What’s new with CRISPR-Cas9?
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Where is the most promising avenue to success in Pharmaceuticals with CRISPR-Cas9?
Author: Larry H. Bernstein, MD, FCAP

2.2.18 CRISPR-Cas9 and Regenerative Medicine, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
http://pharmaceuticalintelligence.com/2015/09/01/where-is-the-most-promising-avenue-to-success-in-pharmaceuticals-with-crispr-cas9/
There has been a rapid development of methods for genetic engineering that is based on an initial work on bacterial resistance to viral invasion. The engineering called RNA inhibition (RNAi) has gone through several stages leading to a more rapid and more specific application with minimal error.
It is a different issue to consider this application with respect to bacterial, viral, fungal, or parasitic invasion than it would be for complex human metabolic conditions and human cancer. The difference is that humans and multi-organ species are well differentiated systems with organ specific genome translation to function.
I would expect to see the use of genomic alteration as most promising in the near term for the enormous battle against antimicrobial, antifungal, and antiparasitic drug resistance. This could well be expected to be a long-term battle because of the invading organisms innate propensity to develop resistance.
A CRISPR/Cas system mediates bacterial innate immune evasion and virulence
Timothy R. Sampson, Sunil D. Saroj, Anna C. Llewellyn, Yih-Ling Tzeng & David S. Weiss
Affiliations, Contributions, Corresponding author
Nature 497, 254–257 (09 May 2013), http://dx.doi.org:/10.1038/nature12048
CRISPR/Cas (clustered regularly interspaced palindromic repeats/CRISPR-associated) systems are a bacterial defence against invading foreign nucleic acids derived from bacteriophages or exogenous plasmids1, 2, 3, 4. These systems use an array of small CRISPR RNAs (crRNAs) consisting of repetitive sequences flanking unique spacers to recognize their targets, and conserved Cas proteins to mediate target degradation5, 6, 7, 8. Recent studies have suggested that these systems may have broader functions in bacterial physiology, and it is unknown if they regulate expression of endogenous genes9, 10. Here we demonstrate that the Cas protein Cas9 of Francisella novicida uses a unique, small, CRISPR/Cas-associated RNA (scaRNA) to repress an endogenous transcript encoding a bacterial lipoprotein. As bacterial lipoproteins trigger a proinflammatory innate immune response aimed at combating pathogens11, 12, CRISPR/Cas-mediated repression of bacterial lipoprotein expression is critical for F. novicida to dampen this host response and promote virulence. Because Cas9 proteins are highly enriched in pathogenic and commensal bacteria, our work indicates that CRISPR/Cas-mediated gene regulation may broadly contribute to the regulation of endogenous bacterial genes, particularly during the interaction of such bacteria with eukaryotic hosts.
http://www.nature.com/nature/journal/v497/n7448/carousel/nature12048-f1.2.jpg
http://www.nature.com/nature/journal/v497/n7448/carousel/nature12048-f2.2.jpg
http://www.nature.com/nature/journal/v497/n7448/carousel/nature12048-f4.2.jpg
Zhang lab unlocks crystal structure of new CRISPR/Cas9 genome editing tool
Paul Goldsmith, 2015 Aug
In a paper published today in Cell researchers from the Broad Institute and University of Tokyo revealed the crystal structure of theStaphylococcus aureus Cas9 complex (SaCas9)—a highly efficient enzyme that overcomes one of the primary challenges to in vivo mammalian genome editing.
First identified as a potential genome-editing tool by Broad Institute core member Feng Zhang and his colleagues (and published by Zhang lab in April 2015), SaCas9 is expected to expand scientists’ ability to edit genomes in vivo. This new structural study will help researchers refine and further engineer this promising tool to accelerate genomic research and bring the technology closer to use in the treatment of human genetic disease.
“SaCas9 is the latest addition to our Cas9 toolbox, and the crystal shows us its blueprint,” said co-senior author Feng Zhang, who in addition to his Broad role, is also an investigator at the McGovern Institute for Brain Research, and an assistant professor at MIT.
The engineered CRISPR-Cas9 system adapts a naturally-occurring system that bacteria use as a defense mechanism against viral infection. The Zhang lab first harnessed this system as an effective genome-editing tool in mammalian cells using the Cas9 enzymes from Streptococcus thermophilus (StCas9) andStreptococcus pyogenes (SpCas9). Now, Zhang and colleagues have detailed the molecular structure of SaCas9, providing scientists with a high-resolution map of this enzyme. By comparing the crystal structure of SaCas9 to the crystal structure of the more commonly-used SpCas9 (published by the Zhang lab in February 2014), the team was able to focus on aspects important to Cas9 function— potentially paving the way to further develop the experimental and therapeutic potential of the CRISPR-Cas9 system.
