Effect of mitochondrial stress on epigenetic modifiers
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Early Mitochondrial Stress Alters Epigenetics, Secures Lifelong Health Benefits
A little adversity builds character, or so the saying goes. True or not, the saying does seem an apt description of a developmental phenomenon that shapes gene expression. While it knows nothing of character, the gene expression apparatus appears to respond well to short-term mitochondrial stress that occurs early in development. In fact, transient stress seems to result in lasting benefits. These benefits, which include improved metabolic function and increased longevity, have been observed in both worms and mice, and may even occur—or be made to occur—in humans.
Gene expression is known to be subject to reprogramming by epigenetic modifiers, but such modifiers generally affect metabolism or lifespan, not both. A new set of epigenetic modifiers, however, has been found to trigger changes that do just that—both improve metabolism and extend lifespan.
Scientists based at the University of California, Berkeley, and the École Polytechnique Fédérale de Lausanne (EPFL) have discovered enzymes that are ramped up after mild stress during early development and continue to affect the expression of genes throughout the animal’s life. When the scientists looked at strains of inbred mice that have radically different lifespans, those with the longest lifespans had significantly higher expression of these enzymes than did the short-lived mice.
“Two of the enzymes we discovered are highly, highly correlated with lifespan; it is the biggest genetic correlation that has ever been found for lifespan in mice, and they’re both naturally occurring variants,” said Andrew Dillin, a UC Berkeley professor of molecular and cell biology. “Based on what we see in worms, boosting these enzymes could reprogram your metabolism to create better health, with a possible side effect of altering lifespan.”
Details of the work, which appeared online April 29 in the journal Cell, are presented in a pair of papers. One paper (“Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity”) resulted from an effort led by Dillin and the EPFL’s Johan Auwerx. The other paper (“Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPRmt”) resulted from an effort led by Dillin and his UC Berkeley colleague Barbara Meyer.
According to these papers, mitochondrial stress activates enzymes in the brain that affect DNA folding, exposing a segment of DNA that contains the 1500 genes involved in the work of the mitochondria. A second set of enzymes then tags these genes, affecting their activation for much or all of the lifetime of the animal and causing permanent changes in how the mitochondria generates energy.
The first set of enzymes—methylases, in particular LIN-65—add methyl groups to the DNA, which can silence promoters and thus suppress gene expression. By also opening up the mitochondrial genes, these methylases set the stage for the second set of enzymes—demethylases, in this case jmjd-1.2 and jmjd-3.1—to ramp up transcription of the mitochondrial genes. When the researchers artificially increased production of the demethylases in worms, all the worms lived longer, a result identical to what is observed after mitochondrial stress.
“By changing the epigenetic state, these enzymes are able to switch genes on and off,” Dillin noted. This happens only in the brain of the worm, however, in areas that sense hunger or satiety. “These genes are expressed in neurons that are sensing the nutritional status of the animal, and these signals emanate out to the periphery to change peripheral metabolism,” he continued.
When the scientists profiled enzymes in short- and long-lived mice, they found upregulation of these genes in the brains of long-lived mice, but not in other tissues or in the brains of short-lived mice. “These genes are expressed in the hypothalamus, exactly where, when you eat, the signals are generated that tell you that you are full. And when you are hungry, signals in that region tell you to go and eat,” Dillin explained said. “These genes are all involved in peripheral feedback.”
Among the mitochondrial genes activated by these enzymes are those involved in the body’s response to proteins that unfold, which is a sign of stress. Increased activity of the proteins that refold other proteins is another hallmark of longer life.
These observations suggest that the reversal of aging by epigenetic enzymes could also take place in humans.
“It seems that, while extreme metabolic stress can lead to problems later in life, mild stress early in development says to the body, ‘Whoa, things are a little bit off-kilter here, let’s try to repair this and make it better.’ These epigenetic switches keep this up for the rest of the animal’s life,” Dillin stated.
Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity
- •H3K27 demethylases jmjd-1.2 and jmjd-3.1 are required for ETC-mediated longevity
- •jmjd-1.2 and jmjd-3.1 extend lifespan and are sufficient for UPRmt activation
- •UPRmt is required for increased lifespan due to jmjd-1.2 or jmjd-3.1 overexpression
- •JMJD expression is correlated with UPRmt and murine lifespan in inbred BXD lines
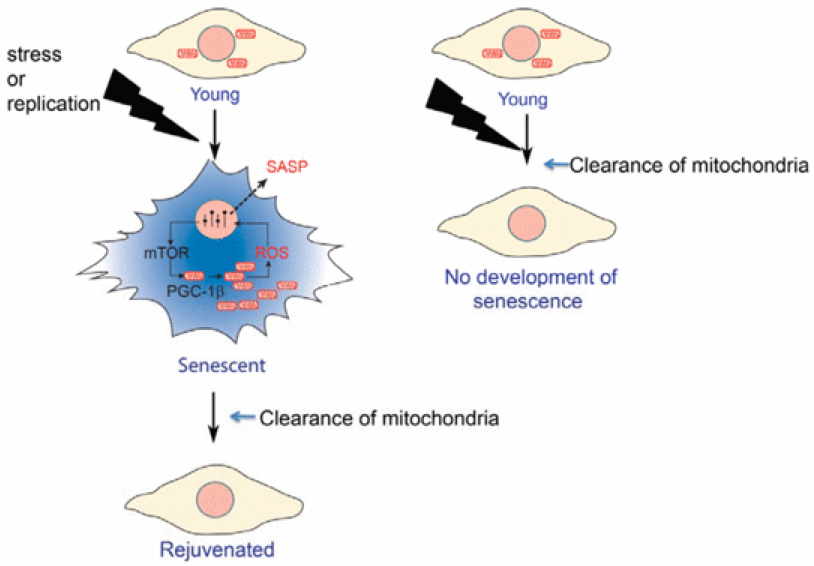
Across eukaryotic species, mild mitochondrial stress can have beneficial effects on the lifespan of organisms. Mitochondrial dysfunction activates an unfolded protein response (UPRmt), a stress signaling mechanism designed to ensure mitochondrial homeostasis. Perturbation of mitochondria during larval development in C. elegans not only delays aging but also maintains UPRmt signaling, suggesting an epigenetic mechanism that modulates both longevity and mitochondrial proteostasis throughout life. We identify the conserved histone lysine demethylases jmjd-1.2/PHF8 and jmjd-3.1/JMJD3 as positive regulators of lifespan in response to mitochondrial dysfunction across species. Reduction of function of the demethylases potently suppresses longevity and UPRmt induction, while gain of function is sufficient to extend lifespan in a UPRmt-dependent manner. A systems genetics approach in the BXD mouse reference population further indicates conserved roles of the mammalian orthologs in longevity and UPRmt signaling. These findings illustrate an evolutionary conserved epigenetic mechanism that determines the rate of aging downstream of mitochondrial perturbations.


 Article Info Publication stage: In Press Corrected Proof
Article Info Publication stage: In Press Corrected Proof- •LIN-65 accumulates in the nucleus in response to mitochondrial stress
- •Mitochondrial stress-induced chromatin changes depend on MET-2 and LIN-65
- •LIN-65 and DVE-1 exhibit interdependence in nuclear accumulation
- •met-2 and atfs-1 act in parallel to affect mitochondrial stress-induced longevity
Organisms respond to mitochondrial stress through the upregulation of an array of protective genes, often perpetuating an early response to metabolic dysfunction across a lifetime. We find that mitochondrial stress causes widespread changes in chromatin structure through histone H3K9 di-methylation marks traditionally associated with gene silencing. Mitochondrial stress response activation requires the di-methylation of histone H3K9 through the activity of the histone methyltransferase met-2 and the nuclear co-factor lin-65. While globally the chromatin becomes silenced by these marks, remaining portions of the chromatin open up, at which point the binding of canonical stress responsive factors such as DVE-1 occurs. Thus, a metabolic stress response is established and propagated into adulthood of animals through specific epigenetic modifications that allow for selective gene expression and lifespan extension
Siddharha Mukherjee won the 2011 Pulitzer Prize in non-fiction for his book, The Emperor of All Maladies. The book has received widespread acclaim among lay audience, physicians, and scientists alike. Last year the book was turned into a special PBS series. But, according to a slew of scientists, we should all be skeptical of his next book scheduled to hit book shelves this month, The Gene, An Intimate History.
Publishing an article on epigenetics in the New Yorker this week–perhaps a selection from his new book–Mukherjee has waltzed into one of the most active scientific debates in all of biology: that of gene regulation, or epigenetics.
Jerry Coyne, the evolutionary biologist known for keeping journalists honest, has published a two part critique of Mukherjee’s New Yorker piece. The first part–wildly tweeted yesterday–is a list of quotes from Coyne’s colleagues and those who have written in to the New Yorker, including two Nobel prize winners, Wally Gilbert and Sidney Altman, offering some very unfriendly sentences.
Wally Gilbert: “The New Yorker article is so wildly wrong that it defies rational analysis.”
