Systems Biology analysis of Transcription Networks, Artificial Intelligence, and High-End Computing Coming to Fruition in Personalized Oncology
Curator: Stephen J. Williams, Ph.D.
In the June 2020 issue of the journal Science, writer Roxanne Khamsi has an interesting article “Computing Cancer’s Weak Spots; An algorithm to unmask tumors’ molecular linchpins is tested in patients”[1], describing some early successes in the incorporation of cancer genome sequencing in conjunction with artificial intelligence algorithms toward a personalized clinical treatment decision for various tumor types. In 2016, oncologists Amy Tiersten collaborated with systems biologist Andrea Califano and cell biologist Jose Silva at Mount Sinai Hospital to develop a systems biology approach to determine that the drug ruxolitinib, a STAT3 inhibitor, would be effective for one of her patient’s aggressively recurring, Herceptin-resistant breast tumor. Dr. Califano, instead of defining networks of driver mutations, focused on identifying a few transcription factors that act as ‘linchpins’ or master controllers of transcriptional networks withing tumor cells, and in doing so hoping to, in essence, ‘bottleneck’ the transcriptional machinery of potential oncogenic products. As Dr. Castilano states
“targeting those master regulators and you will stop cancer in its tracks, no matter what mutation initially caused it.”
It is important to note that this approach also relies on the ability to sequence tumors by RNA-seq to determine the underlying mutations which alter which master regulators are pertinent in any one tumor. And given the wide tumor heterogeneity in tumor samples, this sequencing effort may have to involve multiple biopsies (as discussed in earlier posts on tumor heterogeneity in renal cancer).
As stated in the article, Califano co-founded a company called Darwin-Health in 2015 to guide doctors by identifying the key transcription factors in a patient’s tumor and suggesting personalized therapeutics to those identified molecular targets (OncoTarget™). He had collaborated with the Jackson Laboratory and most recently Columbia University to conduct a $15 million 3000 patient clinical trial. This was a bit of a stretch from his initial training as a physicist and, in 1986, IBM hired him for some artificial intelligence projects. He then landed in 2003 at Columbia and has been working on identifying these transcriptional nodes that govern cancer survival and tumorigenicity. Dr. Califano had figured that the number of genetic mutations which potentially could be drivers were too vast:
A 2018 study which analyzed more than 9000 tumor samples reported over 1.5 million mutations[2]
and impossible to develop therapeutics against. He reasoned that you would just have to identify the common connections between these pathways or transcriptional nodes and termed them master regulators.
A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples
Chen H, Li C, Peng X, et al. Cell. 2018;173(2):386-399.e12.
Abstract
The role of enhancers, a key class of non-coding regulatory DNA elements, in cancer development has increasingly been appreciated. Here, we present the detection and characterization of a large number of expressed enhancers in a genome-wide analysis of 8928 tumor samples across 33 cancer types using TCGA RNA-seq data. Compared with matched normal tissues, global enhancer activation was observed in most cancers. Across cancer types, global enhancer activity was positively associated with aneuploidy, but not mutation load, suggesting a hypothesis centered on “chromatin-state” to explain their interplay. Integrating eQTL, mRNA co-expression, and Hi-C data analysis, we developed a computational method to infer causal enhancer-gene interactions, revealing enhancers of clinically actionable genes. Having identified an enhancer ∼140 kb downstream of PD-L1, a major immunotherapy target, we validated it experimentally. This study provides a systematic view of enhancer activity in diverse tumor contexts and suggests the clinical implications of enhancers.
A diagram of how concentrating on these transcriptional linchpins or nodes may be more therapeutically advantageous as only one pharmacologic agent is needed versus multiple agents to inhibit the various upstream pathways:

From: Khamsi R: Computing cancer’s weak spots. Science 2020, 368(6496):1174-1177.
VIPER Algorithm (Virtual Inference of Protein activity by Enriched Regulon Analysis)
The algorithm that Califano and DarwinHealth developed is a systems biology approach using a tumor’s RNASeq data to determine controlling nodes of transcription. They have recently used the VIPER algorithm to look at RNA-Seq data from more than 10,000 tumor samples from TCGA and identified 407 transcription factor genes that acted as these linchpins across all tumor types. Only 20 to 25 of them were implicated in just one tumor type so these potential nodes are common in many forms of cancer.
Other institutions like the Cold Spring Harbor Laboratories have been using VIPER in their patient tumor analysis. Linchpins for other tumor types have been found. For instance, VIPER identified transcription factors IKZF1 and IKF3 as linchpins in multiple myeloma. But currently approved therapeutics are hard to come by for targets with are transcription factors, as most pharma has concentrated on inhibiting an easier target like kinases and their associated activity. In general, developing transcription factor inhibitors in more difficult an undertaking for multiple reasons.
