Action of Hormones on the Circulation
Writer and Curator: Larry H. Bernstein, MD, FCAP
Introduction
This is perhaps the most difficult piece to write, unexpectedly. I have done a careful search for related material using different search phrases. It is perhaps because of the great complexity of the topic, which is inextricably linked to sepsis, the Systemic Inflammatory Response Syndrome SIRS), and is poised differently than the neural innervation of the hormonal response and circulation, as in the previous piece. In the SIRS mechanism, we find a very large factor in glucocorticoids, the cytokine shower (IL-1, IL-6, TNF-α), and gluconeogenesis, with circulatory changes. In this sequence, it appears that we are focused on the arteriolar and bronchial smooth muscle architecture, the adrenal medulla, vasoconstriction and vasodilation, and another set of peptide interactions. This may be concurrent with the other effects described.
Related articles in Pharmaceutical Intelligence Journal:
Endothelial Function and Cardiovascular Disease
Pathologist and Author: Larry H Bernstein, MD, FCAP
http://pharmaceuticalintelligence.com/2012/10/25/endothelial-function-and-cardiovascular-disease/
Clinical Trials Results for Endothelin System: Pathophysiological role in Chronic Heart Failure, Acute Coronary Syndromes and MI – Marker of Disease Severity or Genetic Determination?
Curator: Aviva Lev-Ari, PhD, RN
Endothelin Receptors in Cardiovascular Diseases: The Role of eNOS Stimulation
Author and Curator of an Investigator Initiated Study: Aviva Lev-Ari, PhD, RN
Inhibition of ET-1, ETA and ETA-ETB, Induction of NO production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): cEPCs Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
Curator of an Investigator Initiated Study: Aviva Lev-Ari, PhD, RN
Cardiovascular Disease (CVD) and the Role of Agent Alternatives in endothelial Nitric Oxide Synthase (eNOS) Activation and Nitric Oxide Production
Curator and Investigator Initiated Study: Aviva Lev-Ari, PhD, RN
Innervation of Heart and Heart Rate
Writer and Curator: Larry H Bernstein, MD, FCAP
http://pharmaceuticalintelligence.com/2015/02/15/innervation-of-heart-and-heart-rate/
αllbβ3 Antagonists As An Example of Translational Medicine Therapeutics
Larry H Bernstein, MD, FCAP, Reporter and curator
Cardiac Contractility & Myocardium Performance: Therapeutic Implications for Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Author, and Content Consultant to e-SERIES A: Cardiovascular Diseases: Justin Pearlman, MD, PhD, FACC
The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Ca2+-Stimulated Exocytosis: The Role of Calmodulin and Protein Kinase C in Ca2+ Regulation of Hormone and Neurotransmitter
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Disruption of Calcium Homeostasis: Cardiomyocytes and Vascular Smooth Muscle Cells: The Cardiac and Cardiovascular Calcium Signaling Mechanism
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Calcium-Channel Blockers, Calcium Release-related Contractile Dysfunction (Ryanopathy) and Calcium as Neurotransmitter Sensor
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Synaptotagmin functions as a Calcium Sensor: How Calcium Ions Regulate the fusion of vesicles with cell membranes during Neurotransmission
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Advanced Topics in Sepsis and the Cardiovascular System at its End Stage
Larry H Bernstein, MD, FCAP
For most comprehensive Bibliography on the Ryanodine receptor calcium release channel complex and for FIGURES illustrating the phenomenon, see
Pharmacol Ther. 2009 August; 123(2): 151–177.
http://dx.doi.org:/10.1016/j.pharmthera.2009.03.006
PMCID: PMC2704947
Ryanodine receptor-mediated arrhythmias and sudden cardiac death
Lynda M. Blayney[low asterisk] and F. Anthony Lai
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2704947/
Oxidized Calcium Calmodulin Kinase and Atrial Fibrillation
Author: Larry H. Bernstein, MD, FCAP and Curator: Aviva Lev-Ari, PhD, RN
Contributions to cardiomyocyte interactions and signaling
Author and Curator: Larry H Bernstein, MD, FCAP and Curator: Aviva Lev-Ari, PhD, RN
Cardiac Contractility & Myocardium Performance: Therapeutic Implications for Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Editor: Justin Pearlman, MD, PhD, FACC, Author and Curator: Larry H Bernstein, MD, FCAP, and Article Curator: Aviva Lev-Ari, PhD, RN
The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Action of hormones on the circulation
Limbic system mechanisms of stress regulation: Hypothalamo-pituitary-adrenocortical axis
James P. Herman, Michelle M. Ostrander, Nancy K. Muelle, Helmer Figueiredo
Prog in Neuro-Psychopharmacol & Biol Psychiatry 29 (2005) 1201 – 1213
http://dx.doi.org:/10.1016/j.pnpbp.2005.08.006
Limbic dysfunction and hypothalamo-pituitary-adrenocortical (HPA) axis dysregulation are key features of affective disorders. The following review summarizes our current understanding of the relationship between limbic structures and control of ACTH and glucocorticoid release, focusing on the hippocampus, medial prefrontal cortex and amygdala. In general, the hippocampus and anterior cingulate/prelimbic cortex inhibit stress-induced HPA activation, whereas the amygdala and perhaps the infralimbic cortex may enhance glucocorticoid secretion. Several characteristics of limbic–HPA interaction are notable: first, in all cases, the role of given limbic structures is both region- and stimulus-specific. Second, limbic sites have minimal direct projections to HPA effector neurons of the paraventricular nucleus (PVN); hippocampal, cortical and amygdalar efferents apparently relay with neurons in the bed nucleus of the stria terminalis, hypothalamus and brainstem to access corticotropin releasing hormone neurons. Third, hippocampal, cortical and amygdalar projection pathways show extensive overlap in regions such as the bed nucleus of the stria terminalis, hypothalamus and perhaps brainstem, implying that limbic information may be integrated at subcortical relay sites prior to accessing the PVN. Fourth, these limbic sites also show divergent projections, with the various structures having distinct subcortical targets. Finally, all regions express both glucocorticoid and mineralocorticoid receptors, allowing for glucocorticoid modulation of limbic signaling patterns. Overall, the influence of the limbic system on the HPA axis is likely the end result of the overall patterning of responses to given stimuli and glucocorticoids, with the magnitude of the secretory response determined with respect to the relative contributions of the various structures.

representations of the HPA axis
Diagrammatic representations of the HPA axis of the rat. HPA responses are initiated by neurosecretory neurons of medial parvocellular paraventricular nucleus (mpPVN), which secretes ACTH secretagogues such as corticotropin releasing hormone (CRH) and arginine vasopressin (AVP) in the hypophysial portal circulation at the level of the median eminence. These secretagogues promote release of ACTH into the systemic circulation, whereby it promotes synthesis and release of glucocorticoids at the adrenal cortex.
When exposed to chronic stress, the HPA axis can show both response Fhabituation_ and response Ffacilitation_. FHabituation_ occurs when the same (homotypic) stressor is delivered repeatedly, and is characterized by progressive diminution of glucocorticoid responses to the stimulus. Systemic administration of a mineralocorticoid receptor antagonist is sufficient to block habituation, implying a role for MR signaling in this process. It should be noted that HPA axis habituation is highly dependent on both the intensity and predictability of the stressful stimulus. FFacilitation_ is observed when animals repeatedly exposed to one stimulus are presented with a novel (heterotypic). In chronically stressed animals, exposure to a novel stimulus results in rise in glucocorticoids that is as large as or greater than that seen in a chronic stress naıve animal. Importantly, facilitation can occur in the context of chronic stress-induced elevations in resting glucocorticoids levels, suggesting that this process involves a bypass or override of negative feedback signals.
Hippocampal regulation of the HPA axis appears to be both region- and stressor-specific. Using a sequential lesion approach, our group has noted that the inhibitory effects of the hippocampus on stress-induced corticosterone release and CRH/AVP mRNA expression are likely subserved by neurons resident in the ventral subiculum-caudotemporal CA1. In addition to spatial specificity, hippocampal regulation of the HPA axis also appears to be specific to certain stress modalities; our studies indicate that ventral subiculum lesions cause elevated glucocorticoid secretion following restraint, open field or elevated plus maze exposure, but not to ether inhalation or hypoxia.
The research posits an intricate topographical organization of prefrontal cortex output to HPA regulatory circuits. The anatomy of medial prefrontal cortex efferents may illuminate this issue. The infralimbic cortex projects extensively to the anterior bed nucleus of the stria terminalis, medial and central amygdala and the nucleus of the solitary tract, all of which are implicated in stress excitation. In contrast, the prelimbic cortex has minimal input to these structures, but projects to the ventrolateral preoptic area, dorsomedial hypothalamus and peri-PVN region, areas implicated in stress inhibition. Thus, the infralimbic and prelimbic/anterior cingulate components of the prefrontal cortex may play very different roles in HPA axis regulation. Like other limbic regions, the influence of the amygdala on the HPA axis is stressor- and region-specific. The medial amygdala shows intense c-fos induction following stressors such as restraint, swimming, predator exposure and social interaction.
Despite the prominent involvement of the hippocampus, medial prefrontal cortex and amygdala in HPA axis regulation, there is limited evidence of direct innervation of the PVN by these structures. Rather, these regions appear to project to a number of basal forebrain, hypothalamic and brainstem cell populations that in turn innervate the medial parvocellular PVN. Thus, in order to access principle stress effector neurons, information from the limbic system requires an intermediary synapse. In the bed nucleus of the stria terminalis and hypothalamus, the majority of these intermediary neurons are GABAergic. For example, the bed nucleus of the stria terminalis, ventrolateral preoptic area, dorsomedial hypothalamic nucleus and peri-PVN region all contain rich populations of neurons expressing the GABA marker glutamatic acid decarboxylase (GAD) 65/67.
The organization of the peri-PVN cell groups is particularly interesting. In the case of the ventral subiculum and to a lesser extent, the medial prefrontal cortex, terminal fields can be observed in the immediate surround of the PVN, corresponding to areas containing substantial numbers of GABA neurons. Importantly, dendrites of PVN neurons are largely confined within the nucleus proper, indicating that limbic afferents are unlikely to interact directly with the PVN neurons themselves. The peri-PVN GABA neurons are activated by glutamate, and likely express glutamate receptor subunits. These neurons also up-regulate GAD65 mRNA following chronic stress, commensurate with involvement in long-term HPA regulation. Injections of a general ionotroptic glutamate receptor antagonist into the PVN surround potentiates glucocorticoid responses to restraint, consistent with blockade of glutamate excitation of these GABA neurons. The data are consistent with an interaction between the excitatory limbic structures and inhibitory PVN-regulatory cells at the level of the PVN surround.
Brainstem stress-modulatory pathways likely relay excitatory information to the PVN. For example, the nucleus of the solitary tract provides both catecholaminergic (norepinephrine) and non-catecholaminergic (e.g., glucagon-like peptide-1 (GLP-1) input to the medial parvocellular. Norepinephrine is released into the PVN following stress and is believed to activate CRH neurons via alpha-1 adrenergic receptors. The role of this pathway is thought to be associated with systemic stressors, as selective destruction of PVN norepinephrine input using anti-dopamine beta hydroxylase-saporin conjugate blocks responses to 2-deoxy-glucose but not restraint. In contrast, blockade of central GLP-1 receptors using exendin 9–36 markedly inhibits responsiveness to both lithium chloride and novelty, suggesting that this non-catecholaminergic cell population may play a more general role in stress integration.
The existence of these putative two-neuron circuits lends important insight into the nature of stress information processing. Anatomical data support the hypothesis that the vast majority of medial prefrontal cortex and ventral subicular inputs to subcortical stress relays are glutamate-containing. As can be appreciated, pyramidal cells of the medial prefrontal cortex and subiculum richly express mRNA encoding vesicular glutamate transporter-1 (VGlut1), a specific marker of glutamate neurons. Combined retrograde tracing/in situ hybridization studies performed in our lab indicate that the vast majority of cortical and hippocampal afferents to PVN-projecting regions (e.g., bed nucleus of the stria terminalis, dorsomedial hypothalamus, ventrolateral medial preoptic area) indeed contain VGlut1, verifying a glutamatergic input to these areas. In contrast, the majority of amygdalar areas implicated in stress regulation express glutamic acid decarboxylase (GAD) 65 or 67 mRNA, suggesting a GABAergic phenotype; indeed, the vast majority of medial and central amygdaloid projections to PVN relays are GABAergic.

representations of limbic stress-integrative pathways from the prefrontal cortex, amygdala and hippocampus
Diagrammatic representations of limbic stress-integrative pathways from the prefrontal cortex, amygdala and hippocampus. The medial prefrontal cortex (mPFC) subsumes neurons of the prelimbic (pl), anterior cingulate (ac) and infralimbic cortices (il), which appear to have different actions on the HPA axis stress response. The pl/ac send excitatory projections (designated as dark circles, filled line with arrows) to regions such as the peri-PVN zone and bed nucleus of the stria terminalis (BST), both of which send direct GABAergic projections to the medial parvocellular PVN (delineated as open circles, dotted lines ending in squares). This two-neuron chain is likely to be inhibitory in nature. In contrast, the infralimbic cortex projects to regions such as the nucleus of the solitary tract (NTS), which sends excitatory projections to the PVN, implying a means of PVN excitation from this cortical region. The ventral subiculum (vSUB) sends excitatory projections to numerous subcortical regions, including the posterior BST, peri-PVN region, ventrolateral region of the medial preoptic area (vlPOA) and ventrolateral region of the dorsomedial hypothalamic nucleus (vlDMH), all of which send GABAergic projections to the PVN and are likely to communicate transsynaptic inhibition. The medial amygdaloid nucleus (MeA) sends inhibitory projections to GABAergic PVN-projecting populations, such as the BST, vlPOA and peri-PVN, eliciting a transsynaptic disinhibition. A similar arrangement likely exists for the central amygdaloid nucleus (CeA), which sends GABAergic outflow to the ventrolateral BST and to a lesser extent, the vlDMH. The CeA also projects to GABAergic neurons in the NTS, which may disinhibit ascending projections to the PVN.
Inotropes and vasopressors: more than haemodynamics!
