Summary and Perspectives: Impairments in Pathological States: Endocrine Disorders, Stress Hypermetabolism and Cancer
Author and Curator: Larry H. Bernstein, MD, FCAP

Article ID #160: Summary and Perspectives: Impairments in Pathological States: Endocrine Disorders, Stress Hypermetabolism and Cancer. Published on 11/9/2014
WordCloud Image Produced by Adam Tubman
This summary is the last of a series on the impact of transcriptomics, proteomics, and metabolomics on disease investigation, and the sorting and integration of genomic signatures and metabolic signatures to explain phenotypic relationships in variability and individuality of response to disease expression and how this leads to pharmaceutical discovery and personalized medicine. We have unquestionably better tools at our disposal than has ever existed in the history of mankind, and an enormous knowledge-base that has to be accessed. I shall conclude here these discussions with the powerful contribution to and current knowledge pertaining to biochemistry, metabolism, protein-interactions, signaling, and the application of the -OMICS to diseases and drug discovery at this time.
The Ever-Transcendent Cell
Deriving physiologic first principles By John S. Torday | The Scientist Nov 1, 2014
http://www.the-scientist.com/?articles.view/articleNo/41282/title/The-Ever-Transcendent-Cell/
Both the developmental and phylogenetic histories of an organism describe the evolution of physiology—the complex of metabolic pathways that govern the function of an organism as a whole. The necessity of establishing and maintaining homeostatic mechanisms began at the cellular level, with the very first cells, and homeostasis provides the underlying selection pressure fueling evolution.
While the events leading to the formation of the first functioning cell are debatable, a critical one was certainly the formation of simple lipid-enclosed vesicles, which provided a protected space for the evolution of metabolic pathways. Protocells evolved from a common ancestor that experienced environmental stresses early in the history of cellular development, such as acidic ocean conditions and low atmospheric oxygen levels, which shaped the evolution of metabolism.
The reduction of evolution to cell biology may answer the perennially unresolved question of why organisms return to their unicellular origins during the life cycle.
As primitive protocells evolved to form prokaryotes and, much later, eukaryotes, changes to the cell membrane occurred that were critical to the maintenance of chemiosmosis, the generation of bioenergy through the partitioning of ions. The incorporation of cholesterol into the plasma membrane surrounding primitive eukaryotic cells marked the beginning of their differentiation from prokaryotes. Cholesterol imparted more fluidity to eukaryotic cell membranes, enhancing functionality by increasing motility and endocytosis. Membrane deformability also allowed for increased gas exchange.
Acidification of the oceans by atmospheric carbon dioxide generated high intracellular calcium ion concentrations in primitive aquatic eukaryotes, which had to be lowered to prevent toxic effects, namely the aggregation of nucleotides, proteins, and lipids. The early cells achieved this by the evolution of calcium channels composed of cholesterol embedded within the cell’s plasma membrane, and of internal membranes, such as that of the endoplasmic reticulum, peroxisomes, and other cytoplasmic organelles, which hosted intracellular chemiosmosis and helped regulate calcium.
As eukaryotes thrived, they experienced increasingly competitive pressure for metabolic efficiency. Engulfed bacteria, assimilated as mitochondria, provided more bioenergy. As the evolution of eukaryotic organisms progressed, metabolic cooperation evolved, perhaps to enable competition with biofilm-forming, quorum-sensing prokaryotes. The subsequent appearance of multicellular eukaryotes expressing cellular growth factors and their respective receptors facilitated cell-cell signaling, forming the basis for an explosion of multicellular eukaryote evolution, culminating in the metazoans.
Casting a cellular perspective on evolution highlights the integration of genotype and phenotype. Starting from the protocell membrane, the functional homolog for all complex metazoan organs, it offers a way of experimentally determining the role of genes that fostered evolution based on the ontogeny and phylogeny of cellular processes that can be traced back, in some cases, to our last universal common ancestor. ….
As eukaryotes thrived, they experienced increasingly competitive pressure for metabolic efficiency. Engulfed bacteria, assimilated as mitochondria, provided more bioenergy. As the evolution of eukaryotic organisms progressed, metabolic cooperation evolved, perhaps to enable competition with biofilm-forming, quorum-sensing prokaryotes. The subsequent appearance of multicellular eukaryotes expressing cellular growth factors and their respective receptors facilitated cell-cell signaling, forming the basis for an explosion of multicellular eukaryote evolution, culminating in the metazoans.
Casting a cellular perspective on evolution highlights the integration of genotype and phenotype. Starting from the protocell membrane, the functional homolog for all complex metazoan organs, it offers a way of experimentally determining the role of genes that fostered evolution based on the ontogeny and phylogeny of cellular processes that can be traced back, in some cases, to our last universal common ancestor.
