Science Teaching in Math and Technology
Larry H. Bernstein, MD, FCAP, Curator
LPBI
2015 Best High Schools for STEM Rankings Methodology
U.S. News looked at 500 public high schools to identify the best in math and science education.
By Robert Morse May 11, 2015
U.S. News & World Report’s Best High Schools for STEM rankings methodology is based on the key principle that students at the Best High Schools for STEM must participate in and pass a robust curriculum of college-level math and science courses. STEM stands for science, technology, engineering and math.
To be included in the U.S. News Best High Schools for STEM rankings, a public high school first had to be listed as a gold medal winner in the 2015 U.S. News Best High Schools rankings. That meant that the top 500 nationally ranked high schools were eligible for the STEM rankings.
Those eligible schools were next judged nationally on their level of math and science participation and success, using Advanced Placement STEM test data for 2013 graduates as the benchmark to conduct the analysis. The U.S. News Best High Schools for STEM rankings methodology does not rely on any data from the U.S. Department of Education.
AP is a College Board program that offers college-level courses at high schools across the country. College Board defines STEM Math as AP courses in Calculus AB, Calculus BC, Computer Science A and Statistics; and STEM Science as AP courses in Biology, Chemistry, Environmental Science, Physics B, Physics C: Electricity and Magnetism and Physics C: Mechanics.
Math and science success at the high school level was assessed by computing a STEM Achievement Index for each school that ranked in the top 500 of the 2015 Best High Schools. The index was based on the percentage of all the AP test-takers in a school’s 2013 graduating class who took and passed college-level AP STEM Math and AP STEM Science tests. The higher a high school scored on the STEM Achievement Index, the better it placed in the Best High Schools for STEM rankings.
The maximum STEM Achievement Index value is 100. No public high school evaluated achieved that top score. The highest index was 98.3.
The first step in the rankings process was to compute the STEM Math Achievement Index. It was derived from two variables. The first was the percentage of AP test-takers in the 2013 graduating class who took at least one AP STEM Math course during high school, which was weighted 25 percent. The second was the percentage of those AP STEM Math test-takers who passed at least one AP STEM Math test during high school, receiving an AP score of 3 or higher. This variable was weighted 75 percent.
The next step was to calculate a STEM Science Achievement Index. Much like the math index, it was derived from the percentage of AP test-takers in the 2013 graduating class who took at least one AP STEM Science course during high school – weighted 25 percent – and the percentage of those AP STEM Science test-takers who passed at least one AP STEM Science test during high school, receiving an AP score of 3 or higher – weighted 75 percent.
This means that the methodology weights students taking AP math and science STEM courses at the high school level at 25 percent and passing those same AP STEM courses at 75 percent. In other words, passing both AP math and science tests was three times as important in the rankings as simply taking AP math and science courses.
The final step in the rankings process was to calculate the overall STEM Achievement Index, a combination of the STEM Math Achievement Index and the STEM Science Achievement Index. Each index was weighted at 50 percent, and then added together to create a composite value that is the STEM Achievement Index score.
The STEM rankings were based on sorting the unrounded – to many decimal places – STEM Achievement Index in descending order, with the top-ranked schools having the highest index values. The STEM Achievement Index was then rounded to the nearest 10th for online publication.
The top 250 high schools that achieved a value of greater than or equal to 66.8 in their STEM Achievement Index scored high enough to be numerically ranked. That high index cutoff point was used since it meant that all the high schools in the STEM rankings had, on average, more than two-thirds of the AP test-takers in their 2013 graduating class take and pass at least one AP STEM Math and one AP STEM Science test.
AP® and Advanced Placement® are registered trademarks of the College Board. Used with permission.
Top 50 Science Teacher Blogs
Bringing the subject of science to life for students is the challenge shared by the teachers who author these 50 amazing and insightful science education blogs. Sharing narratives set within and beyond the classroom walls, these next generation educators embrace technology but are never so dazzled by it that they lose sight of their common goal.
Action-Reaction
Physics Teacher Frank Noschese discusses science education topics like whether Khan Academy is effective at teaching physics, applying Angry Birds in physics lessons, and the idea of pseudoteaching.
Teaching High School Psychology
Teaching High School Psychology is a joint collaboration that explores the deeper lesson of the Stanford Prison Experiment, Gamification and its implications as a behavioral motivator, and opportunities for teaching Operant Conditioning with TV’s Big Bang Theory.
