Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
This portion of the discussion is a series of articles on signaling and signaling pathways. Many of the protein-protein interactions or protein-membrane interactions and associated regulatory features have been referred to previously, but the focus of the discussion or points made were different. I considered placing this after the discussion of proteins and how they play out their essential role, but this is quite a suitable place for a progression to what follows. This is introduced by material taken from Wikipedia, which will be followed by a series of mechanisms and examples from the current literature, which give insight into the developments in cell metabolism, with the later goal of separating views introduced by molecular biology and genomics from functional cellular dynamics that are not dependent on the classic view. The work is vast, and this discussion does not attempt to cover it in great depth. It is the first in a series. This discussion, in particular is a tutorial on signaling transduction that was already available, and relevant. One may note that all of the slides used herein were also used in the previous blog, but in a different construction. I shall tweak the contents, as I find helpful.
Signaling and signaling pathways
Signaling transduction tutorial.
Carbohydrate metabolism
Lipid metabolism
Protein synthesis and degradation
Subcellular structure
Impairments in pathological states: endocrine disorders; stress hypermetabolism; cancer.
Signal Transduction Tutorial
The goal of this tutorial is for you to gain an understanding of how cell signaling occurs in a cell. Upon completion of the tutorial,
you will have a basic understanding signal transduction and
the role of phosphorylation in signal transduction.
You will also have detailed knowledge of
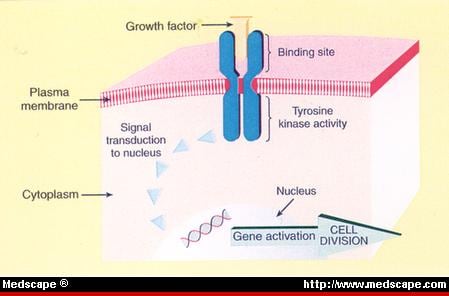
the role of Tyrosine kinases and
G protein-coupled receptors in cell signaling.
Description of Signal Transduction
As living organisms
we are constantly receiving and interpreting signals from our environment.
These signals can come
in the form of light, heat, odors, touch or sound.
The cells of our bodies are also
constantly receiving signals from other cells.
These signals are important to
keep cells alive and functioning as well as
to stimulate important events such as
cell division and differentiation.
Signals are most often chemicals that can be found
in the extracellular fluid around cells.
These chemicals can come
from distant locations in the body (endocrine signaling by hormones), from
nearby cells (paracrine signaling) or can even
be secreted by the same cell (autocrine signaling).
Signaling molecules may trigger any number of cellular responses, including
changing the metabolism of the cell receiving the signal or
result in a change in gene expression (transcription) within the nucleus of the cell or both.
Overview of Cell Signaling
Cell signaling can be divided into 3 stages.
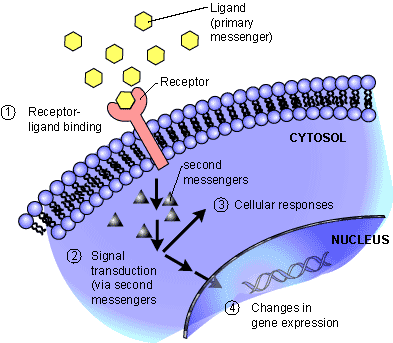
Reception: A cell detects a signaling molecule from the outside of the cell. A signal is detected when the chemical signal (also known as a ligand) binds to a receptor protein on the surface of the cell or inside the cell.
Transduction: When the signaling molecule binds the receptor it changes the receptor protein in some way. This change initiates the process of transduction. Signal transduction is usually a pathway of several steps. Each relay molecule in the signal transduction pathway changes the next molecule in the pathway.
Response: Finally, the signal triggers a specific cellular response.
Signal Transduction – ligand binds to surface receptor
Membrane receptors function by binding the signal molecule (ligand) and causing the production of a second signal (also known as a second messenger) that then causes a cellular response. These types of receptors transmit information from the extracellular environment to the inside of the cell
Intracellular receptors are found inside the cell, either in the cytopolasm or in the nucleus of the target cell (the cell receiving the signal).
