Mitochondrial Damage and Repair under Oxidative Stress
Curator: Larry H Bernstein, MD, FCAP
Keywords: Mitochondria, mitochondrial dysfunction, electron transport chain, mtDNA, oxidative stress, oxidation-reduction, NO, DNA repair, lipid peroxidation, thiols, ROS, RNS, sulfur,base excision repair, ferredoxin.
Summary: The mitochondrion is the energy source for aerobic activity of the cell, but it also has regulatory functions that will be discussed. The mitochondrion has been discussed in other posts at this site. It has origins from organisms that emerged from an anaerobic environment, such as the bogs and marshes, and may be related to the chloroplast. The aerobic cell was an advance in evolutionary development, but despite the energetic advantage of using oxygen, the associated toxicity of oxygen abundance required adaptive changes. Most bacteria that reduce nitrate (producing nitrite, nitrous oxide or nitrogen) are called facultative anaerobes use electron acceptors such as ferric ions, sulfate or carbon dioxide which become reduced to ferrous ions, hydrogen sulfide and methane, respectively, during the oxidation of NADH (reduced nicotinamide adenine dinucleotide is a major electron carrier in the oxidation of fuel molecules).
The underlying problem we are left with is oxidation-reduction reactions that are necessary for catabolic and synthetic reactions, and that cumulatively damage the organism associated with cancer, cardiovascular disease, neurodegerative disease, and inflammatory overload. Aerobic organisms tolerate have evolved mechanisms to repair or remove damaged molecules or to prevent or deactivate the formationof toxic species that lead to oxidative stress and disease. However, the normal balance between production of pro-oxidant species and destruction by the antioxidant defenses is upset in favor of overproduction of the toxic species, which leads to oxidative stress and disease. How this all comes together is the topic of choice.

The transformation of energy is central to mitochondrial function. The system of energetics includes:
- the enzymes of the Kreb’s citric acid or TCA cycle,
- some of the enzymes involved in fatty acid catabolism (β-oxidation), and
- the proteins needed to help regulate these systems,
central to mitochondrial physiology through the production of reducing equivalents. Reducing equivalents are also used for anabolic reactions.
Electron Transport Chain
It also houses the protein complexes involved in the electron transport component of oxidative phosphorylation and proteins involved in substrate and ion transport. The chemical energy contained in both fats and amino acids can also be converted into NADH and FADH2 through mitochondrial pathways. The major mechanism for harvesting energy from fats is β-oxidation; the major mechanism for harvesting energy from amino acids and pyruvate is the TCA cycle. Once the chemical energy has been transformed into NADH and FADH2, these compounds are fed into the mitochondrial respiratory chain.
Under physiological conditions, electrons generally enter either through complex I (NADH-mediated, examined in vitro using substrates such as glutamate/malate) or complex II (FADH2-mediated, examined in vitro using succinate).
Electrons are then sequentially passed through a series of electron carriers.
The progressive transfer of electrons (and resultant proton pumping) converts the chemical energy stored in carbohydrates, lipids, and amino acids into potential energy in the form of the proton gradient. The potential energy stored in this gradient is used to phosphorylate ADP forming ATP.
Redox-Cycling
In redox cycling the reductant is continuously regenerated, thereby providing substrate for the “auto-oxidation” reaction.
When partially oxidized compounds are enzymatically reduced, the auto-oxidative generation of superoxide and other ROS to start again. Several enzymes
- NADPH-cytochrome P450 reductase,
- NADPH-cytochrome b5 reductase [EC 1.6.2.2]
- NADPH-ubiquinone oxidoreductase [EC 1.6.5.3], and
- xanthine oxidase [EC 1.2.3.2]),
can reduce quinones into semiquinones in a single electron process.
The semiquinone can then reduce dioxygen to superoxide during its oxidation to a quinone.
Redox cycling is thought to play a role in carcinogenesis. The naturally occurring estrogen metabolites (the catecholestrogens) have been implicated in hormone-induced cancer, possibly as a result of their redox cycling and production of ROS. It is thought that diethylstilbestrol causes the production of the mutagenic lesion 8-hydroxy-2’deoxyguanosine. It can also cause DNA strand breakage.
Another oxidative reaction that is associated with H2O2 is a significant problem for living organisms as a consequence of the reaction between hydrogen peroxide and oxidizable metals, the Fenton reaction [originally described in the oxidation of an α-hydroxy acid to an α-keto acid in the presence of hydrogen peroxide (or hypochlorite) and low levels of iron salts (Fenton (1876, 1894)).
Chemical Reactions and Biological Significance
The hydroxyl free radical is so aggressive that it will react within 5 (or so) molecular diameters from its site of production. The damage caused by it, therefore, is very site specific. Biological defenses have evolved that reduce the chance that the hydroxyl free radical will be produced to repair damage. An antioxidant would have to occur at the site of hydroxyl free radical production and be at sufficient concentration to be effective.
Some endogenous markers have been proposed as a useful measures of total “oxidative stress” e.g., 8-hydroxy-2’deoxyguanosine in urine. The ideal scavenger
- must be non-toxic,
- have limited or no biological activity,
- readily reach the site of hydroxyl free radical production,
- react rapidly with the free radical, be specific for this radical, and
- neither the scavenger nor its product(s) should undergo further metabolism.
Unlike oxygen, nitrogen does not possess unpaired electrons and is therefore considered diamagnetic. Nitrogen does not possess available d orbitals so it is limited to a valency of 3. In the presence of oxygen, nitrogen can produce Nitric oxide which occurs physiologically with the immune system which, when activated, can produce large quantities of nitric oxide.
Nitric oxide is produced by stepwise oxidation of L-arginine catalyzed by nitric oxide synthase (NOS). Nitric oxide is formed from the guanidino nitrogen of the L-arginine in a reaction that
- consumes five electrons and
- requires flavin adenine dinucleotide (FAD),
- flavin mononucleotide (FMN) tetrahydrobiopterin (BH4), and
- iron protoporphyrin IX as cofactors.
