Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Protein Switches: The Programmable Future of Bio-therapeutics
Curator: Dr. Sudipta Saha, Ph. D.
A PNAS paper entitled “A protein therapeutic modality founded on molecular regulation” presents a pioneering approach to creating protein switches—engineered enzymes that activate only in specific molecular environments. This design introduces a new class of context-dependent therapeutics for precision medicine.
Using domain-insertion techniques, researchers inserted ligand-binding domains into scaffold proteins like β-lactamase. These proteins remain inactive until encountering a specific small molecule, which triggers a conformational change and restores enzymatic activity. This offers precise spatiotemporal control—ideal for minimizing off-target effects.
One key innovation is the systematic insertional mutagenesis that identifies functional switch sites across the protein scaffold. This enables the construction of vast protein libraries, increasing the likelihood of finding optimal switch configurations. Furthermore, the approach is modular—different binding domains and enzymes can be combined to create switches tailored to specific clinical contexts.
These smart proteins can be programmed to respond to cancer biomarkers, metabolite levels, or disease-specific molecular cues. By activating only under disease conditions, they provide a blueprint for next-generation bio-therapeutics—potent, selective, and safer.
The method also opens avenues for drug delivery systems, diagnostics, and biosensors, where conditional activation is critical. Overall, this work represents a conceptual leap in synthetic biology and bioengineering, with implications spanning oncology, infectious disease, and regenerative medicine.
Recent research from University of Alberta is looking at the role of fibrinogen, the substrate of thrombin in regulating a natural defense mechanism in the body. Fibrinogen is a well-known protein that is essential for wound healing and blood clotting in the body. Levels of fibrinogen increase in inflammatory states as part of the acute-phase response.
However, daily variation in plasma fibrinogen levels weakens its potential as a biomarker of cardiovascular risk. The discovery is expected to contribute to enhanced diagnosis and treatments for patients in a variety of diseases ranging from inflammation, to heart failure, to cancer.
Yet, a study published in Scientific Reports in March 2019 show that fibrinogen can also be a natural inhibitor of an enzyme named MMP2, which is important for normal organ development and repair. The researchers believe an essential function of fibrinogen is to allow or disallow the enzyme to carry out its normal functions. Nevertheless, high levels of fibrinogen may disproportionately inhibit MMP2, that could result in arthritic and cardiac disorders.
The researcher highlights the inner workings of the MMP family of enzymes by having a greater understanding of how MMPs are regulated. They hypothesize that abnormal MMP2 activity could be an unwelcome side effect of common medications such as the cholesterol-lowering drugs and the antibiotic doxycycline, both of which are known to inhibit MMPs. They also emphasize that future therapeutic developments must strike a balance between the levels of MMPs and their inhibitors, such as fibrinogen, so that net MMP activity in the body remains at nearly normal levels.
One of the unexpected findings of the Human Genome Project was that over 98% of the human genome does not encode for proteins. Once dismissed as “junk” genomic material, non-protein-coding DNA is now appraised more highly.
Or to be more precise, at least some portions of non-protein-coding DNA are thought to serve important biological functions.
For example, some stretches of DNA give rise to a noncoding but still functional kind of RNA called microRNA. MicroRNAs have increasingly emerged in recent years as key regulators of biological processes and pathways.
During the years since their discovery, a key question in the biology of microRNAs has focused on the number of microRNAs encoded in the genome. Between 1993 and 2015, approximately 1,900 human genome loci were discovered to produce microRNAs and were added to miRBbase, the public database that catalogues and annotates microRNA molecules.
The cataloguing of microRNAs work has been pursued with extra urgency since 2004, the year the connection between microRNAs and human disease was first demonstrated. “When this connection was made, it launched a whole new field,” says Isidore Rigoutsos, Ph.D., professor of pathology, anatomy, and cell biology and director of the Computational Medicine Center at Thomas Jefferson University.
Another Set of MicroRNAs Emerge
“We wanted to know how many microRNA-producing loci really exist in humans,” recalls Dr. Rigoutsos. In a study published in 2015, Dr. Rigoutsos and colleagues analyzed datasets from 1,323 individuals that represented 13 different tissues and identified an additional 3,356 such genomic loci that produce (at least) 3,707 novel microRNs
“We basically tripled the number of locations in the human genome that are now known to encode microRNAs,” asserts Dr. Rigoutsos. Considering that each microRNA regulates up to hundreds of different mRNAs, and that each mRNA is regulated by tens of microRNAs, this finding adds a new layer of complexity to the regulatory dynamics of the human transcriptome.
The newly unveiled microRNAs and previously characterized microRNAs have distinct expression patterns. While 50–60% of the microRNAs previously deposited into the miRBase are expressed in multiple tissues, only about 10% of the newly discovered microRNAs are shared across multiple tissue types. Also, most of the newly found microRNAs show tissue-specific expression.
Using Argonaute CLIP-seq data, Dr. Rigoutsos and colleagues showed that similar percentages of the two sets of microRNAs were in complex with Argonaute proteins. “This shows that these novel microRNAs participate in RNA interference just as frequently as the miRBase microRNAs,” contends Dr. Rigoutsos.
In a comparative analysis between the human microRNA datasets and the chimpanzee, gorilla, orangutan, macaque, mouse, fruit fly, and mouse genomes, Dr. Rigoutsos and colleagues discovered that almost 95% of the newly unveiled microRNAs were primate-specific, and over 56% of them were found only in humans.
“We are seeing many human microRNAs that do not exist in the mouse,” states Dr. Rigoutsos. “This means that the mouse models engineered to capture human disease cannot recapitulate the interactions mediated by these microRNAs.
Interest in IsomiRs Grows
In the years since the biology of microRNAs started receiving increasing attention, the conventional view has been that one microRNA locus generates one microRNA. However, once deep sequencing became widely available, microRNA variants that showed differences at their 5′- or 3′-termini have been described.
“It was initially presumed that these variants were likely the result of the enzyme Dicer not being sufficiently accurate when processing microRNA precursors,” notes Dr. Rigoutsos. Subsequent research revealed that microRNAs are more dynamic than previously thought, with each precursor being able to generate multiple mature microRNA species known as isomiRs.
To gain insight into the biology of isomiRs, Dr. Rigoutsos and colleagues analyzed genomic datasets from 452 individuals participating in the 1000 Genomes Project. The datasets comprised five different populations and two races. In addition, each population was represented by an even number of men and women.
This collection allowed the abundance of microRNA isoforms to be examined with respect to population, gender, and race. “We found that isomiRs have expression profiles that are population-, race-, and gender-dependent,” informs Dr. Rigoutsos.
All the transcriptome data that this analysis was based on came from immortalized B cells. “These are cells that normally are not associated with gender differences, but molecularly we found, in these cells, differences between men and women of the same population and race,” explains Dr. Rigoutsos.
Expanding these observations to disease states, Dr. Rigoutsos and colleagues collected isomiR profiles from tissue affected by breast cancer, and compared them with isomiR profiles from control breast tissue. The investigators found that the isomiR profiles also depend on tissue state (healthy vs. diseased), on disease subtype, and on the patient’s race.
For example, their analysis identified several miR-183-5p isoforms that were upregulated in white triple-negative breast cancer patients compared to control breast samples, but not in black/African-American triple-negative breast cancer patients. In an in vitro phase of this study, three isoforms of this microRNA species were overexpressed in human breast cancer cell lines.
“We found very little overlap in the gene sets that were affected by each of these isoforms,” emphasizes Dr. Rigoutsos. Despite being generated simultaneously by the same locus, each of the three isoforms affected distinct groups of genes, thus exerting different effects on the transcriptome.
“As the relative abundance of these isoforms changes ever so slightly from patient to patient, it will affect the corresponding gene groups slightly differently,” concludes Dr. Rigoutsos. “In the process, it creates a new molecular background in each patient.”
MicroRNAs Point to Therapeutic Strategies against Colorectal Cancer
“We are using microRNAs as modulators to overcome chemotherapy resistance in colorectal cancer,” says Jingfang Ju, Ph.D., associate professor of pathology and co-director of translational research at Stony Brook University School of Medicine. Resistance to chemotherapy is one of the major challenges in the clinical management of malignancies, including colorectal cancer. Chemotherapy is usually unable to eliminate cancer stem cells, which may become even more resistant over time, and several microRNAs have been implicated in this process. “We reasoned that we could provide new modulatory approaches to target this small cell population and allow chemotherapy, radiotherapy, or immunotherapy to eliminate resistant populations or at least prolong long-term survival,” Dr. Ju said.
This image shows how miR-129 may function as a tumor suppressor in colorectal cancer. In this model, which has been proposed by researchers at Stony Brook University’s Translational Research Laboratory, miR-129 suppresses the protein expression of three critical targets—BCL2, TS, and E2F3. Downregulation of BCL2 activates the intrinsic apoptosis pathway by cleaving caspase-9 and caspase-3. Downregulation of TS and E2F3 inhibits cell proliferation by impacting the cell cycle. Consequently, miR-129 exerts a strong antitumor phenotype by induction of apoptosis and impairment of proliferation in tumor cells. [Mihriban Karaayvaz, Haiyan Zhai, Jingfang Ju]
In a retrospective study in which colorectal patient samples were used, Dr. Ju and colleagues revealed that hsa-miR-140-5p expression progressively decreases from normal tissues to primary colorectal cancer tissue, and that it shows a further decrease in liver and lymph node metastases. The experimental overexpression of hsa-miR-140-5p inhibited colorectal cancer stem cell growth by disrupting autophagy, and in a mouse model of disease it abolished tumor formation and metastasis.