Chemical messengers that are hydrophobic or very small (steroid hormones for example) can
pass through the plasma membrane without assistance and
bind these intracellular receptors.
Once bound and activated by the signal molecule,
the activated receptor can initiate a cellular response, such as a
change in gene expression.
Note that this is the first time that change in gene expression is stated. Is the change in gene expression implication of a change in the genetic information – such as – mutation? That does not have to be the case in the normal homeostatic case. This might only be
a change in the rate of a transcription or a suppression of expression through RNA.
responsive to small concentrations of chemical signals and act quickly,
cells often use a multi-step pathway that transmits the signal quickly,
while amplifying the signal to numerous molecules at each step.
Signal transduction cascades amplify the signal output
Steps in the signal transduction pathway often involve
the addition or removal of phosphate groups which results in the activation of proteins.
Enzymes that transfer phosphate groups from ATP to a protein are called protein kinases.
Many of the relay molecules in a signal transduction pathway are protein kinases and
often act on other protein kinases in the pathway. Often
this creates a phosphorylation cascade, where
one enzyme phosphorylates another, which then phosphorylates another protein, causing a chain reaction.
phosphorylation-cascade
Also important to the phosphorylation cascade are
a group of proteins known as protein phosphatases.
Protein phosphatases are enzymes that can rapidly remove phosphate groups from proteins (dephosphorylation) and thus inactivate protein kinases. Protein phosphatases are
the “off switch” in the signal transduction pathway.
Turning the signal transduction pathway off when the signal is no longer present is important
to ensure that the cellular response is regulated appropriately.
Dephosphorylation also makes protein kinases
available for reuse and
enables the cell to respond again when another signal is received.
Kinases are not the only tools used by cells in signal transduction. Small, nonprotein, water-soluble molecules or ions called second messengers (the ligand that binds the receptor is the first messenger) can also
relay signals received by receptors on the cell surface
to target molecules in the cytoplasm or the nucleus.
membrane protein receptor binds with hormone
insulin receptor and and insulin receptor signaling pathway (IRS)
Cell signaling ultimately leads to the regulation of one or more cellular activities. Regulation of gene expression (turning transcription of specific genes on or off) is a common outcome of cell signaling. A signaling pathway may also
In a three part series: Part IIA. CRACKING THE CODE OF HUMAN LIFE: Milestones along the Way Part IIB. CRACKING THE CODE OF HUMAN LIFE: The Birth of BioInformatics & Computational Genomics Part IIC. CRACKING THE CODE OF HUMAN LIFE: Recent Advances in Genomic Analysis and Disease
Part III will conclude with Ubiquitin, it’s Role in Signaling and Regulatory Control. Part I reviewed the huge expansion of the biological research enterprise after the Second World War. It concentrated on the
discovery of cellular structures,
metabolic function, and
creation of a new science of Molecular Biology.
Part II follows the race to delineation of the Human Genome, discovery methods and fundamental genomic patterns that are ancient in both animal and plant speciation. But it explores both the complexity and the systems view of the architecture that underlies and understanding of the genome.
These articles review a web-like connectivity between inter-connected scientific discoveries, as significant findings have led to novel hypotheses and many expectations over the last 75 years. This largely post WWII revolution has driven our understanding of biological and medical processes at an exponential pace owing to successive discoveries of
chemical structure,
the basic building blocks of DNA and proteins,
nucleotide and protein-protein interactions,
protein folding, allostericity,
genomic structure,
DNA replication,
nuclear polyribosome interaction, and
metabolic control.
In addition, the emergence of methods for
copying,
removal,
insertion,
improvements in structural analysis
developments in applied mathematics that have transformed the research framework.
