Funding, Deals & Partnerships: BIOLOGICS & MEDICAL DEVICES; BioMed e-Series; Medicine and Life Sciences Scientific Journal – http://PharmaceuticalIntelligence.com
Eight Subcellular Pathologies driving Chronic Metabolic Diseases – Methods for Mapping Bioelectronic Adjustable Measurements as potential new Therapeutics: Impact on Pharmaceuticals in Use
In this curation we wish to present two breaking through goals:
Goal 1:
Exposition of a new direction of research leading to a more comprehensive understanding of Metabolic Dysfunctional Diseases that are implicated in effecting the emergence of the two leading causes of human mortality in the World in 2023: (a) Cardiovascular Diseases, and (b) Cancer
Goal 2:
Development of Methods for Mapping Bioelectronic Adjustable Measurements as potential new Therapeutics for these eight subcellular causes of chronic metabolic diseases. It is anticipated that it will have a potential impact on the future of Pharmaceuticals to be used, a change from the present time current treatment protocols for Metabolic Dysfunctional Diseases.
According to Dr. Robert Lustig, M.D, an American pediatric endocrinologist. He is Professor emeritus of Pediatrics in the Division of Endocrinology at the University of California, San Francisco, where he specialized in neuroendocrinology and childhood obesity, there are eight subcellular pathologies that drive chronic metabolic diseases.
These eight subcellular pathologies can’t be measured at present time.
In this curation we will attempt to explore methods of measurement for each of these eight pathologies by harnessing the promise of the emerging field known as Bioelectronics.
Unmeasurable eight subcellular pathologies that drive chronic metabolic diseases
Glycation
Oxidative Stress
Mitochondrial dysfunction [beta-oxidation Ac CoA malonyl fatty acid]
Insulin resistance/sensitive [more important than BMI], known as a driver to cancer development
Membrane instability
Inflammation in the gut [mucin layer and tight junctions]
Epigenetics/Methylation
Autophagy [AMPKbeta1 improvement in health span]
Diseases that are not Diseases: no drugs for them, only diet modification will help
Image source
Robert Lustig, M.D. on the Subcellular Processes That Belie Chronic Disease
These eight Subcellular Pathologies driving Chronic Metabolic Diseases are becoming our focus for exploration of the promise of Bioelectronics for two pursuits:
Will Bioelectronics be deemed helpful in measurement of each of the eight pathological processes that underlie and that drive the chronic metabolic syndrome(s) and disease(s)?
IF we will be able to suggest new measurements to currently unmeasurable health harming processes THEN we will attempt to conceptualize new therapeutic targets and new modalities for therapeutics delivery – WE ARE HOPEFUL
In the Bioelecronics domain we are inspired by the work of the following three research sources:
Michael Levin is an American developmental and synthetic biologist at Tufts University, where he is the Vannevar Bush Distinguished Professor. Levin is a director of the Allen Discovery Center at Tufts University and Tufts Center for Regenerative and Developmental Biology. Wikipedia
THE VOICE of Dr. Justin D. Pearlman, MD, PhD, FACC
PENDING
THE VOICE of Stephen J. Williams, PhD
Ten TakeAway Points of Dr. Lustig’s talk on role of diet on the incidence of Type II Diabetes
25% of US children have fatty liver
Type II diabetes can be manifested from fatty live with 151 million people worldwide affected moving up to 568 million in 7 years
A common myth is diabetes due to overweight condition driving the metabolic disease
There is a trend of ‘lean’ diabetes or diabetes in lean people, therefore body mass index not a reliable biomarker for risk for diabetes
Thirty percent of ‘obese’ people just have high subcutaneous fat. the visceral fat is more problematic
there are people who are ‘fat’ but insulin sensitive while have growth hormone receptor defects. Points to other issues related to metabolic state other than insulin and potentially the insulin like growth factors
At any BMI some patients are insulin sensitive while some resistant
Visceral fat accumulation may be more due to chronic stress condition
Fructose can decrease liver mitochondrial function
A methionine and choline deficient diet can lead to rapid NASH development
The following paper in Cells describes the discovery of protein interactors of endoglin, which is recruited to membranes at the TGF-β receptor complex upon TGF-β signaling. Interesting a carbohydrate binding protein, galectin-3, and an E3-ligase, TRIM21, were found to be unique interactors within this complex.
Gallardo-Vara E, Ruiz-Llorente L, Casado-Vela J, Ruiz-Rodríguez MJ, López-Andrés N, Pattnaik AK, Quintanilla M, Bernabeu C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells. 2019 Sep 13;8(9):1082. doi: 10.3390/cells8091082. PMID: 31540324; PMCID: PMC6769930.
Abstract
Endoglin is a 180-kDa glycoprotein receptor primarily expressed by the vascular endothelium and involved in cardiovascular disease and cancer. Heterozygous mutations in the endoglin gene (ENG) cause hereditary hemorrhagic telangiectasia type 1, a vascular disease that presents with nasal and gastrointestinal bleeding, skin and mucosa telangiectases, and arteriovenous malformations in internal organs. A circulating form of endoglin (alias soluble endoglin, sEng), proteolytically released from the membrane-bound protein, has been observed in several inflammation-related pathological conditions and appears to contribute to endothelial dysfunction and cancer development through unknown mechanisms. Membrane-bound endoglin is an auxiliary component of the TGF-β receptor complex and the extracellular region of endoglin has been shown to interact with types I and II TGF-β receptors, as well as with BMP9 and BMP10 ligands, both members of the TGF-β family. To search for novel protein interactors, we screened a microarray containing over 9000 unique human proteins using recombinant sEng as bait. We find that sEng binds with high affinity, at least, to 22 new proteins. Among these, we validated the interaction of endoglin with galectin-3, a secreted member of the lectin family with capacity to bind membrane glycoproteins, and with tripartite motif-containing protein 21 (TRIM21), an E3 ubiquitin-protein ligase. Using human endothelial cells and Chinese hamster ovary cells, we showed that endoglin co-immunoprecipitates and co-localizes with galectin-3 or TRIM21. These results open new research avenues on endoglin function and regulation.
Endoglin is an auxiliary TGF-β co-receptor predominantly expressed in endothelial cells, which is involved in vascular development, repair, homeostasis, and disease [1,2,3,4]. Heterozygous mutations in the human ENDOGLIN gene (ENG) cause hereditary hemorrhagic telangiectasia (HHT) type 1, a vascular disease associated with nasal and gastrointestinal bleeds, telangiectases on skin and mucosa and arteriovenous malformations in the lung, liver, and brain [4,5,6]. The key role of endoglin in the vasculature is also illustrated by the fact that endoglin-KO mice die in utero due to defects in the vascular system [7]. Endoglin expression is markedly upregulated in proliferating endothelial cells involved in active angiogenesis, including the solid tumor neovasculature [8,9]. For this reason, endoglin has become a promising target for the antiangiogenic treatment of cancer [10,11,12]. Endoglin is also expressed in cancer cells where it can behave as both a tumor suppressor in prostate, breast, esophageal, and skin carcinomas [13,14,15,16] and a promoter of malignancy in melanoma and Ewing’s sarcoma [17]. Ectodomain shedding of membrane-bound endoglin may lead to a circulating form of the protein, also known as soluble endoglin (sEng) [18,19,20]. Increased levels of sEng have been found in several vascular-related pathologies, including preeclampsia, a disease of high prevalence in pregnant women which, if left untreated, can lead to serious and even fatal complications for both mother and baby [2,18,19,21]. Interestingly, several lines of evidence support a pathogenic role of sEng in the vascular system, including endothelial dysfunction, antiangiogenic activity, increased vascular permeability, inflammation-associated leukocyte adhesion and transmigration, and hypertension [18,22,23,24,25,26,27]. Because of its key role in vascular pathology, a large number of studies have addressed the structure and function of endoglin at the molecular level, in order to better understand its mechanism of action.
Galectin-3 Interacts with Endoglin in Cells
Galectin-3 is a secreted member of the lectin family with the capacity to bind membrane glycoproteins like endoglin and is involved in the pathogenesis of many human diseases [52]. We confirmed the protein screen data for galectin-3, as evidenced by two-way co-immunoprecipitation of endoglin and galectin-3 upon co-transfection in CHO-K1 cells. As shown in Figure 1A, galectin-3 and endoglin were efficiently transfected, as demonstrated by Western blot analysis in total cell extracts. No background levels of endoglin were observed in control cells transfected with the empty vector (Ø). By contrast, galectin-3 could be detected in all samples but, as expected, showed an increased signal in cells transfected with the galectin-3 expression vector. Co-immunoprecipitation studies of these cell lysates showed that galectin-3 was present in endoglin immunoprecipitates (Figure 1B). Conversely, endoglin was also detected in galectin-3 immunoprecipitates (Figure 1C).
Figure 1. Protein–protein association between galectin-3 and endoglin. (A–C). Co-immunoprecipitation of galectin-3 and endoglin. CHO-K1 cells were transiently transfected with pcEXV-Ø (Ø), pcEXV–HA–EngFL (Eng) and pcDNA3.1–Gal-3 (Gal3) expression vectors. (A) Total cell lysates (TCL) were analyzed by SDS-PAGE under reducing conditions, followed by Western blot (WB) analysis using specific antibodies to endoglin, galectin-3 and β-actin (loading control). Cell lysates were subjected to immunoprecipitation (IP) with anti-endoglin (B) or anti-galectin-3 (C) antibodies, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin or anti-galectin-3 antibodies, as indicated. Negative controls with an IgG2b (B) and IgG1 (C) were included. (D) Protein-protein interactions between galectin-3 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 7.3 µM recombinant human galectin-3/6xHis at the C-terminus (LGALS3), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Figure 2.Galectin-3 and endoglin co-localize in human endothelial cells. Human umbilical vein-derived endothelial cell (HUVEC) monolayers were fixed with paraformaldehyde, permeabilized with Triton X-100, incubated with the mouse mAb P4A4 anti-endoglin, washed, and incubated with a rabbit polyclonal anti-galectin-3 antibody (PA5-34819). Galectin-3 and endoglin were detected by immunofluorescence upon incubation with Alexa 647 goat anti-rabbit IgG (red staining) and Alexa 488 goat anti-mouse IgG (green staining) secondary antibodies, respectively. (A) Single staining of galectin-3 (red) and endoglin (green) at the indicated magnifications. (B) Merge images plus DAPI (nuclear staining in blue) show co-localization of galectin-3 and endoglin (yellow color). Representative images of five different experiments are shown.
