Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Genomic Diagnostics: Three Techniques to Perform Single Cell Gene Expression and Genome Sequencing Single Molecule DNA Sequencing
Curator: Aviva Lev-Ari, PhD, RN
4.2.3 Genomic Diagnostics: Three Techniques to Perform Single Cell Gene Expression and Genome Sequencing Single Molecule DNA Sequencing, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 4: Single Cell Genomics
Ido Amit, AmitLab @Department of Immunology at the Weizmann Institute of Science, Rehovot, Israel
The Amit lab studies the genomic code enabling immune cells to differentiate to specific subtypes and devise a specific response to invading pathogens. Our main focus is to understand how gene regulatory networks activate this code. We develop and apply state of the art high throughputgenomic tools and interdisciplinary approaches to address these critical biological and therapeutic questions. We believe that elucidating the underlying principles of the regulatory code will allow us to impact the future of personalized medicine. We are currently seeking highly motivated individuals who want to join us on this quest.
Basic lessons – “Single-cell genomics will soon be commonplace in basic and applied immunology research.”
It is early days for single-cell genomics. But already, a number of important lessons can be learnt from the experiences of our lab and those of others.
First, it is clear that many of the current categories of immune cells, such as T cells or monocytes, encompass heterogeneous populations. To probe cellular complexity, researchers must therefore cast their nets wide, and try to collect all immune cells within a tissue or region of interest. This is a very different approach from that used with methods based on cell-surface markers, which aim to obtain as pure a sample as possible.
Second, success will depend, in part, on the extent to which researchers preserve the states of cells and the original composition of a tissue. Cell stress or death should be minimized to ensure that tissue preparation does not favour specific cell types. (Some are more sensitive to heat stress, for example, than others.)
Third, bioinformaticians will need to develop scalable and robust algorithms to cope with greater numbers of cells, conflicting or overlapping programs of gene expression and fleeting developmental stages.
Fourth, after researchers have characterized all of the immune cells in a sample, they will need to find molecular markers that can be used to either enrich or deplete certain cell types in further samples. Tissues comprise trillions of cells with myriad molecular characteristics and functions, and the types or states of these cells may vary in abundance by many orders of magnitude. For instance, in the brains of healthy mice, our newly identified population of DAM makes up less than 0.01% of cells15. Thus, repeated unbiased sampling to characterize rare populations will keep on accumulating cells that are not those of interest.
Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, Meinhof K, Chow A, Kim-Shulze S, Wolf A, Medaglia C, Li H, Rytlewski JA, Emerson RO, Solovyov A, Greenbaum BD, Sanders C, Vignali M, Beasley MB, Flores R, Gnjatic S, Pe’er D, Rahman A, Amit I, Merad M.
Cell. 2017 May 4;169(4):750-765.e17. doi: 10.1016/j.cell.2017.04.014.
Mildner A, Chapnik E, Varol D, Aychek T, Lampl N, Rivkin N, Bringmann A, Paul F, Boura-Halfon S, Hayoun YS, Barnett-Itzhaki Z, Amit I, Hornstein E, Jung S.
Eur J Immunol. 2017 May 4. doi: 10.1002/eji.201746987. [Epub ahead of print]
Guillaumet-Adkins A, Rodríguez-Esteban G, Mereu E, Mendez-Lago M, Jaitin DA, Villanueva A, Vidal A, Martinez-Marti A, Felip E, Vivancos A, Keren-Shaul H, Heath S, Gut M, Amit I, Gut I, Heyn H.
Genome Biol. 2017 Mar 1;18(1):45. doi: 10.1186/s13059-017-1171-9.
Bahar Halpern K, Shenhav R, Matcovitch-Natan O, Tóth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, Brandis A, Giladi A, Stokar-Avihail A, David E, Amit I, Itzkovitz S.
Science 04 Nov 2016:
Vol. 354, Issue 6312, pp. 557-558
DOI: 10.1126/science.aak9761
Summary
We each begin life as a single cell harboring a single genome, which—over the course of development—gives rise to the trillions of cells that make up the body. From skin cells to heart cells to neurons of the brain, each bears a copy of the original cell’s genome. But as anyone who has used a copy machine or played the childhood game of “telephone” knows, copies are never perfect. Every cell in an individual actually has a unique genome, an imperfect copy of its cellular ancestor differentiated by inevitable somatic mutations arising from errors in DNA replication and other mutagenic forces (1). Somatic mutation is the fundamental process leading to all genetic diseases, including cancer; every inherited genetic disease also has its origins in such mutation events that occurred in an ancestor’s germline cells. Yet how many and what kinds of somatic mutations accumulate in our cells as we develop and age has long been unknown and a blind spot in our understanding of the origins of genetic disease.
The author of the prize-winning essay, Gilad Evrony, received his undergraduate degree from the Massachusetts Institute of Technology. He served in the Intelligence Division of the Israel Defense Forces and completed an M.D. and Ph.D. at Harvard Medical School, with graduate research in the laboratory of Dr. Christopher Walsh at Boston Children’s Hospital. Dr. Evrony is currently pursuing clinical training in pediatrics at Mount Sinai Hospital and continuing his research developing novel technologies for studying the brain and neuropsychiatric diseases.
1Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, NY 11724, USA.
Abstract
Transcription factors (TFs) are commonly deregulated in the pathogenesis of human cancer and are a major class of cancer cell dependencies. Consequently, targeting of TFs can be highly effective in treating particular malignancies, as highlighted by the clinical efficacy of agents that target nuclear hormone receptors. In this review we discuss recent advances in our understanding of TFs as drug targets in oncology, with an emphasis on the emerging chemical approaches to modulate TF function. The remarkable diversity and potency of TFs as drivers of cell transformation justifies a continued pursuit of TFs as therapeutic targets for drug discovery.
Tumor cells often have aberrant transcription factor expression profiles. The aberration could have its origin at the genomic or transcriptomic level. The potency of transcription factors as drivers of tumorigenesis makes it an attractive therapeutic target for drug discovery.
Disease related changes in proteomics, protein folding, protein-protein interaction, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Disease related changes in proteomics, protein folding, protein-protein interaction
Curator: Larry H. Bernstein, MD, FCAP
LPBI
Frankenstein Proteins Stitched Together by Scientists
The Frankenstein monster, stitched together from disparate body parts, proved to be an abomination, but stitched together proteins may fare better. They may, for example, serve specific purposes in medicine, research, and industry. At least, that’s the ambition of scientists based at the University of North Carolina. They have developed a computational protocol called SEWING that builds new proteins from connected or disconnected pieces of existing structures. [Wikipedia]
Unlike Victor Frankenstein, who betrayed Promethean ambition when he sewed together his infamous creature, today’s biochemists are relatively modest. Rather than defy nature, they emulate it. For example, at the University of North Carolina (UNC), researchers have taken inspiration from natural evolutionary mechanisms to develop a technique called SEWING—Structure Extension With Native-substructure Graphs. SEWING is a computational protocol that describes how to stitch together new proteins from connected or disconnected pieces of existing structures.
“We can now begin to think about engineering proteins to do things that nothing else is capable of doing,” said UNC’s Brian Kuhlman, Ph.D. “The structure of a protein determines its function, so if we are going to learn how to design new functions, we have to learn how to design new structures. Our study is a critical step in that direction and provides tools for creating proteins that haven’t been seen before in nature.”
Traditionally, researchers have used computational protein design to recreate in the laboratory what already exists in the natural world. In recent years, their focus has shifted toward inventing novel proteins with new functionality. These design projects all start with a specific structural “blueprint” in mind, and as a result are limited. Dr. Kuhlman and his colleagues, however, believe that by removing the limitations of a predetermined blueprint and taking cues from evolution they can more easily create functional proteins.
Dr. Kuhlman’s UNC team developed a protein design approach that emulates natural mechanisms for shuffling tertiary structures such as pleats, coils, and furrows. Putting the approach into action, the UNC team mapped 50,000 stitched together proteins on the computer, and then it produced 21 promising structures in the laboratory. Details of this work appeared May 6 in the journal Science, in an article entitled, “Design of Structurally Distinct Proteins Using Strategies Inspired by Evolution.”
“Helical proteins designed with SEWING contain structural features absent from other de novo designed proteins and, in some cases, remain folded at more than 100°C,” wrote the authors. “High-resolution structures of the designed proteins CA01 and DA05R1 were solved by x-ray crystallography (2.2 angstrom resolution) and nuclear magnetic resonance, respectively, and there was excellent agreement with the design models.”
Essentially, the UNC scientists confirmed that the proteins they had synthesized contained the unique structural varieties that had been designed on the computer. The UNC scientists also determined that the structures they had created had new surface and pocket features. Such features, they noted, provide potential binding sites for ligands or macromolecules.
“We were excited that some had clefts or grooves on the surface, regions that naturally occurring proteins use for binding other proteins,” said the Science article’s first author, Tim M. Jacobs, Ph.D., a former graduate student in Dr. Kuhlman’s laboratory. “That’s important because if we wanted to create a protein that can act as a biosensor to detect a certain metabolite in the body, either for diagnostic or research purposes, it would need to have these grooves. Likewise, if we wanted to develop novel therapeutics, they would also need to attach to specific proteins.”
Currently, the UNC researchers are using SEWING to create proteins that can bind to several other proteins at a time. Many of the most important proteins are such multitaskers, including the blood protein hemoglobin.
Histone Mutation Deranges DNA Methylation to Cause Cancer
In some cancers, including chondroblastoma and a rare form of childhood sarcoma, a mutation in histone H3 reduces global levels of methylation (dark areas) in tumor cells but not in normal cells (arrowhead). The mutation locks the cells in a proliferative state to promote tumor development. [Laboratory of Chromatin Biology and Epigenetics at The Rockefeller University]
They have been called oncohistones, the mutated histones that are known to accompany certain pediatric cancers. Despite their suggestive moniker, oncohistones have kept their oncogenic secrets. For example, it has been unclear whether oncohistones are able to cause cancer on their own, or whether they need to act in concert with additional DNA mutations, that is, mutations other than those affecting histone structures.
While oncohistone mechanisms remain poorly understood, this particular question—the oncogenicity of lone oncohistones—has been resolved, at least in part. According to researchers based at The Rockefeller University, a change to the structure of a histone can trigger a tumor on its own.
This finding appeared May 13 in the journal Science, in an article entitled, “Histone H3K36 Mutations Promote Sarcomagenesis Through Altered Histone Methylation Landscape.” The article describes the Rockefeller team’s study of a histone protein called H3, which has been found in about 95% of samples of chondoblastoma, a benign tumor that arises in cartilage, typically during adolescence.
The Rockefeller scientists found that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo.
After the scientists inserted the H3 histone mutation into mouse mesenchymal progenitor cells (MPCs)—which generate cartilage, bone, and fat—they watched these cells lose the ability to differentiate in the lab. Next, the scientists injected the mutant cells into living mice, and the animals developed the tumors rich in MPCs, known as an undifferentiated sarcoma. Finally, the researchers tried to understand how the mutation causes the tumors to develop.
The scientists determined that H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases.
“Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation,” the authors of the Science study wrote. “After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation.”
Essentially, when the H3K36M mutation occurs, the cell becomes locked in a proliferative state—meaning it divides constantly, leading to tumors. Specifically, the mutation inhibits enzymes that normally tag the histone with chemical groups known as methyls, allowing genes to be expressed normally.
In response to this lack of modification, another part of the histone becomes overmodified, or tagged with too many methyl groups. “This leads to an overall resetting of the landscape of chromatin, the complex of DNA and its associated factors, including histones,” explained co-author Peter Lewis, Ph.D., a professor at the University of Wisconsin-Madison and a former postdoctoral fellow in laboratory of C. David Allis, Ph.D., a professor at Rockefeller.
The finding—that a “resetting” of the chromatin landscape can lock the cell into a proliferative state—suggests that researchers should be on the hunt for more mutations in histones that might be driving tumors. For their part, the Rockefeller researchers are trying to learn more about how this specific mutation in histone H3 causes tumors to develop.
“We want to know which pathways cause the mesenchymal progenitor cells that carry the mutation to continue to divide, and not differentiate into the bone, fat, and cartilage cells they are destined to become,” said co-author Chao Lu, Ph.D., a postdoctoral fellow in the Allis lab.
Once researchers understand more about these pathways, added Dr. Lewis, they can consider ways of blocking them with drugs, particularly in tumors such as MPC-rich sarcomas—which, unlike chondroblastoma, can be deadly. In fact, drugs that block these pathways may already exist and may even be in use for other types of cancers.
“One long-term goal of our collaborative team is to better understand fundamental mechanisms that drive these processes, with the hope of providing new therapeutic approaches,” concluded Dr. Allis.
Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape
Missense mutations (that change one amino acid for another) in histone H3 can produce a so-called oncohistone and are found in a number of pediatric cancers. For example, the lysine-36–to-methionine (K36M) mutation is seen in almost all chondroblastomas. Lu et al. show that K36M mutant histones are oncogenic, and they inhibit the normal methylation of this same residue in wild-type H3 histones. The mutant histones also interfere with the normal development of bone-related cells and the deposition of inhibitory chromatin marks.
Several types of pediatric cancers reportedly contain high-frequency missense mutations in histone H3, yet the underlying oncogenic mechanism remains poorly characterized. Here we report that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo. H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases. Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation. After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation. Our findings are mirrored in human undifferentiated sarcomas in which novel K36M/I mutations in H3.1 are identified.
Mitochondria? We Don’t Need No Stinking Mitochondria!
Diagram comparing typical eukaryotic cell to the newly discovered mitochondria-free organism. [Karnkowska et al., 2016, Current Biology 26, 1–11]
The organelle that produces a significant portion of energy for eukaryotic cells would seemingly be indispensable, yet over the years, a number of organisms have been discovered that challenge that biological pretense. However, these so-called amitochondrial species may lack a defined organelle, but they still retain some residual functions of their mitochondria-containing brethren. Even the intestinal eukaryotic parasite Giardia intestinalis, which was for many years considered to be mitochondria-free, was proven recently to contain a considerably shriveled version of the organelle.
Now, an international group of scientists has released results from a new study that challenges the notion that mitochondria are essential for eukaryotes—discovering an organism that resides in the gut of chinchillas that contains absolutely no trace of mitochondria at all.
“In low-oxygen environments, eukaryotes often possess a reduced form of the mitochondrion, but it was believed that some of the mitochondrial functions are so essential that these organelles are indispensable for their life,” explained lead study author Anna Karnkowska, Ph.D., visiting scientist at the University of British Columbia in Vancouver. “We have characterized a eukaryotic microbe which indeed possesses no mitochondrion at all.”
Mysterious Eukaryote Missing Mitochondria
Researchers uncover the first example of a eukaryotic organism that lacks the organelles.
Monocercomonoides sp. PA203VLADIMIR HAMPL, CHARLES UNIVERSITY, PRAGUE, CZECH REPUBLIC
Scientists have long thought that mitochondria—organelles responsible for energy generation—are an essential and defining feature of a eukaryotic cell. Now, researchers from Charles University in Prague and their colleagues are challenging this notion with their discovery of a eukaryotic organism,Monocercomonoides species PA203, which lacks mitochondria. The team’s phylogenetic analysis, published today (May 12) in Current Biology,suggests that Monocercomonoides—which belong to the Oxymonadida group of protozoa and live in low-oxygen environments—did have mitochondria at one point, but eventually lost the organelles.
“This is quite a groundbreaking discovery,” said Thijs Ettema, who studies microbial genome evolution at Uppsala University in Sweden and was not involved in the work.
“This study shows that mitochondria are not so central for all lineages of living eukaryotes,” Toni Gabaldonof the Center for Genomic Regulation in Barcelona, Spain, who also was not involved in the work, wrote in an email to The Scientist. “Yet, this mitochondrial-devoid, single-cell eukaryote is as complex as other eukaryotic cells in almost any other aspect of cellular complexity.”
Charles University’s Vladimir Hampl studies the evolution of protists. Along with Anna Karnkowska and colleagues, Hampl decided to sequence the genome of Monocercomonoides, a little-studied protist that lives in the digestive tracts of vertebrates. The 75-megabase genome—the first of an oxymonad—did not contain any conserved genes found on mitochondrial genomes of other eukaryotes, the researchers found. It also did not contain any nuclear genes associated with mitochondrial functions.
“It was surprising and for a long time, we didn’t believe that the [mitochondria-associated genes were really not there]. We thought we were missing something,” Hampl told The Scientist. “But when the data kept accumulating, we switched to the hypothesis that this organism really didn’t have mitochondria.”
Because researchers have previously not found examples of eukaryotes without some form of mitochondria, the current theory of the origin of eukaryotes poses that the appearance of mitochondria was crucial to the identity of these organisms.
“We now view these mitochondria-like organelles as a continuum from full mitochondria to very small . Some anaerobic protists, for example, have only pared down versions of mitochondria, such as hydrogenosomes and mitosomes, which lack a mitochondrial genome. But these mitochondrion-like organelles perform essential functions of the iron-sulfur cluster assembly pathway, which is known to be conserved in virtually all eukaryotic organisms studied to date.
Yet, in their analysis, the researchers found no evidence of the presence of any components of this mitochondrial pathway.
Like the scaling down of mitochondria into mitosomes in some organisms, the ancestors of modernMonocercomonoides once had mitochondria. “Because this organism is phylogenetically nested among relatives that had conventional mitochondria, this is most likely a secondary adaptation,” said Michael Gray, a biochemist who studies mitochondria at Dalhousie University in Nova Scotia and was not involved in the study. According to Gray, the finding of a mitochondria-deficient eukaryote does not mean that the organelles did not play a major role in the evolution of eukaryotic cells.
To be sure they were not missing mitochondrial proteins, Hampl’s team also searched for potential mitochondrial protein homologs of other anaerobic species, and for signature sequences of a range of known mitochondrial proteins. While similar searches with other species uncovered a few mitochondrial proteins, the team’s analysis of Monocercomonoides came up empty.
“The data is very complete,” said Ettema. “It is difficult to prove the absence of something but [these authors] do a convincing job.”
To form the essential iron-sulfur clusters, the team discovered that Monocercomonoides use a sulfur mobilization system found in the cytosol, and that an ancestor of the organism acquired this system by lateral gene transfer from bacteria. This cytosolic, compensating system allowed Monocercomonoides to lose the otherwise essential iron-sulfur cluster-forming pathway in the mitochondrion, the team proposed.
“This work shows the great evolutionary plasticity of the eukaryotic cell,” said Karnkowska, who participated in the study while she was a postdoc at Charles University. Karnkowska, who is now a visiting researcher at the University of British Columbia in Canada, added: “This is a striking example of how far the evolution of a eukaryotic cell can go that was beyond our expectations.”
“The results highlight how many surprises may await us in the poorly studied eukaryotic phyla that live in under-explored environments,” Gabaldon said.
Ettema agreed. “Now that we’ve found one, we need to look at the bigger picture and see if there are other examples of eukaryotes that have lost their mitochondria, to understand how adaptable eukaryotes are.”
Karnkowska et al., “A eukaryote without a mitochondrial organelle,” Current Biology,doi:10.1016/j.cub.2016.03.053, 2016.
•Monocercomonoides sp. is a eukaryotic microorganism with no mitochondria
•The complete absence of mitochondria is a secondary loss, not an ancestral feature
•The essential mitochondrial ISC pathway was replaced by a bacterial SUF system
The presence of mitochondria and related organelles in every studied eukaryote supports the view that mitochondria are essential cellular components. Here, we report the genome sequence of a microbial eukaryote, the oxymonad Monocercomonoides sp., which revealed that this organism lacks all hallmark mitochondrial proteins. Crucially, the mitochondrial iron-sulfur cluster assembly pathway, thought to be conserved in virtually all eukaryotic cells, has been replaced by a cytosolic sulfur mobilization system (SUF) acquired by lateral gene transfer from bacteria. In the context of eukaryotic phylogeny, our data suggest that Monocercomonoides is not primitively amitochondrial but has lost the mitochondrion secondarily. This is the first example of a eukaryote lacking any form of a mitochondrion, demonstrating that this organelle is not absolutely essential for the viability of a eukaryotic cell.
This method catches a bait protein together with its associated protein partners in virus-like particles that are budded from human cells. Like this, cell lysis is not needed and protein complexes are preserved during purification.
With his feet in both a proteomics lab and an interactomics lab, VIB/UGent professor Sven Eyckerman is well aware of the shortcomings of conventional approaches to analyze protein complexes. The lysis conditions required in mass spectrometry–based strategies to break open cell membranes often affect protein-protein interactions. “The first step in a classical study on protein complexes essentially turns the highly organized cellular structure into a big messy soup”, Eyckerman explains.
Inspired by virus biology, Eyckerman came up with a creative solution. “We used the natural process of HIV particle formation to our benefit by hacking a completely safe form of the virus to abduct intact protein machines from the cell.” It is well known that the HIV virus captures a number of host proteins during its particle formation. By fusing a bait protein to the HIV-1 GAG protein, interaction partners become trapped within virus-like particles that bud from mammalian cells. Standard proteomic approaches are used next to reveal the content of these particles. Fittingly, the team named the method ‘Virotrap’.
The Virotrap approach is exceptional as protein networks can be characterized under natural conditions. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. The researchers showed the method was suitable for detection of known binary interactions as well as mass spectrometry-based identification of novel protein partners.
Virotrap is a textbook example of bringing research teams with complementary expertise together. Cross-pollination with the labs of Jan Tavernier (VIB/UGent) and Kris Gevaert (VIB/UGent) enabled the development of this platform.
Jan Tavernier: “Virotrap represents a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. It is complementary to the arsenal of existing interactomics methods, but also holds potential for other fields, like drug target characterization. We also developed a small molecule-variant of Virotrap that could successfully trap protein partners for small molecule baits.”
