Single Cell Shines Light on Cell Malignant Transformation
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Single Cell Lights Beacon to Cancer Origins
http://www.genengnews.com/gen-news-highlights/single-cell-lights-beacon-to-cancer-origins/81252303/
http://www.nytimes.com/2016/01/29/health/how-skin-cancer-develops-melanoma-zebra-fish.html?ref=todayspaper
For the first time scientists are able to visualize the origin of cancer cells and track their progression through tumorigenesis. [Dr. Leonard Zon, Boston Children’s Hospital].
Conundrums exist in every facet of our daily lives and are often dismissed as either general annoyances or fleeting curiosities. Yet for scientists, solving common scientific enigmas can often lead to a watershed moment within their respective discipline. For instance, researchers have been perplexed for decades as to why the vast number of cells that have cancer genes never morph into cancerous tumors.
Researchers at Boston Children’s Hospital have published results from a new study, which visualized the origins of cancer from the first affected cell and watched its spread within a live animal. The research team is hopeful that their findings could change the way scientists understand melanoma and other cancers, as well as open doors to new therapeutic interventions.
“An important mystery has been why some cells in the body already have mutations seen in cancer, but do not yet fully behave like the cancer,” stated lead author Charles Kaufman, M.D., Ph.D., a postdoctoral fellow in the laboratory of Howard Hughes Medical Institute investigator Leonard Zon, M.D. at Boston Children’s Hospital. “We found that the beginning of cancer occurs after activation of an oncogene or loss of a tumor suppressor, and involves a change that takes a single cell back to a stem cell state.”
The findings from this study were published online today in Science through an article entitled “A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation.”
The Boston researchers tracked the development of melanoma in live zebrafish that had been genetically engineered to have the human cancer mutation BRAFV600E—found in most benign moles—and the p53 tumor suppressor gene removed. Moreover, the fish contained a GFP-crestin gene that would light up if it was turned on, a beacon indicating activation of a genetic program characteristic of stem cells. Typically this program is turned off after embryonic development. Occasionally, however, crestin and other genes in the program turn back on in individual cells.
“Every so often we would see a green spot on a fish,” noted Dr. Zon, who is the director of the Stem Cell Research Program at Boston Children’s, member of the Harvard Stem Cell Institute, and senior author on the current study. “When we followed them, they became tumors 100 percent of the time.”
When Dr. Zon and his colleagues looked to see what was different in these early cancer cells, they found that crestin and the other activated genes are the same ones turned on during zebrafish embryonic development, specifically in the stem cells that give rise to the pigment cells known as melanocytes within a structure called the neural crest.
“What’s cool about this group of genes is that they also get turned on in human melanoma,” Dr. Zon said. “It’s a change in cell fate, back to neural crest status.”
While trying to track these glowing cells in live fish was exceedingly difficult, Dr. Kaufman managed to isolate and characterize 30 fish with a small green glowing cluster of cells. Interestingly, in all 30 cases, the clusters grew into melanomas. In two fish, Dr. Kaufman was able to see a single green-glowing cell and watch it divide to ultimately become a tumor mass.
“It’s estimated that only one in tens or hundreds of millions of cells in a mole eventually become a melanoma,” explained Dr. Kaufman. “Because we can also efficiently breed many fish, we can look for these very rare events. The rarity is very similar in both humans and fish, which suggests that the underlying process of melanoma formation is probably much the same in humans.”
The investigators are optimistic that the results from their study could lead to new genetic tests for suspicious moles to see whether the cells are behaving like neural crest cells, indicating that the stem-cell program has been turned on. Additionally, the scientists are investigating the regulatory elements that turn on the genetic program, often called super-enhancers. These control elements have analogous epigenetic functions in zebrafish and human melanoma, potentially providing druggable targets to stop a mole from becoming cancerous.
“https://vimeo.com/152633925”
A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation
Charles K. Kaufman, …., Richard A. Young, Leonard I. Zon
Science 29 Jan 2016; 351(6272) http://dx.doi.org:/10.1126/science.aad2197
Stem Cell Program and Division of Hematology/Oncology, Children’s Hospital Boston, Howard Hughes Medical Institute, Boston, MA 02115, USA. 2Harvard Stem Cell Institute, Boston, MA 02115, USA.3Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.4Harvard Medical School, Boston, MA 02115, USA
Visualizing the beginnings of melanoma
In cancer biology, a tumor begins from a single cell within a group of precancerous cells that share genetic mutations. Kaufman et al. used a zebrafish melanoma model to visualize cancer initiation (see the Perspective by Boumahdi and Blanpain). They used a fluorescent reporter that specifically lit up neural crest progenitors that are only present during embryogenesis or during adult melanoma tumor formation. The appearance of this tumor correlated with a set of gene regulatory elements, called super-enhancers, whose identification and manipulation may prove beneficial in detecting and preventing melanoma initiation.
Science, this issue p. 10.1126/science.aad3867; see also p. 453
Structured Abstract
INTRODUCTION
The “cancerized field” concept posits that cells in a given tissue sharing an oncogenic mutation are cancer-prone, yet only discreet clones within the field initiate tumors. Studying the process of cancer initiation has remained challenging because of (i) the rarity of these events, (ii) the difficulty of visiualizing initiating clones in living organisms, and (iii) the transient nature of a newly transformed clone emerging before it expands to form an early tumor. A more complete understanding of the molecular processes that regulate cancer initiation could provide important prognostic information about which precancerous lesions are most prone to becoming cancer and also implicate druggable molecular pathways that, when inhibited, may prevent the cancer from ever starting.
RATIONALE
The majority of benign nevi carry oncogenic BRAFV600E mutations and can be considered a cancerized field of melanocytes, but they only rarely convert to melanoma. In an effort to define events that initiate cancer, we used a melanoma model in the zebrafish in which the humanBRAFV600E oncogene is driven by the melanocyte-specific mitfa promoter. When bred into a p53mutant background, these fish develop melanoma tumors over the course of many months. The zebrafish crestin gene is expressed embryonically in neural crest progenitors (NCPs) and is specifically reexpressed only in melanoma tumors, making it an ideal candidate for tracking melanoma from initiation onward.
RESULTS
We developed a crestin:EGFP reporter that recapitulates the embryonic neural crest expression pattern of crestin and its expression in melanoma tumors. We show through live imaging of transgenic zebrafish crestin reporters that within a cancerized field (BRAFV600E–mutant; p53-deficient), a single melanocyte reactivates the NCP state, and this establishes that a fate change occurs at melanoma initiation in this model. Early crestin+ patches of cells expand and are transplantable in a manner consistent with their possessing tumorigenic activity, and they exhibit a gene expression pattern consistent with the NCP identity readout by the crestin reporter. Thecrestin element is regulated by NCP transcription factors, including sox10. Forced sox10overexpression in melanocytes accelerated melanoma formation, whereas CRISPR/Cas9 targeting of sox10 delayed melanoma onset. We show activation of super-enhancers at NCP genes in both zebrafish and human melanomas, identifying an epigenetic mechanism for control of this NCP signature leading to melanoma.
CONCLUSION
This work using our zebrafish melanoma model and in vivo reporter of NCP identity allows us to see cancer from its birth as a single cell and shows the importance of NCP-state reemergence as a key event in melanoma initiation from a field of cancer-prone melanocytes. Thus, in addition to the typical fixed genetic alterations in oncogenes and tumor supressors that are required for cancer development, the reemergence of progenitor identity may be an additional rate-limiting step in the formation of melanoma. Preventing NCP reemergence in a field of cancer-prone melanocytes may thus prove therapeutically useful, and the association of NCP genes with super-enhancer regulatory elements implicates the associated druggable epigenetic machinery in this process.
https://d2ufo47lrtsv5s.cloudfront.net/content/sci/351/6272/aad2197/F1.large.jpg
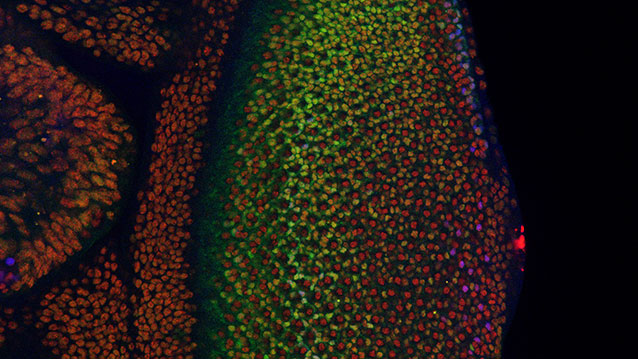
Neural crest reporter expression in melanoma.