Sidney Altman: “I am not aware that there is such a thing as an epigenetic code. It is unfortunate to inflict this article, without proper scientific review, on the audience of the New Yorker.”
The second part is a thorough scientific rebuttal of the Mukherjee piece. It all serves as a great drama about one of the most contested ideas in biology and also as a cautionary tale to journalists, even experienced writers such as Mukherjee, about the dangers of wading into scientific arguments. Readers may remember that a few years ago, science writer, David Dobbs, similarly skated into the same topic with his piece, Die, Selfish Gene, Die, and which raised a similar shitstorm, much of it from Coyne.
Mukherjee’s mistake is in giving credence to only one side of a very fierce debate–that the environment causes changes in the genome which can be passed on; another kind of evolution–as though it were settled science. Either Mukherjee, a physicisan coming off from a successful book and PBS miniseries on cancer, is setting himself up as a scientist, or he has been a truly naive science reporter. If he got this chapter so wrong, what does it mean about an entire book on the gene?
Coyne quotes one of his colleagues who raised some questions about the New Yorker’s science reporting, one particular question we’ve been asking here at Mendelspod. How do we know what we know? Does science now have an edge on any other discipline for being able to create knowledge?
Coyne’s colleague is troubled by science coverage in the New Yorker, and goes so far as to write that the New Yorker has been waging a “war on behalf of cultural critics and literary intellectuals against scientists and technologists.”
From my experience, it’s not quite that tidy. First of all, the New Yorker is the best writing I read each week. Period. Second, I haven’t found their science writing to have the slant claimed in the quote above. For example, most other mainstream outlets–including the New York Times with the Amy Harmon pieces–have given the anti-GMO crowd an equal say in the mistaken search for a “balance” on whether GMOs are harmful. (Remember John Stewart’s criticism of Fox News? That they give a false equivalent between two sides even when there is no equivalent on the other side?)
But the New Yorker has not fallen into this trap on GMOs and most of their pieces on the topic–mainly by Michael Specter–have been decidedly pro science and therefore decided pro GMO.
So what led Mukherjee to play scientist as well as journalist? There’s no question about whether I enjoy his prose. His writing beautifully whisks me away so that I don’t feel that I’m really working to understand. There is a poetic complexity that constantly brings different threads effortlessly together, weaving them into the same light. At one point he uses the metaphor of a web for the genome, with the epigenome being the stuff that sticks to the web. He borrows the metaphor from the Hindu notion of “being”, or jaal.
“Genes form the threads of the web; the detritus that adheres to it transforms every web into a singular being.”
There have been a few writers on Twitter defending Mukherjee’s piece. Tech Review’s Antonio Regalado called Coyne and his colleagues “tedious literalists” who have an “issue with epigenetic poetry.”
At his best, Mukherjee can take us down the sweet alleys of his metaphors and family stories with a new curiosity for the scientific truth. He can hold a mirror up to scientists, or put the spotlight on their work. At their worst, Coyne and his scientific colleagues can reek of a fear of language and therefore metaphor. The always outspoken scientist and author, Richard Dawkins, who made his name by personifying the gene, was quick to personify epigentics in a tweet: “It’s high time the 15 minutes of underserved fame for “epigenetics” came to an overdue end.” Dawkins is that rare scientist who has consistently been as comfortable with rhetoric and language as he is with data.
Hats off to Coyne who reminds us that a metaphor–however lovely–does not some science make. If Mukherjee wants to play scientist, let him create and gather data. If it’s the role of science journalist he wants, let him collect all the science he can before he begins to pour it into his poetry.
Same but Different
How epigenetics can blur the line between nature and nurture.
Annals of Science MAY 2, 2016 ISSUE BY SIDDHARTHA MUKHERJEE
http://www.newyorker.com/magazine/2016/05/02/breakthroughs-in-epigenetics
http://www.newyorker.com/wp-content/uploads/2016/05/160502_r28072-1200.jpg
The author’s mother (right) and her twin are a study in difference and identity. CREDIT: PHOTOGRAPH BY DAYANITA SINGH FOR THE NEW YORKER
October 6, 1942, my mother was born twice in Delhi. Bulu, her identical twin, came first, placid and beautiful. My mother, Tulu, emerged several minutes later, squirming and squalling. The midwife must have known enough about infants to recognize that the beautiful are often the damned: the quiet twin, on the edge of listlessness, was severely undernourished and had to be swaddled in blankets and revived.
The first few days of my aunt’s life were the most tenuous. She could not suckle at the breast, the story runs, and there were no infant bottles to be found in Delhi in the forties, so she was fed through a cotton wick dipped in milk, and then from a cowrie shell shaped like a spoon. When the breast milk began to run dry, at seven months, my mother was quickly weaned so that her sister could have the last remnants.