Kris Gevaert: “Virotrap can also impact our understanding of disease pathways. We were actually surprised to see that this virus-based system could be used to study antiviral pathways, like Toll-like receptor signaling. Understanding these protein machines in their natural environment is essential if we want to modulate their activity in pathology.“
Trapping mammalian protein complexes in viral particles
Cell lysis is an inevitable step in classical mass spectrometry–based strategies to analyse protein complexes. Complementary lysis conditions, in situ cross-linking strategies and proximal labelling techniques are currently used to reduce lysis effects on the protein complex. We have developed Virotrap, a viral particle sorting approach that obviates the need for cell homogenization and preserves the protein complexes during purification. By fusing a bait protein to the HIV-1 GAG protein, we show that interaction partners become trapped within virus-like particles (VLPs) that bud from mammalian cells. Using an efficient VLP enrichment protocol, Virotrap allows the detection of known binary interactions and MS-based identification of novel protein partners as well. In addition, we show the identification of stimulus-dependent interactions and demonstrate trapping of protein partners for small molecules. Virotrap constitutes an elegant complementary approach to the arsenal of methods to study protein complexes.
Proteins mostly exert their function within supramolecular complexes. Strategies for detecting protein–protein interactions (PPIs) can be roughly divided into genetic systems1 and co-purification strategies combined with mass spectrometry (MS) analysis (for example, AP–MS)2. The latter approaches typically require cell or tissue homogenization using detergents, followed by capture of the protein complex using affinity tags3 or specific antibodies4. The protein complexes extracted from this ‘soup’ of constituents are then subjected to several washing steps before actual analysis by trypsin digestion and liquid chromatography–MS/MS analysis. Such lysis and purification protocols are typically empirical and have mostly been optimized using model interactions in single labs. In fact, lysis conditions can profoundly affect the number of both specific and nonspecific proteins that are identified in a typical AP–MS set-up. Indeed, recent studies using the nuclear pore complex as a model protein complex describe optimization of purifications for the different proteins in the complex by examining 96 different conditions5. Nevertheless, for new purifications, it remains hard to correctly estimate the loss of factors in a standard AP–MS experiment due to washing and dilution effects during treatments (that is, false negatives). These considerations have pushed the concept of stabilizing PPIs before the actual homogenization step. A classical approach involves cross-linking with simple reagents (for example, formaldehyde) or with more advanced isotope-labelled cross-linkers (reviewed in ref. 2). However, experimental challenges such as cell permeability and reactivity still preclude the widespread use of cross-linking agents. Moreover, MS-generated spectra of cross-linked peptides are notoriously difficult to identify correctly. A recent lysis-independent solution involves the expression of a bait protein fused to a promiscuous biotin ligase, which results in labelling of proteins proximal to the activity of the enzyme-tagged bait protein6. When compared with AP–MS, this BioID approach delivers a complementary set of candidate proteins, including novel interaction partners7, 8. Such particular studies clearly underscore the need for complementary approaches in the co-complex strategies.
The evolutionary stress on viruses promoted highly condensed coding of information and maximal functionality for small genomes. Accordingly, for HIV-1 it is sufficient to express a single protein, the p55 GAG protein, for efficient production of virus-like particles (VLPs) from cells9, 10. This protein is highly mobile before its accumulation in cholesterol-rich regions of the membrane, where multimerization initiates the budding process11. A total of 4,000–5,000 GAG molecules is required to form a single particle of about 145 nm (ref. 12). Both VLPs and mature viruses contain a number of host proteins that are recruited by binding to viral proteins. These proteins can either contribute to the infectivity (for example, Cyclophilin/FKBPA13) or act as antiviral proteins preventing the spreading of the virus (for example, APOBEC proteins14).
We here describe the development and application of Virotrap, an elegant co-purification strategy based on the trapping of a bait protein together with its associated protein partners in VLPs that are budded from the cell. After enrichment, these particles can be analysed by targeted (for example, western blotting) or unbiased approaches (MS-based proteomics). Virotrap allows detection of known binary PPIs, analysis of protein complexes and their dynamics, and readily detects protein binders for small molecules.
Concept of the Virotrap system
Classical AP–MS approaches rely on cell homogenization to access protein complexes, a step that can vary significantly with the lysis conditions (detergents, salt concentrations, pH conditions and so on)5. To eliminate the homogenization step in AP–MS, we reasoned that incorporation of a protein complex inside a secreted VLP traps the interaction partners under native conditions and protects them during further purification. We thus explored the possibility of protein complex packaging by the expression of GAG-bait protein chimeras (Fig. 1) as expression of GAG results in the release of VLPs from the cells9, 10. As a first PPI pair to evaluate this concept, we selected the HRAS protein as a bait combined with the RAF1 prey protein. We were able to specifically detect the HRAS–RAF1 interaction following enrichment of VLPs via ultracentrifugation (Supplementary Fig. 1a). To prevent tedious ultracentrifugation steps, we designed a novel single-step protocol wherein we co-express the vesicular stomatitis virus glycoprotein (VSV-G) together with a tagged version of this glycoprotein in addition to the GAG bait and prey. Both tagged and untagged VSV-G proteins are probably presented as trimers on the surface of the VLPs, allowing efficient antibody-based recovery from large volumes. The HRAS–RAF1 interaction was confirmed using this single-step protocol (Supplementary Fig. 1b). No associations with unrelated bait or prey proteins were observed for both protocols.
Figure 1: Schematic representation of the Virotrap strategy.
Expression of a GAG-bait fusion protein (1) results in submembrane multimerization (2) and subsequent budding of VLPs from cells (3). Interaction partners of the bait protein are also trapped within these VLPs and can be identified after purification by western blotting or MS analysis (4).
Virotrap for the detection of binary interactions
We next explored the reciprocal detection of a set of PPI pairs, which were selected based on published evidence and cytosolic localization15. After single-step purification and western blot analysis, we could readily detect reciprocal interactions between CDK2 and CKS1B, LCP2 and GRAP2, and S100A1 and S100B (Fig. 2a). Only for the LCP2 prey we observed nonspecific association with an irrelevant bait construct. However, the particle levels of the GRAP2 bait were substantially lower as compared with those of the GAG control construct (GAG protein levels in VLPs; Fig. 2a, second panel of the LCP2 prey). After quantification of the intensities of bait and prey proteins and normalization of prey levels using bait levels, we observed a strong enrichment for the GAG-GRAP2 bait (Supplementary Fig. 2).
…..
Virotrap for unbiased discovery of novel interactions
For the detection of novel interaction partners, we scaled up VLP production and purification protocols (Supplementary Fig. 5 and Supplementary Note 1 for an overview of the protocol) and investigated protein partners trapped using the following bait proteins: Fas-associated via death domain (FADD), A20 (TNFAIP3), nuclear factor-κB (NF-κB) essential modifier (IKBKG), TRAF family member-associated NF-κB activator (TANK), MYD88 and ring finger protein 41 (RNF41). To obtain specific interactors from the lists of identified proteins, we challenged the data with a combined protein list of 19 unrelated Virotrap experiments (Supplementary Table 1 for an overview). Figure 3 shows the design and the list of candidate interactors obtained after removal of all proteins that were found in the 19 control samples (including removal of proteins from the control list identified with a single peptide). The remaining list of confident protein identifications (identified with at least two peptides in at least two biological repeats) reveals both known and novel candidate interaction partners. All candidate interactors including single peptide protein identifications are given in Supplementary Data 2 and also include recurrent protein identifications of known interactors based on a single peptide; for example, CASP8 for FADD and TANK for NEMO. Using alternative methods, we confirmed the interaction between A20 and FADD, and the associations with transmembrane proteins (insulin receptor and insulin-like growth factor receptor 1) that were captured using RNF41 as a bait (Supplementary Fig. 6). To address the use of Virotrap for the detection of dynamic interactions, we activated the NF-κB pathway via the tumour necrosis factor (TNF) receptor (TNFRSF1A) using TNFα (TNF) and performed Virotrap analysis using A20 as bait (Fig. 3). This resulted in the additional enrichment of receptor-interacting kinase (RIPK1), TNFR1-associated via death domain (TRADD), TNFRSF1A and TNF itself, confirming the expected activated complex20.
Figure 3: Use of Virotrap for unbiased interactome analysis
Lysis conditions used in AP–MS strategies are critical for the preservation of protein complexes. A multitude of lysis conditions have been described, culminating in a recent report where protein complex stability was assessed under 96 lysis/purification protocols5. Moreover, the authors suggest to optimize the conditions for every complex, implying an important workload for researchers embarking on protein complex analysis using classical AP–MS. As lysis results in a profound change of the subcellular context and significantly alters the concentration of proteins, loss of complex integrity during a classical AP–MS protocol can be expected. A clear evolution towards ‘lysis-independent’ approaches in the co-complex analysis field is evident with the introduction of BioID6 and APEX25 where proximal proteins, including proteins residing in the complex, are labelled with biotin by an enzymatic activity fused to a bait protein. A side-by-side comparison between classical AP–MS and BioID showed overlapping and unique candidate binding proteins for both approaches7, 8, supporting the notion that complementary methods are needed to provide a comprehensive view on protein complexes. This has also been clearly demonstrated for binary approaches15 and is a logical consequence of the heterogenic nature underlying PPIs (binding mechanism, requirement for posttranslational modifications, location, affinity and so on).
In this report, we explore an alternative, yet complementary method to isolate protein complexes without interfering with cellular integrity. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. This constitutes a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. A comparison of our Virotrap approach with AP–MS shows complementary data, with specific false positives and false negatives for both methods (Supplementary Fig. 7).
The current implementation of the Virotrap platform implies the use of a GAG-bait construct resulting in considerable expression of the bait protein. Different strategies are currently pursued to reduce bait expression including co-expression of a native GAG protein together with the GAG-bait protein, not only reducing bait expression but also creating more ‘space’ in the particles potentially accommodating larger bait protein complexes. Nevertheless, the presence of the bait on the forming GAG scaffold creates an intracellular affinity matrix (comparable to the early in vitro affinity columns for purification of interaction partners from lysates26) that has the potential to compete with endogenous complexes by avidity effects. This avidity effect is a powerful mechanism that aids in the recruitment of cyclophilin to GAG27, a well-known weak interaction (Kd=16 μM (ref. 28)) detectable as a background association in the Virotrap system. Although background binding may be increased by elevated bait expression, weaker associations are readily detectable (for example, MAL—MYD88-binding study; Fig. 2c).
The size of Virotrap particles (around 145 nm) suggests limitations in the size of the protein complex that can be accommodated in the particles. Further experimentation is required to define the maximum size of proteins or the number of protein complexes that can be trapped inside the particles.
….
In conclusion, Virotrap captures significant parts of known interactomes and reveals new interactions. This cell lysis-free approach purifies protein complexes under native conditions and thus provides a powerful method to complement AP–MS or other PPI data. Future improvements of the system include strategies to reduce bait expression to more physiological levels and application of advanced data analysis options to filter out background. These developments can further aid in the deployment of Virotrap as a powerful extension of the current co-complex technology arsenal.
New Autism Blood Biomarker Identified
Researchers at UT Southwestern Medical Center have identified a blood biomarker that may aid in earlier diagnosis of children with autism spectrum disorder, or ASD
In a recent edition of Scientific Reports, UT Southwestern researchers reported on the identification of a blood biomarker that could distinguish the majority of ASD study participants versus a control group of similar age range. In addition, the biomarker was significantly correlated with the level of communication impairment, suggesting that the blood test may give insight into ASD severity.
“Numerous investigators have long sought a biomarker for ASD,” said Dr. Dwight German, study senior author and Professor of Psychiatry at UT Southwestern. “The blood biomarker reported here along with others we are testing can represent a useful test with over 80 percent accuracy in identifying ASD.”
ASD1 – was 66 percent accurate in diagnosing ASD. When combined with thyroid stimulating hormone level measurements, the ASD1-binding biomarker was 73 percent accurate at diagnosis
A Search for Blood Biomarkers for Autism: Peptoids
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impairments in social interaction and communication, and restricted, repetitive patterns of behavior. In order to identify individuals with ASD and initiate interventions at the earliest possible age, biomarkers for the disorder are desirable. Research findings have identified widespread changes in the immune system in children with autism, at both systemic and cellular levels. In an attempt to find candidate antibody biomarkers for ASD, highly complex libraries of peptoids (oligo-N-substituted glycines) were screened for compounds that preferentially bind IgG from boys with ASD over typically developing (TD) boys. Unexpectedly, many peptoids were identified that preferentially bound IgG from TD boys. One of these peptoids was studied further and found to bind significantly higher levels (>2-fold) of the IgG1 subtype in serum from TD boys (n = 60) compared to ASD boys (n = 74), as well as compared to older adult males (n = 53). Together these data suggest that ASD boys have reduced levels (>50%) of an IgG1 antibody, which resembles the level found normally with advanced age. In this discovery study, the ASD1 peptoid was 66% accurate in predicting ASD.
….
Peptoid libraries have been used previously to search for autoantibodies for neurodegenerative diseases19 and for systemic lupus erythematosus (SLE)21. In the case of SLE, peptoids were identified that could identify subjects with the disease and related syndromes with moderate sensitivity (70%) and excellent specificity (97.5%). Peptoids were used to measure IgG levels from both healthy subjects and SLE patients. Binding to the SLE-peptoid was significantly higher in SLE patients vs. healthy controls. The IgG bound to the SLE-peptoid was found to react with several autoantigens, suggesting that the peptoids are capable of interacting with multiple, structurally similar molecules. These data indicate that IgG binding to peptoids can identify subjects with high levels of pathogenic autoantibodies vs. a single antibody.
In the present study, the ASD1 peptoid binds significantly lower levels of IgG1 in ASD males vs. TD males. This finding suggests that the ASD1 peptoid recognizes antibody(-ies) of an IgG1 subtype that is (are) significantly lower in abundance in the ASD males vs. TD males. Although a previous study14 has demonstrated lower levels of plasma IgG in ASD vs. TD children, here, we additionally quantified serum IgG levels in our individuals and found no difference in IgG between the two groups (data not shown). Furthermore, our IgG levels did not correlate with ASD1 binding levels, indicating that ASD1 does not bind IgG generically, and that the peptoid’s ability to differentiate between ASD and TD males is related to a specific antibody(-ies).
ASD subjects underwent a diagnostic evaluation using the ADOS and ADI-R, and application of the DSM-IV criteria prior to study inclusion. Only those subjects with a diagnosis of Autistic Disorder were included in the study. The ADOS is a semi-structured observation of a child’s behavior that allows examiners to observe the three core domains of ASD symptoms: reciprocal social interaction, communication, and restricted and repetitive behaviors1. When ADOS subdomain scores were compared with peptoid binding, the only significant relationship was with Social Interaction. However, the positive correlation would suggest that lower peptoid binding is associated with better social interaction, not poorer social interaction as anticipated.
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
It is interesting that in serum samples from older men, the ASD1 binding is similar to that in the ASD boys. This is consistent with the observation that with aging there is a reduction in the strength of the immune system, and the changes are gender-specific25. Recent studies using parabiosis26, in which blood from young mice reverse age-related impairments in cognitive function and synaptic plasticity in old mice, reveal that blood constituents from young subjects may contain important substances for maintaining neuronal functions. Work is in progress to identify the antibody/antibodies that are differentially binding to the ASD1 peptoid, which appear as a single band on the electrophoresis gel (Fig. 4).
……..
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
Titration of IgG binding to ASD1 using serum pooled from 10 TD males and 10 ASD males demonstrates ASD1’s ability to differentiate between the two groups. (B)Detecting IgG1 subclass instead of total IgG amplifies this differentiation. (C) IgG1 binding of individual ASD (n=74) and TD (n=60) male serum samples (1:100 dilution) to ASD1 significantly differs with TD>ASD. In addition, IgG1 binding of older adult male (AM) serum samples (n=53) to ASD1 is significantly lower than TD males, and not different from ASD males. The three groups were compared with a Kruskal-Wallis ANOVA, H = 10.1781, p<0.006. **p<0.005. Error bars show SEM. (D) Receiver-operating characteristic curve for ASD1’s ability to discriminate between ASD and TD males.
Association between peptoid binding and ADOS and ADI-R subdomains
Higher scores in any domain on the ADOS and ADI-R are indicative of more abnormal behaviors and/or symptoms. Among ADOS subdomains, there was no significant relationship between Communication and peptoid binding (z = 0.04, p = 0.966), Communication + Social interaction (z = 1.53, p = 0.127), or Stereotyped Behaviors and Restrictive Interests (SBRI) (z = 0.46, p = 0.647). Higher scores on the Social Interaction domain were significantly associated with higher peptoid binding (z = 2.04, p = 0.041).
Among ADI-R subdomains, higher scores on the Communication domain were associated with lower levels of peptoid binding (z = −2.28, p = 0.023). There was not a significant relationship between Social Interaction (z = 0.07, p = 0.941) or Restrictive/Repetitive Stereotyped Behaviors (z = −1.40, p = 0.162) and peptoid binding.
Computational Model Finds New Protein-Protein Interactions
Researchers at University of Pittsburgh have discovered 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia.
Using a computational model they developed, researchers at the University of Pittsburgh School of Medicine have discovered more than 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia. The findings, published online in npj Schizophrenia, a Nature Publishing Group journal, could lead to greater understanding of the biological underpinnings of this mental illness, as well as point the way to treatments.
There have been many genome-wide association studies (GWAS) that have identified gene variants associated with an increased risk for schizophrenia, but in most cases there is little known about the proteins that these genes make, what they do and how they interact, said senior investigator Madhavi Ganapathiraju, Ph.D., assistant professor of biomedical informatics, Pitt School of Medicine.
“GWAS studies and other research efforts have shown us what genes might be relevant in schizophrenia,” she said. “What we have done is the next step. We are trying to understand how these genes relate to each other, which could show us the biological pathways that are important in the disease.”
Each gene makes proteins and proteins typically interact with each other in a biological process. Information about interacting partners can shed light on the role of a gene that has not been studied, revealing pathways and biological processes associated with the disease and also its relation to other complex diseases.
Dr. Ganapathiraju’s team developed a computational model called High-Precision Protein Interaction Prediction (HiPPIP) and applied it to discover PPIs of schizophrenia-linked genes identified through GWAS, as well as historically known risk genes. They found 504 never-before known PPIs, and noted also that while schizophrenia-linked genes identified historically and through GWAS had little overlap, the model showed they shared more than 100 common interactors.
“We can infer what the protein might do by checking out the company it keeps,” Dr. Ganapathiraju explained. “For example, if I know you have many friends who play hockey, it could mean that you are involved in hockey, too. Similarly, if we see that an unknown protein interacts with multiple proteins involved in neural signaling, for example, there is a high likelihood that the unknown entity also is involved in the same.”
Dr. Ganapathiraju and colleagues have drawn such inferences on protein function based on the PPIs of proteins, and made their findings available on a website Schizo-Pi. This information can be used by biologists to explore the schizophrenia interactome with the aim of understanding more about the disease or developing new treatment drugs.
Schizophrenia interactome with 504 novel protein–protein interactions
(GWAS) have revealed the role of rare and common genetic variants, but the functional effects of the risk variants remain to be understood. Protein interactome-based studies can facilitate the study of molecular mechanisms by which the risk genes relate to schizophrenia (SZ) genesis, but protein–protein interactions (PPIs) are unknown for many of the liability genes. We developed a computational model to discover PPIs, which is found to be highly accurate according to computational evaluations and experimental validations of selected PPIs. We present here, 365 novel PPIs of liability genes identified by the SZ Working Group of the Psychiatric Genomics Consortium (PGC). Seventeen genes that had no previously known interactions have 57 novel interactions by our method. Among the new interactors are 19 drug targets that are targeted by 130 drugs. In addition, we computed 147 novel PPIs of 25 candidate genes investigated in the pre-GWAS era. While there is little overlap between the GWAS genes and the pre-GWAS genes, the interactomes reveal that they largely belong to the same pathways, thus reconciling the apparent disparities between the GWAS and prior gene association studies. The interactome including 504 novel PPIs overall, could motivate other systems biology studies and trials with repurposed drugs. The PPIs are made available on a webserver, called Schizo-Pi at http://severus.dbmi.pitt.edu/schizo-pi with advanced search capabilities.
Schizophrenia (SZ) is a common, potentially severe psychiatric disorder that afflicts all populations.1 Gene mapping studies suggest that SZ is a complex disorder, with a cumulative impact of variable genetic effects coupled with environmental factors.2 As many as 38 genome-wide association studies (GWAS) have been reported on SZ out of a total of 1,750 GWAS publications on 1,087 traits or diseases reported in the GWAS catalog maintained by the National Human Genome Research Institute of USA3 (as of April 2015), revealing the common variants associated with SZ.4 The SZ Working Group of the Psychiatric Genomics Consortium (PGC) identified 108 genetic loci that likely confer risk for SZ.5 While the role of genetics has been clearly validated by this study, the functional impact of the risk variants is not well-understood.6,7 Several of the genes implicated by the GWAS have unknown functions and could participate in possibly hitherto unknown pathways.8 Further, there is little or no overlap between the genes identified through GWAS and ‘candidate genes’ proposed in the pre-GWAS era.9
Interactome-based studies can be useful in discovering the functional associations of genes. For example,disrupted in schizophrenia 1 (DISC1), an SZ related candidate gene originally had no known homolog in humans. Although it had well-characterized protein domains such as coiled-coil domains and leucine-zipper domains, its function was unknown.10,11 Once its protein–protein interactions (PPIs) were determined using yeast 2-hybrid technology,12 investigators successfully linked DISC1 to cAMP signaling, axon elongation, and neuronal migration, and accelerated the research pertaining to SZ in general, and DISC1 in particular.13 Typically such studies are carried out on known protein–protein interaction (PPI) networks, or as in the case of DISC1, when there is a specific gene of interest, its PPIs are determined by methods such as yeast 2-hybrid technology.