The crestin:EGFP transgene is specifically expressed in melanoma inBRAFV600E/p53 mutant melanoma-prone zebrafish. (Top) A single cell expressingcrestin:EGFP expands into a small patch of cells over the course of 2 weeks, capturing the initiation of melanoma formation (bracket). (Bottom) A fully formed melanoma specifically expresses crestin:EGFP, whereas the rest of the fish remains EGFP-negative.
Neural crest reporter expression in melanoma.
The crestin:EGFP transgene is specifically expressed in melanoma inBRAFV600E/p53 mutant melanoma-prone zebrafish. (Top) A single cell expands into a small patch of cells over the course of 2 weeks, capturing the initiation of melanoma formation (bracket). (Bottom) A fully formed melanoma specifically expresses crestin:EGFP, whereas the rest of the fish remains EGFP-negative.
The “cancerized field” concept posits that cancer-prone cells in a given tissue share an oncogenic mutation, but only discreet clones within the field initiate tumors. Most benign nevi carry oncogenic BRAFV600E mutations but rarely become melanoma. The zebrafish crestin gene is expressed embryonically in neural crest progenitors (NCPs) and specifically reexpressed in melanoma. Live imaging of transgenic zebrafish crestin reporters shows that within a cancerized field (BRAFV600E–mutant; p53-deficient), a single melanocyte reactivates the NCP state, revealing a fate change at melanoma initiation in this model. NCP transcription factors, including sox10, regulate crestin expression. Forced sox10 overexpression in melanocytes accelerated melanoma formation, which is consistent with activation of NCP genes and super-enhancers leading to melanoma. Our work highlights NCP state reemergence as a key event in melanoma initiation.
A Cancer’s Surprise Origins, Caught in Action
Harvard University

http://www.biosciencetechnology.com/sites/biosciencetechnology.com/files/bt1601_harvard_melenoma.jpg
Most of the time skin moles are harmless, but they occasionally turn into melanoma, a life-threatening skin cancer. Leonard Zon and colleagues found that this happens when a single cell regresses back to a stem cell state and starts to divide and invade the surrounding tissue. (Image: Courtesy of Boston Children’s Hospital)
Researchers at Harvard-affiliated Boston Children’s Hospital have, for the first time, visualized the origins of cancer from the first affected cell and watched its spread in a live animal. Their work, published in the Jan. 29 issue of Science, could change the way scientists understand melanoma and other cancers and lead to new, early treatments before the cancer has taken hold.
“An important mystery has been why some cells in the body already have mutations seen in cancer, but do not yet fully behave like the cancer,” said the paper’s first author, Charles Kaufman, a postdoctoral fellow in the Zon Laboratory at Boston Children’s Hospital. “We found that the beginning of cancer occurs after activation of an oncogene or loss of a tumor suppressor, and involves a change that takes a single cell back to a stem cell state.”
That change, Kaufman and colleagues found, involves a set of genes that could be targeted to stop cancer from ever starting.
The study imaged live zebrafish over time to track the development of melanoma. All the fish had the human cancer mutation BRAFV600E — found in most benign moles — and had also lost the tumor suppressor gene p53.
Kaufman and colleagues engineered the fish to light up in fluorescent green if a gene called crestin was turned on — a “beacon” indicating activation of a genetic program characteristic of stem cells. This program normally shuts off after embryonic development, but occasionally, in certain cells and for reasons not yet known, crestin and other genes in the program turn back on.
“Every so often we would see a green spot on a fish,” said Leonard Zon, director of the Stem Cell Research Program at Boston Children’s and senior investigator on the study. “When we followed them, they became tumors 100 percent of the time.”
The cell that caused melanoma
When Kaufman, Zon, and colleagues looked to see what was different about these early cancer cells, they found that crestin and the other activated genes were the same ones turned on during zebrafish embryonic development — specifically, in the stem cells that give rise to the pigment cells known as melanocytes, within a structure called the neural crest.
“What’s cool about this group of genes is that they also get turned on in human melanoma,” said Zon, who is also a member of the Harvard Stem Cell Institute and a Howard Hughes Medical Institute investigator. “It’s a change in cell fate, back to neural crest status.”
Finding these cancer-originating cells was tedious. Wearing goggles and using a microscope with a fluorescent filter, Kaufman examined the fish as they swam around, shooting video with his iPhone. Scanning 50 fish could take two to three hours. In 30 fish, Kaufman spotted a small cluster of green-glowing cells about the size of the head of a Sharpie marker — and in all 30 cases, these spots grew into melanomas. In two cases, he was able to see on a single green-glowing cell and watch it divide and ultimately become a tumor mass.
“It’s estimated that only one in tens or hundreds of millions of cells in a mole eventually becomes a melanoma,” said Kaufman, who is also an instructor at the Harvard-affiliated Dana-Farber Cancer Institute. “Because we can also efficiently breed many fish, we can look for these very rare events. The rarity is very similar in both humans and fish, which suggests that the underlying process of melanoma formation is probably much the same in humans.”
Zon, the Grousbeck Professor of Pediatric Medicine at Harvard Medical School, and Kaufman believe that their findings could lead to a new genetic test for suspicious moles to see whether the cells are behaving like neural crest cells, indicating that the stem-cell program has been turned on. They are also investigating the regulatory elements that turn on the genetic program (known as super-enhancers). These DNA elements have epigenetic functions that are similar in zebrafish and human melanoma, and could potentially be targeted with drugs to stop a mole from becoming cancerous.
A paradigm shift for cancer?
Zon and Kaufman posit a new model for cancer formation, going back to a decades-old concept of “field cancerization.” They propose that normal tissue becomes primed for cancer when oncogenes are activated and tumor suppressor genes are silenced or lost, but that cancer develops only when a cell in the tissue reverts to a more primitive, embryonic state and starts dividing. They believe this model may apply to most if not all cancers, not just melanoma.
The study was supported by the National Institutes of Health, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the Ellison Foundation, the Melanoma Research Alliance, the V Foundation, and the Howard Hughes Medical Institute. Zon is a founder and stockholder of Fate, Inc., and Scholar Rock.
Source: Harvard Gazette
The little fish that could
Zebrafish help HSCI researchers fight human disease

Leonard Zon, MD, could be called HSCI’s most prolific aquarist. The Executive Committee Chair estimates that he has over 300,000 fish spread throughout his laboratories at the Harvard Department of Stem Cell and Regenerative Biology and Boston Children’s Hospital
His collection isn’t very diverse, composed as it is entirely of zebrafish—minnow-like, freshwater, tropical fish that grow about an inch-and-a-half long. But Zon isn’t interested in winning best in show. Instead, he is leading a scientific movement to show that his striped fish could be humanity’s new animal of choice for drug discovery and studying disease.
Not only does the zebrafish require fewer facility resources than science’s go-to organism, the mouse, but fish are quicker to reproduce, easier to experimentally manipulate, and at least 70 percent of human protein-coding genes have analogs in the zebrafish, including those related to skin cancer, muscular dystrophy, and T cell leukemia.
“The zebrafish is now emerging as another powerful organism for the modeling and study of human diseases, and it is conceivable that zebrafish models will complement mouse models in the future,” Zon wrote in the December issue of Trends in Cell Biology, pointing out the accelerating increase in studies on zebrafish, from about 150 in 1995 to over 2,000 in 2013.
As a pediatric oncologist, Zon’s main purpose in using zebrafish is to help cancer patients. He is the first scientist to successfully apply basic research from the zebrafish to develop an FDA-approved treatment for human melanoma, and is now applying similar methods to find therapies for muscle and blood cancers.
One cancer Zon is pursuing is rhabdomyosarcoma, a rare muscle cancer diagnosed primarily in early childhood. Zebrafish, under certain conditions, develop tumors very similar to the human disease. He is currently using zebrafish embryos to identify which genes transform a normal muscle stem cell into a malignant tumor, as well as searching for factors that might suppress the cancer.
“One of the things that’s really interesting in zebrafish is that the embryos are completely transparent and you can watch the tumors invade the normal tissues,”
he said. “That’s a process that you can’t study in any other organism.”
Postdocs in Zon’s lab can expose the zebrafish embryos to multiple chemicals and literally watch changes in the fish’s development. In 2007, this technique led to the discovery of a type of prostaglandin that expands blood stem cells about 300 to 400 percent. Last fall, the prostaglandin found in Zon’s lab passed Phase Ib clinical trials as a therapy that increases the success of cord blood transplants.