Endoglin associates with the cullin-type E3 ligase TRIM21
Figure 3.Protein–protein association between TRIM21 and endoglin. (A–E) Co-immunoprecipitation of TRIM21 and endoglin. A,B. HUVEC monolayers were lysed and total cell lysates (TCL) were subjected to SDS-PAGE under reducing (for TRIM21 detection) or nonreducing (for endoglin detection) conditions, followed by Western blot (WB) analysis using antibodies to endoglin, TRIM21 or β-actin (A). HUVECs lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or negative control antibodies, followed by WB analysis with anti-endoglin (B). C,D. CHO-K1 cells were transiently transfected with pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (E) or pcDNA3.1–HA–hTRIM21 (T) expression vectors, as indicated. Total cell lysates (TCL) were subjected to SDS-PAGE under nonreducing conditions and WB analysis using specific antibodies to endoglin, TRIM21, and β-actin (C). Cell lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or anti-endoglin antibodies, followed by SDS-PAGE under reducing (upper panel) or nonreducing (lower panel) conditions and WB analysis with anti-TRIM21 or anti-endoglin antibodies. Negative controls of appropriate IgG were included (D). E. CHO-K1 cells were transiently transfected with pcDNA3.1–HA–hTRIM21 and pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (FL; full-length), pDisplay–HA–EngEC (EC; cytoplasmic-less) or pDisplay–HA–EngTMEC (TMEC; cytoplasmic-less) expression vectors, as indicated. Cell lysates were subjected to immunoprecipitation with anti-TRIM21, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin antibodies, as indicated. The asterisk indicates the presence of a nonspecific band. Mr, molecular reference; Eng, endoglin; TRIM, TRIM21. (F) Protein–protein interactions between TRIM21 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 5.4 µM recombinant human TRIM21/6xHis at the N-terminus (R052), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Table 1. Human protein-array analysis of endoglin interactors1.
1 Microarrays containing over 9000 unique human proteins were screened using recombinant sEng as a probe. Protein interactors showing the highest scores (Z-score ≥2.0) are listed. GeneBank (https://www.ncbi.nlm.nih.gov/genbank/) and UniProtKB (https://www.uniprot.org/help/uniprotkb) accession numbers are indicated with a yellow or green background, respectively. The cellular compartment of each protein was obtained from the UniProtKB webpage. Proteins selected for further studies (TRIM21 and galectin-3) are indicated in bold type with blue background.
Note: the following are from NCBI Genbank and Genecards on TRIM21
Official Symbol TRIM21provided by HGNC Official Full Name tripartite motif containing 21provided by HGNC Primary source HGNC:HGNC:11312 See related Ensembl:ENSG00000132109MIM:109092;AllianceGenome:HGNC:11312 Gene type protein coding RefSeq status REVIEWED Organism Homo sapiens Lineage Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini; Catarrhini; Hominidae; Homo Also known as SSA; RO52; SSA1; RNF81; Ro/SSA Summary This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008] Expression Ubiquitous expression in spleen (RPKM 15.5), appendix (RPKM 13.2) and 24 other tissues See more Orthologs mouseall NEW Try the new Gene table Try the new Transcript table
This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008]
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression. Enhances the decapping activity of DCP2. Exists as a ribonucleoprotein particle present in all mammalian cells studied and composed of a single polypeptide and one of four small RNA molecules. At least two isoforms are present in nucleated and red blood cells, and tissue specific differences in RO/SSA proteins have been identified. The common feature of these proteins is their ability to bind HY RNAs.2. Involved in the regulation of innate immunity and the inflammatory response in response to IFNG/IFN-gamma. Organizes autophagic machinery by serving as a platform for the assembly of ULK1, Beclin 1/BECN1 and ATG8 family members and recognizes specific autophagy targets, thus coordinating target recognition with assembly of the autophagic apparatus and initiation of autophagy. Acts as an autophagy receptor for the degradation of IRF3, hence attenuating type I interferon (IFN)-dependent immune responses (PubMed:26347139, 16297862, 16316627, 16472766, 16880511, 18022694, 18361920, 18641315, 18845142, 19675099). Represses the innate antiviral response by facilitating the formation of the NMI-IFI35 complex through ‘Lys-63’-linked ubiquitination of NMI (PubMed:26342464). ( RO52_HUMAN,P19474 )
Molecular function for TRIM21 Gene according to UniProtKB/Swiss-Prot
Function:
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression.
Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners
Gallardo-Vara E, Ruiz-Llorente L, Casado-Vela J, Ruiz-Rodríguez MJ, López-Andrés N, Pattnaik AK, Quintanilla M, Bernabeu C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells. 2019 Sep 13;8(9):1082. doi: 10.3390/cells8091082. PMID: 31540324; PMCID: PMC6769930.
Abstract
Endoglin is a 180-kDa glycoprotein receptor primarily expressed by the vascular endothelium and involved in cardiovascular disease and cancer. Heterozygous mutations in the endoglin gene (ENG) cause hereditary hemorrhagic telangiectasia type 1, a vascular disease that presents with nasal and gastrointestinal bleeding, skin and mucosa telangiectases, and arteriovenous malformations in internal organs. A circulating form of endoglin (alias soluble endoglin, sEng), proteolytically released from the membrane-bound protein, has been observed in several inflammation-related pathological conditions and appears to contribute to endothelial dysfunction and cancer development through unknown mechanisms. Membrane-bound endoglin is an auxiliary component of the TGF-β receptor complex and the extracellular region of endoglin has been shown to interact with types I and II TGF-β receptors, as well as with BMP9 and BMP10 ligands, both members of the TGF-β family. To search for novel protein interactors, we screened a microarray containing over 9000 unique human proteins using recombinant sEng as bait. We find that sEng binds with high affinity, at least, to 22 new proteins. Among these, we validated the interaction of endoglin with galectin-3, a secreted member of the lectin family with capacity to bind membrane glycoproteins, and with tripartite motif-containing protein 21 (TRIM21), an E3 ubiquitin-protein ligase. Using human endothelial cells and Chinese hamster ovary cells, we showed that endoglin co-immunoprecipitates and co-localizes with galectin-3 or TRIM21. These results open new research avenues on endoglin function and regulation.
Endoglin is an auxiliary TGF-β co-receptor predominantly expressed in endothelial cells, which is involved in vascular development, repair, homeostasis, and disease [1,2,3,4]. Heterozygous mutations in the human ENDOGLIN gene (ENG) cause hereditary hemorrhagic telangiectasia (HHT) type 1, a vascular disease associated with nasal and gastrointestinal bleeds, telangiectases on skin and mucosa and arteriovenous malformations in the lung, liver, and brain [4,5,6]. The key role of endoglin in the vasculature is also illustrated by the fact that endoglin-KO mice die in utero due to defects in the vascular system [7]. Endoglin expression is markedly upregulated in proliferating endothelial cells involved in active angiogenesis, including the solid tumor neovasculature [8,9]. For this reason, endoglin has become a promising target for the antiangiogenic treatment of cancer [10,11,12]. Endoglin is also expressed in cancer cells where it can behave as both a tumor suppressor in prostate, breast, esophageal, and skin carcinomas [13,14,15,16] and a promoter of malignancy in melanoma and Ewing’s sarcoma [17]. Ectodomain shedding of membrane-bound endoglin may lead to a circulating form of the protein, also known as soluble endoglin (sEng) [18,19,20]. Increased levels of sEng have been found in several vascular-related pathologies, including preeclampsia, a disease of high prevalence in pregnant women which, if left untreated, can lead to serious and even fatal complications for both mother and baby [2,18,19,21]. Interestingly, several lines of evidence support a pathogenic role of sEng in the vascular system, including endothelial dysfunction, antiangiogenic activity, increased vascular permeability, inflammation-associated leukocyte adhesion and transmigration, and hypertension [18,22,23,24,25,26,27]. Because of its key role in vascular pathology, a large number of studies have addressed the structure and function of endoglin at the molecular level, in order to better understand its mechanism of action.
Galectin-3 Interacts with Endoglin in Cells
Galectin-3 is a secreted member of the lectin family with the capacity to bind membrane glycoproteins like endoglin and is involved in the pathogenesis of many human diseases [52]. We confirmed the protein screen data for galectin-3, as evidenced by two-way co-immunoprecipitation of endoglin and galectin-3 upon co-transfection in CHO-K1 cells. As shown in Figure 1A, galectin-3 and endoglin were efficiently transfected, as demonstrated by Western blot analysis in total cell extracts. No background levels of endoglin were observed in control cells transfected with the empty vector (Ø). By contrast, galectin-3 could be detected in all samples but, as expected, showed an increased signal in cells transfected with the galectin-3 expression vector. Co-immunoprecipitation studies of these cell lysates showed that galectin-3 was present in endoglin immunoprecipitates (Figure 1B). Conversely, endoglin was also detected in galectin-3 immunoprecipitates (Figure 1C).
Figure 1. Protein–protein association between galectin-3 and endoglin. (A–C). Co-immunoprecipitation of galectin-3 and endoglin. CHO-K1 cells were transiently transfected with pcEXV-Ø (Ø), pcEXV–HA–EngFL (Eng) and pcDNA3.1–Gal-3 (Gal3) expression vectors. (A) Total cell lysates (TCL) were analyzed by SDS-PAGE under reducing conditions, followed by Western blot (WB) analysis using specific antibodies to endoglin, galectin-3 and β-actin (loading control). Cell lysates were subjected to immunoprecipitation (IP) with anti-endoglin (B) or anti-galectin-3 (C) antibodies, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin or anti-galectin-3 antibodies, as indicated. Negative controls with an IgG2b (B) and IgG1 (C) were included. (D) Protein-protein interactions between galectin-3 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 7.3 µM recombinant human galectin-3/6xHis at the C-terminus (LGALS3), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Figure 2.Galectin-3 and endoglin co-localize in human endothelial cells. Human umbilical vein-derived endothelial cell (HUVEC) monolayers were fixed with paraformaldehyde, permeabilized with Triton X-100, incubated with the mouse mAb P4A4 anti-endoglin, washed, and incubated with a rabbit polyclonal anti-galectin-3 antibody (PA5-34819). Galectin-3 and endoglin were detected by immunofluorescence upon incubation with Alexa 647 goat anti-rabbit IgG (red staining) and Alexa 488 goat anti-mouse IgG (green staining) secondary antibodies, respectively. (A) Single staining of galectin-3 (red) and endoglin (green) at the indicated magnifications. (B) Merge images plus DAPI (nuclear staining in blue) show co-localization of galectin-3 and endoglin (yellow color). Representative images of five different experiments are shown.