Part IIA:
CRACKING THE CODE OF HUMAN LIFE:
Milestones along the Way
A NOVA interview with Francis Collins (NHGRI) (FC), J. Craig Venter (CELERA)(JCV), and Eric Lander (EL). RK: For the past ten years, scientists all over the world have been painstakingly trying to read the tiny instructions buried inside our DNA. And now, finally, the “Human Genome” has been decoded. EL: The genome is a storybook that’s been edited for a couple billion years. The following will address the odd similarity of genes between man and yeast
EL: In the nucleus of your cell the DNA molecule resides that is about 10 angstroms wide curled up, but the amount of curling is limited by the negative charges that repel one another, but there are folds upon folds. If the DNA is stretched the length of the DNA would be thousands of feet. EL: We have known for 2000 years that your kids look a lot like you. Well it’s because you must pass them instructions that give them the eyes, the hair color, and the nose shape they have. RK: Cracking the code of those minuscule differences in DNA that influence health and illness is what the Human Genome Project is all about. Since 1990, scientists all over the world have been involved in the effort to read all three billion As, Ts, Gs, and Cs of human DNA. It took 10 years to find the one genetic mistake that causes cystic fibrosis. Another 10 years to find the gene for Huntington’s disease. Fifteen years to find one of the genes that increase the risk for breast cancer. One letter at a time, painfully slowly… And then came the revolution. In the last ten years the entire process has been computerized. The computations can do a thousand every second and that has made all the difference. EL: This is basically a parts list with a lot of parts. If you take an airplane, a Boeing 777, I think it has like 100,000 parts. If I gave you a parts list for the Boeing 777 in one sense you’d know 100,000 components, screws and wires and rudders and things like that. But you wouldn’t know how to put it together, or why it flies. We now have a parts list, and that’s not enough to understand why it flies.
The Human Genome (Photo credit: dullhunk)
A Quest For Clarity
Tracy Vence is a senior editor of Genome Technology Tracy Vence @GenomeTechMag Projects supported by the US National Institutes of Health will have produced 68,000 total human genomes — around 18,000 of those whole human genomes — through the end of this year, National Human Genome Research Institute estimates indicate. And in his book, The Creative Destruction of Medicine, the Scripps Research Institute’s Eric Topol projects that 1 million human genomes will have been sequenced by 2013 and 5 million by 2014. Daniel MacArthur, a group leader in Massachusetts General Hospital’s Analytic and Translational Genetics Unit estimates that “From a capacity perspective … millions of genomes are not that far off. If you look at the rate that we’re scaling, we can certainly achieve that.” The prospect of so many genomes has brought clinical interpretation into focus. But there is an important distinction to be made between the interpretation of an apparently healthy person’s genome and that of an individual who is already affected by a disease. In an April Science Translational Medicine paper, Johns Hopkins University School of Medicine‘s Nicholas Roberts and his colleagues reported that personal genome sequences for healthy monozygotic twin pairs are not predictive of significant risk for 24 different diseases in those individuals. The researchers concluded that whole-genome sequencing was not likely to be clinically useful. Ambiguities have clouded even the most targeted interpretation efforts.
Technological challenges,
meager sample sizes,
a need for increased,
fail-safe automation and most important
a lack of community-wide standards for the task.
have hampered researchers’ attempts to reliably interpret the clinical significance of genomic variation.
How signals from the cell surface affect transcription of genes in the nucleus.
James Darnell, Jr., MD, Astor Professor, Rockefeller After graduation from Washington University School of Medicine he worked with Francois Jacob at the Pasteur Institute in Paris and served as Vice President for Academic Affairs at Rockefeller in 1990-91. He is the coauthor with S.E. Luria of General Virology and the founding author with Harvey Lodish and David Baltimore of Molecular Cell Biology, now in its sixth edition. His book RNA, Life’s Indispensable Molecule was published in July 2011 by Cold Spring Harbor Laboratory Press. A member of the National Academy of Sciences since 1973, recipient of numerous awards, including the 2003 National Medal of Science, the 2002 Albert Lasker Award. Using interferon as a model cytokine, the Darnell group discovered that cell transcription was quickly changed by binding of cytokines to the cell surface. The bound interferon led to the tyrosine phosphorylation of latent cytoplasmic proteins now called STATs (signal transducers and activators of transcription) that dimerize by
reciprocal phosphotyrosine-SH2 interchange.
accumulate in the nucleus,
bind DNA and drive transcription.