Hendrik Bracht, E Calzia, M Georgieff, J Singer, P Radermacher and JA Russell
British Journal of Pharmacology (2012) 165 2009–2011
http://dx.doi.org:/10.1111/j.1476-5381.2011.01776.x
Circulatory shock is characterized by arterial hypotension requiring fluid resuscitation combined with inotropes and/or vasopressors to correct the otherwise life-threatening impairment of oxygen supply to peripheral tissues. Catecholamines represent the current therapeutic choice, but this standard is only based on empirical clinical experience. Although there is evidence that some catecholamines may be better than others, it is a matter of debate which one may be the most effective and/or the safest for the different situations. In their review in this issue of the British Journal of Pharmacology, Bangash et al. provide an overview of the pharmacology as well as the available clinical data on the therapeutic use of endogenous catecholamines, their synthetic derivatives and a range of other agents (vasopressin and its analogues, PDE inhibitors and levosimendan). The authors point out that, despite well-established receptor pharmacology, the clinical effects of these treatments are poorly understood. Hence, further investigations are essential to determine which catecholamine, or, in a broader sense, which alternative vasopressor and/or inotrope is the most appropriate for a particular clinical condition.
LINKED ARTICLES This article is a commentary on Bangash et al., pp. 2015–2033 of this issue and is commented on by De Backer and Scolletta, pp. 2012–2014 of this issue. To view Bangash et al. visit http://dx.doi.org/10.1111/j.1476-5381.2011.01588.x and to view De Backer and Scolletta visit http://dx.doi.org/10.1111/j.1476-5381.2011.01746.x
In the present issue of the British Journal of Pharmacology, Bangash et al. (2012) review the pharmacology as well as the available clinical data on the therapeutic use of various inotropes and vasopressor agents used for the hemodynamic management of (septic) shock. By definition, circulatory shock is characterized by arterial hypotension that necessitates immediate intervention to maintain the balance of tissue oxygen supply and demand. In practice, the longer and the more frequent periods of hypotension are present in a patient, the less likely is survival, and early aggressive resuscitation is associated with improved outcome. Besides fluid administration to increase the circulating blood volume, in most cases, vasoactive drugs are required to restore an adequate perfusion pressure, and up to now, catecholamines represent the current therapeutic choice. According to their pharmacological profile, catecholamines are traditionally used for their predominant inotropic, vasodilating or constrictor effects.
Clinicians should not forget two fundamental aspects of catecholamine action. First, because of the ubiquitous presence of adrenoceptors, endogenous catecholamines. as well as their synthetic derivatives, have pronounced effects on virtually all tissues (many of which were described several years ago), in particular on the immune system (van der Poll et al., 1996; Flierl et al., 2008), on energy metabolism (Cori and Cori, 1928; Bearn et al., 1951) and on gastrointestinal motility (McDougal and West, 1954). Second, the adrenoceptor density and responsiveness to catecholamines are markedly altered by both the underlying disease and the ongoing catecholamine. Bangash et al. (2012) have to be commended that they not only describe the various endogenous catecholamines and their synthetic derivatives but also thoroughly discuss possible alternatives, such as vasopressin and its analogues, PDE inhibitors and levosimendan.
Inhibitory effects of cortisone and hydrocortisone on human Kv1.5 channel currents
Jing Yu, Mi-Hyeong Park, Su-Hyun Jo
Eur J Pharmacol 746 (2015) 158–166 http://dx.doi.org/10.1016/j.ejphar.2014.11.007
Glucocorticoids are the primary hormones that respond to stress and protect organisms from dangerous situations. The glucocorticoids hydrocortisone and its dormant form, cortisone, affect the cardiovascular system with changes such as increased blood pressure and cardioprotection. Kv1.5 channels play a critical role in the maintenance of cellular membrane potential and are widely expressed in pancreatic β-cells, neurons, myocytes, and smooth muscle cells of the pulmonary vasculature. We examined the electrophysiological effects of both cortisone and hydrocortisone on human Kv1.5 channels expressed in Xenopus oocytes using a two-microelectrode voltage clamp technique. Both cortisone and hydrocortisone rapidly and irreversibly suppressed the amplitude of Kv1.5 channel current with IC50 values of 50.2 + 74.2 μM and 33.4 + 73.2 μM, respectively, while sustained the current trace shape of Kv1.5 current. The inhibitory effect of cortisone on Kv1.5 decreased progressively from – 10mV to +30 mV, while hydrocortisone’s inhibition of the channel did not change across the same voltage range. Both cortisone and hydrocortisone blocked Kv1.5 channel currents in a non-use-dependent manner and neither altered the channel’s steady-state activation or inactivation curves. These results show that cortisone and hydrocortisone inhibited Kv1.5 channel currents differently. Kv1.5 channels were more sensitive to hydrocortisone than to cortisone.
In conclusion, cortisone and hydrocortisones rapidly and irreversibly blocked human Kv1.5 channels expressed in Xenopus oocytes in a closed state without altering activation and inactivation gating. These data provide a possible mechanism for GC effects on the cardiovascular system. The detailed mechanism of the interaction between GCs and human Kv1.5 channels merits further exploration.
Inflammasome and cytokine blocking strategies in autoinflammatory disorders
Monika Moll, Jasmin B. Kuemmerle-Deschner
Clinical Immunology (2013) 147, 242–275 http://dx.doi.org/10.1016/j.clim.2013.04.008
Autoinflammatory disorders are characterized by usually unprovoked recurrent episodes of features of inflammation caused by activation of the innate immune system. Many autoinflammatory disorders – the monogenetic defects in particular – are associated with alterations of inflammasomes. Inflammasomes are complex multimolecular structures, which respond to “danger” signals by activation of cytokines. Among these, IL-1 is the key player of the innate immune response and inflammation. Consequently, IL-1 blocking strategies are specific pathway targeting therapies in autoinflammatory diseases and applied in CAPS, colchicine-resistant FMF, TRAPS, HIDS and DIRA. A number of rare genetic disorders involve inflammasome malfunction resulting in enhanced inflammatory response. IL-1 inhibition to date is the most successful specific therapy in autoinflammatory disorders. Here, current treatment strategies in autoinflammatory disorders are reviewed with a focus on inflammasome and cytokine inhibition.
Autoinflammatory disorders have been defined as “clinical disorders marked by abnormally increased inflammation, mediated predominantly by the cells and molecules of the innate immune system.” This means that in autoinflammatory disorders autoantibodies or antigen related T-cells are usually absent. These are features of the adaptive immune system and found in autoimmune diseases.
In general, autoinflammatory disorders are characterized by a large spectrum of rather non-specific systemic and organ-specific signs and symptoms of inflammation. In some diseases specific symptoms are observed like hearing loss in Muckle–Wells syndrome or CNS-disease in NOMID/CINCA. Most autoinflammatory disorders are associated with high levels of serum amyloid A (SAA) during inflammatory attacks and high risk of life-threatening amyloidosis. In most cases the disease will start in infancy and childhood. Only rarely primary manifestations in adulthood are reported.
Because recurrent fevers have been the most prominent feature of this group of diseases, historically they have been summarized under the term “hereditary periodic fever syndromes”. With the deeper understanding of the underlying pathophysiologic mechanisms on the genetic and cellular level, the more comprehensive term “autoinflammatory syndromes”.
Along with the detection of the genetic origin of the autoinflammatory disorders, the cellular pathomechanism leading to the resulting inflammation has been described. A number of genes, which are affected by mutations in autoinflammatory disorders, encode proteins forming intracellular complexes called inflammasomes. External and endogenous “dangers” are recognized by these “danger sensors” and are able to induce an inflammatory reaction. Microbial components from infectious agents such as LPS, flagellin, lipoteichoic acid from bacteria, peptidoglycan or double-stranded DNA from viruses, or inorganic crystalline structures such as uric acid crystals, display pathogen-associated molecular patterns (PAMPs). These and endogenous damage-associated molecular patterns (DAMPs) like heat-shock proteins, the chromatin-associated protein high-mobility group box 1 (HMGB1), hyaluronan fragments, ATP, uric acid, and DNA which are released with cellular waste and injury stimulate the inflammasome. Also, the myeloid related proteins MRP8 and 14 (also known as S100A8 and S100A9) which are used as biomarkers, belong to the group of DAMPs. In addition to PAMPs and DAMPs, the inflammasome may interact with and be stimulated by proteins such as pyrin, proline–serine–threonine phosphatase interacting protein 1 (PSTPIP1), mevalonate kinase (MK) and NLRP7. All of these may also be altered in structure and function by monogenetic mutations.
As a consequence of inflammasome activation, a large variety of cytokines are produced and released by cells of the innate immune system (monocytes, macrophages, dendritic cells). They include the IL-1 family (IL-1, IL-18, IL-33), the TNF family (TNF-α, LT-α), the IL-6 family (IL-6, IL-11), the IL-17 family (IL-17A, IL-25), and type 1 IFNs (IFN-α, IFN-β). These cytokines play redundant roles depending on the cause and pathway of inflammation in the respective disease. Therefore, therapeutic strategies targeting only one cytokine should be expected to be inadequate to treat inflammatory disorders. However, improvement observed in diabetes mellitus Type 2 after blockade of IL-1 indicates that targeting one cytokine, even in a polygenic, complex inflammatory disorder, may cause beneficial effects. Regarding the inflammatory pathogenesis involved in the disease, Goldbach–Mansky and co-workers have classified the monogenetic autoinflammatory disorders as IL-1 mediated (CAPS and DIRA), partially IL-1 mediated (FMF, HIDS, PAPA) and mediated by other pathways (TRAPS, Blau-syndrome, Majeed’s syndrome, cherubism and IL-10 receptor deficiency).
Intracellular signaling pathways and therapeutic targets in autoinflammatory diseases. In autoinflammatory diseases, complex intracellular pathways lead to activation of the inflammatory response, particularly IL-1β activation and release, but also induction of NFκB and TNFα. Several mechanisms may activate the inflammasome, one crucial step in the IL-1 pathway. These include DAMPs (1), K+-efflux (2), activation of ROS (3) by ATP, anorganic crystals, membrane perturbation and proteases which are released from lysosomes damaged by β-amyloid, and heat shock proteins (4). NFκB may be induced by PAMPs via toll like receptors (5), IL-1β-signaling (6) or UPR (7). Activated NFκB eventually leads to the release of pro-inflammatory cytokines like IL-1, IL-6 and TNFα (8). Most of these steps to activation have been identified as targets for anti-inflammatory therapies, which are either already used in clinical practice or still experimental. IL-1- (a), TNF- (b), and IL-6 (c) inhibition are established safe and effective treatment strategies in many autoinflammatory diseases. Thalidomide (d) probably inhibits activation of IκB and is also part of routine treatment. Still experimental strategies include inhibition of PAMPs (e), DAMPs (f), potassium efflux (g), ROS by antioxidants (h), heat shock proteins (i), or caspase-1 (k). Caspase-inhibitors have entered clinical trials.
Colchicine has been used for the treatment of inflammatory disorders for centuries. Colchicine is effective in gout, but also in Behcet’s disease and FMF, where it is able to prevent amyloidosis. The drug affects many cell types and accumulates preferentially in neutrophils. Although its mode of action is still unclear it has microtubule destabilizing properties which may be part of its effects. Additional effects such as alteration of adhesion molecule expression, chemotaxis, and ROS generation also impact inflammation. Colchicine is generally tolerated well. However gastrointestinal, hematologic, and neuromuscular side-effects occur, when the administered dose is too high.
Inflammasome activation by heat shock proteins may be prevented by direct inhibition of HSP. HSP90 inhibition was effective in reducing gout-like arthritis in an animal model. Targeting caspase-1 (caspase-1-inhibitors) may be a strategy which has even greater potential in the treatment of autoimmune diseases and autoinflammatory disorders. IL-1 converting enzyme/caspase inhibitor VX-765 was able to inhibit IL-β-secretion in LPS-stimulated cells from FCAS and control subjects. A new IL-1 inhibitor, gevokizumab or Xoma 052 has entered clinical pilot trials. Therapeutic targets particularly for the protein-misfolding autoinflammatory diseases could be chemical chaperones and drugs that stimulate autophagy. Also inhibiting the signaling molecules that mediate the UPR activation which causes activation of the innate immune system and exacerbate inflammation could be a target.
To date IL-1 blockade is the most effective therapy in most monogenetic autoinflammatory diseases — in intrinsic and in extrinsic inflammasom-opathies. The most favorable effects are seen in the treatment of cryopyrin associated periodic syndromes like FACS, MWS and CINCA. But IL-1-blockade is also effective in other diseases like DIRA, TRAPS, PFAPA, colchicine-resistant FMF etc. IL-1 inhibition also has a role in multifactorial and common autoinflammatory diseases like diabetes, gout and artherosclerosis.
Endothelin—Biology and disease
Al-karim Khimji, Don C. Rockey
Cellular Signalling 22 (2010) 1615–1625
http://dx.doi.org:/10.1016/j.cellsig.2010.05.002
Endothelins are important mediators of physiological and pathophysiologic processes including cardiovascular disorders, pulmonary disease, renal diseases and many others. Additionally, endothelins are involved in many other important processes such as development, cancer biology, wound healing, and even neurotransmission. Here, we review the cell and molecular biology as well as the prominent pathophysiological aspects of the endothelin system.
Endothelin-1 (ET-1) was originally isolated from porcine aortic endothelial cells and is a 21 amino acid cyclic peptide, with two disulphide bridges joining the cysteine amino acids (positions 1–15 and 3–11) at the N-terminal end and hydrophobic amino acids at the c-terminal end of the peptide (Fig. 1). The C-terminal end contains the amino acids that bind to the receptor, the N-terminal end determines the peptide’s binding affinity to the receptor (see Fig. 1). There appear to be at least 2 other endothelin isoforms including endothelin-2 (ET-2) and endothelin-3 (ET-3), which differ from ET-1 in two and six amino acid residues, respectively.

Endothelin (ET) structure
Endothelin (ET) structure. Endothelin is a 21 amino acid cyclic peptide, with two disulphide bridges joining the cysteine residues at positions 1–15 and 3–11. The C-terminal end containsamino acids that appear tomediate receptor binding,while the N-terminal residues determine the peptide’s binding affinity to the receptor. The amino acids highlighted in black in panels (b) and (c) show differences in ET-2 and ET-3 compared to ET-1. As can be seen, the remainder of the primary sequence of the different family members is identical.

Endothelin-1 biosynthetic pathway
Endothelin-1 biosynthetic pathway. Preproendothelin mRNA is synthesized via transcriptional activation of the preproendothelin gene. The translational product is a 203-amino acid peptide known as preproendothelin, which is cleaved at dibasic sites by furin-like endopeptidases to form big endothelins. These biologically inactive, 37- to 41-amino acid intermediates, are cleaved at Trp21–Val 22 by a family of endothelin-converting enzymes (ECE) to produce mature ET-1. The pathway for endothelin-2 and -3 is presumed to be similar.
