Pathology Insights
Larry H Bernstein, MD, FCAP, Curator
LPBI
Predicting the Prognosis of Lung Cancer: The Evolution of Tumor, Node and Metastasis in the Molecular Age—Challenges and Opportunities
Ramón Rami-Porta; Hisao Asamura; Peter Goldstraw
Transl Lung Cancer Res. 2015;4(4):415-423.
http://www.medscape.com/viewarticle/852315
The tumor, node and metastasis (TNM) classification of malignant tumors was proposed by Pierre Denoit in the mid-20th century to code the anatomic extent of tumors. Soon after, it was accepted by the Union for International Cancer Control and by the American Joint Committee on Cancer, and published in their respective staging manuals. Till 2002, the revisions of the TNM classification were based on the analyses of a database that included over 5,000 patients, and that was managed by Clifton Mountain. These patients originated from North America and almost all of them had undergone surgical treatment. To overcome these limitations, the International Association for the Study of Lung Cancer proposed the creation of an international database of lung cancer patients treated with a wider range of therapeutic modalities. The changes introduced in the 7th edition of the TNM classification of lung cancer, published in 2009, derived from the analysis of an international retrospective database of 81,495 patients. The revisions for the 8th edition, to be published in 2016, will be based on a new retrospective and prospective international database of 77,156 patients, and will mainly concern tumor size, extrathoracic metastatic disease, and stage grouping. These revisions will improve our capacity to indicate prognosis and will make the TNM classification more robust. In the future the TNM classification will be combined with non-anatomic parameters to define prognostic groups to further refine personalized prognosis.
Introduction
Obvious as it may seem, it is important that the readers of this article keep in mind that the tumor, node and metastasis (TNM) classification of lung cancer is no more and no less than a system to code the anatomic extent of the disease. Therefore, by definition, the TNM classification does not include other elements that, while they can help improve our capacity to prognosticate the disease for a given patient, are unrelated to the anatomy of the tumor, i.e., parameters from blood analysis, tumor markers, genetic signatures, comorbidity index, environmental factors, etc. Prognostic indexes combining the TNM classification and other non-anatomic parameters are called, by consensus between the Union for International Cancer Control (UICC) and the American Joint Committee on Cancer (AJCC), prognostic groups to differentiate them from the anatomic stage groupings.
The TNM classification of lung cancer is applied to all histopathological subtypes of non-small cell carcinoma, to small cell carcinoma and to typical and atypical carcinoids. It is governed by general rules[1–3] (Table 1) that apply to all malignancies classified with this system, and by site-specific rules applicable to lung cancer exclusively.[4] There also are recommendations and requirements issued with the objective to classify tumors in a uniform way when their particular characteristics do not fit in the basic rules.[4]
The three components of the classification have several categories that are defined by different descriptors. For lung cancer, those for the T component are based on tumor size, tumor location and involved structures; those for the N, on the absence, presence and location of lymph node metastasis; and those for the M, on the absence, presence and location of distant metastasis. There are optional descriptors that add information on the local aggressiveness of the tumor (differentiation grade, perineural invasion, vascular invasion and lymphatic permeation) all of which have prognostic relevance;[5–8] assess the intensity of the investigation to determine the stage (certainty factor); and assess the residual tumor after therapy (residual tumor).
Origin and Evolution of the TNM Classification for Lung Cancer
The TNM classification was developed by Pierre Denoit in a series of articles published from 1943 to 1952. It was soon adopted by the UICC that published brochures covering several anatomical sites, the lung being included in 1966. Two years later, the UICC published the first edition of the TNM Classification of Malignant Tumors and agreements were reached with the AJCC, created in 1959 as the American Joint Committee for Cancer Staging and End Results Reporting, to consult each other to avoid publication of differing classifications. Since then, the UICC and the AJCC have been responsible for updating and revising the TNM classifications of malignant tumors with the participation of national TNM committees of several countries and taking into account the published reports on the topic. The second to sixth editions of the UICC manual on the TNM Classification of Malignant Tumors and the first to sixth editions of the AJCC Staging Manual included classifications for lung cancer that had been informed by a progressively enlarging database initially collected by Mountain, Carr and Anderson, and subsequently managed by Mountain. Their database originally contained a little over 2,000 patients, but it had grown to more than 5,000 by the time the fifth edition of the TNM classification for lung cancer was published in 1997. The sixth edition was published in 2002 with no modifications.[9]
While the fifth edition of the classification was being printed, the International Workshop on Intrathoracic Staging took place in London, United Kingdom, in October 1996, sponsored by the International Association for the Study of Lung Cancer (IASLC).[10] At that meeting, in the presence of Dr. Mountain, the limitations of the database that had been used to revise the TNM classification for lung cancer were openly discussed. In essence, it was considered that, while the database consisted of a relatively large number of patients, all of them originated from the United States of America, and, therefore, the staging system could not really be called ‘international’, as it was called at that time; and, although all tumors had clinical and pathological classifications, the majority had been treated surgically. So, the database was thought not to be representative of the international community, as there were no patients from other countries; or of the current clinical practice, as there were no patients treated with other therapies. Therefore, an agreement was reached to issue a worldwide call to build a really international database of lung cancer patients treated by all therapeutic modalities. This required the constitution of an International Staging Committee that was approved and given a small amount of funding, to pump-prime, by the IASLC Board in 1998. Subsequently substantial financial support was secured by an unrestricted grant from Eli-Lilly. Cancer Research And Biostatistics (CRAB), a not-for-profit biosciences statistical center in Seattle, was appointed to collect, manage and analyze the new database. The proprietors and managers of known databases were subsequently summoned to attend a series of preparatory meetings to identify potential contributors to the IASLC international database for the purpose of revising the TNM classification of lung cancer.
