Pancreatic Islets
Writer and Curator: Larry H. Bernstein, MD, FCAP
Part I. Endocrine Pancreas
The eclipse and rehabilitation of JJR Macleod, Scotland’s insulin laureate
Bliss, M
Journal of the Royal College of Physicians of Edinburgh 2013; 43(4): 1-8
John JR Macleod (1876-1935,) an Aberdonian Scot who had emigrated to North America, shared the 1923 Nobel Prize with Frederick Banting for their discovery of insulin at the University of Toronto in 1921-22. Macleod finished his career as Regius Professor of Physiology at the University of Aberdeen from 1928 to 1935.Macleod’s posthumous reputation was deeply tarnished by the campaigns against him carried out by his fellow laureate, Banting, and by Banting’s student assistant during the insulin research, Charles Best. Banting’s denigration of Macleod was based on their almost total personality conflict; Best’s was based on a hunger for personal recognition. New research indicates how scarred both men were in their obsessions.The rehabilitation of Macleod’s reputation, begun in 1982 with my book, The Discovery of Insulin, has continued in both scholarly and popular circles. By 2012, the ninetieth anniversary of the discovery of insulin, it had become complete both at the University of Toronto and in Canada.
Almost famous: E. Clark Noble, the common thread in the discovery of insulin and vinblastine
Wright Jr., J.R.
CMAJ 2002; 167 (12), pp. 1391-1396
CLARK NOBLE WAS ONE OF THE FIRST members of the University of Toronto insulin team and came within a coin toss of replacing Charles Best as Frederick Banting’s assistant during the summer of 1921. Noble performed important early studies helping to characterize insulin’s action, and he co-authored many of the original papers describing insulin. Because mass production of insulin from livestock pancreata had proved elusive throughout 1922, J.J.R. Macleod hired Noble during the summer of 1923 to help him test and develop a new method for producing commercial quantities of insulin that Macleod believed would revolutionize insulin production. However, commercial production of insulin from fish proved impractical and was dropped by 1924, as methods to produce large quantities of mammalian insulin had improved very rapidly. Noble later played a small but critical role in the most important Canadian contribution to cancer chemotherapy research: the discovery of vinca alkaloids by his brother Robert Laing Noble. Although one might expect that a physician involved in 2 of Canada’s most important medical discoveries during the 20th century must be famous, such was not Clark Noble’s fate. He died without so much as an obituary in CMAJ.
The Pathophysiology of Diabetes and Cardiovascular Disease
Larry H. Bernstein, MD, FCAP, Reviewer and Curator
and Aviva Lev-Ari, PhD, RN, Curator
This is a multipart article that develops the pathological effects of type-2 diabetes in the progression of a systemic inflammatory disease with a development of neuropathy, and fully developing into cardiovascular disease. It also identifies a systemic relationship to the development of chronic obstructive pulmonary disease. In medical school we were taught that syphilis is the great masquerader. The more we learn about diabetes, we learn about its generalized systemic effects.
Part 1. Role of Autonomic Cardiovascular Neuropathy in Pathogenesis
This article is an abstract only of a related publication of the pathogenesis of autonomic neuropathy in diabetics leading to ischemic heart disease.
The role of autonomic cardiovascular neuropathy in pathogenesis of ischemic heart disease in patients with diabetes mellitus
Subjects: Medicine (General), Medicine, Medicine (General), Health Sciences
Authors: Popović-Pejičić Snježana, Todorović-Đilas Ljiljana, Pantelinac Pavle
Publisher: Društvo lekara Vojvodine Srpskog lekarskog društva
Publication: Medicinski Pregled 2006; 59(3-4): Pp 118-123 (2006)
http://dx.doi.org/10.2298/MPNS0604118P
http://www.doiserbia.nb.rs/img/doi/0025-8105/2006/0025-81050604118P.pdf
Keywords: diabetes mellitus, autonomic nervous system diseases, heart diseases, myocardial ischemia, comorbidity
Introduction. Diabetes is strongly associated with macrovascular complications, among which ischemic heart disease is the major cause of mortality. Autonomic neuropathy increases the risk of complications, which calls for an early diagnosis. The aim of this study was to determine both presence and extent of cardiac autonomic neuropathy, in regard to the type of diabetes mellitus, as well as its correlation with coronary disease and major cardiovascular risk factors. Material and methods. We have examined 90 subjects, classified into three groups, with 30 patients each: those with type 1 diabetes, type 2 diabetes and control group of healthy subjects. All patients underwent cardiovascular tests (Valsalva maneuver, deep breathing test, response to standing, blood pressure response to standing sustained, handgrip test), electrocardiogram, treadmill exercise test and filled out a questionnaire referring to major cardiovascular risk factors: smoking, obesity, hypertension, and dyslipidemia. Results. Our results showed that cardiovascular autonomic neuropathy was more frequent in type 2 diabetes, manifesting as autonomic neuropathy. In patients with autonomic neuropathy, regardless of the type of diabetes, the treadmill test was positive, i.e. strongly correlating with coronary disease. In regard to coronary disease risk factors, the most frequent correlation was found for obesity and hypertension. Discussion. Cardiovascular autonomic neuropathy is considered to be the principal cause of arteriosclerosis and coronary disease. Our results showed that the occurrence of cardiovascular autonomic neuropathy increases the risk of coronary disease due to dysfunction of autonomic nervous system. Conclusions. Cardiovascular autonomic neuropathy is a common complication of diabetes that significantly correlates with coronary disease. Early diagnosis of cardiovascular autonomic neuropathy points to increased cardiovascular risk, providing a basis for preventive and therapeutic measures.
Part 2. a longitudinal cohort study of the cardiovascular experience of individuals at high risk for diabetes
Protocol for ADDITION-PRO: a longitudinal cohort study of the cardiovascular experience of individuals at high risk for diabetes recruited from Danish primary care
Subjects: Public aspects of medicine, Medicine, Public Health, Health Sciences
Authors: Johansen Nanna B, Hansen Anne-Louise S, Jensen Troels M, Philipsen Annelotte, Rasmussen Signe S, Jørgensen Marit E, Simmons Rebecca K, Lauritzen Torsten, Sandbæk Annelli, Witte Daniel R
Publisher: BioMed Central Date of publication: 2012 December
Published in: BMC Public Health 2012; 12(1): 1078
ISSN(s): 1471-2458 Added to DOAJ: 2013-03-12 http://dx.doi.org/10.1186/1471-2458-12-1078 http://www.biomedcentral.com/1471-2458/12/1078
Keywords: Diabetes, Cardiovascular disease, Primary care, Complications, Microvascular, Impaired fasting glucose, Impaired glucose intolerance, Aortic stiffness, Physical activity, Body composition.
Background: Screening programmes for type 2 diabetes inevitably find more individuals at high risk for diabetes than people with undiagnosed prevalent disease. While well established guidelines for the treatment of diabetes exist, less is known about treatment or prevention strategies for individuals found at high risk following screening. In order to make better use of the opportunities for primary prevention of diabetes and its complications among this high risk group, it is important to quantify diabetes progression rates and to examine the development of early markers of cardiovascular disease and microvascular diabetic complications. We also require a better understanding of the mechanisms that underlie and drive early changes in cardiometabolic physiology. The ADDITION-PRO study was designed to address these issues among individuals at different levels of diabetes risk recruited from Danish primary care. Methods/Design: ADDITION-PRO is a population-based, longitudinal cohort study of individuals at high risk for diabetes. 16,136 eligible individuals were identified at high risk following participation in a stepwise screening programme in Danish general practice between 2001 and 2006. All individuals with impaired glucose regulation at screening, those who developed diabetes following screening, and a random sub-sample of those at lower levels of diabetes risk were invited to attend a follow-up health assessment in 2009–2011 (n = 4,188), of whom 2,082 (50%) attended. The health assessment included detailed measurement of anthropometry, body composition, biochemistry, physical activity and cardiovascular risk factors including aortic stiffness and central blood pressure. All ADDITION-PRO participants are being followed for incident cardiovascular disease and death. Discussion: The ADDITION-PRO study is designed to increase understanding of cardiovascular risk and its underlying mechanisms among individuals at high risk of diabetes. Key features of this study include (i) a carefully characterised cohort at different levels of diabetes risk; (ii) detailed measurement of cardiovascular and metabolic risk factors; (iii) objective measurement of physical activity behaviour; and (iv) long-term follow-up of hard clinical outcomes including mortality and cardiovascular disease. Results will inform policy recommendations concerning cardiovascular risk reduction and treatment among individuals at high risk for diabetes. The detailed phenotyping of this cohort will also allow a number of research questions concerning early changes in cardiometabolic physiology to be addressed.
Part 3. Clinical significance of cardiovascular dysmetabolic syndrome
This third part is a description of a longitudinal cohort study of individuals at high-risk for diabetes. Unlike the SSA study, the study is not focused on protein-energy malnutrition. This study also addresses the issue of diabetes insulin resistance leading to cardiovascular dysmetabolic syndrome.
Subjects: Diseases of the circulatory (Cardiovascular) system, Specialties of internal medicine, Internal medicine, Medicine, Cardiovascular, Medicine (General), Health Sciences
Authors: Deedwania Prakash C
Publisher: BioMed Central Date of publication: 2002 January
Published in: Trials 2002; 3: 1(2)
ISSN(s): 1468-6708
Added to DOAJ: 2004-06-03
http://dx.doi.org/10.1186/1468-6708-3-2
Full text: http://cvm.controlled-trials.com/content/3/1/2
Keywords: cardiovascular dysmetabolic syndrome, coronary heart disease, diabetes mellitus, hyperinsulinemia, insulin resistance
Although diabetes mellitus is predominantly a metabolic disorder, recent data suggest that it is as much a vascular disorder. Cardiovascular complications are the leading cause of death and disability in patients with diabetes mellitus. A number of recent reports have emphasized that many patients already have atherosclerosis in progression by the time they are diagnosed with clinical evidence of diabetes mellitus. The increased risk of atherosclerosis and cardiovascular complications in diabetic patients is related to the frequently associated dyslipidemia, hypertension, hyperglycemia, hyperinsulinemia, and endothelial dysfunction.
The evolving knowledge regarding the variety of metabolic, hormonal, and hemodynamic abnormalities in patients with diabetes mellitus has led to efforts designed for early identification of individuals at risk of subsequent disease. It has been suggested that insulin resistance, the key abnormality in type II diabetes, often precedes clinical features of diabetes by 5–6 years.
Careful attention to the criteria described for the cardiovascular dysmetabolic syndrome should help identify those at risk at an early stage. The application of nonpharmacologic as well as newer emerging pharmacologic therapies can have beneficial effects in individuals with cardiovascular dysmetabolic syndrome and/or diabetes mellitus by improving insulin sensitivity and related abnormalities. Early identification and implementation of appropriate therapeutic strategies would be necessary to contain the emerging new epidemic of cardiovascular disease related to diabetes.
Part 4. Waist circumference a good indicator of future risk for type 2 diabetes and cardiovascular disease
Subjects: Public aspects of medicine, Medicine, Public Health, Health Sciences Authors: Siren Reijo, Eriksson Johan G, Vanhanen Hannu
Publisher: BioMed Central Date of publication: 2012 August
Published in: BMC Public Health 2012; 12: 1(631)
ISSN(s): 1471-2458
Added to DOAJ: 2013-03-12
http://dx.doi.org/10.1186/1471-2458-12-631
http://www.biomedcentral.com/1471-2458/12/631
Keywords: Waist circumference, Type 2 diabetes, Cardiovascular disease, Middle-aged men.
Background: Abdominal obesity is a more important risk factor than overall obesity in predicting the development of type 2 diabetes and cardiovascular disease. From a preventive and public health point of view it is crucial that risk factors are identified at an early stage, in order to change and modify behaviour and lifestyle in high risk individuals. Methods: Data from a community based study was used to assess the risk for type 2 diabetes, cardiovascular disease and prevalence of metabolic syndrome in middle-aged men. In order to identify those with increased risk for type 2 diabetes and/or cardiovascular disease sensitivity and specificity analysis were performed, including calculation of positive and negative predictive values, and corresponding 95% CI for eleven different cut-off points, with 1 cm intervals (92 to 102 cm), for waist circumference. Results: A waist circumference ≥94 cm in middle-aged men, identified those with increased risk for type 2 diabetes and/or for cardiovascular disease with a sensitivity of 84.4% (95% CI 76.4% to 90.0%), and a specificity of 78.2% (95% CI 68.4% to 85.5%). The positive predictive value was 82.9% (95% CI 74.8% to 88.8%), and negative predictive value 80.0%, respectively (95% CI 70.3% to 87.1%). Conclusions: Measurement of waist circumference in middle-aged men is a reliable test to identify individuals at increased risk for type 2 diabetes and cardiovascular disease. This measurement should be used more frequently in daily practice in primary care in order to identify individuals at risk and when planning health counselling and interventions.
Part 5. Chronic obstructive pulmonary disease and glucose metabolism: a bitter sweet symphony
Subjects: Diseases of the circulatory (Cardiovascular) system, Specialties of internal medicine, Internal medicine, Medicine, Cardiovascular, Medicine (General), Health Sciences
Authors: Mirrakhimov Aibek E
Publisher: BioMed Central
Date of publication: Oct 2012
ISSN(s): 1475-2840
ADDED to DOAJ: 2013-03-12
Published in: Cardiovascular Diabetology 2012; 11(1):132
Journal Language(s): English Country of publication: United Kingdom
http://dx.doi.org:/10.1186/1475-2840-11-132
Full text: http://www.cardiab.com/content/11/1/132
Keywords: COPD, Dysglycemia, Insulin resistance, Obesity, Metabolic syndrome, Diabetes mellitus endothelial dysfunction, Vasculopathy
Chronic obstructive pulmonary disease, metabolic syndrome and diabetes mellitus are common and underdiagnosed medical conditions. It was predicted that chronic obstructive pulmonary disease will be the third leading cause of death worldwide by 2020. The healthcare burden of this disease is even greater if we consider the significant impact of chronic obstructive pulmonary disease on the cardiovascular morbidity and mortality.
Chronic obstructive pulmonary disease may be considered as a novel risk factor for new onset type 2 diabetes mellitus via multiple pathophysiological alterations such as: inflammation and oxidative stress, insulin resistance, weight gain and alterations in metabolism of adipokines.
On the other hand, diabetes may act as an independent factor, negatively affecting pulmonary structure and function. Diabetes is associated with an increased risk of pulmonary infections, disease exacerbations and worsened COPD outcomes. On the top of that, coexistent OSA may increase the risk for type 2 DM in some individuals.
The current scientific data necessitate a greater outlook on chronic obstructive pulmonary disease and chronic obstructive pulmonary disease may be viewed as a risk factor for the new onset type 2 diabetes mellitus. Conversely, both types of diabetes mellitus should be viewed as strong contributing factors for the development of obstructive lung disease. Such approach can potentially improve the outcomes and medical control for both conditions, and, thus, decrease the healthcare burden of these major medical problems.
The Economic Costs of Diabetes: Is It Time for a New Treatment Paradigm?
Commentary: William H. Herman
Diabetes Care Apr 2013; 36: 775-776
In a series of rigorous and exhaustive descriptive cost analyses conducted over the past decade, the American Diabetes Association (ADA) has documented an inexorable increase in the cost of diabetes in the U.S. and its detrimental impact on productivity. For the 2012 study, the ADA estimated that there were 22.3 million Americans diagnosed with diabetes. These patients incurred $306 billion in direct medical costs, more than 1 of 5 dollars spent on medical care in the U.S. The direct medical costs attributed to diabetes, that is, the costs of medical care for people with diabetes in excess of those that would be expected in the absence of diabetes, were $176 billion or approximately 1 of 8 dollars spent on medical care in the U.S. Americans with diagnosed diabetes have annual medical expenditures that are $7,900 or approximately 2.3 times higher than they would be in the absence of diabetes ($13,700 vs. $5,800). Americans with diabetes also incur $69 billion in costs related to absenteeism, reduced productivity while at work or at home, diabetes-related disability, and premature mortality. The increasing economic burden of diabetes is due in large part to the increase in the number of people with diagnosed diabetes.
Randomized controlled clinical trials have demonstrated that intensive glycemic management can delay the onset of microvascular, neuropathic, and cardiovascular complications in people with both type 1 and type 2 diabetes, and that the benefits of early intensive treatment persist over time. Randomized controlled clinical trials have also demonstrated that blood pressure management (target blood pressure 135/80 mmHg) and lipid management using statin medications can delay or prevent the development of adverse cardiovascular outcomes.
The growing economic and societal burden of diabetes as documented by the ADA in this issue of Diabetes Care highlights the urgent need to implement interventions to delay the development of type 2 diabetes. Both intensive lifestyle and pharmacologic interventions are proven effective and cost-effective. Health policy should support their implementation.
Complimentary societal interventions to delay the onset of type 2 diabetes include school-based health promotion programs and interventions that address advertising, food availability and price, the built and workplace environment, and even tax policy. In addition, early aggressive management of glycemia and cardiovascular risk factors must be implemented for persons diagnosed with diabetes. Increasing access to care, including self management education and nutritional counseling, and ensuring access to necessary treatments and supplies are critical, especially in light of the proven value of early intensive treatment in preventing chronic complications. The cost estimates provided by the ADA from 2002, 2007, and 2012 show that the economic and societal burden of diabetes is growing in the U.S. This trend underscores the importance of prevention and interventions to mitigate the complications of diabetes.
Insulin regulates carboxypeptidase E by modulating translation initiation scaffolding protein eIF4G1 in pancreatic β cells
Liew, C.W., Assmann, A., Templin, A.T., (…), Urano, F., Kulkarni, R.N
2014 Proc National Academy of Sciences USA 111 (22), pp. E2319-E2328
Insulin resistance, hyperinsulinemia, and hyperproinsulinemia occur early in the pathogenesis of type 2 diabetes (T2D). Elevated levels of proinsulin and proinsulin intermediates are markers of β-cell dysfunction and are strongly associated with development of T2D in humans. However, the mechanism(s) underlying β-cell dysfunction leading to hyperproinsulinemia is poorly understood. Here, we show that disruption of insulin receptor (IR) expression in β cells has a direct impact on the expression of the convertase enzyme carboxypeptidase E (CPE) by inhibition of the eukaryotic translation initiation factor 4 gamma 1 translation initiation complex scaffolding protein that is mediated by the key transcription factors pancreatic and duodenal homeobox 1 and sterol regulatory element-binding protein 1, together leading to poor proinsulin processing. Reexpression of IR or restoring CPE expression each independently reverses the phenotype. Our results reveal the identity of key players that establish a previously unknown link between insulin signaling, translation initiation, and proinsulin processing, and provide previously unidentified mechanistic insight into the development of hyperproinsulinemia in insulin-resistant states.
Disruption of growth factor receptor-binding protein 10 in the pancreas enhances β-cell proliferation and protects mice from streptozotocin-induced β-cell apoptosis
Zhang, J., Zhang, N., Liu, M., (…), Lu, X.-Y., Liu, F.
2014 Environmental Science and Technology 48 (9), pp. 5179-5186
It has been reported that organotin compounds such as triphenyltin or tributyltin (TBT) induce diabetes and insulin resistance. However, histopathological effects of organotin compounds on the Islets of Langerhans and exocrine pancreas are still unclear. In the present study, male KM mice were orally administered with TBT (0.5, 5, and 50 µg/kg) once every 3 days. The fasting plasma glucose levels significantly elevated, and the levels of serum insulin or glucagon decreased in the animals treated with TBT for 60 days. In animals treated for 45 days, the number of apoptotic cells in the islets and exocrine pancreas was elevated in a dose-dependent manner. The percentage of proliferating (PCNA-positive) cells was decreased in the islets, while it was increased in exocrine acinar cells. Immunohistochemistry analysis showed that estrogen receptor (ER) and androgen receptor (AR) were present in vascular endothelium, ductal cells, and islet cells, but absent from pancreatic exocrine cells. TBT exposure decreased the production of estradiol and triiodothyronine and elevated the concentration of testosterone, and resulted in a decrease of ERβ expression and an elevation of AR in the pancreas measured by Western blotting. The results suggested that TBT inhibited the proliferation and induced the apoptosis of islet cells via multipathways, causing a decrease of relative islet area in the animals treated for 60 days, which could result in a disruption of glucose homeostasis. The different presence of ERs and AR between the islets and exocrine pancreas might be one of reasons causing different effects on cell proliferation
Pancreatic alpha-cell dysfunction contributes to the disruption of glucose homeostasis and compensatory insulin hypersecretion in glucocorticoid-treated rats
Rafacho, A., Gonçalves-Neto, L.M., Santos-Silva, J.C., (…), Nadal, A., Quesada, I.