Network-based inference of protein activity helps functionalize the genetic landscape of cancer. Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A:. Nature genetics 2016, 48(8):838-847 [3]
Abstract
Identifying the multiple dysregulated oncoproteins that contribute to tumorigenesis in a given patient is crucial for developing personalized treatment plans. However, accurate inference of aberrant protein activity in biological samples is still challenging as genetic alterations are only partially predictive and direct measurements of protein activity are generally not feasible. To address this problem we introduce and experimentally validate a new algorithm, VIPER (Virtual Inference of Protein-activity by Enriched Regulon analysis), for the accurate assessment of protein activity from gene expression data. We use VIPER to evaluate the functional relevance of genetic alterations in regulatory proteins across all TCGA samples. In addition to accurately inferring aberrant protein activity induced by established mutations, we also identify a significant fraction of tumors with aberrant activity of druggable oncoproteins—despite a lack of mutations, and vice-versa. In vitro assays confirmed that VIPER-inferred protein activity outperforms mutational analysis in predicting sensitivity to targeted inhibitors.
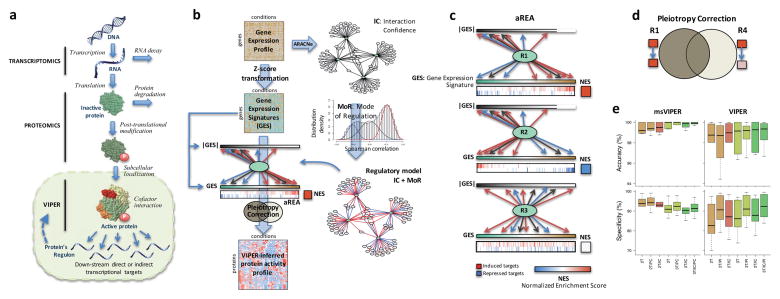
Schematic overview of the VIPER algorithm From: Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A: Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature genetics 2016, 48(8):838-847.
(a) Molecular layers profiled by different technologies. Transcriptomics measures steady-state mRNA levels; Proteomics quantifies protein levels, including some defined post-translational isoforms; VIPER infers protein activity based on the protein’s regulon, reflecting the abundance of the active protein isoform, including post-translational modifications, proper subcellular localization and interaction with co-factors. (b) Representation of VIPER workflow. A regulatory model is generated from ARACNe-inferred context-specific interactome and Mode of Regulation computed from the correlation between regulator and target genes. Single-sample gene expression signatures are computed from genome-wide expression data, and transformed into regulatory protein activity profiles by the aREA algorithm. (c) Three possible scenarios for the aREA analysis, including increased, decreased or no change in protein activity. The gene expression signature and its absolute value (|GES|) are indicated by color scale bars, induced and repressed target genes according to the regulatory model are indicated by blue and red vertical lines. (d) Pleiotropy Correction is performed by evaluating whether the enrichment of a given regulon (R4) is driven by genes co-regulated by a second regulator (R4∩R1). (e) Benchmark results for VIPER analysis based on multiple-samples gene expression signatures (msVIPER) and single-sample gene expression signatures (VIPER). Boxplots show the accuracy (relative rank for the silenced protein), and the specificity (fraction of proteins inferred as differentially active at p < 0.05) for the 6 benchmark experiments (see Table 2). Different colors indicate different implementations of the aREA algorithm, including 2-tail (2T) and 3-tail (3T), Interaction Confidence (IC) and Pleiotropy Correction (PC).
Other articles from Andrea Califano on VIPER algorithm in cancer include:
Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, Cai S, Tu Y, McCoy A, Peoples M, Sun Y, Qiu H, Chang Q, Bristow C, Carugo A, Shao J, Ma X, Harris A, Mundi P, Lau R, Ramamoorthy V, Wu Y, Alvarez MJ, Califano A, Moulder SL, Symmans WF, Marszalek JR, Heffernan TP, Chang JT, Piwnica-Worms H.Sci Transl Med. 2019 Apr 17;11(488):eaav0936. doi: 10.1126/scitranslmed.aav0936.PMID: 30996079
Walsh LA, Alvarez MJ, Sabio EY, Reyngold M, Makarov V, Mukherjee S, Lee KW, Desrichard A, Turcan Ş, Dalin MG, Rajasekhar VK, Chen S, Vahdat LT, Califano A, Chan TA.Cell Rep. 2017 Aug 15;20(7):1623-1640. doi: 10.1016/j.celrep.2017.07.052.PMID: 28813674
Rodriguez-Barrueco R, Yu J, Saucedo-Cuevas LP, Olivan M, Llobet-Navas D, Putcha P, Castro V, Murga-Penas EM, Collazo-Lorduy A, Castillo-Martin M, Alvarez M, Cordon-Cardo C, Kalinsky K, Maurer M, Califano A, Silva JM.Genes Dev. 2015 Aug 1;29(15):1631-48. doi: 10.1101/gad.262642.115. Epub 2015 Jul 30.PMID: 26227964
Master regulators used as breast cancer metastasis classifier.
Lim WK, Lyashenko E, Califano A.Pac Symp Biocomput. 2009:504-15.PMID: 19209726 Free
Additional References
- Khamsi R: Computing cancer’s weak spots. Science 2020, 368(6496):1174-1177.
- Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Liang H: A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173(2):386-399 e312.
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A: Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature genetics 2016, 48(8):838-847.
Other articles of Note on this Open Access Online Journal Include: