Biology, Physiology and Pathophysiology of Heat Shock Proteins
Curation: Larry H. Bernstein, MD, FCAP
Heat Shock Proteins (HSP)
-
Exploring the association of molecular chaperones, heat shock proteins, and the heat shock response in physiological/pathological processes
Hsp70 chaperones: Cellular functions and molecular mechanism
M. P. Mayer, B. Bukau
Cell and Molec Life Sci Mar 2005; 62:670 http://dx.doi.org:/10.1007/s00018-004-4464-6
Hsp70 proteins are central components of the cellular network of molecular chaperones and folding catalysts. They assist a large variety of protein folding processes in the cell by transient association of their substrate binding domain with short hydrophobic peptide segments within their substrate proteins. The substrate binding and release cycle is driven by the switching of Hsp70 between the low-affinity ATP bound state and the high-affinity ADP bound state. Thus, ATP binding and hydrolysis are essential in vitro and in vivo for the chaperone activity of Hsp70 proteins. This ATPase cycle is controlled by co-chaperones of the family of J-domain proteins, which target Hsp70s to their substrates, and by nucleotide exchange factors, which determine the lifetime of the Hsp70-substrate complex. Additional co-chaperones fine-tune this chaperone cycle. For specific tasks the Hsp70 cycle is coupled to the action of other chaperones, such as Hsp90 and Hsp100.
70-kDa heat shock proteins (Hsp70s) assist a wide range of folding processes, including the folding and assembly of newly synthesized proteins, refolding of misfolded and aggregated proteins, membrane translocation of organellar and secretory proteins, and control of the activity of regulatory proteins [1–7]. Hsp70s have thus housekeeping functions in the cell in which they are built-in components of folding and signal transduction pathways, and quality control functions in which they proofread the structure of proteins and repair misfolded conformers. All of these activities appear to be based on the property of Hsp70 to interact with hydrophobic peptide segments of proteins in an ATP-controlled fashion. The broad spectrum of cellular functions of Hsp70 proteins is achieved through
- the amplification and diversification of hsp70genes in evolution, which has generated specialized Hsp70 chaperones,
- co-chaperones which are selectively recruited by Hsp70 chaperones to fulfill specific cellular functions and
- cooperation of Hsp70s with other chaperone systems to broaden their activity spectrum. Hsp70 proteins with their co-chaperones and cooperating chaperones thus constitute a complex network of folding machines.
Protein folding processes assisted by Hsp70
The role of Hsp70s in the folding of non-native proteins can be divided into three related activities: prevention of aggregation, promotion of folding to the native state, and solubilization and refolding of aggregated proteins. In the cellular milieu, Hsp70s exert these activities in the quality control of misfolded proteins and the co- and posttranslational folding of newly synthesized proteins. Mechanistically related but less understood is the role of Hsp70s in the disassembly of protein complexes such as clathrin coats, viral capsids and the nucleoprotein complex, which initiates the replication of bacteriophage λ DNA. A more complex folding situation exists for the Hsp70-dependent control of regulatory proteins since several steps in the folding and activation process of these substrates are assisted by multiple chaperones.
Hsp70 proteins together with their co-chaperones of the J-domain protein (JDP) family prevent the aggregation of non-native proteins through association with hydrophobic patches of substrate molecules, which shields them from intermolecular interactions (‘holder’ activity). Some JDPs such as Escherichia coli DnaJ and Saccharomyces cerevisiae Ydj1 can prevent aggregation by themselves through ATP-independent transient and rapid association with the substrates. Only members of the Hsp70 family with general chaperone functions have such general holder activity.
Hsp70 chaperone systems assist non-native folding intermediates to fold to the native state (‘folder’ activity). The mechanism by which Hsp70-chaperones assist the folding of non-native substrates is still unclear. Hsp70-dependent protein folding in vitro occurs typically on the time scale of minutes or longer. Substrates cycle between chaperone-bound and free states until the ensemble of molecules has reached the native state. There are at least two alternative modes of action. In the first mechanism Hsp70s play a rather passive role. Through repetitive substrate binding and release cycles they keep the free concentration of the substrate sufficiently low to prevent aggregation, while allowing free molecules to fold to the native state (‘kinetic partitioning’). In the second mechanism, the binding and release cycles induce local unfolding in the substrate, e.g. the untangling of a misfolded β-sheet, which helps to overcome kinetic barriers for folding to the native state (‘local unfolding’) [8–11]. The energy of ATP may be used to induce such conformational changes or alternatively to drive the ATPase cycle in the right direction.
Hsp70 in cellular physiology and pathophysiology
Two Hsp70 functions are especially interesting, de novo folding of nascent polypeptides and interaction with signal transduction proteins, and therefore some aspects of these functions shall be discussed below in more detail. Hsp70 chaperones were estimated to assist the de novo folding of 10–20% of all bacterial proteins whereby the dependence on Hsp70 for efficient folding correlated with the size of the protein [1, 2]. Since the average protein size in eukaryotic cells is increased (52 kDa in humans) as compared to bacteria (35 kDa in E. coli) [25], it is to be expected that an even larger percentage of eukaryotic proteins will be in need of Hsp70 during de novo folding. This reliance on Hsp70 chaperones increases even more under stress conditions. Interestingly, mutated proteins [for example mutant p53, cystis fibrosis transmembrane regulator (CFTR) variant ΔF508, mutant superoxid dismutase (SOD) 1] seem to require more attention by the Hsp70 chaperones than the corresponding wild-type protein [26–29]. As a consequence of this interaction the function of the mutant protein can be preserved. Thereby Hsp70 functions as a capacitor, buffering destabilizing mutations [30], a function demonstrated earlier for Hsp90 [31, 32]. Such mutations are only uncovered when the overall need for Hsp70 action exceeds the chaperone capacity of the Hsp70 proteins, for example during stress conditions [30], at certain stages in development or during aging, when the magnitude of stress-induced increase in Hsp70 levels declines [33, 34]. Alternatively, the mutant protein can be targeted by Hsp70 and its co-chaperones to degradation as shown e.g. for CFTRΔF508 and some of the SOD1 mutant proteins [35,36]. Deleterious mutant proteins may then only accumulate when Hsp70 proteins are overwhelmed by other, stress-denatured proteins. Both mechanisms may contribute to pathological processes such as oncogenesis (mutant p53) and neurodegenerative diseases, including amyotrophic, lateral sclerosis (SOD1 mutations), Parkinsonism (α-synuclein mutations), Huntington’s chorea (huntingtin with polyglutamin expansions) and spinocerebellar ataxias (proteins with polyglutamin expansions).
De novo folding is not necessarily accelerated by Hsp70 chaperones. In some cases folding is delayed for different reasons. First, folding of certain proteins can only proceed productively after synthesis of the polypeptide is completed as shown, e.g. for the reovirus lollipop-shaped protein sigma 1 [37]. Second, proteins destined for posttranslational insertion into organellar membranes are prevented from aggregation and transported to the translocation pore [38]. Third, in the case of the caspase-activated DNase (CAD), the active protein is dangerous for the cell and therefore can only complete folding in the presence of its specific inhibitor (ICAD). Hsp70 binds CAD cotranslationally and mediates folding only to an intermediate state. Folding is completed after addition of ICAD, which is assembled into a complex with CAD in an Hsp70-dependent manner [39]. Similar folding pathways may exist also for other potentially dangerous proteins.
As mentioned above Hsp70 interacts with key regulators of many signal transduction pathways controlling cell homeostasis, proliferation, differentiation and cell death. The interaction of Hsp70 with these regulatory proteins continues in activation cycles that also involve Hsp90 and a number of co-chaperones. The regulatory proteins, called clients, are thereby kept in an inactive state from which they are rapidly activated by the appropriate signals. Hsp70 and Hsp90 thus repress regulators in the absence of the upstream signal and guarantee full activation after the signal transduction pathway is switched on [6]. Hsp70 can be titrated away from these clients by other misfolded proteins that may arise from internal or external stresses. Consequently, through Hsp70 disturbances of the cellular system induced by environmental, developmental or pathological processes act on these signal transduction pathways.
In this way stress response and apoptosis are linked to each other. Hsp70 inhibits apoptosis acting on the caspase-dependent pathway at several steps both upstream and downstream of caspase activation and on the caspase-independent pathway. Overproduction of Hsp70 leads to increased resistance against apoptosis-inducing agents such as tumor necrosis factor-α(TNFα), staurosporin and doxorubicin, while downregulation of Hsp70 levels by antisense technology leads to increased sensitivity towards these agents [18, 40]. This observation relates to many pathological processes, such as oncogenesis, neurodegeneration and senescence. In many tumor cells increased Hsp70 levels are observed and correlate with increased malignancy and resistance to therapy. Downregulation of the Hsp70 levels in cancer cells induce differentiation and cell death [41]. Neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s corea and spinocerebellar ataxias are characterized by excessive apoptosis. In several different model systems overexpression of Hsp70 or one of its co-chaperones could overcome the neurodegenerative symptoms induced by expression of a disease-related gene (huntingtin, α-synuclein or ataxin) [20,42]. Senescence in cell culture as well as aging in vivo is correlated with a continuous decline in the ability to mount a stress response [34, 43]. Age-related symptoms and diseases reflect this decreased ability to cope with cellular stresses. Interestingly, centenarians seem to be an exception to the rule, as they show a significant induction of Hsp70 production after heat shock challenge [44].
ATPase domain and ATPase cycle
Substrate binding
The coupling mechanism: nucleotide-controlled opening and closing of the substrate binding cavity
The targeting activity of co-chaperones
J-domain proteins
Bag proteins
Hip, Hop and CHIP
Perspectives
The Hsp70 protein family and their co-chaperones constitute a complex network of folding machines which is utilized by cells in many ways. Despite considerable progress in the elucidation of the mechanistic basis of these folding machines, important aspects remain to be solved. With respect to the Hsp70 proteins it is still unclear whether their activity to assist protein folding relies on the ability to induce conformational changes in the bound substrates, how the coupling mechanism allows ATP to control substrate binding and to what extent sequence variations within the family translate into variations of the mechanism. With respect to the action of co-chaperones we lack a molecular understanding of the coupling function of JDPs and of how co-chaperones target their Hsp70 partner proteins to substrates. Furthermore, it can be expected that more cellular processes will be discovered that depend on the chaperone activity of Hsp70 chaperones.
-
The biochemistry and ultrastructure of molecular chaperones
Structure and Mechanism of the Hsp90 Molecular Chaperone Machinery
Laurence H. Pearl and Chrisostomos Prodromou
Ann Rev of Biochem July 2006;75:271-294
http://dx.doi.org:/10.1146/annurev.biochem.75.103004.142738
Heat shock protein 90 (Hsp90) is a molecular chaperone essential for activating many signaling proteins in the eukaryotic cell. Biochemical and structural analysis of Hsp90 has revealed a complex mechanism of ATPase-coupled conformational changes and interactions with cochaperone proteins, which facilitate activation of Hsp90’s diverse “clientele.” Despite recent progress, key aspects of the ATPase-coupled mechanism of Hsp90 remain controversial, and the nature of the changes, engendered by Hsp90 in client proteins, is largely unknown. Here, we discuss present knowledge of Hsp90 structure and function gleaned from crystallographic studies of individual domains and recent progress in obtaining a structure for the ATP-bound conformation of the intact dimeric chaperone. Additionally, we describe the roles of the plethora of cochaperones with which Hsp90 cooperates and growing insights into their biochemical mechanisms, which come from crystal structures of Hsp90 cochaperone complexes.
-
Properties of heat shock proteins (HSPs) and heat shock factor (HSF)
Heat shock factors: integrators of cell stress, development and lifespan
Malin Åkerfelt,*‡ Richard I. Morimoto,§ and Lea Sistonen*‡
Nat Rev Mol Cell Biol. 2010 Aug; 11(8): 545–555. doi: 10.1038/nrm2938
Heat shock factors (HSFs) are essential for all organisms to survive exposures to acute stress. They are best known as inducible transcriptional regulators of genes encoding molecular chaperones and other stress proteins. Four members of the HSF family are also important for normal development and lifespan-enhancing pathways, and the repertoire of HSF targets has thus expanded well beyond the heat shock genes. These unexpected observations have uncovered complex layers of post-translational regulation of HSFs that integrate the metabolic state of the cell with stress biology, and in doing so control fundamental aspects of the health of the proteome and ageing.
In the early 1960s, Ritossa made the seminal discovery of temperature-induced puffs in polytene chromosomes of Drosophila melanogaster larvae salivary glands1. A decade later, it was shown that the puffing pattern corresponded to a robust activation of genes encoding the heat shock proteins (HSPs), which function as molecular chaperones2. The heat shock response is a highly conserved mechanism in all organisms from yeast to humans that is induced by extreme proteotoxic insults such as heat, oxidative stress, heavy metals, toxins and bacterial infections. The conservation among different eukaryotes suggests that the heat shock response is essential for survival in a stressful environment.
The heat shock response is mediated at the transcriptional level by cis-acting sequences called heat shock elements (HSEs; BOX 1) that are present in multiple copies upstream of the HSP genes3. The first evidence for a specific transcriptional regulator, the heat shock factor (HSF) that can bind to the HSEs and induce HSP gene expression, was obtained through DNA–protein interaction studies on nuclei isolated from D. melanogaster cells4,5. Subsequent studies showed that, in contrast to a single HSF in invertebrates, multiple HSFs are expressed in plants and vertebrates6–8. The mammalian HSF family consists of four members: HSF1,HSF2, HSF3 and HSF4. Distinct HSFs possess unique and overlapping functions (FIG. 1), exhibit tissue-specific patterns of expression and have multiple post-translational modifications (PTMs) and interacting protein partners7,9,10. Functional crosstalk between HSF family members and PTMs facilitates the fine-tuning of HSF-mediated gene regulation. The identification of many targets has further extended the impact of HSFs beyond the heat shock response. Here, we present the recent discoveries of novel target genes and physiological functions of HSFs, which have changed the view that HSFs act solely in the heat shock response. Based on the current knowledge of small-molecule activators and inhibitors of HSFs, we also highlight the potential for pharmacologic modulation of HSF-mediated gene regulation.
Box 1
The heat shock element
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3402356/bin/nihms281610u1.jpg
Heat shock factors (HSFs) act through a regulatory upstream promoter element, called the heat shock element (HSE). In the DNA-bound form of a HSF, each DNA-binding domain (DBD) recognizes the HSE in the major groove of the double helix6. The HSE was originally identified using S1 mapping of transcripts of the Drosophila melanogaster heat shock protein (HSP) genes3 (see the figure; part a). Residues –47 to –66 are necessary for heat inducibility. HSEs in HSP gene promoters are highly conserved and consist of inverted repeats of the pentameric sequence nGAAn132. The type of HSEs that can be found in the proximal promoter regions of HSP genes is composed of at least three contiguous inverted repeats: nTTCnnGAAnnTTCn132–134. The promoters of HSF target genes can also contain more than one HSE, thereby allowing the simultaneous binding of multiple HSFs. The binding of an HSF to an HSE occurs in a cooperative manner, whereby binding of an HSF trimer facilitates binding of the next one135. More recently, Trinklein and colleagues used chromatin immunoprecipitation to enrich sequences bound by HSF1 in heat-shocked human cells to define the HSE consensus sequence. They confirmed the original finding of Xiao and Lis, who identified guanines as the most conserved nucleotides in HSEs87,133 (see the figure; part b). Moreover, in a pair of inverted repeats, a TTC triplet 5′ of a GAA triplet is separated by a pyrimidine–purine dinucleotide, whereas the two nucleotides separating a GAA triplet 5′ from a TTC triplet is unconstrained87. The discovery of novel HSF target genes that are not involved in the heat shock response has rendered it possible that there may be HSEs in many genes other than the HSP genes. Although there are variations in these HSEs, the spacing and position of the guanines are invariable7. Therefore, both the nucleotides and the exact spacing of the repeated units are considered as key determinants for recognition by HSFs and transcriptional activation. Part b of the figure is modified, with permission, from REF. 87 © (2004) The American Society for Cell Biology.
Figure 1 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3402356/bin/nihms281610f1.gif
The mammalian HSF machinery
HSFs as stress integrators
A hallmark of stressed cells and organisms is the increased synthesis of HSPs, which function as molecular chaperones to prevent protein misfolding and aggregation to maintain protein homeostasis, also called proteostasis11. The transcriptional activation of HSP genes is mediated by HSFs (FIG. 2a), of which HSF1 is the master regulator in vertebrates. Hsf1-knockout mouse and cell models have revealed that HSF1 is a prerequisite for the transactivation of HSP genes, maintenance of cellular integrity during stress and development of thermotolerance12–15. HSF1 is constitutively expressed in most tissues and cell types16, where it is kept inactive in the absence of stress stimuli. Thus, the DNA-binding and transactivation capacity of HSF1 are coordinately regulated through multiple PTMs, protein–protein interactions and subcellular localization. HSF1 also has an intrinsic stress-sensing capacity, as both D. melanogaster and mammalian HSF1 can be converted from a monomer to a homotrimer in vitro in response to thermal or oxidative stress17–19.
Figure 2 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3402356/bin/nihms281610f2.gif
Members of the mammalian HSF family
Functional domains
HSFs, like other transcription factors, are composed of functional domains. These have been most thoroughly characterized for HSF1 and are schematically presented in FIG. 2b. The DNA-binding domain (DBD) is the best preserved domain in evolution and belongs to the family of winged helix-turn-helix DBDs20–22. The DBD forms a compact globular structure, except for a flexible wing or loop that is located between β-strands 3 and 4 (REF. 6). This loop generates a protein– protein interface between adjacent subunits of the HSF trimer that enhances high-affinity binding to DNA by cooperativity between different HSFs23. The DBD can also mediate interactions with other factors to modulate the transactivating capacity of HSFs24. Consequently, the DBD is considered as the signature domain of HSFs for target-gene recognition.