Knowledge of human PPI networks is thus valuable for accelerating discovery of protein function, and indeed, biomedical research in general. However, of the hundreds of thousands of biophysical PPIs thought to exist in the human interactome,14,15 <100,000 are known today (Human Protein Reference Database, HPRD16 and BioGRID17 databases). Gold standard experimental methods for the determination of all the PPIs in human interactome are time-consuming, expensive and may not even be feasible, as about 250 million pairs of proteins would need to be tested overall; high-throughput methods such as yeast 2-hybrid have important limitations for whole interactome determination as they have a low recall of 23% (i.e., remaining 77% of true interactions need to be determined by other means), and a low precision (i.e., the screens have to be repeated multiple times to achieve high selectivity).18,19Computational methods are therefore necessary to complete the interactome expeditiously. Algorithms have begun emerging to predict PPIs using statistical machine learning on the characteristics of the proteins, but these algorithms are employed predominantly to study yeast. Two significant computational predictions have been reported for human interactome; although they have had high false positive rates, these methods have laid the foundation for computational prediction of human PPIs.20,21
We have created a new PPI prediction model called High-Confidence Protein–Protein Interaction Prediction (HiPPIP) model. Novel interactions predicted with this model are making translational impact. For example, we discovered a PPI between OASL and DDX58, which on validation showed that an increased expression of OASL could boost innate immunity to combat influenza by activating the RIG-I pathway.22 Also, the interactome of the genes associated with congenital heart disease showed that the disease morphogenesis has a close connection with the structure and function of cilia.23Here, we describe the HiPPIP model and its application to SZ genes to construct the SZ interactome. After computational evaluations and experimental validations of selected novel PPIs, we present here 504 highly confident novel PPIs in the SZ interactome, shedding new light onto several uncharacterized genes that are associated with SZ.
We developed a computational model called HiPPIP to predict PPIs (see Methods and Supplementary File 1). The model has been evaluated by computational methods and experimental validations and is found to be highly accurate. Evaluations on a held-out test data showed a precision of 97.5% and a recall of 5%. 5% recall out of 150,000 to 600,000 estimated number of interactions in the human interactome corresponds to 7,500–30,000 novel PPIs in the whole interactome. Note that, it is likely that the real precision would be higher than 97.5% because in this test data, randomly paired proteins are treated as non-interacting protein pairs, whereas some of them may actually be interacting pairs with a small probability; thus, some of the pairs that are treated as false positives in test set are likely to be true but hitherto unknown interactions. In Figure 1a, we show the precision versus recall of our method on ‘hub proteins’ where we considered all pairs that received a score >0.5 by HiPPIP to be novel interactions. In Figure 1b, we show the number of true positives versus false positives observed in hub proteins. Both these figures also show our method to be superior in comparison to the prediction of membrane-receptor interactome by Qi et al’s.24 True positives versus false positives are also shown for individual hub proteins by our method in Figure 1cand by Qi et al’s.23 in Figure 1d. These evaluations showed that our predictions contain mostly true positives. Unlike in other domains where ranked lists are commonly used such as information retrieval, in PPI prediction the ‘false positives’ may actually be unlabeled instances that are indeed true interactions that are not yet discovered. In fact, such unlabeled pairs predicted as interactors of the hub gene HMGB1 (namely, the pairs HMGB1-KL and HMGB1-FLT1) were validated by experimental methods and found to be true PPIs (See the Figures e–g inSupplementary File 3). Thus, we concluded that the protein pairs that received a score of ⩾0.5 are highly confident to be true interactions. The pairs that receive a score less than but close to 0.5 (i.e., in the range of 0.4–0.5) may also contain several true PPIs; however, we cannot confidently say that all in this range are true PPIs. Only the PPIs predicted with a score >0.5 are included in the interactome.
Computational evaluation of predicted protein–protein interactions on hub proteins: (a) precision recall curve. (b) True positive versus false positives in ranked lists of hub type membrane receptors for our method and that by Qi et al. True positives versus false positives are shown for individual membrane receptors by our method in (c) and by Qi et al. in (d). Thick line is the average, which is also the same as shown in (b). Note:x-axis is recall in (a), whereas it is number of false positives in (b–d). The range of y-axis is observed by varying the threshold from 1.0–0 in (a), and to 0.5 in (b–d).
SZ interactome
By applying HiPPIP to the GWAS genes and Historic (pre-GWAS) genes, we predicted over 500 high confidence new PPIs adding to about 1400 previously known PPIs.
Schizophrenia interactome: network view of the schizophrenia interactome is shown as a graph, where genes are shown as nodes and PPIs as edges connecting the nodes. Schizophrenia-associated genes are shown as dark blue nodes, novel interactors as red color nodes and known interactors as blue color nodes. The source of the schizophrenia genes is indicated by its label font, where Historic genes are shown italicized, GWAS genes are shown in bold, and the one gene that is common to both is shown in italicized and bold. For clarity, the source is also indicated by the shape of the node (triangular for GWAS and square for Historic and hexagonal for both). Symbols are shown only for the schizophrenia-associated genes; actual interactions may be accessed on the web. Red edges are the novel interactions, whereas blue edges are known interactions. GWAS, genome-wide association studies of schizophrenia; PPI, protein–protein interaction.
We have made the known and novel interactions of all SZ-associated genes available on a webserver called Schizo-Pi, at the addresshttp://severus.dbmi.pitt.edu/schizo-pi. This webserver is similar to Wiki-Pi33 which presents comprehensive annotations of both participating proteins of a PPI side-by-side. The difference between Wiki-Pi which we developed earlier, and Schizo-Pi, is the inclusion of novel predicted interactions of the SZ genes into the latter.
Despite the many advances in biomedical research, identifying the molecular mechanisms underlying the disease is still challenging. Studies based on protein interactions were proven to be valuable in identifying novel gene associations that could shed new light on disease pathology.35 The interactome including more than 500 novel PPIs will help to identify pathways and biological processes associated with the disease and also its relation to other complex diseases. It also helps identify potential drugs that could be repurposed to use for SZ treatment.
Functional and pathway enrichment in SZ interactome
When a gene of interest has little known information, functions of its interacting partners serve as a starting point to hypothesize its own function. We computed statistically significant enrichment of GO biological process terms among the interacting partners of each of the genes using BinGO36 (see online at http://severus.dbmi.pitt.edu/schizo-pi).
Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution
Massimo Stefani · Christopher M. Dobson
Abstract The deposition of proteins in the form of amyloid fibrils and plaques is the characteristic feature of more than 20 degenerative conditions affecting either the central nervous system or a variety of peripheral tissues. As these conditions include Alzheimer’s, Parkinson’s and the prion diseases, several forms of fatal systemic amyloidosis, and at least one condition associated with medical intervention (haemodialysis), they are of enormous importance in the context of present-day human health and welfare. Much remains to be learned about the mechanism by which the proteins associated with these diseases aggregate and form amyloid structures, and how the latter affect the functions of the organs with which they are associated. A great deal of information concerning these diseases has emerged, however, during the past 5 years, much of it causing a number of fundamental assumptions about the amyloid diseases to be reexamined. For example, it is now apparent that the ability to form amyloid structures is not an unusual feature of the small number of proteins associated with these diseases but is instead a general property of polypeptide chains. It has also been found recently that aggregates of proteins not associated with amyloid diseases can impair the ability of cells to function to a similar extent as aggregates of proteins linked with specific neurodegenerative conditions. Moreover, the mature amyloid fibrils or plaques appear to be substantially less toxic than the prefibrillar aggregates that are their precursors. The toxicity of these early aggregates appears to result from an intrinsic ability to impair fundamental cellular processes by interacting with cellular membranes, causing oxidative stress and increases in free Ca2+ that eventually lead to apoptotic or necrotic cell death. The ‘new view’ of these diseases also suggests that other degenerative conditions could have similar underlying origins to those of the amyloidoses. In addition, cellular protection mechanisms, such as molecular chaperones and the protein degradation machinery, appear to be crucial in the prevention of disease in normally functioning living organisms. It also suggests some intriguing new factors that could be of great significance in the evolution of biological molecules and the mechanisms that regulate their behaviour.
The genetic information within a cell encodes not only the specific structures and functions of proteins but also the way these structures are attained through the process known as protein folding. In recent years many of the underlying features of the fundamental mechanism of this complex process and the manner in which it is regulated in living systems have emerged from a combination of experimental and theoretical studies [1]. The knowledge gained from these studies has also raised a host of interesting issues. It has become apparent, for example, that the folding and unfolding of proteins is associated with a whole range of cellular processes from the trafficking of molecules to specific organelles to the regulation of the cell cycle and the immune response. Such observations led to the inevitable conclusion that the failure to fold correctly, or to remain correctly folded, gives rise to many different types of biological malfunctions and hence to many different forms of disease [2]. In addition, it has been recognised recently that a large number of eukaryotic genes code for proteins that appear to be ‘natively unfolded’, and that proteins can adopt, under certain circumstances, highly organised multi-molecular assemblies whose structures are not specifically encoded in the amino acid sequence. Both these observations have raised challenging questions about one of the most fundamental principles of biology: the close relationship between the sequence, structure and function of proteins, as we discuss below [3].
It is well established that proteins that are ‘misfolded’, i.e. that are not in their functionally relevant conformation, are devoid of normal biological activity. In addition, they often aggregate and/or interact inappropriately with other cellular components leading to impairment of cell viability and eventually to cell death. Many diseases, often known as misfolding or conformational diseases, ultimately result from the presence in a living system of protein molecules with structures that are ‘incorrect’, i.e. that differ from those in normally functioning organisms [4]. Such diseases include conditions in which a specific protein, or protein complex, fails to fold correctly (e.g. cystic fibrosis, Marfan syndrome, amyotonic lateral sclerosis) or is not sufficiently stable to perform its normal function (e.g. many forms of cancer). They also include conditions in which aberrant folding behaviour results in the failure of a protein to be correctly trafficked (e.g. familial hypercholesterolaemia, α1-antitrypsin deficiency, and some forms of retinitis pigmentosa) [4]. The tendency of proteins to aggregate, often to give species extremely intractable to dissolution and refolding, is of course also well known in other circumstances. Examples include the formation of inclusion bodies during overexpression of heterologous proteins in bacteria and the precipitation of proteins during laboratory purification procedures. Indeed, protein aggregation is well established as one of the major difficulties associated with the production and handling of proteins in the biotechnology and pharmaceutical industries [5].
Considerable attention is presently focused on a group of protein folding diseases known as amyloidoses. In these diseases specific peptides or proteins fail to fold or to remain correctly folded and then aggregate (often with other components) so as to give rise to ‘amyloid’ deposits in tissue. Amyloid structures can be recognised because they possess a series of specific tinctorial and biophysical characteristics that reflect a common core structure based on the presence of highly organised βsheets [6]. The deposits in strictly defined amyloidoses are extracellular and can often be observed as thread-like fibrillar structures, sometimes assembled further into larger aggregates or plaques. These diseases include a range of sporadic, familial or transmissible degenerative diseases, some of which affect the brain and the central nervous system (e.g. Alzheimer’s and Creutzfeldt-Jakob diseases), while others involve peripheral tissues and organs such as the liver, heart and spleen (e.g. systemic amyloidoses and type II diabetes) [7, 8]. In other forms of amyloidosis, such as primary or secondary systemic amyloidoses, proteinaceous deposits are found in skeletal tissue and joints (e.g. haemodialysis-related amyloidosis) as well as in several organs (e.g. heart and kidney). Yet other components such as collagen, glycosaminoglycans and proteins (e.g. serum amyloid protein) are often present in the deposits protecting them against degradation [9, 10, 11]. Similar deposits to those in the amyloidoses are, however, found intracellularly in other diseases; these can be localised either in the cytoplasm, in the form of specialised aggregates known as aggresomes or as Lewy or Russell bodies or in the nucleus (see below).
The presence in tissue of proteinaceous deposits is a hallmark of all these diseases, suggesting a causative link between aggregate formation and pathological symptoms (often known as the amyloid hypothesis) [7, 8, 12]. At the present time the link between amyloid formation and disease is widely accepted on the basis of a large number of biochemical and genetic studies. The specific nature of the pathogenic species, and the molecular basis of their ability to damage cells, are however, the subject of intense debate [13, 14, 15, 16, 17, 18, 19, 20]. In neurodegenerative disorders it is very likely that the impairment of cellular function follows directly from the interactions of the aggregated proteins with cellular components [21, 22]. In the systemic non-neurological diseases, however, it is widely believed that the accumulation in vital organs of large amounts of amyloid deposits can by itself cause at least some of the clinical symptoms [23]. It is quite possible, however, that there are other more specific effects of aggregates on biochemical processes even in these diseases. The presence of extracellular or intracellular aggregates of a specific polypeptide molecule is a characteristic of all the 20 or so recognised amyloid diseases. The polypeptides involved include full length proteins (e.g. lysozyme or immunoglobulin light chains), biological peptides (amylin, atrial natriuretic factor) and fragments of larger proteins produced as a result of specific processing (e.g. the Alzheimer βpeptide) or of more general degradation [e.g. poly(Q) stretches cleaved from proteins with poly(Q) extensions such as huntingtin, ataxins and the androgen receptor]. The peptides and proteins associated with known amyloid diseases are listed in Table 1. In some cases the proteins involved have wild type sequences, as in sporadic forms of the diseases, but in other cases these are variants resulting from genetic mutations associated with familial forms of the diseases. In some cases both sporadic and familial diseases are associated with a given protein; in this case the mutational variants are usually associated with early-onset forms of the disease. In the case of the neurodegenerative diseases associated with the prion protein some forms of the diseases are transmissible. The existence of familial forms of a number of amyloid diseases has provided significant clues to the origins of the pathologies. For example, there are increasingly strong links between the age at onset of familial forms of disease and the effects of the mutations involved on the propensity of the affected proteins to aggregate in vitro. Such findings also support the link between the process of aggregation and the clinical manifestations of disease [24, 25].
The presence in cells of misfolded or aggregated proteins triggers a complex biological response. In the cytosol, this is referred to as the ‘heat shock response’ and in the endoplasmic reticulum (ER) it is known as the ‘unfolded protein response’. These responses lead to the expression, among others, of the genes for heat shock proteins (Hsp, or molecular chaperone proteins) and proteins involved in the ubiquitin-proteasome pathway [26]. The evolution of such complex biochemical machinery testifies to the fact that it is necessary for cells to isolate and clear rapidly and efficiently any unfolded or incorrectly folded protein as soon as it appears. In itself this fact suggests that these species could have a generally adverse effect on cellular components and cell viability. Indeed, it was a major step forward in understanding many aspects of cell biology when it was recognised that proteins previously associated only with stress, such as heat shock, are in fact crucial in the normal functioning of living systems. This advance, for example, led to the discovery of the role of molecular chaperones in protein folding and in the normal ‘housekeeping’ processes that are inherent in healthy cells [27, 28]. More recently a number of degenerative diseases, both neurological and systemic, have been linked to, or shown to be affected by, impairment of the ubiquitin-proteasome pathway (Table 2). The diseases are primarily associated with a reduction in either the expression or the biological activity of Hsps, ubiquitin, ubiquitinating or deubiquitinating enzymes and the proteasome itself, as we show below [29, 30, 31, 32], or even to the failure of the quality control mechanisms that ensure proper maturation of proteins in the ER. The latter normally leads to degradation of a significant proportion of polypeptide chains before they have attained their native conformations through retrograde translocation to the cytosol [33, 34].
….
It is now well established that the molecular basis of protein aggregation into amyloid structures involves the existence of ‘misfolded’ forms of proteins, i.e. proteins that are not in the structures in which they normally function in vivo or of fragments of proteins resulting from degradation processes that are inherently unable to fold [4, 7, 8, 36]. Aggregation is one of the common consequences of a polypeptide chain failing to reach or maintain its functional three-dimensional structure. Such events can be associated with specific mutations, misprocessing phenomena, aberrant interactions with metal ions, changes in environmental conditions, such as pH or temperature, or chemical modification (oxidation, proteolysis). Perturbations in the conformational properties of the polypeptide chain resulting from such phenomena may affect equilibrium 1 in Fig. 1 increasing the population of partially unfolded, or misfolded, species that are much more aggregation-prone than the native state.
Fig. 1 Overview of the possible fates of a newly synthesised polypeptide chain. The equilibrium ① between the partially folded molecules and the natively folded ones is usually strongly in favour of the latter except as a result of specific mutations, chemical modifications or partially destabilising solution conditions. The increased equilibrium populations of molecules in the partially or completely unfolded ensemble of structures are usually degraded by the proteasome; when this clearance mechanism is impaired, such species often form disordered aggregates or shift equilibrium ② towards the nucleation of pre-fibrillar assemblies that eventually grow into mature fibrils (equilibrium ③). DANGER! indicates that pre-fibrillar aggregates in most cases display much higher toxicity than mature fibrils. Heat shock proteins (Hsp) can suppress the appearance of pre-fibrillar assemblies by minimising the population of the partially folded molecules by assisting in the correct folding of the nascent chain and the unfolded protein response target incorrectly folded proteins for degradation.
……
Little is known at present about the detailed arrangement of the polypeptide chains themselves within amyloid fibrils, either those parts involved in the core βstrands or in regions that connect the various β-strands. Recent data suggest that the sheets are relatively untwisted and may in some cases at least exist in quite specific supersecondary structure motifs such as β-helices [6, 40] or the recently proposed µ-helix [41]. It seems possible that there may be significant differences in the way the strands are assembled depending on characteristics of the polypeptide chain involved [6, 42]. Factors including length, sequence (and in some cases the presence of disulphide bonds or post-translational modifications such as glycosylation) may be important in determining details of the structures. Several recent papers report structural models for amyloid fibrils containing different polypeptide chains, including the Aβ40 peptide, insulin and fragments of the prion protein, based on data from such techniques as cryo-electron microscopy and solid-state magnetic resonance spectroscopy [43, 44]. These models have much in common and do indeed appear to reflect the fact that the structures of different fibrils are likely to be variations on a common theme [40]. It is also emerging that there may be some common and highly organised assemblies of amyloid protofilaments that are not simply extended threads or ribbons. It is clear, for example, that in some cases large closed loops can be formed [45, 46, 47], and there may be specific types of relatively small spherical or ‘doughnut’ shaped structures that can result in at least some circumstances (see below).
…..
The similarity of some early amyloid aggregates with the pores resulting from oligomerisation of bacterial toxins and pore-forming eukaryotic proteins (see below) also suggest that the basic mechanism of protein aggregation into amyloid structures may not only be associated with diseases but in some cases could result in species with functional significance. Recent evidence indicates that a variety of micro-organisms may exploit the controlled aggregation of specific proteins (or their precursors) to generate functional structures. Examples include bacterial curli [52] and proteins of the interior fibre cells of mammalian ocular lenses, whose β-sheet arrays seem to be organised in an amyloid-like supramolecular order [53]. In this case the inherent stability of amyloid-like protein structure may contribute to the long-term structural integrity and transparency of the lens. Recently it has been hypothesised that amyloid-like aggregates of serum amyloid A found in secondary amyloidoses following chronic inflammatory diseases protect the host against bacterial infections by inducing lysis of bacterial cells [54]. One particularly interesting example is a ‘misfolded’ form of the milk protein α-lactalbumin that is formed at low pH and trapped by the presence of specific lipid molecules [55]. This form of the protein has been reported to trigger apoptosis selectively in tumour cells providing evidence for its importance in protecting infants from certain types of cancer [55]. ….
Amyloid formation is a generic property of polypeptide chains ….
It is clear that the presence of different side chains can influence the details of amyloid structures, particularly the assembly of protofibrils, and that they give rise to the variations on the common structural theme discussed above. More fundamentally, the composition and sequence of a peptide or protein affects profoundly its propensity to form amyloid structures under given conditions (see below).
Because the formation of stable protein aggregates of amyloid type does not normally occur in vivo under physiological conditions, it is likely that the proteins encoded in the genomes of living organisms are endowed with structural adaptations that mitigate against aggregation under these conditions. A recent survey involving a large number of structures of β-proteins highlights several strategies through which natural proteins avoid intermolecular association of β-strands in their native states [65]. Other surveys of protein databases indicate that nature disfavours sequences of alternating polar and nonpolar residues, as well as clusters of several consecutive hydrophobic residues, both of which enhance the tendency of a protein to aggregate prior to becoming completely folded [66, 67].
……
Precursors of amyloid fibrils can be toxic to cells
It was generally assumed until recently that the proteinaceous aggregates most toxic to cells are likely to be mature amyloid fibrils, the form of aggregates that have been commonly detected in pathological deposits. It therefore appeared probable that the pathogenic features underlying amyloid diseases are a consequence of the interaction with cells of extracellular deposits of aggregated material. As well as forming the basis for understanding the fundamental causes of these diseases, this scenario stimulated the exploration of therapeutic approaches to amyloidoses that focused mainly on the search for molecules able to impair the growth and deposition of fibrillar forms of aggregated proteins. ….
Structural basis and molecular features of amyloid toxicity
The presence of toxic aggregates inside or outside cells can impair a number of cell functions that ultimately lead to cell death by an apoptotic mechanism [95, 96]. Recent research suggests, however, that in most cases initial perturbations to fundamental cellular processes underlie the impairment of cell function induced by aggregates of disease-associated polypeptides. Many pieces of data point to a central role of modifications to the intracellular redox status and free Ca2+ levels in cells exposed to toxic aggregates [45, 89, 97, 98, 99, 100, 101]. A modification of the intracellular redox status in such cells is associated with a sharp increase in the quantity of reactive oxygen species (ROS) that is reminiscent of the oxidative burst by which leukocytes destroy invading foreign cells after phagocytosis. In addition, changes have been observed in reactive nitrogen species, lipid peroxidation, deregulation of NO metabolism [97], protein nitrosylation [102] and upregulation of heme oxygenase-1, a specific marker of oxidative stress [103]. ….
Results have recently been reported concerning the toxicity towards cultured cells of aggregates of poly(Q) peptides which argues against a disease mechanism based on specific toxic features of the aggregates. These results indicate that there is a close relationship between the toxicity of proteins with poly(Q) extensions and their nuclear localisation. In addition they support the hypotheses that the toxicity of poly(Q) aggregates can be a consequence of altered interactions with nuclear coactivator or corepressor molecules including p53, CBP, Sp1 and TAF130 or of the interaction with transcription factors and nuclear coactivators, such as CBP, endowed with short poly(Q) stretches ([95] and references therein)…..
Concluding remarks
The data reported in the past few years strongly suggest that the conversion of normally soluble proteins into amyloid fibrils and the toxicity of small aggregates appearing during the early stages of the formation of the latter are common or generic features of polypeptide chains. Moreover, the molecular basis of this toxicity also appears to display common features between the different systems that have so far been studied. The ability of many, perhaps all, natural polypeptides to ‘misfold’ and convert into toxic aggregates under suitable conditions suggests that one of the most important driving forces in the evolution of proteins must have been the negative selection against sequence changes that increase the tendency of a polypeptide chain to aggregate. Nevertheless, as protein folding is a stochastic process, and no such process can be completely infallible, misfolded proteins or protein folding intermediates in equilibrium with the natively folded molecules must continuously form within cells. Thus mechanisms to deal with such species must have co-evolved with proteins. Indeed, it is clear that misfolding, and the associated tendency to aggregate, is kept under control by molecular chaperones, which render the resulting species harmless assisting in their refolding, or triggering their degradation by the cellular clearance machinery [166, 167, 168, 169, 170, 171, 172, 173, 175, 177, 178].