Paper cited: Nishimasu H et al. “Crystal Structure of Staphylococcus aureus Cas9.” Cell, http://dx.doi.org:/10.1016/j.cell.2015.08.007
Advances in CRISPR-Cas9 genome engineering: lessons learned from RNA interference
Rodolphe Barrangou1,†, Amanda Birmingham2,†, Stefan Wiemann3, Roderick L. Beijersbergen4, Veit Hornung5 and Anja van Brabant Smith2
Nucleic Acids Research, 2015 Mar 23. http:dx.doi.org:/10.1093/nar/gkv226
RNAi and CRISPR-Cas9 have many clear similarities. Indeed, the mechanisms of both use small RNAs with an on-target specificity of ∼18–20 nt. Both methods have been extensively reviewed recently (3–5) so we only highlight their main features here. RNAi operates by piggybacking on the endogenous eukaryotic pathway for microRNA-based gene regulation (Figure 1A). microRNAs (miRNAs) are small, ∼22-nt-long molecules that cause cleavage, degradation and/or translational repression of RNAs with adequate complementarity to them(6).RNAi reagentsfor research aim to exploit the cleavage pathway using perfect complementarity to their targets to produce robust downregulation of only the intended target gene. The CRISPRCas9 system, on the other hand, originates from the bacterial CRISPR-Cas system, which provides adaptive immunity against invading genetic elements (7). Generally, CRISPR-Cas systems provide DNA-encoded (7), RNAmediated (8), DNA- (9) or RNA-targeting(10) sequencespecific targeting. Cas9 is the signature protein for Type II CRISPR-Cas systems (11).
…….
Both RNAi and CRISPR-Cas9 have experienced significant milestones in their technological development, as highlighted in Figure 2 (7–14,16–22,24–51) (highlighted topics have been detailed in recent reviews (2,4,52–58)). The CRISPR-Cas9 milestones to date have mimicked a compressed version of those for RNAi, underlining the practical benefit of leveraging similarities to this well-trodden research path. While RNAi has already influenced many advances in the CRISPR-Cas9 field, other applications of CRISPR-Cas9 have not yet been attained but will likely continue to be inspired by the corresponding advances in the RNAi field (Table 1). Of particular interest are the potential parallels in efficiency, specificity, screening and in vivo/therapeutic applications, which we discuss further below.
Figure2. Timeline of milestones for RNAi and CRISPR-Cas9. Milestones in the RNAi field are noted above the line and milestones in the CRISPR-Cas9 field are noted below the line. These milestones have been covered in depth in recent reviews (2,4,52–29).
Table 1. Summary of improvements in the CRISPR-Cas9 field that can be anticipated by corresponding RNAi advances
more…. see at http://pharmaceuticalintelligence.com/2015/09/01/where-is-the-most-promising-avenue-to-success-in-pharmaceuticals-with-crispr-cas9/
Early Diagnosis
http://pharmaceuticalintelligence.com/tag/research/
Reporter: Stephen J. Williams, Ph.D.
This post contains a curation of all Early Diagnosis posts on this site as well as a curation of the Early Detection Research Network.
Highlights of the accomplishments of the Early Detection Research Network.
A brief list of major EDRN-developed assays that have been adapted for clinical use is described in the table below:
Since its inception in 1999 EDRN has achieved several key milestones, summarized below:
1998 through 2000: Inception and Inauguration of EDRN
……
The European Society for Gene and Cell Therapy and the Spanish Society for Gene and Cell Therapy Collaborative Congress 2013
HUMAN GENE THERAPY XX:A2–A172 (XXXX 2013) ª Mary Ann Liebert, Inc. http://dx.doi.org:/10.1089/hum.2013.2513
Bases of gene therapy in leukemias
C. Bonini Experimental Hematology Unit, Division of Regenerative Medicine, Gene Therapy and Stem Cells,
Program of Immunology, Gene Therapy and Bio-Immunotherapy of Cancer, Leukemia Unit, San Raffaele Scientific Institute, Milan, Italy
Hematopoietic stem cell transplantation from a healthy donor (allo-HSCT) represents the most potent form of cellular adoptive immunotherapy to treat leukemias. During the past decades, allo-HSCT has developed from being an experimental therapy offered to patients with end-stage leukemia into a wellestablished therapeutic option for patients affected by several hematological malignancies. In allo-HSCT, donor T cells are double edge-swords, highly potent against residual tumor cells, but potentially highly toxic, and responsible of the graft versus host disease (GVHD), a major clinical complication of transplantation. Gene transfer technologies can improve the safety (ie: use of suicide genes), and the efficacy (ie: TCR gene transfer, TCR gene editing, CAR gene transfer) of adoptive T-cell therapy in the context of allo-HSCT. The encouraging preclinical and clinical results obtained in these years with genetically engineered T lymphocytes in the treatment of leukemias will be discussed.