Little Miss Hypothesis
Inspiring Kindergarten scientists and giving a too-often neglected subject its due is the aim of Little Miss Hypothesis, where Mrs. Coe chronicles activities with growing brains, harvesting Spirit Garden salads and the development of science centers in a classroom that is home to Bluebonnet the Betta fish and the crab shack’s resident hermits.
Science for Kids
Sue Cahalane shares her passion for teaching science to elementary students in grades PK-4 on Science for Kids with ideas for classroom experiments, tutorials for science lessons, updates on science education news, and photos of students engaged in science activities.
Science Education on the Edge
Chris Ludwig, a high school science teacher from Colorado, writes about improving assessment and instruction in science and education technology.
Teach Science for All
Kirk Robbins shares helpful resources and tools for science teachers including reports, useful websites, and online tools.
Teaching | chemistry
Ellena Bethea, a high school chemistry teacher, writes about grading practices, online tools, and lab activities.
Adventures with the Lower Level
Tracie Schroeder shares her experiences teaching science, teaching methods, and thoughts on learning.
Physics in Flux
Dan Fullerton provides a resource for teachers with details of his successes and failures, technology guides, and physics book reviews.
Think Thank Thunk
On his blog, Think Thank Thunk, Shawn Cornally celebrates the Merlin within every teacher, the need for repackaging education, the debate surrounding Standards Based Grading and the dread of being dull as he chronicles his plight as an educator.
nashworld
Marine biology teacher Sean Nash gets inspired by WordFoto and invites educators to appreciate and aim for “Whoa” moments on his blog, nashworld.
Science Teacher
A science teacher and former pediatrican finds an exemplary model in Dr. Seuss, challenges technophiles to understand deeply, and explains why he has made a tradition of culminating each school year with a field trip to watch horseshoe crabs in the throes of romance.
Teach Science
At Teach Science, Ed Hitchcock muses on the DNA shared by Socrates and explains why science’s greatest appeal is the unexpected.
Quantum Progress
At Quantum Progress, 9th grade Atlanta physics teacher John Burk relives a childhood tradition at Physics Teacher Camp, promotes blogging as a tool for professional development, and ponders why physics buildings never win campus beauty contests.
Pedagogue Padawan
At Pedagogue Padawan, Geoff Schmit shares innovative ideas for using Sudoku to teach Circuit Analysis, Angry Birds as a lesson in holography, and wikispaces as a tool for science projects.
Re:thinking
Re:thinking blends personal reflection with a challenge to rethink school culture and policy as 9th grade teacher Ben Wildeboer finds teachable moments in events like the Japanese quake and explains the importance of “hard fun” for students.
Journey in Technology
At Journey in Technology a Dallas Physics teacher discusses implementing Khan Academy, discovering community and deep connections at Educon, and transforming the pseudoteaching of “cookbook” lab projects into real learning in the classroom.
Always Formative
Jason Buell is a middle school science teacher from California who writes about standards-based grading, education conferences, education books and more.
Stretching Forward
At Stretching Forward, Earth science teacher Janelle Wilson shares experiences from the Space Academy for Educators, discusses class blogging, and shares thoughts on engaging students and parents in science.
Tearing Down Walls
Derrick Willard teaches AP Environmental Science and discusses using social media and online tools to extend lessons outside the walls of the physical classroom.
Teaching Computer Science
Alfred Thompson is a former high school computer science teacher who currently works at Microsoft and writes about computer science education and resources.
A+ Computer Science Blog
High school computer science teacher Stacey Armstrong discusses why computer science is cool, game programming, career options in computer science, and computer science resources.
Teaching CS in Dallas
Kathleen Weaver writes about teaching robotics, Android development, and computer science education topics on her blog.
In Need of a Base Case
This blog discusses the need for change in computer science education, computer science project ideas, and the value of learning computer science.
Hélène Martin
Hélène Martin muses on the power of childhood playthings to fuel tech career ambitions and describes how lost airport luggage is a reminder to look for ways to leverage computing to solve real-life problems in this blog from the perspective of a computer science teacher.
Garth’s CS Education Blog
A computer science and programming teacher at a private school writes about teaching fun and important concepts and preparing students for computer science careers.