Given that the unicellular toolkit is complete with all the traits necessary for forming multicellular organisms (Science, 301:361-63, 2003), it is distinctly possible that metazoans are merely permutations of the unicellular body plan. That scenario would clarify a lot of puzzling biology: molecular commonalities between the skin, lung, gut, and brain that affect physiology and pathophysiology exist because the cell membranes of unicellular organisms perform the equivalents of these tissue functions, and the existence of pleiotropy—one gene affecting many phenotypes—may be a consequence of the common unicellular source for all complex biologic traits. …
The cell-molecular homeostatic model for evolution and stability addresses how the external environment generates homeostasis developmentally at the cellular level. It also determines homeostatic set points in adaptation to the environment through specific effectors, such as growth factors and their receptors, second messengers, inflammatory mediators, crossover mutations, and gene duplications. This is a highly mechanistic, heritable, plastic process that lends itself to understanding evolution at the cellular, tissue, organ, system, and population levels, mediated by physiologically linked mechanisms throughout, without having to invoke random, chance mechanisms to bridge different scales of evolutionary change. In other words, it is an integrated mechanism that can often be traced all the way back to its unicellular origins.
The switch from swim bladder to lung as vertebrates moved from water to land is proof of principle that stress-induced evolution in metazoans can be understood from changes at the cellular level.
http://www.the-scientist.com/Nov2014/TE_21.jpg
A MECHANISTIC BASIS FOR LUNG DEVELOPMENT: Stress from periodic atmospheric hypoxia (1) during vertebrate adaptation to land enhances positive selection of the stretch-regulated parathyroid hormone-related protein (PTHrP) in the pituitary and adrenal glands. In the pituitary (2), PTHrP signaling upregulates the release of adrenocorticotropic hormone (ACTH) (3), which stimulates the release of glucocorticoids (GC) by the adrenal gland (4). In the adrenal gland, PTHrP signaling also stimulates glucocorticoid production of adrenaline (5), which in turn affects the secretion of lung surfactant, the distension of alveoli, and the perfusion of alveolar capillaries (6). PTHrP signaling integrates the inflation and deflation of the alveoli with surfactant production and capillary perfusion. THE SCIENTIST STAFF
From a cell-cell signaling perspective, two critical duplications in genes coding for cell-surface receptors occurred during this period of water-to-land transition—in the stretch-regulated parathyroid hormone-related protein (PTHrP) receptor gene and the β adrenergic (βA) receptor gene. These gene duplications can be disassembled by following their effects on vertebrate physiology backwards over phylogeny. PTHrP signaling is necessary for traits specifically relevant to land adaptation: calcification of bone, skin barrier formation, and the inflation and distention of lung alveoli. Microvascular shear stress in PTHrP-expressing organs such as bone, skin, kidney, and lung would have favored duplication of the PTHrP receptor, since sheer stress generates radical oxygen species (ROS) known to have this effect and PTHrP is a potent vasodilator, acting as an epistatic balancing selection for this constraint.
Positive selection for PTHrP signaling also evolved in the pituitary and adrenal cortex (see figure on this page), stimulating the secretion of ACTH and corticoids, respectively, in response to the stress of land adaptation. This cascade amplified adrenaline production by the adrenal medulla, since corticoids passing through it enzymatically stimulate adrenaline synthesis. Positive selection for this functional trait may have resulted from hypoxic stress that arose during global episodes of atmospheric hypoxia over geologic time. Since hypoxia is the most potent physiologic stressor, such transient oxygen deficiencies would have been acutely alleviated by increasing adrenaline levels, which would have stimulated alveolar surfactant production, increasing gas exchange by facilitating the distension of the alveoli. Over time, increased alveolar distension would have generated more alveoli by stimulating PTHrP secretion, impelling evolution of the alveolar bed of the lung.
This scenario similarly explains βA receptor gene duplication, since increased density of the βA receptor within the alveolar walls was necessary for relieving another constraint during the evolution of the lung in adaptation to land: the bottleneck created by the existence of a common mechanism for blood pressure control in both the lung alveoli and the systemic blood pressure. The pulmonary vasculature was constrained by its ability to withstand the swings in pressure caused by the systemic perfusion necessary to sustain all the other vital organs. PTHrP is a potent vasodilator, subserving the blood pressure constraint, but eventually the βA receptors evolved to coordinate blood pressure in both the lung and the periphery.
Gut Microbiome Heritability
Analyzing data from a large twin study, researchers have homed in on how host genetics can shape the gut microbiome.
By Tracy Vence | The Scientist Nov 6, 2014
Previous research suggested host genetic variation can influence microbial phenotype, but an analysis of data from a large twin study published in Cell today (November 6) solidifies the connection between human genotype and the composition of the gut microbiome. Studying more than 1,000 fecal samples from 416 monozygotic and dizygotic twin pairs, Cornell University’s Ruth Ley and her colleagues have homed in on one bacterial taxon, the family Christensenellaceae, as the most highly heritable group of microbes in the human gut. The researchers also found that Christensenellaceae—which was first described just two years ago—is central to a network of co-occurring heritable microbes that is associated with lean body mass index (BMI). …
Of particular interest was the family Christensenellaceae, which was the most heritable taxon among those identified in the team’s analysis of fecal samples obtained from the TwinsUK study population.
While microbiologists had previously detected 16S rRNA sequences belonging to Christensenellaceae in the human microbiome, the family wasn’t named until 2012. “People hadn’t looked into it, partly because it didn’t have a name . . . it sort of flew under the radar,” said Ley.
Ley and her colleagues discovered that Christensenellaceae appears to be the hub in a network of co-occurring heritable taxa, which—among TwinsUK participants—was associated with low BMI. The researchers also found that Christensenellaceae had been found at greater abundance in low-BMI twins in older studies.