The trimerization of HSFs is mediated by arrays of hydrophobic heptad repeats (HR-A and HR-B) that form a coiled coil, which is characteristic for many Leu zippers6,25 (FIG. 2b). The trimeric assembly is unusual, as Leu zippers typically facilitate the formation of homodimers or heterodimers. Suppression of spontaneous HSF trimerization is mediated by yet another hydrophobic repeat, HR-C26–28. Human HSF4 lacks the HR-C, which could explain its constitutive trimerization and DNA-binding activity29. Positioned at the extreme carboxyl terminus of HSFs is the transactivation domain, which is shared among all HSFs6except for yeast Hsf, which has transactivation domains in both the amino and C termini, and HSF4A, which completely lacks a transactivation domain29–31. In HSF1, the transactivation domain is composed of two modules — AD1 and AD2, which are rich in hydrophobic and acidic residues (FIG. 3a) — that together ensures a rapid and prolonged response to stress32,33. The transactivation domain was originally proposed to provide stress inducibility to HSF1 (REFS 34,35), but it soon became evident that an intact regulatory domain, located between the HR-A and HR-B and the transactivation domain, is essential for the responsiveness to stress stimuli32,33,36,37. Because several amino acids that are known targets for different PTMs reside in the regulatory domain33,38–42, the structure and function of this domain are under intensive investigation.
Figure 3 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3402356/bin/nihms281610f3.gif
HSF1 undergoes multiple PTMs on activation
Regulation of the HSF1 activation–attenuation cycle
The conversion of the inactive monomeric HSF1 to high-affinity DNA-binding trimers is the initial step in the multistep activation process and is a common feature of all eukaryotic HSFs43,44 (FIG. 3b). There is compelling evidence for HSF1 interacting with multiple HSPs at different phases of its activation cycle. For example, monomeric HSF1 interacts weakly with HSP90 and, on stress, HSF1 dissociates from the complex, allowing HSF1 trimerization45,46 (FIG. 3b). Trimeric HSF1 can be kept inactive when its regulatory domain is bound by a multi-chaperone complex of HSP90, co-chaperone p23 (also known as PTGES3) and immunophilin FK506-binding protein 5 (FKBP52; also known as FKBP4)46–51. Elevated levels of both HSP90 and HSP70 negatively regulate HSF1 and prevent trimer formation on heat shock52. Activated HSF1 trimers also interact with HSP70 and the co-chaperone HSP40 (also known as DNAJB1), but instead of suppressing the DNA-binding activity of HSF1, this interaction inhibits its transactivation capacity52–54. Although the inhibitory mechanism is still unknown, the negative feedback from the end products of HSF1-dependent transcription (the HSPs) provides an important control step in adjusting the duration and intensity of HSF1 activation according to the levels of chaperones and presumably the levels of nascent and misfolded peptides.
A ribonucleoprotein complex containing eukaryotic elongation factor 1A (eEF1A) and a non-coding RNA, heat shock RNA-1 (HSR-1), has been reported to possess a thermosensing capacity. According to the proposed model, HSR-1 undergoes a conformational change in response to heat stress and together with eEF1A facilitates trimerization of HSF1 (REF. 55). How this activation mode relates to the other regulatory mechanisms associated with HSFs remains to be elucidated.
Throughout the activation–attenuation cycle, HSF1 undergoes extensive PTMs, including acetylation, phosphorylation and sumoylation (FIG. 3). HSF1 is also a phosphoprotein under non-stress conditions, and the results from mass spectrometry (MS) analyses combined with phosphopeptide mapping experiments indicate that at least 12 Ser residues are phosphorylated41,56–59. Among these sites, stress-inducible phosphorylation of Ser230 and Ser326 in the regulatory domain contributes to the transactivation function of HSF1 (REFS 38,41). Phosphorylation-mediated sumoylation on a single Lys residue in the regulatory domain occurs rapidly and transiently on exposure to heat shock; Ser303 needs to be phosphorylated before a small ubiquitin-related modifier (SUMO) can be conjugated to Lys298 (REF. 39). The extended consensus sequence ΨKxExxSP has been named the phosphorylation-dependent sumoylation motif (PDSM; FIG. 3)40. The PDSM was initially discovered in HSF1 and subsequently found in many other proteins, especially transcriptional regulators such as HSF4, GATA1, myocyte-specific enhancer factor 2A (MEF2A) and SP3, which are substrates for both SUMO conjugation and Pro-directed kinases40,60–62.
Recently, Mohideen and colleagues showed that a conserved basic patch on the surface of the SUMO-conjugating enzyme ubiquitin carrier protein 9 (UBC9; also known as UBE2I) discriminates between the phosphorylated and non-phosphorylated PDSM of HSF1 (REF. 63). Future studies will be directed at elucidating the molecular mechanisms for dynamic phosphorylation and UBC9-dependent SUMO conjugation in response to stress stimuli and establishing the roles of kinases, phosphatases and desumoylating enzymes in the heat shock response. The kinetics of phosphorylation-dependent sumoylation of HSF1 correlates inversely with the severity of heat stress, and, as the transactivation capacity of HSF1 is impaired by sumoylation and this PTM is removed when maximal HSF1 activity is required40, sumoylation could modulate HSF1 activity under moderate stress conditions. The mechanisms by which SUMO modification represses the transactivating capacity of HSF1, and the functional relationship of this PTM with other modifications that HSF1 is subjected to, will be investigated with endogenous substrate proteins.
Phosphorylation and sumoylation of HSF1 occur rapidly on heat shock, whereas the kinetics of acetylation are delayed and coincide with the attenuation phase of the HSF1 activation cycle. Stress-inducible acetylation of HSF1 is regulated by the balance of acetylation by p300–CBP (CREB-binding protein) and deacetylation by the NAD+-dependent sirtuin, SIRT1. Increased expression and activity of SIRT1 enhances and prolongs the DNA-binding activity of HSF1 at the human HSP70.1promoter, whereas downregulation of SIRT1 enhances the acetylation of HSF1 and the attenuation of DNA-binding without affecting the formation of HSF1 trimers42. This finding led to the discovery of a novel regulatory mechanism of HSF1 activity, whereby SIRT1 maintains HSF1 in a state that is competent for DNA binding by counteracting acetylation (FIG. 3). In the light of current knowledge, the attenuation phase of the HSF1 cycle is regulated by a dual mechanism: a dependency on the levels of HSPs that feed back directly by weak interactions with HSF1, and a parallel step that involves the SIRT1-dependent control of the DNA-binding activity of HSF1. Because SIRT1 has been implicated in caloric restriction and ageing, the age-dependent loss of SIRT1 and impaired HSF1 activity correlate with an impairment of the heat shock response and proteostasis in senescent cells, connecting the heat shock response to nutrition and ageing (see below).
HSF dynamics on the HSP70 promoter
For decades, the binding of HSF to the HSP70.1 gene has served as a model system for inducible transcription in eukaryotes. In D. melanogaster, HSF is constitutively nuclear and low levels of HSF are associated with the HSP70promoter before heat shock64–66. The uninduced HSP70 promoter is primed for transcription by a transcriptionally engaged paused RNA polymerase II (RNAP II)67,68. RNAP II pausing is greatly enhanced by nucleosome formation in vitro, implying that chromatin remodelling is crucial for the release of paused RNAP II69. It has been proposed that distinct hydrophobic residues in the transactivation domain of human HSF1 can stimulate RNAP II release and directly interact withBRG1, the ATPase subunit of the chromatin remodelling complex SWI/SNF70,71. Upon heat shock, RNAP II is released from its paused state, leading to the synthesis of a full-length transcript. Rapid disruption of nucleosomes occurs across the entire HSP70 gene, at a rate that is faster than RNAP II-mediated transcription72. The nucleosome displacement occurs simultaneously with HSF recruitment to the promoter in D. melanogaster. Downregulation of HSF abrogates the loss of nucleosomes, indicating that HSF provides a signal for chromatin rearrangement, which is required for HSP70 nucleosome displacement. Within seconds of heat shock, the amount of HSF at the promoter increases drastically and HSF translocates from the nucleoplasm to several native loci, including HSP genes. Interestingly, the levels of HSF occupying the HSP70 promoter reach saturation soon after just one minute65,73.
HSF recruits the co-activating mediator complex to the heat shock loci, which acts as a bridge to transmit activating signals from transcription factors to the basal transcription machinery. The mediator complex is recruited by a direct interaction with HSF: the transactivation domain of D. melanogaster HSF binds to TRAP80(also known as MED17), a subunit of the mediator complex74. HSF probably has other macromolecular contacts with the preinitiation complex as it binds to TATA-binding protein (TBP) and the general transcription factor TFIIB in vitro75,76. In contrast to the rapid recruitment and elongation of RNAP II on heat shock, activated HSF exchanges very slowly at the HSP70 promoter. HSF stays stably bound to DNA in vivo and no turnover or disassembly of transcription activator is required for successive rounds of HSP70 transcription65,68.
Functional interplay between HSFs
Although HSF1 is the principal regulator of the heat shock response, HSF2 also binds to the promoters of HSP genes. In light of our current knowledge, HSF2 strictly depends on HSF1 for its stress-related functions as it is recruited to HSP gene promoters only in the presence of HSF1 and this cooperation requires an intact HSF1 DBD77. Nevertheless, HSF2 modulates, both positively and negatively, the HSF1-mediated inducible expression of HSP genes, indicating that HSF2 can actively participate in the transcriptional regulation of the heat shock response. Coincident with the stress-induced transcription of HSP genes, HSF1 and HSF2 colocalize and accumulate rapidly on stress into nuclear stress bodies (NSBs; BOX 2), where they bind to a subclass of satellite III repeats, predominantly in the human chromosome 9q12 (REFS 78–80). Consequently, large and stable non-coding satellite III transcripts are synthesized in an HSF1-dependent manner in NSBs81,82. The function of these transcripts and their relationship with other HSF1 targets, and the heat shock response in general, remain to be elucidated.
Box 2
Nuclear stress bodies
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3402356/bin/nihms281610u2.jpg
The cell nucleus is highly compartmentalized and dynamic. Many nuclear factors are diffusely distributed throughout the nucleoplasm, but they can also accumulate in distinct subnuclear compartments, such as nucleoli, speckles, Cajal bodies and promyelocytic leukaemia (PML) bodies136. Nuclear stress bodies (NSBs) are different from any other known nuclear bodies137,138. Although NSBs were initially thought to contain aggregates of denatured proteins and be markers of heat-shocked cells, their formation can be elicited by various stresses, such as heavy metals and proteasome inhibitors137. NSBs are large structures, 0.3–3 μm in diameter, and are usually located close to the nucleoli or nuclear envelope137,138. NSBs consist of two populations: small, brightly stained bodies and large, clustered and ring-like structures137.
NSBs appear transiently and are the main site of heat shock factor 1 (HSF1) and HSF2 accumulation in stressed human cells80. HSF1 and HSF2 form a physically interacting complex and colocalize into small and barely detectable NSBs after only five minutes of heat shock, but the intensity and size of NSBs increase after hours of continuous heat shock. HSF1 and HSF2 colocalize in HeLa cells that have been exposed to heat shock for one hour at 42°C (see the figure; confocal microscopy image with HSF1–green fluorescent protein in green and endogenous HSF2 in red). NSBs form on specific chromosomal loci, mainly on q12 of human chromosome 9, where HSFs bind to a subclass of satellite III repeats78,79,83. Stress-inducible and HSF1-dependent transcription of satellite III repeats has been shown to produce non-coding RNA molecules, called satellite III transcripts81,82. The 9q12 locus consists of pericentromeric heterochromatin, and the satellite III repeats provide scaffolds for docking components, such as splicing factors and other RNA-processing proteins139–143.
HSF2 also modulates the heat shock response through the formation of heterotrimers with HSF1 in the NSBs when bound to the satellite III repeats83 (FIG. 4). Studies on the functional significance of heterotrimerization indicate that HSF1 depletion prevents localization of HSF2 to NSBs and abolishes the stress-induced synthesis of satellite III transcripts. By contrast, increased expression of HSF2 leads to its own activation and the localization of both HSF1 and HSF2 to NSBs, where transcription is spontaneously induced in the absence of stress stimuli. These results suggest that HSF2 can incorporate HSF1 into a transcriptionally competent heterotrimer83. It is possible that the amounts of HSF2 available for heterotrimerization with HSF1 influence stress-inducible transcription, and that HSF1–HSF2 heterotrimers regulate transcription in a temporal manner. During the acute phase of heat shock, HSF1 is activated and HSF1–HSF2 heterotrimers are formed, whereas upon prolonged exposures to heat stress the levels of HSF2 are diminished, thereby limiting heterotrimerization83. Intriguingly, in specific developmental processes such as corticogenesis and spermatogenesis, the expression of HSF2 increases spatiotemporarily, leading to its spontaneous activation. Therefore, it has been proposed that HSF-mediated transactivation can be modulated by the levels of HSF2 to provide a switch that integrates the responses to stress and developmental stimuli83 (FIG. 4). Functional relationships between different HSFs are emerging, and the synergy of DNA-binding activities among HSF family members offers an efficient way to control gene expression in a cell- and stimulus-specific manner to orchestrate the differential upstream signalling and target-gene networks.
Figure 4 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3402356/bin/nihms281610f4.gif
Interactions between different HSFs provide distinct functional modes in transcriptional regulation
A new member of the mammalian HSF family, mouse HSF3, was recently identified10. Avian HSF3 was shown to be activated at higher temperatures and with different kinetics than HSF1 (REF. 84), whereas in mice, heat shock induces the nuclear translocation of HSF3 and activation of stress-responsive genes other than HSP genes10. Future experiments will determine whether HSF3 is capable of interacting with other HSFs, potentially through heterocomplex formation. HSF4 has not been implicated in the heat shock response, but it competes with HSF1 for common target genes in mouse lens epithelial cells85, which will be discussed below. It is important to elucidate whether the formation of homotrimers or hetero trimers between different family members is a common theme in HSF-mediated transcriptional regulation.
HSFs as developmental regulators
Evidence is accumulating that HSFs are highly versatile transcription factors that, in addition to protecting cells against proteotoxic stress, are vital for many physioogical functions, especially during development. The initial observations using deletion experiments of the D. melanogaster Hsf gene revealed defective oogenesis and larvae development86. These effects were not caused by obvious changes in HSP gene expression patterns, which is consistent with the subsequent studies showing that basal expression of HSP genes during mouse embryogenesis is not affected by the lack of HSF1 (REF. 13). These results are further supported by genome-wide gene expression studies revealing that numerous genes, not classified as HSP genes or molecular chaperones, are under HSF1-dependent control87,88.
Although mice lacking HSF1 can survive to adulthood, they exhibit multiple defects, such as increased prenatal lethality, growth retardation and female infertility13. Fertilized oocytes do not develop past the zygotic stage when HSF1-deficient female mice are mated with wild-type male mice, indicating that HSF1 is a maternal factor that is essential for early post-fertilization development89. Recently, it was shown that HSF1 is abundantly expressed in maturing oocytes, where it regulates specifically Hsp90α transcription90. The HSF1-deficient oocytes are devoid of HSP90α and exhibit a blockage of meiotic maturation, including delayed G2–M transition or germinal vesicle breakdown and defective asymmetrical division90. Moreover, intra-ovarian HSF1-depleted oocytes contain dysfunctional mitochondria and are sensitive to oxidative stress, leading to reduced survival91. The complex phenotype of Hsf1-knockout mice also demonstrates the involvement of HSF1 in placenta formation, placode development and the immune system15,85,92,93, further strengthening the evidence for a protective function of HSF1 in development and survival.
Both HSF1 and HSF2 are key regulators in the developing brain and in maintaining proteostasis in the central nervous system. Disruption of Hsf1 results in enlarged ventricles, accompanied by astrogliosis, neurodegeneration, progressive myelin loss and accumulation of ubiquitylated proteins in specific regions of the postnatal brain under non-stressed conditions94,95. The expression of HSP25 (also known as HSPB1) and α-crystallin B chain (CRYAB), which are known to protect cells against stress-induced protein damage and cell death, is dramatically decreased in brains lacking HSF1 (REF. 13). In contrast to HSF1, HSF2 is already at peak levels during early brain development in mice and is predominantly expressed in the proliferative neuronal progenitors of the ventricular zone and post-mitotic neurons of the cortical plate96–99. HSF2-deficient mice have enlarged ventricles and defects in cortical lamination owing to abnormal neuronal migration97–99. Incorrect positioning of superficial neurons during cortex formation in HSF2-deficient embryos is caused by decreased expression of the cyclin-dependent kinase 5 (CDK5) activator p35, which is a crucial regulator of the cortical migration signalling pathway100,101. The p35 gene was identified as the first direct target of HSF2 in cortex development99. As correct cortical migration requires the coordination of multiple signalling molecules, it is likely that HSF2, either directly or indirectly, also regulates other components of the same pathway.
Cooperativity of HSFs in development
In adult mice, HSF2 is most abundantly expressed in certain cell types of testes, specifically pachytene spermatocytes and round spermatids102. The cell-specific expression of HSF2 in testes is regulated by a microRNA, miR-18, that directly binds to the 3′ untranslated region (UTR) of HSF2 (J.K. Björk, A. Sandqvist, A.N. Elsing, N. Kotaja and L.S., unpublished observations). Targeting of HSF2 in spermatogenesis reveals the first physiological role for miR-18, which belongs to the oncomir-1 cluster associated mainly with tumour progression103. In accordance with the expression pattern during the maturation of male germ cells, HSF2-null male mice display several abnormal features in spermatogenesis, ranging from smaller testis size and increased apoptosis at the pachytene stage to a reduced amount of sperm and abnormal sperm head shape97,98,104. A genome-wide search for HSF2 target promoters in mouse testis revealed the occupancy of HSF2 on the sex chromosomal multi-copy genes spermiogenesis specific transcript on the Y 2 (Ssty2), Sycp3-like Y-linked (Sly) and Sycp3-like X-linked (Slx), which are important for sperm quality104. Compared with the Hsf2-knockout phenotype, disruption of both Hsf1 and Hsf2 results in a more pronounced phenotype, including larger vacuolar structures, more widely spread apoptosis and a complete lack of mature spermatozoa and male sterility105. The hypo thesis that the activities of HSF1 and HSF2 are intertwined and essential for spermatogenesis is further supported by our results that HSF1 and HSF2 synergistically regulate the sex chromosomal multi-copy genes in post-meiotic round spermatids (M.Å., A. Vihervaara, E.S. Christians, E. Henriksson and L.S., unpublished observations). Given that the sex chromatin mostly remains silent after meiosis, HSF1 and HSF2 are currently the only known transcriptional regulators during post-meiotic repression. These results, together with the earlier findings that HSF2 can also form heterotrimers with HSF1 in testes83, strongly suggest that HSF1 and HSF2 act in a heterocomplex and fine-tune transcription of their common target genes during the maturation of male germ cells.