Endoglin associates with the cullin-type E3 ligase TRIM21
Figure 3.Protein–protein association between TRIM21 and endoglin. (A–E) Co-immunoprecipitation of TRIM21 and endoglin. A,B. HUVEC monolayers were lysed and total cell lysates (TCL) were subjected to SDS-PAGE under reducing (for TRIM21 detection) or nonreducing (for endoglin detection) conditions, followed by Western blot (WB) analysis using antibodies to endoglin, TRIM21 or β-actin (A). HUVECs lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or negative control antibodies, followed by WB analysis with anti-endoglin (B). C,D. CHO-K1 cells were transiently transfected with pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (E) or pcDNA3.1–HA–hTRIM21 (T) expression vectors, as indicated. Total cell lysates (TCL) were subjected to SDS-PAGE under nonreducing conditions and WB analysis using specific antibodies to endoglin, TRIM21, and β-actin (C). Cell lysates were subjected to immunoprecipitation (IP) with anti-TRIM21 or anti-endoglin antibodies, followed by SDS-PAGE under reducing (upper panel) or nonreducing (lower panel) conditions and WB analysis with anti-TRIM21 or anti-endoglin antibodies. Negative controls of appropriate IgG were included (D). E. CHO-K1 cells were transiently transfected with pcDNA3.1–HA–hTRIM21 and pDisplay–HA–Mock (Ø), pDisplay–HA–EngFL (FL; full-length), pDisplay–HA–EngEC (EC; cytoplasmic-less) or pDisplay–HA–EngTMEC (TMEC; cytoplasmic-less) expression vectors, as indicated. Cell lysates were subjected to immunoprecipitation with anti-TRIM21, followed by SDS-PAGE under reducing conditions and WB analysis with anti-endoglin antibodies, as indicated. The asterisk indicates the presence of a nonspecific band. Mr, molecular reference; Eng, endoglin; TRIM, TRIM21. (F) Protein–protein interactions between TRIM21 and endoglin using Bio-layer interferometry (BLItz). The Ni–NTA biosensors tips were loaded with 5.4 µM recombinant human TRIM21/6xHis at the N-terminus (R052), and protein binding was measured against 0.1% BSA in PBS (negative control) or 4.1 µM soluble endoglin (sEng). Kinetic sensorgrams were obtained using a single channel ForteBioBLItzTM instrument.
Table 1. Human protein-array analysis of endoglin interactors1.
1 Microarrays containing over 9000 unique human proteins were screened using recombinant sEng as a probe. Protein interactors showing the highest scores (Z-score ≥2.0) are listed. GeneBank (https://www.ncbi.nlm.nih.gov/genbank/) and UniProtKB (https://www.uniprot.org/help/uniprotkb) accession numbers are indicated with a yellow or green background, respectively. The cellular compartment of each protein was obtained from the UniProtKB webpage. Proteins selected for further studies (TRIM21 and galectin-3) are indicated in bold type with blue background.
Note: the following are from NCBI Genbank and Genecards on TRIM21
This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008]
Expression
Ubiquitous expression in spleen (RPKM 15.5), appendix (RPKM 13.2) and 24 other tissues See more
This gene encodes a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. The encoded protein is part of the RoSSA ribonucleoprotein, which includes a single polypeptide and one of four small RNA molecules. The RoSSA particle localizes to both the cytoplasm and the nucleus. RoSSA interacts with autoantigens in patients with Sjogren syndrome and systemic lupus erythematosus. Alternatively spliced transcript variants for this gene have been described but the full-length nature of only one has been determined. [provided by RefSeq, Jul 2008]
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression. Enhances the decapping activity of DCP2. Exists as a ribonucleoprotein particle present in all mammalian cells studied and composed of a single polypeptide and one of four small RNA molecules. At least two isoforms are present in nucleated and red blood cells, and tissue specific differences in RO/SSA proteins have been identified. The common feature of these proteins is their ability to bind HY RNAs.2. Involved in the regulation of innate immunity and the inflammatory response in response to IFNG/IFN-gamma. Organizes autophagic machinery by serving as a platform for the assembly of ULK1, Beclin 1/BECN1 and ATG8 family members and recognizes specific autophagy targets, thus coordinating target recognition with assembly of the autophagic apparatus and initiation of autophagy. Acts as an autophagy receptor for the degradation of IRF3, hence attenuating type I interferon (IFN)-dependent immune responses (PubMed:26347139, 16297862, 16316627, 16472766, 16880511, 18022694, 18361920, 18641315, 18845142, 19675099). Represses the innate antiviral response by facilitating the formation of the NMI-IFI35 complex through ‘Lys-63’-linked ubiquitination of NMI (PubMed:26342464). ( RO52_HUMAN,P19474 )
Molecular function for TRIM21 Gene according to UniProtKB/Swiss-Prot
Function:
E3 ubiquitin-protein ligase whose activity is dependent on E2 enzymes, UBE2D1, UBE2D2, UBE2E1 and UBE2E2. Forms a ubiquitin ligase complex in cooperation with the E2 UBE2D2 that is used not only for the ubiquitination of USP4 and IKBKB but also for its self-ubiquitination. Component of cullin-RING-based SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes such as SCF(SKP2)-like complexes. A TRIM21-containing SCF(SKP2)-like complex is shown to mediate ubiquitination of CDKN1B (‘Thr-187’ phosphorylated-form), thereby promoting its degradation by the proteasome. Monoubiquitinates IKBKB that will negatively regulates Tax-induced NF-kappa-B signaling. Negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. Mediates the ubiquitin-mediated proteasomal degradation of IgG1 heavy chain, which is linked to the VCP-mediated ER-associated degradation (ERAD) pathway. Promotes IRF8 ubiquitination, which enhanced the ability of IRF8 to stimulate cytokine genes transcription in macrophages. Plays a role in the regulation of the cell cycle progression.
Other Articles in this Open Access Scientific Journal on Galectins and Proteosome Include
Recent genetic studies have identified variants associated with bipolar disorder (BD), but it remains unclear how brain gene expression is altered in BD and how genetic risk for BD may contribute to these alterations. Here, we obtained transcriptomes from subgenual anterior cingulate cortex and amygdala samples from post-mortem brains of individuals with BD and neurotypical controls, including 511 total samples from 295 unique donors. We examined differential gene expression between cases and controls and the transcriptional effects of BD-associated genetic variants. We found two coexpressed modules that were associated with transcriptional changes in BD: one enriched for immune and inflammatory genes and the other with genes related to the postsynaptic membrane. Over 50% of BD genome-wide significant loci contained significant expression quantitative trait loci (QTL) (eQTL), and these data converged on several individual genes, including SCN2A and GRIN2A. Thus, these data implicate specific genes and pathways that may contribute to the pathology of BP.
Gene Expression Markers for Bipolar Disorder Pinpointed
The work was led by researchers at Johns Hopkins’ Lieber Institute for Brain Development. The findings, published this week in Nature Neuroscience, represent the first time that researchers have been able to apply large-scale genetic research to brain samples from hundreds of patients with bipolar disorder (BD). They used 511 total samples from 295 unique donors.
“This is the first deep dive into the molecular biology of the brain in people who died with bipolar disorder—studying actual genes, not urine, blood or skin samples,” said Thomas Hyde of the Lieber Institute and a lead author of the paper. “If we can figure out the mechanisms behind BD, if we can figure out what’s wrong in the brain, then we can begin to develop new targeted treatments of what has long been a mysterious condition.”
Bipolar disorder is characterized by extreme mood swings, with episodes of mania alternating with episodes of depression. It usually emerges in people in their 20s and 30s and remains with them for life. This condition affects approximately 2.8% of the adult American population, or about 7 million people. Patients face higher rates of suicide, poorer quality of life, and lower productivity than the general population. Some estimates put the annual cost of the condition in the U.S. alone at $219.1 billion.
While drugs can be useful in treating BD, many patients find they have bothersome side effects, and for some patients, current medications don’t work at all.
In this study, researchers measured levels of messenger RNA in the brain samples. They observed almost eight times more differentially expressed gene features in the sACC versus the amygdala, suggesting that the sACC may play an especially prominent role—both in mood regulation in general and BD specifically.
In patients who died with BD, the researchers found abnormalities in two families of genes: one containing genes related to the synapse and the second related to immune and inflammatory function.
“There finally is a study using modern technology and our current understanding of genetics to uncover how the brain is doing,” Hyde said. “We know that BD tends to run in families, and there is strong evidence that there are inherited genetic abnormalities that put an individual at risk for bipolar disorder. Unlike diseases such as sickle-cell anemia, bipolar disorder does not result from a single genetic abnormality. Rather, most patients have inherited a group of variants spread across a number of genes.”
“Bipolar disorder, also known as manic-depressive disorder, is a highly damaging and paradoxical condition,” said Daniel R. Weinberger, chief executive and director of the Lieber Institute and a co-author of the study. “It can make people very productive so they can lead countries and companies, but it can also hurl them into the meat grinder of dysfunction and depression. Patients with BD may live on two hours of sleep a night, saving the world with their abundance of energy, and then become so self-destructive that they spend their family’s fortune in a week and lose all friends as they spiral downward. Bipolar disorder also has some shared genetic links to other psychiatric disorders, such as schizophrenia, and is implicated in overuse of drugs and alcohol.”
@MIT Artificial intelligence system rapidly predicts how two proteins will attach: The model called Equidock, focuses on rigid body docking — which occurs when two proteins attach by rotating or translating in 3D space, but their shapes don’t squeeze or bend
Reporter: Aviva Lev-Ari, PhD, RN
This paper introduces a novel SE(3) equivariant graph matching network, along with a keypoint discovery and alignment approach, for the problem of protein-protein docking, with a novel loss based on optimal transport. The overall consensus is that this is an impactful solution to an important problem, whereby competitive results are achieved without the need for templates, refinement, and are achieved with substantially faster run times.
