Lung Cancer Therapy
Curator: Larry H. Bernstein, MD, FCAP
Lung Cancer Targets
http://www.oncotherapynetwork.com/lung-cancer-targets
The enzyme ephrin receptor A2 (EphA2) normally blocks KRAS mutation-driven lung adenocarcinoma tumorigenesis, but a new study shows that EphA2 deletion mutation allows aggressive tumor growth—providing “important therapeutic targets” for this deadly form of lung cancer.
Using shRNA-mediated screening of 4,700 candidate human genes with tumor suppression potential, the team subsequently identified 16 genes in animal models that inhibit or slow KRAS- or TP53-driven tumorigenesis. The loss of EphA2 enhanced KRASG12D-driven lung adenocarcinoma carcinogenesis, they found.1
“We identified several tumor suppressors, including EphA2, loss of which promotes adenocarcinoma in the context of KRASG12D mutation,” the coauthors reported.1 “EphA2 loss promotes cell proliferation by activating ERK MAP kinase signaling and hedgehog signaling pathways, leading to tumorigenesis.”
A KRAS mutation is associated with tumorigenesis in 300 days, in animal models, noted senior author Inder Verma, PhD, Professor of Genetics and the Salk Institute’s Irwin and Joan Jacobs Chair in Exemplary Life Science.
“But without EphA2, the KRAS mutation leads to tumors in half the time, 120 to 150 days,” Dr. Verma noted. “This molecule EphA2 is having a huge effect on restraining cancer growth when KRAS is mutated.”
Up to 20% of all cancers harbor KRAS mutations, and these aberrations are particularly common in lung and colon cancers. EphA2 gene mutations were found in 54 of 230 patients whose lung adenocarcinoma tumor genomes were sequenced in the Cancer Genome Atlas Project, the coauthors noted.
“Oddly, among human lung cancer patients with EPhA2 mutations, around 8% of patients actually have high EphA2 expression,” cautioned coauthor Yifeng Xia, PhD, also at the Salk Institute. “So, in some instances, EphA2 is not suppressing tumors and may be context-dependent. Therefore, we need to carefully evaluate the molecule’s function when designing new therapeutics.”
EphA2 activation suppresses both cell signaling and cell proliferation, the team noted. “We believe that the enzyme might serve as a potential drug target in KRAS-dependent lung adenocarcinoma,” explained lead study author Narayana Yeddula, PhD, a Salk research associate.
Salk Institute for Biological Studies. (2015). Molecular “brake” stifles human lung cancer.
Molecular “brake” stifles human lung cancer
By testing over 4,000 genes in human tumors, a Salk team uncovered an enzyme responsible for suppressing a common and deadly lung cancer
LA JOLLA–Scientists at the Salk Institute have uncovered a molecule whose mutation leads to the aggressive growth of a common and deadly type of lung cancer in humans.
This enzyme, called EphA2, normally polices a gene responsible for tissue growth. But when EphA2 is mutated, the Salk team discovered, cellular systems can run amok and quickly develop tumors. The new work, published the week of November 2, 2015 in PNAS, suggests that EphA2 could be a new target for a subset of lung cancer, which affects nonsmokers as well as smokers, and is the leading cause of cancer-related deaths worldwide.

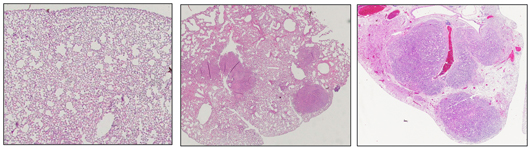
Salk scientists discover a molecule in humans called EphA2 that acts as a tumor suppressor in certain types of lung cancer. This image shows normal mouse lung tissue (left) and lungs with tumors resulting from a KRAS mutation (center). When EphA2 is knocked out, the KRAS mutation results in many more and larger tumors during the same time frame (right). Image: Courtesy of the Salk Institute for Biological Studies
http://www.salk.edu/images/pressrelease/2015/2129-EphA2-verma.jpg
{kind=link}
“Sometimes there are hundreds of mutations in the genes of a patient’s tumors, but you don’t know whether they are drivers of the disease or byproducts,” says senior author Inder Verma, professor of genetics and holder of Salk’s Irwin and Joan Jacobs Chair in Exemplary Life Science. “We found a new way by which to identify cancer suppressor genes and understand how they could be targeted for therapies.”
Two gene mutations in particular are known to spur the growth of human tumors: KRAS and p53. Though both genes have been heavily studied, they are difficult to therapeutically target, so the Salk team decided to look at genes that might police KRAS and p53 instead.
The researchers narrowed in on the 4,700 genes in the human genome related to cellular signaling–specifically, genes that have the ability to tamp down cell growth and proliferation. Then the team adapted a genetic screening technique to quickly and efficiently test the effect of these thousands of genes on tumor development. In animal models, the Salk team found that 16 of these cell-signaling genes produced molecules that had a significant effect on KRAS- and p53-related tumors.
Of these 16 molecules, one especially stood out: the EphA2 enzyme, originally discovered in the lab of another Salk scientist, Tony Hunter. Previously, EphA2’s significance in lung cancer was unclear, but the team discovered that its absence let KRAS-associated tumors grow much more aggressively.
“With a mutation in KRAS, a tumor forms in 300 days. But without EphA2, the KRAS mutation leads to tumors in half the time, 120 to 150 days,” says Verma, who is also an American Cancer Society Professor of Molecular Biology. “This molecule EphA2 is having a huge effect on restraining cancer growth when KRAS is mutated.” Mutated KRAS is a common culprit in approximately 10 to 20 percent of all cancers, particularly colon cancer and human lung cancer.

From left: Salk researchers Yifeng Xia, Eugene Ke, Narayana Yeddula and Inder Verma Image: Courtesy of the Salk Institute for Biological Studies
“Since activating EphA2 led to the suppression of both cell signaling and cell proliferation, we believe that the enzyme might serve as a potential drug target in KRAS-dependent lung adenocarcinoma,” says Narayana Yeddula, a Salk research associate and first author of the paper.