In addition to hsa-miR-140-5p, Dr. Ju and colleagues recently identified hsa-miR-129 and found that it, too, has therapeutic potential. Specifically, they showed that hsa-miR-129 enhanced the sensitivity of colorectal cancer cells to 5-fluorouracil, pointing toward its ability to function as a tumor suppressor.
One of the mechanisms implicated in this process was the ability of miR-192 to inhibit protein translation of several important targets. These include Bcl-2 (B-cell lymphoma 2), a key anti-apoptotic protein; E2F3, a major cell cycle regulator; and thymidylate synthase, an enzyme that is inhibited by 5-fluorouracil.
The NIH recently awarded a $3 million grant to establish the Long Island Bioscience Hub (LIBH), which is part of the NIH’s Research Evaluation and Commercialization Hub (REACH) program and represents a partnership between the Center for Biotechnology, Stony Brook University, Cold Spring Harbor Laboratory, and Brookhaven National Laboratory. One of the technology development grants, as part of the first funding cycle of this initiative, will support a feasibility investigation of hsa-miR-129-based therapeutics in colon cancer, an effort led by Dr. Ju. “We are further exploring this novel mechanism,” states Dr. Ju. “We anticipate conducting pharmacokinetic studies and moving to a clinical trial in the future.”
MicroRNA Insights Gleaned from Host-Virus Interactions
At Mount Sinai Hospital’s Icahn School of Medicine, researchers used a codon-optimized version of VP55 produced from an adenovirus-based vector to study the impact of microRNA deletion on the response to virus infection. This image shows RNA in situ hybridization of fibroblasts expressing VP55 (top left), and that of mock-treated fibroblasts (bottom right). Ribosomal RNA, DNA, and microRNAs (miR-26) are depicted by red, blue (DAPI), and green fluorophores, respectively.
“We observed that when a poxvirus is artificially engineered to encode a microRNA, the microRNA is destroyed along with all the microRNAs from the host cell,” says Benjamin R. tenOever, Ph.D., professor of microbiology at the Icahn School of Medicine, Mount Sinai Hospital. Previously, Dr. tenOever’s group reported that a single vaccinia virus-encoded gene product, VP55, is sufficient to achieve this effect. The group also found that the protein adds nontemplate adenosines to the 3′-end of microRNAs associated with the RNA-induced silencing complex.
biology,” asserts Dr. tenOever.
In a recent study, Dr. tenOever and colleagues used a codon-optimized version of VP55 produced from an adenovirus-based vector to study the impact microRNA deletion would have on our normal response to virus infection. “We found that after administration of the vector and rapid ablation of microRNA expression, there is very little that happens over the first one to two days,” informs Dr. tenOever. During the first 24–48 hours after VP55 delivery, the elimination of cellular microRNAs impacted less than 0.35% of the over 11,000 genes expressed in the cell. After 9 days, however, almost 20% of the genes showed significant changes in expression.
“MicroRNAs are very powerful and influential in controlling the biology of the cell but they do so over the long term,” declares Dr. tenOever. These findings are in agreement with knowledge that has accumulated over the years about microRNA biology, which established that microRNAs play a central role in determining how cells differentiate during development.
“While microRNAs can act on hundreds of mRNAs, their action requires several days of fine-tuning to have long-term consequences,” adds Dr. tenOever. This finding suggests miRNAs are unable to significantly contribute to the acute response to virus infection.
The one exception to this observation was that, even though very few genes were affected in the first 48 hours after VP55 delivery, several genes encoding chemokines were impacted. These included chemokines responsible for recruiting antigen-presenting cells, neutrophils, and other immune cells.
An in vivo analysis of mouse lung tissue 48 hours after vector administration confirmed that several cytokines were specifically upregulated, resulting in immune cell infiltration following the degradation of all microRNAs. These results indicate that the acute viral infection is largely independent of microRNAs, and that microRNAs are primarily involved in the adaptive response to infection and other longer term processes.
MicroRNA Biomarkers Reveal Molecular Pathways of Kidney Damage
“Our approach involves looking at microRNAs from the perspective of biomarkers as a readout for kidney damage,” says Vishal S. Vaidya, Ph.D., associate professor of medicine and environmental health at Brigham and Women’s Hospital, Harvard Medical School, and Harvard T.H. Chan School of Public Health. “At the same time, we are exploring their utility as therapeutics.”
A large number of medications and occupational toxins cause kidney damage, but many tests to assess kidney function and damage are not sufficiently sensitive or specific, opening the need for novel diagnostic strategies. MicroRNAs, which are differentially expressed between healthy and diseased states, are promising as early biomarkers for impaired renal function.
“MicroRNAs can also provide information about which pathways are active and which targets can be druggable,” points out Dr. Vaidya.
In a study that used microRNAs and proteins to provide a combined biomarker signature, Dr. Vaidya and colleagues examined two patient cohorts, one presenting with acetaminophen-induced kidney injury and the other one with cisplatin-induced kidney damage. “Protein biomarkers provide sensitivity, and microRNAs offer mechanistic insight,” explains Dr. Vaidya.
This approach helped visualize metabolic pathways that are altered in the kidney during toxic injury. “The biggest challenge, from a therapeutic perspective, is that microRNAs regulate many mRNAs and, therefore, impact many proteins,” concludes Dr. Vaidya.
Signaling of Immune Response in Colon Cancer, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Signaling of Immune Response in Colon Cancer
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Revised 1/13/2016
STING Protein May Serve as Biomarker for Colorectal and Other Cancers
Scientists at University of Miami Miller School of Medicine’s Sylvester Comprehensive Cancer Center say they have discovered how the stimulator of interferon genes (STING) signaling pathway may play an important role in alerting the immune system to cellular transformation. They believe their finding will shed further light on the immune system’s response to cancer development.
In 2008, Glen N. Barber, Ph.D., leader of the viral oncology program at Sylvester, and professor and chairman of cell biology at the Miller School of Medicine, and colleagues published in Nature (“STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling”) the discovery of STING as a new cellular molecule that recognizes virus and bacteria infection to initiate host defense and immune responses. In the new study, published in Cell Reports (“Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis”), they describe STING’s role in the potential suppression of colorectal cancer.
“Since 2008 we’ve known that STING is crucial for antiviral and antibacterial responses,” said Dr. Barber. “But until now, little had been known about its function in human tumors. In this study we show, for the first time, that STING signaling is repressed in colorectal carcinoma and other cancers, an event which may enable transformed cells to evade the immune system.”
Colorectal cancer currently affects around 1.2 million people in the U.S. and 150,000 new cases are diagnosed every year, making it the third most common cancer in both men and women. Since most colon cancers develop from benign polyps, they can be treated successfully when detected early. However, if the tumor has already spread, survival rates are generally low.
Using disease models of colorectal cancer, the team of Sylvester scientists showed that loss of STING signaling negatively affected the body’s ability to recognize DNA-damaged cells. In particular, certain cytokines that facilitate tissue repair and antitumor priming of the immune system were not sufficiently produced to initiate a significant immune response to eradicate the colorectal cancer.
“We were able to show that impaired STING responses may enable damaged cells to elude the immune system,” continued Dr. Barber. “And if the body doesn’t recognize and attack cancer cells, they will multiply and, ultimately, spread to other parts of the body.”
He and his colleagues suggest evaluating STING signaling as a prognostic marker for the treatment of colorectal as well as other cancers. For example, Dr. Barber’s study showed that cancer cells with defective STING signaling were particularly prone to attack by oncolytic viruses presently being used as cancer therapies.
“Impaired STING responses may enable damaged cells to evade host immunosurveillance processes, although they provide a critical prognostic measurement that could help predict the outcome of effective oncoviral therapy,” wrote the investigators.
This is the first detailed examination of how the stimulator of interferon genes (STING) signaling pathway, discovered by Glen N. Barber, Ph.D., Leader of the Viral Oncology Program at Sylvester Comprehensive Cancer Center, may play an important role in alerting the immune system to cellular transformation.
In 2008, Barber, who is also Professor and Chairman of Cell Biology at the University of Miami Miller School of Medicine, and colleagues published in Nature the discovery of STINGas a new cellular molecule that recognizes virus and bacteria infection to initiate host defense and immune responses. In the new study they describe STING’s role in the potential suppression of colorectal cancer.
“Since 2008 we’ve known that STING is crucial for antiviral and antibacterial responses,” said Barber. “But until now, little had been known about its function in human tumors. In this study we show, for the first time, that STING signaling is repressed in colorectal carcinoma and other cancers, an event which may enable transformed cells to evade the immune system.”
Colorectal cancer currently affects around 1.2 million people in the United States and 150.000 new cases are diagnosed every year, making it the third most common cancer in both men and women. Since most colon cancers develop from benign polyps, they can be treated successfully when detected early. However, if the tumor has already spread, survival rates are generally low.
Using disease models of colorectal cancer, the team of Sylvester scientists showed that loss of STING signaling negatively affected the body’s ability to recognize DNA-damaged cells. In particular, certain cytokines – small proteins important for cell signaling – that facilitate tissue repair and anti-tumor priming of the immune system were not sufficiently produced to initiate a significant immune response to eradicate the colorectal cancer.
“We were able to show that impaired STING responses may enable damaged cells to elude the immune system,” added Barber. “And if the body doesn’t recognize and attack cancer cells, they will multiply and, ultimately, spread to other parts of the body.”