The Blog of Phyz
The Blog of Phyz is California teacher Dean Baird’s platform for debunking “Magnet Boys” and magic wristbands, and touting a 75 cent investment guaranteed to wow even the most cynical student.
Mr. Gonzalez’s Classroom
An Olympic Odyssey customarily culminates the academic year for middle school teacher Alfonso Gonzalez, who explores the challenge of giving terms like “on-task” and “structured learning” 21st century relevance on his blog, Mr. Gonzalez’s Classroom.
Free/Libre Open Source Science Education
Pseudoteaching and trends like the “reverse lecture” are hot topics on Free/Libre Open Source Science Education, where Steve Dickie shares his own innovative methods, including cartooning with GoAnimate and creating his own textbooks.
The Science Classroom
Oklahoma physics teacher Jody Bowie reports on the thrill of seeing students connect classroom lessons in everyday life, explains why everyone needs a whetstone to hone their thinking and divulges his identification with the Wizard in Wicked on The Science Classroom blog.
Jacobs Physics
On his blog Greg Jacobs calls course evaluations brutal but vital and bucks a few trends by advocating daily work and disparaging summer assignments in favor of starting each year “from the ground up”.
New Physics Modeler
Bryan Battaglia explains the appeal of professional conferences, the career changing power of blogging, and reflects that teachers gain as many lessons by year’s end as their students.
Just Call Me Ms Frizzle
Becky offers a distinctive first-year teacher perspective on Just Call Me Ms Frizzle, contrasting the low of leaving the room in frustration with the high of a Friday classroom on its best behavior, along with the challenge of teaching a non-traditional class.
And Yet it Moves
On his blog, And Yet it Moves, Ben Chun explains why problem-solving skills trump smarts, tackles the debate over doing away with honors classes, and challenges the AP curriculum.
Reflections of a Science Teacher
Sandra McCarron dismisses the notion of a rubric for thinking, believes that a successful classroom starts out with a vision and ponders the merits of science fairs that have been sacrificed to make way for education reforms on her blog, Reflections of a Science Teacher.
Hurricane Maine
A veteran teacher discusses ideas in education and technology, interesting articles, and how to make school more like play rather than work.
The Physics of Learning
Doug Smith authors this physics education blog that discusses topics like whether to use iPads in the classroom, the myths of merit pay, and scientific literacy.
Room 611
Mr. Young teaches Earth science and other subjects in Canada and provides insights into class by outlining what is covered in class almost every school day.
Using Blogs in Science Education
Stacey Baker is a high school biology teacher and writes about how to use classroom blogs to help students learn science.
Physics! Blog!
Physics! Blog! shares results of The No Homework Experiment and discusses standards based grading, the goal of testing, and teaching students how to learn from mistakes.
Ideas for Teaching Computer Technology to Kids
A blog sharing ideas and resources for teaching computer technology including robots for computer science education, programming resources, and computer science teaching tools.
MrReid.org
A physics teacher shares interesting science articles like Nobel prize winning sentences, things from movies that cannot exist, and cool science videos.
Teach. Brian. Teach.
Brian discusses what makes for a good science conversation, reflects on teaching, shares observations of students, and explains why it is important to point out when students are having fun doing science.
The Skeptical Teacher
A high school physics teacher discusses science education and promotes critical thinking on his blog.
Physics & Physical Science Demos, Labs, & Projects for High School Teachers
A physics teacher provides a resource for science teachers to share ideas for labs and demonstrations and commentary on what works.
The Art of Teaching Science
Jack Hassard is a professor of science education and explores issues in teaching science, shares resources for science teachers, and discusses why teaching science is important.
SuperFly Physics
At SuperFly Physics, Andy Rundquist shares ideas for teaching physics, fun science experiments, and interesting physics problems.
Newton’s Minions
A physics blog sharing student work, anecdotes from the classroom, thoughts on student assessment, and ideas for teaching complex physics lessons.
Mr. Barlow’s Blog
Mr. Barlow is a high school science teacher and podcaster from Melbourne, Australia who shares interesting science studies, cool science news, and optical illusions at his blog.
http://www.teachercertificationdegrees.com/top-blogs/science-teacher/
What is JASON?
We are a non-profit organization that connects students, in the classroom and out, to real science and exploration to inspire and motivate them to study and pursue careers in Science, Technology, Engineering and Math (STEM).