A 10-year national project called the Cancer Genome Atlas mapped the genomes of hundreds of patients for over 20 different cancers and uncovered a number of related genetic mutations, though the role of these mutations has not been well understood in lung cancer (especially adenocarcinoma, which makes up almost a quarter of all lung cancers). From the Cancer Genome Atlas data, the Salk team found that genetic alterations of EphA2 were detected in 54 out of 230 patients with adenocarcinoma. The team also found, surprisingly, that the loss of EphA2 activated a pathway commonly associated with cancer (dubbed Hedgehog) that promotes tumor growth.
“Oddly, among human lung cancer patients with EphA2 mutations, around 8 percent of patients actually have high EphA2 expression. So, in some instances, EphA2 is not suppressing tumors and may be context-dependent. Therefore, we need to carefully evaluate the molecule’s function when designing new therapeutics,” adds Yifeng Xia, a Salk staff researcher involved in the work.
Other authors on the paper were Eugene Ke of the Salk Institute and Joep Beumer of the Salk Institute and the Hubrecht Institute in the Netherlands.
This work was supported in part by an NIH grant, a Salk Cancer Center Core grant, Ipsen, the H.N. and Frances C. Berger Foundation, and the Leona M. and Harry B. Helmsley Charitable Trust.
SMO Gene Amplification and Activation of the Hedgehog Pathway as Novel Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Drugs in Human Lung Cancer
Carminia Maria Della Corte1, Claudio Bellevicine2, Giovanni Vicidomini3, Donata Vitagliano1, Umberto Malapelle2, et al.
Clin Cancer Res Oct 15, 2015; 21: 4686 http://dx.doi.org:/10.1158/1078-0432.CCR-14-3319
Purpose: Resistance to tyrosine kinase inhibitors (TKI) of EGF receptor (EGFR) is often related to activation of other signaling pathways and evolution through a mesenchymal phenotype.
Experimental Design: Because the Hedgehog (Hh) pathway has emerged as an important mediator of epithelial-to-mesenchymal transition (EMT), we studied the activation of Hh signaling in models of EGFR-TKIs intrinsic or acquired resistance from both EGFR-mutated and wild-type (WT) non–small cell lung cancer (NSCLC) cell lines.
Results: Activation of the Hh pathway was found in both models of EGFR-mutated and EGFR-WT NSCLC cell line resistant to EGFR-TKIs. In EGFR-mutated HCC827-GR cells, we found SMO (the Hh receptor) gene amplification, MET activation, and the functional interaction of these two signaling pathways. In HCC827-GR cells, inhibition of SMO or downregulation of GLI1 (the most important Hh-induced transcription factor) expression in combination with MET inhibition exerted significant antitumor activity.
In EGFR-WT NSCLC cell lines resistant to EGFR inhibitors, the combined inhibition of SMO and EGFR exerted a strong antiproliferative activity with a complete inhibition of PI3K/Akt and MAPK phosphorylation. In addition, the inhibition of SMO by the use of LDE225 sensitizes EGFR-WT NSCLC cells to standard chemotherapy.
Conclusions:This result supports the role of the Hh pathway in mediating resistance to anti-EGFR-TKIs through the induction of EMT and suggests new opportunities to design new treatment strategies in lung cancer. Clin Cancer Res; 21(20); 4686–97. ©2015 AACR.
This article is featured in Highlights of This Issue, p. 4497
Translational Relevance
The amplification of SMO in non–small cell lung cancer (NSCLC) resistant to EGFR-TKIs opens new possibilities of treatment for those patients who failed first-line EGFR-targeted therapies. The synergistic interaction of the Hedgehog (Hh) and MET pathways further support the rationale for a combined therapy with specific inhibitors. In addition, Hh pathway activation is essential for the acquisition of mesenchymal properties and, as such, for the aggressiveness of the disease. Also, in EGFR wild-type NSCLC models, inhibition of Hh, along with inhibition of EGF receptor (EGFR), can revert the resistance to anti-EGFR targeted drugs. In addition, inhibition of the Hh pathway sensitizes EGFR wild-type NSCLC to standard chemotherapy. These data encourage further evaluation of Hh inhibitors as novel therapeutic agents to overcome tyrosine kinase inhibitor (TKI) resistance and to revert epithelial-to-mesenchymal transition (EMT) in NSCLC.
Tyrosine kinase inhibitors (TKI) against the EGF receptor (EGFR) represent the first example of molecularly targeted agents developed in the treatment of non–small cell lung cancer (NSCLC) and are, currently, useful treatments after failure of first-line chemotherapy and, more importantly, for the first-line treatment of patients whose tumors have EGFR-activating gene mutations (1). However, after an initial response, all patients experience disease progression as a result of resistance occurrence. Recognized mechanisms of acquired resistance to anti-EGFR-TKIs in EGFR-mutated NSCLC are METgene amplification or the acquisition of secondary mutations such as the substitution of a threonine with a methionine (T790M) in exon 20 of the EGFR gene itself (2). However, these molecular changes are able to identify only a portion of patients with cancer defined as “non-responders” to EGFR-targeted agents. A number of molecular abnormalities in cancer cells may partly contribute to resistance to anti-EGFR agents (2, 3). Our group and others have shown that epithelial-to-mesenchymal transition (EMT) is a critical event in the metastatic switch and is generally associated with resistance to molecularly targeted agents in NSCLC models (4, 5). EMT is a process characterized by loss of polarity and dramatic remodeling of cell cytoskeleton through loss of epithelial cell junction proteins, such as E-cadherin, and gain of mesenchymal markers, such as vimentin (6). The clinical relevance of EMT and drug insensitivity comes from studies showing an association between epithelial markers and sensitivity to erlotinib in NSCLC cell lines, suggesting that EMT-type cells are resistant to erlotinib (7). In particular, recent data suggest that cancer cells with EMT phenotype demonstrate stem cell–like features and strategies reverting EMT could enhance the therapeutic efficacy of EGFR inhibitors (4, 5).
