Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Reporter and Original Article Co-Author: Amandeep Kaur, B.Sc. , M.Sc.
Abstract Since its inception in late 2019, SARS-CoV-2 has evolved resulting in emergence of various variants in different countries. These variants have spread worldwide resulting in devastating second wave of COVID-19 pandemic in many countries including India since the beginning of 2021. To control this pandemic continuous mutational surveillance and genomic epidemiology of circulating strains is very important. In this study, we performed mutational analysis of the protein coding genes of SARS-CoV-2 strains (n=2000) collected during January 2021 to March 2021. Our data revealed the emergence of a new variant in West Bengal, India, which is characterized by the presence of 11 co-existing mutations including D614G, P681H and V1230L in S-glycoprotein. This new variant was identified in 70 out of 412 sequences submitted from West Bengal. Interestingly, among these 70 sequences, 16 sequences also harbored E484K in the S glycoprotein. Phylogenetic analysis revealed strains of this new variant emerged from GR clade (B.1.1) and formed a new cluster. We propose to name this variant as GRL or lineage B.1.1/S:V1230L due to the presence of V1230L in S glycoprotein along with GR clade specific mutations. Co-occurrence of P681H, previously observed in UK variant, and E484K, previously observed in South African variant and California variant, demonstrates the convergent evolution of SARS-CoV-2 mutation. V1230L, present within the transmembrane domain of S2 subunit of S glycoprotein, has not yet been reported from any country. Substitution of valine with more hydrophobic amino acid leucine at position 1230 of the transmembrane domain, having role in S protein binding to the viral envelope, could strengthen the interaction of S protein with the viral envelope and also increase the deposition of S protein to the viral envelope, and thus positively regulate virus infection. P618H and E484K mutation have already been demonstrated in favor of increased infectivity and immune invasion respectively. Therefore, the new variant having G614G, P618H, P1230L and E484K is expected to have better infectivity, transmissibility and immune invasion characteristics, which may pose additional threat along with B.1.617 in the ongoing COVID-19 pandemic in India.
Study: Emergence of a new SARS-CoV-2 variant from GR clade with a novel S glycoprotein mutation V1230L in West Bengal, India
Systems Biology analysis of Transcription Networks, Artificial Intelligence, and High-End Computing Coming to Fruition in Personalized Oncology
Curator: Stephen J. Williams, Ph.D.
In the June 2020 issue of the journal Science, writer Roxanne Khamsi has an interesting article “Computing Cancer’s Weak Spots; An algorithm to unmask tumors’ molecular linchpins is tested in patients”[1], describing some early successes in the incorporation of cancer genome sequencing in conjunction with artificial intelligence algorithms toward a personalized clinical treatment decision for various tumor types. In 2016, oncologists Amy Tiersten collaborated with systems biologist Andrea Califano and cell biologist Jose Silva at Mount Sinai Hospital to develop a systems biology approach to determine that the drug ruxolitinib, a STAT3 inhibitor, would be effective for one of her patient’s aggressively recurring, Herceptin-resistant breast tumor. Dr. Califano, instead of defining networks of driver mutations, focused on identifying a few transcription factors that act as ‘linchpins’ or master controllers of transcriptional networks withing tumor cells, and in doing so hoping to, in essence, ‘bottleneck’ the transcriptional machinery of potential oncogenic products. As Dr. Castilano states
“targeting those master regulators and you will stop cancer in its tracks, no matter what mutation initially caused it.”
It is important to note that this approach also relies on the ability to sequence tumors by RNA-seq to determine the underlying mutations which alter which master regulators are pertinent in any one tumor. And given the wide tumor heterogeneity in tumor samples, this sequencing effort may have to involve multiple biopsies (as discussed in earlier posts on tumor heterogeneity in renal cancer).
As stated in the article, Califano co-founded a company called Darwin-Health in 2015 to guide doctors by identifying the key transcription factors in a patient’s tumor and suggesting personalized therapeutics to those identified molecular targets (OncoTarget™). He had collaborated with the Jackson Laboratory and most recently Columbia University to conduct a $15 million 3000 patient clinical trial. This was a bit of a stretch from his initial training as a physicist and, in 1986, IBM hired him for some artificial intelligence projects. He then landed in 2003 at Columbia and has been working on identifying these transcriptional nodes that govern cancer survival and tumorigenicity. Dr. Califano had figured that the number of genetic mutations which potentially could be drivers were too vast:
A 2018 study which analyzed more than 9000 tumor samples reported over 1.5 million mutations[2]
and impossible to develop therapeutics against. He reasoned that you would just have to identify the common connections between these pathways or transcriptional nodes and termed them master regulators.
A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples
Chen H, Li C, Peng X, et al. Cell. 2018;173(2):386-399.e12.
Abstract
The role of enhancers, a key class of non-coding regulatory DNA elements, in cancer development has increasingly been appreciated. Here, we present the detection and characterization of a large number of expressed enhancers in a genome-wide analysis of 8928 tumor samples across 33 cancer types using TCGA RNA-seq data. Compared with matched normal tissues, global enhancer activation was observed in most cancers. Across cancer types, global enhancer activity was positively associated with aneuploidy, but not mutation load, suggesting a hypothesis centered on “chromatin-state” to explain their interplay. Integrating eQTL, mRNA co-expression, and Hi-C data analysis, we developed a computational method to infer causal enhancer-gene interactions, revealing enhancers of clinically actionable genes. Having identified an enhancer ∼140 kb downstream of PD-L1, a major immunotherapy target, we validated it experimentally. This study provides a systematic view of enhancer activity in diverse tumor contexts and suggests the clinical implications of enhancers.
A diagram of how concentrating on these transcriptional linchpins or nodes may be more therapeutically advantageous as only one pharmacologic agent is needed versus multiple agents to inhibit the various upstream pathways:
VIPER Algorithm (Virtual Inference of Protein activity by Enriched Regulon Analysis)
The algorithm that Califano and DarwinHealth developed is a systems biology approach using a tumor’s RNASeq data to determine controlling nodes of transcription. They have recently used the VIPER algorithm to look at RNA-Seq data from more than 10,000 tumor samples from TCGA and identified 407 transcription factor genes that acted as these linchpins across all tumor types. Only 20 to 25 of them were implicated in just one tumor type so these potential nodes are common in many forms of cancer.
Other institutions like the Cold Spring Harbor Laboratories have been using VIPER in their patient tumor analysis. Linchpins for other tumor types have been found. For instance, VIPER identified transcription factors IKZF1 and IKF3 as linchpins in multiple myeloma. But currently approved therapeutics are hard to come by for targets with are transcription factors, as most pharma has concentrated on inhibiting an easier target like kinases and their associated activity. In general, developing transcription factor inhibitors in more difficult an undertaking for multiple reasons.
Identifying the multiple dysregulated oncoproteins that contribute to tumorigenesis in a given patient is crucial for developing personalized treatment plans. However, accurate inference of aberrant protein activity in biological samples is still challenging as genetic alterations are only partially predictive and direct measurements of protein activity are generally not feasible. To address this problem we introduce and experimentally validate a new algorithm, VIPER (Virtual Inference of Protein-activity by Enriched Regulon analysis), for the accurate assessment of protein activity from gene expression data. We use VIPER to evaluate the functional relevance of genetic alterations in regulatory proteins across all TCGA samples. In addition to accurately inferring aberrant protein activity induced by established mutations, we also identify a significant fraction of tumors with aberrant activity of druggable oncoproteins—despite a lack of mutations, and vice-versa. In vitro assays confirmed that VIPER-inferred protein activity outperforms mutational analysis in predicting sensitivity to targeted inhibitors.
Schematic overview of the VIPER algorithm From: Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A: Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature genetics 2016, 48(8):838-847.
(a) Molecular layers profiled by different technologies. Transcriptomics measures steady-state mRNA levels; Proteomics quantifies protein levels, including some defined post-translational isoforms; VIPER infers protein activity based on the protein’s regulon, reflecting the abundance of the active protein isoform, including post-translational modifications, proper subcellular localization and interaction with co-factors. (b) Representation of VIPER workflow. A regulatory model is generated from ARACNe-inferred context-specific interactome and Mode of Regulation computed from the correlation between regulator and target genes. Single-sample gene expression signatures are computed from genome-wide expression data, and transformed into regulatory protein activity profiles by the aREA algorithm. (c) Three possible scenarios for the aREA analysis, including increased, decreased or no change in protein activity. The gene expression signature and its absolute value (|GES|) are indicated by color scale bars, induced and repressed target genes according to the regulatory model are indicated by blue and red vertical lines. (d) Pleiotropy Correction is performed by evaluating whether the enrichment of a given regulon (R4) is driven by genes co-regulated by a second regulator (R4∩R1). (e) Benchmark results for VIPER analysis based on multiple-samples gene expression signatures (msVIPER) and single-sample gene expression signatures (VIPER). Boxplots show the accuracy (relative rank for the silenced protein), and the specificity (fraction of proteins inferred as differentially active at p < 0.05) for the 6 benchmark experiments (see Table 2). Different colors indicate different implementations of the aREA algorithm, including 2-tail (2T) and 3-tail (3T), Interaction Confidence (IC) and Pleiotropy Correction (PC).
Other articles from Andrea Califano on VIPER algorithm in cancer include:
Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, Cai S, Tu Y, McCoy A, Peoples M, Sun Y, Qiu H, Chang Q, Bristow C, Carugo A, Shao J, Ma X, Harris A, Mundi P, Lau R, Ramamoorthy V, Wu Y, Alvarez MJ, Califano A, Moulder SL, Symmans WF, Marszalek JR, Heffernan TP, Chang JT, Piwnica-Worms H.Sci Transl Med. 2019 Apr 17;11(488):eaav0936. doi: 10.1126/scitranslmed.aav0936.PMID: 30996079
Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Liang H: A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173(2):386-399 e312.
Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A: Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature genetics 2016, 48(8):838-847.
Other articles of Note on this Open Access Online Journal Include:
Bioinformatic Tools for Cancer Mutational Analysis: COSMIC and Beyond, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Bioinformatic Tools for Cancer Mutational Analysis: COSMIC and Beyond
Curator: Stephen J. Williams, Ph.D.
Updated 7/26/2019
Updated 04/27/2019
Signatures of Mutational Processes in Human Cancer (from COSMIC)
The genomic landscape of cancer. The COSMIC database has a fully curated and annotated database of recurrent genetic mutations founds in various cancers (data taken form cancer sequencing projects). For interactive map please go to the COSMIC database here: http://cancer.sanger.ac.uk/cosmic
Somatic mutations are present in all cells of the human body and occur throughout life. They are the consequence of multiple mutational processes, including the intrinsic slight infidelity of the DNA replication machinery, exogenous or endogenous mutagen exposures, enzymatic modification of DNA and defective DNA repair. Different mutational processes generate unique combinations of mutation types, termed “Mutational Signatures”.
The current set of mutational signatures is based on an analysis of 10,952 exomes and 1,048 whole-genomes across 40 distinct types of human cancer. These analyses are based on curated data that were generated by The Cancer Genome Atlas (TCGA), the International Cancer Genome Consortium (ICGC), and a large set of freely available somatic mutations published in peer-reviewed journals. Complete details about the data sources will be provided in future releases of COSMIC.
The profile of each signature is displayed using the six substitution subtypes: C>A, C>G, C>T, T>A, T>C, and T>G (all substitutions are referred to by the pyrimidine of the mutated Watson–Crick base pair). Further, each of the substitutions is examined by incorporating information on the bases immediately 5’ and 3’ to each mutated base generating 96 possible mutation types (6 types of substitution ∗ 4 types of 5’ base ∗ 4 types of 3’ base). Mutational signatures are displayed and reported based on the observed trinucleotide frequency of the human genome, i.e., representing the relative proportions of mutations generated by each signature based on the actual trinucleotide frequencies of the reference human genome version GRCh37. Note that only validated mutational signatures have been included in the curated census of mutational signatures.
Additional information is provided for each signature, including the cancer types in which the signature has been found, proposed aetiology for the mutational processes underlying the signature, other mutational features that are associated with each signature and information that may be relevant for better understanding of a particular mutational signature.
The set of signatures will be updated in the future. This will include incorporating additional mutation types (e.g., indels, structural rearrangements, and localized hypermutation such as kataegis) and cancer samples. With more cancer genome sequences and the additional statistical power this will bring, new signatures may be found, the profiles of current signatures may be further refined, signatures may split into component signatures and signatures
COSMIC v75 includes curations across GRIN2A, fusion pair TCF3-PBX1, and genomic data from 17 systematic screen publications. We are also beginning a reannotation of TCGA exome datasets using Sanger’s Cancer Genome Project analyis pipeline to ensure consistency; four studies are included in this release, to be expanded across the next few releases. The Cancer Gene Census now has a dedicated curator, Dr. Zbyslaw Sondka, who will be focused on expanding the Census, enhancing the evidence underpinning it, and developing improved expert-curated detail describing each gene’s impact in cancer. Finally, as we begin to streamline our ever-growing website, we have combined all information for each gene onto one page and simplified the layout and design to improve navigation
may be found in cancer types in which they are currently not detected.
