expression datasets of breast cancers.
Tracking down gene patterns of a multi-drug-resistant bacterium
The new study, published Jan. 19 in an open-access paper in mSystems, was more ambitious. It applied ADAGE to a dataset of 950 gene-expression arrays publicly available at the time for the multi-drug-resistant bacteriumPseudomonas aeruginosa. This bacterium is a notorious pathogen in the hospital and in individuals with cystic fibrosis and other chronic lung conditions; it’s often difficult to treat due to its high resistance to standard antibiotic therapies.
The data included only the identities of the roughly 5,000 P. aeruginosa genes and their measured expression levels in each published experiment. The goal was to see if this “unsupervised” learning system could uncover important patterns in P. aeruginosa gene expression and clarify how those patterns change when the bacterium’s environment changes — for example, when in the presence of an antibiotic.
Even though the model built with ADAGE was relatively simple — roughly equivalent to a brain with only a few dozen neurons — it had no trouble learning which sets of P. aeruginosa genes tend to work together or in opposition. To the researchers’ surprise, the ADAGE system also detected differences between the main laboratory strain of P. aeruginosa and strains isolated from infected patients. “That turned out to be one of the strongest features of the data,” Greene said.
“We expect that this approach will be particularly useful to microbiologists researching bacterial species that lack a decades-long history of study in the lab,” said Greene. “Microbiologists can use these models to identify where the data agree with their own knowledge and where the data seem to be pointing in a different direction … and to find completely new things in biology that we didn’t even know to look for.”
Support for the research came from the Gordon and Betty Moore Foundation, the William H. Neukom Institute for Computational Science, the National Institutes of Health, and the Cystic Fibrosis Foundation.
* In 2012, Google-sponsored researchers applied a similar method to randomly selected YouTube images; their system learned to recognize major recurring features of those images — including cats of course.
Abstract of ADAGE-Based Integration of Publicly Available Pseudomonas aeruginosa Gene Expression Data with Denoising Autoencoders Illuminates Microbe-Host Interactions
The increasing number of genome-wide assays of gene expression available from public databases presents opportunities for computational methods that facilitate hypothesis generation and biological interpretation of these data. We present an unsupervised machine learning approach, ADAGE (analysis using denoising autoencoders of gene expression), and apply it to the publicly available gene expression data compendium for Pseudomonas aeruginosa. In this approach, the machine-learned ADAGE model contained 50 nodes which we predicted would correspond to gene expression patterns across the gene expression compendium. While no biological knowledge was used during model construction, cooperonic genes had similar weights across nodes, and genes with similar weights across nodes were significantly more likely to share KEGG pathways. By analyzing newly generated and previously published microarray and transcriptome sequencing data, the ADAGE model identified differences between strains, modeled the cellular response to low oxygen, and predicted the involvement of biological processes based on low-level gene expression differences. ADAGE compared favorably with traditional principal component analysis and independent component analysis approaches in its ability to extract validated patterns, and based on our analyses, we propose that these approaches differ in the types of patterns they preferentially identify. We provide the ADAGE model with analysis of all publicly available P. aeruginosa GeneChip experiments and open source code for use with other species and settings. Extraction of consistent patterns across large-scale collections of genomic data using methods like ADAGE provides the opportunity to identify general principles and biologically important patterns in microbial biology. This approach will be particularly useful in less-well-studied microbial species.
Abstract of Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising autoencoders
Big data bring new opportunities for methods that efficiently summarize and automatically extract knowledge from such compendia. While both supervised learning algorithms and unsupervised clustering algorithms have been successfully applied to biological data, they are either dependent on known biology or limited to discerning the most significant signals in the data. Here we present denoising autoencoders (DAs), which employ a data-defined learning objective independent of known biology, as a method to identify and extract complex patterns from genomic data. We evaluate the performance of DAs by applying them to a large collection of breast cancer gene expression data. Results show that DAs successfully construct features that contain both clinical and molecular information. There are features that represent tumor or normal samples, estrogen receptor (ER) status, and molecular subtypes. Features constructed by the autoencoder generalize to an independent dataset collected using a distinct experimental platform. By integrating data from ENCODE for feature interpretation, we discover a feature representing ER status through association with key transcription factors in breast cancer. We also identify a feature highly predictive of patient survival and it is enriched by FOXM1 signaling pathway. The features constructed by DAs are often bimodally distributed with one peak near zero and another near one, which facilitates discretization. In summary, we demonstrate that DAs effectively extract key biological principles from gene expression data and summarize them into constructed features with convenient properties.
NIH Funds Nine Anti-Microbial Resistance Diagnostic Projects to Deal with ‘Super Bugs’ and Give Clinical Laboratories New Diagnostic Tools to Improve Patient Care
Lab-on-a-chip technology could reduce the time needed to identify infection-causing bacteria and for physicians to prescribe correct antibiotics
Pathology groups and medical laboratories may see their role in the patient-care process grow if researchers succeed in developing culture-independent diagnostic tools that quickly identify bacterial infections as well as pinpoint the antibiotics needed to treat them.
In the battle against antibiotic-resistant infections (AKA “super bugs”) the National Institutes of Health (NIH) is funding nine research projects aimed at thwarting the growing problem of life-threatening infections that no longer are controlled or killed by today’s arsenal of drugs.
Common Practices in Hospitals Leading to Super Bugs
Currently, when infections are suspected in hospitals or other settings where illness can quickly spread, samples are sent to a central medical laboratory where it may take up to three days to determine what germ is causing the infection. Because of that delay, physicians often prescribe broad-spectrum antibiotics based on a patient’s symptoms rather than lab test results, a practice that can lead to the growth of antibiotic-resistant microbes.
“Antimicrobial resistance is a serious global health threat that is undermining our ability to effectively detect, treat, and prevent infections,” said National Institute of Allergy and Infectious Diseases (NIAID) Director Anthony S. Fauci, MD, in a news release. “One way we can combat drug resistance is by developing enhanced diagnostic tests that rapidly identify the bacteria causing an infection and their susceptibility to various antimicrobials. This will help physicians determine the most effective treatments for infected individuals and thereby reduce the use of broad-spectrum antibiotics that can contribute to the drug resistance problem.”
The Centers for Disease Control and Prevention (CDC) estimates that preventing infections and improving antibiotic prescribing could save 37,000 lives from drug-resistant infections over five years.
As Director of the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH), Anthony S. Fauci, MD, (above) leads research to prevent, diagnose, and treat infectious diseases, such as HIV/AIDS, influenza, tuberculosis, malaria, and illness from potential agents of bioterrorism. He serves as one of the key advisors to the White House and U.S. Department of Health and Human Services (HHS) on global AIDS issues. (Photo and caption copyright: NIH Medline Plus.)
Antibiotic resistance has become a major challenge of our time. Common microorganisms that inhabit the skin, mouth and nares, and fecal organisms are transmitted in the hospital setting. Handwashing procedures have had limited benefit. Operating rooms are ventilated and environmentally engineered to minimize transmission intraoperatively. The patient may be immune-compromized. The organisms that are encountered have genetically adapted to the most effective antibiotics at our disposal. even with some risk of secondary toxicity in some cases.
What is Drug Resistance?
Antimicrobial resistance is the ability of microbes, such as bacteria, viruses,
parasites, or fungi, to grow in the presence of a chemical (drug) that would
normally kill it or limit its growth.
Diagram showing the difference between non-resistant bacteria and drug
resistant bacteria.
Drug Resistance difference between non-resistant bacteria and drug resistant bacteria
Diagram showing the difference between non-resistant bacteria and drug
resistant bacteria. Non-resistant bacteria multiply, and upon drug treatment,
the bacteria die. Drug resistant bacteria multiply as well, but upon drug
treatment, the bacteria continue to spread.
Many infectious diseases are increasingly difficult to treat because of
antimicrobial-resistant organisms, including HIV infection, staphylococcal
infection, tuberculosis, influenza, gonorrhea, candida infection, and malaria.
Between 5 and 10 percent of all hospital patients develop an infection. About
90,000 of these patients die each year as a result of their infection, up from
13,300 patient deaths in 1992.
According to the Centers for Disease Control and Prevention (April 2011),
antibiotic resistance in the United States costs an estimated $20 billion a year
in excess health care costs, $35 million in other societal costs and more than 8
million additional days that people spend in the hospital.
Antimicrobial resistance (AMR) is an increasingly serious threat to
global public health. AMR develops when a microorganism (bacteria,
fungus, virus or parasite) no longer responds to a drug to which it
was originally sensitive. This means that standard treatments no
longer work; infections are harder or impossible to control; the risk
of the spread of infection to others is increased; illness and hospital
stays are prolonged, with added economic and social costs; and the
risk of death is greater—in some cases, twice that of patients who
have infections caused by non-resistant bacteria. The problem is so
serious that it threatens the achievements of modern medicine. A
post-antibiotic era—in which common infections and minor
injuries can kill—is a very real possibility for the 21st century.
WHO is developing a global action plan for AMR that
will include:
• development of tools and standards for harmonized
surveillance of ABR in humans, and for integrated
surveillance in food-producing animals and the
food chain;
• elaboration of strategies for population-based
surveillance of AMR and its health and economic
impact; and
• collaboration between AMR surveillance networks
and centres to create or strengthen coordinated
regional and global surveillance.
AMR is a global health security threat that requires
action across government sectors and society as a
whole. Surveillance that generates reliable data is the
essential foundation of global strategies and public
health actions to contain AMR.
Serious infections caused by bacteria that have become resistant
to commonly used antibiotics have become a major global healthcare
problem in the 21st century. They not only are more severe and
require longer and more complex treatments, but they are also
significantly more expensive to diagnose and to treat. Antibiotic
resistance, initially a problem of the hospital setting associated
with an increased number of hospital acquired infections usually
in critically ill and immunosuppressed patients, has now extended
into the community causing severe infections difficult to diagnose
and treat. The molecular mechanisms by which bacteria have
become resistant to antibiotics are diverse and complex. Bacteria
have developed resistance to all different classes of antibiotics
discovered to date. The most frequent type of resistance is
acquired and transmitted horizontally via the conjugation
of a plasmid. In recent times new mechanisms of resistance
have resulted in the simultaneous development of resistance
to several antibiotic classes creating very dangerous multidrug
-resistant (MDR) bacterial strains, some also known as
‘‘superbugs’’. The indiscriminate and inappropriate use of
antibiotics in outpatient clinics, hospitalized patients and
in the food industry is the single largest factor leading to
antibiotic resistance. In recent years, the number of new
antibiotics licensed for human use in different parts of the
world has been lower than in the recent past. In addition,
there has been less innovation in the field of antimicrobial
discovery research and development. The pharmaceutical
industry, large academic institutions or the government are
not investing the necessary resources to produce the next
generation of newer safe and effective antimicrobial drugs.
