Contributions to Cardiomyocyte Interactions and Signaling
Author and Curator: Larry H Bernstein, MD, FCAP
and
Curator: Aviva Lev-Ari, PhD, RN
Introduction
This is Part II of the ongoing research in the Lee Laboratory, concerned with Richard T Lee’s dissection of the underlying problems that will lead to a successful resolution of myocardiocyte regeneration unhampered by toxicity, and having a suffuciently sustained effect for an evaluation and introduction to the clinic. This would be a milestone in the treatment of heart failure, and an alternative to transplantation surgery. This second presentation focuses on the basic science work underpinning the therapeutic investigations. It is work that, if it was unsupported and did not occur because of insufficient funding, the Part I story could not be told.
Cardiomyocyte hypertrophy and degradation of connexin43 through spatially restricted autocrine/paracrine heparin-binding EGF
J Yoshioka, RN Prince, H Huang, SB Perkins, FU Cruz, C MacGillivray, DA Lauffenburger, and RT Lee *
Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA; and Biological Engineering Division, MIT, Cambridge, MA
PNAS 2005; 302(30):10622-10627. http://pnas.org/cgi/doi/10.1073/pnas.0501198102
Growth factor signaling can affect tissue remodeling through autocrine/paracrine mechanisms. Recent evidence indicates that EGF receptor transactivation by heparin-binding EGF (HB-EGF) contributes to hypertrophic signaling in cardiomyocytes. Here, we show that HB-EGF operates in a spatially restricted circuit in the extracellular space within the myocardium, revealing the critical nature of the local microenvironment in intercellular signaling. This highly localized microenvironment of HB-EGF signaling was demonstrated with 3D morphology, consistent with predictions from a computational model of EGF signaling. HB-EGF secretion by a given cardiomyocyte in mouse left ventricles led to cellular hypertrophy and reduced expression of connexin43 in the overexpressing cell and in immediately adjacent cells but not in cells farther away. Thus, HB-EGF acts as an autocrine and local paracrine cardiac growth factor that leads to loss of gap junction proteins within a spatially confined microenvironment. These findings demonstrate how cells can coordinate remodeling with their immediate neighboring cells with highly localized extracellular EGF signaling. Within 3D tissues, cells must coordinate remodeling in response to stress or growth signals, and this communication may occur by direct contact or by secreted signaling molecules. Cardiac hypertrophy is a physiological response that enables the heart to adapt to an initial stress; however, hypertrophy can ultimately lead to the deterioration in cardiac function and an increase in cardiac arrhythmias. Although considerable progress has been made in elucidating the molecular pathogenesis of cardiac hypertrophy, the precise mechanisms guiding the hypertrophic process remain unknown. Recent evidence suggests that myocardial heparin-binding (HB)-epidermal growth factor participates in the hypertrophic response. In cardiomyocytes, hypertrophic stimuli markedly increase expression of the HB-EGF gene, suggesting that HB-EGF can act as an autocrine trophic factor that contributes to cellular growth. HB-EGF is first synthesized as a membrane-anchored form (proHB-EGF), and subsequent ectodo-main shedding at the cell surface releases the soluble form of HB-EGF. Soluble HB-EGF is a diffusible factor that can be captured by the receptors to activate the intracellular EGF receptor signaling cascade. Indeed, EGF receptor (EGFR) transactivation, triggered by shedding of HB-EGF from the cell surface, plays an important role in cardiac hypertrophy resulting from pressure overload in the aortic-banding model. EGFR activation can occur through autocrine and paracrine signaling. In autocrine signaling, a cell produces and responds to the same signaling molecules. Paracrine signaling molecules can target groups of distant cells or act as localized mediators affecting only cells in the immediate environment of the signaling cell. Thus, although locally produced HB-EGF may travel through the extra-cellular space, it may also be recaptured by the EGFR close to the point where it was released from the cell surface. The impact of spatially localized microenvironments of signaling could be extensive heterogeneous tissue remodeling, which can be particularly important in an electrically coupled tissue like myocardium. Interestingly, recent data suggest that EGF can regulate protea-some-dependent degradation of connexin43 (Cx43), a major trans-membrane gap junction protein, in liver epithelial cells, along with a rapid inhibition of cell–cell communication through gap junctions. One of the critical potential myocardial effects of HB-EGF could therefore be to increase degradation of Cx43 and reduce electrical stability of the heart. Reduced content of Cx43 is commonly observed in chronic heart diseases such as hypertrophy, myocardial infarction, and failure. Thus, we hypothesized that HB-EGF signals may operate in a spatially restricted local circuit in the extracellular space. We also hypothesized that HB-EGF secretion by a given cardiomyocyte could create a local remodeling microenvironment of decreased Cx43 within the myocardium. To explore whether HB-EGF signaling is highly spatially constrained, we took advantage of the nonuniform gene transfer to cardiac myocytes in vivo, normally considered a pitfall of gene therapy. We also performed computational modeling to predict HB-EGF dynamics and developed a 3D approach to measure cardiomyocyte hypertrophy.
Results
Autocrine HB-EGF and Cardiomyocyte Growth.
To assess the effects of gene transfer of HB-EGF on cardiomyocyte hypertrophy, cells were infected with adenoviral vectors expressing GFP alone (Ad-GFP) or HB-EGF and GFP (Ad-HB-EGF). At this level of infection, 99% of cardiomyocytes were transduced. The incidence of apoptotic cell death (sub-G1 fraction) was not different between Ad-GFP cells, suggesting that expression of GFP by the adenoviral vector was not cardiotoxic in these conditions. Western analysis by using an anti-HB-EGF antibody confirmed successful gene transfer of HB-EGF in cardiomyocytes (18 ± 5-fold, n = 4, P < 0.01); HB-EGF appeared electrophoretically as several bands from 15 to 30 kDa (Fig. 1A). The strongest band corresponds to the soluble 20-kDa form of HB-EGF. To confirm that Ad-HB-EGF results in cellular hypertrophy, cell size and protein synthesis were measured. Ad-HB-EGF enlarged cardiomyocytes compared with Ad-GFP-infected cells by phase-contrast microscopy (24 ± 10% increase in cell surface area, n = 27, P < 0.05) and with flow cytometry analysis (26 ± 10% increase of Ad-GFP infected cells, P < 0.01, Fig. 1B). Overexpression of HB-EGF increased total protein synthesis in cardiomyocytes as measured by [3H]leucine uptake (34 ± 6% of Ad-GFP, n = 6, P < 0.01, Fig. 1C). Uninfected cells within the same dish (and thus sharing the same culture media) did not develop hypertrophy. Additionally, medium from cultures previously infected with Ad-HB-EGF for 48 h was collected and applied to adenovirus-free cultures. Conditioned medium from Ad-HB-EGF-infected cardiomyocytes failed to stimulate hypertrophy in naive cardiomyocytes (Fig. 1C), and there were no significant differences in cell size between noninfected cells from Ad-GFP and Ad-HB-EGF dishes. These results suggest that HB-EGF acts primarily as an autocrine growth factor in cardiomyocytes in vitro.
Because the dilution factor in culture media is important for autocrine/paracrine signaling, we determined the concentration of soluble HB-EGF in the conditioned medium and the effective concentration to stimulate cardiomyocyte growth. HB-EGF levels in the conditioned medium from Ad-HB-EGF dishes were 258 ± 73 pg/ml (n = 4), whereas HB-EGF levels from Ad-GFP dishes (n = 8) were below the limit of detection (6.7 pg/ml). The addition of 300 pg/ml of exogenous recombinant HB-EGF into fresh media failed to stimulate hypertrophy in cardiomyocytes as measured by [3H]leucine uptake (-12 ± 5.0% compared with control, n = 5,P = not significant), but 2,000 pg/ml of recombinant HB-EGF did result in a significant effect (+24 ± 5.5% compared with control, n = 6, P < 0.05). This comparison implies that the local concentration of autocrine ligand is substantially greater than that indicated by a bulk measurement of conditioned media, consistent with previous experimental and theoretical studies.
Fig. 1. Effects of gene transfer of HB-EGF on rat neonatal cardiomyocyte growth.
(A) Cells were infected with adenoviral vectors expressing GFP (Ad-GFP), or HB-EGF and GFP (Ad-HB-EGF). Western analysis showed the successful gene transfer of HB-EGF. (8) FACS analysis of 5,000 cardiomyocytes demonstrated that overexpression of HB-EGF produced a 26 ± 10% increase in cell size that was significantly greater than the overex-pression of GFP. Bar graphs with errors represent mean ± SEM from three independent experiments. **, P < 0.01 vs. Ad-HB-EGF-nonin-fected cells and Ad-GFP nonin-fected cells. , P < 0.05 vs. Ad-GFP infected cells. (C) Overexpression of HB-EGF resulted in a 34 ± 6% increase in [3H]leucine uptake compared with Ad-GFP (n = 6), whereas conditioned medium from Ad-HB-EGF cells caused an only insignificant increase. **, P < 0.05 vs. Ad-GFP control and conditioned medium Ad-GFP.
Effects of HB-EGF on Cx43 Content in Cultured Cardiomyocytes
Because EGF can induce degradation of the gap junction protein Cx43 in other cells, we then determined whether Cx43 is regulated by HB-EGF in cardiomyocytes. Fig. 2A shows a representative immunoblot from three separate experiments in which Cx43 migrated as three major bands at 46, 43, and 41 kDa, as reported in ref. 16. Overexpression of HB-EGF decreased total Cx43 content (27 ± 11% compared with Ad-GFP, n = 4, P < 0.05) without affecting the intercellular adhesion protein, N-cadherin. The phosphorylation of ERK1/2, an intracellular signaling kinase downstream of EGFR transactivation, was augmented by HB-EGF (3.2 ± 1.0-fold compared with Ad-GFP, n = 4, P < 0.05). Northern analysis showed that HB-EGF did not reduce Cx43 gene expression, suggesting that HB-EGF decreases Cx43 by posttranslational modification (Fig. 2B). AG 1478 (10 iLM), a specific inhibitor of EGFR tyrosine kinase, abolished the effect of HB-EGF on Cx43 (Fig. 2C), indicating that the decrease in Cx43 content depends on EGFR transactivation by HB-EGF. The conditioned medium from Ad-HB-EGF-infected cells did not change expression of Cx43 in naive cells, even though ERK1/2 was slightly activated by the conditioned medium (Fig. 1D). These data are consistent with the hypertrophy data presented above, demonstrating that HB-EGF can act as a predominantly autocrine factor both in hypertrophy and in the reduction of Cx43 content in cardiomyocytes.