To interrogate the effects of Christensenellaceae on host metabolic phenotype, the Ley’s team introduced lean and obese human fecal samples into germ-free mice. They found animals that received lean fecal samples containing more Christensenellaceae showed reduced weight gain compared with their counterparts. And treatment of mice that had obesity-associated microbiomes with one member of the Christensenellaceae family, Christensenella minuta, led to reduced weight gain. …
Ley and her colleagues are now focusing on the host alleles underlying the heritability of the gut microbiome. “We’re running a genome-wide association analysis to try to find genes—particular variants of genes—that might associate with higher levels of these highly heritable microbiota. . . . Hopefully that will point us to possible reasons they’re heritable,” she said. “The genes will guide us toward understanding how these relationships are maintained between host genotype and microbiome composition.”
J.K. Goodrich et al., “Human genetics shape the gut microbiome,” Cell, http://dx.doi.org:/10.1016/j.cell.2014.09.053, 2014.
Light-Operated Drugs
Scientists create a photosensitive pharmaceutical to target a glutamate receptor.
By Ruth Williams | The Scentist Nov 1, 2014
http://www.the-scientist.com/?articles.view/articleNo/41279/title/Light-Operated-Drugs/

light operated drugs MO1
http://www.the-scientist.com/Nov2014/MO1.jpg
The desire for temporal and spatial control of medications to minimize side effects and maximize benefits has inspired the development of light-controllable drugs, or optopharmacology. Early versions of such drugs have manipulated ion channels or protein-protein interactions, “but never, to my knowledge, G protein–coupled receptors [GPCRs], which are one of the most important pharmacological targets,” says Pau Gorostiza of the Institute for Bioengineering of Catalonia, in Barcelona.
Gorostiza has taken the first step toward filling that gap, creating a photosensitive inhibitor of the metabotropic glutamate 5 (mGlu5) receptor—a GPCR expressed in neurons and implicated in a number of neurological and psychiatric disorders. The new mGlu5 inhibitor—called alloswitch-1—is based on a known mGlu receptor inhibitor, but the simple addition of a light-responsive appendage, as had been done for other photosensitive drugs, wasn’t an option. The binding site on mGlu5 is “extremely tight,” explains Gorostiza, and would not accommodate a differently shaped molecule. Instead, alloswitch-1 has an intrinsic light-responsive element.
In a human cell line, the drug was active under dim light conditions, switched off by exposure to violet light, and switched back on by green light. When Gorostiza’s team administered alloswitch-1 to tadpoles, switching between violet and green light made the animals stop and start swimming, respectively.
The fact that alloswitch-1 is constitutively active and switched off by light is not ideal, says Gorostiza. “If you are thinking of therapy, then in principle you would prefer the opposite,” an “on” switch. Indeed, tweaks are required before alloswitch-1 could be a useful drug or research tool, says Stefan Herlitze, who studies ion channels at Ruhr-Universität Bochum in Germany. But, he adds, “as a proof of principle it is great.” (Nat Chem Biol, http://dx.doi.org:/10.1038/nchembio.1612, 2014)
Enhanced Enhancers
The recent discovery of super-enhancers may offer new drug targets for a range of diseases.
By Eric Olson | The Scientist Nov 1, 2014
http://www.the-scientist.com/?articles.view/articleNo/41281/title/Enhanced-Enhancers/
To understand disease processes, scientists often focus on unraveling how gene expression in disease-associated cells is altered. Increases or decreases in transcription—as dictated by a regulatory stretch of DNA called an enhancer, which serves as a binding site for transcription factors and associated proteins—can produce an aberrant composition of proteins, metabolites, and signaling molecules that drives pathologic states. Identifying the root causes of these changes may lead to new therapeutic approaches for many different diseases.
Although few therapies for human diseases aim to alter gene expression, the outstanding examples—including antiestrogens for hormone-positive breast cancer, antiandrogens for prostate cancer, and PPAR-γ agonists for type 2 diabetes—demonstrate the benefits that can be achieved through targeting gene-control mechanisms. Now, thanks to recent papers from laboratories at MIT, Harvard, and the National Institutes of Health, researchers have a new, much bigger transcriptional target: large DNA regions known as super-enhancers or stretch-enhancers. Already, work on super-enhancers is providing insights into how gene-expression programs are established and maintained, and how they may go awry in disease. Such research promises to open new avenues for discovering medicines for diseases where novel approaches are sorely needed.
Super-enhancers cover stretches of DNA that are 10- to 100-fold longer and about 10-fold less abundant in the genome than typical enhancer regions (Cell, 153:307-19, 2013). They also appear to bind a large percentage of the transcriptional machinery compared to typical enhancers, allowing them to better establish and enforce cell-type specific transcriptional programs (Cell, 153:320-34, 2013).
Super-enhancers are closely associated with genes that dictate cell identity, including those for cell-type–specific master regulatory transcription factors. This observation led to the intriguing hypothesis that cells with a pathologic identity, such as cancer cells, have an altered gene expression program driven by the loss, gain, or altered function of super-enhancers.
Sure enough, by mapping the genome-wide location of super-enhancers in several cancer cell lines and from patients’ tumor cells, we and others have demonstrated that genes located near super-enhancers are involved in processes that underlie tumorigenesis, such as cell proliferation, signaling, and apoptosis.