Recent developments in gene therapy of solid tumors
R. Hernandez Division of Gene Therapy and Hepatology,
Universidad de Navarra, Madrid, Spain
Treatment of cancer has been one of the earliest and most frequent applications of gene therapy in experimental medicine. However, this indication entails unique difficulties, especially in the case of solid tumors. Pioneering strategies were aimed to reverse the malignant phenotype or to induce the death of cancer cells by transferring tumor-suppressor genes, inhibiting oncogenes or selectively expressing toxic genes. Proof of principle has been generated in abundant pre-clinical models and in humans. However, clinical efficacy is hampered by the diffi- culty in delivering therapeutic genes to a significant proportion of cancer cells in solid tumors using the currently available vectors. Therefore, current work aims to extend the effect to non-transduced cancer cells. This can be achieved by local or systemic expression of secreted proteins with the ability to block key pathways involved in angiogenesis, cell proliferation and invasion. Recent advances in gene therapy vectors allow sustained expression of transgenes and make these strategies feasible in the clinic. Another attractive option is the stimulation of immune reactions against cancer cells using gene transfer. In this case the therapeutic genes are antigens, cytokines or proteins capable of blocking the immunosuppressive microenvironment of tumors. Adaptation of replication-competent (oncolytic) viruses as vectors for these genes combines the intrinsic immunogenicity of viruses, their capacity to amplify gene expression and their direct lytic effect on cancer cells. In general, the ‘‘immunogene therapy’’ strategies offer the opportunity to destroy primary and distant lesions, especially if they are combined with other treatments that reduce tumor burden. More importantly, vaccination against cancer cells could prevent cancer relapse. Finally, gene and cell therapies are joining forces to improve the efficacy of adoptive cell therapy. Ex vivo gene transfer of natural or chimeric tumor-specific receptors in T lymphocytes enhances the cytotoxic potency of the cells and is expanding the applicability of this promising approach to different tumor types.
Production of vector and genetically modified stem cells
A. Galy and E. de Barbeyrac Genethon, 1
bis rue de l’Internationale, F91002 Evry, France
Hematopoietic gene therapy is currently used to treat a variety of genetic disorders of the blood and immune systems, or metabolic diseases, with promising results. The approach currently relies on the infusion of patient-autologous hematopoietic stem cells that have been subjected to gene-transfer ex vivo with a viral vector of clinical grade, during a short period of culture. The manufacture of such advanced therapy medicinal products for clinical trials should comply with the clinical trials EC directive. Requirements for gene and cell-based medicinal products both apply, therefore a high level of complexity is involved in the development of such products. Hematopoietic cell and gene therapy has many potential indications based on encouraging preclinical and early-phase clinical results. However, somatic cell and gene therapy medicinal products are still in early phases of development and no such product has been registered yet. The standardization of the manufacturing process and characterization of the drug product (i.e. geneticallymodified cells) are important but present challenges. Many aspects, and in particular limited available patient material, complicate a precise characterization of the drug product. On the other hand, clinical-grade gene transfer retroviral vectors are well-characterized starting materials that are described in a pharmacopeia monograph and can be robustly manufactured in successive campaigns of production under GMP conditions. Examples obtained in preclinical and ongoing clinical studies to treat Wiskott Aldrich Syndrome illustrate the vast differences in the level of characterization between the viral vector starting material and the drug product used in hematopoietic gene therapy. Characterization of the products and standardization/ validation of the manufacturing process are the next challenges in the field.
Gammaretro and lentiviral vectors for the gene therapy of X-linked chronic Granulomatous disease
M. Grez Institute for Biomedical Research,
Georg-Speyer-Haus, Frankfurt, Germany
Gene therapy of inherited diseases has provided convincing evidence of therapeutic benefits for many treated patients. In particular, treatment of primary severe congenital immunodeficiencies by gene transfer into hematopoietic stem cells (HSCs) has proven in some cases to be as beneficial as allogeneic stem cell transplantation, the treatment of choice for these diseases if HLA-matched donors are available. We conducted a Phase I clinical trial aimed at the correction of X-CGD, a rare inherited immunodeficiency characterized by severe and life threatening bacterial and fungal infections as well as widespread tissue granuloma formation. Phagocytic cells of CGD patients fail to kill ingested microbes due to a defect in the nicotinamide dinucleotide phosphate (NADPH) oxidase complex resulting in compromised antimicrobial activity. In this clinical trial we used a gammaretroviral vector with strong enhancer-promoter sequences in the long terminal repeats (LTRs) to genetically modify CD34 + cells in two X-CGD patients. After successful reconstitution of phagocytic functions, both patients experienced a clonal outgrowth of gene marked cells caused by vector-mediated insertional activation of proto-oncogenes leading to the development of myeloid malignancies. Moreover, functional correction of gene transduced cells decreased with time, due to epigenetic inactivation of the vector promoter within the LTR, resulting in the accumulation of nonfunctional gene transduced cells. The understanding of the molecular basis of insertional mutagenesis has motivated the development of advanced integrating vectors with equal therapeutic potency but reduced genotoxicity. In particular, the deletion of the enhancer elements within the viral LTR U3 regions has significantly contributed to the reduction of genotoxic effects associated with LTR-driven gammaretroviral vectors. Moreover, the use of tissue specific promoters, which are inactive in stem/progenitor cells but active in terminally differentiated cells, should further increase the safety level of SIN vectors. Based on the aforementioned advancements, we developed SIN gammaretroviral and lentiviral vectors for the safe and effective gene therapy of X-linked CGD. We combined the SIN configuration with an internal promoter, with preferential expression in myeloid cells. However, the introduction of a new vector into the clinic demands a series of sophisticated pre-clinical studies, which are quite challenging in particular within an academic environment. In this presentation we will report on the comprehensive and thorough preclinical efficacy and safety testing of both SIN vectors assessing dosage requirements, therapeutic efficacy, resistance to transgene silencing and genotoxic potential.