Computational Analysis Predicts HB-EGF Autocrine/Paracrine Signaling in Vivo.
Although these in vitro experiments showed HB-EGF as a predominantly autocrine cardiac growth factor, HB-EGF signaling in vivo takes place in a very different environment. Therefore, we sought to determine the extent that soluble HB-EGF may travel in the interstitial space of the myocardium with a simple 2D model of HB-EGF diffusion (Fig. 3A). An approximate geometric representation of myocytes in cross-section is a square (15 x 15 iLm), with each of the corners occupied by a capillary (diameter 5 iLm). The cell shape was chosen so that the extracellular matrix width (0.5 iLm), in which soluble HB-EGF is free to diffuse, was constant around all tissue features. This model geometry is based on a square array of capillaries; although a hexagonal pattern of capillary distribution is commonly accepted, the results are not expected to be substantially different with this simpler construction, because both have four capillaries surrounding each myocyte. The model represents a single central cell that is releasing HB-EGF at a constant rate, Rgen, (approximated from the HB-EGF concentration measurement in conditioned medium) into the extracellular space. HB-EGF then can diffuse throughout this space, or enter a capillary and leave the system. This system is governed by
- the diffusion equation at steady state (DV2C = 0),
- the boundary condition for the ligand producing cell (DVC = Rgen),
- the boundary condition for all other cells (DVC = 0), and
- the capillary boundary condition (DVC = h(C Cblood)).
C denotes HB-EGF concentration, D is the diffusivity constant, h is the mass transfer coefficient, and Cblood is the concentration of HB-EGF in the blood, approximated to be zero. The numerical solution in Fig. 3B illustrates that HB-EGF remained localized around the cell which produced it and did not diffuse farther because of the sink-like effect of the capillaries. The maximum concentration of soluble HB-EGF achieved is 0.27 nM, which is near the threshold level of HB-EGF measured to stimulate cardiomyocyte growth (2,000 pg/ml). Therefore, the central HB-EGF-producing cell only signals to its four adjacent neighbors where the HB-EGF concentration reaches this threshold. However, if the model geometry is altered to reflect a 50% and 150% increase in cross-sectional area in all cells because of hypertrophy, estimated from 1 and 4 weeks of transverse aortic constriction, the maximum concentration achieved increases slightly to 0.29 nM and 0.37 nM, respectively. As the cell width increases, HB-EGF must diffuse farther to reach a capillary, exposing adjacent cells to a higher concentration during hypertrophy. However, no additional cells are exposed to HB-EGF.
Fig. 3. Computational modeling of HB-EGF diffusion in the myocardium.
Red areas represent capillaries, green represents the HB-EGF ligand producing cell, pink represents adjacent cells, and white is an extracellular matrix where HB-EGF is free to diffuse. (A) The model geometry where HB-EGF is generated by the ligand-producing cell at a constant rate, Rgen, and diffuses throughout the extracellular space or enters a capillary and leaves the system with a mass transfer coefficient, h. (B) Numerical solution of the steady-state HB-EGF concentration profile with Rgen = 10 cell-1s-1, D = 0.7 µm2/s, and h = 0.02 µm/s, where concentration is shown by the color scale and height depicted. The maximum concentration achieved with the stated parameters was 0.27 nM from a capillary. Myocyte length was assumed to be 100 µm.
The driving force that determined the extent to which HB-EGF traveled was the rate of HB-EGF transfer into the capillaries and the diffusivity of HB-EGF. The exact mechanism of macromolecule transport into capillaries is unknown; however, it is most likely through diffusion, transcytosis, or a combination of the two. In the case of diffusion, the mass transfer coefficient governing the flux of HB-EGF through the capillary wall is coupled to the diffusivity of HB-EGF, whereas the terms are uncoupled for the case of transcytosis. Therefore, this model assessed transcytosis as a conservative scenario for HB-EGF localization. Parameter perturbation with uncoupled diffusion and capillary mass transfer showed that HB-EGF remained localized around the origin of production and diffused only to immediate neighbors for mass transfer coefficients >0.002 µm/s. For values <0.002 µm/s, HB-EGF diffused distances more than two cells away from the origin. Although the actual mass transfer coefficient of ligands in the size range of HB-EGF is unknown, values for O2 (0.02 µm/s, 0.032 kDa) (19) and LDL (1.7 x 10-5 µm/s, 2,000–3,000 kDa) (20) have been reported, and we assumed HB-EGF is in the upper end of that range due to its small size. HB-EGF also binds to EGFRs, the extracellular matrix, and cell surface heparan sulfate proteoglycans. EGFR binding and internalization could serve to further localize HB-EGF. The number of extracellular binding sites does not affect the steady-state HB-EGF concentration profile if this binding is reversible. However, these binding sites could serve to localize HB-EGF as the cell begins to produce the ligand by slowing the travel of HB-EGF to the capillaries in the approach to the steady state, or as a source of HB-EGF as the cell slows or stops HB-EGF production. At a diffusivity of 0.7 µm2/s (21), HB-EGF traveled only one cell away, but traveled approximately five cells away at 51.8 µm2/s (22), with a peak concentration below the estimated threshold for stimulating.
�Overexpression of HB-EGF Causes Hypertrophy on the Infected Cell and Its Immediate Neighbor in Vivo.
To explore whether HB-EGF signals operate in a spatially restricted local circuit in the in vivo myocardial extracellular space as predicted by computational modeling, adenoviral vectors were injected directly into the left ventricular free wall in 26 male mice (Ad-GFP, n = 12; Ad-HB-EGF, n = 14). Of the 26 mice, 5 (4 Ad-GFP and 1 Ad-HB-EGF) mice died after the surgery. Gene expression was confirmed as positive cellular fluorescence in the presence of GFP, allowing determination of which cells were infected at 7 days (Fig. 4A). Immunohis-tochemical staining revealed that HB-EGF was localized on the Ad-HB-EGF-infected cell membrane or in the extracellular space around the overexpressing cell (Fig. 4A). For comparison, remote cells were defined as noninfected cells far (15–20 cell dimensions) from the adenovirus-infected area and in the same field as infected cells. Conventional 2D cross-sectional analysis blinded to treatment group (Fig. 4B) showed that Ad-GFP-infected cells (n = 102) resulted in no cellular hypertrophy compared with noninfected, adjacent (n = 92), or remote (n = 97) cells (2D myocyte cross-sectional area, 250 ± 7 versus 251 ± 7 or 255 ± 6 µm2, respectively). These data suggest that expression of GFP in these conditions does not cause cellular hypertrophy. However, overexpression of HB-EGF caused hypertrophy in both Ad-HB-EGF-infected cells (a 41 ± 5% increase of Ad-GFP-infected cells, n = 119, P < 0.01) and noninfected adjacent cells (a 33 ± 5% increase of Ad-GFP-adjacent cells, n = 97, P < 0.01) compared with remote cells (n = 109). Because 2D analysis of cardiomyocyte hypertrophy can be influenced by the plane of sectioning, we then developed a 3D histology approach that allowed reconstruction of cardiomyocytes in situ (Fig. 4C). We performed an independent 3D histology analysis of cardiomyocytes to determine cell volumes, blinded to treatment group (Fig. 4B). The volumes of both HB-EGF-infected cells (n = 19, 42,700 ± 4,000 µm3) and their adjacent cells (n = 11, 33,500 ± 3,300 µm3) were significantly greater than volumes of remote cells (n = 13, 18,600 ± 1,700 µm3, P < 0.01 vs. HB-EGF-infected cells and P < 0.05 vs. HB-EGF-adjacent cells, Fig. 4D). In contrast, cells treated with Ad-GFP (n = 12) showed no hypertrophy in the Ad-GFP-adjacent (n = 10) or remote cells (n = 9). These data demonstrate that HB-EGF acts as both an autocrine and local paracrine growth factor within myocardium, as predicted by computational modeling.
Degradation of Cx43 Through Local Autocrine/Paracrine HB-EGF
To determine whether the spatially confined effect of HB-EGF reduces local myocardial Cx43 in vivo, Cx43 was assessed with immunohistochemistry and confocal fluorescence imaging. Cells infected with Ad-HB-EGF had significant decreases in Cx43 immunoreactive signal compared with Ad-GFP cells, consistent with the results of in vitro immunoblotting (Fig. 5A). Quantitative digital image analyses of Cx43 in a total of 22 fields in 6 Ad-HB-EGF hearts and 19 fields in 4 Ad-GFP hearts were analyzed (Fig. 5B). Although Ad-GFP-infected cells showed immunoreactive Cx43 at the appositional membrane, overexpression of HB-EGF increased Cx43 in intracellular vesicle-like components (Fig. 5C), with reduced gap junction plaques (percent Cx43 area per cell area, 52 ± 8% of Ad-GFP control, P < 0.01). These data suggest that reduced expression of Cx43 can be attributed to an increased rate of internalization and degradation in gap junction plaques in cardiomyocytes. Interestingly, HB-EGF secretion by a given cardiomyocyte caused a 37 ± 13% reduction of Cx43 content in its adjacent cells compared with GFP controls (P < 0.05). As degradation of Cx43 may accompany structural changes with marked rearrangement of intercellular connections. In contrast to Cx43, there was no significant difference in total area occupied by N-cadherin immunoreactive signal in between Ad-GFP (n = 19) and Ad-HB-EGF hearts (1.8 ± 0.5-fold compared with Ad-GFP, n = 17, P = not significant), indicating that HB-EGF has a selective effect on Cx43. Taken together, these data show that HB-EGF leads to cardiomyocyte hypertrophy and degradation of Cx43 in the infected cell and its immediately adjacent neighbors because of autocrine/ paracrine signaling. It should be noted, however, that quantifying the Cx43 from immunostaining could be limited by a nonlinear relation between the amount of Cx43 present and the area of staining.
Fig. 4. Effects of gene transfer of HB-EGF on cardiomyocyte hypertrophy in vivo.
(A) Adenoviral vectors (Ad-GFP or Ad-HB-EGF) were injected into the left ventricular free wall in mice. Myocytes were grouped as infected or noninfected on the basis of GFP fluorescence. Overex-pression of HB-EGF was confirmed by im-munohistochemistry. The presented image was pseudocolored with blue from that stained with Alexa Fluor 555 for the presence of HB-EGF. (Scale bars: 20 sm.) (B) 2D cross-sectional area of cardiomyo-cytes was measured in infected and non-infected cells in the same region of the same animal. Overexpression of HB-EGF caused cellular hypertrophy in both infected and adjacent cells. **, P < 0.01 vs. Ad-GFP infected; , P < 0.01 vs. Ad-HB-EGF remote; and §, P < 0.01 vs. Ad-GFP adjacent cells. GFP (infected 102 cells, adjacent 92 cells, and remote 97 cells from 5 mice), and HB-EGF (infected 119 cells, adjacent 97 cells, and remote 109 cells from 7 mice). The 3D histology also revealed cellular hypertrophy in both Ad-HB-EGF-infected cell and its adjacent cell. **, P < 0.01 vs. Ad-GFP infected; , P < 0.01; and *, P < 0.05 vs. Ad-HB-EGF remote cells. GFP (infected 12 cells, adjacent 10 cells, and remote 9 cells), and HB-EGF (infected 19 cells, adjacent 11 cells, and remote 13 cells). Statistical analysis was performed with one-way ANOVA. (C) Sample image of extracted myocytes in three dimensions.
Discussion
We have demonstrated in this study that
HB-EGF secreted by cardiomyocytes leads to cellular growth and reduced expression of the principal ventricular gap junction protein Cx43 in a local autocrine/paracrine manner. Although
proHB-EGF is biologically active as a juxtacrine growth factor that can signal to immediately neighboring cells in a nondiffusible manner,
several studies have revealed the crucial role of metalloproteases in the enzymatic conversion of proHB-EGF to soluble HB-EGF, which binds to and activates the EGFR. Hypertrophic stimuli such as mechanical strain and G protein-coupled receptors agonists mediate cardiac hypertrophy through the shedding of membrane-bound proHB-EGF. Thus, an autocrine/paracrine loop, which requires the diffusible, soluble form of HB-EGF, is necessary for subsequent transactivation of the EGFR to produce the hypertrophic response.
To our knowledge, there have been no previous reports concerning the spatial extent of autocrine/paracrine ligand distribution and signaling in myocardial tissue. A theoretical analysis by Shvartsman et al. predicted, from computational modeling in an idealized cell culture environment, that autocrine ligands may remain highly localized, even within subcellular distances; this prediction has support from experimental data in the EGFR system. In contrast, a theoretical estimate by Francis and Palsson has suggested that cytokines might effectively communicate larger distances, approximated to be 200–300 m from the point of release. However, these studies have all focused on idealized cell culture systems, so our combined experimental and computational investigation here aimed at understanding both in vitro and in vivo situations offers insight.
Our computational model of diffusion in the extracellular space predicts that HB-EGF acts as both an autocrine and spatially restricted paracrine growth factor for neighboring cells. We studied the responses of the signaling cell and its immediate neighbors compared with more distant cells. For a paracrine signal to be delivered to its proper target, the secreted signaling molecules cannot diffuse too far; in vitro experiments, in fact, indicated that HB-EGF acts as a predominantly autocrine signal in cell culture, where diffusion into the medium is relatively unconstrained.
In contrast, in the extracellular space of the myocardium, HB-EGF is localized around the source of production because of tissue geometry, thereby acting in a local paracrine or autocrine manner only. Indeed, our results from in vivo gene transfer demonstrated that both the cell releasing soluble HB-EGF and its surrounding cells undergo hypertrophy. This localized conversation between neighboring cells may allow remodeling to be fine-tuned on a highly spatially restricted level within the myocardium and in other tissues.
Common genetic variation at the IL1RL1locus regulates IL-33/ST2 signaling
JE Ho, Wei-Yu Chen, Ming-Huei Chen, MG Larson, ElL McCabe, S Cheng, A Ghorbani, E Coglianese, V Emilsson, AD Johnson,….. CARDIoGRAM Consortium, CHARGE Inflammation Working Group, A Dehghan, C Lu, D Levy, C Newton-Cheh, CHARGE Heart Failure Working Group, …. JL Januzzi, RT Lee, and TJ Wang J Clin Invest Oct 2013; 123(10):4208-4218. http://dx.doi.org/10.1172/JCI67119
Abstract and Introduction
The suppression of tumorigenicity 2/IL-33 (ST2/IL-33) pathway has been implicated in several immune and inflammatory diseases. ST2 is produced as 2 isoforms. The membrane-bound isoform (ST2L) induces an immune response when bound to its ligand, IL-33. The other isoform is a soluble protein (sST2) that is thought to be a decoy receptor for IL-33 signaling. Elevated sST2 levels in serum are associated with an increased risk for cardiovascular disease. We investigated the determinants of sST2 plasma concentrations in 2,991 Framingham Offspring Cohort participants. While clinical and environmental factors explained some variation in sST2 levels, much of the variation in sST2 production was driven by genetic factors. In a genome-wide association study (GWAS), multiple SNPs within IL1RL1 (the gene encoding ST2) demonstrated associations with sST2 concentrations. Five missense variants of IL1RL1 correlated with higher sST2 levels in the GWAS and mapped to the intracellular domain of ST2, which is absent in sST2. In a cell culture model, IL1RL1 missense variants increased sST2 expression by inducing IL-33 expression and enhancing IL-33 responsiveness (via ST2L). Our data suggest that genetic variation in IL1RL1 can result in increased levels of sST2 and alter immune and inflammatory signaling through the ST2/IL-33 pathway. Suppression of tumorigenicity 2 (ST2) is a member of the IL-1 receptor (IL-1R) family that plays a major role in immune and inflammatory responses. Alternative promoter activation and splicing produces both a membrane-bound protein (ST2L) and a soluble form (sST2). The transmembrane form of ST2 is selectively expressed on Th2- but not Th1-type T cells, and binding of its ligand, IL-33, induces Th2 immune responses. In contrast, the soluble form of ST2 acts as a decoy receptor by sequestering IL-33. The IL-33/ST2 pathway has important immunomodulatory effects. Clinically, the ST2/IL-33 signaling pathway participates in the pathophysiology of a number of inflammatory and immune diseases related to Th2 activation, including asthma, ulcerative colitis, and inflammatory arthritis. ST2 expression is also upregulated in cardiomyocytes in response to stress and appears to have cardioprotective effects in experimental studies. As a biomarker, circulating sST2 concentrations have been linked to worse prognosis in patients with heart failure, acute dyspnea, and acute coronary syndrome, and also predict mortality and incident cardiovascular events in individuals without existing cardiovascular disease. Both sST2 and its transmembrane form are encoded by IL-1R– like 1 (IL1RL1). Genetic variation in this pathway has been linked to a number of immune and inflammatory diseases. The contribution of IL1RL1 locus variants to interindividual variation in sST2 has not been investigated. The emergence of sST2 as an important predictor of cardiovascular risk and the important role outside of the ST2/IL-33 pathway in inflammatory diseases highlight the value of understanding genetic determinants of sST2. The family-based FHS cohort provides a unique opportunity to examine the heritability of sST2 and to identify specific variants involved using a genome-wide association study (GWAS). Thus, we performed a population-based study to examine genetic determinants of sST2 concentrations, coupled with experimental studies to elucidate the underlying molecular mechanisms.
Results
Clinical characteristics of the 2,991 FHS participants are presented in Supplemental Table 1 (supplemental material available online with this article; doi:10.1172/JCI67119DS1). The mean age of participants was 59 years, and 56% of participants were women. Soluble ST2 concentrations were higher in men compared with those in women (P < 0.001). Soluble ST2 concentrations were positively associated with age, systolic blood pressure, body-mass index, antihypertensive medication use, and diabetes mellitus (P < 0.05 for all). Together, these variables accounted for 14% of the variation in sST2 concentrations. The duration of hypertension or diabetes did not materially influence variation in sST2 concentrations. After additionally accounting for inflammatory conditions, clinical variables accounted for 14.8% of sST2 variation.
Heritability of sS72.
The age- and sex-adjusted heritability (h2) of sST2 was 0.45 (P = 5.3 x 10–16), suggesting that up to 45% of the variation in sST2 not explained by clinical variables was attributable to genetic factors. Multivariable adjustment for clinical variables previously shown to be associated with sST2 concentrations (21) did not attenuate the heritability estimate (adjusted h2 = 0.45, P = 8.2 x 10–16). To investigate the influence of shared environmental factors, we examined the correlation of sST2 concentrations among 603 spousal pairs and found no significant correlation (r = 0.05, P = 0.25).
Genetic correlates of sS72.
We conducted a GWAS of circulating sST2 concentrations. Quantile-quantile, Manhattan, and regional linkage disequilibrium plots are shown in Supplemental Figures 1–3. All genome-wide significant SNPs were located in a 400-kb linkage disequilibrium block that included IL1RL1 (the gene encoding ST2), IL1R1, IL1RL2, IL18R1, IL18RAP, and SLC9A4 (Figure 1). Results for 11 genome-wide significant “independent” SNPs, defined as pairwise r2 < 0.2, are shown in Table 1. In aggregate, these 11 “independent” genome-wide significant SNPs across the IL1RL1 locus accounted for 36% of heritability of sST2. In conditional analyses, 4 out of the 11 SNPs remained genome-wide significant, independent of each other (rs950880, rs13029918, rs1420103, and rs17639215), all within the IL1RL1 locus. The most significant SNP (rs950880, P = 7.1 x 10–94) accounted for 12% of the residual interindividual variability in circulating sST2 concentrations. Estimated mean sST2 concentrations were 43% higher in major homozygotes (CC) compared with minor homozygotes (AA). Tree loci outside of the IL1RL1 locus had suggestive associations with sST2 (P < 1 x 10–6) and are displayed in Supplemental Table 3.
In silico association with expression SNPs.
The top 10 sST2 SNPs (among 11 listed in Table 1) were explored in collected gene expression databases. There were 5 genome-wide significant sST2 SNPs associated with gene expression across a variety of tissue types (Table 2). Specifically, rs13001325 was associated with IL1RL1 gene expression (the gene encoding both soluble and transmembrane ST2) in several subtypes of brain tissue (prefrontal cortex, P = 1.95 x 10–12; cerebellum, P = 1.54 x 10–5; visual cortex, P = 1.85 x 10–7). The CC genotype of rs13001325 was associated with a higher IL1RL1 gene expression level as well as a higher circulating sST2 concentration when compared with the TT genotype (Supplemental Figure 4). Other ST2 variants were significantly associated with IL18RAP (P = 8.50 x 10–41, blood) and IL18R1 gene expression (P = 2.99 x 10–12, prefrontal cortex).
In silico association with clinical phenotypes in published data.
The G allele of rs1558648 was associated with lower sST2 concentrations in the FHS (0.88-fold change per G allele, P = 3.94 x 10–16) and higher all-cause mortality (hazard ratio [HR] 1.10 per G allele, 95% CI 1.03–1.16, P = 0.003) in the CHARGE consortium, which observed 8,444 deaths in 25,007 participants during an average follow-up of 10.6 years (22). The T allele of rs13019803 was associated with lower sST2 concentrations in the FHS (0.87-fold change per G allele, P = 5.95 x 10–20), higher mortality in the CHARGE consortium (HR 1.06 per C allele, 95% CI 1.01–1.12, P = 0.03), and higher risk of coronary artery disease (odds ratio 1.06, 95% CI 1.00–1.11, P = 0.035) in the CARDIoGRAM consortium, which included 22,233 individuals with coronary artery disease and 64,762 controls (23). In relating sST2 SNPs to other clinical phenotypes (including blood pressure, body-mass index, lipids, fasting glucose, natriuretic peptides, C-reactive protein, and echocardiographic traits) in previously published studies, we found nominal associations with C-reactive protein for 2 SNPs (Supplemental Table 4).
Putative functional variants.
Using GeneCruiser, we examined nonsynonymous SNPs (nSNPs) (missense variants) that had at least suggestive association with sST2 (P < 1 x 10–4), including SNPs that served as proxies (r2 = 1.0) for nSNPs within the 1000 Genomes Pilot 1 data set (ref. 24 and Table 3). There were 6 missense variants located within the IL1RL1 gene, 5 of which had genome-wide significant associations with sST2 concentrations, including rs6749114 (proxy for rs10192036, Q501K), rs4988956 (A433T), rs10204137 (Q501R), rs10192157 (T549I), rs10206753 (L551S), and rs1041973 (A78AE). Base substitutions and corresponding amino acid changes for these coding mutations are listed in Table 3. In combination, these 6 missense mutations accounted for 5% of estimated heritability, with an effect estimate of 0.23 (standard error [s.e.] 0.02, P = 2.4 x 10–20). When comparing major homozygotes with minor homozygotes, the estimated sST2 concentrations for these missense variants differed by 11% to 15% according to genotype (Supplemental Table 5). In conditional analyses, intracellular and extracellular variants appeared to be independently associated with sST2. For instance, in a model containing rs4988956 (A433T) and rs1041973 (A78E), both SNPs remained significantly associated with sST2 (P = 2.61 x 10–24 and P = 7.67 x 10–15, respectively). In total, missence variants added little to the proportion of sST2 variance explained by the 11 genome-wide significant nonmissense variants listed in Table 1. In relating these 6 missense variants to other clinical phenotypes in large consortia, we found an association with asthma for 4 out of the 6 variants (lowest P = 4.8 x 10–12 for rs10204137) (25).
Homology map of IL1RL1 missense variants and ST2 structure.
Of the 6 missense variants mapping to IL1RL1, 5 were within the cytoplasmic Toll/IL-1R (TIR) domain of the transmembrane ST2 receptor (Figure 2A), and these intracellular variants are thus not part of the circulating sST2 protein. Of these cytoplasmic domain variants, A433T was located within the “box 2” region of sequence conservation, described in the IL-1R1 TIR domain as important for IL-1 signaling . Q501R/K was within a conserved motif called “box 3,” but mutants of IL-1R1 in box 3 did not significantly affect IL-1 signaling in previous experiments (26). Both T549I and L551S were near the C terminus of the transmembrane ST2 receptor and were not predicted to alter signaling function based on previous experiments with the IL-1R . The A78E SNP was located within the extracellular domain of ST2 and is thus present in both the sST2 isoform and the transmembrane ST2 receptor. In models of the ST2/IL-33/IL-1RAcP complex derived from a crystal structure of the IL-1RII/IL-1β/IL-1RAcP complex (protein data bank ID 1T3G and 3O4O), A78E was predicted to be located on a surface loop within the first immunoglobulin-like domain (Figure 2B), distant from the putative IL-33 binding site or the site of interaction with IL-1RAcP. There were 2 rare extracellular variants that were not captured in our GWAS due to low minor allele frequencies (A80E, MAF 0.008; A176T, MAF 0.002). Both were distant from the IL-33 binding site on homology mapping and unlikely to affect IL-33 binding.
Functional effects of IL1RL1 missense variants on sST2 expression and promoter activity.
Since 5 of the IL1RL1 missense variants associated with sST2 levels mapped to the intracellular domain of ST2L and hence are not present on sST2 itself, we hypothesized that these missense variants exert effects via intracellular mechanisms downstream of ST2 transmembrane receptor signaling to regulate sST2 levels. To investigate the effect of IL1RL1 missense variants (identified by GWAS) on sST2 expression, stable cell lines expressing WT ST2L, IL1RL1 variants (A78E, A433T, T549I, Q501K, Q501R, and L551S), and a construct containing the 5 IL1RL1 intracellular domain variants (5-mut) were generated. Expression of ST2L mRNA and protein (detected in membrane fractions) was confirmed (Supplemental Figures 5 and 6). Eight different stable clones in each group were analyzed to reduce bias from clonal selection. Intracellular domain variants (A433T, T549I, Q501K, Q501R, L551S, and 5-mut), but not the extracellular domain variant (A78E), were associated with increased basal sST2 expression when compared with WT expression (P < 0.05 for all, Figure 3A). sST2 expression was highest in the 5-mut construct, suggesting that intracellular ST2L variants cooperatively regulate sST2 levels. This same pattern was consistent across different cell types (U937, Jurkat T, and A549 cells; Supplemental Figure 7). These findings suggest that intracellular domain variants of the transmembrane ST2 receptor may functionally regulate downstream signaling. IL1RL1 transcription may occur via two alternative promoters (proximal vs. distal), which leads to differential expression of the soluble versus membrane-bound ST2 proteins. Similar to the sST2 protein expression results above, the intracellular domain variants, but not the extracellular domain variant, were associated with higher basal proximal promoter activity. Distal promoter activity was also increased for most intracellular domain variants (Supplemental Figure 8).
IL1RL1 intracellular missense variants resulted in higher IL-33 protein levels.
In addition to upregulation of sST2 protein levels, IL1RL1 intracellular missense variants caused increased basal IL-33 protein expression (Figure 3B), suggesting a possible autoregulatory loop whereby IL-33 signaling positively induces sST2 expression. IL-33 induced sST2 protein expression in cells expressing both WT and IL1RL1 missense variants. Interestingly, this effect was particularly pronounced in the A433T and Q501R variants (Supplemental Figure 9A).
Enhanced IL-33 responsiveness is mediated by IL-113 in A433T and Q501R variants.
Interaction among IL-33, sST2, and IL-113.
Inhibition of IL-113 by anti–IL-113 mAb reduced basal expression of sST2 (Supplemental Figure 11A). Blocking of IL-33 by sST2 did not reduce the induction of IL-113 levels by the IL1RL1 variants (Supplemental Figure 11B). Furthermore, inhibition of IL-113 by anti–IL-113 reduced the basal IL-33 levels. IL-33 itself upregulated sST2 levels, which in turn reduced IL-33 levels (Supplemental Figure 11C). Our results revealed that both IL-33 and IL-113 drive sST2 expression and that IL-113 acts as an upstream inducer of IL-33 and maintains IL-33 expression by intracellular IL1RL1 variants (Supplemental Figure 11D). This suggests that IL1RL1 variants upregulated sST2 mainly through IL-33 autoregulation and that the enhanced IL-33 responsiveness by A433T and Q501R was mediated by IL-113 upregulation.
IL1RL1 missense variants modulate ST2 signaling pathways
The effect of IL1RL1 missense variants on known ST2 downstream regulatory pathways, including NF-KB, AP-1/c-Jun, AKT, and STAT3 , was examined in the presence and absence of IL-33 (Figure 4 and Supplemental Figure 12). The IL1RL1 intracellular missense variants (A433T, T549I, Q501K, Q501R, and L551S) were associated with higher basal phospho–NF-KB p65 and phospho–c-Jun levels (Figure 4, A and B). Consistent with enhanced IL-33 responsiveness in A433T and Q501R cells, levels of IL-33–induced NF-KB and c-Jun phosphorylation were enhanced in these 2 variants (Figure 4, B and D). In contrast, A433T and Q501R variants showed lower basal phospho-AKT levels (Figure 4E). ……….. The majority of sST2 gene variants in our study were located within or near IL1RL1, the gene coding for both transmembrane ST2 and sST2. IL1RL1 resides within a linkage disequilibrium block of 400 kb on chromosome 2q12, a region that includes a number of other cytokines, including IL-18 receptor 1 (IL18R1) and IL-18 receptor accessory protein (IL18RAP). Polymorphisms in this gene cluster have been associated previously with a number of immune and inflammatory conditions, including asthma, celiac disease, and type 1 diabetes mellitus . Many of these variants were associated with sST2 concentrations in our analysis (Supplemental Table 6). The immune effects of ST2 are corroborated by experimental evidence: membrane-bound ST2 is selectively expressed on Th2- but not Th1-type T helper cells, and activation of the ST2/IL-33 axis elaborates Th2 responses. In general, the allergic phenotypes above are thought to be Th2-mediated processes, in contrast to atherosclerosis, which appears to be a Th1-driven process.

Figure 2 Models of ST2 illustrate IL1RL1 missense variant locations.
Models of the (A) intracellular TIR domain (ST2-TIR) and the (B) extracellular domain (ST2-ECD) of ST2 (protein data bank codes 3O4O and 1T3G, respectively). Domains of ST2 are shown in yellow, with identified mis-sense SNP positions represented as red spheres and labels. Note that positions 549 and 551 are near the C terminus of ST2, which is not defined in the crystal structure (protein data bank ID 1T3G, shown as dashed black line in A). Arrows point toward the transmembrane domain, which is also not observed in crystal structures.

Figure 3 IL1RL1 intracellular missense variants resulted in higher sST2 and IL-33.
Media from KU812 cells expressing WT and IL1RL1 missense variants were collected for ELISA analysis of (A) sST2, (B) IL-33, and (C) IL-113 levels. Horizontal bars indicate mean values, and symbols represent individual variants. *P < 0.05, **P < 0.01 vs. WT. (D) Effect of anti–IL-113 mAb on IL-33–induced sST2 expression. Dashed line indicates PBS-treated cells as referent group. Error bars represent mean ± SEM from 2 independent experiments. *P < 0.05 vs. IL-33.

Figure 4 IL1RL1 missense variants modulated ST2 signaling pathways.
KU812 cells expressing WT or
IL1RL1 variants were treated with PBS or IL-33. Levels of the following phosphorylated proteins were detected in cell lysates using ELISA: (A and B) phospho-NF-
KB p65; (C and D) phospho-c-Jun activity; and (E and F) phospho-AKT. (A, C, and E) White bars represent basal levels, and (B, D, and F) gray bars represent relative fold increase (compared with PBS-treated group) after IL-33 treatment. *
P < 0.05 vs. WT; **
P < 0.01 vs. PBS-treated group. Dashed line in B, D, and F represents PBS-treated cells as referent group. Error bars represent mean ± SEM from 2 independent experiments.

Figure 5 IL-33–induced sST2 expression is enhanced with mTOR inhibition and occurs via ST2L-dependent signaling.
(A) sST2 mRNA expression in KU812 cells after treatment with DMSO, IL-33, or IL-33 plus signal inhibitors (wortmannin, LY294002, rapamycin, PD98059, SP60125, BAY11-7082, or SR11302). (B) ST2L mRNA and (C) sST2 mRNA expression in KU812 cells treated with PBS (white columns), rapamycin (rapa), anti-ST2 mAb, IL-33, IL-33 plus anti-ST2, IL-33 plus rapamycin, IL-33 plus rapamycin plus anti-ST2 mAb, or rapamycin plus anti-ST2. (D) IL33 mRNA expression in KU812 cells after treatment with DMSO, signal inhibitors, IL-33 plus signal inhibitors, and IL-1n plus signal inhibitors. *P < 0.05 vs. PBS-treated group; #P < 0.05 vs. IL-33–treated group; &P < 0.05 vs. IL-1n–treated group. Error bars represent mean ± SEM from 2 independent experiments. (E) A schematic model illustrating the regulation of sST2 expression by IL1RL1 missense variants through enhanced induction of IL-33 via enhanced NF-KB and AP-1 signaling and enhanced IL-33 responsiveness via increasing ST2L expression.
Quantitating subcellular metabolism with multi-isotope imaging mass spectrometry
ML Steinhauser, A Bailey, SE Senyo, C Guillermier, TS Perlstein, AP Gould, RT Lee, and CP Lechene
Department of Medicine, Divisions of Cardiovascular Medicine & Genetics, Brigham and Women’s Hospital, Harvard Medical School & Harvard Stem Cell Institute Division of Physiology and Metabolism, Medical Research Council National Institute for Medical Research, Mill Hill, London, UK National Resource for Imaging Mass Spectroscopy
Nature 2012;481(7382): 516–519. http://dx. do.org/10.1038/nature10734
Mass spectrometry with stable isotope labels has been seminal in discovering the dynamic state of living matter, but is limited to bulk tissues or cells. We developed multi-isotope imaging mass spectrometry (MIMS) that allowed us to view and measure stable isotope incorporation with sub-micron resolution. Here we apply MIMS to diverse organisms, including Drosophila, mice, and humans. We test the “immortal strand hypothesis,” which predicts that during asymmetric stem cell division chromosomes containing older template DNA are segregated to the daughter destined to remain a stem cell, thus insuring lifetime genetic stability. After labeling mice with 15N-thymidine from gestation through post-natal week 8, we find no 15N label retention by dividing small intestinal crypt cells after 4wk chase. In adult mice administered 15N-thymidine pulse-chase, we find that proliferating crypt cells dilute label consistent with random strand segregation. We demonstrate the broad utility of MIMS with proof-of-principle studies of lipid turnover in Drosophila and translation to the human hematopoietic system. These studies show that MIMS provides high-resolution quantitation of stable isotope labels that cannot be obtained using other techniques and that is broadly applicable to biological and medical research. MIMS combines ion microscopy with secondary ion mass spectrometry (SIMS), stable isotope reporters, and intensive computation (Supplemental Fig 1). MIMS allows imaging and measuring stable isotope labels in cell domains smaller than one micron cubed. We tested the potential of MIMS to quantitatively track DNA labeling with 15N-thymidine in vitro. In proliferating fibroblasts, we detected label incorporation within the nucleus by an increase in the 15N/14N ratio above natural ratio (Fig 1a). The labeling pattern resembled chromatin with either stable isotope-tagged thymidine or thymidine analogs (Fig 1b). We measured dose-dependent incorporation of 15N-thymidine over three orders of magnitude (Fig 1d, Supplemental Fig 2). We also tracked fibroblast division after a 24-hour label-free chase (Fig 1d,e, Supplemental Fig 3). Cells segregated into two populations, one indistinguishable from control cells suggesting no division, the other with halving of label, consistent with one division during chase. We found similar results by tracking cell division in vivo in the small intestine (Fig 1f,g, Supplemental Figs 4–6). We measured dose-dependent 15N-thymidine incorporation within nuclei of actively dividing crypt cells (Fig 1g, Supplemental Fig 4), down to a dose of 0.1µg/ g (Supplemental Fig 2). The cytoplasm was slightly above natural ratio, likely due to low level soluble 15N-thymidine or mitochondrial incorporation (Supplemental Fig 2). We measured halving of label with each division during label-free chase (Supplemental Fig 6). We then tested the “immortal strand hypothesis,” a concept that emerged from autoradiographic studies and that predicted long-term label retaining cells in the small intestinal crypt. It proposes that asymmetrically dividing stem cells also asymmetrically segregate DNA, such that older template strands are retained by daughter cells that will remain stem cells and newer strands are passed to daughters committed to differentiation (Supplemental Fig 7)5,6. Modern studies continue to argue both for or against the hypothesis, leading to the suggestion that definitive resolution of the debate will require a new experimental approach. Although prior evidence suggests a concentration of label-retaining cells in the +4 anatomic position, we searched for DNA label retention irrespective of anatomic position or molecular identity. We labeled mice with 15N-thymidine for the first 8 wks of life when intestinal stem cells are proposed to form. After a 4-wk chase, mice received bromodeoxyuridine (BrdU) for 24h prior to sacrifice to identify proliferating cells(Fig 2a, Supplemental Fig 8: Exp 1), specifically crypt base columnar (CBC) cells and transit amplifying cells (TA) (Supplemental Fig 9), which cycle at a rate of one and two times per 24h, respectively (Supplemental Fig 10). All crypt cell nuclei were highly labeled upon completion of 15N-thymidine; after a four-week chase, however, we found no label retention by non-Paneth crypt cells (Fig 2b–f; n=3 mice, 136 crypts analysed). 15N-labeling in BrdU−/15N+ Paneth and mesenchymal cells was equivalent to that measured at pulse completion (Fig2b,c) suggesting quiescence during the chase (values above 15N/14N natural ratio: Paneth pulse=107.8 +/− 5.0% s.e.m. n=51 vs Paneth pulse-chase=96.3+/−2.8% s.e.m. n=218; mesenchymal pulse=92.0+/−5.0% s.e.m. n=89 vs mesenchymal pulse-chase=90.5+/ −2.2% s.e.m. n=543). The number of randomly selected crypt sections was sufficient to detect a frequency as low as one label-retaining stem cell per crypt irrespective of anatomic location within the crypt. Because each anatomic level contains approximately 16 circumferentially arrayed cells, a 2-dimensional analysis captures approximately 1/8th of the cells at each anatomic position (one on each side of the crypt; Supplemental Fig 9a). Therefore, assuming only 1 label-retaining stem cell per crypt we should have found 17 label-retaining cells in the 136 sampled crypts (1/8th of 136); we found 0 (binomial test p<0.0001). The significance of this result held after lowering the expected frequency of label-retaining cells by 25% to account for the development of new crypts, a process thought to continue into adulthood. In three additional experiments, using shorter labeling periods and including in utero development, we also found no label-retaining cells in the crypt other than Paneth cells (Supplemental Fig 8, Exps 2–4).
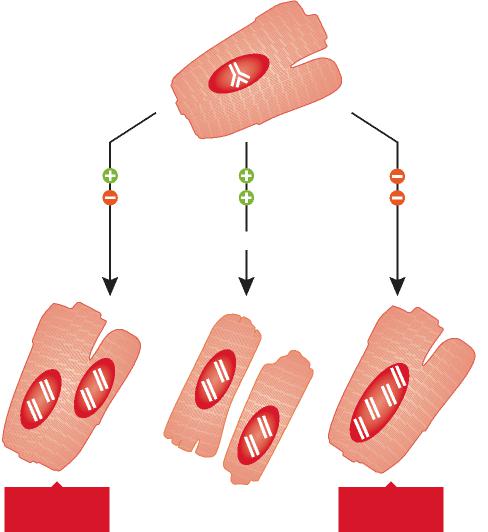
In recent years, several protocols have been developed experimentally in an attempt to identify novel therapeutic interventions aiming at the reduction of infarct size and prevention of short and long term negative ventricular remodeling following ischemic myocardial injury. Three main strategies have been employed and a significant amount of work is being conducted to determine the most effective form of action for acute ischemic heart failure. The delivery of bone marrow progenitor cells (BMCs) has been highly controversial, but recent clinical data have shown improvement in ventricular performance and clinical outcome. These observations have not changed the nature of the debate concerning the efficacy of this cell category for the human disease and the mechanisms involved in the impact of BMCs on cardiac structure and function. Whether BMCs transdifferentiate and acquire the cardiomyocyte lineage has faced strong opposition and data in favor and against this possibility have been reported. However, this is the only cell class which has been introduced in the treatment of heart failure in patients and large clinical trials are in progress.