Super-enhancers cover stretches of DNA that are 10- to 100-fold longer and about 10-fold less abundant in the genome than typical enhancer regions.
Genome-wide association studies (GWAS) have found that disease- and trait-associated genetic variants often occur in greater numbers in super-enhancers (compared to typical enhancers) in cell types involved in the disease or trait of interest (Cell, 155:934-47, 2013). For example, an enrichment of fasting glucose–associated single nucleotide polymorphisms (SNPs) was found in the stretch-enhancers of pancreatic islet cells (PNAS, 110:17921-26, 2013). Given that some 90 percent of reported disease-associated SNPs are located in noncoding regions, super-enhancer maps may be extremely valuable in assigning functional significance to GWAS variants and identifying target pathways.
Because only 1 to 2 percent of active genes are physically linked to a super-enhancer, mapping the locations of super-enhancers can be used to pinpoint the small number of genes that may drive the biology of that cell. Differential super-enhancer maps that compare normal cells to diseased cells can be used to unravel the gene-control circuitry and identify new molecular targets, in much the same way that somatic mutations in tumor cells can point to oncogenic drivers in cancer. This approach is especially attractive in diseases for which an incomplete understanding of the pathogenic mechanisms has been a barrier to discovering effective new therapies.
Another therapeutic approach could be to disrupt the formation or function of super-enhancers by interfering with their associated protein components. This strategy could make it possible to downregulate multiple disease-associated genes through a single molecular intervention. A group of Boston-area researchers recently published support for this concept when they described inhibited expression of cancer-specific genes, leading to a decrease in cancer cell growth, by using a small molecule inhibitor to knock down a super-enhancer component called BRD4 (Cancer Cell, 24:777-90, 2013). More recently, another group showed that expression of the RUNX1 transcription factor, involved in a form of T-cell leukemia, can be diminished by treating cells with an inhibitor of a transcriptional kinase that is present at the RUNX1 super-enhancer (Nature, 511:616-20, 2014).
Fungal effector Ecp6 outcompetes host immune receptor for chitin binding through intrachain LysM dimerization
Andrea Sánchez-Vallet, et al. eLife 2013;2:e00790 http://elifesciences.org/content/2/e00790#sthash.LnqVMJ9p.dpuf

LysM effector
http://img.scoop.it/ZniCRKQSvJOG18fHbb4p0Tl72eJkfbmt4t8yenImKBVvK0kTmF0xjctABnaLJIm9
While host immune receptors
- detect pathogen-associated molecular patterns to activate immunity,
- pathogens attempt to deregulate host immunity through secreted effectors.
Fungi employ LysM effectors to prevent
- recognition of cell wall-derived chitin by host immune receptors
Structural analysis of the LysM effector Ecp6 of
- the fungal tomato pathogen Cladosporium fulvum reveals
- a novel mechanism for chitin binding,
- mediated by intrachain LysM dimerization,
leading to a chitin-binding groove that is deeply buried in the effector protein.
This composite binding site involves
- two of the three LysMs of Ecp6 and
- mediates chitin binding with ultra-high (pM) affinity.
The remaining singular LysM domain of Ecp6 binds chitin with
- low micromolar affinity but can nevertheless still perturb chitin-triggered immunity.
Conceivably, the perturbation by this LysM domain is not established through chitin sequestration but possibly through interference with the host immune receptor complex.
Mutated Genes in Schizophrenia Map to Brain Networks
From www.nih.gov – Sep 3, 2013
Previous studies have shown that many people with schizophrenia have de novo, or new, genetic mutations. These misspellings in a gene’s DNA sequence
- occur spontaneously and so aren’t shared by their close relatives.
Dr. Mary-Claire King of the University of Washington in Seattle and colleagues set out to
- identify spontaneous genetic mutations in people with schizophrenia and
- to assess where and when in the brain these misspelled genes are turned on, or expressed.
The study was funded in part by NIH’s National Institute of Mental Health (NIMH). The results were published in the August 1, 2013, issue of Cell.
The researchers sequenced the exomes (protein-coding DNA regions) of 399 people—105 with schizophrenia plus their unaffected parents and siblings. Gene variations
that were found in a person with schizophrenia but not in either parent were considered spontaneous.
The likelihood of having a spontaneous mutation was associated with
- the age of the father in both affected and unaffected siblings.
Significantly more mutations were found in people
- whose fathers were 33-45 years at the time of conception compared to 19-28 years.
Among people with schizophrenia, the scientists identified
- 54 genes with spontaneous mutations
- predicted to cause damage to the function of the protein they encode.
The researchers used newly available database resources that show
- where in the brain and when during development genes are expressed.
The genes form an interconnected expression network with many more connections than
- that of the genes with spontaneous damaging mutations in unaffected siblings.
The spontaneously mutated genes in people with schizophrenia
- were expressed in the prefrontal cortex, a region in the front of the brain.
The genes are known to be involved in important pathways in brain development. Fifty of these genes were active
- mainly during the period of fetal development.
“Processes critical for the brain’s development can be revealed by the mutations that disrupt them,” King says. “Mutations can lead to loss of integrity of a whole pathway,
not just of a single gene.”
These findings support the concept that schizophrenia may result, in part, from
- disruptions in development in the prefrontal cortex during fetal development.