The endothelin peptides are produced through a set of complex molecular processes. Preproendothelins are synthesized via transcriptional activation of the preproendothelin gene, which is regulated by c-fos and c-jun, nuclear factor-1, AP-1 and GATA-2. The translational product is a 203-amino acid peptide known as preproendothelin which is cleaved at dibasic sites by furin-like endopeptidases to form big endothelins. These biologically inactive 37- to 41-amino acid intermediates are cleaved at Trp21–Val 22 by a family of endothelin-converting enzymes (ECE) to produce mature ET-1.
Three isoforms of ECE have been reported, namely ECE-1, ECE-2 and ECE-3; ECE-1 and ECE-2 are most prominent. (Endothelin receptors are widely distributed in many different tissues and cells, there is a marked difference in cell and tissue distribution patterns between the two receptor subtypes i.e. ETA and ETB. [ET Receptors: Endothelial cells -ETB Vascular tone, clearance of circulating ET-1]). ECEs belong to the M13 group of proteins—which is a family that includes neutral endopeptidases, kell blood group antigens (Kell), a peptide from phosphate regulating gene (PEX), X-converting enzyme (XCE), “secreted” endopeptidases, and the ECEs. M13 family members contain type II integral membrane proteins with zinc metalloprotease activity, and their function is inhibited by phosphoramidon. Four variants of ECE-1 have been reported in humans, namely ECE-1a, ECE-1b, ECE-1c and ECE-1d which are a result of alternate splicing of ECE-1mRNA. ECE-1 appears to be localized in the plasma cell membrane and its optimal activity is atpH7; it processes big ETs both intracellularly and on the cell surface. It is distributed predominantly in smooth muscle cells. ECE-1 can also hydrolyze other proteins including bradykinin, substance P, and insulin. ECE-2 is localized to the trans-Golgi network and is expressed abundantly in neural tissues and endothelial cells. Its optimal activity is at pH5; the acidic activity marks ECE-2 as an intracellular enzyme. Substrate selectivity experiments indicate that both ECE-1 and ECE-2 show preference for big ET-1 over big ET-2 or big ET-3.
Although there has been controversy about the precise repertoire of endothelin receptors, it appears that the endothelins exert their actions through two major receptor subtypes known as ETA and ETB receptors. ETA and ETB receptors belong to the superfamily of G-protein coupled receptors and contain seven transmembrane domains of 22–26 hydrophobic amino acids among approximately 400 total amino acids. The ETA receptor is found predominantly in smooth muscle cells and cardiac muscles, whereas the ETB receptor is abundantly expressed in endothelial cells.
ET-1 signaling is extremely complicated and ET receptor activation leads to diverse cellular responses through interaction in a chain of pathways that includes the G-protein-activated cell surface receptor, coupling G-proteins and phospholipase (PLC) pathway and other G protein-activated effectors. In one of the canonical signaling pathways, ETA induced activation of phospholipase C leads to the formation of inositol triphosphate and diacylglcerol from phosphatidylinositol. Inositol 1,4,5 triphosphate (IP3) then diffuses to specific receptors on the endoplasmic reticulum and releases stored Ca2+ into the cytosol. This causes a rapid elevation in intracellular Ca2+, which in turn causes cellular contraction and then vasoconstriction; the vasoconstrictive effects of ET persist despite dissociation of ET-1 from the receptor, perhaps because the levels of intracellular calcium remain elevated or because endothelin signaling pathways remain activated for prolonged time periods.

Endothelin signaling – smooth muscle cells
Endothelin signaling – smooth muscle cells. ET receptor stimulation leads to diverse cellular responses in a chain of pathways that include the G protein bg activation. This is followed by activation of a variety of different downstream cascades. For example, shown on the left, ETA induced activation of phosphatidyl inositol specific phospholipase C (PI-PLC) leads to the formation of inositol triphosphate (IP3) and diacylglcerol (DAG) from phosphoinositol 4,5 bisphosphate (PIP2). Inositol 1, 4, 5 triphosphate (IP3) then diffuses to specific receptors on the endoplasmic reticulum and releases stored Ca2+ into the cytosol. This causes a rapid elevation in intracellular Ca2+, which in turn causes cellular contraction

Endothelin signaling – endothelial cells.
Endothelin signaling – endothelial cells. ET-1 stimulates NO production in endothelial cells by activation of endothelial cell NO synthase (eNOS). This occurs via ET-1’s activation of the ET-B receptor and the PI3-K/Akt pathway, which in turn stimulates phosphorylation of eNOS, with subequent conversion of L-arginine to L-citrulline and at the same time, generating NO. In addition shear stress, G-protein coupled receptors (GPCR), transient receptor potential channel (TRPC) and receptor tyrosine kinase (RTK) are also activators of eNOS. As a result, NO diffuses to stellate cell, where it directly activates the heme moiety of soluble guanylate cyclase, leading to the production of cyclic GMP. Intracellular cyclic GMP leads to activation of protein kinase G (PKG) resulting in relaxation of stellate cells – offsetting ET’s contractile effect on stellate cells.
The plasma levels of endothelin do not correlate with either the presence of essential hypertension or its severity, presumably, due to the fact that endothelin appears to be biologically active in a paracrine or autocrine fashion (i.e., rather than in an endocrine fashion. Systemic administration of ET-1 in low doses produces a modest increase in blood pressure which is normalized by selective ETA receptor blockade. In experimental models, long-term infusion with ET-1 leads to stroke and renal injury, which can be prevented with long-term administration of selective ETA receptor antagonists. Apart from its direct vasoconstrictor effects, mediated by smooth muscle cell contraction in the arterial system, ET-1 also indirectly enhances the vasoconstrictor effects of other neurohumoral and endocrine factors and may potentiate essential hypertension via this mechanism. For example, ET-1 induces conversion of angiotensin I to angiotensin II in in vitro models and stimulates adrenal synthesis of epinephrine and aldosterone. Thus there is cross-talk between the endothelin and renin–angiotensin–aldosterone systems—to synergistically act to facilitate vasoconstriction. In aggregate, the data suggest that dysregulation of the endothelin system contributes to multisystem complications of hypertension such as progressive renal disease, cerebrovascular diseases, atherosclerosis, and cardiac disease.
ET-1 in the renal system is synthesized in vascular endothelial cells and epithelial cells of the collecting ducts. Both ET receptors are present in renal vasculature and epithelial cells where ETB is the predominant receptor type. Renal vasculature is relatively more sensitive to the vasoconstrictive effects of ET-1 than any other vasculature and it causes constriction of both afferent and efferent renal arterioles.
ET-1 administration in humans significantly reduces renal blood flow, glomerular filtration rate and urine volume. In addition to its hemodynamic effects, ET-1 system is also involved in salt and water reabsorption, acid-base balance, promotion of mesangial cell growth and activation of inflammatory cells. ET-1 has been implicated in the pathophysiology of acute renal injury, chronic renal failure as well as renal remodeling. Transgenic mice overexpressing ET-1 develop glomerulosclerosis, interstitial fibrosis and reduced renal function. Increased ET-1 and ET receptor upregulation has been described in various animal models of acute renal injury and also in patients with chronic renal failure. Additionally, plasma ET-1 levels have been shown to correlate with the severity of chronic renal failure.
ET-1 is produced and released by airway epithelial cells, macrophages, and pulmonary vascular endothelial cells. Endothelin receptors are similarly widely distributed in airway smooth muscle cells, the pulmonary vasculature, and in the autonomic neuronal network lining tracheal muscles. ET-1 has a potent bronchoconstrictor effect. In animal models, intravenous ET-1 injection led to a dose-dependent increase in airway resistance. The increase in airway resistance is in part due to enhanced production of thromboxanes with subsequent activation of thromboxane receptors and smooth muscle cell proliferation. The ET system has been emphasized in a number of pulmonary disorders, including asthma, cryptogenic fibrosing alveolitis, and pulmonary hypertension. Increased lung vasculature ET-1 immunoreactivity has been reported in both animals and patients with pulmonary hypertension and increases in ET-1 immunoreactivity correlate with the degree of pulmonary vascular resistance, disorders such as pulmonary hypertension, myocardial infarction, heart failure, neoplasia, vascular disorders, wound healing, and many others.
Endothelin and endothelin antagonism: Roles in cardiovascular health and disease
Praveen Tamirisa, William H. Frishman, and Anil Kumar
Am Heart J 1995;130:601-10
Endothelin is a naturally occurring polypeptide substance with potent vasoconstrictive actions. It was originally described as endotensin or endothelial contracting factor in 1985 by Hickey et al., who reported on the finding of a potent stable vasoconstricting substance produced by cultured endothelial cells. Subsequently, Yanagisawa et al. isolated and purified the substance from the supernatant of cultured porcine aortic and endothelial
cells and then went on to prepare its complementary deoxyribonucleic acid (cDNA). This substance was renamed endothelin.
Endothelin is the most potent vasoconstrictor known to date. Its chemical structure is closely related to certain neurotoxins (sarafotoxins) produced by scorpions and the burrowing asp (Atractaspis engaddensis). Endothelins have now been isolated in various cell lines from several organisms. They are now considered to be autocoids or cytokines 4 because of their wide distribution, their expression during ontogeny and adult life, their primary role as intracellular factors, and the complexity of their biologic effects.
The superfamily of endothelins and sarafotoxins have two main branches with four members each. Endothelin is a polypeptide consisting of 21 amino acids. There are three closely related isoforms endothelin-1, endothelin-2, and endothelin-3 (ET1, ET2, and ET3, respectively), which differ in a few of the amino acid constituents. The fourth member, called ET4 or vasoactive intestinal constrictor, is considered to be the murine form ofET2. The endothelin molecules have several conserved amino acids, including the last six carboxyl (C)-terminal amino acids and four cysteine residues, which form two intrachain disulfide bonds between residues 1 and 15 and 3 and 11. These residues may have biologic implications particularly in relation to three dimensional structure and function. The main differences in the endothelin isopeptides reside in their amino (N)-terminal segments. There is a very high degree of sequence similarity between the two branches (approximately 60%) and within the constituent members of a branch (71% to 95%).
Endothelin has been demonstrated to be produced from endothelial and nonendothelial cells. The synthesis of endothelins parallels that of the various peptide hormones in that a precursor polypeptide is sequentially cleaved to generate the active form. Recently, endothelin-converting enzyme (ECE) was cloned. ECE acts at an essential step in the production of active forms of endothelins. The fully formed molecule is then broken down into inactive peptides by as yet uncharacterized proteases. Some candidates are the lysosomal protective protein (deamidase) and enkephalinase (neutral endopeptidase EC 24.11). The regulation of endothelin production occurs predominantly at the levels of transcription and translation. No storage
vesicles containing endothelin have been identified. The genes for the various endothelin isoforms have been sequenced and are found to be scattered in different chromosomes. Current evidence suggests that they arose from a common ancestor by exon duplication.
| Factors known to release endothelinThrombinTransforming growth factor-~Arginine vasopressinHypoxia
Phorbol ester Glucose Angiotensin II Cyclosporin Insulinlike growth factor Bombesin Cortisol Low-density lipeprotein cholesterol Hypercholesterolemia Changes in shear stress on vascular wall |
| Receptor affinities | |
| Receptor | Affinity |
| ETA | ET1 > ET2 > ET3 |
| ETB | ET1 = ET2 = ET3 |
| ETC | ET3 > ET1 |

Intracellular signal transduction pathways activated by endothelins (ETs)
Intracellular signal transduction pathways activated by endothelins (ETs). Activated ET receptor stimulates phospholipase C (PLC) and phospholipase A2 (PLA2). Activated ET receptor also stimulates voltage-dependent calcium channels (VDC) and probably receptor-operated calcium channel (ROC). Inositol triphosphate (IP3) elicits release of calcium ion from caffeine-sensitive calcium store. Protein kinase C (PKC) activated by diacylglycerol (DG) sensitizes contractile apparatus. Increased concentration of intracellular free calcium ion ([Ca2+]i induces contraction. Cyclooxygenase products (prostacyclin [PGI2], prostaglandin E2 [PGE2], and thromboxane A2 [TXA2]) modify contraction. G, G protein; IP2, inositol biphosphate; IP3, inositol triphosphate; PIP2, phosphatidyl inositol biphosphate. (From Masaki T et al. Circulation 1991;84: 1460.)
Systemic hypertension. Endothelin is the most potent vasoconstrictor known to date and has an exceptionally long duration of physiologic action. The influence of endothelin in maintaining normal blood pressure and its role in the cause of systemic hypertension remain unclear. Intravenous injections of endothelin in animals cause a transient decrease in systolic blood pressure (ETB) followed by a prolonged pressor response (ETA). The vasoconstrictor action is mediated by ETA receptors in the vascular smooth muscle, whereas the predominant vasodilation effect is mediated by the ETB receptors on the endothelial cells that cause release of prostacyclin and nitric oxide. Therefore the overall predominant hemodynamic effect of endothelin in a given organ depends on the receptor type being stimulated, its location, and its relative abundance.
Angiotensin II has been found to increase endothelin concentrations in vitro from endo thelial cells, suggesting one mechanism by which angiotensin-converting-enzyme (ACE) inhibition could function in vivo. ACE inhibitors also can indirectly interfere with endothelin: increased concentrations of bradykinin decrease endothelin release (by acting through bradykinin 2 receptors, stimulation of which cause increased nitric oxide release). ACE inhibitors can cause regression of intimal hyperplasia, whereas other antihypertensive drugs are ineffective in this regard.
Myocardial ischemia. Myocardial ischemia can enhance the release of endothelin by cardiomyocytes and increase its vasoactive effects. Infusion of the ET1 isoform directly into the coronary circulation of animals results in the development of myocardial infarction, with impaired ventricular functioning and the development of arrhythmias. Endothelin has been shown to lower the threshold for ventricular fibrillation in dogs. An increase in ET1 has been observed in cardiac tissue after experimental myocardial infarction in rats, and pretreatment with an antiendothelin ϒ-globulin in this model can reduce infarct size by as much as 40%. Infusion of ETA receptor antagonist drugs before an ischemic insult can also reduce infarct size in animals.
Plasma endothelin concentrations can predict hemodynamic complications in patients with myocardial infarction. Patients with the highest plasma endothelin concentrations after myocardial infarction have the highest creatine phosphokinase (CPK) and CPK MB-isoenzyme concentrations and the lowest angiographically determined ejection fractions.