The Future of the TNM Classification
The TNM classification of lung cancer is the most consistent and solid prognosticator of the disease, but it does not explain the whole prognosis because prognosis is multifactorial. In addition to the anatomic extent of the tumor, patient and environmental factors also count. Prognosis also is dynamic, as it may be different at the time of diagnosis, after treatment or at recurrence.[71] In the TNM classification, tumor resection plays an important role as it defines pathological staging and may modify the prognostic assessment based on clinical staging. Other than that, the TNM classification does not include blood analyses, tumor markers, genetic characteristic of the tumor or environmental factors that may account for the differences in survival among similar tumors in different geographic areas.
In order to make progress to indicate a more personalized prognosis, instead of a prognosis based on cohorts of patients with tumors of similar anatomic extent, the IASLC Staging and Prognosis Factors Committee decided to expand its activities to the study of non-anatomic prognostic factors. Therefore, in the third phase of the IASLC Lung Cancer Staging Project, the activities of the committee will be directed to further refine the TNM classification and to find available factors that can be combined with tumor staging to define prognostic groups. To some extent, this already was done with the analyses of the database used for the 7th edition. Prognostic groups with statistically significant differences were defined by combining anatomic tumor extent and very simple clinical variables, such as performance status, gender, and age. These prognostic groups were defined for clinically and pathologically staged tumors, and for small-cell and non-small cell lung cancers.[22,23]
The database used for the 8th edition includes several non-anatomical elements related to the patient, the tumor and the environment that may help refine prognosis at clinical and pathological staging.[69]Due to the limitations of the previous databases, future revisions of the TNM classification will need to be more balanced in terms of therapeutic modalities, and better populated with patients from underrepresented geographical areas, such as Africa, India, Indonesia, North, Central and South America, and South East Asia. The data contributed in the future will have to be complete regarding the TNM descriptors, and preferably prospective. The more robust the TNM, the more important its contribution to the prognostic groups.
To achieve all of the above, international collaboration is essential. Those interested in participating in this project should send an email expressing their interest to information@crab.org, stating ‘IASLC staging project’ in the subject of the email. The IASLC Staging and Prognostic Factors Committee has been very touched by the overwhelming generosity of colleagues around the world who have contributed cases to inform the 7th and the 8th editions of the TNM classification of lung cancer. We continue to count on their collaboration to further revise future editions and to define prognostic groups that will eventually allow a more personalized indication of prognosis.
Sarcomas are a rare and heterogeneous group of tumors. The last decade has witnessed extensive efforts to understand the pathobiology of many aggressive sarcoma types. In parallel, we have also begun to unravel the complex gene regulation processes mediated by microRNAs (miRNAs) in sarcomas and other cancers, discovering that microRNAs have critical roles in the majority of both oncogenic and tumor suppressor signaling networks. Expression profiles and a greater understanding of the biologic roles of microRNAs and other noncoding RNAs have considerably expanded our current knowledge and provided key pathobiological insights into many sarcomas, and helped identify novel therapeutic targets. The limited number of sarcoma patients in each sarcoma type and their heterogeneity pose distinct challenges in translating this knowledge into the clinic. It will be critical to prioritize these novel targets and choose those that have a broad applicability. A small group of microRNAs have conserved roles across many types of sarcomas and other cancers. Therapies that target these key microRNA-gene signaling and regulatory networks, in combination with standard of care treatment, may be the pivotal component in significantly improving treatment outcomes in patients with sarcoma or other cancers.
Sarcomas are a heterogenous group of tumors that account for ~200 000 cancers worldwide each year (~1% of all human malignant tumors); however, they represent a disproportionately high 15% of all pediatric malignant tumors.[1,2] Sarcomas comprise over 50 subtypes that can broadly be classified into bone and soft-tissue sarcomas that are generally based on the cell and/or tissue type.[3] The vast majority of sarcomas fall into the soft-tissue group, primarily affecting connective tissues such as muscle (smooth and skeletal), fat, and blood vessels. Bone sarcomas are relatively rare, representing only ~20% of all diagnosed sarcomas (~0.2% of all cancers). Even within a specific subtype, sarcomas are highly heterogenous making them diagnostically and therapeutically challenging. Several sarcoma types are genetically characterized by chromosomal translocations or DNA copy number alterations, both of which are used as diagnostic markers.[2,4,5]
The four main types of bone sarcomas are defined by their histology, cell of origin (when known), clinical features, and site distribution—osteosarcoma, Ewing’s sarcoma, chondrosarcoma, and chordoma. The most common primary bone malignancy, osteosarcoma, predominantly affects children and young adults and is characterized by undifferentiated bone-forming proliferating cells.[6] Ewing’s sarcoma, another aggressive pediatric malignancy, usually arises in growing bone and is genetically characterized by a fusion of EWS–FLI1 oncoproteins that act as gain-of-function transcriptional regulators.[7] Chondrosarcoma is itself a heterogenous group of malignant bone tumors arising from the malignant transformation of cartilage-producing cells, frequently with mutations in IDH1/2 and COL2A1.[8,9] Chordoma is an aggressive, locally invasive cancer that typically arises from bones in the base of the skull and along the spine. It is characterized, in part, by its abnormal expression of transcription factor T, which is normally only expressed during embryonic development or in the testes.[10]
Soft-tissue sarcomas are also primarily defined by their histology, cell of origin, and, in some cases, by characteristic genetic translocation events. Rhabdomyosarcoma is a malignant skeletal-muscle derived tumor comprised of two main histological subtypes, embryonal and alveolar, is one of the most common childhood soft-tissue sarcomas, accounting for 6–8% of all pediatric tumors.[11] Liposarcoma is the most common soft-tissue cancer overall, accounting for 20% of adult sarcoma cases. It originates in deep-tissue fat cells and is characterized primarily by amplification of the 12q chromosomal region.[12] Other common soft-tissue sarcomas include angiosarcomas, fibrosarcomas, gastrointestinal stromal tumors, and synovial sarcomas, each with their own unique genetic signature.