Misfolded and aggregated species are likely to owe their toxicity to the exposure on their surfaces of regions of proteins that are buried in the interior of the structures of the correctly folded native states. The exposure of large patches of hydrophobic groups is likely to be particularly significant as such patches favour the interaction of the misfolded species with cell membranes [44, 83, 89, 90, 91, 93]. Interactions of this type are likely to lead to the impairment of the function and integrity of the membranes involved, giving rise to a loss of regulation of the intracellular ion balance and redox status and eventually to cell death. In addition, misfolded proteins undoubtedly interact inappropriately with other cellular components, potentially giving rise to the impairment of a range of other biological processes. Under some conditions the intracellular content of aggregated species may increase directly, due to an enhanced propensity of incompletely folded or misfolded species to aggregate within the cell itself. This could occur as the result of the expression of mutational variants of proteins with decreased stability or cooperativity or with an intrinsically higher propensity to aggregate. It could also occur as a result of the overproduction of some types of protein, for example, because of other genetic factors or other disease conditions, or because of perturbations to the cellular environment that generate conditions favouring aggregation, such as heat shock or oxidative stress. Finally, the accumulation of misfolded or aggregated proteins could arise from the chaperone and clearance mechanisms becoming overwhelmed as a result of specific mutant phenotypes or of the general effects of ageing [173, 174].
The topics discussed in this review not only provide a great deal of evidence for the ‘new view’ that proteins have an intrinsic capability of misfolding and forming structures such as amyloid fibrils but also suggest that the role of molecular chaperones is even more important than was thought in the past. The role of these ubiquitous proteins in enhancing the efficiency of protein folding is well established [185]. It could well be that they are at least as important in controlling the harmful effects of misfolded or aggregated proteins as in enhancing the yield of functional molecules.
Nutritional Status is Associated with Faster Cognitive Decline and Worse Functional Impairment in the Progression of Dementia: The Cache County Dementia Progression Study1
Nutritional status may be a modifiable factor in the progression of dementia. We examined the association of nutritional status and rate of cognitive and functional decline in a U.S. population-based sample. Study design was an observational longitudinal study with annual follow-ups up to 6 years of 292 persons with dementia (72% Alzheimer’s disease, 56% female) in Cache County, UT using the Mini-Mental State Exam (MMSE), Clinical Dementia Rating Sum of Boxes (CDR-sb), and modified Mini Nutritional Assessment (mMNA). mMNA scores declined by approximately 0.50 points/year, suggesting increasing risk for malnutrition. Lower mMNA score predicted faster rate of decline on the MMSE at earlier follow-up times, but slower decline at later follow-up times, whereas higher mMNA scores had the opposite pattern (mMNA by time β= 0.22, p = 0.017; mMNA by time2 β= –0.04, p = 0.04). Lower mMNA score was associated with greater impairment on the CDR-sb over the course of dementia (β= 0.35, p < 0.001). Assessment of malnutrition may be useful in predicting rates of progression in dementia and may provide a target for clinical intervention.
Shared Genetic Risk Factors for Late-Life Depression and Alzheimer’s Disease
Background: Considerable evidence has been reported for the comorbidity between late-life depression (LLD) and Alzheimer’s disease (AD), both of which are very common in the general elderly population and represent a large burden on the health of the elderly. The pathophysiological mechanisms underlying the link between LLD and AD are poorly understood. Because both LLD and AD can be heritable and are influenced by multiple risk genes, shared genetic risk factors between LLD and AD may exist. Objective: The objective is to review the existing evidence for genetic risk factors that are common to LLD and AD and to outline the biological substrates proposed to mediate this association. Methods: A literature review was performed. Results: Genetic polymorphisms of brain-derived neurotrophic factor, apolipoprotein E, interleukin 1-beta, and methylenetetrahydrofolate reductase have been demonstrated to confer increased risk to both LLD and AD by studies examining either LLD or AD patients. These results contribute to the understanding of pathophysiological mechanisms that are common to both of these disorders, including deficits in nerve growth factors, inflammatory changes, and dysregulation mechanisms involving lipoprotein and folate. Other conflicting results have also been reviewed, and few studies have investigated the effects of the described polymorphisms on both LLD and AD. Conclusion: The findings suggest that common genetic pathways may underlie LLD and AD comorbidity. Studies to evaluate the genetic relationship between LLD and AD may provide insights into the molecular mechanisms that trigger disease progression as the population ages.
Association of Vitamin B12, Folate, and Sulfur Amino Acids With Brain Magnetic Resonance Imaging Measures in Older Adults: A Longitudinal Population-Based Study
Importance Vitamin B12, folate, and sulfur amino acids may be modifiable risk factors for structural brain changes that precede clinical dementia.
Objective To investigate the association of circulating levels of vitamin B12, red blood cell folate, and sulfur amino acids with the rate of total brain volume loss and the change in white matter hyperintensity volume as measured by fluid-attenuated inversion recovery in older adults.
Design, Setting, and Participants The magnetic resonance imaging subsample of the Swedish National Study on Aging and Care in Kungsholmen, a population-based longitudinal study in Stockholm, Sweden, was conducted in 501 participants aged 60 years or older who were free of dementia at baseline. A total of 299 participants underwent repeated structural brain magnetic resonance imaging scans from September 17, 2001, to December 17, 2009.
Main Outcomes and Measures The rate of brain tissue volume loss and the progression of total white matter hyperintensity volume.
Results In the multi-adjusted linear mixed models, among 501 participants (300 women [59.9%]; mean [SD] age, 70.9 [9.1] years), higher baseline vitamin B12 and holotranscobalamin levels were associated with a decreased rate of total brain volume loss during the study period: for each increase of 1 SD, β (SE) was 0.048 (0.013) for vitamin B12 (P < .001) and 0.040 (0.013) for holotranscobalamin (P = .002). Increased total homocysteine levels were associated with faster rates of total brain volume loss in the whole sample (β [SE] per 1-SD increase, –0.035 [0.015]; P = .02) and with the progression of white matter hyperintensity among participants with systolic blood pressure greater than 140 mm Hg (β [SE] per 1-SD increase, 0.000019 [0.00001]; P = .047). No longitudinal associations were found for red blood cell folate and other sulfur amino acids.
Conclusions and Relevance This study suggests that both vitamin B12 and total homocysteine concentrations may be related to accelerated aging of the brain. Randomized clinical trials are needed to determine the importance of vitamin B12supplementation on slowing brain aging in older adults.
Notes from Kurzweill
This vitamin stops the aging process in organs, say Swiss researchers
A potential breakthrough for regenerative medicine, pending further studies
Improved muscle stem cell numbers and muscle function in NR-treated aged mice: Newly regenerated muscle fibers 7 days after muscle damage in aged mice (left: control group; right: fed NR). (Scale bar = 50 μm). (credit: Hongbo Zhang et al./Science) http://www.kurzweilai.net/images/improved-muscle-fibers.png
EPFL researchers have restored the ability of mice organs to regenerate and extend life by simply administering nicotinamide riboside (NR) to them.
NR has been shown in previous studies to be effective in boosting metabolism and treating a number of degenerative diseases. Now, an article by PhD student Hongbo Zhang published in Science also describes the restorative effects of NR on the functioning of stem cells for regenerating organs.
As in all mammals, as mice age, the regenerative capacity of certain organs (such as the liver and kidneys) and muscles (including the heart) diminishes. Their ability to repair them following an injury is also affected. This leads to many of the disorders typical of aging.
Mitochondria —> stem cells —> organs
To understand how the regeneration process deteriorates with age, Zhang teamed up with colleagues from ETH Zurich, the University of Zurich, and universities in Canada and Brazil. By using several biomarkers, they were able to identify the molecular chain that regulates how mitochondria — the “powerhouse” of the cell — function and how they change with age. “We were able to show for the first time that their ability to function properly was important for stem cells,” said Auwerx.
Under normal conditions, these stem cells, reacting to signals sent by the body, regenerate damaged organs by producing new specific cells. At least in young bodies. “We demonstrated that fatigue in stem cells was one of the main causes of poor regeneration or even degeneration in certain tissues or organs,” said Zhang.
How to revitalize stem cells
Which is why the researchers wanted to “revitalize” stem cells in the muscles of elderly mice. And they did so by precisely targeting the molecules that help the mitochondria to function properly. “We gave nicotinamide riboside to 2-year-old mice, which is an advanced age for them,” said Zhang.
“This substance, which is close to vitamin B3, is a precursor of NAD+, a molecule that plays a key role in mitochondrial activity. And our results are extremely promising: muscular regeneration is much better in mice that received NR, and they lived longer than the mice that didn’t get it.”
Parallel studies have revealed a comparable effect on stem cells of the brain and skin. “This work could have very important implications in the field of regenerative medicine,” said Auwerx. This work on the aging process also has potential for treating diseases that can affect — and be fatal — in young people, like muscular dystrophy (myopathy).
So far, no negative side effects have been observed following the use of NR, even at high doses. But while it appears to boost the functioning of all cells, it could include pathological ones, so further in-depth studies are required.
Abstract of NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice
Adult stem cells (SCs) are essential for tissue maintenance and regeneration yet are susceptible to senescence during aging. We demonstrate the importance of the amount of the oxidized form of cellular nicotinamide adenine dinucleotide (NAD+) and its impact on mitochondrial activity as a pivotal switch to modulate muscle SC (MuSC) senescence. Treatment with the NAD+ precursor nicotinamide riboside (NR) induced the mitochondrial unfolded protein response (UPRmt) and synthesis of prohibitin proteins, and this rejuvenated MuSCs in aged mice. NR also prevented MuSC senescence in the Mdx mouse model of muscular dystrophy. We furthermore demonstrate that NR delays senescence of neural SCs (NSCs) and melanocyte SCs (McSCs), and increased mouse lifespan. Strategies that conserve cellular NAD+ may reprogram dysfunctional SCs and improve lifespan in mammals.
Discriminating the gene target of a distal regulatory element from other nearby transcribed genes is a challenging problem with the potential to illuminate the causal underpinnings of complex diseases. We present TargetFinder, a computational method that reconstructs regulatory landscapes from diverse features along the genome. The resulting models accurately predict individual enhancer–promoter interactions across multiple cell lines with a false discovery rate up to 15 times smaller than that obtained using the closest gene. By evaluating the genomic features driving this accuracy, we uncover interactions between structural proteins, transcription factors, epigenetic modifications, and transcription that together distinguish interacting from non-interacting enhancer–promoter pairs. Most of this signature is not proximal to the enhancers and promoters but instead decorates the looping DNA. We conclude that complex but consistent combinations of marks on the one-dimensional genome encode the three-dimensional structure of fine-scale regulatory interactions.
The cell is the basic unit of biology and protein expression drives cellular function. Tracking protein expression in single cells enables the study of cellular pathways and behavior, but requires methodologies sensitive enough to detect low numbers of protein molecules with a wide dynamic range to distinguish unique cells and quantify population distributions. This study presents an ultrasensitive and automated approach for quantifying phenotypic responses with single cell resolution using single molecule array (Simoa) technology. We demonstrate how prostate specific antigen (PSA) expression varies over several orders of magnitude between single prostate cancer cells, and how PSA expression shifts with genetic drift. Single cell Simoa intduces a straightforward process that is capable of detecting both high and low protein expression levels. This technique could be useful for understanding fundamental biology and may eventually enable both earlier disease detection and targeted therapy.
Quanterix’s proprietary Simoa™ technology (named for single molecule array) is based upon the isolation of individual immunocomplexes on paramagnetic beads using standard ELISA reagents. The main difference between Simoa and conventional immunoassays lies in the ability to trap single molecules in femtoliter-sized wells, allowing for a “digital” readout of each individual bead to determine if it is bound to the target analyte or not.
The digital nature of the technique allows an average of 1000x sensitivity increase over conventional assays with CVs <10%.
B. Tens of thousands of beads – with or without immunoconjugate – are mixed with enzyme substrate and loaded into individual femtoliter-sized wells.
C. The microwells are sealed with oil.
D. Fluorophore concentration in the small sample volume of wells containing the target analyte rapidly reach detectable limits using conventional fluorescence imaging and can be digitally counted.
E. The percentage of beads containing labelled immunocomplexes can be computed at low concentration because they follow a Poisson distribution; at higher concentrations the intensity of the aggregate signal provides an analog measurement.
The cerebrospinal fluid (CSF) amyloid-β (Aβ42) peptide is an important biomarker for Alzheimer’s disease (AD). Variability in measured Aβ42 concentrations at different laboratories may be overcome by standardization and establishing traceability to a reference system. Candidate certified reference materials (CRMs) are validated herein for this purpose.
METHODS:
Commutability of 16 candidate CRM formats was assessed across five CSF Aβ42 immunoassays and one mass spectrometry (MS) method in a set of 48 individual clinical CSF samples. Promising candidate CRM formats (neat CSF and CSF spiked with Aβ42) were identified and subjected to validation across eight (Elecsys, EUROIMMUN, IBL, INNO-BIA AlzBio3, INNOTEST, MSD, Simoa, and Saladax) immunoassays and the MS method in 32 individual CSF samples. Commutability was evaluated by Passing-Bablok regression and the candidate CRM termed commutable when found within the prediction interval (PI). The relative distance to the regression line was assessed.
RESULTS:
The neat CSF candidate CRM format was commutable for almost all method comparisons, except for the Simoa/MSD, Simoa/MS and MS/IBL where it was found just outside the 95% PI. However, the neat CSF was found within 5% relative distance to the regression line for MS/IBL, between 5% and 10% for Simoa/MS and between 10% and 15% for Simoa/MSD comparisons.
CONCLUSIONS:
The neat CSF candidate CRM format was commutable for 33 of 36 method comparisons, only one comparison more than expected given the 95% PI acceptance limit. We conclude that the neat CSF candidate CRM can be used for value assignment of the kit calibrators for the different Aβ42 methods.
Cerebral β-amyloidosis is induced by inoculation of Aβ seeds into APP transgenic mice, but not into App−/− (APP null) mice. We found that brain extracts from APP null mice that had been inoculated with Aβ seeds up to 6 months previously still induced β-amyloidosis in APP transgenic hosts following secondary transmission. Thus, Aβ seeds can persist in the brain for months, and they regain propagative and pathogenic activity in the presence of host Aβ.
Ultra-sensitive bead-based Simoa technology detects residual human Aβ in hippocampal extracts of inoculated App−/− mice.
Aβ seed-containing APP23 brain extract or seed-negative WT mouse brain extract was injected into the hippocampus of App−/− mice. The injected hippocampi were isolated 1, 30, 60, or 180 dpi (see Figures 1 and 2).
Induced amyloid lesions are partly congophilic and surrounded by activated microglia and dystrophic boutons.
(a) Congo red-positive amyloid deposits induced in the dentate gyrus were surrounded by Iba1-positive microglia (black). (b) Congo red-positive plaque with surrounding hypertrophic microglial cell bodies and processes at higher magnification
Glial fibrillary acidic protein is a body fluid biomarker for glial pathology in human disease
• Reviewing 45 years of Glial fibrillary acidic protein (Gfap).
•Gfap discovered in multiple sclerosis brain tissue.
•From Gfap genetics to post-translational modifications.
•Ninety-nine ways to quantify Gfap and related immune phenomena.
•Emergence of Gfap as a body fluid biomarker in human disease.
This review on the role of glial fibrillary acidic protein (GFAP) as a biomarker for astroglial pathology in neurological diseases provides background to protein synthesis, assembly, function and degeneration. Qualitative and quantitative analytical techniques for the investigation of human tissue and biological fluid samples are discussed including partial lack of parallelism and multiplexing capabilities. Pathological implications are reviewed in view of immunocytochemical, cell-culture and genetic findings. Particular emphasis is given to neurodegeneration related to autoimmune astrocytopathies and to genetic gain of function mutations. The current literature on body fluid levels of GFAP in human disease is summarised and illustrated by disease specific meta-analyses. In addition to the role of GFAP as a diagnostic biomarker for chronic disease, there are important data on the prognostic value for acute conditions. The published evidence permits to classify the dominant GFAP signatures in biological fluids. This classification may serve as a template for supporting diagnostic criteria of autoimmune astrocytopathies, monitoring disease progression in toxic gain of function mutations, clinical treatment trials (secondary outcome and toxicity biomarker) and provide prognostic information in neurocritical care if used within well defined time-frames.
CSF and Plasma Amyloid-b Temporal Profiles and Relationships with Neurological Status and Mortality after Severe Traumatic Brain Injury
by Stefania Mondello, Andras Burk, Pal Barzo, Jeff Randall, Gail Provuncher, David Hanlon, David Wilson, Firas Kobeissy & Andreas Jeromin
The role of amyloid-b (Ab) neuropathology and its significant changes in biofluids after traumatic braininjury (TBI) is still debated. We used ultrasensitive digital ELISA approach to assess amyloid-b1-42 (Ab42) concentrations and time-course in cerebrospinal fluid (CSF) and in plasma of patients with severe TBI and
investigated their relationship to injury characteristics, neurological status and clinical outcome. We found decreased CSF Ab42 levels in TBI patients acutely after injury with lower levels in patients who died 6 months post-injury than in survivors. Conversely, plasma Ab42 levels were significantly increased in TBI
with lower levels in patients who survived. A trend analysis showed that both CSF and plasma Ab42 levels strongly correlated with mortality. A positive correlation between changes in CSF Ab42 concentrations and neurological status as assessed by Glasgow Coma Scale (GCS) was identified. Our results suggest that determination of Ab42 may be valuable to obtain prognostic information in patients with severe TBI as well as in monitoring the response of the brain to injury.
Plasma tau levels in Alzheimer’s disease
Henrik Zetterberg, David Wilson, Ulf Andreasson, Lennart Minthon, Kaj Blennow, Jeffrey Randall and Oskar Hansson
Efforts to find reliable blood biomarkers for Alzheimer’s disease (AD) in a highly warranted clinical laboratory test have met with little success. There is no clear change in plasma ß-amyloid in AD, and assays for the axonal injury marker tau have been hampered by a lack of analytical sensitivity for accurate measurement in blood samples[1]. Here, the results of a novel ultra-sensitive assay for tau in peripheral blood are reported. Publication: Alzheimer’s Research & Therapy 2013, 5:9.
Efforts to find reliable blood biomarkers for Alzheimer’s disease (AD) in a highly warranted clinical laboratory test have met with little success. There is no clear change in plasma β-amyloid in AD, and assays for the axonal injury marker tau have been hampered by a lack of analytical sensitivity for accurate measurement in blood samples [1]. Here, the results of a novel ultra-sensitive assay for tau in peripheral blood are reported.
We have developed an ultra-sensitive assay for tau in peripheral blood [2]. In brief, the assay is based on digital array technology [3] and uses the Tau5 monoclonal antibody for capture (Covance, Princeton, NJ, USA) and HT7 and BT2 monoclonal antibodies for detection (Pierce, now part of Thermo Fisher Scientific Inc., Waltham, MA, USA). This combination reacts with both normal and phosphorylated tau with epitopes in the mid-region of the molecule, making the assay sensitive to all known tau isoforms. The calibrator was recombinant tau 381 (EMD Millipore Corporation, Billerica, MA, USA). To minimize matrix effects, all samples were diluted 1:4 in phosphate-buffered saline with 2% bovine serum albumin diluent prior to assay. The limit of detection of the assay, which requires 30 μL of plasma, is 0.02 pg/mL [2], which is more than 1,000-fold more sensitive than conventional immunoassays.
Here, we assess the association of plasma tau levels with AD in a cross-sectional study of 54 patients with AD dementia [4], 75 patients with mild cognitive impairment (MCI) [5], and 25 cognitively normal controls (Table 1). All participants were recruited at the specialized memory clinic at Skåne University Hospital in Malmö, Sweden, and underwent extensive clinical evaluation, including cerebrospinal fluid (CSF) sampling by lumbar puncture, in addition to venipuncture and collection of blood in ethylenediaminetetraacetic acid (EDTA) tubes for plasma preparation by centrifugation within 15 minutes from sampling. Plasma samples were aliquoted into cryo tubes and stored at -80°C pending analysis, which was performed on one occasion by using one batch of reagents with an average coefficient of variation of 9.7% for triplicate measurements of each sample. The patients with MCI were cognitively stable for an average of 101 months (n = 36) or developed AD dementia (n = 35) or other types of dementias – vascular dementia (n = 3) and semantic dementia (n = 1) – during follow-up. The study was approved by the regional ethics committee at Lund University and complied with the Declaration of Helsinki. Informed consent was obtained from all study participants.
Tau levels in plasma were significantly higher in AD patients compared with both controls and MCI patients (Figure 1a). MCI patients who developed AD during follow-up had tau levels similar to those of patients with stable MCI and cognitively normal controls (Figure 1b). There was no correlation between tau levels in plasma and CSF in any diagnostic group (Figure 1c).
Elevated tau levels in plasma from patients with Alzheimer’s disease (AD). (a) Plasma levels of tau are elevated in patients with AD compared with cognitively normal controls and patients with mild cognitive impairment (MCI). (b) MCI patients who developed AD (MCI-AD) during follow-up had baseline tau levels similar to those of patients with stable MCI (SMCI). (c) There was no correlation between tau levels in plasma and cerebrospinal fluid (CSF) in any diagnostic group. Thin horizontal lines in panels (a) and (b) indicate medians. A nonparametric Kruskal-Wallis test followed by Mann-Whitney was performed to test for statistical significance. Spearman’s rank correlation coefficient was used to assess the relationship between plasma and CSF tau concentrations in panel (c), where open circles, gray squares, and black triangles represent AD, MCI, and controls, respectively.