This pathway has proved to be of wide importance with seven STATs now known in mammals that take part in a wide variety of developmental and homeostatic events in all multicellular animals. Crystallographic analysis defined functional domains in the STATs, and current attention is focused on two areas:
how the STATs complete their cycle of activation and inactivation, which requires regulated tyrosine dephosphorylation; and how
persistent activation of STAT3 that occurs in a high proportion of many human cancers contributes to blocking apoptosis in cancer cells.
Current efforts are devoted to inhibiting STAT3 with modified peptides that can enter cells.
Cell cycle regulation and the cellular response to genotoxic stress
Stephen J Elledge, PhD, Gregor Mendel Professor of Genetics and Medicine, Investigator, Howard Hughes Medical Institute, Harvard Medical School As a postdoctoral fellow at Stanford working on eukaryotic homologous recombination, he serendipitously found a family of genes known as ribonucleotide reductases. He subsequently showed that
these genes are activated by DNA damage and
could serve as tools to help scientists dissect the signaling pathways
through which cells sense and respond to DNA damage and replication stress.
At Baylor College of Medicine he made a second major breakthrough with the discovery of the cyclin-dependent kinase 2 gene (Cdk2), which
controls the G1-to-S cell cycle transition,
an entry checkpoint for the cell proliferation cycle and
a critical regulatory step in tumorigenesis.
From there, using a novel “two-hybrid” cloning method he developed, Elledge and Wade Harper, PhD, proceeded to
isolate several members of the Cdk2-inhibitory family.
Their discoveries included the p21 and p57 genes, mutations in the latter (responsible for Beckwith-Wiedemann syndrome), characterized by somatic overgrowth and increased cancer risk. Elledge is also recognized for his work in understanding
proteome remodeling through ubiquitin-mediated proteolysis.
they identified F-box proteins that regulate protein degradation in the cell by
binding to specific target protein sequences and then
marking them with ubiquitin for destruction by the cell’s proteasome machinery.
This breakthrough resulted in
the elucidation of the cullin ubiquitin ligase family,
which controls regulated protein stability in eukaryotes.
Elledge’s recent research has focused on the cellular mechanisms underlying DNA damage detection and cancer using genetic technologies. In collaboration with Cold Spring Harbor Laboratory researcher Gregory Hannon, PhD, Elledge has generated complete human and mouse short hairpin RNA (shRNA) libraries for genome-wide loss-of-function studies. Their efforts have led to
the identification of a number of tumor suppressor proteins
genes upon which cancer cells uniquely depend for survival.
This work led to the development of the “non-oncogene addiction” concept. This is noted as follows:
proteome remodeling through ubiquitin-mediated proteolysis
F-box proteins regulate protein degradation in the cell by binding to specific target protein sequences
and then marking them with ubiquitin for destruction by the cell’s proteasome machinery
elucidation of the cullin ubiquitin ligase family, which controls regulated protein stability in eukaryotes
Playing the dual roles of inventor and investigator, Elledge developed original techniques to define
what drives the cell cycle and
how cells respond to DNA damage.
By using these tools, he and his colleagues have identified multiple genes involved in cell-cycle regulation.
Elledge’s work has earned him many awards, including a 2001 Paul Marks Prize for Cancer Research and a 2003 election to the National Academy of Sciences. In his Inaugural Article (1), published in this issue of PNAS, Elledge and his colleagues describe the function of Fbw7, a protein involved in controlling cell proliferation (see below). Elledge studied the error-prone DNA repair mechanism in E-Coli (Escherichia coli) called SOSmutagenesis for his PhD thesis at MIT. His work identified and described
the regulation of a group of enzymes now known as error-prone polymerases,
the first members of which were the umuCD genes in E. coli.
It was then that he developed a new cloning tool. Elledge invented a technique that allowed him to approach future cloning problems of this type with great rapidity. With the new technique, “you could make large libraries in lambda that behave like plasmids. We called them `phasmid’ vectors, like plasmid and phage together”. The phasmid cloning method was an early cornerstone for molecular biology research.
