Disease Disablers
Larry H. Bernstein, MD, FCAP, Curator
LPBI

Disease Disablers, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
The Gene Hackers
A powerful new technology enables us to manipulate our DNA more easily than ever before.
BY MICHAEL SPECTER
http://www.newyorker.com/magazine/2015/11/16/the-gene-hackers

At thirty-four, Feng Zhang is the youngest member of the core faculty at the Broad Institute of Harvard and M.I.T. He is also among the most accomplished. In 1999, while still a high-school student, in Des Moines, Zhang found a structural protein capable of preventing retroviruses like H.I.V. from infecting human cells. The project earned him third place in the Intel Science Talent Search, and he applied the fifty thousand dollars in prize money toward tuition at Harvard, where he studied chemistry and physics. By the time he received his doctorate, from Stanford, in 2009, he had shifted gears, helping to create optogenetics, a powerful new discipline that enables scientists to use light to study the behavior of individual neurons.
Zhang decided to become a biological engineer, forging tools to repair the broken genes that are responsible for many of humanity’s most intractable afflictions. The following year, he returned to Harvard, as a member of the Society of Fellows, and became the first scientist to use a modular set of proteins, called TALEs, to control the genes of a mammal. “Imagine being able to manipulate a specific region of DNA . . . almost as easily as correcting a typo,” one molecular biologist wrote, referring to TALEs, which stands for transcription activator-like effectors. He concluded that although such an advance “will probably never happen,” the new technology was as close as scientists might get.
Having already helped assemble two critical constituents of the genetic toolbox used in thousands of labs throughout the world, Zhang was invited, at the age of twenty-nine, to create his own research team at the Broad. One day soon after his arrival, he attended a meeting during which one of his colleagues mentioned that he had encountered a curious region of DNA in some bacteria he had been studying. He referred to it as a CRISPR sequence.
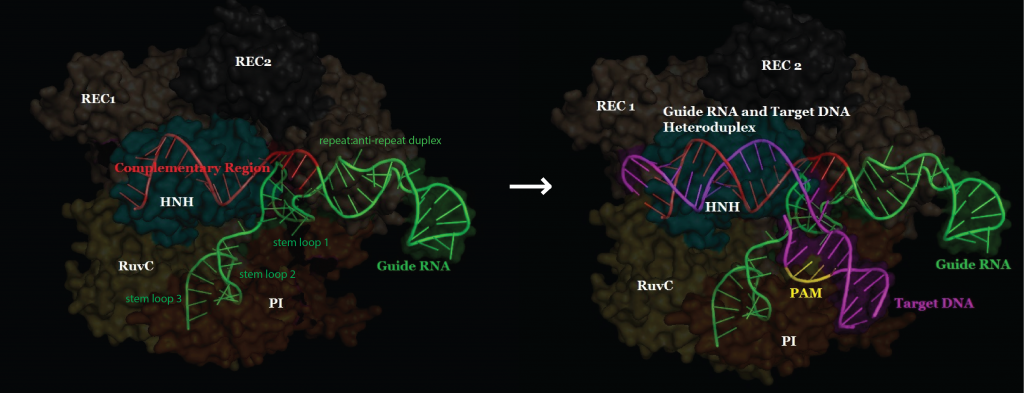
“I had never heard that word,” Zhang told me recently as we sat in his office, which looks out across the Charles River and Beacon Hill. Zhang has a perfectly round face, its shape accentuated by rectangular wire-rimmed glasses and a bowl cut. “So I went to Google just to see what was there,” he said. Zhang read every paper he could; five years later, he still seemed surprised by what he found. CRISPR, he learned, was a strange cluster of DNA sequences that could recognize invading viruses, deploy a special enzyme to chop them into pieces, and use the viral shards that remained to form a rudimentary immune system. The sequences, identical strings of nucleotides that could be read the same way backward and forward, looked like Morse code, a series of dashes punctuated by an occasional dot. The system had an awkward name—clustered regularly interspaced short palindromic repeats—but a memorable acronym.
CRISPR has two components. The first is essentially a cellular scalpel that cuts DNA. The other consists of RNA, the molecule most often used to transmit biological information throughout the genome. It serves as a guide, leading the scalpel on a search past thousands of genes until it finds and fixes itself to the precise string of nucleotides it needs to cut. It has been clear at least since Louis Pasteur did some of his earliest experiments into the germ theory of disease, in the nineteenth century, that the immune systems of humans and other vertebrates are capable of adapting to new threats. But few scientists had considered the possibility that single bacterial cells could defend themselves in the same way. The day after Zhang heard about CRISPR, he flew to Florida for a genetics conference. Rather than attend the meetings, however, he stayed in his hotel room and kept Googling. “I just sat there reading every paper on CRISPR I could find,” he said. “The more I read, the harder it was to contain my excitement.”