2014 Journal of Biological Chemistry 289 (9), pp. 6028-604
In α-cells, syntaxin (Syn)-1A interacts with SUR1 to inhibit ATP-sensitive potassium channels (KATP channels). PIP2 binds the Kir6.2 subunit to open KATP channels. PIP2 also modifies Syn-1A clustering in plasma membrane (PM) that may alter Syn-1A actions on PM proteins like SUR1. Here, we assessed whether the actions of PIP 2 on activating KATP channels is contributed by sequestering Syn-1A from binding SUR1. In vitro binding showed that PIP 2 dose-dependently disrupted Syn-1A·SUR1 complexes, corroborated by an in vivo Forster resonance energy transfer assay showing disruption of SUR1-(-EGFP)/Syn-1A(-mCherry) interaction along with increased Syn-1A cluster formation. Electrophysiological studies of rat α-cells, INS-1, and SUR1/Kir6.2-expressing HEK293 cells showed that PIP2 dose-dependent activation of KATP currents was uniformly reduced by Syn-1A. To unequivocally distinguish between PIP2 actions on Syn-1A and Kir6.2, we employed several strategies. First, we showed that PIP 2-insensitive Syn-1A-5RK/A mutant complex with SUR1 could not be disrupted by PIP2, consequently reducing PIP2 activation of KATP channels. Next, Syn-1A·SUR1 complex modulation of KATP channels could be observed at a physiologically low PIP 2 concentration that did not disrupt the Syn-1A·SUR1 complex, compared with higher PIP2 concentrations acting directly on Kir6.2. These effects were specific to PIP2 and not observed with physiologic concentrations of other phospholipids. Finally, depleting endogenous PIP 2 with polyphosphoinositide phosphatase synaptojanin-1, known to disperse Syn-1A clusters, freed Syn-1A from Syn-1A clusters to bind SUR1, causing inhibition of KATP channels that could no longer be further inhibited by exogenous Syn-1A. These results taken together indicate that PIP2 affects islet β-cell KATP channels not only by its actions on Kir6.2 but also by sequestering Syn-1A to modulate Syn-1A availability and its interactions with SUR1 on PM.
Aging and sleep deprivation induce the unfolded protein response in the pancreas: Implications for metabolism
Naidoo, N., Davis, J.G., Zhu, J., (…), Agarwal, B., Baur, J.A.
2014 Aging Cell 13 (1), pp. 131-141
Sleep disruption has detrimental effects on glucose metabolism through pathways that remain poorly defined. Although numerous studies have examined the consequences of sleep deprivation (SD) in the brain, few have directly tested its effects on peripheral organs. We examined several tissues in mice for induction of the unfolded protein response (UPR) following acute SD. In young animals, we found a robust induction of BiP in the pancreas, indicating an active UPR. At baseline, pancreata from aged animals exhibited a marked increase in a pro-apoptotic transcription factor, CHOP, that was amplified by SD, whereas BiP induction was not observed, suggesting a maladaptive response to cellular stress with age. Acute SD increased plasma glucose levels in both young and old animals. However, this change was not overtly related to stress in the pancreatic beta cells, as plasma insulin levels were not lower following acute SD. Accordingly, animals subjected to acute SD remained tolerant to a glucose challenge. In a chronic SD experiment, young mice were found to be sensitized to insulin and have improved glycemic control, whereas aged animals became hyperglycemic and failed to maintain appropriate plasma insulin concentrations. Our results show that both age and SD cooperate to induce the UPR in pancreatic tissue. While changes in insulin secretion are unlikely to play a major role in the acute effects of SD, CHOP induction in pancreatic tissues suggests that chronic SD may contribute to the loss or dysfunction of endocrine cells and that these effects may be exacerbated by normal aging
Bayesian total internal reflection fluorescence correlation spectroscopy reveals hIAPP-induced plasma membrane domain organization in live cells
Guo, S.-M., Bag, N., Mishra, A., Wohland, T., Bathe, M.
2014 Biophysical Journal 106 (1), pp. 190-200
Amyloid fibril deposition of human islet amyloid polypeptide (hIAPP) in pancreatic islet cells is implicated in the pathogenesis of type II diabetes. A growing number of studies suggest that small peptide aggregates are cytotoxic via their interaction with the plasma membrane, which leads to membrane permeabilization or disruption. A recent study using imaging total internal reflection-fluorescence correlation spectroscopy (ITIR-FCS) showed that monomeric hIAPP induced the formation of cellular plasma membrane microdomains containing dense lipids, in addition to the modulation of membrane fluidity. However, the spatial organization of microdomains and their temporal evolution were only partially characterized due to limitations in the conventional analysis and interpretation of imaging FCS datasets. Here, we apply a previously developed Bayesian analysis procedure to ITIR-FCS data to resolve hIAPP-induced microdomain spatial organization and temporal dynamics. Our analysis enables the visualization of the temporal evolution of multiple diffusing species in the spatially heterogeneous cell membrane, lending support to the carpet model for the association mode of hIAPP aggregates with the plasma membrane. The presented Bayesian analysis procedure provides an automated and general approach to unbiased model-based interpretation of imaging FCS data, with broad applicability to resolving the heterogeneous spatial-temporal organization of biological membrane systems.
SMAD2 disruption in mouse pancreatic beta cells leads to islet hyperplasia and impaired insulin secretion due to the attenuation of ATP-sensitive K + channel activity
Nomura, M., Zhu, H.-L., Wang, L., (…), Takayanagi, R., Teramoto, N.
2014 Diabetologia 57 (1), pp. 157-166
Aims/hypothesis: The TGF-β superfamily of ligands provides important signals for the development of pancreas islets. However, it is not yet known whether the TGF-β family signalling pathway is required for essential islet functions in the adult pancreas. Methods: To identify distinct roles for the downstream components of the canonical TGF-β signalling pathway, a Cre-loxP system was used to disrupt SMAD2, an intracellular transducer of TGF-β signals, in pancreatic beta cells (i.e. Smad2-β- knockout [KO] mice). The activity of ATP-sensitive K+ channels (KATP channels) was recorded in mutant beta cells using patch-clamp techniques. Results: The Smad2-β-KO mice exhibited defective insulin secretion in response to glucose and overt diabetes. Interestingly, disruption of SMAD2 in β-cells was associated with a striking islet hyperplasia and increased pancreatic insulin content, together with defective glucose-responsive insulin secretion. The activity of KATP channels was decreased in mutant β-cells. Conclusions/interpretation: These results suggest that in the adult pancreas, TGF-β signalling through SMAD2 is crucial for not only the determination of beta cell mass but also the maintenance of defining features of mature pancreatic beta cells, and that this involves modulation of KATP channel activity.
Disruption of protein-tyrosine phosphatase 1B expression in the pancreas affects β-cell function
Liu, S., Xi, Y., Bettaieb, A., (…), Kulkarni, R.N., Haj, F.G.
2014 Endocrinology 155 (9), pp. 3329-3338
Protein-tyrosine phosphatase 1B (PTP1B) is a physiological regulator of glucose homeostasis and energy balance. However, the role of PTP1B in pancreatic endocrine function remains largely unknown. To investigate the metabolic role of pancreatic PTP1B, we generated mice with pancreas PTP1B deletion (panc-PTP1B KO). Mice were fed regular chow or a high-fat diet, and metabolic parameters, insulin secretion and glucose tolerance were determined. On regular chow, panc-PTP1B KO and control mice exhibited comparable glucose tolerance whereas aged panc-PTP1B KO exhibited mild glucose intolerance. Furthermore, high-fat feeding promoted earlier impairment of glucose tolerance and attenuated glucose-stimulated insulin secretion in panc-PTP1B KO mice. The secretory defect in glucose-stimulated insulin secretion was recapitulated in primary islets ex vivo, suggesting that the effects were likely cell-autonomous. At the molecular level, PTP1B deficiency in vivo enhanced basal and glucose-stimulated tyrosyl phosphorylation of EphA5 in islets. Consistently, PTP1B overexpression in the glucose-responsive MIN6 β-cell line attenuated EphA5 tyrosyl phosphorylation, and substrate trapping identified EphA5 as a PTP1B substrate. In summary, these studies identify a novel role forPTP1Bin pancreatic endocrine function.
Fluorescence recovery after photobleaching reveals regulation and distribution of connexin36 gap junction coupling within mouse islets of Langerhans
Farnsworth, N.L., Hemmati, A., Pozzoli, M., Benninger, R.K.P.
2014 Journal of Physiology 592 (20), pp. 4431-4446
Key points: Gap junctions provide electrical coupling that is critical to the function of pancreatic islets. Disruptions to connexin36 (Cx36) have been suggested to occur in diabetes. No accurate and non-invasive technique has yet been established to quantify changes in Cx36 gap junction coupling in the intact islet. This study developed fluorescence recovery after photobleaching (FRAP) as a non-invasive technique for quantifying Cx36 gap junction coupling in living islets. The study identified treatments that modulate gap junction coupling, confirmed that the cellular distribution of coupling throughout the islet is highly heterogeneous and confirmed that β-cells and β-cells do not form functional Cx36 gap junctions. This technique will enable future studies to examine the regulation of Cx36 gap junction coupling and its disruption in diabetes, and to uncover potential novel therapeutic targets associated with gap junction coupling. The pancreatic islets are central to the maintenance of glucose homeostasis through insulin secretion. Glucose-stimulated insulin secretion is tightly linked to electrical activity in β-cells within the islet. Gap junctions, composed of connexin36 (Cx36), form intercellular channels between β-cells, synchronizing electrical activity and insulin secretion. Loss of gap junction coupling leads to altered insulin secretion dynamics and disrupted glucose homeostasis. Gap junction coupling is known to be disrupted in mouse models of pre-diabetes. Although approaches to measure gap junction coupling have been devised, they either lack cell specificity, suitable quantification of coupling or spatial resolution, or are invasive. The purpose of this study was to develop fluorescence recovery after photobleaching (FRAP) as a technique to accurately and robustly measure gap junction coupling in the islet. The cationic dye Rhodamine 123 was used with FRAP to quantify dye diffusion between islet β-cells as a measure of Cx36 gap junction coupling. Measurements in islets with reduced Cx36 verified the accuracy of this technique in distinguishing between distinct levels of gap junction coupling. Analysis of individual cells revealed that the distribution of coupling across the islet is highly heterogeneous. Analysis of several modulators of gap junction coupling revealed glucose- and cAMP-dependent modulation of gap junction coupling in islets. Finally, FRAP was used to determine cell population specific coupling, where no functional gap junction coupling was observed between β-cells and β-cells in the islet. The results of this study show FRAP to be a robust technique which provides the cellular resolution to quantify the distribution and regulation of Cx36 gap junction coupling in specific cell populations within the islet. Future studies utilizing this technique may elucidate the role of gap junction coupling in the progression of diabetes and identify mechanisms of gap junction regulation for potential therapies.
Glucocorticoid treatment and endocrine pancreas function: Implications for glucose homeostasis, insulin resistance and diabetes
Rafacho, A., Ortsäter, H., Nadal, A., Quesada, I.
2014 Journal of Endocrinology 223 (3), pp. R49-R62
Glucocorticoids (GCs) are broadly prescribed for numerous pathological conditions because of their anti-inflammatory, antiallergic and immunosuppressive effects, among other actions. Nevertheless, GCs can produce undesired diabetogenic side effects through interactions with the regulation of glucose homeostasis. Under conditions of excess and/or long-term treatment, GCs can induce peripheral insulin resistance (IR) by impairing insulin signalling, which results in reduced glucose disposal and augmented endogenous glucose production. In addition, GCs can promote abdominal obesity, elevate plasma fatty acids and triglycerides, and suppress osteocalcin synthesis in bone tissue. In response to GC-induced peripheral IR and in an attempt to maintain normoglycaemia, pancreatic β-cells undergo several morphofunctional adaptations that result in hyperinsulinaemia. Failure of β-cells to compensate for this situation favours glucose homeostasis disruption, which can result in hyperglycaemia, particularly in susceptible individuals. GC treatment does not only alter pancreatic β-cell function but also affect them by their actions that can lead to hyperglucagonaemia, further contributing to glucose homeostasis imbalance and hyperglycaemia. In addition, the release of other islet hormones, such as somatostatin, amylin and ghrelin, is also affected by GC administration. These undesired GC actions merit further consideration for the design of improved GC therapies without diabetogenic effects. In summary, in this review, we consider the implication of GC treatment on peripheral IR, islet function and glucose homeostasis.
β-Cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment
Halban, P.A., Polonsky, K.S., Bowden, D.W., (…), Sussel, L., Weir, G.C.
2014 Journal of Clinical Endocrinology and Metabolism 99 (6), pp. 1983-1992
OBJECTIVE: This article examines the foundation of β-cell failure in type 2 diabetes (T2D) and suggests areas for future research on the underlying mechanisms that may lead to improved prevention and treatment. RESEARCH DESIGN AND METHODS: A group of experts participated in a conference on 14-16 October 2013 cosponsored by the Endocrine Society and the American Diabetes Association. A writing group prepared this summary and recommendations. RESULTS: The writing group based this article on conference presentations, discussion, and debate. Topics covered include genetic predisposition, foundations of β-cell failure, natural history of β-cell failure, and impact of therapeutic interventions. CONCLUSIONS: β-Cell failure is central to the development and progression of T2D. It antedates and predicts diabetes onset and progression, is in part genetically determined, and often can be identified with accuracy even though current tests are cumbersome and not well standardized. Multiple pathways underlie decreased β-cell function and mass, some of which may be shared and may also be a consequence of processes that initially caused dysfunction. Goals for future research include to 1) impact the natural history of β-cell failure; 2) identify and characterize genetic loci for T2D; 3) target β-cell signaling, metabolic, and genetic pathways to improve function/mass; 4) develop alternative sources of β-cells for cell-based therapy; 5) focus on metabolic environment to provide indirect benefit to β-cells; 6) improve understanding of the physiology of responses to bypass surgery; and 7) identify circulating factors and neuronal circuits underlying the axis of communication between the brain and β-cells.
Metabolic effects of sleep disruption, links to obesity and diabetes
Nedeltcheva, A.V., Scheer, F.A.J.L
2014 Current Opinion in Endocrinology, Diabetes and Obesity 21 (4), pp. 293-298
Purpose of Review: To highlight the adverse metabolic effects of sleep disruption and to open ground for research aimed at preventive measures. This area of research is especially relevant given the increasing prevalence of voluntary sleep curtailment, sleep disorders, diabetes, and obesity. Recent Findings: Epidemiological studies have established an association between decreased self-reported sleep duration and an increased incidence of type 2 diabetes (T2D), obesity, and cardiovascular disease. Experimental laboratory studies have demonstrated that decreasing either the amount or quality of sleep decreases insulin sensitivity and decreases glucose tolerance. Experimental sleep restriction also causes physiological and behavioral changes that promote a positive energy balance. Although sleep restriction increases energy expenditure because of increased wakefulness, it can lead to a disproportionate increase in food intake, decrease in physical activity, and weight gain. SUMMARY: Sleep disruption has detrimental effects on metabolic health. These insights may help in the development of new preventive and therapeutic approaches against obesity and T2D based on increasing the quality and/or quantity of sleep. Video abstract http://links.lww.com/COE/A6.
Impaired proteostasis: Role in the pathogenesis of diabetes mellitus
Jaisson, S., Gillery, P.
2014 Diabetologia 57 (8), pp. 1517-1527
In living organisms, proteins are regularly exposed to ‘molecular ageing’, which corresponds to a set of non-enzymatic modifications that progressively cause irreversible damage to proteins. This phenomenon is greatly amplified under pathological conditions, such as diabetes mellitus. For their survival and optimal functioning, cells have to maintain protein homeostasis, also called ‘proteostasis’. This process acts to maintain a high proportion of functional and undamaged proteins. Different mechanisms are involved in proteostasis, among them degradation systems (the main intracellular proteolytic systems being proteasome and lysosomes), folding systems (including molecular chaperones), and enzymatic mechanisms of protein repair. There is growing evidence that the disruption of proteostasis may constitute a determining event in pathophysiology. The aim of this review is to demonstrate how such a dysregulation may be involved in the pathogenesis of diabetes mellitus and in the onset of its long-term complications.
Influence of miRNA in insulin signaling pathway and insulin resistance: Micro-molecules with a major role in type-2 diabetes
Chakraborty, C., Doss, C.G.P., Bandyopadhyay, S., Agoramoorthy, G.
2014 Wiley Interdisciplinary Reviews: RNA 5 (5), pp. 697-712
The prevalence of type-2 diabetes (T2D) is increasing significantly throughout the globe since the last decade. This heterogeneous and multifactorial disease, also known as insulin resistance, is caused by the disruption of the insulin signaling pathway. In this review, we discuss the existence of various miRNAs involved in regulating the main protein cascades in the insulin signaling pathway that affect insulin resistance. The influence of miRNAs (miR-7, miR-124α, miR-9, miR-96, miR-15α/β, miR-34α, miR-195, miR-376, miR-103, miR-107, and miR-146) in insulin secretion and beta (β) cell development has been well discussed. Here, we highlight the role of miRNAs in different significant protein cascades within the insulin signaling pathway such as miR-320, miR-383, miR-181β with IGF-1, and its receptor (IGF1R); miR-128α, miR-96, miR-126 with insulin receptor substrate (IRS) proteins; miR-29, miR-384-5p, miR-1 with phosphatidylinositol 3-kinase (PI3K); miR-143, miR-145, miR-29, miR-383, miR-33α/β miR-21 with AKT/protein kinase B (PKB) and miR-133α/β, miR-223, miR-143 with glucose transporter 4 (GLUT4). Insulin resistance, obesity, and hyperlipidemia (high lipid levels in the blood) have a strong connection with T2D and several miRNAs influence these clinical outcomes such as miR-143, miR-103, and miR-107, miR-29α, and miR-27β. We also corroborate from previous evidence how these interactions are related to insulin resistance and T2D. The insights highlighted in this review will provide a better understanding on the impact of miRNA in the insulin signaling pathway and insulin resistance-associated diagnostics and therapeutics for T2D
Genetic disruption of sod1 gene causes glucose intolerance and impairs β-cell function
Muscogiuri, G., Salmon, A.B., Aguayo-Mazzucato, C., (…), Van Remmen, H., Musi, N.
2013 Diabetes 62 (12), pp. 4201-4207
Oxidative stress has been associated with insulin resistance and type 2 diabetes. However, it is not clear whether oxidative damage is a cause or a consequence of the metabolic abnormalities present in diabetic subjects. The goal of this study was to determine whether inducing oxidative damage through genetic ablation of superoxide dismutase 1 (SOD1) leads to abnormalities in glucose homeostasis. We studied SOD1-null mice and wild-type (WT) littermates. Glucose tolerance was evaluated with intraperitoneal glucose tolerance tests. Peripheral and hepatic insulin sensitivity was quantitated with the euglycemic-hyperinsulinemic clamp. β-Cell function was determined with the hyperglycemic clamp and morphometric analysis of pancreatic islets. Genetic ablation of SOD1 caused glucose intolerance, which was associated with reduced in vivo β-cell insulin secretion and decreased b-cell volume. Peripheral and hepatic insulin sensitivity were not significantly altered in SOD1-null mice. High-fat diet caused glucose intolerance in WT mice but did not further worsen the glucose intolerance observed in standard chow-fed SOD1-null mice. Our findings suggest that oxidative stress per se does not play a major role in the pathogenesis of insulin resistance and demonstrate that oxidative stress caused by SOD1 ablation leads to glucose intolerance secondary to β-cell dysfunction.
VHL-mediated disruption of Sox9 activity compromises β-cell identity and results in diabetes mellitus
Puri, S., Akiyama, H., Hebrok, M.
2013 Genes and Development 27 (23), pp. 2563-2575
Precise functioning of the pancreatic β cell is paramount to whole-body glucose homeostasis, and β-cell dysfunction contributes significantly to diabetes mellitus. Using transgenic mouse models, we demonstrate that deletion of the von Hippel-Lindau (Vhlh) gene (encoding an E3 ubiquitin ligase implicated in, among other functions, oxygen sensing in pancreatic β cells) is deleterious to canonical β-cell gene expression. This triggers erroneous expression of factors normally active in progenitor cells, including effectors of the Notch, Wnt, and Hedgehog signaling cascades. Significantly, an up-regulation of the transcription factor Sox9, normally excluded from functional β cells, occurs upon deletion of Vhlh. Sox9 plays important roles during pancreas development but does not have a described role in the adult β cell. β-Cell-specific ectopic expression of Sox9 results in diabetes mellitus from similar perturbations in β-cell identity. These findings reveal that assaults on the β cell that impact the differentiation state of the cell have clear implications toward our understanding of diabetes mellitus
Second generation antipsychotic-induced type 2 diabetes: A role for the muscarinic M3 receptor
Weston-Green, K., Huang, X.-F., Deng, C.
2013 CNS Drugs 27 (12), pp. 1069-1080
Second generation antipsychotics (SGAs) are widely prescribed to treat various disorders, most notably schizophrenia and bipolar disorder; however, SGAs can cause abnormal glucose metabolism that can lead to insulin-resistance and type 2 diabetes mellitus side-effects by largely unknown mechanisms. This review explores the potential candidature of the acetylcholine (ACh) muscarinic M3 receptor (M3R) as a prime mechanistic and possible therapeutic target of interest in SGA-induced insulin dysregulation. Studies have identified that SGA binding affinity to the M3R is a predictor of diabetes risk; indeed, olanzapine and clozapine, SGAs with the highest clinical incidence of diabetes side-effects, are potent M3R antagonists. Pancreatic M3Rs regulate the glucose-stimulated cholinergic pathway of insulin secretion; their activation on β-cells stimulates insulin secretion, while M3R blockade decreases insulin secretion. Genetic modification of M3Rs causes robust alterations in insulin levels and glucose tolerance in mice. Olanzapine alters M3R density in discrete nuclei of the hypothalamus and caudal brainstem, regions that regulate glucose homeostasis and insulin secretion through vagal innervation of the pancreas. Furthermore, studies have demonstrated a dynamic sensitivity of hypothalamic and brainstem M3Rs to altered glucometabolic status of the body. Therefore, the M3R is in a prime position to influence glucose homeostasis through direct effects on pancreatic β-cells and by potentially altering signaling in the hypothalamus and brainstem. SGA-induced insulin dysregulation may be partly due to blockade of central and peripheral M3Rs, causing an initial disruption to insulin secretion and glucose homeostasis that can progressively lead to insulin resistance and diabetes during chronic treatment.