Elledge began working on homologous recombination in postdoctoral fellowship at Stanford University, an important niche in the field of eukaryotic genetics. Working with the yeast genome, Elledge searched for rec A, a gene that allows DNA to recombine homologously. Although he never located rec A, he discovered a family of genes known as ribonucleotide reductases (RNRs), which are involved in DNA production. Rec A and RNRs share the same last 4 amino acids, which caused an antibody crossreaction in one of Elledge’s experiments. Initially disappointed with the false positives in his hunt for rec A, Elledge was later delighted with his luck. He found that
RNRs are turned on by DNA damage, and
these genes are regulated by the cell cycle.
Prior to leaving Stanford, Elledge attended a talk at the University of California, San Francisco, by Paul Nurse, a leader in cell-cycle research who would later win the 2001 Nobel Prize in medicine. Nurse described his success in isolating the homolog of a key human cell-cycle kinase gene, Cdc2, by using a mutant strain of yeast (8). Although Nurse’s methods were primitive, Elledge was struck by the message he carried: that
cell-cycle regulation was functionally conserved, and
many human genes could be isolated by looking for complimentary genes in yeast.
Elledge then took advantage of his past successes in building phasmid vectors to build a versatile human cDNA library that could be expressed in yeast. After setting up a laboratory at Baylor, he introduced this library into yeast, screening for complimentary cell-cycle genes. He quickly identified the same Cdc2 gene isolated by Nurse. However, Elledge also discovered a related gene known as Cdk2. Elledge subsequently found that
Cdk2 controlled the G1 to S cell-cycle transition, a step that often goes awry in cancer. These results were published in the EMBO Journal in 1991.
He then continued to use
RNRs to perform genetic screens to
identify genes involved in sensing and responding to DNA damage.
He subsequently worked out the
signal transduction pathways in both yeast and humans that recognize damaged DNA and replication problems.
These “checkpoint” pathways are central to the
prevention of genomic instability and a key to understanding tumorigenesis.
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 29, 2003.
Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein.
The mammalian F-box protein Fbw7 and its Caenorhabditis elegans counterpart Sel-10 have been implicated in
the ubiquitin-mediated turnover of cyclin E
as well as the Notch Lin-12 family of transcriptional activators. Both unregulated
Notch and cyclin E
promote tumorigenesis, and
inactivate mutations in human
Fbw7 studies suggest that it may be a tumor suppressor. To generate an in vivo system to assess the consequences of such unregulated signaling, we generated mice deficient for Fbw7. Fbw7-null mice die around 10.5 days post coitus because of a combination of deficiencies in hematopoietic and vascular development and heart chamber mutations. The absence of Fbw7 results in elevated levels of cyclin E, concurrent with inappropriate DNA replication in placental giant trophoblast cells. Moreover, the levels of both Notch 1 and Notch 4 intracellular domains were elevated, leading to stimulation of downstream transcriptional pathways involving Hes1, Herp1, and Herp2. These data suggest essential functions for Fbw7 in controlling cyclin E and Notch signaling pathways in the mouse.
Science as an Adventure
Ubiquitins
Prof. Avram Hershko – Science as an Adventure Prof. Avram Hershko shared the 2004 Nobel Prize in Chemistry with Aaron Ciechanover and Irwin Rose for “for the discovery of ubiquitin-mediated protein degradation.”
Nipam Patel is a professor in the Departments of Molecular and Cell Biology and Integrative Biology at UC Berkeley and runs a research laboratory that studies the role, during embryonic development, of homeotic genes (the genetic switches described in this feature). “Ghost in Your Genes” focuses on epigenetic “switches” that turn genes “on” or “off.” But not all switches are epigenetic; some are genetic. That is, other genes within the chromosome turn genes on or off. In an animal’s embryonic stage, these gene switches play a predominant role in laying out the animal’s basic body plan and perform other early functions;
the epigenome begins to take over during the later stages of embryogenesis.