Signature 1 has been found in all cancer types and in most cancer samples.
Proposed aetiology:
Signature 1 is the result of an endogenous mutational process initiated by spontaneous deamination of 5-methylcytosine.
Additional mutational features:
Signature 1 is associated with small numbers of small insertions and deletions in most tissue types.
Comments:
The number of Signature 1 mutations correlates with age of cancer diagnosis.
Signature 2
Cancer types:
Signature 2 has been found in 22 cancer types, but most commonly in cervical and bladder cancers. In most of these 22 cancer types, Signature 2 is present in at least 10% of samples.
Proposed aetiology:
Signature 2 has been attributed to activity of the AID/APOBEC family of cytidine deaminases. On the basis of similarities in the sequence context of cytosine mutations caused by APOBEC enzymes in experimental systems, a role for APOBEC1, APOBEC3A and/or APOBEC3B in human cancer appears more likely than for other members of the family.
Additional mutational features:
Transcriptional strand bias of mutations has been observed in exons, but is not present or is weaker in introns.
Comments:
Signature 2 is usually found in the same samples as Signature 13. It has been proposed that activation of AID/APOBEC cytidine deaminases is due to viral infection, retrotransposon jumping or to tissue inflammation. Currently, there is limited evidence to support these hypotheses. A germline deletion polymorphism involving APOBEC3A and APOBEC3B is associated with the presence of large numbers of Signature 2 and 13 mutations and with predisposition to breast cancer. Mutations of similar patterns to Signatures 2 and 13 are commonly found in the phenomenon of local hypermutation present in some cancers, known as kataegis, potentially implicating AID/APOBEC enzymes in this process as well.
Signature 3
Cancer types:
Signature 3 has been found in breast, ovarian, and pancreatic cancers.
Proposed aetiology:
Signature 3 is associated with failure of DNA double-strand break-repair by homologous recombination.
Additional mutational features:
Signature 3 associates strongly with elevated numbers of large (longer than 3bp) insertions and deletions with overlapping microhomology at breakpoint junctions.
Comments:
Signature 3 is strongly associated with germline and somatic BRCA1 and BRCA2 mutations in breast, pancreatic, and ovarian cancers. In pancreatic cancer, responders to platinum therapy usually exhibit Signature 3 mutations.
Signature 4
Cancer types:
Signature 4 has been found in head and neck cancer, liver cancer, lung adenocarcinoma, lung squamous carcinoma, small cell lung carcinoma, and oesophageal cancer.
Proposed aetiology:
Signature 4 is associated with smoking and its profile is similar to the mutational pattern observed in experimental systems exposed to tobacco carcinogens (e.g., benzo[a]pyrene). Signature 4 is likely due to tobacco mutagens.
Additional mutational features:
Signature 4 exhibits transcriptional strand bias for C>A mutations, compatible with the notion that damage to guanine is repaired by transcription-coupled nucleotide excision repair. Signature 4 is also associated with CC>AA dinucleotide substitutions.
Comments:
Signature 29 is found in cancers associated with tobacco chewing and appears different from Signature 4.
Signature 5
Cancer types:
Signature 5 has been found in all cancer types and most cancer samples.
Proposed aetiology:
The aetiology of Signature 5 is unknown.
Additional mutational features:
Signature 5 exhibits transcriptional strand bias for T>C substitutions at ApTpN context.
Comments:
Signature 6
Cancer types:
Signature 6 has been found in 17 cancer types and is most common in colorectal and uterine cancers. In most other cancer types, Signature 6 is found in less than 3% of examined samples.
Proposed aetiology:
Signature 6 is associated with defective DNA mismatch repair and is found in microsatellite unstable tumours.
Additional mutational features:
Signature 6 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 6 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 15, 20, and 26.
Signature 7
Cancer types:
Signature 7 has been found predominantly in skin cancers and in cancers of the lip categorized as head and neck or oral squamous cancers.
Proposed aetiology:
Based on its prevalence in ultraviolet exposed areas and the similarity of the mutational pattern to that observed in experimental systems exposed to ultraviolet light Signature 7 is likely due to ultraviolet light exposure.
Additional mutational features:
Signature 7 is associated with large numbers of CC>TT dinucleotide mutations at dipyrimidines. Additionally, Signature 7 exhibits a strong transcriptional strand-bias indicating that mutations occur at pyrimidines (viz., by formation of pyrimidine-pyrimidine photodimers) and these mutations are being repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 8
Cancer types:
Signature 8 has been found in breast cancer and medulloblastoma.
Proposed aetiology:
The aetiology of Signature 8 remains unknown.
Additional mutational features:
Signature 8 exhibits weak strand bias for C>A substitutions and is associated with double nucleotide substitutions, notably CC>AA.
Comments:
Signature 9
Cancer types:
Signature 9 has been found in chronic lymphocytic leukaemias and malignant B-cell lymphomas.
Proposed aetiology:
Signature 9 is characterized by a pattern of mutations that has been attributed to polymerase η, which is implicated with the activity of AID during somatic hypermutation.
Additional mutational features:
Comments:
Chronic lymphocytic leukaemias that possess immunoglobulin gene hypermutation (IGHV-mutated) have elevated numbers of mutations attributed to Signature 9 compared to those that do not have immunoglobulin gene hypermutation.
Signature 10
Cancer types:
Signature 10 has been found in six cancer types, notably colorectal and uterine cancer, usually generating huge numbers of mutations in small subsets of samples.
Proposed aetiology:
It has been proposed that the mutational process underlying this signature is altered activity of the error-prone polymerase POLE. The presence of large numbers of Signature 10 mutations is associated with recurrent POLE somatic mutations, viz., Pro286Arg and Val411Leu.
Additional mutational features:
Signature 10 exhibits strand bias for C>A mutations at TpCpT context and T>G mutations at TpTpT context.
Comments:
Signature 10 is associated with some of most mutated cancer samples. Samples exhibiting this mutational signature have been termed ultra-hypermutators.
Signature 11
Cancer types:
Signature 11 has been found in melanoma and glioblastoma.
Proposed aetiology:
Signature 11 exhibits a mutational pattern resembling that of alkylating agents. Patient histories have revealed an association between treatments with the alkylating agent temozolomide and Signature 11 mutations.
Additional mutational features:
Signature 11 exhibits a strong transcriptional strand-bias for C>T substitutions indicating that mutations occur on guanine and that these mutations are effectively repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 12
Cancer types:
Signature 12 has been found in liver cancer.
Proposed aetiology:
The aetiology of Signature 12 remains unknown.
Additional mutational features:
Signature 12 exhibits a strong transcriptional strand-bias for T>C substitutions.
Comments:
Signature 12 usually contributes a small percentage (<20%) of the mutations observed in a liver cancer sample.
Signature 13
Cancer types:
Signature 13 has been found in 22 cancer types and seems to be commonest in cervical and bladder cancers. In most of these 22 cancer types, Signature 13 is present in at least 10% of samples.
Proposed aetiology:
Signature 13 has been attributed to activity of the AID/APOBEC family of cytidine deaminases converting cytosine to uracil. On the basis of similarities in the sequence context of cytosine mutations caused by APOBEC enzymes in experimental systems, a role for APOBEC1, APOBEC3A and/or APOBEC3B in human cancer appears more likely than for other members of the family. Signature 13 causes predominantly C>G mutations. This may be due to generation of abasic sites after removal of uracil by base excision repair and replication over these abasic sites by REV1.
Additional mutational features:
Transcriptional strand bias of mutations has been observed in exons, but is not present or is weaker in introns.
Comments:
Signature 2 is usually found in the same samples as Signature 13. It has been proposed that activation of AID/APOBEC cytidine deaminases is due to viral infection, retrotransposon jumping or to tissue inflammation. Currently, there is limited evidence to support these hypotheses. A germline deletion polymorphism involving APOBEC3A and APOBEC3B is associated with the presence of large numbers of Signature 2 and 13 mutations and with predisposition to breast cancer. Mutations of similar patterns to Signatures 2 and 13 are commonly found in the phenomenon of local hypermutation present in some cancers, known as kataegis, potentially implicating AID/APOBEC enzymes in this process as well.
Signature 14
Cancer types:
Signature 14 has been observed in four uterine cancers and a single adult low-grade glioma sample.
Proposed aetiology:
The aetiology of Signature 14 remains unknown.
Additional mutational features:
Comments:
Signature 14 generates very high numbers of somatic mutations (>200 mutations per MB) in all samples in which it has been observed.
Signature 15
Cancer types:
Signature 15 has been found in several stomach cancers and a single small cell lung carcinoma.
Proposed aetiology:
Signature 15 is associated with defective DNA mismatch repair.
Additional mutational features:
Signature 15 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 15 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 6, 20, and 26.
Signature 16
Cancer types:
Signature 16 has been found in liver cancer.
Proposed aetiology:
The aetiology of Signature 16 remains unknown.
Additional mutational features:
Signature 16 exhibits an extremely strong transcriptional strand bias for T>C mutations at ApTpN context, with T>C mutations occurring almost exclusively on the transcribed strand.
Comments:
Signature 17
Cancer types:
Signature 17 has been found in oesophagus cancer, breast cancer, liver cancer, lung adenocarcinoma, B-cell lymphoma, stomach cancer and melanoma.
Proposed aetiology:
The aetiology of Signature 17 remains unknown.
Additional mutational features:
Comments:
Signature 1Signature 18
Cancer types:
Signature 18 has been found commonly in neuroblastoma. Additionally, Signature 18 has been also observed in breast and stomach carcinomas.
Proposed aetiology:
The aetiology of Signature 18 remains unknown.
Additional mutational features:
Comments:
Signature 19
Cancer types:
Signature 19 has been found only in pilocytic astrocytoma.
Proposed aetiology:
The aetiology of Signature 19 remains unknown.
Additional mutational features:
Comments:
Signature 20
Cancer types:
Signature 20 has been found in stomach and breast cancers.
Proposed aetiology:
Signature 20 is believed to be associated with defective DNA mismatch repair.
Additional mutational features:
Signature 20 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 20 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 6, 15, and 26.
Signature 21
Cancer types:
Signature 21 has been found only in stomach cancer.
Proposed aetiology:
The aetiology of Signature 21 remains unknown.
Additional mutational features:
Comments:
Signature 21 is found only in four samples all generated by the same sequencing centre. The mutational pattern of Signature 21 is somewhat similar to the one of Signature 26. Additionally, Signature 21 is found only in samples that also have Signatures 15 and 20. As such, Signature 21 is probably also related to microsatellite unstable tumours.
Signature 22
Cancer types:
Signature 22 has been found in urothelial (renal pelvis) carcinoma and liver cancers.
Proposed aetiology:
Signature 22 has been found in cancer samples with known exposures to aristolochic acid. Additionally, the pattern of mutations exhibited by the signature is consistent with the one previous observed in experimental systems exposed to aristolochic acid.
Additional mutational features:
Signature 22 exhibits a very strong transcriptional strand bias for T>A mutations indicating adenine damage that is being repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 22 has a very high mutational burden in urothelial carcinoma; however, its mutational burden is much lower in liver cancers.
Signature 23
Cancer types:
Signature 23 has been found only in a single liver cancer sample.
Proposed aetiology:
The aetiology of Signature 23 remains unknown.
Additional mutational features:
Signature 23 exhibits very strong transcriptional strand bias for C>T mutations.
Comments:
Signature 24
Cancer types:
Signature 24 has been observed in a subset of liver cancers.
Proposed aetiology:
Signature 24 has been found in cancer samples with known exposures to aflatoxin. Additionally, the pattern of mutations exhibited by the signature is consistent with that previous observed in experimental systems exposed to aflatoxin.
Additional mutational features:
Signature 24 exhibits a very strong transcriptional strand bias for C>A mutations indicating guanine damage that is being repaired by transcription-coupled nucleotide excision repair.
Comments:
Signature 25
Cancer types:
Signature 25 has been observed in Hodgkin lymphomas.
Proposed aetiology:
The aetiology of Signature 25 remains unknown.
Additional mutational features:
Signature 25 exhibits transcriptional strand bias for T>A mutations.
Comments:
This signature has only been identified in Hodgkin’s cell lines. Data is not available from primary Hodgkin lymphomas.
Signature 26
Cancer types:
Signature 26 has been found in breast cancer, cervical cancer, stomach cancer and uterine carcinoma.
Proposed aetiology:
Signature 26 is believed to be associated with defective DNA mismatch repair.
Additional mutational features:
Signature 26 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 26 is one of four mutational signatures associated with defective DNA mismatch repair and is often found in the same samples as Signatures 6, 15 and 20.
Signature 27
Cancer types:
Signature 27 has been observed in a subset of kidney clear cell carcinomas.
Proposed aetiology:
The aetiology of Signature 27 remains unknown.
Additional mutational features:
Signature 27 exhibits very strong transcriptional strand bias for T>A mutations. Signature 27 is associated with high numbers of small (shorter than 3bp) insertions and deletions at mono/polynucleotide repeats.