It didn’t take Zhang or other scientists long to realize that, if nature could turn these molecules into the genetic equivalent of a global positioning system, so could we. Researchers soon learned how to create synthetic versions of the RNA guides and program them to deliver their cargo to virtually any cell. Once the enzyme locks onto the matching DNA sequence, it can cut and paste nucleotides with the precision we have come to expect from the search-and-replace function of a word processor. “This was a finding of mind-boggling importance,” Zhang told me. “And it set off a cascade of experiments that have transformed genetic research.”
With CRISPR, scientists can change, delete, and replace genes in any animal, including us. Working mostly with mice, researchers have already deployed the tool to correct the genetic errors responsible for sickle-cell anemia, muscular dystrophy, and the fundamental defect associated with cystic fibrosis. One group has replaced a mutation that causes cataracts; another has destroyed receptors that H.I.V. uses to infiltrate our immune system.
The potential impact of CRISPR on the biosphere is equally profound. Last year, by deleting all three copies of a single wheat gene, a team led by the Chinese geneticist Gao Caixia created a strain that is fully resistant to powdery mildew, one of the world’s most pervasive blights. In September, Japanese scientists used the technique to prolong the life of tomatoes by turning off genes that control how quickly they ripen. Agricultural researchers hope that such an approach to enhancing crops will prove far less controversial than using genetically modified organisms, a process that requires technicians to introduce foreign DNA into the genes of many of the foods we eat.
The technology has also made it possible to study complicated illnesses in an entirely new way. A few well-known disorders, such as Huntington’s disease and sickle-cell anemia, are caused by defects in a single gene. But most devastating illnesses, among them diabetes, autism, Alzheimer’s, and cancer, are almost always the result of a constantly shifting dynamic that can include hundreds of genes. The best way to understand those connections has been to test them in animal models, a process of trial and error that can take years. CRISPR promises to make that process easier, more accurate, and exponentially faster.
Inevitably, the technology will also permit scientists to correct genetic flaws in human embryos. Any such change, though, would infiltrate the entire genome and eventually be passed down to children, grandchildren, great-grandchildren, and every subsequent generation. That raises the possibility, more realistically than ever before, that scientists will be able to rewrite the fundamental code of life, with consequences for future generations that we may never be able to anticipate. Vague fears of a dystopian world, full of manufactured humans, long ago became a standard part of any debate about scientific progress. Yet not since J. Robert Oppenheimer realized that the atomic bomb he built to protect the world might actually destroy it have the scientists responsible for a discovery been so leery of using it.
For much of the past century, biology has been consumed with three essential questions: What does each gene do? How do we find the genetic mutations that make us sick? And how can we overcome them? With CRISPR, the answers have become attainable, and we are closing in on a sort of grand unified theory of genetics. “I am not sure what a Golden Age looks like,” Winston Yan, a member of Zhang’s research team, told me one day when I was with him in the lab, “but I think we are in one.”
At least since 1953, when James Watson and Francis Crick characterized the helical structure of DNA, the central project of biology has been the effort to understand how the shifting arrangement of four compounds—adenine, guanine, cytosine, and thymine—determines the ways in which humans differ from each other and from everything else alive. CRISPR is not the first system to help scientists pursue that goal, but it is the first that anyone with basic skills and a few hundred dollars’ worth of equipment can use.
“CRISPR is the Model T of genetics,” Hank Greely told me when I visited him recently, at Stanford Law School, where he is a professor and the director of the Center for Law and the Biosciences. “The Model T wasn’t the first car, but it changed the way we drive, work, and live. CRISPR has made a difficult process cheap and reliable. It’s incredibly precise. But an important part of the history of molecular biology is the history of editing genes.”
Scientists took the first serious step toward controlling our genes in the early nineteen-seventies, when they learned to cut chains of DNA by using proteins called restriction enzymes. Suddenly, genes from organisms that would never have been able to mate in nature could be combined in the laboratory. But those initial tools were more hatchet than scalpel, and, because they could recognize only short stretches within the vast universe of the human genome, the editing was rarely precise. (Imagine searching through all of Shakespeare for Hamlet’s soliloquy on suicide, relying solely on the phrase “to be.” You’d find the passage, but only after landing on several hundred unrelated citations.)
When the first draft of the Human Genome Project was published, in 2001, the results were expected to transform our understanding of life. In fundamental ways, they have; the map has helped researchers locate thousands of genes associated with particular illnesses, including hundreds that cause specific types of cancer. To understand the role that those genes play in the evolution of a disease, however, and repair them, scientists need to turn genes on and off systematically and in many combinations. Until recently, though, altering even a single gene took months or years of work.
That began to change with the growing use of zinc fingers, a set of molecular tools that, like CRISPR clusters, were discovered by accident. In 1985, scientists studying the genetic code of the African clawed frog noticed a finger-shaped protein wrapped around its DNA. They soon figured out how to combine that tenacious grip with an enzyme that could cut the DNA like a knife. Two decades later, geneticists began using TALEs, which are made up of proteins secreted by bacteria. But both engineering methods are expensive and cumbersome. Even Zhang, who published the first report on using TALEs to alter the genes of mammals, realized that the system was little more than an interim measure. “It is difficult to use,” he told me. “I had to assign a graduate student just to make the proteins and test them before I could begin to use them in an experiment. The procedure was not easy.”