We embed exciting STEM professionals and cutting-edge research into award-winning, standards-aligned in and out-of-school curricula. Live webcasts connect students with inspirational STEM role models. Student materials include reading selections with read-to-me functionality, inquiry-based labs, videos, and online games. For teachers and informal educators, we provide lesson plans, assessments, and comprehensive professional development programs.
http://www.jason.org/sites/default/files/images/rotators/website%20klein%20feature.jpg
Ten Websites for Science Teachers
Originally Published: February 7, 2012 | Updated: October 10, 2014
www.nsta.org/about/awards.aspx
http://www.edutopia.org/sites/default/files/styles/feature_image_breakpoints_theme_edutopia_desktop_1x/public/slates/Science_Teachers_0.jpg?itok=i6yFGrRK
We all know that the web is full of excellent web resources for science teachers and students. However, unless you live on the web, finding the best websites can become quite a challenge. This isn’t a “Top Ten” list — instead, it is a list of websites that I either use on a regular basis or just find interesting. From teaching resources for the nature of science and authentic field journals to wacky videos about numbers, I am sure that you will find something in the following list the works for you!
1) Understanding Science
UC Berkeley’s Understanding Science website is a “must use” for all science teachers. It is a great resource for learning more about the process of science. The resource goes much deeper than the standard “PHEOC” model of the scientific method by emphasizing peer review, the testing of ideas, a science flowchart and “what is science?” checklist. Understanding Science also provides a variety of teaching resources including case studies of scientific discoveries and lesson plans for every grade level.
2) Field Research Journals
The Field Book Project from the National Museum of Natural History and the Smithsonian Institution Archives intends to create a “one stop” archive for field research journals and other documentation. You can find plenty of examples from actual field research journals for your classes.
3) Evolution
Berkeley’s Understanding Evolution website is the precursor to their Understanding Science efforts. The Understanding Evolution website provides a plethora of resources, news items and lessons for teaching about evolution. Lessons provide appropriate “building blocks” to help students at any grade level work towards a deeper understanding of evolution. The Evo 101 tutorial provides a great overview of the science behind evolution and the multiple lines of evidence that support the theory.
4) PhET Simulations
PhET from the University of Colorado provides dozens of fantastic simulations for physics, chemistry and biology. The website also includes a collection of teacher contributed activities, lab experiences, homework assignments and conceptual questions that can be used with the simulations.
5) Earth Exploration
The Earth Exploration Toolbook provides a series of activities, tools and case studies for using data sets with your students.
6) EdHead Interactives
Edheads is an organization that provides engaging web simulations and activities for kids. Current activities focus on simulated surgical procedures, cell phone design (with market research), simple and compound machines, and weather prediction.
7) Plant Mentors
Do you teach about plants? Check out Planting Science to connect your middle or high school students to science mentors and a collaborative inquiry project. From the project:
Planting Science is a learning and research resource, bringing together students, plant scientists, and teachers from across the nation. Students engage in hands-on plant investigations, working with peers and scientist mentors to build collaborations and to improve their understanding of science.
8) Periodic Table of Videos
Check out The Periodic Table of Videos for a wide array of videos about the elements and other chemistry topics.
9) More Videos!
Students can read and watch video about 21 Smithsonian scientistsincluding a volcano watcher, fossil hunter, art scientist, germinator and zoo vet.
10) Even More Videos!
How many videos were watched on YouTube in 2010? If you said 22 billion, you are sort of correct… Those 22 billion views only represent the number of times education videos were watched! In addition to this list of science and math YouTube channels, here are two of my favorites:
- SciShow is all about teaching scientific concepts in an accessible and easy-to-understand manner. This channel includes a variety of short (3 minute) and long (10 minute) videos. New videos are released weekly.
- Former BBC journalist Brady Haran is crazy about math and science. If you love numbers, you will love his Numberphile channel, dedicated to exploring the stories behind numbers.
- And let’s close with a particularly good SciShow on Climate Change:
https://youtu.be/M2Jxs7lR8ZI
Best High Schools
http://www.usnews.com/education/best-high-schools/national-rankings
|
School for the Talented and Gifted
1201 EAST EIGHTH ST
DALLAS, TX 75203
Dallas Independent School District
GOLD Medal |
15:1
Near National Avg
253 Students
17 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #2 |
BASIS Scottsdale
11440 NORTH 136TH ST
SCOTTSDALE, AZ 85259
BASIS Schools Inc.