The Hedgehog (Hh) signaling cascade has emerged as an important mediator of cancer development and metastatic progression. The Hh signaling pathway is composed of the ligands sonic, Indian, and desert hedgehog (Shh, Ihh, Dhh, respectively) and the cell surface molecules Patched (PTCH) and Smoothened (SMO). In the absence of Hh ligands, PTCH causes suppression of SMO; however, upon ligand binding to PTCH, SMO protein leads to activation of the transcription factor GLI1, which in turn translocates into the nucleus, leading to the expression of Hh induced genes (8). The Hh signaling pathway is normally active in human embryogenesis and in tissue repair, as well as in cancer stem cell renewal and survival. This pathway is critical for lung development and its aberrant reactivation has been implicated in cellular response to injury and cancer growth (9–11). Indeed, increased Hh signaling has been demonstrated in bronchial epithelial cells exposed to cigarette smoke extraction. In particular, the activation of this pathway happens at an early stage of carcinogenesis when cells acquire the ability to growth in soft agar and as tumors when xenografted in immunocompromised mice. Treatment with Hh inhibitors at this stage can cause complete regression of tumors (12). Overexpression of Hh signaling molecules has been demonstrated in NSCLC compared with adjacent normal lung parenchyma, suggesting an involvement in the pathogenesis of this tumor (13, 14).
Reactivation of the Hh pathway with induction of EMT has been implicated in the carcinogenesis of several cancer types (15). Inhibition of the Hh pathway can reverse EMT and is associated with enhanced tumor sensitivity to cytotoxic agents (16). Recently, upregulation of the Hh pathway has been demonstrated in the NSCLC cell line A549, concomitantly with the acquisition of a TGFβ1-induced EMT phenotype with increased cell motility and invasion (17).
The aim of the present work was to study the role of the Hh signaling pathway as mechanism of resistance to EGFR-TKIs in different models of NSCLC.
Activation of Hh signaling pathway in NSCLC cell lines with resistance to EGFR-TKIs
We established an in vitro model of acquired resistance to the EGFR-TKI gefitinib using the EGFR exon 19 deletion mutant (delE746-A750) HCC827 human NSCLC cell line by continuous culturing these cells in the presence of increasing doses of gefitinib. HCC827 cells, which were initially sensitive to gefitinib treatment (in vitro IC50 ∼ 80 nmol/L), became resistant (HCC827-GR cells) after 12 months of continuous treatment with IC50 > 20 μmol/L. This cell line was also cross-resistant to erlotinib and to the irreversible EGFR kinase inhibitor BIBW2992 (afatinib; data not shown). Sequencing of the EGFR gene in gefitinib-resistant HCC827-GR cells showed the absence of EGFRT790M mutation (data not shown). After the establishment of HCC827-GR cells, we characterized their resistant phenotype by protein expression analysis. While the activation of EGFR resulted efficiently inhibited by gefitinib treatment both in HCC827 and in HCC827-GR cells, phosphorylation of AKT and MAPK proteins persisted in HCC827-GR cells despite the inhibition of the upstream EGFR (Fig. 1A).

Figure 1.
Activation of Hh signaling pathway in NSCLC cell lines resistant to EGFR-TKIs. A, Western blot analysis of EGFR and of downstream signaling pathways in parental EGFR-mutated human lung adenocarcinoma HCC827 cells and in their gefitinib-resistant derivative (HCC827-GR). β-Actin was included as a loading control. B, Western blot analysis of Hh pathway, MET, and selected epithelial- and mesenchymal-related proteins in a panel of EGFR-TKI–sensitive (HCC827, H322, and Calu-3) and -resistant (HCC827-GR, H1299, Calu-3 ER, H460) NSCLC cell lines. β -Actin was included as a loading control. C, FISH analysis of gain in MET andSMO gene copy number in HCC827 and HCC827-GR. D, top, GLI-driven luciferase expression in HCC827 and HCC827-GR cells before and after depletion of GLI1 in both cell lines; bottom, evidence of GLI1 mRNA downregulation by siRNA. β-Actin was included as a loading control. E, MTT cell proliferation assays in HCC827-GR and PC9 cancer cell transfected with an empty vector or SMO expression plasmid with the indicated concentrations of gefitinib for 3 days. Bottom, Western blotting for evaluation of SMO after transfection.
HCC827-GR cells exhibited a mesenchymal phenotype with increased ability to invade, to migrate, and to grow in an anchorage-independent manner (Fig. 2A–C). Therefore, we next examined whether HCC827-GR cell line exhibits molecular changes known to occur during the EMT. Indeed, we found expression of vimentin and SLUG proteins and loss of E-cadherin protein expression in gefitinib-resistant cells as compared with gefitinib-sensitive cells (Fig. 1B). Although activation of the AXL kinase and NF-κB (20–22) have been described as known mechanisms of EGFR-TKI resistance, the analysis of their activation status resulted not significantly different among our cell lines. However, further studies are needed to explore a potential cooperation of AXL and NF-κB with Hh signaling.

Figure 2.
Activation of Hh signaling pathway mediates resistance to EGFR-TKIs in EGFR-dependent NSCLC cell lines. A, invasion assay. B, migration assay, C, anchorage-independent colony formation in soft agar. D, cell proliferation measured with the MTT assay in parental human lung adenocarcinoma HCC827 cells and in HCC827-GR derivative. The results are the average ± SD of 3 independent experiments, each done in triplicate.