Zhang’s obsession with science began in middle school, when his mother prodded him to attend a Saturday-morning class in molecular biology. “I was thirteen and had no idea what molecular biology was,” he said one evening as we walked across the M.I.T. campus on the way to the fiftieth-anniversary celebration of the Department of Brain and Cognitive Sciences, where Zhang is also a faculty member. “It really opened my imagination.” His parents, both engineers, moved the family to Iowa when he was eleven. They stayed largely because they thought he would get a better education in the United States than in China.
In 1997, when Zhang was fifteen, he was offered an internship in a biosafety facility at the Des Moines Human Gene Therapy Research Institute—but he was told that federal law prohibited him from working in a secure lab until he was sixteen. “So I had to wait,” he said. On his birthday, Zhang went to the lab and met the scientists. “I was assigned to a man who had a Ph.D. in chemistry but trained as a molecular biologist,” he continued. “He had a lot of passion for science, and he had a very big impact on me and my research.” On his first day, Zhang spent five hours in the lab, and nearly as much time every day after school until he graduated.
Zhang is unusually reserved, and he speaks in low, almost sleepy tones. I asked him if he considered himself to be mellow, a characteristic rarely associated with prize-winning molecular biologists. “You came to the lab meeting, right?” he replied. Earlier that morning, I had caught the tail end of a weekly meeting that Zhang holds for his group. I watched as he gently but relentlessly demolished a presentation given by one of the people on his team. When I mentioned it to one of the scientists who was at the meeting, he responded, “That was nothing. You should have been there from the start.”
At his Saturday-morning classes, Zhang learned how to extract DNA from cells and determine the length of each sequence. But that isn’t what he remembers best. “They showed us ‘Jurassic Park,’ ” he said, his voice moving up a register. “And it was amazing to me. The teacher explained the different scientific concepts in the movie, and they all seemed completely feasible.”
We had reached the cocktail party, a tepid affair crowded with men in khakis and women wearing sensible shoes. Zhang left after barely twenty minutes and headed back to the lab. He retains his position on the cognitive-sciences faculty, because he hopes that his research will help neuroscientists study the brain in greater detail. He told me that when he was young he had a friend who suffered from serious depression, and he had been surprised to find that there was almost no treatment available. It spurred a lasting interest in psychiatry. “People think you are weak if you are depressed,” he said. “It is still a common prejudice. But many people suffer from problems we cannot begin to address. The brain is still the place in the universe with the most unanswered questions.”
The Broad Institute was founded, in 2003, by the entrepreneur Eli Broad and his wife, Edythe, to foster research into the molecular components of life and their connections to disease. One afternoon in Zhang’s laboratory, Winston Yan offered to walk me through the mechanics of using CRISPR to edit a gene. “We need to be able to break DNA in a very precise place in the genome,” he said as I watched him at work. He swivelled in his chair and pointed to a row of vials that contained DNA samples to be analyzed and edited. Yan, a thin, bespectacled man, wore black laboratory gloves and a white Apple Watch; he clapped his hands and shrugged, as if to suggest that the work was simple.
Ordering the genetic parts required to tailor DNA isn’t as easy as buying a pair of shoes from Zappos, but it seems to be headed in that direction. Yan turned on the computer at his lab station and navigated to an order form for a company called Integrated DNA Technologies, which synthesizes biological parts. “It takes orders online, so if I want a particular sequence I can have it here in a day or two,” he said. That is not unusual. Researchers can now order online almost any biological component, including DNA, RNA, and the chemicals necessary to use them. One can buy the parts required to assemble a working version of the polio virus (it’s been done) or genes that, when put together properly, can make feces smell like wintergreen. In Cambridge, I.D.T. often makes same-day deliveries. Another organization, Addgene, was established, more than a decade ago, as a nonprofit repository that houses tens of thousands of ready-made sequences, including nearly every guide used to edit genes with CRISPR. When researchers at the Broad, and at many other institutions, create a new guide, they typically donate a copy to Addgene.
The RNA that CRISPR relies upon to guide the molecular scalpel to its target is made of twenty base pairs. Humans have twenty thousand genes, and twenty base pairs occupy roughly the same percentage of space in a single gene as would one person standing in a circle that contained the entire population of the United States. CRISPR is better at locating specific genes than any other system, but it isn’t perfect, and sometimes it cuts the wrong target. Yan would order a ready-made probe from Addgene. When it arrives, he pairs it with a cutting enzyme and sends it to the designated gene.
Yan joined Zhang’s lab just before what he described as “the CRISPR craze” began. But, he added, the technology has already transformed the field. “For many years, there was a reductionist approach to genetics,” he said. “A kind of wishful thinking: ‘We will find the gene that causes cancer or the gene that makes you prone to heart disease.’ It is almost never that simple.”