GOLD Medal |
N/A
N/A
698 Students
N/A Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #3 |
Thomas Jefferson High School for Science and Technology
6560 BRADDOCK RD
ALEXANDRIA, VA 22312
Fairfax County Public Schools
GOLD Medal |
17:1
Near National Avg
1,846 Students
111 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #4 |
Gwinnett School of Mathematics, Science and Technology
970 MCELVANEY LN
LAWRENCEVILLE, GA 30044
Gwinnett County Public Schools
GOLD Medal |
18:1
Near National Avg
851 Students
48 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
|
School of Science and Engineering Magnet
1201 EAST EIGHTH ST
DALLAS, TX 75203
Dallas Independent School District
GOLD Medal |
16:1
Near National Avg
386 Students
24 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
|
|
|
|
| #6 |
Carnegie Vanguard High School
1501 TAFT
HOUSTON, TX 77019
Houston Independent School District
GOLD Medal |
17:1
Near National Avg
590 Students
34 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #7 |
Academic Magnet High School
5109A WEST ENTERPRISE ST
NORTH CHARLESTON, SC 29405
Charleston County School District
GOLD Medal |
14:1
Near National Avg
610 Students
44 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #8 |
University High School
9419 WEST VAN BUREN ST
TOLLESON, AZ 85353
Tolleson Union High School District
GOLD Medal |
34:1
Larger than National Avg
460 Students
14 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #9 |
Lamar Academy
1009 NORTH 10TH ST
MCALLEN, TX 78501
Mcallen Independent School District
GOLD Medal |
6:1
Smaller than National Avg
106 Students
19 Teachers |
100.0
Above National Avg
100% Tested (IB)
100% Passed (IB) |
| #10 |
Gilbert Classical Academy High School
55 NORTH GREENFIELD RD
GILBERT, AZ 85234
Gilbert Unified District
GOLD Medal |
11:1
Smaller than National Avg
220 Students
20 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #11 |
The High School of American Studies at Lehman College
2925 GOULDEN AVE
BRONX, NY 10468
New York City Public Schools
GOLD Medal |
16:1
Near National Avg
393 Students
25 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #12 |
American Indian Public High School
3637 MAGEE AVE
OAKLAND, CA 94619
Oakland Unf
GOLD Medal |
19:1
Larger than National Avg
243 Students
13 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #13 |
International Studies Charter High School
2480 SW 8TH ST
MIAMI, FL 33135
Miami-Dade County Public Schools
GOLD Medal |
13:1
Near National Avg
359 Students
27 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #14 |
High School for Dual Language and Asian Studies
350 GRAND ST
NEW YORK, NY 10002
New York City Public Schools
GOLD Medal |
16:1
Near National Avg
392 Students
25 Teachers |
100.0
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #15 |
Northside College Preparatory High School
5501 NORTH KEDZIE AVE
CHICAGO, IL 60625
Chicago Public Schools
GOLD Medal |
14:1
Near National Avg
1,069 Students
74 Teachers |
99.7
Above National Avg
100% Tested (AP®)
100% Passed (AP®) |
| #16 |
Oxford Academy
5172 ORANGE AVE
CYPRESS, CA 90630
Anaheim Union High
GOLD Medal |
30:1
Larger than National Avg
1,152 Students
38 Teachers |
99.5
Above National Avg
100% Tested (AP®)
99% Passed (AP®) |
| #17 |
University High School
421 NORTH ARCADIA BLVD
TUCSON, AZ 85711
Tucson Unified School District
GOLD Medal |
21:1
Larger than National Avg
934 Students
44 Teachers |
99.3
Above National Avg
100% Tested (AP®)
99% Passed (AP®) |
| #18 |
Pacific Collegiate School
255 SWIFT ST
SANTA CRUZ, CA 95060
Santa Cruz County Office Of Education
GOLD Medal |
19:1
Larger than National Avg
515 Students
28 Teachers |
99.1
Above National Avg
100% Tested (AP®)
99% Passed (AP®) |
| #19 |
Biotechnology High School
5000 KOZLOSKI RD
FREEHOLD, NJ 07728
Monmouth County Vocational School District
GOLD Medal |
14:1
Near National Avg
311 Students
23 Teachers |
99.1
Above National Avg
100% Tested (IB)
99% Passed (IB) |
| #20 |
High Technology High School
765 NEWMAN SPRINGS RD
LINCROFT, NJ 07738
Monmouth County Vocational School District