A similar screen led to a major paper last November, in which Zon and fellow HSCI Executive Committee member Amy Wagers, PhD, showed that the same chemicals that stimulate muscle development in zebrafish can also be used to differentiate human stem cells into muscle cells in the laboratory, an historically challenging task that, now overcome, makes muscle cell therapy a more realistic possibility.
“This research demonstrates that over 300 million years of evolution, the pathways used in the fish are conserved through vertebrates all the way up to the human,” Zon said.
His passion for and success with zebrafish has helped make the animal a staple in HSCI faculty member laboratories across Boston, and an unexpected symbol for stem cell research.
Human muscle stem cell therapy gets help from zebrafish
Harvard Stem Cell Scientists have discovered that the same chemicals that stimulate muscle development in zebrafish can also be used to differentiate human stem cells into muscle cells in the laboratory, an historically challenging task that, now overcome, makes muscle cell therapy a more realistic clinical possibility.
The work, published this week in the journal Cell, began with a discovery by Boston Children’s Hospital researchers, led by Leonard Zon, MD, and graduate student Cong (Tony) Xu, who tested 2,400 different chemicals in cultures of zebrafish embryo cells to determine if any could increase the numbers of muscle cells formed. Using fluorescent reporter fish in which muscle cells were visible during their creation, the researchers found six chemicals that were very effective at promoting muscle formation.

Zon shared his results with Harvard Department of Stem Cell and Regenerative Biologyprofessor Amy Wagers, PhD, and Mohammadsharif Tabebordbar, a graduate student in her laboratory, who tested the six chemicals in mice. One of the six, called forskolin, was found to increase the numbers of muscle stem cells from mice that could be obtained when these cells were grown in laboratory dishes. Moreover, the cultured cells successfully integrated into muscle when transplanted back into mice.
Inspired by the successful application of these chemicals in mice, Salvatore Iovino, PhD, a joint postdoctoral fellow in the Wagers lab and the lab of C. Ronald Kahn, MD, at the Joslin Diabetes Center, investigated whether the chemicals would also affect human cells and found that a combination of three chemicals, including forskolin, could induce differentiation of human induced pluripotent stem (iPS) cells, made by reprogramming skin cells. Exposure of iPS cells to these chemicals converted them into skeletal muscle, an outcome the Wagers and Kahn labs had been striving to achieve for years using conventional methods. When transplanted into a mouse, the human iPS-derived muscle cells also contributed to muscle repair, offering early promise that this protocol could provide a route to muscle stem cell therapy in humans.
The interdisciplinary, cross-laboratory collaboration between Zon, Wagers, and Kahn highlights the advantage of open exchange between researchers. “If we had done this screen directly on human iPS cells, it would have taken at least 10 times as long and cost 100 times as much,” said Wagers. “The zebrafish gave us a big advantage here because it has a fast generation time, rapid development, and can be easily and relatively cheaply screened in a culture dish.”
“This research demonstrates that over 300 million years of evolution, the pathways used in the fish are conserved through vertebrates all the way up to the human,” said Wagers’ fellow HSCRB professor Leonard Zon, chair of the Harvard Stem Cell InstituteExecutive Committee and director of the stem cell program at Boston Children’s Hospital. “We can now make enough human muscle progenitors in a dish to allow us to model diseases of the muscle lineage, like Duchenne muscular dystrophy, conduct drug screens to find chemicals that correct those disease, and in the long term, efficiently transplant muscle stem cells into a patient.”
In a similar biomedical application, Kahn, who is chief academic officer at the Joslin, plans to apply the new ability to quickly produce muscle stem cells for diabetes research. His lab will generate iPS-derived muscle cells from people who are at risk for diabetes and people who have diabetes to identify alterations that lead to insulin resistance in the muscle.
Going forward, Zon plans to apply this platform of cross-species discovery to other stem cell lines, including those involved in blood and eye development. “We have a new system to use to study tissue development, and it’s not just muscle that can be studied, every single organ can be studied in the zebrafish system,” he said.
The research was funded by the Harvard Stem Cell Institute, the Howard Hughes Medical Institute, the National Institutes of Health, the Novo Nordisk Diabetes Center, and the Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center.
Photo:Newly created muscle progenitor cells (green) and muscle fibers (red) in a zebrafish embryo. (credit: Zon Lab)
Dynamics and heterogeneity of a fate determinant during transition towards cell differentiation
Nicolás Peláez
Department of Molecular Biosciences, Northwestern University, Evanston, United States; Department of Chemical and Biological Engineering, Howard Hughes Medical Institute, University Northwestern, Evanston, United States
Contribution: Conception and design; Acquisition of data; Analysis and interpretation of data; Drafting or revising the article
No competing interests declared
” data-author-inst=”NorthwesternUniversityUnitedStates|HowardHughesMedicalInstituteUniversityNorthwesternUnitedStates”>Nicolás Peláez, et al. eLife 2015;4:e08924 http://dx.doi.org/10.7554/eLife.08924.001
Yan is an ETS-domain transcription factor responsible for maintaining Drosophila eye cells in a multipotent state. Yan is at the core of a regulatory network that determines the time and place in which cells transit from multipotency to one of several differentiated lineages. Using a fluorescent reporter for Yan expression, we observed a biphasic distribution of Yan in multipotent cells, with a rapid inductive phase and slow decay phase. Transitions to various differentiated states occurred over the course of this dynamic process, suggesting that Yan expression level does not strongly determine cell potential. Consistent with this conclusion, perturbing Yan expression by varying gene dosage had no effect on cell fate transitions. However, we observed that as cells transited to differentiation, Yan expression became highly heterogeneous and this heterogeneity was transient. Signals received via the EGF Receptor were necessary for the transience in Yan noise since genetic loss caused sustained noise. Since these signals are essential for eye cells to differentiate, we suggest that dynamic heterogeneity of Yan is a necessary element of the transition process, and cell states are stabilized through noise reduction.
As animal cells develop, they pass through different states to mature into specific cell types. For some cells, this development depends on the cell’s ability to switch between two stable states, a property called bistability.
Many bistable systems operate during development and often feature proteins called transcription factors that regulate a few cell states in specific tissues. These proteins activate or repress a range of target genes, and cells can adjust the levels of the transcription factors to bring about transitions between states. In fruit flies, two transcription factors, called Yan and Pnt, regulate numerous processes throughout development.
In the developing embryo of a fruit fly, Yan and Pnt are regulated by signals induced by a receptor called EGFR. When EGFR is activated, Pnt is produced and Yan is degraded. When this receptor is not activated, Yan is produced and represses Pnt. Mathematical modelling of these interactions has concluded that this is a bistable system: that is, cells should either have high levels of Yan and low levels Pnt, or low Yan levels and high Pnt levels. However, larval eye cells first activate, and then turn off, both proteins together. This argues against bistability and raises questions about how these proteins regulate cell fate transitions in the eye, and perhaps in other organs.
To investigate this question, Peláez et al. tagged Yan with a fluorescent marker to track its activity in the eyes of fruit fly larvae as they develop. A combination of fluorescence-based microscopy and an automated imaging analysis system were then used to score fluorescence in thousands of individual eye cells and assess the changes in the levels of Yan over time. This approach revealed some unexpected results.
Yan levels were seen to vary in both immature and maturing cells. Thus eye cells transition between states as Yan levels increase rapidly, suggesting the need for a mechanism that is distinct from bistability. Peláez et al. suggest that larger changes in the seemingly random fluctuations in the levels of the transcription factor (also known as “expression noise”) might play a role in this mechanism. In particular, Yan expression noise briefly peaks as cells transition to a more mature state, and the transient nature of this ‘noisy’ response requires the activation of EGFR.
One possible explanation for these observations is that Yan’s effect on cell states depends on this variability in its levels, which might prime cells to change states when they receive another signal. These findings also raise many questions for future studies to explore, including how this increase in the noise level of Yan is triggered as cells begin their transitions towards specific cell types.
Introduction
Cells within complex organisms exist in different states that confer specific functionalities to each cell. Cellular states can be organized into cyclic cascades such as the G1-S-G2-M cell cycle, or into linear cascades as observed in cell differentiation. A common feature of cells that undergo state transitions is the apparent irreversibility of the transitions even when such transitions are triggered by transient stimuli. Modeling of these transitions often assumes system bistability, in which cells can be resting in one of two stable states.