Barber and his colleagues suggest evaluating STING signaling as a prognostic marker for the treatment of colorectal as well as other cancers. For example, Barber’s study showed that cancer cells with defective STING signaling were particularly prone to attack by oncolytic viruses presently being used as cancer therapies. Alternate studies with colleagues have also shown that activators of STING signaling are potent stimulators of anti-tumor immune responses. Collectively, the control of STING signaling may have important implications for cancer development as well as cancer treatment.
Every step you take: STING pathway key to tumor immunity
A recently discovered protein complex known as STING plays a crucial role in detecting the presence of tumor cells and promoting an aggressive anti-tumor response by the body’s innate immune system, according to two separate studies published in the Nov. 20 issue of the journal Immunity.
The studies, both from University of Chicago-based research teams, have major implications for the growing field of cancer immunotherapy. The findings show that when activated, the STING pathway triggers a natural immune response against the tumor. This includes production of chemical signals that help the immune system identify tumor cells and generate specific killer T cells. The research also found that targeted high-dose radiation therapy dials up the activation of this pathway, which promotes immune-mediated tumor control.
These findings could “enlarge the fraction of patients who respond to immunotherapy with prolonged control of the tumor,” according to a commentary on the papers by the University of Verona’s Vincenzo Bronte, MD. “Enhancing the immunogenicity of their cancers might expand the lymphocyte repertoire that is then unleashed by interference with checkpoint blockade pathways,” such as anti-PD-1.
STING, short for STimulator of INterferon Genes complex, is a crucial part of the process the immune system relies on to detect threats — such as infections or cancer cells — that are marked by the presence of DNA that is damaged or in the wrong place, inside the cell but outside the nucleus.
Detection of such “cytosolic” DNA initiates a series of interactions that lead to the STING pathway. Activating the pathway triggers the production of interferon-beta, which in turn alerts the immune system to the threat, helps the system detect cancerous or infected cells, and ultimately sends activated T cells into the battle.
“We have learned
“Innate immune sensing via the host STING pathway is critical for tumor control by checkpoint blockade,” Gajewski’s team noted in their paper. They found promising drugs known as checkpoint inhibitors — such as anti PD-1 or anti PD-L1, which can take the brakes off of an immune response — were not effective in STING-deficient mice. New agents that stimulate the STING pathway are being developed as potential cancer therapeutics.
A second University of Chicago team, led by cancer biologistYang-Xin Fu, MD, PhD, professor of pathology, and Ralph Weichselbaum, MD, chairman of radiation and cellular oncology and co-director of the Ludwig Center for Metastasis Research, found that high-dose radiation therapy not only kills targeted cancer cells but the resulting DNA damage drives a systemic immune response.
a great deal recently about what we call checkpoints, the stumbling blocks that prevent the immune system from ultimately destroying cancers,” said Thomas Gajewski, MD, PhD, professor of medicine and pathology at the University of Chicago and senior author of one of the studies. “Blockade of immune checkpoints, such as with anti-PD-1, is therapeutic in a subset of patients, but many individuals still don’t respond. Understanding the role of the STING pathway provides insights into how we can ‘wake up’ the immune response against tumors. This can be further boosted by checkpoint therapies.”
The two published studies, he said, help move this approach forward.
In a series of experiments in mice, both research teams found tumor cell-derived DNA could initiate an immune response against cancers. But when tested in mice that lacked a functional gene for STING, the immune system did not effectively respond.
“This result unifies traditional studies of DNA damage with newly identified DNA sensing of immune responses,” Fu said.
“This is a previously unknown mechanism,” Weichselbaum added.
In mice that lacked STING, however, there was no therapeutic immune response. The induction of interferons by radiation and consequent cancer cell killing, they conclude, depends on STING-pathway signaling.
They did find that combining cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), an earlier step in the STING pathway, with radiation, could greatly enhance the antitumor efficacy of radiation.
“This opens a new avenue to develop STING-related agonists for patients with radiation-resistant cancers,” Fu said.
STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors
• Spontaneous T cell responses against tumors require the host STING pathway in vivo
• Tumor-derived DNA can induce type I interferon production via STING
• Tumor DNA can be identified in host APCs in the tumor microenvironment in vivo
Summary
Spontaneous T cell responses against tumors occur frequently and have prognostic value in patients. The mechanism of innate immune sensing of immunogenic tumors leading to adaptive T cell responses remains undefined, although type I interferons (IFNs) are implicated in this process. We found that spontaneous CD8+ T cell priming against tumors was defective in mice lacking stimulator of interferon genes complex (STING), but not other innate signaling pathways, suggesting involvement of a cytosolic DNA sensing pathway. In vitro, IFN-β production and dendritic cell activation were triggered by tumor-cell-derived DNA, via cyclic-GMP-AMP synthase (cGAS), STING, and interferon regulatory factor 3 (IRF3). In the tumor microenvironment in vivo, tumor cell DNA was detected within host antigen-presenting cells, which correlated with STING pathway activation and IFN-β production. Our results demonstrate that a major mechanism for innate immune sensing of cancer occurs via the host STING pathway, with major implications for cancer immunotherapy.
Immunity Erratum STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors
Seng-Ryong Woo, Mercedes B. Fuertes, Leticia Corrales, Stefani Spranger, Michael J. Furdyna, Michael Y.K. Leung, Ryan Duggan, Ying Wang, Glen N. Barber, Katherine A. Fitzgerald, Maria-Luisa Alegre, and Thomas F. Gajewski* *Correspondence: tgajewsk@medicine.bsd.uchicago.eduhttp://dx.doi.org/10.1016/j.immuni.2014.12.015 (Immunity 41, 830–842; November 20, 2014)
The original Figure 3C accidentally contained a duplicated panel in the bright-field column, third row down, and this has now been replaced with the correct data. The change does not alter the conclusions of the paper. This mistake has now been corrected online, and the authors regret the error.
Cytosolic DNA Sensors (CDSs): a STING in the tail – Review
The innate immune system provides the first line of defense against infectious pathogens and serves to limit their early proliferation. It is also vital in priming and activating the adaptive immune system.
Innate immune detection of intracellular DNA derived from viruses and invasive bacteria is important to initiate an effective protective response. This crucial step depends on cytosolic DNA sensors (CDSs), which upon activation trigger the production of type I interferons (IFNs) and the induction of IFN-responsive genes and proinflammatory chemokines.
Although the identity of these CDSs is not fully uncovered, much progress has been made in understanding the signaling pathways triggered by these sensors.
Cytosolic DNA-mediated production of type I IFNs (mainly IFN-β) requires the transcription factor IFN regulatory factor 3 (IRF3), which is activated upon phosphorylation by TANK-binding-kinase-1 (TBK1) [1].
STING in DNA sensing
Recently, a new molecule, STING (stimulator of IFN genes), has been shown to be essential for the TBK1-IRF3- dependent induction of IFN-β by transfected DNA ligands and intracellular DNA produced by pathogens after infection [2, 3].
STING (also known as MITA, MPYS and ERIS) is a transmembrane protein that resides in the endoplasmic reticulum (ER) [2-6]. In response to cytosolic DNA, STING forms dimers and translocates from the ER to the Golgi then to punctate cytosolic structures where it colocalizes with TBK-1, leading to the phosphorylation of IRF3.
How STING stimulates TBK1-dependent IRF3 activation was recently elucidated by Tanaka and Chen. They found that, upon cytosolic DNA sensing, the C-terminal tail of STING acts as a scaffold protein to promote the phosphorylation of IRF3 by TBK1 [7].
STING in the host response to intracellular pathogens. Linking type I IFN response and autophagy for better defense
Until very recently, STING in addition to its role as an adaptor protein was also thought to function as a sensor of cyclic dinucleotides.
Burdette et al. first demonstrated that STING binds directly to the bacterial molecule cyclic diguanylate monophosphate (c-di-GMP) [8]. This finding was confirmed by several teams who examined the structure of STING bound to c-di-GMP [9-11], including Cheng and colleagues, however their data suggest that STING is not the primary sensor of c-di-GMP [12]. Rather, they indicate that DDX41, an identified CDS, functions as a direct receptor for cyclic dinucleotides upstream of STING. The authors hypothesized that DDX41 binds to c-di-GMP then forms a complex with STING to activate the IFN response.
STING induces autophagy
Exciting new developments reveal that STING participates in another aspect of innate immunity, autophagy.
Autophagy plays a critical role in host defense responses to pathogens by promoting the elimination of microbes that enter into the cytosol by their sequestration into autophagosomes and their delivery to the lysosome.
Recent studies have reported that DNA viruses and intracellular bacteria induce autophagy and that this process is dependent on cytosolic genomic DNA and STING [13-15]. Robust induction of autophagy was also observed after transfection of various double stranded (ds) DNA species, such as poly(dA:dT), poly(dG:dC) or plasmid DNA, but not single stranded (ss) DNA, dsRNA or ssRNA [16].
Interestingly, activated STING was shown to relocate to unidentified membrame-bound compartments where it colocalizes with LC3, a hallmark of autophagy, and ATg9a. The latter protein was reported to regulate the interaction between STING and TBK1 after dsDNA stimulation [16]. The E3 ubiquitin ligases TRIM56 and TRIM32
were also found to regulate STING by mediating its dimerization through K63-linked ubiquitination [17, 18].
Several cytosolic DNA sensors upstream of STING have been proposed. DNA-dependent activator of IRFs (DAI) was the first CDS discovered based on the ability of transfected poly(dA:dT) to induce IFN-β [19]. However, the role of DAI has been shown to be very cell-type specific and cells derived from DAI-deficient mice responded normally to dsDNA ligands [20].