GOLD Medal |
13:1
Near National Avg
280 Students
22 Teachers |
98.5
Above National Avg
99% Tested (AP®)
99% Passed (AP®) |
|
|
|
|
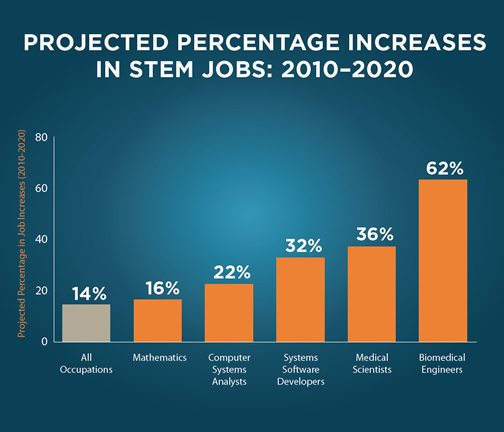
The United States has developed as a global leader, in large part, through the genius and hard work of its scientists, engineers, and innovators. In a world that’s becoming increasingly complex, where success is driven not only by what you know, but by what youcan do with what you know, it’s more important than ever for our youth to be equipped with the knowledge and skills to solve tough problems, gather and evaluate evidence, and make sense of information. These are the types of skills that students learn by studying science, technology, engineering, and math—subjects collectively known as STEM.
Yet today, few American students pursue expertise in STEM fields—and we have an inadequate pipeline of teachers skilled in those subjects. That’s why President Obama has set a priority of increasing the number of students and teachers who are proficient in these vital fields.

http://www.ed.gov/sites/default/files/stem-infographic.jpg
The need
All young people should be prepared to think deeply and to think well so that they have the chance to become the innovators, educators, researchers, and leaders who can solve the most pressing challenges facing our nation and our world, both today and tomorrow. But, right now, not enough of our youth have access to quality STEM learning opportunities and too few students see these disciplines as springboards for their careers.expand/collapse
The goals
President Obama has articulated a clear priority for STEM education: within a decade, American students must “move from the middle to the top of the pack in science and math.” The Obama Administration also is working toward the goal of fairness between places, where an equitable distribution of quality STEM learning opportunities and talented teachers can ensure that all students have the chance to study and be inspired by science, technology, engineering, and math—and have the chance to reach their full potential.expand/collapse
The plan
The Committee on STEM Education (CoSTEM), comprised of 13 agencies—including all of the mission-science agencies and the Department of Education—are facilitating a cohesive national strategy, with new and repurposed funds, to increase the impact of federal investments in five areas: 1.) improving STEM instruction in preschool through 12th grade; 2.) increasing and sustaining public and youth engagement with STEM; 3.) improving the STEM experience for undergraduate students; 4.) better serving groups historically underrepresented in STEM fields; and 5.) designing graduate education for tomorrow’s STEM workforce.expand/collapse
Supporting Teachers and Students in STEM
At the Department of Education, we share the President’s commitment to supporting and improving STEM education. Ensuring that all students have access to high-quality learning opportunities in STEM subjects is a priority, demonstrated by the fact that dozens of federal programs have made teaching and learning in science, technology, engineering, and math a critical component of competitiveness for grant funding. Just this year, for the very first time, the Department announced that its Ready-to-Learn Television grant competition would include a priority to promote the development of television and digital media focused on science.
The Department’s Race to the Top-District program supports educators in providing students with more personalized learning—in which the pace of and approach to instruction are uniquely tailored to meet students’ individual needs and interests—often supported by innovative technologies. STEM teachers across the country also are receiving resources, support, training, and development through programs like Investing in Innovation (i3), the Teacher Incentive Fund, the Math and Science Partnershipsprogram, Teachers for a Competitive Tomorrow, and the Teacher Quality Partnerships program.