Animal cells frequently utilize transcription factors to enforce a given state, and transitions to another state are driven by increasing or decreasing the levels of these transcription factors (Yao et al., 2008;Kueh et al., 2013; Laslo et al., 2006; Park et al., 2012). The Rb-E2F pathway generates bistability in E2F expression, which dictates the transition from G1 to S phase (Yao et al., 2008). Expression of the transcription factor PU.1 determines lymphoid versus myeloid hematopoietic cell lineages (Kueh et al., 2013; Laslo et al., 2006). Adipocyte differentiation is controlled by differential expression of C/EBP and PPARγ proteins (Park et al., 2012). Regulation by positive feedback is a hallmark of bistable systems, and in all of the above cases, the transcription factors act in one or more positive feedback circuits. In some systems, positive feedback is generated by two transcription factors that mutually repress each other’s expression. In these scenarios, cells in one state continually express a transcription factor that represses a second transcription factor, and when these cells transit to another state, they continually express the second transcription factor, which represses its antagonist. Fate restriction in hematopoietic, neural, pancreatic, and muscle cell lineages is regulated by such double-negative feedback circuits (Briscoe et al., 2000; Laslo et al., 2006; Olguin et al., 2007; Schaffer et al., 2010).
Most examples of this type of bistable mechanism have transcription factors that act specifically within a handful of cell states limited to a single tissue or organ system. One remarkable exception to this rule is found in Drosophila. There, two ETS-domain transcription factors act in a wide assortment of cell types across the body and across the life cycle. Yan and Pointed (Pnt) act downstream of signals mediated by receptor tyrosine kinases (RTKs) that regulate cell differentiation, migration, and division in tissues ranging from ovarian follicular cells, dorsal and ventral neuroectoderm, embryonic mesoderm, the embryonic trachea, and the post-embryonic compound eye (Dumstrei et al., 1998;Gabay et al., 1996; Halfon et al., 2000; Jurgens et al., 1984; Morimoto et al., 1996; O’Neill et al., 1994; Ohshiro et al., 2002; Yao et al., 2008) It is thought that Yan and Pnt control such diverse cell states by acting in concert with tissue-specific transcription factors to regulate transcription of appropriate target genes. For instance, transcription of even-skipped occurs in mesoderm only if Yan/Pnt act in combination with Tinman and Twist proteins (Halfon et al., 2000), whereas transcription of prospero (pros) in the eye only occurs if Yan/Pnt act in combination with Lozenge, Sine Oculis, and Glass proteins (Hayashi et al., 2008; Xu et al., 2000).
Several tissues show mutually exclusive expression of Yan and Pnt, suggestive of cross-repression (Boisclair Lachance et al., 2014). The best characterized is the embryonic ventral ectoderm, where ventral-most cells express Pnt and more lateral cells express Yan (Gabay et al., 1996). This pattern is established by secretion of a ligand for the EGF Receptor (EGFR) from the ventral midline (Golembo et al., 1996). EGFR activation in nearby ventral cells induces the Ras-MAPK pathway to express Pnt and degrade Yan (Gabay et al., 1996; Melen et al., 2005). Cells with insufficient EGFR activation express Yan, which represses Pnt. Mathematical modeling has described this as a bistable system in which cells are either in a High Yan/Low Pnt state or a Low Yan/High Pnt state (Graham et al., 2010; Melen et al., 2005). Transition from one state to the other is ultrasensitive to the strength of EGFR activation (Melen et al., 2005).
Paradoxically, other Drosophila tissues show co-expression of Yan and Pnt (Boisclair Lachance et al., 2014). The larval eye is one such tissue. Retinal progenitor cells initiate expression of both proteins, and when they transit to differentiated photoreceptor fates, these cells reduce expression of both proteins. In contrast, when retinal progenitor cells transit to differentiated cone cell fates, they maintain their expression of both proteins. These observations are at odds with long-standing genetic studies that support a standard bistable mechanism acting in the eye (Lai and Rubin, 1992;O’Neill et al., 1994; Rebay and Rubin, 1995). Thus, new approaches to studying these transitions in the eye are needed.
Here, we have adopted a systems-level approach to study Yan dynamics in the larval eye. A yellow fluorescent protein (YFP) — based isoform of Yan was developed as a reporter for Yan protein levels. Fluorescence-based microscopic imaging of cells was coupled with automated high-throughput image analysis to score fluorescence in each cell and annotate the data in a quantitative and unbiased fashion. Yan exhibits monostability, both in progenitor and differentiating cells, with Yan levels varying in cells in either state. Cell state transitions occur independent of absolute Yan concentrations, suggesting that some other mechanism allows Yan to regulate transitions. One such mechanism might be the noise in Yan levels, which undergoes a transient spike as cells begin to transition to differentiated states. Loss of EGFR signaling, which prevents cells from differentiating, causes these cells to have prolonged noisy Yan expression, and suggests that Yan noise is key for cell state transitions in the eye.
Results
The compound eye epithelium is established during embryogenesis as an internal disc of cells called the eye imaginal disc (Wolff and Ready, 1993). During the larval phase of the life cycle, the disc grows in size by asynchronous cell division. During the final 50 hr of the larval phase, a morphogenetic furrow (MF) moves across the eye disc from posterior to anterior (Figure 1A,B). All cells arrest in G1 phase within five cell diameters anterior to the furrow, and then as the furrow passes through them, periodic clusters of cells express the proneural gene atonal (Jarman et al., 1994). Atonal expression is subsequently restricted to one cell per cluster, which becomes the R8 photoreceptor. Each R8 cell then secretes an EGFR ligand that activates the receptor in neighboring cells and causes them to transit from multipotent progenitor to differentiated states (Figure 1C) (Freeman, 1996). Transitions occur in a sequence of symmetric pairs of multipotent progenitor cells that differentiate into R2/R5, R3/R4, and R1/R6 photoreceptors (Figure 1C) (Wolff and Ready, 1993). Thereafter, a single progenitor transits to a R7 photoreceptor fate followed by two pairs of cells, C1/C2 and C3/C4, that differentiate into cone cells. These cone cells are non-neuronal and form the simple lens that overlies each cluster of eight photoreceptors. The furrow induces the nearly simultaneous differentiation of a column of R8 cells, with repeated column inductions producing approximately 800 units or ommatidia as the furrow moves across the eye.
Figure 1.Development and patterning of the compound eye.
(A) Differentiation is initiated in the developing eye by the MF, which moves across the eye epithelium. On the furrow’s posterior side, G1-arrested progenitor cells undergo differentiation (light blue). On the anterior side, progenitor cells are still proliferating (dark blue). The large grey rectangle outlines the region that was analyzed for Yan levels; the small rectangle corresponds to the developmental sequence outlined in panel C (B) A maximal projection of Yan-YFP fluorescence in an eye. Bar = 100 μm. (C) Top, an apical view of the sequential differentiation of eight photoreceptor (R1-R8) and four cone cell types (C1-C4) from multipotent progenitor cells (grey) in an ommatidium. Arrows denote inductive signal transmitted from the R8 to activate EGFR on nearby cells. Bottom, a cross-section view through an eye disc adapted after Wolff and Ready (1993). Note the progenitor cell nuclei are basally positioned, and as they transition into a differentiated state, their nuclei migrate apically. C1/C2 cells are positioned anterior and posterior in the ommatidium while C3/C4 cells are positioned equatorial and polar in the ommatidium. (D) Top, an optical slice of H2Av-mRFP fluorescence in an eye disc at a plane that bisects progenitor cell nuclei. Bottom, the same optical slice imaged for Yan-YFP fluorescence. Bars = 8 μm
A central tenet of the bistable model of cell differentiation in the eye posits that differentiation is marked by a transition from high Yan protein levels in multipotent progenitor cells to low Yan levels in differentiating cells (Graham et al., 2010). Formulation of this model stemmed from studies of R7 cell differentiation, the final photoreceptor recruited to each ommatidium. Reduced Yan causes inappropriate expression of the R7 determinant pros and ectopic R7 cells in yan hypomorphic mutants (Kauffmann et al., 1996; Lai and Rubin, 1992). Conversely, a Yan isoform that is resistant to MAPK-dependent degradation, blocks R7 differentiation and pros expression (Kauffmann et al., 1996; Rebay and Rubin, 1995).