The next morning, I walked over to the Broad’s new Stanley Building and rode the elevator to the top floor, where I emptied my pockets, put on a mask and gown, and slipped booties over my shoes. Then I passed through an air chamber that was sealed with special gaskets and had a fan blowing continuously to keep out foreign microbes. I entered the vivarium, a long, clean floor that looked like a combination of research unit and hospital ward. The vivarium, which opened last year, provides thousands of mice with some of the world’s most carefully monitored accommodations.
Despite our growing knowledge of the way that cancer develops in human cells, mutations can’t be studied effectively in a petri dish, and, since the late nineteen-eighties, genetically modified mice have served as the standard proxy. What cures (or kills) a mouse won’t necessarily have the same effect on a human, but the mouse genome is surprisingly similar to our own, and the animals are cheap and easy to maintain. Like humans, and many other mammals, mice develop complex diseases that affect the immune system and the brain. They get cancer, atherosclerosis, hypertension, and diabetes, among other chronic illnesses. Mice also reproduce every three weeks, which allows researchers to follow several generations at once. Typically, technicians would remove a stem cell from the mouse, then edit it in a lab to produce a particular gene or to prevent the gene from working properly. After putting the stem cell back into the developing embryo of the mouse, and waiting for it to multiply, they can study the gene’s effect on the animal’s development. The process works well, but it generally allows for the study of only one characteristic in one gene at a time.
The vivarium at the Broad houses an entirely different kind of mouse, one that carries the protein Cas9 (which stands for CRISPR-associated nuclease) in every cell. Cas9, the part of the CRISPR system that acts like a genetic scalpel, is an enzyme. When scientists originally began editing DNA with CRISPR, they had to inject both the Cas9 enzyme and the probe required to guide it. A year ago, Randall Platt, another member of Zhang’s team, realized that it would be possible to cut the CRISPR system in two. He implanted the surgical enzyme into a mouse embryo, which made it a part of the animal’s permanent genome. Every time a cell divided, the Cas9 enzyme would go with it. In other words, he and his colleagues created a mouse that was easy to edit. Last year, they published a study explaining their methodology, and since then Platt has shared the technique with more than a thousand laboratories around the world.
The “Cas9 mouse” has become the first essential tool in the emerging CRISPR arsenal. With the enzyme that acts as molecular scissors already present in every cell, scientists no longer have to fit it onto an RNA guide. They can dispatch many probes at once and simply make mutations in the genes they want to study.
To demonstrate a potential application for cancer research, the team used the Cas9 mouse to model lung adenocarcinoma, the most common form of lung cancer. Previously, scientists working with animal models had to modify one gene at a time or cross-breed animals to produce a colony with the needed genetic modifications. Both processes were challenging and time-consuming. “Now we can activate CRISPR directly in the cells we’re interested in studying, and modify the genome in whatever way we want,” Platt said, as he showed me around the vivarium. We entered a small exam room with a commanding view of Cambridge. I watched as a technician placed a Cas9 mouse in a harness inside a biological safety cabinet. Then, peering through a Leica microscope, she used a fine capillary needle to inject a single cell into the mouse’s tail.
“And now we have our model,” Platt said, explaining that the mouse had just received an injection that carried three probes, each of which was programmed to carry a mutation that scientists believe is associated with lung cancer. “The cells will carry as many mutations as we want to study. That really is a revolutionary development.”
“In the past, this would have taken the field a decade, and would have required a consortium,” Platt said. “With CRISPR, it took me four months to do it by myself.” In September, Zhang published a report, in the journal Cell, describing yet another CRISPR protein, called Cpf1, that is smaller and easier to program than Cas9.
The lab employs a similar approach to studying autism. Recent experiments suggest that certain psychiatric conditions can be caused by just a few malfunctioning neurons out of the trillions in every brain. Studying the way neurons function within the brain is difficult. But by re-creating, in the lab, genetic mutations that others have linked to autism and schizophrenia Zhang’s team has been able to investigate faulty neurons that may play a role in those conditions.
As the price of sequencing plunges, cancer clinics throughout the United States have begun to study their patients’ tumors in greater detail. Tumors are almost never uniform; one may have five mutations or fifty, which means, essentially, that every cancer is a specific, personal disease. Until CRISPR became available, the wide genetic variations in cancer cells often made it hard to develop effective treatments.
“What I love most about the CRISPR process is that you can take any cancer-cell line, knock out every gene, and identify every one of the cell’s Achilles’ heels,” Eric Lander, the fifty-eight-year-old director of the Broad, told me recently. Lander, who was among the leaders of the Human Genome Project, said that he had never encountered a more promising research tool. “You can also use CRISPR to systematically study the ways that a cancer cell can escape from a treatment,” he said. “That should make it possible to build a comprehensive road map for cancer.”
Lander went on to say that each vulnerability of a tumor might be attacked by a single drug. But cancer cells elude drugs in many ways, and, to succeed, a therapy may need to block them all. That strategy has proved effective for infectious diseases like AIDS. “Remember the pessimism about H.I.V.,” he said, referring to the early years of the AIDS epidemic, when a diagnosis was essentially a death sentence. Eventually, virologists developed a series of drugs that interfere with the virus’s ability to replicate. The therapy became truly successful, however, only when those drugs, working together, could block the virus completely.