Keywords:protein complexes, protein structure, rigid body docking, SE(3) equivariance, graph neural networks
Abstract: Protein complex formation is a central problem in biology, being involved in most of the cell’s processes, and essential for applications such as drug design or protein engineering. We tackle rigid body protein-protein docking, i.e., computationally predicting the 3D structure of a protein-protein complex from the individual unbound structures, assuming no three-dimensional flexibility during binding. We design a novel pairwise-independent SE(3)-equivariant graph matching network to predict the rotation and translation to place one of the proteins at the right location and the right orientation relative to the second protein. We mathematically guarantee that the predicted complex is always identical regardless of the initial placements of the two structures, avoiding expensive data augmentation. Our model approximates the binding pocket and predicts the docking pose using keypoint matching and alignment through optimal transport and a differentiable Kabsch algorithm. Empirically, we achieve significant running time improvements over existing protein docking software and predict qualitatively plausible protein complex structures despite not using heavy sampling, structure refinement, or templates.
One-sentence Summary: We perform rigid protein docking using a novel independent SE(3)-equivariant message passing mechanism that guarantees the same resulting protein complex independent of the initial placement of the two 3D structures.
MIT researchers created a machine-learning model that can directly predict the complex that will form when two proteins bind together. Their technique is between 80 and 500 times faster than state-of-the-art software methods, and often predicts protein structures that are closer to actual structures that have been observed experimentally.
This technique could help scientists better understand some biological processes that involve protein interactions, like DNA replication and repair; it could also speed up the process of developing new medicines.
“Deep learning is very good at capturing interactions between different proteins that are otherwise difficult for chemists or biologists to write experimentally. Some of these interactions are very complicated, and people haven’t found good ways to express them. This deep-learning model can learn these types of interactions from data,” says Octavian-Eugen Ganea, a postdoc in the MIT Computer Science and Artificial Intelligence Laboratory (CSAIL) and co-lead author of the paper.
Ganea’s co-lead author is Xinyuan Huang, a graduate student at ETH Zurich. MIT co-authors include Regina Barzilay, the School of Engineering Distinguished Professor for AI and Health in CSAIL, and Tommi Jaakkola, the Thomas Siebel Professor of Electrical Engineering in CSAIL and a member of the Institute for Data, Systems, and Society. The research will be presented at the International Conference on Learning Representations.
Significance of the Scientific Development by the @MIT Team
EquiDock wide applicability:
Our method can be integrated end-to-end to boost the quality of other models (see above discussion on runtime importance). Examples are predicting functions of protein complexes [3] or their binding affinity [5], de novo generation of proteins binding to specific targets (e.g., antibodies [6]), modeling back-bone and side-chain flexibility [4], or devising methods for non-binary multimers. See the updated discussion in the “Conclusion” section of our paper.
Advantages over previous methods:
Our method does not rely on templates or heavy candidate sampling [7], aiming at the ambitious goal of predicting the complex pose directly. This should be interpreted in terms of generalization (to unseen structures) and scalability capabilities of docking models, as well as their applicability to various other tasks (discussed above).
Our method obtains a competitive quality without explicitly using previous geometric (e.g., 3D Zernike descriptors [8]) or chemical (e.g., hydrophilic information) features [3]. Future EquiDock extensions would find creative ways to leverage these different signals and, thus, obtain more improvements.
Novelty of theory:
Our work is the first to formalize the notion of pairwise independent SE(3)-equivariance. Previous work (e.g., [9,10]) has incorporated only single object Euclidean-equivariances into deep learning models. For tasks such as docking and binding of biological objects, it is crucial that models understand the concept of multi-independent Euclidean equivariances.
All propositions in Section 3 are our novel theoretical contributions.
We have rewritten the Contribution and Related Work sections to clarify this aspect.
Footnote [a]: We have fixed an important bug in the cross-attention code. We have done a more extensive hyperparameter search and understood that layer normalization is crucial in layers used in Eqs. 5 and 9, but not on the h embeddings as it was originally shown in Eq. 10. We have seen benefits from training our models with a longer patience in the early stopping criteria (30 epochs for DIPS and 150 epochs for DB5). Increasing the learning rate to 2e-4 is important to speed-up training. Using an intersection loss weight of 10 leads to improved results compared to the default of 1.
Bibliography:
[1] Protein-ligand blind docking using QuickVina-W with inter-process spatio-temporal integration, Hassan et al., 2017
[2] GNINA 1.0: molecular docking with deep learning, McNutt et al., 2021
[3] Protein-protein and domain-domain interactions, Kangueane and Nilofer, 2018
[4] Side-chain Packing Using SE(3)-Transformer, Jindal et al., 2022
[5] Contacts-based prediction of binding affinity in protein–protein complexes, Vangone et al., 2015
[6] Iterative refinement graph neural network for antibody sequence-structure co-design, Jin et al., 2021
[7] Hierarchical, rotation-equivariant neural networks to select structural models of protein complexes, Eismann et al, 2020
[8] Protein-protein docking using region-based 3D Zernike descriptors, Venkatraman et al., 2009
[9] SE(3)-transformers: 3D roto-translation equivariant attention networks, Fuchs et al, 2020
[10] E(n) equivariant graph neural networks, Satorras et al., 2021
[11] Fast end-to-end learning on protein surfaces, Sverrisson et al., 2020
The Map of human proteins drawn by artificial intelligence and PROTAC (proteolysis targeting chimeras) Technology for Drug Discovery
Curators: Dr. Stephen J. Williams and Aviva Lev-Ari, PhD, RN
UPDATED on 11/5/2021
Introducing Isomorphic Labs
I believe we are on the cusp of an incredible new era of biological and medical research. Last year DeepMind’s breakthrough AI system AlphaFold2 was recognised as a solution to the 50-year-old grand challenge of protein folding, capable of predicting the 3D structure of a protein directly from its amino acid sequence to atomic-level accuracy. This has been a watershed moment for computational and AI methods for biology. Building on this advance, today, I’m thrilled to announce the creation of a new Alphabet company – Isomorphic Labs – a commercial venture with the mission to reimagine the entire drug discovery process from the ground up with an AI-first approach and, ultimately, to model and understand some of the fundamental mechanisms of life.
For over a decade DeepMind has been in the vanguard of advancing the state-of-the-art in AI, often using games as a proving ground for developing general purpose learning systems, like AlphaGo, our program that beat the world champion at the complex game of Go. We are at an exciting moment in history now where these techniques and methods are becoming powerful and sophisticated enough to be applied to real-world problems including scientific discovery itself. One of the most important applications of AI that I can think of is in the field of biological and medical research, and it is an area I have been passionate about addressing for many years. Now the time is right to push this forward at pace, and with the dedicated focus and resources that Isomorphic Labs will bring.
An AI-first approach to drug discovery and biology The pandemic has brought to the fore the vital work that brilliant scientists and clinicians do every day to understand and combat disease. We believe that the foundational use of cutting edge computational and AI methods can help scientists take their work to the next level, and massively accelerate the drug discovery process. AI methods will increasingly be used not just for analysing data, but to also build powerful predictive and generative models of complex biological phenomena. AlphaFold2 is an important first proof point of this, but there is so much more to come. At its most fundamental level, I think biology can be thought of as an information processing system, albeit an extraordinarily complex and dynamic one. Taking this perspective implies there may be a common underlying structure between biology and information science – an isomorphic mapping between the two – hence the name of the company. Biology is likely far too complex and messy to ever be encapsulated as a simple set of neat mathematical equations. But just as mathematics turned out to be the right description language for physics, biology may turn out to be the perfect type of regime for the application of AI. What’s next for Isomorphic Labs This is just the beginning of what we hope will become a radical new approach to drug discovery, and I’m incredibly excited to get this ambitious new commercial venture off the ground and to partner with pharmaceutical and biomedical companies. I will serve as CEO for Isomorphic’s initial phase, while remaining as DeepMind CEO, partially to help facilitate collaboration between the two companies where relevant, and to set out the strategy, vision and culture of the new company. This will of course include the building of a world-class multidisciplinary team, with deep expertise in areas such as AI, biology, medicinal chemistry, biophysics, and engineering, brought together in a highly collaborative and innovative environment. (We are hiring!) As pioneers in the emerging field of ‘digital biology’, we look forward to helping usher in an amazingly productive new age of biomedical breakthroughs. Isomorphic’s mission could not be a more important one: to use AI to accelerate drug discovery, and ultimately, find cures for some of humanity’s most devastating diseases.
AI research lab DeepMind has created the most comprehensive map of human proteins to date using artificial intelligence. The company, a subsidiary of Google-parent Alphabet, is releasing the data for free, with some scientists comparing the potential impact of the work to that of the Human Genome Project, an international effort to map every human gene.
Proteins are long, complex molecules that perform numerous tasks in the body, from building tissue to fighting disease. Their purpose is dictated by their structure, which folds like origami into complex and irregular shapes. Understanding how a protein folds helps explain its function, which in turn helps scientists with a range of tasks — from pursuing fundamental research on how the body works, to designing new medicines and treatments. “the culmination of the entire 10-year-plus lifetime of DeepMind” Previously, determining the structure of a protein relied on expensive and time-consuming experiments. But last year DeepMind showed it can produce accurate predictions of a protein’s structure using AI software called AlphaFold. Now, the company is releasing hundreds of thousands of predictions made by the program to the public. “I see this as the culmination of the entire 10-year-plus lifetime of DeepMind,” company CEO and co-founder Demis Hassabis told The Verge. “From the beginning, this is what we set out to do: to make breakthroughs in AI, test that on games like Go and Atari, [and] apply that to real-world problems, to see if we can accelerate scientific breakthroughs and use those to benefit humanity.”
Two examples of protein structures predicted by AlphaFold (in blue) compared with experimental results (in green). Image: DeepMind
There are currently around 180,000 protein structures available in the public domain, each produced by experimental methods and accessible through the Protein Data Bank. DeepMind is releasing predictions for the structure of some 350,000 proteins across 20 different organisms, including animals like mice and fruit flies, and bacteria like E. coli. (There is some overlap between DeepMind’s data and pre-existing protein structures, but exactly how much is difficult to quantify because of the nature of the models.) Most significantly, the release includes predictions for 98 percent of all human proteins, around 20,000 different structures, which are collectively known as the human proteome. It isn’t the first public dataset of human proteins, but it is the most comprehensive and accurate.