Recently, expression of Shh and activation of the Hh pathway have been correlated to the TGFβ-induced EMT in A549 lung cancer cells (17). To investigate the expression profile of Hh signaling components in this in vitro model of acquired resistance to anti-EGFR–TKIs, we performed Western blot analysis for Shh, GLI1, 2, 3, SMO, and PTCH in HCC827-GR cells. While Shh levels did not differ between HCC827 and HCC827-GR cells, a significantly increased expression of SMO and GLI1 was found in HCC827-GR cells as compared with parental cells (Fig. 1B). No differences in the levels of GLI2 and 3 were observed (data not shown). Of interest, also PTCH protein levels resulted increased in HCC827-GR cells. This is of relevance, as PTCH is a target gene of GLI1 transcriptional activity and increased PTCH levels indicate activation of Hh signaling. We further analyzed expression and activation of MET, as a known mechanism of acquired resistance to anti-EGFR drugs in NSCLC. Indeed, MET phosphorylation resulted strongly activated in HCC827-GR cells (Fig. 1B). Analysis of the MET ligand levels, HGF, by ELISA assay, did not evidence any significant difference in conditioned media of our cells (data not shown). As previous studies have demonstrated MET gene amplification in NSCLC cell lines with acquired resistance to gefitinib (23), we evaluated MET gene copy number by FISH analysis and D-PCR in HCC827 and in HCC827-GR cell lines. The mean MET gene copy number was similar between gefitinib-sensitive and gefitinib-resistant HCC827 cell line (Fig. 1C).
Of interest, while we were working to these experiments, data on SMO gene amplification in EGFR-mutated NSCLC patients with acquired resistance to anti-EGFR targeted drugs were reported on rebiopsies performed at progression, revealing SMO amplification in 2 of 16 patients (12.5%; ref. 24). For this reason, we evaluated by FISH SMO gene copy number in HCC827-GR cells, in which the mean SMO gene copy number was 4-fold higher than that of parental HCC827 cells, indicating SMO gene amplification (Fig. 1C).
We further analyzed the expression and the activation of these molecules on a larger panel of EGFR-WT NSCLC cell lines, including NSCLC cells sensitive to EGFRTKIs, such as H322 and Calu-3 cells, NSCLC cell lines with intrinsic resistance to EGFR-TKIs, such as H1299 and H460 cells and Calu-3 ER (erlotinib-resistant) cells, which represents an in vitromodel of acquired resistance to erlotinib obtained from Calu-3 cells (refs. 4, 18; Supplementary Table S1). As shown in Fig. 1B, similarly to HCC827-GR cells, the Hh signaling pathway resulted in activation of these NSCLC models of intrinsic or acquired resistance to EGFR-TKI.
To further investigate the presence of specific mutations in the Hh pathway components, we sequenced DNA from our panel of NSCLC cell lines by Ion Torrent NGS; results indicated the absence of specific mutations in Hh-related genes (data not shown).
Because GLI1 is a transcription factor, we tested the functional significance of increased expression of this gene in the EGFR-sensitive and -resistant cell lines, using a GLI1-responsive promoter within a luciferase reporter expression vector (Fig. 1D). Analysis of luciferase activity of HCC827-GR cells revealed a 6- to 7-fold increase in GLI-responsive promoter activity as compared with HCC827 cells (P < 0.001), suggesting that transcriptional activity of GLI1 is significantly higher in gefitinib-resistant HCC827-GR cells. Furthermore, depletion of GLI1 protein expression by transfection with a GLI1-specific siRNA expression vector led to approximately 65% decrease in GLI1-driven promoter activity in HCC827-GR (P < 0.01; Fig. 1D). To determine whether SMO expression may promote resistance to gefitinib, 2 cell lines harboring the mutated EGFR gene, HCC827 and PC9 cells, and the sensitive EGFR-WT cell line Calu-3, were transiently transfected with an SMO expression plasmid. When treated with gefitinib, transfected cells exhibited a partial loss of sensitivity to the EGFR inhibition (Fig. 1E).
Activation of Hh signaling pathway mediates resistance to EGFR-TKIs in EGFR-dependent NSCLC cell lines
As previously mentioned, HCC827-GR cells acquired expression of vimentin and SLUG and loss of E-cadherin when compared with gefitinib-sensitive HCC827 cancer cells along with an increased ability to invade, migrate, and form colonies in semisolid medium (Fig. 2A–C). We next evaluated whether the Hh pathway activation was necessary for gefitinib acquired resistance by genetically or by pharmacologically inhibiting Hh components in the HCC827-GR cell line. Knockdown of GLI1 by a GLI1siRNA approach had a very little effect on HCC827-GR cells. However, when gefitinib treatment (1 μmol/L) was performed in HCC827-GR cells after GLI1 blockade, invasion, migration, and colony-forming capabilities were significantly inhibited (Fig. 2A–C). Next, we evaluated the effects of 2 small-molecule inhibitors of SMO, such as LDE225 and vismodegib. Treatment with LDE225 (1 μmol/L;Fig. 2A–D) or with vismodegib (1 μmol/L; data not shown) alone did not significantly affect the viability and the invasion and migration abilities of HCC827-GR cells. Combined treatment with gefitinib and LDE225 (1 μmol/L) or vismodegib (1 μmol/L) caused inhibition of these parameters in HCC827-GR cells (Fig. 2A–C).
Taken together, these data show that Hh activation is required for acquisition of gefitinib resistance in HCC827-GR cells.
As overexpression and activation of MET was found in HCC827-GR cells, we evaluated whether inhibition of MET phosphorylation by PHA-665752 could restore gefitinib sensitivity in this model. Although abrogation of MET signaling in combination with the inhibition of EGFR signaling marginally affected gefitinib sensitivity of HCC827-GR cells, surprisingly, inhibition of MET synergistically enhanced the effects of Hh inhibition in HCC827-GR cells (Fig. 2A–D) in terms of invasion, migration, colony-forming, and proliferation abilities, indicating a significant synergism between these 2 signaling pathways. The triple inhibition of EGFR, SMO, and MET did not result in any additional antiproliferative effects (data not shown).