Progress and challenges of in vivo gene transfer with AAV vectors
F. Mingozzi1,2 1 Genethon, Evry, France; 2
University Pierre and Marie Curie, Paris, France
In vivo gene replacement for the treatment of an inherited disease is one of the most compelling concepts in modern medicine. Adeno-associated virus (AAV) vectors have been extensively used for this purpose and have shown therapeutic efficacy in a range of animal models. The translation of preclinical results to the clinic was initially slow, but early studies in humans helped defining the roadblocks to successful therapeutic gene transfer in vivo, which are highly depending on the target tissue, the route of vector delivery, and the specific disease. The development of strategies to overcome these limitations allowed achieving long-term expression of donated genes at therapeutic levels in patients with inherited retinal disorders, hemophilia B and other diseases. The recent market approval of Glybera, an AAV vector-based gene therapy product for lipoprotein lipase deficiency, further con- firmed the potential of AAV vectors as a therapeutic platform, raising hopes for the development of in vivo gene transfer treatments for many additional inherited and acquired diseases.
Glybera approval: a road map for advanced therapies in the orphan space
H. Petry
uniQure, Amsterdam, Netherlands
Glybera, is a gene therapy product based on the use of recombinant adeno-associated virus for gene delivery. It is designed for patients with Lipoprotein Lipase Deficiency (LPLD). On November 2, 2012, the European Commission approved the marketing authorisation for Glybera as a treatment for LPLD, under exceptional circumstances, in all 27 EU member states. Glybera is intended to treat patients with lipoprotein lipase deficiency. LPLD is caused by errors in the gene that codes for the protein lipoprotein lipase (LPL). LPL has a central role in fat metabolism. Non-functional LPL can lead to pancreatitis attacks, the most sever phenotype of this disease. The presentation will cover a summary of the clinical development, as well as a summary of the regulatory process. In addition post approval commitments will be discussed and their importance to follow up on the long term safety and efficacy of the this gene therapy product.
Phase Ib/IIa, escalating dose, single blind, clinical trial to assess the safety of the intravenous administration of expanded allogeneic adipose-derived mesenchymal stem cells (eASCs) to refractory rheumatoid arthritis (RA) patients
L. Dorrego
Tigenix, Madrid, Spain
Advanced therapies are emerging and fast-growing biotechnology sector paves the way for new, highly promising treatment opportunities for European patients. TiGenix is a leading European cell therapy company a marketed product for cartilage repair, and a strong pipeline with advanced clinical stage allogeneic adult stem cell programs for the treatment of autoimmune and inflammatory diseases. TiGenix has developed an innovative trial design in the stem cell area for treating refractory rheumatoid arthritis (RA) using expanded allogeneic adipose-derived mesenchymal stem cells (eASCS). The multicenter, randomized, double blind, placebocontrolled Phase IIa trial enrolled 53 patients with active refractory rheumatoid arthritis (mean time since diagnosis 15 years), who failed to respond to at least two biologics (mean previous treatment with 3 or more disease-modifying antirheumatic drugs and 3 or more biologics). The study design was based on a threecohort dose-escalating protocol. For both the low and medium dose regimens 20 patients received active treatment versus 3 patients on placebo; for the high dose regimen 6 patients received active treatment versus 1 on placebo. Patients were dosed at day 1, 8, and 15 and were followed up monthly over a six-month period. Follow-up consisted of a detailed monthly workup of all patients measuring all pre-defined parameters. The aim was to evaluate the safety, tolerability and optimal dosing over the full 6 months of the trial, as well as exploring therapeutic activity. Twenty five Spanish sites participated in this clinical trial. Coordinating Investigator: Dr. Jose´ Marı´a Alvaro-Gracia
Induction of multi-, pluri- and totipotency
H.R. Scho¨ler
Department Cell and Developmental Biology, Max Planck Institute for Molecular Biomedicine, Muenster, 48149, Germany