Islet amyloid polypeptide toxicity and membrane interactions
Cao, P., Abedini, A., Wang, H., (…), Schmidt, A.M., Raleigh, D.P.
2013 Proc National Academy of Sciences USA 110 (48), pp. 19279-19284
Islet amyloid polypeptide (IAPP) is responsible for amyloid formation in type 2 diabetes and contributes to the failure of islet cell transplants, however the mechanisms of IAPP-induced cytotoxicity are not known. Interactions with model anionic membranes are known to catalyze IAPP amyloid formation in vitro. Human IAPP damages anionic membranes, promoting vesicle leakage, but the features that control IAPP-membrane interactions and the connection with cellular toxicity are not clear. Kinetic studies with wild type IAPP and IAPP mutants demonstrate that membrane leakage is induced by prefibrillar IAPP species and continues over the course of amyloid formation, correlating additional membrane disruption with fibril growth. Analyses of a set of designed mutants reveal that membrane leakage does not require the formation of α-sheet or α-helical structures. A His-18 to Arg substitution enhances leakage, whereas replacement of all of the aromatic residues via a triple leucine mutant has no effect. Biophysical measurements in conjunction with cytotoxicity studies show that nonamyloidogenic rat IAPP is as effective as human IAPP at disrupting standard anionic model membranes under conditions where rat IAPP does not induce cellular toxicity. Similar results are obtained with more complex model membranes, including ternary systems that contain cholesterol and are capable of forming lipid rafts. A designed point mutant, I26P-IAPP; a designed double mutant, G24P, I26P-IAPP; a double N-methylated variant; and pramlintide, a US Food and Drug Administration-approved IAPP variant all induce membrane leakage, but are not cytotoxic, showing that there is no one-to-one relationship between disruption of model membranes and induction of cellular toxicity.
Diabetes and beta cell function: From mechanisms to evaluation and clinical implications
Cernea, S., Dobreanu, M.
2013 Biochemia Medica 23 (3), pp. 266-280
Diabetes is a complex, heterogeneous condition that has beta cell dysfunction at its core. Many factors (e.g. hyperglycemia/glucotoxicity, lipotoxicity, autoimmunity, inflammation, adipokines, islet amyloid, incretins and insulin resistance) influence the function of pancreatic beta cells. Chronic hyperglycemia may result in detrimental effects on insulin synthesis/secretion, cell survival and insulin sensitivity through multiple mechanisms: gradual loss of insulin gene expression and other beta-cell specific genes; chronic endoplasmic reticulum stress and oxidative stress; changes in mitochondrial number, morphology and function; disruption in calcium homeostasis. In the presence of hyperglycemia, prolonged exposure to increased free fatty acids result in accumulation of toxic metabolites in the cells (“lipotoxicity”), finally causing decreased insulin gene expression and impairment of insulin secretion. The rest of the factors/mechanisms which impact on the course of the disease are also discusses in detail. The correct assessment of beta cell function requires a concomitant quantification of insulin secretion and insulin sensitivity, because the two variables are closely interrelated. In order to better understand the fundamental pathogenetic mechanisms that contribute to disease development in a certain individual with diabetes, additional markers could be used, apart from those that evaluate beta cell function. The aim of the paper was to overview the relevant mechanisms/factors that influence beta cell function and to discuss the available methods of its assessment. In addition, clinical considerations are made regarding the therapeutical options that have potential protective effects on beta cell function/mass by targeting various underlying factors and mechanisms with a role in disease progression.
The PACAP-regulated gene selenoprotein T is abundantly expressed in mouse and human β-cells and its targeted inactivation impairs glucose tolerance
Prevost, G., Arabo, A., Jian, L., (…), Pattou, F., Anouar, Y
2013 Endocrinology 154 (10), pp. 3796-3806
Selenoproteins are involved in the regulation of redox status, which affects several cellular processes, including cell survival and homeostasis. Considerable interest has arisen recently concerning the role of selenoproteins in the regulation of glucose metabolism. Here, we found that selenoprotein T (SelT), a new thioredoxin-like protein of the endoplasmic reticulum, is present at high levels in human and mouse pancreas as revealed by immunofluorescence and quantitative PCR. Confocal immunohistochemistry studies revealed that SelT is mostly confined to insulin- and somatostatin-producing cells in mouse and human islets. To elucidate the role of SelT in β-cells, we generated, using a Cre-Lox strategy, a conditional pancreatic β-cell SelT-knockout C57BL/6J mice (SelT-insKO) in which SelT gene disruption is under the control of the rat insulin promoter Cre gene. Glucose administration revealed that male SelT-insKO mice display impaired glucose tolerance. Although insulin sensitivity was not modified in the mutant mice, the ratio of glucose to insulin was significantly higher in the SelT-insKO mice compared with wild-type littermates, pointing to a deficit in insulin production/secretion in mutant mice. In addition, morphometric analysis showed that islets from SelT-insKO mice were smaller and that their number was significantly increased compared with islets from their wild-type littermates. Finally, we found that SelT is up-regulated by pituitary adenylate cyclase-activating polypeptide (PACAP) in β-pancreatic cells and that SelT could act by facilitating a feed-forward mechanism to potentiate insulin secretion induced by the neuropeptide. Our findings are the first to show that the PACAP-regulated SelT is localized in pancreatic α- and β-cells and is involved in the control of glucose homeostasis
SIRT1 deacetylates FOXA2 and is critical for Pdx1 transcription and β-cell formation
Wang, R.-H., Xu, X., Kim, H.-S., Xiao, Z., Deng, C.-X.
2013 International Journal of Biological Sciences 9 (9), pp. 934-946
Pancreas duodenum homeobox 1 (PDX1) is essential for pancreas development and β-cell formation; however more studies are needed to clearly illustrate the precise mechanism regarding spatiotemporal regulation of Pdx1 expression during β-cell formation and development. Here, we demonstrate that SIRT1, FOXA2 and a number of proteins form a protein complex on the promoter of the Pdx1 gene. SIRT1 and PDX1 are expressed in the same set of cells during β-cell differentiation and maturation. Pancreas-specific disruption of SIRT1 diminished PDX1 expression and impaired islet development. Consequently, SIRT1 mutant mice develop progressive hyperglycemia, glucose intolerance, and insulin insufficiency, which directly correlate with the extent of SIRT1 deletion. We further show that SIRT1 interacts with and deacetylates FOXA2 on the promoter of the Pdx1gene, and positively regulates its transcription. These results uncover an essential role of SIRT1 in β-cell formation by maintaining expression of PDX1 and its downstream genes, and identify pancreas-specific SIRT1 mutant mice as a relevant model for studying insulin insufficiency.
NOX, NOX who is there? The contribution of NADPH oxidase one to beta cell dysfunction
Taylor-Fishwick, D.A.
2013 Frontiers in Endocrinology 4 (APR), Article 40
Predictions of diabetes prevalence over the next decades warrant the aggressive discovery of new approaches to stop or reverse loss of functional beta cell mass. Beta cells are recognized to have a relatively high sensitivity to reactive oxygen species (ROS) and become dysfunctional under oxidative stress conditions. New discoveries have identified NADPH oxidases in beta cells as contributors to elevated cellular ROS. Reviewed are recent reports that evidence a role for NADPH oxidase-1 (NOX-1) in β-cell dysfunction. NOX-1 is stimulated by inflammatory cytokines that are elevated in diabetes. First, regulation of cytokine-stimulated NOX-1 expression has been linked to inflammatory lipid mediators derived from 12-lipoxygenase activity. For the first time in beta cells these data integrate distinct pathways associated with beta cell dysfunction. Second, regulation of NOX-1 in
β-cells involves feed-forward control linked to elevated ROS and Src-kinase activation. This potentially results in unbridled ROS generation and identifies candidate targets for pharmacologic intervention. Third, consideration is provided of new, first-in-class, selective inhibitors of NOX-1. These compounds could have an important role in assessing a disruption of NOX-1/ROS signaling as a new approach to preserve and protect beta cell mass in diabetes.
Retinoblastoma tumor suppressor protein in pancreatic progenitors controls α- and β-cell fate
Cai, E.P., Wu, X., Schroer, S.A., (…), Zacksenhaus, E., Woo, M.
2013 Proc National Academy of Sciences USA 110 (36), pp. 14723-14728
Pancreatic endocrine cells expand rapidly during embryogenesis by neogenesis and proliferation, but during adulthood, islet cells have a very slow turnover. Disruption of murine retinoblastoma tumor suppressor protein (Rb) in mature pancreatic β-cells has a limited effect on cell proliferation. Here we show that deletion of Rb during embryogenesis in islet progenitors leads to an increase in the neurogenin 3-expressing precursor cell population, which persists in the postnatal period and is associated with increased β-cell mass in adults. In contrast, Rb-deficient islet precursors, through repression of the cell fate factor aristaless related homeobox, result in decreased β-cell mass. The opposing effect on survival of Rb-deficient β- and β-cells was a result of opposing effects on p53 in these cell types. As a consequence, loss of Rb in islet precursors led to a reduced α- to β-cell ratio, leading to improved glucose homeostasis and protection against diabetes.
Statin therapy and new-onset diabetes: Molecular mechanisms and clinical relevance
Banach, M., Malodobra-Mazur, M., Gluba, A., (…), Rysz, J., Dobrzyn, A.
2013 Current Pharmaceutical Design 19 (27), pp. 4904-4912
Despite positive effects on the plasma lipid profile and vascular events, statin use is associated with various side effects. Among these, statins might cause a disruption of a number of regulatory pathways including insulin signaling. This may affect insulin sensitivity, pancreatic beta-cell function and adipokine secretion. The statin-associated risk of new-onset diabetes (NOD) appears to be a dose-dependent class effect. It still remains unclear whether statin treatment is associated with increased risk of NOD in the general population or if there are groups of individuals at particular risk. However, according to the available data it seems that cardiovascular (CV) benefits in high-risk individuals strongly favor statin therapy since it outweighs other risks. Whether statins should be used for primary prevention among patients with a relatively low baseline CV risk is still questionable, however the results of primary prevention trials have shown reductions in mortality in this population. Thus, there is a need for randomized, placebo-controlled statin studies with carefully selected groups of patients and NOD as a key end point in order to resolve queries concerning this issue.
Basement membrane extract preserves islet viability and activity in vitro by up-regulating α3 integrin and its signal
Miao, G., Zhao, Y., Li, Y., (…), Li, J., Wei, J
2013 Pancreas 42 (6), pp. 971-976
OBJECTIVE: Survival of transplanted islets is limited partly because of the disruption of the islet basement membrane (BM) occurring during isolation. We hypothesized that the embedment of BM extract (BME) could induce a viable cell mass and prolong islet functionality before transplantation. METHODS: A special reconstituted BME that solidifies into a gel at 37 C was used to embed isolated islets in this study. The strategy was used to re-establish the interaction between the islets and peri-islet BM. RESULTS: Islets embedded in BME showed lower caspase-3 levels and higher Akt activity than those in suspension. Moreover, we found for the first time that the expression of β3 integrin and focal adhesion kinase (FAK) and FAK activity was up-regulated in islets after BME embedment. The reverse effect was observed on islet apoptosis when islets rescued from a 24-hour suspension culture were embedded in BME for the next 24 hours. In addition, expression of pancreatic duodenal homeobox factor-1 and phospho-extracellular signal-regulated kinase 1/2 was partially preserved, suggesting the positive effect of BME on islet development. CONCLUSIONS: These results indicate that BME embedment of islets can up-regulate the expression of β3 integrin and its signal transduction, which may improve islet viability.
Involvement of the Clock Gene Rev-erb alpha in the Regulation of Glucagon Secretion in Pancreatic Alpha-Cells
Vieira, E., Marroquí, L., Figueroa, A.C., (…), Gomis, R., Quesada, I.
2013 PLoS ONE 8 (7), e6993
Disruption of pancreatic clock genes impairs pancreatic β-cell function, leading to the onset of diabetes. Despite the importance of pancreatic α-cells in the regulation of glucose homeostasis and in diabetes pathophysiology, nothing is known about the role of clock genes in these cells. Here, we identify the clock gene Rev-erbα as a new intracellular regulator of glucagon secretion. Rev-erbα down-regulation by siRNA (60-70% inhibition) in alphaTC1-9 cells inhibited low-glucose induced glucagon secretion (p<0.05) and led to a decrease in key genes of the exocytotic machinery. The Rev-erbα agonist GSK4112 increased glucagon secretion (1.6 fold) and intracellular calcium signals in αTC1-9 cells and mouse primary alpha-cells, whereas the Rev-erbα antagonist SR8278 produced the opposite effect. At 0.5 mM glucose, alphaTC1-9 cells exhibited intrinsic circadian Rev-erbα expression oscillations that were inhibited by 11 mM glucose. In mouse primary alpha-cells, glucose induced similar effects (p<0.001). High glucose inhibited key genes controlled by AMPK such as Nampt, Sirt1 and PGC-1 alpha in alphaTC1-9 cells (p<0.05). AMPK activation by metformin completely reversed the inhibitory effect of glucose on Nampt-Sirt1-PGC-1 alpha and Rev-erb alpha. Nampt inhibition decreased Sirt1, PGC-1 alpha and Rev-erb alpha mRNA expression (p<0.01) and glucagon release (p<0.05). These findings identify Rev-erb alpha as a new intracellular regulator of glucagon secretion via AMPK/Nampt/Sirt1 pathway.
Bmal1 and β-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress- induced β-cell failure in mice
Lee, J., Moulik, M., Fang, Z., (…), Moore, D.D., Yechoor, V.K.
2013 Molecular and Cellular Biology 33 (11), pp. 2327-2338
Circadian disruption has deleterious effects on metabolism. Global deletion of Bmal1, a core clock gene, results in β-cell dysfunction and diabetes. But it is unknown if this is due to loss of cell-autonomous function of Bmal1 in β cells. To address this, we generated mice with β-cell clock disruption by deleting Bmal1 in β cells (β-Bmal1-/-). β-Bmal1-/- mice develop diabetes due to loss of glucose-stimulated insulin secretion (GSIS). This loss of GSIS is due to the accumulation of reactive oxygen species (ROS) and consequent mitochondrial uncoupling, as it is fully rescued by scavenging of the ROS or by inhibition of uncoupling protein 2. The expression of the master antioxidant regulatory factor Nrf2 (nuclear factor erythroid 2-related factor 2) and its targets, Sesn2, Prdx3, Gclc, and Gclm, was decreased in β-Bmal1-/- islets, which may contribute to the observed increase in ROS accumulation. In addition, by chromatin immunoprecipitation experiments, we show that Nrf2 is a direct transcriptional target of Bmal1. Interestingly, simulation of shift work-induced circadian misalignment in mice recapitulates many of the defects seen in Bmal1-deficient islets.
Thus, the cell-autonomous function of Bmal1 is required for normal β-cell function by mitigating oxidative stress and serves to preserve β-cell function in the face of circadian misalignment.
A common landscape for membraneactive peptides
Last, N.B., Schlamadinger, D.E., Miranker, A.D.
2013 Protein Science 22 (7), pp. 870-882
Three families of membrane-active peptides are commonly found in nature and are classified according to their initial apparent activity. Antimicrobial peptides are ancient components of the innate immune system and typically act by disruption of microbial membranes leading to cell death. Amyloid peptides contribute to the pathology of diverse diseases from Alzheimer’s to type II diabetes. Preamyloid states of these peptides can act as toxins by binding to and permeabilizing cellular membranes. Cell-penetrating peptides are natural or engineered short sequences that can spontaneously translocate across a membrane. Despite these differences in classification, many similarities in sequence, structure, and activity suggest that peptides from all three classes act through a small, common set of physical principles. Namely, these peptides alter the Brownian properties of phospholipid bilayers, enhancing the sampling of intrinsic fluctuations that include membrane defects. A complete energy landscape for such systems can be described by the innate membrane properties, differential partition, and the associated kinetics of peptides dividing between surface and defect regions of the bilayer. The goal of this review is to argue that the activities of these membrane-active families of peptides simply represent different facets of what is a shared energy landscape.
Membrane disordering is not sufficient for membrane permeabilization by islet amyloid polypeptide: Studies of IAPP(20-29) fragments
Brender, J.R., Heyl, D.L., Samisetti, S., (…), Pesaru, R.R., Ramamoorthy, A.
2013 Physical Chemistry Chemical Physics 15 (23), pp. 8908-8915
A key factor in the development of type II diabetes is the loss of insulin-producing beta-cells. Human islet amyloid polypeptide protein (human-IAPP) is believed to play a crucial role in this process by forming small aggregates that exhibit toxicity by disrupting the cell membrane. The actual mechanism of membrane disruption is complex and appears to involve an early component before fiber formation and a later component associated with fiber formation on the membrane. By comparing the peptide-lipid interactions derived from solid-state NMR experiments of two IAPP fragments that cause membrane disordering to IAPP derived peptides known to cause significant early membrane permeabilization, we show here that membrane disordering is not likely to be sufficient by itself to cause the early membrane permeabilization observed by IAPP, and may play a lesser role in IAPP membrane disruption than expected.
Downregulation of Fas activity rescues early onset of diabetes in c-KitWv/+ mice
Feng, Z.-C., Riopel, M., Li, J., Donnelly, L., Wang, R.
2013 American Journal of Physiology – Endocrinology and Metabolism 304 (6), pp. E557-E565
c-Kit and its ligand stem cell factor (SCF) are important for β-cell survival and maturation; meanwhile, interactions between the Fas receptor (Fas) and Fas ligand are capable of triggering β-cell apoptosis. Disruption of c-Kit signaling leads to severe loss of β-cell mass and function with upregulation of Fas expression in c-KitWv/++ mouse islets, suggesting that there is a critical balance between c-Kit and Fas activation in β-cells. In the present study, we investigated the interrelationship between c-Kit and Fas activation that mediates β-cell survival and function. We generated double mutant, c-KitWv/++;Faslpr/lpr (Wv-/-), mice to study the physiological and functional role of Fas with respect to β-cell function in c-KitWv/++ mice. Isolated islets from these mice and the INS-1 cell line were used. We observed that islets in c-KitWv/++ mice showed a significant increase in β-cell apoptosis along with upregulated p53 and Fas expression. These results were verified in vitro in INS-1 cells treated with SCF or c-Kit siRNA combined with a p53 inhibitor and Fas siRNA. In vivo, Wv-/- mice displayed improved β-cell function, with significantly enhanced insulin secretion and increased β-cell mass and proliferation compared with Wv+/+ mice. This improvement was associated with downregulation of the Fas-mediated caspase-dependent apoptotic pathway and upregulation of the cFlip/NF-?B pathway. These findings demonstrate that a balance between the c-Kit and Fas signaling pathways is critical in the regulation of β-cell survival and function.
Study Suggests Genetic Susceptibility to T2D May Have Shifted with Human Migration
May 24, 2013 By a GenomeWeb staff reporter
NEW YORK (GenomeWeb News) – The apparent genetic risk for type 2 diabetes seems to vary between human populations from different parts of the world, new research suggests, with populations in Africa and East Asia showing particularly pronounced differences in T2D susceptibility.
A pair of papers appearing online — both led by investigators at Stanford University — outline the approaches and analyses used to reach that conclusion.
For the first study, published in PLOS Genetics, researchers trolled through data on more than 1,000 individuals from around the world who’d been genotyped for the Human Genome Diversity Panel project. Patterns in that data revealed geography or population-related differentiation in the genetic risk associated with certain diseases.
“We demonstrated that differences in genetic risk for multiple diseases go well beyond what is expected by genetic drift,” the study authors noted. “In addition, using a human population phylogenetic tree allowed us to elucidate a substructure of worldwide relationships.”
In the East Asian population, for instance, the team saw diminished genetic risk for both T2D and pancreatic cancer. On the other hand, individuals of African ancestry appeared to be more apt to carry T2D risk alleles, results of the analysis suggest, pointing to possible migration-related shifts in genetic susceptibility to T2D.
For their PLOS Genetics analysis, the researchers used data for 1,043 individuals genotyped for the HGDP to delve into the genetic risk associated with more than 100 diseases, including T2D.
Because the individuals hailed from 51 different populations around the world, the group was able to get a glimpse at relationships between these genetic risk contributors and human migration and population patterns.
From that data, investigators saw at least 11 conditions for which risk variant profiles differed across human populations, researchers reported, including ulcerative colitis, bladder cancer, lupus, and inflammatory bowel disease.
For T2D, that genetic differentiation appeared to correspond with population patterns stemming from human migrations out of Africa and into other parts of the world. For instance, the analysis indicated that genetic risk for T2D dips in East Asian populations but tends to be elevated in populations from Africa — particularly the Mandinka population, which appeared to be at highest genetic risk of T2D.
