Lipid Metabolism
Reporter and Curator: Larry H. Bernstein, MD, FCAP
This is fourth of a series of articles, lipid metabolism, that began with signaling and signaling pathways. These discussion lay the groundwork to proceed in later discussions that will take on a somewhat different approach. These are critical to develop a more complete point of view of life processes. I have indicated that many of the protein-protein interactions or protein-membrane interactions and associated regulatory features have been referred to previously, but the focus of the discussion or points made were different. The role of lipids in circulating plasma proteins as biomarkers for coronary vascular disease can be traced to the early work of Frederickson and the classification of lipid disorders. The very critical role of lipids in membrane structure in health and disease has had much less attention, despite the enormous importance, especially in the nervous system.
- Signaling and signaling pathways
- Signaling transduction tutorial.
- Carbohydrate metabolism
3.1 Selected References to Signaling and Metabolic Pathways in Leaders in Pharmaceutical Intelligence
- Lipid metabolism
- Protein synthesis and degradation
- Subcellular structure
- Impairments in pathological states: endocrine disorders; stress hypermetabolism; cancer.
Lipid Metabolism
http://www.elmhurst.edu/~chm/vchembook/622overview.html
Overview of Lipid Catabolism:
The major aspects of lipid metabolism are involved with
- Fatty Acid Oxidationto produce energy or
- the synthesis of lipids which is called Lipogenesis.
The metabolism of lipids and carbohydrates are related by the conversion of lipids from carbohydrates. This can be seen in the diagram. Notice the link through actyl-CoA, the seminal discovery of Fritz Lipmann. The metabolism of both is upset by diabetes mellitus, which results in the release of ketones (2/3 betahydroxybutyric acid) into the circulation.

metabolism of fats
http://www.elmhurst.edu/~chm/vchembook/images/590metabolism.gif
The first step in lipid metabolism is the hydrolysis of the lipid in the cytoplasm to produce glycerol and fatty acids.
Since glycerol is a three carbon alcohol, it is metabolized quite readily into an intermediate in glycolysis, dihydroxyacetone phosphate. The last reaction is readily reversible if glycerol is needed for the synthesis of a lipid.
The hydroxyacetone, obtained from glycerol is metabolized into one of two possible compounds. Dihydroxyacetone may be converted into pyruvic acid, a 3-C intermediate at the last step of glycolysis to make energy.
In addition, the dihydroxyacetone may also be used in gluconeogenesis (usually dependent on conversion of gluconeogenic amino acids) to make glucose-6-phosphate for glucose to the blood or glycogen depending upon what is required at that time.
Fatty acids are oxidized to acetyl CoA in the mitochondria using the fatty acid spiral. The acetyl CoA is then ultimately converted into ATP, CO2, and H2O using the citric acid cycle and the electron transport chain.
There are two major types of fatty acids – ω-3 and ω-6. There are also saturated and unsaturated with respect to the existence of double bonds, and monounsaturated and polyunsatured. Polyunsaturated fatty acids (PUFAs) are important in long term health, and it will be seen that high cardiovascular risk is most associated with a low ratio of ω-3/ω-6, the denominator being from animal fat. Ω-3 fatty acids are readily available from fish, seaweed, and flax seed. More can be said of this later.
Fatty acids are synthesized from carbohydrates and occasionally from proteins. Actually, the carbohydrates and proteins have first been catabolized into acetyl CoA. Depending upon the energy requirements, the acetyl CoA enters the citric acid cycle or is used to synthesize fatty acids in a process known as LIPOGENESIS.
The relationships between lipid and carbohydrate metabolism are
summarized in Figure 2.