Beginning as a fertilized single egg that egg becomes many different kinds of cells. Altogether, multicellular organisms like humans have thousands of differentiated cells. Each is optimized for use in the brain, the liver, the skin, and so on. Remarkably, the DNA inside all these cells is exactly the same. What makes the cells differ from one another is that different genes in that DNA are either turned on or off in each type of cell.
Take a typical cell, such as a red blood cell. Each gene within that cell has a coding region that encodes the information used to make a particular protein. (Hemoglobin shuttles oxygen to the tissues and carbon dioxide back out to the lungs—or gills, if you’re a fish.) But another region of the gene, called “regulatory DNA,” determines whether and when the gene will be expressed, or turned on, in a particular kind of cell. This precise transcribing of genes is handled by proteins known as transcription factors, which bind to the regulatory DNA, thereby generating instructions for the coding region.
One important class of transcription factors is encoded by the so called homeotic, or Hox, genes. Found in all animals, Hox genes act to “regionalize” the body along the embryo’s anterior-to-posterior (head-to-tail) axis. In a fruit fly, for example, Hox genes lay out the various main body segments—the head, thorax, and abdomen. Amazingly, all animals, from fruit flies to mice to people, rely on the same basic Hox-gene complex. Using different-colored antibody stains, we can see exactly where and to what degree Hox genes are expressed. Each Hox gene is expressed in a specific region along the anterior-to-posterior axis of the embryo.
A fly’s body has three main divisions: head, thorax, and abdomen. We’ll focus on the thorax, which itself has three main segments. In a normal adult fly, the second thoracic segment features a pair of wings, while the third thoracic segment has a pair of small, balloon-shaped structures called halteres. A modified second wing, the haltere serves as a flight stabilizer. In order for the pair of wings and the pair of halteres (as well as all other parts of the fly) to develop properly, the fly’s suite of
Hox genes must be expressed in a precise way and at precise times.
During development, the fly’s two wings grow from a structure in the larva known as the wing imaginal disk. (An imago is an insect in its final, adult state.) The haltere grows from the larval haltere imaginal disk. Remember the Ubx Hox gene? Using staining again, we can detect the gene product of Ubx. This reveals that
the Ubx gene is naturally “off” in the wing disk—
and is “on” in the haltere disk.
Now you’ll see what happens when the Ubx gene—just one of a large number of Hox genes—is turned off in the haltere disk. What if a genetic mutation caused the Ubx gene to be turned off, during the larval stage, in the third thoracic segment, the segment that normally produces the haltere? Instead of a pair of halteres, the fly has a second set of wings. With the switch of that single Hox gene, Ubx, from on to off, the third thoracic segment becomes an additional second thoracic segment and the pair of halteres became a second pair of wings. This illustrates the remarkable ability of transcription factors like Ubx to control patterning as well as cell type during development.
ENCODE
A. Data Suggests “Gene” Redefinition
As part of a huge collaborative effort called ENCODE (Encyclopedia of DNA Elements), a research team led by Cold Spring Harbor Laboratory (CSHL) Professor Thomas Gingeras, PhD, publishes a genome-wide analysis of RNA messages, called transcripts, produced within human cells. Their analysis—one component of a massive release of research results by ENCODE teams from 32 institutes in 5 countries, with 30 papers appearing in 3 different high-level scientific journals—shows that three-quarters of the genome is capable of being transcribed. This indicates that nearly all of our genome is dynamic and active. It stands in marked contrast to consensus views prior to ENCODE’s comprehensive research efforts, which suggested that
only the small protein-encoding fraction of the genome was transcribed.
The vast amount of data generated with advanced technologies by Gingeras’ group and others in the ENCODE project changes the prevailing understanding of what defines a gene. The current outstanding question concerns
the nature and range of those functions. It is thought that these
“non-coding” RNA transcripts act something like components of a giant, complex switchboard, controlling a network of many events in the cell by
regulating the processes of
replication,
transcription
and translation
– that is, the copying of DNA and the making of proteins is based on information carried by messenger RNAs. With the understanding that so much of our DNA can be transcribed into RNA comes the realization that there is much less space between what we previously thought of as genes, Gingeras points out.