The results of this study have several important implications. First, plasma tau levels are elevated in AD but with overlapping ranges across diagnostic groups. This overlap diminishes the utility of plasma tau as a diagnostic test. However, further studies are needed to evaluate plasma tau as a first-in-line screening tool (for example, in the primary care setting and perhaps together with other markers in a biomarker panel). Second, normal plasma tau levels in the MCI stage of AD suggest that plasma tau is a late marker, requiring substantial axonal injury before increasing to abnormal levels. In this context, other neurodegenerative diseases (for example, Creutzfeldt-Jakob disease) as well as acute conditions (for example, stroke and brain trauma) should be tested. Third, the lack of correlation of tau levels in plasma and CSF suggests that steady-state concentrations of tau in these two body fluids are differentially regulated. In our earlier study of patients with hypoxic brain injury following cardiac arrest, tau was rapidly (within 24 hours) cleared from blood in patients with good neurological outcome [2], indicating potent clearance mechanisms for this marker in the bloodstream. This may obscure any correlation with CSF tau levels, which stay elevated for weeks following an acute neurological insult [6].
Researchers Use CRISPR-based Method to Track RNA In Vivo
A research team led by researchers from the University of California has modified the CRISPR/Cas9 system to demonstrate the ability to track specific RNA sequences and processes in vivo.
As described in a paper published today in Cell, the investigators were able to use their system to visualize specific RNA molecules accumulating in stress granules — dense aggregations of proteins and RNA that form in the cytosol in response to cellular stress and have been linked to neurodegenerative disorders such as amyotrophic lateral sclerosis.
They also found that they could use Cas9 to target an mRNA without altering mRNA abundance or the amount of translated protein.
“We are just beginning to see the implications of genome engineering using the CRISPR technology, but many diseases, including cancer and autism, are linked to problems with another fundamental biological molecule: RNA,” Gene Yeo, senior study author and an associate professor at the University of California, San Diego, said in a statement.
The researchers began their project based on a modification attempted in the lab of co-author Jennifer Doudna from the University of California, Berkeley. Inthat study, the researchers found that it was possible to design a protospacer adjacent motif (PAM) as part of an oligonucleotide (PAMmer) which binds to the single-stranded RNA, allowing Cas9 to efficiently recognize and cleave RNA rather than DNA (RCas9). The researchers determined that with a few further modifications, they could use this method to not only recognize RNA instead of DNA but actually track its movements through cells.
Previously, researchers have attempted to use molecular beacons to track RNA sequences, however, these are limited to imaging applications and are difficult to deliver into cells. Researchers have also attempted to use aptamers to enable RNA tracking in living cells, but these are limited in the number of RNA sequences that they can recognize.
CRISPR/Cas9, however, has thus far proved extremely useful in the genome engineering field and the research team thought that it would be an ideal base to create a better RNA tracking tool.
To prove their concept, the team tested whether a dead Cas9 (dCas9) that was tagged with the fluorescent protein mCherry and contained a nuclear localization signal could be co-exported from the nucleus with a messenger RNA in the presence of a single-guide RNA (sgRNA) and PAMmer designed to recognize that specific mRNA.
The experiment succeeded and the researchers were also able to observe accumulation of ACTB, CCNA2, and TFRC mRNAs in RNA granules that correlated with fluorescence in situ hybridization visualization using image analysis software.
Once they had established that their method was effective, the researchers showed that they could use the sgRNA and PAMmer targeting sequences to track mRNA trafficking to stress granules.
The researchers demonstrated that they could take time-resolved measurements of ACTB mRNA trafficking to stress granules over a period of 30 minutes. They noted in the paper that RCas9 was capable of measuring the association of CCNA2 and TFRC mRNA trafficking to stress granules, as well.
Based on their results, the investigators believe they have established RCas9 as a means to track RNA in living cells in a programmable manner that doesn’t require genetically encoded tags.
“One potential application of this technique is to track RNA transport in diseased neurons over time in order to identify the molecular features of these diseases and support the developments of therapies,” David Nelles, first author on the study and a researcher at the University of California, San Diego, said in a statement. “Just as CRISPR-Cas9 is making genetic engineering accessible to any scientists with access to basic equipment, RNA-targeted Cas9 may support countless other efforts for studying the role of RNA processing in disease or for identifying drugs that reverse defects in RNA processing.”
Programmable RNA Tracking in Live Cells with CRISPR/Cas9
David A. Nelles, Mark Y. Fang, Mitchell R. O’Connell, Jia L. Xu, Sebastian J. Markmiller, Jennifer A. Doudna, Gene W. Yeo
Clustered regularly-interspaced short palindromic repeats (CRISPRs) form the basis of adaptive immune systems in bacteria and archaea by encoding CRISPR RNAs that guide CRISPR-associated (Cas) nucleases to invading genetic material (Wiedenheft et al., 2012). Cas9 from the type II CRISPR system ofS. pyogenes has been repurposed for genome engineering in eukaryotic organisms (Hwang et al., 2013, Li et al., 2013a, Mali et al., 2013, Nakayama et al., 2013, Sander and Joung, 2014, Yang et al., 2014) and is rapidly proving to be an efficient means of DNA targeting for other applications such as gene expression modulation (Qi et al., 2013) and imaging (Chen et al., 2013). Cas9 and its associated single-guide RNA (sgRNA) require two critical features to target DNA: a short DNA sequence of the form 5′-NGG-3′ (where “N” = any nucleotide) known as the protospacer adjacent motif (PAM) and an adjacent sequence on the opposite DNA strand that is antisense to the sgRNA. By supporting DNA recognition with specificity determined entirely by a short spacer sequence within the sgRNA, CRISPR/Cas9 provides uniquely flexible and accessible manipulation of the genome. Manipulating cellular RNA content, in contrast, remains problematic. Whereas there exist robust means of attenuating gene expression via RNAi and antisense oligonucleotides, other critical aspects of post-transcriptional gene expression regulation such as subcellular trafficking, alternative splicing or polyadenylation, and spatiotemporally restricted translation are difficult to measure in living cells and are largely intractable.
Analogous to the assembly of zinc finger nucleases (Urnov et al., 2010) and transcription activator-like effector nucleases (TALEN) to recognize specific DNA sequences, efforts to recognize specific RNA sequences have focused on engineered RNA-binding domains. Pumilio and FBF homology (PUF) proteins carry well-defined modules capable of recognizing a single base each and have supported successful targeting of a handful of transcripts for imaging and other manipulations (Filipovska et al., 2011, Ozawa et al., 2007, Wang et al., 2009). PUF proteins can be fused to arbitrary effector domains to alter or tag target RNAs, but PUFs must be redesigned and validated for each RNA target and can only recognize eight contiguous bases, which does not allow unique discrimination in the transcriptome. Molecular beacons are self-quenched synthetic oligonucleotides that fluoresce upon binding to target RNAs and allow RNA detection without construction of a target-specific protein (Sokol et al., 1998). But molecular beacons must be microinjected to avoid the generation of excessive background signal associated with endosome-trapped probes and are limited to imaging applications. An alternative approach to recognition of RNA substrates is to introduce RNA aptamers into target RNAs, enabling specific and strong association of cognate aptamer-binding proteins such as the MS2 coat protein (Fouts et al., 1997). This approach has enabled tracking of RNA localization in living cells over time with high sensitivity (Bertrand et al., 1998) but relies upon laborious genetic manipulation of the target RNA and is not suitable for recognition of arbitrary RNA sequences. Furthermore, insertion of exogenous aptamer sequence has the potential to interfere with endogenous RNA functions. Analogous to CRISPR/Cas9-based recognition of DNA, programmable RNA recognition based on nucleic acid specificity alone without the need for genetic manipulation or libraries of RNA-binding proteins would greatly expand researchers’ ability to modify the mammalian transcriptome and enable transcriptome engineering.
Although the CRISPR/Cas9 system has evolved to recognize double-stranded DNA, recent in vitro work has demonstrated that programmable targeting of RNAs with Cas9 is possible by providing the PAM as part of an oligonucleotide (PAMmer) that hybridizes to the target RNA (O’Connell et al., 2014). By taking advantage of the Cas9 target search mechanism that relies on PAM sequences (Sternberg et al., 2014), a mismatched PAM sequence in the PAMmer/RNA hybrid allows exclusive targeting of RNA and not the encoding DNA. The high affinity and specificity of RNA recognition by Cas9 in cell-free extracts and the success of genome targeting with Cas9 indicate the potential of CRISPR/Cas9 to support programmable RNA targeting in living cells.
To assess the potential of Cas9 as a programmable RNA-binding protein in live cells, we used a modified sgRNA scaffold with improved expression and Cas9 association (Chen et al., 2013) with a stabilized PAMmer oligonucleotide that does not form a substrate for RNase H. We measured the degree of nuclear export of a nuclear localization signal-tagged nuclease-deficient Cas9-GFP fusion and demonstrate that the sgRNA alone is sufficient to promote nuclear export of Cas9 without influencing the abundance of the targeted mRNA or encoded protein. In order to evaluate whether RNA-targeting Cas9 (RCas9) signal patterns correspond with an established untagged RNA-labeling method, we compared distributions of RCas9 and fluorescence in situ hybridization (FISH) targeting ACTB mRNA. We observed high correlation among FISH and RCas9 signal that was dependent on the presence of a PAMmer, indicating the importance of the PAM for efficient RNA targeting. RNA trafficking and subcellular localization are critical to gene expression regulation and reaction to stimuli such as cellular stress. To address whether RCas9 allows tracking of RNA to oxidative stress-induced RNA/protein accumulations called stress granules, we measured ACTB, TFRC, and CCNA2 mRNA association with stress granules in cells subjected to sodium arsenite. Finally, we demonstrated the ability of RCas9 to track trafficking of ACTB mRNA to stress granules over time in living cells. This work establishes the ability of RCas9 to bind RNA in live cells and sets the foundation for manipulation of the transcriptome in addition to the genome by CRISPR/Cas9.
(A) Components required for RNA-targeting Cas9 (RCas9) recognition of mRNA include a nuclear localization signal-tagged nuclease-inactive Cas9 fused to a fluorescent protein such as GFP, a modified sgRNA with expression driven by the U6 polymerase III promoter, and a PAMmer composed of DNA and 2′-O-methyl RNA bases with a phosphodiester backbone. The sgRNA and PAMmer are antisense to adjacent regions of the target mRNA whose encoding DNA does not carry a PAM sequence. After formation of the RCas9/mRNA complex in the nucleus, the complex is exported to the cytoplasm.
(B) RCas9 nuclear co-export with GAPDH mRNA. The RCas9 system was delivered to U2OS cells with a sgRNA and PAMmer targeting the 3′ UTR of GAPDH or sgRNA and PAMmer targeting a sequence from λ bacteriophage that should not be present in human cells (“N/A”). Cellular nuclei are outlined with a dashed white line. Scale bars represent 5 microns.
(C) Fraction of cells with cytoplasmic RCas9 signal. Mean values ± SD (n = 50).
(D) A plasmid carrying the Renilla luciferase open reading frame with a β-globin 3′ UTR containing a target site for RCas9 and MS2 aptamer. A PEST protein degradation signal was appended to luciferase to reveal any translational effects of RCas9 binding to the mRNA.
(E) RNA immunoprecipitation of EGFP after transient transfection of the RCas9 system in HEK293T cells targeting the luciferase mRNA compared to non-targeting sgRNA and PAMmer or EGFP alone. Mean values ± SD (n = 3).
(F and G) Renilla luciferase mRNA (F) and protein (G) abundances were compared among the targeting and non-targeting conditions. Mean values ± SD (n = 4).
p values are calculated by Student’s t test, and one, two, and three asterisks represent p values less than 0.05, 0.01, and 0.001, respectively. See also Figure S1.
•RNA-targeting Cas9 (RCas9) enabled recognition of endogenous, unmodified mRNAs
•RCas9 did not influence mRNA abundance or amount of translated protein
•Subcellular distribution of RCas9 was highly correlated with RNA-FISH
•RCas9 revealed trafficking of mRNAs to stress granules in live cells
Summary
RNA-programmed genome editing using CRISPR/Cas9 from Streptococcus pyogenes has enabled rapid and accessible alteration of specific genomic loci in many organisms. A flexible means to target RNA would allow alteration and imaging of endogenous RNA transcripts analogous to CRISPR/Cas-based genomic tools, but most RNA targeting methods rely on incorporation of exogenous tags. Here, we demonstrate that nuclease-inactive S. pyogenesCRISPR/Cas9 can bind RNA in a nucleic-acid-programmed manner and allow endogenous RNA tracking in living cells. We show that nuclear-localized RNA-targeting Cas9 (RCas9) is exported to the cytoplasm only in the presence of sgRNAs targeting mRNA and observe accumulation ofACTB, CCNA2, and TFRC mRNAs in RNA granules that correlate with fluorescence in situ hybridization. We also demonstrate time-resolved measurements of ACTB mRNA trafficking to stress granules. Our results establish RCas9 as a means to track RNA in living cells in a programmable manner without genetically encoded tags.
Correlation of RNA-Targeting Cas9 Signal Distributions with an Established Untagged RNA Localization Measurement
Tracking RNA Trafficking to Stress Granules over Time
Effective RNA recognition by Cas9 in living cells while avoiding perturbation of the target transcript relies on careful design of the PAMmer and delivery of Cas9 and its cognate guide RNA to the appropriate cellular compartments. Binding of Cas9 to nucleic acids requires two critical features: a PAM DNA sequence and an adjacent spacer sequence antisense to the Cas9-associated sgRNA. By separating the PAM and sgRNA target among two molecules (the PAMmer oligonucleotide and the target mRNA) that only associate in the presence of a target mRNA, RCas9 allows recognition of RNA while avoiding the encoding DNA. To avoid unwanted degradation of the target RNA, the PAMmer is composed of a mixed 2′OMe RNA and DNA that does not form a substrate for RNase H. Further, the sgRNA features a modified scaffold that removes partial transcription termination sequences and a modified structure that promotes association with Cas9 (Chen et al., 2013). Other CRISPR/Cas systems have demonstrated RNA binding in bacteria (Hale et al., 2009, Sampson et al., 2013) or eukaryotes (Price et al., 2015), although these systems cannot discriminate RNA from DNA targets, feature RNA-targeting rules that remain unclear, or rely on large protein complexes that may be difficult to reconstitute in mammalian cells.
In this work, we demonstrate RCas9-based recognition of GAPDH, ACTB,CCNA2, and TFRC mRNAs in live cells. Because the U6-driven sgRNA is largely restricted to the nucleus, the NLS-tagged dCas9 allows association with its sgRNA and subsequent interaction with the target mRNA before nuclear co-export with the target mRNA. As an initial experiment, we evaluated the potential of RNA recognition with Cas9 by targeting GAPDH mRNA and evaluating degree of nuclear export of dCas9-mCherry (Figure 1B). Robust cytoplasmic localization of dCas9-mCherry in the presence of a sgRNA-targeting GAPDH mRNA compared to nuclear retention in the presence of a non-targeting sgRNA indicated that Cas9 association with the mRNA was sufficiently stable to support co-export from the nucleus.
RCas9 as an RNA-imaging reagent requires that RNA recognition by RCas9 does not interfere with normal RNA metabolism. Here, we show that RCas9 binding within the 3′ UTR of Renilla luciferase does not affect its mRNA abundance and translation (Figures 1F and 1G). The utility of RCas9 for imaging and other applications hinges on the recognition of endogenous transcripts, so we evaluated the influence of RCas9 targeting on GAPDH and ACTB mRNAs and observed no significant differences among the mRNA and protein abundances by western blot analysis and qRT-PCR (Figure S1). These results indicate that RCas9 targeting these 3′ UTRs does not perturb the levels of mRNA or encoded protein.
We also evaluated the ability of RCas9 to reveal RNA localization by comparing RCas9 signal patterns to FISH. We utilized a FISH probe set composed of tens of singly labeled probes targeting ACTB mRNA and compared FISH signal distributions to a single dCas9-GFP/sgRNA/PAMmer that recognizes the ACTBmRNA. Our findings indicate that the sgRNA primarily determines the degree of overlap among the FISH and RCas9 signals whereas the PAMmer plays a significant but secondary role. Importantly, in contrast to other untagged RNA localization determination methods such as FISH and molecular beacons, RCas9 is compatible with tracking untagged RNA localization in living cells and can be delivered rapidly to cells using established transfection methods. We also note that the distribution of ACTB mRNA was visualized using a single EGFP tag per transcript, and higher-sensitivity RNA tracking or single endogenous RNA molecule visualization may be possible in the future with RCas9 targeting multiple sites in a transcript or with a multiply tagged dCas9 protein.
Stress granules are translationally silent mRNA and protein accumulations that form in response to cellular stress and are increasingly thought to be involved with neurodegeneration (Li et al., 2013b). There are limited means that can track the movement of endogenous RNA to these structures in live cells (Bertrand et al., 1998). In addition to ACTB mRNA, we demonstrate that RCas9 is capable of measuring association of CCNA2 and TFRC mRNA trafficking to stress granules (Figure 3A). Upon stress induction with sodium arsenite, we observed that 50%, 39%, and 23% of stress granules featured overlapping RCas9 foci when targeting ACTB, TFRC, and CCNA2 mRNAs, respectively (Figure 3C). This result correlates with the expression levels of these transcripts (Figure S3) asACTB is expressed about 8 and 11 times more highly than CCNA2 and TFRC, respectively. We also observed that RCas9 is capable of tracking RNA localization over time as ACTB mRNA is trafficked to stress granules over a period of 30 min (Figure 3B). We noted a dependence of RCas9 signal accumulation in stress granules on stressor concentration (Figure 3D). This approach for live-cell RNA tracking stands in contrast to molecular beacons and aptamer-based RNA-tracking methods, which suffer from delivery issues and/or require alteration of the target RNA sequence via incorporation of RNA tags.
Future applications of RCas9 could allow the measurement or alteration of RNA splicing via recruitment of split fluorescent proteins or splicing factors adjacent to alternatively spliced exons. Further, the nucleic-acid-programmable nature of RCas9 lends itself to multiplexed targeting (Cong et al., 2013) and the use of Cas9 proteins that bind orthogonal sgRNAs (Esvelt et al., 2013) could support distinct activities on multiple target RNAs simultaneously. It is possible that the simple RNA targeting afforded by RCas9 could support the development of sensors that recognize specific healthy or disease-related gene expression patterns and reprogram cell behavior via alteration of gene expression or concatenation of enzymes on a target RNA (Delebecque et al., 2011, Sachdeva et al., 2014). Efforts toward Cas9 delivery in vivo are underway (Dow et al., 2015,Swiech et al., 2015, Zuris et al., 2015), and these efforts combined with existing oligonucleotide chemistries (Bennett and Swayze, 2010) could support in vivo delivery of the RCas9 system for targeted modulation of many features of RNA processing in living organisms.
RNA is subject to processing steps that include alternative splicing, nuclear export, subcellular transport, and base or backbone modifications that work in concert to regulate gene expression. The development of a programmable means of RNA recognition in order to measure and manipulate these processes has been sought after in biotechnology for decades. This work is, to our knowledge, the first demonstration of nucleic-acid-programmed RNA recognition in living cells with CRISPR/Cas9. By relying upon a sgRNA and PAMmer to determine target specificity, RCas9 supports versatile and unambiguous RNA recognition analogous to DNA recognition afforded by CRISPR/Cas9. The diverse applications supported by DNA-targeted CRISPR/Cas9 range from directed cleavage, imaging, transcription modulation, and targeted methylation, indicating the utility of both the native nucleolytic activity of Cas9 as well as the range of activities supported by Cas9-fused effectors. In addition to providing a flexible means to track this RNA in live cells, future developments of RCas9 could include effectors that modulate a variety of RNA-processing steps with applications in synthetic biology and disease modeling or treatment.
Single-cell RNA sequencing (scRNA-seq) is a recent technology that enables fine-grained discovery of cellular subtypes and specific cell states. It routinely uses machine learning methods, such as feature learning, clustering, and classification, to assist in uncovering novel information from scRNA-seq data. However, current methods are not well suited to deal with the substantial amounts of noise that is created by the experiments or the variation that occurs due to differences in the cells of the same type. Here, we develop a new hybrid approach, Deep Unsupervised Single-cell Clustering (DUSC), that integrates feature generation based on a deep learning architecture with a model-based clustering algorithm, to find a compact and informative representation of the single-cell transcriptomic data generating robust clusters. We also include a technique to estimate an efficient number of latent features in the deep learning model. Our method outperforms both classical and state-of-the-art feature learning and clustering methods, approaching the accuracy of supervised learning. We applied DUSC to single-cell transcriptomics dataset obtained from a triple-negative breast cancer tumor to identify potential cancer subclones accentuated by copy-number variation and investigate the role of clonal heterogeneity. Our method is freely available to the community and will hopefully facilitate our understanding of the cellular atlas of living organisms as well as provide the means to improve patient diagnostics and treatment.
Jenny Mjösberg and Rickard Sandberg are principal investigators at Karolinska Institutet’s Department of Medicine, Huddinge and Department of Cell and Molecular Biology, respectively. Credit: Stefan Zimmerman.
A relatively newly discovered group of immune cells known as ILCs have been examined in detail in a new study published in the journal Nature Immunology. By analysing the gene expression in individual tonsil cells, scientists at Karolinska Institutet have found three previously unknown subgroups of ILCs, and revealed more about how these cells function in the human body.
Innate lymphoid cells (ILCs) are a group of immune cells that have only relatively recently been discovered in humans. Most of current knowledge about ILCs stems from animal studies of e.g. inflammation or infection in the gastrointestinal tract. There is therefore an urgent need to learn more about these cells in humans.
Previous studies have shown that ILCs are important for maintaining the barrier function of the mucosa, which serves as a first line of defence against microorganisms in the lungs, intestines and elsewhere. However, while there is growing evidence to suggest that ILCs are involved in diseases such as inflammatory bowel disease, asthma and intestinal cancer, basic research still needs to be done to ascertain exactly what part they play.
Two research groups, led by Rickard Sandberg and Jenny Mjösberg, collaborated on a study of ILCs from human tonsils. To date, three main groups of human ILCs are characterized. In this present study, the teams used a novel approach that enabled them to sort individual tonsil cells and measure their expression across thousands of genes. This way, the researchers managed to categorise hundreds of cells, one by one, to define the types of ILCs found in the human tonsils.