Tulu and Bulu grew up looking strikingly similar: they had the same freckled skin, almond-shaped face, and high cheekbones, unusual among Bengalis, and a slight downward tilt of the outer edge of the eye, something that Italian painters used to make Madonnas exude a mysterious empathy. They shared an inner language, as so often happens with twins; they had jokes that only the other twin understood. They even smelled the same: when I was four or five and Bulu came to visit us, my mother, in a bait-and-switch trick that amused her endlessly, would send her sister to put me to bed; eventually, searching in the half-light for identity and difference—for the precise map of freckles on her face—I would realize that I had been fooled.
But the differences were striking, too. My mother was boisterous. She had a mercurial temper that rose fast and died suddenly, like a gust of wind in a tunnel. Bulu was physically timid yet intellectually more adventurous. Her mind was more agile, her tongue sharper, her wit more lancing. Tulu was gregarious. She made friends easily. She was impervious to insults. Bulu was reserved, quieter, and more brittle. Tulu liked theatre and dancing. Bulu was a poet, a writer, a dreamer.
….. more
Why are identical twins alike? In the late nineteen-seventies, a team of scientists in Minnesota set out to determine how much these similarities arose from genes, rather than environments—from “nature,” rather than “nurture.” Scouring thousands of adoption records and news clips, the researchers gleaned a rare cohort of fifty-six identical twins who had been separated at birth. Reared in different families and different cities, often in vastly dissimilar circumstances, these twins shared only their genomes. Yet on tests designed to measure personality, attitudes, temperaments, and anxieties, they converged astonishingly. Social and political attitudes were powerfully correlated: liberals clustered with liberals, and orthodoxy was twinned with orthodoxy. The same went for religiosity (or its absence), even for the ability to be transported by an aesthetic experience. Two brothers, separated by geographic and economic continents, might be brought to tears by the same Chopin nocturne, as if responding to some subtle, common chord struck by their genomes.
One pair of twins both suffered crippling migraines, owned dogs that they had named Toy, married women named Linda, and had sons named James Allan (although one spelled the middle name with a single “l”). Another pair—one brought up Jewish, in Trinidad, and the other Catholic, in Nazi Germany, where he joined the Hitler Youth—wore blue shirts with epaulets and four pockets, and shared peculiar obsessive behaviors, such as flushing the toilet before using it. Both had invented fake sneezes to diffuse tense moments. Two sisters—separated long before the development of language—had invented the same word to describe the way they scrunched up their noses: “squidging.” Another pair confessed that they had been haunted by nightmares of being suffocated by various metallic objects—doorknobs, fishhooks, and the like.
The Minnesota twin study raised questions about the depth and pervasiveness of qualities specified by genes: Where in the genome, exactly, might one find the locus of recurrent nightmares or of fake sneezes? Yet it provoked an equally puzzling converse question: Why are identical twins different? Because, you might answer, fate impinges differently on their bodies. One twin falls down the crumbling stairs of her Calcutta house and breaks her ankle; the other scalds her thigh on a tipped cup of coffee in a European station. Each acquires the wounds, calluses, and memories of chance and fate. But how are these changes recorded, so that they persist over the years? We know that the genome can manufacture identity; the trickier question is how it gives rise to difference.
….. more
But what turns those genes on and off, and keeps them turned on or off? Why doesn’t a liver cell wake up one morning and find itself transformed into a neuron? Allis unpacked the problem further: suppose he could find an organism with two distinct sets of genes—an active set and an inactive set—between which it regularly toggled. If he could identify the molecular switches that maintain one state, or toggle between the two states, he might be able to identify the mechanism responsible for cellular memory. “What I really needed, then, was a cell with these properties,” he recalled when we spoke at his office a few weeks ago. “Two sets of genes, turned ‘on’ or ‘off’ by some signal.”
more…
“Histones had been known as part of the inner scaffold for DNA for decades,” Allis went on. “But most biologists thought of these proteins merely as packaging, or stuffing, for genes.” When Allis gave scientific seminars in the early nineties, he recalled, skeptics asked him why he was so obsessed with the packing material, the stuff in between the DNA. …. A skein of silk tangled into a ball has very different properties from that same skein extended; might the coiling or uncoiling of DNA change the activity of genes?
In 1996, Allis and his research group deepened this theory with a seminal discovery. “We became interested in the process of histone modification,” he said. “What is the signal that changes the structure of the histone so that DNA can be packed into such radically different states? We finally found a protein that makes a specific chemical change in the histone, possibly forcing the DNA coil to open. And when we studied the properties of this protein it became quite clear that it was also changing the activity of genes.” The coils of DNA seemed to open and close in response to histone modifications—inhaling, exhaling, inhaling, like life.