fattyacidspiral
http://www.elmhurst.edu/~chm/vchembook/images/620fattyacidspiral.gif
Energy Production Fatty Acid Oxidation:
“Visible” ATP:
In the fatty acid spiral, there is only one reaction which directly uses ATP and that is in the initiating step. So this is a loss of ATP and must be subtracted later.
A large amount of energy is released and restored as ATP during the oxidation of fatty acids. The ATP is formed from both the fatty acid spiral and the citric acid cycle.
Connections to Electron Transport and ATP:
One turn of the fatty acid spiral produces ATP from the interaction of the coenzymes FAD (step 1) and NAD+ (step 3) with the electron transport chain. Total ATP per turn of the fatty acid spiral is:
Electron Transport Diagram – (e.t.c.)
Step 1 – FAD into e.t.c. = 2 ATP
Step 3 – NAD+ into e.t.c. = 3 ATP
Total ATP per turn of spiral = 5 ATP
In order to calculate total ATP from the fatty acid spiral, you must calculate the number of turns that the spiral makes. Remember that the number of turns is found by subtracting one from the number of acetyl CoA produced. See the graphic on the left bottom.
Example with Palmitic Acid = 16 carbons = 8 acetyl groups
Number of turns of fatty acid spiral = 8-1 = 7 turns
ATP from fatty acid spiral = 7 turns and 5 per turn = 35 ATP.
This would be a good time to remember that single ATP that was needed to get the fatty acid spiral started. Therefore subtract it now.
NET ATP from Fatty Acid Spiral = 35 – 1 = 34 ATP
Review ATP Summary for Citric Acid Cycle:The acetyl CoA produced from the fatty acid spiral enters the citric acid cycle. When calculating ATP production, you have to show how many acetyl CoA are produced from a given fatty acid as this controls how many “turns” the citric acid cycle makes.Starting with acetyl CoA, how many ATP are made using the citric acid cycle? E.T.C = electron transport chain
| Step |
ATP produced |
| 7 |
1 |
| Step 4 (NAD+ to E.T.C.) |
3 |
| Step 6 (NAD+ to E.T.C.) |
3 |
| Step10 (NAD+ to E.T.C.) |
3 |
| Step 8 (FAD to E.T.C.) |
2 |
| NET |
12 ATP |
|
ATP Summary for Palmitic Acid – Complete Metabolism:The phrase “complete metabolism” means do reactions until you end up with carbon dioxide and water. This also means to use fatty acid spiral, citric acid cycle, and electron transport as needed.Starting with palmitic acid (16 carbons) how many ATP are made using fatty acid spiral? This is a review of the above panel E.T.C = electron transport chain
| Step |
ATP (used -) (produced +) |
| Step 1 (FAD to E.T.C.) |
+2 |
| Step 4 (NAD+ to E.T.C.) |
+3 |
| Total ATP |
+5 |
| 7 turns |
7 x 5 = 35 |
| initial step |
-1 |
| NET |
34 ATP |
The fatty acid spiral ends with the production of 8 acetyl CoA from the 16 carbon palmitic acid.
Starting with one acetyl CoA, how many ATP are made using the citric acid cycle? Above panel gave the answer of 12 ATP per acetyl CoA.
E.T.C = electron transport chain
| Step |
ATP produced |
| One acetyl CoA per turn C.A.C. |
+12 ATP |
| 8 Acetyl CoA = 8 turns C.A.C. |
8 x 12 = 96 ATP |
| Fatty Acid Spiral |
34 ATP |
| GRAND TOTAL |
130 ATP |
|
Fyodor Lynen
Feodor Lynen was born in Munich on 6 April 1911, the son of Wilhelm Lynen, Professor of Mechanical Engineering at the Munich Technische Hochschule. He received his Doctorate in Chemistry from Munich University under Heinrich Wieland, who had won the Nobel Prize for Chemistry in 1927, in March 1937 with the work: «On the Toxic Substances in Amanita». in 1954 he became head of the Max-Planck-Institut für Zellchemie, newly created for him as a result of the initiative of Otto Warburg and Otto Hahn, then President of the Max-Planck-Gesellschaft zur Förderung der Wissenschaften.
Lynen’s work was devoted to the elucidation of the chemical details of metabolic processes in living cells, and of the mechanisms of metabolic regulation. The problems tackled by him, in conjunction with German and other workers, include the Pasteur effect, acetic acid degradation in yeast, the chemical structure of «activated acetic acid» of «activated isoprene», of «activated carboxylic acid», and of cytohaemin, degradation of fatty acids and formation of acetoacetic acid, degradation of tararic acid, biosynthesis of cysteine, of terpenes, of rubber, and of fatty acids.
In 1954 Lynen received the Neuberg Medal of the American Society of European Chemists and Pharmacists, in 1955 the Liebig Commemorative Medal of the Gesellschaft Deutscher Chemiker, in 1961 the Carus Medal of the Deutsche Akademie der Naturforscher «Leopoldina», and in 1963 the Otto Warburg Medal of the Gesellschaft für Physiologische Chemie. He was also a member of the U>S> National Academy of Sciences, and shared the Nobel Prize in Physiology and Medicine with Konrad Bloch in 1964, and was made President of the Gesellschaft Deutscher Chemiker (GDCh) in 1972.
This biography was written at the time of the award and first published in the book series Les Prix Nobel. It was later edited and republished in Nobel Lectures, and shortened by myself.
The Pathway from “Activated Acetic Acid” to the Terpenes and Fatty Acids
My first contact with dynamic biochemistry in 1937 occurred at an exceedingly propitious time. The remarkable investigations on the enzyme chain of respiration, on the oxygen-transferring haemin enzyme of respiration, the cytochromes, the yellow enzymes, and the pyridine proteins had thrown the first rays of light on the chemical processes underlying the mystery of biological catalysis, which had been recognised by your famous countryman Jöns Jakob Berzelius. Vitamin B2 , which is essential to the nourishment of man and of animals, had been recognised by Hugo Theorell in the form of the phosphate ester as the active group of an important class of enzymes, and the fermentation processes that are necessary for Pasteur’s “life without oxygen”
had been elucidated as the result of a sequence of reactions centered around “hydrogen shift” and “phosphate shift” with adenosine triphosphate as the phosphate-transferring coenzyme. However, 1,3-diphosphoglyceric acid, the key substance to an understanding of the chemical relation between oxidation and phosphorylation, still lay in the depths of the unknown. Never-
theless, Otto Warburg was on its trail in the course of his investigations on the fermentation enzymes, and he was able to present it to the world in 1939.
This was the period in which I carried out my first independent investigation, which was concerned with the metabolism of yeast cells after freezing in liquid air, and which brought me directly into contact with the mechanism of alcoholic fermentation. This work taught me a great deal, and yielded two important pieces of information.
- The first was that in experiments with living cells, special attention must be given to the permeability properties of the cell membranes, and
- the second was that the adenosine polyphosphate system plays a vital part in the cell,
- not only in energy transfer, but
- also in the regulation of the metabolic processes.
.