James E. Darnell’s “Reflections”
A brief history of the discovery of RNA and its role in transcription — peppered with career advice
By Joseph P. Tiano
James Darnell begins his Journal of Biological Chemistry “Reflections” article by saying, “graduate students these days
- have to swim in a sea virtually turgid with the daily avalanche of new information and
- may be momentarily too overwhelmed to listen to the aging.
I firmly believe how we learned what we know can provide useful guidance for how and what a newcomer will learn.” Considering his remarkable discoveries in
- RNA processing and eukaryotic transcriptional regulation
spanning 60 years of research, Darnell’s advice should be cherished. In his second year at medical school at Washington University School of Medicine in St. Louis, while
studying streptococcal disease in Robert J. Glaser’s laboratory, Darnell realized he “loved doing the experiments” and had his first “career advancement event.”
He and technician Barbara Pesch discovered that in vivo penicillin treatment killed streptococci only in the exponential growth phase and not in the stationary phase. These
results were published in the Journal of Clinical Investigation and earned Darnell an interview with Harry Eagle at the National Institutes of Health.
Darnell arrived at the NIH in 1956, shortly after Eagle shifted his research interest to developing his minimal essential cell culture medium, still used. Eagle, then studying cell metabolism, suggested that Darnell take up a side project on poliovirus replication in mammalian cells in collaboration with Robert I. DeMars. DeMars’ Ph.D.
adviser was also James Watson’s mentor, so Darnell met Watson, who invited him to give a talk at Harvard University, which led to an assistant professor position
at the MIT under Salvador Luria. A take-home message is to embrace side projects, because you never know where they may lead: this project helped to shape
his career.
Darnell arrived in Boston in 1961. Following the discovery of DNA’s structure in 1953, the world of molecular biology was turning to RNA in an effort to understand how
proteins are made. Darnell’s background in virology (it was discovered in 1960 that viruses used RNA to replicate) was ideal for the aim of his first independent lab:
exploring mRNA in animal cells grown in culture. While at MIT, he developed a new technique for purifying RNA along with making other observations
- suggesting that nonribosomal cytoplasmic RNA may be involved in protein synthesis.
When Darnell moved to Albert Einstein College of Medicine for full professorship in 1964, it was hypothesized that heterogenous nuclear RNA was a precursor to mRNA.
At Einstein, Darnell discovered RNA processing of pre-tRNAs and demonstrated for the first time
- that a specific nuclear RNA could represent a possible specific mRNA precursor.
In 1967 Darnell took a position at Columbia University, and it was there that he discovered (simultaneously with two other labs) that
- mRNA contained a polyadenosine tail.
The three groups all published their results together in the Proceedings of the National Academy of Sciences in 1971. Shortly afterward, Darnell made his final career move
four short miles down the street to Rockefeller University in 1974.
Over the next 35-plus years at Rockefeller, Darnell never strayed from his original research question: How do mammalian cells make and control the making of different
mRNAs? His work was instrumental in the collaborative discovery of
- splicing in the late 1970s and
- in identifying and cloning many transcriptional activators.
Perhaps his greatest contribution during this time, with the help of Ernest Knight, was
- the discovery and cloning of the signal transducers and activators of transcription (STAT) proteins.
And with George Stark, Andy Wilks and John Krowlewski, he described
- cytokine signaling via the JAK-STAT pathway.
Darnell closes his “Reflections” with perhaps his best advice: Do not get too wrapped up in your own work, because “we are all needed and we are all in this together.”

Darnell Reflections – James_Darnell
http://www.asbmb.org/assets/0/366/418/428/85528/85529/85530/8758cb87-84ff-42d6-8aea-96fda4031a1b.jpg
Recent findings on presenilins and signal peptide peptidase
By Dinu-Valantin Bălănescu

γ-secretase and SPP
Fig. 1 from the minireview shows a schematic depiction of γ-secretase and SPP
http://www.asbmb.org/assets/0/366/418/428/85528/85529/85530/c2de032a-daad-41e5-ba19-87a17bd26362.png
GxGD proteases are a family of intramembranous enzymes capable of hydrolyzing
- the transmembrane domain of some integral membrane proteins.
The GxGD family is one of the three families of
- intramembrane-cleaving proteases discovered so far (along with the rhomboid and site-2 protease) and
- includes the γ-secretase and the signal peptide peptidase.
Although only recently discovered, a number of functions in human pathology and in numerous other biological processes
- have been attributed to γ-secretase and SPP.
Taisuke Tomita and Takeshi Iwatsubo of the University of Tokyo highlighted the latest findings on the structure and function of γ-secretase and SPP
in a recent minireview in The Journal of Biological Chemistry.
- γ-secretase is involved in cleaving the amyloid-β precursor protein, thus producing amyloid-β peptide,
the main component of senile plaques in Alzheimer’s disease patients’ brains. The complete structure of mammalian γ-secretase is not yet known; however,
Tomita and Iwatsubo note that biochemical analyses have revealed it to be a multisubunit protein complex.
- Its catalytic subunit is presenilin, an aspartyl protease.
In vitro and in vivo functional and chemical biology analyses have revealed that
- presenilin is a modulator and mandatory component of the γ-secretase–mediated cleavage of APP.