Ever since the discovery of oncogenes, the primary emphasis in cancer research has been on understanding the role of proteins and protein-coding genes. However, the percent of the genome dedicated to coding genes is small compared with noncoding regions. The last decade has seen a surge of interest in these noncoding regions with small noncoding RNAs such as microRNAs (miRNAs) gaining particular prominence. These small RNAs have critical roles in tumor formation and progression. Understanding their roles in sarcoma will lead to new therapeutic targets and diagnostic biomarkers, opening the door to a greater understanding of the molecular mechanisms of all cancers.
miRNAs are evolutionarily conserved, small, noncoding RNA molecules of 18–24 nucleotides in length at maturity that can control gene function through mRNA degradation, translational inhibition, or chromatin-based silencing mechanisms.[13] Each miRNA can potentially regulate hundreds of targets via a ‘seed’ sequence of ~5–8 nucleotides at the 5′ end of the mature miRNA. miRNAs bind to complementary sequences in the 3′-untranslated regions (3′-UTRs) of target mRNA molecules, leading to either translational repression or transcriptional degradation.[14] The short seed sequence length and relatively low stringency requirement for these miRNA–3′-UTR interactions allow a single miRNA to potentially regulate hundreds of genes.[15] Small changes in the expression level of a few miRNAs can therefore have a dramatic biological impact, particularly when dysregulated. miRNA expression profiles can be used to distinguish between closely related soft-tissue sarcoma subtypes and may provide a more consistent diagnosis than histological inspection.[16–18]
miRNAs have critical roles in the majority of canonical cellular signaling networks and their dysregulation is implicated in many cancers including breast cancer, colon cancer, gastric cancer, lung cancer, and sarcomas.[19,20] Dysregulation of miRNA expression may result from a variety of factors, including abnormal cellular stimuli, genetic mutations, epigenetic alterations, copy number variations, and chromosomal fusions. Because miRNAs act as critical regulator molecules in a variety of signaling pathways and regulatory networks, their dysregulation can be amplified across the entire signaling network.[21–24] Selected miRNAs and targets that have critical regulatory roles in sarcoma and other cancers are summarized in Table 1 .
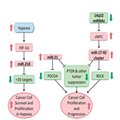
The p53 signaling pathway is one of the most highly studied cellular signaling networks. It actively induces apoptosis in response to DNA damage and oncogene activation and is therefore a key tumor suppressor pathway.[25] Germline mutations in TP53 are strongly associated with the development of soft-tissue sarcomas, osteosarcoma, and are the underlying cause of Li–Fraumeni Syndrome, a familial clustering of early-onset tumors including sarcomas.[26,27] It is estimated that over 50% of human tumors harbor a TP53 mutation but over 80% of tumors have dysfunctional p53 signaling.[28,29] It is only within the last 10 years that researchers have started uncovering the roles of miRNAs in mediating p53’s activity and resulting pro-apoptotic signals (Figure 1). miRNA dysregulation could be a key factor in the ~30% of tumors with dysfunctional p53 signaling that lack an apparent TP53 mutation.

Figure 1.
http://img.medscape.com/article/852/145/852145-fig1.jpg
p53–miRNA interaction network. p53 interacts with the Drosha complex and promotes the processing of pri-miRNA to pre-miRNA. Although p53 directly or indirectly regulates hundreds of miRNAs, for clarity, only selected cancer-relevant miRNAs are shown. miRNAs and proteins in red are upregulated by p53. miRNAs and proteins in green are downregulated by p53. miRNAs in gray are not known to be directly regulated by p53, they are included because they target p53 regulators MDM2 and/or MDM4. miRNA, microRNA.
Like other transcription factors, p53 exerts its function primarily through transcriptional regulation of target genes that contain p53 response elements in their promoters. p53 also regulates the post-transcriptional maturation of miRNAs by interacting with the Drosha processing complex, promoting the processing of primary miRNAs to precursor miRNAs.[30] In addition to protein-coding genes, many miRNA genes also contain p53 regulatory sites in their promoter regions. Large-scale screens have revealed many different miRNAs directly regulated by p53 including miR-22-3p, miR-34a, miR-125a/b, miR-182, and miR-199a-3p.[31] Some of these miRNAs, such as miR-34a and miR-199a-3p, function themselves as tumor suppressors via the regulation of genes involved in cell cycle, cell proliferation, and even of itself.[32–34] Although some p53-targeted miRNAs form a feedback loop, translationally and transcriptionally inhibiting the TP53 gene (e.g., miR-22-3p, miR-34a, and miR-125b), others target, or are predicted to target, p53 repressors such as MDM2 and/or MDM4 (miR-199a-3p, miR-661).[31,33,35,36] It is impossible to fully understand the regulation of the p53 signaling network without considering the role of these miRNAs.