The same approach has proved successful in treating tuberculosis. Lander is convinced that it will also work for many cancers: “With triple-drug therapy,” for H.I.V., “we reached an inflection point: we were losing badly, and one day suddenly we were winning.”
He stood up and walked across the office toward his desk, then pointed at the wall and described his vision for the future of cancer treatment. “There will be an enormous chart,” he said. “Well, it will be electronic, and it will contain the therapeutic road map of every trick that cancer cells have—how they form, all the ways you can defeat them, and all the ways they can escape and defeat a treatment. And when we have that we win. Because every cancer cell starts naïve. It doesn’t know what we have waiting in the freezer for it. Infectious diseases are a different story; they share their knowledge as they spread. They learn from us as they move from person to person. But every person’s cancer starts naïve. And this is why we will beat it.”
Developing any technology as complex and widely used as CRISPR invariably involves contributions from many scientists. Patent fights over claims of discovery and licensing rights are common. Zhang, the Broad Institute, and M.I.T. are now embroiled in such a dispute with Jennifer Doudna and the University of California; she is a professor of chemistry and of molecular biology at Berkeley. By 2012, Doudna, along with Emmanuelle Charpentier, a medical microbiologist who studies pathogens at the Helmholtz Centre for Infection Research, in Germany, and their lab teams, demonstrated, for the first time, that CRISPR could edit purified DNA. Their paper was published that June. In January of 2013, though, Zhang and George Church, a professor of genetics at both Harvard Medical School and M.I.T., published the first studies demonstrating that CRISPR could be used to edit human cells. Today, patents are generally awarded to the first people to file—in this case, Doudna and Charpentier. But Zhang and the Broad argued that the earlier success with CRISPR had no bearing on whether the technique would work in the complex organisms that matter most to scientists looking for ways to treat and prevent diseases.
Zhang was awarded the patent, but the University of California has requested an official reassessment, and a ruling has not yet been issued. Both he and Doudna described the suit to me as “a distraction” that they wished would go away. Both pledged to release all intellectual property to researchers without charge (and they have). But both are also involved in new companies that intend to develop CRISPR technology as therapies, as do many pharmaceutical firms and other profit-seeking enterprises.
CRISPR research is becoming big business: venture-capital firms are competing with one another to invest millions, and any patent holder would have the right to impose licensing fees. Whoever wins stands to make a fortune. Other achievements are also at stake, possibly including a Nobel Prize. (Doudna’s supporters have described her as America’s next female Nobel Prize winner, and at times the publicity war seems a bit like the battles waged by movie studios during Academy Award season.) Last year, the National Science Foundation presented Zhang with its most prestigious award, saying that his fundamental research “moves us in the direction” of eliminating schizophrenia, autism, and other brain disorders. A few months later, Doudna and Charpentier received three million dollars each for the Breakthrough Prize, awarded each year for scientific achievement. The prize was established, in 2012, by several Silicon Valley billionaires who are seeking to make science a more attractive career path. The two women also appeared on Time’s annual list of the world’s hundred most influential people.
In fact, neither group was involved in the earliest identification of CRISPR or in the first studies to demonstrate how it works. In December, 1987, biologists at the Research Institute of Microbial Diseases, in Osaka, Japan, published the DNA sequence of a gene taken from the common intestinal bacterium E. coli. Those were early days in the genomic era, and thousands of labs around the world had embarked on similar attempts to map the genes of species ranging from fruit flies to humans. In an effort to better understand how this particular gene functioned, the Japanese scientists also sequenced some of the DNA that surrounded it. When they examined the data, they were surprised to see cellular structures that none of them recognized: they had no idea what to make of the strange phenomenon, but they took note of it, writing in the final sentence of their report, published in the Journal of Bacteriology, that the “biological significance of these sequences is not known.”
The mystery remained until 2005, when Francisco Mojica, a microbiologist at the University of Alicante, who had long sought to understand CRISPR, decided to compare its DNA with the DNA of tens of thousands of similar organisms. What he saw amazed him: every unknown sequence turned out to be a fragment of DNA from an invading virus.
The pace of research quickened. In 2007, Rodolphe Barrangou and Philippe Horvath, microbiologists then working for Danisco, the Danish food company, had noticed that some of its yogurt cultures were routinely destroyed by viruses and others were not. They decided to find out why. The scientists infected the microbe Streptococcus thermophilus, which is widely used to make yogurt, with two viruses. Most of the bacteria died, but those which survived had one property in common: they all contained CRISPR molecules to defend them.
“No single person discovers things anymore,” George Church told me when we met in his office at Harvard Medical School. “The whole patent battle is silly. There has been much research. And if anybody should be making a fuss about this I should be making a fuss. But I am not doing that, because I don’t think it matters. They are all nice people. They are all doing important work. It’s a tempest in a teapot.”