Genetic studies have identified three other components required for γ-secretase activity:
- nicastrin,
- anterior pharynx defective 1 and
- presenilin enhancer 2.
By coexpression of presenilin with the other three components, the authors managed to
- reconstitute γ-secretase activity.
Tomita and Iwatsubo determined using the substituted cysteine accessibility method and by topological analyses, that
- the catalytic aspartates are located at the center of the nine transmembrane domains of presenilin,
- by revealing the exact location of the enzyme’s catalytic site.
The minireview also describes in detail the formerly enigmatic mechanism of γ-secretase mediated cleavage.
SPP, an enzyme that cleaves remnant signal peptides in the membrane
- during the biogenesis of membrane proteins and
- signal peptides from major histocompatibility complex type I,
- also is involved in the maturation of proteins of the hepatitis C virus and GB virus B.
Bioinformatics methods have revealed in fruit flies and mammals four SPP-like proteins,
- two of which are involved in immunological processes.
By using γ-secretase inhibitors and modulators, it has been confirmed
- that SPP shares a similar GxGD active site and proteolytic activity with γ-secretase.
Upon purification of the human SPP protein with the baculovirus/Sf9 cell system,
- single-particle analysis revealed further structural and functional details.
HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection
From www.pnas.org – Sep 3, 2013 4:24 PM
Experimental and computational evidence suggests that
- HLAs preferentially bind conserved regions of viral proteins, a concept we term “targeting efficiency,” and that
- this preference may provide improved clearance of infection in several viral systems.
To test this hypothesis, T-cell responses to A/H1N1 (2009) were measured from peripheral blood mononuclear cells obtained from a household cohort study
performed during the 2009–2010 influenza season. We found that HLA targeting efficiency scores significantly correlated with
- IFN-γ enzyme-linked immunosorbent spot responses (P = 0.042, multiple regression).
A further population-based analysis found that the carriage frequencies of the alleles with the lowest targeting efficiencies, A*24,
- were associated with pH1N1 mortality (r = 0.37, P = 0.031) and
- are common in certain indigenous populations in which increased pH1N1 morbidity has been reported.
HLA efficiency scores and HLA use are associated with CD8 T-cell magnitude in humans after influenza infection.
The computational tools used in this study may be useful predictors of potential morbidity and
- identify immunologic differences of new variant influenza strains
- more accurately than evolutionary sequence comparisons.
Population-based studies of the relative frequency of these alleles in severe vs. mild influenza cases
- might advance clinical practices for severe H1N1 infections among genetically susceptible populations.
Metabolomics in drug target discovery
J D Rabinowitz et al.
Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ.
Cold Spring Harbor Symposia on Quantitative Biology 11/2011; 76:235-46.
http://dx.doi.org:/10.1101/sqb.2011.76.010694
Most diseases result in metabolic changes. In many cases, these changes play a causative role in disease progression. By identifying pathological metabolic changes,
- metabolomics can point to potential new sites for therapeutic intervention.
Particularly promising enzymatic targets are those that
- carry increased flux in the disease state.
Definitive assessment of flux requires the use of isotope tracers. Here we present techniques for
- finding new drug targets using metabolomics and isotope tracers.
The utility of these methods is exemplified in the study of three different viral pathogens. For influenza A and herpes simplex virus,
- metabolomic analysis of infected versus mock-infected cells revealed
- dramatic concentration changes around the current antiviral target enzymes.
Similar analysis of human-cytomegalovirus-infected cells, however, found the greatest changes
- in a region of metabolism unrelated to the current antiviral target.
Instead, it pointed to the tricarboxylic acid (TCA) cycle and
- its efflux to feed fatty acid biosynthesis as a potential preferred target.
Isotope tracer studies revealed that cytomegalovirus greatly increases flux through
- the key fatty acid metabolic enzyme acetyl-coenzyme A carboxylase.
- Inhibition of this enzyme blocks human cytomegalovirus replication.
Examples where metabolomics has contributed to identification of anticancer drug targets are also discussed. Eventual proof of the value of
- metabolomics as a drug target discovery strategy will be
- successful clinical development of therapeutics hitting these new targets.
Related References
Use of metabolic pathway flux information in targeted cancer drug design. Drug Discovery Today: Therapeutic Strategies 1:435-443, 2004.
Detection of resistance to imatinib by metabolic profiling: clinical and drug development implications. Am J Pharmacogenomics. 2005;5(5):293-302. Review. PMID: 16196499
Medicinal chemistry, metabolic profiling and drug target discovery: a role for metabolic profiling in reverse pharmacology and chemical genetics.
Mini Rev Med Chem. 2005 Jan;5(1):13-20. Review. PMID: 15638788 [PubMed – indexed for MEDLINE] Related citations
Development of Tracer-Based Metabolomics and its Implications for the Pharmaceutical Industry. Int J Pharm Med 2007; 21 (3): 217-224.