“East Asians definitely get diabetes,” Stanford University’s Atul Butte, senior author on the study, said in a statement.
Nevertheless, he added, it’s possible that there are population-specific differences in the risk alleles and genetic pathways involved, potentially producing somewhat distinct forms of the disease.
Those involved in the study noted that additional, follow-up research is needed, including whole-genome sequencing analysis, which can offer a look at larger structural variants contributing to disease risk in different populations, for instance.
But if findings from the current analysis hold in future studies, that may ultimately prompt a shift in researchers’ understanding of T2D and the factors contributing to it.
“Other fields of medicine have undergone a radical rethinking in disease taxonomy,” Butte said in a statement, “but this has not happened yet for diabetes, one of the world’s public health menaces.”
“If these are separate diseases at a molecular level, we need to try to understand that,” he added.
A related study in the journal Diabetes Care, also by Stanford’s Butte and his colleagues, touched on the consequences of such genetic differences. That work highlighted apparent clinical differences in T2D-related traits — particularly in insulin resistance and insulin response — in African, East Asian, and Caucasian populations.
More generally, Butte and his colleagues put together a so-called “Genetic Risk World Map” to tie together the information generated from their study of disease risk genetics in the context of human migration. The resource is available online through a Stanford website.
Use of pioglitazone in the treatment of diabetes: effect on cardiovascular risk
Authors: Zou C, Hu H
Published Date: 25 July 2013; 9: 429 – 433
DOI: http://dx.doi.org/10.2147/VHRM.S34421
Pioglitazone and other thiazolidinediones (TZDs) initially showed great promise as unique receptor-mediated oral therapy for type 2 diabetes, but a host of serious side effects, primarily cardiovascular, have limited their utility. It is crucial at this point to perform a risk–benefit analysis to determine what role pioglitazone should play in our current treatment of type 2 diabetes and where the future of this class of drugs is headed. This review provides a comprehensive overview of the present literature. Clinical data currently available indicate that pioglitazone is an effective and generally well-tolerated treatment option for use in patients with type 2 diabetes. Pioglitazone can still reduce adverse cardiovascular risk.
Glucophage, Glucophage XR
In a US double-blind clinical study of GLUCOPHAGE in patients with type 2 diabetes, a total of 141 patients received GLUCOPHAGE therapy (up to 2550 mg per day) and 145 patients received placebo. Adverse reactions reported in greater than 5% of the GLUCOPHAGE patients, and that were more common in GLUCOPHAGE- than placebo-treated patients are reported.
The following adverse reactions were reported in ≥ 1.0% to ≤ 5.0% of GLUCOPHAGE patients and were more commonly reported with GLUCOPHAGE than placebo:
abnormal stools, – myalgia, – lightheaded, – dyspnea,
the following adverse reactions were reported in ≥ 1.0% to ≤ 5.0% of GLUCOPHAGE XR patients and were more commonly reported with GLUCOPHAGE XR than placebo
dizziness, – More common
Metabolic side effects have included lactic acidosis, which is a potentially fatal metabolic complication. The incidence of lactic acidosis has been about 1.5 cases per 10,000 patient years. The risk of lactic acidosis has been particularly high in patients with underlying renal insufficiency. Cases of lactic acidosis occurring in patients with normal renal function have been rarely reported.
- Signs and symptoms of severe acidosis may include bradycardia (lactic acidosis)
- lactic acid concentration, serum electrolytes, blood pH
High-Fructose Corn Syrup Linked to Diabetes
By Brenda Goodman, MA WebMD Health News
Reviewed By Louise Chang, MD
In a study published in the journal Global Health, researchers compared the average availability of high-fructose corn syrup to rates of diabetes in 43 countries.
About half the countries in the study had little or no high-fructose corn syrup in their food supply. In the other 20 countries, high-fructose corn syrup in foods ranged from about a pound a year per person in Germany to about 55 pounds each year per person in the United States.
The researchers found that countries using high-fructose corn syrup had rates of diabetes that were about 20% higher than countries that didn’t mix the sweetener into foods. Those differences remained even after researchers took into account data for differences in body size, population, and wealth.
But couldn’t that mean that people in countries that used more high-fructose corn syrup were just eating more sugar or more total calories?
The researchers say no: There were no overall differences in total sugars or total calories between countries that did and didn’t use high-fructose corn syrup, suggesting that there’s an independent relationship between high-fructose corn syrup and diabetes.
“It raises a lot of questions about fructose,” says researcher Michael I. Goran, PhD, co-director of the Diabetes and Obesity Research Institute at the Keck School of Medicine at the University of Southern California, in Los Angeles. Although the study found an association, it doesn’t establish a cause/effect relationship.
Genetic association of ADIPOQ gene variants with type 2 diabetes, obesity and serum adiponectin levels in south Indian population.
Ramya K; Ayyappa KA; Ghosh S; Mohan V; Radha V
Gene 2013 Dec 15;532(2):253-62 (ISSN: 1879-0038)
OBJECTIVE: To investigate the genetic association of eight variants of the adiponectin gene with type 2 diabetes mellitus (T2DM), obesity and serum adiponectin level in the south Indian population. METHODS: The study comprised of 1100 normal glucose tolerant (NGT) and 1100 type 2 diabetic, unrelated subjects randomly selected from the Chennai Urban Rural Epidemiology Study (CURES), in southern India. Fasting serum adiponectin
levels were measured by radioimmunoassay. The variants were screened by polymerase chain reaction-restriction fragment length polymorphism. Linkage disequilibrium was estimated from the estimates of haplotype frequencies. RESULTS: Of the 8 variants, four SNPs namely, +276 G/T (rs1501299), -4522 C/T (rs822393), -11365 C/G (rs266729), and +712 G/A (rs3774261) were significantly associated with T2DM in our study population. The -3971 A/G (rs822396) and -11391 G/A (rs17300539) SNPs’ association with T2DM diabetes was mediated through obesity (where the association with type 2 diabetes was lost after adjusting for BMI). There was an independent
association of +276 G/T (rs1501299) and -3971 A/G (rs822396) SNPs with generalized obesity and +349 A/G (rs2241767) with central obesity. Four SNPs, -3971 A/G (rs822396), +276 G/T (rs1501299), -4522 C/T (rs822393) and Y111H T/C (rs17366743) were significantly associated with hypoadiponectinemia. The haplotypes GCCATGAAT and AGCGTGGGT conferred lower risk of T2DM in this south Indian population. CONCLUSION: The adiponectin gene variants and haplotype contribute to the genetic risk towards the development of type 2 diabetes, obesity and hypoadiponectinemia in the south Indianpopulation. [ 2013.].
Association of family history of type 2 diabetes mellitus with markers of endothelial dysfunction in South Indian population.
Dhananjayan R; Malati T; Brindha G; Kutala VK
Indian J Biochem Biophys 2013 Apr;50(2):93-8 (ISSN: 0301-1208)
Studies indicate that risk for type 2 diabetes mellitus (T2D) or cardiovascular disease is detectable in childhood, though these disorders may not emerge until adulthood. This study was aimed to assess the markers of endothelial dysfunction in patients with the family history of T2D from South Indian population. A total of 450 subjects were included in the study comprising Group I (n = 200) of T2D, Group II (n = 200) of age- and sex-matched healthy controls, Group III (n = 25) of children of T2D patients and Group IV (n = 25) of children of healthy controls. Results showed that intimal medial thickening (IMT) was significantly higher in T2D patients, compared with control subjects with no family history of diabetes. The fasting plasma glucose, glycated hemoglobin, serum total cholesterol, triglyceride, LDL-cholesterol, apolipoprotein B (ApoB) and high-sensitive C-reactive protein (hsCRP) levels were significantly increased, whereas HDL-cholesterol and serum nitrite levels were significantly decreased in T2D patients. However, children of T2D patients who were not diabetic did not show significant increase in the IMT, as compared to those of healthy controls. In conclusion, the present study demonstrate that IMT was significantly higher in the T2D patients and increased with age and family history. The increased levels of lipids, hsCRP, IMT and decreased nitrite levels might contribute to the risk of endothelial dysfunction in patients with T2D. However, further studies are warranted with other biomarkers of endothelial dysfunction in T2D patients with increased sample size.
Hemoglobin A1c variability as an independent correlate of cardiovascular disease in patients with type 2 diabetes: a cross-sectional analysis of the renal insufficiency and cardiovascular events (RIACE) Italian multicenter study.
Penno G; Solini A; Zoppini G; Orsi E; Fondelli C; Zerbini G; Morano S; and
Renal Insufficiency and Cardiovascular Events (RIACE) Study Group.
Cardiovasc Diabetol 2013;12:98 (ISSN: 1475-2840)
BACKGROUND: Previous reports have clearly indicated a significant relationship between hemoglobin (Hb) A1c change from one visit to the next and microvascular complications, especially nephropathy (albuminuria and albuminuric chronic kidney disease, CKD). In contrast, data on macrovascular disease are less clear. This study was aimed at examining the association of HbA1c variability with cardiovascular disease (CVD) in the large cohort of subjects with type 2 diabetes from the Renal Insufficiency and Cardiovascular Events (RIACE) Italian Multicenter Study. METHODS: Serial (3-5) HbA1c values obtained during the 2-year period preceding recruitment, including that obtained at the enrolment, were available from 8,290 subjects from 9 centers (out of 15,773 patients from 19 centers). Average HbA1c and HbA1c variability were calculated as the intra-individual mean (HbA1c-MEAN) and standard deviation (HbA1c-SD), respectively, of 4.52 0.76 values. Prevalent CVD, total and by vascular bed, was assessed from medical history by recording previous documented major acute events. Diabetic retinopathy (DR) was assessed by dilated fundoscopy. CKD was defined based on albuminuria, as measured by immunonephelometry or immunoturbidimetry, and estimated glomerular filtration rate, as calculated from serum creatinine. RESULTS: HbA1c-MEAN, but not HbA1c-SD, was significantly higher (P <0.0001) in subjects with history of any CVD (n. 2,133, 25.7%) than in those without CVD (n. 6,157, 74.3%). Median and interquartile range were 7.78 (7.04-8.56) and 7.49 (6.81-8.31), respectively, for HbA1c-MEAN, and 0.47 (0.29-0.75) and 0.46 (0.28-0.73), respectively, for HbA1c-SD. Logistic regression analyses showed that HbA1c-MEAN, but not HbA1c-SD (and independent of it), was a significant correlate of any CVD. Similar findings were observed in subjects with versus those without any coronary or cerebrovascular event or myocardial infarction. Conversely, none of these measures were associated with stroke, whereas both correlated with any lower limb vascular event and HbA1c-SD alone with ulceration/gangrene. All these associations were independent of known CVD risk factors and microvascular complications (DR and CKD). CONCLUSIONS: In patients with type 2 diabetes, HbA1c variability has not a major impact on macrovascular complications, at variance with average HbA1c, an opposite finding as compared with microvascular disease, and particularly nephropathy. TRIAL REGISTRATION: ClinicalTrials.Gov NCT00715481.
Genetic association of adiponectin gene polymorphisms (+45T/G and +10211T/G) with type 2 diabetes in North Indians.
Saxena M; Srivastava N; Banerjee M
Diabetes Metab Syndr 2012 Apr-Jun;6(2):65-9 (ISSN: 1878-0334)
Adiponectin (ADIPOQ) is an abundant protein hormone which belongs to a family of so-called adipokines. It is expressed mostly by adipocytes and is an important regulator of lipid and glucose metabolism. It was shown that decreased serum adiponectin concentration indicated insulin resistance and type 2 diabetes (T2DM) with the risk of cardiovascular complications. The fact that adiponectin is an insulin-sensitizing hormone with anti-diabetic, anti-inflammatory and anti-atherogenic properties, we proposed to study the association of ADIPOQ gene polymorphisms in subjects with T2DM. DNA was isolated from venous blood samples, quantified and subjected to Polymerase Chain Reaction-Restriction Fragment Length Polymorphism (PCR-RFLP) using suitable primers and restriction endonucleases. Adiponectin levels were measured in serum using ELISA. The genotypic, allelic and carriage rate frequencies distribution in patients and controls were analyzed by PSAW software (ver. 17.0). Odd ratios (OR) with 95% confidence interval (CI) were determined to describe the strength of association by logistic regression model. Out of the two polymorphisms studied, +10211T/G showed significant association (P=0.042), the ‘G’ allele association being highly significant (P=0.022). Further analysis showed that individuals with ‘GG’ haplotype were at increased risk of T2DM up to 15.5 times [P=0.015, OR (95% CI); 15.558 (1.690-143.174)]. The present study showed that the ‘G’ allele of ADIPOQ gene (+10211T/G) plays a prominent role with respect to T2DM susceptibility in North-Indian population. [Copyright 2012 Diabetes India. Published by Elsevier Ltd. All rights reserved.].
Association of RAGE gene polymorphism with vascular complications in Indian type 2 diabetes mellitus patients [In Process Citation]
Tripathi AK; Chawla D; Bansal S; Banerjee BD; Madhu SV; Kalra OP
Diabetes Res Clin Pract 2014 Mar;103(3):474-81 (ISSN: 1872-8227)
AIMS: The study was designed to evaluate the association of -374T/A and -429T/C polymorphism in the promoter region and Gly82Ser polymorphism in exon 3 region of RAGE gene with diabetic vascular complications in Indian population. METHODS: We screened 603 subjects which includes 176 healthy controls, 140 type 2 diabetes mellitus (T2DM) subjects without any vascular complications (DM), 152 T2DM subjects with microvascular complications (DM-micro) and 135 T2DM subjects with macrovascular complications (DM-macro) for -374T/A, -429T/C and Gly82Ser polymorphisms of RAGE gene. DNA isolated from the enrolled subjects were genotyped by PCR-RFLP. Logistic regression analysis was used to evaluate the association of single nucleotide polymorphisms (SNPs). RESULTS: The -429 T/C and Gly82Ser RAGE polymorphisms were found to be significantly associated with the development of macrovascular and microvascular complications, respectively, in T2DM subjects while -374A allele showed reduced risk towards the development of macrovascular complications. Further, -429T/C, -374T/A and Gly82Ser haplotype analysis revealed association of CTG haplotype with development of macrovascular complications while haplotype TAG was observed to be significantly protective towards development of macrovascular complications in T2DM subjects (OR=0.617, p=0.0202). CONCLUSIONS: Our data indicates significant association of RAGE SNPs and haplotypes with vascular complications in North Indian T2DM subjects.
Clinical profile and complications of childhood- and adolescent-onset type 2 diabetes seen at a diabetes center in south India.
Amutha A; Datta M; Unnikrishnan R; Anjana RM; Mohan V
Diabetes Technol Ther 2012 Jun;14(6):497-504 (ISSN: 1557-8593)
OBJECTIVE: This study describes the clinical characteristics of childhood- and adolescent-onset type 2 diabetes mellitus (CAT2DM) seen at a diabetes center in southern India. RESEARCH DESIGN AND METHODS: Between January 1992 and December 2009, 368 CAT2DM patients were registered. Anthropometric measurements were done using standardized techniques. Biochemical investigations included C-peptide measurements and glutamic acid decarboxylase antibody assay wherever feasible. Retinopathy was diagnosed by retinal photography; microalbuminuria, if urinary albumin excretion was between 30 and 299vmg/1/4g of creatinine; nephropathy, if urinary albumin excretion was (yen)300vmg/1/4g; and neuropathy, if vibration perception threshold on biothesiometry was (yen)20vV. RESULTS: The proportion of CAT2DM patients, expressed as percentage of total patients registered at our center, rose from 0.01% in 1992 to 0.35% in 2009 (P <0.001). Among the 368 cases of CAT2DM, 96 (26%) were diagnosed before the age of 15 years. The mean age at first visit and age at diagnosis of the CAT2DM subjects were 22.29.7 and 16.12.5 years, respectively. Using World Health Organization growth reference charts, 56% of boys and 50.4% of girls were > 85(th) percentile of body mass index for age. Prevalence rates of retinopathy, microalbuminuria, nephropathy, and neuropathy were 26.7%, 14.7%, 8.4%, and 14.2%, respectively. Regression analysis revealed female gender, body mass index > 85(th) percentile, parental history of diabetes, serum cholesterol, and blood pressure to be associated with earlier age at onset of CAT2DM. CONCLUSIONS: CAT2DM appears to be increasing in urban India, and the prevalence of microvascular complications is high. Female predominance is seen at younger ages.
Variants of the adiponectin gene and diabetic microvascular complications in patients with type 2 diabetes.
Choe EY; Wang HJ; Kwon O; Kim KJ; Kim BS; Lee BW; Ahn CW; et al.
Metabolism 2013 May;62(5):677-85 (ISSN: 1532-8600)
OBJECTIVE: The aim of this study was to examine the association between common polymorphisms of the adiponectin gene (ADIPOQ) and microvascular complications in patients with type 2 diabetes mellitus (T2DM). RESEARCH DESIGN AND METHODS: Rs2241766 and rs1501299 of ADIPOQ were genotyped in 708 patients with T2DM. Fundus photography, nerve conducting velocity, and urine analysis were performed to check for the presence of microvascular complications including diabetic nephropathy, retinopathy and neuropathy. RESULTS: The prevalence of diabetic nephropathy tended to be different according to rs2241766 genotype (p=0.057) and the GG genotype of rs2241766 was associated with diabetic nephropathy [urine albumin/creatinine ratio (UACR) greater than 30 mg/g] after adjusting for age, sex, body mass index, duration of diabetes, HDL-cholesterol, smoking status, and blood pressure (odds ratio=1.96; 95% confidence interval=1.01-3.82, p=0.049). Also, the G allele of rs2241766 demonstrated a trend to be associated with an increase in UACR (p=0.087). Rs2241766 genotype was not associated with diabetic retinopathy (p=0.955) and neuropathy (p=0.104) or any diabetic microvascular complications (p=0.104). There was no significant association between the rs1501299 genotype of ADIPOQ and the prevalence of diabetic retinopathy and neuropathy or any diabetic microvascular complications even after adjustment. CONCLUSION: These data suggest that the GG genotype at rs2241766 is implicated in the pathogenesis of risk for diabetic nephropathy defined as UACR greater than 30 mg/day in patients with T2DM. [Copyright 2013 Elsevier Inc. All rights reserved.].
The prevalence of presarcopenia in Asian Indian individuals with and without type 2 diabetes.
Anbalagan VP; Venkataraman V; Pradeepa R; Deepa M; Anjana RM; Mohan V
Diabetes Technol Ther 2013 Sep;15(9):768-75 (ISSN: 1557-8593)
OBJECTIVE: This study compared the skeletal muscle mass and prevalence of presarcopenia between Asian Indian individuals with and without type 2 diabetes. SUBJECTS AND METHODS: Participants with type 2 diabetes (n=76) and age- and sex-matched controls without diabetes (n=76) were drawn from the Chennai Urban Rural Epidemiological Study (CURES), which was carried out on a representative sample of Chennai City in South India. Skeletal muscle mass was estimated by dual-energy X-ray absorptiometry, and skeletal muscle mass index (SMI) was calculated by dividing the appendicular skeletal muscle mass by the square of the individual’s height in meters and expressed as kg/m. Presarcopenia was defined as an SMI of 7.26 kg/m2 for males and 5.5 kg/m2 for females. Biochemical and anthropometric measurements were done using standardized procedures. RESULTS: The 152 participants included 68 women (44.7%). Mean age was 449 years (range, 28-67 years), and the mean body mass index (BMI) was 25.73.8 kg/m2. The prevalence rates of presarcopenia among individuals with and without diabetes were 39.5% and 15.8%, respectively (P=0.001). The mean SMI values were significantly lower in those with diabetes (6.841.02 kg/m2 compared with participants without diabetes (7.281.01 kg/m2) (P=0.009). SMI showed a positive correlation with BMI and waist circumference but a negative correlation with age, fasting plasma glucose, glycated hemoglobin, and low-density lipoprotien cholesterol in the total study population. Logistic regression analysis showed that diabetes was independently associated with presarcopenia (P=0.001). CONCLUSIONS: Prevalence of presarcopenia is higher among Asian Indian subjects with type 2 diabetes compared with age- and sex-matched participants without diabetes.
Increased risk of type 2 diabetes with ascending social class in urban South Indians is explained by obesity: The Chennai urban rural epidemiology study (CURES-116).
Skar M; Villumsen AB; Christensen DL; Petersen JH; Deepa M; Anjana RM; et al.
Indian J Endocrinol Metab 2013 Nov;17(6):1084-9 (ISSN: 2230-8210)
AIM: The aim of this study is to determine the factors responsible for differences in the prevalence of diabetes mellitus (DM) in subjects of different social class in an urban South Indian population. MATERIALS AND METHODS: Analyses were based on the cross-sectional data from the Chennai Urban Rural Epidemiology Study of 1989 individuals, aged (yen)20 years. Entered in the analyses were information obtained by self-report on (1) household income; (2) family history of diabetes; (3) physical activity; (4) smoking status; (5) alcohol consumption. Biochemical, clinical and anthropometrical measurements were performed and included in the analyses. Social class was classified based on income as low (Rs. <2000) intermediate (Rs. 2000-5000`) and high (Rs. 5000-20000). RESULTS: The prevalence rates of DM were 12.0%, 18.4% and 21.7% in low, intermediate and high social class, respectively (P < 0.001). A significant increase in the risk of diabetes was found with ascending social class (Intermediate class: Odds ratio [OR], 1.7 [confidence interval [CI], 1.2-2.3]; High class: OR, 2.0 [CI-1.4-2.9]). The multivariable adjusted logistic regression analysis revealed that the effect of social class on the risk of diabetes remained significant (P = 0.016) when age, family history of diabetesand blood pressure were included. However, with the inclusion of abdominal obesity in the model, the significant effect of social class disappeared (P = 0.087). CONCLUSION: An increased prevalence of DM was found in the higher social class in this urban South Indian population, which is explained by obesity.