The pluripotent and multipotent states of stem cells are governed by the expression of few, specific transcription factors forming a highly interconnected regulatory network with more numerous, widely expressed transcription factors. When the set of master transcription factors comprising Oct4, Sox2, Klf4, and Myc is expressed ectopically in somatic cells, this network organizes itself to support a pluripotent cell state. But when Oct4 is replaced by Brn4, another POU transcription factor, fibroblasts are converted into multipotent neural stem cells. These two transcription factors appear to play distinct but interdependent roles in remodelling gene expression by influencing the local chromatin status during reprogramming. Furthermore, structural analysis of Oct4 bound to DNA shows that the Oct4 linker—a region connecting the two POU domains of Oct4—is exposed to the surface, and we therefore postulate that it recruits key epigenetic players onto Oct4 target genes during reprogramming. The role of Oct4 in defining totipotency and inducing pluripotency during embryonic development remains unclear, however. We genetically eliminated maternal Oct4 using a Cre/ lox approach and found no effect on the establishment of totipotency, as shown by the generation of live pups. After complete inactivation of both maternal and zygotic Oct4 expression, the embryos still formed Oct4-GFP– and Nanog–expressing inner cell masses, albeit nonpluripotent, indicating that Oct4 is not a determinant for the pluripotent cell lineage separation. Interestingly, Oct4-deficient oocytes were able to reprogram fibroblasts into pluripotent cells. Our results indicate that, in contrast to its crucial role in the maintenance of pluripotency, maternal Oct4 is crucial for neither the establishment of totipotency in embryos, nor the induction of pluripotency in somatic cells using oocytes.
Reprogramming in vivo is possible and generates a new type of iPS
M. Serrano
Spanish National Cancer Research Center (CNIO), Madrid, Spain
Reprogramming into induced pluripotent stem cells (iPSCs) has opened new therapeutic opportunities, however, little is known about the possibility of in vivo reprogramming within tissues. We have generated transgenic mice with inducible expression of the four Yamanaka factors. Interestingly, transitory induction of the reprogramming factors results in teratomas emerging from multiple organs, thereby, implying that full reprogramming can occur in vivo. Analyses of the stomach, intestine, pancreas and kidney reveal groups of dedifferentiated cells that express the pluripotency marker NANOG, indicative of in situ reprogramming. Also, by bone marrow transplantation, we demonstrate that hematopoietic cells can also be reprogrammed in vivo. Remarkably, induced reprogrammable mice also present circulating iPSCs in the blood. These in vivo-generated iPSCs can be purified and grown (in the absence of further induction of the reprogramming factors). Strikingly, at the transcriptome level, the in vivo-generated iPSCs are closer to embryonic stem cells (ESCs) than to standard in vitro-generated iPSCs. Moreover, in vivo-iPSCs efficiently contribute to the trophectoderm lineage, suggesting that they achieve a more plastic or primitive state than ESCs. Finally, in vivo-iPSCs show an unprecedented capacity to form embryo-like structures upon intraperitoneal injection, including the three germ layers of the proper embryo and extraembryonic tissues, such as extraembryonic ectoderm and yolk sac-like with associated embryonic erythropoiesis. These capacities are absent in ESCs or in standard in vitro-iPSCs. In summary, in vivo-iPSCs represent a more primitive or plastic state than ESCs or in vitro-iPSCs. These discoveries could be relevant for future applications of reprogramming in regenerative medicine.
Sleeping Beauty transpsons for molecular medicine
J.C. Izpisua
Belmonte Salk Institute for Biological Studies, La Jolla, CA, USA
The development of gene-editing technologies in combination with the generation of patient-specific induced pluripotent stem cells (iPSCs) represents the merge of both the stem cell and gene therapy fields. Novel gene-editing technologies in combination with iPSCs derivation methodologies open the possibility not only for direct gene therapy but also for the replenishment of loss and/or defective cell populations with gene-corrected cells. We will present recent examples developed in our laboratory to illustrate some of the different approaches being undertaken in these fields.