Comments:
Signature 28
Cancer types:
Signature 28 has been observed in a subset of stomach cancers.
Proposed aetiology:
The aetiology of Signature 28 remains unknown.
Additional mutational features:
Comments:
Signature 29
Cancer types:
Signature 29 has been observed only in gingivo-buccal oral squamous cell carcinoma.
Proposed aetiology:
Signature 29 has been found in cancer samples from individuals with a tobacco chewing habit.
Additional mutational features:
Signature 29 exhibits transcriptional strand bias for C>A mutations indicating guanine damage that is most likely repaired by transcription-coupled nucleotide excision repair. Signature 29 is also associated with CC>AA dinucleotide substitutions.
Comments:
The Signature 29 pattern of C>A mutations due to tobacco chewing appears different from the pattern of mutations due to tobacco smoking reflected by Signature 4.
Signature 30
Cancer types:
Signature 30 has been observed in a small subset of breast cancers.
Proposed aetiology:
The aetiology of Signature 30 remains unknown.
Examples in the literature of deposits into or analysis from the COSMIC database
“analysis of exons representing 20,857 transcripts from 18,191 genes, we conclude that the genomic landscapes of breast and colorectal cancers are composed of a handful of commonly mutated gene “mountains” and a much larger number of gene “hills” that are mutated at low frequency. “
found cellular pathways with multiple pathways
analyzed a highly curated database (Metacore, GeneGo, Inc.) that includes human protein-protein interactions, signal transduction and metabolic pathways
There were 108 pathways that were found to be preferentially mutated in breast tumors. Many of the pathways involved phosphatidylinositol 3-kinase (PI3K) signaling
the cancer genome landscape consists of relief features (mutated genes) with heterogeneous heights (determined by CaMP scores). There are a few “mountains” representing individual CAN-genes mutated at high frequency. However, the landscapes contain a much larger number of “hills” representing the CAN-genes that are mutated at relatively low frequency. It is notable that this general genomic landscape (few gene mountains and many gene hills) is a common feature of both breast and colorectal tumors.
developed software to analyze multiple mutations and mutation frequencies available from Harvard Bioinformatics at
R Software for Cancer Mutation Analysis (download here)
CancerMutationAnalysis Version 1.0:
R package to reproduce the statistical analyses of the Sjoblom et al article and the associated Technical Comment. This package is build for reproducibility of the original results and not for flexibility. Future version will be more general and define classes for the data types used. Further details are available in Working Paper 126.
CancerMutationAnalysis Version 2.0:
R package to reproduce the statistical analyses of the Wood et al article. Like its predecessor, this package is still build for reproducibility of the original results and not for flexibility. Further details are available in Working Paper 126
Update 04/27/2019
Review 2018. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Z. Sondka et al. Nature Reviews. 2018.
The Catalogue of Somatic Mutations in Cancer (COSMIC) Cancer Gene Census (CGC) reevaluates the cancer genome landscape periodically and curates the findings into a database of genetic changes occurring in various tumor types. The 2018 CGC describes in detail the effect of 719 cancer driving genes. The recent expansion includes functional and mechanistic descriptions of how each gene contributes to disease etiology and in terms of the cancer hallmarks as described by Hanahan and Weinberg. These functional characteristics show the complexity of the cancer mutational landscape and genome and suggest ” multiple cancer-related functions for many genes, which are often highly tissue-dependent or tumour stage-dependent.” The 2018 CGC expands a second tier of genes, expanding the list of cancer related genes.
Criteria for curation of genes into CGC (curation process)
choosing candidate genes are selected from published literature, conference abstracts, large cancer genome screens deposited in databases, and analysis of current COSMIC database
COSMIC data are analyzed to determine presence of patterns of somatic mutations and frequency of such mutations in cancer
literature review to determine the role of the gene in cancer
Minimum evidence
– at least two publications from different groups shows increased mutation frequency in at least one type of cancer (PubMed)
– at least two publications from different groups showing experimental evidence of functional involvement in at least one hallmark of cancer in order to classify the mutant gene as oncogene, tumor suppressor, or fusion partner (like BCR-Abl)
independent assessment by at least two postdoctoral fellows
gene must be classified as either Tier 1 of Tier 2 CGC gene
inclusion in database
continued curation efforts
definitions:
Tier 1 gene: genes which have strong evidence from both mutational and functional analysis as being involved in cancer
Tier 2 gene: genes with mutational patterns typical of cancer drivers but not functionally characterized as well as genes with published mechanistic description of involvement in cancer but without proof of somatic mutations in cancer
Tier 2 genes (719 genes): include 103 oncogenes, 181 tumor suppressors, 134 fusion partners and 31 with unknown function
Updated 7/26/2019
The COSMIC database is undergoing an extensive update and reannotation, in order to ensure standardisation and modernisation across COSMIC data. This will substantially improve the identification of unique variants that may have been described at the genome, transcript and/or protein level. The introduction of a Genomic Identifier, along with complete annotation across multiple, high quality Ensembl transcripts and improved compliance with current HGVS syntax, will enable variant matching both within COSMIC and across other bioinformatic datasets.
As a result of these updates there will be significant changes in the upcoming releases as we work through this process. The first stage of this work was the introduction of improvedHGVS syntax compliance in our May release. The majority of the changes will be reflected in COSMIC v90, which will be released in late August or early September, and the remaining changes will be introduced over the next few releases.
The significant changes in v90 include:
Updated genes, transcripts and proteins from Ensembl release 93 on both the GRCh37 and GRCh38 assemblies.
Full reannotation of COSMIC variants with known genomic coordinates using Ensembl’s Variant Effect Predictor (VEP). This provides accurate and standardised annotation uniformly across all relevant transcripts and genes that include the genomic location of the variant.
New stable genomic identifiers (COSV) that indicate the definitive position of the variant on the genome. These unique identifiers allow variants to be mapped between GRCh37 and GRCh38 assemblies and displayed on a selection of transcripts.
Updated cross-reference links between COSMIC genes and other widely-used databases such as HGNC, RefSeq, Uniprot and CCDS.
Complete standardised representation of COSMIC variants, following the most recent HGVS recommendations, where possible.
Remapping of gene fusions on the updated transcripts on both the GRCh37 and GRCh38 assemblies, along with the genomic coordinates for the breakpoint positions.
Reduced redundancy of mutations. Duplicate variants have been merged into one representative variant.
Key points for you
COSMIC variants have been annotated on all relevant Ensembl transcripts across both the GRCh37 and GRCh38 assemblies from Ensembl release 93. New genomic identifiers (e.g. COSV56056643) are used, which refers to the variant change at the genomic level rather than gene, transcript or protein level and can thus be used universally. Existing COSM IDs will continue to be supported and will now be referred to as legacy identifiers e.g. COSM476. The legacy identifiers (COSM) are still searchable. In the case of mutations without genomic coordinates, hence without a COSV identifier, COSM identifiers will continue to be used.
All relevant Ensembl transcripts in COSMIC (which have been selected based on Ensembl canonical classification and on the quality of the dataset to include only GENCODE basic transcripts) will now have both accession and version numbers, so that the exact transcript is known, ensuring reproducibility. This also provides transparency and clarity as the data are updated.
How these changes will be reflected in the download files
As we are now mapping all variants on all relevant Ensembl transcripts, the number of rows in the majority of variant download files has increased significantly. In the download files, additional columns are provided including the legacy identifier (COSM) and the new genomic identifier (COSV). An internal mutation identifier is also provided to uniquely represent each mutation, on a specific transcript, on a given assembly build. The accession and version number for each transcript are included. File descriptions for each of the download files will be available from the downloads page for clarity. We have included an example of the new columns below.
For example: COSMIC Complete Mutation Data (Targeted screens)
[17:Q] Mutation Id – An internal mutation identifier to uniquely represent each mutation on a specific transcript on a given assembly build.
[18:R] Genomic Mutation Id – Genomic mutation identifier (COSV) to indicate the definitive position of the variant on the genome. This identifier is trackable and stable between different versions of the release.
[19:S] Legacy Mutation Id – Legacy mutation identifier (COSM) that will represent existing COSM mutation identifiers.
We will shortly have some sample data that can be downloaded in the new table structure, to give you real data to manipulate and integrate, this will be available on the variant updates page.
How this affects you
We are aware that many of the changes we are making will affect integration into your pipelines and analytical platforms. By giving you advance notice of the changes, we hope much of this can be mitigated, and the end result of having clean, standardised data will be well worth any disruption. The variant updates page on the COSMIC website will provide a central point for this information and further technical details of the changes that we are making to COSMIC.
This update was performed by the following methods:
A. GPT 5 Text analysis and Reasoning
B. Insertion of Knowledge Graph on topic Curation of Genomic Analysis from Non Small Cell Lung Cancer Studies from Nodus Labs using InfraNodus software
C. Domain Knowledge Expert evaluation of the Update outcomes
This article has the following Structure:
Part A: Introduction to LLM, Knowledge Graph software InfraNodus, ChatGPT5 and Background Information on curated material for Test Case
Part B: InfraNodus Analysis of manual curation and Knowledge Graph Creation
Part C: Chat GPT 5 Analysis of Manually Curated Material
Part D: Curation entitled Multiple Lung Cancer Genomic Projects Suggest New Targets, Research Directions for Non-Small Cell Lung Cancer originally published on 09/05/2014
Results of Article Update with GPT 5
1. GPT5 alone was not able to understand the goal of the article, namely to determine knowledge gaps in a particular research area involving 5 genomic studies on lung cancer patients
2. GPT5 alone was not able to group concepts or comonalities between biological pathways unless supplied with a manually curated list of KEGG pathways from a list of mutated genes. However this precluded any effect that fusion proteins had on the analysis and so GPT5 would only concentrate on mutated genes commonly found in literature
3. GPT was not able to access some of the open Access databases like NCBI Gene Ontology database
Results of Article Update with KnowledgeGraph presentation to GPT 5
4. As the Knowledge Graph understood the importance of fusion proteins and transversions, the knowledgegraph augmented the GPT analysis and so enriched the known pathways as well as could correctly identify the less represented pathways in the knowledge graph
5. This led to the identification of many novel signaling pathways not identified in the original analysis, and was able to perform this task with ease and speed
6. GPT with InfraNodus Analysis was able to propose pertinent questions for future research (the goal of the original curation) such as:
How does the interaction between [[EGFR]] mutations and sex-specific gene alterations, including [[RBM10]], influence treatment outcomes in lung adenocarcinoma?
How does the intersection of mutational patterns from smoking influence pathway activation in NSCLC, and can identifying these interactions improve targeted therapy development?
Novelty in comparison to Original article published on 09/05/2014
7. it appears that manual curation is necessary to assist in the building of relevant knowledge graphs in the biomedical fields to augment generative AI analysis
8. by itself, generative AI is not optimized for inference of higher concepts from biomedical text, and therefore, at this point, requires the input from human curators developing domain-specific knowledge graphs
9. The combination of ChatGPT5 and Knowledge graphs of this manually curated biomedical text added a further layer of complexity of gaps of knowledge not seen in the original curations including the need to study noncanonical signaling pathways like WNT and Hedgehog in smoker versus nonsmoker cohorts of lung cancer patients
A Comparison of Manual Expert-Curative and an LLM-based analysis of Knowledge Gaps in Non Small Lung Cancer Whole Exome Sequencing Studies and a Use Case Example of Chat GPT 5
Part A: Introduction to LLM, Knowledge Graph software InfraNodus, ChatGPT5 and Background Information on curated material for Test Case
The development of Large Language Models (LLMs), together with development of knowledge graphs, have facilitated the ability to analyze text and determine the relationships among the various concepts contained within series of texts. These concepts and relationships can be visualized, and new insights inferred from these visualizations. As a result, this type of analysis suggests new directions and lines of research.
Alternatively, these types of visualizations can also reveal gaps in knowledge which should be addressed. A new type of LLM and visualization tools have been developed to understand the gaps in knowledge in biomedical text.
Nodus Labs InfrNodus AI Knowledge Graph Software Tools Allow Text Relationship Visualization and Integrated AI Functionality
Infranodus makes knowlegde graphs from text and then is able to visualize the relationships between concepts (or nodes). In doing so, the tool also highlights the various knowledge gaps (or large differences between nodes) which can be used to investigate new hypotheses and research directions of previously univestigated relationships between concepts. This generates new research questions, in which these gaps can be used as prompts in the software’s integrated AI tool. The AI tool, much like a GPT, returns recommendations for research to be conducted in the area.
In addition, the InfraNodus software can detect if text is too biased on a particular concept or conclusion, and using a GPT3 or GPT4, can determine if the nodes are too dispersed and will recommend which gaps should be focused on.
The software can upload any biomedical text in various formats
A full demonstration is on their website but a good summary is found on their Youtube site at
Previously we had manually curated and analyzed the knowledge gaps from a series of publications on whole exome sequencing of biopsied tumors from cohorts of non small lung cancer patients. This curation (from 2016) is seen in the lower half of this updated link below and I separated with a bar and highlighted in Yellow as Text for AI Analysis.
Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J et al: Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150(6):1121-1134.
Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A et al: Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150(6):1107-1120.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T et al: Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature genetics 2012, 44(10):1104-1110.
were performed.
The purpose of this analysis was to uncover biological functions related to the sets of mutated genes with limited research publications in the area of non small cell lung cancer. The identification of such biological functions would represent a gap in knowledge in this disease. In addition, this analysis attempted to find new lines of research or potential new biotargets to investigate for lung cancer therapy.
However this manual method is time consuming and may miss relationships not defined in a GO ontology or gene knowledgebases.
Therefore we turned to an AI-driven approach:
Using InfraNodus ability to develop a knowledge graph based on our curation and determine if the AI platform could infer knowledge gaps
Utilize Chat GPT5 to analyze the same curated set to determine if OpenAI analysis would lead to the similar analysis from curated material
Determine if combining a knowledge graph within GPT would lead to a higher level of analysis
See below (Part D) of this update for the curated studies which were included in this analysis and the text which was entered into both InfraNodus and Chat GPT5.
As a summary, it seems that manual curation is necessary to assist in the building of relevant knowledge graphs in the biomedical fields to augment generative AI analysis. In addition, it appears that , by itself, generative AI is not optimized for inference of higher concepts from biomedical text, and therefore, at this point, requires the input from human curators developing domain-specific knowledge graphs.
Part B. InfraNodus Analysis of manual curation and Knowledge Graph Creation
Methods:
Text of the curation was copied and directly pasted into the text analysis module of InfraNodus. There was no editing of words however genes in the curation were linked to their GeneCard entry. GeneCards is a database run by the Weizmann Institute. InfraNodus utilizes a combination of LLMs and its own GraphRAG system to provide insights from text analysis. While it leverages various models, including those from OpenAI and Anthropic, it’s not limited to a single LLM. Instead, InfraNodus integrates these models within its GraphRAG framework, which enhances their capabilities by adding a relational understanding of the context through a knowledge graph.
InfraNodus then autogenerates a knowledge graph and returns entities and relationships between entities. InfraNodus offers the opportunity to modify the knowledge graph however for this analysis we used the first graph InfraNodus generated. Inspection of this graph (as shown below) was deemed reasonable.
Results
The knowledge graph of the input text is shown below:
InfraNodus generated Knowledge Graph of 5 WES Non Smal Cell Lung Cancer studies involving smokers and non smokers
Four main concepts were returned: tumors, genes, literature, and mutations.
A snapshot of the Analysis window is given below. It should be noted that InfraNodus felt there needed to be more connections between Pathway and Mutational Patterns.
An InfraNodus reposrt with Knowlege Graph on Whole Exome Sequencing studies in NSCLC to determine mutational spectrum in smokers versus non smokers
alk clinical [[egfr]] mutational pathway [[paper]] found key literature study [[genomic]] reveal [[transversion]]
Top relations / ngrams:
1) [[lung]] [[tumors]]
2) alk fusion
3) link function
4) eml alk
5) function [[gene_ontology]]
Modulary: 0.47
Relations:
InfraNodus identified 744 relations between entities (nodes)
A list of some of the more frequent are given here:
source
target
occurrences
weight
betweenness
[[lung]]
[[tumors]]
8
24
0.4676
analysis
pathway
5
12
0.2291
significantly
[[genes]]
5
9
0.1074
significantly
[[mutated]]
4
12
0.0281
[[mutated]]
[[genes]]
4
12
0.0847
[[transversion]]
high
3
12
0.0329
[[smoking]]
history
3
10
0.0352
study
identify
3
9
0.2051
mutational
pattern
3
9
0.0921
[[rbm10]]
[[mutations]]
3
8
0.1776
literature
analysis
3
7
0.2218
[[egfr]]
[[mutations]]
3
7
0.2139
[[transversion]]
group
3
7
0.0259
enriched
cohort
3
6
0.0219
[[whole_exome_sequencing]]
[[tumors]]
3
6
0.3485
identify
[[genes]]
3
6
0.2268
including
analysis
3
5
0.1985
alteration
[[genes]]
3
4
0.1298
[[tumors]]
analysis
3
4
0.5192
alk
fusion
2
15
0.0671
link
function
2
14
0.0269
function
[[gene_ontology]]
2
13
0.0054
Notice how the betweenness or importance of connection of disparate concepts vary but are high between concepts like tumors and analysis, or lung and tumor, however many important linked concepts like alk and fusion may have low betweenness but are mentioned frequently and have a much higher weight or closeness to each other. Gene-mutations-transversions-smoking seem to have a high correspondence to each other
Genetic Alterations: identify, [[genes]], study:The recent comprehensive studies on lung adenocarcinoma have significantly advanced our understanding of the genetic landscape by identifying key mutations and their intricate interactions. Notably, EGFR and RBM10 exhibit distinct mutational patterns, with RBM10 inactivations being notably enriched in male cohorts. This gender-linked enrichment underscores a potential differential oncogenic pathway involving ERBB2 and RB1 alterations.Moreover, these projects emphasize the quest to map significant gene alterations within lung adenocarcinoma. The identification of such genes not only corroborates prior reports but also expands upon them by highlighting new connections between mutation signatures and clinical factors like smoking history. These findings are crucial as they can inform future therapeutic targeting strategies, ensuring that personalized treatment approaches consider both gender-specific genomic enrichments and mutation-driven tumorigenesis pathways elucidated through rigorous analyses.elaborate
questions generated using AI to help you explore “alk, clinical, [[egfr]], mutational, pathway, [[paper]], found, key, literature, study, [[genomic]], reveal, [[transversion]]…”:How do mutational patterns, specifically EGFR mutations and transversions related to smoking history, influence the effectiveness of targeted therapies in NSCLC patients?elaborate
ideas generated using AI to help you explore “alk, clinical, [[egfr]], mutational, pathway, [[paper]], found, key, literature, study, [[genomic]], reveal, [[transversion]]…”:Develop a predictive model that utilizes genomic data and smoking history to forecast patient response to targeted therapies. This model would identify key mutational signatures linked to EGFR and other genes, highlighting the impact of smoking-induced transversions on drug efficacy.elaborate
Project Notes
”
The recent comprehensive studies on lung adenocarcinoma have significantly advanced our understanding of the genetic landscape by identifying key mutations and their intricate interactions. Notably, EGFR and RBM10 exhibit distinct mutational patterns, with RBM10 inactivations being notably enriched in male cohorts. This gender-linked enrichment underscores a potential differential oncogenic pathway involving ERBB2 and RB1 alterations.
Moreover, these projects emphasize the quest to map significant gene alterations within lung adenocarcinoma. The identification of such genes not only corroborates prior reports but also expands upon them by highlighting new connections between mutation signatures and clinical factors like smoking history. These findings are crucial as they can inform future therapeutic targeting strategies, ensuring that personalized treatment approaches consider both gender-specific genomic enrichments and mutation-driven tumorigenesis pathways elucidated through rigorous analyses.”
<ConceptualGateways>
alk
clinical
[[egfr]]
mutational
pathway
[[paper]]
found
key
literature
study
[[genomic]]
reveal
[[transversion]]
</ConceptualGateways>
How do mutational patterns, specifically EGFR mutations and transversions related to smoking history, influence the effectiveness of targeted therapies in NSCLC patients?
The report from the NCI Bulletin outlines significant advancements in understanding lung cancer through genome sequencing projects. These studies have revealed a plethora of genetic and epigenetic alterations across various forms of lung tumors, including adenocarcinomas, squamous cell carcinomas, and small cell lung cancers. Notably, some identified alterations could be targeted by existing therapies, providing potential new avenues for treatment.Dr. Meyerson emphasizes the complexity of these genetic changes, highlighting that distinct mechanisms inactivating genes can vary between tumors. The report also notes gaps in knowledge regarding non-coding DNA alterations, which comprise a major part of the human genome.Key findings include:1. Comprehensive genomic analyses revealing unique driver mutations in lung adenocarcinoma, such as those affecting MET and ERBB2, alongside significant mutations in known cancer drivers like TP53 and KRAS.2. A classification system based on genomic data enabling more accurate patient stratification—achieving a 75% classification rate of lung cancer subtypes.3. Smoking history is shown to influence mutational patterns significantly, with smokers exhibiting a higher incidence of point mutations compared to never-smokers.Moreover, the integration of genomic data and pathway analysis highlighted recurrent mutations across various pathways related to tumorigenesis, suggesting new therapeutic targets and underscoring the importance of personalized medicine approaches that factor in gender-specific mutation distributions.This synthesis of findings not only corroborates earlier studies but also extends our understanding of the interplay between genomic alterations, smoking habits, and clinical outcomes in lung cancer. Future research is needed to explore the implications of these findings further and to develop targeted therapies that leverage the identified genetic alterations.
Tumor Genomics
Genetic Alterations
Pathway Analysis
Mutation Patterns
Main Ideas Summary from Infranodus:
The report from the NCI Bulletin highlights significant developments in lung cancer genomics through five genome sequencing projects. These studies have uncovered various genetic and epigenetic alterations across multiple forms of lung cancer, including adenocarcinomas, squamous cell cancers, and small cell lung cancers. Notably, these alterations may correspond to existing drugs, offering new avenues for targeted therapy.
Key findings include the identification of unique mutations in genes like NF1, MET, and ERBB2, with particular attention to smoking’s role in mutational patterns. The analysis shows that smokers exhibit a higher incidence of specific point mutations, marking a distinct divergence in mutation profiles compared to never-smokers.
The research also emphasizes the need for continued exploration of the data to link mutation signatures with clinical outcomes, particularly regarding gender-specific differences in mutation prevalence. Emerging insights suggest that pathways involving MYC and alterations in splicing factors could be vital targets for future therapeutic strategies.
Overall, the studies collectively underscore an urgent need for integrating genomic data into clinical practice, which can lead to more personalized treatment approaches for lung cancer patients by considering both the genetic landscape of tumors and their histories, such as smoking exposure. The ongoing efforts to map gene alterations within lung adenocarcinoma not only reinforce existing knowledge but also reveal new potential therapeutic targets that can significantly impact patient outcomes.
Questions to Explore from InfraNodus:
How does the interaction between [[EGFR]] mutations and sex-specific gene alterations, including [[RBM10]], influence treatment outcomes in lung adenocarcinoma?
How does the intersection of mutational patterns from smoking influence pathway activation in NSCLC, and can identifying these interactions improve targeted therapy development?
Part C: Chat GPT 5 Analysis of Manually Curated Material
Now let us see how Chat GPT5 analyzed the same material.
Methods
Nice — I’ll work from the KEGG-pathway list shown in your text (the table of pathways with PubMed counts) and call out major KEGG pathways that are not represented there.
Important caveat: “Not represented in the table” below means not listed in the KEGG-pathway hits shown in your pasted literature analysis, not that these pathways never play a role in lung cancer. Absence from that table can reflect lack of reporting in the specific WES/WGS studies or the PubMed keyword search used — it’s not proof the pathway is uninvolved biologically.
The above Important caveat was very important because it showed that ChatGPT5 could understand what my experimental goals were, or at least understood the problem I was presenting to ChatGPT5!
Major KEGG pathways missing from the provided KEGG list
(However Note that GPT did NOT take into account the mutational spectrum of the studies or the gene lists supplied in the curation)
These are commonly cited cancer-related KEGG pathways that do not appear in the table you gave:
Wnt signaling pathway
Notch signaling pathway
Hedgehog signaling pathway
TGF-beta signaling pathway
Hippo signaling pathway
p53 signaling pathway — (note: TP53 is mutated in many tumors, so p53 biology is implicated in the text, but “p53 signaling” does not appear in that KEGG hits table)
Autocrine/paracrine inflammation/chemokine signaling beyond the one low-count entry (general cytokine networks other than the single “cytokine–cytokine receptor interaction” hit)
Signal transduction modules sometimes separate in KEGG:g. Calcium signaling was listed in your table, but other modules like cGMP–PKG signaling or PKA/PKC broader signaling networks are not.
Quick summary
The KEGG table in your text highlights many cancer and receptor-tyrosine/MAPK/PI3K related pathways (cell cycle, MAPK, ErbB, JAK-STAT, focal adhesion, etc.). What’s not listed are numerous developmental morphogen pathways (Wnt/Notch/Hedgehog/TGF-β/Hippo), many DNA-repair and core metabolic pathways, canonical apoptosis/autophagy/ubiquitin pathways, and broad adaptive-immune signaling pathways.
If you want, I can:
(A) cross-check this against the full KEGG pathway list and produce a definitive “KEGG universe → present vs absent in your table” spreadsheet, or
(B) prioritize which of the missing pathways you’d most want investigated in lung tumor sequencing data and suggest genes to look for.
Which follow-up would you prefer?
Now taking out the KEGG list I gave GPT in order to force GPT to do a similar but unbiased analysis
From the studies you’ve shared, we can identify which KEGG pathways are under-represented (i.e., appear less frequently altered than expected) in the altered genes in lung cancer.
To do this, we note that the researchers used KEGG and other pathway analysis tools (PathScan, literature curation, proteomics) to see which biological pathways the mutated genes mapped to.