Cooperation between Hh and MET signaling pathways in mediating resistance to EGFR-TKI in EGFR-dependent NSCLC cell lines
To study the role of Hh pathway in the regulation of key signaling mediators downstream of the EGFR and to explore the interaction between Hh and MET pathways, we further characterized the effects of Hh inhibition alone and in combination with EGFR or MET inhibitor on the intracellular signaling by Western blotting. As illustrated in Fig. 3A, treatment of HCC827-GR cells with the SMO inhibitor LDE225, gefitinib or with the MET inhibitor PHA-665772, for 72 hours, did not affect total MAPK and AKT protein levels and activation. A marked decrease of the activated form of both proteins was observed only when LDE225 was combined with PHA-665772, at greater level than inhibition of EGFR and MET, suggesting that the Hh pathway cooperates with MET to the activation of both MAPK and AKT signaling pathways. In addition, vimentin expression, induced during the acquisition of gefitinib resistance, was significantly decreased after Hh inhibition, suggesting that the Hh pathway represents a key mediator of EMT in this model. The combination of MET and Hh inhibitors strongly induced cleavage of the 113-kDa PARP to the 89-kDa fragment, indicating an enhanced programmed cell death.

Figure 3.
Cooperation between Hh and MET signaling pathways in mediating resistance to EGFR-TKIs in HCC827-GR cells. A, Western blot analysis of Hh, MET, and EGFR activation and their downstream pathways activation following treatment with the indicated concentration LDE225 and PHA-556752 on HCC827-GR NSCLC cell line. β-Actin was included as a loading control. B, co-immunoprecipitation for the interaction between MET and SMO. Whole-cell extracts from HCC827 and HCC827-GR cells untreated or treated with LDE225 or/and PHA556752 were immunoprecipitated (IP) with anti- SMO (top) or anti-MET (bottom). The immunoprecipitates were subjected to Western blot analysis (WB) with indicated antibodies. Control immunoprecipitation was done using control mouse preimmune serum (PS). C, GLI-driven luciferase expression in HCC827-GR cells during treatment with gefitinib, LDE225, PHA-556752, or their combinations. D, co-immunoprecipitation for the interaction between SUFU and GLI1. Whole-cell extracts from HCC827 and HCC827-GR cells untreated or treated with LDE225 or/and PHA556752 were immunoprecipitated (IP) with anti-GLI1 (top) or anti-SUFU (bottom) antibodies. The immunoprecipitates were subjected to Western blot analysis with indicated antibodies. Control immunoprecipitation was done using control mouse PS.
Of interest, the inhibition of SMO by LDE225 also reduced the activated, phosphorylated form of MET (Fig. 3A), revealing an interaction between SMO and MET receptors. To address this issue, we hypothesized a direct interplay between both receptors. SMO immunoprecipitates from HCC827-GR cells showed greater MET binding than that from the parental HCC827 cells (Fig. 3B). As MET has been demonstrated to interact with HER3 to mediate resistance to EGFR inhibitors (25), we explored the expression of HER3 in SMO immunoprecipitates. Protein expression analysis did not show any association with HER3; similar results were obtained with EGFR protein expression analysis in the immunoprecipitates (data not shown).
The increased SMO/MET heterodimerization observed in HCC827-GR cells was partially reduced by the inhibition of SMO or MET with LDE225 or PHA-665752, respectively, and to a greater extent with the combined treatment (Fig. 3B). These results support the hypothesis that Hh and MET pathways interplay at level of their receptors.
To study whether the cooperation between these 2 pathways appears also at a downstream level, and considering that, as shown in Fig. 3A, MET inhibition partially reduces the levels of GLI1 and PTCH proteins, we analyzed luciferase expression of GLI1 reporter vector in HCC827-GR cells after treatment with LDE225, PHA-665752, or both. As shown in Fig. 3C, transcriptional activity of GLI1 resulted strongly decreased by the combined treatment. In particular, treatment with single-agent LDE225 did not abrogate the transcriptional activity of GLI1 suggesting a GLI1 noncanonical activation. In addition, single-agent PHA-665752 reduced GLI1-dependent signal, suggesting a role for MET in GLI1 regulation. To better investigate these findings, we hypothesized that MET can regulate GLI1 activity through its nuclear translocation. We, therefore, analyzed the binding ability of SUFU, a known cytoplasmic negative regulator of GLI1, following treatment of HCC827-GR cells with LDE225 and/or PHA-665752. Indeed, interaction between SUFU and GLI1 was markedly decreased in HCC827-GR cells as compared with HCC827 cells (Fig. 3D), which further confirmed the role of the activation of Hh pathway in this gefitinib-resistant NSCLC model. Furthermore, while combined treatment with LDE225 and PHA-665752 strongly increased the binding between GLI1 and SUFU, suggesting an inhibitory effect on GLI1 activity, also treatment with the MET inhibitor PHA-665752 alone favored the interaction of GLI1 with SUFU (Fig. 3D), indicating a role of MET on the activation of GLI1. This phenomenon could be a consequence of the decreased interplay between SMO and MET receptors or the effect of a direct regulation of GLI1 by MET.