Use of metabolic pathway flux information in anticancer drug design. Ernst Schering Found Symp Proc. 2007;(4):189-203. Review. PMID: 18811058
Pharmacological targeting of glucagon and glucagon-like peptide 1 receptors has different effects on energy state and glucose homeostasis in diet-induced obese mice. J Pharmacol Exp Ther. 2011 Jul;338(1):70-81. http://dx.doi.org:/10.1124/jpet.111.179986. PMID: 21471191
Single valproic acid treatment inhibits glycogen and RNA ribose turnover while disrupting glucose-derived cholesterol synthesis in liver as revealed by the
[U-C(6)]-d-glucose tracer in mice. Metabolomics. 2009 Sep;5(3):336-345. PMID: 19718458
Metabolic Pathways as Targets for Drug Screening, Metabolomics, Dr Ute Roessner (Ed.), ISBN: 978-953-51-0046-1, InTech, Available from: http://www.intechopen.com/books/metabolomics/metabolic-pathways-as-targets-for-drug-screening
Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 2013 Jul;27(7):2845-54.
http://dx.doi.org:/10.1096/fj.12-216929. PMID: 23515442
Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery
Drug Discov. Today 19 (2014), 171–182 http://dx.doi.org:/10.1016/j.drudis.2013.07.014
Highlights
- We now have metabolic network models; the metabolome is represented by their nodes.
- Metabolite levels are sensitive to changes in enzyme activities.
- Drugs hitchhike on metabolite transporters to get into and out of cells.
- The consensus network Recon2 represents the present state of the art, and has predictive power.
- Constraint-based modelling relates network structure to metabolic fluxes.
Metabolism represents the ‘sharp end’ of systems biology, because changes in metabolite concentrations are
- necessarily amplified relative to changes in the transcriptome, proteome and enzyme activities, which can be modulated by drugs.
To understand such behaviour, we therefore need (and increasingly have) reliable consensus (community) models of
- the human metabolic network that include the important transporters.
Small molecule ‘drug’ transporters are in fact metabolite transporters, because
- drugs bear structural similarities to metabolites known from the network reconstructions and
- from measurements of the metabolome.
Recon2 represents the present state-of-the-art human metabolic network reconstruction; it can predict inter alia:
(i) the effects of inborn errors of metabolism;
(ii) which metabolites are exometabolites, and
(iii) how metabolism varies between tissues and cellular compartments.
However, even these qualitative network models are not yet complete. As our understanding improves
- so do we recognise more clearly the need for a systems (poly)pharmacology.
Introduction – a systems biology approach to drug discovery
It is clearly not news that the productivity of the pharmaceutical industry has declined significantly during recent years
- following an ‘inverse Moore’s Law’, Eroom’s Law, or
- that many commentators, consider that the main cause of this is
- because of an excessive focus on individual molecular target discovery rather than a more sensible strategy
- based on a systems-level approach (Fig. 1).

drug discovery science
Figure 1.
The change in drug discovery strategy from ‘classical’ function-first approaches (in which the assay of drug function was at the tissue or organism level),
with mechanistic studies potentially coming later, to more-recent target-based approaches where initial assays usually involve assessing the interactions
of drugs with specified (and often cloned, recombinant) proteins in vitro. In the latter cases, effects in vivo are assessed later, with concomitantly high levels of attrition.
Arguably the two chief hallmarks of the systems biology approach are:
(i) that we seek to make mathematical models of our systems iteratively or in parallel with well-designed ‘wet’ experiments, and
(ii) that we do not necessarily start with a hypothesis but measure as many things as possible (the ’omes) and
- let the data tell us the hypothesis that best fits and describes them.
Although metabolism was once seen as something of a Cinderella subject,
- there are fundamental reasons to do with the organisation of biochemical networks as
- to why the metabol(om)ic level – now in fact seen as the ‘apogee’ of the ’omics trilogy –
- is indeed likely to be far more discriminating than are
- changes in the transcriptome or proteome.
The next two subsections deal with these points and Fig. 2 summarises the paper in the form of a Mind Map.

metabolomics and systems pharmacology
http://ars.els-cdn.com/content/image/1-s2.0-S1359644613002481-gr2.jpg
Metabolic Disease Drug Discovery— “Hitting the Target” Is Easier Said Than Done
David E. Moller, et al. http://dx.doi.org:/10.1016/j.cmet.2011.10.012
Despite the advent of new drug classes, the global epidemic of cardiometabolic disease has not abated. Continuing
- unmet medical needs remain a major driver for new research.
Drug discovery approaches in this field have mirrored industry trends, leading to a recent
- increase in the number of molecules entering development.
However, worrisome trends and newer hurdles are also apparent. The history of two newer drug classes—
- glucagon-like peptide-1 receptor agonists and
- dipeptidyl peptidase-4 inhibitors—
illustrates both progress and challenges. Future success requires that researchers learn from these experiences and
- continue to explore and apply new technology platforms and research paradigms.
The global epidemic of obesity and diabetes continues to progress relentlessly. The International Diabetes Federation predicts an even greater diabetes burden (>430 million people afflicted) by 2030, which will disproportionately affect developing nations (International Diabetes Federation, 2011). Yet
- existing drug classes for diabetes, obesity, and comorbid cardiovascular (CV) conditions have substantial limitations.
Currently available prescription drugs for treatment of hyperglycemia in patients with type 2 diabetes (Table 1) have notable shortcomings. In general,
Therefore, clinicians must often use combination therapy, adding additional agents over time. Ultimately many patients will need to use insulin—a therapeutic class first introduced in 1922. Most existing agents also have
- issues around safety and tolerability as well as dosing convenience (which can impact patient compliance).