The Sleeping Beauty transposon system for molecular medicine
Z. Ivics
Paul Ehrlich Institute, Langen, Germany
Non-viral gene transfer approaches typically result in only short-lived transgene expression in primary cells, due to the lack of nuclear maintenance of the vector over time and cell division. The development of efficient and safe non-viral vectors armed with an integrating feature would thus greatly facilitate clinical gene therapy studies. The latest generation transposon technology based on the Sleeping Beauty (SB) transposon may potentially overcome some of these limitations. SB was recently shown to provide efficient stable gene transfer and sustained transgene expression in primary cell types, including human hematopoietic progenitors, mesenchymal stem cells, muscle stem/progenitor cells (myoblasts), iPSCs and T cells. The first-in-man clinical trial has been launched to use redirected T cells engineered with SB for gene therapy of B cell lymphoma. In addition, an EU FP7 project was recently initiated with the aim of replacing degenerated retinal pigment epithelial cells with cells that have been genetically modified by SB gene vectors ex vivo to produce an anti-angiogenic and neuroprotective factor for the potential treatment of patients suffering from age-related macular degeneration.
X-reactivation impacts human iPSC differentiation potential towards blood
N-B. Woods
Lund’s Stem Cell Center, Lund University, Sweden
To determine novel key regulators that direct ES/iPS cell differentiation to hematopoietic lineages, we compared the gene expression profiles of multiple iPS cell lines with differential blood forming capacity. We generated multiple iPS cell lines from amniotic fluid derived mesenchymal stromal cells (AFiPS) which differentiated towards hematopoietic lineages using our standardized and highly reproducible differentiation protocol. Of the 9 AF-iPS cell lines derived from an individual female patient, the average efficiency of CD45 + hematopoietic cells was 14.2 + / – 9% (range 1.6 to 26.3%). To elucidate the possible reasons for this diversity in efficiency, we grouped the AF-iPS cell lines on the basis of lowest and highest blood differentiation capacity and compared their gene expression pro- files by microarray. We found very few changes above 1.5-fold, but interestingly, among the 11 genes that were over-expressed in the AF-iPSC lines with poor blood differentiation efficiency, 10 were located on X chromosome, and the remaining one reported to be involved in Notch signalling. A combination of cumulative sum analysis and the location of differentially expressed genes on the X chromosome identified putative regions of reactivation at multiple, but distinct locations. The possibility of X-reactivation in these female lines was reinforced further where lower levels of XIST were seen in AF-iPSC lines shown to have low blood forming potential, however only half of the iPS cell lines with high blood differentiation capacity showed normal XIST expression when compared to the amniotic fluid mesenchymal starting cell material. To determine whether the block in differentiation was tissue specific we tested the differentiation capacity of the AF-iPSC lines towards neuronal lineages. Intriguingly, we found neural cell differentiation was not hampered within all lines with poor blood potential suggesting that the over-expression of genes as a consequence of X-reactivation can impart a specific negative effect on differentiation towards the blood lineages from pluripotency stage, while not having an effect on neuronal cell development. To further define the source of this block, we have begun working knocking down the overexpressed genes on X chromosome in lines with poor blood differentiation potential to determine whether the efficiency can be increased (or fully rescued) with one, or a combination of these 11 candidate genes. These results have implications for the identification and selection of female iPS lines suitable for therapeutic purposes. I will also discuss the identification of three new factors for improving blood lineage potential of iPS cells lines.
DLL4/Notch1 signaling is required for endothelial-tohematopoietic transition in a hESC model of human embryonic hematopoiesis
V. Ayllon1 , V. Ramos-Mejı´a1 , P.J. Real1 , O. Navarro-Montero1 , T. Romero1 , C. Bueno1,2, P. Menendez1,2,3 1
GENyO, Centre for Genomics & Oncological Research: Pfizer/ University of Granada / Andalusian Government, Granada, Spain; 2 Josep Carreras Leukemia Research Institute and Cell Therapy Program of University of Barcelona, Barcelona, Spain; 3 ICREA: Institucio´ Catalana de Reserca i Estudis Avanc¸ats, Catalunya Government, Spain
Notch signaling is essential for definitive embryonic hematopoiesis, but little is known on how Notch regulates hematopoiesis in early human embryonic development. Here we analyzed the contribution of Notch signaling to human embryonic hematopoietic differentiation using hESCs. We determined the expression of Notch receptors and ligands during hematopoietic differentiation of hESCs and found that expression of the Notch ligand DLL4 strongly parallels the emergence of bipotent hematoendothelial progenitors (HEPs). Co-cultures of hESCs with OP9-DLL4 cells demonstrated that DLL4 has a dual role in hematopoietic differentiation: during HEPs specification untimely DLL4-mediated Notch activation is detrimental for HEPs generation; however, once HEPs are specified, activation of Notch by DLL4 enhances hematopoietic commitment of these HEPs. We determined by flow cytometry that in hESCs differentiation, DLL4 is only expressed in a subpopulation of HEPs. Gene expression profiling of DLL4high and DLL4low/- HEPs showed that these two subpopulations already exhibit a distinct transcriptome program which determines their differentiation commitment: DLL4high HEPs are highly enriched in endothelial genes, while DLL4low/- HEPs display a clear hematopoietic transcriptional signature. Single cell cloning analysis of these two populations confirmed that DLL4high HEPs are enriched in committed endothelial precursors, while DLL4low/- HEPs contain committed hematopoietic progenitors. Confocal microscopy analysis of whole embryoid bodies revealed that DLL4high HEPs are located in close proximity to DLL4low/- HEPs, and at the base of clusters of CD45 + cells forming structures that resemble AGM hematopoietic clusters found in mouse embryos. Moreover, we found active Notch1 in clusters of emerging CD45 + cells. Overall, our data indicate that DLL4 regulates blood formation from hESCs, with DLL4high HEPs enriched in endothelial potential, whereas DLL4low/- HEPs are transcriptional and functionally committed to hematopoietic development. We propose a model for human embryonic hematopoiesis in which DLL4low/- HEPs receive a signal from DLL4high HEPs to activate Notch1, to undergo an endothelial-to-hematopoietic transition and differentiate into CD45 + hematopoietic cells, resembling what occurs in mouse AGM hematopoietic clusters.