In many cases, large pharmaceutical companies have terminated
their anti-infective research programs altogether due to economic
reasons. The potential negative consequences of all these events
are relevant because they put society at risk for the spread of
potentially serious MDR bacterial infections.
Structural and biological studies on bacterial nitric oxide synthase
inhibitors
JK Holden, H Li, Q Jing, S Kang, J Richo, RB Silverman, TL Poulos
Significance: Nitric oxide (NO) produced by bacterial nitric oxide
synthase has recently been shown to protect the Gram-positive
pathogens Bacillus anthracis and Staphylococcus aureus from
antibiotics and oxidative stress. Using Bacillus subtilis as a model
system, we identified two NOS inhibitors that work in conjunction
with an antibiotic to kill B. subtilis. Moreover, comparison of inhibitor-bound crystal structures between the bacterial NOS and mammalian
NOS revealed an unprecedented mode of binding to the bacterial NOS
that can be further exploited for future structure-based drug design.
Overall, this work is an important advance in developing inhibitors
against gram-positive pathogens.
Summary: Nitric oxide (NO) produced by bacterial NOS functions as a
cytoprotective agent against oxidative stress in Staphylococcus aureus,
Bacillus anthracis, and Bacillus subtilis. The screening of several NOS-selective inhibitors uncovered two inhibitors with potential antimicrobial
properties. These two compounds impede the growth of B. subtilis under
oxidative stress, and crystal structures show that each compound exhibits
a unique binding mode. Both compounds serve as excellent leads for the
future development of antimicrobials against bacterial NOS-containing
bacteria. http://dx.doi.org/10.1073/pnas.1314080110
Speciation of clinically significant coagulase negative Staphylococci
and their antibiotic resistant patterns in a tertiary care hospital
PR Vysakh, S Kandasamy and RM Prabhavathi
Int.J.Curr.Microbiol.App.Sci (2015) 4(1): 704-709 http://www.ijcmas.com
Human skin and mucus membrane has Coagulase Negative Staphylococci
(CoNS) as the indigenous flora. CoNS had become an important agent for
nosocomial infections accounting for about 9%. These infections are
difficult to treat because of the risk factors and the multiple drug resistance
nature of these organisms. The study was undertaken to identify the
prevalence of clinical isolates of CoNS, their speciation and to determine
the antibiotic sensitivity/resistant patterns of CoNS. A total of 490 isolates
were collected from different samples and subjected to biochemical
characterization and antimicrobial screening using conventional
microbiological methods. 165 isolates were identified as CoNS. 23% of
CoNS were isolated from blood, 30% from post-operative wound infections,
23% from pus, 18% from urine, 3% from body fluids (CSF, ascitic fluid etc)
and 3% from CVP tips. The antibiotic sensitivity revealed 81% resistance
to Penicillin,32% resistance to Cefoxitin, 27% resistance to Cefazolin,
55% resistance to Erythromycin, 22% to Clindamycin and 35% to
Cotrimoxazole and with no resistance to Vancomycin, Linezolid and
Ciprofloxacin. The increased recognition of CoNS and emergence of
drug resistance among them demonstrates the need to consider them
as a potent pathogen and to devise laboratory procedure to identify
and to determine the prevalence and antibiotic resistant patterns of CoNS.
Resistance to rifampicin (RIF) is a broad subject covering not just the
mechanism of clinical resistance, nearly always due to a genetic change
in the b subunit of bacterial RNA polymerase (RNAP), but also how
studies of resistant polymerases have helped us understand the structure
of the enzyme, the intricacies of the transcription process and its role
in complex physiological pathways. This review can only scratch the
surface of these phenomena. The identification, in strains of
Escherichia coli, of the positions within b of the mutations determining
resistance is discussed in some detail, as are mutations in organisms
that are therapeutic targets of RIF, in particular Mycobacterium
tuberculosis. Interestingly, changes in the same three codons of
the consensus sequence occur repeatedly in unrelated RIF-resistant
(RIFr) clinical isolates of several different single mutation
predominates in mycobacteria. The utilization of our knowledge of
these mutations to develop rapid screening tests for detecting resistance
is briefly discussed. Cross-resistance among rifamycins has been a topic
of controversy; current thinking is that there is no difference in the
susceptibility of RNAP mutants to RIF, rifapentine and rifabutin.
Also summarized are intrinsic RIF resistance and other resistance
mechanisms.
Multi-drug resistance, inappropriate initial antibiotic therapy and
mortality in Gram negative severe sepsis and septic shock: A
retrospective cohort study
MD Zilberberg, AF Shorr, ST Micek, C Vazquez-Guillamet, MH Kollef
Critical Care 2014, 18:596 http://dx.doi.org:/10.1186/s13054-014-0596-8
http://ccforum.com/content/18/6/596
Introduction
The impact of in vitro resistance on initially appropriate antibiotic therapy
(IAAT) remains unclear. We elucidated the relationship between non-IAAT
and mortality, and between IAAT and multi-drug resistance (MDR) in
sepsis due to Gram-negative bacteremia (GNS).
Methods
We conducted a single-center retrospective cohort study of adult intensive
care unit patients with bacteremia and severe sepsis/septic shock caused by
a gram-negative (GN) organism. We identified the following MDR pathogens:
MDR P. aeruginosa, extended spectrum beta lactamase and carbapenemase-
producing organisms. IAAT was defined as exposure within 24 hours of
infection onset to antibiotics active against identified pathogens based on
in vitro susceptibility testing. We derived logistic regression models to
examine a) predictors of hospital mortality and b) impact of MDR on
non-IAAT. Proportions are presented for categorical variables, and
median values with interquartile ranges (IQR) for continuous
variables.
Results
Out of 1,064 patients with GNS, 351 (29.2%) did not survive
hospitalization. Non-survivors were older (66.5 (55, 73.5)
versus 63 (53, 72) years, P =0.036), sicker (Acute Physiology and
Chronic Health Evaluation II (19 (15, 25) versus 16 (12, 19),
P <0.001), and more likely to be on pressors (odds ratio (OR) 2.79,
95% confidence interval (CI) 2.12 to 3.68), mechanically ventilated
(OR 3.06, 95% CI 2.29 to 4.10) have MDR (10.0% versus 4.0%,
P <0.001) and receive non-IAAT (43.4% versus 14.6%, P <0.001).
In a logistic regression model, non-IAAT was an independent
predictor of hospital mortality (adjusted OR 3.87, 95% CI 2.77 to
5.41). In a separate model, MDR was strongly associated with
the receipt of non-IAAT (adjusted OR 13.05, 95% CI 7.00 to 24.31).
Conclusions
MDR, an important determinant of non-IAAT, is associated with
a three-fold increase in the risk of hospital mortality. Given the
paucity of therapies to cover GN MDRs, prevention and
development of new agents are critical.
Phenotypic and molecular characteristics of methicillin-resistant
Staphylococcus aureus isolates from Ekiti State, Nigeria
OA Olowe, OO Kukoyi, SS Taiwo, O Ojurongbe, OO Opaleye, et al.
Infection and Drug Resistance 2013:6 87–92 http://dx.doi.org/10.2147/IDR.S48809
Introduction: The characteristics and antimicrobial resistance profiles
of Staphylococcus aureus differs according to geographical regions and
in relation to antibiotic usage. The aim of this study was to determine
the biochemical characteristics of the prevalent S. aureus from Ekiti State,
Nigeria, and to evaluate three commonly used disk diffusion methods
(cefoxitin, oxacillin, and methicillin) for the detection of methicillin
resistance in comparison with mecA gene detection by polymerase chain
reaction.
Materials and methods: A total of 208 isolates of S. aureus recovered
from clinical specimens were included in this study. Standard
microbiological procedures were employed in isolating the strains.
Susceptibility of each isolate to methicillin (5 μg), oxacillin (1 μg),
and cefoxitin (30 μg) was carried out using the modified Kirby–Bauer/
Clinical and Laboratory Standard Institute disk diffusion technique.
They were also tested against panels of antibiotics including vancomycin.
The conventional polymerase chain reaction method was used to detect
the presence of the mecA gene.
Results: Phenotypic resistance to methicillin, oxacillin, and cefoxitin
were 32.7%, 40.3%, and 46.5%, respectively. The mecA gene was detected
in 40 isolates, giving a methicillin-resistant S. aureus (MRSA) prevalence
of 19.2%. The S. aureus isolates were resistant to penicillin (82.7%) and
tetracycline (65.4%), but largely susceptible to erythromycin (78.8%
sensitive), pefloxacin (82.7%), and gentamicin (88.5%). When compared
to the mecA gene as the gold standard for MRSA detection, methicillin,
oxacillin, and cefoxitin gave sensitivity rates of 70%, 80%, and 100%,
and specificity rates of 76.2%, 69.1%, and 78.5% respectively.
Conclusion: When compared with previous studies employing mecA
polymerase chain reaction for MRSA detection, the prevalence of 19.2%
reported in Ekiti State, Nigeria in this study is an indication of gradual rise
in the prevalence of MRSA in Nigeria. A cefoxitin (30 μg) disk diffusion test
is recommended above methicillin and oxacillin for the phenotypic detection
of MRSA in clinical laboratories.
Direct sequencing for rapid detection of multidrug resistant Mycobacterium
tuberculosis strains in Morocco
F Zakham, I Chaoui, AH Echchaoui, F Chetioui, M Driss Elmessaoudi, et al.
Infection and Drug Resistance 2013:6 207–213 http://dx.doi.org/10.2147/IDR.S47724
Background: Tuberculosis (TB) is a major public health problem with high
mortality and morbidity rates, especially in low-income countries.
Disturbingly, the emergence of multidrug resistant (MDR) and extensively
drug resistant (XDR) TB cases has worsened the situation, raising concerns
of a future epidemic of virtually untreatable TB. Indeed, the rapid diagnosis
of MDR TB is a critical issue for TB management. This study is an attempt to
establish a rapid diagnosis of MDR TB by sequencing the target fragments of
the rpoB gene which linked to resistance against rifampicin and the katG gene
and inhA promoter region, which are associated with resistance to isoniazid.
Methods: For this purpose, 133 sputum samples of TB patients from Morocco
were enrolled in this study. One hundred samples were collected from new
cases, and the remaining 33 were from previously treated patients (drug
relapse or failure, chronic cases) and did not respond to anti-TB drugs after
a sufficient duration of treatment. All samples were subjected to rpoB, katG
and pinhA mutation analysis by polymerase chain reaction and DNA sequencing.