miR-34a has emerged as a critical and conserved member of the p53 signaling pathway. miR-34a is downregulated in osteosarcoma tumor samples and, in conjunction with other miRNAs, regulates p53-mediated apoptosis in human osteosarcoma cell lines.[32,33,37] The gene encoding miR-34a contains a conserved p53-binding site and is upregulated in response to cellular damage in a p53-dependent manner.[37,38] Protein-coding members of the p53 signaling pathway are well-liked targets for anticancer therapeutic development efforts and miRNAs may prove equally effective. In a preclinical model of lung cancer, therapeutic delivery of a miR-34a mimic specifically downregulated miR-34a-target genes and resulted in slower tumor growth. When combined with a siRNA targeting Kras, this small RNA combination therapy resulted in tumor regression.[39] miRNAs such as miR-34a, miR-125b, and miR-199a-3p also mediate p53’s regulation of other key signaling pathways such as the IGF-1/PI3K/AKT/mTOR signaling network. Activation of the AKT network due to downregulation of PTEN (a negative regulator of AKT) by miR-21 or miR-221 or by alternate activation of AKT is a common mechanism underlying many different types of cancer.[40–43] The induction of cell growth, migration, invasion, and metastasis resulting from the upregulation of either miR-21 or miR-221 is seen across different tumor types.[41,44–50] Dysregulation of these miRNAs is a common factor in sarcomas and other tumors. Understanding their mechanisms of action in sarcoma could lead to broadly useful cancer therapeutics.
In prospective analyses that could be models for other sarcoma studies with sufficient numbers of patient samples, Thayanithy et al[19] and Maire et al[23] each analyzed collections of osteosarcoma tissues and compared them with either normal bone or osteoblasts. They each found a set of consistently downregulated miRNAs localized to the 14q32 region.[19,23] Targeting predictions performed by Thayanithy et al[19] identified a subset of four miRNAs as potential regulators of cMYC. One of the many roles of cMYC is to promote the expression of the miR-17–92 family, a known oncogenic cluster that has been observed to be highly expressed in many cancer types including osteosarcoma, leiomyosarcoma, and alveolar rhabdomyosarcoma.[51–57] Restoring the expression of the four 14q32 miRNAs increased apoptosis of SAOS-2 cells, an effect that was attenuated either by overexpression of a cMYC construct lacking the 3′UTR or by ectopic expression of the miR-17–92 cluster.[19] Although the 14q32 region is dysregulated across many different cancer types, this pattern of dysregulation appears to be a hallmark of osteosarcoma, which is particularly interesting due to the heterogenous nature of osteosarcomas and provides an extremely attractive common therapeutic target.
One particular challenge with these types of expression profiling studies is that the cell-of-origin for a particular sarcoma subtype may not be definitely established. Another challenge is the scarcity of patient samples, particularly for the rare sarcoma subtypes. As a result, there have only been a limited number of studies designed to comprehensively profile miRNA expression in various sarcoma subtypes and to compare those expression profiles with the corresponding normal tissues or cell lines. These studies were reviewed recently in Drury et al[20] and Subramanian and Kartha.[58]
Owing to the scarcity of frozen sarcoma tissue samples, it is tempting to study sarcoma cells in vitro, using either primary or immortalized cell cultures. Studies performed in culture are less expensive and more accessible; however, the cell lines used must be chosen with care and may not truly represent the tumors. Any results derived from cultured cells must be interpreted with caution and validated in vivo when possible. A tumor cell’s microenvironment has a profound effect on gene expression and cell metabolism and culturing for even short periods of time can result in large changes in gene/miRNA expression.[59] Three-dimensional cultures can provide more physiological relevant in vitro models of individual tumors (eg, spheroid cultures) or multi-layered epithelial tissues (eg, organotypic cultures using extracellular matrix proteins, fibroblasts, and/or artificial matrix components) vs the previous standard two dimensional culture model.[60,61]
Complicating the analysis of these miRNA expression changes is the fact that many miRNAs showing differential expression in multiple different studies do not have a consistent direction of change and/or a consistent role (tumor suppressor vs tumor promoter). This likely reflects both random chance observational differences and different tissue biology reflected in different regulatory networks. Elucidation of the regulatory roles played by miRNAs in these networks in their appropriate biological contexts may provide suitable upstream targets for more effective treatment of sarcomas. Recent advances in sequencing and downstream bioinformatics techniques provide the tools to efficiently examine these questions.