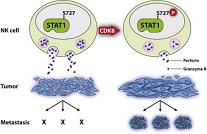
Researchers Investigate Importance of STAT1 Phosphorylation in NK Cells
“If we can stop CDK8 from inactivating STAT1 in NK cells, we could stimulate tumor surveillance and thus possibly have a new handle on treating cancer, harnessing the body’s own weapons against malignant cells.” –Dr. Eva Maria Putz.
http://www.regenerativemedicine.net/NewsletterArchives.asp?qEmpID=8422&qCat=WN

http://www.regenerativemedicine.net/images/Newsletter/uvmpic%20rs.jpg
Mammals contain cells whose primary function is to kill other cells in the body. The so-called Natural Killer (NK) cells are highly important in defending our bodies against viruses or even cancer. Scientists at the University of Veterinary Medicine, Vienna (Vetmeduni Vienna) provide evidence that NK cell activity can be influenced by phosphorylating a protein (STAT1) in NK cells. The results, which could be of immediate therapeutic relevance, were recently published.
Since its discovery in the early 1990s, the protein STAT1 (Signal Transducer and Activator of Transcription 1) has been found to be central in passing signals across immune cells, ensuring that our bodies react quickly and appropriately to threats from viruses or other pathogens. Animals without STAT1 are also prone to develop cancer, suggesting that STAT1 is somehow involved in protection against malignant cells. The STAT1 protein is known to be phosphorylated on at least two positions: phosphorylation of a particular tyrosine (tyr-701) is required for the protein to enter the cell nucleus (where it exerts its effects), while subsequent phosphorylation of a serine residue alters the way it interacts with other proteins, thereby affecting its function.
Natural Killer (NK) cells are among the first cells to respond to infections by viruses or to attack malignant cells when tumors develop. When they detect cells to be targeted, they produce a number of proteins, such as granzyme B and perforin, which enter infected cells and destroy them from within. Clearly, the lethal activity must be tightly controlled to prevent NK cells from running wild and destroying healthy cells or tissues. How is this done?
Eva Maria Putz and colleagues at the Institute of Pharmacology and Toxicology of the University of Veterinary Medicine, Vienna (Vetmeduni) have now investigated the importance of STAT1 phosphorylation in NK cells. The researchers found that when a particular serine residue (ser-727) in the STAT1 protein is mutated, NK cells produce far higher amounts of granzyme B and perforin and are far more effective at killing a wide range of tumor cells. Mice with the correspondingly mutated Stat1 gene are far less likely to develop melanoma, leukemia, or metastasizing breast cancer. On the other hand, when the same serine residue is phosphorylated, the NK cells are less able to kill infected or cancerous cells.
The Vetmeduni researchers have accumulated a body of evidence to suggest that the cyclin-dependent kinase CDK8 phosphorylates STAT1 on serine 727. Surprisingly, this phosphorylation does not require prior phosphorylation of the activating tyrosine residue, at least in NK cells. Instead, it seems to represent a way in which the lethal activity of the NK cells is kept in check. Putz is keen to note the potential significance of the finding. As she says, “If we can stop CDK8 from inactivating STAT1 in NK cells, we could stimulate tumor surveillance and thus possibly have a new handle on treating cancer, harnessing the body’s own weapons against malignant cells.”
Illustration: Inhibition of NK cells by phosphorylation of STAT1-Serin 727 mediated by CDK8. –Eva-Maria Putz/Vetmeduni Vienna.
