Curator: Ritu Saxena, Ph.D.
Melanoma
Melanoma represents approximately 4% of human skin cancers, yet accounts for approximately 80% of deaths from cutaneous neoplasms. It remains one of the most common types of cancer among young adults. Melanoma is recognized as the most common fatal skin cancer with its incidence rising to 15 fold in the past 40 years in the United States. Melanoma develops from the malignant transformation of melanocytes, the pigment-producing cells that reside in the basal epidermal layer in human skin. (Greenlee RT, et al, Cancer J Clin. Jan-Feb 2001;51(1):15-36 ; Weinstock MA, et al, Med Health R I. Jul 2001;84(7):234-6). Classic clinical signs of melanoma include change in color, recent enlargement, nodularity, irregular borders, and bleeding. Cardinal signs of melanoma are sometimes referred to by the mnemonic ABCDEs (asymmetry, border irregularity, color, diameter, elevation) (Chudnovsky Y, et al. J Clin Invest, 1 April 2005; 115(4): 813–824).
Clinical characteristics
Melanoma primarily affects fair-haired and fair-skinned individuals, and those who burn easily or have a history of severe sunburn are at higher risk than their darkly pigmented, age-matched controls. The exact mechanism and wavelengths of UV light that are the most critical remain controversial, but both UV-A (wavelength 320–400 nm) and UV-B (290–320 nm) have been implicated (Jhappan C, et al, Oncogene, 19 May 2003;22(20):3099-112). Case-control studies have identified several risk factors in populations susceptible to developing melanoma. MacKie RM et al (1989) stated that the relative risk of cutaneous melanoma is estimated from the four strongest risk factors identified by conditional logistic regression. These factors are
- total number of benign pigmented naevi above 2 mm diameter;
- freckling tendency;
- number of clinically atypical naevi (over 5 mm diameter and having an irregular edge, irregular pigmentation, or inflammation); and
- a history of severe sunburn at any time in life.
Use of this risk-factor chart should enable preventive advice for and surveillance of those at greatest risk (MacKie RM, et al, Lancet 26 Aug1989;2(8661):487-90).
Cutaneous melanoma can be subdivided into several subtypes, primarily based on anatomic location and patterns of growth (Table 1).

Table 1: Clinical Classification of Melanoma (Chudnovsky Y, et al, 2005)
The genetics of melanoma
As in many cancers, both genetic predisposition and exposure to environmental agents are risk factors for melanoma development. Many studies conducted over several decades on benign and malignant melanocytic lesions as well as melanoma cell lines have implicated numerous genes in melanoma development and progression.

Table 2: Genes involved in Melanoma (Chudnovsky Y, et al, 2005)
Apart from the risk factors such as skin pigmentation, freckling, and so on, another significant risk factor is ‘strong family history of melanoma’. Older case-control studies of patients with familial atypical mole-melanoma (FAMM) syndrome suggested an elevated risk of ∼434-to 1000-fold over the general population (Greene MH, et al, Ann Intern Med, Apr 1985;102(4):458-65). A more recent meta-analysis of family history found that the presence of at least one first-degree relative with melanoma increases the risk by 2.24-fold (Gandini S, et al, Eur J Cancer, Sep 2005;41(14):2040-59). Genetic studies of melanoma-prone families have given important clues regarding melanoma susceptibility loci.
CDKN2A, the familial melanoma locus
CDKN2A is located at chromosome 9p21 and is composed of 4 exons (E) – 1α, 1β, 2, and 3. LOH or mutations at this locus cosegregated with melanoma susceptibility in familial melanoma kindred and 9p21 mutations have been observed in different cancer cell lines. The locus encodes two tumor suppressors via alternate reading frames, INK4 (p16INK4a) and ARF (p14ARF). INK4A and ARF encode alternative first exons, 1α and 1β respectively and different promoters. INK4A is translated from the splice product of E1α, E2, and E3, while ARF is translated from the splice product of E1β, E2, and E3. Second exons of the two proteins are shared and two translated proteins share no amino acid homology.
INK4A is the founding member of the INK4 (Inhibitor of cyclin-dependent kinase 4) family of proteins and inhibits the G1 cyclin-dependent kinases (CDKs) 4/6, which phosphorylate and inactivate the retinoblastoma protein (RB), thereby allowing for S-phase entry. Thus, loss of INK4K function promotes RB inactivation through hyperphosphorylation, resulting in unconstrained cell cycle progression.
ARF (Alternative Reading Frame) protein of the locus inhibits HDM2-mediated ubiquitination and subsequent degradation of p53. Thus, loss of ARF inactivates another tumor suppressor, p53. The loss of p53 impairs mechanisms that normally target genetically damaged cells for cell cycle arrest and/or apoptosis, which leads to proliferation of damaged cells. Loss of CDKN2A therefore contributes to tumorigenesis by disruption of both the pRB and p53 pathways.