If they want, scientists can download the entire human proteome for themselves, says AlphaFold’s technical lead John Jumper. “There is a HumanProteome.zip effectively, I think it’s about 50 gigabytes in size,” Jumper tells The Verge. “You can put it on a flash drive if you want, though it wouldn’t do you much good without a computer for analysis!” “anyone can use it for anything” After launching this first tranche of data, DeepMind plans to keep adding to the store of proteins, which will be maintained by Europe’s flagship life sciences lab, the European Molecular Biology Laboratory (EMBL). By the end of the year, DeepMind hopes to release predictions for 100 million protein structures, a dataset that will be “transformative for our understanding of how life works,” according to Edith Heard, director general of the EMBL. The data will be free in perpetuity for both scientific and commercial researchers, says Hassabis. “Anyone can use it for anything,” the DeepMind CEO noted at a press briefing. “They just need to credit the people involved in the citation.” The benefits of protein folding
Understanding a protein’s structure is useful for scientists across a range of fields. The information can help design new medicines, synthesize novel enzymes that break down waste materials, and create crops that are resistant to viruses or extreme weather. Already, DeepMind’s protein predictions are being used for medical research, including studying the workings of SARS-CoV-2, the virus that causes COVID-19. “it will definitely have a huge impact for the scientific community” New data will speed these efforts, but scientists note it will still take a lot of time to turn this information into real-world results. “I don’t think it’s going to be something that changes the way patients are treated within the year, but it will definitely have a huge impact for the scientific community,” Marcelo C. Sousa, a professor at the University of Colorado’s biochemistry department, told The Verge. Scientists will have to get used to having such information at their fingertips, says DeepMind senior research scientist Kathryn Tunyasuvunakool. “As a biologist, I can confirm we have no playbook for looking at even 20,000 structures, so this [amount of data] is hugely unexpected,” Tunyasuvunakool told The Verge. “To be analyzing hundreds of thousands of structures — it’s crazy.”
Notably, though, DeepMind’s software produces predictions of protein structures rather than experimentally determined models, which means that in some cases further work will be needed to verify the structure. DeepMind says it spent a lot of time building accuracy metrics into its AlphaFold software, which ranks how confident it is for each prediction.
Example protein structures predicted by AlphaFold. Image: DeepMind Predictions of protein structures are still hugely useful, though. Determining a protein’s structure through experimental methods is expensive, time-consuming, and relies on a lot of trial and error. That means even a low-confidence prediction can save scientists years of work by pointing them in the right direction for research. Helen Walden, a professor of structural biology at the University of Glasgow, tells The Verge that DeepMind’s data will “significantly ease” research bottlenecks, but that “the laborious, resource-draining work of doing the biochemistry and biological evaluation of, for example, drug functions” will remain. Sousa, who has previously used data from AlphaFold in his work, says for scientists the impact will be felt immediately. “In our collaboration we had with DeepMind, we had a dataset with a protein sample we’d had for 10 years, and we’d never got to the point of developing a model that fit,” he says. “DeepMind agreed to provide us with a structure, and they were able to solve the problem in 15 minutes after we’d been sitting on it for 10 years.”
Why protein folding is so difficult
Proteins are constructed from chains of amino acids, which come in 20 different varieties in the human body. As any individual protein can be comprised of hundreds of individual amino acids, each of which can fold and twist in different directions, it means a molecule’s final structure has an incredibly large number of possible configurations. One estimate is that the typical protein can be folded in 10^300 ways — that’s a 1 followed by 300 zeroes.
Protein folding has been a “grand challenge” of biology for decades
Because proteins are too small to examine with microscopes, scientists have had to indirectly determine their structure using expensive and complicated methods like nuclear magnetic resonance and X-ray crystallography. The idea of determining the structure of a protein simply by reading a list of its constituent amino acids has been long theorized but difficult to achieve, leading many to describe it as a “grand challenge” of biology. In recent years, though, computational methods — particularly those using artificial intelligence — have suggested such analysis is possible. With these techniques, AI systems are trained on datasets of known protein structures and use this information to create their own predictions.
DeepMind’s AlphaFold software has significantly increased the accuracy of computational protein-folding, as shown by its performance in the CASP competition. Image: DeepMind Many groups have been working on this problem for years, but DeepMind’s deep bench of AI talent and access to computing resources allowed it to accelerate progress dramatically. Last year, the company competed in an international protein-folding competition known as CASP and blew away the competition. Its results were so accurate that computational biologist John Moult, one of CASP’s co-founders, said that “in some sense the problem [of protein folding] is solved.”
DeepMind’s AlphaFold program has been upgraded since last year’s CASP competition and is now 16 times faster. “We can fold an average protein in a matter of minutes, most cases seconds,” says Hassabis.
@@@@@@@
The company also released the underlying code for AlphaFold last week as open-source, allowing others to build on its work in the future.
@@@@@@@
Liam McGuffin, a professor at Reading University who developed some of the UK’s leading protein-folding software, praised the technical brilliance of AlphaFold, but also noted that the program’s success relied on decades of prior research and public data. “DeepMind has vast resources to keep this database up to date and they are better placed to do this than any single academic group,” McGuffin told The Verge. “I think academics would have got there in the end, but it would have been slower because we’re not as well resourced.”
Why does DeepMind care?
Many scientists The Verge spoke to noted the generosity of DeepMind in releasing this data for free. After all, the lab is owned by Google-parent Alphabet, which has been pouring huge amounts of resources into commercial healthcare projects. DeepMind itself loses a lot of money each year, and there have been numerousreportsof tensions between the company and its parent firm over issues like research autonomy and commercial viability.
Hassabis, though, tells The Verge that the company always planned to make this information freely available, and that doing so is a fulfillment of DeepMind’s founding ethos. He stresses that DeepMind’s work is used in lots of places at Google — “almost anything you use, there’s some of our technology that’s part of that under the hood” — but that the company’s primary goal has always been fundamental research. “There’s many ways value can be attained.”
“The agreement when we got acquired is that we are here primarily to advance the state of AGI and AI technologies and then use that to accelerate scientific breakthroughs,” says Hassabis. “[Alphabet] has plenty of divisions focused on making money,” he adds, noting that DeepMind’s focus on research “brings all sorts of benefits, in terms of prestige and goodwill for the scientific community. There’s many ways value can be attained.” Hassabis predicts that AlphaFold is a sign of things to come — a project that shows the huge potential of artificial intelligence to handle messy problems like human biology.
“I think we’re at a really exciting moment,” he says. “In the next decade, we, and others in the AI field, are hoping to produce amazing breakthroughs that will genuinely accelerate solutions to the really big problems we have here on Earth.”
Abstract PROTACs-induced targeted protein degradation has emerged as a novel therapeutic strategy in drug development and attracted the favor of academic institutions, large pharmaceutical enterprises (e.g., AstraZeneca, Bayer, Novartis, Amgen, Pfizer, GlaxoSmithKline, Merck, and Boehringer Ingelheim, etc.), and biotechnology companies. PROTACs opened a new chapter for novel drug development. However, any new technology will face many new problems and challenges. Perspectives on the potential opportunities and challenges of PROTACs will contribute to the research and development of new protein degradation drugs and degrader tools.
Although PROTAC technology has a bright future in drug development, it also has many challenges as follows: (1) Until now, there is only one example of PROTAC reported for an “undruggable” target; (18) more cases are needed to prove the advantages of PROTAC in “undruggable” targets in the future. (2) “Molecular glue”, existing in nature, represents the mechanism of stabilized protein–protein interactions through small molecule modulators of E3 ligases. For instance, auxin, the plant hormone, binds to the ligase SCF-TIR1 to drive recruitment of Aux/IAA proteins and subsequently triggers its degradation. In addition, some small molecules that induce targeted protein degradation through “molecular glue” mode of action have been reported. (21,22) Furthermore, it has been recently reported that some PROTACs may actually achieve target protein degradation via a mechanism that includes “molecular glue” or via “molecular glue” alone. (23) How to distinguish between these two mechanisms and how to combine them to work together is one of the challenges for future research. (3) Since PROTAC acts in a catalytic mode, traditional methods cannot accurately evaluate the pharmacokinetics (PK) and pharmacodynamics (PD) properties of PROTACs. Thus, more studies are urgently needed to establish PK and PD evaluation systems for PROTACs. (4) How to quickly and effectively screen for target protein ligands that can be used in PROTACs, especially those targeting protein–protein interactions, is another challenge. (5) How to understand the degradation activity, selectivity, and possible off-target effects (based on different targets, different cell lines, and different animal models) and how to rationally design PROTACs etc. are still unclear. (6) The human genome encodes more than 600 E3 ubiquitin ligases. However, there are only very few E3 ligases (VHL, CRBN, cIAPs, and MDM2) used in the design of PROTACs. How to expand E3 ubiquitin ligase scope is another challenge faced in this area.
PROTAC technology is rapidly developing, and with the joint efforts of the vast number of scientists in both academia and industry, these problems shall be solved in the near future.