Allis walked me to his lab, a fluorescent-lit space overlooking the East River, divided by wide, polished-stone benches. A mechanical stirrer, whirring in a corner, clinked on the edge of a glass beaker. “Two features of histone modifications are notable,” Allis said. “First, changing histones can change the activity of a gene without affecting the sequence of the DNA.” It is, in short, formally epi-genetic, just as Waddington had imagined. “And, second, the histone modifications are passed from a parent cell to its daughter cells when cells divide. A cell can thus record ‘memory,’ and not just for itself but for all its daughter cells.”
…..
The New Yorker screws up big time with science: researchers criticize the Mukherjee piece on epigenetics
Jerry Coyne
https://whyevolutionistrue.wordpress.com/2016/05/05/the-new-yorker-screws-up-big-time-with-science-researchers-criticize-the-mukherjee-piece-on-epigenetics/
Abstract: This is a two part-post about a science piece on gene regulation that just appeared in the New Yorker. Today I give quotes from scientists criticizing that piece; tomorrow I’ll present a semi-formal critique of the piece by two experts in the field.
esterday I gave readers an assignment: read the new New Yorkerpiece by Siddhartha Mukherjee about epigenetics. The piece, called “Same but different” (subtitle: “How epigenetics can blur the line between nature and nurture”) was brought to my attention by two readers, both of whom praised it. Mukherjee, a physician, is well known for writing the Pulitzer-Prize-winning book (2011) The Emperor of All Maladies: A Biography of Cancer. (I haven’t read it yet, but it’s on my list.) Mukherjee has a new book that will be published in May: The Gene: An Intimate History. As I haven’t seen it, the New Yorker piece may be an excerpt from this book.
Everyone I know who has read The Emperor of All Maladies gives it high praise. I wish I could say the same for Mukherjee’s New Yorker piece. When I read it at the behest of the two readers, I found his analysis of gene regulation incomplete and superficial. Although I’m not an expert in that area, I knew that there was a lot of evidence that regulatory proteins called “transcription factors”, and not “epigenetic markers” (see discussion of this term tomorrow) or modified histones—the factors emphasized by Mukherjee—played hugely important roles in gene regulation. The speculations at the end of the piece about “Lamarckian evolution” via environmentally induced epigenetic changes in the genome were also unfounded, for we have no evidence for that kind of adaptive evolution. Mukherjee does, however, mention that lack of evidence, though I wish he’d done so more strongly given that environmental modification of DNA bases is constantly touted as an important and neglected factor in evolution.
Unbeknownst to me, there was a bit of a kerfuffle going on in the community of scientists who study gene regulation, with many of them finding serious mistakes and omissions in Mukherjee’s piece. There appears to have been some back-and-forth emailing among them, and several wrote letters to the New Yorker, urging them to correct the misconceptions, omissions, and scientific errors in “Same but different.” As I understand it, both Mukherjee and the New Yorker simply batted these criticisms away, and, as far as I know, will not publish any corrections. So today and tomorrow I’ll present the criticisms here, just so they’ll be on the record.
Because Mukherjee writes very well, and because even educated laypeople won’t know the story of gene regulation revealed over the last few decades, they may not see the big lacunae in his piece. It is, then, important to set matters straight, for at least we should know what science has told us about how genes are turned on and off. The criticism of Mukherjee’s piece, coming from scientists who really are experts in gene regulation, shows a lack of care on the part of Mukherjee and theNew Yorker: both a superficial and misleading treatment of the state of the science, and a failure of the magazine to properly vet this piece (I have no idea whether they had it “refereed” not just by editors but by scientists not mentioned in the piece).
Let me add one thing about science and the New Yorker. I believe I’ve said this before, but the way the New Yorker treats science is symptomatic of the “two cultures” problem. This is summarized in an email sent me a while back by a colleague, which I quote with permission:
The New Yorker is fine with science that either serves a literary purpose (doctors’ portraits of interesting patients) or a political purpose (environmental writing with its implicit critique of modern technology and capitalism). But the subtext of most of its coverage (there are exceptions) is that scientists are just a self-interested tribe with their own narrative and no claim to finding the truth, and that science must concede the supremacy of literary culture when it comes to anything human, and never try to submit human affairs to quantification or consilience with biology. Because the magazine is undoubtedly sophisticated in its writing and editing they don’t flaunt their postmodernism or their literary-intellectual proprietariness, but once you notice it you can make sense of a lot of their material.