While analyzing immune responses to dsDNA regions derived from vaccinia virus (VACV-70) or Herpes simplex virus 1 (HSV-60) genomes, Unterholzner et al. identified IFI16 as a DNA binding protein mediating IFN-β induction [21]. Interestingly, IFI16 belongs to a new family of pattern recognition receptors that contain the pyrin and HIN domain (PYHIN), termed AIM2-like receptors (ALRs).
AIM2 is a STING-independent cytosolic DNA sensor that forms an inflammasome with ASC to trigger caspase-1 activation and the secretion of the proinflammatory cytokines IL-1β and IL-18 [20].
Members of the DExD/H-box helicase superfamily have also been reported to function as cytosolic DNA sensors. While DHX36 and DHX9 were identified as STING-independent but MyD88-dependent sensors of CpG-containing DNA in plasmacytoid dendritic cells, DDX41 was found to bind various dsDNA ligands and localize with STING to promote IFN-β expression [22]. Other CDSs have been reported to function independently of STING: RNA Pol III, LRRFIP1 and Ku70 [20].
Unlike cytosolic RNA sensors (RIG-I, MDA-5), which detect structural moieties specific to pathogen RNA, such as 5’-triphosphates, it is not clear whether cytosolic DNA sensors can recognize any particular structural motif of DNA that would discriminate between self and non-self. This suggests that CDSs may have a role not only in anti-microbial innate immune responses but also in autoimmunity. A multitude of CDSs have been described but whether they are all true receptors remains an open question.
1. Stetson DB & Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 24(1):93-103.
2. Ishikawa H. & Barber GN., 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 455(7213):674-8.
3. Ishikawa H. et al., 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 461(7265):788-92.
4. Zhong B. et al., 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 29(4):538-50.
5. Jin L. et al., 2008. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 28(16):5014-26.
UV Light Potentiates STING (Stimulator of Interferon Genes)-dependent Innate Immune Signaling through Deregulation of ULK1 (Unc51-like Kinase 1).
The mechanism by which ultraviolet (UV) wavelengths of sunlight trigger or exacerbate the symptoms of the autoimmune disorder lupus erythematosus is not known but may involve a role for the innate immune system. Here we show that UV radiation potentiates STING (stimulator of interferon genes)-dependent activation of the immune signaling transcription factor interferon regulatory factor 3 (IRF3) in response to cytosolic DNA and cyclic dinucleotides in keratinocytes and other human cells. Furthermore, we find that modulation of this innate immune response also occurs with UV-mimetic chemical carcinogens and in a manner that is independent of DNA repair and several DNA damage and cell stress response signaling pathways. Rather, we find that the stimulation of STING-dependent IRF3 activation by UV is due to apoptotic signaling-dependent disruption of ULK1 (Unc51-like kinase 1), a pro-autophagic protein that negatively regulates STING. Thus, deregulation of ULK1 signaling by UV-induced DNA damage may contribute to the negative effects of sunlight UV exposure in patients with autoimmune disorders.
STING and the innate immune response to nucleic acids in the cytosol
Cytosolic detection of pathogen-derived nucleic acids is critical for the initiation of innate immune defense against diverse bacterial, viral and eukaryotic pathogens. Conversely, inappropriate responses to cytosolic nucleic acids can produce severe autoimmune pathology. The host protein STING has been identified as a central signaling molecule in the innate immune response to cytosolic nucleic acids. STING seems to be especially critical for responses to cytosolic DNA and the unique bacterial nucleic acids called ‘cyclic dinucleotides’. Here we discuss advances in the understanding of STING and highlight the many unresolved issues in the field.
The detection of pathogen-derived nucleic acids is a central strategy by which the innate immune system senses microbes to then initiate protective responses1. Conversely, inappropriate recognition of self nucleic acids can result in debilitating autoimmune diseases such as systemic lupus erythematosus2. It is therefore important to understand the molecular basis of the detection of nucleic acids by the innate immune system. Studies have established that nucleic acids derived from extracellular sources are sensed mainly by endosomal Toll-like receptors (TLRs), such as TLR3, TLR7 and TLR9, whereas cytosolic nucleic acids are detected independently of TLRs by a variety of less-well-characterized mechanisms1.
Studies have identified STING (‘stimulator of interferon genes’; also known as TMEM173, MPYS, MITA and ERIS) as a critical signaling molecule in the innate response to cytosolic nucleic-acid ligands. STING was first described as a protein that interacts with major histocompatibility complex class II molecules3, but the relevance of this interaction remains unclear. Subsequent studies have instead focused on the role of STING in the transcriptional induction of type I interferons and coregulated genes in response to nucleic acids in the cytosol. Several groups have independently isolated STING by screening for proteins able to induce interferon-B (IFN-B) when overexpressed4–6. Studies of STING-deficient mice have subsequently confirmed the essential role of STING in innate responses to cytosolic nucleic-acid ligands, particularly double-stranded DNA (dsDNA) and unique bacterial nucleic acids called ‘cyclic dinucleotides’7–9. Several studies have also linked STING to the interferon response to cytosolic RNA5–7, but this has not been found consistently7,8,10,11; thus, we focus here on the role of STING in response to DNA and cyclic dinucleotides.
Protein Stimulator of interferon genes protein
Gene TMEM173
Organism Homo sapiens (Human)
Facilitator of innate immune signaling that acts as a sensor of cytosolic DNA from bacteria and viruses and promotes the production of type I interferon (IFN-alpha and IFN-beta). Innate immune response is triggered in response to non-CpG double-stranded DNA from viruses and bacteria delivered to the cytoplasm. Acts by recognizing and binding cyclic di-GMP (c-di-GMP), a second messenger produced by bacteria, and cyclic GMP-AMP (cGAMP), a messenger produced in response to DNA virus in the cytosol: upon binding of c-di-GMP or cGAMP, autoinhibition is alleviated and TMEM173/STING is able to activate both NF-kappa-B and IRF3 transcription pathways to induce expression of type I interferon and exert a potent anti-viral state. May be involved in translocon function, the translocon possibly being able to influence the induction of type I interferons. May be involved in transduction of apoptotic signals via its association with the major histocompatibility complex class II (MHC-II). Mediates death signaling via activation of the extracellular signal-regulated kinase (ERK) pathway. Essential for the induction of IFN-beta in response to human herpes simplex virus 1 (HHV-1) infection. Exhibits 2′,3′ phosphodiester linkage-specific ligand recognition. Can bind both 2′-3′ linked cGAMP and 3′-3′ linked cGAMP but is preferentially activated by 2′-3′ linked cGAMP (PubMed:26300263)
Stimulator of interferon genes protein (IPR029158)
Transmembrane protein 173, also known as stimulator of interferon genes protein (STING) or endoplasmic reticulum interferon stimulator (ERIS), is a transmembrane adaptor protein which is involved in innate immune signalling processes. It induces expression of type I interferons (IFN-alpha and IFN-beta) via the NF-kappa-B and IRF3, pathways in response to non-self cytosolic RNA and dsDNA [PMID: 18724357, PMID: 19776740,PMID: 18818105, PMID: 19433799].
A new periodic table presents a systematic, ordered view of protein assembly, providing a visual tool for understanding biological function. [EMBL-EBI / Spencer Phillips]
Move over Mendeleev, there’s a new periodic table in science. Unlike the original periodic table, which organized the chemical elements, the new periodic table organizes protein complexes, or more precisely, quaternary structure topologies. Though there are other differences between the old and new periodic tables, they share at least one important feature—predictive power.
When Mendeleev introduced his periodic table, he predicted that when new chemical elements were discovered, they would fill his table’s blank spots. Analogous predictions are being ventured by the scientific team that assembled the new periodic table. This team, consisting of scientists from the Wellcome Genome Campus and the University of Cambridge, asserts that its periodic table reveals the regions of quaternary structure space that remain to be populated.
The periodic table of protein complexes not only offers a new way of looking at the enormous variety of structures that proteins can build in nature, it also indicates which structures might be discovered next. Moreover, it could point protein engineers toward entirely novel structures that never occurred in nature, but could be engineered.
The new table appeared December 11 in the journal Science, in an article entitled, “Principles of assembly reveal a periodic table of protein complexes.” The “principles of assembly” referenced in this title amount to three basic assembly types: dimerization, cyclization, and heteromeric subunit addition. In dimerization, one protein complex subunit doubles, and becomes two; in cyclization, protein complex subunits from a ring of three or more; and in heteromeric subunit addition, two different proteins bind to each other.
These steps, repeated in different combinations, gives rise to enormous number of proteins of different kinds. “Evolution has given rise to a huge variety of protein complexes, and it can seem a bit chaotic,” explained Joe Marsh, Ph.D., formerly of the Wellcome Genome Campus and now of the MRC Human Genetics Unit at the University of Edinburgh. “But if you break down the steps proteins take to become complexes, there are some basic rules that can explain almost all of the assemblies people have observed so far.”
The authors of the Science article noted that many protein complexes assemble spontaneously via ordered pathways in vitro, and these pathways have a strong tendency to be evolutionarily conserved. “[There] are strong similarities,” the authors added, “between protein complex assembly and evolutionary pathways, with assembly pathways often being reflective of evolutionary histories, and vice versa. This suggests that it may be useful to consider the types of protein complexes that have evolved from the perspective of what assembly pathways are possible.”
To explore this rationale, the authors examined the fundamental steps by which protein complexes can assemble, using electrospray mass spectrometry experiments, literature-curated assembly data, and a large-scale analysis of protein complex structures. Ultimately, they derived their approach to explaining the observed distribution of known protein complexes in quaternary structure space. This approach, they insist, provides a framework for understanding their evolution.