PROTACs have opened a new chapter for the development of new drugs and novel chemical knockdown tools and brought unprecedented opportunities to the industry and academia, which are mainly reflected in the following aspects: (1) Overcoming drug resistance of cancer. In addition to traditional chemotherapy, kinase inhibitors have been developing rapidly in the past 20 years. (12) Although kinase inhibitors are very effective in cancer therapy, patients often develop drug resistance and disease recurrence, consequently. PROTACs showed greater advantages in drug resistant cancers through degrading the whole target protein. For example, ARCC-4 targeting androgen receptor could overcome enzalutamide-resistant prostate cancer (13) and L18I targeting BTK could overcome C481S mutation. (14) (2) Eliminating both the enzymatic and nonenzymatic functions of kinase. Traditional small molecule inhibitors usually inhibit the enzymatic activity of the target, while PROTACs affect not only the enzymatic activity of the protein but also nonenzymatic activity by degrading the entire protein. For example, FAK possesses the kinase dependent enzymatic functions and kinase independent scaffold functions, but regulating the kinase activity does not successfully inhibit all FAK function. In 2018, a highly effective and selective FAK PROTAC reported by Craig M. Crews’ group showed a far superior activity to clinical candidate drug in cell migration and invasion. (15) Therefore, PROTAC can expand the druggable space of the existing targets and regulate proteins that are difficult to control by traditional small molecule inhibitors. (3) Degrade the “undruggable” protein target. At present, only 20–25% of the known protein targets (include kinases, G protein-coupled receptors (GPCRs), nuclear hormone receptors, and iron channels) can be targeted by using conventional drug discovery technologies. (16,17) The proteins that lack catalytic activity and/or have catalytic independent functions are still regarded as “undruggable” targets. The involvement of Signal Transducer and Activator of Transcription 3 (STAT3) in the multiple signaling pathway makes it an attractive therapeutic target; however, the lack of an obviously druggable site on the surface of STAT3 limited the development of STAT3 inhibitors. Thus, there are still no effective drugs directly targeting STAT3 approved by the Food and Drug Administration (FDA). In November 2019, Shaomeng Wang’s group first reported a potent PROTAC targeting STAT3 with potent biological activities in vitro and in vivo. (18) This successful case confirms the key potential of PROTAC technology, especially in the field of “undruggable” targets, such as K-Ras, a tricky tumor target activated by multiple mutations as G12A, G12C, G12D, G12S, G12 V, G13C, and G13D in the clinic. (19) (4) Fast and reversible chemical knockdown strategy in vivo. Traditional genetic protein knockout technologies, zinc-finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), or CRISPR-Cas9, usually have a long cycle, irreversible mode of action, and high cost, which brings a lot of inconvenience for research, especially in nonhuman primates. In addition, these genetic animal models sometimes produce phenotypic misunderstanding due to potential gene compensation or gene mutation. More importantly, the traditional genetic method cannot be used to study the function of embryonic-lethal genes in vivo. Unlike DNA-based protein knockout technology, PROTACs knock down target proteins directly, rather than acting at the genome level, and are suitable for the functional study of embryonic-lethal proteins in adult organisms. In addition, PROTACs provide exquisite temporal control, allowing the knockdown of a target protein at specific time points and enabling the recovery of the target protein after withdrawal of drug treatment. As a new, rapid and reversible chemical knockdown method, PROTAC can be used as an effective supplement to the existing genetic tools. (20)
Peroxisome proliferator-activated receptor (PPAR-gamma) Receptors Activation: PPARγ transrepression for Angiogenesis in Cardiovascular Disease and PPARγ transactivation for Treatment of Diabetes
New avenues for research in membrane biology reveals the mobility of protein at work
Curator and Reporter: Dr. Premalata Pati, Ph.D., Postdoc
Membrane proteins(MPs) are proteins that exist in the plasma membrane and conduct a variety of biological functions such as ion transport, substrate transport, and signal transduction. MPs undergo function-related conformational changes on time intervals spanning from nanoseconds to seconds. Many MP structures have been solved thanks to recent developments in structural biology, particularly in single-particle cryo-Electron Microscopy (cryo-EM). Obtaining time-resolved dynamic information on MPs in their membrane surroundings, on the other hand, remains a significant difficulty.
OmpG (Open state) in a fully hydrated dimyristoylphosphatidylcholine (DMPC) bilayer. The protein is shown in light green cartoon. Lipids units are depicted in yellow, while their phosphate and choline groups are illustrated as orange and green van der Waals spheres, respectively. Potassium and chloride counterions are shown in green and purple, respectively. A continuous and semi-transparent cyan representation is used for water. https://static-content.springer.com/esm/art%3A10.1038%2Fs41467-021-24660-1/MediaObjects/41467_2021_24660_MOESM1_ESM.pdf
Weill Cornell Medicine (WCM) researchers have found that they can record high-speed protein movements while linking them to function. The accomplishment should allow scientists to examine proteins in more depth than ever before, and in theory, it should allow for the development of drugs that work better by hitting their protein targets much more effectively.
The researchers utilized High-Speed Atomic Force Microscopy (HS-AFM) to record the rapid motions of a channel protein and published in a report in Nature Communications on July 16. Such proteins generally create channel or tube-like structures in cell membranes, which open to allow molecules to flow under particular conditions. The researchers were able to record the channel protein’s rapid openings and closings with the same temporal resolution as single channel recordings, a typical technique for recording the intermittent passage of charged molecules through the channel.
Senior author Simon Scheuring, professor of physiology and biophysics in anesthesiology at WCM, said,
There has been a significant need for a tool like this that achieves such a high bandwidth that it can ‘see’ the structural variations of molecules as they work.
Researchers can now produce incredibly detailed photographs of molecules using techniques like X-ray crystallography and electron microscopy, showing their structures down to the atomic scale. The average or dominant structural positionings, or conformations, of the molecules, are depicted in these “images,” which are often calculated from thousands of individual photos. In that way, they’re similar to the long-exposure still photos from the dawn of photography.
Many molecules, on the other hand, are flexible and always-moving machinery rather than fixed structures. Scientists need to generate videos, not still photos, to reveal how such molecules move as they work, to see how their motion translates to function to catch their critical functional conformations, which may only exist for a brief moment. Current techniques for dynamic structural imaging, on the other hand, have several drawbacks, one of which being the requirement for fluorescent tags to be inserted on the molecules being photographed in many cases.
Scheuring and his lab were early adopters of the tag-free HS-AFM approach for studying molecular dynamics. The technology, which can photograph molecules in a liquid solution similar to a genuine cellular environment, employs an extremely sensitive probe, similar to a record player’s stylus, to feel its way over a molecule and therefore build up a picture of its structure. Standard HS-AFM isn’t quick enough to capture the high-speed dynamics of many proteins, but Scheuring and colleagues have developed a modified version, HS-AFM height spectroscopy(HS-AFM-HS), that works much faster by collecting dynamic changes in only one dimension: height.
The researchers used HS-AFM-HS to record the opening and closing of a relatively simple channel protein, OmpG, found in bacteria and widely studied as a model channel protein in the new study, led by the first author Raghavendar Reddy Sanganna Gari, a postdoctoral research associate in Scheuring’s laboratory. They were able to monitor OmpG gating at an effective rate of roughly 20,000 data points per second, seeing how it transitioned from open to closed states or vice versa as the acidity of the surrounding fluid varied.
More significantly, they were able to correlate structural dynamics with functional dynamics in a membrane protein of this size for the first time in a partnership with Crina Nimigean, professor of physiology and biophysics in anesthesiology, and her group at WCM.
The demonstration opens the door for a wider application of this method in basic biology and drug development.
Sanganna Gari stated,
We’re now in an exciting period of HS-AFM technology, for example using this technique to study how some drugs modulate the structural dynamics of the channel proteins they target.
Main Source
Technique reveals proteins moving as they work. By Jim Schnabel in Cornell Chronicle, August 16, 2021.
Cryo-EM disclosed how the D614G mutation changes SARS-CoV-2 spike protein structure.
Reporter: Dr. Premalata Pati, Ph.D., Postdoc
SARS-CoV-2, the virus that causes COVID-19, has had a major impact on human health globally; infecting a massive quantity of people around 136,046,262 (John Hopkins University); causing severe disease and associated long-term health sequelae; resulting in death and excess mortality, especially among older and prone populations; altering routine healthcare services; disruptions to travel, trade, education, and many other societal functions; and more broadly having a negative impact on peoples physical and mental health.
It’s need of the hour to answer the questions like what allows the variants of SARS-CoV-2 first detected in the UK, South Africa, and Brazil to spread so quickly? How can current COVID-19 vaccines better protect against them?
Bing Chen, HMS professor of pediatrics at Boston Children’s, and colleagues analyzed the changes in the structure of the spike proteins with the genetic change by D614G mutation by all three variants. Hence they assessed the structure of the coronavirus spike protein down to the atomic level and revealed the reason for the quick spreading of these variants.
This model shows the structure of the spike protein in its closed configuration, in its original D614 form (left) and its mutant form (G614). In the mutant spike protein, the 630 loop (in red) stabilizes the spike, preventing it from flipping open prematurely and rendering SARS-CoV-2 more infectious.
Fig. 1. Cryo-EM structures of the full-length SARS-CoV-2 S protein carrying G614.
(A) Three structures of the G614 S trimer, representing a closed, three RBD-down conformation, an RBD-intermediate conformation and a one RBD-up conformation, were modeled based on corresponding cryo-EM density maps at 3.1-3.5Å resolution. Three protomers (a, b, c) are colored in red, blue and green, respectively. RBD locations are indicated. (B) Top views of superposition of three structures of the G614 S in (A) in ribbon representation with the structure of the prefusion trimer of the D614 S (PDB ID: 6XR8), shown in yellow. NTD and RBD of each protomer are indicated. Side views of the superposition are shown in fig. S8.
The mutant spikes were imaged by Cryo-Electron microscopy (cryo-EM), which has resolution down to the atomic level. They found that the D614G mutation (substitution of in a single amino acid “letter” in the genetic code for the spike protein) makes the spike more stable as compared with the original SARS-CoV-2 virus. As a result, more functional spikes are available to bind to our cells’ ACE2 receptors, making the virus more contagious.
Fig. 2. Cryo-EM revealed how the D614G mutation changes SARS-CoV-2 spike protein structure.
Say the original virus has 100 spikes,” Chen explained. “Because of the shape instability, you may have just 50 percent of them functional. In the G614 variants, you may have 90 percent that is functional. So even though they don’t bind as well, the chances are greater and you will have an infection
Forthcoming directions by Bing Chen and Team
The findings suggest the current approved COVID-19 vaccines and any vaccines in the works should include the genetic code for this mutation. Chen has quoted:
Since most of the vaccines so far—including the Moderna, Pfizer–BioNTech, Johnson & Johnson, and AstraZeneca vaccines are based on the original spike protein, adding the D614G mutation could make the vaccines better able to elicit protective neutralizing antibodies against the viral variants
Chen proposes that redesigned vaccines incorporate the code for this mutant spike protein. He believes the more stable spike shape should make any vaccine based on the spike more likely to elicit protective antibodies. Chen also has his sights set on therapeutics. He and his colleagues are further applying structural biology to better understand how SARS-CoV-2 binds to the ACE2 receptor. That could point the way to drugs that would block the virus from gaining entry to our cells.