Left ventricular function and congestive heart failure. Endothelin exhibits potent inotropic activity in isolated hearts, cardiac muscle strips, isolated cells, and instrumented intact animals. High-affinity receptors for endothelin have been demonstrated in the atria and the ventricles. Intravenous administration of the ET1 isoform produces delayed prolonged augmentation of left ventricular performance in addition to its biphasic vasoactive effects of transient vasodilation followed by sustained vasocontraction.
Endothelin is a potent secretogogue of atrial natriuretic factor, which is a naturally occurring antagonist of endothelin. The ETA receptor appears to mediate endothelin’s actions of vasoconstriction and the stimulation of atrial natriuretic factor secretion, and the ETB receptor mediates endothelin-induced vasodilation and activation of the renin-angiotensin-aldosterone system. Urinary water excretion is mediated through both receptors, but sodium excretion is mediated through the ETA receptor.
Increased concentrations of endothelin described in patients with congestive heart failure are predictive of increased mortality risk. It also has been suggested that increased concentrations of endothelin may play an important role in the increased systemic vascular resistance observed in congestive heart failure.
There is early clinical evidence that treatment with ETA receptor antagonists and ECE inhibitors can influence favorably the course of human heart failure. ACE inhibitors may also benefit patients with heart failure because of their antiendothelin actions.
Pulmonary hypertension. Expression of ET1 in the lung has been studied by immunocytochemistry and hybridization in situ in specimens from patients with pulmonary hypertension of primary or secondary causes. In contrast to normal lung, specimens from patients with pulmonary hypertension exhibit abundant ET2 immunostaining, particularly over endothelium of markedly hypertrophied muscular pulmonary arteries and plexogenic lesions. Endothelin has been suggested as a potent vasoconstrictor and growth-promoting factor in the pathophysiologic pathophysiologic mechanisms of pulmonary hypertension.
Ventricular and vascular hypertrophy. Endothelin increases DNA synthesis in vascular smooth-muscle ceils, cardiomyocytes, fibroblasts, glial cells, mesangial cells, and other cells; causes expression of protooncogenes; causes cell proliferation; and causes hypertrophy. It acts in synergy with various factors such as transforming growth factor, epidermal growth factor, platelet-derived growth factor, basic fibroblast growth factor and insulin to potentiate cellular transformation and replication. This synergy suggests that all of these factors act through common pathways involving PKC and cyclic adenosine monophosphate. Endothelin per se may not be a direct mediator of angiogenesis but may function as a comitogenic factor.
Neointima formation after vascular wall trauma. The efficacy of coronary angioplasty is limited by the high incidence of restenosis. ET1 induces cultured vascular smooth-muscle cell proliferation by activation of the ETA-receptor subtype, a response that normally is attenuated by an intact, functional endothelium. In addition, ET1 also induces the expression and release of several protooncogenes and growth factors that modulate smooth-muscle cell migration, proliferation, and matrix formulation. In addition to inhibiting smooth-muscle cell proliferation in vitro, endothelin-receptor antagonism with SB 209670 ameliorates the degree of neointima formation observed after rat carotid artery angioplasty. The observations raise the possibility that ET1 antagonists will serve as novel therapeutic agents in the control of restenosis.
| Nonspecific endothelin antagonists ECE inhibitorsAngiotensin-converting-enzyme inhibitorsAngiotensin II receptor blocking agentsCalcium-entry blocking agentsPotassium-channel opening agentsAdenosineNitroglycerin |
SUMMARY
Endothelin is the most potent mammalian vasoconstrictor yet discovered. Its three isoforms play leading roles in regulating vascular tone and causing mitogenesis. The isoforms bind to two major receptor subtypes (ETA and ETB), which mediate a wide variety of physiologic actions in several organ systems. Endothelin may also be a disease marker or an etiologic factor in ischemic heart disease, atherosclerosis, congestive heart failure, renal failure, myocardial and vascular wall hypertrophy, systemic hypertension, pulmonary hypertension, and subarachnoid hemorrhage. Specific and nonspecific receptor antagonists and ECE inhibitors that have been developed interfere with endothelin’s function. Many available cardiovascular therapeutic agents, such as angiotensin-converting-enzyme inhibitors, calcium-entry blocking drugs, and nitroglycerin, also may interfere with endothelin release or may modify its activity. The endothelin antagonists have great potential as agents for use in the treatment of a wide spectrum of disease entities and as biologic probes for understanding the actions of endothelin in human beings.
Endothelin receptor antagonists
Sophie Motte, Kathleen McEntee, Robert Naeije
Pharmacology & Therapeutics 110 (2006) 386 – 414
http://dx.doi.org:/10.1016/j.pharmthera.2005.08.012
Endothelin receptor antagonists (ERAs) have been developed to block the effects of endothelin-1 (ET-1) in a variety of cardiovascular conditions. ET-1 is a powerful vasoconstrictor with mitogenic or co-mitogenic properties, which acts through the stimulation of 2 subtypes of receptors [endothelin receptor subtype A (ETA) and endothelin receptor subtype B (ETB) receptors]. Endogenous ET-1 is involved in a variety of conditions including systemic and pulmonary hypertension (PH), congestive heart failure (CHF), vascular remodeling (restenosis, atherosclerosis), renal failure, cancer, and cerebrovascular disease. The first dual ETA/ETB receptor blocker, bosentan, has already been approved by the Food and Drug Administration for the treatment of pulmonary arterial hypertension (PAH). Trials of endothelin receptor antagonists in heart failure have been completed with mixed results so far. Studies are ongoing on the effects of selective ETA antagonists or dual ETA/ETB antagonists in lung fibrosis, cancer, and subarachnoid hemorrhage. While non-peptidic ET-1 receptor antagonists suitable for oral intake with excellent bioavailability have become available, proven efficacy is limited to pulmonary hypertension, but it is possible that these agents might find a place in the treatment of several cardiovascular and non-cardiovascular diseases in the coming future.
Proposed mechanism by which ET-1 triggers vasoconstriction and vascular remodeling. Activation of G-protein-coupled endothelin receptors leads to stimulation of phospholipase C (PLC) which hydrolyses phosphatidyl inositol biphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol (DAG). DAG opens receptor-operated Ca++ channels (ROC) while IP3 induces Ca++ mobilization from the sarcoplasmic reticulum (SR) and opens store-operated Ca++ channels (SOC) directly or indirectly by store depletion to further increase cytosolic Ca++. This Ca++ increase may also trigger Ca++ release from the SR through ryanodine receptors. Depolarization induced by the opening of non-selective cationic channels (NSCC) via ET-1 and Ca++-activated Cl[1] channels as well as by the inhibition of voltage-gated K+ channels (Kv), opens voltage-dependent Ca++ channels (VDCC) to further increase the Ca++ entry across the plasma membrane. The cytosolic Ca++ increase may also activate Na/H exchangers resulting in alkalinization of the cells and promoting Ca++ influx by activating the Na/Ca exchanger. In addition, the elevated cytosolic Ca++ concentrations and DAG activate the protein kinase C and thus promote cell cycle progression by the Ca++/calmodulin complex (Ca++/CaM) and induction of proto-oncogenes. The intracellular signaling cascade induced by activation of ETB receptor is similar to the ETA receptor one, in stimulating the activation of PLC, generating IP3 and DAG and mobilizing of calcium. However, the PLA2 is also activated via ETB receptors to release prostaglandins (PG) and thromboxane A2 (TXA2).
Endothelin-1 increases isoprenaline-enhanced cyclic AMP levels in cerebral cortex
Marıa J. Perez-Alvareza, MC Calcerrada, F Hernandez, RE Catalan, AM Martınez
Regulatory Peptides 88 (2000) 41–46 PII: S0167-0115(99)00118-4
We examined the effect of ET-1 on cyclic AMP levels in rat cerebral cortex. The peptide caused a concentration-dependent increase of [3 H] cyclic AMP accumulation after 10 min of treatment. This effect was due to adenosine accumulation since it was inhibited by the treatment with adenosine deaminase. ET-1, apart from being able to increase cyclic AMP, also potentiated the cyclic AMP generated by isoprenaline in the presence of adenosine deaminase. Experiments performed in the presence of BQ-123 or BQ-788, specific ETA or ETB receptor antagonists respectively indicated that ET was the receptor involved. This effect was dependent on extracellular and B intracellular calcium concentration. These findings suggest that ET-1 plays a modulatory role in cyclic AMP generation systems in cerebral cortex.
Endothelins And Asthma
Roy G. Goldie and Peter J. Henry
Life Sciences I999; 65(1), pp. I-15, PI1 SOO24-3205(98)00614-6
In the decade since endothelin-1 (ET-l) and related endogenous peptides were first identified as vascular endothelium-derived spasmogens, with potential pathophysiological roles in vascular diseases, there has been a significant accumulation of evidence pointing to mediator roles in obstructive respiratory diseases such as asthma. Critical pieces of evidence for this concept include the fact that ET-l is an extremely potent spasmogen in human and animal airway smooth muscle and that it is synthesised in and released from the bronchial epithelium. Importantly, symptomatic asthma involves a marked enhancement of these processes, whereas asthmatics treated with anti-inflammatory glucocorticoids exhibit reductions in these previously elevated indices. Despite this profile, a causal link between ET-l and asthma has not been definitively established. This review attempts to bring together some of the evidence suggesting the potential mediator roles for ET-l in this disease.
Endothelial Cell Peroxisome Proliferator–Activated Receptor ϒ Reduces Endotoxemic Pulmonary Inflammation and Injury
Aravind T. Reddy, SP Lakshmi, JM Kleinhenz, RL Sutliff, CM Hart, and R. Reddy
J Immunol 2012; 189:5411-5420
http://www.jimmunol.org/content/189/11/5411
Bacterial endotoxin (LPS)-mediated sepsis involves severe, dysregulated inflammation that injures the lungs and other organs Bacterial endotoxin (LPS)-mediated sepsis involves severe, dysregulated inflammation that injures the lungs and other organs, often fatally. Vascular endothelial cells are both key mediators and targets of LPS-induced inflammatory responses. The nuclear hormone receptor peroxisome proliferator–activated receptor ϒ (PPARϒ) exerts anti-inflammatory actions in various cells, but it is unknown whether it modulates inflammation through actions within endothelial cells. To determine whether PPARϒ acts within endothelial cells to diminish endotoxemic lung inflammation and injury, we measured inflammatory responses and mediators in mice with endothelial-targeted deletion of PPARϒ. Endothelial cell PPARϒ (ePPARϒ) knockout exacerbated LPS-induced pulmonary inflammation and injury as shown by several measures, including infiltration of inflammatory cells, edema, and production of reactive oxygen species and proinflammatory cytokines, along with upregulation of the LPS receptor TLR4 in lung tissue and increased activation of its downstream signaling pathways. In isolated LPS-stimulated endothelial cells in vitro, absence of PPARϒ enhanced the production of numerous inflammatory markers. We hypothesized that the observed in vivo activity of the ligand-activated ePPARϒ may arise, in part, from nitrated fatty acids (NFAs), a novel class of endogenous PPARϒ ligands.
Supporting this idea, we found that treating isolated endothelial cells with physiologically relevant concentrations of the endogenous NFA 10-nitro-oleate reduced LPS-induced expression of a wide range of inflammatory markers in the presence of PPARϒ, but not in its absence, and also inhibited neutrophil mobility in a PPARϒ-dependent manner. Our results demonstrate a key protective role of ePPARϒ against endotoxemic injury and a potential ePPARϒ-mediated anti-inflammatory role for NFAs.
Endothelins in health and disease
Rahman Shah
European Journal of Internal Medicine 18 (2007) 272–282
http://dx.doi.org:/10.1016/j.ejim.2007.04.002
Endothelins are powerful vasoconstrictor peptides that also play numerous other roles. The endothelin (ET) family consists of three peptides produced by a variety of tissues. Endothelin-1 (ET-1) is the principal isoform produced by the endothelium in the human cardiovascular system, and it exerts its actions through binding to specific receptors, the so-called type A (ETA) and type B (ETB) receptors. ET-1 is primarily a locally acting paracrine substance that appears to contribute to the maintenance of basal vascular tone. It is also activated in several diseases, including congestive heart failure, arterial hypertension, atherosclerosis, endothelial dysfunction, coronary artery diseases, renal failure, cerebrovascular disease, pulmonary arterial hypertension, and sepsis. Thus, ET-1 antagonists are promising new agents. They have been shown to be effective in the management of primary pulmonary hypertension, but disappointing in heart failure. Clinical trials are needed to determine whether manipulation of the ET system will be beneficial in other diseases.
The production of ET receptors is affected by several factors. Hypoxia, cyclosporine, epidermal growth factor, basic fibroblast growth factor, cyclic AMP, and estrogen upregulate ETA receptors in some tissues, and C-type natriuretic hormone, angiotensin II, and perhaps basic fibroblast growth factor up-regulate ETB receptors. In contrast, the endothelins, angiotensin II, platelet-derived growth factor, and transforming growth factor down-regulate ETA receptors, whereas cyclic AMP and catecholamines down-regulate ETB receptors.
The ETA receptor contains 427 amino acids and binds with the following affinity: ET-1N>T-2>ET-3. It is predominantly expressed in vascular smooth muscle cells and cardiac myocytes. Its interaction with ET-1 results in vasoconstriction and cell proliferation. In contrast, the ETB receptor contains 442 amino acids and binds all endothelins with equal affinity. It is predominantly expressed on vascular endothelial cells and is linked to an inhibitory G protein. Activation of ETB receptors stimulates the release of NO and prostacyclin, prevents apoptosis, and inhibits ECE-1 expression in endothelial cells. ETB receptors also mediate the pulmonary clearance of circulating ET-1 and the re-uptake of ET-1 by endothelial cells.
All three endothelins cause transient endothelium dependent vasodilatation before the development of constriction, though this is most apparent for ET-1. Endothelins induce vasodilatation via the endothelial cell ETB receptors through generation of endothelium-derived dilator substances (Fig. 3), including nitric oxide (NO), which perhaps acts by physiologically antagonizing ETA receptor mediated vasoconstriction. The transient early vasodilator actions of the endothelins are attenuated by NO synthase inhibitors. Additionally, ET-1 increases generation of prostacyclin by cultured endothelial cells, whereas cyclo-oxygenase inhibitors potentiate ET-1-induced constriction, suggesting that vasodilator prostaglandins play a similar modulatory role.