Prevalence of inflammatory markers (high-sensitivity C-reactive protein, nuclear factor-(ordM)B, and adiponectin) in Indian patients with type 2 diabetes mellitus with and without macrovascular complications.
Misra DP; Das S; Sahu PK
Metab Syndr Relat Disord 2012 Jun;10(3):209-13 (ISSN: 1557-8518)
BACKGROUND: Atherosclerosis is more prevalent in subjects with diabetes mellitus. Recent evidence suggests that diabetic atherosclerosis is not simply a disease of hyperlipidemia, but is also an inflammatory disorder. Our aim was to study the prevalence of inflammatory markers such as high-sensitivity C-reactive protein (hsCRP), adiponectin, and nuclear factor-(ordM)B (NF-(ordM)B) expression, in peripheral blood mononuclear cells in Indian patients with type 2 diabetes mellitus (T2DM) with and without macrovascular disease (MVD). METHODS: A total of 29 consecutive cases of T2DM with proven MVD (group A), 28 matched cases without MVD (group B), and 14 healthy controls (group C) were evaluated for the clinical parameters fasting blood glucose (FBG), 2-h postprandial blood glucose (PPBG), glycosylated hemoglobin (HbA1c), lipid profile, and the above-mentioned inflammatory markers. RESULTS: Diabetic subjects with T2DM had higher hsCRP and NF-(ordM)B expression and lower values of adiponectin compared to healthy controls. Group A had significantly higher serum hsCRP than group B (P=0.0001) despite comparable values of BMI, FBG, 2-h PPBG, HbA1c, and lipid parameters. Group A had significantly higher serum hsCRP and NF-(ordM)B expression and significantly lower levels of adiponectin than group C (P=0.0001, 0.007, and 0.02, respectively). In Group A, serum adiponectin negatively correlated with NF-(ordM)B expression. In Group B, adiponectin values correlated negatively with both FBG and 2-h PPBG. CONCLUSIONS: Indian subjects with T2DM with or without MVD had higher hsCRP and lower adiponectin values as compared to healthy controls, whereas hsCRP was significantly higher in those with MVD, suggesting that our patients with T2DM were in a proinflammatory state.
Adiponectin G276T gene polymorphism is associated with cardiovascular disease in Japanese patients with type 2 diabetes.
Katakami N; Kaneto H; Matsuoka TA; Takahara M; Maeda N; Shimizu I; et al.
Atherosclerosis 2012 Feb;220(2):437-42 (ISSN: 1879-1484)
OBJECTIVE: Adiponectin has anti-atherogenic properties and reduced serum adiponectin levels are associated with cardiovascular disease (CVD). In this study, we examined the relationship between CVD and adiponectin (ADIPOQ) gene G276T polymorphism that is associated with serum adiponectin level in a large cohort of type 2 diabetic patients. RESEARCH DESIGN AND METHODS: We enrolled 2637 Japanese type 2 diabetic subjects (males, 61.1%; age, 54.97.9 years old), determined their genotypes regarding ADIPOQ G276T polymorphisms, and evaluated the association between this polymorphism and the prevalence of CVD (myocardial infarction and/or cerebral infarction). RESULTS: The prevalence of CVD tended to be higher as the number of G alleles increased [GG (9.5%), GT (6.8%), TT (5.6%), p value for trend=0.0059] and was significantly higher in the subjects with GG genotype compared to those with GT or TT genotype (9.5% vs. 6.6%, p=0.0060). Multiple logistic regression analyses revealed that the number of G alleles (Odds ratio (OR)=1.49 with 95%CI 1.09-2.05, p=0.0125) and GG genotype (OR=1.66 with 95%CI 1.13-2.43, p=0.0098) were significantly associated with CVD even after adjustment for conventional risk factors. Interestingly, the presence of obesity further and significantly increased the risk of CVD in the subjects with GG genotype (OR=1.67 with 95%CI 1.14-2.44, p=0.0090) but not in the subjects with TT or GT genotype (OR=1.17 with 95%CI 0.73-1.89, NS). CONCLUSIONS: It is likely that the G allele of the ADIPOQ G276T polymorphism is a susceptibility allele for CVD in Japanese type 2 diabetic patients, especially when they accompany obesity. [Copyright 2011 Elsevier Ireland Ltd. All rights reserved.].
A comprehensive investigation of variants in genes encoding adiponectin (ADIPOQ) and its receptors (ADIPOR1/R2), and their association with serum adiponectin, type 2 diabetes, insulin resistance and the metabolic syndrome.
Peters KE; Beilby J; Cadby G; Warrington NM; Bruce DG; Davis WA; et al.
BMC Med Genet 2013;14:15 (ISSN: 1471-2350)
BACKGROUND: Low levels of serum adiponectin have been linked to central obesity, insulin resistance, metabolic syndrome, and type 2 diabetes. Variants in ADIPOQ, the gene encoding adiponectin, have been shown to influence serum adiponectin concentration, and along with variants in theadiponectin receptors (ADIPOR1 and ADIPOR2) have been implicated in metabolic syndrome and type 2 diabetes. This study aimed to comprehensively investigate the association of common variants in ADIPOQ, ADIPOR1 and ADIPOR2 with serum adiponectin and insulin resistance syndromes in a large cohort of European-Australian individuals. METHODS: Sixty-four tagging single nucleotide polymorphisms in ADIPOQ, ADIPOR1 and ADIPOR2 were genotyped in two general population cohorts consisting of 2,355 subjects, and one cohort of 967 subjects with type 2 diabetes. The association of tagSNPs with outcomes were evaluated using linear or logistic modelling. Meta-analysis of the three cohorts was performed by random-effects modelling. RESULTS: Meta-analysis revealed nine genotyped tagSNPs in ADIPOQ significantly associated with serum adiponectinacross all cohorts after adjustment for age, gender and BMI, including rs10937273, rs12637534, rs1648707, rs16861209, rs822395, rs17366568, rs3774261, rs6444175 and rs17373414. The results of haplotype-based analyses were also consistent. Overall, the variants in the ADIPOQ gene explained <5% of the variance in serum adiponectin concentration. None of the ADIPOR1/R2 tagSNPs were associated with serum adiponectin. There was no association between any of the genetic variants and insulin resistance or metabolic syndrome. A multi-SNP genotypic risk score for ADIPOQ alleles revealed an association with 3 independent SNPs, rs12637534, rs16861209, rs17366568 and type 2 diabetes after adjusting foradiponectin levels (OR=0.86, 95% CI=(0.75, 0.99), P=0.0134). CONCLUSIONS: Genetic variation in ADIPOQ, but not its receptors, was associated with altered serum adiponectin. However, genetic variation in ADIPOQ and its receptors does not appear to contribute to the risk of insulin resistance or metabolic syndrome but did for type 2
diabetes in a European-Australian population.
Autophagy: Protection Against T2D?
By Salynn Boyles, Contributing Writer,
MedPage Today Published: Jul 27, 2014 | Updated: Jul 28, 2014
The cellular regulatory system known as autophagy appeared to play a key role in preventing type 2 diabetes by protecting insulin-secreting beta cells from the accumulation of toxic amylin oligomers, researchers reported.
Findings from three independent research teams, published online in the Journal of Clinical Investigation, suggested autophagy boosting therapies could prove to be a novel approach for type 2 diabetes prevention.
Autophagy — derived from the Greek words for “self” (auto) and “to eat” (phagein) — describes the controlled disposal of damaged organelles within the cell. This cell-cleaning process is increasingly being recognized as a potential protective mechanism against many diseases, including Parkinson’s disease, amyotrophic lateral sclerosis, and Alzheimer’s disease.
Earlier studies found autophagy to be important for normal beta-cell functionand autophage activity to be increased in beta cells from patients with type 2 diabetes.
The studies provide new insight into how beta cells are normally protected against amylin (IAPP) toxic oligomers, wrote Dhananjay Gupta, PhD, and Jack L. Leahy, MD, of the University of Vermont in Burlington in an accompanying editorial.
Action Points:
- Autophagy appeared to play a key role in preventing type 2 diabetes by protecting insulin-secreting beta cells from the accumulation of toxic amylin oligomers.
- Note that the studies suggest that autophagy — controlled disposal of damaged organelles within the cell — boosting therapies could prove to be a novel approach for type 2 diabetes prevention.
Autophagy – continued
IAPP: Co-Expressed With Insulin
Type 2 diabetes is characterized by loss of beta-cell, beta-cell dysfunction, and increased beta-cell apoptosis. Islet pathology in type 2 diabetes is also characterized by accumulation of extracellular islet amyloid derived from islet amyloid polypeptide (IAPP).
“IAPP is a 37-amino acid protein co-expressed and secreted by pancreatic [beta cells] along with insulin,” wrote Peter Butler, MD, from the University of California Los Angeles, and colleagues. “While the extracellular islet amyloid is relatively inert, intracellular membrane-permeant toxic oligomers of IAPP that form within [beta cells in type 2 diabetes] are thought to induce [beta-cell dysfunction and apoptosis].”
In contrast to the human form of IAPP (h-IAPP), which forms toxic membrane-permeant oligomers, the rodent form of IAPP (r-IAPP) is nonamyloidogenic and nontoxic due to proline substitutions. Transgenic expression of h-IAPP in [beta cells] of rodents may lead to development of diabetes as a consequence of [beta-cell] apoptosis and formation of intracellular IAPP oligomers comparable to those found in humans with type 2 diabetes.
In earlier in vitro studies, the authors reported that enhancement of autophagy was protective while attenuated lysosomal degradation rendered beta cells more vulnerable to h-IAPP-induced apoptosis.
In the current study, the researchers determined that beta-cell IAPP content is regulated by autophagy through p62-dependent lysosomal degradation.
“Induction of high levels of human IAPP in mouse [beta cells] resulted in accumulation of this amyloidogenic protein as relatively inert fibrils with cytosolic p62-positive inclusions, which temporarily averts formation of toxic oligomers,” they wrote.
Mice hemizygous for transgenic expression of human IAPP did not develop diabetes. But the loss of beta cell-specific autophagy in the mice induced diabetes as a result of the accumulation of toxic human IAPP oligomers and loss of beta-cell mass, the researchers noted.
“In human IAPP-expressing mice that lack [beta-cell] autophagy, increased oxidative damage and loss of an antioxidant-protective pathway appeared to contribute to increased [beta- cell] apoptosis,” they wrote. “These findings indicate that autophagy/lysosomal degradation defends [beta cells] against proteotoxicity induced by oligomerization-prone human IAPP.”
‘Enhance the Toxic Potential of h-IAPP’
In a separate study, Yoshio Fujitani, PhD, of Juntendo University, Tokyo, and colleagues, examined the pathogenic role of human-IAPP and its relation to autophagy in h-IAPP-knock-in mice.
In animals fed a standard diet, h-IAPP had no toxic effects on beta-cell function. However, h-IAPP-knock-in mice did not exhibit a high-fat diet-induced compensatory increase in beta-cell mass, which was due to limited beta-cell proliferation and enhanced beta-cell apoptosis, the researchers wrote.
Expression of h-IAPP in mice with a beta-cell-specific autophagy defect resulted in substantial deterioration of glucose tolerance and dispersed cytoplasmic expression of p62-associated toxic oligomers, which were otherwise sequestrated within p62-positive inclusions.
“Together, our results indicate that increased insulin resistance in combination with reduced autophagy may enhance the toxic potential of h-IAPP and enhance [beta-cell] dysfunction and progression of type 2 diabetes,” the researchers noted.
Autophagy Enhancers
In the third paper, Myung-Shik Lee, MD, PhD, of the Sungkyunkwan University School of Medicine in Seoul, and colleagues, studied transgenic mice with beta cell-specific expression of h-IAPP to evaluate the contribution of autophagy in type 2 diabetes-associated accumulation of h-IAPP.
In mice with beta-cell-specific expression of h-IAPP, a deficiency in autophagy resulted in development of overt diabetes, which was not observed in mice expressing h-IAPP alone or lacking autophagy alone. Lack of autophagy in h-IAPP-expressing animals also resulted in h-IAPP oligomer and amyloid accumulation in pancreatic islets, leading to increased death and decreased mass of beta cells.
“Expression of h-IAPP in purified monkey islet cells or a murine [beta cell] line resulted in pro-h-IAPP dimer formation, while dimer formation was absent or reduced dramatically in cells expressing either nonamyloidogenic mouse-IAPP or nonfibrillar mutant h-IAPP,” the researchers wrote. “In autophagy-deficient cells, accumulation of pro-h-IAPP dimers increased markedly, and pro-h-IAPP trimers were detected in the detergent-insoluble fraction.”
Enhancement of autophagy also improved the metabolic profile of h-IAPP-expressing mice fed a high-fat diet.
“These results suggest that autophagy promotes clearance of amyloidogenic h-IAPP, autophagy deficiency exacerbates pathogenesis of human [type 2 diabetes], and autophagy enhancers have therapeutic potential for islet amyloid accumulation-associated human [type 2 diabetes],” the researchers concluded.
Building on Previous Work
Gupta and Leahy noted that all three research teams generated human IAPP-expressing mice with a beta-cell-specific deficiency of the autophagy indicator ATG7, and all three found that autophagy-dependent packaging of monomeric or unprocessed IAPP dimers or trimers into p62-associated vacuoles allowed autophagosomes to dispose of these molecules, keeping them nontoxic.
Each team showed the activity of this detoxification system to be increased when a high-fat diet was fed to the mice with hyperexpression of h-IAPP.
The studies build on previous work and the findings that don’t discern – “how and when during the course of type 2 diabetes development this autophagy-dependent detoxification system might be overcome, allowing toxic IAPP oligomers to form.”
“There are many additional mechanisms that have been proposed for [beta-cell] dysfunction and death in type 2 diabetes, including ER stress, oxidative stress, and autoimmune damage, all of which have been linked to IAPP toxicity,” they wrote. “While it is tempting to try and connect the dots through a single, unified mechanism, all of these proposed pathways of [beta-cell] dysfunction have been recapitulated and extensively studied in rodent models of diabetogenic systems, such as high-fat feeding and partial pancreatectomy, or through genetic modification.”
Given the absence of rodent IAPP oligomerization, these mechanisms of reduced beta-cell function clearly do not require IAPP activation, they noted.
These papers have implications for the study of target therapies for type 2 diabetes based on the common link to T2D and IAPP oligomerization.
“Patients with type 2 diabetes have an increased risk of Alzheimer’s disease, suggesting a common pathogenesis,” they wrote. Disordered neuronal autophagy, described in Alzheimer’s, with alteration in the clearance of amyloidogenic proteins may be a tie between these two diseases
They concluded that acceptance of the hypothesis that IAPP oligomer formation and subsequent plaque development are a major cause of type 2 diabetes will require a better understanding of
- when this mechanism is activated and
- what modulates its destructive potential.
“These current studies may shift the focus away from
- the biology of how IAPP oligomers cause [beta cell] destruction
- to probing for defects within the protective system against the formation of toxic IAPP oligomers,” they wrote.
Part 2. Pancreatic Islet Cell Dysfunction
N-terminal fragment of probrain natriuretic peptide is associated with diabetes microvascular complications in type 2 diabetes
Kumiko Hamano, Ikue Nakadaira, Jun Suzuki, Megumi Gonai
Vascular Health and Risk Management 2014:10 585–589
http://dx.doi.org/10.2147/VHRM.S67753
Aim/introduction: Circulating levels of N-terminal fragment of probrain natriuretic peptide (NT-proBNP) are established as a risk factor for cardiovascular disease and mortality in patients with diabetes, as well as in the general population. We sought to examine the possibility of NT-proBNP as a biomarker of microvascular complications in patients with type 2 diabetes. Materials and methods: In total, 277 outpatients with type 2 diabetes were consecutively enrolled as a hospital cohort. Two hundred and seventeen of these patients (132 males; mean age, 63.4 years) were designated as cases with any of the diabetic complications (retinopathy, neuropathy, nephropathy, ischemic heart disease, strokes, peripheral artery disease), and 60 (42 males; mean age, 54.1 years) were set as controls without clinical evidence of diabetic complications. Diabetic complications were evaluated by medical record and routine laboratory examinations. NT-proBNP was measured and investigated with regard to the associations with diabetic complications. Results: Mean NT-proBNP levels were significantly higher in patients with any of the diabetic complications (59 versus 33 pg/mL; P,0.0001). In logistic regression analysis, NT-proBNP levels .79 pg/mL, which was the highest tertile, were independently associated with a 5.04 fold increased risk of all complications (P,0.0051) compared to the lowest tertile (NT-proBNP levels ,31 pg/mL). Odd ratios of cardiovascular disease and nephropathy, neuropathy, and retinopathy were 9.33, 6.23, 6.6 and 13.78 respectively, in patients with NT-proBNP values in the highest tertile (.79 pg/mL), independently of age, sex, duration of diabetes or other risk factors, such as body mass index or hemoglobin A1c. In addition, NT-proBNP levels were associated with surrogate markers of atherosclerosis, such as brachial-ankle pulse wave velocity (r=0.449, P,0.0001) and left ventricular hypertrophy (r=0.212, P,0.001). Conclusion: In this hospital-based cohort of type 2 diabetes, the NT-proBNP levels were associated with systemic atherosclerosis and comorbid diabetic microvascular as well as macrovascular complications. It is useful to stratify high-risk diabetic patients by measuring NT-proBNP and to start comprehensive care for preventing the progression of diabetic complications. It is necessary to elucidate the underlying mechanism for the progression of diabetic complications represented by an elevation of NT-proBNP and to demonstrate the ability of NT-proBNP as a predictive global biomarker for diabetic complications in Japanese type 2 diabetic patients.
How are patients with type 2 diabetes and renal disease monitored and managed? Insights from the observational OREDIA study
Alfred Penfornis, J F Blicklé, B Fiquet, S Quéré, S Dejager
Vascular Health and Risk Management 2014:10 341–352
http://dx.doi.org/10.2147/VHRM.S60312
Background and aim: Chronic kidney disease (CKD) is frequent in type 2 diabetes mellitus (T2DM), and therapeutic management of diabetes is more challenging in patients with renal impairment (RI). The place of metformin is of particular interest since most scientific societies now recommend using half the dosage in moderate RI and abstaining from use in severe RI, while the classic contraindication with RI has not been removed from the label. This study aimed to assess the therapeutic management, in particular the use of metformin, of T2DM patients with CKD in real life. Methods: This was a French cross-sectional observational study: 3,704 patients with T2DM diagnosed for over 1 year and pharmacologically treated were recruited in two cohorts (two-thirds were considered to have renal disease [CKD patients] and one-third were not [non-CKD patients]) by 968 physicians (81% general practitioners) in 2012. Results: CKD versus non-CKD patients were significantly older with longer diabetes history, more diabetic complications, and less strict glycemic control (mean glycated hemoglobin [HbA1c] 7.5% versus 7.1%; 25% of CKD patients had HbA1c $8% versus 15% of non-CKD patients). Fifteen percent of CKD patients had severe RI, and 66% moderate RI. Therapeutic management of T2DM was clearly distinct in CKD, with less use of metformin (62% versus 86%) but at similar mean daily doses (∼2 g/d). Of patients with severe RI, 33% were still treated with metformin, at similar doses. For other oral anti-diabetics, a distinct pattern of use was seen across renal function (RF): use of sulfonylureas (32%, 31%, and 20% in normal RF, moderate RI, and severe RI, respectively) and DPP4-i (dipeptidyl peptidase-4 inhibitors) (41%, 36%, and 25%, respectively) decreased with RF, while that of glinides increased (8%, 14%, and 18%, respectively). CKD patients were more frequently treated with insulin (40% versus 16% of non-CKD patients), and use of insulin increased with deterioration of RF (19%, 39%, and 61% of patients with normal RF, moderate RI, and severe RI, respectively). Treatment was modified at the end of the study-visit in 34% of CKD patients, primarily to stop or reduce metformin. However, metformin was stopped in only 40% of the severe RI patients. Conclusion: Despite a fairly good detection of CKD in patients with T2DM, RI was insufficiently taken into account for adjusting anti-diabetic treatment.