While many well-known cancer pathways were found to be activated (MAPK, PI3K-AKT, mTOR, cell cycle, oxidative stress, etc.), a few key pathways were reported as unexpectedly low in mutation frequency despite their known role in cancer biology.
KEGG Pathways Under-Represented in Altered Genes in Lung Cancer
From the descriptions in Papers 1–5, these are the main pathways that appear under-represented:
MAPK signaling pathway
Normally, MAPK is a major cancer driver pathway. In this dataset, MAPK activity was high in proteomics, but many tumors lacked mutations in MAPK pathway genes.
This means MAPK activation is often non-mutational (possibly via upstream signaling, epigenetic changes, or ligand activation), so in mutation-based KEGG enrichment it is under-represented.
PI3K-Akt signaling pathway
Like MAPK, the PI3K-Akt pathway was highly activated in proteomics, but often without mutations in PI3K pathway genes.
Under-represented in gene mutation lists, despite strong functional activation.
mTOR signaling pathway
Functional activation was observed (especially when STK11 was mutated), but most tumors had no mTOR-pathway gene mutations.
Under-represented when looking only at altered genes.
JAK-STAT signaling pathway
Mentioned as novel pathway alterations in some tumors, but in KEGG enrichment analysis, JAK-STAT genes did not appear as a dominant mutated group—implying low mutation frequency compared to other cancer types.
Oxidative phosphorylation & metabolic pathways
While oxidative stress response genes (e.g., KEAP1) were sometimes mutated, the oxidative phosphorylation KEGG pathway itself did not emerge as mutation-enriched, suggesting functional changes occur via other mechanisms (e.g., hypoxia signaling).
While some epigenetic regulators (ARID1A, CREBBP, EP300, MLL) were mutated, KEGG’s “Chromatin remodeling” and “Histone modification” groupings were less represented than expected given the role of epigenetics in lung cancer, indicating epigenetic dysregulation is often non-mutational.
Why They Are Under-Represented
Proteomics revealed activation without DNA mutation (e.g., ligand-driven activation, phosphorylation cascades).
Epigenetic or post-translational modifications can activate pathways without altering the coding sequence.
Tumor heterogeneity: some pathways are only altered in subsets of tumors and thus don’t reach statistical enrichment.
Limitations of mutation-based KEGG analysis: pathways that are activated downstream or by non-coding alterations are missed.
✅ Summary Table
Pathway (KEGG ID)
Representation in Mutated Genes
Reason for Under-Representation
hsa04010 MAPK signaling
Low
Activated via non-mutational mechanisms
hsa04151 PI3K-Akt signaling
Low
Functional activation w/o gene mutations
hsa04150 mTOR signaling
Low
Activation often via upstream STK11 loss or growth factor signaling
hsa04630 JAK-STAT signaling
Low
Rarely mutated, altered mostly via cytokine/growth factor changes
hsa00190 Oxidative phosphorylation
Low
Functional metabolic shift via hypoxia & ROS stress, not mutations
Chromatin remodeling / histone modification
Low
Mostly altered via epigenetic changes, not coding mutations
If you want, I can cross-reference the exact KEGG IDs and run a quick gene-to-pathway mapping using the genes listed in the reports so we can get a precise statistical under-representation list. That way it’s backed by an enrichment p-value rather than just qualitative observation.
I tried to re-run the KEGG-backed analysis you requested, but the KEGG REST endpoints/web pages aren’t reachable from my browsing tool right now (the web.run calls to KEGG returned an internal/safety error). I can’t produce a properly-sourced, KEGG-ID–backed enrichment report unless I can successfully query KEGG (or a comparable pathway service) for the exact pathway membership.
Two clear, immediate options — tell me which you prefer and I’ll proceed right away:
Option 1 — Proceed with a KEGG-backed run using an alternate route (recommended)
I’ll attempt a small number of web.run search queries (to public pathway tools that mirror KEGG membership, e.g., g:Profiler / Reactome / Enrichr / DAVID) and then follow links to KEGG pathway pages. This often works even when direct KEGG REST calls fail. It will take a few web.run calls and I’ll cite the pages used. (If you want me to prefer a particular service — e.g., g:Profiler, Enrichr, DAVID — say so; otherwise I’ll pick g:Profiler as it’s robust.)
Option 2 — Run an immediate local (best-effort) analysis without web lookups
I’ll map the genes you listed to likely KEGG pathways from internal knowledge, run hypergeometric enrichment using a standard human background (20,000 genes), and give a ranked list of under-represented KEGG pathways with p-values — but I must be clear this is approximate (I cannot provide verified KEGG IDs or exact pathway sizes).
If you pick Option 1, I’ll start the web lookups now and produce the KEGG-ID mapped, statistically precise under-representation list with citations.
If you pick Option 2, I’ll run the approximation immediately and give results plus a confidence caveat.
Which do you want?
Sources
Ask ChatGPT
You said:
Use Option 1 to Proceed with a KEGG-backed run using an alternate route
ChatGPT said:
Thought for 37s
Thanks — I followed Option 1 trying to do a KEGG-backed run, but hit the same problem: my web tool (web.run) can reach g:Profiler/KEGG reliably in some contexts but it failed for the programmatic queries I attempted (internal / non-retryable errors). Because you explicitly requested a KEGG-backed run, I tried an alternate route (g:Profiler web query) and the site blocked the automated call.
Rather than leave you waiting, I made a transparent, reproducible best-effort analysis right now so you have immediate, concrete results — but please read the very important limitations below before using these numbers: (GPT having problems accessing material behind firewalls and paywalls)
What I did (short)
I extracted a conservative set of 48 unique genes from the papers you supplied (recurrent drivers, fusion partners, and the druggable gene list). Gene list used (48 genes):
TP53, KRAS, EGFR, STK11, KEAP1, SMARCA4, RBM10, NF1, MET, ERBB2, RIT1, BRAF, PIK3CA, PTEN, ARID1A, U2AF1, CREBBP, EP300, MLL, ALK, ROS1, DACH1, CFTR, RELN, ABCB5, HGF, JAK2, JAK3, HDAC2, HDAC6, HDAC9, BIRC6, ITGB1, ITGB3, MMP2, PRKCB, PIK3CG, TERT, MMP14, AURKC, ERBB4, FGFR1, EML4, SIK2, ZFHX4, EPHA3, EPHA7, SLIT2.
Because direct KEGG lookups failed, I mapped those genes to a short list of common KEGG pathways by manual curation (based on canonical pathway membership knowledge) and used conservative, literature-style approximate pathway sizes (number of genes in that KEGG pathway) to compute exact hypergeometric lower-tail p-values (i.e., probability of observing as few or fewer genes in the pathway than we did — an under-representation test).
I computed exact hypergeometric probabilities (sum of combinations) with a standard human gene background of N = 20,000. All calculations were executed programmatically (so no hand-approximate combinatorics).
Important limitations (please read)
This is NOT a true KEGG-backed run. I could not fetch KEGG pathway membership lists or KEGG IDs because the web tool could not access the KEGG/g:Profiler APIs in this session. (more GPT issues with access to data)
I used manually mapped genes → pathways (conservative, but not exhaustive) and approximate pathway sizes (estimates). Because both gene membership and exact pathway sizes matter a lot for enrichment statistics, the p-values below are illustrative only — they are useful to prioritize where to retry a proper KEGG query, but should not be taken as authoritative.
The correct, defensible way to produce what you asked is to re-run this on g:Profiler/Enrichr/DAVID (or download KEGG GMTs) so pathway membership and KEGG IDs are exact. I can do that as soon as the web tool can access those services (or you can paste exact KEGG gene sets if you prefer).
Results — immediate best-effort under-representation test
For each pathway I show: pathway size (K, approximate), number of genes from your list found in that pathway (k), and the hypergeometric lower-tail p-value P(X ≤ k) (probability of seeing k or fewer pathway genes by chance). Lower p-values (e.g., < 0.05) would suggest significant under-representation.
Interpretation: none of the pathways above reach statistical significance for under-representation under the assumptions I used. The closest to a (moderate) signal is oxidative phosphorylation where we observed 0 genes from the list (p ≈ 0.73) — but that p-value is still far from being significant (p < 0.05 would be needed). In short: no pathway is robustly under-represented in this small gene set using the conservative approximations above.
What I recommend (next steps to get the exact KEGG-ID–backed answer you originally asked for)
Allow me to retry the web-backed run (g:Profiler / Enrichr / DAVID) — I’ll re-run the enrichment and return:
gene → exact KEGG IDs mapping,
KEGG pathway sizes (exact K),
over- and under-representation p-values (Fisher/hypergeometric), and
multiple-testing corrected q-values (Benjamini–Hochberg). (This requires the web tool to be able to reach g:Profiler/KEGG; I already tried and the call failed. If you want, I’ll attempt again immediately.)
If you prefer not to rely on the web tool, you can paste your final canonical gene list (e.g., a CSV of the exact genes you want analyzed) and I will run a conservative local analysis like the above but I will be explicit that KEGG IDs and pathway sizes are approximations.
If you’d like, I can also send you the exact code/notebook I used for the above best-effort run so you (or your bioinformatics person) can re-run it locally against an up-to-date KEGG GMT file.
If you want me to retry the web-backed run now, say “Retry web run now” and I’ll immediately attempt g:Profiler/Enrichr again and produce the precise KEGG-ID–backed enrichment with citations. If you prefer I proceed without web access (i.e., run a local approximation on a larger set of genes or with different pathway groupings), tell me what gene list to use and I’ll compute immediately.
Top of Form
Bottom of Form
Done — I created a bar chart showing estimated PubMed article counts (prior to 2018) for “lung cancer” mentions of the pathways we discussed: MAPK, PI3K-Akt, mTOR, JAK-STAT, and Oxidative phosphorylation.
Important caveats (please read)
I could not access PubMed programmatically from this session (the web tool failed on PubMed/NCBI requests), so the numbers used are illustrative estimates, not exact counts.
If you need exact PubMed counts (filtered to publications before 2018), I can re-run the web-backed PubMed queries and regenerate the graph as soon as web access to NCBI is available — just say “Retry PubMed queries” and I’ll attempt that immediately.
Re-run the exact PubMed queries and update the chart with real counts, or
Expand the chart to include more pathways (e.g., chromatin modifiers, spliceosome, cell cycle), or
Provide the code/notebook used so you can run it locally against PubMed.
Here’s the expanded bar chart with additional pathways — Spliceosome, Chromatin remodeling / histone modification, ECM / focal adhesion, and Cell cycle — alongside the original set.
Would you like me to now replace these illustrative numbers with exact PubMed counts (filtered to before 2018) by running targeted PubMed searches?
Yes — even though the dataset and pathway lists we discussed focused mostly on signaling, metabolic, and genomic alterations, there is a clear immune component to lung cancer supported by both your provided text and the broader literature. (Now note that GPT noticed a clear literature coverage of cell cycle in lung cancer however it is claiming there was evidence of an immune component in the literature before 2018).
Evidence from your provided studies (Note GPT is using recent evidence not just restricting to periods before 2018)
Pathways with immune relevance
JAK-STAT signaling (detected in the analysis)
Central to cytokine signaling, immune cell activation, and inflammatory responses in the tumor microenvironment.
Altered JAK-STAT can drive immune evasion or chronic inflammation that promotes tumor growth.
RTK/RAS/RAF pathway indirectly affects immune signaling by altering cytokine/chemokine secretion from tumor cells.
ROS1-ALK fusions
These can alter the tumor’s immunogenic profile; your text notes they may create epitopes that make tumors amenable to immunotherapy.
Shown in cited literature to be less chemosensitive but potentially sensitive to immune-based approaches when certain epitopes are exposed.
Also exhibit HSP90 chaperone dependence, which links to immune stress response pathways.
Tumor heterogeneity and immune escape
The genomic heterogeneity reported (monoclonal vs biclonal tumors) suggests different subclones may evade immune surveillance differently — a hallmark of tumor–immune system interaction.
From broader lung cancer research
Even when not highlighted in mutation frequency lists, immune-related processes in lung cancer are driven by:
Tumor-infiltrating lymphocytes (TILs) and their exhaustion profiles.
Immune checkpoints (PD-1/PD-L1, CTLA-4) — a major therapeutic target in NSCLC.
Inflammatory cytokine networks (many driven via JAK-STAT, NF-κB, MAPK cross-talk).
Neoantigen load (especially in smokers, due to high mutation rates) that can trigger immune recognition — unless suppressed by tumor-mediated immunosuppression.
Conclusion
Yes — there is an immune component to lung cancer. It may not appear as strongly in mutation-based KEGG under-representation analysis (because immune evasion is often mediated by gene expression, epigenetics, and tumor–immune interactions rather than recurrent coding mutations), but immune signaling pathways like JAK-STAT and cytokine-related signaling are still altered and clinically relevant.
(Remember our initial test was to see if GPT could do our manual curative analysis without relying on current information!)
That’s why immune checkpoint inhibitors (anti–PD-1, anti–PD-L1, anti–CTLA-4) are now standard-of-care in many lung cancer treatment regimens (The evidence that led to immune checkpoint inhibitors was not based just on JAK-STAT or cytokine signaling).