HSF1 and HSF4 are required for the maintenance of sensory organs, especially when the organs are exposed to environmental stimuli for the first time after birth85,88. During the early postnatal period, Hsf1-knockout mice display severe atrophy of the olfactory epithelium, increased accumulation of mucus and death of olfactory sensory neurons88. Although lens development in HSF4-deficient mouse embryos is normal, severe abnormalities, including inclusion-like structures in lens fibre cells, appear soon after birth and the mice develop cataracts85,106,107. Intriguingly, inherited severe cataracts occurring in Chinese and Danish families have been associated with a mutation in the DBD of HSF4 (REF. 108). In addition to the established target genes, Hsp25, Hsp70 and Hsp90, several new targets for HSF1 and HSF4, such as crystallin γF (Crygf), fibroblast growth factor 7 (Fgf7) and leukaemia inhibitory factor (Lif) have been found to be crucial for sensory organs85,88. Furthermore, binding of either HSF1 or HSF4 to the Fgf7 promoter shows opposite effects on gene expression, suggesting competitive functions between the two family members85. In addition to the proximal promoters, HSF1, HSF2 and HSF4 bind to other genomic regions (that is, introns and distal parts of protein-coding genes in mouse lens), and there is also evidence for either synergistic interplay or competition between distinct HSFs occupying the target-gene promoters109. It is possible that the different HSFs are able to compensate for each other to some extent. Thus, the identification of novel functions and target genes for HSFs has been a considerable step forward in understanding their regulatory mechanisms in development.
HSFs and lifespan
The lifespan of an organism is directly linked to the health of its tissues, which is a consequence of the stability of the proteome and functionality of its molecular machineries. During its lifetime, an organism constantly encounters environmental and physiological stress and requires an efficient surveillance of protein quality to prevent the accumulation of protein damage and the disruption of proteostasis. Proteotoxic insults contribute to cellular ageing, and numerous pathophysiological conditions, associated with impaired protein quality control, increase prominently with age11. From studies on the molecular basis of ageing, in which a wide range of different model systems and experimental strategies have been used, the insulin and insulin-like growth factor 1 receptor (IGF1R) signalling pathway, which involves the phosphoinositide 3-kinase (PI3K) and AKT kinases and the Forkhead box protein O (FOXO) transcription factors (such as DAF-16 in Caenorhabditis elegans), has emerged as a key process. The downregulation of HSF reduces the lifespan and accelerates the formation of protein aggregates in C. elegans carrying mutations in different components of the IGF1R-mediated pathway. Conversely, inhibition of IGF1R signalling results in HSF activation and promotes longevity by maintaining proteostasis110,111. These results have prompted many laboratories that use other model organisms to investigate the functional relationship between HSFs and the IGF1R signalling pathway.
The impact of HSFs on the lifespan of whole organisms is further emphasized by a recent study, in which proteome stability was examined during C. elegansageing112. The age-dependent misfolding and downregulation of distinct metastable proteins, which display temperature-sensitive missense mutations, was examined in different tissues. Widespread failure in proteostasis occurred rapidly at an early stage of adulthood, coinciding with the severely impaired heat shock response and unfolded protein response112. The age-dependent collapse of proteostasis could be restored by overexpression of HSF and DAF-16, strengthening the evidence for the unique roles of these stress-responsive transcription factors to prevent global instability of the proteome.
Limited food intake or caloric restriction is another process that is associated with an enhancement of lifespan. In addition to promoting longevity, caloric restriction slows down the progression of age-related diseases such as cancer, cardiovascular diseases and metabolic disorders, stimulates metabolic and motor activities, and increases resistance to environmental stress stimuli113. To this end, the dynamic regulation of HSF1 by the NAD+-dependent protein deacetylase SIRT1, a mammalian orthologue of the yeast transcriptional regulator Sir2, which is activated by caloric restriction and stress, is of particular interest. Indeed, SIRT1 directly deacetylates HSF1 and keeps it in a state that is competent for DNA binding. During ageing, the DNA-binding activity of HSF1 and the amount of SIRT1 are reduced. Consequently, a decrease in SIRT1 levels was shown to inhibit HSF1 DNA-binding activity in a cell-based model of ageing and senescence42. Furthermore, an age-related decrease in the HSF1 DNA-binding activity is reversed in cells exposed to caloric restriction114. These results indicate that HSF1 and SIRT1 function together to protect cells from stress insults, thereby promoting survival and extending lifespan. Impaired proteostasis during ageing may at least partly reflect the compromised HSF1 activity due to lowered SIRT1 expression.
Impact of HSFs in disease
The heat shock response is thought to be initiated by the presence of misfolded and damaged proteins, and is thus a cell-autonomous response. When exposed to heat, cells in culture, unicellular organisms, and cells in a multicellular organism can all trigger a heat shock response autonomously115–117. However, it has been proposed that multicellular organisms sense stress differently to isolated cells. For example, the stress response is not properly induced even if damaged proteins are accumulated in neurodegenerative diseases like Huntington’s disease and Parkinson’s disease, suggesting that there is an additional control of the heat shock response at the organismal level118. Uncoordinated activation of the heat shock response in cells in a multicellular organism could cause severe disturbances of interactions between cells and tissues. In C. elegans, a pair of thermosensory neurons called AFDs, which sense and respond to temperature, regulate the heat shock response in somatic tissues by controlling HSF activity119,120. Moreover, the heat shock response in C. elegans is influenced by the metabolic state of the organism and is reduced under conditions that are unfavourable for growth and reproduction121. Neuronal control may therefore allow organisms to coordinate the stress response of individual cells with the varying metabolic requirements in different tissues and developmental stages. These observations are probably relevant to diseases of protein misfolding that are highly tissue-specific despite the often ubiquitous expression of damaged proteins and the heat shock response.
Elevated levels of HSF1 have been detected in several types of human cancer, such as breast cancer and prostate cancer122,123. Mice deficient in HSF1 exhibit a lower incidence of tumours and increased survival than their wild-type counterparts in a classical chemical skin carcinogenesis model and in a genetic model expressing an oncogenic mutation of p53. Similar results have been obtained in human cancer cells lines, in which HSF1 was depleted using an RNA interference strategy124. HSF1 expression is likely to be crucial for non-oncogene addiction and the stress phenotype of cancer cells, which are attributes given to many cancer cells owing to their high intrinsic level of proteotoxic and oxidative stress, frequent spontaneous DNA damage and aneuploidy125. Each of these features may disrupt proteostasis, raising the need for efficient chaperone and proteasome activities. Accordingly, HSF1 would be essential for the survival of cancer cells that experience constant stress and develop non-oncogene addiction.
HSFs as therapeutic targets
Given the unique role of HSF1 in stress biology and proteostasis, enhanced activity of this principal regulator during development and early adulthood is important for the stability of the proteome and the health of the cell. However, HSF1 is a potent modifier of tumorigenesis and, therefore, a potential target for cancer therapeutics125. In addition to modulating the expression of HSF1, the various PTMs of HSF1 that regulate its activity should be considered from a clinical perspective. As many human, age-related pathologies are associated with stress and misfolded proteins, several HSF-based therapeutic strategies have been proposed126. In many academic and industrial laboratories, small molecule regulators of HSF1 are actively being searched for (see Supplementary information S1 (table)). For example, celastrol, which has antioxidant properties and is a natural compound derived from the Celastreace family of plants, activates HSF1 and induces HSP expression with similar kinetics to heat shock, and could therefore be a potential candidate molecule for treating neurodegenerative diseases127,128. In a yeast-based screen, a small-molecule activator of human HSF1 was found and named HSF1A129. HSF1A, which is structurally distinct from the other known activators, activates HSF1 and enhances chaperone expression, thereby counteracting protein misfolding and cell death in polyQ-expressing neuronal precursor cells129. Triptolide, also from the Celastreace family of plants, is a potent inhibitor of the transactivating capacity of HSF1 and has been shown to have beneficial effects in treatments of pancreatic cancer xenografts130,131. These examples of small-molecule regulators of HSF1 are promising candidates for drug discovery and development. However, the existence of multiple mammalian HSFs and their functional interplay should also be taken into consideration when planning future HSF-targeted therapies.
Concluding remarks and future perspectives
HSFs were originally identified as specific heat shock-inducible transcriptional regulators of HSP genes, but now there is unambiguous evidence for a wide variety of HSF target genes that extends beyond the molecular chaperones. The known functions governed by HSFs span from the heat shock response to development, metabolism, lifespan and disease, thereby integrating pathways that were earlier strictly divided into either cellular stress responses or normal physiology.
Although the extensive efforts from many laboratories focusing on HSF biology have provided a richness of understanding of the complex regulatory mechanisms of the HSF family of transcription factors, several key questions remain. For example, what are the initial molecular events (that is, what is the ‘thermometer’) leading to the multistep activation of HSFs? The chromatin-based interaction between HSFs and the basic transcription machinery needs further investigation before the exact interaction partners at the chromatin level can be established. The activation and attenuation mechanisms of HSFs require additional mechanistic insights, and the roles of the multiple signal transduction pathways involved in post-translational regulation of HSFs are only now being discovered and are clearly more complex than anticipated. Although still lacking sufficient evidence, the PTMs probably serve as rheostats to allow distinct forms of HSF-mediated regulation in different tissues during development. Further emphasis should therefore be placed on understanding the PTMs of HSFs during development, ageing and different protein folding diseases. Likewise, the subcellular distribution of HSF molecules, including the mechanism by which HSFs shuttle between the cytoplasm and the nucleus, remains enigmatic, as do the movements of HSF molecules in different nuclear compartments such as NSBs.
Most studies on the impact of HSFs in lifespan and disease have been conducted with model organisms such as D. melanogaster and C. elegans, which express a single HSF. The existence of multiple members of the HSF family in mammals warrants further investigation of their specific and overlapping functions, including their extended repertoire of target genes. The existence of multiple HSFs in higher eukaryotes with different expression patterns suggests that they may have functions that are triggered by distinct stimuli, leading to activation of specific target genes. The impact of the HSF family in the adaptation to diverse biological environments is still poorly understood, and future studies are likely to broaden the prevailing view of HSFs being solely stress-inducible factors. To this end, the crosstalk between distinct HSFs that has only recently been uncovered raises obvious questions about the stoichiometry between the components in different complexes residing in different cellular compartments, and the mechanisms by which the factors interact with each other. Interaction between distinct HSF family members could generate new opportunities in designing therapeutics for protein-folding diseases, metabolic disorders and cancer.
-
Role in the etiology of cancer
Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo
Dan Tang,1 Md Abdul Khaleque,2 Ellen L. Jones,1 Jimmy R. Theriault,2 Cheng Li,3 Wing Hung Wong,3 Mary Ann Stevenson,2 and Stuart K. Calderwood1,2,4
Cell Stress Chaperones. 2005 Mar; 10(1): 46–58. doi: 10.1379/CSC-44R.1
Heat shock proteins (HSPs) are thought to play a role in the development of cancer and to modulate tumor response to cytotoxic therapy. In this study, we have examined the expression of hsf and HSP genes in normal human prostate epithelial cells and a range of prostate carcinoma cell lines derived from human tumors. We have observed elevated expressions of HSF1, HSP60, and HSP70 in the aggressively malignant cell lines PC-3, DU-145, and CA-HPV-10. Elevated HSP expression in cancer cell lines appeared to be regulated at the post–messenger ribonucleic acid (mRNA) levels, as indicated by gene chip microarray studies, which indicated little difference in heat shock factor (HSF) or HSP mRNA expression between the normal and malignant prostate cell lines. When we compared the expression patterns of constitutive HSP genes between PC-3 prostate carcinoma cells growing as monolayers in vitro and as tumor xenografts growing in nude mice in vivo, we found a marked reduction in expression of a wide spectrum of the HSPs in PC-3 tumors. This decreased HSP expression pattern in tumors may underlie the increased sensitivity to heat shock of PC-3 tumors. However, the induction by heat shock of HSP genes was not markedly altered by growth in the tumor microenvironment, and HSP40, HSP70, and HSP110 were expressed abundantly after stress in each growth condition. Our experiments indicate therefore that HSF and HSP levels are elevated in the more highly malignant prostate carcinoma cells and also show the dominant nature of the heat shock–induced gene expression, leading to abundant HSP induction in vitro or in vivo.
Heat shock proteins (HSPs) were first discovered as a cohort of proteins that is induced en masse by heat shock and other chemical and physical stresses in a wide range of species (Lindquist and Craig 1988; Georgopolis and Welch 1993). The HSPs (Table 1) have been subsequently characterized as molecular chaperones, proteins that have in common the property of modifying the structures and interactions of other proteins (Lindquist and Craig 1988; Beckmann et al 1990;Gething and Sambrook 1992; Georgopolis and Welch 1993; Netzer and Hartl 1998). Molecular chaperone function dictates that the HSP often interact in a stoichiometric, one-on-one manner with their substrates, necessitating high intracellular concentrations of the proteins (Lindquist and Craig 1988; Georgopolis and Welch 1993). As molecules that shift the balance from denatured, aggregated protein conformation toward ordered, functional conformation, HSPs are particularly in demand when the protein structure is disrupted by heat shock, oxidative stress, or other protein-damaging events (Lindquist and Craig 1988;Gething and Sambrook 1992; Georgopolis and Welch 1993). The HSP27, HSP40,HSP70, and HSP110 genes have therefore evolved a highly efficient mechanism for mass synthesis during stress, with powerful transcriptional activation, efficient messenger ribonucleic acid (mRNA) stabilization, and selective mRNA translation (Voellmy 1994). HSP27, HSP70, HSP90, and HSP110 increase to become the dominantly expressed proteins after stress (Hickey and Weber 1982; Landry et al 1982; Li and Werb 1982; Subjeck et al 1982; Henics et al 1999) (Zhao et al 2002). Heat shock factor (HSF) proteins have been shown to interact with the promoters of many HSP genes and ensure prompt transcriptional activation in stress and equally precipitous switch off after recovery (Sorger and Pelham 1988; Wu 1995). The hsf gene family includes HSF1 (hsf1), the molecular coordinator of the heat shock response, as well as 2 less well-characterized genes, hsf2 and hsf4(Rabindran et al 1991; Schuetz et al 1991) (Nakai et al 1997). In addition to the class of HSPs induced by heat, cells also contain a large number of constitutively expressed HSP homologs, which are also listed in Table 1. The constitutive HSPs are found in a variety of multiprotein complexes containing both HSPs and cofactors (Buchner 1999). These include HSP10-HSP60 complexes that mediate protein folding and HSP70- and HSP90-containing complexes that are involved in both generic protein-folding pathways and in specific association with regulatory proteins within the cell (Netzer and Hartl 1998). HSP90 plays a particularly versatile role in cell regulation, forming complexes with a large number of cellular kinases, transcription factors, and other molecules (Buchner 1999; Grammatikakis et al 2002).
Table 1 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1074571/bin/i1466-1268-10-1-46-t01.jpg
Heat shock protein family genes studied by microchip array analysis
Many tumor types contain high concentrations of HSP of the HSP28, HSP70, and HSP90 families compared with adjacent normal tissues (Ciocca et al 1993; Yano et al 1999; Cornford et al 2000; Strik et al 2000; Ricaniadis et al 2001; Ciocca and Vargas-Roig 2002). We have concentrated here on HSP gene expression in prostate carcinoma. The progression of prostatic epithelial cells to the fully malignant, metastatic phenotype is a complex process and involves the expression of oncogenes as well as escape from androgen-dependent growth and survival (Cornford et al 2000). There is a molecular link between HSP expression and tumor progression in prostate cancer in that HSP56, HSP70, and HSP90 regulate the function of the androgen receptor (AR) (Froesch et al 1998; Grossmann et al 2001). Escape from AR dependence during tumorigenesis may involve altered HSP-AR interactions (Grossmann et al 2001). The role of HSPs in tumor development may also be related to their function in the development of tolerance to stress (Li and Hahn 1981). Thermotolerance is induced in cells preconditioned by mild stress coordinately with the expression of high HSP levels (Landry et al 1982; Li and Werb 1982; Subjeck et al 1982). Elevated HSP expression appears to be a factor in tumor pathogenesis, and, among other mechanisms, this may involve the ability of individual HSPs to block the pathways of apoptosis and permit malignant cells to arise despite the triggering of apoptotic signals during transformation (Volloch and Sherman 1999). De novo HSP expression may also afford protection of cancer cells from treatments such as chemotherapy and hyperthermia by thwarting the proapoptotic influence of these modalities (Gabai et al 1998; Hansen et al 1999; Blagosklonny 2001; Asea et al 2001; Van Molle et al 2002). The mechanisms underlying HSP induction in tumor cells are not known but may reflect the genetic alterations accompanying malignancy or the disordered state of the tumor microenvironment, which would be expected to lead to cellular stress.
