Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Two pediatric siblings with recurrent multifocal glioblastoma multiforme (GBM) refractory to current standard therapies exhibited “remarkable and durable” responses to immune checkpoint inhibition with single-agent nivolumab (Opdivo), researchers said.
Following pre-clinical testing in 37 biallelic mismatch repair deficiency (bMMRD) cancers, a regimen of 3 mg/kg nivolumab every 2 weeks resulted in clinically significant responses and a profound radiologic response, Uri Tabori, MD, of The Hospital for Sick Children, Toronto, Ontario, Canada, and colleagues reported in the Journal of Clinical Oncology.
The 6-year-old white female patient and her 3.5-year-old brother resumed normal schooling and daily activities after 9 and 5 months of therapy, respectively, the researchers said.
“This observation is especially encouraging because these children are still clinically stable, whereas most relapsed pediatric GBMs will progress within 1 to 2 months despite salvage treatment, and survival is usually 3 to 6 months post-recurrence. It also highlights the utility of germline predisposition in guiding novel treatment options — in this case, immunotherapy — for cancer treatment.”
Findings from this lab study and small case series report may have implications for GBM as well as for other hypermutant cancers arising from primary (genetic predisposition) or secondary MMRD, the researchers said. “Given the increasing availability of commercial sequencing platforms, analysis of mutation burden and neoantigens can play a role in transforming treatment of these patients.”
Still, they added that these results, while encouraging, need to be validated in multinational prospective clinical trials of these “universally lethal” bMMRD-driven hypermutant cancers.
“Sometimes very small studies can yield meaningful results,” Robert Fenstermaker, MD, of Roswell Park Cancer Institute in Buffalo, N.Y., told MedPage Today via email. “Although anecdotal, the results of this study are quite encouraging because they tend to confirm current theory about immunotherapy for glioblastoma.”
Although these kinds of clinical responses to single-agent drug therapy in GBM are uncommon and the results may not be broadly applicable to all glioblastoma patients, this paper “is of much greater importance than just these few cases,” Fenstermaker emphasized. “The excellent responses in these particular cases suggest that an immune checkpoint inhibitor (nivolumab) may have enabled the immune system to respond fully.”
This “very small case series” report of a “compelling clinical experience” is a “fascinating and beautiful example of how mechanistic insight can be linked to rationally designed clinical applications — in turn, stimulating new downstream ideas,” Stephanie Weiss, MD, a radiation oncologist at Fox Chase Cancer Center in Philadelphia, commented in an email.
“This series also tests ‘proof of principle,’ that bMMRD tumors are hypermutated and associated with a high neoantigen load, and therefore may respond much like other immune checkpoint inhibitor-sensitive tumors. In this sense, the results reveal a tantalizing glimpse into the disease process of at least a subset of GBMs and can guide high-quality study of novel treatment for GBM.”
For the study, Tabori and colleagues performed exome sequencing and neoantigen prediction on 37 bMMRD-associated tumors, including 21 GBMs, and compared them with childhood and adult brain neoplasms.
The bMMRD GBMs were found to be hypermutant and to have an extremely strong neoantigen load — up to 16 times higher than the signature commonly seen in known immune checkpoint inhibitors (P<.001).
The female patient, diagnosed with a left parietal GBM, underwent near-total resection and focal irradiation over 6.5 weeks. After a clinical remission lasting 3 months, surveillance MRI revealed recurrence in the initial tumor bed and a second lesion in the left temporal lobe.
Six months earlier, the index patient’s brother had been diagnosed with a right frontoparietal GBM and treated with surgery, focal irradiation, and temozolomide (Temodal). Ten months after diagnosis, surveillance MRI revealed an asymptomatic diffuse multinodular GBM recurrence.
When given nivolumab as a last-resort therapeutic agent, both children initially experienced serious symptoms that on imaging mimicked tutor progression. After symptomatic management and observation, both stabilized, and follow-up imaging demonstrated significant improvement in tumor-related abnormalities.