“In addition, it can contribute considerably to the prediction and modeling of quaternary structures by specifying which topologies are most likely to be adopted by a complex with a given stoichiometry, potentially providing constraints for multi-subunit docking and hybrid methods,” the authors concluded. “Lastly, it could help in the bioengineering of protein complexes by identifying which topologies are most likely to be stable, and thus which types of essential interfaces need to be engineered.”
The rows and columns of the periodic table of the elements, called periods and groups, were originally determined by each element’s atomic mass and chemical properties, later by atomic number and electron configuration. In contrast, the rows and columns of the periodic table of protein complexes correspond to the number of different subunit types and the number of times these subunits are repeated. The new table is not, it should be noted, periodic in the same sense as the periodic table of the elements. It is in principle open-ended.
Although there are no theoretical limitations to quaternary structure topology space in either dimension, the abridged version of the table presented in the Science article can accommodate the vast majority of known structures. Moreover, when the table’s creators compared the large variety of countenanced topologies to observed structures, they found that about 92% of known protein complex structures were compatible with their model.
“Despite its strong predictive power, the basic periodic table model does not account for about 8% of known protein complex structures,” the authors conceded. “More than half of these exceptions arise as a result of quaternary structure assignment errors.
“A benefit of this approach is that it highlights likely quaternary structure misassignments, particularly by identifying nonbijective complexes with even subunit stoichiometry. However, this still leaves about 4% of known structures that are correct but are not compatible with the periodic table.” The authors added that the exceptions to their model are interesting in their own right, and are the subject of ongoing studies.
The Periodic Table of Protein Complexes, published today in Science, offers a new way of looking at the enormous variety of structures that proteins can build in nature, which ones might be discovered next, and predicting how entirely novel structures could be engineered. Created by an interdisciplinary team led by researchers at the Wellcome Genome Campus and the University of Cambridge, the Table provides a valuable tool for research into evolution and protein engineering.
Different ballroom dances can be seen as an endless combination of a small number of basic steps. Similarly, the ‘dance’ of protein complex assembly can be seen as endless variations on dimerization (one doubles, and becomes two), cyclisation (one forms a ring of three or more) and subunit addition (two different proteins bind to each other). Because these happen in a fairly predictable way, it’s not as hard as you might think to predict how a novel protein would form.
“We’re bringing a lot of order into the messy world of protein complexes,” explains Sebastian Ahnert of the Cavendish Laboratory at the University of Cambridge, a physicist who regularly tangles with biological problems. “Proteins can keep go through several iterations of these simple steps, , adding more and more levels of complexity and resulting in a huge variety of structures. What we’ve made is a classification based on these underlying principles that helps people get a handle on the complexity.”
The exceptions to the rule are interesting in their own right, adds Sebastian, as are the subject of on-going studies.
“By analysing the tens of thousands of protein complexes for which three-dimensional structures have already been experimentally determined, we could see repeating patterns in the assembly transitions that occur – and with new data from mass spectrometry we could start to see the bigger picture,” says Joe.
“The core work for this study is in theoretical physics and computational biology, but it couldn’t have been done without the mass spectrometry work by our colleagues at Oxford University,” adds Sarah Teichmann, Research Group Leader at the European Bioinformatics Institute (EMBL-EBI) and the Wellcome Trust Sanger Institute. “This is yet another excellent example of how extremely valuable interdisciplinary research can be.”
The assembly of proteins into complexes is crucial for most biological processes. The three-dimensional structures of many thousands of homomeric and heteromeric protein complexes have now been determined, and this has had a broad impact on our understanding of biological function and evolution. Despite this, the organizing principles that underlie the great diversity of protein quaternary structures observed in nature remain poorly understood, particularly in comparison with protein folds, which have been extensively classified in terms of their architecture and evolutionary relationships.
RATIONALE
In this work, we sought a comprehensive understanding of the general principles underlying quaternary structure organization. Our approach was to consider protein complexes in terms of their assembly. Many protein complexes assemble spontaneously via ordered pathways in vitro, and these pathways have a strong tendency to be evolutionarily conserved. Furthermore, there are strong similarities between protein complex assembly and evolutionary pathways, with assembly pathways often being reflective of evolutionary histories, and vice versa. This suggests that it may be useful to consider the types of protein complexes that have evolved from the perspective of what assembly pathways are possible.
RESULTS
We first examined the fundamental steps by which protein complexes can assemble, using electrospray mass spectrometry experiments, literature-curated assembly data, and a large-scale analysis of protein complex structures. We found that most assembly steps can be classified into three basic types: dimerization, cyclization, and heteromeric subunit addition. By systematically combining different assembly steps in different ways, we were able to enumerate a large set of possible quaternary structure topologies, or patterns of key interfaces between the proteins within a complex. The vast majority of real protein complex structures lie within these topologies. This enables a natural organization of protein complexes into a “periodic table,” because each heteromer can be related to a simpler symmetric homomer topology. Exceptions are mostly the result of quaternary structure assignment errors, or cases where sequence-identical subunits can have different interactions and thus introduce asymmetry. Many of these asymmetric complexes fit the paradigm of a periodic table when their assembly role is considered. Finally, we implemented a model based on the periodic table, which predicts the expected frequencies of each quaternary structure topology, including those not yet observed. Our model correctly predicts quaternary structure topologies of recent crystal and electron microscopy structures that are not included in our original data set.
CONCLUSION
This work explains much of the observed distribution of known protein complexes in quaternary structure space and provides a framework for understanding their evolution. In addition, it can contribute considerably to the prediction and modeling of quaternary structures by specifying which topologies are most likely to be adopted by a complex with a given stoichiometry, potentially providing constraints for multi-subunit docking and hybrid methods. Lastly, it could help in the bioengineering of protein complexes by identifying which topologies are most likely to be stable, and thus which types of essential interfaces need to be engineered.
Protein assembly steps lead to a periodic table of protein complexes and can predict likely quaternary structure topologies.
Three main assembly steps are possible: cyclization, dimerization, and subunit addition. By combining these in different ways, a large set of possible quaternary structure topologies can be generated. These can be arranged on a periodic table that describes most known complexes and that can predict previously unobserved topologies.
Classification of protein structure has had a broad impact on our understanding of biological function and evolution, yet this work has largely focused on individual protein domains and their pairwise interactions. In contrast, the assembly of individual polypeptides into protein complexes, which are ubiquitous in cells, has received comparatively little attention. The periodic table of protein complexes is a new framework for analysis of complexes based on the principles of self-assembly. This reveals that sequence-identical subunits almost always have identical assembly roles within a complex and allows us to unify the vast majority of complexes of known structure (~32,000) into about 120 topologies. This facilitates the exhaustive enumeration of unobserved protein complex topologies and has significant practical applications for quaternary structure prediction, modelling and engineering.
Chloroplast genomes encode ∼37 proteins that integrate into the thylakoid membrane. The mechanisms that target these proteins to the membrane are largely unexplored. We used ribosome profiling to provide a comprehensive, high-resolution map of ribosome positions on chloroplast mRNAs in separated membrane and soluble fractions in maize seedlings. The results show that translation invariably initiates off the thylakoid membrane and that ribosomes synthesizing a subset of membrane proteins subsequently become attached to the membrane in a nucleaseresistant fashion. The transition from soluble to membraneattached ribosomes occurs shortly after the first transmembrane segment in the nascent peptide has emerged from the ribosome. Membrane proteins whose translation terminates before emergence of a transmembrane segment are translated in the stroma and targeted to the membrane posttranslationally. These results indicate that the first transmembrane segment generally comprises the signal that links ribosomes to thylakoid membranes for cotranslational integration. The sole exception is cytochrome f, whose cleavable N-terminal cpSecA-dependent signal sequence engages the thylakoid membrane cotranslationally. The distinct behavior of ribosomes synthesizing the inner envelope protein CemA indicates that sorting signals for the thylakoid and envelope membranes are distinguished cotranslationally. In addition, the fractionation behavior of ribosomes in polycistronic transcription units encoding both membrane and soluble proteins adds to the evidence that the removal of upstream ORFs by RNA processing is not typically required for the translation of internal genes in polycistronic chloroplast mRNAs.
Significance Proteins in the chloroplast thylakoid membrane system are derived from both the nuclear and plastid genomes. Mechanisms that localize nucleus-encoded proteins to the thylakoid membrane have been studied intensively, but little is known about the analogous issues for plastid-encoded proteins. This genome-wide, high-resolution analysis of the partitioning of chloroplast ribosomes between membrane and soluble fractions revealed that approximately half of the chloroplast encoded thylakoid proteins integrate cotranslationally and half integrate posttranslationally. Features in the nascent peptide that underlie these distinct behaviors were revealed by analysis of the position on each mRNA at which elongating ribosomes first become attached to the membrane.
Structures of the HIN Domain:DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor
Electrostatic attraction underlies innate dsDNA recognition by the HIN domains
Both OB folds and the linker between them engage the dsDNA backbone
An autoinhibited state of AIM2 is activated by DNA that liberates the PYD domain
DNA serves as an oligomerization platform for the inflammasome assembly
Summary
Recognition of DNA by the innate immune system is central to antiviral and antibacterial defenses, as well as an important contributor to autoimmune diseases involving self DNA. AIM2 (absent in melanoma 2) and IFI16 (interferon-inducible protein 16) have been identified as DNA receptors that induce inflammasome formation and interferon production, respectively. Here we present the crystal structures of their HIN domains in complex with double-stranded (ds) DNA. Non-sequence-specific DNA recognition is accomplished through electrostatic attraction between the positively charged HIN domain residues and the dsDNA sugar-phosphate backbone. An intramolecular complex of the AIM2 Pyrin and HIN domains in an autoinhibited state is liberated by DNA binding, which may facilitate the assembly of inflammasomes along the DNA staircase. These findings provide mechanistic insights into dsDNA as the activation trigger and oligomerization platform for the assembly of large innate signaling complexes such as the inflammasomes.