In January, the team showed that a structurally engineered “decoy” ACE2 protein binds to SARS-CoV-2 200 times more strongly than the body’s own ACE2. The decoy potently inhibited the virus in cell culture, suggesting it could be an anti-COVID-19 treatment. Chen is now working to advance this research into animal models.
Main Source:
Abstract
Substitution for aspartic acid by glycine at position 614 in the spike (S) protein of severe acute respiratory syndrome coronavirus 2 appears to facilitate rapid viral spread. The G614 strain and its recent variants are now the dominant circulating forms. We report here cryo-EM structures of a full-length G614 S trimer, which adopts three distinct prefusion conformations differing primarily by the position of one receptor-binding domain. A loop disordered in the D614 S trimer wedges between domains within a protomer in the G614 spike. This added interaction appears to prevent premature dissociation of the G614 trimer, effectively increasing the number of functional spikes and enhancing infectivity, and to modulate structural rearrangements for membrane fusion. These findings extend our understanding of viral entry and suggest an improved immunogen for vaccine development.
Comparing COVID-19 Vaccine Schedule Combinations, or “Com-COV” – First-of-its-Kind Study will explore the Impact of using eight different Combinations of Doses and Dosing Intervals for Different COVID-19 Vaccines
Intellia announced in its fourth-quarter earnings report that Novartis had ended development of sickle cell treatment OTQ923/HIX763. (Getty Images)
Novartis will no longer develop an ex vivo sickle cell disease program that was part of an older deal with Intellia, and the gene editing biotech’s CEO John Leonard, M.D., thinks he knows why.
“We’ve always believed that the future lies with the in vivo approaches, and that’s been a focus of the work that we do,” Leonard said. “I’m sure they looked at the ex vivo space and may have had some of the same realizations that we had some years ago.”
Leonard, of course, said he wasn’t completely sure why Novartis opted to cut the program, but noted that the Big Pharma is undergoing a broad pipeline reorganization.
Novartis confirmed just that in an emailed statement to Fierce Biotech, saying that the program was discontinued for strategic reasons. The overall partnership with Intellia remains intact, however, the spokesperson said.
Intellia announced in its fourth-quarter earnings report Thursday that the Swiss pharma ended development of OTQ923/HIX763 this month.
The therapy uses autologous, ex vivo, CRISPR-edited hematopoietic stem cells to target fetal hemoglobin for treating sickle cell disease. Novartis initiated dosing on a phase 1/2 trial for the Intellia-partnered program in 2021.
Intellia has both types of candidate in its pipeline, but the in vivo list is longer and more advanced, with NTLA-2001 in transthyretin (ATTR) amyloidosis leading the pack.
Novartis and Intellia have had a cell therapy partnership since January 2015, which was three months after Intellia launched from Atlas Venture and Caribou Biosciences. The agreement was revised in 2018 to expand to ex vivo development of cell therapies using certain ocular stem cells. At that time, Intellia received a $10 million payment, but other financial details of the agreement have not been disclosed. Novartis gained the rights to opt in on one or more programs, while Intellia earned the right to use the pharma’s lipid nanoparticle technology for all genome editing applications in both in vivo and ex vivo settings.
Intellia, working with its partner Regeneron, has shown over the past year that CRISPR/Cas9 in vivo gene editing can cause high, seemingly durable levels of gene knockdown in humans. While questions about the Intellia data, and the concept more broadly, remain unanswered, there is now early evidence that the approach may be effective and, as importantly, safe. Precision is one of a clutch of companies barreling toward the clinic in the wake of Intellia, and the potential of its Arcus platform to provide greater precision and versatility than CRISPR/Cas9 and zinc finger nuclease has now attracted a suitor.
To add to its in vivo capabilities, Novartis is set to pay $50 million in cash to partner with Precision. The deal also features a $25 million equity investment priced at $2.01 per share, a 20% premium over the recent average for the stock, as well as up to $1.4 billion in milestones, research funding and royalties ranging from the mid-single-digit to low-double-digit percentages.
Alnylam Announces First-Ever FDA Approval of an RNAi Therapeutic, ONPATTRO™ (patisiran) for the Treatment of the Polyneuropathy of Hereditary Transthyretin-Mediated Amyloidosis in Adults
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
$The authors would like to note that the first eight authors are listed alphabetically.
Abstract
During its first month, the recently emerged 2019 Wuhan novel coronavirus (2019-nCoV) has already infected many thousands of people in mainland China and worldwide and took hundreds of lives. However, the swiftly spreading virus also caused an unprecedentedly rapid response from the research community facing the unknown health challenge of potentially enormous proportions. Unfortunately, the experimental research to understand the molecular mechanisms behind the viral infection and to design a vaccine or antivirals is costly and takes months to develop. To expedite the advancement of our knowledge we leverage the data about the related coronaviruses that is readily available in public databases, and integrate these data into a single computational pipeline. As a result, we provide a comprehensive structural genomics and interactomics road-maps of 2019-nCoV and use these information to infer the possible functional differences and similarities with the related SARS coronavirus. All data are made publicly available to the research community at http://korkinlab.org/wuhan
Figure 2. Structurally characterized non-structural proteins of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. The structurally resolved part of wNsp7 is sequentially identical to its homolog.
Figure 3. Structurally characterized structural proteins and an ORF of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. Highlighted in yellow are novel protein inserts found in wS.
Figure 4. Structurally characterized intra-viral and host-viral protein-protein interaction complexes of 2019-nCoV. Human proteins (colored in orange) are identified through their gene names. For each intra-viral structure, the number of subunits involved in the interaction is specified.
Figure 5. Evolutionary conservation of functional sites in 2019-nCoV proteins. A. Fully conserved protein binding sites (PBS, light orange) of wNsp12 in its interaction with wNsp7 and wNsp8 while other parts of the protein surface shows mutations (magenta); B. Both major monoclonal antibody binding site (light orange) and ACE2 receptor binding site (dark green) of wS are heavily mutated (binding site mutations are shown in red) compared to the same binding sites in other coronaviruses; mutations not located on the two binding sites are shown in magenta; C. Nearly intact protein binding site (light orange) of wNsp (papain-like protease PLpro domain) for its putative interaction with human ubiquitin-aldehyde (binding site mutations for the only two residues are shown in red, non-binding site mutations are shown in magenta); D. Fully conserved inhibitor ligand binding site (LBS, green) for wNsp5; non-binding site mutations are shown in magenta.
According to the World Health Organization, coronaviruses make up a large family of viruses named for the crown-like spikes found on their surface (Figure 1). They carry their genetic material in single strands of RNA and cause respiratory problems and fever. Like HIV, coronaviruses can be transmitted between animals and humans. Coronaviruses have been responsible for the Severe Acute Respiratory Syndrome (SARS) pandemic in the early 2000s and the Middle East Respiratory Syndrome (MERS) outbreak in South Korea in 2015. While the most recent coronavirus, COVID-19, has caused international concern, accessible and inexpensive sequencing is helping us understand COVID-19 and respond to the outbreak quickly.
Figure 1. Coronaviruses with the characteristic spikes as seen under a microscope.
First studies that explore genetic susceptibility to COVID-19 are now being published. The first results indicate that COVID-19 infects cells using the ACE2 cell-surface receptor. Genetic variants in the ACE2 receptor gene are thus likely to influence how effectively COVID-19 can enter the cells in our bodies. Researchers hope to discover genetic variants that confer resistance to a COVID-19 infection, similar to how some variants in the CCR5 receptor gene make people immune to HIV. At Nebula Genomics, we are monitoring the latest COVID-19 research and will add any relevant discoveries to the Nebula Research Library in a timely manner.
The Role of Genomics in Responding to COVID-19
Scientists in China sequenced COVID-19’s genome just a few weeks after the first case was reported in Wuhan. This stands in contrast to SARS, which was discovered in late 2002 but was not sequenced until April of 2003. It is through inexpensive genome-sequencing that many scientists across the globe are learning and sharing information about COVID-19, allowing us to track the evolution of COVID-19 in real-time. Ultimately, sequencing can help remove the fear of the unknown and allow scientists and health professionals to prepare to combat the spread of COVID-19.
Next-generation DNA sequencing technology has enabled us to understand COVID-19 is ~30,000 bases long. Moreover, researchers in China determined that COVID-19 is also almost identical to a coronavirus found in bats and is very similar to SARS. These insights have been critical in aiding in the development of diagnostics and vaccines. For example, the Centers for Disease Control and Prevention developed a diagnostic test to detect COVID-19 RNA from nose or mouth swabs.
Moreover, a number of different government agencies and pharmaceutical companies are in the process of developing COVID-19 vaccines to stop the COVID-19 from infecting more people. To protect humans from infection inactivated virus particles or parts of the virus (e.g. viral proteins) can be injected into humans. The immune system will recognize the inactivated virus as foreign, priming the body to build immunity against possible future infection. Of note, Moderna Inc., the National Institute of Allergy and Infectious Diseases, and Coalition for Epidemic Preparedness Innovations identified a COVID-19 vaccine candidate in a record 42 days. This vaccine will be tested in human clinical trials starting in April.
For more information about COVID-19, please refer to the World Health Organization website.