Quantifying Yan dynamics
To quantitatively test the bistable model, we used BAC recombineering to insert fast-fold yellow fluorescent protein (YFP) in-frame at the carboxy-terminus of the yan coding sequence (Webber et al., 2013). The Yan-YFP transgene fully complemented null mutations in the endogenous yan gene and completely restored normal eye development (Figure 1—figure supplement 1), demonstrating that the YFP tag does not compromise Yan function and that all essential genomic regulatory sequences are included. The pattern of Yan-YFP protein expression was qualitatively similar to that of endogenous Yan (Figure 1—figure supplement 1).
We used histone His2Av-mRFP fluorescence in fixed specimens to mark all eye cell nuclei for automated segmentation (Figure 1D—figure supplement 2). Nuclei were manually classified into the different cell types of the eye, which is possible because every cell undergoing differentiation can be unambiguously identified by its position and nuclear morphology without the need of cell-specific markers (Ready et al., 1976; Tomlinson, 1985; Tomlinson and Ready, 1987; Wolff and Ready, 1993). To validate our identification of all cell types using this method, we cross-checked with specific cell-specific markers, and found that our classification was accurate over 98% of the time (Figure 1—figure supplement 3). Cells were scored for nuclear Yan-YFP concentration and their exact position within 3D coordinate space of each fixed eye sample (Figure 1—figure supplement 2). Yan-YFP protein levels were normalized to His2Av-mRFP, which provided some control over measurement variation (Figure 1—figure supplement 4). We then mapped each cell’s spatial position in the x-y plane of the eye disc onto time. Two reasons made this possible. First, the furrow moves at an approximately constant velocity forming one column of R8 cells every two hours (Basler and Hafen, 1989; Campos-Ortega and Hofbauer, 1977). Second, each column of R8 cells induces the other cell fates at a constant rate (Lebovitz and Ready, 1986). Thus even in a fixed specimen, the temporal dynamics of cell state transitions are visible in the repetitive physical organization of ommatidia relative to the furrow (Figure 1C). Together these features allow the estimation of time based on a cell’s position relative to the furrow (Figure 2—figure supplement 1). This can be applied repeatedly to hundreds of cells in a single sample, creating a composite picture of the dynamics (Figure 2B–F). Although the developmental progression of an individual cell cannot be measured by this approach, it nevertheless provides a dynamic view of hundreds of cells across a developing epithelium as they respond to signaling processes. From this information, individual cell behaviors can be reconstructed and modeled.

http://cdn-site.elifesciences.org/content/elife/4/e08924/F6.medium.gif
Figure 2.Dynamics of Yan-YFP in eye cells.
(A) Average time at which initiation of differentiation is first detected by apical migration of committing cell nuclei. Time zero is set to when R8 differentiation initiates. Differentiation proceeds over a time course after commitment is initiated (horizontal arrows) (B) Yan-YFP fluorescence in multipotent progenitor cells. We fit a Hill function (blue curve) to the inductive phase and an exponential decay (black curve) to the decay phase. (C-F) Scatter plots of Yan-YFP levels in individual cells for R2/R5 (C), R3/R4 (D), R7 (E), and C3/C4 (F) cells. These are overlaid with scatter plots of Yan-YFP in multipotent cells at times preceding and coincident with the appearance of the relevant differentiated cells. Note the similar Yan-YFP levels between multipotent cells and differentiating cells at their first appearance. (G) Moving averages of Yan-YFP levels for multipotent and photoreceptor cells. Gaps between the multipotent and photoreceptor curves are due to the window size for line averaging. (H) Moving averages of Yan-YFP in multipotent and cone cells.
Yan-YFP expression in multipotent progentior cells was biphasic (Figure 2B). Cells anterior to the furrow expressed a basal level of Yan-YFP, but this level dramatically increased in cells immediately anterior to the furrow, starting eight hours before the first R8 cells were identifiable. Approximately eight hours after R8 definition, Yan-YFP levels peaked, and thereafter gradually decayed until Yan-YFP approached its basal level again. The results are surprising in two ways. First, Yan-YFP is not maintained at a stable steady state within progenitor cells, which would have been predicted by the bistable model. Rather, its dynamics are reminiscent of monostable responses to transient stimuli, with a single basal steady state. Second, at the peak of Yan-YFP expression, there is remarkably large heterogeneity in Yan-YFP levels across cells.
We also followed Yan-YFP dynamics in cells as they transited into a differentiated state and thereafter. Again, the results did not follow the expectations predicted by the bistable model. First, progenitors transited to a differentiated state at levels of Yan-YFP that varied, depending upon the type of differentiated state being adopted (Figure 2C–H). Cells entering the R3/R4 and R1/R6 states began with Yan-YFP levels that were approximately two-fold greater than cells entering the R2/R5 states. Cells entering the R7 state were intermediate between these two extremes. Despite these differences at the transition point, Yan-YFP levels decayed to a similar basal steady state irrespective of the photoreceptor type, and this basal level was at least three-fold lower than that which the progenitor cells achieved (Figure 2G). Thus, rather than the single high Yan progenitor state previously modeled, our results suggest a dynamic range of high Yan states from which different cell fates are specified according to the spatio-temporal sequence of differentiation.
We noted that for most photoreceptors, it took approximately 20 hr for Yan-YFP to decay to the basal steady state (Figure 2G), significantly longer than had been previously modeled (Graham et al., 2010). Since expression of several neural-specific genes is detected 2–8 hr after the transition (Tomlinson and Ready, 1987; Van Vactor et al., 1988), the slower than anticipated Yan decay indicates that early differentiation does not require cells to have assumed a basal steady-state level of Yan-YFP.
The last group of progenitors to differentiate form the non-neuronal cone cells. We also measured Yan-YFP in those cells. Yan-YFP dynamics in cone cells were more similar to progenitor cells over the same time period (Figure 2F,H). This behavior was in contrast to photoreceptors, which exhibited different decay dynamics from progenitor cells. Thus, accelerated degradation of Yan-YFP is not essential for cells to transition to all retinal cell states.
EGFR-ras signaling regulates Yan-YFP dynamics
The bistable model posits that the switch from one state to another is triggered by a signal that progenitor cells receive though the EGFR protein. Given the unanticipated Yan-YFP dynamics, we wanted to ask whether and how they were influenced by EGFR signaling. EGFR null mutants are inviable; however, a temperature sensitive (ts) allele of EGFR enables controlled inactivation of the RTK’s activity (Kumar et al., 1998). We grew EGFRts mutant larvae at a restrictive temperature for 18 hr before analyzing effects on Yan-YFP. Surprisingly, progenitor cells exhibited biphasic expression of Yan-YFP over time, but the amplitude of the pulse in expression was significantly reduced (Figure 3A). This suggests that EGFR signaling contributes to the stimuli that induce the Yan-YFP peak. To further test this hypothesis, we misexpressed a constitutively active form of Ras protein in eye cells. The peak of Yan-YFP in progenitors was now prolonged (Figure 3B). Together, these results suggest that EGFR-Ras signaling stimulates the transient appearance of Yan-YFP in progenitor cells, and that the decline in Yan-YFP within older progenitor cells is linked to a loss of signal reception by these cells over time.

http://cdn-site.elifesciences.org/content/elife/4/e08924/F8.medium.gif
Figure 3.EGFR/Ras and Pnt regulate Yan-YFP levels.
(A–D) Moving averages of Yan-YFP in different cell types. Shown with shading is the standard error of the mean for each moving average. (A,C) Wildtype and EGFRts mutants incubated at the non-permissive temperature and analyzed for progenitors (A) and R2/R5 cells (C). (B,D) Wildtype and sev>Rasv12 mutants were analyzed for progenitors (B) and R2/R5 cells (D). (E) Optical slice through progenitor cell nuclei in a disc that contains clones ofpnt2 mutant cells. Left, fluorescence of RFP, which positively marks wildtype cells and not pnt2 mutant cells. Middle, Yan-YFP fluorescence, showing reduced levels in pnt2mutant clones. Arrows highlight apoptotic nuclei. Right, merged image with Yan-YFP in green and RFP in purple. Clone borders are outlined. (F,G) Moving averages of Yan-YFP in R3/R4 cells that ectopically express PntP1 (F) or PntP2-VP16 (G) due to LongGMR-Gal4 driving the UAS transgenes. Since PntP2 requires MAPK phosphorylation to become transcriptionally active, we misexpressed a VP16 fusion of PntP2. PntP1 is constitutively active (Brunner et al., 1994; Gabay et al., 1996).