The paper
E.W.J. Wallace et al., “Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress,” Cell, 162:1286-98, 2015.
Like cooking an egg, heating up a purified protein enough will denature it, destroying the 3-D structure key to its functionality. The protein unfolds in a one-way trip to a fried state. Previous studies of this phenomenon in cells often used exogenous proteins, which clumped together in response to heat and were largely degraded by the cell’s internal cleanup machinery—a set of molecular chaperones known as heat-shock proteins—if the cell survived.
“That, and other examples, had convinced people that what they were seeing inside cells, the clumps of proteins, represented a disaster—these giant piles of damaged proteins shoved together inside the cell so they can ultimately be cleaned up,” reflects Allan Drummond, a molecular biologist at the University of Chicago. “Nobody had really looked systematically at what happens to the proteins that are native to the cell.”
To do so, Drummond and colleagues tagged proteins in yeast cells with a set of stable isotope labels. They subjected the cells to temperatures that would stress, but not kill, them and then flash-froze them within minutes to capture a snapshot of protein aggregation at different intervals. The researchers then analyzed the clumped proteins using mass spectrometry.
Drummond’s group identified 982 proteins in the yeast cells, 177 of which aggregated in the cytosol and nucleus after heat shock. However, the researchers’ isotope labels revealed something unexpected: aggregated proteins were unclumping and returning to their previous states during the cell’s recovery period. “We had no cases that we were able to detect where proteins go into aggregates and then are degraded,” says Drummond.
“It’s really challenging this long-held assumption that heat stress causes terminal aggregation. They’ve shown that we get these aggregates or assemblies that are reversible and that may actually be pro-survival,” says Kevin Morano, a microbiologist at the University of Texas Health Science Center at Houston who studies cellular responses to stress. “It is paradigm-shifting in a sense. We thought they were destroyed.” But in fact, the researchers found, the yeast cells began growing again without making substantial numbers of new proteins, suggesting that the proteins coming out of the aggregates were still functional.
Drummond’s group figures that heat stress, and the ensuing aggregation, may trigger the cell to produce more of its molecular chaperones to aid in recovery. The team observed a three-protein complex, needed to synthesize chaperones, that was active even while clumped with other proteins. “There are many different things that aggregation seems to be doing,” says Drummond. “It’s stopping the synthesis of most proteins, but promoting synthesis of a small set of proteins that are called in response to heat shock.”
Postdoc Edward Wallace, the lead author on the study, says halting the production of most other proteins may protect newly synthesized ones. Before they have folded into their mature shapes, new proteins are more susceptible to heat shock—and may still be degraded following stress. “We speculate that [the aggregated complexes] are remodeling protein synthesis, stopping the majority of new proteins from being made, and thus preventing the aggregation of these newly synthesized proteins,” says Wallace.
Editor’s Note (December 2): The sub-headline for this article was changed to emphasize the finding that most proteins regain functionality.
Reversible, Specific, Active Aggregates of Endogenous Proteins Assemble upon Heat Stress
Edward W.J. Wallace, Jamie L. Kear-Scott, Evgeny V. Pilipenko, Michael H. Schwartz,…, Edoardo M. Airoldi, Tao Pan, Bogdan A. Budnik, D. Allan Drummond
Cell 10 Sept 2015; 162(6):1286–1298 DOI: http://dx.doi.org/10.1016/j.cell.2015.08.041
•Mass spectrometry quantifies aggregation of endogenous proteins during heat stress
•Aggregates form rapidly in specific subcellular compartments
•Endogenous protein aggregates are disassembled without degradation during recovery
•In vitro, a heat-aggregated enzyme complex retains activity and fidelity
Summary
Heat causes protein misfolding and aggregation and, in eukaryotic cells, triggers aggregation of proteins and RNA into stress granules. We have carried out extensive proteomic studies to quantify heat-triggered aggregation and subsequent disaggregation in budding yeast, identifying >170 endogenous proteins aggregating within minutes of heat shock in multiple subcellular compartments. We demonstrate that these aggregated proteins are not misfolded and destined for degradation. Stable-isotope labeling reveals that even severely aggregated endogenous proteins are disaggregated without degradation during recovery from shock, contrasting with the rapid degradation observed for many exogenous thermolabile proteins. Although aggregation likely inactivates many cellular proteins, in the case of a heterotrimeric aminoacyl-tRNA synthetase complex, the aggregated proteins remain active with unaltered fidelity. We propose that most heat-induced aggregation of mature proteins reflects the operation of an adaptive, autoregulatory process of functionally significant aggregate assembly and disassembly that aids cellular adaptation to thermal stress.
The reprogramming of fibroblasts to induced pluripotent stem cells raises the possibility that somatic cells could be directly reprogrammed to cardiac progenitor cells (CPCs). The present study aimed to assess highly efficient protein-based approaches to reduce or eliminate the genetic manipulations to generate CPCs for cardiac regeneration therapy. A combination of QQ-reagent-modified Gata4, Hand2, Mef2c, and Tbx5 and three cytokines rapidly and efficiently reprogrammed human dermal fibroblasts (HDFs) into CPCs. This reprogramming process enriched trimethylated histone H3 lysine 4, monoacetylated histone H3 lysine 9, and Baf60c at the Nkx2.5 cardiac enhancer region by the chromatin immunoprecipitation quantitative polymerase chain reaction assay. Protein-induced CPCs transplanted into rat hearts after myocardial infarction improved cardiac function, and this was related to differentiation into cardiomyocyte-like cells. These findings demonstrate that the highly efficient protein-transduction method can directly reprogram HDFs into CPCs. This protein reprogramming strategy lays the foundation for future refinements both in vitro and in vivo and might provide a source of CPCs for regenerative approaches.
Significance
The findings from the present study have demonstrated an efficient protein-transduction method of directly reprogramming fibroblasts into cardiac progenitor cells. These results have great potential in cell-based therapy for cardiovascular diseases.
The protein lifecycle is regulated by mRNA expression, translation,
and degradation. Image courtesy of Broad Communications.
Cellular protein levels are dictated by the net balance of mRNA expression (the type of RNA that provides genetic information for proteins), protein synthesis, and protein degradation. While changes in protein levels are commonly inferred from measuring changes in mRNA levels (due to the difficulties involved in measuring protein levels), it’s not often clear whether determining RNA levels is actually a good proxy for measuring protein levels.
In their recent article in the journal Science, Broad Institute researchers working in core member Aviv Regev’s and institute member Nir Hacohen’s laboratories, along with the Broad’s Proteomics Platform led by Steve Carr describe a quantitative genomic model that lets them explain the abundance of proteins in cells based on mRNA expression, translation, and degradation. They performed their study in mouse dendritic cells stimulated with LPS, a component of bacteria.
While previous studies had looked at global levels of regulation in rapidly-dividing, unstimulated cells, this work focuses on understanding how much of the change in protein levels is due to a change in mRNA expression, translation, and degradation in specific genes and classes of genes in response to a stimulus – in this case, LPS. For example, would the changes in levels of one class of proteins be mostly driven by changes in the levels of the mRNAs that encode them? On the other hand, would changes in the levels of other groups of proteins occur without changes in mRNA, but rather due to faster translation or slower degradation of the protein? These were the type of questions the scientists were interested in.
Explains co-first author Marko Jovanovic, “Can we, in a dynamic system, integrate RNA and protein life cycle data? People rarely do this, and never systematically. Can we really make a global model of gene expression where we know, in the end, how much each type of regulatory layer is contributing to each gene? You can get a global answer too, but straight percentages of global contribution of RNA levels and the protein life cycle to final protein levels was not my goal. My question was really, do we see that certain classes of genes are controlled one way and certain other classes another way and therefore gain new regulatory insight?”
Since changes in protein levels are not as dramatic and fast as changes in RNA levels, one of the greatest challenges they faced in their study was distinguishing actual signal from noise. Co-first author Michael Rooney explains how they tackled this problem: “While the quantitative accuracy of mass spectrometry has grown tremendously, we realized that statistical strategies for handling stochastic and systematic errors in the data would still be critical to getting correct results. As a first step, we developed a generative statistical model for the data. This allowed us to leverage the entire time course in a manner that was robust to missing values and stochastic variation. Second, we saw that the contribution of translation might be over-estimated if we allowed translation rates and protein levels to be calculated from the same experimental system, because in such a case they would both be confounded by the same systematic errors, making them appear more similar than they actually are. This led us to the novel strategy of creating biological replicates prepared by distinct peptide library protocols.”
In this way, the team was able to robustly build a dynamic model in which the mRNA synthesis rate, the translation rate and the protein degradation rate change over time. Based on this model, it was possible to predict how much of each of the three types of regulation are contributing to the change in the level of each protein and from that measure both globally, per gene class, and per gene, the relative contributions of each type of regulation.
Analyzing the LPS-stimulated dendritic cells, the researchers found that overall mRNA expression dominates the regulation strategies, accounting for up to 90% of the fold changes in protein level variation. This is a significant increase from their pre-stimulation measurements showing regulation of mRNA expression contributing 60-70%, translation 15-25%, and degradation also 10-20%.