Results: Molecular analysis showed that seven strains were isoniazid-
monoresistant and 17 were rifampicin-monoresistant. MDR TB strains were
identified in nine cases (6.8%). Among them, eight were traditionally
diagnosed as critical cases, comprising four chronic and four drug-relapse
cases. The last strain was isolated from a new case. The most recorded
mutation in the rpoB gene was the substitution TCG . TTG at codon 531
(Ser531 Leu), accounting for 46.15%. Significantly, the only mutation found
in the katG gene was at codon 315 (AGC to ACC) with a Ser315Thr amino acid
change. Only one sample harbored mutation in the inhA promoter region
and was a point mutation at the −15p position (C . T). Conclusion: The
polymerase chain reaction sequencing approach is an accurate and rapid
method for detection of drug-resistant TB in clinical specimens, and could
be of great interest in the management of TB in critical cases to adjust the
treatment regimen and limit the emergence of MDR and XDR strains.
Limiting and controlling carbapenem-resistant Klebsiella pneumoniae
L Saidel-Odes, A Borer.
Infection and Drug Resistance 2014:7 9–14 http://dx.doi.org/10.2147/IDR.S44358
Carbapenem-resistant Klebsiella pneumoniae (CRKP) is resistant to
almost all antimicrobial agents, is associated with substantial morbidity
and mortality, and poses a serious threat to public health. The ongoing
worldwide spread of this pathogen emphasizes the need for immediate
intervention. This article reviews the global spread and risk factors for
CRKP colonization/infection, and provides an overview of the strategy
to combat CRKP dissemination.
Staphylococcus aureus – antimicrobial resistance and the immuno-
compromised child
J Chase McNeil
Infection and Drug Resistance 2014:7 117–127 http://dx.doi.org/10.2147/IDR.S39639
Children with immunocompromising conditions represent a unique
group for the acquisition of antimicrobial resistant infections due to
their frequent encounters with the health care system, need for empiric
antimicrobials, and immune dysfunction. These infections are further
complicated in that there is a relative paucity of literature on the clinical
features and management of Staphylococcus aureus infections in
immunocompromised children. The available literature on the clinical
features, antimicrobial susceptibility, and management of S. aureus
infections in immunocompromised children is reviewed. S. aureus
infections in children with human immunodeficiency virus (HIV) are
associated with higher HIV viral loads and a greater degree of CD4 T-cell
suppression. In addition, staphylococcal infections in children with HIV
often exhibit a multidrug resistant phenotype. Children with cancer have
a high rate of S. aureus bacteremia and associated complications. Increased
tolerance to antiseptics among staphylococcal isolates from pediatric
oncology patients is an emerging area of research. The incidence of S. aureus
infections among pediatric solid organ transplant recipients varies
considerably by the organ transplanted; in general however, staphylococci
figure prominently among infections in the early post-transplant period.
Staphylococcal infections are also prominent pathogens among children
with a number of immunodeficiencies, notably chronic granulomatous
disease. Significant gaps in knowledge exist regarding the epidemiology
and management of S. aureus infection in these vulnerable children.
selected Staphylococcus aureus mechanisms for immune evasion.
Figure 1 A schematic depiction of selected Staphylococcus aureus
mechanisms for immune evasion.
Notes: Cna interacts with C1q preventing formation of the C1qrs complex.
ClfA and SdrE each promote Factor I mediated conversion of C3b to iC3b.
Protein A is depicted binding to the Fc region of IgG preventing immunoglobulin
opsonization.
Abbreviations: ClfA, staphylococcal clumping factor A; Cna, collagen adhesin;
IgG, immunoglobulin G; PVL, Panton–Valentine leukocidin; SdrE, S. aureus
surface protein.
The Future of Antibiotics and Resistance
B Spellberg, JG Bartlett, and DN Gilbert
N Engl J Med Jan 24, 2013; 368(4): 299-302 http://dx.doi.org:/ 10.1056/NEJMp1215093
In its recent annual report on global risks, the World Economic
Forum (WEF) concluded that “arguably the greatest
risk . . . to human health comes in the form of antibiotic-resistant
bacteria. We live in a bacterial world where we will never be able
to stay ahead of the mutation curve. A test of our resilience is
how far behind the curve we allow ourselves to fall.”
The WEF report underscores the facts that antibiotic resistance
and the collapse of the antibiotic research and-development
pipeline continue to worsen despite our ongoing efforts on
current fronts. If we’re to develop countermeasures that
have lasting effects, new ideas that complement traditional
approaches will be needed.
Resistance is primarily the result of bacterial adaptation to eons
of antibiotic exposure. What are the fundamental implications of
this reality? First, in addition to antibiotics’ curative power, their
use naturally selects for preexisting resistant populations of bacteria
in nature. Second, it is not just “inappropriate” antibiotic use
that selects for resistance. Rather, the speed with which resistance
spreads is driven by microbial exposure to all antibiotics, whether
appropriately prescribed or not. Thus, even if all inappropriate
antibiotic use were eliminated, antibiotic-resistant infections
would still occur (albeit at lower frequency). Third, after billions
of years of evolution, microbes have most likely invented
antibiotics against every biochemical target that can be attacked
— and, of necessity, developed resistance mechanisms
to protect all those biochemical targets.
Remarkably, resistance was found even to synthetic antibiotics
that did not exist on earth until the 20th century. These results
underscore a critical reality: antibiotic resistance already exists,
widely disseminated in nature, to drugs we have not yet invented.
Table **
Interventions to Address the Antibiotic-Resistance Crisis.*
Intervention Status Preventing infection
and resistance
“Self-cleaning” hospital rooms; Some commercially available
automated disinfectant application but require clinical validation;
through misting, vapor, radiation, etc. more needed
Novel drug-delivery systems to replace Basic science and
IV catheters; regenerative-tissue technology conceptual stages
to replace prosthetics; superior, noninvasive
ventilation strategies
Improvement of population health and Implementation
health care systems to reduce admissions research stage
to hospitals and skilled nursing facilities
Niche vaccines to prevent resistant Basic and clinical
bacterial infections development stage
Refilling antibiotic pipeline by aligning
economic and regulatory approaches
Models in place, expansion needed in number Government or nonprofit grants
and scope; new nonprofit corporations and contracts to defray R&D costs
needed and establish nonprofits
to develop antibiotics
Institution of novel approval pathways Proposed, legislative
(e.g., Limited Population Antibiotic and regulatory
Drug proposal) action needed
Preserving available antibiotics,
slowing resistance
Public reporting of antibiotic-use data as a Policy action needed to
basis for benchmarking and reimbursement develop and implement
Development of and reimbursement for Basic and applied research
rapid diagnostic and biomarker tests to and policy action and
enable appropriate use of antibiotics policy action needed
Elimination of use of antibiotics to Legislation proposed
promote livestock growth
New waste-treatment strategies; One strategy approaching
targeted chemical or biologic clinical trials
degradation of antibiotics in waste
Studies to define shortest effective Some trials completed
courses of antibiotics for infections
Developing microbe-attacking Preclinical, proof-of-
treatments with diminished principle stage
potential to drive resistance
Immune-based therapies, such
as infusion of monoclonal antibodies
and white cells that kill microbes
Antibiotics or biologic agents that
don’t kill bacteria but alter their ability
to trigger inflammation or cause disease
Developing treatments attacking host Preclinical, proof-of-principle stage
targets rather than microbial targets to
avoid selective pressure driving resistance
Direct moderation of host inflammation
in response to infection (e.g., cytokine
agonists or antagonists, PAMP receptor
agonists)
Sequestration of host nutrients to
prevent microbial access to nutrients
Probiotics that compete with microbial
growth
* IV denotes intravenous, PAMP pathogen-associated molecular
pattern, and R&D research and development
Antibiotic-Resistant Bugs Appear to Use Universal Ribosome-Stalling Mechanism
Researchers at St. Louis University say they have discovered new information
about how antibiotics like azithromycin stop staph infections, and why staph
sometimes becomes resistant to drugs. The team, led by Mee-Ngan F. Yap, Ph.D.,
believe their evidence suggests a universal, evolutionary mechanism by which
the bacteria elude this kind of drug, offering scientists a way to improve the
effectiveness of antibiotics to which bacteria have become resistant. Their
study (“Sequence selectivity of macrolide-induced translational attenuation”)
was published in PNAS.
Staphylococcus aureus is a strain of bacteria that frequently has become
resistant to antibiotics, a development that has been challenging for doctors
and dangerous for patients with severe infections. Dr. Yap and her research
team studied staph that had been treated with the antibiotic azithromycin and
learned two things: One, it turns out that the antibiotic isn’t as effective as was
previously thought. And two, the process that the bacteria use to evade the
antibiotic appears to be an evolutionary mechanism that the bacteria developed
in order to delay genetic replication when beneficial.
Genomic epidemiology of a protracted hospital outbreak caused by multidrug-
resistant Acinetobacter baumannii in Birmingham, England
MR Halachev, J Z-M Chan, CI Constantinidou, N Cumley, C Bradley, et al.
Genome Medicine 2014, 6:70 http://genomemedicine.com/content/6/11/70
Background: Multidrug-resistant Acinetobacter baumannii commonly causes
hospital outbreaks. However, within an outbreak, it can be difficult to identify
the routes of cross-infection rapidly and accurately enough to inform infection
control. Here, we describe a protracted hospital outbreak of multidrug-resistant
A. baumannii, in which whole-genome sequencing (WGS) was used to obtain
a high-resolution view of the relationships between isolates.
Methods: To delineate and investigate the outbreak, we attempted to genome-
sequence 114 isolates that had been assigned to the A. baumannii complex
by the Vitek2 system and obtained informative draft genome sequences from
102 of them. Genomes were mapped against an outbreak reference sequence
to identify single nucleotide variants (SNVs).
Results: We found that the pulsotype 27 outbreak strain was distinct from all
other genome-sequenced strains. Seventy-four isolates from 49 patients
could be assigned to the pulsotype 27 outbreak on the basis of genomic
similarity, while WGS allowed 18 isolates to be ruled out of the outbreak.
Among the pulsotype 27 outbreak isolates, we identified 31 SNVs and seven
major genotypic clusters. In two patients, we documented within-host diversity,
including mixtures of unrelated strains and within-strain clouds of SNV diversity.
By combining WGS and epidemiological data, we reconstructed potential
transmission events that linked all but 10 of the patients and confirmed links
between clinical and environmental isolates. Identification of a contaminated
bed and a burns theatre as sources of transmission led to enhanced
environmental decontamination procedures.
Conclusions: WGS is now poised to make an impact on hospital infection
prevention and control, delivering cost-effective identification of routes of
infection within a clinically relevant timeframe and allowing infection control
teams to track, and even prevent, the spread of drug-resistant hospital pathogens.
Understanding of antibiotic resistance in Streptococcus pneumoniae has been
hindered by the low frequency of recombination events in bacteria and thus the
presence of large linked haplotype blocks, which preclude identification of
causative variants. A recent study combining a large number of genomes of
resistant phenotypes has given an insight into the evolving resistance to
β-lactams, providing the first large-scale identification of candidate variants
underlying resistance.
Additional sources:
A Simple Method for Assessment of MDR Bacteria for Over-Expressed
Efflux Pumps
M Martins, MP McCusker, M Viveiros, I Couto, S Fanning, .., L Amaral
The Open Microbiology Journal, 2013, 7, 1-5
Identification of Efflux Pump-mediated Multidrug-resistant
Bacteria by the Ethidium Bromide-agar Cartwheel Method
M MARTINS, M VIVEIROS, I COUTO,, SS COSTA, .., L AMARAL
in vivo 25: 171-178 (2011)
Efflux Pumps that Bestow Multi-Drug Resistance of Pathogenic
Gram negative Bacteria
Amaral L, Spengler G, Martins A and Molnar J
Biochem Pharmacol 2013; 2(3):119 http://dx.doi.org/10.4172/2167-0501.1000119
graphical abstract
An Instrument-free Method for the Demonstration
of Efflux Pump Activity of Bacteria
M MARTINS, B SANTOS, A MARTINS, M VIVEIROS, I COUTO,
A CRUZ, THE MANAGEMENT COMMITTEE MEMBERS
OF COST B16 OF THE EUROPEAN COMMISSION/
EUROPEAN SCIENCE FOUNDATION,…, J MOLNAR, S FANNING
and LEONARD AMARAL
in vivo 20: 657-664 (2006)
Potential Therapy of Multidrug-resistant and Extremely
Drug-resistant Tuberculosis with Thioridazine
LEONARD AMARAL and JOSEPH MOLNAR
in vivo 26: 231-236 (2012)
Inhibitors of efflux pumps of Gram-negative bacteria
inhibit Quorum Sensing
Leonard Amaral, Joseph Molnar
Open Journal of Pharmacology, 2012, 2-2
Tel Aviv – By sequencing the DNA of bacteria resistant to viral toxins, scientists have identified novel proteins capable of stymieing growth in pathogenic, antibiotic-resistant bacteria.
Today’s arsenal of antibiotics is ineffective against some emerging strains of antibiotic-resistant pathogens. Novel inhibitors of bacterial growth therefore need to be found. One way is looking into the viruses that infect bacteria.
Key to the new initiative is the concept of fighting bacteria from within, rather than using an external chemical to batter through the bacterial cell wall. the basis of the new weapon is viral. In order to select an appropriate viral protein, researchers undertook a comprehensive screening exercise in order to identify proteins in viruses that are known to infect bacteria (bacteriophages). Bacteriophages occur abundantly in the biosphere, with different virions, genomes and lifestyles. The review was so comprehensive that it took almost three years to complete.
The screening was achieved through the use of high-throughput DNA sequencing. This is the process of determining the precise order of nucleotides within a DNA molecule. By using this advanced genetic method, the scientists identified mutations in bacterial genes that resisted the toxicity of growth inhibitors produced by bacterial viruses. Through this, a new, tiny protein was found. The protein is termed “growth inhibitor gene product (Gp) 0.6”.
Later testing found that the protein specifically targets and inhibits the activity of a protein essential to bacterial cells. The bacterial protein affected has the function of holding the microbe’s cell wall together. Without this protein functioning correctly, the cell bursts open from within and the bacterium dies.
Revealing bacterial targets of growth inhibitors encoded by bacteriophage T7
Shahar Molshanski-Mora, Ido Yosefa, Ruth Kiroa, Rotem Edgara, Miriam Manora, Michael Gershovitsb, Mia Lasersonb, Tal Pupkob, and Udi Qimrona,1
Author Affiliations
Edited* by Sankar Adhya, National Institutes of Health, National Cancer Institute, Bethesda, MD, and approved November 24, 2014 (received for review July 13, 2014)
Significance
Antibiotic resistance of pathogens is a growing threat to human health, requiring immediate action. Identifying new gene products of bacterial viruses and their bacterial targets may provide potent tools for fighting antibiotic-resistant strains. We show that a significant number of phage proteins are inhibitory to their bacterial host. DNA sequencing was used to map the targets of these proteins. One particular target was a key cytoskeleton protein whose function is impaired following the phage protein’s expression, resulting in bacterial death. Strikingly, in over 70 y of extensive research into the tested bacteriophage, this inhibition had never been characterized. We believe that the presented approach may be broadened to identify novel, clinically relevant bacteriophage growth inhibitors and to characterize their targets.
Abstract
Today’s arsenal of antibiotics is ineffective against some emerging strains of antibiotic-resistant pathogens. Novel inhibitors of bacterial growth therefore need to be found. The target of such bacterial-growth inhibitors must be identified, and one way to achieve this is by locating mutations that suppress their inhibitory effect. Here, we identified five growth inhibitors encoded by T7 bacteriophage. High-throughput sequencing of genomic DNA of resistant bacterial mutants evolving against three of these inhibitors revealed unique mutations in three specific genes. We found that a nonessential host gene, ppiB, is required for growth inhibition by one bacteriophage inhibitor and another nonessential gene, pcnB, is required for growth inhibition by a different inhibitor. Notably, we found a previously unidentified growth inhibitor, gene product (Gp) 0.6, that interacts with the essential cytoskeleton protein MreB and inhibits its function. We further identified mutations in two distinct regions in the mreB gene that overcome this inhibition. Bacterial two-hybrid assay and accumulation of Gp0.6 only in MreB-expressing bacteria confirmed interaction of MreB and Gp0.6. Expression of Gp0.6 resulted in lemon-shaped bacteria followed by cell lysis, as previously reported for MreB inhibitors. The described approach may be extended for the identification of new growth inhibitors and their targets across bacterial species and in higher organisms.
New funding to fight antibiotic resistance SPECIAL
By Tim Sandle
This week the White House stated that it will double the amount of federal funding put aside to combat and preventing antibiotic resistance. The sum stands at greater than $1.2 billion.
U.S. Senator Sherrod Brown has been campaigning across the U.S. about the risks related to antibiotic-resistant infections for several years. Such infections affect more than two million U.S. citizens each year. The issue is not only of importance in one country for the growing menace of antibiotic resistance is, arguably, the single biggest threat faced by the world’s population. Moreover, emerging antimicrobial resistance and the growing shortage of effective antibiotic drugs is widely regarded as a crisis that jeopardizes patient safety and public health.
Senator Brown has welcomed the increased spending, although he also feels that more action is required. “To combat antibiotic resistance, it’s important that we leverage the best in medical expertise, stewardship, and technological innovation,” Brown has told Digital Journal.
He went on to add: “This unprecedented proposal underscores the importance of taking a comprehensive, wide-ranging approach to tackle this issue. I look forward to continuing to work with federal agencies, research institutions, and health care providers to combat this threat to America’s health.”
In 2014, Brown proposed the Strategies to Address Antimicrobial Resistance (STAAR) Act. The aim of this legislation was to boost the federal response to antibiotic resistance through promoting prevention and control. Other measures included: tracking drug-resistant bacteria; supporting enhanced research efforts; and improving the development, use, and stewardship of antibiotics. The Act would have provided an opportunity to bring multiple federal and non-governmental partners together to protect the public health from these drug-resistant bugs.
The Act, reported by Digital Journal, did not get through, despite the recent announcement of increased federal spending. Senator Brown argues that more preventative measures are needed. For this reason he plans to reintroduce similar legislation this year.
The STAAR Act would:
Promote prevention through public health partnerships at the U.S. Centers for Disease Control and Prevention (CDC) and local health departments;
Track resistant bacteria by making data collection better and requiring better reporting;
Improve the use of antibiotics by educating health care facilities on appropriate antibiotic use;
Enhance leadership and accountability in antibiotic resistance by reauthorizing a task force and coordinating agency efforts;
Support research by directing the National Institutes of Health (NIH) to work with other agencies and experts to create a strategic plan to address the problem.
Senator takes on antibiotic resistant organisms SPECIAL
By Tim Sandle Apr 16, 2014 in Science
Washington – With so-called “super bugs” on the rise, U.S. Sen. Sherrod Brown (D-OH) has introduced a bill aimed at slowing down the rate of antibiotic resistant microorganisms.
Senator Brown has introduced the Strategies to Address Antimicrobial Resistance (STAAR) Act. This is legislation aimed at combating antimicrobial resistance. In presenting the Act, Brown called for greater Federal attention to the growth of antibiotic-resistant infections, which affect more than two million Americans each year.
Brown is aiming for the STAAR Act to provide an opportunity to bring multiple federal and non-governmental partners together to protect the public health from these drug-resistant bugs.
Senator Brown contacted Digital Journal to explain more. In explaining the basis to the Act, Brown said: “Each year more than 23,000 Americans die from bacterial infections that are resistant to antibiotics.”
Antimicrobial resistance describes the ability of a microorganism to resist the action of antimicrobial drugs. In some instances some microorganisms are naturally resistant to particular antimicrobial agents; in other instances, the genes of non-disease-causing bacteria can be transferred to pathogenic bacteria, leading to patterns of clinically significant antibiotic resistance. Since the 1990s antibiotic resistance has been of concern for scientists and health policy makers.
Looking at the reasons for this, Brown explained that: “Antibiotics and other antimicrobial drugs have been a victim of their own success. We have used these drugs so widely and for so long that the microbes they are designed to kill have adapted to them, making the drugs less effective.”
Considering this in the context of his Act, Brown added: “We need a comprehensive strategy to address antimicrobial resistance. That is why I am introducing the STAAR Act, which would revitalize efforts to combat super bugs.”
Emerging antimicrobial resistance and the growing shortage of effective antibiotic drugs is widely regarded as a crisis that jeopardizes patient safety and public health. Once confined to hospitals, drug-resistant microbes, such as multi-drug-resistant Staphylococcus aureus (MRSA), are now striking down healthy, non-hospitalized citizens. This includes both the young and old, adults and children. These infections are painful, difficult to treat, and have become a silent epidemic in communities and hospitals across the U.S. (according to CDC).
Brown hopes that the STAAR Act will help strengthen the federal response to antimicrobial resistance by placing more of an emphasis on federal antimicrobial resistance surveillance, prevention and control, and research efforts.
In addition the Senator hopes that the Act will strengthen coordination within both Department of Health and Human Services (HHS) agencies as well as across other federal departments that are important to addressing antimicrobial resistance and enable opportunities to address this issue globally.
By providing for a more comprehensive and coordinated approach to the antimicrobial resistance crisis, it would seem that the STAAR Act represents a critical first step toward resolving what has become a major public health crisis.
Cancer is an inescapable fact of life. All of us with either die from it or know someone who will. Cancer is so prevalent because it isn’t a disease in the way a flu or a cold is. No outside force or germ is needed to cause cancer (although it can). It arises from the very way we are put together. Most of the genes that are needed for multicellular life have been found to be associated with cancer. Cancer is a result of our natural genetic machinery that has been built up over billions of years breaking down over time.