Human embryonic stem cells (ESCs) have repeatedly been utilized in animal models to restore the acutely infarcted myocardium, but limited cell engraftment, modest ability to generate vascular structures, teratoma formation and the apparent transient beneficial effects on cardiac hemodynamics have questioned the current feasibility of this approach clinically. Tremendous efforts are being performed to reduce the malignant tumorigenic potential of ESCs and promote their differentiation into cardiomyocytes with the expectation that these extremely powerful cells may be applied to human beings in the future. Additionally, the study of ESCs may provide unique understanding of the mechanisms of embryonic development that may lead to therapeutic interventions in utero and the correction of congenital malformations.
The recognition that a pool of primitive cells with the characteristics of stem cells resides in the myocardium and that these cells form myocytes, ECs and SMCs has provided a different perspective of the biology of the heart and mechanisms of cardiac homeostasis and tissue repair. Regeneration implies that dead cells are replaced by newly formed cells restoring the original structure of the organ. In adulthood, this process occurs during physiological cell turnover, in the absence of injury. However, myocardial damage interferes with recapitulation of cell turnover and restitutio ad integrum of the organ. Because of the inability of the adult heart to regenerate itself after infarction, previous studies have promoted tissue repair by injecting exogenously expanded CPCs in proximity of the necrotic myocardium or by activating resident CPCs through the delivery of growth factors known to induce cell migration and differentiation. These strategies have attenuated ventricular dilation and the impairment in cardiac function and in some cases have decreased animal mortality.
Although various subsets of CPCs have been used to reconstitute the infarcted myocardium and different degrees of muscle mass regeneration have been obtained, in all cases the newly formed cardiomyocytes possessed fetal-neonatal characteristics and failed to acquire the adult cell phenotype. In the current study, to enhance myocyte growth and differentiation, we have introduced cell therapy together with the delivery of self-assembly peptide nanofibers to provide a specific and prolonged local myocardial release of IGF-1. IGF-1 increases CPC growth and survival in vitro and in vivo and this effect resulted here in a major increase in the formation of cardiomyocytes and coronary vessels, decreasing infarct size and restoring partly cardiac performance. This therapeutic approach was superior to the administration of CPCs or NF-IGF-1 only. Combination therapy appeared to be additive; it promoted myocardial regeneration through the activation and differentiation of resident and exogenously delivered CPCs. Additionally, the strategy implemented here may be superior to the utilization of BMCs for cardiac repair. CPCs are destined to form myocytes, and vascular SMCs and ECs and, in contrast to BMCs, do not have to transdifferentiate to acquire cardiac cell lineages. Transdifferentiation involves chromatin reorganization with activation and silencing of transcription factors and epigenetic modifications.
Selected References
- Hsieh PC, Davis ME, Gannon J, MacGillivray C, Lee RT. Controlled delivery of PDGF-BB for myocardial protection using injectable self-assembling peptide nanofibers. J Clin Invest 2006;116:237–248. [PubMed: 16357943]
- Davis ME, Hsieh PC, Takahashi T, Song Q, Zhang S, Kamm RD, Grodzinsky AJ, Anversa P, Lee RT. Local myocardial insulin-like growth factor 1 (IGF-1) delivery with biotinylated peptide nanofibers improves cell therapy for myocardial infarction. Proc Natl Acad Sci USA 2006;103:8155–8160. [PubMed: 16698918]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003;114:763–776. [PubMed: 14505575]
- Rota M, Padin-Iruegas ME, Misao Y, De Angelis A, Maestroni S, Ferreira-Martins J, Fiumana E, Rastaldo R, Arcarese ML, Mitchell TS, Boni A, Bolli R, Urbanek K, Hosoda T, Anversa P, Leri A, Kajstura J. Local activation or implantation of cardiac progenitor cells rescues scarred infarcted myocardium improving cardiac function. Circ Res 2008;103:107–116. [PubMed: 18556576]

Figure 2. Cardiac anatomy.
(A and B) Cardiac weights and infarct size. R and L correspond, respectively, to the number of myocytes remaining and lost after infarction. (C–G) LV dimensions. Sham-operated: SO. *Indicates P<0.05 vs SO; **vs untreated infarcts (UN); †vs infarcts treated with CPCs; ‡vs infarcts treated with NF-IGF-1.

Figure 3. Ventricular function.
Combination therapy (CPC-NF-IGF-1) attenuated the most the negative impact of myocardial infarction on cardiac performance. See Figure 2 for symbols.
Endothelial Cells Promote Cardiac Myocyte Survival and Spatial Reorganization: Implications for Cardiac Regeneration
Daria A. Narmoneva, Rada Vukmirovic, Michael E. Davis, Roger D. Kamm, and Richard T. Lee
Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, and the Division of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA
Circulation. 2004 August 24; 110(8): 962–968. http://dx.doi.org/10.1161/01.CIR.0000140667.37070.07
Background
Endothelial-cardiac myocyte (CM) interactions play a key role in regulating cardiac function, but the role of these interactions in CM survival is unknown. This study tested the hypothesis that endothelial cells (ECs) promote CM survival and enhance spatial organization in a 3-dimensional configuration.
Methods and Results
Microvascular ECs and neonatal CMs were seeded on peptide hydrogels in 1 of 3 experimental configurations:
- CMs alone,
- CMs mixed with ECs (coculture), or
- CMs seeded on preformed EC networks (prevascularized).
Capillary-like networks formed by ECs promoted marked CM reorganization along the EC structures, in contrast to limited organization of CMs cultured alone. The presence of ECs markedly inhibited CM apoptosis and necrosis at all time points. In addition, CMs on preformed EC networks resulted in significantly less CM apoptosis and necrosis compared with simultaneous EC-CM seeding (P<0.01, ANOVA). Furthermore, ECs promoted synchronized contraction of CMs as well as connexin 43 expression.
Conclusions
These results provide direct evidence for a novel role of endothelium in survival and organization of nearby CMs. Successful strategies for cardiac regeneration may therefore depend on establishing functional CM-endothelium interactions.
Keywords: endothelium; cardiomyopathy; heart failure; tissue
Introduction
Recent studies suggest that the mammalian heart possesses some ability to regenerate itself through several potential mechanisms, including generation of new cardiomyocytes (CMs) from extracardiac progenitors, CM proliferation, or fusion with stem cells with subsequent hybrid cell division. These mechanisms are insufficient to regenerate adequate heart tissue in humans, although some vertebrates can regenerate large volumes of injured myocardium.
Several approaches in cell transplantation and cardiac tissue engineering have been investigated as potential treatments to enhance cardiac function after myocardial injury. Implantation of skeletal muscle cells, bone marrow cells, embryonic stem cell-derived CMs, and myoblasts can enhance cardiac function. Cell-seeded grafts have been used instead of isolated cells for in vitro cardiac tissue growth or in vivo transplantation. These grafts can develop a high degree of myocyte spatial organization, differentiation, and spontaneous and coordinated contractions. On implantation in vivo, cardiac grafts can integrate into the host tissue and neovascularization can develop. However, the presence of scar tissue and the death of cells in the graft can limit the amount of new myocardium formed, most likely due to ischemia. Therefore, creating a favorable environment to promote survival of transplanted cells and differentiation of progenitor cells remains one of the most important steps in regeneration of heart tissue.
One of the key factors for myocardial regeneration is revascularization of damaged tissue. In the normal heart, there is a capillary next to almost every CM, and endothelial cells (ECs) outnumber cardiomyocytes by ≈3:1. Developmental biology experiments reveal that myocardial cell maturation and function depend on the presence of endocardial endothelium at an early stage. Experiments with inactivation or overexpression of vascular endothelial growth factor (VEGF) demonstrated that at later stages, either an excess or a deficit in blood vessel formation results in lethality due to cardiac dysfunction. Both endocardium and myocardial capillaries have been shown to modulate cardiac performance, rhythmicity, and growth. In addition, a recent study showed the critical importance of CM-derived VEGF in paracrine regulation of cardiac morphogenesis. These findings and others highlight the significance of interactions between CMs and endothelium for normal cardiac function. However, little is known about the specific mechanisms for these interactions, as well as the role of a complex, 3-dimensional organization of myocytes, ECs, and fibroblasts in the maintenance of healthy cardiac muscle.
The critical relation of CMs and the microvasculature suggests that successful cardiac regeneration will require a strategy that promotes survival of both ECs and CMs. The present study explored the hypothesis that ECs (both as preexisting capillary-like structures and mixed with myocytes at the time of seeding) promote myocyte survival and enhance spatial reorganization in a 3-dimensional configuration. The results demonstrate that CM interactions with ECs markedly decrease myocyte death and show that endothelium may be important not only for the delivery of blood and oxygen but also for the formation and maintenance of myocardial structure.
Methods
- Three-Dimensional Culture
- Immunohistochemistry and Cell Death Assays
- Evaluation of Contractile Areas
Results
- EC-CM Interactions Affect Myocyte Reorganization
- ECs Improve Survival of CMs
- Preformed Endothelial Networks Promote Coordinated, Spontaneous Contractions
- ECs Promote Cx43 Expression
EC-CM Interactions Affect Myocyte Reorganization
To explore interactions between CMs and ECs in 3-dimensional culture, we used peptide hydrogels, a tissue engineering scaffold. Cells seeded on the surface of the hydrogel attach and then migrate into the hydrogel. When CMs alone were used, cells attached on day 1 and then formed small clusters of cells at days 3 and 7 (Figure 1). In contrast, when CMs were seeded together with ECs, cells formed interconnected linear networks, as commonly seen with ECs in 3-dimensional culture environments, with increasing spatial organization from day 1 to day 7 (Figure 1).
Figure 1. ECs promote CM reorganization.
When CMs were cultured alone (left column), they aggregated into sparse clusters. When CMs were cultured with ECs (center), cells organized into capillary-like networks. There was no difference in morphological appearance between coculture or prevascularized cultures (not shown) and ECs alone (right column). Bar=100 μm. Abbreviations are as defined in text.