Here, we have examined expression of hsf and HSP genes in immortalized normal human prostate epithelial cells and a range of prostate carcinoma cells obtained from human tumors at the mRNA and protein levels. Our aim was to determine whether hsf-HSP expression profiles are conserved in cells that express varying degrees of malignancy, under resting conditions and after heat and ionizing radiation. In addition, we have compared HSP expression profiles of a metastatic human prostate carcinoma cell line growing either in monolayer culture or as a tumor xenograft in nude mice. These studies were prompted by findings in our laboratory that prostate carcinoma cells are considerably more sensitive to heat-induced apoptosis in vivo growing as tumors compared with similar cells growing in tissue culture in vitro. Our studies show that, although the hsf-HSP expression profiles are similar in normal and malignant prostate-derived cells at the mRNA level, expression at the protein level was very different. HSF1 and HSP protein expression was highest in the 3 aggressively metastatic prostate cancer cell lines (PC-3, DU-145, and CA-HPV-10). Although the gene expression patterns of constitutive HSP differ enormously in PC-3 cells in vitro and in xenografts in vivo, stress induction of HSP genes is not markedly altered by exposure to the tumor microenvironment, indicating the hierarchical rank of the stress response that permits it to override other forms of regulation. ……
The experiments described here are largely supportive of the notion that HSP gene expression and HSF activity and expression are increased in more advanced stages of cancer (Fig 4). The most striking finding in the study was the elevation of HSF1 and HSP levels in aggressively malignant prostate carcinoma cell lines (Fig 4). It is significant that these changes in HSF and HSP levels would not have been predicted from microarray studies of HSF (Fig 3) and HSP (Fig 1) mRNA levels. The increased HSF levels observed in the metastatic prostate carcinoma cell lines in particular appear to be due to altered regulation of either mRNA translation or protein turnover (or both) (Figs 3 and and4).4). Although we do not at this stage know the mechanisms involved, 1 candidate could be differential activity of the proteosome in the metastatic cell lines: both HSF1 and HSF2 are targets for proteosomal degradation (Mathew et al 1998). Despite these differences in HSP expression between cells of varying degrees of malignancy under growth conditions, stress caused a major shift in HSP gene expression and activation of HSP40-1, HSP70-1A, HSP70-1B, HSP70-6 (HSP70B), DNA-J2–like, and HSP105 in all cells (Fig 2). Even in LnCap cells with minimal HSF1 and HSF2 expression, heat-inducible HSP70 protein expression was observed (Fig 4). Interestingly, we observed minimal induction of the HSP70B gene in LnCap cells: because the HSP70B promoter is known to be almost exclusively induced by stress through the HSE in its promoter, the findings may suggest that a mechanism for HSP70 induction alternative to HSF1 activation may be operative in LnCap cells (Schiller et al 1988). Increased HSP expression in cancer patients has been shown to signal a poor response to treatment by a number of modalities, suggesting that HSP expression is involved with development of resistance to treatment in addition to being involved in the mechanisms of malignant progression (Ciocca et al 1993, Cornford et al 2000; Yamamoto et al 2001; Ciocca and Vargas-Roig 2002;Mese et al 2002). In addition, subpopulations of LnCap-derived cells, selected for enhanced capacity to metastasize, have been shown to express elevated levels of HSF1, HSP70, and HSP27 compared with nonselected controls (Hoang et al 2000). This may be highly significant because our studies indicate minimal levels of HSF1 and HSP in the poorly metastatic parent LnCap cells (Figs 1 and and4).4). Previous studies have also indicated that elevated HSP70 expression occurs at an early stage in cellular immortalization from embryonic stem cells (Ravagnan et al 2001). We had to use immortalized prostatic epithelial cells for our normal controls and may have missed a very early change in HSP expression during the immortalization process.
As indicated by the kinetic studies (Figs 5–7), HSPs are activated at a number of regulatory levels by stress in addition to transcriptional activation, and these may include stress-induced mRNA stabilization, differential translation, and protein stabilization (Hickey and Weber 1982; Zhao et al 2002). HSF1 activity and HSP expression appear to be subject to differential regulation by a number of pathways at normal temperatures but are largely independent of such regulation when exposed to heat shock, which overrides constitutive regulation and permits prompt induction of this emergency response.
Growth of PC-3 cells in vivo as tumor xenografts was accompanied by a marked decrease in constitutive HSP expression (Figs 8 and and11).11). Decreased HSP expression was part of a global switch in gene expression that accompanies the switch of PC-3 cells from growth as monolayers in tissue culture to growth as tumors in vivo (D. Tang and S.K. Calderwood, in preparation). Many reports indicate changes in a wide range of cellular properties as cells grow as tumors, and these properties may reflect the remodeling of gene expression patterns. These changes may reflect adaptation to the chemical nature of the tumor microenvironment and the alterations in cell-cell interaction in growth as a tumor in vivo. Our studies also indicate the remarkable sturdiness of the heat shock response that remains intact in the PC-3 cells growing in vivo despite the global rearrangements in other gene expressions mentioned above (Figs 10 and and1111).
The elevation in HSF1 and HSP levels in cancer shown in our studies and in those of others and its association with a poor prognosis and inferior response to therapy suggests the strategy of targeting HSP in cancer therapy. Treatment with HSP70 antisense oligonucleotides, for instance, can cause tumor cell apoptosis on its own and can synergize with heat shock in cell killing (Jones et al 2004). Indeed, it has been shown that antagonizing heat-inducible HSP expression with quercitin, a bioflavonoid drug that inhibits HSF1 activation, or by using antisense oligonucleotides directed against HSP70 mRNA further sensitizes PC-3 cells to heat-induced apoptosis in vitro and leads to tumor regression in vivo (Asea et al 2001, Lepchammer et al 2002; Jones et al 2004) (A. Asea et al, personal communication). The strategy of targeting HSP expression or function in cancer cells may thus be indicated. Such a strategy might prove particularly effective because constitutive HSP expression is reduced in tumors, and this might be related to increased killing of PC-3 tumor cells by heat (Fig 12).
-
Molecular chaperones in aging
Aging and molecular chaperones
Csaba So˝ti*, Pe´ter Csermely
Exper Geront 2003; 38:1037–1040 http://195.111.72.71/docs/pcs/03exger.pdf
Chaperone function plays a key role in sequestering damaged proteins and in repairing proteotoxic damage. Chaperones are induced by environmental stress and are called as stress or heat shock proteins. Here, we summarize the current knowledge about protein damage in aged organisms, about changes in proteolytic degradation, chaperone expression and function in the aging process, as well as the involvement of chaperones in longevity and cellular senescence. The role of chaperones in aging diseases, such as in Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and in other neurodegenerative diseases as well as in atherosclerosis and in cancer is discussed. We also describe how the balance between chaperone requirement and availability becomes disturbed in aged organisms, or in other words, how chaperone overload develops. The consequences of chaperone overload are also outlined together with several new research strategies to assess the functional status of chaperones in the aging process.
Molecular chaperones Chaperones are ubiquitous, highly conserved proteins (Hartl, 1996), either assisting in the folding of newly synthesized or damaged proteins in an ATP-dependent active process or working in an ATP-independent passive mode sequestering damaged proteins for future refolding or digestion. Environmental stress leads to proteotoxic damage. Damaged, misfolded proteins bind to chaperones, and liberate the heat shock factor (HSF) from its chaperone complexes. HSF is activated and transcription of chaperone genes takes place (Morimoto, 2002). Most chaperones, therefore, are also called stress or (after the archetype of experimental stress) heat shock proteins (Hsp-s).
Aging proteins—proteins of aging organisms During the life-span of a stable protein, various posttranslational modifications occur including backbone and side chain oxidation, glycation, etc. In aging organisms, the disturbed cellular homeostasis leads to an increased rate of protein modification: in an 80-year old human, half of all proteins may become oxidized (Stadtman and Berlett, 1998). Susceptibility to various proteotoxic damages is mainly increased due to dysfunction of mitochondrial oxidation of starving yeast cells (Aguilaniu et al., 2001). In prokaryotes, translational errors result in folding defects and subsequent protein oxidation (Dukan et al., 2000), which predominantly takes place in growth arrested cells (Ballesteros et al., 2001). Additionally, damaged signalling networks loose their original stringency, and irregular protein phosphorylation occurs (e.g.: the Parkinson disease-related a-synuclein also becomes phosphorylated, leading to misfolding and aggregation; Neumann et al., 2002).
Aging protein degradation Irreversibly damaged proteins are recognized by chaperones, and targeted for degradation. Proteasome level and function decreases with aging, and some oxidized, aggregated proteins exert a direct inhibition on proteasome activity. Chaperones also aid in lysosomal degradation. The proteolytic changes are comprehensively reviewed by Szweda et al. (2002). Due to the degradation defects, damaged proteins accumulate in the cells of aged organisms, and by aggregation may cause a variety of protein folding diseases (reviewed by So˝ti and Csermely, 2002a).
Aging chaperones I: defects in chaperone induction Damaged proteins compete with the HSF in binding to the Hsp90-based cytosolic chaperone complex, which may contribute to the generally observed constitutively elevated chaperone levels in aged organisms (Zou et al., 1998; So˝ti and Csermely, 2002b). On the contrary, the majority of the reports showed that stress-induced synthesis of chaperones is impaired in aged animals. While HSF activation does not change, DNA binding activity may be reduced during aging (Heydari et al., 2000). A number of signaling events use an overlapping network of chaperones not only to establish the activation-competent state of different transcription factors (e.g. steroid receptors), but also as important elements in the attenuation of respective responses. HSF transcriptional activity is also negatively influenced by higher levels of chaperones (Morimoto, 2002). Differential changes of these proteins in various organisms and tissues may lead to different extents of (dys)regulation. More importantly, the cross-talk between different signalling pathways through a shared pool of chaperones may have severe consequences during aging when the cellular conformational homeostasis is deranged (see below).
Aging chaperones II: defects in chaperone function Direct studies on chaperone function in aged organisms are largely restricted to a-crystallin having a decreased activity in aged human lenses (Cherian and Abraham, 1995; Cherian-Shaw et al., 1999). In a recent study, an initial test of passive chaperone function of whole cytosols was assessed showing a decreased chaperone capacity in aged rats compared to those of young counterparts (Nardai et al., 2002). What can be the mechanism behind these deleterious changes in chaperone function? Chaperones may also be prone to oxidative damage, as GroEL is preferentially oxidized in growth-arrested E. coli (Dukan and Nystro¨m, 1999). Macario and Conway de Macario (2002) raised the idea of ‘sick chaperones’ in aged organisms in a recent review. Indeed, chaperones are interacting with a plethora of other proteins (Csermely, 2001a), which requires rather extensive binding surfaces. These exposed areas may make chaperones a preferential target for proteotoxic damage: chaperones may behave as ‘suicide proteins’ during aging, sacrificing themselves instead of ‘normal’ proteins. The high abundance of chaperones (which may constitute more than 5% of cellular proteins), and their increased constitutive expression in aged organisms makes them a good candidate for this ‘altruistic courtesy.’ It may be especially true for mitochondrial Hsp60, the role of which would deserve extensive studies.
Aging chaperones III: defects in capacity, the chaperone overload Another possible reason of decreased chaperone function is chaperone overload (Csermely, 2001b). In aging organisms, the balance between misfolded proteins and available free chaperones is grossly disturbed: increased protein damage, protein degradation defects increase the amount of misfolded proteins, while chaperone damage, inadequate synthesis of molecular chaperones and irreparable folding defects (due to posttranslational changes) decrease the amount of available free chaperones. Chaperone overload occurs, where the need for chaperones may greatly exceed the available chaperone capacity (Fig. 1). Under these conditions, the competition for available chaperones becomes fierce and the abundance of damaged proteins may disrupt the folding assistance to other chaperone targets, such as: (1) newly synthesized proteins; (2) ‘constantly damaged’ (mutant) proteins; and (3) constituents of the cytoarchitecture (Csermely, 2001a). This may cause defects in signal transduction, protein transport, immune recognition, cellular organization as well as the appearance of previously buffered, hidden mutations in the phenotype of the cell (Csermely, 2001b). Chaperone overload may significantly decrease the robustness of cellular networks, as well as shift their function towards a more stochastic behavior. As a result of this, aging cells become more disorganized, their adaptation is impaired.
Fig. 1. Chaperone overload: a shift in the balance between misfolded proteins and available free chaperones in aging organisms. The accumulation of chaperone substrates along with an impaired chaperone function may exhaust the folding assistance to specific chaperone targets and leads to deterioration in vital processes. Chaperone overload may significantly decrease the robustness of cellular networks, and compromise the adaptative responses. See text for details.
Senescent cells and chaperones The involvement of chaperones in aging at the cellular level is recently reviewed (So˝ti et al., 2003). Non-dividingsenescent-peripheral cells tend to have increased chaperone levels (Verbeke et al., 2001), and cannot preserve the induction of several chaperones (Liu et al., 1989), similarly to cells from aged animals. Activation and binding of HSF to the heat shock element is decreased in aged cells (Choi et al., 1990). Interestingly, cellular senescence seems to unmask a proteasomal activity leading to the degradation of HSF (Bonelli et al., 2001). Chaperone induction per se seems to counteract senescence. Repeated mild heat shock (a kind of hormesis) has been reported to delay fibroblast aging (Verbeke et al., 2001), though it does not seem to extend replicative lifespan. A major chaperone, Hsp90 is required for the correct function of telomerase, an important enzyme to extend the life-span of cells (Holt et al., 1999). Mortalin (mtHsp70/Grp75), a member of the Hsp70 family, produces opposing phenotypic effects related to its localization. In normal cells, it is pancytoplasmically distributed, and its expression causes senescence. Its upregulation and perinuclear distribution, however, is connected to transformation, probably via p53 inactivation. Mortalin also induces life-span extension in human fibroblasts or in C. elegans harboring extra copies of the orthologous gene (Kaul et al., 2002).
Aging organisms and chaperones: age-related diseases Unbalanced chaperone requirement and chaperone capacity in aged organisms helps the accumulation of aggregated proteins, which often cause folding diseases, mostly of the nervous system, due to the very limited proliferation potential of neurons. Over expression of chaperones often delays the onset or diminishes the symptoms of the disease (So˝ti and Csermely, 2002b). Other aging diseases, such as atherosclerosis and cancer are also related to chaperone action. Here space limitation precludes a detailed description of these rapidly developing fields, however, numerous recent reviews were published on these subjects, where the interested readers may find a good summary and several hints for further readings (Ferreira and Carlos, 2002; Neckers, 2002; Sarto et al., 2000; Wick and Xu, 1999).
Chaperones and Longevity
Increased chaperone induction leads to increased longevity (Tatar et al., 1997). Moreover, a close correlation exists between stress resistance and longevity in several long-lived C. elegans and Drosophila mutants (Lithgow and Kirkwood, 1996). As the other side of the same coin, damaged HSF has been found as an important gene to cause accelerated aging in C. elegans (Garigan et al., 2002). Caloric restriction, the only effective experimental manipulation known to retard aging in rodents and primates (Ramsey et al., 2000), restores age-impaired chaperone induction, while reversing the age-induced changes in constitutive Hsp levels (see So˝ti and Csermely, 2002a,b). These examples confirm the hypothesis that a better adaptation capacity to various stresses greatly increases the chances to reach longevity. 10. Conclusions and perspectives Aging can be defined as a multicausal process leading to a gradual decay of self-defensive mechanisms, and an exponential accumulation of damage at the molecular, cellular and organismal level. The protein oxidation, damage, misfolding and aggregation together with the simultaneously impaired function and induction of chaperones in aged organisms disturb the balance between chaperone requirement and availability. There are several important aspects for future investigation of this field: † the measurement of active chaperone function (i.e. chaperone-assisted refolding of damaged proteins) in cellular extracts does not have a well-established method yet; † we have no methods to measure free chaperone levels; † among the consequences of chaperone overload, changes in signal transduction, protein transport, immune recognition and cellular organization have not been systematically measured and/or related to the protein folding homeostasis of aging organisms and cells.
-
Extracellular HSPs in inflammation and immunity
Cutting Edge: Heat Shock Protein (HSP) 60 Activates the Innate Immune Response: CD14 Is an Essential Receptor for HSP60 Activation of Mononuclear Cells1
Amir Kol,* Andrew H. Lichtman,† Robert W. Finberg,‡ Peter Libby,*† and Evelyn A. Kurt-Jones2‡
J Immunol 2000; 164: 13–17. https://www.researchgate.net/profile/Robert_Finberg/publication/12696457_Cutting_Edge_Heat_Shock_Protein_(HSP)_60_Activates_the_Innate_Immune_Response_CD14_Is_an_Essential_Receptor_for_HSP60_Activation_of_Mononuclear_Cells/links/53ee00460cf23733e80b21c0.pdf
Heat shock proteins (HSP), highly conserved across species, are generally viewed as intracellular proteins thought to serve protective functions against infection and cellular stress. Recently, we have reported the surprising finding that human and chlamydial HSP60, both present in human atheroma, can activate vascular cells and macrophages. However, the transmembrane signaling pathways by which extracellular HSP60 may activate cells remains unclear. CD14, the monocyte receptor for LPS, binds numerous microbial products and can mediate activation of monocytes/macrophages and endothelial cells, thus promoting the innate immune response. We show here that human HSP60 activates human PBMC and monocyte-derived macrophages through CD14 signaling and p38 mitogen-activated protein kinase, sharing this pathway with bacterial LPS. These findings provide further insight into the molecular mechanisms by which extracellular HSP may participate in atherosclerosis and other inflammatory disorders by activating the innate immune system.
There is increasing interest in the role of nontraditional mediators of inflammation in atherosclerosis (1). Recent studies from our laboratory have shown that chlamydial and human heat shock protein 60 (HSP60)3 colocalize in human atheroma (2), and either HSP60 induces adhesion molecule and cytokine production by human vascular cells and macrophages, in a pattern similar to that induced by Escherichia coli LPS (3, 4). These results suggested that HSP60 and LPS might share similar signaling mechanisms. CD14 is the major high-affinity receptor for bacterial LPS on the cell membrane of mononuclear cells and macrophages (5, 6). In addition to LPS, CD14 functions as a signaling receptor for other microbial products, including peptidoglycan from Gram-positive bacteria and mycobacterial lipoarabinomann (7, 8). CD14 is considered a pattern recognition receptor for microbial Ags and, with Toll-like receptor (TLR) proteins, an important mediator of innate immune responses to infection (9–14). We have examined the role of CD14 in the response of human monocytes and macrophages to HSP60. …..
HSP may play a central role in the innate immune response to microbial infections. Because both microbes and stressed or injured host cells produce abundant HSP (36), and dying cells likely release these proteins, it is conceivable that HSP furnish signals that inform the innate immune system of the presence of infection and cell damage. The findings reported here, that human HSP60 induces IL-6 production by mononuclear cells and macrophages via the CD14, supports this hypothesis, suggesting that human HSP60 may act together with LPS or other microbial products to provoke innate immune responses.