Cancer is not only a result of evolutionary processes, cancer itselffollows evolutionary theory as it grows. The immune system places a selective pressure on cancer cells, keeping it in check until the cancer evolves a way to avoid it and surpass it in a process known as immunoediting. Cancers face selective pressures in the microenvironments in which they grow. Due to the fast growth of cancer cells, they suck up oxygen in the tissues, causing wildly fluctuating oxygen levels as the body tries to get oxygen to the tissues. This sort of situation is bad for normal tissues and so it is for cancer, at least until they evolve and adapt. At some point, some cancer cells will develop the ability to use what is called aerobic glycolysis to make the ATP we use for energy. Ordinarily, our cells only use glycolysis when they run out of oxygen because aerobic respiration (aka oxidative phosphorylation) is far more efficient. Cancer cells, on the other hand, learn to use glycolysis all the time, even in the presence of abundant oxygen. They may not grow as quickly when there is plenty of oxygen, but they are far better than normal cells at hypoxic, or low oxygen, conditions, which they create by virtue of their metabolism. Moreover, they are better at taking up nutrients because many of the metabolic pathways for aerobic respiration also influence nutrient uptake, so shifting those pathways to nutrient uptake rather than metabolism ensures cancer cells get first pick of any nutrients in the area. The Warburg Effect, as this is called, works by selective pressures hindering those cells that can’t do so and favoring those that can. Because cancer cells have loose genetic controls and they are constantly dividing, the cancer population can evolve, whereas the normal cells cannot.
Evolutionary theory can also be used to track cancer as it metastasizes. If a person has several tumors, it is possible to take biopsies of each one and use standard cladistic programs that are normally used to determine evolutionary relationships between organisms to find which tumor is the original tumor. If the original tumor is not one of those biopsied, it will tell you where the cancer originated within the body. You can thus track the progression of cancer throughout a person’s body. Expanding on this, one can even track the effect of cancer through its effects on how organisms interact within ecosystems, creating its own evolutionary stamp on the environment as its effects radiate throughout the ecosystem.
I’ve talked about cancer at decent length (although I could easily go one for many more pages) because it is less well publicly known than some of the other ways that evolutionary theory helps us out in medicine. The increasing resistance of bacteria and viruses to antibiotics is well known. Antibiotic resistance follows standard evolutionary processes, with the result that antibiotic resistant bacteria are expected to kill 10 million people a year by 2050. People have to get a new flu shot every year because the flu viruses are legion and they evolve rapidly to bypass old vaccinations. If we are to accurately predict how the viruses may adapt and properly prepare vaccines for the coming year, evolutionary theory must be taken into account. Without it, the vaccines are much less likely to be effective. Evolutionary studies have pointed out important changes in the Ebola virus and how those changes areaffecting its lethality, which will need to be taken into account for effective treatments. Tracking the origins of viruses, like the avian flu or swine flu, gives us information that will be useful in combating them or even stopping them at their source before they become a problem.
An Overview of Clinical Microbiology, Classification, and Antimicrobial Resistance
Author and Curator: Larry H. Bernstein, MD, FCAP
Classification of Microbiota
Introduction to Overview of Microbiology
This is a contribution to a series of pieces on the history of biochemistry, molecular biology, physiology and medicine in the 20th century. Here I describe the common microbial organisms encountered in the clinical laboratory, the method of their collection, plating, culture and identification, and antibiotic sensitivity testing and resistant strains.
I may begin with the recognition that there are common strains in the environment that are not pathogenic, and there are pathogenic bacteria.
In addition, there are bacteria that coexist in the body habitat under specific conditions so that we are able to map the types expected to location, such as, skin, mouth and nasal cavities, the colon, the vagina and urinary system. Meningitides occur as a result of extension from the nasal cavity to the brain. When bacteria invade the circulation, it is referred to as septicemia, and the bacteria can cause valvular heart damage.
Bacteriology can be traced to origins in the 19th century. The clinical features of localized infection are classically referred to as redness, heat, a raised lesion (pustule), and exudate (serous or purulent – watery or cellular). This not only holds for a focal lesion (as skin), but also for pneumonia, urinary infection, and genital. It may be accompanied by cough, or bloody cough and wheezing, or by an unclear urine. In the case of septicemia, there is fever, and there may be seizures or delirium.
Collection and handling of specimens
Specimens are collected by sterile technique by a nurse or physician and sent to a lab as a swab, or as a blood specimen. In the case of a febrile illness, blood cultures may be obtained from opposite arms, and another an hour later. This is related to the possible cyclical seeding of bacteria into the circulation. If the specimen is collected from a site of infection, a swab may be put onto a glass slide for gram staining. The specimen collected is sent to the laboratory.
We may consider syphilis and tuberculosis special cases that I’ll set aside. I shall not go into virology either, although I may referred to smallpox, influenza, polio, HIV under epidemic. The first step in identification is the Gram stain, developed in the 19th century. Organisms of the skin are Gram positive and appear blue on staining. They are cocci, or circular, organized in characteristic clusters (staphylococcus, streptococcus) or in pairs (diplococci, eg. Pneumococcus), and if from the intestine (enterococcus). If they are elongated rods, they might be coliform. If they stain red, they are Gram negative. Gram negative rods are coliform, and are enterobacteriaceae. Meningococci are Gram negative cocci. So we have certain information about these organisms before we plate them for growth.
Laboratory growth characteristics
The specimen is applied to an agar plate with a metal rod applicator, or perhaps onto more than one agar plate. The agar plate contains a growth media or a growth inhibitor that is more favorable to certain species than to others. The bacteria are grown at 37o C in an incubator and colonies develop that are white or nonwhite, and they are smooth or wrinkled. The appearance of the colonies is characteristic for certain strains. If there is no contamination, all of the colonies look the same. The next step is to:
Gram stain from a colony
Transfer samples from the colony to a series of growth media that identify presence or absence of specific nutrient requirements for growth (which is presumed from the prior findings).
In addition, the colony samples are grown on an agar to which is applied antibiotic tabs. The tabs either allow or repress growth. It wa some 50 years ago that the infectious disease physician and microbiologist Abraham Braude would culture the bacteria on agar plates that had a gradient of antibiotic to check for concentration that would inhibit growth.
Principles of Diagnosis (Extracts)
By John A. Washington
The clinical presentation of an infectious disease reflects the interaction between the host and the microorganism. This interaction is affected by the host immune status and microbial virulence factors. Signs and symptoms vary according to the site and severity of infection. Diagnosis requires a composite of information, including history, physical examination, radiographic findings, and laboratory data.
Microbiologic Examination
Direct Examination and Techniques: Direct examination of specimens reveals gross pathology. Microscopy may identify microorganisms. Immunofluorescence, immuno-peroxidase staining, and other immunoassays may detect specific microbial antigens. Genetic probes identify genus- or species-specific DNA or RNA sequences.
Culture: Isolation of infectious agents frequently requires specialized media. Nonselective (noninhibitory) media permit the growth of many microorganisms. Selective media contain inhibitory substances that permit the isolation of specific types of microorganisms.
Microbial Identification: Colony and cellular morphology may permit preliminary identification. Growth characteristics under various conditions, utilization of carbohydrates and other substrates, enzymatic activity, immunoassays, and genetic probes are also used.
Serodiagnosis: A high or rising titer of specific IgG antibodies or the presence of specific IgM antibodies may suggest or confirm a diagnosis.
Antimicrobial Susceptibility: Microorganisms, particularly bacteria, are tested in vitro to determine whether they are susceptible to antimicrobial agents.
Diagnostic medical microbiology is the discipline that identifies etiologic agents of disease. The job of the clinical microbiology laboratory is to test specimens from patients for microorganisms that are, or may be, a cause of the illness and to provide information (when appropriate) about the in vitro activity of antimicrobial drugs against the microorganisms identified (Fig. 1).
Laboratory procedures used in confirming a clinical diagnosis of infectious disease with a bacterial etiology
A variety of microscopic, immunologic, and hybridization techniques have been developed for rapid diagnosis
techniques have been developed for rapid diagnosis
From: Chapter 10, Principles of Diagnosis
Medical Microbiology. 4th edition.
Baron S, editor.
Galveston (TX): University of Texas Medical Branch at Galveston; 1996.
For immunologic detection of microbial antigens, latex particle agglutination, coagglutination, and enzyme-linked immunosorbent assay (ELISA) are the most frequently used techniques in the clinical laboratory. Antibody to a specific antigen is bound to latex particles or to a heat-killed and treated protein A-rich strain of Staphylococcus aureus to produce agglutination (Fig. 10-2). There are several approaches to ELISA; the one most frequently used for the detection of microbial antigens uses an antigen-specific antibody that is fixed to a solid phase, which may be a latex or metal bead or the inside surface of a well in a plastic tray. Antigen present in the specimen binds to the antibody as inFig. 10-2. The test is then completed by adding a second antigen-specific antibody bound to an enzyme that can react with a substrate to produce a colored product. The initial antigen antibody complex forms in a manner similar to that shown inFigure 10-2. When the enzyme-conjugated antibody is added, it binds to previously unbound antigenic sites, and the antigen is, in effect, sandwiched between the solid phase and the enzyme-conjugated antibody. The reaction is completed by adding the enzyme substrate.
agglutination test ch10f2
Figure 2 Agglutination test in which inert particles (latex beads or heat-killed S aureus Cowan 1 strain with protein A) are coated with antibody to any of a variety of antigens and then used to detect the antigen in specimens or in isolated bacteria
Genetic probes are based on the detection of unique nucleotide sequences with the DNA or RNA of a microorganism. Once such a unique nucleotide sequence, which may represent a portion of a virulence gene or of chromosomal DNA, is found, it is isolated and inserted into a cloning vector (plasmid), which is then transformed into Escherichia coli to produce multiple copies of the probe. The sequence is then reisolated from plasmids and labeled with an isotope or substrate for diagnostic use. Hybridization of the sequence with a complementary sequence of DNA or RNA follows cleavage of the double-stranded DNA of the microorganism in the specimen.
The use of molecular technology in the diagnoses of infectious diseases has been further enhanced by the introduction of gene amplication techniques, such as the polymerase chain reaction (PCR) in which DNA polymerase is able to copy a strand of DNA by elongating complementary strands of DNA that have been initiated from a pair of closely spaced oligonucleotide primers. This approach has had major applications in the detection of infections due to microorganisms that are difficult to culture (e.g. the human immunodeficiency virus) or that have not as yet been successfully cultured (e.g. the Whipple’s disease bacillus).
Solid media, although somewhat less sensitive than liquid media, provide isolated colonies that can be quantified if necessary and identified. Some genera and species can be recognized on the basis of their colony morphologies.
In some instances one can take advantage of differential carbohydrate fermentation capabilities of microorganisms by incorporating one or more carbohydrates in the medium along with a suitable pH indicator. Such media are called differential media (e.g., eosin methylene blue or MacConkey agar) and are commonly used to isolate enteric bacilli. Different genera of the Enterobacteriaceae can then be presumptively identified by the color as well as the morphology of colonies.
Culture media can also be made selective by incorporating compounds such as antimicrobial agents that inhibit the indigenous flora while permitting growth of specific microorganisms resistant to these inhibitors. One such example is Thayer-Martin medium, which is used to isolate Neisseria gonorrhoeae. This medium contains vancomycin to inhibit Gram-positive bacteria, colistin to inhibit most Gram-negative bacilli, trimethoprim-sulfamethoxazole to inhibit Proteus species and other species that are not inhibited by colistin and anisomycin to inhibit fungi. The pathogenic Neisseria species, N gonorrhoeae and N meningitidis, are ordinarily resistant to the concentrations of these antimicrobial agents in the medium.
Infection of the bladder (cystitis) or kidney (pyelone-phritis) is usually accompanied by bacteriuria of about ≥ 104 CFU/ml. For this reason, quantitative cultures (Fig. 10-3) of urine must always be performed. For most other specimens a semiquantitative streak method (Fig. 10-3) over the agar surface is sufficient. For quantitative cultures, a specific volume of specimen is spread over the agar surface and the number of colonies per milliliter is estimated.
Identification of bacteria (including mycobacteria) is based on growth characteristics (such as the time required for growth to appear or the atmosphere in which growth occurs), colony and microscopic morphology, and biochemical, physiologic, and, in some instances, antigenic or nucleotide sequence characteristics. The selection and number of tests for bacterial identification depend upon the category of bacteria present (aerobic versus anaerobic, Gram-positive versus Gram-negative, cocci versus bacilli) and the expertise of the microbiologist examining the culture. Gram-positive cocci that grow in air with or without added CO2 may be identified by a relatively small number of tests. The identification of most Gram-negative bacilli is far more complex and often requires panels of 20 tests for determining biochemical and physiologic characteristics.
Antimicrobial susceptibility tests are performed by either disk diffusion or a dilution method. In the former, a standardized suspension of a particular microorganism is inoculated onto an agar surface to which paper disks containing various antimicrobial agents are applied. Following overnight incubation, any zone diameters of inhibition about the disks are measured. An alternative method is to dilute on a log2 scale each antimicrobial agent in broth to provide a range of concentrations and to inoculate each tube or, if a microplate is used, each well containing the antimicrobial agent in broth with a standardized suspension of the microorganism to be tested. The lowest concentration of antimicrobial agent that inhibits the growth of the microorganism is the minimal inhibitory concentration.
Classification Principles
This Week’s Citation Classic®_______ Sneath P H A & Sokal R R.
Numerical taxonomy: the principles and practice of
numerical classification. San Francisco: Freeman, 1973. 573 p.
[Medical Research Council Microbial Systematics Unit, Univ. Leicester, England
and Dept. Ecology and Evolution, State Univ. New York, Stony Brook, NY]
Numerical taxonomy establishes classification
of organisms based on their similarities. It utilizes
many equally weighted characters and employs
clustering and similar algorithms to yield
objective groupings. It can beextended to give
phylogenetic or diagnostic systems and can be
applied to many other fields of endeavour.
Mathematical Foundations of Computer Science 1998
Lecture Notes in Computer Science Volume 1450, 1998, pp 474-482
Date: 28 May 2006
Positive Turing and truth-table completeness for NEXP are incomparable 1998
Levke Bentzien
The truth-table method [matrix method] is one of the decision procedures for sentence logic (q.v., §3.2). The method is based on the fact that the truth value of a compound formula of sentence logic, construed as a truth-function, is determined by the truth values of its arguments (cf. “Sentence logic” §2.2). To decide whether a formula A is a tautology or not, we list all possible combinations of truth values to the variables in A: A is a tautology if it takes the value truth under each assignment.
Using ideas introduced by Buhrman et al. ([2], [3]) to separate various completeness notions for NEXP = NTIME (2poly), positive Turing complete sets for NEXP are studied. In contrast to many-one completeness and bounded truth-table completeness with norm 1 which are known to coincide on NEXP ([3]), whence any such set for NEXP is positive Turing complete, we give sets A and B such that
A is ≤ bT(2) P -complete but not ≤ posT P -complete for NEXP
B is ≤ posT P -complete but not ≤ tt P -complete for NEXP. These results come close to optimality since a further strengthening of (1), as was done by Buhrman in [1] for EXP = DTIME(2poly), seems to require the assumption NEXP = co-NEXP.
Computability and Models
The University Series in Mathematics 2003, pp 1-10
Truth-Table Complete Computably Enumerable Sets
Marat M. Arslanov
We prove a truth-table completeness criterion for computably enumerable sets.
The authors research was partially supported by Russian Foundation of Basic Research, Project 99-01-00830, and RFBR-INTAS, Project 97-91-71991.
TRUTH TABLE CLASSIFICATION AND IDENTIFICATION*
EUGENE W. RYPKA
Department of Microbiology, Lovelace Foundation for Medical Education and Research,
Albuquerque, N.M. 87108, U.S.A.
Space life sciences 1971-12-1; 3(2): pp 135-156 http://dx.doi.org:/10.1007/BF00927988
(Received 15 July, 1971)
Abstract. A logical basis for classification is that elements grouped together and higher categories of elements should have a high degree of similarity with the provision that all groups and categories be disjoint to some degree. A methodology has been developed for constructing classifications automatically that gives
nearly instantaneous correlations of character patterns of organisms with time and clusters with apparent similarity. This means that automatic numerical identification will always construct schemes from which disjoint answers can be obtained if test sensitivities for characters are correct. Unidentified organisms are recycled through continuous classification with reconstruction of identification schemes. This process is
cyclic and self-correcting. The method also accumulates and analyzes data which updates and presents a more accurate biological picture.
Syndromic classification: A process for amplifying information using S-clustering
Rypka’s[1] method[2] utilizes the theoretical and empirical separatory equations shown below to perform the task of optimal classification. The method finds the optimal order of the fewest attributes, which in combination define a bounded class of elements.
Application of the method begins with construction of an attribute-valued system in truth table[3] or spreadsheet form with elements listed in the left most column beginning in the second row. Characteristics[4] are listed in the first row beginning in the second column with the code name of the data in the upper left most cell. The values which connect each characteristic with each element are placed in the intersecting cells. Selecting appropriate characteristics to universally define the class of elements may be the most difficult part for the classifier of utilizing this method.
The elements are first sorted in descending order according to their truth table value, which is calculated from the existing sequence and value of characteristics for each element. Duplicate truth table values or multisets for the entire bounded class reveal either the need to eliminate duplicate elements or the need to include additional characteristics.
An empirical separatory value is calculated for each characteristic in the set and the characteristic with the greatest empirical separatory value is exchanged with the characteristic which occupies the most significant attribute position.
Next the second most significant characteristic is found by calculating an empirical separatory value for each remaining characteristic in combination with the first characteristic. The characteristic which produces the greatest separatory value is then exchanged with the characteristic which occupies the second most significant attribute position.
Next the third most significant characteristic is found by calculating an empirical separatory value for each remaining characteristic in combination with the first and second characteristics. The characteristic which produces the greatest empirical separatory value is then exchanged with the characteristic which occupies the third most significant attribute position. This procedure may continue until all characteristics have been processed or until one hundred percent separation of the elements has been achieved.
A larger radix will allow faster identification by excluding a greater percentage of elements per characteristic. A binary radix for instance excludes only fifty percent of the elements per characteristic whereas a five-valued radix excludes eighty percent of the elements per characteristic.[5] What follows is an elucidation of the matrix and separatory equations.[6]
Computational Example
Bounded Class Data
Bounded Class Dimensions
G = 28 – 28 elements – i = 0…G-1[1]
C = 10 – 10 characteristics or attributes – j = 0…C-1
V = 5 – 5 valued logic – l = 0…V-1
Order of Elements
Count multisets
Squared multiset Counts
Separatory Values
max(T) = 309 = S8 = highest initial separatory value
Notes
Mathcad’s ORIGIN function applies to all arrays such that if more than one array is being used and one array requires a zero origin then the other arrays must use a zero origin with all variables being adapted as well.
Rypka’s Method Edit
Rypka’s[1] method[2] utilizes the theoretical and empirical separatory equations shown below to perform the task of optimal classification. The method finds the optimal order of the fewest attributes, which in combination define a bounded class of elements.
Application of the method begins with construction of an attribute-valued system in truth table[3] or spreadsheet form with elements listed in the left most column beginning in the second row. Characteristics[4] are listed in the first row beginning in the second column with the title of the attributes in the upper left most cell. Normally the file name of the data is given the title of the element class. The values which connect each characteristic with each element are placed in the intersecting cells. Selecting characteristics which all elements share may be the most difficult part of creating a database which can utilizing this method.
The elements are first sorted in descending order according to their truth table value, which is calculated from the existing sequence and value of characteristics for each element. Duplicate truth table values or multisets for the entire bounded class reveal either the need to eliminate duplicate elements or the need to include additional characteristics.
An empirical separatory value is calculated for each characteristic in the set and the characteristic with the greatest empirical separatory value is exchanged with the characteristic which occupies the most significant attribute position.
Next the second most significant characteristic is found by calculating an empirical separatory value for each remaining characteristic in combination with the first characteristic. The characteristic which produces the greatest separatory value is then exchanged with the characteristic which occupies the second most significant attribute position.
Next the third most significant characteristic is found by calculating an empirical separatory value for each remaining characteristic in combination with the first and second characteristics. The characteristic which produces the greatest empirical separatory value is then exchanged with the characteristic which occupies the third most significant attribute position. This procedure may continue until all characteristics have been processed or until one hundred percent separation of the elements has been achieved.
A larger radix will allow faster identification by excluding a greater percentage of elements per characteristic. A binary radix for instance excludes only fifty percent of the elements per characteristic whereas a five-valued radix excludes eighty percent of the elements per characteristic.[5] What follows is an elucidation of the matrix and separatory equations.[6]
Syndromic Classification: A Process for Amplifying Information Using S-Clustering
Eugene W. Rypka, PhD
University of New Mexico, Albuquerque, New Mexico, USA
Statistics Editor: Marcello Pagano, PhD
Harvard School of Public Health, Boston, Massachusetts, USA
Nutrition 1996; 12(11/12): 827-829
In a previous issue of Nutrition, Drs. Bernstein and Pleban’ use the method of S-clustering to aid in nutritional classification of patients directly on-line. Classification of this type is called primary or syndromic classification.* It is created by a process called separatory (S-) clustering (E. Rypka, unpublished observations). The authors use S-clustering in Table I. S-clustering extracts features (analytes, variables) from endogenous data that amplify or maximize structural information to create classes of patients (pathophysiologic events) which are the most disjointed or separable. S-clustering differs from other classificatory methods because it finds in a database a theoretic- or more- number of variables with the required variety that map closest to an ideal, theoretic, or structural information standard. In Table I of their article, Bernstein and Pleban’ indicate there would have to be 3 ’ = 243 rows to show all possible patterns. In Table II of this article, I have used a 33 = 27 row truth table to convey the notion of mapping amplified information to an ideal, theoretic standard using just the first three columns. Variables are scaled for use in S-clustering.
A Survey of Binary Similarity and Distance Measures
Seung-Seok Choi, Sung-Hyuk Cha, Charles C. Tappert
SYSTEMICS, CYBERNETICS AND INFORMATICS 2010; 8(1): 43-48
The binary feature vector is one of the most common
representations of patterns and measuring similarity and
distance measures play a critical role in many problems
such as clustering, classification, etc. Ever since Jaccard
proposed a similarity measure to classify ecological
species in 1901, numerous binary similarity and distance
measures have been proposed in various fields. Applying
appropriate measures results in more accurate data
analysis. Notwithstanding, few comprehensive surveys
on binary measures have been conducted. Hence we
collected 76 binary similarity and distance measures used
over the last century and reveal their correlations through
the hierarchical clustering technique.
This paper is organized as follows. Section 2 describes
the definitions of 76 binary similarity and dissimilarity
measures. Section 3 discusses the grouping of those
measures using hierarchical clustering. Section 4
concludes this work.
Historically, all the binary measures observed above have
had a meaningful performance in their respective fields.
The binary similarity coefficients proposed by Peirce,
Yule, and Pearson in 1900s contributes to the evolution
of the various correlation based binary similarity
measures. The Jaccard coefficient proposed at 1901 is
still widely used in the various fields such as ecology and
biology. The discussion of inclusion or exclusion of
negative matches was actively arisen by Sokal & Sneath
in during 1960s and by Goodman & Kruskal in 1970s.
Polyphasic Taxonomy of the Genus Vibrio: Numerical Taxonomy of Vibrio cholerae, Vibrio
parahaemolyticus, and Related Vibrio Species
R. R. COLWELL
JOURNAL OF BACTERIOLOGY, Oct. 1970; 104(1): 410-433
A set of 86 bacterial cultures, including 30 strains of Vibrio cholerae, 35 strains of
V. parahaemolyticus, and 21 representative strains of Pseudomonas, Spirillum,
Achromobacter, Arthrobacter, and marine Vibrio species were tested for a total of 200
characteristics. Morphological, physiological, and biochemical characteristics were
included in the analysis. Overall deoxyribonucleic acid (DNA) base compositions
and ultrastructure, under the electron microscope, were also examined. The taxonomic
data were analyzed by computer by using numerical taxonomy programs
designed to sort and cluster strains related phenetically. The V. cholerae strains
formed an homogeneous cluster, sharing overall S values of >75%. Two strains,
V. cholerae NCTC 30 and NCTC 8042, did not fall into the V. cholerae species
group when tested by the hypothetical median organism calculation. No separation
of “classic” V. cholerae, El Tor vibrios, and nonagglutinable vibrios was observed.
These all fell into a single, relatively homogeneous, V. cholerae species cluster.
PJ. parahaemolyticus strains, excepting 5144, 5146, and 5162, designated members
of the species V. alginolyticus, clustered at S >80%. Characteristics uniformly
present in all the Vibrio species examined are given, as are also characteristics and
frequency of occurrence for V. cholerae and V. parahaemolyticus. The clusters formed
in the numerical taxonomy analyses revealed similar overall DNA base compositions,
with the range for the Vibrio species of 40 to 48% guanine plus cytosine. Generic
level of relationship of V. cholerae and V. parahaemolyticus is considered
dubious. Intra- and intergroup relationships obtained from the numerical taxonomy
studies showed highly significant correlation with DNA/DNA reassociation data.
A Numerical Classification of the Genus Bacillus
By FERGUS G . PRIEST, MICHAEL GOODFELLOW AND CAROLE TODD
Journal of General Microbiology (1988), 134, 1847-1882.
Three hundred and sixty-eight strains of aerobic, endospore-forming bacteria which included type and reference cultures of Bacillus and environmental isolates were studied. Overall similarities of these strains for 118 unit characters were determined by the SSMS,, and Dp coefficients and clustering achieved using the UPGMA algorithm. Test error was within acceptable limits. Six cluster-groups were defined at 70% SSM which corresponded to 69% Sp and 48-57% SJ.G roupings obtained with the three coefficients were generally similar but there were some changes in the definition and membership of cluster-groups and clusters, particularly with the SJ coefficient. The Bacillus strains were distributed among 31 major (4 or more strains), 18 minor (2 or 3 strains) and 30 single-member clusters at the 83% SsMle vel. Most of these clusters can be regarded as taxospecies. The heterogeneity of several species, including Bacillus breuis, B. circulans, B. coagulans, B. megateriun, B . sphaericus and B . stearothermophilus, has been indicated and the species status of several taxa of hitherto uncertain validity confirmed. Thus on the basis of the numerical phenetic and appropriate (published) molecular genetic data, it is proposed
that the following names be recognized; BacillusJlexus (Batchelor) nom. rev., Bacillus fusiformis (Smith et al.) comb. nov., Bacillus kaustophilus (Prickett) nom. rev., Bacilluspsychrosaccharolyticus (Larkin & Stokes) nom. rev. and Bacillus simplex (Gottheil) nom. rev. Other phenetically well-defined taxospecies included ‘ B. aneurinolyticus’, ‘B. apiarius’, ‘B. cascainensis’, ‘B. thiaminolyticus’ and three clusters of environmental isolates related to B . firmus and previously described as ‘B. firmus-B. lentus intermediates’. Future developments in the light of the numerical phenetic data are discussed.
Numerical Classification of Bacteria
Part II. * Computer Analysis of Coryneform Bacteria (2)
Comparison of Group-Formations Obtained on Two
Different Methods of Scoring Data
By Eitaro MASUOan d Toshio NAKAGAWA
[Agr. Biol. Chem., 1969; 33(8): 1124-1133.
Sixty three organisms selected from 12 genera of bacteria were subjected to numerical analysis. The purpose of this work is to examine the relationships among 38 coryneform bacteria included in the test organisms by two coding methods-Sneath’s and Lockhart’s systems-, and to compare the results with conventional classification. In both cases of codification, five groups and one or two single item(s) were found in the resultant classifications. Different codings brought, however, a few distinct differences in some groups , especially in a group of sporogenic bacilli or lactic-acid bacteria. So far as the present work concerns, the result obtained on Lockhart’s coding rather than that obtained on Sneath’s coding resembled the conventional classification. The taxonomic positions of corynebacteria were quite different from those of the conventional classification, regardless
of which coding method was applied.
Though animal corynebacteria have conventionally been considered to occupy the
taxonomic position neighboring to genera Arthrobacter and Cellulornonas and regarded to be the nucleus of so-called “coryneform bacteria,’ the present work showed that many of the corynebacteria are akin to certain mycobacteria rather than to the organisms belonging to the above two genera.
Numerical Classification of Bacteria
Part III. Computer Analysis of “Coryneform Bacteria” (3)
Classification Based on DNA Base Compositions
By EitaroM ASUaOnd ToshioN AKAGAWA
Agr. Biol. Chem., 1969; 33(11): 1570-1576
It has been known that the base compositions of deoxyribonucleic acids (DNA) are
quite different from organism to organism. A pertinent example of this diversity is
found in bacterial species. The base compositions of DNA isolated from a wide variety
of bacteria are distributed in a range from 25 to 75 GC mole-percent (100x(G+C)/
(A+T+G+C)).1) The usefulness of the information of DNA base composition for
the taxonomy of bacteria has been emphasized by several authors. Lee et al.,” Sueoka,” and Freese) have speculated on the evolutionary significance of microbial DNA base composition. They pointed out that closely related microorganisms generally showed similar base compositions of DNA, and suggested that phylogenetic relationship should be reflected in the GC content.
In the present paper are compared the results of numerical classifications of 45
bacteria based on the two different similarity matrices: One representing the overall
similarities of phenotypic properties, the other representing the similarities of GC contents.
Advanced computational algorithms for microbial community analysis using massive 16S rRNA
sequence data
Y Sun, Y Cai, V Mai, W Farmerie, F Yu, J Li and S Goodison
Nucleic Acids Research, 2010; 38(22): e205 http://dx.doi.org:/10.1093/nar/gkq872
With the aid of next-generation sequencing technology, researchers can now obtain millions of microbial signature sequences for diverse applications ranging from human epidemiological studies to global ocean surveys. The development of advanced computational strategies to maximally extract pertinent information from massive nucleotide data has become a major focus of the bioinformatics community. Here, we describe a novel analytical strategy including discriminant and topology analyses that enables researchers to deeply investigate the hidden world of microbial communities, far beyond basic microbial diversity estimation. We demonstrate the utility of our
approach through a computational study performed on a previously published massive human gut 16S rRNA data set. The application of discriminant and
topology analyses enabled us to derive quantitative disease-associated microbial signatures and describe microbial community structure in far more detail than previously achievable. Our approach provides rigorous statistical tools for sequence based studies aimed at elucidating associations between known or unknown organisms and a variety of physiological or environmental conditions.
What is Drug Resistance?
Antimicrobial resistance is the ability of microbes, such as bacteria, viruses, parasites, or fungi, to grow in the presence of a chemical (drug) that would normally kill it or limit its growth.
Diagram showing the difference between non-resistant bacteria and drug resistant bacteria.
Credit: NIAID
DrugResistance difference between non-resistant bacteria and drug resistant bacteria
Diagram showing the difference between non-resistant bacteria and drug resistant bacteria. Non-resistant bacteria multiply, and upon drug treatment, the bacteria die. Drug resistant bacteria multiply as well, but upon drug treatment, the bacteria continue to spread.
Between 5 and 10 percent of all hospital patients develop an infection. About 90,000 of these patients die each year as a result of their infection, up from 13,300 patient deaths in 1992.
According to the Centers for Disease Control and Prevention (April 2011), antibiotic resistance in the United States costs an estimated $20 billion a year in excess health care costs, $35 million in other societal costs and more than 8 million additional days that people spend in the hospital.
Resistance to Antibiotics: Are We in the Post-Antibiotic Era?