Unique gene expression profiles
Rickard Sandberg, credit: Stefan Zimmerman,
“We used cluster analyses to demonstrate that ILCs congregate into ILC1, ILC2, ILC3 and NK cells, based on their unique gene expression profiles,” says Professor Sandberg at Karolinska Institutet’sDepartment of Cell and Molecular Biology, and the Stockholm branch of Ludwig Cancer Research. “Our analyses also discovered the expression of numerous genes of previously unknown function in ILCs, highlighting that these cells are likely doing more than what we previously knew.”
By analysing the gene expression profiles (or transcriptome) of individual cells, the researchers found that one of the formerly known main groups could be subdivided.
Jenny Mjösberg, credit: Stefan Zimmerman.
“We’ve identified three new subgroups of ILC3s that evince different gene expression patterns and that differ in how they react to signalling molecules and in their ability to secrete proteins,” says Dr Mjösberg at Karolinska Institutet’s Department of Medicine in Huddinge, South Stockholm. “All in all, our study has taught us a lot about this relatively uncharacterised family of cells and our data will serve as an important resource for other researchers.”
The study was financed by grants from a number of bodies, including the Swedish Research Council, the Swedish Cancer Society, the EU Framework Programme for Research and Innovation, the Swedish Society for Medical Research, the Swedish Foundation for Strategic Research and Karolinska Institutet.
January 20, 2016 | An adult mouse’s brain, an object not much bigger than the last joint of your pinky finger, contains around 75 million neurons. At the Allen Institute for Brain Science in Seattle, the Mouse Cell Types program, led by Hongkui Zeng, is trying to figure out just how many varieties of neurons make up this vast complex, and what makes each one unique.
Zeng’s research focuses on the primary visual cortex, a tiny sliver of the brain where signals from the eyes are processed and interpreted. Because vision is a relatively well-defined process, it’s thought to be a good model for connecting the behavior of individual neurons to larger brain functions.
“You really can’t understand a system until you understand its parts,” says Bosiljka Tasic, a founding member of the Mouse Cell Types program.
To a shocking extent, those parts are still a mystery. Many supposed cell types are based on little more than what you can see through a microscope: a neuron’s shape, or the pattern of rootlike dendrites extending from its body. These morphological traits, though important, are hard to see in full, and even harder to track methodically across thousands or millions of cells.
This month, Zeng’s team published a study in Nature Neuroscience that takes advantage of new technological developments to get a fine-grained look at the molecular toolkits of single neurons. Using newly refined methods to isolate single cells, Zeng’s lab collected over 1,600 brain cells from the visual cortexes of adult mice, intact and in good shape for sequencing. With advances in highly parallel, unbiased RNA sequencing, the group was able to measure each cell’s entire “transcriptome”―the array of RNA molecules that indicate which genes are actively producing proteins―at a depth that reveals even the scarcest RNA traces.
“We think this is probably the most comprehensive survey of a cortical area,” says Tasic, who co-led the study with her colleague Vilas Menon. “Many studies that are coming out now do very shallow sequencing… We wanted to go deeper.” With a median of 8.7 million sequencing reads per cell, the authors discovered a wealth of new RNA markers that define discrete groups of neurons. Some of these markers suggest that known cell types in the brain can be split into smaller sub-categories. A few even stake out rare types of neurons that may be new to science.
Yet the data collected for this study also confirms that the brain’s biology is neither tidy nor easy to unravel.
“There is this obsession in the field, and in many other areas of biology, that people always want cleanliness and discreteness,” Tasic says. Instead, her efforts to classify neurons have shown that “types” can be slippery, and many cells straddle the line between closely related groups. As projects like this one seek to redefine cell types for the genomics age, scientists will have to face these ambiguities and consider what they can tell us about the nature of the brain.
Patterns within Patterns
Whole transcriptomes provide an impressive amount of data with which to organize cells, but that data is hard to interpret in an unbiased way. “We’re trying, in some sense, to solve two problems simultaneously,” says Vilas Menon, co-lead author of the paper. “We’re trying to cluster the genes, and also to cluster the cells.”
To disentangle these problems, the team performed an iterative analysis. First, their software looked for RNA markers that diverged most widely between different cells, using those markers to sort all the cells in the study into large clusters. Then, they wiped the slate clean, looking for brand-new markers within each cluster to split the cells step by step into smaller groups. The smallest possible divisions, in which no new RNA markers could strongly distinguish cells from one another, became the group’s proposed “cell types.”
The researchers used two different computational methods to define clusters, but both revealed the same basic hierarchy of types. “In general, the higher level splits correspond to what’s already known for these broad classes of neurons,” says Menon. For instance, the first split simply divided all the neurons in their data from a handful of other cell types present in the brain, like the glial cells that support the brain’s physical structure. The second split separated GABAergic cells, which mostly damp down chemical signals in the brain, from glutamatergic cells, which mostly spark and amplify signals.
Beyond this point, the patterns became more revealing. Within the glutamatergic cells, for example, later clustering tended to split neurons according to how deeply they were embedded in the cortex. A mouse’s primary visual cortex is organized in six layers, and the Allen Institute’s transcriptome data suggests that the neurons in each layer may be closely related to one another, or have similar functions that require the same genes to be activated. Yet the GABAergic cells did not split out so naturally by layer, implying that their development may follow very different rules.
At the narrowest levels of clustering, the genes that defined cell types sometimes came as complete surprises. Within a group of GABAergic neurons known for producing high levels of the hormone somatostatin, the authors found a subtype of cells expressing an additional gene called Chodl. “Nobody has ever heard of this marker Chodl,” says Tasic. “But it’s the most beautiful pattern you’ve ever seen, because it’s only in that cell type. This is the beauty of transcriptomics.”
With luck, genes like Chodl will provide new clues to the roles of specific cell types. If no other neurons make use of this gene, it’s reasonable to think it may have a very specialized function. But even if that’s not the case, highly unique markers like Chodl are invaluable for studying neurons more closely, letting scientists design new molecular and genetic tools to target single cell types for follow-up research.
“I see this as a first step in allowing us to selectively manipulate cell types,” says Tasic. “And then you can do all sorts of things to those cells. You can label them specifically, and study their morphology. You can perturb them. You can inactivate them. I think this will be the way to truly understand what these different cells do.”
Mountains and Ridges
“Technically, this is a very impressive achievement,” says Joshua Sanes, a neurobiologist at the Harvard Center for Brain Science. “It’s using a really nice combination of state-of-the-art methods to address what, to me, is a big problem in neurobiology.”
Like the researchers at the Allen Institute, Sanes is interested in the problem of defining cell types. (Both his group and Hongkui Zeng’s receive funding from the national BRAIN Initiative, which has provided grants for big data-gathering projects to attack this question.) It’s a vexing issue, both because it requires such an immense amount of data to address, and because biology again and again rejects easy categories.
To Sanes, one of the most interesting aspects of Tasic and Menon’s paper is their decision to point out neurons with traits of more than one cell type. Unlike other groups that may exclude ambiguous data from analysis, the Allen Institute accepted cells with “intermediate” transcriptomes as important findings of their study. In some cases―most notably, a class of glutamatergic neurons in layer four of the cortex―these intermediate cells are so abundant that two or more supposedly separate “types” almost seem to merge together.
“That could mean that, although some cells are in types, there’s a certain amount of slipperiness,” says Sanes. “It’s been pretty hard to define neurons in a way that will help research move forward.”
It’s possible that some classes of neurons don’t exist in discrete types at all, but include a spectrum of cells expressing different mixes of the same genes. Or transcriptomes may just not be the best way to define cell types―because neurons of the same type change their RNA arsenals depending on their stage of development, or the chemical signals they’re responding to.
“Some parts of the overall phenotypic landscape may have features of a continuum,” says Tasic, but that doesn’t mean that her group’s proposed cell types are not useful ways of thinking about neurobiology. “If there are two mountains that are connected by a ridge, there are still two mountains. The fact that you have a ridge is fine. Maybe that’s biology.”
From Rosetta Stones to Searchable Databases
Tasic, Menon, and their colleagues identified 49 cell types altogether, but the number is less important than the process that produced it. Almost certainly, there are still new cell types to discover, and perhaps further divisions within the types the Allen Institute has identified.
“I think it’s extremely unlikely they’ve gotten all the types,” says Sanes. “It’s terrific, but it’s not like you should think of this as a complete catalogue.” To isolate single neurons, the Allen Institute used a method called FACS, which relies on sampling many different strains of transgenic mice to collect both abundant and rare cell types. The authors agree that this approach leaves open the possibility that some rare types were not sampled, and future studies will use different methods of capturing single cells, adding yet more data to the mix. (At his lab, Sanes is working with a new method called Drop-seq, which the Allen Institute also plans to adopt.)
For work like this to be meaningful, it’s not necessary for the Allen Institute to come up with a complete encyclopedia of cell types on its own. What is essential is that the data be made easily available to neuroscientists everywhere, to compare with their own studies and gradually refine with new discoveries.
Today, this is far from assured. A lot of research on cell types is only available through journal articles, and there are few standards for formatting data so it can be shared and understood across institutions. This is apparent in some of the detective work that Zeng’s team did to see if their proposed cell types matched any previously identified types. Tasic, Menon, and colleagues trawled through the scientific literature looking for what they called “Rosetta stones,” unique molecular features that could clearly be seen in their own transcriptome data.
In the future, this work could be made almost automatic, especially as objective data types like RNA sequencing information become more common. Just a few weeks ago, many of the first recipients of BRAIN Initiative grants―including both Zeng and Sanes―met in Bethesda, Md., to discuss plans for sharing neurobiological data, and ways to make that data more uniform and searchable.
“I think the BRAIN Initiative has been helpful in drawing attention and funding,” says Sanes. “The NIH is doing everything it can to ensure data sharing, and I think the community is going along with that well.”
In the meantime, Zeng’s group has released their raw transcriptome data to GEO, an NIH-supported database of RNA information, and made an annotated version of their data available online on the Allen Institute website. Tasic and Menon hope that outside researchers will use these resources to design more detailed studies of specific neuron types. Neuroscience is still in the earliest stages of data gathering, but to truly understand the brain, scientists will eventually have to make the leap into exploring function, cell type by cell type.
“We can find genes that are differentially expressed at the level of the whole brain, but we really don’t know what these genes do,” Tasic says. “Once you see that this gene is expressed in a specific type, you can formulate a hypothesis.”
Adult mouse cortical cell taxonomy revealed by single cell transcriptomics
Nervous systems are composed of various cell types, but the extent of cell type diversity is poorly understood. We constructed a cellular taxonomy of one cortical region, primary visual cortex, in adult mice on the basis of single-cell RNA sequencing. We identified 49 transcriptomic cell types, including 23 GABAergic, 19 glutamatergic and 7 non-neuronal types. We also analyzed cell type–specific mRNA processing and characterized genetic access to these transcriptomic types by many transgenic Cre lines. Finally, we found that some of our transcriptomic cell types displayed specific and differential electrophysiological and axon projection properties, thereby confirming that the single-cell transcriptomic signatures can be associated with specific cellular properties.
Cell types summary and relationships.close
(a–c) Constellation diagrams showing core and intermediate cells for all cell types. Core cells (N = 1,424 total, 664 GABAergic, 609 glutamatergic, 151 non-neuronal) are represented by colored disks
MIT researchers engineered a microfluidic device that traces detailed family histories for several generations of cells descended from one “ancestor.” [Video: Melanie Gonick/MIT (additional video courtesy of the Manalis Lab)]
Scientists at MIT said they can now trace detailed family histories for several generations of cells descended from one ancestor after combining RNA sequencing with a novel device that isolates single cells and their progeny. This technique, which can track changes in gene expression as cells differentiate, could be particularly useful for studying how stem cells or immune cells mature, noted the researchers, adding that it could also shed light on how cancer develops.
“Existing methods allow for snapshot measurements of single-cell gene expression, which can provide very in-depth information. What this new approach offers is the ability to track cells over multiple generations and put this information in the context of a cell’s lineal history,” says Robert Kimmerling, a graduate student in biological engineering and the lead author of a paper (“A microfluidic platform enabling single-cell RNA-seq of multigenerational lineages”) describing the technique in Nature Communications.
The new method incorporates single-cell RNA-seq, which sequences a single cell’s transcriptome and reveals which genes are being transcribed inside a cell at a given point in time. This helps scientists understand, for example, what makes a skin cell so different from a heart cell even though the cells share the same DNA.
“Scientists have well-established methods for resolving diverse subsets of a population, but one thing that’s not very well worked out is how this diversity is generated. That’s the key question we were targeting: how a single founding cell gives rise to very diverse progeny,” points out Kimmerling.
To try to answer that question, the researchers designed a microfluidic device that traps first an individual cell and then all of its descendants. The device has several connected channels, each of which has a trap that can capture a single cell. After the initial cell divides, its daughter cells flow further along the device and get trapped in the next channel. The researchers showed that they can capture up to five generations of cells this way and keep track of their relationships.
To get the cells off the chip, the researchers temporarily reverse the direction of the fluid flowing across the chip, allowing them to remove the cells one at a time to perform single-cell RNA-seq.
In this study, the team captured and sequenced T cells that—when they encounter a cell infected with a virus or bacterium—create effector T cells, which seek and destroy infected cells, as well as memory T cells that retain a “memory” of the encounter and circulate in the body indefinitely in case of a subsequent encounter.
“A single founding cell can give rise to both effector and memory cell subtypes, but how that diversity is generated isn’t very clear,” explain Kimmerling.
The scientists analyzed RNA from recently activated T cells and two subsequent generations. When comparing genes with functions related to T-cell activation and differentiation, they found that sister cells produced from the same division event are much more similar in their gene expression profiles than two unrelated cells. They also found that “cousin” cells, which have the same “grandmother,” are more similar than unrelated cells. This suggests unique, family-specific transcriptional profiles for single T cells.
The researchers hope that future studies with this device could help to resolve the long-standing debate over how T cells differentiate into effector cells and memory cells. One theory is that the distinction occurs as early as the first T cell division following activation, while a competing theory suggests that the distinction happens later on, as a result of changes in the cells’ microenvironment.
To address this question, the researchers believe they would need to analyze the development of T cells taken from a mouse that had been exposed to a foreign pathogen, providing a useful model of T cell activation in response to infection.
Roeder – the coactivator OCA-B, the first cell-specific coactivator, discovered by Roeder in 1992, is unique to immune system B cells
Larry H Bernstein, MD, FCAP, Curator
Leaders in Pharmaceutical Intelligence
The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes
Unkyu Kim*, Xiao-Feng Qin†, Shiaoching Gong†, Sean Stevens*, Yan Luo*, Michel Nussenzweig† & Robert G. Roeder*
Nature 383, 542 – 547 (10 October 1996); http://dx.doi.org:/10.1038/383542a0 * Laboratory of Biochemistry and Molecular Biology, and † Laboratory of Molecular Immunology, Howard Hughes Medical Institute, The Rockefeller University, New York, New York 10021, USA
OCA-B was initially identified as a B-cell-restricted coactivator that functions with octamer binding transcription factors (Oct-1 and Oct-2) to mediate efficient cell type-specific transcription of immunoglobulin promoters in vitro1–3. Subsequent cloning studies led to identification of the coactivator as a single poly-peptide, designated either as OCA-B (ref. 3), OBF-1 (ref. 4) or Bob-1 (ref. 5). OCA-B itself does not bind to DNA directly, but interacts with either Oct-1 or Oct-2 to potentiate transcriptional activation1–5. To determine the biological role of OCA-B, we generated OCA-B-deficient mice by gene targeting. Mice lacking OCA-B undergo normal antigen-independent, B-cell differentiation, including appropriate expression of both immunoglobulin genes and other early B-cell-restricted genes. However, antigen-dependent maturation of B cells is greatly affected. The pro- liferative response to surface IgM crosslinking is impaired, and there is a severe deficiency in the production of secondary immunoglobulin isotypes including IgGl, IgG2a, IgG2b, IgG3, IgA and IgE in OCA-B-deficient B cells. This defect is not due to a failure of the isotype switching process, but rather to reduced levels of transcription from normally switched immunoglobulin heavy-chain loci. In accord with the defective isotype production, germinal centre formation is absent in these mutant mice.
References
1.
Pierani, A., Heguy, A., Fujii, H. & Roeder, R. G. Mol. Cell. Biol.10, 6204−6215 (1990). | PubMed | ChemPort |
2.
Luo, Y., Fujii, H., Gerster, T. & Roeder, R. G. Cell71, 231−241 (1992). | Article | PubMed | ISI | ChemPort |
3.
Luo, Y. & Roeder, R. G. Mol. Cell. Biol.15, 4115−4124 (1995). | PubMed | ISI | ChemPort |
4.
Strubin, M., Newell, J. W. & Matthias, P. Cell80, 497−506 (1995). | Article | PubMed | ISI | ChemPort |
5.
Gstaiger, M., Knoepfel, L., Georgiev, O., Schaffner, W. & Hovens, C. M. Nature373, 360−362 (1995). | Article | PubMed | ISI | ChemPort |
Bottaro, A. et al.EMBO J.13, 665−674 (1994). | PubMed | ISI | ChemPort |
24.
Cogne, M. et al.Cell77, 737−747 (1994). | Article | PubMed | ISI |
25.
Gong, S. & Nussenzweig, M. C. Science272, 411−414 (1996). | PubMed | ISI | ChemPort |
26.
Qin, X. F. et al.EMBO J.13, 5967−5976 (1994). | PubMed | ISI | ChemPort |
27.
Li, S. C. et al.Int. Immunol.6, 491−497 (1994). | PubMed | ChemPort |
Cloning, Functional Characterization, and Mechanism of Action of the B-Cell-Specific Transcriptional Coactivator
Oca-B Yan Luo & Robert G. Roeder*
Laboratory of Biochemistry and Molecular Biology, The Rockefeller University, New York, New York 10021
Molecular And Cellular Biology, Aug. 1995; 15(8):4115–4124 0270-7306/95/ http://www.ncbi.nlm.nih.gov/pmc/articles/PMC230650/pdf/154115.pdf
Biochemical purification and cognate cDNA cloning studies have revealed that the previously described transcriptional coactivator OCA-B consists of a 34- or 35-kDa polypeptide with sequence relationships to known coactivators that function by protein-protein interactions. Studies with a recombinant protein have proved that a single OCA-B polypeptide is the main determinant for B-cell-specific activation of immunoglobulin (Ig) promoters and provided additional insights into its mechanism of action. Recombinant OCA-B can function equally well with Oct-1 or Oct-2 on an Ig promoter, but while corresponding POU domains are sufficient for OCA-B interaction, and for octamer-mediated transcription of a histone H2B promoter, an additional Oct-1 or Oct-2 activation domain(s) is necessary for functional synergy with OCA-B. Further studies show that Ig promoter activation by Oct-1 and OCA-B requires still other general (USA-derived) cofactors and also provide indirect evidence that distinct Oct-interacting cofactors regulate H2B transcription.
A novel B cell-derived coactivator potentiates the activation of immunoglobulin promoters by octamer-binding transcription factors
Yan Luo, Hiroshi Fujii, Thomas Gerster, Robert G. Roeder
Laboratory of Biochemistry and Molecular Biology The Rockefeller University New York, New York 10021 USA
★Present address: Department of Biochemistry, Niigata University School of Medicine, Niigata 951, Japan.
A novel B cell-restricted activity, required for high levels of octamer/Oct-dependent transcription from an immunoglobulin heavy chain (IgH) promoter, was detected in an in vitro system consisting of HeLa cell-derived extracts complemented with fractionated B cell nuclear proteins. The factor responsible for this activity was designated Oct coactivator from B cells (OCA-B). OCA-B stimulates the transcription from an IgH promoter in conjunction with either Oct-1 or Oct-2 but shows no significant effect on the octamer/Oct-dependent transcription of the ubiquitously expressed histone H2B promoter and the transcription of USF- and Sp1-regulated promoters. Taken together, our results suggest that OCA-B is a tissue-, promoter-, and factor-specific coactivator and that OCA-B may be a major determinant for B cell-specific activation of immunoglobulin promoters. In light of the evidence showing physical and functional interactions between Oct factors and OCA-B, we propose a mechanism of action for OCA-B and discuss the implications of OCA-B for the transcriptional regulation of other tissue-specific promoters.
Identification of transcription coactivator OCA-B dependent genes involved in antigen-dependent B cell differentiation by cDNA array analyses
Unkyu Kim*, Rachael Siegel*, Xiaodi Ren*, Cary S. Gunther*, Terry Gaasterland†, and Robert G. Roeder*‡
*Laboratory of Biochemistry and Molecular Biology and †Laboratory of Computational Genomics, The Rockefeller University, 1230 York Avenue, New York, NY 10021
The tissue-specific transcriptional coactivator OCA-B is required for antigen-dependent B cell differentiation events, including germinal center formation. However, the identity of OCA-B target genes involved in this process is unknown. This study has used large-scale cDNA arrays to monitor changes in gene expression patterns that accompany mature B cell differentiation. B cell receptor ligation alone induces many genes involved in B cell expansion, whereas B cell receptor and helper T cell costimulation induce genes associated with B cell effector function. OCA-B expression is induced by both B cell receptor ligation alone and helper T cell costimulation, suggesting that OCA-B is involved in B cell expansion as well as B cell function. Accordingly, several genes involved in cell proliferation and signaling, such as Lck, Kcnn4, Cdc37, cyclin D3, B4galt1, and Ms4a11, have been identified as OCA-B-dependent genes. Further studies on the roles played by these genes in B cells will contribute to an understanding of B cell differentiation.
Identification and Characterization of a Novel OCA-B Isoform: Implications for a Role in B Cell Signaling Pathways
OCA-B is a B lymphocyte–specific transcription coactivator that mediates tissue- and stage-restricted transcription of immunoglobulin genes. Earlier genetic studies revealed that OCA-B is essential for germinal center formation and production of secondary immunoglobulin isotypes. Biochemically purified OCA-B contains p35 and p34 isoforms, and a further analysis has now revealed that p35 is derived from a newly found isoform, p40. More importantly, it has been found that p35 is myristoylated in vivo and that this leads to dramatic changes (including localization to membrane compartments) in its properties. These results suggest that the p35 isoform of OCA-B has functions distinct from those of the nuclear p34 and that it might be a component of a signaling pathway that is required for late-stage B cell development.