To establish whether preformed endothelial networks enhanced the organization of myocytes, we also seeded ECs 1 day before myocytes were added. These ECs formed similar interconnected networks in the absence of myocytes; preforming the vascular network did not lead to significant differences in morphology (data not shown). Furthermore, to exclude the possibility that the increasing cell density of added ECs caused the spatial organization, we also performed control experiments with varying numbers and combinations of cells; there was no effect of doubling or halving cell numbers, indicating that the spatial organization effect was specifically due to ECs. To establish that both myocytes and ECs were forming networks together, we performed immunofluorescence studies with specific antibodies, as well as analysis of cross sections of CM-EC cocultures, whereby cells were labeled with CellTracker dyes before seeding. Immunofluorescent staining demonstrated that >95% of CMs were present within these networks, suggesting that CMs preferentially migrate to or survive better near ECs (Figure 2).
Figure 2. CMs appear on outside of endothelial networks.
CMs appear on outside of endothelial networks. High-magnification, double-immunofluorescence image of structures formed in EC-CM coculture at day 7 demonstrating CMs (sarcomeric actinin, red) spread on top of ECs (von Willebrand factor, green) with no myocytes present outside structure. Bar=100 μm. Abbreviations are as defined in text.
The analysis of cross sections demonstrated the presence of what appeared to be EC-derived, tubelike structures (Figure 3), with myocytes spread on the outer part of the capillary wall. Along with the capillary-like structures, clusters of intermingled cells (both myocytes and ECs) not containing the lumen were also observed (not shown). However, when the lumen was present, ECs were always on the inner side and myocytes on the outer side of the structure.
Figure 3. ECs form tubelike structures with myocytes spreading on outer wall.
Cross section of paraffin-embedded sample of 3-day coculture of myocytes (red) and ECs (green) incubated in CellTracker dye before seeding on hydrogel. Bar=50 μm. Abbreviations are as defined in text.
In CM-fibroblast cocultures, cells rapidly (within 24 hours) formed large clusters consisting of cells of both types (not shown). At later time points, fibroblast proliferation resulted in their migration outside the clusters and spreading on the hydrogel without any pattern. However, in contrast to EC-CM cocultures, CMs remained in the clusters and demonstrated only limited spreading. Immunofluorescent staining revealed that there was no orientation of myocytes relative to the fibroblasts in the clusters. In cultures with EC-conditioned medium, myocyte morphology and spatial organization remained similar to those of myocyte controls.
ECs Improve Survival of CMs
To test the hypothesis that ECs promote CM survival, we assessed apoptosis and necrosis in the 3-dimensional cultures. Quantitative analyses of CMs positive for TUNEL and necrosis staining demonstrated significantly decreased myocyte apoptosis and necrosis when cultured with ECs, compared with CM-only cultures (Figure 4, P<0.01). This effect was observed at all 3 time points, although the decreased necrosis was most pronounced at day 1. In addition, CMs seeded on the preformed EC networks had a lower rate of apoptosis at day 1 relative to same-time seeding cultures (P<0.05, post hoc test), suggesting that early EC-CM interactions provided by the presence of well-attached and prearranged ECs may further promote CM survival. In contrast to the ECs, cardiac fibroblasts did not affect myocyte survival (P>0.05, Figure 4), with ratios for myocyte apoptosis and necrosis in the myocyte-fibroblast cocultures being similar to those for myocyte-only controls. However, addition of EC-conditioned medium resulted in a significant decrease in apoptosis and necrosis ratios of myocytes (P<0.01). Interestingly, the effect of conditioned medium on myocyte necrosis was similar in magnitude to the effect of ECs, whereas myocyte apoptosis ratios in the conditioned-medium group were only partially decreased compared with those in the presence of ECs. These results suggest that the prosurvival effect of ECs on CMs may not only be merely due to the local interactions between myocytes and ECs during myocyte attachment but may also involve direct signaling between myocytes and ECs.
Figure 4. ECs prolong survival of CMs
Top, dual immunostaining of CMs and EC-myocyte prevascularized groups at day 3 in culture, with TUNEL-positive cells in red; green indicates sarcomeric actinin; blue, DAPI. Bottom, presence of ECs decreased CM apoptosis and necrosis, both in coculture conditions and when cultures were prevascularized by seeding with ECs 1 day before CMs (mean±SD, P<0.01). EC-conditioned medium decreased myocyte apoptosis and necrosis (P<0.01), whereas fibroblasts did not have any effect (P>0.05). *Different from myocytes alone; **different from EC-myocyte coculture and pre-vascularized. Bar=100 μm. Abbreviations are as defined in text.
Preformed Endothelial Networks Promote Coordinated, Spontaneous Contractions
In the prevascularized group with preformed vascular structures, synchronized, spontaneous contractions of large areas (Figure 5, top panels) were detected as early as days 2 to 3after seeding, in contrast to the coculture group, wherein such contractions were observed on days 6 to 7. In CM-only cultures, beating of separate cells and small cell clusters was also detected at days 2 to 3, similar to that in the prevascularized group. However, the average area of synchronized beating at day 3 in the myocyte-only group (3.5±0.5×102 μm2) was nearly 3 orders of magnitude smaller than the synchronously contracting area in the prevascularized group (4.3±2.5×105 μm2, mean±SD, n=5). These data suggest that ECs promote synchronized CM contraction, particularly when vascular networks are already formed.
Figure 5. ECs promote large-scale, synchronized contraction of CMs.
Left, phase-contrast video of beating areas in CM-only and prevascularized groups (day 3). Right, motion analysis of video showing regions of synchronized contractions (connected areas in purple are contracting synchronously) and nonmoving areas in blue. Bars=100 μm. Abbreviations are as defined in text.
ECs Promote Cx43 Expression
Staining for Cx43 showed striking differences in the distribution pattern of this gap junction protein between EC-CM cocultures and CMs cultured alone. In myocyte-only cultures, Cx43 expression was barely detectable at day 1 (not shown); at days 3 and 7, Cx43 expression was sparse throughout the cell clusters (Figure 6). In the presence of ECs (in both coculture and prevascularized groups), Cx43 staining was evident at day 1, both between ECs and distributed among CMs. As early as day 3 in culture, patches of localized junction-like Cx43, in addition to diffuse staining, were observed for myocytes in the coculture group (Figure 6). In the prevascularized group at day 3, wherein spontaneous contractions were already observed, more junction-like patches of Cx43 were observed compared with the coculture group, indicating electrical connections between myocytes (Figure 6). In addition to junctions between myocytes, there was also evidence of Cx43 localized at the interface between ECs and myocytes (Figure 6) detected in both the coculture group (at day 7) and the preculture group (as early as day 3). When myocytes and myocyte-EC coculture groups were cultured for 3 days with or without addition of 100 ng/mL of neutralizing anti-mouse VEGF antibody (R&D Systems), we observed no differences in either apoptosis or Cx43 staining between VEGF antibody-containing cultures and controls.
Figure 6. ECs promote Cx43 expression
Cultures at 3 days immunostained for Cx43 (red) and anti-sarcomeric actinin (green); nuclei are stained with DAPI (blue). For CMs alone (left), Cx43 staining is diffuse and sparse, with no evidence of gap junctions; for coculture (center), both diffuse (yellow arrow) and patchlike (thin, white arrow) Cx43 staining is observed; for prevascularized (right), increased patchlike staining indicates presence of gap junctions. Thick arrow-heads indicate junctions between myocytes and ECs. Bar=50 μm. Abbreviations are as defined in text.






Endothelial-Cardiomyocyte Interactions in Cardiac Development and Repair: Implications for Cardiac Regeneration
Patrick C.H. Hsieh, Michael E. Davis, Laura K. Lisowski, and Richard T. Lee
Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA
Annu Rev Physiol. PMC 2009 September 30
The ongoing molecular conversation between endothelial cells and cardiomyocytes is highly relevant to the recent excitement in promoting cardiac regeneration. The ultimate goal of myocardial regeneration is to rebuild a functional tissue that closely resembles mature myocardium, not just to improve systolic function transiently. Thus, regenerating myocardium will require rebuilding the vascular network along with the cardiomyocyte architecture. Here we review evidence demonstrating crucial molecular interactions between endothelial cells and cardiomyocytes. We first discuss endothelial-cardiomyocyte interactions during embryonic cardiogenesis, followed with morphological and functional characteristics of endothelial-cardiomyocyte interactions in mature myocardium. Finally, we consider strategies exploiting endothelial-cardiomyocyte interplay for cardiac regeneration.
Signaling from Cardiomyocytes to Endothelial Cells
The examples of neuregulin-1, NF1, and PDGF-B demonstrate that signals from endothelial cells regulate the formation of primary myocardium. Similarly, signaling from myocardial cells to endothelial cells is also required for cardiac development. Two examples of myocardial-to-endothelial signaling are vascular endothelial growth factor (VEGF)-A and angiopoietin-1.
VASCULAR ENDOTHELIAL GROWTH FACTOR-A
VEGF-A is a key regulator of angiogenesis during embryogenesis. In mice, a mutation in VEGF-A causes endocardial detachment from an underdeveloped myocardium. A mutation in VEGF receptor-2 (or Flk-1) also results in failure of the endocardium and myocardium to develop (18). Furthermore, cardiomyocyte-specific deletion of VEGF-A results in defects in vasculogenesis/angiogenesis and a thinned ventricular wall, further confirming reciprocal signaling from the myocardial cell to the endothelial cell during cardiac development. Interestingly, this cardiomyocyte-selective VEGF-A-deletion mouse has underdeveloped myocardial microvasculature but preserved coronary artery structure, implying a different signaling mechanism for vasculogenesis/angiogenesis in the myocardium and in the epicardial coronary arteries.
Cardiomyocyte-derived VEGF-A also inhibits cardiac endocardial-to-mesenchymal transformation. This process is essential in the formation of the cardiac cushions and requires delicate control of VEGF-A concentration. A minimal amount of VEGF initiates endocardial-to-mesenchymal transformation, whereas higher doses of VEGF-A terminate this transformation. Interestingly, this cardiomyocyte-derived VEGF-A signaling for endocardial-to-mesenchymal transformation may be controlled by an endothelial-derived feedback mechanism through the calcineurin/NFAT pathway (24), demonstrating the importance of endothelial-cardiomyocyte interactions for cardiac morphogenesis.