Inflammation and immunity can contribute to the pathogenesis and complications of atherosclerosis (37). Moreover, the search for novel risk factors for atherosclerosis has revived the concept that microbial products might substantially contribute to the inflammatory reaction in the atheromatous vessel wall (38, 39). We have shown that chlamydial HSP60 colocalizes with human HSP60 in the macrophages of human atheroma (2). Therefore, bacterial and human HSP60, released from dying or injured cells during atherogenesis (40) or myocardial injury (41), may further promote local inflammation and possibly activate the innate immune system. Previous reports that immunization with mycobacterial HSP65 enhances atheroma formation in rabbits (42), have suggested an important role for HSPs in atherogenesis, particularly because the high degree of homology between HSPs of the same m.w. among different species might stimulate autoimmunity (43).
In conclusion, our findings, that CD14 mediates cellular activation induced by human HSP60 provide further insight into the molecular mechanisms by which HSP may activate the innate immune system and participate in atherogenesis and other inflammatory disorders.
DAMPs, PAMPs and alarmins: all we need to know about danger
Marco E. Bianchi1
J. Leukoc. Biol. 81: 1–5; 2007. http://aerozon.ru/documents/publications/37_Bianche.pdf
Multicellular animals detect pathogens via a set of receptors that recognize pathogen associated molecular patterns (PAMPs). However, pathogens are not the only causative agents of tissue and cell damage: trauma is another one. Evidence is accumulating that trauma and its associated tissue damage are recognized at the cell level via receptor-mediated detection of intracellular proteins released by the dead cells. The term “alarmin” is proposed to categorize such endogenous molecules that signal tissue and cell damage. Intriguingly, effector cells of innate and adaptive immunity can secrete alarmins via nonclassical pathways and often do so when they are activated by PAMPs or other alarmins. Endogenous alarmins and exogenous PAMPs therefore convey a similar message and elicit similar responses; they can be considered subgroups of a larger set, the damage associated molecular patterns (DAMPs).
Multicellular animals must distinguish whether their cells are alive or dead and detect when microorganisms intrude, and have evolved surveillance/defense/repair mechanisms to this end. How these mechanisms are activated and orchestrated is still incompletely understood, and I will argue that that these themes define a unitary field of investigation, of both basic and medical interest.
A complete system for the detection, containment, and repair of damage caused to cells in the organism requires warning signals, cells to respond to them via receptors and signaling pathways, and outputs in the form of physiological responses. Classically, a subset of this system has been recognized and studied in a coherent form: pathogen-associated molecular patterns (PAMPs) are a diverse set of microbial molecules which share a number of different recognizable biochemical features (entire molecules or, more often, part of molecules or polymeric assemblages) that alert the organism to intruding pathogens [1]. Such exogenous PAMPs are recognized by cells of the innate and acquired immunity system, primarily through toll-like receptors (TLRs), which activate several signaling pathways, among which NF-kB is the most distinctive. As a result, some cells are activated to destroy the pathogen and/or pathogen-infected cells, and an immunological response is triggered in order to produce and select specific T cell receptors and antibodies that are best suited to recognize the pathogen on a future occasion. Most of the responses triggered by PAMPs fall into the general categories of inflammation and immunity.
However, pathogens are not the only causative agents of tissue and cell damage: trauma is another one. Tissues can be ripped, squashed, or wounded by mechanical forces, like falling rocks or simply the impact of one’s own body hitting the ground. Animals can be wounded by predators. In addition, tissues can be damaged by excessive heat (burns), cold, chemical insults (strong acids or bases, or a number of different cytotoxic poisons), radiation, or the withdrawal of oxygen and/or nutrients. Finally, humans can also be damaged by specially designed drugs, such as chemotherapeutics, that are meant to kill their tumor cells with preference over their healthy cells. Very likely, we would not be here to discuss these issues if evolution had not incorporated in our genetic program ways to deal with these damages, which are not caused by pathogens but are nonetheless real and common enough. Tellingly, inflammation is also activated by these types of insults. A frequently quoted reason for the similarity of the responses evoked by pathogens and trauma is that pathogens can easily breach wounds, and infection often follows trauma; thus, it is generally effective to respond to trauma as if pathogens were present. In my opinion, an additional reason is that pathogens and trauma both cause tissue and cell damage and thus trigger similar responses.
None of these considerations is new; however, a new awareness of the close relationship between trauma- and pathogenevoked responses emerged from the EMBO Workshop on Innate Danger Signals and HMGB1, which was held in February 2006 in Milano (Italy); many of the findings presented at the meeting are published in this issue of the Journal of Leukocyte Biology. At the end of the meeting, Joost Oppenheim proposed the term “alarmin” to differentiate the endogenous molecules that signal tissue and cell damage. Together, alarmins and PAMPs therefore constitute the larger family of damage-associated molecular patterns, or DAMPs.
Extranuclear expression of HMGB1 has been involved in a number of pathogenic conditions: sepsis [44], arthritis [45, 46], atherosclerosis [10], systemic lupus erythematosus (SLE) [47], cancer [48] and hepatitis [49, this issue]. Uric acid has been known to be the aethiologic agent for gout since the 19th century. S100s may be involved in arthritis [31, this issue] and psoriasis [50]. However, although it is clear that excessive alarmin expression might lead to acute and chronic diseases, the molecular mechanisms underlying these effects are still largely unexplored.
The short list of alarmins presented above is certainly both provisional and incomplete and serves only as an introduction to the alarmin concept and to the papers published in this issue of JLB. Other molecules may be added to the list, including cathelicidins, defensins and eosinophil-derived neurotoxin (EDN) [51], galectins [52], thymosins [53], nucleolin [54], and annexins [55; and 56, this issue]; more will emerge with time. Eventually, the concept will have to be revised and adjusted to the growing information. Indeed, I have previously argued that any misplaced protein in the cell can signal damage [57], and Polly Matzinger has proposed that any hydrophobic surface (“Hyppo”, or Hydrophobic protein part) might act as a DAMP [58]. As most concepts in biology, the alarmin category serves for our understanding and does not correspond to a blueprint or a plan in the construction of organisms. Biology proceeds via evolution, and evolution is a tinkerer or bricoleur, finding new functions for old molecules. In this, the reuse of cellular components as signals for alerting cells to respond to damage and danger, is a prime example.
-
Role of heat shock and the heat shock response in immunity and cancer
Heat Shock Proteins: Conditional Mediators of Inflammation in Tumor Immunity
Stuart K. Calderwood,1,* Ayesha Murshid,1 and Jianlin Gong1
Front Immunol. 2012; 3: 75. doi: 10.3389/fimmu.2012.00075
Heat shock protein (HSP)-based anticancer vaccines have undergone successful preclinical testing and are now entering clinical trial. Questions still remain, however regarding the immunological properties of HSPs. It is now accepted that many of the HSPs participate in tumor immunity, at least in part by chaperoning tumor antigenic peptides, introducing them into antigen presenting cells such as dendritic cells (DC) that display the antigens on MHC class I molecules on the cell surface and stimulate cytotoxic lymphocytes (CTL). However, in order for activated CD8+ T cells to function as effective CTL and kill tumor cells, additional signals must be induced to obtain a sturdy CTL response. These include the expression of co-stimulatory molecules on the DC surface and inflammatory events that can induce immunogenic cytokine cascades. That such events occur is indicated by the ability of Hsp70 vaccines to induce antitumor immunity and overcome tolerance to tumor antigens such as mucin1. Secondary activation of CTL can be induced by inflammatory signaling through Toll-like receptors and/or by interaction of antigen-activated T helper cells with the APC. We will discuss the role of the inflammatory properties of HSPs in tumor immunity and the potential role of HSPs in activating T helper cells and DC licensing.
Heat shock protein, vaccine, inflammation, antigen presentation
Heat shock proteins (HSP) were first discovered as a group of polypeptides whose level of expression increases to dominate the cellular proteome after stress (Lindquist and Craig, 1988). These increases in HSPs synthesis correlate with a marked resistance to potentially toxic stresses such as heat shock (Li and Werb,1982). The finding that such proteins have extracellular immune functions suggested that, as highly abundant intracellular proteins they could be prime candidates as danger signals to the immune response (Srivastava and Amato,2001). There are several human HSP gene families with known immune significance and their classification is reviewed in Kampinga et al. (2009). These include the HSPA (Hsp70) family, which includes the HPA1A and HSPA1B genes encoding the two major stress-inducible Hsp70s, that together are often referred to as Hsp72. When referring to Hsp70 in this chapter, we generally refer to the products of these two genes. The Hsp70 family also includes two other members with immune function – HSPA8 and HSPA5 genes, whose protein products are known as Hsc70 the major constitutive Hsp70 family member and Grp78, a key ER-resident protein. In addition two more Hsp70 related genes have immune significance and these include HSPH2 (Hsp110) and HSPH4 the ER-resident class H protein Grp170. The Hsp90 family also has major functions in tumor immunity and these include HSPC2 and HSPC3, which encode the major cytoplasmic proteins Hsp90a and Hsp90b, and HSPC4 that encodes ER chaperone Grp94. In addition, the product of the HSPD1 gene, the mitochondrial chaperone Hsp60 has some immunological functions. Mice have been shown to encode orthologs of each of these genes (Kampinga et al., 2009).
It has been suggested that many of the HSPs have the property of damage associated molecular patterns (DAMPs), inducers of sterile inflammation and innate immunity (Kono and Rock, 2008). The additional discovery that intracellular HSPs function as molecular chaperones and can bind to a wide spectrum of intracellular polypeptides further indicated that they could play a broad role in the immune response and might mediate both innate immunity due to their status as DAMPs and adaptive immunity by chaperoning antigens.
Heat shock proteins are currently employed as vaccines in cancer immunotherapy (Tamura et al., 1997; Murshid et al., 2011a). The rationale behind the approach is that if HSPs can be extracted from tumor tissue bound to the polypeptides which they chaperone during normal metabolism, they may retain antigenic peptides specific to the tumor (Noessner et al., 2002; Srivastava, 2002; Wang et al., 2003; Enomoto et al., 2006; Gong et al., 2010). Indeed, vaccines based on Hsp70, Hsp90, Grp94, Hsp110, and Grp170 polypeptide complexes have been used successfully to immunize mice to a range of tumor types and Hsp70 and Grp94 vaccines have undergone recent clinical trials (rev: Murshid et al., 2011a). These effects of the HSP vaccines on tumor immunity appear to be mediated largely to the associated, co-isolated tumor polypeptides, although in the case of Grp94 this question is still controversial and tumor regression was observed in mice treated with the chaperone devoid of its peptide binding domain (Udono and Srivastava, 1993; Srivastava, 2002; Nicchitta, 2003; Chandawarkar et al., 2004; Nicchitta et al.,2004). Use of such HSP vaccines is potentially a powerful approach to tumor immunotherapy as the majority of the antigenic repertoire of most individual tumor cells is unknown (Srivastava and Old, 1988; Srivastava, 1996). Individual cancer cells are likely to take a lone path in accumulating a spectrum of random mutations. Although some mutations are functional, permitting cells to become transformed and to progress into a highly malignant state, many such changes are likely to be passenger mutations not required to drive tumor growth (Srivastava and Old, 1988; Srivastava, 1996). Some of these individual mutant sequences will be novel antigenic epitopes and together with the few known shared tumor antigens comprise an “antigenic fingerprint” for each individual tumor (Srivastava,1996). Accumulation of mutations in cancer appears to be related to, and may drive the increases in HSPs observed in many tumors (Kamal et al., 2003; Whitesell and Lindquist, 2005; Trepel et al., 2010). As the mutant conformations of tumor proteins are “locked in” due to the covalent nature of the alterations, cancer cells appear to be under permanent proteotoxic stress and rich in HSP expression (Ciocca and Calderwood, 2005). For tumor immunology these conditions may offer a therapeutic opportunity as individual HSPs, whose expression is expanded in cancer will chaperone a cross-section of the “antigenic fingerprint” of the individual tumors (Murshid et al., 2011a). This approach was first utilized by Srivastava (2000, 2006) and led to the development of immunotherapy using HSP–peptide complexes.
In addition to using HSP–peptide complexes extracted from tumors, in cases where tumor antigens are known, these can be directly loaded onto purified or recombinant HSPs and the complex used as a vaccine. This procedure has been carried out successfully in the case of the “large HSPs,” Hsp110 and Grp170 (Manjili et al., 2002, 2003). A variant of this approach employs the molecular engineering of tumor antigens in order to produce molecular chaperone-fusion genes which encode products in which the HSP is fused covalently to the antigen. The fusion proteins are then employed as vaccines. This approach was pioneered by Young et al. who showed that a fusion between mycobacterial Hsp70 and ovalbumin could induced cytotoxic lymphocytes (CTL) in mice with the capacity to kill Ova-expressing cancer cells (Suzue et al., 1997). The vaccines could be used effectively without adjuvant and adjuvant properties were ascribed to the molecular chaperone component of the fusion protein. Subsequent studies have confirmed the utility of the approach in targeting common tumor antigens such as the melanoma antigen Mage3 (Wang et al., 2009).
HSPs and Immunosurveillance in Cancer
The question next arises as to the role of endogenous HSPs, with or without bound antigens in immunosurveillance of cancer cells. Although the immune system can recognize tumor antigens and generate a CTL response, most cancers evade immune cell killing by a range of strategies (van der Bruggen et al., 1991; Pardoll,2003). These include the down-regulation of surface MHC class I molecules by individual tumor cells and release of immunosuppressive IL-10 by tumors (Moller and Hammerling, 1992; Chouaib et al., 2002). Tumors in vivo also appear to attract a range of hematopoietic cells with immunosuppressive action including regulatory CD4+CD25+FoxP3+ T cells (Treg), M2 macrophages, myeloid-derived suppressor cells (MDSC) and some classes of natural killer cells (Pekarek et al.,1995; Terabe et al., 2005; Mantovani et al., 2008; Marigo et al., 2008). The tumor milieu also contain a small fraction of cells of mesenchymal origin identified by surface fibroblast activation protein-a (FAP cells) that suppress antitumor immune responses (Kraman et al., 2010). Endogenous tumor HSPs may also participate in immune suppression. Although the majority of the HSPs function as intracellular molecular chaperones, a fraction of these proteins can be released from cells even under unstressed conditions and may participate in immune functions (rev: Murshid and Calderwood, 2012). Intracellular Hsp70 can be actively secreted from tumor cells in either free form or packaged into lipid-bounded structures called exosomes (Mambula and Calderwood, 2006b; Chalmin et al., 2010). In addition Hsp70 and Hsp90 can also be found associated with the surfaces of tumor cells where they can function as molecular chaperones or as recognition structures for immune cells (Sidera et al., 2008; Qin et al., 2010; Multhoff and Hightower, 2011). As Hsp70 was shown in a number of earlier studies to be pro-inflammatory due to its interaction with pattern recognition receptors such as Toll-like receptors 2 and 4 (TLR2 and TLR4), these findings might suggest, as mentioned above, that Hsp70 released by tumors could be pro-inflammatory and possess the properties of DAMPs (Asea et al., 2000, 2002; Vabulas et al., 2002). However, subsequent studies indicated that a portion of the TLR4 activation detected in the earlier reports, involving exposure of monocytes, macrophages, or dendritic cells (DC) to HSPs in vitro may be due to trace contamination with bacterial pathogen associated molecular patterns (PAMPs), potent TLR activators (Tsan and Gao,2004). In spite of these drawbacks, an overwhelming amount of evidence now seems to indicate the interaction of Hsp70 and other HSPs with TLRs (particularly TLR4) in vivo – in a wide range of physiological and pathological conditions, leading to acute inflammation in many conditions (Chase et al., 2007; Wheeler et al., 2009; see Appendix for a full list of references). Thus both TLR2 and TLR4 appear to be important components of inflammatory responses to Hsp70 under many pathophysiological conditions. In cancer therapy it has been shown that autoimmunity can be triggered in mice through necrotic killing of melanocytes engineered to overexpress Hsp70; such treatment led to the concomitant immune destruction of B16 melanoma tumors that share patterns of antigen expression with the killed melanocytes (Sanchez-Perez et al., 2006). Hsp70 appears to play an adjuvant role in this form of therapy through its interaction with TLR4 and induction of the cytokine TNF-a (Sanchez-Perez et al., 2006). However, despite these findings it has also been shown that depletion of Hsp70 in cancer cells can, in the absence of other treatments lead to tumor regression by inducing antitumor immunity (Rerole et al., 2011). This effect appears to be due to the secretion by cancer cells of immunosuppressive exosomes containing Hsp70 that activate MDSC and lead to local immunosuppression (Chalmin et al., 2010). Under normal circumstances therefore, release of endogenous Hsp70 into the extracellular microenvironment may be a component of the tumor defenses against immunosurveillance. Extracellular Hsp60 has also been shown be immunomodulatory and can increase levels of FoxP3 Treg in vitro and suppress T cell-mediated immunity (de Kleer et al., 2010; Aalberse et al., 2011).
The pro-inflammatory properties of extracellular HSPs may be more evident underin vivo situations particularly in the context of tissue damage (Sanchez-Perez et al.,2006). For instance when elevated temperatures were used to boost Hsp70 release from Lewis Lung carcinoma cells in vivo, antitumor immunity was activated along with release of chemokines CCL2, CCL5, and CCL10, in a TLR4-dependent manner, leading to attraction of DC and T cells into the tumor (Chen et al., 2009). Thus under resting conditions, the tumor milieu appears to be a specialized immunosuppressive environment, rich in inhibitory cells such as Treg, MDSC, and M2 macrophages and inaccessible to “exhausted” CD8+ T cells that often fail to penetrate the tumor microcirculation. However, under inflammatory conditions involving necrotic cell killing of tumor cells, extracellular HSPs may be able to amplify the anticancer immune response, intracellular HSPs may be released to further increase such a response and CTL may triggered to penetrate the tumor milieu, inducing antigen-specific cancer cell killing (Evans et al., 2001; Mambula and Calderwood, 2006a; Sanchez-Perez et al., 2006; Chen et al., 2009).