It has been proposed that ET-1 can affect vascular tone indirectly through its effect on the sympathetic nervous system, and it has been shown that that ET-1 may increase peripheral sympathetic activity through postsynaptic potentiation of the effects of norepinephrine. While in vitro low concentrations of ET-1 potentiate the effects of other vasoconstrictor hormones, including norepinephrine and serotonin, these findings have not been confirmed in vivo in the forearm resistance bed of healthy subjects. In addition to its action on vascular vasomotion, ET-1 is thought to be a mediator in the vascular remodeling process. It seems that ET-1 interactions with the renin–angiotensin–aldosterone system play a significant role in this remodeling process.

Vascular actions of endothelin-1
Vascular actions of endothelin-1. Modified from – Galie N, Manes A, Branzi A; The endothelin system in pulmonary arterial hypertension. Cardiovasc Res 2004;61:227–37.
ET-1 appears to have a diverse role as a modulator of vascular tone and growth and as a mediator in many cardiovascular and non-cardiovascular diseases. To date, no disease entity, however, has been attributed solely to an abnormality in ET-1. Yet, ET-1 receptor antagonists have been studied in clinical trials involving a wide spectrum of cardiovascular diseases, though the only proven efficacy has been in patients with PAH.
Learning points
- Endothelins are powerful vasoconstrictors and major regulators of vascular tone.
- The endothelin (ET) family consists of three peptides (ET-1 ∼60%, ET-2 ∼30%, and ET-3 ∼10%) produced by a variety of tissues.
- ET-1 is the principal isoform produced by the endothelium in the human cardiovascular system and appears to be foremost a locally acting paracrine substance rather than a circulating endocrine hormone.
- Several human studies suggest that circulating ET-1 levels, which are elevated in heart failure and pulmonary hypertension, correlate with the prognosis of the disease.
- ET-1 antagonists have been shown to be effective in the management of primary pulmonary hypertension, but disappointing in heart failure.
- Clinical trials are needed to investigate the role of ET-1 receptor antagonists for other conditions, as ET-1 levels have been shown to be elevated in arterial hypertension, atherosclerosis, endothelial dysfunction, coronary artery disease, renal failure, cerebrovascular disease, and sepsis.
In Vitro Stability and Intestinal Absorption Characteristics of Hexapeptide Endothelin Receptor Antagonists
Hyo-kyung Han, BH Stewart, AM Doherty, WL Cody and GL Amidon
Life Sciences. I998; 63(18), pp. 1599-1609. PI1 SOO24-3205(98)00429-9
Endothelins are potent vasoconstrictor peptides which have a wide range of tissue distribution and three receptor subtypes (ETA ETB and ETC). Among the linear hexapeptide ETA / ETB receptor antagonists, PD 145065 (Ac-D-Bhg-L-Leu-L-Asp-L-Ile-L-Ile-L-Trp, Bhg = (10,ll -dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-Gly) and PD 156252 (Ac-o-Bhg-L-Leu-L-Asp-L-Ile-(N-methyl)-L-Ile-L-Trp) were selected to evaluate the metabolic stability and intestinal absorption in the absence and/or in the presence of protease inhibitors. In vitro stability of both compounds was investigated in fresh plasma, lumenal perfusate, intestinal and liver homogenates. PD 156252 was more stable than PD 145065 in intestinal tissue homogenate (63.4% vs. 20.5% remaining) and liver homogenate (74.4% vs. 35.5 % remaining), while both compounds showed relatively good stability in the fresh plasma (94.5% vs. 86.7% remaining) and lumenal perfusate (85.8% vs. 72.3% remaining). The effect of protease inhibitors on the degradation of PD 145065 and PD 156252 was also investigated. Amastatin, thiorphan, chymostatin and the mixture of these three inhibitors were effective in reducing the degradation of both compounds. The pharmacokinetic parameters of PD 156252, calculated by using a non-compartmental model, were 6.95 min (terminal half-life), 191 mL (Vss), and 25.5 mL/min (Cltot) after intravenous administration in rats. The intestinal absorption of PD 156252 in rats was evaluated in the absence and/or in the presence of protease inhibitors. The results indicate that the major elimination pathway of PD 156252 appears to be the biliary excretion and protease inhibitors increase the intestinal absorption of PD 156252 through increasing metabolic stability.
Inhibitory and facilitatory presynaptic effects of endothelin on sympathetic cotransmission in the rat isolated tail artery
Violeta N. Mutafova-Yambolieva & David P. Westfall
British Journal of Pharmacology (1998) 123, 136 – 142
1 The present study was undertaken to determine the modulatory effects of the endothelin peptides on the neurogenically-induced release of endogenous noradrenaline (NA) and the cotransmitter adenosine 5′-triphosphate (ATP) from the sympathetic nerves of endothelium-free segments of the rat isolated tail artery. The electrical field stimulation (EFS, 8 Hz, 0.5 ms, 3 min) evoked over¯ow of NA and ATP, in the absence of endothelins, was 0.035+0.002 pmol mg71 tissue and 0.026+0.002 pmol mg71 tissue, respectively.
2 Endothelin-1 (ET-1; 1 ± 30 nM) significantly reduced the EFS evoked overflow of both NA and ATP. The maximum inhibitory effect was produced by a peptide concentration of 10 nM, the amount of NA overflow being 0.020+0.002 pmol mg71 and that of ATP overflow 0.015+0.001 pmol mg71. Higher peptide concentrations (100 and 300 nM) reversed the EFS-evoked overflow of NA to control levels and that of ATP to above control levels. The inhibitory effect of ET-1 (10 nM) was resistant to the selective ETA receptor antagonist cyclo-D-Trp-D-Asp(ONa)-Pro-D-Val-Leu (BQ-123) but was prevented by ETB receptor desensitization with sarafotoxin S6c (StxS6c) or by ETB receptor blockade with N, cis-2,6-dimethyl-piperidinocarbonyl-L-gmethylleucyl-D-1-methoxycarbonyl-tryptophanyl-D-norleucine (BQ-788).
3 StxS6c, upon acute application, exerted a dual effect on transmitter release. At concentrations of 0.001 ± 0.3 nM the peptide significantly reduced the EFS-evoked NA overflow, whereas at concentrations of 1 ± 10 nM it caused a significant increase in the evoked overflow of both ATP and NA. Both the maximum inhibitory effect of StxS6c at a concentration of 0.003 nM approximately 85% reduction of NA overflow and 40% of ATP overflow) and the maximum facilitatory effect of the peptide at a concentration of 3 nM (approximately 400% increase of ATP overflow and 200% of NA overflow) were completely antagonized by either BQ-788 or by StxS6c-induced ETB receptor desensitization.
4 ET-3 (10 ± 100 nM) did not aect the EFS evoked overflow of either ATP or NA, but at a concentration of 300 nM significantly potentiated the release of both transmitters (0.118+ 0.02 pmol mg71 tissue ATP overflow and .077+0.004 pmol mg71 NA overflow). This effect was prevented either by BQ-123 or by BQ-788.
5 In summary, the endothelin peptides exerted both facilitatory and inhibitory effects on the neurogenically-induced release of the sympathetic cotransmitters ATP and NA in the rat tail artery. Both transmitters were modulated in parallel indicating that the endothelins do not differentially modulate the release of NA and ATP in this tissue.
Involvement of the central adrenomedullin peptides in the baroreflex
Meghan M. Taylo, Cynthia A. Keown, Willis K. Samson
Regulatory Peptides 112 (2003) 87– 93
http://dx.doi.org:/10.1016/S0167-0115(03)00026-0
The peptides derived from post-translational processing of preproadreno-medullin are produced in and act on areas of the autonomic nervous system important for blood pressure regulation. We examined the role of endogenous, brain-derived adrenomedullin (AM) and proadrenomedullin N-terminal 20 peptide (PAMP) in the central nervous system arm of the baroreflex by using passive immunoneutralization to block the actions of the endogenous peptides. Our results indicate that the preproadrenomedullin-derived peptides do not play a role in sensing changes in blood pressure (baroreflex sensitivity), but the adrenomedullin peptides do regulate the speed with which an animal returns to a normal, stable blood pressure. These findings suggest that endogenous, brain-derived AM and PAMP participate in the regulation of autonomic activity in response to baroreceptor activation and inactivation.
Pharmacological characterization of cardiovascular responses induced by endothelin-1 in the perfused rat heart
Keiji Kusumoto, A Fujiwara, S Ikeda, T Watanabe, M Fujino
Eur J Pharmacology 296 (1996) 65-74 SSDI 0014-2999(95)00680-X
The effects of the endothelin receptor antagonist TAK-044 (cyclo[D-α-aspartyl-3-[(4-phenylpiperazin-l-yl)carbonyl]-L-alanyl-L-α-aspartyl-D-2-(2-thienyl)-glycyl-L-leucyl-D-tryptophyl] disodium salt) and BQ-123 (cyclo[D-Asp-Pro-D-VaI-Leu-D-Trp]) were studied in the rat heart to characterize the receptor subtypes responsible for the cardiovascular actions of endothelin-1. Endothelin-1 induced a transient decrease and subsequent increase in perfusion pressure in perfused rat hearts, and increased left ventricular developed pressure. TAK-044 diminished these endothelin-l-induced responses (100 pmol/heart) with IC50 values of 140, 57 and 1.3 nM, respectively. BQ-123 (1-30/µM) partially inhibited the endothelin-l-induced hypertension (30-40%) in the rat heart, and failed to inhibit the hypotension. The positive inotropic effect of endothelin-1 was abolished by BQ-123. Neither indomethacin (10/µM) nor N’°-nitro-L-arginine methyl ester (100/pM) attenuated the endothelin-l-induced hypotension. TAK-044 and BQ-123 attenuated the positive inotropic effect of endothelin-1 in rat papillary muscles. In rat cardiac membrane fractions, TAK-044 and BQ-123 inhibited [125I]endothelin-1 binding to endothelin ET A receptors with IC50 values of 0.39 + 0.6 and 36 + 9 nM, respectively, whereas only TAK-044 potently blocked the endothelin ET B receptor subtype (IC50 value: 370 + 180 nM). These results suggest that endothelin-1 modulates cardiovascular functions in the rat heart by activating both endothelin ET A and endothelin ET B receptors, all of which are sensitive to TAK-044.
Molecular Pharmacology and Pathophysiological Significance of Endothelin
Katsutoshi Goto, Hiroshi Hama and Yoshitoshi Kasuya
Jp J Pharmacol 1996; 72: 261-290
Since the discovery of the most potent vasoconstrictor peptide, endothelin, in 1988, explosive investigations have rapidly clarified much of the basic pharmacological, biochemical and molecular biological features of endothelin, including the presence and structure of isopeptides and their genes (endothelin- 1, -2 and -3), regulation of gene expression, intracellular processing, specific endothelia converting enzyme (ECE), receptor subtypes (ETA and ETB), intracellular signal transduction following receptor activation, etc. ECE was recently cloned, and its structure was shown to be a single transmembrane protein with a short intracellular N-terminal and a long extracellular C-terminal that contains the catalytic domain and numerous N-glycosylation sites. In addition to acute contractile or secretory actions, endothelin has been shown to exert long-term proliferative actions on many cell types. In this case, intracellular signal transduction appears to converge to activation of mitogen-activated protein kinase. As a recent dramatic advance, a number of non-peptide and orally active receptor antagonists have been developed. They, as well as current peptide antagonists, markedly accelerated the pace of investigations into the true pathophysiological roles of endogenous endothelin-1 in mature animals.
The discovery of endothelin in 1988 soon triggered explosive investigations of a worldwide scale, presumably due to its unusual characteristics; i.e., marked potency and long-lasting pressor actions. As a result, most of the basic problems concerned with the science of endothelin have rapidly been solved; e.g., features and regulations of the expression of endothelin genes, biosynthetic pathways including characterization and cloning of endothelin converting enzyme, pharmacological, biochemical and molecular-biological identification of endothelin receptor subtypes, intracellular signal transduction following receptor activation, and discovery of various receptor agonists and antagonists. In addition to its potent cardiovascular actions, endothelin-1 shows a wide variety of biological effects, including contraction of nonvascular smooth muscle (intestinal, tracheal, broncheal, mesangial, bladder, uterine and prostatic smooth muscle), stimulation of neuropeptides, pituitary hormone and atrial natriuretic peptide release and aldosterone biosynthesis, modulation of neurotransmitter release, and increase of bone resorption. Furthermore, endothelin-1 has mitogenic properties and causes proliferation and hypertrophy of a number of cell types, including vascular smooth muscle cells, cardiac myocytes, mesangial cells, bronchial smooth muscle cells and fibroblasts. Endothelin-1 also induces the expression of several protooncogenes (c fos, C -Jun, c-myc, etc.).
These actions, whereby endothelin- 1 might influence the development of cellular hypertrophy/hyperplasia, are of potential significance in pathophysiological conditions associated with long-term changes in cardiovascular tissues, e.g., hypertension, myocardial infarction, chronic heart failure, vascular restenosis following balloon angioplasty, and atherosclerosis. These pathophysiological conditions are usually associated with increased plasma levels of endothelin-1, although the correlation is relatively poor. Nevertheless, a considerable increase in the tissue content of endothelin-1 has been gradually uncovered in many cases of these conditions. Even if the concentration of endothelin-1 at the cell surface is not high enough to induce contraction, it is well known that subthreshold concentrations of endothelin will enhance or potentiate the contraction produced by other vasoconstrictors (e.g., norepinephrine, serotonin, angiotensin II), indicating the existence of cross-talk among various vasoactive substances. Another important cross-talk among these substances may be mutual enhancement or inhibition of their expression in various tissues. In addition to these interactions, the true physiological and/or pathophysiological roles of each of the endothelin family peptide and receptor subtypes remain to be investigated.
Hydrogen Sulfide and Endothelium-Dependent Vasorelaxation
Jerzy Bełtowski, and Anna Jamroz-Wiśniewska
Molecules 2014, 19, 21183-21199; http://dx.doi.org:/10.3390/molecules191221183
In addition to nitric oxide and carbon monoxide, hydrogen sulfide (H2S), synthesized enzymatically from L-cysteine or L-homocysteine, is the third gasotransmitter in mammals. Endogenous H2S is involved in the regulation of many physiological processes, including vascular tone. Although initially it was suggested that in the vascular wall H2S is synthesized only by smooth muscle cells and relaxes them by activating ATP-sensitive potassium channels, more recent studies indicate that H2S is synthesized in endothelial cells as well. Endothelial H2S production is stimulated by many factors, including acetylcholine, shear stress, adipose tissue hormone leptin, estrogens and plant flavonoids. In some vascular preparations H2S plays a role of endothelium-derived hyperpolarizing factor by activating small and intermediate-conductance calcium-activated potassium channels. Endothelial H2S signaling is up-regulated in some pathologies, such as obesity and cerebral ischemia-reperfusion. In addition, H2S activates endothelial NO synthase and inhibits cGMP degradation by phosphodiesterase thus potentiating the effect of NO-cGMP pathway. Moreover, H2S-derived polysulfides directly activate protein kinase G. Finally, H2S interacts with NO to form nitroxyl (HNO)—a potent vasorelaxant. H2S appears to play an important and multidimensional role in endothelium-dependent vasorelaxation.