Serious infections caused by bacteria that have become resistant to commonly used antibiotics have become a major global healthcare problem in the 21st century. They not only are more severe and require longer and more complex treatments, but they are also significantly more expensive to diagnose and to treat. Antibiotic resistance, initially a problem of the hospital setting associated with an increased number of hospital acquired infections usually in critically ill and immunosuppressed patients, has now extended into the community causing severe infections difficult to diagnose and treat. The molecular mechanisms by which bacteria have become resistant to antibiotics are diverse and complex. Bacteria have developed resistance to all different classes of antibiotics discovered to date. The most frequent type of resistance is acquired and transmitted horizontally via the conjugation of a plasmid. In recent times new mechanisms of resistance have resulted in the simultaneous development of resistance to several antibiotic classes creating very dangerous multidrug-resistant (MDR) bacterial strains, some also known as ‘‘superbugs’’. The indiscriminate and inappropriate use of antibiotics in outpatient clinics, hospitalized patients and in the food industry is the single largest factor leading to antibiotic resistance. The pharmaceutical industry, large academic institutions or the government are not investing the necessary resources to produce the next generation of newer safe and effective antimicrobial drugs. In many cases, large pharmaceutical companies have terminated their anti-infective research programs altogether due to economic reasons. The potential negative consequences of all these events are relevant because they put society at risk for the spread of potentially serious MDR bacterial infections.
Targeting the Human Macrophage with Combinations of Drugs and Inhibitors of Ca2+ and K+ Transport to Enhance the Killing of Intracellular Multi-Drug Resistant M. tuberculosis (MDR-TB) – a Novel, Patentable Approach to Limit the Emergence of XDR-TB
Marta Martins
Recent Patents on Anti-Infective Drug Discovery, 2011, 6, 000-000
The emergence of resistance in Tuberculosis has become a serious problem for the control of this disease. For that reason, new therapeutic strategies that can be implemented in the clinical setting are urgently needed. The design of new compounds active against mycobacteria must take into account that Tuberculosis is mainly an intracellular infection of the alveolar macrophage and therefore must maintain activity within the host cells. An alternative therapeutic approach will be described in this review, focusing on the activation of the phagocytic cell and the subsequent killing of the internalized bacteria. This approach explores the combined use of antibiotics and phenothiazines, or Ca2+ and K+ flux inhibitors, in the infected macrophage. Targeting the infected macrophage and not the internalized bacteria could overcome the problem of bacterial multi-drug resistance. This will potentially eliminate the appearance of new multi-drug resistant tuberculosis (MDR-TB) cases and subsequently prevent the emergence of extensively-drug resistant tuberculosis (XDR-TB). Patents resulting from this novel and innovative approach could be extremely valuable if they can be implemented in the clinical setting. Other patents will also be discussed such as the treatment of TB using immunomodulator compounds (for example: betaglycans).
Medical illustration of Streptococcus pneumonia. [CDC]
Streptococcus pneumonia
It appears that S. pneumoniae has even more personalities, each associated with a different proclivity toward invasive, life-threatening disease. In fact, any of six personalities may emerge depending on the action of a single genetic switch.
To uncover the switch, an international team of scientists conducted a study in genomics, but they looked beyond nucleotide polymorphisms or accessory regions as possible phenotype-shifting mechanisms. Instead, they focused on the potential of restriction-modification (RM) systems to mediate gene regulation via epigenetic changes.
Scientists representing the University of Leicester, Griffith University’s Institute for Glycomics, theUniversity of Adelaide, and Pacific Biosciences realized that the S. pneumoniae genome contains two Type I, three Type II, and one Type IV RM systems. Of these, only the DpnI Type II RM system had been described in detail. Switchable Type I systems had been described previously, but these reports did not provide evidence for differential methylation or for phenotypic impact.
As it turned out, the Type I system embodied a mechanism capable of randomly changing the bacterium’s characteristics into six alternative states. The mechanism’s details were presented September 30 in Nature Communications, in an article entitled, “A random six-phase switch regulates pneumococcal virulence via global epigenetic changes.”
“The underlying mechanism for such phase variation consists of genetic rearrangements in a Type I restriction-modification system (SpnD39III),” wrote the authors. “The rearrangements generate six alternative specificities with distinct methylation patterns, as defined by single-molecule, real-time (SMRT) methylomics.”
Eradication of multidrug-resistant A. baumanniii in burn wounds by antiseptic pulsed electric field.
A Golberg, GF Broelsch, D Vecchio,S Khan, MR Hamblin, WG Austen, Jr, RL Sheridan, ML Yarmush.
Emerging bacterial resistance to multiple drugs is an increasing problem in burn wound management. New non-pharmacologic interventions are needed for wound disinfection. Here we report on a novel physical method for disinfection: antiseptic pulsed electric field (PEF) applied externally to the infected wounds. In an animal model, we show that PEF can reduce the load of multidrug resistant Acinetobacter baumannii present in a full thickness burn wound by more than four orders of magnitude, as detected by bioluminescence imaging. Furthermore, using a finite element numerical model, we demonstrate that PEF provides non-thermal, homogeneous, full thickness treatment for the burn wound, thus, overcoming the limitation of treatment depth for many topical antimicrobials. These modeling tools and our in vivo results will be extremely useful for further translation of the PEF technology to the clinical setting. We believe that PEF, in combination with systemic antibiotics, will synergistically eradicate multidrug-resistant burn wound infections, prevent biofilm formation and restore natural skin microbiome. PEF provides a new platform for infection combat in patients, therefore it has a potential to significantly decreasing morbidity and mortality.
Golberg, A. & Yarmush, M. L. Nonthermal irreversible electroporation: fundamentals, applications, and challenges. IEEE Trans Biomed Eng 60, 707-14 (2013).
Mechanisms Of Antibiotic Resistance In Salmonella: Efflux Pumps, Genetics, Quorum Sensing And Biofilm Formation.
Martins M, McCusker M, Amaral L, Fanning S
Perspectives in Drug Discovery and Design 02/2011; 8:114-123.
In Salmonella the main mechanisms of antibiotic resistance are mutations in target genes (such as DNA gyrase and topoisomerase IV) and the over-expression of efflux pumps. However, other mechanisms such as changes in the cell envelope; down regulation of membrane porins; increased lipopolysaccharide (LPS) component of the outer cell membrane; quorum sensing and biofilm formation can also contribute to the resistance seen in this microorganism. To overcome this problem new therapeutic approaches are urgently needed. In the case of efflux-mediated multidrug resistant isolates, one of the treatment options could be the use of efflux pump inhibitors (EPIs) in combination with the antibiotics to which the bacteria is resistant. By blocking the efflux pumps resistance is partly or wholly reversed, allowing antibiotics showing no activity against the MDR strains to be used to treat these infections. Compounds that show potential as an EPI are therefore of interest, as well as new strategies to target the efflux systems. Quorum sensing (QS) and biofilm formation are systems also known to be involved in antibiotic resistance. Consequently, compounds that can disrupt or inhibit these bacterial “communication systems” will be of use in the treatment of these infections.
Role of Phenothiazines and Structurally Similar Compounds of Plant Origin in the Fight against Infections by Drug Resistant Bacteria
Phenothiazines have their primary effects on the plasma membranes of prokaryotes and eukaryotes. Among the components of the prokaryotic plasma membrane affected are efflux pumps, their energy sources and energy providing enzymes, such as ATPase, and genes that regulate and code for the permeability aspect of a bacterium. The response of multidrug and extensively drug resistant tuberculosis to phenothiazines shows an alternative therapy for its treatment. Many phenothiazines have shown synergistic activity with several antibiotics thereby lowering the doses of antibiotics administered for specific bacterial infections. Trimeprazine is synergistic with trimethoprim. Flupenthixol (Fp) has been found to be synergistic with penicillin and chlorpromazine (CPZ); in addition, some antibiotics are also synergistic. Along with the antibacterial action described in this review, many phenothiazines possess plasmid curing activities, which render the bacterial carrier of the plasmid sensitive to antibiotics. Thus, simultaneous applications of a phenothiazine like TZ would not only act as an additional antibacterial agent but also would help to eliminate drug resistant plasmid from the infectious bacterial cells.
Multidrug Efflux Pumps Described for Staphylococcus aureus
a n.d.: The family of transporters to which SepA belongs is not elucidated to date.
b n.i.: The transporter has no regulator identified to date.
QACs: quaternary ammonium compounds
The importance of efflux pumps in bacterial antibiotic resistance
A. Webber and L. J. V. Piddock
Journal of Antimicrobial Chemotherapy (2003) 51, 9–11 http://dx.doi.org:/10.1093/jac/dkg050Efflux pumps are transport proteins involved in the extrusion of toxic substrates (including virtually all classes of clinically relevant antibiotics) from within cells into the external environment. These proteins are found in both Gram-positive and -negative bacteria as well as in eukaryotic organisms. Pumps may be specific for one substrate or may transport a range of structurally dissimilar compounds (including antibiotics of multiple classes); such pumps can be associated with multiple drug resistance (MDR). In the prokaryotic kingdom there are five major families of efflux transporter: MF (major facilitator), MATE (multidrug and toxic efflux), RND (resistance-nodulation-division), SMR (small multidrug resistance) and ABC (ATP binding cassette). All these systems utilize the proton motive force as an energy source. Advances in DNA technology have led to the identification of members of the above families. Transporters that efflux multiple substrates, including antibiotics, have not evolved in response to the stresses of the antibiotic era. All bacterial genomes studied contain efflux pumps that indicate their ancestral origins. It has been estimated that ∼5–10% of all bacterial genes are involved in transport and a large proportion of these encode efflux pumps.
The efflux pump
Multidrug-resistance efflux pumps — not just for resistance
Laura J. V. Piddock
Nature Reviews | Microbiology | Aug 2006; 4: 629
It is well established that multidrug-resistance efflux pumps encoded by bacteria can confer clinically relevant resistance to antibiotics. It is now understood that these efflux pumps also have a physiological role(s). They can confer resistance to natural substances produced by the host, including bile, hormones and host defense molecules. In addition, some efflux pumps of the resistance nodulation division (RND) family have been shown to have a role in the colonization and the persistence of bacteria in the host. Here, I present the accumulating evidence that multidrug-resistance efflux pumps have roles in bacterial pathogenicity and propose that these pumps therefore have greater clinical relevance than is usually attributed to them.


















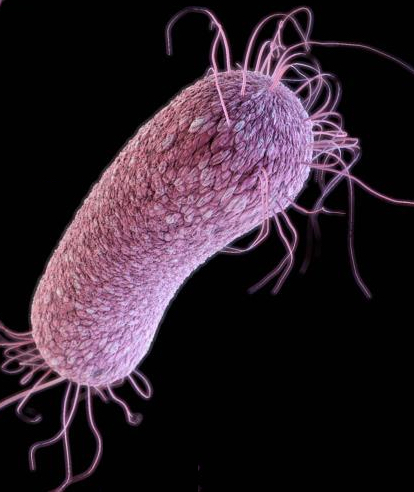{kind=link}
{kind=link}
{kind=link}
{kind=link}