Effects of the combined treatment with LDE225 and gefitinib or PHA-665752 on HCC827-GR tumor xenografts
We finally investigated the in vivo antitumor activity of Hh inhibition by LDE225, alone and in combination with gefitinib or with the MET inhibitor in nude mice bearing HCC827-GR cells. Treatment with gefitinib, as single agent, did not cause any change in tumor size as compared with control untreated mice, confirming that the in vitro model of gefitinib resistance is valid also in vivo. Treatment with LDE225 or with PHA-665752 as single agents caused a decrease in tumor size even stronger than that observed in vitro, suggesting a major role of these drugs on tumor microenvironment. However, combined treatments, such as LDE225 plus gefitinib or LDE225 plus PHA-665752, significantly suppressed HCC827-GR tumor growth with a major activity of LDE225 plus PHA-665752 combination. Indeed at 21 days from the starting of treatment, the mean tumor volumes in mice bearing HCC827-GR tumor xenografts and treated with LDE225 plus gefitinib or with LDE225 plus PHA-665752 were 24% and 2%, respectively, as compared with control untreated mice (Fig. 4A). Figure 4B shows changes in tumor size from baseline in the 6 groups of treatment. A total of eight mice for each treatment group were considered. Combined treatment of LDE225 plus gefitinib caused objective responses in 5 of 8 mice (62.5%). Of interest, the most active treatment combination was LDE225 plus PHA-665752 with complete responses in 8 of 8 mice (100%).

Figure 4.
Effects of the combined treatment with LDE225 and gefitinib or PHA-665752 on HCC827-GR tumor xenografts. A, athymic nude mice were injected subcutaneously into the dorsal flank with 107 HCC827-GR cancer cells. After 7 to 10 days (average tumor size, 75 mm3), mice were treated as indicated in Materials and Methods for 3 weeks. HCC827-GR xenografted mice received only vehicle (control group), gefitinib (100 mg/kg daily orally by gavage), LDE225 (20 mg/kg intraperitoneally three times a week), PHA-665752 (25 mg/kg intraperitoneally twice a week), or their combination. Data represent the average (±SD). The Student t test was used to compare tumor sizes among different treatment groups at day 21 following the start of treatment. B, waterfall plot representing the change in tumor size from baseline in the 6 groups of treatment. A total of 8 mice for each treatment group were evaluated. C, effects of combined LDE225 and PHA-665752 on expression of MET, PTCH, and vimentin. Tissues were stained with hematoxylin and eosin (H&E). Representative section from each condition.
We then studied the effects of gefitinib, LDE225, PHA-665752, and their combinations on the expression of PTCH, MET, and vimentin in tumor xenografts biopsies from mice of each group of treatment (Fig. 4C and Supplementary Table S2). We measured PTCH expression, as it represents a direct marker of Hh activation. While vimentin staining was particularly intense in control and gefitinib-treated tumors, treatment with LDE225 alone and in combination with PHA-665752 significantly reduced the intensity of the staining further confirming the role of Hh inhibition on the reversal of mesenchymal phenotype. Of interest, MET immunostaining resulted in a consistent nuclear positivity: this particular localization has been described as a marker of poor outcome and tendency to a mesenchymal phenotype (26). Although the combination of LDE225 and gefitinib resulted in a significant reduction of tumor growth with a concomitant reduction in staining intensity of vimentin, the combination of LDE225 and PHA-665752 was the most effective treatment, with 8 of 8 (100%) mice having a complete response in their tumors. In fact, histologic evaluations of these tumors found only fibrosis and no viable cancer cells. According to Western blot analysis of protein extracts harvested from the HCC827-GR xenograft tumors, the levels of phospho-EGFR, phospho-MET, and GLI1 resulted in a decrease after treatment with the respective inhibitor. Interestingly, the combined treatment with LDE225 and PHA-665752 resulted in a stronger inhibition of phospho-MAPK and phospho-AKT (Supplementary Fig. S1).
Role of the Hh pathway in mediating resistance to EGFR inhibitors in EGFR-WT NSCLC
As shown in Fig. 1B, although H1299, H460, and Calu-3 ER lacked SMO amplification (data not shown), these cells displayed Hh pathway activation. We further conducted luciferase expression analysis that showed a 8- to 9-fold increase in GLI1-dependent promoter activity in these lines as compared with EGFR inhibitor–sensitive H322 and Calu-3 cells, suggesting that transcriptional activity of GLI1 is higher in EGFR-TKI–resistant EGFR-WT NSCLC lines (Supplementary Fig. S2A). Similar to HCC827-GR cells, these cells showed also activation of MET. However, as reported in previous studies (4), MET inhibition alone or in combination with EGFR inhibition or with SMO inhibition resulted ineffective in inhibiting cancer cell proliferation and survival (data not shown).
We therefore tested the effects of Hh inhibition, by silencing GLI1 or by using LDE225, alone and/or in combination with erlotinib. Although knockdown of GLI1 or treatment with LDE225 (1 μmol/L) did not significantly affect NSCLC cell viability, combined treatment with erlotinib restored sensitivity to erlotinib (Supplementary Fig. S2B).
In addition, H1299, Calu-3 ER, and H460 cells exhibited significantly higher invasive and migratory abilities than H322 and Calu-3 cells and inhibition of Hh pathway significantly reduced these abilities. Collectively, these results suggest that Hh pathway activation mediates the acquisition of mesenchymal properties in EGFR-WT lung adenocarcinoma cells with erlotinib resistance (Supplementary Fig. S2B–S2D).
We next evaluated the effects of LDE225 alone and/or in combination with erlotinib on the activation of downstream pathways. Erlotinib treatment result was unable to decrease the phosphorylation levels of AKT and MAPK in H1299 and Calu-3 ER cells (Fig. 5A). However, when LDE225 was combined with erlotinib, a strong inhibition of AKT and MAPK activation was observed in these EGFR inhibitor–resistant cells (Fig. 5A). Furthermore, flow cytometric analysis revealed that combined treatment with both erlotinib and LDE225 significantly enhanced the apoptotic cell percentage to 65% and 70% (P < 0.001) in H1299 and Calu-3 ER cells, respectively (Fig. 5B), confirmed by the induction of PARP cleavage after the combined treatment (Fig. 5A). These findings suggest that Hh pathway drives proliferation and survival signals in NSCLC cells in which EGFR is blocked by erlotinib, and only the inhibition of both pathways can induce strong antiproliferative and proapoptotic effects. The in vitro synergism between EGFR and SMO was confirmed alsoin vivo. Combination of erlotinib and LDE225 significantly suppressed growth of Calu-3 ER xenografted tumors in nude mice (Supplementary Fig. S1F).