From the moment that manipulating genes became possible, many people, including some of those involved in the experiments, were horrified by the idea of scientists in lab coats rearranging the basic elements of life. In 1974, David Baltimore, the pioneering molecular biologist, who was then at M.I.T., and Paul Berg, of Stanford, both of whom went on to win a Nobel Prize for their research into the fundamentals of viral genetics, called for a moratorium on gene-editing research until scientists could develop safety principles for handling organisms that contained recombinant DNA. That meeting, which took place in 1975, at a conference center in Asilomar, California, has come to be regarded as biotechnology’s Constitutional Convention.
Roughly a hundred and fifty participants, most of them scientists, gathered to discuss ways to limit the risks of accidentally releasing genetically modified organisms. At the time, the possibility of creating “designer babies”—a prospect that, no matter how unlikely, is attached to almost everything written or said about CRISPR—was too remote to consider. Nevertheless, the technology seemed frightening. In Cambridge, home to both M.I.T. and Harvard, the city council nearly banned such research altogether. The work went on, but decoding sequences of DNA wasn’t easy. “In 1974, thirty base pairs”—thirty rungs on the helical ladder of the six billion nucleotides that make up our DNA—“was a good year’s work,” George Church told me. Now the same work would take seconds.
At least for the foreseeable future, CRISPR’s greatest impact will lie in its ability to help scientists rapidly rewrite the genomes of animal and plant species. In laboratories, agricultural companies have already begun to use CRISPR to edit soybeans, rice, and potatoes in an effort to make them more nutritious and more resistant to drought. Scientists might even be able to edit allergens out of foods like peanuts.
Normally, it takes years for genetic changes to spread through a population. That is because, during sexual reproduction, each of the two versions of any gene has only a fifty per cent chance of being inherited. But a “gene drive”—which is named for its ability to propel genes through populations over many generations—manages to override the traditional rules of genetics. A mutation made by CRISPR on one chromosome can copy itself in every generation, so that nearly all descendants would inherit the change. A mutation engineered into a mosquito that would block the parasite responsible for malaria, for instance, could be driven through a large population of mosquitoes within a year or two. If the mutation reduced the number of eggs produced by that mosquito, the population could be wiped out, along with any malaria parasites it carried.
Kevin Esvelt, an evolutionary biologist at Harvard, was the first to demonstrate how gene drives and CRISPR could combine to alter the traits of wild populations. Recently, he has begun to study the possibility of using the technology to eliminate Lyme disease by rewriting the genes of mice in the wild. Lyme disease is caused by a bacterium and transmitted by ticks, and more than eighty-five per cent of the time they become infected after biting a mouse. Once exposed, however, some mice naturally acquire resistance or immunity. “My idea is to take the existing genes that confer resistance to Lyme and make sure that all mice have the most effective version,’’ Esvelt said. To do that, scientists could encode the most protective genes next to the CRISPR system and force them to be passed on together. Esvelt stressed that such an approach would become possible only after much more research and a lengthy series of public discussions on the risks and benefits of the process.
The promise of CRISPR research becomes more evident almost every month. Recently, Church reported that he had edited sixty-two genes simultaneously in a pig cell. The technique, if it proves accurate and easy to repeat, could help alleviate the constant shortage of organ donors in the U.S. For years, scientists have tried to find a way to use pig organs for transplants, but a pig’s DNA is filled with retroviruses that have been shown in labs to infect human cells. Church and his colleagues discovered that those viruses share a common genetic sequence. He deployed CRISPR to their exact locations and snipped them out of the genome. In the most successful of the experiments, the CRISPR system deleted all sixty-two of the retroviruses embedded in the pig’s DNA. Church then mixed those edited cells with human cells in the laboratory, and none became infected.
While CRISPR will clearly make it possible to alter our DNA, serious risks remain. Jennifer Doudna has been among the most vocal of those calling for caution on what she sees as the inevitable march toward editing human genes. “It’s going to happen,” she told me the first time we met, in her office at Berkeley. “As a research tool, CRISPR could hardly be more valuable—but we are far from the day when it should be used in a clinical setting.” Doudna was a principal author of a letter published in Science this spring calling for a temporary research moratorium. She and others have organized a conference to discuss the ethics of editing DNA, a sort of Asilomar redux. The conference, to be attended by more than two hundred scientists—from the U.S., England, and China, among other countries—will take place during the first week of December at the National Academy of Sciences, in Washington.