HSP-Based Anticancer Vaccines
It is apparent that a number of HSP types, conjugated to peptide complexes (HSP.PC) from cancer cells form effective bases for immunotherapy approaches with unique properties, as mentioned above (Calderwood et al., 2008; Murshid et al., 2011a). The immunogenicity of most HSP.PC appears to involve the ability of the HSPs to sample the tumor “antigenic fingerprint,” deliver the antigens to antigen presenting cells (APC) such as DC and stimulate activation of CTL (Tamura et al., 1997; Singh-Jasuja et al., 2000b; Wang et al., 2003; Murshid et al.,2010). A number of studies show that HSPs can chaperone tumor antigens and deliver them to the appropriate destination – MHC class I molecules on the DC surface (Singh-Jasuja et al., 2000a,b; Srivastava and Amato, 2001; Delneste et al.,2002; Enomoto et al., 2006; Gong et al., 2009). In addition, Hsp70 has been shown to chaperone viral antigenic peptides and increase cross priming of human CTL under ex vivo conditions (Tischer et al., 2011). However, it is still far from clear how the process of HSP-mediated cross priming unfolds. For instance, the CD8+ expressing DC subpopulation in lymph nodes is regarded as the primary cross-presenting APC (Heath and Carbone, 2009). It is not however currently known whether the CD8+ DC subset or other peripheral or lymph-node resident, DC interact with HSP vaccines to induce cross presentation. HSPs appear to be able to enter APC, such as mouse bone marrow derived DC (BMDC) and human DC in a receptor-mediated manner (Basu et al., 2001; Delneste et al., 2002; Gong et al.,2009; Murshid et al., 2010). However, no unique endocytosing HSP receptor has emerged and HSP–antigen complexes appear instead to be taken up by proteins with “scavenger” function such as LOX-1, SRECI, and CD91 that can each take up a wide range of extracellular ligands (Basu et al., 2001; Delneste et al., 2002; Theriault et al., 2006; Murshid et al., 2010). A pathway for Hsp90–peptide (Hsp90.PC) uptake has been characterized in mouse BMDC by scavenger receptor SRECI (Murshid et al., 2010). SRECI is able to mediate the whole process of Hsp90.PC endocytosis, trafficking through the cytoplasm to the sites of antigen processing and presentation of antigens to CD8+ T lymphocytes on MHC class I molecules (Murshid et al., 2010). This process is known as antigen cross presentation (Kurts et al., 2010). It is not currently clear what the relative contribution to antigen cross presentation of the various HSP receptors might be under in vivo conditions. It may be that each receptor class contributes to an individual aspect of CTL activation by HSP peptide complexes although a definitive understanding may await studies in mice deficient in each receptor class.
HSPs and CTL Programming
It is evident that that HSPs can mediate antigen cross presentation and activate CD8+ T lymphocytes. However, presentation of tumor antigens by DC is not sufficient for CTL programming and, in the absence of co-stimulatory molecules and innate immunity, the “helpless” CD8+ cells will cease to proliferate abundantly and will most likely undergo apoptosis (Schurich et al., 2009; Kurts et al., 2010). One mechanism for enhancing CTL programming involves activation of the TLR pathways that lead to synthesis of co-stimulatory molecules (Rudd et al.,2009; Yamamoto and Takeda, 2010). The co-stimulatory molecules, including CD80 and CD86 then become expressed on the DC cell surface and amplify the signals induced by binding of the T cell receptor on CD8+ T cells to MHC class I peptide complexes on the presenting DC (Parra et al., 1995; Rudd et al., 2009). This process is important in pathogen infection in which microbially derived antigens are encountered in the presence of inflammatory PAMPs that can activate innate immune transcriptional networks. Originally it had been thought that HSPs could provide analogous stimulation through their suspected activity as DAMPs and their inbuilt ability to trigger innate immunity through TLR2 and TLR4 on DC (Asea et al., 2000, 2002; Vabulas et al., 2002). (The potential role of HSPs as DAMPs has been the subject of a recent review: van Eden et al., 2012). Subsequent studies on the capacity of HSPs to bind TLRs do not indicate avid binding of Hsp70 to either TLR2 or TLR4 when expressed in cells deficient in HSP receptors in vitro (Theriault et al., 2006). In vivo however, TLR signaling is essential for Hsp70 vaccine-induced tumor cell killing. Studies of tumor-bearing mice treated with an Hsp70 vaccine in vivo indicated that vaccine function is depleted by knockout of the TLR signaling intermediate Myd88 and completely abrogated by double knockout of TLR2 and TLR4 (Gong et al., 2009). These findings were somewhat complicated by the fact that TLR4 is involved in upstream regulation of the expression of Hsp70 receptor SRECI, but do strongly implicate a role for these receptors in amplifying immune signaling by Hsp70 vaccines and Hsp70-based immunotherapy (Sanchez-Perez et al., 2006; Gong et al., 2009). It is still not clear to what degree HSPs are capable of providing a sturdy DC maturing signal through TLR2/TLR4. The potency of HSP anticancer vaccines could potentially be improved by addition of PAMPs such as CpG DNA shown to activate TLR9, or double stranded RNA that can activate TLR3 (Murshid et al., 2011a). As mentioned, one contradictory factor in the earlier studies was that, although TLR2 and TLR4 are required for a sturdy Hsp70 vaccine-mediated immune response, direct binding of Hsp70 to these receptors was not observed (Theriault et al., 2006; Gong et al., 2009; Murshid et al., 2012). A rationale for these findings might be that HSPs can activate TLR signaling indirectly through primary binding to established HSP receptors such as LOX-1 and SRECI which secondarily recruit and activate the TLRs (Murshid et al., 2011b). Both of these scavenger receptors bind to TLR2 upon stimulation and activate TLR2-based signaling (Jeannin et al., 2005; A. Murshid and SK Calderwood, in preparation). In addition, we have found that Hsp90–SRECI complexes move to the lipid raft compartment of the cell, an environment highly enriched in TLR2 and TLR4 (Triantafilou et al., 2002; Murshid et al., 2010).
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3342006/bin/fimmu-03-00075-g001.jpg
Heat shock protein–peptide complexes extracted from tumor cells interact with endocytosing receptors (HSP-R) such as SRECI or signaling receptors (TLR) such as TLR4 on DC. SREC1 mediates uptake and intracellular processing of antigens and the presentation of resulting peptides on surface MHC class I and MHC class II proteins. MHC class II receptor–peptide complexes then bind to T cell receptors on CD4+ cells. One consequence of binding is interaction of CD40 ligand on the MHC class II cell with CD40 on the DC leading to the licensing interaction that results in enhanced expression of co-stimulatory proteins on the DC cell surface. The licensed DC may then interact with CD8+ T cells through T cell interaction with MHC class I peptide complexes. This effect will be enhanced by simultaneous interaction of CD80 or Cd86 co-stimulatory complexes on the DC with CD28 on the CD8+ cells, leading to effective CD8+ CTL that can lyse tumor cells. T cell programming can also be amplified by signals emanating from activated TLR that can boost levels of CD80 and CD86 as well as inflammatory cytokines (not shown).
Hsp70, Cell Damage, and Inflammation
The question of whether Hsp70 acts as DAMP and could by itself induce an inflammatory response in cancer patients in vivo is still open. However, some recent studies by Vile et al. using a gene therapy approach may shed some light on the inflammatory role of Hsp70 in tumor therapy. In this approach, as mentioned above, normal murine tissues were engineered to express high Hsp70 levels then subjected to treatments that lead to necrotic killing. The aim was to stimulate an autoimmune response that could lead to bystander immune killing of tumor cells that share the antigenic repertoire as the killed normal cells (Sanchez-Perez et al.,2006). In the initial studies, normal melanocytes were preloaded with Hsp70 plasmids and then necrotic cell death was triggered (Daniels et al., 2004). This treatment led to T cell-mediated immune killing of syngeneic B16 melanoma cells transplanted at a distant site in the mouse, presumably in response to antigens shared by the killed normal melanocytes and melanoma cell (Daniels et al., 2004). This effect only occurred when melanocytes were induced to undergo necrosis and Hsp70 levels were elevated, indicating a role for high levels of Hsp70 in the tumor specific immune response. Interestingly, these conditions did not lead to a prolonged autoimmune response, an effect mediated by the induction of a delayed Treg response (Srivastava, 2003; Daniels et al., 2004). It is notable that some early studies of chaperone-based tumor vaccines in animal models demonstrated a primary CTL response to tumors in response to treatment followed by delayed activation of a Treg reaction, and that chaperone levels must be carefully titrated for effective induction of tumor immunity (Udono and Srivastava, 1993; Liu et al.,2009). The role of Hsp70 in autoimmune rejection of tumors was also investigated in prostate cancer (Kottke et al., 2007). Ablation of normal prostate cells by necrotic killing with fusogenic viruses in the absence of Hsp70 elevation led to the induction of the cytokines IL-10 and TGF-b in the mouse prostate and a Treg response. However, when Hsp70 levels were elevated in these cells, IL-10, TGF-b, and IL-6 were induced simultaneously, the IL-6 component leading to further induction of IL-17, a profound Th17 response and tumor rejection (Kottke et al.,2007). Thus elevated levels of Hsp70, presumably released from cells undergoing necrosis can influence the local cytokine patterns and lead to an inflammatory statein vivo. Interestingly, these results seem to be tissue specific as inflammatory killing of pancreatic cells even in the presence of elevated Hsp70 did not provoke IL-6 release, a Th17 response or tumor rejection and the Treg response dominated under these conditions (Kottke et al., 2009). Thus the role of Hsp70 in tissue inflammation and tumor rejection seems to require elevated concentrations of extracellular chaperones, significant levels of necrotic cell killing, and tissue specific cytokine release.
Conclusion
- Earlier studies investigating HSP vaccines considered such structures to be the “Swiss penknives” of immunology able to deliver antigens directly to APC and confer a maturing signal that could render DC able to effectively program CTL (Srivastava and Amato, 2001; Noessner et al., 2002). It is well established now that Hsp70, Hsp90, Hsp110, and GRP170 can chaperone tumor antigens and activate antigen cross presentation (Murshid et al., 2011a). In addition, HSPs were thought to be DAMPs with ability to strongly activate TLR signaling and innate immunity (Asea et al., 2000). However, although there is compelling evidence to indicate that Hsp70, for instance can interact with TLR4 under a number of pathological situations (see Appendix, Sanchez-Perez et al., 2006), it remains unclear whether free Hsp70 binds directly to the Toll-like receptor and induces innate immunity in the absence of other treatments in vitro(Tsan and Gao, 2004).
- Elevated levels of extracellular HSPs appear to have the capacity to amplify the effects of inflammatory signals emanating from necrotic cells in vivoin a TLR4-dependent manner (Daniels et al., 2004; Sanchez-Perez et al., 2006; Kottke et al., 2007). In the presence of cell injury and death, elevated levels of Hsp70 appear to increase the production of inflammatory signals that involve cytokines such as IL-6 and IL-17 and lead to a specific T cell-mediated immune response to tumor cells sharing antigens with the dying cells (Kottke et al., 2007). The mechanisms involved in these processes are not clear although one possibility is that HSPs can induce the engulfment of necrotic cells. Hsp70 has been shown to increase bystander engulfment of a variety of structures (Wang et al., 2006a,b). In addition, tumor cells treated with elevated temperatures release inflammatory chemokines in an Hsp70 and TLR4-dependent mechanisms and this effect may be significant in CTL programming and tumor cell killing (Chen et al., 2009). Our studies indicate that CTL induction by Hsp70 vaccines in vivo has an absolute requirement for TLR2 and TLR4 suggesting that at least in vivo HSPs can trigger innate immunity through TLR signaling (Gong et al., 2009).
- HSPs appear also to be able to direct antigen presentation through the class II pathway in DC and may stimulate T helper cells (Gong et al., 2009). It may thus be possible that HSPs participate in DC licensing and reinforce CTL programming during exposure to HSP vaccines. Future studies will address these questions.
- A further interesting consideration is whether HSPs released from untreated tumor cells enhance or depress tumor immunity. One initial study shows that Hsp70 released from tumor cells in exosomes can strongly decrease tumor immunity through effects on MDSC (Chalmin et al., 2010). Further studies will be required to make a definitive statement on these questions.
-
Protein aggregation disorders and HSP expression
Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1
Christopher J. Cummings1,5, Michael A. Mancini3, Barbara Antalffy4, Donald B. DeFranco7, Harry T. Orr8 & Huda Y. Zoghbi1,2,6
Nature Genetics 19, 148 – 154 (1998) http://dx.doi.org:/10.1038/502
Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant neurodegenerative disorder caused by expansion of a polyglutamine tract in ataxin-1. In affected neurons of SCA1 patients and transgenic mice, mutant ataxin-1 accumulates in a single, ubiquitin-positive nuclear inclusion. In this study, we show that these inclusions stain positively for the 20S proteasome and the molecular chaperone HDJ-2/HSDJ. Similarly, HeLa cells transfected with mutant ataxin-1 develop nuclear aggregates which colocalize with the 20S proteasome and endogenous HDJ-2/HSDJ. Overexpression of wild-type HDJ-2/HSDJ in HeLa cells decreases the frequency of ataxin-1 aggregation. These data suggest that protein misfolding is responsible for the nuclear aggregates seen in SCA1, and that overexpression of a DnaJ chaperone promotes the recognition of a misfolded polyglutamine repeat protein, allowing its refolding and/or ubiquitin-dependent degradation.
Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease
Andreas Wyttenbach, Jenny Carmichael, Jina Swartz, Robert A. Furlong, Yolanda Narain, Julia Rankin, and David C. Rubinsztein*
https://www.researchgate.net/profile/David_Rubinsztein/publication/24447892_Effects_of_heat_shock_heat_shock_protein_40_(HDJ2)_and_proteasome_inhibition_on_protein_aggregation_in_cellular_models_of_Huntington’s_disease/links/00b7d528b80aab69bb000000.pdf
Huntington’s disease (HD), spinocerebellar ataxias types 1 and 3 (SCA1, SCA3), and spinobulbar muscular atrophy (SBMA) are caused by CAGypolyglutamine expansion mutations. A feature of these diseases is ubiquitinated intraneuronal inclusions derived from the mutant proteins, which colocalize with heat shock proteins (HSPs) in SCA1 and SBMA and proteasomal components in SCA1, SCA3, and SBMA. Previous studies suggested that HSPs might protect against inclusion formation, because overexpression of HDJ-2yHSDJ (a human HSP40 homologue) reduced ataxin-1 (SCA1) and androgen receptor (SBMA) aggregate formation in HeLa cells. We investigated these phenomena by transiently transfecting part of huntingtin exon 1 in COS-7, PC12, and SH-SY5Y cells. Inclusion formation was not seen with constructs expressing 23 glutamines but was repeat length and time dependent for mutant constructs with 43–74 repeats. HSP70, HSP40, the 20S proteasome and ubiquitin colocalized with inclusions. Treatment with heat shock and lactacystin, a proteasome inhibitor, increased the proportion of mutant huntingtin exon 1-expressing cells with inclusions. Thus, inclusion formation may be enhanced in polyglutamine diseases, if the pathological process results in proteasome inhibition or a heat-shock response. Overexpression of HDJ-2yHSDJ did not modify inclusion formation in PC12 and SH-SY5Y cells but increased inclusion formation in COS-7 cells. To our knowledge, this is the first report of an HSP increasing aggregation of an abnormally folded protein in mammalian cells and expands the current understanding of the roles of HDJ-2yHSDJ in protein folding.
-
Hsp70 in blood cell differentiation.
Apoptosis Versus Cell Differentiation -Role of Heat Shock Proteins HSP90, HSP70 and HSP27
David Lanneau, Aurelie de Thonel, Sebastien Maurel, Celine Didelot, and Carmen Garrido
Prion. 2007 Jan-Mar; 1(1): 53–60. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2633709/
Heat shock proteins HSP27, HSP70 and HSP90 are molecular chaperones whose expression is increased after many different types of stress. They have a protective function helping the cell to cope with lethal conditions. The cytoprotective function of HSPs is largely explained by their anti-apoptotic function. HSPs have been shown to interact with different key apoptotic proteins. As a result, HSPs can block essentially all apoptotic pathways, most of them involving the activation of cystein proteases called caspases. Apoptosis and differentiation are physiological processes that share many common features, for instance, chromatin condensation and the activation of caspases are frequently observed. It is, therefore, not surprising that many recent reports imply HSPs in the differentiation process. This review will comment on the role of HSP90, HSP70 and HSP27 in apoptosis and cell differentiation. HSPs may determine de fate of the cells by orchestrating the decision of apoptosis versus differentiation.
Key Words: apoptosis, differentiation, heat shock proteins, chaperones, cancer cells, anticancer drugs
Introduction
Stress or heat shock proteins (HSPs) were first discovered in 19621 as a set of highly conserved proteins whose expression was induced by different kinds of stress. It has subsequently been shown that most HSPs have strong cytoprotective effects and behave as molecular chaperones for other cellular proteins. HSPs are also induced at specific stages of development, differentiation and during oncogenesis.2 Mammalian HSPs have been classified into five families according to their molecular size: HSP100, HSP90, HSP70, HSP60 and the small HSPs. Each family of HSPs is composed of members expressed either constitutively or regulated inducibly, and/or targeted to different sub-cellular compartments. The most studied HSPs are HSP90, the inducible HSP70 (also called HSP72) and the small heat shock protein HSP27.