GPCR modulation by RAMPs
Debbie L. Hay, David R. Poyner, Patrick M. Sexton
Pharmacology & Therapeutics 109 (2006) 173 – 197
http://dx.doi.org:/10.1016/j.pharmthera.2005.06.015
Our conceptual understanding of the molecular architecture of G-protein coupled receptors (GPCRs) has transformed over the last decade. Once considered as largely independent functional units (aside from their interaction with the G-protein itself), it is now clear that a single GPCR is but part of a multifaceted signaling complex, each component providing an additional layer of sophistication. Receptor activity modifying proteins (RAMPs) provide a notable example of proteins that interact with GPCRs to modify their function. They act as pharmacological switches, modifying GPCR pharmacology for a particular subset of receptors. However, there is accumulating evidence that these ubiquitous proteins have a broader role, regulating signaling and receptor trafficking. This article aims to provide the reader with a comprehensive appraisal of RAMP literature and perhaps some insight into
the impact that their discovery has had on those who study GPCRs.
RAMPs were first identified during attempts to expression clone a receptor for the neuropeptide calcitonin gene related peptide (CGRP; McLatchie et al., 1998). Historical evidence had suggested that CGRP acted through a GPCR, as its binding had proven sensitive to GTP analogues and stimulation of various tissues and cells led to the accumulation of cAMP, suggesting activation of a Gs-coupled GPCR. However, attempts to clone such a receptor proved difficult. A putative canine CGRP receptor, RDC-1, was identified in 1995, but the original findings have not been replicated and current IUPHAR guidelines do not consider this receptor a genuine CGRP receptor (Kapas & Clark, 1995; Poyner et al., 2002). Shortly afterward, a further orphan receptor (CL, a close homologue of the calcitonin receptor) was shown to be activated by CGRP when transfected into HEK293 cells (Aiyar et al., 1996). This finding posed something of a conundrum since earlier attempts to examine the function of this receptor (or its rat homologue) in Cos 7 cells had not given positive results with CGRP.
Given the apparent functionality of the human CL receptor in HEK293 cells, the rat homologue was also transfected into this cell type and now responded to CGRP (Han et al., 1997). The authors speculated that there was a factor present in HEK293 cells that conferred high affinity for CGRP on the receptor.
In 1998, McLatchie and colleagues confirmed this speculation and provided new insights into the way that GPCRs and their pharmacology can be regulated (McLatchie et al., 1998). It was discovered that a novel family of single transmembrane domain proteins, termed RAMPs, was required for functional expression of CL at the cell surface, explaining why it had been so difficult to observe CGRP binding or function when CL was transfected into cells lacking RAMP expression (Fluhmann et al., 1995; Han et al., 1997; McLatchie et al., 1998). RAMPs were first identified from a library derived from SK-N-MC cells, cells known to express CGRP receptors. An expression-cloning strategy was utilized, whereby an SK-N-MC cDNA library was transcribed and the corresponding cRNA was used for injection into Xenopus oocytes. Cystic
fibrosis transmembrane regulator chloride conductance, a reporter for cAMP formation, was strongly potentiated by a single cRNA pool (in the presence of CGRP). Subsequently, a single cDNA encoding a 148-amino-acid protein comprising RAMP1 was isolated. The structure of the protein was unexpected, as it was not a GPCR and it did not respond to CGRP in mammalian cells. Thus, it was postulated that RAMP1 might potentiate CGRP receptors. A CL/RAMP1 co-transfection experiment supported this hypothesis.
CGRP/AM on the outside of the cell and did not simply act as anchoring/chaperone proteins for CL. RAMPs therefore provide a novel mechanism for modulating receptor–ligand specificity. The unique pharmacological profiles supported by RAMPs are discussed in later sections.
Fig. (not shown). CGRP1 receptor-specific small molecule antagonists. The small molecule antagonist BIBN4096 BS (brown) is a specific antagonist of the CGRP1 receptor, acting at the interface between RAMP1 and the CL receptor to inhibit CGRP action. At least part of the binding affinity for BIBN4096 BS arises from interaction with Trp74 (red) of RAMP1. In contrast, antagonists that bind principally to the CL component of the complex will not discriminate between different CL/RAMP complexes.
The classic function attributed to RAMPs is their ability to switch the pharmacology of CL, thus providing a novel mechanism for modulating receptor specificity. Thus, the CL/RAMP1 complex is a high affinity CGRP receptor, but in the presence of RAMP2, CL specificity is radically altered, the related peptide AM being recognized with the highest affinity and the affinity for CGRP being reduced ¨100-fold. While AM is the highest affinity peptide, CGRP is recognized with moderate, rather than low affinity. Indeed, depending on the species and the form of CGRP (h vs. a), the separation between the 2 peptides can be as little as 10-fold (Hay et al., 2003a). This may particularly be true if receptor components of mixed species are used. The detailed pharmacology of the CGRP and AM receptors formed by RAMP interaction with CL has recently been reviewed (Born et al., 2002; Poyner et al., 2002; Hay et al., 2004; Kuwasako et al., 2004).
Fig. (not shown). The broadening spectrum of RAMP–receptor interactions. RAMPs can interact with multiple receptor partners. All RAMPs interact with the calcitonin receptor-like receptor (CL-R), the calcitonin receptor (CTR), and the VPAC1 receptor, while the glucagon and PTH1 receptors interact with RAMP2, the PTH2 receptor with RAMP3, and the calcium sensing receptor (CalS-R) with RAMP1 or RAMP3. The consequence of RAMP interaction varies. For the CL and CalS receptors, RAMPs play a chaperone role, allowing cell surface expression. For the CL and calcitonin receptors, RAMP interaction leads to novel receptor binding phenotypes . There is also evidence that RAMP interaction will modify signaling, and this has been seen for the VPAC1–RAMP2 heterodimer and for calcitonin receptor/RAMP complexes. In many instances, however, the consequence of RAMP interaction has yet to be defined.
Overall, the distribution data presented so far are supportive of the hypothesis that RAMP and CL or calcitonin receptor combinations are able to account for the observed CGRP, AM, and AMY pharmacology. A salient point for CGRP receptors relates to the cerebellum, where the lack of CL mRNA in some studies despite abundant CGRP binding has prompted speculation of alternative CGRP receptors (Oliver et al., 2001; Chauhan et al., 2003). Nevertheless, this apparent lack is study dependent and CL has been identified in cerebellum in other studies.
Some consideration has been given to the potential role that RAMPs may have in modifying receptor behaviors other than ligand binding pharmacology. An additional functional consequence might be that of alteration of receptor signaling characteristics.
While there is currently little evidence for signaling modifications of CL-based receptors in association with RAMPs, a completely different paradigm is evident for the VPAC1 receptor. This receptor has strong interactions with all 3 RAMPs, but its pharmacology, in terms of agonist binding, does not appear to be modified by their presence. On the other hand, there was a clear functional consequence of RAMP2 overexpression with the VPAC1 receptor where PI hydrolysis was specifically augmented relative to cAMP, which did not change. The potency of the response (EC50 of vasoactive intestinal peptide) was not altered, but the maximal PI hydrolysis response was elevated in the presence of RAMP2 . It has been suggested that this may reflect a change in compartmentalization of the receptor signaling complex. Such augmentation was not evident for the interaction of the VPAC1 receptor with RAMP1 or RAMP3; in these cases, the outcome of heterodimerization may be more subtle or involve the modification of different receptor parameters such as trafficking.
RAMPs transformed our understanding of how receptor pharmacology can be modulated and provided a novel mechanism for generating receptor subtypes within a subset of family B GPCRs. Their role has now broadened and they have been shown to interact with several other family B GPCRs, in 1 case modifying signaling parameters. There is now evidence to suggest that their interactions also reach into family C, and possibly family A, GPCRs, indicating that their function may not be restricted to modulation of a highly specific subset of receptors. Indeed, many aspects of RAMP function remain poorly understood, and the full extent of their action remains to be explored.
Receptor activity modifying proteins
Patrick M. Sexton, Anthony Albiston, Maria Morfis, Nanda Tilakaratne
Cellular Signalling 13 (2001) 73-83 PII: S0898-6568(00)00143-1
Our understanding of G protein-coupled receptor (GPCR) function has recently expanded to encompass novel protein interactions that underlie both cell-surface receptor expression and the exhibited phenotype. The most notable examples are those involving receptor activity modifying proteins (RAMPs). RAMP association with the calcitonin (CT) receptor-like receptor (CRLR) traffics this receptor to the cell surface where individual RAMPs dictate the expression of unique phenotypes. A similar function has been ascribed to RAMP interaction with the CT receptor (CTR) gene product. This review examines
our current state of knowledge of the mechanisms underlying RAMP function.
It is now evident that RAMPs can interact with receptors other than CRLR. Expression of amylin receptor phenotypes requires the coexpression of
RAMPs with the CTR gene product. However, as seen in CRLR, the phenotype engendered by individual RAMPs was distinct. In COS-7 or rabbit aortic endothelial cells (RAECs), RAMP1 and RAMP3 induced amylin receptors that differ in their affinity for CGRP, while RAMP2 was relatively ineffective in inducing amylin receptor phenotype. RAMP2 can also induce an amylin receptor phenotype, which is distinct from either the RAMP1- or RAMP3-induced receptors. However, the efficacy of RAMP2 was highly dependent upon the cellular background and the isoform of CTR used in the study.
In humans, the major CTR variants differ by the presence or absence of a 16 amino acid insert in the first intracellular domain, with the insert negative isoform (hCTRI1ÿ) being the most commonly expressed form and the variant used for initial studies with RAMPs. Unlike hCTRI1ÿ, cotransfection of the hCTRI1+ variant with any of the RAMPs into COS-7 cells caused strong induction of amylin receptor phenotype. The hCTR isoforms differ in their ability to activate signaling pathways (presumably due to an effect on G protein coupling) and to internalize in response to agonist treatment, which may suggest a role for G proteins in the ability of RAMPs to alter receptor phenotype.
There are at least three potential consequences of RAMP interaction with its associating receptors. The first is trafficking of receptor protein from an intracellular compartment to the cell surface. The second is an alteration in
the terminal glycosylation of the receptor, and the third is alteration of receptor phenotype, presumably through a direct or indirect effect on the ligand-binding site.

potential actions of RAMPs
Schematic diagram illustrating potential actions of RAMPs. (A) RAMPs facilitate the trafficking of CRLR from an intracellular compartment to the cell surface. (B) RAMP1 (but not RAMP2 or RAMP3) modifies the terminal glycosylation
of CRLR. (C) The cell surface RAMP1±CRLR complex is a Type 1 CGRP receptor, which displays a 1:1 stoichiometry. (D,E) Cell surface RAMP2±CRLR and RAMP3±CRLR complexes are adrenomedullin receptors. (F,G) For at least RAMP1 and RAMP3, RAMPs form stable homodimers, although the function
of these complexes is unknown. (H) Unlike CRLR, the CTR gene product is trafficked to the cell surface in the absence of RAMPs, where it displays classical CTR phenotype. (I,J) RAMP1± and RAMP3±CTR complexes form distinct amylin receptors. RAMP2 can also generate a separate amylin receptor phenotype (not illustrated). (C ±E,I,J) RAMPs are trafficked with either receptor to the plasma membrane. (K) For all three RAMP±CRLR complexes, agonist treatment causes clathrin-mediated internalization of both CRLR and RAMP.
(L) The majority of the internalized complex is targeted to the lysosomal-degradation pathway.
The data from Zumpe et al. suggest that RAMP2 interacts more weakly with the hCTRI1ÿ than RAMP1, and that the affinity of this interaction derives principally from the transmembrane domain/C-terminus (Ct) of the RAMPs. As RAMP3 induces an amylin receptor phenotype in COS-7 cells where RAMP2 is relatively weak, it is inferred that RAMP3 interaction with the hCTRI1ÿ is probably greater than that of RAMP2. Nonetheless, this has not been examined empirically. Given the recent data suggesting a potential role for G protein coupling in expression of RAMP-induced phenotype, it is also possible that the strength of RAMP interaction is, at least partially, dictated by receptor-G protein or RAMP-G protein interaction.
The discovery of RAMPs has led to a greater understanding of the nature of receptor diversity. However, although much progress has been made into elucidating the molecular mechanism of RAMP action, emerging data continue to open up new areas for investigation. These include identification of other RAMP-interacting receptors, understanding of the role of specific G proteins in RAMP-receptor function and the potential importance of RAMP regulation in disease progression. It also seems likely that the RAMP-receptor interface can provide a useful target for future drug development.
Cardiovascular endothelins: Essential regulators of cardiovascular homeostasis
Friedrich Brunner, C Bras-Silva, AS Cerdeira, AF Leite-Moreira
Pharmacology & Therapeutics 111 (2006) 508 – 531
http://dx.doi.org:/10.1016/j.pharmthera.2005.11.001
The endothelin (ET) system consists of 3 ET isopeptides, several isoforms of activating peptidases, and 2 G-protein-coupled receptors, ETA and ETB, that are linked to multiple signaling pathways. In the cardiovascular system, the components of the ET family are expressed in several tissues, notably the vascular endothelium, smooth muscle cells, and cardiomyocytes. There is general agreement that ETs play important physiological roles in the regulation of normal cardiovascular function, and excessive generation of ET isopeptides has been linked to major cardiovascular pathologies, including hypertension and heart failure. However, several recent clinical trials with ET receptor antagonists were disappointing.
In the present review, the authors take the stance that ETs are mainly and foremost essential regulators of cardiovascular function, hence that antagonizing normal ET actions, even in patients, will potentially do more harm than good. To support this notion, we describe the predominant roles of ETs in blood vessels, which are (indirect) vasodilatation and ET clearance from plasma and interstitial spaces, against the background of the subcellular mechanisms mediating these effects. Furthermore, important roles of ETs in regulating and adapting heart functions to different needs are addressed, including recent progress in understanding the effects of ETs on diastolic function, adaptations to changes in preload, and the interactions between endocardial-derived ET-1 and myocardial pump function. Finally, the potential dangers (and gains) resulting from the suppression of excessive generation or activity of ETs occurring in some cardiovascular pathological states, such as hypertension, myocardial ischemia, and heart failure, are discussed.