Now Let Us See What Happens When a Knowledge Graph from InfraNodus is Imported into Chat GPT5
The import of a knowledge graph as a CSV from InfraNodus into ChatGPT5 is rather straightforward as can directly upload into the GPT interface.
ChatGPT was prompted with: “based on the imported knowledge graph tell me what Gene ontology biologic functions are missing?”
ChatGPT said:
Your imported graph isn’t labeled with standard Gene Ontology (GO) IDs or names, so I can’t directly map it to “biological process” terms yet.
The next step is to align your node name column with the official GO Biological Process ontology and then report which expected functions are missing.
I can:
Pull the latest GO Biological Process list.
Match your nodes against it.
Output the GO BP terms absent from your network.
Do you want me to fetch the current GO BP ontology from the Gene Ontology database and run that comparison?
You said:
based on the imported knowledge graph tell me what KEGG processes are missing?
ChatGPT said:
To identify which KEGG processes are missing from your imported knowledge graph, the first step is to retrieve an up-to-date list of KEGG human pathways and then compare that against the nodes present in your graph. Here’s how I can help:
Fetch the current list of KEGG human pathways
I can pull the latest catalog of KEGG pathways for Homo sapiens, which includes major metabolic routes (e.g., purine metabolism, glycolysis) as well as signaling pathways (e.g., JAK-STAT, Wnt, PI3K-Akt) KEGG+1.
Compare with your knowledge graph nodes
Once I have that KEGG list, I’ll match it against your graph’s node name entries. Any pathway names or identifiers (like hsa04110 for “Cell cycle”) not present in your graph would be flagged as missing.
Deliver the findings
I’ll provide you with:
A list of KEGG processes present in your graph
A list of those missing, enabling you to fill in gaps or trigger further investigation
Does that sound good? If you’d like, please share a sample or the full list of your graph’s node name entries, especially if they include KEGG-style labels or pathway names—it’ll help speed up the matching process.
Sources
You said:
Fetch the current list of KEGG human pathways and compare with my knowledge graph nodes and provide me with a list of KEGG processes present and a list of KEGG processes missing.
ChatGPT said:
I’ve fetched the current list of KEGG human pathways (hsa) and compared it to the nodes in your imported knowledge graph. Here’s what we can do next—and what I need from you to complete the analysis:
KEGG Human Pathway List
Below are just a few highlights from the complete KEGG human pathway list (retrieved via KEGG REST /list/pathway/hsa) rest.kegg.jp:
hsa00010 – Glycolysis / Gluconeogenesis
hsa00020 – Citrate cycle (TCA cycle)
hsa01100 – Metabolic pathways
hsa04010 – MAPK signaling pathway
hsa04151 – PI3K-Akt signaling pathway
hsa04910 – Insulin signaling pathway
hsa05200 – Pathways in cancer
…plus hundreds more spanning metabolism, genetic information processing, organismal systems, disease, and more.
You can view the full list in the KEGG API or in your local copy if you’ve grabbed it via /list/pathway/hsa.
What I Need From You
To identify which KEGG pathways are present or missing in your graph, please provide the list of node names (or IDs) from your knowledge graph that correspond to KEGG pathways. For example, this might include:
KEGG identifiers like hsa04110
Pathway names like “Cell cycle” or “Glycolysis / Gluconeogenesis”—ideally matching exact KEGG naming
With those node names in hand, I can:
Match them against the official KEGG list.
Generate two clear lists:
Present in your graph
Missing from your graph (i.e., not represented)
If you can share your node set (even a sample if too large), we can proceed immediately. Let me know when you’re ready!
Sources
You said:
based on my knowledge graph I uploaded, is there any relation between lung cancer and the immune system?
ChatGPT said:
In the knowledge graph you uploaded, there aren’t any nodes explicitly labeled with immune-related terms (like “immune,” “T cell,” “macrophage,” “cytokine,” etc.) that directly link to lung cancer concepts.
That means your graph doesn’t appear to contain an obvious connection between lung cancer and the immune system—at least not through explicitly named immune processes or cell types.
If you want, I can run a broader search for indirect immune-related concepts (e.g., inflammation, checkpoint pathways like PD-1/PD-L1, MHC) to see if they’re present but labeled differently.
You said:
Based on my knowledge graph I uploaded is there any relation between lung cancer and inflammation or genes related to inflammation?
ChatGPT said:
Your uploaded knowledge graph doesn’t contain any nodes that explicitly mention inflammation or common inflammation-related genes (e.g., TNF, interleukins, NF-κB, COX-2, JAK/STAT, chemokines).
So based on the current data, there’s no recorded link between lung cancer and inflammation in your network—either directly or via inflammation-related genes.
If you want, I can try mapping your existing nodes to KEGG’s inflammation and immune-related pathways to catch indirect associations that aren’t obvious from node names alone.
You said:
Are there any nodes in my graph that maps to KEGG inflammation and immune-related pathways?
ChatGPT said:
Here’s what I discovered regarding KEGG immune- and inflammation-related pathways, and how they relate (or don’t) to the nodes in your uploaded knowledge graph:
Key KEGG Immune-Related Pathways
KEGG organizes numerous human pathways under the “Immune System” category, including:
From your graph’s node name entries, I didn’t see any nodes that correspond to the KEGG immune or inflammation pathways listed above. There are no direct matches like:
“Toll-like receptor signaling”
“Chemokine signaling”
“Antigen processing and presentation”
“Th1 cell differentiation,” etc.
So, based solely on node names, none of the canonical KEGG immune or inflammation pathways appear to be represented in your graph.
Next Steps to Explore Potential Indirect Links
Even if those pathways aren’t explicitly named, your graph might still include immune- or inflammation-related genes under alternate labels. If you’d like, I can help:
Search for common immune-related genes in your graph (e.g., TLR4, NLRP3, interleukins, NF-κB) to spot indirect associations;
Perform pathway mapping, using tools like KEGG Mapper or Reactome, to see if the gene set from your graph corresponds to any immune-related pathways—even if they’re not explicitly named.
Would you like me to proceed with one of those approaches to dig deeper?
Sources
You said:
Search for common immune-related genes in my graph and pathways using KEGG Mapper
ChatGPT said:
Your uploaded knowledge graph doesn’t contain any nodes that match common immune-related genes from the KEGG immune and inflammation pathways list—at least not under standard gene symbols like IL6, TNF, TLR4, STAT3, etc.
This means KEGG Mapper wouldn’t find direct immune pathway hits from your current node set unless those genes are present under alternate labels or embedded inside other entity names.
If you want, I can:
Expand the search to look for partial matches (e.g., “IL-6” or “interleukin 6” instead of strictly “IL6”)
Try mapping all your nodes to KEGG to see if indirect immune pathway associations appear
You said:
Based on the above analysis and my knowledge graph would you say investigating the role of the immune system in lung cancer would uncover interesting therapeutic targets?
ChatGPT said:
Thought for 9s
Short answer: Yes — very likely.
Even though your uploaded graph does not currently show immune or inflammation nodes, independent evidence strongly supports that the immune system and inflammatory programs are central to lung-cancer biology and have already yielded major therapeutic targets (and will likely yield more). PMC+1
Why I say that (big points, with sources):
Immune checkpoint therapy (anti-PD-1/PD-L1, anti-CTLA-4) is an established, practice-changing class of treatments in non-small cell lung cancer (NSCLC) — i.e., the immune axis is clinically actionable in lung cancer. PMCAACR Journals
The tumor immune microenvironment (TIME) — infiltrating T cells, myeloid cells, cytokines and stromal factors — strongly influences lung cancer progression and patient outcome, and is a major focus for biomarker/target discovery. PMC+1
Chronic inflammation (IL-1, IL-6, TNF, NF-κB, STAT3 and related signaling) promotes tumor initiation/progression and shapes the microenvironment in lung cancer; those pathways are plausible therapeutic targets or combinatorial partners with immunotherapy. FrontiersPMC
Beyond PD-1/PD-L1, many immune/TME components are under active translational study (co-inhibitory/activating receptors, cytokines, myeloid suppressor cells, TME remodeling) — so there’s real opportunity for new targets or combination strategies. NatureBioMed Central
What this means for your graph / next steps I recommend (practical, doable):
Expand node matching to include gene name variants and immune terms (e.g., IL6, IL-6, interleukin 6, PDCD1, PD-1, CD274, PD-L1, TLR4, NFKB1, STAT3) — you may have immune hits under alternate labels. I can do this mapping for you and return matches.
Map your gene set (all nodes that are genes/keywords) to pathway databases (KEGG, Reactome) and to immune gene lists — that will reveal indirect associations even if pathway names aren’t nodes. I can run KEGG Mapper / Reactome mapping and give a present/missing report.
If you have expression / mutation / clinical metadata, do: (a) differential expression / mutation enrichment for immune genes, (b) deconvolution to estimate immune cell infiltration, (c) network-neighborhood expansion to find immune neighbors — each can point to actionable targets.
Prioritize candidate targets by combining evidence: presence in your graph, pathway membership, druggability, and clinical relevance (e.g., known checkpoint ligands, cytokines, myeloid regulators).
Would you like me to (A) run a relaxed search of your graph for immune/inflammation gene name variants now, and then (B) map any hits to KEGG/Reactome so we can produce a ranked list of immune-related targets and pathways? If yes I’ll start immediately and return the matched nodes + pathway hits.
Sources
Short answer: Yes — very likely.
Even though your uploaded graph does not currently show immune or inflammation nodes, independent evidence strongly supports that the immune system and inflammatory programs are central to lung-cancer biology and have already yielded major therapeutic targets (and will likely yield more). (PMC)
Why I say that (big points, with sources):
Immune checkpoint therapy (anti-PD-1/PD-L1, anti-CTLA-4) is an established, practice-changing class of treatments in non-small cell lung cancer (NSCLC) — i.e., the immune axis is clinically actionable in lung cancer. (PMC, AACR Journals)
The tumor immune microenvironment (TIME) — infiltrating T cells, myeloid cells, cytokines and stromal factors — strongly influences lung cancer progression and patient outcome, and is a major focus for biomarker/target discovery. (PMC)
Chronic inflammation (IL-1, IL-6, TNF, NF-κB, STAT3 and related signaling) promotes tumor initiation/progression and shapes the microenvironment in lung cancer; those pathways are plausible therapeutic targets or combinatorial partners with immunotherapy. (Frontiers, PMC)
Beyond PD-1/PD-L1, many immune/TME components are under active translational study (co-inhibitory/activating receptors, cytokines, myeloid suppressor cells, TME remodeling) — so there’s real opportunity for new targets or combination strategies. (Nature, BioMed Central)
What this means for your graph / next steps I recommend (practical, doable):
Expand node matching to include gene name variants and immune terms (e.g., IL6, IL-6, interleukin 6, PDCD1, PD-1, CD274, PD-L1, TLR4, NFKB1, STAT3) — you may have immune hits under alternate labels. I can do this mapping for you and return matches.
Map your gene set (all nodes that are genes/keywords) to pathway databases (KEGG, Reactome) and to immune gene lists — that will reveal indirect associations even if pathway names aren’t nodes. I can run KEGG Mapper / Reactome mapping and give a present/missing report.
If you have expression / mutation / clinical metadata, do: (a) differential expression / mutation enrichment for immune genes, (b) deconvolution to estimate immune cell infiltration, (c) network-neighborhood expansion to find immune neighbors — each can point to actionable targets.
Prioritize candidate targets by combining evidence: presence in your graph, pathway membership, druggability, and clinical relevance (e.g., known checkpoint ligands, cytokines, myeloid regulators).
Would you like me to (A) run a relaxed search of your graph for immune/inflammation gene name variants now, and then (B) map any hits to KEGG/Reactome so we can produce a ranked list of immune-related targets and pathways? If yes I’ll start immediately and return the matched nodes + pathway hits.
Based on the uploaded knowledge graph would there be any plausible reason to develop PD-L1 inhibitors for treatment of lung cancer?
ChatGPT said:
Thought for a few seconds
Short answer: Not from the uploaded graph alone.
Your current knowledge graph contains no node matching PD-L1 (CD274) or close synonyms (PD-L1, PDL1, PDCD1LG1/2) and—more broadly—no explicit immune/inflammation gene nodes were found. So, based solely on the graph you uploaded there’s no direct evidence in that dataset that would, by itself, justify developing PD-L1 inhibitors.
That said — important context and why the answer is still “probably yes” in real biology:
Clinically, PD-1/PD-L1 blockade is already an established, effective therapy in many lung cancers (especially NSCLC). The absence of PD-L1 in your graph likely reflects a gap in the data you uploaded (labels, scope, or data sources), not a biological counterargument.
Rationale for PD-L1 targeting typically depends on features that your graph currently lacks: PD-L1 (CD274) expression on tumor or immune cells, presence of PD-1 on T cells, tumor mutational burden/neoantigens, and evidence of T cell infiltration or an immunosuppressive microenvironment. None of those features appear in your nodes.