The B cell–restricted function of immunoglobulin (Ig) promoters is mediated mainly by an octamer element (5′-ATGCAAAT-3′) that is conserved in virtually all Ig heavy (H) and light (L) chain gene promoters, as well as in some Ig enhancers (reviewed by Staudt and Lenardo, 1990). However, this same element is also a key central element for transcription of differentially regulated genes that include ubiquitously expressed small nuclear RNA genes (snRNA) and cell cycle-regulated histone H2B genes (reviewed inLuo et al., 1992). The regulatory functions of octamer elements, therefore, are likely dependent on transcription factors that bind this DNA sequence. The well-characterized octamer binding transcription factors include the ubiquitous Oct-1 and the B cell–enriched Oct-2, both of which belong to the POU family and share a conserved DNA binding structure called the POU domain (reviewed by Herr et al. 1988 and Wegner et al. 1993). It was originally thought that Oct-2 would account for the tissue-specific activity of Ig promoters, whereas Oct-1 would facilitate transcription of the ubiquitously expressed genes regulated through octamer elements (e.g., snRNA and histone H2B genes) Staudt et al. 1986, Cockerill and Klinken 1990 and Murphy et al. 1992. However, subsequent biochemical Pierani et al. 1990 and Luo et al. 1992 and genetic (Corcoran et al., 1993)analyses clearly demonstrated that this was not the case. Instead, the promoter specificity was shown to be due to an Oct-1 interacting factor called OCA-B (Luo et al., 1992), and the purification of related p35 and p34 isoforms with apparently equivalent activity in vitro (Luo and Roeder, 1995) set the stage for further studies of the structure and function of OCA-B.
Subsequent to the biochemical identification of OCA-B and its mechanism of action, cognate cDNAs were cloned using both biochemical (Luo and Roeder, 1995) and genetic screening Gstaiger et al. 1995 and Strubin et al. 1995 methods. Analyses of recombinant OCA-B (p34) function in cell-free systems and in transfection assays confirmed both physical and functional interactions with Oct-1 and Oct-2 (via their POU domains) on Ig promoters Gstaiger et al. 1995, Luo and Roeder 1995 and Strubin et al. 1995 and led to the definition of an N-terminal OCA-B domain that interacts with the Oct POU domainCepek et al. 1996, Gstaiger et al. 1996, Babb et al. 1997 and Chaseman et al. 1999 and a C-terminal activation domain that acts synergistically with Oct activation domains to recruit additional coactivators (Luo et al., 1998).
The physiological roles of OCA-B were further investigated by genetic disruption of OCA-B expression in mice Kim et al. 1996, Nielson et al. 1996 and Schubart et al. 1996. These studies showed that, although not required for early B cell development, OCA-B functions are essential both for germinal center formation and for efficient secondary Ig isotype production (including IgGs, IgA, and IgE). In accordance with the biochemical function of OCA-B in activating Ig promoter transcription, it has been found that the decrease of secondary antibody production in OCA-B-deficient mice is largely due to reduced levels of transcription from normally switched IgH chain loci, rather than a reduced capacity for class switching events per se Kim et al. 1996 and Schubart et al. 1996. Recent results further demonstrated that OCA-B plays an essential role in efficient transcription from switched IgH loci by directly regulating 3′ IgH enhancer function in conjunction with Oct-1 or Oct-2 Tang and Sharp 1999 and Stevens et al. 2000b. On the other hand, the lack of germinal center formation in OCA-B-deficient mice cannot be explained by reduced Ig isotype production, since these are two independent events in B cell development (Vajdy et al., 1995). Therefore, OCA-B may regulate germinal center formation by activating the expression of other target genes or by mediating signal pathways that in turn trigger a specific genetic program. At least two lines of evidence support this idea: (1) B cells lacking OCA-B are defective in the proliferative response to surface IgM cross-linking (Kim et al., 1996); (2) OCA-B expression, which is very low in early B cells but high in activated B cells in vivo, can be dramatically and synergistically induced in naive B cells by B cell stimuli (CD40L, Ig cross-linking, and IL4) that are required for germinal center formation (Qin et al., 1998).
Our findings in this report raise the possibility that OCA-B may be directly involved in B cell signaling pathways through novel mechanisms. We report the presence of a novel isoform of OCA-B (p40) that results from utilization of an upstream alternative translation initiation codon and that serves as a precursor to the p35 isoform of OCA-B. Relative to the conventional p34 OCA-B isoform, p35 shows distinct protein modification, subcellular localization, and transcriptional coactivator properties. The unique features of p35 suggest a novel function for this molecule in signal transduction.
Synergism with the Coactivator OBF-1 (OCA-B, BOB-1) Is Mediated by a Specific POU Dimer Configuration
Alexey Tomilin1, 2, #, Attila Reményi2, 4, #, Katharina Lins1, Hanne Bak2, Sebastian Leidel1, Gerrit Vriend3, Matthias Wilmanns4, Hans R Schöler1, 2
Cell Dec 2000; 103(6):853–864 doi:10.1016/S0092-8674(00)00189-6
POU domain proteins contain a bipartite DNA binding domain divided by a flexible linker that enables them to adopt various monomer configurations on DNA. The versatility of POU protein operation is additionally conferred at the dimerization level. The POU dimer formed on the PORE (ATTTGAAATGCAAAT) can recruit the transcriptional coactivator OBF-1, whereas POU dimers formed on the consensus MORE (ATGCATATGCAT) or on MOREs from immunoglobulin heavy chain promoters (AT[G/A][C/A]ATATGCAA) fail to interact. An interaction with OBF-1 is precluded since the same Oct-1 residues that form the MORE dimerization interface are also used for OBF-1/Oct-1 interactions on the PORE. Our findings provide a paradigm of how specific POU dimer assemblies can differentially recruit a coregulatory activity with distinct transcriptional readouts.
Development of multicellular organisms is characterized by an intricate series of genetic and epigenetic events that generate the complex adult body from the unicellular zygote. A refined and sophisticated regulatory network that is established during embryogenesis reflects the complexity of organisms. Although embryonic development is a multistep process characterized by the sequential activation and repression of many genes, only a relatively small number of transcription factors are responsible for regulating the expression of developmental genes. This diversity in transcriptional control by a limited array of transcription factors is achieved through a complex network of interactions between these proteins and specific DNA sequences found in promoters and enhancers of developmental genes. The primary structure of these DNA elements defines the composition and architecture of the transcriptional activation complexes that ultimately control gene expression in the appropriate temporo-spatial context of the developing organism. For example, nonsteroid members of the nuclear receptor superfamily that possess a zinc-finger DNA binding domain operate by binding to the hormone response elements (HREs). HREs consist of two minimal core hexad sequences, AGGTCA, which can be configured into various functional motifs. The orientation and spacing between these two hexamers as well as subtle differences in their sequence dictate the identity and the mode (monomer, hetero-, or homodimer) of nuclear receptor binding that results in diverse effects on transcription (Mangelsdorf and Evans 1995).
The operation of members of the POU domain family of transcription factors is also highly dependent on the nature of cognate DNA elements. The 160 amino-acid-long DNA binding domain of these proteins is composed of two structurally independent subdomains: the POU-type homeodomain (POU-homeo or POUH), and the POU-specific domain (POUS) that are connected by a flexible linker region (27 and 36). POU domain proteins demonstrate impressive versatility in how they regulate transcription. This is due to several, often interdependent, factors: (1) flexible amino acid–base interaction, (2) variable orientation, spacing, and positioning of DNA-tethered POU subdomains relative to each other, (3) posttranslational modification, and (4) interaction with heterologous proteins (Herr and Cleary 1995).
POU domain proteins are able to bind to DNA cooperatively, thus conferring additional functional variability. The homo- and heterodimerization of Oct-1 and Oct-2 on immunoglobulin (Ig) heavy chain promoters (VH) provided evidence of cooperativity, with a yet unknown dimer arrangement (13, 16 and 23). The cis-elements are considered to consist of low-affinity heptamer and high-affinity octamer sites separated by two nucleotides (AT).
The pituitary-specific POU domain protein Pit-1 binds to DNA either as a homodimer or as a heterodimer with Oct-1 (Voss et al. 1991). Crystallographic studies determined the structure of a Pit-1 homodimer assembled on the synthetic motif ATGTATATACAT (referred to here as PitD) that had been derived from the natural Pit-1 cognate element within the prolactin gene promoter (ATATATATTCAT) (Jacobson et al. 1997). The structure of the Pit-1 POUS and POUH domains, and their docking onto DNA, are very similar to that observed in the cocrystal of the Oct-1 POU domain monomer with the octamer site (ATGCAAAT, Klemm et al. 1994). The Oct-1 POUS domain recognizes the ATGC subsite whereas the Pit-1 POUS domain binds to the sequence ATAC. However, the latter subsite lies on the opposite strand and, as a consequence, the orientation of POUS relative to the POUH domain is inverted (Jacobson et al. 1997).
Another mechanism outlining cooperative DNA binding by POU proteins was recently determined during the course of an Oct-4 target gene characterization (Botquin et al. 1998). The P alindromic O ct factor R ecognition E lement (PORE), ATTTGAAATGCAAAT (15 bp), of the Osteopontin (OPN) enhancer interacts with an Oct-4 dimer, thereby mediating strong transcriptional activation in preimplantation mouse embryos. Homo- and heterodimerization of other Oct factors like Oct-1 and Oct-6 on the PORE has also been demonstrated.
The aforementioned examples provide evidence of the various ways in which POU domain proteins are able to cooperatively bind to substrate DNA. The particular mode of binding employed is primarily defined by the DNA sequence. To address the question of whether diversity in cooperative binding is reflected in transcriptional regulation, we have assessed and compared the ability of two different types of POU dimers to interact with the coactivator OBF-1 (OCA-B, Bob-1). This coactivator synergistically interacts with Oct-1 and Oct-2 monomers bound to the octamer motif (18, 9, 17 and 33). We have investigated one type of POU dimer that is formed on the PORE and another that is formed on another palindromic DNA motif called MORE (M ore P ORE), ATGCATATGCAT. The data presented in this study provide an example of how POU domain molecules that bind to DNA in the same stoichiometry but in different configurations can differentially recruit a transcriptional coactivator to the promoter resulting in differential transcriptional activation.
B-cell-specific Coactivator OCA-B: Biochemical Aspects, Role in B-Cell Development and Beyond
This e-Book is a comprehensive review of recent Original Research on METABOLOMICS and related opportunities for Targeted Therapy written by Experts, Authors, Writers. This is the first volume of the Series D: e-Books on BioMedicine – Metabolomics, Immunology, Infectious Diseases. It is written for comprehension at the third year medical student level, or as a reference for licensing board exams, but it is also written for the education of a first time baccalaureate degree reader in the biological sciences. Hopefully, it can be read with great interest by the undergraduate student who is undecided in the choice of a career. The results of Original Research are gaining value added for the e-Reader by the Methodology of Curation.The e-Book’s articles have been published on the Open Access Online Scientific Journal, since April 2012. All new articles on this subject, will continue to be incorporated, as published with periodical updates.
We invite e-Readers to write an Article Reviews on Amazon for this e-Book on Amazon.
All forthcoming BioMed e-Book Titles can be viewed at:
Leaders in Pharmaceutical Business Intelligence, launched in April 2012 an Open Access Online Scientific Journal is a scientific, medical and business multi expert authoring environment in several domains of life sciences, pharmaceutical, healthcare & medicine industries. The venture operates as an online scientific intellectual exchange at their website http://pharmaceuticalintelligence.com and for curation and reporting on frontiers in biomedical, biological sciences, healthcare economics, pharmacology, pharmaceuticals & medicine. In addition the venture publishes a Medical E-book Series available on Amazon’s Kindle platform.
Analyzing and sharing the vast and rapidly expanding volume of scientific knowledge has never been so crucial to innovation in the medical field. WE are addressing need of overcoming this scientific information overload by:
delivering curation and summary interpretations of latest findings and innovations on an open-access, Web 2.0 platform with future goals of providing primarily concept-driven search in the near future
providing a social platform for scientists and clinicians to enter into discussion using social media
compiling recent discoveries and issues in yearly-updated Medical E-book Series on Amazon’s mobile Kindle platform
This curation offers better organization and visibility to the critical information useful for the next innovations in academic, clinical, and industrial research by providing these hybrid networks.
Table of Contents forMetabolic Genomics & Pharmaceutics, Vol. I
Chapter 1: Metabolic Pathways
Chapter 2: Lipid Metabolism
Chapter 3: Cell Signaling
Chapter 4: Protein Synthesis and Degradation
Chapter 5: Sub-cellular Structure
Chapter 6: Proteomics
Chapter 7: Metabolomics
Chapter 8: Impairments in Pathological States: Endocrine Disorders; Stress
Hypermetabolism and Cancer
Chapter 9: Genomic Expression in Health and Disease
Summary and Perspectives: Impairments in Pathological States: Endocrine Disorders, Stress Hypermetabolism and Cancer
Author and Curator: Larry H. Bernstein, MD, FCAP
Article ID #160: Summary and Perspectives: Impairments in Pathological States: Endocrine Disorders, Stress Hypermetabolism and Cancer. Published on 11/9/2014
WordCloud Image Produced by Adam Tubman
This summary is the last of a series on the impact of transcriptomics, proteomics, and metabolomics on disease investigation, and the sorting and integration of genomic signatures and metabolic signatures to explain phenotypic relationships in variability and individuality of response to disease expression and how this leads to pharmaceutical discovery and personalized medicine. We have unquestionably better tools at our disposal than has ever existed in the history of mankind, and an enormous knowledge-base that has to be accessed. I shall conclude here these discussions with the powerful contribution to and current knowledge pertaining to biochemistry, metabolism, protein-interactions, signaling, and the application of the -OMICS to diseases and drug discovery at this time.
Both the developmental and phylogenetic histories of an organism describe the evolution of physiology—the complex of metabolic pathways that govern the function of an organism as a whole. The necessity of establishing and maintaining homeostatic mechanisms began at the cellular level, with the very first cells, and homeostasis provides the underlying selection pressure fueling evolution.
While the events leading to the formation of the first functioning cell are debatable, a critical one was certainly the formation of simple lipid-enclosed vesicles, which provided a protected space for the evolution of metabolic pathways. Protocells evolved from a common ancestor that experienced environmental stresses early in the history of cellular development, such as acidic ocean conditions and low atmospheric oxygen levels, which shaped the evolution of metabolism.
The reduction of evolution to cell biology may answer the perennially unresolved question of why organisms return to their unicellular origins during the life cycle.
As primitive protocells evolved to form prokaryotes and, much later, eukaryotes, changes to the cell membrane occurred that were critical to the maintenance of chemiosmosis, the generation of bioenergy through the partitioning of ions. The incorporation of cholesterol into the plasma membrane surrounding primitive eukaryotic cells marked the beginning of their differentiation from prokaryotes. Cholesterol imparted more fluidity to eukaryotic cell membranes, enhancing functionality by increasing motility and endocytosis. Membrane deformability also allowed for increased gas exchange.
Acidification of the oceans by atmospheric carbon dioxide generated high intracellular calcium ion concentrations in primitive aquatic eukaryotes, which had to be lowered to prevent toxic effects, namely the aggregation of nucleotides, proteins, and lipids. The early cells achieved this by the evolution of calcium channels composed of cholesterol embedded within the cell’s plasma membrane, and of internal membranes, such as that of the endoplasmic reticulum, peroxisomes, and other cytoplasmic organelles, which hosted intracellular chemiosmosis and helped regulate calcium.
As eukaryotes thrived, they experienced increasingly competitive pressure for metabolic efficiency. Engulfed bacteria, assimilated as mitochondria, provided more bioenergy. As the evolution of eukaryotic organisms progressed, metabolic cooperation evolved, perhaps to enable competition with biofilm-forming, quorum-sensing prokaryotes. The subsequent appearance of multicellular eukaryotes expressing cellular growth factors and their respective receptors facilitated cell-cell signaling, forming the basis for an explosion of multicellular eukaryote evolution, culminating in the metazoans.
Casting a cellular perspective on evolution highlights the integration of genotype and phenotype. Starting from the protocell membrane, the functional homolog for all complex metazoan organs, it offers a way of experimentally determining the role of genes that fostered evolution based on the ontogeny and phylogeny of cellular processes that can be traced back, in some cases, to our last universal common ancestor. ….
As eukaryotes thrived, they experienced increasingly competitive pressure for metabolic efficiency. Engulfed bacteria, assimilated as mitochondria, provided more bioenergy. As the evolution of eukaryotic organisms progressed, metabolic cooperation evolved, perhaps to enable competition with biofilm-forming, quorum-sensing prokaryotes. The subsequent appearance of multicellular eukaryotes expressing cellular growth factors and their respective receptors facilitated cell-cell signaling, forming the basis for an explosion of multicellular eukaryote evolution, culminating in the metazoans.
Casting a cellular perspective on evolution highlights the integration of genotype and phenotype. Starting from the protocell membrane, the functional homolog for all complex metazoan organs, it offers a way of experimentally determining the role of genes that fostered evolution based on the ontogeny and phylogeny of cellular processes that can be traced back, in some cases, to our last universal common ancestor.
Given that the unicellular toolkit is complete with all the traits necessary for forming multicellular organisms (Science, 301:361-63, 2003), it is distinctly possible that metazoans are merely permutations of the unicellular body plan. That scenario would clarify a lot of puzzling biology: molecular commonalities between the skin, lung, gut, and brain that affect physiology and pathophysiology exist because the cell membranes of unicellular organisms perform the equivalents of these tissue functions, and the existence of pleiotropy—one gene affecting many phenotypes—may be a consequence of the common unicellular source for all complex biologic traits. …
The cell-molecular homeostatic model for evolution and stability addresses how the external environment generates homeostasis developmentally at the cellular level. It also determines homeostatic set points in adaptation to the environment through specific effectors, such as growth factors and their receptors, second messengers, inflammatory mediators, crossover mutations, and gene duplications. This is a highly mechanistic, heritable, plastic process that lends itself to understanding evolution at the cellular, tissue, organ, system, and population levels, mediated by physiologically linked mechanisms throughout, without having to invoke random, chance mechanisms to bridge different scales of evolutionary change. In other words, it is an integrated mechanism that can often be traced all the way back to its unicellular origins.
The switch from swim bladder to lung as vertebrates moved from water to land is proof of principle that stress-induced evolution in metazoans can be understood from changes at the cellular level.
A MECHANISTIC BASIS FOR LUNG DEVELOPMENT: Stress from periodic atmospheric hypoxia (1) during vertebrate adaptation to land enhances positive selection of the stretch-regulated parathyroid hormone-related protein (PTHrP) in the pituitary and adrenal glands. In the pituitary (2), PTHrP signaling upregulates the release of adrenocorticotropic hormone (ACTH) (3), which stimulates the release of glucocorticoids (GC) by the adrenal gland (4). In the adrenal gland, PTHrP signaling also stimulates glucocorticoid production of adrenaline (5), which in turn affects the secretion of lung surfactant, the distension of alveoli, and the perfusion of alveolar capillaries (6). PTHrP signaling integrates the inflation and deflation of the alveoli with surfactant production and capillary perfusion. THE SCIENTIST STAFF
From a cell-cell signaling perspective, two critical duplications in genes coding for cell-surface receptors occurred during this period of water-to-land transition—in the stretch-regulated parathyroid hormone-related protein (PTHrP) receptor gene and the β adrenergic (βA) receptor gene. These gene duplications can be disassembled by following their effects on vertebrate physiology backwards over phylogeny. PTHrP signaling is necessary for traits specifically relevant to land adaptation: calcification of bone, skin barrier formation, and the inflation and distention of lung alveoli. Microvascular shear stress in PTHrP-expressing organs such as bone, skin, kidney, and lung would have favored duplication of the PTHrP receptor, since sheer stress generates radical oxygen species (ROS) known to have this effect and PTHrP is a potent vasodilator, acting as an epistatic balancing selection for this constraint.
Positive selection for PTHrP signaling also evolved in the pituitary and adrenal cortex (see figure on this page), stimulating the secretion of ACTH and corticoids, respectively, in response to the stress of land adaptation. This cascade amplified adrenaline production by the adrenal medulla, since corticoids passing through it enzymatically stimulate adrenaline synthesis. Positive selection for this functional trait may have resulted from hypoxic stress that arose during global episodes of atmospheric hypoxia over geologic time. Since hypoxia is the most potent physiologic stressor, such transient oxygen deficiencies would have been acutely alleviated by increasing adrenaline levels, which would have stimulated alveolar surfactant production, increasing gas exchange by facilitating the distension of the alveoli. Over time, increased alveolar distension would have generated more alveoli by stimulating PTHrP secretion, impelling evolution of the alveolar bed of the lung.
This scenario similarly explains βA receptor gene duplication, since increased density of the βA receptor within the alveolar walls was necessary for relieving another constraint during the evolution of the lung in adaptation to land: the bottleneck created by the existence of a common mechanism for blood pressure control in both the lung alveoli and the systemic blood pressure. The pulmonary vasculature was constrained by its ability to withstand the swings in pressure caused by the systemic perfusion necessary to sustain all the other vital organs. PTHrP is a potent vasodilator, subserving the blood pressure constraint, but eventually the βA receptors evolved to coordinate blood pressure in both the lung and the periphery.
Gut Microbiome Heritability
Analyzing data from a large twin study, researchers have homed in on how host genetics can shape the gut microbiome.
By Tracy Vence | The Scientist Nov 6, 2014
Previous research suggested host genetic variation can influence microbial phenotype, but an analysis of data from a large twin study published in Cell today (November 6) solidifies the connection between human genotype and the composition of the gut microbiome. Studying more than 1,000 fecal samples from 416 monozygotic and dizygotic twin pairs, Cornell University’s Ruth Ley and her colleagues have homed in on one bacterial taxon, the family Christensenellaceae, as the most highly heritable group of microbes in the human gut. The researchers also found that Christensenellaceae—which was first described just two years ago—is central to a network of co-occurring heritable microbes that is associated with lean body mass index (BMI). …
Of particular interest was the family Christensenellaceae, which was the most heritable taxon among those identified in the team’s analysis of fecal samples obtained from the TwinsUK study population.
While microbiologists had previously detected 16S rRNA sequences belonging to Christensenellaceae in the human microbiome, the family wasn’t named until 2012. “People hadn’t looked into it, partly because it didn’t have a name . . . it sort of flew under the radar,” said Ley.