We next examined the effects of EGFR and Ras on Yan-YFP dynamics in cells as they differentiate. The bistable model predicts that EGFR is required for the loss of Yan-YFP in photoreceptors. Indeed,EGFRts mutant R2/R5 cells delayed their initial decline in Yan-YFP levels (Figure 3C). Conversely, misexpression of constitutively active Ras caused a premature decline in Yan-YFP (Figure 3D). These results are consistent with EGFR-Ras stimulating the degradation of Yan-YFP as cells switch their states. However, Yan-YFP dropped to below-normal levels in EGFRts mutant R2/R5 cells (Figure 3C). These complex effects suggest a dual role for EGFR signaling in photoreceptors. In the first few hours as cells transit to a photoreceptor state, EGFR stimulates the accelerated decay of Yan-YFP. Thereafter, EGFR inhibits the decay of Yan-YFP in a manner that might be related to that role that EGFR plays in boosting Yan-YFP levels in progenitor cells.
The source of EGFR ligand originates from the R8 cell (Freeman, 1996). If this diffusive ligand is responsible for controlling Yan-YFP levels in other photoreceptors as they are recruited to an ommatidium, we would predict a correlation between Yan-YFP levels in differentiating cells and their distances from adjacent R8 cells. Indeed, at the times when R2/R5 cells differentiate (~0–15 hr), their Yan-YFP levels are positively correlated (p<0.01) with their physical distance to the nearest R8 cells (Figure 3—figure supplement 1). These correlations are absent in EGFRts mutants, providing evidence that R8 cells act through EGFR to control Yan-YFP dynamics in differentiating cells.
Pnt regulates Yan dynamics
Pnt proteins have been hypothesized to cross-repress Yan expression, and this double negative feedback loop is thought to be necessary for bistability (Graham et al., 2010; Shilo, 2014). At odds with this view, Pnt and Yan proteins are co-expressed in progenitor and differentiating cells of the eye (Boisclair Lachance et al., 2014). Since Pnt proteins act downstream of many RTKs including EGFR, we wondered if Pnt mediated the positive effects of EGFR-Ras signaling on Yan-YFP in progenitor cells. Null mutants of the pnt gene are embryonic inviable; therefore we generated clones of eye cells that were null mutant for pnt in an otherwise wildtype animal. Mutant progenitor cells showed a reduction in Yan-YFP levels as they progressed through their biphasic expression pattern (Figure 3E). Thus, Pnt possibly mediates the positive effect of EGFR signaling on Yan-YFP expression in progenitors. We also wished to know if Pnt mediates the complex effects of EGFR in differentiating photoreceptors. Pnt proteins are rapidly cleared in photoreceptors (Boisclair Lachance et al., 2014) and so loss-of-function mutant analysis would be uninformative. Therefore, we overexpressed PntP1 or constitutively-active PntP2 in cells as they transited into a photoreceptor state and beyond. The early phase of Yan-YFP decay was accelerated while the later phase of Yan-YFP decay was inhibited (Figure 3F,G). These complex effects are precisely the opposite to those caused by loss of EGFR signaling, as would be expected if Pnt mediated EGFR’s complex effects on Yan-YFP dynamics in photoreceptor cells.
Cells are indifferent to absolute levels of Yan
The bistable model predicts that different cell states depend upon discrete absolute concentration of Yan present inside cells. To test this idea, we varied the number of Yan-YFP gene copies. In general, protein output is proportional to gene copy number in Drosophila (Lucchesi and Rawls, 1973). We increased Yan-YFP copy number from its normal diploid number to tetraploid, and monitored Yan-YFP in progenitors and differentiating cells. As expected, four copies caused a higher steady-state level of Yan-YFP in progenitor cells, though this increase was less than two-fold (Figure 4A). The amplitude of the Yan-YFP pulse was also increased as progenitor cells aged. Strikingly, as four-copy progenitor cells transited to a differentiated state, the onset of Yan-YFP decay occurred at the same time as it occurred for two-copy cells (Figure 4A–C). Yan-YFP levels were much greater in four-copy cells compared to two-copy cells making their transit into the identical cell states. To confirm that absolute Yan-YFP concentration had little effect on cell state transitions, we examined expression of a direct target of Yan in R7 cells: the pros gene (Kauffmann et al., 1996; Xu et al., 2000). Expression was monitored with an antibody specific for Pros protein. We observed at most a one hour delay in the onset of Pros expression in R7 cells containing four copies of Yan-YFP (Figure 4D), far less than the ten-hour delay predicted if absolute concentration of Yan-YFP dictated when Pros expression begins (Figure 4D).
Figure 4.Cell state transitions are unaffected by Yan-YFP gene copy number.
(A–C) Moving averages of Yan-YFP in eye discs containing two versus four copies of the Yan-YFP transgene. (A) R2/R5 and progenitor cells. (B) R3/R4 and progenitor cells. (C) R7 and progenitor cells. (D) Moving averages of Yan-YFP and Pros proteins in R7 cells containing either two vs. four copies of the Yan-YFP transgene.
Possibly, absolute concentration of Yan is unimportant when a cell transits to a different state, but the switch to a constant basal Yan level is robustly maintained regardless of starting concentration. An examination of Yan-YFP decay in photoreceptor cells makes that possibility less likely; Yan-YFP decays to different basal levels in two- versus four-copy differentiated cells (Figure 4A–C). To further test this notion, we fit the data to several plausible functional forms. We found that exponentially decaying functions systematically best-fit to the data (Figure 4—figure supplement 1). Thus, for each cell state we fit an exponential decay function to its Yan-YFP temporal profile (Figure 4—figure supplement 2). From these fits, we derived the average decay rate constants and half-lives of Yan-YFP for cells carrying two, four, or six copies of yan. As expected, we found that Yan-YFP half-life was different between progenitors and differentiating photoreceptors (Figure 5—figure supplement 1). The half-life in photoreceptors was two-fold lower than in progenitors, accounting for the more rapid loss of Yan-YFP in the former cells. Strikingly, Yan-YFP half-life was not significantly affected byyan copy number within either progenitor or photoreceptor cells (Figure 5). Thus, Yan-YFP concentration only affected its decay rate as a first-order function, implying that there is no higher order mechanism to accelerate Yan-YFP decay when cells contain greater concentrations of Yan-YFP.
http://cdn-site.elifesciences.org/content/elife/4/e08924/F13.medium.gif
Figure 5.Yan protein half-life is largely unaffected by yan gene copy number.
Exponential decay of Yan-YFP levels. Note the robustness of Yan-YFP half-lives across replicates and yan gene copy number. Note also how half-lives are nearly twice as long for progenitor cells versus photoreceptor neurons.
As a final test of the effects of Yan-YFP levels on cell states, we allowed 4X and 6X yan animals to complete eye development and then examined the external patterning of the fully differentiated compound eye. The compound eyes were completely normal in appearance (Figure 5—figure supplement 2), implying that differentiation was unaffected by the absolute concentrations of Yan inside eye cells.
Yan expression noise spikes during cell state transitions
Our results indicate that Yan’s effects on retinal cell states are not dictated by uniform and stable concentrations of Yan protein. One potential explanation is that Yan’s effect on cell states actually depends on variability in Yan protein levels. A growing body of evidence is pointing to the importance of transient fluctuations in expression of factors to control cell states (Cahan and Daley, 2013;Torres-Padilla and Chambers, 2014). Key regulators of stem cells fluctuate in abundance over time, and this is correlated with stem cells existing in multiple connected microstates, with the overall structure of the population remaining in a stable pluripotent macrostate (MacArthur and Lemischka, 2013). Heterogeneity among cells is not simply due to independent noise in expression of individual genes but is due to fluctuating networks of regulatory genes (Chang et al., 2008; Kumar et al., 2014). Such fluctuations appear to be stochastic in nature, priming cells to transit into differentiated states with a certain probability that is guided by extrinsic signals.
Our data revealed considerable heterogeneity in the level of Yan-YFP among cells at similar developmental times (Figure 2B). To quantify the noise, we used developmental time to bin cells of similar age, and measured detrended fluctuations of Yan-YFP by calculating residuals to the fitted function and normalizing binned residuals to the function (Goldberger et al., 2002). Progenitor cells showed a spike in Yan-YFP noise as they began to induce Yan-YFP expression (Figure 6A). The noise spike was short-lived (approximately 17 hr), and noise thereafter returned to a basal level with secondary minor spikes. The major peak in noise magnitude coincided with the time at which R8 cells are formed.
Figure 6. Noise in Yan-YFP expression is highly dynamic.