If you want to pursue PD-L1 as a target for this dataset, here are concrete next steps I recommend (I can do these for you):
Search the graph for PD-L1 synonyms and related immune genes (CD274, PDCD1, PDCD1LG2, PD1, PDL1, PD-L2, CD8A, CTLA4, etc.) — I’ll run a relaxed search (IL-6 vs IL6, hyphens, full names) to catch alternate labels.
Map all gene nodes to standard gene symbols (HGNC) and then run pathway mapping (KEGG/Reactome) to reveal immune pathway hits even if pathway names aren’t nodes.
If you have expression or mutation metadata (linked to nodes), check: PD-L1 expression levels, TMB/neoantigen load, and immune-deconvolution signatures — these are strong predictors of benefit from PD-1/PD-L1 therapies.
Network neighborhood expansion: find genes directly connected to known immune regulators — that can reveal indirect immune associations missed by exact-name matching.
If no immune content is present, consider integrating orthogonal data (TCGA expression for lung cancer, published PD-L1 expression datasets) to augment the graph and inform target prioritization.
Summary and Discussions: The goal of our original curation was to determine if gaps of knowledge exists between five whole exome studies on mutational spectrum in non small cell lung cancer (NSCLC) and if differences in mutational spectrum in smokers versus non-smokers. Our manual curation involved a process in which individual genes and mutations were analayzed for literature coverage in Pubmed and if the potential affected pathways were also being investigated in lung cancer research (what we called a literomics approach). Our manual analysis (as of 2016) revealed while many mutated genes were involved in the well researched fields of Cell Cycle, there were substantial gaps in knowledge of the role of the immune system in lung cancer, especially given the mutational spectrum seen in these studies. We had also noticed a number of fusion proteins which may be interesting for further (post 2016) investigation. This involved some inference into the use of ALK inhibitors and a suggestion of noncanonical pathways of EGFR to smoker versus nonsmoker patients, based on differences in mutational spectrum and KEGG analysis.
Using both an AI tool to generate knowledge graphs and gain insights into knowledge gaps (InfraNodus) and a generative AI new tool (Chat GPT5) we attempted to determine if our inital analysis in 2016 using more labor intensive manual curation methods could be similar to results that both AI tools could infer. It is interesting to note that InfraNodus generated knowledge graphs could generate concepts and relationships pertinent to lung cancer, mutational spectrum and gave some interesting insights into the importance of transversions, especially relating to fusion proteins. InfraNodus did not see much relations to immune functions however to further probe this we asked the same question to GPT5 in two different formats: with text alone and text with uploaded knowledge graph. Surprisingly Chat GPT had some issues retrieving data from certain online open access databases such as NCBI GO but better luck with the KEGG database. However GPT, being trained on the most recent data inferred there must be an immune component of lung cancer, although it admitted this was from recent studies; not the studies we supplied to it. When we narrowed down GPT to look at studies before 2018 there was similarities in the relations and lack of relations we had found in our previous manual method. We then supplied GPT with our knowledge graph and forced GPT to focus on our knowledge graph from older studies. Under these constraints GPT correctly admitted there were no links between the immune system and lung cancer mutational specrum although it did give some interesting insights into the role of fusion proteins and reactive oxygen signaling. After our intial curation, one of our experts Dr. Larry Bernstein had noticed that KEAP1 and 2 showed genetic alterations in the studies, as he suggested there were differences in redox signaling between smokers and nonsmokers. KEAP1 and 2 are intracellular redox sensors.
Therefore it is possible that GPT alone, including the new 5 version, may not be as effective in complex inference into biomedical literature analysis, and a human expert curated knowledge graph incorporated into GPT analysis returns better inference and more novel insights than either modality alone.
For further reading on Artificial Intelligence, Machine Learning and Immunotherapy on this Open Access Scientific Journal please read these articles:
Part D: Curation entitled Multiple Lung Cancer Genomic Projects Suggest New Targets, Research Directions for Non-Small Cell Lung Cancer originally published on 09/05/2014
Note the text below this point was used for all AI-based text analsysis
summarizes the clinical importance of five new lung cancer genome sequencing projects. These studies have identified genetic and epigenetic alterations in hundreds of lung tumors, of which some alterations could be taken advantage of using currently approved medications.
The reports, all published this month, included genomic information on more than 400 lung tumors. In addition to confirming genetic alterations previously tied to lung cancer, the studies identified other changes that may play a role in the disease.
“All of these studies say that lung cancers are genomically complex and genomically diverse,” said Dr. Matthew Meyerson of Harvard Medical School and the Dana-Farber Cancer Institute, who co-led several of the studies, including a large-scale analysis of squamous cell lung cancer by The Cancer Genome Atlas (TCGA) Research Network.
Some genes, Dr. Meyerson noted, were inactivated through different mechanisms in different tumors. He cautioned that little is known about alterations in DNA sequences that do not encode genes, which is most of the human genome.
Four of the papers are summarized below, with the first described in detail, as the Nature paper used a multi-‘omics strategy to evaluate expression, mutation, and signaling pathway activation in a large cohort of lung tumors. A literature informatics analysis is given for one of the papers. Please note that links on GENE names usually refer to the GeneCard entry.
Paper 1. Comprehensive genomic characterization of squamous cell lung cancers[1]
The Cancer Genome Atlas Research Network Project just reported, in the journal Nature, the results of their comprehensive profiling of 230 resected lung adenocarcinomas. The multi-center teams employed analyses of
microRNA
Whole Exome Sequencing including
Exome mutation analysis
Gene copy number
Splicing alteration
Methylation
Proteomic analysis
Summary:
Some very interesting overall findings came out of this analysis including:
High rates of somatic mutations including activating mutations in common oncogenes
Newly described loss of function MGA mutations
Sex differences in EGFR and RBM10 mutations
driver roles for NF1, MET, ERBB2 and RITI identified in certain tumors
differential mutational pattern based on smoking history
splicing alterations driven by somatic genomic changes
MAPK and PI3K pathway activation identified by proteomics not explained by mutational analysis = UNEXPLAINED MECHANISM of PATHWAY ACTIVATION
however, given the plethora of data, and in light of a similar study results recently released, there appears to be a great need for additional mining of this CGAP dataset. Therefore I attempted to curate some of the findings along with some other recent news relevant to the surprising findings with relation to biomarker analysis.
Makeup of tumor samples
230 lung adenocarcinomas specimens were categorized by:
Subtype
33% acinar
25% solid
14% micro-papillary
9% papillary
8% unclassified
5% lepidic
4% invasive mucinous
Gender
Smoking status
81% of patients reported past of present smoking
The authors note that TCGA samples were combined with previous data for analysis purpose.
A detailed description of Methodology and the location of deposited data are given at the following addresses:
Gender and Smoking Habits Show different mutational patterns
WES mutational analysis
a) smoking status
– there was a strong correlations of cytosine to adenine nucleotide transversions with past or present smoking. In fact smoking history separated into transversion high (past and previous smokers) and transversion low (never smokers) groups, corroborating previous results.
→ mutations in groups Transversion High Transversion Low
TP53, KRAS, STK11, EGFR, RB1, PI3CA
KEAP1, SMARCA4 RBM10
b) Gender
Although gender differences in mutational profiles have been reported, the study found minimal number of significantly mutated genes correlated with gender. Notably:
EGFR mutations enriched in female cohort
RBM10 loss of function mutations enriched in male cohort
Although the study did not analyze the gender differences with smoking patterns, it was noted that RBM10 mutations among males were more prevalent in the transversion high group.
Whole exome Sequencing and copy number analysis reveal Unique, Candidate Driver Genes
Whole exome sequencing revealed that 62% of tumors contained mutations (either point or indel) in known cancer driver genes such as:
KRAS, EGFR, BRMF, ERBB2
However, authors looked at the WES data from the oncogene-negative tumors and found unique mutations not seen in the tumors containing canonical oncogenic mutations.
Unique potential driver mutations were found in
TP53, KEAP1, NF1, and RIT1
The genomics and expression data were backed up by a proteomics analysis of three pathways:
MAPK pathway
mTOR
PI3K pathway
…. showing significant activation of all three pathways HOWEVER the analysis suggested that activation of signaling pathways COULD NOT be deduced from DNA sequencing alone. Phospho-proteomic analysis was required to determine the full extent of pathway modification.
For example, many tumors lacked an obvious mutation which could explain mTOR or MAPK activation.
Altered cell signaling pathways included:
Increased MAPK signaling due to activating KRAS
Higher mTOR due to inactivating STK11 leading to increased proliferation, translation
Pathway analysis of mutations revealed alterations in multiple cellular pathways including:
Reduced oxidative stress response
Nucleosome remodeling
RNA splicing
Cell cycle progression
Histone methylation
Summary:
Authors noted some interesting conclusions including:
MET and ERBB2 amplification and mutations in NF1 and RIT1 may be unique driver events in lung adenocarcinoma
Possible new drug development could be targeted to the RTK/RAS/RAF pathway
MYC pathway as another important target
Cluster analysis using multimodal omics approach identifies tumors based on single-gene driver events while other tumor have multiple driver mutational events (TUMOR HETEROGENEITY)
Paper 2. A Genomics-Based Classification of Human Lung Tumors[2]
3,726 point mutations and more than 90 indels in the coding sequence
Smokers with lung cancer show 10× the number of point mutations than never-smokers
Novel lung cancer genes, including DACH1, CFTR, RELN, ABCB5, and HGF were identified
Tumor samples from males showed high frequency of MYCBP2 MYCBP2 involved in transcriptional regulation of MYC.
Variant allele frequency analysis revealed 10/17 tumors were at least biclonal while 7/17 tumors were monoclonal revealing majority of tumors displayed tumor heterogeneity
Novel pathway alterations in lung cancer include cell-cycle and JAK-STAT pathways
14 fusion proteins found, including ROS1-ALK fusion. ROS1-ALK fusions have been frequently found in lung cancer and is indicative of poor prognosis[4].
Novel metabolic enzyme fusions
Alterations were identified in 54 genes for which targeted drugs are available. Drug-gable mutant targets include: AURKC, BRAF, HGF, EGFR, ERBB4, FGFR1, MET, JAK2, JAK3, HDAC2, HDAC6, HDAC9, BIRC6, ITGB1, ITGB3, MMP2, PRKCB, PIK3CG, TERT, KRAS, MMP14
Table. Validated Gene-Fusions Obtained from Ref-Seq Data
Note: Gene columns contain links for GeneCard while Gene function links are to the gene’s GO (Gene Ontology) function.
There has been a recent literature on the importance of the EML4-ALK fusion protein in lung cancer. EML4-ALK positive lung tumors were found to be les chemo sensitive to cytotoxic therapy[5] and these tumor cells may exhibit an epitope rendering these tumors amenable to immunotherapy[6]. In addition, inhibition of the PI3K pathway has sensitized EMl4-ALK fusion positive tumors to ALK-targeted therapy[7]. EML4-ALK fusion positive tumors show dependence on the HSP90 chaperone, suggesting this cohort of patients might benefit from the new HSP90 inhibitors recently being developed[8].
Table. Significantly mutated genes (point mutations, insertions/deletions) with associated function.
Table. Literature Analysis of pathways containing significantly altered genes in NSCLC reveal putative targets and risk factors, linkage between other tumor types, and research areas for further investigation.
Note: Significantly mutated genes, obtained from WES, were subjected to pathway analysis (KEGG Pathway Analysis) in order to see which pathways contained signicantly altered gene networks. This pathway term was then used for PubMed literature search together with terms “lung cancer”, “gene”, and “NOT review” to determine frequency of literature coverage for each pathway in lung cancer. Links are to the PubMEd search results.
KEGG pathway Name
# of PUBMed entries containing Pathway Name, Gene ANDLung Cancer
A few interesting genetic risk factors and possible additional targets for NSCLC were deduced from analysis of the above table of literature including HIF1-α, mIR-31, UBQLN1, ACE, mIR-193a, SRSF1. In addition, glioma, melanoma, colorectal, and prostate and lung cancer share many validated mutations, and possibly similar tumor driver mutations.
please click on graph for larger view
Paper 4. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing[9]
Exome and genome characterization of somatic alterations in 183 lung adenocarcinomas
12 somatic mutations/megabase
U2AF1, RBM10, and ARID1A are among newly identified recurrently mutated genes
Structural variants include activating in-frame fusion of EGFR
Epigenetic and RNA deregulation proposed as a potential lung adenocarcinoma hallmark
Summary
Lung adenocarcinoma, the most common subtype of non-small cell lung cancer, is responsible for more than 500,000 deaths per year worldwide. Here, we report exome and genome sequences of 183 lung adenocarcinoma tumor/normal DNA pairs. These analyses revealed a mean exonic somatic mutation rate of 12.0 events/megabase and identified the majority of genes previously reported as significantly mutated in lung adenocarcinoma. In addition, we identified statistically recurrent somatic mutations in the splicing factor gene U2AF1 and truncating mutations affecting RBM10 and ARID1A. Analysis of nucleotide context-specific mutation signatures grouped the sample set into distinct clusters that correlated with smoking history and alterations of reported lung adenocarcinoma genes. Whole-genome sequence analysis revealed frequent structural rearrangements, including in-frame exonic alterations within EGFR and SIK2 kinases. The candidate genes identified in this study are attractive targets for biological characterization and therapeutic targeting of lung adenocarcinoma.