Figure (not shown): Synthesis of ET and its regulation. The release of active ET-1 is controlled via regulation of gene transcription and/or endothelin converting enzyme activity. ET-1 synthesis is stimulated by several factors, of which hypoxia seems to be the most potent in humans (see text). ET-1 formation is down-regulated by activators of the NO/cGMP pathway and other factors.
Figure (not shown): Vascular actions of ET. In healthy blood vessels, the main action of ET-1 is indirect vasodilatation mediated by ETB receptors located on endothelial cells. Their activation generates a Ca2+ signal via PLC that turns on the generation of NO, prostacyclin, adrenomedullin, and other mediators that are powerful relaxants of smooth muscle. On the other hand, binding of ET-1 to ETA receptors located on smooth muscle cells will lead to vascular contraction (physiological effect) and/or wall thickening, inflammation, and tissue remodeling (pathological effects). These latter effects may partly be mediated by vascular ETB2 receptors in certain disease states. Smooth muscle cell signaling involves DAG formation, PKC activation, and extracellular Ca2+ recruited via different cation channels. The specificity of the cellular response resides at the level of G proteins, that is, G-as or G-aq in the case of ETA, G-ai or G-aq for ETB.

signal transduction mechanisms involved in ET-1-mediated positive (left) and negative (right) inotropic effects
Summary of proposed signal transduction mechanisms involved in ET-1-mediated positive (left) and negative (right) inotropic effects. Left: Stimulation of ETA receptors causes Gq protein-directed activation of PLC, formation of IP3 and DAG, and activation of NHE-1. Increased contractile force is the result of (i) Ca2+ release from the sarco(endo)plasmic reticulum, (ii) sensitization of cardiac myofilaments to Ca2+ due to cellular alkalosis, and (iii) increased Ca2+ influx through the NCX operating in reverse mode. The contribution of voltage-gated L-type Ca2+ channels to the systolic Ca2+ transient is unknown, as is the role of myocyte ETB2 receptors. Right: The ET receptor subtypes mediating negative inotropic effects are poorly known. Two main signaling mechanisms involve (i) inhibition of adenylyl cyclase (AC), guided by a G protein, of unknown binding preference, which results in decreased levels of cAMP; (ii) cGMP-mediated activation of phosphatases that dephosphorylate putative targets resulting from cAMP/protein kinase A (PKA) activation. Other kinases like PKC and PKG have also been implicated in accentuated force antagonism.
Adrenomedullin (11–26): a novel endogenous hypertensive peptide isolated from bovine adrenal medulla
Kazuo Kitamuraa,*, Eizaburo Matsuia, Jhoji Katoa, Fumi Katoha
Peptides 22 (2001) 1713–1718 PII: S0196-9781(01)00529-0
Adrenomedullin (AM) is a potent hypotensive peptide originally isolated from pheochromocytoma tissue. Both the ring structure and the C-terminal amide structure of AM are essential for its hypotensive activity. We have developed an RIA which recognizes the ring structure of human AM. Using this RIA, we have characterized the molecular form of AM in bovine adrenal medulla. Gel filtration chromatography revealed that three major peaks of immunoreactive AM existed in the adrenal medulla. The peptide corresponding to Mr 1500 Da was further purified to homogeneity. The peptide was determined to be AM (11–26) which has one intramolecular disulfide bond. Amino acid sequences of bovine AM and its precursor were deduced from the analyses of cDNA encoding bovine AM precursor. The synthetic AM (11–26) produced dose-dependent strong pressor responses in unanesthetized rats in vivo. The hypertensive activity lasted about one minute, and a dose dependent increase in heart rate was also observed. The present data indicate that AM (11–26) is a major component of immunoreactive AM in bovine adrenal medulla and shows pressor activity.
The pressor effect of AM(11–26) was examined by methods similar to those reported for Neuropeptide Y.
We have established a sensitive RIA system using a monoclonal antibody which recognizes the ring structure of human AM. Human AM antiserum recognized the peptide with high affinity at a final dilution of 1:2,800,000. The half maximal inhibition of radioiodinated ligand binding by human AM was observed at 10 fmol/tube. From 1 to 128 fmol/tube of AM was measurable by this RIA system. The intra- and inter-assay coefficients of variance were less than 6% and 9%, respectively. This RIA had 100% cross-reactivity with human AM(13–31), (1–25), (1–52)Gly and AM(1–52)CONH2, but less than 1% cross-reactivity with rat AM.
Sephadex G-50 gel-filtration of strongly basic peptide extract (SP-III) in bovine adrenal medulla identified three major peaks of immunoreactive AM. One emerged at the identical position of authentic AM, the other two unknown peaks were eluted later at molecular weights estimated to be 3000 and 1500 Da, respectively. The peptide corresponding to Mr 1500 Da was further purified.
The purified peptide (20 pmol) was subjected to a gas phase sequencer, and the amino acid sequence was determined up to the 16th residue, which was found to be C terminus . It was found that the purified peptide was AM (11–26). The structure of AM (11–26) was confirmed by chromatographic comparison with native AM (11–26) as well as a synthetic AM (11–26), which has one intramolecular disulfide bond.
3 clones were isolated, and the clone designated pBAM-2, which harbored the longest insert of 1,438 base, was used for sequencing. The bovine AM cDNA contained a single open reading frame encoding a putative 188 amino acid polypeptide. The first 21-residue peptide is thought to be a signal peptide. The bovine AM propeptide contains three signals of dibasic amino acid sequences, Lys-Arg or Arg-Arg. The first Lys-Arg followed proadrenomedullin N-terminal 20 peptide (PAMP) sequences. AM is located between the second signal of Lys-Arg and the third signal of Arg-Arg. Gly residues, which are donors of C-terminal amide structure of PAMP and AM, are found before the first and third signal of Lys-Arg and Arg-Arg. Bovine AM consists of 52 amino acids and is identical to human AM with exception of four amino acids. Bovine PAMP consists of 20 amino acids and is identical to human PAMP with exception of one amino acid. The present cDNA sequence encoding bovine AM precursor is almost identical to those of the reported AM cDNA sequences from bovine aortic endothelial cells. However, a difference in one amino acid was found in the sequences of signal peptide. In addition, three different residues of nucleotides were found in the noncoding region of cDNA encoding bovine preproadreno-medullin.
AM(11–26) elicited a potent hypertensive effects in unanesthetized rats.
When AM(11–26) at 20 nmol/kg was injected i.v., the maximum increase of mean blood pressure was 50 7.1 mmHg. Similarly, the synthetic AM(11–26) produced dose-dependent strong pressor responses in unanesthetized rats in vivo. (Blood pressure increase; F(3, 20 = 13.845, P < 0.0001). Injection of saline did not affects blood pressure and heart rate. The hypertensive activity lasted about 70 s, and a dose dependent increase of heart rate was also observed (Heart rate increase; F(3, 20) = 6.151, P = 0.0039).
We have isolated and characterized bovine AM(11–26) from bovine adrenal medulla as an endogenous peptide. The hallmark biological effects of AM are vasodilation and hypotensive effects in the vascular systems of most species. The mature form of AM has one ring structure formed by an intramolecular disulfide bond and a C terminal amide structure, both of which are essential for the hypotensive and other biological activities of AM. Watanabe et al. reported that the synthetic N-terminal fragment of human AM, AM (1–25)COOH and other related peptides, show vasopressor activity in anesthetized rats. The present purification and characterization of AM(11–26) indicate that the ring structure of AM may function as a biologically active endogenous peptide. The peptide corresponding to Mr 1,500 Da was further purified to homogeneity.
The purified peptide was found to be AM(11–26) which has one intramolecular disulfide bond. The structure of AM(11–26) was confirmed by chromatographic comparison with native AM(11–26) as well as a synthetic specimen which was prepared according to the determined sequence. The structure of bovine AM and related peptides were determined by cDNA analysis encoding bovine AM. Bovine AM consists of 52 amino acids whose sequence is identical to the human sequences with the exception of four amino acids. Furthermore, according to the cDNA analysis and chromatographic comparison of the synthetic AM(11–26) and purified AM, is now determined to be cystine. It should be noted that the structure of bovine AM(11–26) is identical to human AM(11–26).
It is well known that many peptide hormones and neuropeptides are processed from larger, biologically inactive precursors by the specific processing enzyme. It usually recognizes pairs of basic amino acids, processing signals, such as primarily Lys-Arg and Arg-Arg. AM (11–26) is not flanked by such a processing signal, but it was reproducibly observed in bovine adrenal medulla peptide extract. The molar ratio of AM(11–26)/AM was estimated to be 40%. The ratio varied from 5% to 50% according to the individual specimen, but the minor peak corresponding to 1,500 Da was reproducibly observed, suggesting that AM(11–26) is an endogenous peptide. It is likely that AM(11–26) is biosynthesized from AM or AM precursor by a specific enzyme.
In contrast to AM, synthetic bovine AM(11–26) caused potent hypertensive effects in unanesthetized rats. The hypertensive activity of AM(11–26) seems to be comparable to that of AM(1–25) as reported by Watanabe et al. It was unexpected that AM(11–26) would cause a dose dependent increase of heart rate in unanesthetized rats because vasopressor activity normally causes bradycardia through baroreceptor activation. The hypertensive mechanism is not fully understood, but it has been reported that the vasopressor effect of AM(1–25) might be caused by the release of endogenous catecholamine. We speculate that the released catecholamine counters the baroreceptor function resulting in an increased heart rate and blood pressure. It is possible that AM(11–26) participates in blood pressure control as an endogenous peptide.
A review of the biological properties and clinical implications of adrenomedullin and proadrenomedullin N-terminal 20 peptide (PAMP), hypotensive and vasodilating peptides.
Tanenao Eto
Peptides 22 (2001) 1693–1711 PII: S0196-9781(01)00513-7
Adrenomedullin (AM), identified from pheochromocytoma and having 52 amino acids, elicits a long-lasting vasodilatation and diuresis. AM is mainly mediated by the intracellular adenylate cyclase coupled with cyclic adenosine monophosphate (cAMP) and nitric oxide (NO) -cyclic guanosine monophosphate (cGMP) pathway through its specific receptor. The calcitonin receptor-like receptor (CLCR) and receptor-activity modifying protein (RAMP) 2 or RAMP3 models have been proposed as the candidate receptor. AM is produced mainly in cardiovascular tissues in response to stimuli such as shear stress and stretch, hormonal factors and cytokines. Recently established AM knockout mice lines revealed that AM is essential for development of vitelline vessels of embryo. Plasma AM levels elevate in cardiovascular diseases such as heart failure, hypertension and septic shock, where AM may play protective roles through its characteristic biological activities. Human AM gene delivery improves hypertension, renal function, cardiac hypertrophy and nephrosclerosis in the hypertensive rats. AM decreases cardiac preload and afterload and improves cardiac contractility and diuresis in patients with heart failure and hypertension. Advances in gene engineering and receptor studies may contribute to further understandings of biological implication and therapeutic availability of AM.
AM acts as a circulating hormone as well as elicits multiple biological activities in a paracrine or autocrine manner. Among them the most characteristic biological activity of AM is a very powerful hypotensive activity caused by dilatation of resistance vessels. A sensitive and specific radioimmunoassay demonstrated that AM circulates in blood and occurs in a variety of tissues. Plasma AM levels elevate in various diseases including cardiovascular and renal disorders or septic shock. Thus, AM may be involved in pathophysiological processes in these diseases, especially in disorders controlling circulation and body fluid. In this short review, the history of AM and proadrenomedullin N-terminal 20 peptide (PAMP) will be reviewed with special references to biological properties and function, receptors, gene engineering and clinical viewpoints. This review includes oral presentations from the aforementioned symposium; some of which have not yet been published. These unpublished oral presentations are quoted in this paper from the abstracts of this symposium.
Preproadrenomedullin, which consists of 185 amino acids and contains a 21-amino acid signal peptide, is processed to synthesize proadrenomedullin and finally AM. In the proadrenomedullin, a unique twenty amino acid sequence followed by a typical amidation signal known as Gly-Lys-Arg, is included in the N-terminal region. This novel 20 residues peptide with carboxyl terminus of Arg-CONH2 is also present in vivo and is termed “proadrenomedullin N-terminal 20 peptide (PAMP).” PAMP elicits a potent hypotensive activity in anesthetized rats.
Although widely distributed in the adenophypophysis and the neural lobe of pituitary glands, AM and PAMP occur in cell-specific, but not overlapping, patterns in the anterior pituitary. This cell-specific expression of each peptide may be explained by differences in posttranslational processing of AM gene. As such, potential pituitary specific transcription factor binding sites, gonadotropic-specific element (GSE) and a binding site for steroidogenic factor-l (SF-1) are found in the 5flanking region of human and mouse AM gene. SF-1 is a member of the steroid receptor superfamily that has been shown necessary for gonadotrope differentiation within the pituitary. In addition, one putative binding sequence of Pit-1 has been reported in mouse AM gene promoter position.
A specific AM binding protein (AMBP-1) in human plasma was isolated and the purified protein was identified as human complement factor H. AM and factor H interaction may interfere with the radioimmunoassay quantification of circulating AM. Factor H enhances AM-mediated induction of cAMP in fibroblast; augments the AM-mediated growth of a cancer cell line; and suppresses the bactericidal capability of AM on Escherichia coli. Conversely, AM influences the complement regulatory function of factor H by enhancing the cleavage of C3b via factor I. The augmentation of AM actions indicates that AMBP may facilitate the binding of AM to its receptor. In addition, the existence of AMBP suggests that large amounts of AM may circulate bound to this plasma protein.
In rat vascular smooth muscle cells, the CGRP, CGRP1 receptor antagonist, competitively inhibits the intracellular accumulation of cAMP induced by AM. Vasodilation of the rat mesenteric vascular bed elicited by AM and CGRP is also blocked by CGRP. Similar effects of CGRP are observed in the isolated rat heart and its microvasculature. Thus, CGRP1 receptor can mediate some effects of AM, but AM has a low affinity at CGRP2 receptor. Two distinct AM labeled bands with a molecular weight of 120 and 70 kDa was reported in the cultured rat vascular smooth muscle cell membrane. Therefore, the binding specificity and characteristics of the AM receptor may differ regionally by organ or tissue.