Because we know that learning happens everywhere—both inside and outside of formal school settings—the Department’s 21st Century Community Learning Centers program is collaborating with NASA, the National Park Service, and the Institute of Museum and Library Services to bring high-quality STEM content and experiences to students from low-income, high-need schools. This initiative has made a commitment to Native-American students, providing about 350 young people at 11 sites across six states with out-of-school STEM courses focused on science and the environment.
And in higher education, the Hispanic-Serving Institutions-STEM program is helping to increase the number of Hispanic students attaining degrees in STEM subjects.
This sampling of programs represents some of the ways in which federal resources are helping to assist educators in implementing effective approaches for improving STEM teaching and learning; facilitating the dissemination and adoption of effective STEM instructional practices nationwide; and promoting STEM education experiences that prioritize hands-on learning to increase student engagement and achievement.
Learn more
How Do I Find…?
Information About…

seri_scores
http://i.livescience.com/images/i/000/017/835/i02/seri_scores.png?1310070712
A new ranking of how well the United States’ schools are preparing students for science and engineering careers shows that although there’s a small number of high performers, most states are doing a poor job of educating students in these subjects.
According to the ranking of schools teaching kindergarten through 12th grade, Massachusetts leads the pack with a score of 4.82 on a scale of 1 to 5, while Mississippi trails behind as “worst in the United States” with a 1.11 score. Twenty-one states in total, including California, earned what the ranking classified as “below average” or “far below average” scores, and only 10 states earned scores above the national average of 2.82.
Read Full Post »

 and
and 








 The schematic on Slide 10 can be found in
The schematic on Slide 10 can be found in 






 On slide 20 the traditional Ti casting is compared with Ti 3D printing from the powders. The advantage of 3D method is low cost and high productivity. This link
On slide 20 the traditional Ti casting is compared with Ti 3D printing from the powders. The advantage of 3D method is low cost and high productivity. This link  Slide 21 For 3D Bioploter made by EnvisionTec we notice the usage of materials such as metal followed by post-processing sintering, Hydroxylapatite, TCP, Titanium. Using a preciptation method the machine can handle Chitosan, Collagen, 2-component system of the two possible combination: Alginate, Fibrin, PU, and Silicone. More details in
Slide 21 For 3D Bioploter made by EnvisionTec we notice the usage of materials such as metal followed by post-processing sintering, Hydroxylapatite, TCP, Titanium. Using a preciptation method the machine can handle Chitosan, Collagen, 2-component system of the two possible combination: Alginate, Fibrin, PU, and Silicone. More details in 





















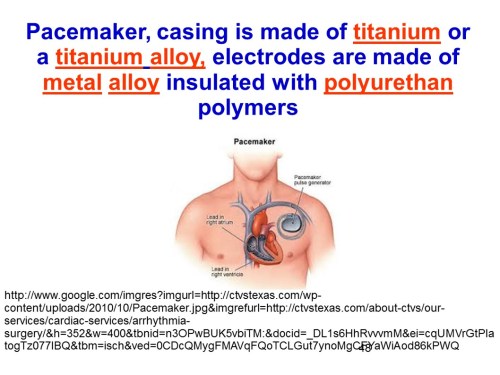

 The insulin pump shown in slide 50 is a schematic of the pump controlled electronically by a control algorithm device, a sensor, an electronic receiver that connects with an iPhone through an wireless channel.
The insulin pump shown in slide 50 is a schematic of the pump controlled electronically by a control algorithm device, a sensor, an electronic receiver that connects with an iPhone through an wireless channel.
 Slide 52 describes how easily available bio-compatible metal powders
Slide 52 describes how easily available bio-compatible metal powders
 Slide 54 shows conclusions regarding the hardware of the presentation, in which: 6) there are two types of metal 3D printing hardware for medical applications: Selective Laser Melting / Selective Laser Sintering, and 3D Bioploter (metal powder mixed with binder and further thermal treatment to remove binder and sinter the metallic matrix in a solid object that can be used as a replacement. Thank you for your attention!
Slide 54 shows conclusions regarding the hardware of the presentation, in which: 6) there are two types of metal 3D printing hardware for medical applications: Selective Laser Melting / Selective Laser Sintering, and 3D Bioploter (metal powder mixed with binder and further thermal treatment to remove binder and sinter the metallic matrix in a solid object that can be used as a replacement. Thank you for your attention!










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}