The problem w/ visionaries is that we don’t recognize them in a timely manner (too late) Ralph Baric @UNCpublichealth and Vineet Menachery deserve recognition for being 5 yrs ahead of #COVID19https://nature.com/articles/nm.3985…@NatureMedicinehttps://pnas.org/content/113/11/3048…@PNASNews via @hoondy
Senior, A.W., Evans, R., Jumper, J. et al.Improved protein structure prediction using potentials from deep learning. Nature577, 706–710 (2020). https://doi.org/10.1038/s41586-019-1923-7
Abstract
Protein structure prediction can be used to determine the three-dimensional shape of a protein from its amino acid sequence1. This problem is of fundamental importance as the structure of a protein largely determines its function2; however, protein structures can be difficult to determine experimentally. Considerable progress has recently been made by leveraging genetic information. It is possible to infer which amino acid residues are in contact by analysing covariation in homologous sequences, which aids in the prediction of protein structures3. Here we show that we can train a neural network to make accurate predictions of the distances between pairs of residues, which convey more information about the structure than contact predictions. Using this information, we construct a potential of mean force4 that can accurately describe the shape of a protein. We find that the resulting potential can be optimized by a simple gradient descent algorithm to generate structures without complex sampling procedures. The resulting system, named AlphaFold, achieves high accuracy, even for sequences with fewer homologous sequences. In the recent Critical Assessment of Protein Structure Prediction5 (CASP13)—a blind assessment of the state of the field—AlphaFold created high-accuracy structures (with template modelling (TM) scores6 of 0.7 or higher) for 24 out of 43 free modelling domains, whereas the next best method, which used sampling and contact information, achieved such accuracy for only 14 out of 43 domains. AlphaFold represents a considerable advance in protein-structure prediction. We expect this increased accuracy to enable insights into the function and malfunction of proteins, especially in cases for which no structures for homologous proteins have been experimentally determined7. https://doi.org/10.1038/s41586-019-1923-7
The scientific community has galvanised in response to the recent COVID-19 outbreak, building on decades of basic research characterising this virus family. Labs at the forefront of the outbreak response shared genomes of the virus in open access databases, which enabled researchers to rapidly develop tests for this novel pathogen. Other labs have shared experimentally-determined and computationally-predicted structures of some of the viral proteins, and still others have shared epidemiological data. We hope to contribute to the scientific effort using the latest version of our AlphaFold system by releasing structure predictions of several under-studied proteins associated with SARS-CoV-2, the virus that causes COVID-19. We emphasise that these structure predictions have not been experimentally verified, but hope they may contribute to the scientific community’s interrogation of how the virus functions, and serve as a hypothesis generation platform for future experimental work in developing therapeutics. We’re indebted to the work of many other labs: this work wouldn’t be possible without the efforts of researchers across the globe who have responded to the COVID-19 outbreak with incredible agility.
Knowing a protein’s structure provides an important resource for understanding how it functions, but experiments to determine the structure can take months or longer, and some prove to be intractable. For this reason, researchers have been developing computational methods to predict protein structure from the amino acid sequence. In cases where the structure of a similar protein has already been experimentally determined, algorithms based on “template modelling” are able to provide accurate predictions of the protein structure. AlphaFold, our recently published deep learning system, focuses on predicting protein structure accurately when no structures of similar proteins are available, called “free modelling”. We’ve continued to improve these methods since that publication and want to provide the most useful predictions, so we’re sharing predicted structures for some of the proteins in SARS-CoV-2 generated using our newly-developed methods.
It’s important to note that our structure prediction system is still in development and we can’t be certain of the accuracy of the structures we are providing, although we are confident that the system is more accurate than our earlier CASP13 system. We confirmed that our system provided an accurate prediction for the experimentally determined SARS-CoV-2 spike protein structure shared in the Protein Data Bank, and this gave us confidence that our model predictions on other proteins may be useful. We recently shared our results with several colleagues at the Francis Crick Institute in the UK, including structural biologists and virologists, who encouraged us to release our structures to the general scientific community now. Our models include per-residue confidence scores to help indicate which parts of the structure are more likely to be correct. We have only provided predictions for proteins which lack suitable templates or are otherwise difficult for template modeling. While these understudied proteins are not the main focus of current therapeutic efforts, they may add to researchers’ understanding of SARS-CoV-2.
Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them.
Interested researchers can download the structures here, and can read more technical details about these predictions in a document included with the data. The protein structure predictions we’re releasing are for SARS-CoV-2 membrane protein, protein 3a, Nsp2, Nsp4, Nsp6, and Papain-like proteinase (C terminal domain). To emphasise, these are predicted structures which have not been experimentally verified. Work on the system continues for us, and we hope to share more about it in due course.
DeepMind has shared its results with researchers at the Francis Crick Institute, a biomedical research lab in the UK, as well as offering it for download from its website.
“Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them,” it said. [ALA added bold face]
There are 93,090 cases of COVID-19, and 3,198 deaths, spread across 76 countries, according to the latest report from the World Health Organization at time of writing. ®
MHC content – The spike protein is thought to be the key to binding to cells via the angiotensin II receptor, the major mechanism the immune system uses to distinguish self from non-self
Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies
Syed Faraz Ahmed 1,† , Ahmed A. Quadeer 1, *,† and Matthew R. McKay 1,2, *
1 Department of Electronic and Computer Engineering, The Hong Kong University of Science and
Technology, Hong Kong, China; sfahmed@connect.ust.hk
2 Department of Chemical and Biological Engineering, The Hong Kong University of Science and
Received: 9 February 2020; Accepted: 24 February 2020; Published: 25 February 2020
Abstract:
The beginning of 2020 has seen the emergence of COVID-19 outbreak caused by a novel coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). There is an imminent need to better understand this new virus and to develop ways to control its spread. In this study, we sought to gain insights for vaccine design against SARS-CoV-2 by considering the high genetic similarity between SARS-CoV-2 and SARS-CoV, which caused the outbreak in 2003, and leveraging existing immunological studies of SARS-CoV. By screening the experimentally determined SARS-CoV-derived B cell and T cell epitopes in the immunogenic structural proteins of SARS-CoV, we identified a set of B cell and T cell epitopes derived from the spike (S) and nucleocapsid (N) proteins that map identically to SARS-CoV-2 proteins. As no mutation has been observed in these identified epitopes among the 120 available SARS-CoV-2 sequences (as of 21 February 2020), immune targeting of these epitopes may potentially offer protection against this novel virus. For the T cell epitopes, we performed a population coverage analysis of the associated MHC alleles and proposed a set of epitopes that is estimated to provide broad coverage globally, as well as in China. Our findings provide a screened set of epitopes that can help guide experimental efforts towards the development of vaccines against SARS-CoV-2.
Re: Protein structure prediction has been done for ages…
Not quite, Natural Selection does not measure methods, it measures outputs, usually at the organism level.
Sure correct folding is necessary for much protein function and we have prions and chaperone proteins to get it wrong and right.
The only way NS measures methods and mechanisms is if they are very energetically wasteful. But there are some very wasteful ones out there. Beta-Catenin at the end of point of Wnt signalling comes particularly to mind.
“Determining the structure of the virus proteins might also help in developing a molecule that disrupts the operation of just those proteins, and not anything else in the human body.”
Well it might, but predicting whether a ‘drug’ will NOT interact with any other of the 20000+ protein in complex organisms is well beyond current science. If we could do that we could predict/avoid toxicity and other non-mechanism related side-effects & mostly we can’t.
There are 480 structures on PDBe resulting from a search on ‘coronavirus,’ the top hits from MERS and SARS. PR stunt or not, they did win the most recent CASP ‘competition’, so arguably it’s probably our best shot right now – and I am certainly not satisfied that they have been sufficiently open in explaining their algorithms though I have not checked in the last few months. No one is betting anyone’s health on this, and it is not like making one wrong turn in a series of car directions. Latest prediction algorithms incorporate contact map predictions, so it’s not like a wrong dihedral angle sends the chain off in the wrong direction. A decent model would give something to run docking algorithms against with a series of already approved drugs, then we take that shortlist into the lab. A confirmed hit could be an instantly available treatment, no two year wait as currently estimated. [ALA added bold face]
Re: these structure predictions have not been experimentally verified
Naaaah. Can’t possibly be a stupid marketing stunt.
Well yes, a good possibility. But it can also be trying to build on the open-source model of putting it out there for others to build and improve upon. Essentially opening that “peer review” to a larger audience quicker. [ALA added bold face]
What bothers me, besides the obvious PR stunt, is that they say this prediction is licensed. How can a prediction from software be protected by, I presume, patents? And if this can be protected without even verifying which predictions actually work, what’s to stop someone spitting out millions of random, untested predictions just in case they can claim ownership later when one of them is proven to work? [ALA added bold face]
AI-predicted protein structures could unlock vaccine for Wuhan coronavirus… if correct… after clinical trials It’s not quite DeepMind’s ‘Come with me if you want to live’ moment, but it’s close, maybe
Experimentally derived by a group of scientists at the University of Texas at Austin and the National Institute of Allergy and Infectious Diseases, an agency under the US National Institute of Health. They both feature a “Spike protein structure.”
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Recent research from University of Alberta is looking at the role of fibrinogen, the substrate of thrombin in regulating a natural defense mechanism in the body. Fibrinogen is a well-known protein that is essential for wound healing and blood clotting in the body. Levels of fibrinogen increase in inflammatory states as part of the acute-phase response.
However, daily variation in plasma fibrinogen levels weakens its potential as a biomarker of cardiovascular risk. The discovery is expected to contribute to enhanced diagnosis and treatments for patients in a variety of diseases ranging from inflammation, to heart failure, to cancer.
Yet, a study published in Scientific Reports in March 2019 show that fibrinogen can also be a natural inhibitor of an enzyme named MMP2, which is important for normal organ development and repair. The researchers believe an essential function of fibrinogen is to allow or disallow the enzyme to carry out its normal functions. Nevertheless, high levels of fibrinogen may disproportionately inhibit MMP2, that could result in arthritic and cardiac disorders.
The researcher highlights the inner workings of the MMP family of enzymes by having a greater understanding of how MMPs are regulated. They hypothesize that abnormal MMP2 activity could be an unwelcome side effect of common medications such as the cholesterol-lowering drugs and the antibiotic doxycycline, both of which are known to inhibit MMPs. They also emphasize that future therapeutic developments must strike a balance between the levels of MMPs and their inhibitors, such as fibrinogen, so that net MMP activity in the body remains at nearly normal levels.

















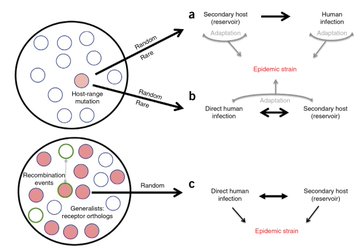
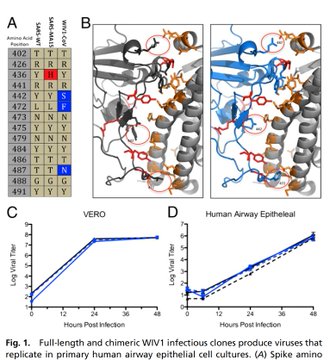

Paper in collection COVID-19 SARS-CoV-2 preprints from medRxiv and bioRxiv