HSP90 is a constitutively abundant chaperone that makes up 1–2% of cytosolic proteins. It is an ATP-dependent chaperone that accounts for the maturation and functional stability of a plethora of proteins termed HSP90 client proteins. In mammals, HSP90 comprises 2 homologue proteins (HSP90α and HSP90β) encoded by separated but highly conserved genes that arose through duplication during evolution.3 Most studies do not differentiate between the two isoforms because for a long time they have been considered as having the same function in the cells. However, recent data and notably out-of-function experiments indicate that at least some functions of the beta isoform are not overlapped by HSP90α’s functions.4 HSP70, like HSP90, binds ATP and undergoes a conformational change upon ATP binding, needed to facilitate the refolding of denatured proteins. The chaperone function of HSP70 is to assist the folding of newly synthesized polypeptides or misfolded proteins, the assembly of multi-protein complexes and the transport of proteins across cellular membranes.5,6 HSP90 and HSP70 chaperone activity is regulated by co-chaperones like Hip, CHIP or Bag-1 that increase or decrease their affinity for substrates through the stabilization of the ADP or ATP bound state. In contrast to HSP90 and HSP70, HSP27 is an ATP-independent chaperone, its main chaperone function being protection against protein aggregation.7 HSP27 can form oligomers of more than 1000 Kda. The chaperone role of HSP27 seems modulated by its state of oligomerization, the multimer being the chaperone competent state.8 This oligomerization is a very dynamic process modulated by the phosphorylation of the protein that favors the formation of small oligomers. Cell-cell contact and methylglyoxal can also modulate the oligomerization of the protein.9
It is now well accepted that HSPs are important modulators of the apoptotic pathway. Apoptosis, or programmed cell death, is a type of death essential during embryogenesis and, latter on in the organism, to assure cell homeostasis. Apoptosis is also a very frequent type of cell death observed after treatment with cytotoxic drugs.10 Mainly, two pathways of apoptosis can be distinguished, although cross-talk between the two signal transducing cascades exists (Fig. 1). The extrinsic pathway is triggered through plasma membrane proteins of the tumor necrosis factor (TNF) receptor family known as death receptors, and leads to the direct activation of the proteases called caspases, starting with the receptor-proximal caspase-8. The intrinsic pathway involves intracellular stress signals that provoke the permeabilization of the outer mitochondrial membrane, resulting in the release of pro-apoptotic molecules normally confined to the inter-membrane space. Such proteins translocate from mitochondria to the cytosol in a reaction that is controlled by Bcl-2 and Bcl-2-related proteins.11 One of them is the cytochrome c, which interacts with cytosolic apoptosis protease-activating factor-1 (Apaf-1) and pro-caspase-9 to form the apoptosome, the caspase-3 activation complex.12Apoptosis inducing factor (AIF) and the Dnase, EndoG, are other mitochondria intermembrane proteins released upon an apoptotic stimulus. They translocate to the nucleus and trigger caspase-independent nuclear changes.13,14 Two additional released mitochondrial proteins, Smac/Diablo and Htra2/Omi, activate apoptosis by neutralizing the inhibitory activity of the inhibitory apoptotic proteins (IAPs) that associate with and inhibit caspases15 (Fig. 1).
Figure 1 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2633709/figure/F1/
Modulation of apoptosis and differentiation by HSP90, HSP70 and HSP27. In apoptosis (upper part), HSP90 can inhibit caspase (casp.) activation by its interaction with Apaf1. HSP90 stabilizes proteins from the survival signaling including RIP, Akt and …
Apoptosis and differentiation are two physiological processes that share different features like chromatin condensation or the need of caspase activity.16 It has been demonstrated in many differentiation models that the activation of caspases is preceded by a mitochondrial membrane depolarization and release of mitochondria apoptogenic molecules.17,18 This suggests that the mitochondrial-caspase dependent apoptotic pathway is a common intermediate for conveying apoptosis and differentiation. Timing, intensity and cellular compartmentalization might determine whether a cell is to die or differentiate. HSPs might be essential to orchestrate this decision. This review will describe the role of HSP90, HSP70 and HSP27 in apoptosis and cell differentiation.
HSP27, HSP70 and HSP90 are Anti-Apoptotic Proteins
Overexpression of HSP27, HSP70 or HSP90 prevents apoptosis triggered by various stimuli, including hyperthermia, oxidative stress, staurosporine, ligation of the Fas/Apo-1/CD95 death receptor or anticancer drugs.2,19–21 Downregulation or inhibition of HSP27, HSP70 or HSP90 have been shown to be enough to sensitize a cell to apoptosis, proving that endogenous levels of those chaperones seem to be sufficiently high to control apoptosis.22–24 It is now known that these chaperones can interact with key proteins of the apoptotic signaling pathways (Fig. 1).
HSP90: A survival protein through its client proteins.
HSP90 client proteins include a number of signaling proteins like ligand-dependent transcription factors and signal transducing kinases that play a role in the apoptotic process. Upon binding and hydrolysis of ATP, the conformation of HSP90 changes and the client protein, which is no longer chaperoned, is ubiquitinated and degraded by the proteasome.25
A function for HSP90 in the serine/threonine protein kinase Akt pathway was first suggested by studies using an HSP90 inhibitor that promoted apoptosis in HEK293T and resulted in suppressed Akt activity.26 A direct interaction between Akt and HSP90 was reported later.27 Binding of HSP90 protects Akt from protein phosphatase 2A (PP2A)-mediated dephosphorylation.26 Phosphorylated Akt can then phosphorylate the Bcl-2 family protein Bad and caspase-9 leading to their inactivation and to cell survival.28,29 But Akt has been also shown to phosphorylate IkB kinase, which results in promotion of NFkB-mediated inhibition of apoptosis.30 When the interaction HSP90/Akt was prevented by HSP90 inhibitors, Akt was dephosphorylated and destabilized and the likelihood of apoptosis increased.27 Additional studies showed that another chaperone participates in the Akt-HSP90 complex, namely Cdc37.26 Together this complex protects Akt from proteasome degradation. In human endothelial cells during high glucose exposure, apoptosis can be prevented by HSP90 through augmentation of the protein interaction between eNOS and HSP90 and recruitment of the activated Akt.31 HSP90 has also been shown to interact with and stabilize the receptor interacting protein (RIP). Upon ligation of TNFR-1, RIP-1 is recruited to the receptor and promotes the activation of NFκB and JNK. Degradation of RIP-1 in the absence of HSP90 precludes activation of NFκB mediated by TNFα and sensitizes cells to apoptosis.32 Another route by which HSP90 can affect NFκB survival activity is via the IKK complex.33 The HSP90 inhibitor geldanamycin prevents TNF-induced activation of IKK, highlighting the role of HSP90 in NFκB activation. Some other HSP90 client proteins through which this chaperone could participate in cell survival are p5334 and the transcription factors Her2 and Hif1α.35,36
But the anti-apoptotic role of HSP90 can also be explained by its effect and interaction with proteins not defined as HSP90 client proteins (i.e., whose stability is not regulated by HSP90). HSP90 overexpression in human leukemic U937 cells can prevent the activation of caspases in cytosolic extracts treated with cytochrome c probably because HSP90 can bind to Apaf-1 and inhibit its oligomerization and further recruitment of procaspase-9.37
Unfortunately, most studies do not differentiate between HSP90α and HSP90β. It has recently been demonstrated in multiple myeloma, in which an over expression of HSP90 is necessary for cell survival, that depletion of HSP90β by siRNA is sufficient to induce apoptosis. This effect is strongly increased when also HSP90α is also depleted,23 suggesting different and cooperating anti-apoptotic properties for HSP90α and HSP90β. Confirming this assumption, in mast cells, HSP90β has been shown to associate with the anti-apoptotic protein Bcl-2. Depletion of HSP90β with a siRNA or inhibion of HSP90 with geldanamycin inhibits HSP90β interaction with Bcl-2 and results in cytochrome c release, caspase activation and apoptosis.38
In conclusion, HSP90 anti-apoptotic functions can largely be explained by its chaperone role assuring the stability of different proteins. Recent studies suggest that the two homologue proteins, HSP90α and HSP90β, might have different survival properties. It would be interesting to determine whether HSP90α and HSP90β bind to different client proteins or bind with different affinity.
HSP70: A quintessential inhibitor of apoptosis.
HSP70 loss-of-function studies demonstrated the important role of HSP70 in apoptosis. Cells lacking hsp70.1 and hsp70.3, the two genes that code for inductive HSP70, are very sensitive to apoptosis induced by a wide range of lethal stimuli.39Further, the testis specific isoform of HSP70 (hsp70.2) when ablated, results in germ cell apoptosis.40 In cancer cells, depletion of HSP70 results in spontaneous apoptosis.41
HSP70 has been shown to inhibit the apoptotic pathways at different levels (Fig. 1). At the pre-mitochondrial level, HSP70 binds to and blocks c-Jun N-terminal Kinase (JNK1) activity.42,43 Confirming this result, HSP70 deficiency induces JNK activation and caspase-3 activation44 in apoptosis induced by hyperosmolarity. HSP70 also has been shown to bind to non-phosphorylated protein kinase C (PKC) and Akt, stabilizing both proteins.45
At the mitochondrial level, HSP70 inhibits Bax translocation and insertion into the outer mitochondrial membrane. As a consequence, HSP70 prevents mitochondrial membrane permeabilization and release of cytochrome c and AIF.46
At the post-mitochondrial level HSP70 has been demonstrated to bind directly to Apaf-1, thereby preventing the recruitment of procaspase-9 to the apoptosome.47However, these results have been contradicted by a study in which the authors demonstrated that HSP70 do not have any direct effect on caspase activation. They explain these contradictory results by showing that it is a high salt concentration and not HSP70 that inhibits caspase activation.48
HSP70 also prevents cell death in conditions in which caspase activation does not occur.49 Indeed, HSP70 binds to AIF, inhibits AIF nuclear translocation and chromatin condensation.39,50,51 The interaction involves a domain of AIF between aminoacids 150 and 228.52 AIF sequestration by HSP70 has been shown to reduce neonatal hypoxic/ischemic brain injury.53 HSP70 has also been shown to associate with EndoG and to prevent DNA fragmentation54 but since EndoG can form complexes with AIF, its association with HSP70 could involve AIF as a molecular bridge.
HSP70 can also rescue cells from a later phase of apoptosis than any known survival protein, downstream caspase-3 activation.55 During the final phases of apoptosis, chromosomal DNA is digested by the DNase CAD (caspase activated DNase), following activation by caspase-3. The enzymatic activity and proper folding of CAD has been reported to be regulated by HSP70.56
At the death receptors level, HSP70 binds to DR4 and DR5, thereby inhibiting TRAIL-induced assembly and activity of death inducing signaling complex (DISC).57 Finally, HSP70 has been shown to inhibit lysosomal membrane permeabilization thereby preventing cathepsines release, proteases also implicated in apoptosis.58,59
In conclusion, HSP70 is a quintessential regulator of apoptosis that can interfere with all main apoptotic pathways. Interestingly, the ATP binding domain of HSP70 is not always required. For instance, while the ATPase function is needed for the Apaf-150 and AIF binding,51 it is dispensable for JNK60 or GATA-161binding/protection. In this way, in erythroblasts, in which HSP70 blocks apoptosis by protecting GATA-1 from caspase-3 cleavage, a HSP70 mutant that lacks the ATP binding domain of HSP70 is as efficient as wild type HSP70 in assuring the protection of erythroblasts.61
HSP27: An inhibitor of caspase activation.
HSP27 depletion reports demonstrate that HSP27 essentially blocks caspase-dependent apoptotic pathways. Small interefence targeting HSP27 induces apoptosis through caspase-3 activation.62,63 This may be consequence of the association of HSP27 with cytochrome c in the cytosol, thereby inhibiting the formation of the caspase-3 activation complex as demonstrated in leukemia and colon cancer cells treated with different apoptotic stimuli.64–66 This interaction involves amino-acids 51 and 141 of HSP27 and do not need the phosphorylation of the protein.65 In multiple myeloma cells treated with dexamethasone, HSP27 has also been shown to interact with Smac.67
HSP27 can also interfere with caspase activation upstream of the mitochondria.66This effect seems related to the ability of HSP27 to interact and regulate actin microfilaments dynamics. In L929 murine fibrosarcoma cells exposed to cytochalasin D or staurosporine, overexpressed HSP27 binds to F-actin68preventing the cytoskeletal disruption, Bid intracellular redistribution and cytochrome c release66 (Fig. 1). HSP27 has also important anti-oxidant properties. This is related to its ability to uphold glutathione in its reduced form,69 to decrease reactive oxygen species cell content,19 and to neutralize the toxic effects of oxidized proteins.70 These anti-oxidant properties of HSP27 seem particularly relevant in HSP27 protective effect in neuronal cells.71
HSP27 has been shown to bind to the kinase Akt, an interaction that is necessary for Akt activation in stressed cells. In turn, Akt could phosphorylate HSP27, thus leading to the disruption of HSP27-Akt complexes.72 HSP27 also affects one downstream event elicited by Fas/CD95. The phosphorylated form of HSP27 directly interacts with Daxx.73 In LNCaP tumor cells, HSP27 has been shown to induce cell protection through its interaction with the activators of transcription 3 (Stat3).74 Finally, HSP27 protective effect can also be consequence of its effect favouring the proteasomal degradation of certain proteins under stress conditions. Two of the proteins that HSP27 targets for their ubiquitination/proteasomal degradation are the transcription factor nuclear factor κB (NFκB) inhibitor IκBα and p27kip1. The pronounced degradation of IkBα induced by HSP27 overexpression increases NFκB dependent cell survival75 while that of p27kip1facilitates the passage of cells to the proliferate phases of the cellular cycle. As a consequence HSP27 allows the cells to rapidly resume proliferation after a stress.76
Therefore, HSP27 is able to block apoptosis at different stages because of its interaction with different partners. The capacity of HSP27 to interact with one or another partner seems to be determined by the oligomerization/phosphorylation status of the protein, which, at its turn, might depend on the cellular model/experimental conditions. We have demonstrated in vitro and in vivo that for HSP27 caspase-dependent anti-apoptotic effect, large non-phosphorylated oligomers of HSP27 were the active form of the protein.77 Confirming these results, it has recently been demonstrated that methylglyoxal modification of HSP27 induces large oligomers formation and increases the anti-apoptotic caspase-inhibitory properties of HSP27.78 In contrast, for HSP27 interaction with the F-actin and with Daxx, phosphorylated and small oligomers of HSP27 were necessary73,79 and it is its phosphorylated form that protects against neurotoxicity.80
HSP27, HSP70 and HSP90 and Cell Differentiation
Under the prescribed context of HSPs as powerful inhibitors of apoptosis, it is reasonable to assume that an increase or decrease in their expression might modulate the differentiation program. The first evidence of the role of HSPs in cell differentiation comes from their tightly regulated expression at different stages of development and cell differentiation. For instance during the process of endochondrial bone formation, they are differentially expressed in a stage-specific manner.81 In addition, during post-natal development, time at which extensive differentiation takes place, HSPs expression is regulated in neuronal and non-neuronal tissues.82 In hemin-induced differentiation of human K562 erythroleukemic cells, genes coding for HSPs are induced.83
In leukemic cells HSP27 has been described as a pre-differentiation marker84because its induction occurs early during differentiation.85–88 HSP27 expression has also been suggested as a differentiation marker for skin keratinocytes89 and for C2C12 muscle cells.90 This role for HSP27 in cell differentiation might be related to the fact that HSP27 expression increases as cells reach the non proliferative/quiescent phases of the cellular cycle (G0/G1).19,76
Subcellular localization is another mechanism whereby HSPs can determine whether a cell is to die or to differentiate. We, and others, have recently demonstrated the essential function of nuclear HSP70 for erythroid differentiation. During red blood cells’ formation, HSP70 and activated caspase-3 accumulate in the nucleus of the erythroblast.91 HSP70 directly associates with GATA-1 protecting this transcription factor required for erythropoiesis from caspase-3 cleavage. As a result, erythroblats continue their differentiation process instead of dying by apoptosis.61 HSP70, during erythropoiesis in TF-1 cells, have been shown to bind to AIF and thereby to block AIF-induced apoptosis, thus allowing the differentiation of erythroblasts to proceed.18
HSP90 has been required for erythroid differentiation of leukemia K562 cells induced by sodium butyrate92 and for DMSO-differentiated HL-60 cells. Regulation of HSP90 isoforms may be a critical event in the differentiation of human embryonic carcinoma cells and may be involved in differentiation into specific cell lineages.93 This effect of HSP90 in cell differentiation is probably because multiple transduction proteins essential for differentiation are client proteins of HSP90 such as Akt,94 RIP32 or Rb.95 Loss of function studies confirm that HSP90 plays a role in cell differentiation and development. In Drosophila melanogaster, point mutations of HSP83 (the drosophila HSP90 gene) are lethal as homozygotes. Heteterozygous mutant combinations produce viable adults with the same developmental defect: sterility.96 In Caenorhabditis elegans, DAF-21, the homologue of HSP90, is necessary for oocyte development.97 In zebrafish, HSP90 is expressed during normal differentiation of triated muscle fibres. Disruption of the activity of the proteins or the genes give rise to failure in proper somatic muscle development.98 In mice, loss-of-function studies demonstrate that while HSP90α loss-of-function phenotype appears to be normal, HSP90β is lethal. HSP90β is essential for trophoblasts differentiation and thereby for placenta development and this function can not be performed by HSP90α.4
HSP90 inhibitors have also been used to study the role of HSP90 in cell differentiation. These inhibitors such as the benzoquinone ansamycin geldanamycin or its derivative the 17-allylamino-17-demethoxygeldanamycin (17-AAG), bind to the ATP-binding “pocket” of HSP90 with higher affinity than natural nucleotides and thereby HSP90 chaperone activity is impaired and its client proteins are degraded. As could be expected by the reported role of HSP90 in cell differentiation, inhibitors of HSP90 block C2C12 myoblasts differentiation.99 In cancer cells and human leukemic blasts, 17-AAG induces a retinoblastoma-dependent G1 block. These G1 arrested cells do not differentiate but instead die by apoptosis.100
However, some reports describe that inhibitors of HSP90 can induce the differentiation process. In acute myeloid leukemia cells, 17-AAG induced apoptosis or differentiation depending on the dose and time of the treatment.101Opposite effects on cell differentiation and apoptosis are also obtained with the HSP90 inhibitor geldanamycin: in PC12 cells it induced apoptosis while in murin neuroblastoma N2A cells it induced differentiation.102 In breast cancer cells, 17-AAG-induced G1 block is accompanied by differentiation followed by apoptosis.103 The HSP90 inhibitor PU3, a synthetic purine that like 17-AAG binds with high affinity to the ATP “pocket” of HSP90, caused breast cancer cells arrest in G1 phase and differentiation.104
These contradictory reports concerning the inhibitors of HSP90 and cell differentiation could be explained if we consider that these drugs, depending on the experimental conditions, can have some side effects more or less independent of HSP90. Another possibility is that these studies do not differentiate between the amount of HSP90α and HSP90β inhibited. It is presently unknown whether HSP90 inhibitors equally block both isoforms, HSP90α and HSP90β. It not known neither whether post-translational modifications of HSP90 (acetylation, phosphorylation.) can affect their affinity for the inhibitors. HSP90α has been reported to be induced by lethal stimuli while the HSP90β can be induced by growth factors or cell differentiating signals.105 Mouse embryos out-of-function studies clearly show the role of HSP90β in the differentiation process and, at least for HSP90β role in embryo cell differentiation, there is not an overlap with HSP90α functions. Therefore, we can hypothesized that it can be the degree of inhibition of HSP90β by the HSP90 inhibitors that would determine whether or not there is a blockade of the differentiation process. This degree of inhibition of the different HSP90 isoforms might be conditioned by their cellular localization and their post-translational modifications. It should be noted, however, that the relative relevance of HSP90β in the differentiation process might depend on the differentiation model studied.