Figure 1: Genetic encoding and mechanism of action of INK4A and ARF.
(Chudnovsky Y, et al, 2005)
RAF and RAS pathways
A genetic hallmark of melanoma is the presence of activating mutations in the oncogenes BRAF and NRAS, which are present in 70% and 15% of melanomas, respectively, and lead to constitutive activation of mitogen-activated protein kinase (MAPK) pathway signaling. However, molecules that inhibit MAPK pathway–associated kinases, like BRAF and MEK, have shown only limited efficacy in the treatment of metastatic melanoma. Thus, a deeper understanding of the cross talk between signaling networks and the complexity of melanoma progression should lead to more effective therapy.
NRAS mutations activate both effector pathways, Raf-MEK-ERK and PI3K-Akt in melanoma. The Raf-MEK-ERK pathway may also be activated via mutations in the BRAF gene. In a subset of melanomas, the ERK kinases have been shown to be constitutively active even in the absence of NRAS or BRAF mutations. The PI3K-Akt pathway may be activated through loss or mutation of the tumor suppressor gene PTEN, occurring in 30–50% of melanomas, or through gene amplification of the AKT3 isoform. Activation of ERK and/or Akt3 promotes the development of melanoma by various mechanisms, including stimulation of cell proliferation and enhanced resistance to apoptosis.
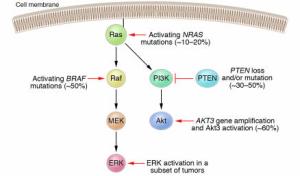
Figure 2: Schematic of the canonical Ras effector pathways Raf-MEK-ERK and PI3K-Akt in melanoma.
Curtin et al (2005) compared genome-wide alterations in the number of copies of DNA and mutational status of BRAF and NRAS in 126 melanomas from four groups in which the degree of exposure to ultraviolet light differs: 30 melanomas from skin with chronic sun-induced damage and 40 melanomas from skin without such damage; 36 melanomas from palms, soles, and subungual (acral) sites; and 20 mucosal melanomas. Significant differences were observed in number of copies of DNA and mutation frequencies in BRAF among the four groups of melanomas. Eighty-one percent of the melanomas on skin without sun-induced damaged had mutations in BRAF or NRAS. Melanomas with wild-type BRAF or NRAS frequently had increases in the number of copies of the genes for cyclin-dependent kinase 4 (CDK4) and cyclin D1 (CCND1), downstream components of the RAS-BRAF pathway. Thus, the genetic alterations identified in melanomas at different sites and with different levels of sun exposure indicate that there are distinct genetic pathways in the development of melanoma and implicate CDK4 and CCND1 as independent oncogenes in melanomas without mutations in BRAF or NRAS. (Curtin JA, et al, N Engl J Med, 17 Nov 2005;353(20):2135-47).
Genetic Heterogeneity of Melanoma
Melanoma exhibits molecular heterogeneity with markedly distinct biological and clinical behaviors. Lentigo maligna melanomas, for example, are indolent tumors that develop over decades on chronically sun-exposed area such as the face. Acral lentigenous melanoma, or the other hand, develops on sun-protected regions, tend to be more aggressive. Also, transcription profiling has provided distinct molecular subclasses of melanoma. It is also speculated that alterations at the DNA and RNA and the non-random nature of chromosomal aberrations may segregate melanoma tumors into subtypes with distinct clinical behaviors.
The melanoma gene atlas
Whole-genome screening technologies such as spectral karyotype analysis and array-CGH have identified many recurrent nonrandom chromosomal structural alterations, particularly in chromosomes 1, 6, 7, 9, 10, and 11 (Curtin JA, et al, N Engl J Med, 17 Nov 2005;353(20):2135-47); however, in most cases, no known or validated targets have been linked to these alterations.
In A systematic high-resolution genomic analysis of melanocytic genomes, array-CGH profiles of 120 melanocytic lesions, including 32 melanoma cell lines, 10 benign melanocytic nevi, and 78 melanomas (primary and metastatic) by Chin et al (2006) revealed a level of genomic complexity not previously appreciated. In total, 435 distinct copy number aberrations (CNAs) were defined among the metastatic lesions, including 163 recurrent, high-amplitude events. These include all previously described large and focal events (e.g., 1q gain, 6p gain/6q loss, 7 gain, 9p loss, and 10 loss). Genomic complexity observed in primary and benign nevi melanoma is significantly less than that observed in metastatic melanoma (Figure 3) (Chin L, et al, Genes Dev. 15 Aug 2006;20 (16):2149-2182).

Figure 3: Genome comparisons of melanocyte lesions (Chin L, et al, 2006)
Thus, genomic profiling of various melanoma progression types could reveal important information regarding genetic events those likely drive as metastasis and possibly, reveal provide cues regarding therapy targeted against melanoma.
Reference:
- Greenlee RT, et al, Cancer J Clin. Jan-Feb 2001;51(1):15-36
- Weinstock MA, et al, Med Health R I. Jul 2001;84(7):234-6
- Chudnovsky Y, et al. J Clin Invest, 1 April 2005; 115(4): 813–824
- Jhappan C, et al, Oncogene, 19 May 2003;22(20):3099-112
- MacKie RM, et al, Lancet 26 Aug1989;2(8661):487-90)
- Gandini S, et al, Eur J Cancer, Sep 2005;41(14):2040-59)
- Curtin JA, et al, N Engl J Med, 17 Nov 2005;353(20):2135-47
- Chin L, et al, Genes Dev. 15 Aug 2006;20 (16):2149-2182
Related articles on Melanoma on this Open Access Online Scientific Journal, include the following:
Thymosin alpha1 and melanoma Author/Editor- Tilda Barliya, Ph.D.
A New Therapy for Melanoma Reporter- Larry H Bernstein, M.D.
Melanoma: Molecule in Immune System Could Help Treat Dangerous Skin Cancer Reporter: Prabodh Kandala, Ph.D.
Why Braf inhibitors fail to treat melanoma. Reporter: Prabodh Kandala, Ph.D.
Read Full Post »


 Article Info
Article Info





{kind=link}