The primary product of NOS activity may be the nitroxyl anion that is then converted to nitric oxide by electron acceptors.
NOS cDNAs show homology with the cytochrome P450 reductase family. Based on molecular genetics there appears to be at least three distinct forms of NOS:
- A Ca2+/calmodulin-requiring constitutive enzyme (c-NOS; ncNOS or type I)
- A calcium-independent inducible enzyme (i-NOS; type II), which is primarily involved in the mediation of the cellular immune response; and
- A second Ca2+/calmodulin-requiring constitutive enzyme found in aortic and umbilical endothelia (ec-NOS or type III)
This has been discussed extensively in this series of posts. Recently, a mitochondrial form of the enzyme, which appears to be similar to the endothelial form, has been found in brain and liver tissue. Although the exact role of nitric oxide in the mitochondrion remains elusive, it may play a role in the regulation of cytochrome oxidase.
Nitric Oxide
Nitric oxide appears to regulate its own production through a negative feedback loop. The binding of nitric oxide to the heme prosthetic group of NOS inhibits this enzyme, and c-NOS and ec-NOS are much more sensitive to this regulation than i-NOS. It appears that in the brain, NO can regulate its own synthesis and therefore the neurotransmission process.
- On the one hand, inhibition of ec-NOS will prevent the cytotoxicity associated with excessive nitric oxide production.
- On the other, the insensitivity of i-NOS to nitric oxide will enable high levels of nitric oxide to be produced for cytotoxic effects.
Endogenous inhibitors of NOS (guanidino-substituted derivatives of arginine) occur in vivo as a result of post-translational modification of protein contained arginine residues by S-adenosylmethionine. The dimethylarginines (NG,NG-dimethyl-L-arginine and NG,N’G-dimethyl-L-arginine) occurs in tissue proteins, plasma, and urine of humans and they are thought to act as both regulators of NOS activity and reservoirs of arginine for the synthesis of nitric oxide.
It has been calculated that even though membrane makes up about 3% of the total tissue volume, 90% of the reaction of nitric oxide with oxygen occurs within this compartment. Thus the membrane is an important site for nitric oxide chemistry.
There are two major aspects to nitric oxide chemistry.
- It can undergo single electron oxidation and reduction reactions producing nitrosonium and nitroxyl
- Having a single unpaired electron in its π*2p molecular orbital it will react readily with other molecules that also have unpaired electrons, such as free radicals and transition metals.
Examples of the reaction of nitric oxide with radical species include:
- Nitric oxide will react with oxygen to form the peroxynitrite (nitrosyldioxyl) radical (ONO2)
- and with superoxide to form the powerful oxidizing and nitrating agent, peroxynitrite anion (ONO2-). Peroxynitrite causes damage to many important biomolecules
Importance:
- nitrosothiols that are important in the regulation of blood pressure terminates lipid peroxidation
- 3-nitrosotyrosine and/or 4-O-nitrosotyrosine can affect the activity of enzymes that utilize tyrosyl radicals
- rapidly reacts with oxyhemoglobin, the primary route of its destruction in vivo
- the reaction between nitric oxide and transition metal complexes
During the last reaction a “ligand” bond is formed (the unpaired electron of nitric oxide is partially transferred to the metal cation),
resulting in a nitrosated (nitrosylated) complex.
For example, such complexes can be formed with free iron ions,
iron bound to heme or iron located in iron-sulfur clusters.
Ligand formation allows nitric oxide to act as a signal, activating some enzymes while inhibiting others. Thus, the binding of nitric oxide to the Fe (II)-heme of guanylate (guanalyl) cyclase [GTP-pyrophosphate lyase: cyclizing] is the signal transduction mechanism. Guanylate cyclase exists as cytosolic and membrane-bound isozymes.
Thiol-Didulfide Redox Couple
The thiol-disulfide redox couple is very important to oxidative metabolism. For example, GSH is a reducing cofactor for glutathione peroxidase, an antioxidant enzyme responsible for the destruction of hydrogen peroxide.
The importance of the antioxidant role of the thiol-disulfide redox couple:
Thiols and disulfides can readily undergo exchange reactions, forming mixed disulfides. Thiol-disulfide exchange is biologically very important. For example,
- GSH can react with protein cystine groups and influence the correct folding of proteins.
- GSH may also play a direct role in cellular signaling through thiol-disulfide exchange reactions with membrane bound receptor proteins
- the insulin receptor complex)
- transcription factors (e.g., nuclear factor κB)
- and regulatory proteins in cells
Conditions that alter the redox status of the cell can have important consequences on cellular function.
The generation of ROS by redox cycling is only one possible explanation for the action of many drugs. Rifamycin not only owes its activity to ROS generation but also to its ability to block bacterial RNA synthesis as well. Quinones (and/or semiquinones) can also form adducts with nucleophiles, especially thiols. These adducts may act as toxins directly or indirectly through the inhibition of key enzymes (e.g., by reacting with essential cysteinyl residues) or the depletion of GSH.
DNA Adduct Formation
By far the most intense research in this field has been directed towards the chemistry and biology of DNA adduct formation. Attack of the free bases and nucleosides by pro-oxidants can yield a wide variety of adducts and DNA-protein cross-links. Such attack usually occurs
- at the C-4 and C-8 position of purines and
- C-5 and C-6 of pyrimidines.
Hydroxyl free radical-induced damage to purine bases and nucleosides can proceed through a C-8-hydroxy N-7 radical intermediate, and then either undergo oxidation with the production of an 8-hydroxy purine, or reduction, probably by cellular thiols, followed by ring opening and the formation of FAPy (formamido-pyrimidine) metabolites (hydroxyl free radical-induced damage to guanosine). Although most research has focused on 8-hydroxy-purine adducts a growing number of publications are attempting to measure the FAPy derivative.
Nitrosation of the Amines of the Nucleic Acid Bases.
Primary aromatic amines produce deaminated products, while secondary amines form N-nitroso compounds.
Formation of Peroxynitrite from Nitric Oxide.