Ley and her colleagues discovered that Christensenellaceae appears to be the hub in a network of co-occurring heritable taxa, which—among TwinsUK participants—was associated with low BMI. The researchers also found that Christensenellaceae had been found at greater abundance in low-BMI twins in older studies.
To interrogate the effects of Christensenellaceae on host metabolic phenotype, the Ley’s team introduced lean and obese human fecal samples into germ-free mice. They found animals that received lean fecal samples containing more Christensenellaceae showed reduced weight gain compared with their counterparts. And treatment of mice that had obesity-associated microbiomes with one member of the Christensenellaceae family, Christensenella minuta, led to reduced weight gain. …
Ley and her colleagues are now focusing on the host alleles underlying the heritability of the gut microbiome. “We’re running a genome-wide association analysis to try to find genes—particular variants of genes—that might associate with higher levels of these highly heritable microbiota. . . . Hopefully that will point us to possible reasons they’re heritable,” she said. “The genes will guide us toward understanding how these relationships are maintained between host genotype and microbiome composition.”
The desire for temporal and spatial control of medications to minimize side effects and maximize benefits has inspired the development of light-controllable drugs, or optopharmacology. Early versions of such drugs have manipulated ion channels or protein-protein interactions, “but never, to my knowledge, G protein–coupled receptors [GPCRs], which are one of the most important pharmacological targets,” says Pau Gorostiza of the Institute for Bioengineering of Catalonia, in Barcelona.
Gorostiza has taken the first step toward filling that gap, creating a photosensitive inhibitor of the metabotropic glutamate 5 (mGlu5) receptor—a GPCR expressed in neurons and implicated in a number of neurological and psychiatric disorders. The new mGlu5 inhibitor—called alloswitch-1—is based on a known mGlu receptor inhibitor, but the simple addition of a light-responsive appendage, as had been done for other photosensitive drugs, wasn’t an option. The binding site on mGlu5 is “extremely tight,” explains Gorostiza, and would not accommodate a differently shaped molecule. Instead, alloswitch-1 has an intrinsic light-responsive element.
In a human cell line, the drug was active under dim light conditions, switched off by exposure to violet light, and switched back on by green light. When Gorostiza’s team administered alloswitch-1 to tadpoles, switching between violet and green light made the animals stop and start swimming, respectively.
The fact that alloswitch-1 is constitutively active and switched off by light is not ideal, says Gorostiza. “If you are thinking of therapy, then in principle you would prefer the opposite,” an “on” switch. Indeed, tweaks are required before alloswitch-1 could be a useful drug or research tool, says Stefan Herlitze, who studies ion channels at Ruhr-Universität Bochum in Germany. But, he adds, “as a proof of principle it is great.” (Nat Chem Biol, http://dx.doi.org:/10.1038/nchembio.1612, 2014)
To understand disease processes, scientists often focus on unraveling how gene expression in disease-associated cells is altered. Increases or decreases in transcription—as dictated by a regulatory stretch of DNA called an enhancer, which serves as a binding site for transcription factors and associated proteins—can produce an aberrant composition of proteins, metabolites, and signaling molecules that drives pathologic states. Identifying the root causes of these changes may lead to new therapeutic approaches for many different diseases.
Although few therapies for human diseases aim to alter gene expression, the outstanding examples—including antiestrogens for hormone-positive breast cancer, antiandrogens for prostate cancer, and PPAR-γ agonists for type 2 diabetes—demonstrate the benefits that can be achieved through targeting gene-control mechanisms. Now, thanks to recent papers from laboratories at MIT, Harvard, and the National Institutes of Health, researchers have a new, much bigger transcriptional target: large DNA regions known as super-enhancers or stretch-enhancers. Already, work on super-enhancers is providing insights into how gene-expression programs are established and maintained, and how they may go awry in disease. Such research promises to open new avenues for discovering medicines for diseases where novel approaches are sorely needed.
Super-enhancers cover stretches of DNA that are 10- to 100-fold longer and about 10-fold less abundant in the genome than typical enhancer regions (Cell, 153:307-19, 2013). They also appear to bind a large percentage of the transcriptional machinery compared to typical enhancers, allowing them to better establish and enforce cell-type specific transcriptional programs (Cell, 153:320-34, 2013).
Super-enhancers are closely associated with genes that dictate cell identity, including those for cell-type–specific master regulatory transcription factors. This observation led to the intriguing hypothesis that cells with a pathologic identity, such as cancer cells, have an altered gene expression program driven by the loss, gain, or altered function of super-enhancers.
Sure enough, by mapping the genome-wide location of super-enhancers in several cancer cell lines and from patients’ tumor cells, we and others have demonstrated that genes located near super-enhancers are involved in processes that underlie tumorigenesis, such as cell proliferation, signaling, and apoptosis.
Super-enhancers cover stretches of DNA that are 10- to 100-fold longer and about 10-fold less abundant in the genome than typical enhancer regions.
Genome-wide association studies (GWAS) have found that disease- and trait-associated genetic variants often occur in greater numbers in super-enhancers (compared to typical enhancers) in cell types involved in the disease or trait of interest (Cell, 155:934-47, 2013). For example, an enrichment of fasting glucose–associated single nucleotide polymorphisms (SNPs) was found in the stretch-enhancers of pancreatic islet cells (PNAS, 110:17921-26, 2013). Given that some 90 percent of reported disease-associated SNPs are located in noncoding regions, super-enhancer maps may be extremely valuable in assigning functional significance to GWAS variants and identifying target pathways.
Because only 1 to 2 percent of active genes are physically linked to a super-enhancer, mapping the locations of super-enhancers can be used to pinpoint the small number of genes that may drive the biology of that cell. Differential super-enhancer maps that compare normal cells to diseased cells can be used to unravel the gene-control circuitry and identify new molecular targets, in much the same way that somatic mutations in tumor cells can point to oncogenic drivers in cancer. This approach is especially attractive in diseases for which an incomplete understanding of the pathogenic mechanisms has been a barrier to discovering effective new therapies.
Another therapeutic approach could be to disrupt the formation or function of super-enhancers by interfering with their associated protein components. This strategy could make it possible to downregulate multiple disease-associated genes through a single molecular intervention. A group of Boston-area researchers recently published support for this concept when they described inhibited expression of cancer-specific genes, leading to a decrease in cancer cell growth, by using a small molecule inhibitor to knock down a super-enhancer component called BRD4 (Cancer Cell, 24:777-90, 2013). More recently, another group showed that expression of the RUNX1 transcription factor, involved in a form of T-cell leukemia, can be diminished by treating cells with an inhibitor of a transcriptional kinase that is present at the RUNX1 super-enhancer (Nature, 511:616-20, 2014).
detect pathogen-associated molecular patterns to activate immunity,
pathogens attempt to deregulate host immunity through secreted effectors.
Fungi employ LysM effectors to prevent
recognition of cell wall-derived chitin by host immune receptors
Structural analysis of the LysM effector Ecp6 of
the fungal tomato pathogen Cladosporium fulvum reveals
a novel mechanism for chitin binding,
mediated by intrachain LysM dimerization,
leading to a chitin-binding groove that is deeply buried in the effector protein.
This composite binding site involves
two of the three LysMs of Ecp6 and
mediates chitin binding with ultra-high (pM) affinity.
The remaining singular LysM domain of Ecp6 binds chitin with
low micromolar affinity but can nevertheless still perturb chitin-triggered immunity.
Conceivably, the perturbation by this LysM domain is not established through chitin sequestration but possibly through interference with the host immune receptor complex.
Mutated Genes in Schizophrenia Map to Brain Networks From www.nih.gov – Sep 3, 2013
Previous studies have shown that many people with schizophrenia have de novo, or new, genetic mutations. These misspellings in a gene’s DNA sequence
occur spontaneously and so aren’t shared by their close relatives.
Dr. Mary-Claire King of the University of Washington in Seattle and colleagues set out to
identify spontaneous genetic mutations in people with schizophrenia and
to assess where and when in the brain these misspelled genes are turned on, or expressed.
The study was funded in part by NIH’s National Institute of Mental Health (NIMH). The results were published in the August 1, 2013, issue of Cell.
The researchers sequenced the exomes (protein-coding DNA regions) of 399 people—105 with schizophrenia plus their unaffected parents and siblings. Gene variations
that were found in a person with schizophrenia but not in either parent were considered spontaneous.
The likelihood of having a spontaneous mutation was associated with
the age of the father in both affected and unaffected siblings.
Significantly more mutations were found in people
whose fathers were 33-45 years at the time of conception compared to 19-28 years.
Among people with schizophrenia, the scientists identified
54 genes with spontaneous mutations
predicted to cause damage to the function of the protein they encode.
The researchers used newly available database resources that show
where in the brain and when during development genes are expressed.
The genes form an interconnected expression network with many more connections than
that of the genes with spontaneous damaging mutations in unaffected siblings.
The spontaneously mutated genes in people with schizophrenia
were expressed in the prefrontal cortex, a region in the front of the brain.
The genes are known to be involved in important pathways in brain development. Fifty of these genes were active
mainly during the period of fetal development.
“Processes critical for the brain’s development can be revealed by the mutations that disrupt them,” King says. “Mutations can lead to loss of integrity of a whole pathway,
not just of a single gene.”
These findings support the concept that schizophrenia may result, in part, from
disruptions in development in the prefrontal cortex during fetal development.
James E. Darnell’s “Reflections”
A brief history of the discovery of RNA and its role in transcription — peppered with career advice
By Joseph P. Tiano
James Darnell begins his Journal of Biological Chemistry “Reflections” article by saying, “graduate students these days
have to swim in a sea virtually turgid with the daily avalanche of new information and
may be momentarily too overwhelmed to listen to the aging.
I firmly believe how we learned what we know can provide useful guidance for how and what a newcomer will learn.” Considering his remarkable discoveries in
RNA processing and eukaryotic transcriptional regulation
spanning 60 years of research, Darnell’s advice should be cherished. In his second year at medical school at Washington University School of Medicine in St. Louis, while
studying streptococcal disease in Robert J. Glaser’s laboratory, Darnell realized he “loved doing the experiments” and had his first “career advancement event.”
He and technician Barbara Pesch discovered that in vivo penicillin treatment killed streptococci only in the exponential growth phase and not in the stationary phase. These
results were published in the Journal of Clinical Investigation and earned Darnell an interview with Harry Eagle at the National Institutes of Health.
Darnell arrived at the NIH in 1956, shortly after Eagle shifted his research interest to developing his minimal essential cell culture medium, still used. Eagle, then studying cell metabolism, suggested that Darnell take up a side project on poliovirus replication in mammalian cells in collaboration with Robert I. DeMars. DeMars’ Ph.D.
adviser was also James Watson’s mentor, so Darnell met Watson, who invited him to give a talk at Harvard University, which led to an assistant professor position
at the MIT under Salvador Luria. A take-home message is to embrace side projects, because you never know where they may lead: this project helped to shape
his career.
Darnell arrived in Boston in 1961. Following the discovery of DNA’s structure in 1953, the world of molecular biology was turning to RNA in an effort to understand how
proteins are made. Darnell’s background in virology (it was discovered in 1960 that viruses used RNA to replicate) was ideal for the aim of his first independent lab:
exploring mRNA in animal cells grown in culture. While at MIT, he developed a new technique for purifying RNA along with making other observations
suggesting that nonribosomal cytoplasmic RNA may be involved in protein synthesis.
When Darnell moved to Albert Einstein College of Medicine for full professorship in 1964, it was hypothesized that heterogenous nuclear RNA was a precursor to mRNA.
At Einstein, Darnell discovered RNA processing of pre-tRNAs and demonstrated for the first time
that a specific nuclear RNA could represent a possible specific mRNA precursor.
In 1967 Darnell took a position at Columbia University, and it was there that he discovered (simultaneously with two other labs) that
mRNA contained a polyadenosine tail.
The three groups all published their results together in the Proceedings of the National Academy of Sciences in 1971. Shortly afterward, Darnell made his final career move
four short miles down the street to Rockefeller University in 1974.
Over the next 35-plus years at Rockefeller, Darnell never strayed from his original research question: How do mammalian cells make and control the making of different
mRNAs? His work was instrumental in the collaborative discovery of
splicing in the late 1970s and
in identifying and cloning many transcriptional activators.
Perhaps his greatest contribution during this time, with the help of Ernest Knight, was
the discovery and cloning of the signal transducers and activators of transcription (STAT) proteins.
And with George Stark, Andy Wilks and John Krowlewski, he described
cytokine signaling via the JAK-STAT pathway.
Darnell closes his “Reflections” with perhaps his best advice: Do not get too wrapped up in your own work, because “we are all needed and we are all in this together.”
GxGD proteases are a family of intramembranous enzymes capable of hydrolyzing
the transmembrane domain of some integral membrane proteins.
The GxGD family is one of the three families of
intramembrane-cleaving proteases discovered so far (along with the rhomboid and site-2 protease) and
includes the γ-secretase and the signal peptide peptidase.
Although only recently discovered, a number of functions in human pathology and in numerous other biological processes
have been attributed to γ-secretase and SPP.
Taisuke Tomita and Takeshi Iwatsubo of the University of Tokyo highlighted the latest findings on the structure and function of γ-secretase and SPP
in a recent minireview in The Journal of Biological Chemistry.
γ-secretase is involved in cleaving the amyloid-β precursor protein, thus producing amyloid-β peptide,
the main component of senile plaques in Alzheimer’s disease patients’ brains. The complete structure of mammalian γ-secretase is not yet known; however,
Tomita and Iwatsubo note that biochemical analyses have revealed it to be a multisubunit protein complex.
Its catalytic subunit is presenilin, an aspartyl protease.
In vitro and in vivo functional and chemical biology analyses have revealed that
presenilin is a modulator and mandatory component of the γ-secretase–mediated cleavage of APP.
Genetic studies have identified three other components required for γ-secretase activity:
nicastrin,
anterior pharynx defective 1 and
presenilin enhancer 2.
By coexpression of presenilin with the other three components, the authors managed to
reconstitute γ-secretase activity.
Tomita and Iwatsubo determined using the substituted cysteine accessibility method and by topological analyses, that
the catalytic aspartates are located at the center of the nine transmembrane domains of presenilin,
by revealing the exact location of the enzyme’s catalytic site.
The minireview also describes in detail the formerly enigmatic mechanism of γ-secretase mediated cleavage.
SPP, an enzyme that cleaves remnant signal peptides in the membrane
during the biogenesis of membrane proteins and
signal peptides from major histocompatibility complex type I,
also is involved in the maturation of proteins of the hepatitis C virus and GB virus B.
Bioinformatics methods have revealed in fruit flies and mammals four SPP-like proteins,
two of which are involved in immunological processes.
By using γ-secretase inhibitors and modulators, it has been confirmed
that SPP shares a similar GxGD active site and proteolytic activity with γ-secretase.
Upon purification of the human SPP protein with the baculovirus/Sf9 cell system,
single-particle analysis revealed further structural and functional details.
HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection
Experimental and computational evidence suggests that
HLAs preferentially bind conserved regions of viral proteins, a concept we term “targeting efficiency,” and that
this preference may provide improved clearance of infection in several viral systems.
To test this hypothesis, T-cell responses to A/H1N1 (2009) were measured from peripheral blood mononuclear cells obtained from a household cohort study
performed during the 2009–2010 influenza season. We found that HLA targeting efficiency scores significantly correlated with
A further population-based analysis found that the carriage frequencies of the alleles with the lowest targeting efficiencies, A*24,
were associated with pH1N1 mortality (r = 0.37, P = 0.031) and
are common in certain indigenous populations in which increased pH1N1 morbidity has been reported.
HLA efficiency scores and HLA use are associated with CD8 T-cell magnitude in humans after influenza infection.
The computational tools used in this study may be useful predictors of potential morbidity and
identify immunologic differences of new variant influenza strains
more accurately than evolutionary sequence comparisons.
Population-based studies of the relative frequency of these alleles in severe vs. mild influenza cases
might advance clinical practices for severe H1N1 infections among genetically susceptible populations.
Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ.
Cold Spring Harbor Symposia on Quantitative Biology 11/2011; 76:235-46. http://dx.doi.org:/10.1101/sqb.2011.76.010694
Most diseases result in metabolic changes. In many cases, these changes play a causative role in disease progression. By identifying pathological metabolic changes,
metabolomics can point to potential new sites for therapeutic intervention.
Particularly promising enzymatic targets are those that
carry increased flux in the disease state.
Definitive assessment of flux requires the use of isotope tracers. Here we present techniques for
finding new drug targets using metabolomics and isotope tracers.
The utility of these methods is exemplified in the study of three different viral pathogens. For influenza A and herpes simplex virus,
metabolomic analysis of infected versus mock-infected cells revealed
dramatic concentration changes around the current antiviral target enzymes.
Similar analysis of human-cytomegalovirus-infected cells, however, found the greatest changes
in a region of metabolism unrelated to the current antiviral target.
Instead, it pointed to the tricarboxylic acid (TCA) cycle and
its efflux to feed fatty acid biosynthesis as a potential preferred target.
Isotope tracer studies revealed that cytomegalovirus greatly increases flux through
the key fatty acid metabolic enzyme acetyl-coenzyme A carboxylase.
Inhibition of this enzyme blocks human cytomegalovirus replication.
Examples where metabolomics has contributed to identification of anticancer drug targets are also discussed. Eventual proof of the value of
metabolomics as a drug target discovery strategy will be
successful clinical development of therapeutics hitting these new targets.
Related References
Use of metabolic pathway flux information in targeted cancer drug design. Drug Discovery Today: Therapeutic Strategies 1:435-443, 2004.
Detection of resistance to imatinib by metabolic profiling: clinical and drug development implications. Am J Pharmacogenomics. 2005;5(5):293-302. Review. PMID: 16196499
Medicinal chemistry, metabolic profiling and drug target discovery: a role for metabolic profiling in reverse pharmacology and chemical genetics.
Mini Rev Med Chem. 2005 Jan;5(1):13-20. Review. PMID: 15638788 [PubMed – indexed for MEDLINE] Related citations
Development of Tracer-Based Metabolomics and its Implications for the Pharmaceutical Industry. Int J Pharm Med 2007; 21 (3): 217-224.
Use of metabolic pathway flux information in anticancer drug design. Ernst Schering Found Symp Proc. 2007;(4):189-203. Review. PMID: 18811058
Pharmacological targeting of glucagon and glucagon-like peptide 1 receptors has different effects on energy state and glucose homeostasis in diet-induced obese mice. J Pharmacol Exp Ther. 2011 Jul;338(1):70-81. http://dx.doi.org:/10.1124/jpet.111.179986. PMID: 21471191
Single valproic acid treatment inhibits glycogen and RNA ribose turnover while disrupting glucose-derived cholesterol synthesis in liver as revealed by the
[U-C(6)]-d-glucose tracer in mice. Metabolomics. 2009 Sep;5(3):336-345. PMID: 19718458
Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 2013 Jul;27(7):2845-54. http://dx.doi.org:/10.1096/fj.12-216929. PMID: 23515442
Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery
The change in drug discovery strategy from ‘classical’ function-first approaches (in which the assay of drug function was at the tissue or organism level),
with mechanistic studies potentially coming later, to more-recent target-based approaches where initial assays usually involve assessing the interactions
of drugs with specified (and often cloned, recombinant) proteins in vitro. In the latter cases, effects in vivo are assessed later, with concomitantly high levels of attrition.
Arguably the two chief hallmarks of the systems biology approach are:
(i) that we seek to make mathematical models of our systems iteratively or in parallel with well-designed ‘wet’ experiments, and
(ii) that we do not necessarily start with a hypothesis but measure as many things as possible (the ’omes) and
let the data tell us the hypothesis that best fits and describes them.
Although metabolism was once seen as something of a Cinderella subject,
there are fundamental reasons to do with the organisation of biochemical networks as
to why the metabol(om)ic level – now in fact seen as the ‘apogee’ of the ’omics trilogy –
is indeed likely to be far more discriminating than are
changes in the transcriptome or proteome.
The next two subsections deal with these points and Fig. 2 summarises the paper in the form of a Mind Map.
Despite the advent of new drug classes, the global epidemic of cardiometabolic disease has not abated. Continuing
unmet medical needs remain a major driver for new research.
Drug discovery approaches in this field have mirrored industry trends, leading to a recent
increase in the number of molecules entering development.
However, worrisome trends and newer hurdles are also apparent. The history of two newer drug classes—
glucagon-like peptide-1 receptor agonists and
dipeptidyl peptidase-4 inhibitors—
illustrates both progress and challenges. Future success requires that researchers learn from these experiences and
continue to explore and apply new technology platforms and research paradigms.
The global epidemic of obesity and diabetes continues to progress relentlessly. The International Diabetes Federation predicts an even greater diabetes burden (>430 million people afflicted) by 2030, which will disproportionately affect developing nations (International Diabetes Federation, 2011). Yet
existing drug classes for diabetes, obesity, and comorbid cardiovascular (CV) conditions have substantial limitations.
Currently available prescription drugs for treatment of hyperglycemia in patients with type 2 diabetes (Table 1) have notable shortcomings. In general,
Therefore, clinicians must often use combination therapy, adding additional agents over time. Ultimately many patients will need to use insulin—a therapeutic class first introduced in 1922. Most existing agents also have
issues around safety and tolerability as well as dosing convenience (which can impact patient compliance).
Pharmacometabolomics, also known as pharmacometabonomics, is a field which stems from metabolomics,
the quantification and analysis of metabolites produced by the body.
It refers to the direct measurement of metabolites in an individual’s bodily fluids, in order to









![Ultra-sensitive bead-based Simoa technology detects residual human A[beta] in hippocampal extracts of inoculated App-/- mice.](https://i0.wp.com/www.nature.com/neuro/journal/v18/n11/carousel/nn.4117-SF1.jpg?resize=245%2C200)


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
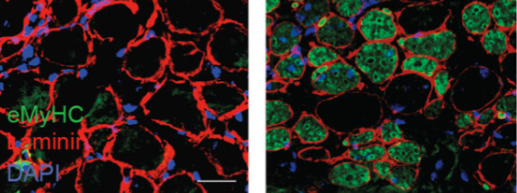{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}