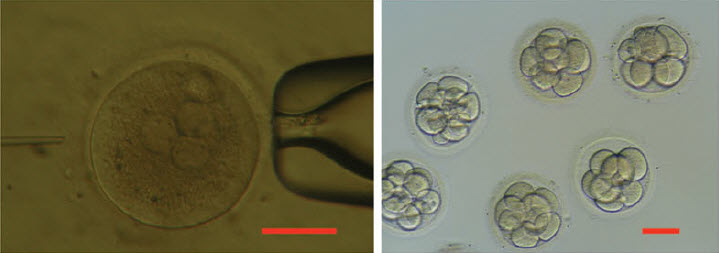
Until April, the ethical debate over the uses of CRISPR technology in humans was largely theoretical. Then a group at Sun Yat-sen University, in southern China, attempted to repair, in eighty-six human embryos, the gene responsible for betathalassemia, a rare but often fatal blood disorder. If those disease genes, and genes that cause conditions like cystic fibrosis, could be modified successfully in a fertilized egg, the alteration could not only protect a single individual but eventually eliminate the malady from that person’s hereditary lineage. Given enough time, the changes would affect all of humanity. The response to the experiment was largely one of fear and outrage. The Times carried the story under the headline “CHINESE SCIENTISTS EDIT GENES OF HUMAN EMBRYOS, RAISING CONCERNS.”
Critics called the experiment irresponsible and suggested that the scientists had violated an established code of conduct. “This paper demonstrates the enormous safety risks that any such attempt would entail, and underlines the urgency of working to forestall other such efforts,” Marcy Darnovsky, of the Center for Genetics and Society, told National Public Radio when the report was published. “The social dangers of creating genetically modified human beings cannot be overstated.”
There seems to be little disagreement about that. But the Chinese researchers were not trying to create genetically modified humans. They were testing the process, and every CRISPR researcher I spoke to considered the experiment to have been well planned and carried out with extraordinary care. The scientists also agreed that the results were illuminating. “That was an ethical paper, and a highly responsible project,’’ Lander told me. “What did they do? They took triploid zygotes’’—a relatively common genetic aberration—“from I.V.F. clinics. They deliberately chose those because they knew no human could ever develop from them. And what did the paper say? ‘Boy, we see problems everywhere.’ That was good science, and it was cautionary.”
Fewer than half the embryos were edited successfully, and, of those, most retained none of the new DNA that was inserted into the genes. The experiment, which was published in the Beijing-based journal Protein & Cell, demonstrated clearly that the day when scientists could safely edit humans is far off. The CRISPR system also made unintended cuts and substitutions, the potential effects of which are unknown. In other cases, it made the right changes in some cells of the embryo but not in all of them, which could cause other problems. “These authors did a very good job, pointing out the challenges,” Dieter Egli, a stem-cell researcher at Columbia University, said when the study was published. “They say themselves that this type of technology is not ready for any kind of application.”
Doudna agreed that the Chinese experiment yielded valuable results. She is fifty-one, and has been at Berkeley since 2002, when she and her husband, the biochemist Jamie Cate, were offered joint appointments to the departments of chemistry and molecular and cell biology. Their offices are next to each other, with the same commanding view of San Francisco Bay and the Golden Gate Bridge. Doudna’s work, unlike that of the scientists at the Broad, has been focussed on molecules, not mammalian genetics. For years, she has been leading investigations into the shape, structure, and capabilities of RNA, and in 2011 Charpentier asked for her help in exploring the mechanism of CRISPR. Doudna is tall, with graying blond hair and piercing blue eyes. She grew up in Hawaii, where her parents were academics; when it was time for college, she decided to leave the island and study in California, at Pomona. She earned her doctorate at Harvard and then moved on to Yale. “I have always been a bit of a restless soul,” she said. “I may spend too much time wondering what comes next.”
Doudna is a highly regarded biochemist, but she told me that not long ago she considered attending medical school or perhaps going into business. She said that she wanted to have an effect on the world and had begun to fear that the impact of her laboratory research might be limited. The promise of her work on CRISPR, however, has persuaded her to remain in the lab. She told me that she was constantly amazed by its potential, but when I asked if she had ever wondered whether the powerful new tool might do more harm than good she looked uncomfortable. “I lie in bed almost every night and ask myself that question,” she said. “When I’m ninety, will I look back and be glad about what we have accomplished with this technology? Or will I wish I’d never discovered how it works?”
Her eyes narrowed, and she lowered her voice almost to a whisper. “I have never said this in public, but it will show you where my psyche is,” she said. “I had a dream recently, and in my dream”—she mentioned the name of a leading scientific researcher—“had come to see me and said, ‘I have somebody very powerful with me who I want you to meet, and I want you to explain to him how this technology functions.’ So I said, Sure, who is it? It was Adolf Hitler. I was really horrified, but I went into a room and there was Hitler. He had a pig face and I could only see him from behind and he was taking notes and he said, ‘I want to understand the uses and implications of this amazing technology.’ I woke up in a cold sweat. And that dream has haunted me from that day. Because suppose somebody like Hitler had access to this—we can only imagine the kind of horrible uses he could put it to.”
Nobody is going to employ CRISPR technology to design a baby, let alone transform the genetic profile of humanity, anytime soon. Even if scientists become capable of editing human embryos, it would take years for the genetically modified baby to grow old enough to reproduce—and then many generations for the alteration to disseminate throughout the population.