This investigation aroused by interest in problems of metabolic regulation, which led me to the investigation of the Pasteur effects, and has remained with me to the present day.
My subsequent concern with the problem of the acetic acid metabolism arose from my stay at Heinrich Wieland’s laboratory. Workers here had studied the oxidation of acetic acid by yeast cells, and had found that though most of the acetic acid undergoes complete oxidation, some remains in the form of succinic and citric acids.
The explanation of these observations was provided-by the Thunberg-Wieland process, according to which two molecules of acetic acid are dehydrogenated to succinic acid, which is converted back into acetic acid via oxaloacetic acid, pyruvic acid, and acetaldehyde, or combines at the oxaloacetic acid stage with a further molecule of acetic acid to form citric acid (Fig. 1). However, an experimental check on this view by a Wieland’s student Robert Sonderhoffs brought a surprise. The citric acid formed when trideuteroacetic acid was supplied to yeast cells contained the expected quantity of deuterium, but the succinic acid contained only half of the four deuterium atoms required by Wieland’s scheme.
This investigation aroused by interest in problems of metabolic regulation, which led me to the investigation of the Pasteur effects, and has remained with me to the present day. My subsequent concern with the problem of the acetic acid metabolism arose from my stay at Heinrich Wieland’s laboratory. Workers here had studied the oxidation of acetic acid by yeast cells, and had found that though most of the acetic acid undergoes complete oxidation, some remains in the form of succinic and citric acid
The answer provided by Martius was that citric acid is in equilibrium with isocitric acid and is oxidised to cr-ketoglutaric acid, the conversion of which into succinic acid had already been discovered by Carl Neuberg (Fig. 1).
It was possible to assume with fair certainty from these results that the succinic acid produced by yeast from acetate is formed via citric acid. Sonderhoff’s experiments with deuterated acetic acid led to another important discovery.
In the analysis of the yeast cells themselves, it was found that while the carbohydrate fraction contained only insignificant quantities of deuterium, large quantities of heavy hydrogen were present in the fatty acids formed and in the sterol fraction. This showed that
- fatty acids and sterols were formed directly from acetic acid, and not indirectly via the carbohydrates.
As a result of Sonderhoff’s early death, these important findings were not pursued further in the Munich laboratory.
- This situation was elucidated only by Konrad Bloch’s isotope experiments, on which he reports.
My interest first turned entirely to the conversion of acetic acid into citric acid, which had been made the focus of the aerobic degradation of carbohydrates by the formulation of the citric acid cycle by Hans Adolf Krebs. Unlike Krebs, who regarded pyruvic acid as the condensation partner of acetic acid,
- we were firmly convinced, on the basis of the experiments on yeast, that pyruvic acid is first oxidised to acetic acid, and only then does the condensation take place.
Further progress resulted from Wieland’s observation that yeast cells that had been “impoverished” in endogenous fuels by shaking under oxygen were able to oxidise added acetic acid only after a certain “induction period” (Fig. 2). This “induction period” could be shortened by addition of small quantities of a readily oxidisable substrate such as ethyl alcohol, though propyl and butyl alcohol were also effective. I explained this by assuming that acetic acid is converted, at the expense of the oxidation of the alcohol, into an “activated acetic acid”, and can only then condense with oxalacetic acid.
In retrospect, we find that I had come independently on the same group of problems as Fritz Lipmann, who had discovered that inorganic phosphate is indispensable to the oxidation of pyruvic acid by lactobacilli, and had detected acetylphosphate as an oxidation product. Since this anhydride of acetic acid and phosphoric acid could be assumed to be the “activated acetic acid”.
I learned of the advances that had been made in the meantime in the investigation of the problem of “activated acetic acid”. Fritz Lipmann has described the development at length in his Nobel Lecture’s, and I need not repeat it. The main advance was the recognition that the formation of “activated acetic acid” from acetate involved not only ATP as an energy source, but also the newly discovered coenzyme A, which contains the vitamin pantothenic acid, and that “activated acetic acid” was probably an acetylated coenzyme A.
http://www.nobelprize.org/nobel_prizes/medicine/laureates/1964/lynen-bio.html
http://onlinelibrary.wiley.com/store/10.1002/anie.201106003/asset/image_m/mcontent.gif?v=1&s=1e6dc789dfa585fe48947e92cc5dfdcabd8e2677
Fyodor Lynen
Lynen’s most important research at the University of Munich focused on intermediary metabolism, cholesterol synthesis, and fatty acid biosynthesis. Metabolism involves all the chemical processes by which an organism converts matter and energy into forms that it can use. Metabolism supplies the matter—the molecular building blocks an organism needs for the growth of new tissues. These building blocks must either come from the breakdown of molecules of food, such as glucose (sugar) and fat, or be built up from simpler molecules within the organism.
Cholesterol is one of the fatty substances found in animal tissues. The human body produces cholesterol, but this substance also enters the body in food. Meats, egg yolks, and milk products, such as butter and cheese, contain cholesterol. Such organs as the brain and liver contain much cholesterol. Cholesterol is a type of lipid, one of the classes of chemical compounds essential to human health. It makes up an important part of the membranes of each cell in the body. The body also uses cholesterol to produce vitamin D and certain hormones.
All fats are composed of an alcohol called glycerol and substances called fatty acids. A fatty acid consists of a long chain of carbon atoms, to which hydrogen atoms are attached. There are three types of fatty acids: saturated, monounsaturated, and polyunsaturated.
Living cells manufacture complicated chemical compounds from simpler substances through a process called biosynthesis. For example, simple molecules called amino acids are put together to make proteins. The biosynthesis of both fatty acids and cholesterol begins with a chemically active form of acetate, a two-carbon molecule. Lynen discovered that the active form of acetate is a coenzyme, a heat-stabilized, water-soluble portion of an enzyme, called acetyl coenzyme A. Lynen and his colleagues demonstrated that the formation of cholesterol begins with the condensation of two molecules of acetyl coenzyme A to form acetoacetyl coenzyme A, a four-carbon molecule.
http://science.howstuffworks.com/dictionary/famous-scientists/biologists/feodor-lynen-info.htm