Pharmacometabolomics, also known as pharmacometabonomics, is a field which stems from metabolomics,
- the quantification and analysis of metabolites produced by the body.
It refers to the direct measurement of metabolites in an individual’s bodily fluids, in order to
- predict or evaluate the metabolism of pharmaceutical compounds, and
- to better understand the pharmacokinetic profile of a drug.
Alternatively, pharmacometabolomics can be applied to measure metabolite levels
- following the administration of a pharmaceutical compound, in order to
- monitor the effects of the compound on certain metabolic pathways(pharmacodynamics).
This provides detailed mapping of drug effects on metabolism and
- the pathways that are implicated in mechanism of variation of response to treatment.
In addition, the metabolic profile of an individual at baseline (metabotype) provides information about
- how individuals respond to treatment and highlights heterogeneity within a disease state.
All three approaches require the quantification of metabolites found
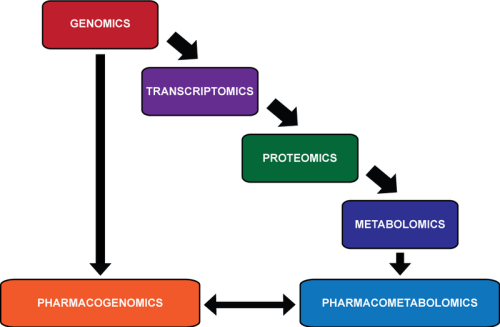
relationship between -OMICS
http://upload.wikimedia.org/wikipedia/commons/thumb/e/eb/OMICS.png/350px-OMICS.png
Pharmacometabolomics is thought to provide information that
Looking at the characteristics of an individual down through these different levels of detail, there is an
- increasingly more accurate prediction of a person’s ability to respond to a pharmaceutical compound.
- the genome, made up of 25 000 genes, can indicate possible errors in drug metabolism;
- the transcriptome, made up of 85,000 transcripts, can provide information about which genes important in metabolism are being actively transcribed;
- and the proteome, >10,000,000 members, depicts which proteins are active in the body to carry out these functions.
Pharmacometabolomics complements the omics with
- direct measurement of the products of all of these reactions, but with perhaps a relatively
- smaller number of members: that was initially projected to be approximately 2200 metabolites,
but could be a larger number when gut derived metabolites and xenobiotics are added to the list. Overall, the goal of pharmacometabolomics is
- to more closely predict or assess the response of an individual to a pharmaceutical compound,
- permitting continued treatment with the right drug or dosage
- depending on the variations in their metabolism and ability to respond to treatment.
Pharmacometabolomic analyses, through the use of a metabolomics approach,
- can provide a comprehensive and detailed metabolic profile or “metabolic fingerprint” for an individual patient.
Such metabolic profiles can provide a complete overview of individual metabolite or pathway alterations,
This approach can then be applied to the prediction of response to a pharmaceutical compound
- by patients with a particular metabolic profile.
Pharmacometabolomic analyses of drug response are
Pharmacogenetics focuses on the identification of genetic variations (e.g. single-nucleotide polymorphisms)
- within patients that may contribute to altered drug responses and overall outcome of a certain treatment.
The results of pharmacometabolomics analyses can act to “inform” or “direct”
This concept has been established with two seminal publications from studies of antidepressants serotonin reuptake inhibitors
- where metabolic signatures were able to define a pathway implicated in response to the antidepressant and
- that lead to identification of genetic variants within a key gene
- within the highlighted pathway as being implicated in variation in response.
These genetic variants were not identified through genetic analysis alone and hence
- illustrated how metabolomics can guide and inform genetic data.
en.wikipedia.org/wiki/Pharmacometabolomics
Benznidazole Biotransformation and Multiple Targets in Trypanosoma cruzi Revealed by Metabolomics
Andrea Trochine, Darren J. Creek, Paula Faral-Tello, Michael P. Barrett, Carlos Robello
Published: May 22, 2014 http://dx.doi.org:/10.1371/journal.pntd.0002844
The first line treatment for Chagas disease, a neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi,
- involves administration of benznidazole (Bzn).
Bzn is a 2-nitroimidazole pro-drug which requires nitroreduction to become active. We used a
- non-targeted MS-based metabolomics approach to study the metabolic response of T. cruzi to Bzn.
Parasites treated with Bzn were minimally altered compared to untreated trypanosomes, although the redox active thiols
- trypanothione,
- homotrypanothione and
- cysteine
were significantly diminished in abundance post-treatment. In addition, multiple Bzn-derived metabolites were detected after treatment.
These metabolites included reduction products, fragments and covalent adducts of reduced Bzn
- linked to each of the major low molecular weight thiols:
- trypanothione,
- glutathione,
- g-glutamylcysteine,
- glutathionylspermidine,
- cysteine and
- ovothiol A.
Bzn products known to be generated in vitro by the unusual trypanosomal nitroreductase, TcNTRI,
- were found within the parasites,
- but low molecular weight adducts of glyoxal, a proposed toxic end-product of NTRI Bzn metabolism, were not detected.
Our data is indicative of a major role of the
- thiol binding capacity of Bzn reduction products
- in the mechanism of Bzn toxicity against T. cruzi.
Read Full Post »

























 shows hESCs...")
 is a s...")




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}