. . . Obviously there are exceptions – Atul Gawande is consistently superb – but as soon as you notice it, their guild war on behalf of cultural critics and literary intellectuals against scientists, technologists, and analytic scholars becomes apparent.
…. more
Researchers criticize the Mukherjee piece on epigenetics: Part 2
Trigger warning: Long science post!
Yesterday I provided a bunch of scientists’ reactions—and these were big names in the field of gene regulation—to Siddhartha Mukherjee’s ill-informed piece in The New Yorker, “Same but different” (subtitle: “How epigenetics can blur the line between nature and nurture”). Today, in part 2, I provide a sentence-by-sentence analysis and reaction by two renowned researchers in that area. We’ll start with a set of definitions (provided by the authors) that we need to understand the debate, and then proceed to the critique.
Let me add one thing to avoid confusion: everything below the line, including the definition (except for my one comment at the end) was written by Ptashne and Greally.
by Mark Ptashne and John Greally
Introduction
Ptashne is The Ludwig Professor of Molecular Biology at the Memorial Sloan Kettering Cancer Center in New York. He wrote A Genetic Switch, now in its third edition, which describes the principles of gene regulation and the workings of a ‘switch’; and, with Alex Gann, Genes and Signals, which extends these principles and ideas to higher organisms and to other cellular processes as well. John Greally is the Director of the Center for Epigenomics at the Albert Einstein College of Medicine in New York.
The New Yorker (May 2, 2016) published an article entitled “Same But Different” written by Siddhartha Mukherjee. As readers will have gathered from the letters posted yesterday, there is a concern that the article is misleading, especially for a non-scientific audience. The issue concerns our current understanding of “gene regulation” and how that understanding has been arrived at.
First some definitions/concepts:
Gene regulation refers to the “turning on and off of genes”. The primary event in turning a gene “on” is to transcribe (copy) it into messenger RNA (mRNA). That mRNA is then decoded, usually, into a specific protein. Genes are transcribed by the enzyme called RNA polymerase.
Development: the process in which a fertilized egg (e.g., a human egg) divides many times and eventually forms an organism. During this process, many of the roughly 23,000 genes of a human are turned “on” or “off” in different combinations, at different times and places in the developing organism. The process produces many different cell types in different organs (e.g. liver and brain), but all retain the original set of genes.
Transcription factors: proteins that bind to specific DNA sequences near specific genes and turn transcription of those genes on and off. A transcriptional ‘activator’, for example, bears two surfaces: one binds a specific sequence in DNA, and the other binds to, and thereby recruits to the gene, protein complexes that include RNA polymerase. It is widely acknowledged that the identity of a cell in the body depends on the array of transcription factors present in the cell, and the cell’s history. RNA molecules can also recognize specific genomic sequences, and they too sometimes work as regulators. Neither transcription factors nor these kinds of RNA molecules – the fundamental regulators of gene expression and development – are mentioned in the New Yorker article.
Signals: these come in many forms (small molecules like estrogen, larger molecules (often proteins such as cytokines) that determine the ability of transcription factors to work. For example, estrogen binds directly to a transcription factor (the estrogen receptor) and, by changing its shape, permits it to bind DNA and activate transcription.
“Memory”: a dividing cell can (often does) produce daughters that are identical, and that express identical genes as does the mother cell. This occurs because the transcription factors present in the mother cell are passively transmitted to the daughters as the cell divides, and they go to work in their new contexts as before. To make two different daughters, the cell must distribute its transcription factors asymmetrically.
Positive Feedback: An activator can maintain its own expression by positive feedback. This requires, simply, that a copy of the DNA sequence to which the activator binds is present near its own gene. Expression of the activator then becomes self-perpetuating. The activator (of which there now are many copies in the cell) activates other target genes as it maintains its own expression. This kind of ‘memory circuit’, first described in bacteria, is found in higher organisms as well. Positive feedback can explain how a fully differentiated cell (that is, a cell that has reached its developmental endpoint) maintains its identity.
Nucleosomes: DNA in higher organisms (eukaryotes) is wrapped, like beads on a string, around certain proteins (called histones), to form nucleosomes. The histones are subject to enzymatic modifications: e.g., acetyl, methyl, phosphate, etc. groups can be added to these structures. In bacteria there are no nucleosomes, and the DNA is more or less ‘naked’.