For two decades, microarray gene chips containing synthetic oligonucleotides whose sequences are designed to be representative of thousands of genes have allowed researchers to perform simultaneous expression analysis of thousands of RNA transcripts in a single reaction.[62–65] Gene expression profiling has been used to characterize and classify a wide range of sarcomas, in some cases providing a diagnostic resolution more accurate than histological examination.[66–72] With the advent of high-throughput RNA-Seq, sarcoma researchers are now able to prospectively analyze the differential expression of small RNAs, such as miRNAs, without prior knowledge of their sequence.[73,74] RNA-Seq also allows for the prospective identification of novel genomic rearrangements resulting from gene fusions or premature truncations that may be of particular interest to cancer researchers.[75,76] These data are highly quantitative and digital in nature, allowing for a dynamic range that is theoretically only limited by the sequencing depth and approaches the estimated range within the cell itself.[77] Marguerat and Bähler[78] provide a basic overview of the different RNA-Seq technologies and their differences from array-based technologies.[78]
Several groups have taken advantage of these technologies to create miRNA expression profiles for a number of different sarcomas in an effort to find both common sarcoma oncomirs and to discover unique miRNA signatures that could be used in diagnosis, prognosis, and novel therapeutic development. Renner et al[18] used a microarray-based miRNA screen, followed by qRT-PCR verification, to analyze the expression of 1146 known miRNAs across a collection of 76 primary soft-tissue sarcoma samples representing eight different subtypes and across a panel of 15 sarcoma cell lines. In addition to identifying overrepresented miRNAs synovial sarcomas (miR-200 family) and liposarcomas (miR-9) compared with other sarcomas and adipose tissue, respectively, their results revealed a high degree of co-expression of 63 miRNAs clustering in the chromosomal region 14q32.[18] The most comprehensive sarcoma miRNA data set has been published by Sarver et al[79] who profiled miRNA expression in over 300 sarcoma primary tumor tissue samples representing 22 different sarcoma types. These data form the basis for the web-accessible comprehensive Sarcoma microRNA Expression Database (SMED) database, which has tools that allows users to query specific sarcoma types and/or specific miRNAs.[79]
Integrative miRNA–mRNA analysis using a tool such as Ingenuity Pathway Analysis (Qiagen) or GeneSpring (Agilent) allows for more biologically relevant results by highlighting miRNA–mRNA pairs that are linked not only by predicted targeting interactions but whose expression levels are inversely correlated (i.e., as miRNA expression increases one would expect the target mRNA levels to decrease). For example, out of 177 differentially expressed miRNAs in osteosarcoma cell lines vs normal bone, an integrated miRNA–mRNA analysis highlighted two particularly interesting miRNA/mRNA pairs (miR-9/TGFBR2 and miR-29/p85α regulatory subunit of PI3K) that were dysregulated.[44]
It is important to note that the general consensus is that there is often no single ‘correct’ method to analyze miRNA expression data. Different experimental and bioinformatics techniques may reveal different aspects in the data that can be further investigated and experimentally validated. All of these experiments, whether performed at the bench or systems biology, contribute to our greater understanding of sarcoma biology and the central role of dysregulated miRNA–gene networks as drivers of tumor formation and progression.
miRNAs are part of a larger family of noncoding RNAs including long noncoding RNAs (lncRNAs) and competing endogenous RNAs (ceRNAs) that deserve to be evaluated for therapeutic potential in sarcomas with broader applicability to other cancer types. Just like miRNAs, lncRNAs are widely expressed in tissue-specific patterns that are highly disrupted in cancer.[80] As their name implies, ceRNAs compete for their common miRNA targets and influence their expression, which has an indirect effect on the protein-coding genes, such as PTEN, regulated by those miRNAs.[81,82] We have just begun to unravel the role of lncRNAs and ceRNAs in cancer development and progression but recent results hint at yet another layer of complexity and genetic control in tumor biology.
The lessons learned from carcinomas, leukemias, and lymphomas will be helpful in understanding the pathobiology of sarcomas and the insights gained from sarcoma biology may form the foundation for therapeutics to treat a wide range of other cancers. Recent studies have shown miRNAs are very stable in blood serum and plasma, and extensive efforts are underway to develop circulating miRNA-based diagnostic and prognostic markers. Major technical challenges in developing circulating miRNA-based markers still need to be addressed, including standardization of pre-analytical, analytical, and post-analytical methods for effective reproducibility. For example, miR-16, which is used in the normalization of miRNA expression in serum/plasma is also found in red blood cells; thus, any hemolysis during sample collection could significantly affect the downstream expression data analysis.
Cancers do not exist in isolation inside the body and extensive research has been performed on how tumor-derived proteins adapt their microenvironment to provide more favorable conditions for tumor growth and development. Recent studies have shown that miRNAs also have a major role in modulating tumor microenvironment. Although most miRNAs are found inside the cell, a significant number of miRNAs are encapsulated in exosomes that can be used as a delivery system to send miRNAs from one cell to another, allowing tumor cells to modulate gene expression in surrounding tissues.[83,84] Exosome and miRNA-mediated cross talk between sarcoma tumor cells and the surrounding stromal cells is a new and exciting avenue of research and the potential for novel therapeutics is high.
Sarcomas are a diverse collection of rare cancers with proportionally limited resources for research and development of novel treatments. It is therefore crucial that potential therapeutic targets are prioritized and novel therapeutic agents carefully selected for clinical trials to succeed. Extensive studies in preclinical models will be required; however, there are also challenges in the development of appropriate in vitro and in vivo model systems that accurately reflect the different sarcoma types. Sarcomas, such as osteosarcoma, leiomyosarcoma, and angiosarcoma are very heterogeneous in nature, making it unlikely that therapies targeting specific genomic mutations will be successful. Even if specific targets were to be identified it would still be a challenge to develop clinical trials based on the small number of patients harboring those specific mutations. Coordinated efforts such as the Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) and its associated preclinical and clinical trial consortiums will help unravel novel miRNA–mRNA interactions and their significance as potential therapeutic targets.