Efficacy and safety of insulin glargine added to a fixed-dose combination of metformin and a dipeptidyl peptidase-4 inhibitor: results of the GOLD observational study
Jochen Seufert, Katrin Pegelow, Peter Bramlage
Vascular Health and Risk Management 2013:9 711–717
http://dx.doi.org/10.2147/VHRM.S54362
Background: For patients with type 2 diabetes who are uncontrolled on a combination of two oral antidiabetic agents, addition of the long-acting basal insulin glargine is a well established treatment option. However, data on the efficacy and safety of a combination of metformin, a dipeptidyl peptidase-4 (DPP-4) inhibitor, and insulin glargine are limited in real-world settings. Therefore, the aim of this study was to analyze blood glucose control, rates of hypoglycemia and body weight in a large cohort of patients with type 2 diabetes treated with this combination therapy in real practice. Methods: This noninterventional, multicenter, prospective, observational trial with a follow-up of 20 weeks enrolled insulin-naïve patients who had been on a stable fixed dose of metformin and a DPP-4 inhibitor for at least 3 months, and had a glycosylated hemoglobin (HbA1c) between 7.5% and 10%. Patients were selected at the investigators’ discretion for initiation of insulin glargine at baseline. A total of 1,483 patients were included, of whom 1,262 were considered to be the efficacy set. Primary efficacy parameters were HbA1c and fasting plasma glucose. Secondary outcome measures included achievement of glycemic targets, body weight, rates of hypoglycemia, and other safety parameters, as well as resource consumption. Results: Upon initiation of insulin glargine, mean HbA1c decreased from 8.51% to 7.36% (−1.15%±0.91%; 95% confidence interval [CI] −1.20 to −1.10). An HbA1c level ,6.5% was achieved in 8.2% of patients and a level ,7.0% in 31.5%. Mean fasting plasma glucose decreased from 174±47 mg/dL to 127±31 mg/dL (−47.3±44.1 mg/dL; 95% CI −49.8 to −44.8). In 11.9% of patients, a fasting plasma glucose level ,100 mg/dL was achieved. Bodyweight decreased on average by 0.98±3.90 kg (95% CI 1.19–0.76). Hypoglycemia (blood glucose #70 mg/dL) was observed in 29 patients (2.30%), of whom six (0.48%) had nocturnal hypoglycemia and four (0.32%) had documented severe events (blood glucose ,56 mg/dL). Conclusion: The results of this observational study show that insulin glargine, when added to a fixed-dose combination of metformin and a DPP-4 inhibitor, resulted in a significant and clinically relevant improvement of glycemic control. Importantly, this intervention did not interfere with the action of the DPP-4 inhibitors, resulting in neutral effects on weight and low rates of hypoglycemia. We conclude that this treatment intensification approach may be useful, efficient, and safe in daily clinical practice for patients with type 2 diabetes.
Long-term insulin glargine therapy in type 2 diabetes mellitus: a focus on cardiovascular outcomes
Joshua J Joseph, Thomas W Donner
Vascular Health and Risk Management 2015:11 107–116
http://dx.doi.org/10.2147/VHRM.S50286
Cardiovascular disease is the leading cause of mortality in type 2 diabetes mellitus. Hyperinsulinemia is associated with increased cardiovascular risk, but the effects of exogenous insulin on cardiovascular disease progression have been less well studied. Insulin has been shown to have both cardioprotective and atherosclerosis-promoting effects in laboratory animal studies. Long-term clinical trials using insulin to attain improved diabetes control in younger type 1 and type 2 diabetes patients have shown improved cardiovascular outcomes. Shorter trials of intensive diabetes control with high insulin use in higher risk patients with type 2 diabetes have shown either no cardiovascular benefit or increased all cause and cardiovascular mortality. Glargine insulin is a basal insulin analog widely used to treat patients with type 1 and type 2 diabetes. This review focuses on the effects of glargine on cardiovascular outcomes. Glargine lowers triglycerides, leads to a modest weight gain, causes less hypoglycemia when compared with intermediate-acting insulin, and has a neutral effect on blood pressure. The Outcome Reduction With Initial Glargine Intervention (ORIGIN trial), a 6.2 year dedicated cardiovascular outcomes trial of glargine demonstrated no increased cardiovascular risk.
Visceral obesity is not an independent risk factor of mortality in subjects over 65 years
Frédérique Thomas, Bruno Pannier, Athanase Benetos, Ulrich M Vischer
Vascular Health and Risk Management 2013:9 739–745
http://dx.doi.org/10.2147/VHRM.S49922
The aim of the study was to determine the role of obesity evaluated by body mass index (BMI), waist circumference (WC), and their combined effect on all-cause mortality according to age and related risk factors. This study included 119,090 subjects (79,325 men and 39,765 women), aged from 17 years to 85 years, who had a general health checkup at the Centre d’Investigations Préventives et Cliniques, Paris, France. The mean follow-up was 5.6±2.4 years. The prevalence of obesity, defined by WC and BMI categories, was determined according to age groups (< 55, 55–65, > 65 years). All-cause mortality according to obesity and age was determined using Cox regression analysis, adjusted for related risk factors and previous cardiovascular events.
For the entire population, WC adjusted for BMI, an index of central obesity, was strongly associated with mortality, even after adjustment for hypertension, dyslipidemia, and diabetes. The prevalence of obesity increased with age, notably when defined by WC. Nonetheless, the association between WC adjusted for BMI and mortality was not observed in subjects .65 years old (hazard ratio [HR] =1.010, P=NS) but was found in subjects < 55 (HR =1.030,
P < 0.0001) and 55–65 years old (HR =1.023, P,0.05). By contrast, hypertension
(HR =1.31, P < 0.05), previous cardiovascular events (HR =1.98, P < 0.05), and smoking (HR =1.33, P < 0.05) remained associated with mortality even after
age 65.
In conclusion, WC adjusted for BMI is strongly and independently associated with all-cause mortality before 65 years of age, after taking into account the associated risk factors. This relationship disappears in subjects
> 65 years of age, suggesting a differential impact of visceral fat deposition according to age.
Insulin degludec/insulin aspart combination for the treatment of type 1 and type 2 diabetes
Angela Dardano, Cristina Bianchi, Stefano Del Prato, Roberto Miccoli
Vascular Health and Risk Management 2014:10 465–475
http://dx.doi.org/10.2147/VHRM.S40097
Glycemic control remains the major therapeutic objective to prevent or delay the onset and progression of complications related to diabetes mellitus. Insulin therapy represents a cornerstone in the treatment of diabetes and has been used widely for achieving glycemic goals. Nevertheless, a large portion of the population with diabetes does not meet the internationally agreed glycemic targets. Moreover, insulin treatment, especially if intensive, may be associated with emergency room visits and hospitalization due to hypoglycemic events. Therefore, fear of hypoglycemia or hypoglycemic events represents the main barriers to the attainment of glycemic targets. The burden associated with multiple daily injections also remains a significant obstacle to initiating and maintaining insulin therapy. The most attractive insulin treatment approach should meet the patients’ preference, rather than demanding patients to change or adapt their lifestyle. Insulin degludec/insulin aspart (IDegAsp) is a new combination, formulated with ultra-long-acting insulin degludec and rapid-acting insulin aspart, with peculiar pharmacological features, clinical efficacy, safety, and tolerability. IDegAsp provides similar, noninferior glycemic control to a standard basal–bolus regimen in patients with type 1 diabetes mellitus, with additional benefits of significantly lower episodes of hypoglycemia (particularly nocturnal) and fewer daily insulin injections. Moreover, although treatment strategy and patients’ viewpoint are different in type 1 and type 2 diabetes, trial results suggest that IDegAsp may be an appropriate and reasonable option for initiating insulin therapy in patients with type 2 diabetes inadequately controlled on maximal doses of conventional oral agents. This paper will discuss the role of IDegAsp combination as a novel treatment option in diabetic patients.
UCP2 Regulates the Glucagon Response to Fasting and Starvation
Emma M. Allister, Christine A. Robson-Doucette, Kacey J. Prentice, et al.
Diabetes Feb 22, 2013; p 1-11. http://dx.doi.org:/10.2337/db12-0981
http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0981/-/DC1
Glucagon is important for maintaining euglycemia during fasting/starvation, and abnormal glucagon secretion is associated with type 1 and type 2 diabetes; however, the mechanisms of hypoglycemia-induced glucagon secretion are poorly understood. We previously demonstrated that global deletion of mitochondrial uncoupling protein 2 (UCP22/2) in mice impaired glucagon secretion from isolated islets. Therefore, UCP2 may contribute to the regulation of hypoglycemia-induced glucagon secretion, which is supported by our current finding that UCP2 expression is increased in nutrient-deprived murine and human islets. Further to this, we created a-cell–specific UCP2 knockout (UCP2AKO) mice, which we used to demonstrate that blood glucose recovery in response to hypoglycemia is impaired owing to attenuated glucagon secretion. UCP2-deleted a-cells have higher levels of intracellular reactive oxygen species (ROS), due to enhanced mitochondrial coupling, which translated into defective stimulus/secretion coupling. The effects of UCP2 deletion were mimicked by the UCP2 inhibitor genipin on both murine and human islets and also by application of exogenous ROS, confirming that changes in oxidative status and electrical activity directly reduce glucagon secretion. Therefore, a-cell UCP2 deletion perturbs the fasting/hypoglycemic glucagon response and shows that UCP2 is necessary for normal a-cell glucose sensing and the maintenance of euglycemia.
Main points:
- UCP2 is efficiently deleted specifically from islet a-cells of UCP2AKO mice.
- α-Cell UCP2 deletion reduces glucagon secretion in vivo
- UCP2AKO mice display normal glucose tolerance and GLP-1 secretion
- α-Cell UCP2 deletion reduces the gluconeogenic response of the liver and switches fatty acid usage during a prolonged fast
- UCP2 expression is increased after nutrient depletion and glucagon secretion from UCP2AKO islets was impaired.
- UCP2AKO α-cells display enhanced hyperpolarization of ΔψCm and increased superoxide levels
- UCP2AKO α-cells have more depolarized plasma membranes and reduced intracellular calcium
- UCP2 is required for normal glucagon secretion in response to hypoglycemia
Management of Diabetes Mellitus: Could Simultaneous Targeting of Hyperglycemia and Oxidative Stress Be a Better Panacea?
Omotayo O. Erejuwa
Int. J. Mol. Sci. 2012, 13, 2965-2972; http://dx.doi.org:/10.3390/ijms13032965
Oxidative stress is defined as an “imbalance between oxidants and antioxidants in favor of the oxidants, potentially leading to damage”. It is implicated in the pathogenesis and complications of diabetes mellitus. The role of oxidative stress is more definite in the pathogenesis of type 2 diabetes mellitus than in type 1 diabetes mellitus. In regard to diabetic complications, there is compelling evidence in support of the role of oxidative stress in both types of diabetes mellitus. Evidence suggests that elevated reactive oxygen species (ROS), which causes oxidative stress, accumulate in certain micro milieu or tissues (such as retina and kidney) where they cause damage or toxicity. In diabetes mellitus, oxidative stress is enhanced through various sources such as hyperglycemia, dyslipidemia, hyperinsulinemia, insulin resistance, impaired antioxidant defense network, uncoupling of ROS-generating enzymes, elevated level of leptin and sedentary lifestyle.
A number of mechanisms or pathways by which hyperglycemia, the major contributing factor of increased ROS production, causes tissue damage or diabetic complications have been identified. These include: hyperglycemia-enhanced polyol pathway; hyperglycemia-enhanced formation of advanced glycation endproducts (AGEs); hyperglycemia-activated protein kinase C (PKC) pathway; hyperglycemia-enhanced hexosamine pathway; and hyperglycemia-activated Poly-ADP ribose polymerase (PARP) pathway. These pathways are activated or enhanced by hyperglycemia-driven mitochondrial superoxide overproduction. Even though oxidative stress plays an important role in its pathogenesis and complications, unlike other diseases characterized by oxidative stress, diabetes mellitus is unique. Its cure (restoration of euglycemia, e.g., via pancreas transplants) does not prevent oxidative stress and diabetic complications. This is very important because hyperglycemia exacerbates oxidative stress which is linked to diabetic complications]. Theoretically, restoration of euglycemia should prevent oxidative stress and diabetic complications. However, this is not the case.
The primary aim of the current management of diabetes mellitus is to achieve and/or maintain a glycated hemoglobin level of ≤6.5%. However, recent evidence indicates that intensive treatment of hyperglycemia is characterized by increased weight gain, severe hypoglycemia and higher mortality. Besides, evidence suggests that it is difficult to achieve and/or maintain optimal glycemic control in many diabetic patients; and that the benefits of intensively-treated hyperglycemia are restricted to microvascular complications only. In view of these adverse effects and limitations of intensive treatment of hyperglycemia in preventing diabetic complications, which is linked to oxidative stress, this commentary proposes a hypothesis that “simultaneous targeting of hyperglycemia and oxidative stress” could be more effective than “intensive treatment of hyperglycemia” in the management of diabetes mellitus.
The Relationship between Inflammation, Oxidative Stress, and Metabolic Risk Factors in Type 2 Diabetic Patients
Fatemeh Azizi Soleiman, N Pahlavani, H Rasad, O Sadeghi, MR Gohari
Iranian Journal Of Diabetes And Obesity 2013; 5(4): 151-156
Increased production of free radicals due to the imbalance between free radicals and antioxidants load may reduce antioxidants levels, partial clearing of free radicals, and cause oxidation of lipids, sugars, proteins and nucleic acids which eventually leads to widespread pathological consequences of diabetes. One of the factors that facilitate formation of atherosclerosis in diabetes is oxidative stress.
Objective: Globally, 3-5.2 percent of people suffer from diabetes which is one of the most serious metabolic disorders resulting in an increase in inflammatory biomarkers e.g. interleukin-6, tumor necrosis factor-alpha, and C-reactive protein. The aim of this study was to investigate the relationship between inflammation, oxidative stress and fasting blood glucose, lipid profile and anthropometric parameters in patients with type 2 diabetes. Material and methods: This study was conducted as a cross sectional study in Tehran through 2009-2010 on 45 men and women aged 35-65 years old with type 2 diabetes. Blood glucose, lipid profile, C-reactive protein, and malonedialdehyde were measured. Independent sample T-test and linear regression analysis were used. Results: Fasting blood glucose, malonedialdehyde, total cholesterol and body mass index were higher in women than in men; but there was no difference between two sexes in other factors. Malonedialdehyde, neither directly or after adjustment for sex was related to fasting blood glucose, total cholesterol, triglycerides and anthropometric indices (weight, body mass index, and body fat mass). Conclusion: This study showed that oxidative stress had no relationship with blood glucose, lipid profile, and anthropometric index, but inflammation was related to glycemia, body mass index, and fat mass. Control of inflammation and oxidative stress is necessary for accelerating treatment process and preventing complications due to them.
This study showed that in diabetic patients, oxidative stress which was measured by MDA, was not significantly associated with fasting blood glucose, lipid profile and anthropometric parameters. However, fasting plasma glucose, body mass index and body fat mass were significant predictors of the inflammatory factor, CRP.
Oxidative Stress as an Underlying Contributor in the Development of Chronic Complications in Diabetes Mellitus
Suziy de M. Bandeira, Lucas José S. da Fonseca, Glaucevane da S. Guedes, et al.
Int. J. Mol. Sci. 2013, 14, 3265-3284; http://dx.doi.doi:/10.3390/ijms14023265
The high prevalence of diabetes mellitus and its increasing incidence worldwide, coupled with several complications observed in its carriers, have become a public health issue of great relevance. Chronic hyperglycemia is the main feature of such a disease, being considered the responsible for the establishment of micro and macrovascular complications observed in diabetes. Several efforts have been directed in order to better comprehend the pathophysiological mechanisms involved in the course of this endocrine disease. Recently, numerous authors have suggested that excess generation of highly reactive oxygen and nitrogen species is a key component in the development of complications invoked by hyperglycemia. Overproduction and/or insufficient removal of these reactive species result in vascular dysfunction, damage to cellular proteins, membrane lipids and nucleic acids, leading different research groups to search for biomarkers which would be capable of a proper and accurate measurement of the oxidative stress (OS) in diabetic patients, especially in the presence of chronic complications.
In the face of this scenario, the present review briefly addresses the role of hyperglycemia in OS, considering basic mechanisms and their effects in diabetes mellitus, describes some of the more commonly used biomarkers of oxidative/nitrosative damage and includes selected examples of studies which evaluated OS biomarkers in patients with diabetes, pointing to the relevance of such biological components in general oxidative stress status of diabetes mellitus carriers.
The role of FOXO1 in β‑cell failure and type 2 diabetes mellitus
Tadahiro Kitamura
Nat. Rev. Endocrinol. 2013; 9, 615–623
http://dx.doi.org:/10.1038/nrendo.2013.157
Over the past two decades, insulin resistance has been considered essential to the etiology of type 2 diabetes mellitus (T2DM). However, insulin resistance does not lead to T2DM unless it is accompanied by pancreatic β‑cell dysfunction, because healthy β cells can compensate for insulin resistance by increasing in number and functional output. Furthermore, β‑cell mass is decreased in patients with diabetes mellitus, suggesting a primary role for β‑cell dysfunction in the pathogenesis of T2DM. The dysfunction of β cells can develop through various mechanisms, including oxidative, endoplasmic reticulum or hypoxic stress, as well as via induction of cytokines; these processes lead to apoptosis, uncontrolled autophagy and failure to proliferate. Transdifferentiation between β cells and α cells occurs under certain pathological conditions, and emerging evidence suggests that β‑cell dedifferentiation or transdifferentiation might account for the reduction in β‑cell mass observed in patients with severe T2DM. FOXO1, a key transcription factor in insulin signaling, is implicated in these mechanisms. This Review discusses advances in our understanding of the contribution of FOXO1 signaling to the development of β‑cell failure in T2DM.
Selective peroxisome proliferator-activated receptor g (PPARg) modulation as a strategy for safer therapeutic PPARg activation
Linda Slanec Higgins and Alex M DePaoli
Am J Clin Nutr 2010;91(suppl):267S–72S.
http://dx.doi.org:/10.3945/ajcn.2009.28449E
Peroxisome proliferator-activated receptor c (PPARc) is a clinically validated target for treatment of insulin resistance. PPARc activation by full agonists such as thiazolidinediones has shown potent and durable glucose-lowering activity in patients with type 2 diabetes without the concern for hypoglycemia or gastrointestinal toxicities associated with some other medications used to treat this disease. However, thiazolidinediones are linked to safety and tolerability issues such as weight gain, fluid retention, edema, congestive heart failure, and bone fracture. Distinctive properties of PPARc provide the opportunity for selective modulation of the receptor such that desirable therapeutic effects may be attained without the unwanted effects of full activation. PPARc is a nuclear receptor that forms a complex with coreceptor RXR and a cell type– and cell state– specific array of coregulators to control gene transcription. PPARc affinity for these components, and hence transcriptional response, is determined by the conformational changes induced by ligand binding within a complex pocket with multiple interaction points. This molecular mechanism thereby offers the opportunity for selective modulation. A desirable selective PPARc modulator profile would include high-affinity interaction with the PPARc-binding pocket in a manner that leads to retention of the insulin-sensitizing activity that is characteristic of full agonists as well as mitigation of the effects leading to increased adiposity, fluid retention, congestive heart failure, and bone fracture. Examples of endogenous and synthetic selective PPARc modulator (SPPARM) ligands have been identified. SPPARM drug candidates are being tested clinically and provide support for this strategy.
Predicting response to incretin-based therapy
Sanjay Kalra, Bharti Kalra, Rakesh Sahay, Navneet Agrawal
Research and Reports in Endocrine Disorders 2011:1 11–19
http://dx.doi.org:/10.2147/RRED.S16282
There are two important incretin hormones, glucose-dependent insulin tropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1). The biological activities of GLP-1 include stimulation of glucose-dependent insulin secretion and insulin biosynthesis, inhibition of glucagon secretion and gastric emptying, and inhibition of food intake. GLP-1 appears to have a number of additional effects in the gastrointestinal tract and central nervous system. Incretin based therapy includes GLP-1 receptor agonists like human GLP-1 analogs (liraglutide) and exendin-4 based molecules (exenatide), as well as DPP-4 inhibitors like sitagliptin, vildagliptin and saxagliptin. Most of the published studies showed a significant reduction in HbA1c using these drugs. A critical analysis of reported data shows that the response rate in terms of target achievers of these drugs is average. One of the first actions identified for GLP-1 was the glucose-dependent stimulation of insulin secretion from islet cell lines. Following the detection of GLP-1 receptors on islet beta cells, a large body of evidence has accumulated illustrating that GLP-1 exerts multiple actions on various signaling pathways and gene products in the β cell. GLP-1 controls glucose homeostasis through well-defined actions on the islet β cell via stimulation of insulin secretion and preservation and expansion of β cell mass. In summary, there are several factors determining the response rate to incretin therapy. Currently minimal clinical data is available to make a conclusion. Key factors appear to be duration of diabetes, obesity, presence of autonomic neuropathy, resting energy expenditure, plasma glucagon levels and plasma free fatty acid levels. More clinical evidence is required to identify the factors affecting response rate to incretin therapy.
Regulation of Large Conductance Ca2+-activated K+ (BK) Channel β1 Subunit Expression by Muscle RING Finger Protein 1 in Diabetic Vessels
Fu Yi, Huan Wang, Qiang Chai, Xiaoli Wang, et al.
J. Biol. Chem. 2014, 289: 10853-10864
http://dx.doi.org:/10.1074/jbc.M113.520940
Background: Impaired BK channel function in diabetic vessels is associated with decreased BK channel[1]1 subunit (BK-β1) expression. Results: Muscle RING finger protein 1 (MuRF1) physically interacts with BK-β1 and accelerates BK-β1 proteolysis. Conclusion: Increased MuRF1 expression is a novel mechanism underlying diabetic BK channelopathy and vasculopathy. Significance: MuRF1 is a potential therapeutic target of BK channel dysfunction and vascular complications in diabetes.