Two more RAMP proteins, RAMP2 and RAMP3, were discovered from database searches. These proteins share approximately 30% homology with RAMP1. Co-expression of RAMP2 or RAMP3 with CRLR appears to constitute AM receptor. RAMP2 and RAMP3 are indistinguishable in terms of AM binding. The RAMPs are required to transport CRLR to the plasma membrane. RAMP1 presents CRLR as a mature glycoprotein at the cell surface to form a CGRP receptor. However, receptors transported by RAMP2 or RAMP3 are core glycosylated and then become AM receptors. Three putative N-glycosylation sites Asn 60, Asn 112 and Asn 117 are present in the amino-terminal extracellular domain of the human CRLR. When the glycosylation of a myc-tagged CRLR was inhibited, specific 125I-CGRP and -AM binding were blocked in parallel. Substitution of the Asn 117 by threonine abolished CGRP and AM binding in the face of intact N-glycosylation and cell surface expression. RAMPs are accessory proteins of CTR and CRLR at the cell surface where they define AM, amylin, calcitonin and CGRP specificity.
The receptor component protein (RCP) was cloned on the basis of its ability to potentiate the endogenous Xenopus oocyte CGRP receptor. RCP is a cytosolic protein with no similarity to RAMPs, consists of a hydrophobic 146 amino acids and is obtained from the Corti organ of guinea pig. RCF plays an essential role for signal-transduction of CGRP and AM, and interacts with CRLR directly within the cells. Thus, a functional AM or CGRP receptor seems to consist of at least three proteins: CRLR, RAMP and RCP, coupling the receptor to the intracellular signal-transduction pathway.
By using a chimera of the CRLR and green fluorescent protein (GFP), the study demonstrated that CRLR-GFP failed to generate responses to CGRP or AM without RAMP2 or RAMP3 in HEK 293 cells. When coexpressed with RAMP2 or RAMP3, CRLR-GFP appeared on the cell membrane and activated an intracellular cAMP production and calcium mobilization. Agonist-mediated internalization of CRLR-GFP was observed in RAMP1/CGRP or AM, RAMP2/AM, and RAMP3/AM, which occurred with similar kinetics, indicating the existence of ligand-specific regulation of CRLR internalization by RAMPs.
The discovery of RAMPs has promoted our understandingthat some of the biological activities of AM are blocked by CGRP receptor antagonist, whereas other biological activities are blocked only by AM receptor antagonist, which indicates the possible existence of AM receptor in dual nature. RAMP association with CRLR traffics this receptor to the cell surface where individual RAMPs dictate the expression of unique phenotypes such as CGRP receptor or AM receptors. Apart from receptor trafficking and glycosylation, the RAMPs may interact directly with the receptors in the cell surface modifying their affinities for the ligands.
Since AM was discovered by monitoring the elevating activity of cAMP in rat platelets, cAMP appears to be its major second messenger. Dose-dependent intracellular production of cAMP induced by AM has been confirmed in various tissues and cells. Moreover, information on the role of NO in alternative signal-transduction pathways for AM is available.
The vasodilating effect of AM is reduced by the blockade of NO synthetase activity with NG-nitro-L-arginine methylester (L-NAME), indicating that NO may at least partly contribute to the AM-induced vasodilation. However, the degree of NO contribution to vasodilation varies depending upon the organ or tissue and the species. NO synthetase inhibitor in the pulmonary vascular beds of rat significantly attenuates the AM-induced vasodilation, but it does not occur in cats. Thus, NO seems to be an important AM mediator despite regional and interspecies variation.
In bovine aortic endothelial cells, AM increases intracellular ionic calcium (Ca2+) and causes the accumulation of cAMP. This increase in intracellular Ca2+ may be involved in the activation of phospholipase C, thereby producing inducible NO synthetase and subsequently NO. NO transferred to medial smooth muscle cells may activate cGMP-mediating smooth muscle cells vasodilatation. In contrast, AM lowers both cytosolic Ca2+ and Ca2+ sensitivity in smooth muscle cells of pig coronary arteries and intracellular Ca2+ in rat renal arterial smooth muscle cells.
Among the multi-functional properties of AM, the most characteristic one is an intensive, long-lasting hypotension that is dose-dependent in humans, rats, rabbits, dogs, cats and sheep. AM dilates resistance vessels in the kidneys, brain, lung, hindlimbs in animals as well as in the mesentery. Moreover, AM elicits relaxation of ring preparations of the aorta and cerebral arteries. An i.v. injection of human AM to conscious sheep causes a dose dependent fall of blood pressure, an increase in heart rate and cardiac output with a small reduction in stroke volume, as well as a marked decrease in total peripheral resistance. Coronary blood flow increases in parallel with the increase in coronary conductance. These cardiovascular responses return to the control level by 40 min after the injection.
The low-dose infusion of AM administered to conscious sheep on a low-salt diet antagonizes the vasopressor actions of administered angiotensin II while stimulating cardiac output and heart rate. AM may control cardiovascular homeostasis in part through antagonism of the vasopressor action of angiotensin II. AM inhibits the secretion of endothelin-1 from the vascular endothelial cells and proliferation of vascular smooth muscle cells. In the cultured cardiomyocytes as well as cardiac fibroblasts, AM inhibits protein synthesis in these cells in an autocrine or a paracrine manner, which may result in modulating the cardiac growth. AM inhibits bronchial constriction induced by acetylcholine or histamine in a dose-dependent manner, indicating the important role of AM on airway function and its usefulness for the management of bronchial asthma. AM inhibits secretion of aldosterone from the adrenal cortex. When infused directly into the adrenal arterial supply of conscious sheep, AM directly inhibits the acute stimulation of aldosterone by angiotensin II, KCl and ACTH while not affecting basal or chronic aldosterone secretion or cortisol secretion stimulated by ACTH. AM co-exists in insulin-producing cells and it inhibits insulin secretion dose-dependently in isolated rat islets.
The N-terminal region of preproadrenomedullin, the precursor of AM, contains a unique 20-residue sequence followed by Gly-Lys-Arg, a typical amidation signal, which was termed as proadrenomedullin N-terminal 20 peptide (PAMP). PAMP was purified from porcine adrenal medulla and human pheochromo-cytoma by using radioimmunoassay for the peptide and its complete amino acid sequence was determined. In addition to the original form of PAMP [1–20], PAMP [9–20] has recently been purified from the bovine adrenal medulla. The amino acid sequences of both forms of PAMP are identical to amino acid sequences deduced by cDNA analysis and their carboxyl terminus of Arg is amidated. The distribution of PAMP is similar to that of human AM, due to the fact that PAMP as well as human AM is biosynthesized from an AM precursor.
AM is processed from its precursor, proadrenomedullin, as the intermediate or immature form, AM-glycine (AM[1–52]-COOH, immature AM). Subsequently, immature AM is converted to the biologically active mature form, AM [1–52]-CONH2 (mature AM) by enzymatic amidation. The AM circulating in the human blood stream (total AM), thus, consists of both mature AM and immature AM. In earlier studies, plasma AM levels were measured by using radioimmunoassay recognizing the entire AM molecule (AM [1–52]), which reflects plasma total AM levels, as previously described.
In healthy volunteers severe exercise elevates the plasma AM levels with an increase in plasma norepinephrine and exaggerated sympathetic nerve activity. In heart transplant recipients, maximal exercise induces an increase in plasma AM that is inversely related to mean blood pressure. AM, therefore, may participate in blood pressure regulation during exercise even after heart transplantation.
When compared with healthy controls, the plasma AM levels are increased in patients with a variety of diseases: congestive heart failure, myocardial infarction, renal diseases, hypertensive diseases, diabetes mellitus, acute phase of stroke, and septic shock.
Adrenomedullin and central cardiovascular regulation
Meghan M. Taylor, Willis K. Samson
Peptides 22 (2001) 1803–1807 PII: S0196-9781(01)00522-8
Adrenomedullin gene products have been localized to neurons in brain that innervate sites known to be important in the regulation of cardiovascular function. Those sites also have been demonstrated to possess receptors for the peptide and central administrations of adrenomedullin (AM) and proadrenomedullin N-terminal 20 peptide (PAMP) elevate blood pressure and heart rate in both conscious and anesthetized animals. The accumulated evidence points to a role of the sympathetic nervous system in these cardiovascular effects. These sympathostimulatory actions of AM and PAMP have been hypothesized to be cardioprotective in nature and to reflect the central nervous system (CNS) equivalent of the direct cardiostimulatory effects of the peptides in the periphery. This review summarizes the most recent data on the CNS actions of the adrenomedullin gene-derived peptides and suggests future strategies for the elucidation of the physiologic relevance of the already demonstrated, pharmacologic actions of these peptides.
Adrenomedullin and related peptides: receptors and accessory proteins
Roman Muff, Walter Born, Jan A. Fischer
Peptides 22 (2001) 1765–1772 PII: S0196-9781(01)00515-0
Adrenomedullin (AM), α- and β-calcitonin gene-related peptide (CGRP), amylin and calcitonin (CT) are structurally and functionally related peptides. The structure of a receptor for CT (CTR) was elucidated in 1991 through molecular cloning, but the structures of the receptors for the other three peptides had yet to be elucidated. The discovery of receptor-activity-modifying proteins (RAMP) 1 and -2 and their co-expression with an orphan receptor, calcitonin receptor-like receptor (CRLR) has led to the elucidation of functional CGRP and AM receptors, respectively. RAMP1 and -3 which are co-expressed with CTR revealed two amylin receptor isotypes. Molecular interactions between CRLR and RAMPs are involved in their transport to the cell surface. Heterodimeric complexes between CRLR or CTR and RAMPs are required for ligand recognition.
| Pharmacological profiles of receptors of the adrenomedullin peptidefamily | |
| AMR | AM>CGRP>>amylin=CT |
| CTR | CT>amylin>>CGRP=AM |
| CGRPR | CGRP>AM>>amylin=CT |
| AmylinR | AmylinsCTCGRP>>hCT>AM |
Specific AM binding sites have been identified in many tissues including the heart, blood vessels, lung and spleen. Based on pharmacological evidence two receptor isotypes have been distinguished, for instance in rat astrocytes and NG108–15 cells. One AM receptor isotype recognizes CGRP and CGRP(8–37). The other receptor isotype specific for the AM ligand and antagonized by AM(22–52) does not recognize CGRP to any great extent. Both isotypes of the receptors have been shown to interact poorly with amylin and CT (Table). Biological actions of AM include vaso- and bronchodilation, and CNS transmitted inhibition of water intake.
CGRP receptors are widely distributed in the nervous and cardiovascular systems. To date, two isotypes have been described. On pharmacological evidence, CGRP1 receptors, such as those identified in human SK-N-MC neuroblastoma cells, recognize intact CGRP and CGRP(8–37) with similar potency, unlike a linear analog lacking the disulfide bridge. CGRP2 receptors,
on the other hand, interact with the linear analog but not with CGRP(8–37). These CGRP receptor isotypes cross-react with AM to some extent, but only minimally with amylin and CT. CGRP shares potent vasodilatory actions with AM, and has chronotropic and inotropic actions in the heart. The ionotropic actions are indirectly brought about via activation of the sympathetic nervous system. There is evidence to suggest the existence of α- or β-CGRP preferring receptor isotypes in both the central nervous system and peripheral tissues.
RAMP1, -2 and -3 are widely expressed, suggesting that RAMPs may have
important functions beyond those of the adrenomedullin family of receptors. To this end, RAMP1 and -3 are thought to reduce cell surface expression of angiotensin (AT) AT1 and AT2 receptors.
RAMP2 and CRLR are expressed in vascular smooth muscle cells, and RAMP1 expression was increased by dexamethasone. Moreover, increased levels of RAMP2 and CRLR were observed in the kidney and heart of rats with obstructive nephropathy and congestive heart failure, respectively. RAMP2
and CRLR levels were reduced, and RAMP3 levels were increased during lipopolysaccharide induced sepsis in rats.
The GABAB receptor 1 is retained as an immature glycoprotein in the cytosol unless co-expressed with GABAB receptor 2 isotype. Heterodimers of fully functional opioid receptors δ and κ result in a novel receptor displaying binding and functional properties distinct from those of the δ or κ receptors alone. Heterodimerization therefore facilitates receptor expression and defines ligand specificity also in G protein-coupled receptor families A and C. Moreover, heterodimers of metabotropic glutamate 1receptor (family C) and adenosine A1 receptors (family A) have been observed. As yet there is no evidence for homo or heterodimerization of family B receptors. Cysteines conserved in the extracellular N-terminal domain in all the receptors of family B and RAMPs suggest that RAMPs are truncated forms of receptors that interact as heterodimers with CRLR and CTR.
The discovery of RAMPs in combination with CRLR and CTR has led to the molecular identification of CGRP1, CGRP/amylin, AM and amylin receptor complexes. The physiological advantage of heterodimers between seven transmembrane domain receptors and the RAMPs required for the functional expression of the adrenomedullin, CGRP and amylin receptors remains to be demonstrated.
Angiotensin II, From Vasoconstrictor to Growth Factor: A Paradigm Shift
Sasa Vukelic, Kathy K. Griendling
Circ Res. 2014;114:754-757
http://dx.doi.org:/10.1161/CIRCRESAHA.114.303045
Angiotensin II (Ang II) is today considered as one of the essential factors in the pathophysiology of cardiovascular disease, producing acute hemodynamic and chronic pleiotropic effects. Although now it is widely accepted that these chronic effects are important, Ang II was initially considered only a short-acting, vasoactive hormone. This view was modified a quarter of a century ago when Dr Owens and his group published an article in Circulation Research with initial evidence that Ang II can act as a growth factor that regulates cell hypertrophy. They showed in a series of elegant experiments that Ang II promotes hypertrophy and hyperploidy of cultured rat aortic smooth muscle cells. However, Ang II had no effect on hyperplasia. These findings led to a paradigm shift in our understanding of the roles of growth factors and vasoactive substances in cardiovascular pathology and helped to redirect basic and clinical renin–angiotensin system research during the next 25 years. Ang II is now known to be a pleiotropic hormone that uses multiple signaling pathways to influence most processes that contribute to the development and progression of cardiovascular diseases, ranging from hypertrophy, endothelial dysfunction, cardiac remodeling, fibrosis, and inflammation to oxidative stress.
























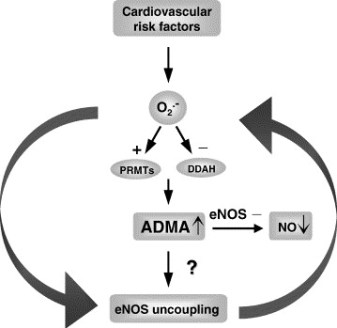



 and Protein C Pat...")