The full ENCODE Consortium data sets can be freely accessed through
the ENCODE project portal as well as at the University of California at Santa Cruz genome browser,
the National Center for Biotechnology Information, and
the European Bioinformatics Institute.
Topic threads that run through several different papers can be explored via the ENCODE microsite page at http://Nature.com/encode. Date: September 5, 2012 Source: Cold Spring Harbor Laboratory
1000 Genomes Project Team Reports on Variation Patterns
(from Phase I Data) October 31, 2012 GenomeWeb
In a study appearing online today in Nature, members of the 1000 Genomes Project Consortium presented an integrated haplotype map representing the genomic variation present in more than 1,000 individuals from 14 human populations. Using data on 1,092 individuals tested by
low-coverage whole-genome sequencing,
deep exome sequencing, and/or
dense genotyping,
the team looked at the nature and extent of the rare and common variation present in the genomes of individuals within these populations. In addition to population-specific differences in common variant profiles, for example, the researchers found distinct rare variant patterns within populations from different parts of the world — information that is expected to be important in interpreting future disease studies. They also encountered a surprising number of the variants that are expected to impact gene function, such as
non-synonymous changes,
loss-of-function variants, and, in some cases,
potentially damaging mutations.
ENCODE was designed to pick up where the Human Genome Project left off. Although that massive effort revealed the blueprint of human biology, it quickly became clear that the instruction manual for reading the blueprint was sketchy at best. Researchers could identify in its 3 billion letters many of the regions that code for proteins, but they make up little more than 1% of the genome, contained in around 20,000 genes. ENCODE, which started in 2003, is a massive data-collection effort designed to catalogue the
‘functional’ DNA sequences,
learn when and in which cells they are active and
trace their effects on how the genome is
packaged,
regulated and
read.
After an initial pilot phase, ENCODE scientists started applying their methods to the entire genome in 2007. That phase came to a close with the publication of 30 papers, in Nature, Genome Research and Genome Biology. The consortium has assigned some sort of function to roughly 80% of the genome, including
more than 70,000 ‘promoter’ regions — the sites, just upstream of genes, where proteins bind to control gene expression —
and nearly 400,000 ‘enhancer’ regions that regulate expression of distant genes (see page 57)1. But the job is far from done.
proteins interact with the DNA to control gene expression.
Overall, the Encode data define regulatory switches that are scattered all over the three billion nucleotides of the genome. In fact, the data suggests,
the regions that lie between gene-coding sequences contain a wealth of previously unrecognized functional elements,Including
nonprotein-coding RNA transcribed sequences,
transcription factor binding sites,
chromatin structural elements, and
DNA methylation sites.
The combined results suggest that 95% of the genome lies within 8 kb of a DNA-protein interaction, and 99% lies within 1.7 kb of at least one of the biochemical events, the researchers say. Importantly, given the complex three-dimensional nature of DNA, it’s also apparent that
a regulatory element for one gene may be located quite some ‘linear’ distance from the gene itself.
“The information processing and the intelligence of the genome reside in the regulatory elements,” explains Jim Kent, director of the University of California, Santa Cruz Genome Browser project and head of the Encode Data Coordination Center. “With this project, we probably went from understanding less than 5% to now around 75% of them.” The ENCODE results also identified SNPs within regulatory regions that are associated with a range of diseases, providing new insights into the roles that
noncoding DNA plays in disease development.
“As much as nine out of 10 times, disease-linked genetic variants are not in protein-coding regions,” comments Mike Pazin, Encode program director at the National Human Genome Research Institute. “Far from being junk DNA, this regulatory DNA clearly makes important contributions to human disease.”
Other Related Articles on this Open Access Online Scientific Journal, include the following:
Impact of evolutionary selection on functional regions: The imprint of evolutionary selection on ENCODE regulatory elements is manifested between species and within human populations s Saha




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}