Targeting common miRNA–gene oncogenic or tumor suppressor networks goes after the common denominator underlying many of these cancers. Key regulatory molecules in sarcoma are highly likely to have similar roles in leukemias and lymphomas, for instance, and vice versa. For example, oncogenic activation of STAT3 strongly promotes the expression of miR-135b in lymphoma, resulting in increased angiogenesis and tumor growth.[85] miR-135b is widely overexpressed in sarcomas and STAT3 may be having a similar transcriptional regulatory role, indicating that STAT3 inhibitors could be an effective supplemental therapy in sarcomas.[86] Interestingly, p53 promotes the transcription of miR-125b, which can directly target both STAT3 and p53 transcription. This finely balanced regulatory network is frequently dysregulated in osteosarcoma and Ewing’s sarcoma.[87,88] In retinoblastoma, STAT3 activation is associated with upregulation of the miR-17-92 cluster via a positive feedback loop and inhibition of STAT3-suppressed retinoblastoma proliferation, providing further evidence that STAT3 may be an attractive therapeutic target in many cancers.[89] The dysregulation of key signaling molecules such as the p53 and STAT3 along with their associated signaling networks are a common feature across most cancer types implying that advances in understanding of sarcoma biology may be highly impactful in more frequently occurring solid tumors and lymphomas.
Certain miRNAs appear to be common players across many types of sarcomas and other cancers and their dysregulation contributes to the development of the hallmarks of cancer (Figure 2). miR-210, a key modulator of many downstream pathways involved in the hypoxic response, is upregulated under hypoxic conditions in most solid tumors, including soft-tissue sarcomas, osteosarcoma, renal cancer, and breast cancer.[90] A recent meta-analysis demonstrated that the elevated expression of miR-210 is a prognostic indicator for disease-free, progression-free, and relapse-free survival in a variety of cancer patients.[91] Perhaps the most consistently upregulated miRNA across all tumor types is the anti-apoptotic miR-21, which directly targets the tumor suppressor PDCD4.[92] Levels of miR-21 correlate with cancer progression and patient prognosis.[93]
Figure 2.

http://img.medscape.com/article/852/145/852145-thumb2.png
Conserved miRNA-tumor suppressor signaling networks in cancer. These miRNAs and tumor suppressors are involved in other network and signaling pathway interactions, such as the p53 signaling network; this figure highlights selected critical conserved pathways.
Human Papillomavirus Oncogenic mRNA Testing for Cervical Cancer Screening
Jennifer L. Reid, PhD; Thomas C. Wright Jr, MD; Mark H. Stoler, MD; Jack Cuzick, PhD; Philip E. Castle, PhD; Janel Dockter; Damon Getman, PhD; Cristina Giachetti, PhD
Am J Clin Pathol. 2015;144(3):473-483.
http://www.medscape.com/viewarticle/850740
Objectives: This study determined the longitudinal clinical performance of a high-risk human papillomavirus (HR-HPV) E6/E7 RNA assay (Aptima HPV [AHPV]; Hologic, San Diego, CA) compared with an HR-HPV DNA assay (Hybrid Capture 2 [HC2]; Qiagen, Gaithersburg, MD) as an adjunctive method for cervical cancer screening.
Methods: Women 30 years or older with a negative result for intraepithelial lesions or malignancy cytology (n = 10,860) positive by AHPV and/or HC2 assays and randomly selected women negative by both assays were referred to colposcopy at baseline. Women without baseline cervical intraepithelial neoplasia (CIN) grade 2 or higher (CIN2+) continued into the 3-year follow-up.
Results: The specificity of AHPV for CIN2 or lower was significantly greater at 96.3% compared with HC2 specificity of 94.8% (P < .001). Estimated sensitivities and risks for detection of CIN2+ were similar between the two assays. After 3 years of follow-up, women negative by either human papillomavirus test had a very low risk of CIN2+ (<0.3%) compared with CIN2+ risk in women with positive AHPV results (6.3%) or positive HC2 results (5.1%).
Conclusions: These results support the use of AHPV as a safe and effective adjunctive cervical cancer screening method.
Introduction
Cervical cancer is one of the most frequent cancers in women worldwide, accounting for approximately 530,000 new cases and 275,000 deaths annually.[1] Countries with well-organized screening programs using conventional Papanicolaou (Pap) stain cytology have experienced substantially reduced mortality from the disease in the past 5 decades.[2–4] Despite this advance, the relatively low sensitivity and reproducibility of both conventional Pap smear and liquid-based cytology screening methods have prompted investigation into identifying adjunctive methods with Pap cytology for improving detection of cervical neoplasia.[5–9]
Infection with 14 high-risk human papillomavirus (HR-HPV) genotypes (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68) is associated with almost all cases of cervical precancer, defined as cervical intraepithelial neoplasia (CIN) grade 2 (CIN2), grade 3 (CIN3), and cancer.[10] Addition of HR-HPV nucleic acid testing to a cervical cytology screening regimen offers higher sensitivity and negative predictive value (NPV) for detection of cervical precancer and cancer compared with cytology alone, especially in older women.[11–15] For this reason, HR-HPV nucleic acid testing is recommended as an adjunctive test to cytology to assess the presence of HR-HPV types in women 30 years of age or older.[16] In this context, HR-HPV testing guides patient management by identifying women at elevated risk for CIN2 or higher (CIN2+) but, importantly, also reassures women who are negative for HR-HPV of their extremely low cancer risk.[17–19]
First-generation HR-HPV molecular tests used for adjunctive cervical cancer screening function by detecting viral genomic DNA in cellular samples from the uterine cervix. However, because the presence of HR-HPV in the female genital tract is common and often transient in nature,[20,21] and most cervical HPV infections resolve without becoming cancerous,[22,23] HR-HPV DNA-based test methods yield only moderate specificity for detection of high-grade cervical disease.[12,24] This leads to unnecessary follow-up and referral of patients to colposcopy, increasing the physical and emotional burdens on patients and elevating health care costs.