What appeared to be regulated more substantially by the protein lifecycle (translation, degradation) were highly expressed genes. And, looking at changes in the number of protein molecules rather than just the relative fold changes in pre- versus post-stimulated cells, what emerges is that post-stimulation, regulation at the level of the protein lifecycle begins to dominate.
The findings lead to a model for the LPS-stimulated system in which protein expression associated with functions critical for a dendritic cell-specific functions is taken care of by regulation at the level of RNA expression. However, the readjustment of the pre-existing proteome when the cells enter a new state (for example, in response to pathogen stimulation) is controlled via regulation of the protein life cycle (translation, degradation) rather than RNA expression.
“We termed this the ‘cupcake model’,” says Jovanovic. “You have to forgive me, this is my European view on how I see people buy cupcakes. They go into the store and choose the cupcake based on the icing, so the icing is kind of the identity of the cupcake. So from one cupcake to another you are basically changing the icing. In our model, the identity of cell states is adjusted by mRNA regulation so mRNA regulation is basically contributing to the icing. However, there’s also the cake part. The cake part is often specifically adjusted to the icing on top of the cupcake. The cake part, analogous to “housekeeping genes’, also needs to change and this is mainly through the protein life cycle. I’m very biased because I don’t like the icing on cupcakes, just the cake part, and so in the same vein, I wanted to know more about how the protein lifecycle contributes to gene expression. I think people have focused too much on the icing. “
So, mRNA changes drive new cell state identity. Protein lifecycle regulation drives readjustment of preexisting “housekeeping genes” such as those encoding ribosomes and factors involved in metabolism to adjust the cell to its new state.
This approach is extensible to test the regulation of gene expression in other perturbed systems as well, and allows researchers for the first time to assess the relative contributions of each of the three levels of protein level regulation – mRNA expression, translation, and degradation – in any perturbed system.
One of the unexpected findings of the Human Genome Project was that human chromosomes contain only 20,000–25,000 protein-encoding genes, fewer than had been anticipated, …
Researchers are working on novel adaptations of HTRF-based assays, as well as their combination with other types of assays, to characterize complex disease pathways that may present multiple drug targets for disease therapy. [iStock/ponsulak]
At the 6th Cisbio HTRF symposium, “Charting the Course of Drug Discovery” held recently in Brewster, MA, investigators described how homogeneous time-resolved fluorescence (HTRF®) continues to expand and improve upon the repertoire of available bioassay formats for basic research and drug discovery. Participants described applications of these assays as integral components in studies ranging from identification of allosteric modulators as potential drugs to determination of critical components in protein-modifying biochemical pathways as new drug targets.
A form of time-resolved fluorescence energy transfer (TR-FRET) technology, HTRF brings together the sensitivity of fluorescence with the homogeneous nature of FRET and the low background of time resolution. As in other FRET systems, HTRF uses two fluorophores—a donor and an acceptor that transfer energy when in close proximity to each other. Excitation of the donor molecule by an energy source such as a laser causes the emission of light waves at donor-specific wave lengths.
If the donor and acceptor are not within proximity to each other, the donor is excited but no energy transfer occurs and no acceptor emission occurs. Dual-wavelength detection reduces buffer and media interference, and the final signal is proportional to the extent of product formation.
The HTRF assay can be miniaturized into 384- and 1536-well plate formats, which proponents say, can save reagent costs and minimize quantities of limited target and compound material used in the assay. This assay technology has been applied to many antibody-based assays, including GPCR signaling (cAMP and IP-One), kinase, cytokine, biomarker, and bioprocess (antibody and protein production), as well as assays for protein-protein, protein-peptide, and protein-DNA/RNA interactions.
Unlike traditional TR-FRET systems that employ fluorophores such as fluorescein and rhodamine that are characterized by immediate and transient emissions, HTRF-specific donors such as europium and terbium cryptate emit relatively long-lived fluorescence upon excitation. Conversely, acceptor molecules rapidly emit fluorescence.
Thus, the nonspecific short-lived background fluorescence that occurs in FRET assays can be reduced by introducing a time delay ranging from 50-150 microseconds between the initial donor excitation and measurement. In HTRF, therefore, if the donor and acceptor molecules are not within proximity, only donor emissions are detected following a time delay.
Participants at the symposium focused on novel adaptations of HTRF-based assays, as well as their combination with other types of assays, to characterize convoluted disease pathways that may present multiple drug targets for disease therapy, especially neurodegenerative disorders. In particular, several presenters noted its use in addressing what the conference keynote speaker, Terrance Kenakin, Ph.D., of the University of North Carolina, characterized as “The Perfect Storm” of pharmacology, receptor allostery, and biased signaling. Strictly defined, allosteric molecules regulate proteins by binding to the molecule at a site other than the protein’s active site.
With regard to the seven transmembrane receptors (7TMRs) also known as G protein-coupled receptors, Dr. Kenakin noted that GPCRs comprise the largest class of receptors in the human genome and are common targets for therapeutics. Originally identified as mediators of 7TMR desensitization, β-arrestins (arrestin 2 and arrestin 3), for example, are now recognized as true adaptor proteins that transduce signals to multiple effector pathways. The introduction of molecular dynamics coupled with new assays, including HTRF, he said, opened new vistas for 7TMRs as therapeutic entities. Specifically, probe-dependent allosteric vectors oriented toward the cell cytosol provided fertile ground for new 7TMR drugs in the form of ligand-producing biased signaling.
Discovering and Characterizing Allosteric Modulators
Positive and negative allosteric modulators (PAMs and NAMs) of GPCRs have emerged as a novel and highly desirable class of compounds, particularly in potential treatment for mental disorders, and for metabolic, neurodegenerative, and neuromuscular diseases. Advocates say they offer some distinct advantages over conventional competitive compounds, including the potential for fine-tuning of GPCR signaling and the promise to address formerly intractable targets.
Introduced to the market in 2010 for the treatment of secondary hyperparathyroidism in adult patients with chronic kidney disease on dialysis, Cinacalcet, a PAM, activates the calcium-sensing receptor that functions as the principal regulator of parathyroid hormone secretion. Cinacalcet is the first clinically administered allosteric modulator acting on a GPCR, and provided a proof-of-concept for future development of allosteric modulators on other GPCR drug targets..
Hayley Jones and Jeff Jerman, both of Medical Research Council Technology (MRCT) in the U.K., talked about the characterization of novel PAMs for the dopamine 1 receptor. Although preclinical and clinical data have validated this receptor as a target for drugs to improve cognitive impairment in schizophrenia, Jones noted that, to date, attempts to clinically develop agonists have failed.
She and her colleagues have approached this problem by targeting D1R via PAM saying that in contrast to “direct” orthosteric D1R agonists, PAMS potentially offer advantages, including physiological spatiotemporal control of dopamine function by enhancing the effect of its endogenous ligand and avoiding over stimulation by self-limiting effects.
The investigators said they had configured a cell-based HTRF assay to screen a subset of an MRCT compound library using CHO cells that transiently express the human receptor. Inclusion of a submaximal concentration of dopamine in the assays facilitated simultaneous detection of both PAMs and agonists, allowing them to identify novel D1R activators.
Michelle Arkin, Ph.D., of the University of California, San Francisco, focuses her research on developing small molecule modulators of allosterically regulated enzymes and protein complexes as potential drug leads. Neurodegenerative diseases such as Alzheimer’s and other “taopathies” are characterized by formation of intracellular tangles comprised of aggregated tau proteins. Previous studies have shown that the protein actyltransferase p300 acetylates tau at several sites, competing with ubiquitination and thereby inhibiting tau degradation.
Dr. Arkin and colleagues developed a high-throughput screen using HTRF to identify p300 inhibitors, designing a suite of counter screens and secondary assays to validate hits. Based on previous findings that the protease caspase-6 clips tau at specific sites and that truncated tau forms are associated with disease progression, the investigators developed selective caspase-6 inhibitors.
HTRF assays demonstrated, she said, that small molecule compounds inhibit caspase-6 mediated cleavage of tau in cell lysates, concluding that the combination of HTRF enzymatic and biophysical assay formats allow characterization of inhibitors of proteins that may be involved in tauopathy progression.
Lack of Suitable Assays
Martha Kimos, biochemist at the Lieber Institute, noted that the discovery of novel catehechol-o methyltransferase (COMT) inhibitors for use in the treatment of Parkinson ’s disease has been limited due to lack of suitable assays for high-throughput screening. COMT inhibitors like entacapone and tolcapone prolong the action of levodopa by preventing its demethylation by COMT.
Kimos and her colleagues developed an HTRF assay involving an enzymatic step that uses membrane-bound human COMT as an enzyme substrate and an assay step that measures s-adenosyl-L-homcysteine (SAH) as an enzymatic reaction product. To directly measure SAH release, an anti- SAH antibody labeled with terbium cryptate and a SAH-d2 tracer were used. The SAH released by the enzymatic reaction competes with the SAH-d2 labeled leading to a decrease of the HTRF signal. The assay, the researchers said, showed good potency for tolcapone, with a high degree of translation between data in fluorescence ratio and data in terms of SAH produced, and suitable for kinetic studies, including Km determination.
At Pfizer USA, Richard Frisbee, a scientist in the hit discovery and lead profiling (HDLP) department, and colleagues have focused on the development of HTS whole blood assays using HTRF, particularly to monitor anti-inflammatory drug potency. They noted that traditional whole-blood formats such as ELISAs for detecting cytokines require multiple assay plate manipulations, including wash steps and incubation steps, have limited throughput, and are relatively time consuming.