Fyodor Lynen
SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver
Jay D. Horton1,2, Joseph L. Goldstein1 and Michael S. Brown1
1Department of Molecular Genetics, and
2Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA
J Clin Invest. 2002;109(9):1125–1131.
http://dx.doi.org:/10.1172/JCI15593
Lipid homeostasis in vertebrate cells is regulated by a family of membrane-bound transcription factors designated sterol regulatory element–binding proteins (SREBPs). SREBPs directly activate the expression of more than 30 genes dedicated to the synthesis and uptake of cholesterol, fatty acids, triglycerides, and phospholipids, as well as the NADPH cofactor required to synthesize these molecules (1–4). In the liver, three SREBPs regulate the production of lipids for export into the plasma as lipoproteins and into the bile as micelles. The complex, interdigitated roles of these three SREBPs have been dissected through the study of ten different lines of gene-manipulated mice. These studies form the subject of this review.
SREBPs: activation through proteolytic processing
SREBPs belong to the basic helix-loop-helix–leucine zipper (bHLH-Zip) family of transcription factors, but they differ from other bHLH-Zip proteins in that they are synthesized as inactive precursors bound to the endoplasmic reticulum (ER) (1, 5). Each SREBP precursor of about 1150 amino acids is organized into three domains: (a) an NH2-terminal domain of about 480 amino acids that contains the bHLH-Zip region for binding DNA; (b) two hydrophobic transmembrane–spanning segments interrupted by a short loop of about 30 amino acids that projects into the lumen of the ER; and (c) a COOH-terminal domain of about 590 amino acids that performs the essential regulatory function described below.
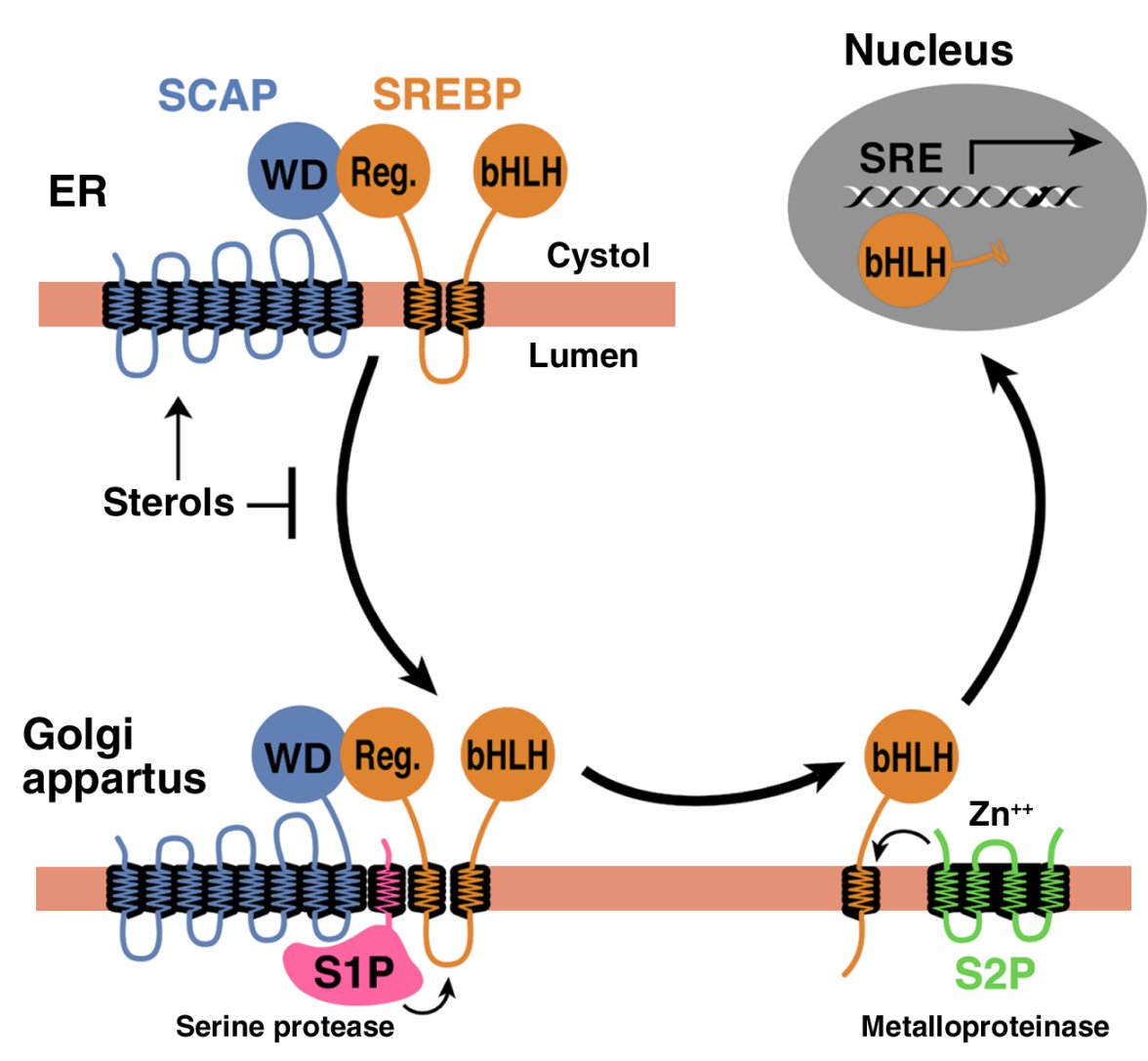
In order to reach the nucleus and act as a transcription factor, the NH2-terminal domain of each SREBP must be released from the membrane proteolytically (Figure 1). Three proteins required for SREBP processing have been delineated in cultured cells, using the tools of somatic cell genetics (see ref. 5for review). One is an escort protein designated SREBP cleavage–activating protein (SCAP). The other two are proteases, designated Site-1 protease (S1P) and Site-2 protease (S2P). Newly synthesized SREBP is inserted into the membranes of the ER, where its COOH-terminal regulatory domain binds to the COOH-terminal domain of SCAP (Figure 1).
Figure 1

Model for the sterol-mediated proteolytic release of SREBPs from membranes JCI0215593.f1
Model for the sterol-mediated proteolytic release of SREBPs from membranes. SCAP is a sensor of sterols and an escort of SREBPs. When cells are depleted of sterols, SCAP transports SREBPs from the ER to the Golgi apparatus, where two proteases, Site-1 protease (S1P) and Site-2 protease (S2P), act sequentially to release the NH2-terminal bHLH-Zip domain from the membrane. The bHLH-Zip domain enters the nucleus and binds to a sterol response element (SRE) in the enhancer/promoter region of target genes, activating their transcription. When cellular cholesterol rises, the SCAP/SREBP complex is no longer incorporated into ER transport vesicles, SREBPs no longer reach the Golgi apparatus, and the bHLH-Zip domain cannot be released from the membrane. As a result, transcription of all target genes declines. Reprinted from ref. 5 with permission.
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/large/JCI0215593.f1.jpg
SCAP is both an escort for SREBPs and a sensor of sterols. When cells become depleted in cholesterol, SCAP escorts the SREBP from the ER to the Golgi apparatus, where the two proteases reside. In the Golgi apparatus, S1P, a membrane-bound serine protease, cleaves the SREBP in the luminal loop between its two membrane-spanning segments, dividing the SREBP molecule in half (Figure 1). The NH2-terminal bHLH-Zip domain is then released from the membrane via a second cleavage mediated by S2P, a membrane-bound zinc metalloproteinase. The NH2-terminal domain, designated nuclear SREBP (nSREBP), translocates to the nucleus, where it activates transcription by binding to nonpalindromic sterol response elements (SREs) in the promoter/enhancer regions of multiple target genes.
Figure 1
When the cholesterol content of cells rises, SCAP senses the excess cholesterol through its membranous sterol-sensing domain, changing its conformation in such a way that the SCAP/SREBP complex is no longer incorporated into ER transport vesicles. The net result is that SREBPs lose their access to S1P and S2P in the Golgi apparatus, so their bHLH-Zip domains cannot be released from the ER membrane, and the transcription of target genes ceases (1, 5). The biophysical mechanism by which SCAP senses sterol levels in the ER membrane and regulates its movement to the Golgi apparatus is not yet understood. Elucidating this mechanism will be fundamental to understanding the molecular basis of cholesterol feedback inhibition of gene expression.
SREBPs: two genes, three proteins
The mammalian genome encodes three SREBP isoforms, designated SREBP-1a, SREBP-1c, and SREBP-2. SREBP-2 is encoded by a gene on human chromosome 22q13. Both SREBP-1a and -1c are derived from a single gene on human chromosome 17p11.2 through the use of alternative transcription start sites that produce alternate forms of exon 1, designated 1a and 1c (1). SREBP-1a is a potent activator of all SREBP-responsive genes, including those that mediate the synthesis of cholesterol, fatty acids, and triglycerides. High-level transcriptional activation is dependent on exon 1a, which encodes a longer acidic transactivation segment than does the first exon of SREBP-1c. The roles of SREBP-1c and SREBP-2 are more restricted than that of SREBP-1a. SREBP-1c preferentially enhances transcription of genes required for fatty acid synthesis but not cholesterol synthesis. Like SREBP-1a, SREBP-2 has a long transcriptional activation domain, but it preferentially activates cholesterol synthesis (1). SREBP-1a and SREBP-2 are the predominant isoforms of SREBP in most cultured cell lines, whereas SREBP-1c and SREBP-2 predominate in the liver and most other intact tissues (6).
When expressed at higher than physiologic levels, each of the three SREBP isoforms can activate all enzymes indicated in Figure 2, which shows the biosynthetic pathways used to generate cholesterol and fatty acids. However, at normal levels of expression, SREBP-1c favors the fatty acid biosynthetic pathway and SREBP-2 favors cholesterologenesis. SREBP-2–responsive genes in the cholesterol biosynthetic pathway include those for the enzymes HMG-CoA synthase, HMG-CoA reductase, farnesyl diphosphate synthase, and squalene synthase. SREBP-1c–responsive genes include those for ATP citrate lyase (which produces acetyl-CoA) and acetyl-CoA carboxylase and fatty acid synthase (which together produce palmitate [C16:0]). Other SREBP-1c target genes encode a rate-limiting enzyme of the fatty acid elongase complex, which converts palmitate to stearate (C18:0) (ref.7); stearoyl-CoA desaturase, which converts stearate to oleate (C18:1); and glycerol-3-phosphate acyltransferase, the first committed enzyme in triglyceride and phospholipid synthesis (3). Finally, SREBP-1c and SREBP-2 activate three genes required to generate NADPH, which is consumed at multiple stages in these lipid biosynthetic pathways (8) (Figure 2).
Figure 2