Read more…
University of Veterinary Medicine, Vienna News Release (09/06/13)
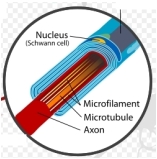
Important Step in Development of Artificial Nerves via Regenerative Medicine
The new cells successfully regenerated axons and extended their growth farther across nerve cell gaps toward damaged nerve stumps, with healthier vascularity.
http://www.regenerativemedicine.net/NewsletterArchives.asp?qEmpID=277&qCat=WN

http://www.regenerativemedicine.net/images/Newsletter/schwann%20cell2.jpg
A study carried out by researchers at the Kyoto University School of Medicine has shown that when transplanted bone marrow cells (BMCs) containing adult stem cells are protected by a 15mm silicon tube and nourished with bio-engineered materials, they successfully help regenerate damaged nerves. The research may provide an important step in developing artificial nerves.
“We focused on the vascular and neurochemical environment within the tube,” said Tomoyuki Yamakawa, MD, the study’s lead author. “We thought that BMCs containing adult stem cells, with the potential to differentiate into bone, cartilage, fat, muscle, or neuronal cells, could survive by obtaining oxygen and nutrients, with the result that rates of cell differentiation and regeneration would improve.”
Nourished with bioengineered additives, such as growth factors and cell adhesion molecules, the BMCs after 24 weeks differentiated into cells with characteristics of Schwann cells – a variety of neural cell that provides the insulating myelin around the axons of peripheral nerve cells. The new cells successfully regenerated axons and extended their growth farther across nerve cell gaps toward damaged nerve stumps, with healthier vascularity.
“The differentiated cells, similar to Schwann cells, contributed significantly to the promotion of axon regeneration through the tube,” explained Yamakawa. “This success may be a further step in developing artificial nerves.”
Grafting self-donated (autologous) nerve cells to damaged nerves has been widely practiced and considered the “gold standard.” However, autologous cells for transplant are in limited supply. Allologous cells, donated by other individuals, require the host to take heavy immunosuppressant drugs.
Translating dosage compensation to trisomy 21
Authors: Jun Jiang, Yuanchun Jing, Gregory J. Cost, Jen-Chieh Chiang, Heather J. Kolpa, Allison M. Cotton, Dawn M. Carone, Benjamin R. Carone, David A. Shivak, Dmitry Y. Guschin, Jocelynn R. Pearl, Edward J. Rebar, Meg Byron, Philip D. Gregory, Carolyn J. Brown, Fyodor D. Urnov, Lisa L. Hall, & Jeanne B. Lawrence
Down’s syndrome is a common disorder with enormous medical and social costs, caused by trisomy for chromosome 21. We tested the concept that gene imbalance across an extra chromosome can be de facto corrected by manipulating a single gene, XIST (the X-inactivation gene). Using genome editing with zinc finger nucleases, we inserted a large, inducible XIST transgene into the DYRK1A locus on chromosome 21, in Down’s syndrome pluripotent stem cells. The XIST non-coding RNA coats chromosome 21 and triggers stable heterochromatin modifications, chromosome-wide transcriptional silencing and DNA methylation to form a ‘chromosome 21 Barr body’. This provides a model to study human chromosome inactivation and creates a system to investigate genomic expression changes and cellular pathologies of trisomy 21, free from genetic and epigenetic noise. Notably, deficits in proliferation and neural rosette formation are rapidly reversed upon silencing one chromosome 21. Successful trisomy silencing in vitro also surmounts the major first step towards potential development of ‘chromosome therapy’.
Source: Nature; (07/17/13)
New article reviews latest advances in magnetic particle tracking in cell therapy
http://www.news-medical.net/news/20151027/New-article-reviews-latest-advances-in-magnetic-particle-tracking-in-cell-therapy.aspx
A new article published in Regenerative Medicine reviews the latest advances in magnetic particle tracking in cell therapy, a potentially groundbreaking strategy in disease treatment and regenerative medicine.
Cell therapy is one of the most promising avenues for regenerative medicine, however, its success is restricted by a number of limitations, such as inefficient delivery and retention of the therapeutic cells at the target organ, difficulties in monitoring the safety and efficacy of the therapy, in addition to issues obtaining and maintaining therapeutic cell phenotypes.
In a review by a group from the UCL Centre for Advanced Biomedical Imaging team (London, UK), emerging and established magnetic particle-based techniques for targeting, imaging and stimulating cells in vivo are discussed, in addition to potential benefits of their application in cell-based regenerative medicine therapies the clinic.
“The magnetic control of stem cells inside the body is a fascinating and promising concept for treatment of a vast range of diseases” commented Mark Lythgoe, director of the Centre for Advanced Biomedical Imaging at UCL. “Using microscopic nanomagnets we now have the potential to image, guide and activate therapeutic cells, combining therapy and diagnosis – theranostics – creating a novel type of dual imaging/therapy’
Commissioning Editor for Regenerative Medicine, Elena Conroy, added: “This timely review provides a much needed update on the different methods by which researchers can track cells with magnetic particles and how these can be used for cell therapy. I strongly believe that this will be of great use to cell biologists in both regenerative medicine and other research areas.”
Read Full Post »





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}