But there are long-term consequences to consider. Modern medicine already shapes our genome, by preserving genes that might otherwise have been edited out of our genome by natural selection. Today, millions of people suffer from myopia, and many of them are legally blind. Were it not for the invention of glasses, which have turned poor eyesight largely into a nuisance rather than an existential threat, the genes responsible for myopia might be less prevalent than they are today. The same could be said about many infectious diseases, and even chronic conditions like diabetes.
Humans also carry genes that protect us from one disease but increase our susceptibility to others, and it’s impossible to predict the impact of changing all or even most of them. The AIDS virus often enters our blood cells through a protein called CCR5. One particular genetic variant of that protein, called the Delta32 mutation, prevents H.I.V. from locking onto the cell. If every person carried that mutation, nobody would get AIDS. So why not introduce that mutation into the human genome? Several research teams are working to develop drugs that do that in people who have already been infected.
Yet it’s important to note that, while such a procedure would prevent H.I.V. infection, it would also elevate our susceptibility to West Nile virus. Today, that trade-off may seem worth the risk, but there’s no way of knowing whether it would be true seven or ten generations from now. For example, sickle cells, which cause anemia, evolved as a protection against malaria; the shape of the cell blocks the spread of the parasite. If CRISPR technology had been available two hundred thousand years ago, it might have seemed sensible to edit sickle cells into the entire human population. But the results would have been devastating.
“This is a little bit like geoengineering,” Zhang told me, referring to attempts to deliberately alter the climate to offset damages associated with global warming. “Once you go down that path, it may not be so reversible.”
George Church disagrees. “It strikes me as a fake argument to say that something is irreversible,” he told me. “There are tons of technologies that are irreversible. But genetics is not one of them. In my lab, we make mutations all the time and then we change them back. Eleven generations from now, if we alter something and it doesn’t work properly we will simply fix it.”
In 1997, Scottish scientists shocked the world by announcing that they had cloned a lamb, which they named Dolly. Scores of journalists (including me) descended on Edinburgh, and wrote that the achievement, while wondrous, also carried the ominous implication that scientists had finally pried open Pandora’s box. Many articles about cloning and the value of human life were published. Evil people and dictators would clone themselves, their children, their pets. A new class of humans would arise.
Eighteen years later, the closest we have come to cloning a person was a failed attempt at a monkey, in 2007. Nobody spends much time worrying about it today. In Cambridge this summer, one of the researchers at the Broad told me that he and Louise Brown, the first success of in-vitro fertilization, were both born in 1978. “Did that set off an uproar?” he asked. It did. Even seven years earlier, James Watson had written, in The Atlantic, that the coming era of designer babies might overwhelm us all. Today, though, with more than five million children on earth born through in-vitro fertilization, that particular furor, too, seems to have passed.
CRISPR technology offers a new outlet for the inchoate fear of tinkering with the fundamentals of life. There are many valid reasons to worry. But it is essential to assess both the risks and the benefits of any new technology. Most people would consider it dangerous to fundamentally alter the human gene pool to treat a disease like AIDS if we could cure it with medicine or a vaccine. But risks always depend on the potential result. If CRISPR helps unravel the mysteries of autism, contributes to a cure for a form of cancer, or makes it easier for farmers to grow more nutritious food while reducing environmental damage, the fears, like the many others before them, will almost certainly disappear.
www.youtube.com/embed/EMLHtywaqSY?feature=player_embedded
Read Full Post »





 Article Info
Article Info































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
It’s not simply based in a desire to have super-children – remember that the PRC did everything in it’s power to crush the traditional clan system in China.
Rather, it’s based on fundamentally different values when it comes to what it means to be a human. In China, traditional religious notions mingle with Atheist practicality to produce a culture which thinks it can judge people as superior or inferior. The west shared this point of view until very recently, relatively.
The revulsion against the Nazis was motivated by a disgust towards the unnatural and the synthetic – hypocritically, despite having eugenics programs of their own which continued after the fall of Nazi Germany, the rest of the world used Germany as a sacrifice to Gaia. To make matters even muddier, Operation Paperclip assured that the USA was infected by the German elite.
Just like the Nazis, the Chinese are motivated by lofty ideals of the perfect human. The world at large doesn’t condemn or punish them for their political repression, their work camps, or their censorship. Germany didn’t apologize to gays until 2001, and it still hasn’t apologized to trade unionists. Instead the world condemns the Nazis and Chinese for trying to make a perfect person.
This has nothing to do with human rights or dignity, and everything to do with social conservatism and a ‘nature is best; god gave you cancer’ mentality. Our biology determines the reality we experience, and how we can interact with that reality. Sentimentalism demands that we all feel the same – or else there is no empathy, as in the modern west.
All this ‘ethical debate’ amounts to is a way to prevent individuals from pursuing their own biological destinies. To muddy the waters, and tie together human rights and state contorol – as if you can’t have one without the other.
Western humanism is being left in the dust – in a few decades, the average westerner isn’t going to be in the running for anything but a Darwin Award. Regulation will have driven everyone with any ambition or imagination further east and west – to China and the pacific ocean. Note seasteading.