Peroxynitrite shows complex reactivity
- with DNA initiating DNA strand breakage, oxidation (e.g., formation of 8-hydroxyguanine, 8-OH2’dG, (5-hydroxymethyl)-uracil, and FAPyGua),
- nitration (e.g., 8-nitroguanine), and
- deamination of bases.
Peroxynitrite can also promote the production of lipid peroxidation related active carbonyls and cause the activation of NAD+ ADP-ribosyltransferase.
Modification of Guanine
Although all DNA bases can be oxidatively damaged, it is the modification of guanine that is the most frequent. 8OH2’dG is the most abundant DNA adduct. This can affect its hydrogen bonding between base-pairs. These base-pair substitutions are usually found clustered into areas called “hot spots”. Guanine normally binds to cytosine.
8OH2’dG, however, can form hydrogen bonds with adenine. The formation of 8OH2’dG in DNA can therefore result in a G→T transversion.
8-Hydroxyguanine was also shown to induce codon 12 activation of c-Ha-ras and K-ras in mammalian systems. G→T transversions are also the most frequent hot spot mutations found in the p53 supressor gene which is associated with human tumors.
Other mechanisms by which ROS/RNS can lead to mutations have been
proposed. Direct mechanisms include:
- conformational changes in the DNA template that reduces the accuracy of replication by DNA polymerases
- altered methylation of cytosine that affects gene control
Indirect mechanisms include:
- Oxidative damage to proteins, including DNA polymerases and repair enzymes.
- Damage to lipids causes the production of mutagenic carbonyl compounds
- Misalignment mutagenesis (“slippery DNA”)

DNA Mismatch Repair 5 (Photo credit: Allen Gathman)
Repair of ROS/RNS-induced DNA Damage
The repair of damaged DNA is an ongoing and continuous process involving a
number of repair enzymes. Damaged DNA appears to be mended by two major mechanisms:
- base excision repair (BER) and
- nucleotide excision repair (NER)
Isolated DNA is found to contain low levels of damaged bases, so it appears that these repair processes are not completely effective.
Base Excision Repair
BER is first started by DNA glycosylases which recognize specific base
modifications (e.g., 8OH2’dG). For example,
- Formamido-pyrimidine-DNA glycosylase (Fpg protein) recognizes damaged purines such as 8-oxoguanine and FAPyGua.
- Damaged pyrimidines are recognized by endonuclease III, which acts as both a glycosylase and AP endonuclease.
- Glycosylases cleave the N-glycosylic bond between the damaged base and the sugar
Following the glycosylase step, AP endonucleases then remove the 3′-deoxyribose moiety by cleavage of the phosphodiester bonds thereby generating a 3’-hydroxyl group that can then be extended by DNA polymerase.
The final step in mending damaged DNA is the rejoining of the free ends of DNA by a DNA ligase. It also appears that the presence of 8-oxoguanine modified bases in DNA is not only a result of ROS attack on this macromolecule. Oxidized nucleosides and nucleotides from free cellular pools can also be incorporated into DNA by polymerases and cause AT to CG base substitution mutations.
Mitochondrial DNA Repair
The mitochondrion genome encodes the various complexes of the electron transport chain, but contains no genetic information for DNA repair enzymes. These enzymes must be obtained from the nucleus. As mitochondria are continuously producing DNA damaging pro-oxidant species, effective DNA repair mechanisms must exist within the mitochondrial matrix in order for these organelles to function. Mitochondria have a short existence, and excessively damaged mitochondria will be quickly removed. Mitochondria contain many BER enzymes and are proficient at repair, but they do not appear to repair damaged DNA by NER mechanisms.
Single Strand DNA Damage and PARP Activation
Single strand DNA breakage activates NAD+ ADP-ribosyltransferase (PARP). PARP is a protein-modifying, nucleotide-polymerizing enzyme and is found at high levels in the nucleus. Activated PARP
- cleaves NAD+ into ADP-ribose and nicotinamide
- then attaches the ADP-ribose units to a variety of nuclear proteins (including histones and its own automodification domain).
- then polymerizes the initial ADP-ribose modification with other ADP-ribose units to form the nucleic acid-like polymer, poly (ADP) ribose.
PARP only appears to be involved with BER and not NER. In BER PARP does not appear to play a direct role but rather it probably helps by keeping the chromatin in a conformation that enables other repair enzymes to be effective. It may also provide temporary protection to DNA molecules while it is being repaired. Conflicting evidence suggests that PARP may not be an important DNA repair enzyme as cells from a PARP knockout mouse model have normal repair characteristics.
Activation of PARP can be dangerous to the cell. For each mole of ADP-ribose transferred, one mole of NAD+ is consumed, and through the regeneration of NAD+ four ATP molecules are wasted. Thus the activation of PARP can rapidly deplete a cell’s energy store and even lead to cell death. Some researchers suggest that this may be one mechanism whereby cells with excessive DNA damage are effectively removed. However, a variety of diseases may involve PARP overactivation including
- circulatory shock,
- CNS injury,
- diabetes,
- drug-induced cytotoxicity, and
- inflammation.
The Indirect Pathway.
This (mutation) pathway does not involve oxidative damage to the protein per se. This process involves oxidative damage to the DNA molecule encoding the protein. Thus pro-oxidants can cause changes in the base sequence of the DNA molecule. If such base modification is in a coding region of DNA (exon) and not corrected, the DNA molecule may be transcribed incorrectly. Translation of the mutant mRNA can result in a mutant protein containing a wrong amino acid in its primary sequence. If this modified amino acid occurs in an essential part of the protein (e.g., the active site of an enzyme or a portion that alters folding), the function of that protein may be impaired. Fortunately, unlike modified DNA
that can pass from cell to cell during mitosis thereby continuing the production of mutant protein, damage to a protein is non-replicating and stops with its destruction.
The Direct Pathway
This (post-translational) pathway involves the action of a pro-oxidant on a protein resulting in
- modification of amino acid residues,
- the formation of carbonyl adducts,
- cross-linking and
- polypeptide chain fragmentation.