Moving averages of Yan-YFP levels and noise (detrended fluctuations) for (A) progenitors, (B) R2/R5, (C) R3/R4, (D) R1/R6, and (E) R7 cells. (F) Comparative noise dynamics for all cells analyzed in (A–E). (G) Moving averages of Yan-YFP noise (coefficient of variation) in R2/R5 cells sampled from wildtype and EGFRts mutant eyes at the non-permissive temperature. Shown with shading is the standard error of the mean for each moving average. (H) Moving averages of Yan-YFP noise (coefficient of variation) in R2/R5 cells sampled from wildtype and sev>Rasv12 mutant eyes. Shown with shading is the standard error of the mean for each moving average.
Photoreceptor cells showed a large spike in Yan-YFP noise as they began to differentiate (Figure 6B–E). The magnitude of each noise peak varied with the photoreceptor cell state; R3 and R4 cells exhibited the greatest amplitude in noise (Figure 6F). These noise spikes showed a distinct temporal relationship, with spikes coinciding with the times at which individual cell states were switched (Figure 6F). Thus, the noise spikes are not a simple consequence of a global stimulus synchronously affecting noise in all cells. Thereafter, all cells reduced Yan-YFP noise to a basal level that was comparable to basal noise in the progenitor cells. However, each cell type exhibited a secondary minor spike at 30–35 hr, which might reflect a synchronous stimulus.
Detrended fluctuation is one method to measure expression variability, but it can suffer from distortion if the model fitting is not adequately weighted. Therefore, we also calculated the coefficient of variation, that is, the standard deviation of Yan-YFP intensity within a sliding window divided by its mean. This method yielded noise profiles with transient spikes for each cell type that was consistent with calculations using detrended fluctuation (Figure 6—figure supplements 1 and 2). However, while the coefficient of variation yielded results that varied strongly with bin width, the detrended fluctuations yielded profiles that were generally robust against changes in bin width (Figure 6—figure supplement 1).
To rule out the possibility that these unexpectedly dynamic features of Yan-YFP were caused by its transgenic origins or fusion with YFP, we compared Yan-YFP dynamics to those of endogenous Yan protein that was bound with an anti-Yan antibody. The profiles of Yan-YFP protein levels and noise were highly similar to endogenous Yan protein levels and noise, in both multipotent and differentiating cells (Figure 6—figure supplement 3). Thus, transient spikes of expression heterogeneity are a fundamental feature of Yan protein.
Since Yan regulates Pros expression in R7 cells, it was possible that Pros showed a transient noise spike as a consequence. Therefore, we measured Pros protein heterogeneity and found that its dynamics did not resemble that of Yan (Figure 6—figure supplement 4). Instead, Pros noise was high starting at the onset of expression, and thereafter gradually declined as Pros protein levels increased in R7 cells. We conclude that noise spikes are not a general feature of gene expression in the developing eye but might reflect unique roles of Yan in mediating cell state transitions.
We wondered what might cause these spikes in Yan-YFP noise during cell state transitions. Because EGFR signaling is important for regulating Yan-YFP concentration during these transitions (Figure 3), we analyzed Yan-YFP noise when EGFR signaling was inhibited in EGFRts mutant animals raised at a non-permissive temperature. The noise spike in progenitor cells was not significantly affected by loss of EGFR signaling (data not shown). We also examined the effects of EGFRts on noise in differentiating photoreceptor cells. Interestingly, noise increased at the normal time of transition but the elevated noise did not quickly drop to basal levels (Figure 6G). Rather, high noise was extended for an additional 10 to 15 hr. Conversely, misexpressing constitutively active Ras within differentiating cells caused a premature dropdown in Yan-YFP noise (Figure 6H). These results indicate that EGFR/Ras signaling is required for the rapid drop in Yan-YFP noise after it has peaked, creating a transient spike.
Discussion
This study relied upon a set of methods that enable systems-level analysis of Yan expression dynamics in a developing animal tissue. Transgene recombineering was used to insert YFP into a genomic rescue fragment of the yan gene, which fully replaced endogenous yan. Yan-YFP protein was quantified in thousands of individual cells by fluorescence confocal microscopy and automated cell segmentation analysis. Based on the unique features of Drosophila eye development, every analyzed cell was assigned an age, and composites of cells across a time-spectrum of ages were derived. This allowed us to reconstruct the temporal dynamics of Yan protein expression in cells as they transited from one state to another or were maintained in a given state. The fact that both Yan concentration and noise were easily measured using our approach indicates that it provides a powerful method for studying how other molecular determinants regulate cell states.
Contrary to what is currently believed, the expression of Yan in progenitor cells has many hallmarks of monostability. A stable basal state exists in cells anterior to the furrow, and when the furrow passes, Yan rises and falls to form a biphasic profile (Figure 7A). If cells transit towards differentiation, then the fall in Yan is accelerated but the fundamental biphasic profile is preserved. This monostable-like behavior is not like a behavior where progenitor cells exist in a high Yan stable-state and switch to a low Yan stable-state when they transit towards differentiation (Figure 7A). Other lines of evidence also point away from a bistable switch mechanism. Yan reaches its basal steady state many hours after cells have adopted their differentiated photoreceptor state and are executing specialized gene expression programs. Thus, Yan levels are variable at the time when cells actually become differentiated.

http://cdn-site.elifesciences.org/content/elife/4/e08924/F21.medium.gif
Figure 7.Summary of analysis.
(A) Top: a hypothetical bistable behavior would be where Yan is in a stable high state within progenitor cells and in a stable low state within differentiated cells. Bottom: the observed behavior of Yan appears monostable, with both progenitors and differentiated cells in unstable Yan states. (B) Heterogeneity in Yan expression is maximal when progenitor cells enter a transition state that resolves to a more homogeneous differentiated state, dependent upon EGFR signaling. This heterogeneous transition state may be a primary mechanism for Yan’s effect on cells, independent of the absolute Yan concentration within progenitors.
…..-> end
Illuminating a Cancer’s Origins
Researchers have developed a technique to visualize the origin of melanoma in zebrafish, throwing light on a genetic switch for cancer.
Many cells containing cancer-associated genes never become tumors. Using a fluorescent reporter to highlight cells activating stem cell-like patterns of gene expression, researchers at Boston Children’s Hospital have now developed a technique to visualize the origins of tumors in zebrafish. The team’s findings were published last week (January 29) in Science.
“An important mystery has been why some cells in the body already have mutations seen in cancer, but do not yet fully behave like the cancer,” Charles Kaufman of Boston Children’s Hospital said in a statement. “We found that the beginning of cancer occurs after activation of an oncogene or loss of a tumor suppressor, and involves a change that takes a single cell back to a stem cell state.”
The researchers focused on a gene present in vertebrates called crestin. This gene is expressed normally only in neural crest progenitor cells, which give rise to a number of other cell types during embryogenesis. However the gene is also expressed in melanoma tumor cells, making it a clear marker of the onset of cancer.
Injecting a fluorescent crestin reporter into transparent zebrafish carrying mutations in melanoma genes, the researchers found they were able to visualize the moment that precancerous cells activated stem cell-like patterns of gene expression.
“Every so often we would see a green spot on a fish,” said study coauthor Leonard Zon of Boston Children’s Hospital in the statement. “When we followed them, they became tumors 100 percent of the time.”
“It’s a significant advance in the field,” Ze’ev Ronai, the scientific director of the Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, told The New York Times. “They provide a pretty strong demonstration that that pathway is correct.”
The team will now focus on the pathways regulating the switch in gene expression, according to the statement—research that Kornelia Polyak of the Dana-Farber Cancer Institute told the Times would be essential to develop drugs targeting melanoma.
“What is the stimulus that allows a single cell out of millions to become cancerous?” she asked. “What is the trigger and why does it happen so rarely? Why, why is this happening?”
Fluorescence, nucleic acid strands help show computing operations inside a living cell
Using strands of nucleic acid and a fluorescence reporter, scientists at the Georgia Institute of Technology (Georgia Tech; Atlanta, GA) and colleagues have demonstrated basic computing operations inside a living mammalian cell. The research could lead to an artificial sensing system that could control a cell’s behavior in response to such stimuli as the presence of toxins or the development of cancer.
Related: Non-damaging, light-emitting nanoprobes enable long-term study of living cells
The research uses DNA strand displacement, a technology that has been widely used outside of cells for the design of molecular circuits, motors, and sensors. Researchers modified the process to provide both “AND” and “OR” logic gates able to operate inside the living cells and interact with native messenger RNA (mRNA).