The large conductance Ca2+-activated K+ (BK) channel, expressed abundantly in vascular smooth muscle cells (SMCs), is a key determinant of vascular tone. BK channel activity is tightly regulated by its accessory β1 subunit (BK-β1). However, BK channel function is impaired in diabetic vessels by increased ubiquitin/proteasome-dependent BK-β1 protein degradation. Muscle RING finger protein 1 (MuRF1), a muscle-specific ubiquitin ligase, is implicated in many cardiac and skeletal muscle diseases. However, the role of MuRF1 in the regulation of vascular BK channel and coronary function has not been examined. In this study, we hypothesized that MuRF1 participated in BK-β1 proteolysis, leading to the down-regulation of BK channel activation and impaired coronary function in diabetes. Combining patch clamp and molecular biological approaches, we found that MuRF1 expression was enhanced, accompanied by reduced BK-β1 expression, in high glucose-cultured human
coronary SMCs and in diabetic vessels. Knockdown of MuRF1 by siRNA in cultured human SMCs attenuated BK-β1 ubiquitination and increased BK-β1 expression, whereas adenoviral expression of MuRF1 in mouse coronary arteries reduced BK-β1 expression and diminished BK channel-mediated vasodilation. Physical interaction between the N terminus of BK-β1 and the coiled-coil domain of MuRF1 was demonstrated by pulldown assay. Moreover, MuRF1 expression was regulated by NF-κB. Most importantly, pharmacological inhibition of proteasome and NF-κB activities preserved BK-β1 expression and BK-channel-mediated coronary vasodilation in diabetic mice. Hence, our results provide the first evidence that the up-regulation of NF-κB-dependent MuRF1 expression is a novel mechanism that leads to BK channelopathy and vasculopathy in diabetes.
The origin of circulating CD36 in type 2 diabetes
MJ Alkhatatbeh, AK Enjeti, S Acharya, RF Thorne, and LF Lincz
Nutrition and Diabetes (2013) 3, e59; http://dx.doi.org:/10.1038/nutd.2013.1
Objective: Elevated plasma levels of the fatty acid transporter, CD36, have been shown to constitute a novel biomarker for type 2 diabetes mellitus (T2DM). We recently reported such circulating CD36 to be entirely associated with cellular microparticles (MPs) and aim here to determine the absolute levels and cellular origin(s) of these CD36 + MPs in persons with T2DM. Design: An ex vivo case-control study was conducted using plasma samples from 33 obese individuals with T2DM (body mass index (BMI) =39.9±6.4 kgm2; age=57±9 years; 18 male:15 female) and age- and gender-matched lean and obese non-T2DM controls (BMI =23.6±1.8 kgm2 and 33.5±5.9 kgm2, respectively). Flow cytometry was used to analyse surface expression of CD36 together with tissue-specific markers: CD41, CD235α, CD14, CD105 and phosphatidyl serine on plasma MPs. An enzyme-linked immunosorbent assay was used to quantify absolute CD36 protein concentrations. Results: CD36 + MP levels were significantly higher in obese people with T2DM (P<0.00001) and were primarily derived from erythrocytes (CD235α + = 35.8±14.6%); although this did not correlate with hemoglobin A1c. By contrast, the main source of CD36 + MPs in non-T2DM individuals was endothelial cells (CD105 + = 40.9±8.3% and 33.9±8.3% for lean and obese controls, respectively). Across the entire cohort, plasma CD36 protein concentration varied from undetectable to 22.9 µgml-1 and was positively correlated with CD36 +MPs measured by flow cytometry (P=0.0006) but only weakly associated with the distribution of controls and T2DM (P=0.021). Multivariate analysis confirmed that plasma CD36 + MP levels were a much better biomarker for diabetes than CD36 protein concentration (P=0.009 vs P=0.398, respectively). Conclusions: Both the levels and cellular profile of CD36 + MPs differ in T2DM compared with controls, suggesting that these specific vesicles could represent distinct biological vectors contributing to the pathology of the disease.
A Novel High-Throughput Assay for Islet Respiration Reveals Uncoupling of Rodent and Human Islets
Jakob D. Wikstrom, Samuel B. Sereda, Linsey Stiles, Alvaro Elorza, et al.
PLoS ONE 7(5): e33023. http://dx.doi.org:/10.1371/journal.pone.0033023
Background: The pancreatic beta cell is unique in its response to nutrient by increased fuel oxidation. Recent studies have demonstrated that oxygen consumption rate (OCR) may be a valuable predictor of islet quality and long term nutrient responsiveness. To date, high-throughput and user-friendly assays for islet respiration are lacking. The aim of this study was to develop such an assay and to examine bioenergetic efficiency of rodent and human islets. Methodology/Principal Findings: The XF24 respirometer platform was adapted to islets by the development of a 24-well plate specifically designed to confine islets. The islet plate generated data with low inter-well variability and enabled stable measurement of oxygen consumption for hours. The F1F0 ATP synthase blocker oligomycin was used to assess uncoupling while rotenone together with myxothiazol/antimycin was used to measure the level of non-mitochondrial respiration. The use of oligomycin in islets was validated by reversing its effect in the presence of the uncoupler FCCP. Respiratory leak averaged to 59% and 49% of basal OCR in islets from C57Bl6/J and FVB/N mice, respectively. In comparison, respiratory leak of INS-1 cells and C2C12 myotubes was measured to 38% and 23% respectively. Islets from a cohort of human donors showed a respiratory leak of 38%, significantly lower than mouse islets. Conclusions/Significance: The assay for islet respiration presented here provides a novel tool that can be used to study islet mitochondrial function in a relatively high-throughput manner. The data obtained in this study shows that rodent islets are less bioenergetically efficient than human islets as well as INS1 cells.
Refeeding and metabolic syndromes: two sides of the same coin
OA Obeid, DH Hachem and JJ Ayoub
Nutrition & Diabetes (2014) 4, e120; http://dx.doi.org:/10.1038/nutd.2014.21
Refeeding syndrome describes the metabolic and clinical changes attributed to aggressive rehabilitation of malnourished subjects. The metabolic changes of refeeding are related to hypophosphatemia, hypokalemia, hypomagnesemia, sodium retention and hyperglycemia, and these are believed to be mainly the result of increased insulin secretion following high carbohydrate intake. In the past few decades, increased consumption of processed food (refined cereals, oils, sugar and sweeteners, and so on) lowered the intake of several macrominerals (mainly phosphorus, potassium and magnesium). This seems to have compromised the postprandial status of these macrominerals, in a manner that mimics low grade refeeding syndrome status. At the pathophysiological level, this condition favored the development of the different components of the metabolic syndrome. Thus, it is reasonable to postulate that metabolic syndrome is the result of long term exposure to a mild refeeding syndrome.
HSP72 protects against obesity-induced insulin resistance
Jason Chung, Anh-Khoi Nguyen, Darren C. Henstridge, Anna G. Holmes, et al.
PNAS Feb 5, 2008; 105(5): 1739–1744
http://www.pnas.org/cgi/doi/10.1073/pnas.0705799105
Patients with type 2 diabetes have reduced gene expression of heat shock protein (HSP) 72, which correlates with reduced insulin sensitivity. Heat therapy, which activates HSP72, improves clinical parameters in these patients. Activation of several inflammatory signaling proteins such as c-jun amino terminal kinase (JNK), inhibitor of B kinase, and tumor necrosis factor-β, can induce insulin resistance, but HSP 72 can block the induction of these molecules in vitro. Accordingly, we examined whether activation of HSP72 can protect against the development of insulin resistance. First, we show that obese, insulin resistant humans have reduced HSP72 protein expression and increased JNK phosphorylation in skeletal muscle. We next used heat shock therapy, transgenic overexpression, and pharmacologic means to overexpress HSP72 either specifically in skeletal muscle or globally in mice. Herein, we show that regardless of the means used to achieve an elevation in HSP72 protein, protection against diet- or obesity induced hyperglycemia, hyperinsulinemia, glucose intolerance, and insulin resistance was observed. This protection was tightly associated with the prevention of JNK phosphorylation. These findings identify an essential role for HSP72 in blocking inflammation and preventing insulin resistance in the context of genetic obesity or high-fat feeding.
pH-responsive modulation of insulin aggregation and structural transformation of the aggregates
Ekaterina Smirnova, I Safenkova, V Stein-Margolina, V Shubin, et al.
Biochimie 109 (2015) 49e59
http://dx.doi.org/10.1016/j.biochi.2014.12.006
Over the past two decades, much information has appeared on electrostatically driven molecular mechanisms of protein self-assembly and formation of aggregates of different morphology, varying from soluble amorphous structures to highly-ordered amyloid-like fibrils. Protein aggregation represents a special tool in biomedicine and biotechnology to produce biological materials for a wide range of applications. This has awakened interest in identification of pH-triggered regulators of transformation of aggregation-prone proteins into structures of higher order. The objective of the present study is to elucidate the effects of low-molecular-weight biogenic agents on aggregation and formation of supramolecular structures of human recombinant insulin, as a model therapeutic protein. Using dynamic light scattering, turbidimetry, circular dichroism, fluorescence spectroscopy, atomic force microscopy, transmission electron microscopy, and nuclear magnetic resonance, we have demonstrated that the amino acid L-arginine (Arg) has the striking potential to influence insulin aggregation propensity. It was shown that modification of the net charge of insulin induced by changes in the pH level of the incubation medium results in dramatic changes in the interaction of the protein with Arg. We have revealed the dual effects of Arg, highly dependent on the pH level of the solution e suppression or acceleration of the aggregation of insulin at pH 7.0 or 8.0, respectively. These effects can be regulated by manipulating the pH of the environment. The results of this study may be of interest for development of appropriate drug formulations and for the more general insight into the functioning of insulin in living systems, as the protein is known to release by exocytosis from pancreatic beta cells in a pH-dependent manner.
Human β-cell proliferation by promoting Wnt signaling
Carol Wilson
Original article Aly, H. et al. A novel strategy to increase the proliferative potential of adult human β-cells while maintaining their differentiated phenotype. PLoS ONE 2013; 8, e66131
Nature Reviews Endocrinology 2013; 9, 502
http://dx.doi.org:/10.1038/nrendo.2013.130
Islet transplantation for patients with type 1 diabetes mellitus typically requires 2–4 donors for one recipient, whereas use of one donor would minimize the risk of immune rejection. Proliferation of adult β cells in vitro could hold the key to providing one donor for one recipient.
“In previous studies, we found that activation of the Wnt/GSK-3/β-catenin pathway by pharmacologic inhibition of GSK-3 in combination with nutrient activation of mTOR, modestly enhanced human β-cell proliferation in vitro,” says lead researcher Haytham Aly of the Washington University School of Medicine in St. Louis, MO, USA. “However, expansion of human islets was associated with a loss of insulin content and secretory function.”
In the current study, the researchers aimed to engage canonical and noncanonical Wnt signalling at the receptor level to increase the proliferation of human β cells in vitro, without losing the capacity of the cells to produce and secrete insulin.
The researchers treated cadaver-derived intact human islets with a conditioned medium from L cells that constitutively produce Wnt-3a, R-spondin-3 and Noggin. A similar medium had previously enabled successful proliferation of mouse colonic intestinal epithelial cells. The researchers added inhibitors of ROCK and RhoA to this medium to augment cell survival.
The conditioned medium with the inhibitors lead to ~20-fold proliferation of the human β cells above that with glucose alone. Crucially, treatment with this conditioned medium did not impair glucose-stimulated insulin secretion or decrease insulin content of the cells.
“This novel strategy has clear potential for use in the in vitro expansion of human islets and the subsequent treatment of impaired β-cell functional mass in type 1 diabetes mellitus and type 2 diabetes mellitus,” concludes Aly.
Betatrophin—inducing β-cell expansion to treat diabetes mellitus?
Elisabeth Kugelberg
Original article Yi, P. et al. Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell http://dx.doi.org:/10.1016/j.cell.2013.04.008
Nature Reviews Endocrinology 2013; 9, 379; http://dx.doi.org:/10.1038/nrendo.2013.98
Betatrophin, a newly identified hormone, increases the production and expansion of insulin-secreting β cells in mice, research from Harvard University suggests.
When insulin resistance develops, pancreatic β cells undergo an expansion in mass and proliferation to compensate for increasing insulin needs. To date, the mechanisms regulating β-cell replication are unclear.
Yi et al. developed a mouse model of insulin resistance using the insulin receptor antagonist S961. Subcutaneous injections of the S961 peptide into mice led to dose-dependent, instant β-cell proliferation and hyperglycemia.
Microarray analysis revealed that a highly conserved mammalian gene, betatrophin, was upregulated fourfold in liver and threefold in white adipose tissue cells in response to the acute peripheral insulin resistance induced by S961.
Yi and coworkers found that Betatrophin encodes a secreted protein that can be detected in human plasma. Intravenous injection of betatrophin-expressing constructs into mice resulted in a 17-fold higher β-cell proliferation rate compared with control vectors, and ultimately led to increased islet size and insulin content, with improvements in glucose tolerance, in betatrophin-injected animals.
The mechanisms of action of betatrophin are still unknown, and the next step is to test the effects of recombinant betatrophin protein on β-cell mass. The authors conclude that the identification of betatrophin and its control of β-cell proliferation opens a new door to possible diabetes therapy.
Blocking RANKL signaling might prevent T2DM
Carol Wilson
Original article Kiechl, S. et al. Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat. Med.
http://dx.doi.org:/10.1038/nm.3084
Nature Reviews Endocrinology 2013; 9, 188;
http://dx.doi.org:/10.1038/nrendo.2013.43
Blockade of receptor activator of nuclear factor κB ligand (RANKL) signaling in hepatocytes protects against type 2 diabetes mellitus (T2DM), report researchers.
“It is well known that activation of nuclear factor κB (NF-κB) in the liver is a crucial event in the development of hepatic insulin resistance and T2DM,” explains lead author Stefan Kiechl of the Medical University of Innsbruck, Austria. “RANKL, a member of the tumour necrosis factor superfamily, is a potent activator of NF-κB, and its receptor RANK is expressed on liver cells. We, thus, hypothesized that RANKL is involved in hepatic NF-κB activation, leading to T2DM.”
The researchers studied the association between serum levels of soluble RANKL and osteoprotegerin and subsequent risk of developing T2DM in 844 men and women without T2DM aged 40–79 years. Soluble RANKL was assessed because it has been shown to be functionally active.
During follow-up, between 1990 and 2005, 78 individuals of the cohort developed T2DM. Baseline levels of soluble RANKL between individuals who had and had not developed T2DM differed considerably: risk of T2DM was elevated in the group with the top tertile T2DM of concentrations of soluble RANKL compared with the group with the bottom tertile (OR 4.06, 95% CI 2.01–8.20). Adjustment for lifestyle factors and body composition did not significantly affect the risk association. Interestingly, although concentrations of osteoprotegerin were not elevated preceding T2DM onset, as they were for soluble RANKL, increased levels were found in individuals after disease occurrence.
In a series of mouse models in which RANKL signaling was downregulated systemically or in the liver, the investigators showed that hepatic insulin sensitivity and plasma glucose concentrations improved with blockade of RANKL signaling. In one such experiment, mice with a hepatocyte-specific Rank knockout were fed a high-fat diet for 4 weeks. These mice did not develop insulin resistance, whereas control mice did.
The investigators note that medications for T2DM already available, such as metformin, lower RANKL activity in bone and might also lower RANKL activity in the liver. They speculate that RANKL antagonism could be a yet unknown.
SFRP4—a biomarker for islet dysfunction?
Carol Wilson
Original article Mahdi, T. et al. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab. http://doi.org:/10.1016/j.cmet.2012.10.009
Secreted frizzled-related protein 4 (SFRP4) reduces insulin secretion and is a potential biomarker for islet dysfunction in type 2 diabetes mellitus (T2DM), report researchers.
Mahdi et al. discovered these insights into the pathophysiology of T2DM by the analysis of global gene expression in human pancreatic islets. The researchers identified a group of co-expressed genes (also called a gene co-expression module) associated with T2DM, reduced insulin secretion and elevated HbA1c levels after analysing global microarray expression data from human islets of 48 individuals, including 10 with T2DM. This module was enriched for IL-1-related genes.
The investigators identified SFRP4 as a gene highly expressed in islets from patients with T2DM. The protein encoded by SFRP4 is an extracellular regulator of the Wnt pathway, and has roles in tissue development, cancer and phosphate metabolism. Further study revealed that the expression and release of SFRP4 from islets was stimulated by IL-1β. Furthermore, elevated systemic SFRP4 levels led to reduced glucose tolerance as a result of decreased islet expression of voltage-gated Ca2+ channels and supressed insulin exocytosis.
Interestingly, levels of SFRP4 were elevated in serum of patients a few years before they developed T2DM, which indicates that this protein has potential to be used as a biomarker for T2DM. The researchers also point out that their data suggest that SFRP4 could be a therapeutic target for the treatment of islet dysfunction.
Add-on to metformin in T2DM —linagliptin or glimepiride?
Mikkel Christensen and Filip K. Knop
Nat. Rev. Endocrinol. 2012; 8, 576–578 http://dx.doi.org:/10.1038/nrendo.2012.163
Dipeptidyl peptidase 4 (DPP4) inhibitors, also known as gliptins, are a rapidly expanding class of oral antidiabetic drugs for the treatment of type 2 diabetes mellitus (T2DM). Since 2006, five DPP4 inhibitors have reached the market and, because they can be administered orally and have an almost impeccable safety profile, these drugs have gained widespread use in the treatment of T2DM. The DPP4 inhibitor linagliptin was approved in 2011 by the FDA and the European Medicines Agency (EMA) for use in patients with T2DM as second-line therapy to add on to metformin either alone or in combination with another second-line treatment.
The UK Prospective Diabetes Study trial showed that sulphonylurea treatment was more effective than metformin treatment after 1 year in terms of reducing HbA1c levels; however, after 6 years of treatment, the effectiveness of sulphonylurea treatment declined and metformin treatment was more effective. A decline in the effectiveness of the sulphonylurea treatment over time could be due to sulphonylureas inducing stress and possibly causing apoptosis in β cells. However, in the trial by Gallwitz et al. the sustained efficacies of the add-on treatments with linagliptin and glimepiride were similar after 2 years.
The inhibitors of DPP4 enhance glucose-dependent insulin secretion and could even augment the counter-regulatory glucagon response to hypoglycemia. DPP4 inhibition generally has a neutral effect upon body weight.
The study by Gallwitz et al. included patients whose plasma glucose levels were near-normal whilst they were receiving metformin monotherapy (baseline level 6–7 mmol/l), which could result in increased occurrence of hypoglycemia. Treating patients whose blood glucose levels were, by many standards, already adequately controlled with metformin with a drug known to be associated with inducing hypoglycemia would be expected to increase the frequency of hypoglycemia in this group, inflating the differences in the frequency of this event between the group receiving linagliptin and that receiving glimepiride.
The most groundbreaking findings in the study by Gallwitz et al. are related to cardiovascular outcomes. Although the study was not adequately powered to detect subtle differences in cardiovascular event frequency, significantly fewer patients who received linagliptin than glimepiride experienced major cardiovascular events (12 versus 26 individuals, respectively). This difference was driven by fewer patients experiencing nonfatal myocardial infarctions and nonfatal strokes in the linagliptin-treated group than in the glimepiride-treated group (9 versus 21 individuals, respectively).
Clinicians are responsible for selecting a suitable second-line treatment for patients with type 2 diabetes mellitus when metformin monotherapy fails. New evidence could aid clinicians in deciding between one of the most commonly used second-line agents, glimepiride, and the recently approved dipeptidyl peptidase 4 inhibitor linagliptin.
Relation of Mitochondrial Oxygen Consumption in Peripheral Blood Mononuclear Cells to Vascular Function in Type 2 Diabetes Mellitus
Mor-Li Hartman, Orian S. Shirihai, Monika Holbrook, Guoquan Xu, et al.
Vasc Med. 2014 February ; 19(1): 67–74. http://dx.doi.org:/10.1177/1358863X14521315.
Recent studies have shown mitochondrial dysfunction and increased production of reactive
oxygen species in peripheral blood mononuclear cells (PBMC’s) and endothelial cells from patients with diabetes mellitus. Mitochondria oxygen consumption is coupled to ATP production and also occurs in an uncoupled fashion during formation of reactive oxygen species by components of the electron transport chain and other enzymatic sites. We therefore hypothesized that diabetes would be associated with higher total and uncoupled oxygen consumption in PBMC’s that would correlate with endothelial dysfunction. We developed a method to measure oxygen consumption in freshly isolated PBMC’s and applied it to 26 patients with type 2 diabetes mellitus and 28 non-diabetic controls. Basal (192±47 vs. 161±44 pMoles/min, P=0.01), uncoupled (64±16 vs. 53±16 pMoles/min, P=0.007), and maximal (795±87 vs. 715±128 pMoles/min, P=0.01) oxygen consumption rates were higher in diabetic patients compared to controls. There were no significant correlations between oxygen consumption rates and endothelium-dependent flow-mediated dilation measured by vascular ultrasound. Non-endothelium-dependent nitroglycerin-mediated dilation was lower in diabetics (10.1±6.6 vs. 15.8±4.8%, P=0.03) and correlated with maximal oxygen consumption (R= −0.64, P=0.001). In summary, we found that diabetes mellitus is associated with a pattern of mitochondrial oxygen consumption consistent with higher production of reactive oxygen species. The correlation between oxygen consumption and nitroglycerin-mediated dilation may suggest a link between mitochondrial dysfunction and vascular smooth muscle cell dysfunction that merits further study. Finally, the described method may have utility for assessment of mitochondrial function in larger scale observational and interventional studies in humans.