To summarize, we can hypothesize that the role in the differentiation process of a chaperone will be determined by its transient expression, subcellular redistribution and/or post-translational modifications induced at a given stage by a differ- entiation factor. How can HSPs affect the differentiation process? First by their anti-apoptotic role interfering with caspase activity, we and other authors have shown that caspase activity was generally required for cell differentiation.16,17Therefore, HSPs by interfering with caspase activity at a given moment, in a specific cellular compartment, may orchestrate the decision differentiation versus apoptosis. In this way, we have recently shown that HSP70 was a key protein to orchestrate this decision in erythroblasts.61 Second, HSPs may affect the differentiation process by regulating the nuclear/cytosolic shuttling of proteins that take place during differentiation. For instance, c-IAP1 is translocated from the nucleus to the cytosol during differentiation of hematopoietic and epithelial cells, and we have demonstrated that HSP90 is needed for this c-IAP1 nuclear export.106It has also been shown that, during erythroblast differentiation, HSP70 is needed to inhibit AIF nuclear translocation.18 Third, in the case of HSP90, the role in the differentiation process could be through certain of its client proteins, like RIP or Akt, whose stability is assured by the chaperone.
Repercussions and Concluding Remarks
The ability of HSPs to modulate the fate of the cells might have important repercussions in pathological situations such as cancer. Apoptosis, differentiation and oncogenesis are very related processes. Defaults in differentiation and/or apoptosis are involved in many cancer cells’ aetiology. HSPs are abnormally constitutively high in most cancer cells and, in clinical tumors, they are associated with poor prognosis. In experimental models, HSP27 and HSP70 have been shown to increase cancer cells’ tumorigenicty and their depletion can induce a spontaneous regression of the tumors.24,107 Several components of tumor cell-associated growth and survival pathways are HSP90 client proteins. These qualities have made HSPs targets for anticancer drug development. Today, although many research groups and pharmaceutical companies look for soluble specific inhibitors of HSP70 and HSP27, only specific soluble inhibitors of HSP90 are available for clinical trials. For some of them (17-AAG) phase II clinical trials are almost finished.108 However, considering the new role of HSP90β in cell differentiation, it seems essential to re-evaluate the functional consequences of HSP90 blockade.
Differential expression of heat shock protein 70 (hsp70) in …
by D Lang – 2000 Journal of Leukocyte Biology www.jleukbio.org/content/68/5/729.long
Differential expression of heat shock protein 70 (hsp70) in human monocytes … Induction of hsp70 in different cell lines also increases the resistance to … (NO), oxidative stress, chemotherapeutic agents, ceramide, or radiation []. ….. and type-2 cytokines in the regulation of human monocyte apoptosis Blood 90,1618-1625.
Cell Death and Disease – Do not stress, just differentiate …
Nature Jan 29, 2015 by C Boudesco – 2015 – Related articles
– The concept that cell differentiation needs a specific pattern of HSPs was first … shock, suggesting a specific role for HSPs in red blood cell formation. … Conversely, HSP70, the well-described role of which is to assist the … Trinklein ND et al Cell Stress Chaperones 2004; 9: 21–28.
Cell Death and Differentiation – Pharmacological induction …
Nature by ZN Demidenko – 2006 – Related articles
Nov 25, 2005 – Pharmacological induction of Hsp70 protects apoptosis-prone cells from …. GA did not cause cleavage of caspase-9 and PARP in HL60 cells …
HSP90 and HSP70: Implication in Inflammation Processes …
by M Sevin – 2015 – Related articles www.hindawi.com/journals/mi/2015/970242/
Sep 27, 2015 – In Bcr-abl leukaemia cells, the expression of the protein HSP70 is also elevated ….. GATA-1 protein level during erythroid cell differentiation,” Blood, vol. …. Cdc37 and Hsp90,” Molec Cell 2002; 9(2): 401–410
HSF-1 activates the ubiquitin proteasome system to promote non-apoptotic developmental cell death inC. elegans
-
Shai Shaham
 :
: </div>”>Shai Shaham
eLife 2016;5:e12821 http://dx.doi.org/10.7554/eLife.12821
Apoptosis is a prominent metazoan cell death form. Yet, mutations in apoptosis regulators cause only minor defects in vertebrate development, suggesting that another developmental cell death mechanism exists. While some non-apoptotic programs have been molecularly characterized, none appear to control developmental cell culling. Linker-cell-type death (LCD) is a morphologically conserved non-apoptotic cell death process operating in Caenorhabditis elegans and vertebrate development, and is therefore a compelling candidate process complementing apoptosis. However, the details of LCD execution are not known. Here we delineate a molecular-genetic pathway governing LCD in C. elegans. Redundant activities of antagonistic Wnt signals, a temporal control pathway, and mitogen-activated protein kinase kinase signaling control heat shock factor 1 (HSF-1), a conserved stress-activated transcription factor. Rather than protecting cells, HSF-1 promotes their demise by activating components of the ubiquitin proteasome system, including the E2 ligase LET-70/UBE2D2 functioning with E3 components CUL-3, RBX-1, BTBD-2, and SIAH-1. Our studies uncover design similarities between LCD and developmental apoptosis, and provide testable predictions for analyzing LCD in vertebrates. http://dx.doi.org/10.7554/eLife.12821.001
eLife digest
Embryos make numerous new cells as they develop, but also destroy many cells to remove the faulty ones and to ensure that tissues grow to the right size and shape. This deliberate form of cell death must be precisely regulated to prevent too many cells or healthy cells, from being destroyed. Understanding the molecular mechanisms that govern cell death is therefore important for understanding normal development and also human disease.
One well-studied process that leads to cell death is called apoptosis. This process carefully dismantles and breaks down the components of a cell, but does not seem to account for all cell death that occurs during animal development. Recently another developmental cell-death pathway, called the linker-cell-type death, was discovered in a small roundworm called Caenorhabditis elegans. This pathway appears to work in mammalian cells as well, and may help to break down nerve fibers that are not needed. However, many of this pathway’s component parts remained unknown.
Kinet, Malin et al. have now used a combination of genetics and cell biology in C. elegans to uncover the components of linker-cell-type death and to investigate how they interact. The results of these studies revealed a hierarchy of genetic interactions that governs this pathway in C. elegans. One protein called HSF-1 plays a particularly important role. This protein is a transcription factor and it binds to, and regulates, the activities of various genes. HSF-1 usually works in cells to protect them from stress, but Kinet, Malin et al. showed that it instead promotes linker-cell-type death by activating a molecular machine, called the proteasome, that breaks down proteins. The experiments also revealed two proteins (called BTBD-2 and SIAH-1) that may be important for shuttling specific proteins for degradation by the proteasome.
Three signalling pathways that regulate important developmental processes also regulate the activation of linker-cell-type death. Kinet, Malin et al. propose that these signalling pathways do so by working together to activate HSF-1, which in turn activates the genes that lead to the destruction of cells by the proteasome.
A future challenge is to understand in more detail how the more recently discovered cell death pathway actually kills cells. Further work could also explore how HSF-1, a protein that normally protects cells, is transformed into a cell-killing protein. DOI: http://dx.doi.org/10.7554/eLife.12821.002
A new pathway for non-apoptotic cell death
The results presented here allow us to construct a model for the initiation and execution of LCD in C. elegans (Figure 7). The logic of the LCD pathway may be similar to that of developmental apoptotic pathways. In C. elegans and Drosophila, where the control of specific cell deaths has been primarily examined, cell lineage or fate determinants control the expression of specific transcription factors that then impinge on proteins regulating caspase activation (Fuchs and Steller, 2011). Likewise, LCD is initiated by redundant determinants that require a transcription factor to activate protein degradation genes.

https://elife-publishing-cdn.s3.amazonaws.com/12821/elife-12821-fig7-v3-480w.jpg
Figure 7. Model for linker cell death.
Green, upstream regulators. Orange, HSF-1. Purple, proteolytic components. DOI: http://dx.doi.org/10.7554/eLife.12821.016
Our data suggest that three partially redundant signals control LCD initiation. The antagonistic Wnt pathways we describe may provide positional information to the linker cell, as the relevant ligands are expressed only near the region where the linker cell dies. The LIN-29 pathway, which controls timing decisions during the L4-adult molt, may ensure that LCD takes place only at the right time. Finally, while the TIR-1/SEK-1 pathway could act constitutively in the linker cell, it may also respond to specific cues from neighboring cells. Indeed, MAPK pathways are often induced by extracellular ligands. We propose that these three pathways, together, trigger activation of HSF-1. Our data support a model in which HSF-1 is present in two forms, HSF-1LC, promoting LCD, and HSF-1HS, protecting cells from stresses, including heat shock. We postulate that the redundant LCD initiation pathways tip the balance in favor of HSF-1LC, allowing this activity to bind to promoters and induce transcription of key LCD effectors, including LET-70/UBE2D2 and other components of the ubiquitin proteasome system (UPS), functioning through E3 ligase complexes consisting of CUL-3, RBX-1, BTBD-2, and SIAH-1.
Importantly, the molecular identification of LCD components and their interactions opens the door to testing the impact of this cell death pathway on vertebrate development. For example, monitoring UBE2D2 expression during development could reveal upregulation in dying cells. Likewise, genetic lesions in pathway components we identified may lead to a block in cell death. Double mutants in apoptotic and LCD genes would allow testing of the combined contributions of these processes.
The proteasome and LCD
As is the case with caspase proteases that mediate apoptosis (Pop and Salvesen, 2009), how the UPS induces LCD is not clear, and remains an exciting area of future work. That loss of BTBD-2, a specific E3 ligase component, causes extensive linker cell survival suggests that a limited set of targets may be required for LCD. Previous work demonstrated that BTBD2, the vertebrate homolog of BTBD-2, interacts with topoisomerase I (Khurana et al., 2010; Xu et al., 2002), raising the possibility that this enzyme may be a relevant target, although other targets may exist.
The UPS has been implicated in a number of cell death processes in which it appears to play a general role in cell dismantling, most notably, perhaps, in intersegmental muscle remodeling during metamorphosis in moths (Haas et al., 1995). However, other studies suggest that the UPS can have specific regulatory functions, as with caspase inhibition by IAP E3 ligases (Ditzel et al., 2008).
During Drosophila sperm development, caspase activity is induced by the UPS to promote sperm individualization, a process that resembles cytoplasm-specific activation of apoptosis (Arama et al., 2007). While C. elegans caspases are dispensible for LCD, it remains possible that they participate in linker cell dismantling or serve as a backup in case the LCD program fails.
Finally, the proteasome contains catalytic domains with target cleavage specificity reminiscent of caspases; however, inactivation of the caspase-like sites does not, alone, result in overt cellular defects (Britton et al., 2009), suggesting that this activity may be needed to degrade only specific substrates. Although the proteasome generally promotes proteolysis to short peptides, site-specific cleavage of proteins by the proteasome has been described (Chen et al., 1999). It is intriguing to speculate, therefore, that caspases and the proteasome may have common, and specific, targets in apoptosis and LCD.
A pro-death developmental function for HSF-1
Our discovery that C. elegans heat-shock factor, HSF-1, promotes cell death is surprising. Heat-shock factors are thought to be protective proteins, orchestrating the response to protein misfolding induced by a variety of stressors, including elevated temperature. Although a role for HSF1 has been proposed in promoting apoptosis of mouse spermatocytes following elevated temperatures (Nakai et al., 2000), it is not clear whether this function is physiological. In this context, HSF1 induces expression of the gene Tdag51 (Hayashida et al., 2006). Both pro- and anti-apoptotic activities have been attributed to Tdag51 (Toyoshima et al., 2004), and which is activated in sperm is not clear. Recently, pathological roles for HSF1 in cancer have been detailed (e.g. Mendillo et al., 2012), but in these capacities HSF1 still supports cell survival.
Developmental functions for HSF1 have been suggested in which HSF1 appears to act through transcriptional targets different from those of the heat-shock response (Jedlicka et al., 1997), although target identity remains obscure. Here, we have shown that HSF-1 has at least partially non-overlapping sets of stress-induced and developmental targets. Indeed, typical stress targets of HSF-1, such as the small heat-shock gene hsp-16.49 as well as genes encoding larger chaperones, likehsp-1, are not expressed during LCD, whereas let-70, a direct transcriptional target for LCD, is not induced by heat shock. Interestingly, the yeast let-70 homologs ubc4 and ubc5 are induced by heat shock (Seufert and Jentsch, 1990), supporting a conserved connection between HSF and UBE2D2-family proteins. However, the distinction between developmental and stress functions is clearly absent in this single-celled organism, raising the possibility that this separation of function may be a metazoan innovation.
What distinguishes the stress-related and developmental forms of HSF-1? One possibility is that whereas the stress response appears to be mediated by HSF-1 trimerization, HSF-1 monomers or dimers might promote LCD roles. Although this model would nicely account for the differential activities in stress responses and LCD of the HSF-1(R145A) transgenic protein, which would be predicted to favor inactivation of a larger proportion of higher order HSF-1 complexes, the identification of conserved tripartite HSEs in the let-70 and rpn-3 regulatory regions argues against this possibility. Alternatively, selective post-translational modification of HSF-1 could account for these differences. In mammals, HSF1 undergoes a variety of modifications including phosphorylation, acetylation, ubiquitination, and sumoylation (Xu et al., 2012), which, depending on the site and modification, stimulate or repress HSF1 activity. In this context, it is of note that p38/MAPK-mediated phosphorylation of HSF1 represses its stress-related activity (Chu et al., 1996), and the LCD regulator SEK-1 encodes a MAPKK. However, no single MAPK has been identified that promotes LCD (E.S.B., M.J.K. unpublished results), suggesting that other mechanisms may be at play.
Our finding that POP-1/TCF does not play a significant role in LCD raises the possibility that Wnt signaling exerts direct control over HSF-1 through interactions with β-catenin. However, we have not been able to demonstrate physical interactions between these proteins to date (M.J.K, unpublished results).
Finally, a recent paper (Labbadia and Morimoto, 2015) demonstrated that in young adult C. elegans, around the time of LCD, global binding of HSF-1 to its stress-induced targets is reduced through changes in chromatin modification. Remarkably, we showed that chromatin regulators play a key role in let-70 induction and LCD (J.A.M., M.J.K and S.S., manuscript in preparation), suggesting, perhaps, that differences in HSF-1 access to different loci may play a role in distinguishing its two functions.
LCD and neurodegeneration
Previous studies from our lab raised the possibility that LCD may be related to degenerative processes that promote vertebrate neuronal death. Nuclear crenellation is evident in dying linker cells and in degenerating cells in polyQ disease (Abraham et al., 2007) and the TIR-1/Sarm adapter protein promotes LCD in C. elegans as well as degeneration of distal axonal segments following axotomy in Drosophila and vertebrates (Osterloh et al., 2012). The studies we present here, implicating the UPS and heat-shock factor in LCD, also support a connection with neurodegeneration. Indeed, protein aggregates found in cells of patients with polyQ diseases are heavily ubiquitylated (Kalchman et al., 1996). Chaperones also colocalize with protein aggregates in brain slices from SCA patients, and HSF1 has been shown to alleviate polyQ aggregation and cellular demise in both polyQ-overexpressing flies and in neuronal precursor cells (Neef et al., 2010). While the failure of proteostatic mechanisms in neurodegenerative diseases is generally thought to be a secondary event in their pathogenesis, it is possible that this failure reflects the involvement of a LCD-like process, in which attempts to engage protective measures instead result in activation of a specific cell death program.









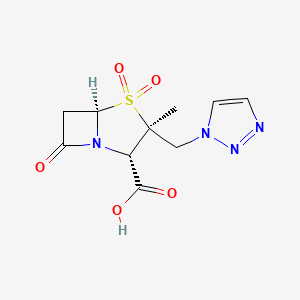

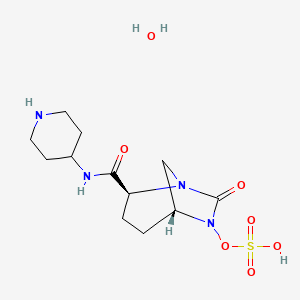
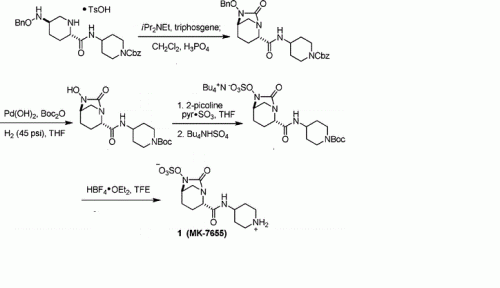



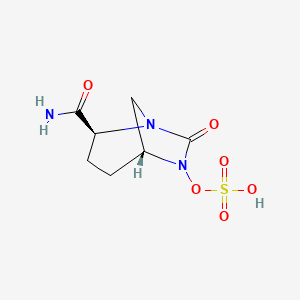




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}