major metabolic intermediates in the pathways for synthesis of cholesterol, fatty acids, and triglycerides JCI0215593.f2
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/large/JCI0215593.f2.jpg
Genes regulated by SREBPs. The diagram shows the major metabolic intermediates in the pathways for synthesis of cholesterol, fatty acids, and triglycerides. In vivo, SREBP-2 preferentially activates genes of cholesterol metabolism, whereas SREBP-1c preferentially activates genes of fatty acid and triglyceride metabolism. DHCR, 7-dehydrocholesterol reductase; FPP, farnesyl diphosphate; GPP, geranylgeranyl pyrophosphate synthase; CYP51, lanosterol 14α-demethylase; G6PD, glucose-6-phosphate dehydrogenase; PGDH, 6-phosphogluconate dehydrogenase; GPAT, glycerol-3-phosphate acyltransferase.
Genes regulated by SREBPs. The diagram shows the major metabolic intermediates in the pathways for synthesis of cholesterol, fatty acids, and triglycerides. In vivo, SREBP-2 preferentially activates genes of cholesterol metabolism, whereas SREBP-1c preferentially activates genes of fatty acid and triglyceride metabolism. DHCR, 7-dehydrocholesterol reductase; FPP, farnesyl diphosphate; GPP, geranylgeranyl pyrophosphate synthase; CYP51, lanosterol 14α-demethylase; G6PD, glucose-6-phosphate dehydrogenase; PGDH, 6-phosphogluconate dehydrogenase; GPAT, glycerol-3-phosphate acyltransferase.
Knockout and transgenic mice
Ten different genetically manipulated mouse models that either lack or overexpress a single component of the SREBP pathway have been generated in the last 6 years (9–16). The key molecular and metabolic alterations observed in these mice are summarized in Table 1.
Table 1
Alterations in hepatic lipid metabolism in gene-manipulated mice overexpressing or lacking SREBPs
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/small/JCI0215593.t1.gif
Knockout mice that lack all nSREBPs die early in embryonic development. For instance, a germline deletion of S1p, which prevents the processing of all SREBP isoforms, results in death before day 4 of development (15, 17). Germline deletion of Srebp2 leads to 100% lethality at a later stage of embryonic development than does deletion of S1p (embryonic day 7–8). In contrast, germline deletion of Srebp1, which eliminates both the 1a and the 1c transcripts, leads to partial lethality, in that about 15–45% of Srebp1–/– mice survive (13). The surviving homozygotes manifest elevated levels of SREBP-2 mRNA and protein (Table 1), which presumably compensates for the loss of SREBP-1a and -1c. When the SREBP-1c transcript is selectively eliminated, no embryonic lethality is observed, suggesting that the partial embryonic lethality in the Srebp1–/– mice is due to the loss of the SREBP-1a transcript (16).
To bypass embryonic lethality, we have produced mice in which all SREBP function can be disrupted in adulthood through induction of Cre recombinase. For this purpose, loxP recombination sites were inserted into genomic regions that flank crucial exons in the Scap or S1p genes (so-called floxed alleles) (14, 15). Mice homozygous for the floxed gene and heterozygous for a Cre recombinase transgene, which is under control of an IFN-inducible promoter (MX1-Cre), can be induced to delete Scap or S1p by stimulating IFN expression. Thus, following injection with polyinosinic acid–polycytidylic acid, a double-stranded RNA that provokes antiviral responses, the Cre recombinase is produced in liver and disrupts the floxed gene by recombination between the loxP sites.
Cre-mediated disruption of Scap or S1p dramatically reduces nSREBP-1 and nSREBP-2 levels in liver and diminishes expression of all SREBP target genes in both the cholesterol and the fatty acid synthetic pathways (Table 1). As a result, the rates of synthesis of cholesterol and fatty acids fall by 70–80% in Scap- and S1p-deficient livers.
In cultured cells, the processing of SREBP is inhibited by sterols, and the sensor for this inhibition is SCAP (5). To learn whether SCAP performs the same function in liver, we have produced transgenic mice that express a mutant SCAP with a single amino acid substitution in the sterol-sensing domain (D443N) (12). Studies in tissue culture show that SCAP(D443N) is resistant to inhibition by sterols. Cells that express a single copy of this mutant gene overproduce cholesterol (18). Transgenic mice that express this mutant version of SCAP in the liver exhibit a similar phenotype (12). These livers manifest elevated levels of nSREBP-1 and nSREBP-2, owing to constitutive SREBP processing, which is not suppressed when the animals are fed a cholesterol-rich diet. nSREBP-1 and -2 increase the expression of all SREBP target genes shown in Figure 2, thus stimulating cholesterol and fatty acid synthesis and causing a marked accumulation of hepatic cholesterol and triglycerides (Table 1). This transgenic model provides strong in vivo evidence that SCAP activity is normally under partial inhibition by endogenous sterols, which keeps the synthesis of cholesterol and fatty acids in a partially repressed state in the liver.
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/small/JCI0215593.t1.gif
Function of individual SREBP isoforms in vivo
To study the functions of individual SREBPs in the liver, we have produced transgenic mice that overexpress truncated versions of SREBPs (nSREBPs) that terminate prior to the membrane attachment domain. These nSREBPs enter the nucleus directly, bypassing the sterol-regulated cleavage step. By studying each nSREBP isoform separately, we could determine their distinct activating properties, albeit when overexpressed at nonphysiologic levels.
Overexpression of nSREBP-1c in the liver of transgenic mice produces a triglyceride-enriched fatty liver with no increase in cholesterol (10). mRNAs for fatty acid synthetic enzymes and rates of fatty acid synthesis are elevated fourfold in this tissue, whereas the mRNAs for cholesterol synthetic enzymes and the rate of cholesterol synthesis are not increased (8). Conversely, overexpression of nSREBP-2 in the liver increases the mRNAs only fourfold. This increase in cholesterol synthesis is even more remarkable when encoding all cholesterol biosynthetic enzymes; the most dramatic is a 75-fold increase in HMG-CoA reductase mRNA (11). mRNAs for fatty acid synthesis enzymes are increased to a lesser extent, consistent with the in vivo observation that the rate of cholesterol synthesis increases 28-fold in these transgenic nSREBP-2 livers, while fatty acid synthesis increases one considers the extent of cholesterol overload in this tissue, which would ordinarily reduce SREBP processing and essentially abolish cholesterol synthesis (Table 1).
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/small/JCI0215593.t1.gif
We have also studied the consequences of overexpressing SREBP-1a, which is expressed only at low levels in the livers of adult mice, rats, hamsters, and humans (6). nSREBP-1a transgenic mice develop a massive fatty liver engorged with both cholesterol and triglycerides (9), with heightened expression of genes controlling cholesterol biosynthesis and, still more dramatically, fatty acid synthesis (Table 1). The preferential activation of fatty acid synthesis (26-fold increase) relative to cholesterol synthesis (fivefold increase) explains the greater accumulation of triglycerides in their livers. The relative representation of the various fatty acids accumulating in this tissue is also unusual. Transgenic nSREBP-1a livers contain about 65% oleate (C18:1), markedly higher levels than the 15–20% found in typical wild-type livers (8) — a result of the induction of fatty acid elongase and stearoyl-CoA desaturase-1 (7). Considered together, the overexpression studies indicate that both SREBP-1 isoforms show a relative preference for activating fatty acid synthesis, whereas SREBP-2 favors cholesterol.
The phenotype of animals lacking the Srebp1 gene, which encodes both the SREBP-1a and -1c transcripts, also supports the notion of distinct hepatic functions for SREBP-1 and SREBP-2 (13). Most homozygous SREBP-1 knockout mice die in utero. The surviving Srebp1–/– mice show reduced synthesis of fatty acids, owing to reduced expression of mRNAs for fatty acid synthetic enzymes (Table 1). Hepatic nSREBP-2 levels increase in these mice, presumably in compensation for the loss of nSREBP-1. As a result, transcription of cholesterol biosynthetic genes increases, producing a threefold increase in hepatic cholesterol synthesis (Table 1).
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/small/JCI0215593.t1.gif
The studies in genetically manipulated mice clearly show that, as in cultured cells, SCAP and S1P are required for normal SREBP processing in the liver. SCAP, acting through its sterol-sensing domain, mediates feedback regulation of cholesterol synthesis. The SREBPs play related but distinct roles: SREBP-1c, the predominant SREBP-1 isoform in adult liver, preferentially activates genes required for fatty acid synthesis, while SREBP-2 preferentially activates the LDL receptor gene and various genes required for cholesterol synthesis. SREBP-1a and SREBP-2, but not SREBP-1c, are required for normal embryogenesis.
Transcriptional regulation of SREBP genes
Regulation of SREBPs occurs at two levels — transcriptional and posttranscriptional. The posttranscriptional regulation discussed above involves the sterol-mediated suppression of SREBP cleavage, which results from sterol-mediated suppression of the movement of the SCAP/SREBP complex from the ER to the Golgi apparatus (Figure 1). This form of regulation is manifest not only in cultured cells (1), but also in the livers of rodents fed cholesterol-enriched diets (19).
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/small/JCI0215593.f1.gif
The transcriptional regulation of the SREBPs is more complex. SREBP-1c and SREBP-2 are subject to distinct forms of transcriptional regulation, whereas SREBP-1a appears to be constitutively expressed at low levels in liver and most other tissues of adult animals (6). One mechanism of regulation shared by SREBP-1c and SREBP-2 involves a feed-forward regulation mediated by SREs present in the enhancer/promoters of each gene (20, 21). Through this feed-forward loop, nSREBPs activate the transcription of their own genes. In contrast, when nSREBPs decline, as in Scap or S1p knockout mice, there is a secondary decline in the mRNAs encoding SREBP-1c and SREBP-2 (14, 15).
Three factors selectively regulate the transcription of SREBP-1c: liver X-activated receptors (LXRs), insulin, and glucagon. LXRα and LXRβ, nuclear receptors that form heterodimers with retinoid X receptors, are activated by a variety of sterols, including oxysterol intermediates that form during cholesterol biosynthesis (22–24). An LXR-binding site in the SREBP-1c promoter activates SREBP-1c transcription in the presence of LXR agonists (23). The functional significance of LXR-mediated SREBP-1c regulation has been confirmed in two animal models. Mice that lack both LXRα and LXRβ express reduced levels of SREBP-1c and its lipogenic target enzymes in liver and respond relatively weakly to treatment with a synthetic LXR agonist (23). Because a similar blunted response is found in mice that lack SREBP-1c, it appears that LXR increases fatty acid synthesis largely by inducing SREBP-1c (16). LXR-mediated activation of SREBP-1c transcription provides a mechanism for the cell to induce the synthesis of oleate when sterols are in excess (23). Oleate is the preferred fatty acid for the synthesis of cholesteryl esters, which are necessary for both the transport and the storage of cholesterol.
LXR-mediated regulation of SREBP-1c appears also to be one mechanism by which unsaturated fatty acids suppress SREBP-1c transcription and thus fatty acid synthesis. Rodents fed diets enriched in polyunsaturated fatty acids manifest reduced SREBP-1c mRNA expression and low rates of lipogenesis in liver (25). In vitro, unsaturated fatty acids competitively block LXR activation of SREBP-1c expression by antagonizing the activation of LXR by its endogenous ligands (26). In addition to LXR-mediated transcriptional inhibition, polyunsaturated fatty acids lower SREBP-1c levels by accelerating degradation of its mRNA (27). These combined effects may contribute to the long-recognized ability of polyunsaturated fatty acids to lower plasma triglyceride levels.
SREBP-1c and the insulin/glucagon ratio
The liver is the organ responsible for the conversion of excess carbohydrates to fatty acids to be stored as triglycerides or burned in muscle. A classic action of insulin is to stimulate fatty acid synthesis in liver during times of carbohydrate excess. The action of insulin is opposed by glucagon, which acts by raising cAMP. Multiple lines of evidence suggest that insulin’s stimulatory effect on fatty acid synthesis is mediated by an increase in SREBP-1c. In isolated rat hepatocytes, insulin treatment increases the amount of mRNA for SREBP-1c in parallel with the mRNAs of its target genes (28, 29). The induction of the target genes can be blocked if a dominant negative form of SREBP-1c is expressed (30). Conversely, incubating primary hepatocytes with glucagon or dibutyryl cAMP decreases the mRNAs for SREBP-1c and its associated lipogenic target genes (30, 31).
In vivo, the total amount of SREBP-1c in liver and adipose tissue is reduced by fasting, which suppresses insulin and increases glucagon levels, and is elevated by refeeding (32, 33). The levels of mRNA for SREBP-1c target genes parallel the changes in SREBP-1c expression. Similarly, SREBP-1c mRNA levels fall when rats are treated with streptozotocin, which abolishes insulin secretion, and rise after insulin injection (29). Overexpression of nSREBP-1c in livers of transgenic mice prevents the reduction in lipogenic mRNAs that normally follows a fall in plasma insulin levels (32). Conversely, in livers of Scap knockout mice that lack all nSREBPs in the liver (14) or knockout mice lacking either nSREBP-1c (16) or both SREBP-1 isoforms (34), there is a marked decrease in the insulin-induced stimulation of lipogenic gene expression that normally occurs after fasting/refeeding. It should be noted that insulin and glucagon also exert a posttranslational control of fatty acid synthesis though changes in the phosphorylation and activation of acetyl-CoA carboxylase. The posttranslational regulation of fatty acid synthesis persists in transgenic mice that overexpress nSREBP-1c (10). In these mice, the rates of fatty acid synthesis, as measured by [3H]water incorporation, decline after fasting even though the levels of the lipogenic mRNAs remain high (our unpublished observations).
Taken together, the above evidence suggests that SREBP-1c mediates insulin’s lipogenic actions in liver. Recent in vitro and in vivo studies involving adenoviral gene transfer suggest that SREBP-1c may also contribute to the regulation of glucose uptake and glucose synthesis. When overexpressed in hepatocytes, nSREBP-1c induces expression of glucokinase, a key enzyme in glucose utilization. It also suppresses phosphoenolpyruvate carboxykinase, a key gluconeogenic enzyme (35, 36).
SREBPs in disease
Many individuals with obesity and insulin resistance also have fatty livers, one of the most commonly encountered liver abnormalities in the US (37). A subset of individuals with fatty liver go on to develop fibrosis, cirrhosis, and liver failure. Evidence indicates that the fatty liver of insulin resistance is caused by SREBP-1c, which is elevated in response to the high insulin levels. Thus, SREBP-1c levels are elevated in the fatty livers of obese (ob/ob) mice with insulin resistance and hyperinsulinemia caused by leptin deficiency (38, 39). Despite the presence of insulin resistance in peripheral tissues, insulin continues to activate SREBP-1c transcription and cleavage in the livers of these insulin-resistant mice. The elevated nSREBP-1c increases lipogenic gene expression, enhances fatty acid synthesis, and accelerates triglyceride accumulation (31, 39). These metabolic abnormalities are reversed with the administration of leptin, which corrects the insulin resistance and lowers the insulin levels (38).
Metformin, a biguanide drug used to treat insulin-resistant diabetes, reduces hepatic nSREBP-1 levels and dramatically lowers the lipid accumulation in livers of insulin-resistant ob/ob mice (40). Metformin stimulates AMP-activated protein kinase (AMPK), an enzyme that inhibits lipid synthesis through phosphorylation and inactivation of key lipogenic enzymes (41). In rat hepatocytes, metformin-induced activation of AMPK also leads to decreased mRNA expression of SREBP-1c and its lipogenic target genes (41), but the basis of this effect is not understood.
The incidence of coronary artery disease increases with increasing plasma LDL-cholesterol levels, which in turn are inversely proportional to the levels of hepatic LDL receptors. SREBPs stimulate LDL receptor expression, but they also enhance lipid synthesis (1), so their net effect on plasma lipoprotein levels depends on a balance between opposing effects. In mice, the plasma levels of lipoproteins tend to fall when SREBPs are either overexpressed or underexpressed. In transgenic mice that overexpress nSREBPs in liver, plasma cholesterol and triglycerides are generally lower than in control mice (Table 1), even though these mice massively overproduce fatty acids, cholesterol, or both. Hepatocytes of nSREBP-1a transgenic mice overproduce VLDL, but these particles are rapidly removed through the action of LDL receptors, and they do not accumulate in the plasma. Indeed, some nascent VLDL particles are degraded even before secretion by a process that is mediated by LDL receptors (42). The high levels of nSREBP-1a in these animals support continued expression of the LDL receptor, even in cells whose cholesterol concentration is elevated. In LDL receptor–deficient mice carrying the nSREBP-1a transgene, plasma cholesterol and triglyceride levels rise tenfold (43).
Mice that lack all SREBPs in liver as a result of disruption of Scap or S1p also manifest lower plasma cholesterol and triglyceride levels (Table 1).
http://dm5migu4zj3pb.cloudfront.net/manuscripts/15000/15593/small/JCI0215593.t1.gif
In these mice, hepatic cholesterol and triglyceride synthesis is markedly reduced, and this likely causes a decrease in VLDL production and secretion. LDL receptor mRNA and LDL clearance from plasma is also significantly reduced in these mice, but the reduction in LDL clearance is less than the overall reduction in VLDL secretion, the net result being a decrease in plasma lipid levels (15). However, because
humans and mice differ substantially with regard to LDL receptor expression, LDL levels, and other aspects of lipoprotein metabolism,
it is difficult to predict whether human plasma lipids will rise or fall when the SREBP pathway is blocked or activated.
SREBPs in liver: unanswered questions
The studies of SREBPs in liver have exposed a complex regulatory system whose individual parts are coming into focus. Major unanswered questions relate to the ways in which the transcriptional and posttranscriptional controls on SREBP activity are integrated so as to permit independent regulation of cholesterol and fatty acid synthesis in specific nutritional states. A few clues regarding these integration mechanisms are discussed below.
Whereas cholesterol synthesis depends almost entirely on SREBPs, fatty acid synthesis is only partially dependent on these proteins. This has been shown most clearly in cultured nonhepatic cells such as Chinese hamster ovary cells. In the absence of SREBP processing, as when the Site-2 protease is defective, the levels of mRNAs encoding cholesterol biosynthetic enzymes and the rates of cholesterol synthesis decline nearly to undetectable levels, whereas the rate of fatty acid synthesis is reduced by only 30% (44). Under these conditions, transcription of the fatty acid biosynthetic genes must be maintained by factors other than SREBPs. In liver, the gene encoding fatty acid synthase (FASN) can be activated transcriptionally by upstream stimulatory factor, which acts in concert with SREBPs (45). The FASN promoter also contains an LXR element that permits a low-level response to LXR ligands even when SREBPs are suppressed (46). These two transcription factors may help to maintain fatty acid synthesis in liver when nSREBP-1c is low.