Figure 5.
Activation of Hh signaling pathway mediates resistance to EGFR-TKI in EGFR-WT NSCLC cell lines. A, Western blot analysis of EGFR and its downstream pathways activation, including PARP cleaved form, following treatment with the indicated concentration LDE225 and erlotinib on Calu-3, Calu-3 ER, and H1299 NSCLC cell line. β-Actin was included as a loading control. B, apoptosis was evaluated as described in Supplementary Materials and Methods with annexin V staining in Calu-3, Calu-3-GR, and H1299 cancer cells, which were treated with the indicated concentration LDE225 and erlotinib. Columns, mean of 3 identical wells of a single representative experiment; bars, top 95% confidence interval; ***, P < 0.001 for comparisons between cells treated with drug combination and cells treated with single agent.
Hh pathway inhibition sensitizes EGFR-WT NSCLC cell lines to standard chemotherapy
To extend our preclinical observations, we further investigated the effects of Hh pathway inhibition on sensitivity of EGFR-WT NSCLC cells to standard chemotherapy used in this setting and mostly represented by cisplatin.
To investigate the role of the Hh pathway in mediating resistance also to chemotherapy, we evaluated the efficacy of cisplatin and Hh inhibition treatment alone or in combination on the colony-forming ability in semisolid medium of H1299 and H460 cell lines (Fig. 6). Treatment with cisplatin alone resulted in a dose-dependent inhibition of colony formation with an IC50 value of 13 and 11 μmol/L for H1299 and H460 cells, respectively. However, when combined with LDE225, the treatment resulted in a significant synergistic antiproliferative effect in both NSCLC cell lines (Fig. 6). Together, these results indicate that treatment of EGFR-WT NSCLC cells with Hh inhibitors could improve sensitivity of NSCLCs to standard chemotherapy.

Figure 6.
Hh pathway inhibition sensitizes EGFR-WT NSCLC cell lines to standard chemotherapy. Anchorage-independent colony formation in soft agar in human lung adenocarcinoma H1299 and H460. The results are the average ± SD of 3 independent experiments, each done in triplicate. For defining the effect of the combined drug treatments, any potentiation was estimated by multiplying the percentage of cells remaining by each individual agent. The synergistic index was calculated as previously described (19). In the following equations, A and B are the effects of each individual agent and AB is the effect of the combination. Subadditivity was defined as %AB/(%A%B) < 0.9; additivity was defined as %AB/(%A%B) = 0.9–1.0; and supra-additivity was defined as %AB/(%A%B) > 1.0.
Resistance to currently available anticancer drugs represents a major clinical challenge for the treatment of patients with advanced NSCLC. Our previous works (4, 18) reported that whereas EGFR-TKI–sensitive NSCLC cell lines express the well-established epithelial markers, cancer cell lines with intrinsic or acquired resistance to anti-EGFR drugs express mesenchymal characteristics, including the expression of vimentin and a fibroblastic scattered morphology. This transition plays a critical role in tumor invasion, metastatic dissemination, and the acquisition of resistance to therapies such as EGFR inhibitors. Among the various molecular pathways, the Hh signaling cascade has emerged as an important mediator of cancer development and progression (8). The Hh signaling pathway is active in human embryogenesis and tissue repair in cancer stem cell renewal and survival and is critical for lung development. Its aberrant reactivation has been implicated in cellular response to injury and cancer growth (9–11). Indeed, increased Hh signaling has been demonstrated by cigarette smoke extraction exposure in bronchial epithelial cells (12). In particular, the activation of this pathway correlated with the ability to growth in soft agar and in mice as xenograft and treatment with Hh inhibitors showed regression of tumors at this stage (12). Overexpression of Hh signaling molecules has been demonstrated in NSCLC compared with adjacent normal lung parenchyma, suggesting an involvement in the pathogenesis of this tumor (13, 14).
Recently, alterations of the SMO gene (mutation, amplification, mRNA overexpression) were found in 12.2% of tumors of The Cancer Genome Atlas (TCGA) lung adenocarcinomas by whole-exome sequencing (27). The incidence of SMO mutations was 2.6% and SMO gene amplifications were found in 5% of cases. SMO mutations and amplification strongly correlated with SHH gene dysregulation (P < 0.0001). In a small case report series, 3 patients with NSCLC with Hh pathway activation had been treated with the SMO inhibitor LDE225 with a significant reduction in tumor burden, suggesting that Hh pathway alterations occur in NSCLC and could be an actionable and valuable therapeutic target (27). Recently, upregulation of Shh, both at the mRNA and at the protein levels, was demonstrated in the A549 NSCLC cell line, concomitantly with the acquisition of a TGFβ1-induced EMT phenotype (17, 28, 29) and mediated increased cell motility, invasion, and tumor cell aggressiveness (30, 31).
In the present study, SMO gene amplification has been identified for the first time as a novel mechanism of acquired resistance to EGFR-TKI in EGFR-mutant HCC827-GR NSCLC cells. These data are in agreement with the results of a cohort of patients with EGFR-mutant NSCLC that were treated with EGFR-TKIs (24). Giannikopoulus and colleagues have demonstrated the presence of SMO gene amplification in tumor biopsies taken at occurrence of resistance to EGFR-TKIs in 2 of 16 patients (24). In both cases, theMET gene was also amplified. In this respect, although the MET gene was not amplified in HCC827-GR cells, we found a significant functional and structural interaction between MET and Hh pathways in these cells. In fact, the combined inhibition of both SMO and MET exerted a significant antiproliferative and proapoptotic effect in this model, demonstrated by tumor regressions with complete response in 100% of HCC827-GR tumors xenografted in nude mice.