“Epigenetic modifications”: please don’t worry about the word ”epigenetic”; it is misused in any case. What Mukherjee refers to by this term are the histone modifications mentioned above, and a modification to DNA itself: the addition of methyl groups. Keep in mind that the organisms that have taught us the most about development – flies (Drosophila) and worms (C. elegans)—do not have the enzymes required for DNA methylation. That does not mean that DNA methylation cannot do interesting things in humans, for example, but it is obviously not at the heart of gene regulation.
Specificity Development requires the highly specific sequential turning on and off of sets of genes. Transcription factors and RNA supply this specificity, but enzymes that impart modifications to histones cannot: every nucleosome (and hence every gene) appears the same to the enzyme. Thus such enzymes cannot pick out particular nucleosomes associated with particular genes to modify. Histone modifications might be imagined to convey ‘memory’ as cells divide – but there are no convincing indications that this happens, nor are there molecular models that might explain why they would have the imputed effects.
Analysis and critique of Mukherjee’s article
The picture we have just sketched has taken the combined efforts of many scientists over 50 years to develop. So what, then, is the problem with the New Yorker article?
There are two: first, the picture we have just sketched, emphasizing the primary role of transcription factors and RNA, is absent. Second, that picture is replaced by highly dubious speculations, some of which don’t make sense, and none of which has been shown to work as imagined in the article.
(Quotes from the Mukherjee article are indented and in plain text; they are followed by comments, flush left and in bold, by Ptashne and Greally.)
In 1978, having obtained a Ph.D. in biology at Indiana University, Allis began to tackle a problem that had long troubled geneticists and cell biologists: if all the cells in the body have the same genome, how does one become a nerve cell, say, and another a blood cell, which looks and functions very differently?
The problems referred to were recognized long before 1978. In fact, these were exactly the problems that the great French scientists François Jacob and Jacques Monod took on in the 1950s-60s. In a series of brilliant experiments, Jacob and Monod showed that in bacteria, certain genes encode products that regulate (turn on and off) specific other genes. Those regulatory molecules turned out to be proteins, some of which respond to signals from the environment. Much of the story of modern biology has been figuring out how these proteins – in bacteria and in higher organisms – bind to and regulate specific genes. Of note is that in higher organisms, the regulatory proteins look and act like those in bacteria, despite the fact that eukaryotic DNA is wrapped in nucleosomes whereas bacterial DNA is not. We have also learned that certain RNA molecules can play a regulatory role, a phenomenon made possible by the fact that RNA molecules, like regulatory proteins, can recognize specific genomic sequences.
In the nineteen-forties, Conrad Waddington, an English embryologist, had proposed an ingenious answer: cells acquired their identities just as humans do—by letting nurture (environmental signals) modify nature (genes). For that to happen, Waddington concluded, an additional layer of information must exist within a cell—a layer that hovered, ghostlike, above the genome. This layer would carry the “memory” of the cell, recording its past and establishing its future, marking its identity and its destiny but permitting that identity to be changed, if needed. He termed the phenomenon “epigenetics”—“above genetics.”
This description greatly misrepresents the original concept. Waddington argued that development proceeds not by the loss (or gain) of genes, which would be a “genetic” process, but rather that some genes would be selectively expressed in specific and complex cellular patterns as development proceeds. He referred to this intersection of embryology (then called “epigenesis”) and genetics as “epigenetic”.We now understand that regulatory proteins work in combinations to turn on and off genes, including their own genes, and that sometimes the regulatory proteins respond to signals sent by other cells. It should be emphasized that Waddington never proposed any “ghost-like” layer of additional information hovering above the gene. This is a later misinterpretation of a literal translation of the term epigenetics, with “epi-“ meaning “above/upon” the genetic information encoded in DNA sequence. Unfortunately, this new and pervasive definition encompasses all of transcriptional regulation and is of no practical value.
…..more
By 2000, Allis and his colleagues around the world had identified a gamut of proteins that could modify histones, and so modulate the activity of genes. Other systems, too, that could scratch different kinds of code on the genome were identified (some of these discoveries predating the identification of histone modifications). One involved the addition of a chemical side chain, called a methyl group, to DNA. The methyl groups hang off the DNA string like Christmas ornaments, and specific proteins add and remove the ornaments, in effect “decorating” the genome. The most heavily methylated parts of the genome tend to be dampened in their activity.
It is true that enzymes that modify histones have been found—lots of them. A striking problem is that, after all this time, it is not at all clear what the vast majority of these modifications do. When these enzymatic activities are eliminated by mutation of their active sites (a task substantially easier to accomplish in yeast than in higher organisms) they mostly have little or no effect on transcription. It is not even clear that histones are the biologically relevant substrates of most of these enzymes.