A test approved by the US Food and Drug Administration (FDA) for detection of HR-HPV E6/E7 messenger RNA (mRNA) (Aptima HPV [AHPV]; Hologic, San Diego, CA) has shown higher specificity with similar sensitivity for detection of CIN2+ compared with HPV DNA-based tests in patients referred for colposcopy due to an abnormal Pap smear result as well as in a screening setting.[25–30] Expression of mRNA from viral E6 and E7 oncogenes is highly associated with the development of CIN,[31,32] and extensive investigation into the role of E6 and E7 oncoproteins in the human papillomavirus (HPV) life cycle has revealed that the expression of the corresponding oncogenes is necessary and sufficient for cell immortalization, neoplastic transformation, and the development of invasive cancer.[33–35]
To confirm and extend the previous evidence on the clinical utility of HR-HPV oncogenic mRNA testing in a US population-based setting, the clinical performance of AHPV was evaluated as an adjunctive method for cervical cancer screening in women aged 30 years or older with negative for intraepithelial lesions or malignancy (NILM) cytology results from routine Pap testing in a pivotal, prospective, multicenter US clinical study including 3 years of follow-up (the Clinical Evaluation of Aptima mRNA [CLEAR] study). We report herein the results from this study.
1 of 4
Figure 1.

Clinical evaluation of Aptima mRNA study participant disposition. aReasons for withdrawal: did not meet eligibility criteria (70); Pap volume insufficient for AHPV testing (117); specimen expired or unsuitable for testing (190); specimen lost (58); noncompliant site (320); other reasons (26). bReasons for withdrawal: collection site did not participate in follow-up (243); subject terminated participation (37); participant had hysterectomy (22); participant not eligible (17); participant treated prior to CIN2+ diagnosis (8); other reasons (4). AHPV, Aptima HPV (Hologic, San Diego, CA); ASC-US, atypical squamous cells of unknown significance; CIN2+, cervical intraepithelial neoplasia grade 2 or higher; HC2, Hybrid Capture 2 (Qiagen, Gaithersburg, MD); HPV, human papillomavirus; NILM, negative for intraepithelial lesions or malignancy; Pap, Papanicolaou test.
HPV Testing
Baseline PreservCyt specimens (1-mL aliquot) were tested with the AHPV (Hologic) on both the automated Tigris DTS System and Panther System. Results from the two systems were similar; Panther System results are presented here. AHPV is a target amplification assay that uses transcription-mediated amplification to detect the E6/E7 oncogene mRNA of 14 HR-HPV genotypes (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68).
HPV and Disease Prevalence
Cervical disease and HPV status are shown in Table 2 for the baseline evaluation and cumulatively after 3 years of follow-up. Of the 10,860 evaluable participants with NILM cytology at baseline, 512 were positive for AHPV, yielding a prevalence of 4.7% for HR-HPV E6/E7 oncogenic mRNA, whereas prevalence of HR-HPV DNA was 6.5% among 10,229 women with HC2 results. A total of 845 HPV RNA-positive or DNA-positive women and 556 randomly selected HPV-negative women were referred to colposcopy at baseline (Figure 1).
At baseline, the percentage of colposcopy attendance was similar between HPV-positive (62%, n = 526) and randomly selected HPV-negative (61%, n = 339) women with 29 cases of CIN1, nine cases of CIN2, eight cases of CIN3, and three cases of adenocarcinoma in situ (AIS) identified (Table 2). Four of the CIN2 cases and two of the AIS cases were identified based on an ECC biopsy specimen only.
In total, 6,271 women completed the 3-year follow-up with a known disease status (Table 2). Of these, 6,098 (97.2%) women had normal (negative) disease status, and 56 (0.9%) had low-grade lesions (CIN1). In addition to the 20 women with CIN2+ identified at baseline, 15 (0.2%) women had CIN2 and 12 (0.2%) women had CIN3 identified during follow-up, with two cases identified from an ECC biopsy specimen only.
Of the 27 women with CIN2+ identified during follow-up, two had CIN1 at baseline, with CIN3 identified during year 1. Ten women had no disease found at baseline, with five cases of CIN2+ identified during year 1, one case of CIN2+ identified during year 2, and four cases of CIN2+ identified during year 3. The remaining 15 women with CIN2+ identified during follow-up did not have a baseline colposcopy; among them, two cases of CIN2+ were identified during year 1, six cases of CIN2+ during year 2, and seven cases of CIN2+ during year 3.
AHPV Assay Performance
Baseline risk and prevalence estimates adjusted for verification bias are provided in Table 3. The prevalence of CIN2+ was 0.9% in the overall population. CIN2+ occurred in 4.5% (95% CI, 2.7%-7.4%) of women with positive AHPV results and in 0.6% (95% CI, 0.2%–1.9%) of women with negative AHPV results, yielding a relative risk of 7.5 (95% CI, 2.1–26.3). This indicates that women with a positive AHPV result are at significantly greater risk of CIN2+ than women with a negative AHPV result. The CIN2+ relative risk obtained for the HC2 test at baseline was similar (7.3; 95% CI, 1.6–33.5). For CIN3+ diagnosis, the overall prevalence was 0.4%. The AHPV relative risk was 24.9 (95% CI, 2.0–307.0), again with a similar relative risk for HC2 (21.0; 95% CI, 1.0–423.8).