Musashi expression in b-cells coordinates insulin expression, apoptosis and proliferation in response to endoplasmic reticulum stress in diabetes
M Szabat, TB Kalynyak, GE Lim, KY Chu, YH Yang, A Asadi, BK Gage, et al.
Cell Death and Disease (2011) 2, e232
http://dx.doi.org:/10.1038/cddis.2011.119
Diabetes is associated with the death and dysfunction of insulin-producing pancreatic b-cells. In other systems, Musashi genes regulate cell fate via Notch signaling, which we recently showed regulates b-cell survival. Here we show for the first time that human and mouse adult islet cells express mRNA and protein of both Musashi isoforms, as well Numb/Notch/Hes/neurogenin-3 pathway components. Musashi expression was observed in insulin/glucagon double-positive cells during human fetal development and increased during directed differentiation of human embryonic stem cells (hESCs) to the pancreatic lineage. De-differentiation of b-cells with activin A increased Msi1 expression. Endoplasmic reticulum (ER) stress increased Msi2 and Hes1, while it decreased Ins1 and Ins2 expression, revealing a molecular link between ER stress and b-cell dedifferentiation in type 2 diabetes. These effects were independent of changes in Numb protein levels and Notch activation. Overexpression of MSI1 was sufficient to increase Hes1, stimulate proliferation, inhibit apoptosis and reduce insulin expression, whereas Msi1 knockdown had the converse effects on proliferation and insulin expression. Overexpression of MSI2 resulted in a decrease in MSI1 expression. Taken together, these results demonstrate overlapping, but distinct roles for Musashi-1 and Musashi-2 in the control of insulin expression and b-cell proliferation. Our data also suggest that Musashi is a novel link between ER stress and the compensatory b-cell proliferation and the loss of b-cell gene expression seen in specific phases of the progression to type 2 diabetes.
Cooperation between brain and islet in glucose homeostasis and diabetes
Michael W. Schwartz, RJ Seeley, MH Tschöp, SC Woods, et al.
Nature 7 Nov 2013; 503: 59–66 http://dx.doi.org/10.1038/nature12709
Although a prominent role for the brain in glucose homeostasis was proposed by scientists in the nineteenth century, research throughout most of the twentieth century focused on evidence that the function of pancreatic islets is both necessary and sufficient to explain glucose homeostasis, and that diabetes results from defects of insulin secretion, action or both. However, insulin-independent mechanisms, referred to as ‘glucose effectiveness’, account for roughly 50% of overall glucose disposal, and reduced glucose effectiveness also contributes importantly to diabetes pathogenesis. Although mechanisms underlying glucose effectiveness are poorly understood, growing evidence suggests that the brain can dynamically regulate this process in ways that improve or even normalize glycaemia in rodent models of diabetes. Here we present evidence of a brain-centred glucoregulatory system (BCGS) that can lower blood glucose levels via both insulin-dependent and -independent mechanisms, and propose a model in which complex and highly coordinated interactions between the BCGS and pancreatic islets promote normal glucose homeostasis. Because activation of either regulatory system can compensate for failure of the other, defects in both may be required for diabetes to develop. Consequently, therapies that target the BCGS in addition to conventional approaches based on enhancing insulin effects may have the potential to induce diabetes remission, whereas targeting just one typically does not.
The traditional view holds that diabetes arises as a consequence of damage to, and ultimately failure of, beta-cell function. We propose a two-component model in which failure of glucose homeostasis can begin after initial impairment.

Schematic illustrations of brain- and islet-centred glucoregulatory systems
Schematic illustrations of brain- and islet-centred glucoregulatory systems
The BCGS is proposed to regulate tissue glucose metabolism and plasma glucose levels via mechanisms that are both insulin dependent (for example, by regulating tissue insulin sensitivity) and insulin independent

Proposed contributions of defective brain- and islet-centred glucoregulatory systems to T2D pathogenesis
Proposed contributions of defective brain- and islet-centred glucoregulatory systems to T2D pathogenesis
Insulin’s discovery: New insights on its ninetieth birthday
Jesse Roth, Sana Qureshi, Ian Whitford, Mladen Vranic, et al.
Diabetes Metab Res Rev 2012; 28: 293–304
http://dx.doi.org:/10.1002/dmrr.2300
2012 marks the 90th year since the purification of insulin and the miraculous rescue from death of youngsters with type 1 diabetes. In this review, we highlight several previously unappreciated or unknown events surroundingthe discovery.
(i) We remind readers of the essential contributions of each of the four discoverers – Banting, Macleod, Collip, and Best.
(ii) Banting and Best (each with his own inner circle) worked not only to accrue credit for himself but also to minimize credit to the other discoverers.
(iii) Banting at the time of the insulin research was very likely suffering from post-traumatic stress disorder (PTSD) that originated during his heroic service as a surgeon in World War I on the Western Front in 1918, including an infected shrapnel wound that threatened amputation of his arm. His war record along with the newly discovered evidence of a suicide threat goes along with his paranoia, combativeness, alcohol excess, and depression, symptoms we associate with PTSD.
(iv) Banting’s eureka idea, ligation of the pancreatic duct to preserve the islets, while it energized the early research, was unnecessary and was bypassed early.
(v) Post discovery,Macleod uncovered many features of insulin action that he summarized in his 1925 Nobel Lecture.Macleod closed by raising the question – what is the mechanism of insulin action in the body? – a challenge that attracted many talented investigators but remained unanswered until the latter third of the 20th century.
Genetic Variants Associated With Glycine Metabolism and Their Role in Insulin Sensitivity and Type 2 Diabetes
Weijia Xie, Andrew R. Wood, Valeriya Lyssenko, Michael N. Weedon, et al.
Diabetes 2013; 62:2141–2150 http://dx.doi.org:/10.2337/db12-0876
Circulating metabolites associated with insulin sensitivity may represent useful biomarkers, but their causal role in insulin sensitivity and diabetes is less certain. We previously identified novel metabolites correlated with insulin sensitivity measured by the hyperinsulinemic-euglycemic clamp. The top-ranking metabolites were in the glutathione and glycine biosynthesis pathways. We aimed to identify common genetic variants associated with metabolites in these pathways and test their role in insulin sensitivity and type 2 diabetes. With 1,004 nondiabetic individuals from the RISC study, we performed a genome-wide association study (GWAS) of 14 insulin sensitivity–related metabolites and one metabolite ratio. We replicated our results in the Botnia study (n = 342). We assessed the association of these variants with diabetes-related traits in GWAS meta-analyses (GENESIS [including RISC, EUGENE2, and Stanford], MAGIC, and DIAGRAM). We identified four associations with three metabolites—glycine (rs715 at CPS1), serine (rs478093 at PHGDH), and betaine (rs499368 at SLC6A12; rs17823642 at BHMT)—and one association signal with glycine-to-serine ratio (rs1107366 at ALDH1L1). There was no robust evidence for association between these variants and insulin resistance or diabetes. Genetic variants associated with genes in the glycine biosynthesis pathways do not provide consistent evidence for a role of glycine in diabetes related traits.
Fractalkine (CX3CL1), a new factor protecting b-cells against TNFa
Sabine Rutti, Caroline Arous, Domitille Schvartz, Katharina Timper, et al.
MOLMET164_proof ■ 14 Aug 2014 ■ 1/11
http://dx.doi.org/10.1016/j.molmet.2014.07.007
Objective: We have previously shown the existence of a muscleepancreas intercommunication axis in which CX3CL1 (fractalkine), a CX3C chemokine produced by skeletal muscle cells, could be implicated. It has recently been shown that the fractalkine system modulates murine β-cell function. However, the impact of CX3CL1 on human islet cells especially regarding a protective role against cytokine-induced apoptosis remains to be investigated. Methods: Gene expression was determined using RNA sequencing in human islets, sorted β- and non-β-cells. Glucose-stimulated insulin secretion (GSIS) and glucagon secretion from human islets was measured following 24 h exposure to 1e50 ng/ml CX3CL1. GSIS and specific protein phosphorylation were measured in rat sorted β-cells exposed to CX3CL1 for 48 h alone or in the presence of TNFα (20 ng/ml). Rat and human β-cell apoptosis (TUNEL) and rat β-cell proliferation (BrdU incorporation) were assessed after 24 h treatment with increasing concentrations of CX3CL1. Results: Both CX3CL1 and its receptor CX3CR1 are expressed in human islets. However, CX3CL1 is more expressed in non-β-cells than in b-cells while its receptor is more expressed in β-cells. CX3CL1 decreased human (but not rat) β-cell apoptosis. CX3CL1 inhibited human islet glucagon secretion stimulated by low glucose but did not impact human islet and rat sorted β-cell GSIS. However, CX3CL1 completely prevented the adverse effect of TNFa on GSIS and on molecular mechanisms involved in insulin granule trafficking by restoring the phosphorylation (Akt, AS160, paxillin) and expression (IRS2, ICAM-1, Sorcin, PCSK1) of key proteins involved in these processes. Conclusions: We demonstrate for the first time that human islets express and secrete CX3CL1 and CX3CL1 impacts them by decreasing glucagon secretion without affecting insulin secretion. Moreover, CX3CL1 decreases basal apoptosis of human β-cells. We further demonstrate that CX3CL1 protects β-cells from the adverse effects of TNFa on their function by restoring the expression and phosphorylation of key proteins of the insulin secretion pathway.
Heart Failure, Saxagliptin and Diabetes Mellitus: Observations from the SAVOR – TIMI 53 Randomized Trial
Benjamin M. Scirica; Eugene Braunwald; Itamar Raz, and SAVOR-TIMI 53 Steering Committee and Investigators
Circulation. Sept 4, 2014 http://dx.doi.org:/10.1161/CIRCULATIONAHA.114.010389
Background—Diabetes and heart failure frequently coexist. However, few diabetes trials have prospectively evaluated and adjudicated heart failure as an endpoint. Methods and Results—16,492 patients with type 2 diabetes and a history of, or at risk for, cardiovascular events were randomized to saxagliptin or placebo (mean followup-2.1 years). The primary endpoint was the composite of cardiovascular death, myocardial infarction, or ischemic stroke. Hospitalization for heart failure was a predefined component of the secondary endpoint. Baseline NT-proBNP was measured in 12,301 patients. More patients treated with saxagliptin (289, 3.5%) were hospitalized for heart failure compared to placebo (228, 2.8%) (HR 1.27; 95%CI 1.07-1.51; p=0.007). Corresponding rates at 12-months were 1.9% vs.1.3% (HR 1.46, 95%CI 1.15-1.88, p=0.002, with no significant difference thereafter time-varying interaction
p=0.017). Subjects at greatest risk for hospitalization for heart failure had prior heart failure, EGFR < 60 ml/min and/or elevated baseline levels of NT-proBNP. There was no evidence of heterogeneity between NT-proBNP and saxagliptin (p for interaction=0.46), though the absolute risk excess for heart failure with saxagliptin was greatest in the highest NT-proBNP quartile (2.1%). Even in patients at high-risk for hospitalization for heart failure, the risk of the primary and secondary endpoints were similar between treatment groups. Conclusions—In the context of balanced primary and secondary endpoints, saxagliptin treatment was associated with an increased risk for hospitalization for heart failure. This increase in risk was highest among patients with elevated levels of natriuretic peptides, prior heart failure, or chronic kidney disease.
Angiotensin 1–7 improves insulin sensitivity by increasing skeletal muscle glucose uptake in vivo
Omar Echeverría-Rodríguez, Leonardo Del Valle-Mondragón, Enrique Hong
Peptides 51 (2014) 26– 30 http://dx.doi.org/10.1016/j.peptides.2013.10.022
The renin–angiotensin system (RAS) regulates skeletal muscle insulin sensitivity through different mechanisms. The overactivation of the ACE (angiotensin-converting enzyme)/Ang (angiotensin) II/AT1R (Ang IItype 1 receptor) axis has been associated with the development of insulin resistance, whereas the stimulation of the ACE2/Ang 1–7/MasR (Mas receptor) axis improves insulin sensitivity. The in vivo mechanismsby which this axis enhances skeletal muscle insulin sensitivity are scarcely known. In this work, we investigated whether rat soleus muscle expresses the ACE2/Ang 1–7/MasR axis and determined the effect ofAng 1–7 on rat skeletal muscle glucose uptake in vivo. Western blot analysis revealed the expression ofACE2 and MasR, while Ang 1–7 levels were detected in rat soleus muscle by capillary zone electrophoresis. The euglycemic clamp exhibited that Ang 1–7 by itself did not promote glucose transport, but itincreased insulin-stimulated glucose disposal in the rat. In a similar manner, captopril (an ACE inhibitor) enhanced insulin-induced glucose uptake and this effect was blocked by the MasR antagonist A-779. Our results show for the first time that rat soleus muscle expresses the ACE2/Ang 1–7/MasR axis of the RAS,and Ang 1–7 improves insulin sensitivity by enhancing insulin-stimulated glucose uptake in rat skeletal muscle in vivo. Thus, endogenous (systemic and/or local) Ang 1–7 could regulate insulin-mediated glucose transport in vivo.
Evolving concepts in advanced glycation, diabetic nephropathy, and diabetic vascular disease
George Jerums, S Panagiotopoulos, J Forbes, T Osicka, and Mark Cooper
Archives of Biochemistry and Biophysics 419 (2003) 55–62
http://dx.doi.org:/10.1016/j.abb.2003.08.017
Advanced glycation endproducts (AGEs) have been postulated to play a role in the development of both nephropathy and large vessel disease in diabetes. However, it is still not clear which AGE subtypes play a pathogenetic role and which of several AGE receptors mediate AGE effects on cells. This review summarises the renoprotective effect of inhibitors of AGE formation, including aminoguanidine, and of cross-link breakers, including ALT-711, on experimental diabetic nephropathy and on mesenteric vascular hypertrophy. It also demonstrates similar effects of aminoguanidine and ramipril (an angiotensin converting enzyme inhibitor) on fluorescent and immunoassayable AGE levels, renal protein kinase C activity, nitrotyrosine expression, lysosomal function, and protein handling in experimental diabetes. These findings indicate that inhibition of the renin angiotensin system blocks both upstream and downstream pathways leading to tissue injury. We postulate that the chemical pathways leading to advanced glycation endproduct formation and the renin angiotensin systems may interact through the generation of free radicals, induced both by glucose and angiotensin II. There is also evidence to suggest that AGE-dependent pathways may play a role in the development of tubulointerstitial fibrosis in the diabetic kidney. This effect is mediated through RAGE and is TGF-b and CTGF-dependent.
Preconditioning with Associated Blocking of Ca2+ Inflow Alleviates Hypoxia-Induced Damage to Pancreatic β-Cells
Zuheng Ma, Noah Moruzzi, Sergiu-Bogdan Catrina, Ingrid Hals, et al.
PLoS ONE 8(7): e67498. http://dx.doi.org:/10.1371/journal.pone.0067498
Objective: Beta cells of pancreatic islets are susceptible to functional deficits and damage by hypoxia. Here we aimed to characterize such effects and to test for and pharmacological means to alleviate a negative impact of hypoxia. Methods and Design: Rat and human pancreatic islets were subjected to 5.5 h of hypoxia after which functional and viability parameters were measured subsequent to the hypoxic period and/or following a 22 h re-oxygenation period. Preconditioning with diazoxide or other agents was usually done during a 22 h period prior to hypoxia. Results: Insulin contents decreased by 23% after 5.5 h of hypoxia and by 61% after a re-oxygenation period. Preconditioning with diazoxide time-dependently alleviated these hypoxia effects in rat and human islets. Hypoxia reduced proinsulin biosynthesis (3H-leucine incorporation into proinsulin) by 35%. Preconditioning counteracted this decrease by 91%. Preconditioning reduced hypoxia-induced necrosis by 40%, attenuated lowering of proteins of mitochondrial complexes I–IV and enhanced stimulation of HIF-1-alpha and phosphorylated AMPK proteins. Preconditioning by diazoxide was abolished by co-exposure to tolbutamide or elevated potassium (i.e. conditions which increase Ca2+ inflow). Preconditioning with nifedipine, a calcium channel blocker, partly reproduced effects of diazoxide. Both diazoxide and nifedipine moderately reduced basal glucose oxidation whereas glucose-induced oxygen consumption (tested with diazoxide) was unaffected. Preconditioning with diaxoxide enhanced insulin contents in transplants of rat islets to nondiabetic rats and lowered hyperglycemia vs. non-preconditioned islets in streptozotocin-diabetic rats. Preconditioning of human islet transplants lowered hyperglycemia in streptozotocin-diabetic nude mice.
Conclusions:
1) Prior blocking of Ca2+ inflow associates with lesser hypoxia-induced damage,
2) preconditioning affects basal mitochondrial metabolism and accelerates activation of hypoxia-reactive and potentially protective factors,
3) results indicate that preconditioning by K+-ATP-channel openers has therapeutic potential for islet transplantations.














































{kind=link}
{kind=link}
{kind=link}
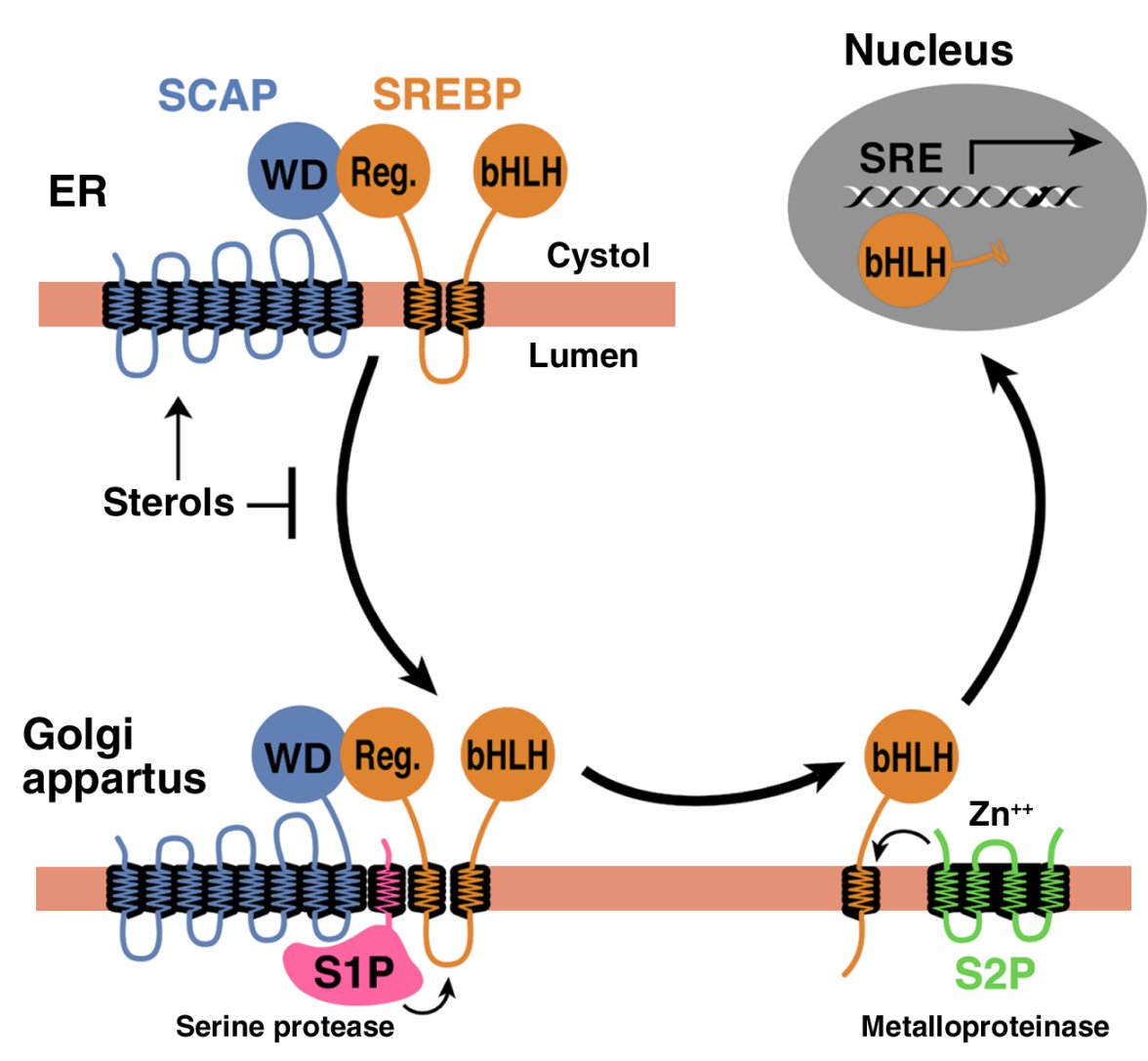{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}