ANGIOPOIETIN-1
—Another mechanism of cardiomyocyte control of endothelial cells during cardiac development is the angiopoietin-Tie-2 system. Both angiopoietin-1 and angiopoietin-2 may bind to Tie-2 receptors in a competitive manner, but with opposite effects: Angiopoietin-1 activates the Tie-2 receptor and prevents vascular edema, whereas angiopoietin-2 blocks Tie-2 phosphorylation and increases vascular permeability. During angiogenesis/vasculogenesis, angiopoietin-1 is produced primarily by pericytes, and Tie-2 receptors are expressed on endothelial cells. Angiopoietin-1 regulates the stabilization and maturation of neovasculature; genetic deletion of angiopoietin-1 or Tie-2 causes a defect in early vasculogenesis/angiogenesis and is lethal.
Cardiac endocardium is one of the earliest vascular components (along with the dorsal aorta and yolk sac vessels) and the adult heart can be regarded as a fully vascularized organ, angiopoietin-Tie-2 signaling may also be required for early cardiac development. Indeed, mice with mutations in Tie-2 have underdeveloped endocardium and myocardium. These Tie-2 knockout mice display defects in the endocardium but have normal vascular morphology at E10.5, suggesting that the endocardial defect is the fundamental cause of death. In addition, a recent study showed that overexpression, and not deletion, of angiopoietin-1 from cardiomyocytes caused embryonic death between E12.5-15.5 due to cardiac hemorrhage. The mice had defects in the endocardium and myocardium and lack of coronary arteries, suggesting that, as with VEGF-A, a delicate control of angiopoietin-1 concentration is critical for early heart development.
ENDOTHELIAL-CARDIOMYOCYTE INTERACTIONS IN NORMAL CARDIAC FUNCTION
Cardiac Endothelial Cells Regulate Cardiomyocyte Contraction
The vascular endothelium senses the shear stress of flowing blood and regulates vascular smooth muscle contraction. It is therefore not surprising that cardiac endothelial cells—the endocardial endothelial cells as well as the endothelial cells of intramyocardial capillaries— regulate the contractile state of cardiomyocytes. Autocrine and paracrine signaling molecules released or activated by cardiac endothelial cells are responsible for this contractile response (Figure 2).
NITRIC OXIDE
Three different nitric oxide synthase isoenzymes synthesize nitric oxide (NO) from L-arginine. The neuronal and endothelial NO synthases (nNOS and eNOS, respectively) are expressed in normal physiological conditions, whereas the inducible NO synthase is induced by stress or cytokines. Like NO in the vessel, which causes relaxation of vascular smooth muscle, NO in the heart affects the onset of ventricular relaxation, which allows for a precise optimization of pump function beat by beat. Although NO is principally a paracrine effector secreted by cardiac endothelial cells, cardiomyocytes also express both nNOS and eNOS. Endothelial expression of eNOS exceeds that in cardiomyocytes by greater than 4:1. Cardiomyocyte autocrine eNOS signaling can regulate β-adrenergic and muscarinic control of contractile state.
Barouch et al. demonstrated that cardiomyocyte nNOS and eNOS may have opposing effects on cardiac structure and function. Using mice with nNOS or eNOS deficiency, they found that nNOS and eNOS have not only different localization in cardiomyocytes but also opposite effects on cardiomyocyte contractility; eNOS localizes to caveolae and inhibits L-type Ca2+ channels, leading to negative inotropy, whereas nNOS is targeted to the sarcoplasmic reticulum and facilitates Ca2+ release and thus positive inotropy (31). These results demonstrate that spatial confinement of different NO synthase isoforms contribute independently to the maintenance of cardiomyocyte structure and phenotype.
As indicated above, mutation of neuregulin or either of two of its cognate receptors, erbB2 and erbB4, causes embryonic death during mid-embryogenesis due to aborted development of myocardial trabeculation . Neuregulin also appears to play a role in fully developed myocardium. In adult mice, cardiomyocyte-specific deletion of erbB2 leads to dilated cardiomyopathy. Neuregulin from endothelial cells may induce a negative inotropic effect in isolated rabbit papillary muscles. This suggests that, along with NO, the neuregulin signaling pathway acts as an endothelial-derived regulator of cardiac inotropism. In fact, the negative inotropic effect of neuregulin may require NO synthase because L-NMMA, an inhibitor of NO synthase, significantly attenuates the negative inotropy of neuregulin.
Studies to date indicate that cardiac regeneration in mammals may be feasible, but the response is inadequate to preserve myocardial function after a substantial injury. Thus, understanding how normal myocardial structure can be regenerated in adult hearts is essential. It is clear that endothelial cells play a role in cardiac morphogenesis and most likely also in survival and function of mature cardiomyocytes. Initial attempts to promote angiogenesis in myocardium were based on the premise that persistent ischemia could be alleviated. However, it is also possible that endothelial-cardiomyocyte interactions are essential in normal cardiomyocyte function and for protection from injury. Understanding the molecular and cellular mechanisms controlling these cell-cell interactions will not only enhance our understanding of the establishment of vascular network in the heart but also allow the development of new targeted therapies for cardiac regeneration by improving cardiomyocyte survival and maturation.

Figure 1. Endothelial-cardiomyocyte assembly in adult mouse myocardium.
Normal adult mouse myocardium is stained with intravital perfusion techniques to demonstrate cardiomyocyte (outlined in red) and capillary (green; stained with isolectin-fluorescein) assembly. Nuclei are blue (Hoechst). Original magnification: 600X

Intramyocardial Fibroblast – Myocyte Communication
Rahul Kakkar, M.D. and Richard T. Lee, M.D.
From the Cardiology Division, Massachusetts General Hospital and the Cardiovascular Division, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Boston, MA
Circ Res. 2010 January 8; 106(1): 47–57. http://dx.doi.org/10.1161/CIRCRESAHA.109.207456
Cardiac fibroblasts have received relatively little attention compared to their more famous neighbors, the cardiomyocytes. Cardiac fibroblasts are often regarded as the “spotters”, nonchalantly watching the cardiomyocytes do the real weight-lifting, and waiting for a catastrophe that requires their actions. However, emerging data now reveal the fibroblast as not only a critical player in the response to injury, but also as an active participant in normal cardiac function.
Interest in cardiac fibroblasts has grown with the recognition that cardiac fibrosis is a prominent contributor to diverse forms of myocardial disease. In the early 1990’s, identification of angiotensin receptors on the surface of cardiac fibroblasts linked the renin-angiotensin-aldosterone system directly with pathologic myocardial and matrix extracellular remodeling. Fibroblasts were also revealed as a major source of not only extracellular matrix, but the proteases that regulate and organize matrix. New research has uncovered paracrine and well as direct cell-to-cell interactions between fibroblasts and their cardiomyocyte neighbors, and cardiac fibroblasts appear to be dynamic participants in ventricular physiology and pathophysiology.
This review will focus on several aspects of fibroblast-myocyte communication, including mechanisms of paracrine communication. Ongoing efforts at regeneration of cardiac tissue focus primarily on increasing the number of cardiomyocytes in damaged myocardium. Although getting cardiomyocytes into myocardium is an important goal, understanding intercellular paracrine communication between different cell types, including endothelial cells but also fibroblasts, may prove crucial to regenerating stable myocardium that responds to physiological conditions appropriately.
An area of active research in cardiovascular therapeutics is the attempt to engineer, ex vivo, functional myocardial tissue that may be engrafted onto areas of injured ventricle. Recent data suggests that the inclusion of cardiac fibroblasts in three-dimensional cultures greatly enhances the stability and growth of the nascent myocardium. Cardiac fibroblasts when included in polymer scaffolds seeded with myocytes and endothelial cells have the ability to promote and stabilize vascular structures. Naito and colleagues constructed three dimensional cultures of neonatal rat cell isolates on collagen type I and Matrigel (a basement membrane protein mixture), and isolates of a mixed cell population versus a myocyte-enriched population were compared. The mixed population cultures, which contained a higher fraction of cardiac fibroblasts than the myocyte-enriched cultures, displayed improved contractile force generation and greater inotropic response despite an equivalent overall cell number. Greater vascularity was also seen in the mixed-pool cultures.(160) Building on this, Nichol and colleagues demonstrated that in a self-assembling nanopeptide scaffold, embedded rat neonatal cardiomyocytes exhibit greater cellular alignment and reduced apoptosis when cardiac fibroblasts were included in the initial culture. A similar result was noted when polymer scaffolds were pre-treated with cardiac fibroblasts before myocyte seeding, suggesting a persistent paracrine effect. These data reinforce the concept that engineering functional myocardium, either in situ or ex vivo will require attention to the nature of cell-cell interactions, including fibroblasts.
To date, a broad initial sketch of cardiac fibroblast-myocyte interactions has been drawn. Future studies in this field will better describe these interactions. How do multiple paracrine factors interact to produce a cohesive and coordinated communication scheme? What are the changes in coordinated bidirectional signaling that during development promotes myocyte progenitor proliferation but have different roles in the adult? Might fibroblasts actually be required for improved cardiac repair and regeneration?
Recent studies have begun to apply genetic and cellular fate-mapping techniques to document the origins of cardiac fibroblasts, the dynamic nature of their population, and how that population may be in flux during time of injury or pressure overload. It is crucial to define on a more specific molecular basis the origins and fates of cardiac fibroblasts. Do fibroblasts that have been resident within the ventricle since development fundamentally differ from those that arise from endothelial transition or that infiltrate from the bone marrow during adulthood? Do fibroblasts with these different origins behave differently or take on different roles in the face of ventricular strain or injury?
Our understanding of the nature of the cardiac fibroblast is evolving from the concept of the fibroblast as a bystander that causes unwanted fibrosis to the picture of a more complex role of fibroblasts in the healthy as well as diseased heart. The pathways used by cardiac fibroblasts to communicate with their neighboring myocytes are only partially described, but the data to date indicate that these pathways will be important for cardiac repair and regeneration.

Figure 2. Paracrine bidirectional cardiac fibroblast-myocyte crosstalk
Under biomechanical overload, cardiac fibroblasts and myocytes respond to an altered environment via multiple mechanisms including integrin-extracellular matrix interactions and renin-angiotensin-aldosterone axis activation. Cardiac fibroblasts increase synthesis of matrix proteins and secrete a variety of paracrine factors that can stimulate myocyte hypertrophy. Cardiac myocytes similarly respond by secreting a conglomerate of factors. Hormones such as TGFβ1, FGF-2, and the IL-6 family members LIF and CT-1 have all been implicated in this bidirectional fibroblast-myocyte hormonal crosstalk.
Read Full Post »



