Cumulative absolute and relative risks for AHPV and HC2 over the 3-year follow-up period for HPV-positive and HPV-negative women are shown in Table 4. Women with an HPV-negative result with either test had very low cervical disease risk after 3 years of follow-up (<0.3%). Comparatively, 5% to 6% of women with an HPV-positive result had CIN2+ and 3% to 4% had CIN3+, with overall cumulative absolute and relative risks slightly higher for the AHPV assay than for HC2. Younger women aged 30 to 39 years who were HPV positive had twice the prevalence of disease but a similar increase in relative risk of cervical disease compared with HPV-positive women 40 years and older (Table 4). Risk of cervical disease in HPV-negative women did not vary by age group.
Figure 2 and Figure 3 show the cumulative absolute risk of CIN2+ and CIN3+, respectively, by year according to AHPV or HC2 positivity status at baseline. Both assays show a similar trend, with consistent slightly higher risk for the AHPV assay each year.
Figure 2.
Cumulative absolute risk of cervical intraepithelial neoplasia grade 2 or higher (CIN2+) by year. AHPV, Aptima HPV (Hologic, San Diego, CA); HC2, Hybrid Capture 2 (Qiagen, Gaithersburg, MD).
Figure 3.
Cumulative absolute risk of cervical intraepithelial neoplasia grade 3 or higher (CIN3+) by year. AHPV, Aptima HPV (Hologic, San Diego, CA); HC2, Hybrid Capture 2 (Qiagen, Gaithersburg, MD).
After 3 years of follow-up, the specificity of AHPV for CIN2 or lower was 96.3% (95% CI, 95.8%-96.7%), significantly greater (P < .001) compared with HC2 specificity of 94.8% (95% CI, 94.3%-95.4%) Table 5. AHPV specificity for CIN3 or lower (96.2%; 95% CI, 95.5%–96.5%) was also significantly greater (P < .001) than HC2 specificity (94.7%; 95% CI, 94.1%-95.2%). Estimated sensitivities for detection of CIN2+ and CIN3+ were similar between the two assays (P = .219 and P = 1.0, respectively). For detection of CIN2+, AHPV sensitivity was 55.3% (95% CI, 41.2%-68.6%), and HC2 sensitivity was 63.6% (95% CI, 48.9%-76.2%). For CIN3+ detection, AHPV sensitivity was 78.3% (95% CI, 58.1%-90.3%), and HC2 sensitivity was 81.8% (95% CI, 61.5%-92.7%) (Table 5).
Discussion
This study presents the results of a 3-year longitudinal evaluation of the AHPV assay as an adjunctive method for screening women 30 years and older who have NILM Pap cytology results. Consistent with previously published data,[28,29] these results demonstrate that HR-HPV oncogenic E6/E7 mRNA testing has a sensitivity similar to an HR-HPV DNA-based test for detection of CIN2+ and CIN3+ and slightly, but significantly, improved specificity compared with HR-HPV DNA testing for both end points. We found that use of AHPV as an adjunctive method for HPV-induced cervical disease screening provided disease detection capability similar to HC2 while reducing the false-positive rate (from 5.2% to 3.7%) relative to the HPV DNA-based test. Reduction of HPV detection in women without cervical disease minimizes the anxiety and burden associated with spurious positive HPV molecular test results in women with NILM cytology, decreases health care costs, and reduces unnecessary follow-up procedures, thereby improving the safety of cervical cancer screening (unnecessary colposcopy is considered a significant “harm” in the recent American Cancer Society guidelines[16]).
Importantly, we show that women with a NILM cytology result who also had a positive AHPV result are approximately 24 times more likely to have CIN2+ disease after 3 years than women with a negative AHPV result. This risk increased to approximately 68-fold for detection of CIN3+ disease. Similar but slightly lower risk estimates were obtained with HC2, demonstrating comparable accuracy of the AHPV and HC2 for identifying participants with CIN2+ and CIN3+ in this respect.
After 3 years of follow-up, women in this study who were HPV negative at baseline using any test method had very low risk for CIN2+ (<0.3%), a result similar to previously published studies with HC2.[42,43] These findings reinforce evidence from previous studies showing that HR-HPV nucleic acid testing should be performed as an adjunctive test to routine Pap for cervical cancer screening of women 30 years or older to increase sensitivity of disease detection.[28] Correspondingly, compared with annual cytology-only screening, this study supports longer screening intervals for women negative for both abnormal cytology and HPV E6/E7 mRNA, due to the high NPV and low risk of disease afforded by this screening algorithm for 3 years following a test-negative baseline visit. Extension of cervical cancer screening intervals following negative HPV and cytology test results in women 30 years or older is a key recommendation of current US screening guidelines from both the American Cancer Society and the US Preventive Services Task Force.[16]
Conversely, since the positive predictive value of any HPV test in women with NILM cytology is low, additional AHPV testing to detect persistent HR-HPV infection during follow-up care in women with an initial AHPV-positive result is likely a better option than direct referral to colposcopy. Alternatively, genotyping with referral for HPV 16– or HPV 18–positive women can optimize referral and minimize loss to follow-up.[44]
Read Full Post »


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}