In the ensuing decade, Allis wrote enormous, magisterial papers in which a rich cast of histone-modifying proteins appear and reappear through various roles, mapping out a hatchwork of complexity. . . These protein systems, overlaying information on the genome, interacted with one another, reinforcing or attenuating their signals. Together, they generated the bewildering intricacy necessary for a cell to build a constellation of other cells out of the same genes, and for the cells to add “memories” to their genomes and transmit these memories to their progeny. “There’s an epigenetic code, just like there’s a genetic code,” Allis said. “There are codes to make parts of the genome more active, and codes to make them inactive.”
By ‘epigenetic code’ the author seems to mean specific arrays of nucleosome modifications, imparted over time and cell divisions, marking genes for expression. This idea has been tested in many experiments and has been found not to hold.
….. and more
Larry H. Bernstein, MD, FCAP
I hope that this piece brings greater clarity to the discussion. I have heard the use of the term “epigenetics” for over a decade. The term was never so clear. I think that the New Yorker article was a reasonable article for the intended audience. It was not intended to clarify debates about a mechanism for epigenetic based changes in evolutionary science. I think it actually punctures the “classic model” of the cell depending only on double stranded DNA and transcription, which deflates our concept of the living cell. The concept of epigenetics was never really formulated as far as I have seen, and I have done serious work in enzymology and proteins at a time that we did not have the technology that exists today. I have considered with the critics that protein folding, protein misfolding, protein interactions with proximity of polar and nonpolar groups, and the regulatory role of microRNAs that are not involved in translation, and the evolving concept of what is “dark (noncoding) DNA” lend credence to the complexity of this discussion. Even more interesting is the fact that enzymes (and isoforms of enzymes) have a huge role in cellular metabolic differences and in the function of metabolic pathways. What is less understood is the extremely fast reactions involved in these cellular reactions. These reactions are in my view critical drivers. This is brought out by Erwin Schroedinger in the book What is Life? which infers that there can be no mathematical expression of life processes.

 and
and 







 These conclusions do not mean that the brain does not remain malleable, even in old age. Any mentally effortful new experience, such as learning a language, acquiring a motor skill, navigating in a new environment, and, yes, playing commercially available computer games, will produce changes in those neural systems that support acquisition of the new skill. For example, there may be an increase in the number of synapses, the number of neurons and supporting cells, or a strengthening of the connections among them. This type of brain plasticity is possible throughout the life span, though younger brains seem to have an advantage over the older ones. It would be appropriate to conclude from such work that the potential to learn new skills remains intact throughout the life span. However at this point it is not appropriate to conclude that training-induced changes go significantly beyond the learned skills, that they affect broad abilities with real-world relevance, or that they generally promote “brain health”.
These conclusions do not mean that the brain does not remain malleable, even in old age. Any mentally effortful new experience, such as learning a language, acquiring a motor skill, navigating in a new environment, and, yes, playing commercially available computer games, will produce changes in those neural systems that support acquisition of the new skill. For example, there may be an increase in the number of synapses, the number of neurons and supporting cells, or a strengthening of the connections among them. This type of brain plasticity is possible throughout the life span, though younger brains seem to have an advantage over the older ones. It would be appropriate to conclude from such work that the potential to learn new skills remains intact throughout the life span. However at this point it is not appropriate to conclude that training-induced changes go significantly beyond the learned skills, that they affect broad abilities with real-world relevance, or that they generally promote “brain health”. In a balanced evaluation of brain games, we also need to keep in mind opportunity costs. Time spent playing the games is time not spent reading, socializing, gardening, exercising, or engaging in many other activities that may benefit cognitive and physical health of older adults. Given that the effects of playing the games tend to be task-specific, it may be advisable to train an activity that by itself comes with benefits for everyday life. Another drawback of publicizing computer games as a fix to deteriorating cognitive performance is that it diverts attention and resources from prevention efforts. The promise of a magic bullet detracts from the message that cognitive vigor in old age, to the extent that it can be influenced by the lives we live, reflects the long-term effects of a healthy and active lifestyle.
In a balanced evaluation of brain games, we also need to keep in mind opportunity costs. Time spent playing the games is time not spent reading, socializing, gardening, exercising, or engaging in many other activities that may benefit cognitive and physical health of older adults. Given that the effects of playing the games tend to be task-specific, it may be advisable to train an activity that by itself comes with benefits for everyday life. Another drawback of publicizing computer games as a fix to deteriorating cognitive performance is that it diverts attention and resources from prevention efforts. The promise of a magic bullet detracts from the message that cognitive vigor in old age, to the extent that it can be influenced by the lives we live, reflects the long-term effects of a healthy and active lifestyle.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}