They reported that they had developed a sandwich immunoassay protocol that measures cytokine production in human whole blood in a 384-well format, describing key elements of the assay, including nanoliter spotting of test compounds, miniaturized blood/reagent transfer, and optimized assay incubations. Development of a relatively convenient assay to monitor compound potency in whole blood can facilitate they said, the prediction of compound doses required for therapeutic efficacy.
Inhibiting the enzyme γ-secretase, which converts amyloid precursor protein to β-amyloid , thus preventing its accumulation in the brain, has been a goal of drug developers.
Most recently, Bristol-Myers Squibb elected to discontinue development of its inhibitor candidate avagacestat into Phase III trials after disappointing Phase II results. BMS remains in the hunt for drugs to treat Alzheimer’s disease. Despite clinical failures of its and other companies’ other gamma secretase inhibitors, researchers continue to search for next-generation compounds they believe may succeed.
At BMS, Dave Harden, Ph.D., principal scientist and team leader, biochemical screening in the leads discovery and optimization group, has developed novel assays to identify molecules that inhibit secretase by measuring multiple amyloid beta species in cell supernatant. He and his team have capitalized on terbium cryptate’s properties as a donor fluorophore in HTRF, that has different photophysical properties compared to the donor fluor europium. These properties afford the opportunity to measure more than one interaction within a well due to the multiple emission spectra observed upon excitation. It can therefore serve as a donor fluorophore to green-emitting fluors because it has multiple emission peaks including one at 490 nm as well as the typically used 665 nm (red) emission.
Dr. Harden and colleagues, in order to “enhance” their screening practices by expanding well information content, enabled two color multiplexed HTRF in multiple settings in large (>1 MM well) screening campaigns. This approach, they reported, successfully identified mechanistically distinct gamma secretase inhibitors by measuring multiple amyloid beta peptide species in cell supernatants. This, and several other examples, the presenters said, demonstrated the power of multiplexed HTRF in maximizing screening outcomes.
Across the board, meeting presenters demonstrated the flexibility of HTRF assays and their adaptability to multiple research settings. The scientists pointed out that the assays yielded values consistent with other assay results using less versatile and convenient assays formats.
A large part of the world’s population is undernourished by the standards of Western Europe and North America. Scientists and nonscientists alike recognize as one of the major challenges of our time the problem of how to ensure that the production and distribution of food keep pace with the increasing number of mouths to be fed. In the world as a whole the most widespread and serious dietary deficiency is that of protein. This fact emerges clearly from the reports of the expert committees of WHO and FAO (World Health Organization, 1951, 1953). Nevertheless, many protein chemists, even those associated with medical research, may not realize the extent and severity of protein malnutrition, because it occurs chiefly in the technically underdeveloped countries far from where they work.
Dietary histories and response to treatment point to deficiency of total protein as the primary cause of the clinical syndrome kwashiorkor. The level of calorie intake has an important influence on the pattern of the disease. Deficiency of one or more specific amino acids, or amino acid imbalances in the diet, may perhaps be responsible for some of the symptoms and signs, particularly those whose incidence varies from one part of the world to another. All these variations on a theme are covered by the general term protein malnutrition. The onset is often precipitated by the added burden of diarrhea, infection, and parasitic infestation. The nutritional state influences the resistance to infection, and conversely the presence of an infection affects the state of nutrition. A further contributory factor may be the psychological upheaval in the child when the next baby in the family is born. At the root of all these causes lie poverty, ignorance, and disruption of the family life.
The planning of preventive measures cannot be effective unless it is based on some knowledge of the magnitude of the problem to be tackled. At a very rough estimate, in some countries perhaps 10% of the children suffer from severe protein malnutrition at some age between birth and 4 years. The marginal deficiency states must be much more common, Clinical signs and biochemical changes are of little value in diagnosing the early case; a deficit in body weight still seems to be the best criterion. Prevention ideally would be by greater production and consumption of animal protein, and by the increased use of skim milk and of surplus fish at present often wasted. However, animal protein is likely to remain scarce and expensive. Plant sources are being investigated with a view to encouraging not only domestic production, but also the production on an industrial scale of cheap foodstuffs rich in protein. A preventive program that is nutritionally sound may fail if account is not taken of local food habits, traditions, and customs. Protein requirements are affected by the quality of protein, the intake of calories, and by the state of the body (growth, the presence of disease, etc.). The maintenance requirement and the amount required for growth in children can be estimated, but the requirement for health is still unknown. For the time being, the allowances of protein recommended for people in the world as a whole are based empirically on the known physiological requirement with an arbitrarily added wide margin of safety.
The absorption of nitrogen is remarkably efficient even in severely malnourished infants. In general the nitrogen of plant proteins is less well absorbed than that of milk. When a baby receives a diet in which the protein is derived entirely from vegetabIe sources, incomplete absorption of nitrogen may play a significant part in the production of protein malnutrition. The malnourished baby who responds to treatment is able to retain and utilize nitrogen very efficiently; there is no evidence of any impairment in the mechanisms of protein synthesis. It is possible, however, that these mechanisms may be irreversibly damaged in babies who die, and that this may be the cause of death. The level of calorie intake has an important influence on the efficiency of utilization of nitrogen. An adequate calorie intake promotes conservation of nitrogen in the body as a whole when supplies of protein are short, but this protective effect may not be exerted equally in all organs. In this way the level of calorie intake may modify the pattern of protein depletion. A greater than normal calorie intake is needed for the restoration of depleted protein stores.
The discussion of protein metabolism in protein malnutrition has been purposely limited to a narrow field-to studies made on man, and to the few animal experiments that have a direct bearing on those studies. For technical reasons most of the work discussed relates to plasma proteins. There is a conflict of evidence between results obtained in man and animals about the effect of protein depletion or a low protein diet on the rate of catabolism of plasma albumin. It is of great importance to settle this point. A priori there seems no reason why the rate of protein catabolism should be affected by nutritional state. Preliminary studies with radioactive methionine in infants suggest, as working hypotheses, that in protein malnutrition there may be an increase in the reutilization of amino acids liberated by tissue catabolism, and an apparent concentration of protein synthesis in the more essential organs at the expense of the less essential. There is some experimental support for both these ideas, but further work is badly needed. The concept of protein stores or reserve protein is based entirely on dynamic and not on chemical considerations. It is suggested that the essential difference between a “labile” and a “fixed” protein is a difference in turnover rate. An attempt is made to show that the changes produced by protein depletion in the protein content of organs such as liver and muscle are a necessary consequence of the metabolic characteristics of proteins in those organs. There may be no need to invoke the help of homeostatic or compensatory regulations to explain the changes found in protein depletion.
Aging and growth are processes during which some metabolic adjustments must take place. It is believed that it may be better to regard the changes which are found in protein malnutrition in a similar light: as evidence of an alteration in functional pattern, rather than of damage or disease. Protein malnutrition in man has two aspects-a practical and a theoretical one. From the practical point of view it is an extremely common disease with a high mortality, and there is every reason to believe that it will become more common unless urgent preventive measures are taken. Theoretically it raises many questions that are of interest in relation to other branches of medicine and biochemistry. It is believed that the two aspects are linked, and that progress towards prevention is still impeded by our lack of basic knowledge as well as by our failure to apply what is already known. In protein malnutrition there is no sharp line between health and disease. The simple concept of specific deficiency diseases that grew from the discovery of vitamins is not applicable. We have to go back instead to the ideas of an earlier era, when nutrition was regarded as a branch of physiology, concerned with the functions, fate, and metabolic interrelationships of the major nutrients.
It is a characteristic of protein metabolism that nitrogen balance can be maintained at many different levels of protein intake. These different steady states are achieved by adjustments of the amount and distribution of proteins in the body as a whole, in organs, and in cells. It is believed that these changes in amount and distribution of proteins must result in alterations of metabolic pattern, with a gradation of change from an optimum, which cannot be defined, to a state of irreversible breakdown incompatible with life. In the intermediate stages function is modified and efficiency perhaps impaired. It seems possible that variations in diet, and particularly in the amount and quality of the protein, may underlie many of the differences in incidence and symptomatology of disease which are gradually being uncovered in different parts of the world.









 Thursday 15 October 2015, 12:00–13:00
Thursday 15 October 2015, 12:00–13:00







{kind=link}
{kind=link}
{kind=link}
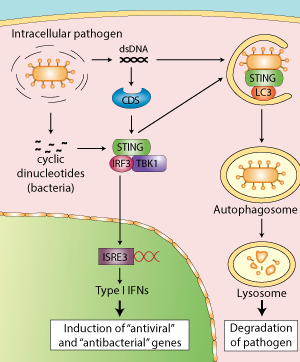{kind=link}
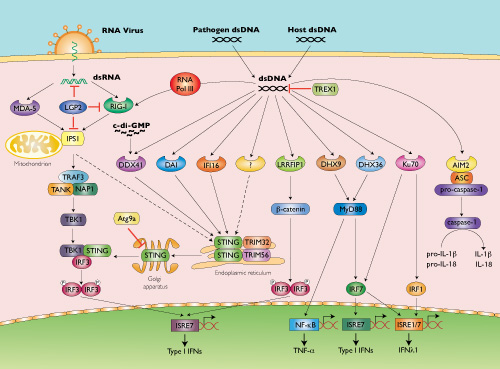{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}