The tools they developed could provide a foundation for biocomputers able to sense, analyze, and modulate molecular information at the cellular level. Philip Santangelo, an associate professor in the Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory University, says the devices could sense an aberrant RNA, for instance, and then shut down cellular translation or induce cell death.
Strand displacement reactions are the biological equivalent of the switches or gates that form the foundation for silicon-based computing. They can be programmed to turn on or off in response to an external stimuli such as a molecule. An “AND” gate, for example, would switch when both conditions were met, while an “OR” gate would switch when either condition was met.
In the switches the researchers used, a fluorescence reporter molecule and its complementary quenching molecule were placed side-by-side to create an “off” mode. Binding of RNA in one of the strands then displaced a portion of nucleic acid, separating the molecules and allowing generation of a signal that created an “on” mode. Two “on” modes on adjacent nucleic acid strands created an “AND” gate.

http://www.bioopticsworld.com/content/dam/bow/online-articles/2016/01/nucleicacid_web.jpg
Image shows activation of “AND” gates in cells as observed by fluorescence microscopy. (Credit: Chiara Zurla, Georgia Tech)
Georg Seelig, assistant professor of computer science and engineering and electrical engineering at the University of Washington, says that in the longer term, the research team wants to expand this technology to create circuits with many inputs, such as those they have constructed in cell-free settings.
The researchers used ligands designed to bind to specific portions of the nucleic acid strands, which can be created as desired and produced by commercial suppliers.
Challenges in the research team’s work were getting the devices into the cells without triggering the switches, providing operation rapid enough to be useful and not killing the human cell lines that the researchers used in the lab.
The nucleic acid computers ultimately operated as desired, and the next step is to use their switching to trigger the production of signaling chemicals that would prompt the desired reaction from the cells. Cellular activity is normally controlled by the production of proteins, so the nucleic acid switches will have to be given the ability to produce enough signaling molecules to induce a change.
Cells, of course, already know how to sense toxic molecules and the development malignant tendencies, and to then take action. But those safeguards can be turned off by viruses or cancer cells that know how to circumvent natural cellular processes.
Full details of the work appear in the journal Nature Nanotechnology; for more information, please visit http://dx.doi.org/10.1038/nnano.2015.278.
Computing in mammalian cells with nucleic acid strand exchange
Benjamin Groves, Yuan-Jyue Chen, Chiara Zurla, Sergii Pochekailov, Jonathan L. Kirschman, Philip J. Santangelo & Georg Seelig
Nature Nanotechnology(2015) http://dx.doi.org:/10.1038/nnano.2015.278
DNA strand displacement has been widely used for the design of molecular circuits, motors, and sensors in cell-free settings. Recently, it has been shown that this technology can also operate in biological environments, but capabilities remain limited. Here, we look to adapt strand displacement and exchange reactions to mammalian cells and report DNA circuitry that can directly interact with a native mRNA. We began by optimizing the cellular performance of fluorescent reporters based on four-way strand exchange reactions and identified robust design principles by systematically varying the molecular structure, chemistry and delivery method. Next, we developed and tested AND and OR logic gates based on four-way strand exchange, demonstrating the feasibility of multi-input logic. Finally, we established that functional siRNA could be activated through strand exchange, and used native mRNA as programmable scaffolds for co-localizing gates and visualizing their operation with subcellular resolution.
Subject terms: DNA computing, RNA nanotechnology
Figure 1: Empirical design parameters determine in-cell performance.close
Decisions made at the design level, such as the choice of gate architecture, nucleic acid modifications and delivery method have a strong impact on reaction kinetics, stability against nuclease degradation and subcellular localization..
aurelianu2007 commented on Single Cell Shines Light on Cell Malignant Transformation
Single Cell Shines Light on Cell Malignant Transformation Larry H. Bernstein, MD, FCAP, Curator LPBI Single Cell …
The lack of oxygen inhibits the transport of protons and thereby causes a decrease in membrane potential. Cell survival under conditions of mild hypoxia is mediated by phosphoinositide-3 kinase (PIK3) using severe hypoxia or anoxia, and then cells initiate a
cascade of events that lead to apoptosis. After DNA damage, a very important regulator of apoptosis is the p53 protein. This tumor suppressor gene has mutations in over 60% of human tumors and acts as a suppressor of cell division. The growth-suppressive effects of p53 are considered to be mediated through the transcriptional trans-activation activity of the protein. In addition to the maturational state of the clonal tumor, the prognosis of patients with CLL is dependent of genetic changes within the neoplastic cell population.
The genetic changes can be identified by fluorescent probes to chromosomal using a technique referred to as fluorescent in situ hybridization (FISH). Chromosomal evaluation using FISH can identify certain chromosomal abnormalities of CLL that have prognostic significance. Deletion of part of the short arm of chromosome 17 (del 17 q), with target the cell cycle regulating protein p53, is particularly deleterious. This abnormality is found in 10% of patients with CCL and has a pour
prognosis. Deletion of long arm of chromosome 13(del 13q) is the most common geneticabnormality in CLL with roughly 50% of patients exhibiting this effect. These patients have the best prognosis and most will live many years without the need for therapy. Agents damaging DNA may increase the expression of p53 and its trans-activation activity, suggesting that p53 acts to protect cells against the accumulation of mutants and their subsequent conversion to a malignant
status.
Protein p53, in its normal form, acts in stopping the cell division whenever damage to a cell’s DNA is detected, thus giving the cells the possibility of repairing DNA before the errors would duplicate and be passed on to the daughter cells. Antibodies to human p53 have been detected in patients with cancer. These antibodies are highly specific for malignant diseases and are rarely detected in healthy donors or patients having benign diseases. This immune response is correlated with the presence of a p53 gene mutation, leading to the accumulation of an ineffectivep53 protein in tumor cells[ 8] with either tridimensional structure;Over-expression of normal p53 protein can result either in G1 arrest, mediated by p21 protein [ or in the induction of apoptosis [ 9 ]. Also hypoxia itself ca also prevent apoptosis by inducing the expression of the anti apoptotic protein IAP-2. A typical response to the hypoxic environment, by hypoxia inducible factor 1, [ 6] for example, is expression of insulin-independent GLUT [5] triggered by HIF 1α [6] insuring maximum glucose uptake for glycolytic ATP generation.
Mechanism of Tumor Suppressing Gene Uncovered
|
Published: Tuesday, January 26, 2016
Last Updated: Tuesday, January 26, 2016 |
|
| The most commonly mutated gene in cancer,p53, works to prevent tumor formation by keeping mobile elements in check that otherwise lead to genomic instability, UT Southwestern Medical Center researchers have found. |
The p53 gene long has been known to suppress tumor formation, but the mechanisms behind this function – and why disabling the gene allows tumors to form – were not fully understood.
Findings from the study answer some of these questions and could one day lead to new ways of diagnosing and treating cancer, said the study’s senior author, Dr. John Abrams, Professor of Cell Biology at UT Southwestern.
The investigators found that normal p53 gene action restrains transposons, mobile genetic elements called retroelements that can make copies of themselves and move to different positions on chromosomes. But, they discovered, when p53 is disabled by mutation, dramatic eruptions of these mobile elements occur. The study revealed that in mice with cancer and in human samples of two types of cancer (Wilms’ tumors and colon tumors) disabled for p53, transposons became very active.
In a healthy state, certain mechanisms work to keep these retroelements quiet and inactive, explained Dr. Abrams. One of those mechanisms is p53 action. Conversely, when p53 is mutated, retroelements can erupt.
“If you take the gene away, transposons can wreak havoc throughout the genome by causing it to become highly dysregulated, which can lead to disease,” Dr. Abrams said. “Our findings help explain why cancer genomes are so much more fluid and destabilized than normal genomes. They also provide a novel framework for understanding how normal cells become tumors.”
Although much more research is needed, Dr. Abrams said, the potential clinical implications of the team’s findings are significant.
“Understanding how p53 prevents tumors raises the prospect of therapeutic interventions to correct cases in which p53 is disabled,” Dr. Abrams said. “If retroelements are at the heart of certain p53-driven cancers, finding ways to suppress them could potentially allow us to prevent those cancers or intervene to keep them from progressing.”
This understanding also could lead to advances in diagnosing some cancers through biomarkers related to p53 and transposon activity.
“One possibility is that perhaps blood or urine tests could detect dysregulated retroelements that could be indicative of certain types of cancer,” Dr. Abrams said.
|
Read Full Post »























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}