Another mechanism of differential regulation is seen in the ability of cholesterol to block the processing of SREBP-2, but not SREBP-1, under certain metabolic conditions. This differential regulation has been studied most thoroughly in cultured cells such as human embryonic kidney (HEK-293) cells. When these cells are incubated in the absence of fatty acids and cholesterol, the addition of sterols blocks processing of SREBP-2, but not SREBP-1, which is largely produced as SREBP-1a in these cells (47). Inhibition of SREBP-1 processing requires an unsaturated fatty acid, such as oleate or arachidonate, in addition to sterols (47). In the absence of fatty acids and in the presence of sterols, SCAP may be able to carry SREBP-1 proteins, but not SREBP-2, to the Golgi apparatus. Further studies are necessary to document this apparent independent regulation of SREBP-1 and SREBP-2 processing and to determine its mechanism.
Acknowledgments
Support for the research cited from the authors’ laboratories was provided by grants from the NIH (HL-20948), the Moss Heart Foundation, the Keck Foundation, and the Perot Family Foundation. J.D. Horton is a Pew Scholar in the Biomedical Sciences and is the recipient of an Established Investigator Grant from the American Heart Association and a Research Scholar Award from the American Digestive Health Industry.
References
- Brown, MS, Goldstein, JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997. 89:331-340.
View this article via: PubMed
- Horton, JD, Shimomura, I. Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis. Curr Opin Lipidol 1999. 10:143-150.
View this article via: PubMed
- Edwards, PA, Tabor, D, Kast, HR, Venkateswaran, A. Regulation of gene expression by SREBP and SCAP. Biochim Biophys Acta 2000. 1529:103-113.
View this article via: PubMed
- Sakakura, Y, et al. Sterol regulatory element-binding proteins induce an entire pathway of cholesterol synthesis. Biochem Biophys Res Commun 2001. 286:176-183.
View this article via: PubMed
- Goldstein, JL, Rawson, RB, Brown, MS. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem Biophys 2002. 397:139-148.
View this article via: PubMed
- Shimomura, I, Shimano, H, Horton, JD, Goldstein, JL, Brown, MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 1997. 99:838-845.
View this article via: JCI.org PubMed
- Moon, Y-A, Shah, NA, Mohapatra, S, Warrington, JA, Horton, JD. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J Biol Chem 2001. 276:45358-45366.
View this article via: PubMed
- Shimomura, I, Shimano, H, Korn, BS, Bashmakov, Y, Horton, JD. Nuclear sterol regulatory element binding proteins activate genes responsible for entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J Biol Chem 1998. 273:35299-35306.
View this article via: PubMed
- Shimano, H, et al. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest 1996. 98:1575-1584.
View this article via: JCI.org PubMed
- Shimano, H, et al. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997. 99:846-854.
View this article via: JCI.org PubMed
- Horton, JD, et al. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest 1998. 101:2331-2339.
View this article via: JCI.org PubMed
- Korn, BS, et al. Blunted feedback suppression of SREBP processing by dietary cholesterol in transgenic mice expressing sterol-resistant SCAP(D443N). J Clin Invest 1998. 102:2050-2060.
View this article via: JCI.org PubMed
- Shimano, H, et al. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J Clin Invest 1997. 100:2115-2124.
View this article via: JCI.org PubMed
- Matsuda, M, et al. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev 2001. 15:1206-1216.
View this article via: PubMed
- Yang, J, et al. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci USA 2001. 98:13607-13612.
View this article via: PubMed
Liang, G, et al. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 2002. 277:9520-9528.
http://www.jci.org/articles/view/15593
Structural Biochemistry/Lipids/Membrane Lipids
< Structural Biochemistry | Lipids
Membrane proteins rely on their interaction with membrane lipids to uphold its structure and maintain its functions as a protein. For membrane proteins to purify and crystallize, it is essential for the membrane protein to be in the appropriate lipid environment. Lipids assist in crystallization and stabilize the protein and provide lattice contacts. Lipids can also help obtain membrane protein structures in a native conformation. Membrane protein structures contain bound lipid molecules. Biological membranes are important in life, providing permeable barriers for cells and their organelles. The interaction between membrane proteins and lipids facilitates basic processes such as respiration, photosynthesis, transport, signal transduction and motility. These basic processes require a diverse group of proteins, which are encoded by 20-30% of an organism’s annotated genes.
There exist a great number of membrane lipids. Specifically, eukaryotic cells have a very complex collection of lipids that rely on many of the cell’s resources for its synthesis. Interactions between proteins and lipids can be very specific. Specific types of lipids can make a structure stable, provide control in insertion and folding processes, and help to assemble multisubunit complexes or supercomplexes, and most importantly, can significantly affect a membrane protein’s functions. Protein and lipid interactions are not sufficiently tight, meaning that lipids are retained during membrane protein purification. Since cellular membranes are fluid arrangements of lipids, some lipids affect interesting changes to membrane due to their characteristics. Glycosphigolipids and cholesterol tend to form small islands within the membranes, called lipid rafts, due to their physical properties. Some proteins also tend to cluster in lipid raft, while others avoid being in lipid rafts. However, the existence of lipid rafts in cells seems to be transitory.
Recent progress in determining membrane protein structure has brought attention to the importance of maintaining a favorable lipid environment so proteins to crystallize and purify successfully. Lipids assist in crystallization by stabilizing the protein fold and the relationships between subunits or monomers. The lipid content in protein-lipid detergent complexes can be altered by adjusting solubilisation and purification protocols, also by adding native or non-native lipids.
There are three type of membrane lipids: 1. Phospholipids: major class of membrane lipids. 2. glycolipids. 3. Cholesterols. Membrane lipids were started with eukaryotes and bacteria.
http://en.wikibooks.org/wiki/Structural_Biochemistry/Lipids/Membrane_Lipids
Types of Membrane Lipids
Lipids are often used as membrane constituents. The three major classes that membrane lipids are divided into are phospholipids, glycolipids, and cholesterol. Lipids are found in eukaryotes and bacteria. Although the lipids in archaea have many features that are related to the membrane formation that is similar with lipids of other organisms, they are still distinct from one another. The membranes of archaea differ in composition in three major ways. Firstly, the nonpolar chains are joined to a glycerol backbone by ether instead of esters, allowing for more resistance to hydrolysis. Second, the alkyl chains are not linear, but branched and make them more resistant to oxidation. The ability of archaeal lipids to resist hydrolysis and oxidation help these types of organisms to withstand the extreme conditions of high temperature, low pH, or high salt concentration. Lastly, the stereochemistry of the central glycerol is inverted. Membrane lipids have an extensive repertoire, but they possess a critical common structural theme in which they are amphipathic molecules, meaning they contain both a hydrophilic and hydrophobic moiety.
Membrane lipids are all closed bodies or boundaries separating substituent parts of the cell. The thickness of membranes is usually between 60 and 100 angstroms. These bodies are constructed from non-covalent assemblies. Their polar heads align with each other and their non-polar hydrocarbon tails align as well. The resulting stability is credited to hydrophobic interaction which proves to be quite stable due to the length of their hydrocarbon tails.
Membrane Lipids
Lipid Vesicles
Lipid vesicles, also known as liposomes, are vesicles that are essentially aqueous vesicles that are surrounded by a circular phospholipid bilayer. Like the other phospholipid structures, they have the hydrocarbon/hydrophobic tails facing inward, away from the aqueous solution, and the hydrophilic heads facing towards the aqueous solution. These vesicles are structures that form enclosed compartments of ions and solutes, and can be utilized to study the permeability of certain membranes, or to transfer these ions or solutes to certain cells found elsewhere.
Liposomes as vesicles can serve various clinical uses. Injecting liposomes containing medicine or DNA (for gene therapy) into patients is a possible method of drug delivery. The liposomes fuse with other cells’ membranes and therefore combine their contents with that of the patient’s cell. This method of drug delivery is less toxic than direct exposure because the liposomes carry the drug directly to cells without any unnecessary intermediate steps.
Because of the hydrophobic interactions among several phospholipids and glycolipids, a certain structure called the lipid bilayer or bimolecular sheet is favored. As mentioned earlier, phospholipids and glycolipids have both hydrophilic and hydrophobic moieties; thus, when several phospholipids or glycolipids come together in an aqueous solution, the hydrophobic tails interact with each other to form a hydrophobic center, while the hydrophilic heads interact with each other forming a hydrophilic coating on each side of the bilayer.
http://upload.wikimedia.org/wikibooks/en/b/ba/Liposome_final%2A.png
http://upload.wikimedia.org/wikibooks/en/f/fa/Membrane_bilayer.jpg