Such changes often result in altered protein conformation and/or activity. Proteins will produce a variety of carbonyl products when exposed to metal-based systems (metal/ascorbate and metal/hydrogen peroxide) in vitro. For example, histidine yields aspartate, asparagine and 2-oxoimidazoline, while proline produces glutamate, pyroglutamate, 4-hydroxyproline isomers, 2-pyrrolidone and γ-aminobutyric acid. Metal-based systems and other pro-oxidant conditions can oxidize methionine to its sulfoxide.
This portion of the presentation is endebted to THE HANDBOOK OF REDOX
BIOCHEMISTRY, Ian N. Acworth, August 2003, esa. (inacworth@esainc.com).
We shall now identify more recent work related to this presentation.
Oxygen and Oxidative Stress
The reduction of oxygen to water proceeds via one electron at a time. In the mitochondrial respiratory chain, Complex IV (cytochrome oxidase) retains all partially reduced intermediates until full reduction is achieved. Other redox centres in the electron transport chain, however, may leak electrons to oxygen, partially reducing this molecule to superoxide anion (O2_•). Even though O2_• is not a strong oxidant, it is a precursor of most other reactive oxygen species, and it also becomes involved in the propagation of oxidative chain reactions. Despite the presence of various antioxidant defences, the mitochondrion appears to be the main intracellular source of these oxidants. This review describes the main mitochondrial sources of reactive species and the antioxidant defences that evolved to prevent oxidative damage in all the mitochondrial compartments.
Reactive oxygen species (ROS) is a phrase used to describe a variety of molecules and free radicals (chemical species with one unpaired electron) derived from molecular oxygen. Molecular oxygen in the ground state is a bi-radical, containing two unpaired electrons in the outer shell (also known as a triplet state).
Since the two single electrons have the same spin, oxygen can only react with one electron at a time and therefore it is not very reactive with the two electrons in a chemical bond.
On the other hand, if one of the two unpaired electrons is excited and changes its spin, the resulting species (known as singlet oxygen) becomes a powerful oxidant as the two electrons with opposing spins can quickly react with other pairs of electrons, especially double bonds.
The formation of OH• is catalysed by reduced transition metals, which in turn may be re-reduced by O2 -•, propagating this process. In addition, O2-• may react with other radicals including nitric oxide (NO•) in a reaction controlled by the rate of diffusion of both radicals. The product, peroxynitrite, is also a very powerful oxidant. The oxidants derived from NO• have been recently called reactive nitrogen species (RNS).
‘Oxidative stress’ is an expression used to describe various deleterious processes resulting from an imbalance between the excessive formation of ROS and/or RNS and limited antioxidant defences.
- Whilst small fluctuations in the steady-state concentration of these oxidants may actually play a role in intracellular signalling,
- uncontrolled increases in the steady-state concentrations of these oxidants lead to free radical mediated chain reactions
which indiscriminately target
- proteins,
- lipids,
- polysaccharides.
In vivo, O2-• is produced both enzymatically and nonenzymatically.
Enzymatic sources include
- NADPH oxidases located on the cell membrane of
- polymorphonuclear cells,
- macrophages and
- endothelial cells and
- cytochrome P450-dependent oxygenases.
The proteolytic conversion of xanthine dehydrogenase to xanthine oxidase provides another enzymatic source of both O2 -• and H2O2 (and therefore constitutes a source of OH•) and has been proposed to mediate deleterious processes in vivo.
Given the highly reducing intramitochondrial environment, various respiratory components, including flavoproteins, iron–sulfur clusters and ubisemiquinone, are thermodynamically capable of transferring one electron to oxygen. Moreover, most steps in the respiratory chain involve single-electron reactions, further favouring the monovalent reduction of oxygen. On the other hand, the mitochondrion possesses various antioxidant defences designed to eliminate both O2- • and H2O2.
The rate of O2 -• formation by the respiratory chain is controlled primarily by mass action, increasing both when electron flow slows down (increasing the concentration of electron donors, R•) and when the concentration of oxygen increases (eqn (1); Turrens et al. 1982).
d[O2]/dt = k [O2] [R•].
The energy released as electrons flow through the respiratory chain is converted into a H+ gradient through the inner mitochondrial membrane (Mitchell, 1977). This gradient, in turn, dissipates through the ATP synthase complex (Complex V) and is responsible for the turning of a rotor-like protein complex required for ATP synthesis. In the absence of ADP,
- the movement of H+ through ATP synthase ceases and
- the H+ gradient builds up
- causing electron flow to slow down and
- the respiratory chain to become more reduced (State IV respiration).
Mitochondrial Antioxidant Defences
The deleterious effects resulting from the formation of ROS in the mitochondrion are, to a large extent, prevented by various antioxidant systems. Superoxide is enzymatically converted to H2O2 by a family of metalloenzymes called superoxide dismutases (SOD). Since O2-• may either reduce transition metals, which in turn can react with H2O2 producing OH• or spontaneously react with NO• to produce peroxynitrite, it is important to maintain the steady-state concentration of O2-• at the lowest possible level. Thus, although the dismutation of O2-• to H2O2 and O2 can also occur spontaneously, the role of SODs is to increase the rate of the reaction to that of a diffusion-controlled process.
The mitochondrial matrix contains a specific form of SOD, with manganese in the active site, which eliminates the O2 -• formed in the matrix or on the inner side of the inner membrane. The expression of MnSOD is further induced by agents that cause oxidative stress, including radiation and hyperoxia, in a process mediated by the oxidative activation of the nuclear transcription factor NFkB .
Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552(2): 335–344. DOI: 10.1113/jphysiol.2003.049478. http://www.jphysiol.org

Reactive Oxygen Species and Control of Apoptosis
Reactive oxygen species (ROS) are products of normal metabolism and xenobiotic exposure, and depending on their concentration, ROS can be beneficial or harmful to cells and tissues.
- At physiological low levels, ROS function as “redox messengers” in intracellular signaling and regulation, whereas
- excess ROS induce oxidative modification of cellular macromolecules, inhibit protein function, and promote cell death.
Additionally, various redox systems, such as
- the glutathione,
- thioredoxin, and
- pyridine nucleotide redox couples,
- NADPH and antioxidant defense
- NAD+ and the function of sirtuin proteins
participate in cell signaling and modulation of cell function, including apoptotic cell death. Cell apoptosis is initiated by extracellular and intracellular signals via two main pathways,
- the death receptor and
- the mitochondria-mediated pathways.
ROS and JNK-mediated apoptotic signaling
GSH redox status and apoptotic signaling
Various pathologies can result from oxidative stress-induced apoptotic signaling that is consequent to
- ROS increases and/or antioxidant decreases,
- disruption of intracellular redox homeostasis, and
- irreversible oxidative modifications of lipid, protein, or DNA.
We focus on several key aspects of ROS and redox mechanisms in apoptotic signaling and highlight the gaps in knowledge and potential avenues for further investigation. A full understanding of the redox control of apoptotic initiation and execution could underpin the development of therapeutic interventions targeted at oxidative stress-associated disorders.
Circu, M. L.; Aw, T. Y., Reactive oxygen species, cellular redox systems, and apoptosis, Free Radic. Biol. Med. 2010. FRB-10057; pp 14. doi:10.1016/j.freeradbiomed.2009.12.022
Assembly of Iron-sulfur (FeyS) Clusters
Iron-sulfur (FeyS) cluster-containing proteins catalyze a number of electron transfer and metabolic reactions. The components and molecular mechanisms involved in the assembly of the FeyS clusters have been identified only partially. In eukaryotes, mitochondria have been proposed to execute a crucial task in the generation of intramitochondrial and extramitochondrial FeyS proteins. Herein, we identify the essential ferredoxin Yah1p of Saccharomyces cerevisiae mitochondria as a central component of the FeyS protein biosynthesis machinery. Depletion of Yah1p by regulated gene expression resulted in a
30-fold accumulation of iron within mitochondria,
similar to what has been reported for other components involved in FeyS protein biogenesis. Yah1p was shown to be required for the assembly of FeyS proteins both inside mitochondria and in the cytosol. Apparently, at least one of the steps of FeyS cluster biogenesis within mitochondria requires reduction by ferredoxin. Our findings lend support to the idea of a primary function of mitochondria in the biosynthesis of FeyS proteins outside the organelle. To our knowledge, Yah1p is the first member of the ferredoxin family for which a function in FeyS cluster formation has been established. A similar role may be predicted for the bacterial homologs that are encoded within iron-sulfur cluster assembly (isc) operons of prokaryotes.
H Lange, A Kaut, G Kispal, and R Lill. A mitochondrial ferredoxin is essential for biogenesis of cellular iron-sulfur proteins. PNAS 2000; 97(3): 1050–1055.
DNA Charge Transport
Damaged bases in DNA are known to lead to errors in replication and transcription, compromising the integrity of the genome. The authors proposed a model where repair proteins containing redoxactive [4Fe-4S] clusters utilize DNA charge transport (CT) as a first step in finding lesions. In this model, the population of sites to search is reduced by a localization of protein in the vicinity of lesions. Here, we examine this model using single-molecule atomic force microscopy (AFM). XPD, a 5′-3′ helicase involved in nucleotide
excision repair, contains a [4Fe-4S] cluster and exhibits a DNA bound redox potential that is physiologically relevant.
In AFM studies, they observe the redistribution of XPD onto kilobase DNA strands containing a single base mismatch, which is not a specific substrate for XPD but, like a lesion, inhibits CT. They also provide evidence for DNA-mediated signaling between XPD and Endonuclease III (EndoIII), a base excision repair glycosylase that also contains a [4Fe-4S] cluster.
- When XPD and EndoIII are mixed together, they coordinate in relocalizing onto the mismatched strand.
- However, when a CT-deficient mutant of either repair protein is combined with the CT-proficient repair partner, no relocalization occurs.
The data presented here indicate that XPD, an archaeal protein from the NER pathway, may cooperate with other proteins that are proficient at DNA CT to localize in the vicinity of damage. XPD, a superfamily 2 DNA helicase with 5′-3′ polarity, is a component of TFIIH that is essential for repair of bulky lesions generated by exogenous sources such as UV light and chemical carcinogens. XPD contains a conserved [4Fe-4S] cluster suggested to be conformationally controlled by ATP binding and hydrolysis.
Mutations in the iron-sulfur domain of XPD can lead to diseases including TTD and XP, yet the function of the [4Fe-4S] cluster appears to be unknown.
Electrochemical studies have shown that when BER proteins MutY and EndoIII bind to DNA, their [4Fe-4S] clusters are activated toward one electron oxidation. XPD exhibits a DNA-bound midpoint potential similar to that of EndoIII and MutY when bound to DNA (approximately 80 mV vs. NHE), indicative of a possible role for the [4Fe-4S] cluster in DNA-mediated CT.
For EndoIII we have also already determined a direct correlation between the ability of proteins to redistribute in the vicinity of mismatches as measured by AFM, and the CT proficiency of the proteins measured electrochemically. Thus, we may utilize single-molecule AFM as a tool to probe the redistribution of proteins in the vicinity of base lesions and in so doing, the proficiency of the protein to carry out DNA CT.
Here we show that, like the BER protein EndoIII, XPD, involved both in transcription and NER, redistributes in the vicinity of a lesion. Importantly, this ability to relocalize is associated with the ability of XPD to carry out DNA CT. The mutant L325V is defective in its ability to carry out DNA CTand this XPD mutant also does not redistribute effectively onto the mismatched strand.
These data not only indicate a general link between the ability of a repair protein to carry out DNA CT and its ability to redistribute onto DNA strands near lesions but also provide evidence for coordinated DNA CT between different repair proteins in their search for damage in the genome. These data also provide evidence that two different repair proteins, each containing a [4Fe-4S] cluster at similar DNA bound potential, can communicate with one another through DNA-mediated CT.
Sontz PA, Mui TP, Fuss JO, Tainer JA, and Barton JK. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. PNAS 2012; 109(6):1856–1861. doi:10.1073/pnas.1120063109/-/ DCSupplemental. http://www.pnas.org/lookup/suppl/
Janus Bifron
The signaling function of mitochondria is considered with a special emphasis on their role in the regulation of redox status of the cell, possibly determining a number of pathologies including cancer and aging. The review summarizes the transport role of mitochondria in energy supply to all cellular compartments (mitochondria as an electric cable in the cell), the role of mitochondria in plastic metabolism of the cell including synthesis of
- heme,
- steroids,
- iron-sulfur clusters, and
- reactive oxygen and nitrogen species.
Mitochondria also play an important role in the Ca2+-signaling and the regulation of apoptotic cell death. Knowledge of mechanisms responsible for apoptotic cell death is important for the strategy for prevention of unwanted degradation of postmitotic cells such as cardiomyocytes and neurons.
In accordance with P. Mitchell’s chemiosmotic concept, vectorial transmembrane transfer of electrons and protons is accompanied by generation of electrochemical difference of proton electrochemical potential on the inner mitochondrial membrane; its utilization by ATP synthase induces conformational rearrangements resulting in ATP synthesis from ADP and inorganic phosphate. Details of the mechanism responsible for ATP synthesis are given elsewhere.
Membrane potential (DY) generated across the inner mitochondrial membrane is the component of the transmembrane electrochemical potential of H+ ions (DμH+), which provides ATP synthesis together with the concentration component (DpH). Maintenance of constant membrane potential is a vitally important precondition for functioning of mitochondria and the cell. Under conditions of limited supply of the cell with oxygen (hypoxia) and inability to carry out aerobic ATP synthesis, mitochondria become ATP consumers (rather than generators) and ATP is hydrolyzed by mitochondrial ATPase, and this is accompanied by generation of membrane potential.
Redox homeostasis, i.e. the sum of redox components (including proteins, low molecular weight redox components such as NAD/NADH, flavins, coenzymes Q, oxidized and reduced substrates, etc.) is one of important preconditions for normal cell functioning.

Single-strand and double-strand DNA damage (Photo credit: Wikipedia)
Mitochondria generate such potent regulators of redox potential as
- superoxide anion,
- hydrogen peroxide,
- nitric oxide,
- peroxynitrite, etc.
They are actively involved in regulation of cell redox potential and consequently
- control proteolysis,
- activation of transcription,
- changes in mitochondrial DNA (mDNA),
- cell metabolism, and
- cell differentiation.
Zorov DB, Isaev NK, Plotnikov EY, Zorova LD, et al. The Mitochondrion as Janus Bifrons. Biochemistry (Moscow) 2007; 72(10): 1115-1126. ISSN 0006-2979.
DOI: 10.1134/S0006297907100094

Structure of the human mitochondrial genome. (Photo credit: Wikipedia)
Gene Expression Associated with Oxidoreduction and Mitochondria
The naked mole-rat (Heterocephalus glaber) is a long-lived, cancer resistant rodent and there is a great interest in identifying the adaptations responsible for these and other of its unique traits. We employed RNA sequencing to compare liver gene expression profiles between naked mole-rats and wild-derived mice. Our results indicate that genes associated with oxidoreduction and mitochondria were expressed at higher relative levels in naked mole-rats. The largest effect is nearly
300-fold higher expression of epithelial cell adhesion molecule (Epcam), a tumour-associated protein.
Also of interest are the
- protease inhibitor, alpha2-macroglobulin (A2m), and the
- mitochondrial complex II subunit Sdhc,
both ageing-related genes found strongly over-expressed in the naked mole-rat.
These results hint at possible candidates for specifying species differences in ageing and cancer, and in particular suggest complex alterations in mitochondrial and oxidation reduction pathways in the naked mole-rat. Our differential gene expression analysis obviated the need for a reference naked mole-rat genome by employing a combination of Illumina/Solexa and 454 platforms for transcriptome sequencing and assembling transcriptome contigs of the non-sequenced species. Overall, our work provides new research foci and methods for studying the naked mole-rat’s fascinating characteristics.
C Yu, Y Li, A Holmes, K Szafranski, CG Faulkes, et al. RNA Sequencing Reveals Differential Expression of Mitochondrial and Oxidation reduction Genes in the Long-Lived Naked Mole-Rat When Compared to Mice. PLoS ONE 2011; 6(11): 1-9. e26729. http://www.plosone.org
The complete set of viable deletion strains in Saccharomyces cerevisiae was screened for sensitivity of mutants to five oxidants to identify cell functions involved in resistance to oxidative stress. This screen identified a unique set of mainly constitutive functions providing the first line of defense against a particular oxidant; these functions are very dependent on the nature of the oxidant. Most of these functions are distinct from those involved in repair and recovery from damage, which are generally induced in response to stress, because there was little correlation between mutant sensitivity and
the reported transcriptional response to oxidants of the relevant gene. The screen identified 456 mutants sensitive to at least one of five different types of oxidant, and these were ranked in order of sensitivity. Many genes identified were not previously known to have a role in resistance to reactive oxygen species. These encode functions including
- protein sorting,
- ergosterol metabolism,
- autophagy, and
- vacuolar acidification.
two mutants were sensitive to all oxidants examined,
12 were sensitive to at least four,
Different oxidants had very different spectra of deletants that were sensitive. These findings highlight the specificity of cellular responses to different oxidants:
- No single oxidant is representative of general oxidative stress.
- Mitochondrial respiratory functions were overrepresented in mutants sensitive to H2O2, and
- vacuolar protein-sorting mutants were enriched in mutants sensitive to diamide.
Core functions required for a broad range of oxidative-stress resistance include
- transcription,
- protein trafficking, and
- vacuolar function.
GW Thorpe, CS Fong, N Alic, VJ Higgins, and IW Dawes. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: Oxidative-stress-response genes. PNAS 2004;101: 6564–6569. http://www.pnas.org cgi doi 10.1073 pnas.0305888101
Subcellular Thiol Redox State in Complex I Deficiency
Isolated complex I deficiency is the most common enzymatic defect of the oxidative phosphorylation (OXPHOS) system, causing a wide range of clinical phenotypes. Th authers reported before that the rates at which reactive oxygen species (ROS)-sensitive dyes are converted into their fluorescent oxidation products are markedly increased in cultured skin fibroblasts of patients with nuclear-inherited isolated complex I deficiency.
Using videoimaging microscopy we show here that these cells also display a marked increase in NAD(P)H autofluorescence. Linear regression analysis revealed a negative correlation with the residual complex I activity and a positive correlation with the oxidation rates of the ROS sensitive dyes (5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein and hydroethidine for a large cohort of 10 patient cell lines.
On the other hand, video-imaging microscopy of cells selectively expressing reduction-oxidation sensitive GFP1 in either the mitochondrial matrix or cytosol showed the absence of any detectable change in thiol redox state. In agreement with this result, neither the glutathione nor the glutathione disulfide content differed significantly between patient and healthy fibroblasts.
Finally, video-rate confocal microscopy of cells loaded with C11-BODIPY581/591 demonstrated that the extent of lipid peroxidation, which is regarded as a measure of oxidative damage, was not altered in patient fibroblasts. Our results indicate that fibroblasts of patients with isolated complex I deficiency maintain their thiol redox state despite marked increases in ROS production.
S Verkaart, WJH Koopman, J Cheek, SE van Emst-de Vries. Mitochondrial and cytosolic thiol redox state are not detectably altered in isolated human NADH:ubiquinone oxidoreductase deficiency. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease 2007; 1772(9): 1041. DOI : 10.1016/j.bbadis.2007.05.004
- Mitochodrial mtDNA and Cancer
- Mitochondrial research has recently been driven by the
identification of mitochondria-associated diseases and
the role of mitochondria in apoptosis.
Moreover, mitochondria have been implicated in the process of carcinogenesis because of their vital role in
- energy production,
- nuclear-cytoplasmic signal integration and
- control of metabolic pathways.
At some point during neoplastic transformation, there is an increase in reactive oxygen species (ROS), which damage the mitochondrial genome. This accelerates the somatic mutation rate of mitochondrial DNA.
Mitochondrial characteristics
There are several biological characteristics which cast mitochondria and, in particular, the mitochondrial genome, as a biological tool for early detection and monitoring of neoplasia and its potential progression. These vital characteristics are important in cancer research, as not all neoplasias become malignant. Mitochondria are archived in the cytoplasm of the ovum and as such do not recombine.
This genome has an accelerated mutation rate, by comparison with the nucleus, and accrues somatic mutations in tumour tissue. Moreover, mitochondrial DNA (mtDNA) has a high copy number in comparison with the nuclear archive of DNA. There are potentially thousands of mitochondrial genomes per cell, which enables detection of important biomarkers, even at low levels. In addition, mtDNA can be heteroplasmic, which means that disease-associated mutations occur in a subset of the genomes.
The presence of heteroplasmy is an indication of disease and is found in many human tumours. Identification of low levels of heteroplasmy may allow unprecedented early identification and monitoring of neoplastic progression to malignancy.
Coding for just 13 enzyme complex subunits, 22 transfer RNAs and two ribosomal RNAs, the mitochondrial genome is packaged in a compact 16,569 base pair (bp) circular molecule. These products participate in the critical electron transport process of ATP production. Collectively, mitochondria generate 80 per cent of the chemical fuel which fires cellular metabolism.
As a result, nuclear investment in the mitochondria is high — that is, several thousand nuclear genes control this organelle in order to accomplish the complex interactions required to maintain a network of pathways, which coordinate energy demand and supply.
It has been proposed that these mutations may serve as an early indication of potential cancer development and may represent a means for tracking tumour progression.
Does this provide a potential utility in that these mutations may be used for the identification and monitoring of neoplasia and malignant transformation where appropriate body fluids or non-invasive tissue access is available for mtDNA recovery? Specifically discussed are:
- prostate,
- breast,
- colorectal,
- skin and
- lung cancers
There are many important questions yet to be addressed: such as
- the relationship between mtDNA and the actual disease;
- are mutations causative or merely a reflection of nuclear instability?
- And, are these processes independent events?
Alterations in the non-coding D-loop suggest genome instability;
however, as studies focus more on the coding regions of the
mitochondrial genome,
Particularly in the case of nonsynonymous mutations in the genes
contributing products to the electron transport process, metabolic
implications are evident. Moreover, mutations in mitochondrial
transfer RNAs indicate the possibility of a global mitochondrial
translational shut down.
RL Parr, GD Dakubo, RE Thayer, K McKenney, MA Birch-Machin. Mitochondrial DNA as a potential tool for early cancer detection. HUMAN GENOMICS 2006; 2(4). 252–257.
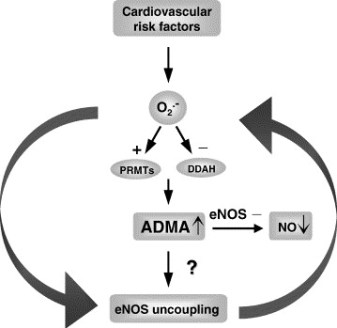
Mitochondrial DNA (mtDNA) is particularly prone to oxidation due to the lack of histones and a deficient mismatch repair system. This explains an increased mutation rate of mtDNA that results in heteroplasmy, e.g., the coexistence of the mutant and wild-type mtDNA molecules within the same mitochondrion. Hyperglycemia is a key risk factor not only for diabetes-related disease, but also for cardiovascular and all-cause mortality. One can assume an increase in the risk of cardiovascular disease by 18% for each unit (%) glycated hemoglobin HbA1c. In the Glucose Tolerance in Acute Myocardial Infarction study of patients with acute coronary syndrome, abnormal glucose tolerance was the strongest independent predictor of subsequent cardiovascular complications and death. In the Asian Pacific Study, fasting plasma glucose was shown to be an independent predictor of cardiovascular events up to a level of 5.2 mmol/L.
Glucose level fluctuations and hyperglycemia are triggers for inflammatory responses via increased mitochondrial superoxide production and endoplasmic reticulum stress. Inflammation leads to insulin resistance and β-cell dysfunction, which further aggravates hyperglycemia. The molecular pathways that integrate hyperglycemia, oxidative stress, and diabetic vascular complications have been most clearly described in the pathogenesis of endothelial dysfunction, which is considered as the first step in atherogenesis according to the response to injury hypothesis.
- In diabetes mellitus,
- glycotoxicity,
- advanced oxidative stress,
- collagen cross-linking, and
- accumulation of lipid peroxides
in foam macrophage cells and arterial wall cells may significantly
- decrease the mutation threshold,
- endothelial dysfunction,
- promoting atherosclerosis.
Alterations in mitochondrial DNA (mtDNA), known as homoplasmic and heteroplasmic mutations, may influence mitochondrial OXPHOS capacity, and in turn contribute to the magnitude of oxidative stress in micro- and macrovascular networks in diabetic patients.
The authors critically consider the impact of mtDNA mutations on the pathogenesis of cardiovascular diabetic complications.
Mutation Threshhold
Although cells may harbor mutant mtDNA, the expression of disease is dependent on the percent of alleles bearing mutations. Modeling confirms that an upper threshold level might exist for mutations beyond which the mitochondrial population collapses, with a subsequent decrease in ATP. This decrease in ATP results in the phenotypic expression of disease. It is estimated that in many patients with clinical manifestations of mitochondrial disorders, the proportion of mutant DNA exceeds 50%.
For the MELAS (mitochondrial encephalopathy, lactic acidosis and stroke-like syndrome)-causing mutation m.3243 A>G in the mitochondrial gene encoding tRNALeu, which is also associated with diabetes plus deafness, a strong correlation between the level of mutational heteroplasmy and documented disease has been found. Increased percentages of mutant mtDNA in muscle cells (up to 71%) can lead to mitochondrial myopathy. Levels of heteroplasmy of over 80% may lead to recurrent stroke and mutation levels of 95% have been associated with MELAS.
Regardless of the type of mutation or the level of heteroplasmy in affected mitochondria, unrepaired damage leads to a decrease in ATP, which in turn causes the phenotypic manifestation of disease. The manifestation of disease not only depends on the ATP level but also on the tissue affected. Various tissues have differing levels of demand on OXPHOS capacity. To evaluate a tissue threshold, Leber’s hereditary optic neuropathy can be used as a model for mitochondrial neurodegenerative disease. For neural and skeletal muscle tissues, the tissue threshold should be as high as or higher than 90% of
damaged (mutated) mtDNA. To induce mitochondrial malfunctions, the tissue threshold of the cardiac muscle is estimated to be significantly lower (approximately 64%-67%). In chronic vascular disease such as atherosclerosis, a mutation threshold in the affected vessel wall (e.g., in the postmortem aortic atherosclerotic plaques) was observed to be significantly lower. For example, for mutations m.3256 C>T, m.12315 G>A, m.15059 G>A, and m.15315 G>A, the heteroplasmy range of 18%-66% in the atherosclerotic lesions was 2-3.5-fold that in normal vascular tissue.
Mitochondrial stress and insulin resistance
- Mitochondrial damage precedes the development of atherosclerosis and tracks the extent of the lesion in apoE-null mice, and
- mitochondrial dysfunction caused by heterozygous deficiency of a superoxide dismutase increases atherosclerosis and vascular mitochondrial damage in the same model.
Blood vessels destined to develop atherosclerosis may be characterized by inefficient ATP production due to the uncoupling of respiration and OXPHOS. Blood vessels have regions of hypoxia, which lower the ratio of state 3 (phosphorylating) to state 4 (nonphosphorylating) respiration. Human atherosclerotic lesions have been known for decades to be deficient in essential fatty acids, a condition that causes respiratory uncoupling and atherosclerosis.
The finding by Kokaze et al. helps to explain, at least in part, the anti-atherogenic effect of the allele m. 5178A due to its relation with the favorable lipid profile. The nucleotide change causes leucine-to-methionine substitution at codon 237 (Leu-237Met) of the NADH dehydrogenase subunit 2 located in the loop between 7th and 8th transmembrane domains of the mitochondrial protein. Given that this methionine residue is exposed at the surface of respiratory Complex I, this residue may be available as an efficient oxidant scavenger. Complex I
- accepts electrons from NADH,
- transfers them to ubiquinone, and
- uses the energy released to pump protons across the mitochondrial inner membrane.
Thus, the Leu237Met replacement in the ND2 subunit might have a protective effect against oxidative damage to mitochondria.
Most fatty acid oxidation, which is promoted by peroxisome proliferator-activated receptor α (PPARα) activation, occurs in the mitochondria. Mitochondrial effects could explain why PPARα- deficient mice are protected from diet-induced insulin resistance and atherosclerosis as well as glucocorticoid induced insulin resistance and hypertension. Caloric restriction,
- improves features of insulin resistance,
- increases mitochondrial biogenesis and, surprisingly,
- enhances the efficiency of ATP production.
Dysfunctional mitochondria in cultured cells can be rescued by transfer of mitochondria from adult stem cells, raising the possibility of restoration of normal bioenergetics in the vasculature to treat atherosclerosis associated with insulin resistance.
Chistiakov DA, Sobenin IA, Bobryshev YV, Orekhov AN. Mitochondrial dysfunction and mitochondrial DNA mutations in atherosclerotic complications in diabetes. World J Cardiol 2012; 4(5): 148-156. ISSN 1949-8462 (online). doi:10.4330/wjc.v4.i5.148. http://www.wjgnet.com/1949-8462/full/v4/i5/148.htm
Read Full Post »

 impact on the vasculature")




 Bhowmick NA.
Bhowmick NA. 


")




")
")