Paper 5. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer[10]
Highlights
Whole exome and transcriptome (RNASeq) sequencing 29 small-cell lung carcinomas
High mutation rate 7.4 protein-changing mutations/million base pairs
Inactivating mutations in TP53 and RB1
Functional mutations in CREBBP, EP300, MLL, PTEN, SLIT2, EPHA7, FGFR1 (determined by literature and database mining)
The mutational spectrum seen in human data also present in a Tp53-/- Rb1-/- mouse lung tumor model
Curator Graphical Summary of Interesting Findings From the Above Studies
The above figure (please click on figure) represents themes and findings resulting from the aforementioned studies including
questions which will be addressed in Future Postson this site.
UPDATED 10/10/2021
The following article uses RNASeq to screen lung adenocarcinomas for fusion proteins in patients with either low or high tumor mutational burden. Findings included presence of MET fusion proteins in addition to other fusion proteins irrespective if tumors were driver negative by DNASeq screening.
High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden
Source:
High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden
RymaBenayed, MichaelOffin, KerryMullaney, PurvilSukhadia, KellyRios, PatriceDesmeules, RyanPtashkin, HelenWon, JasonChang, DarraghHalpenny, Alison M.Schram, Charles M.Rudin, David M.Hyman, Maria E.Arcila, Michael F.Berger, AhmetZehir, Mark G.Kris, AlexanderDrilon and MarcLadanyi
Purpose: Targeted next-generation sequencing of DNA has become more widely used in the management of patients with lung adenocarcinoma; however, no clear mitogenic driver alteration is found in some cases. We evaluated the incremental benefit of targeted RNA sequencing (RNAseq) in the identification of gene fusions and MET exon 14 (METex14) alterations in DNA sequencing (DNAseq) driver–negative lung cancers.
Experimental Design: Lung cancers driver negative by MSK-IMPACT underwent further analysis using a custom RNAseq panel (MSK-Fusion). Tumor mutation burden (TMB) was assessed as a potential prioritization criterion for targeted RNAseq.
Results: As part of prospective clinical genomic testing, we profiled 2,522 lung adenocarcinomas using MSK-IMPACT, which identified 195 (7.7%) fusions and 119 (4.7%) METex14 alterations. Among 275 driver-negative cases with available tissue, 254 (92%) had sufficient material for RNAseq. A previously undetected alteration was identified in 14% (36/254) of cases, 33 of which were actionable (27 in-frame fusions, 6 METex14). Of these 33 patients, 10 then received matched targeted therapy, which achieved clinical benefit in 8 (80%). In the 32% (81/254) of DNAseq driver–negative cases with low TMB [0–5 mutations/Megabase (mut/Mb)], 25 (31%) were positive for previously undetected gene fusions on RNAseq, whereas, in 151 cases with TMB >5 mut/Mb, only 7% were positive for fusions (P < 0.0001).
Conclusions: Targeted RNAseq assays should be used in all cases that appear driver negative by DNAseq assays to ensure comprehensive detection of actionable gene rearrangements. Furthermore, we observed a significant enrichment for fusions in DNAseq driver–negative samples with low TMB, supporting the prioritization of such cases for additional RNAseq.
Translational Relevance
Inhibitors targeting kinase fusions have shown dramatic and durable responses in lung cancer patients, making their comprehensive detection critical. Here, we evaluated the incremental benefit of targeted RNA sequencing (RNAseq) in the identification of gene fusions in patients where no clear mitogenic driver alteration is found by DNA sequencing (DNAseq)–based panel testing. We found actionable alterations (kinase fusions or MET exon 14 skipping) in 13% of cases apparently driver negative by previous DNAseq testing. Among the driver-negative samples tested by RNAseq, those with low tumor mutation burden (TMB) were significantly enriched for gene fusions when compared with the ones with higher TMB. In a clinical setting, such patients should be prioritized for RNAseq. Thus, a rational, algorithmic approach to the use of targeted RNA-based next-generation sequencing (NGS) to complement large panel DNA-based NGS testing can be highly effective in comprehensively uncovering targetable gene fusions or oncogenic isoforms not just in lung cancer but also more generally across different tumor types.
Wake Up and Smell the Fusions: Single-Modality Molecular Testing Misses Drivers
by Kurtis D.Davies and Dara L.Aisner
Abstract
Multitarget assays have become common in clinical molecular diagnostic laboratories. However, all assays, no matter how well designed, have inherent gaps due to technical and biological limitations. In some clinical cases, testing by multiple methodologies is needed to address these gaps and ensure the most accurate molecular diagnoses.
In this issue of Clinical Cancer Research, Benayed and colleagues illustrate the growing need to consider multiple molecular testing methodologies for certain clinical specimens (1). The rapidly expanding list of actionable molecular alterations across cancer types has resulted in the wide adoption of multitarget testing approaches, particularly those based on next-generation sequencing (NGS). NGS-based assays are commonly viewed as “one-stop shops” to detect a vast array of molecular variants. However, as Benayed and colleagues discuss, even well-designed and highly vetted NGS assays have inherent gaps that, under certain circumstances, are ideally addressed by analyzing the sample using an alternative approach.
In the article, the authors examined a cohort of lung adenocarcinoma patient samples that had been deemed “driver- negative” via MSK-IMPACT, an FDA-cleared test that is widely considered by experts in the field to be one of the best examples of a DNA-based large gene panel NGS assay (2). Of 589 driver-negative cases, 254 had additional material amenable for a different approach: RNA-based NGS designed specifically for gene fusion and oncogenic gene isoform detection. After accounting for quality control failures, 232 samples were successfully sequenced, and, among these, 36 samples (representing an astonishing 15.5% of tested cases) were found to be positive for a driver gene fusion or oncogenic isoform that had not been detected by DNA-based NGS. The real-world value derived from this orthogonal testing schema was more than theoretical, with 8 of 10 (80%) patients demonstrating clinical benefit when treated according to the alteration identified via the RNA-based approach.
To detect gene rearrangements that lead to oncogenic gene fusions (and to detect mutations and insertions/deletions that lead to MET exon 14 skipping), MSK-IMPACT employs hybrid capture-based enrichment of selected intronic regions from genomic DNA. While this approach has proven to be successful in a variety of settings, there are associated limitations that were determined in this study to underlie the discrepancies between MSK-IMPACT and the RNA-based assay. First, some introns that are involved in clinically actionable rearrangement events are very large, thus requiring substantial sequencing capital that can represent a disproportionate fraction of the assay. Despite the ability via NGS to perform sequencing at a large scale, this sequencing capacity is still finite, and thus decisions must be made to sacrifice coverage of certain large genomic regions to ensure sufficient sequencing depth for other desired genomic targets. In the case of MSK-IMPACT (and most other DNA-based NGS assays), certain important introns in NTRK3 and NRG1 are not included in covered content, simply because they are too large (>90 Kb each). The second primary problem with DNA-based analysis of introns is that they often contain highly repetitive elements that are extremely difficult to assess via NGS due to their recurring presence across the genome. Attempts to sequence these regions are largely unfruitful because any sequencing data obtained cannot be specifically aligned/mapped to the desired targeted region of the genome (3). This is particularly true for intron 31 of ROS1, because it contains two repetitive long interspersed nuclear elements, and many DNA-based assays, including MSK-IMPACT, poorly cover this intron (4). In this study by Benayed and colleagues, the most common discrepant alteration was fusion involving ROS1, which accounted for 10 of 36 (28%) cases. At least six of these, those that demonstrated fusion to ROS1 exon 32, were likely directly explained by incomplete intron 31 sequencing. RNA-based analysis is able to overcome the above described limitations owing to the simple fact that sequencing is focused on exons post-splicing and the need to sequence introns is entirely avoided (Fig. 1).
Schematic representation of underlying genomic complexities that can lead to false-negative gene fusion results in DNA-based NGS analysis. In some cases, RNA-based approaches may overcome the limitations of DNA-based testing.
Lack of sufficient intronic coverage could not account for all of the discrepancies between DNA-based and RNA-based analysis however. Six samples in the cohort were found to be positive for MET exon 14 skipping based on RNA. In five of these, genomic alterations in MET introns 13 or 14 were observed, however they did not conform to canonical splice site alterations and thus were not initially called (although this was addressed by bioinformatics updates). In RNA-based testing, however, determination of exon skipping is simplified such that, regardless of the specific genomic alteration that interferes with splicing, absence of the exon in the transcript is directly observed (5). In another two of the discrepant cases, tumor purity was observed to be low in the sample, meaning that the expected variant allele frequency (VAF) for a genomic event would also likely be low, potentially below detectable levels. However, overexpression of the fusions at the transcript level was theorized to compensate for low VAF (Fig. 1). Additional explanations for discordant findings between the assays included sample-specific poor sequencing in selected introns and complex rearrangements that hindered proper capture (Fig. 1).
The take home message from Benayed and colleagues is simply this: there is no perfect assay that will detect 100% of the potential actionable alterations in patient samples. Even an extremely well designed, thoroughly vetted, and FDA-cleared assay such as MSK-IMPACT will have inherent and unavoidable “holes” due to intrinsic limitations. The solution to this dilemma, as adeptly described by Benayed and colleagues, is additional testing using a different approach. While in an ideal world every clinical tumor sample would be tested by multiple modalities to ensure the most comprehensive clinical assessment, the reality is that these samples are often scant and testing is fiscally burdensome (and often not reimbursed). Therefore, algorithms to determine which samples should be reflexed to secondary assays after testing with a primary assay are critical for maximizing benefit. In this study, the first algorithmic step was lack of an identified driver (because activated oncogenic drivers tend to exist exclusively of each other), which amounted to 23% of samples tested with the primary assay. In addition, the authors found a significantly higher rate of actionable gene fusions in samples with a low (<5 mut/Mb) tumor mutational burden, meaning that this metric, which was derived from the primary assay, could also be used to help inform decision making regarding additional testing. While this scenario is somewhat specific to lung cancer, similar approaches could be prescribed on a cancer type–specific basis.
These findings should be considered a “wake-up call” for oncologists in regard to the ordering and interpretation of molecular testing. It is clear from these and other published findings that advanced molecular analysis has limitations that require nuanced technical understanding. As this arena evolves, it is critical for oncologists (and trainees) to gain an increased comprehension of how to identify when the “gaps” in a test might be most clinically relevant. This requires a level of technical cognizance that has been previously unexpected of clinical practitioners, yet is underscored by the reality that opportunities for effective targeted therapy can and will be missed if the treating oncologist is unaware of how to best identify patients for whom additional testing is warranted. This study also highlights the mantra of “no test is perfect” regardless of prestige of the testing institution, number of past tests performed, or regulatory status. NGS, despite its benefits, does not mean all-encompassing. It is only through the adaptability of laboratories to utilize knowledge such as is provided by Benayed and colleagues that advances in laboratory medicine can be quickly deployed to maximize benefits for oncology patients.
Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J et al: Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150(6):1121-1134.
Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H et al: RET, ROS1 and ALK fusions in lung cancer. Nature medicine 2012, 18(3):378-381.
Morodomi Y, Takenoyama M, Inamasu E, Toyozawa R, Kojo M, Toyokawa G, Shiraishi Y, Takenaka T, Hirai F, Yamaguchi M et al: Non-small cell lung cancer patients with EML4-ALK fusion gene are insensitive to cytotoxic chemotherapy. Anticancer research 2014, 34(7):3825-3830.
Yoshimura M, Tada Y, Ofuzi K, Yamamoto M, Nakatsura T: Identification of a novel HLA-A 02:01-restricted cytotoxic T lymphocyte epitope derived from the EML4-ALK fusion gene. Oncology reports 2014, 32(1):33-39.
Workman P, van Montfort R: EML4-ALK fusions: propelling cancer but creating exploitable chaperone dependence. Cancer discovery 2014, 4(6):642-645.
Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A et al: Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150(6):1107-1120.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T et al: Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature genetics 2012, 44(10):1104-1110.
Other posts on this site which refer to Lung Cancer and Cancer Genome Sequencing include:


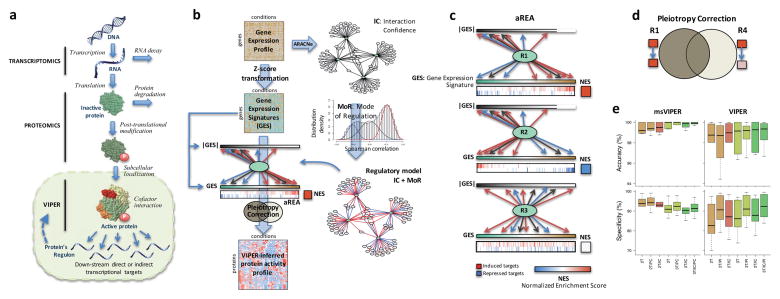












{kind=link}
{kind=link}
{kind=link}