Fenstermaker said that important next steps lie ahead, such as combining immune checkpoint inhibitors with specific cancer vaccines designed to immunize patients with glioblastomas other than this rare hypermutated type. “There are a number of prospective vaccines currently in the glioblastoma drug pipeline that would be candidates for this kind of approach,” he told MedPage Today. Examples include SurVaxM, NeoVax, HSPPC-96, and various dendritic cell vaccines.
In addition, newer genomic techniques are being developed that could make it possible to create a personalized profile of the mutant proteins in a given patient’s tumor, he noted. “One can imagine combining such a personalized vaccine against these mutant proteins together with an immune checkpoint inhibitor. Such a combination might result in many more responses like the ones seen in this small study.”
PD-1 Blockade in Tumors with Mismatch-Repair Deficiency
Somatic mutations have the potential to encode “non-self” immunogenic antigens. We hypothesized that tumors with a large number of somatic mutations due to mismatch-repair defects may be susceptible to immune checkpoint blockade.
We conducted a phase 2 study to evaluate the clinical activity of pembrolizumab, an anti–programmed death 1 immune checkpoint inhibitor, in 41 patients with progressive metastatic carcinoma with or without mismatch-repair deficiency. Pembrolizumab was administered intravenously at a dose of 10 mg per kilogram of body weight every 14 days in patients with mismatch repair–deficient colorectal cancers, patients with mismatch repair–proficient colorectal cancers, and patients with mismatch repair–deficient cancers that were not colorectal. The coprimary end points were the immune-related objective response rate and the 20-week immune-related progression-free survival rate.
The immune-related objective response rate and immune-related progression-free survival rate were 40% (4 of 10 patients) and 78% (7 of 9 patients), respectively, for mismatch repair–deficient colorectal cancers and 0% (0 of 18 patients) and 11% (2 of 18 patients) for mismatch repair–proficient colorectal cancers. The median progression-free survival and overall survival were not reached in the cohort with mismatch repair–deficient colorectal cancer but were 2.2 and 5.0 months, respectively, in the cohort with mismatch repair–proficient colorectal cancer (hazard ratio for disease progression or death, 0.10 [P<0.001], and hazard ratio for death, 0.22 [P=0.05]). Patients with mismatch repair–deficient noncolorectal cancer had responses similar to those of patients with mismatch repair–deficient colorectal cancer (immune-related objective response rate, 71% [5 of 7 patients]; immune-related progression-free survival rate, 67% [4 of 6 patients]). Whole-exome sequencing revealed a mean of 1782 somatic mutations per tumor in mismatch repair–deficient tumors, as compared with 73 in mismatch repair–proficient tumors (P=0.007), and high somatic mutation loads were associated with prolonged progression-free survival (P=0.02).
This study showed that mismatch-repair status predicted clinical benefit of immune checkpoint blockade with pembrolizumab. (Funded by Johns Hopkins University and others; ClinicalTrials.gov number, NCT01876511.)
Purpose Recurrent glioblastoma multiforme (GBM) is incurable with current therapies. Biallelic mismatch repair deficiency (bMMRD) is a highly penetrant childhood cancer syndrome often resulting in GBM characterized by a high mutational burden. Evidence suggests that high mutation and neoantigen loads are associated with response to immune checkpoint inhibition.
Patients and Methods We performed exome sequencing and neoantigen prediction on 37 bMMRD cancers and compared them with childhood and adult brain neoplasms. Neoantigen prediction bMMRD GBM was compared with responsive adult cancers from multiple tissues. Two siblings with recurrent multifocal bMMRD GBM were treated with the immune checkpoint inhibitor nivolumab.
Results All malignant tumors (n = 32) were hypermutant. Although bMMRD brain tumors had the highest mutational load because of secondary polymerase mutations (mean, 17,740 ± standard deviation, 7,703), all other high-grade tumors were hypermutant (mean, 1,589 ± standard deviation, 1,043), similar to other cancers that responded favorably to immune checkpoint inhibitors. bMMRD GBM had a significantly higher mutational load than sporadic pediatric and adult gliomas and all other brain tumors (P < .001). bMMRD GBM harbored mean neoantigen loads seven to 16 times higher than those in immunoresponsive melanomas, lung cancers, or microsatellite-unstable GI cancers (P < .001). On the basis of these preclinical data, we treated two bMMRD siblings with recurrent multifocal GBM with the anti–programmed death-1 inhibitor nivolumab, which resulted in clinically significant responses and a profound radiologic response.