Liposome_

Membrane_bilayer
Evidence Report/Technology Assessment Number 89
Effects of Omega-3 Fatty Acids on Lipids and Glycemic Control in Type II Diabetes and the Metabolic Syndrome and on Inflammatory Bowel Disease, Rheumatoid Arthritis, Renal Disease, Systemic Lupus Erythematosus, and Osteoporosis
Prepared for:
Agency for Healthcare Research and Quality
U.S. Department of Health and Human Services
540 Gaither Road
Rockville, MD 20850
http://www.ahrq.gov
Contract No. 290-02-0003
Chapter 1. Introduction
This report is one of a group of evidence reports prepared by three Agency for Healthcare Research and Quality (AHRQ)-funded Evidence-Based Practice Centers (EPCs) on the role of omega-3 fatty acids (both from food sources and from dietary supplements) in the prevention or treatment of a variety of diseases. These reports were requested and funded by the Office of Dietary Supplements, National Institutes of Health. The three EPCs – the Southern California EPC (SCEPC, based at RAND), the Tufts-New England Medical Center (NEMC) EPC, and the University of Ottawa EPC – have each produced evidence reports. To ensure consistency of approach, the three EPCs collaborated on selected methodological elements, including literature search strategies, rating of evidence, and data table design.
The aim of these reports is to summarize the current evidence on the effects of omega-3 fatty acids on prevention and treatment of cardiovascular diseases, cancer, child and maternal health, eye health, gastrointestinal/renal diseases, asthma, immune- mediated diseases, tissue/organ transplantation, mental health, and neurological diseases and conditions. In addition to informing the research community and the public on the effects of omega-3 fatty acids on various health conditions, it is anticipated that the findings of the reports will also be used to help define the agenda for future research.
This report focuses on the effects of omega-3 fatty acids on immune- mediated diseases, bone metabolism, and gastrointestinal/renal diseases. Subsequent reports from the SCEPC will focus on cancer and neurological diseases and conditions.
This chapter provides a brief review of the current state of knowledge about the metabolism, physiological functions, and sources of omega-3 fatty acids.
The Recognition of Essential Fatty Acids
Dietary fat has long been recognized as an important source of energy for mammals, but in the late 1920s, researchers demonstrated the dietary requirement for particular fatty acids, which came to be called essential fatty acids. It was not until the advent of intravenous feeding, however, that the importance of essential fatty acids was widely accepted: Clinical signs of essential fatty acid deficiency are generally observed only in patients on total parenteral nutrition who received mixtures devoid of essential fatty acids or in those with malabsorption syndromes.
These signs include dermatitis and changes in visual and neural function. Over the past 40 years, an increasing number of physiological functions, such as immunomodulation, have been attributed to the essential fatty acids and their metabolites, and this area of research remains quite active.1, 2
Fatty Acid Nomenclature
The fat found in foods consists largely of a heterogeneous mixture of triacylglycerols (triglycerides)–glycerol molecules that are each combined with three fatty acids. The fatty acids can be divided into two categories, based on chemical properties: saturated fatty acids, which are usually solid at room temperature, and unsaturated fatty acids, which are liquid at room temperature. The term “saturation” refers to a chemical structure in which each carbon atom in the fatty acyl chain is bound to (saturated with) four other atoms, these carbons are linked by single bonds, and no other atoms or molecules can attach; unsaturated fatty acids contain at least one pair of carbon atoms linked by a double bond, which allows the attachment of additional atoms to those carbons (resulting in saturation). Despite their differences in structure, all fats contain approximately the same amount of energy (37 kilojoules/gram, or 9 kilocalories/gram).
The class of unsaturated fatty acids can be further divided into monounsaturated and polyunsaturated fatty acids. Monounsaturated fatty acids (the primary constituents of olive and canola oils) contain only one double bond. Polyunsaturated fatty acids (PUFAs) (the primary constituents of corn, sunflower, flax seed and many other vegetable oils) contain more than one double bond. Fatty acids are often referred to using the number of carbon atoms in the acyl chain, followed by a colon, followed by the number of double bonds in the chain (e.g., 18:1 refers to the 18-carbon monounsaturated fatty acid, oleic acid; 18:3 refers to any 18-carbon PUFA with three double bonds).
PUFAs are further categorized on the basis of the location of their double bonds. An omega or n notation indicates the number of carbon atoms from the methyl end of the acyl chain to the first double bond. Thus, for example, in the omega-3 (n-3) family of PUFAs, the first double bond is 3 carbons from the methyl end of the molecule. The trivial names, chemical names and abbreviations for the omega-3 fatty acids are detailed in Table 1.1. Finally, PUFAs can be categorized according to their chain length. The 18-carbon n-3 and n-6 short-chain PUFAs are precursors to the longer 20- and 22-carbon PUFAs, called long-chain PUFAs (LCPUFAs).
Fatty Acid Metabolism
Mammalian cells can introduce double bonds into all positions on the fatty acid chain except the n-3 and n-6 position. Thus, the short-chain alpha- linolenic acid (ALA, chemical abbreviation: 18:3n-3) and linoleic acid (LA, chemical abbreviation: 18:2n-6) are essential fatty acids.
No other fatty acids found in food are considered ‘essential’ for humans, because they can all be synthesized from the short chain fatty acids.
Following ingestion, ALA and LA can be converted in the liver to the long chain, more unsaturated n-3 and n-6 LCPUFAs by a complex set of synthetic pathways that share several enzymes (Figure 1). LC PUFAs retain the original sites of desaturation (including n-3 or n-6). The omega-6 fatty acid LA is converted to gamma-linolenic acid (GLA, 18:3n-6), an omega- 6 fatty acid that is a positional isomer of ALA. GLA, in turn, can be converted to the longerchain omega-6 fatty acid, arachidonic acid (AA, 20:4n-6). AA is the precursor for certain classes of an important family of hormone- like substances called the eicosanoids (see below).
The omega-3 fatty acid ALA (18:3n-3) can be converted to the long-chain omega-3 fatty acid, eicosapentaenoic acid (EPA; 20:5n-3). EPA can be elongated to docosapentaenoic acid (DPA 22:5n-3), which is further desaturated to docosahexaenoic acid (DHA; 22:6n-3). EPA and DHA are also precursors of several classes of eicosanoids and are known to play several other critical roles, some of which are discussed further below.
The conversion from parent fatty acids into the LC PUFAs – EPA, DHA, and AA – appears to occur slowly in humans. In addition, the regulation of conversion is not well understood, although it is known that ALA and LA compete for entry into the metabolic pathways.
Physiological Functions of EPA and AA
As stated earlier, fatty acids play a variety of physiological roles. The specific biological functions of a fatty acid are determined by the number and position of double bonds and the length of the acyl chain.
Both EPA (20:5n-3) and AA (20:4n-6) are precursors for the formation of a family of hormone- like agents called eicosanoids. Eicosanoids are rudimentary hormones or regulating – molecules that appear to occur in most forms of life. However, unlike endocrine hormones, which travel in the blood stream to exert their effects at distant sites, the eicosanoids are autocrine or paracrine factors, which exert their effects locally – in the cells that synthesize them or adjacent cells. Processes affected include the movement of calcium and other substances into and out of cells, relaxation and contraction of muscles, inhibition and promotion of clotting, regulation of secretions including digestive juices and hormones, and control of fertility, cell division, and growth.3
The eicosanoid family includes subgroups of substances known as prostaglandins, leukotrienes, and thromboxanes, among others. As shown in Figure 1.1, the long-chain omega-6 fatty acid, AA (20:4n-6), is the precursor of a group of eicosanoids that include series-2 prostaglandins and series-4 leukotrienes. The omega-3 fatty acid, EPA (20:5n-3), is the precursor to a group of eicosanoids that includes series-3 prostaglandins and series-5 leukotrienes. The AA-derived series-2 prostaglandins and series-4 leukotrienes are often synthesized in response to some emergency such as injury or stress, whereas the EPA-derived series-3 prostaglandins and series-5 leukotrienes appear to modulate the effects of the series-2 prostaglandins and series-4 leukotrienes (usually on the same target cells). More specifically, the series-3 prostaglandins are formed at a slower rate and work to attenuate the effects of excessive levels of series-2 prostaglandins. Thus, adequate production of the series-3 prostaglandins seems to protect against heart attack and stroke as well as certain inflammatory diseases like arthritis, lupus, and asthma.3.
EPA (22:6 n-3) also affects lipoprotein metabolism and decreases the production of substances – including cytokines, interleukin 1ß (IL-1ß), and tumor necrosis factor a (TNF-a) – that have pro-inflammatory effects (such as stimulation of collagenase synthesis and the expression of adhesion molecules necessary for leukocyte extravasation [movement from the circulatory system into tissues]).2 The mechanism responsible for the suppression of cytokine production by omega-3 LC PUFAs remains unknown, although suppression of omega-6-derived eicosanoid production by omega-3 fatty acids may be involved, because the omega-3 and omega-6 fatty acids compete for a common enzyme in the eicosanoid synthetic pathway, delta-6 desaturase.
DPA (22:5n-3) (the elongation product of EPA) and its metabolite DHA (22:6n-3) are frequently referred to as very long chain n-3 fatty acids (VLCFA). Along with AA, DHA is the major PUFA found in the brain and is thought to be important for brain development and function. Recent research has focused on this role and the effect of supplementing infant formula with DHA (since DHA is naturally present in breast milk but not in formula).
Dietary Sources and Requirements
Both ALA and LA are present in a variety of foods. LA is present in high concentrations in many commonly used oils, including safflower, sunflower, soy, and corn oil. ALA is present in some commonly used oils, including canola and soybean oil, and in some leafy green vegetables. Thus, the major dietary sources of ALA and LA are PUFA-rich vegetable oils. The proportion of LA to ALA as well as the proportion of those PUFAs to others varies considerably by the type of oil. With the exception of flaxseed, canola, and soybean oil, the ratio of LA to ALA in vegetable oils is at least 10 to 1. The ratios of LA to ALA for flaxseed, canola, and soy are approximately 1: 3.5, 2:1, and 8:1, respectively; however, flaxseed oil is not typically consumed in the North American diet. It is estimated that on average in the U.S., LA accounts for 89% of the total PUFAs consumed, and ALA accounts for 9%. Another estimate suggests that Americans consume 10 times more omega-6 than omega-3 fatty acids.4 Table 1.2 shows the proportion of omega 3 fatty acids for a number of foods.
Syntheis and Degradation

Source of Acetyl CoA for Fatty Acid Synthesis

step 1


ACP (acyl carrier protein)

synthesis requires acetyl CoA from citrate shuttle

conversion to fatty acyl co A in cytoplasm

ACP (acyl carrier protein)

FA synthesis not exactly reverse of catabolism

Fatty Acid Synthase

complete FA synthesis

Desaturation

Elongation and Desaturation of Fatty Acids

release of FAs from adiposites

Fatty acid beta oxidation and Krebs cycle produce NAD, NADH, FADH2

ketone bodies

metabolism of ketone bodies

Arachidonoyl-mimicking

Arachidonate pathways

arachidonic acid derivatives

major metabolic intermediates in the pathways for synthesis of cholesterol, fatty acids, and triglycerides

Model for the sterol-mediated proteolytic release of SREBPs from membrane

hormone regulation

insulin receptor and and insulin receptor signaling pathway (IRS)

islet brain glucose signaling

Fish source

omega FAs

Excessive omega 6s

omega 6s

diet and cancer

Patients at risk of FA deficiency

PPAR role
PPAR role

Omega 6_3 pathways

n3 vs n6 PUFAs

triene-teraene ratio

arachidonic acid, leukotrienes, PG and thromboxanes

Cox 2 and cancer

Lipidomics of atherosclerotic plaques

Effect of TPN on EFAD

benefits of omega 3s

food consumption
Read Full Post »



































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}