Several MET inhibitors have been evaluated in phase II/III clinical studies in patients with NSCLC, with controversial results. Most probably, blocking MET receptor alone is not enough to revert the resistant phenotype, as it is implicated in several intracellular interactions, and the best way to overcome resistance to anti-EGFR-TKIs is a combined approach, with Hh pathway inhibitors.
In the context of EMT, Zhang and colleagues demonstrated that AXL activation drives resistance in erlotinib-resistant subclones derived from HCC827, independently from MET activation in the same subclone, and that its inhibition is sufficient to restore erlotinib sensitivity by inhibiting downstream signal MAPK, AKT, and NF-κB (21). In addition, Bivona and colleagues described in 3 HCC827 erlotinib-resistant subclones increased RELA phosphorylation, a marker of NF-κB activation, in the absence of MET upregulation, and demonstrated that NF-κB inhibition enhanced erlotinib sensitivity, independently from AKT or MAPK inhibition (22). Differently, we detected Hh and MET hyperactivation in our resistance model HCC827-GR without a clear increase in AXL and NF-κB activation.
Although the level of activation of AXL and NF-κB did not result in contribution to resistance in our model, further studies are needed to explore a potential cooperation of AXL and NF-κB with Hh signaling.
In a preclinical model, the evolution of resistance can depend strictly from the selective activation of specific pathways, whereas different mechanisms can occur simultaneously in patients with NSCLC, due to tumor heterogeneity. Thus, all data regarding EFGFR-TKIs resistance have to be considered equally valid.
We further extended the evaluation of the Hh pathway to NSCLC cell lines harboring the wild-type EGFR gene and demonstrated that Hh is selectively activated in NSCLC cells with intrinsic or acquired resistance to EGFR inhibition and occurred in the context of EMT.
To further validate these data, we blocked SMO or downregulated GLI1 RNA expression in NSCLC cells that had undergone EMT, and this resulted in resensitization of NSCLC cells to erlotinib and loss of vimentin expression, indicating an mesenchymal-to-epithelial transition promoted by the combined inhibition of EGFR and Hh. Inhibition of the Hh pathway alone was not sufficient to reverse drug resistance but required concomitant EGFR inhibition to block AKT and MAPK activation and to restore apoptosis, indicating that the prosurvival PI3K/AKT pathway and the mitogenic RAS/RAF/MEK/MAPK pathways likely represent the level of interaction of EGFR and Hh signals.
In EGFR-WT NSCLC models, the role of MET amplification/activation is less clear, and in our experience, its inhibition did not increase the antitumor activity of SMO inhibitors.
In addition, Hh inhibition contributed to increase the response to cisplatin treatment which is the standard chemotherapeutic option used in EGFR-WT NSCLC patients and in EGFR-mutated patients after progression on first-line EGFR-TKI, thus representing a valid contribution to achieve a better disease control in those patients without oncogenic activation or after progression on molecularly targeted agents.
Collectively, the results of the present study provide experimental evidence that activation of the Hh pathway, through SMO amplification, is a potential novel mechanism of acquired resistance in EGFR-mutated NSCLC patients that occurs concomitantly with MET activation, and the combined inhibition of these 2 pathways exerts a significant antitumor activity. In light of these results, screening of SMO alteration is strongly recommended in EGFR-mutated NSCLC patients with acquired resistance to EGFR-TKIs at first progression.
– See more at: http://www.cancernetwork.com/lung-cancer
Pembrolizumab Offers Survival Benefit in NSCLC
http://www.cancernetwork.com/lung-cancer/pembrolizumab-offers-survival-benefit-nsclc
The maker of pembrolizumab, a programmed death 1 (PD-1) inhibitor (Keytruda; Merck), announced phase II/III trial results showing that the drug resulted in better overall survival (OS) and progression-free survival (PFS) compared with docetaxel in patients with non–small-cell lung cancer (NSCLC). The study included only patients who had failed prior systemic therapy and whose tumors expressed programmed death ligand 1 (PD-L1). – See more at: http://www.cancernetwork.com/lung-cancer/pembrolizumab-offers-survival-benefit-nsclc#sthash.NUIqYKmi.dpuf
“The results from this trial provide part of a growing body of evidence supporting the potential of Keytruda in the treatment of NSCLC,” said Merck’s president, Roger M. Perlmutter, MD, PhD, in a press release.
The KEYNOTE-010 trial results have not yet been presented or published. The study compared two doses of pembrolizumab (2 mg/kg and 10 mg/kg) with docetaxel in 1,034 patients. All had progressed following treatment with platinum-containing systemic therapy, and all had tumors expressing PD-L1.
According to Merck’s release, pembrolizumab was associated with longer OS, in both the 2-mg/kg and 10-mg/kg dose groups, compared with docetaxel. This survival benefit was seen both in a subgroup of patients with PD-L1 expression tumor proportion scores of 50% or higher, as well as in all enrolled patients (all had a score of 1% or higher).
Both doses also resulted in longer PFS vs docetaxel in the 50% or higher group; this was not statistically significant in the full cohort of patients.
Pembrolizumab received an accelerated approval from the US Food and Drug Administration (FDA) in early October (at the 2-mg/kg dose level). The FDA noted in a press release that the most common side effects in a safety cohort of 550 patients included fatigue, dyspnea, and decreased appetite.
Earlier this year, results of a phase I study of pembrolizumab yielded promising survival outcomes. The median OS in that cohort of 495 patients was 12.0 months, and there was an overall response rate to the drug of 19.4%; this was higher in patients who had not received any previous treatment.
Pembrolizumab is not the first immunotherapy agent approved for the treatment of NSCLC. The FDA granted approval to nivolumab (Opdivo; Bristol-Myers Squibb) for the treatment of metastatic squamous NSCLC in March, and the indication was expanded to advanced non-squamous NSCLC in October.
– See more at: http://www.cancernetwork.com/lung-cancer/pembrolizumab-offers-survival-benefit-nsclc#sthash.NUIqYKmi.dpuf
Leave a Reply