Conclusion This report of initial and durable responses of recurrent GBM to immune checkpoint inhibition may have implications for GBM in general and other hypermutant cancers arising from primary (genetic predisposition) or secondary MMRD.
Glioblastoma multiforme (GBM) is a highly malignant brain tumor and the most common cause of death among children with CNS neoplasms.1 Despite primary management, which consists of surgical resection followed by radiation therapy and chemotherapy, most GBMs will recur, resulting in rapid death. Patients with recurrent disease have a particularly poor prognosis, with a median survival of fewer than 6 months; no effective therapies currently exist.
In contrast to adult CNS malignancies, a significant proportion of childhood brain tumors occur in the context of cancer predisposition syndromes.2 Pediatric GBMs are associated with germline mutations in TP53 (Li-Fraumeni syndrome)1 and the mismatch repair (MMR) genes (biallelic MMR deficiency syndrome [bMMRD]).3 Patients with bMMRD are unique in both the molecular events that lead to GBM formation and opportunities for innovative management of these tumors to possibly improve survival.
bMMRD is caused by homozygous germline mutations in one of the four MMR genes (PMS2, MLH1, MSH2, and MSH6) and is arguably the most penetrant cancer predisposition syndrome, with 100% of biallelic mutation carriers developing cancers in the first two decades of life. These are most commonly malignant gliomas, hematologic malignancies, and GI cancers.3,4 Understanding the relationship between the bMMRD somatic mutational landscape and tumor biology can lead to development of novel therapies and improved patient outcomes.
bMMRD GBMs harbor the highest mutation load among human cancers.5 Combined germline mutations in the MMR genes and somatic mutations in DNA polymerase result in complete ablation of proofreading during DNA replication and underpin this phenomenon. bMMRD GBMs, in contrast to other childhood cancers and adult MMR-proficient gliomas, exhibit a molecular signature characterized by single-nucleotide changes present in exponentially higher numbers. An important characteristic of non-bMMRD cancers exhibiting high mutation loads—subsets of malignant melanomas and lung, bladder, and microsatellite-unstable GI cancers—is responsiveness to immune checkpoint inhibitors.6–9
Checkpoint inhibitors target the immunomodulatory effect of CTLA-4 (cytotoxic T lymphocyte–associated protein 4) and programmed death-1 (PD-1)/programmed death-ligand 1, restoring effector T-cell function and antitumor activity. Recent reports have shown that patients whose tumors bear a high mutation load and/or definedtumor-associated antigen (neoantigen) signatures derive enhanced clinical benefit from checkpoint inhibitor therapy.10
Nivolumab is an anti–PD-1–directed immune checkpoint inhibitor approved for use in the treatment of non–small-cell lung cancer11and melanoma and under clinical investigation in multiple adult and pediatric tumors.12,13 However, this response is currently unknown in bMMRD-associated cancers and the uniformly lethal GBM.
Fig 1. Clinical and molecular features of the biallelic mismatch repair (MMR) deficiency (bMMRD) family. (A) Pedigree of the family with both bMMRD-affected children (solid square and circle). Both siblings presented with glioblastoma multiforme (GBM), whereas parents remained unaffected, as observed in other bMMRD families. (B) Immunohistochemistry staining of the index patient’s GBM for the four MMR genes: MSH2, MSH6,MLH1, and PMS2. A PMS2-negative stain in both tumor and normal cells prompted subsequent genetic testing that confirmed the diagnosis of bMMRD. NF1, neurofibromatosis type 1. http://jco.ascopubs.org/content/early/2016/03/17/JCO.2016.66.6552/F1.small.gif
To examine whether immune checkpoint inhibitors would be applicable for bMMRD cancers, we surveyed the extent of hypermutation across bMMRD tumors form various tissues. Exome sequencing of 37 cancers collected from the bMMRD consortium revealed that all malignant tumors (n = 32) were hypermutant. Although bMMRD brain tumors had the highest mutational load resulting from secondary polymerase mutations (mean, 17,740 ± standard deviation [SD], 7,703), all other high-grade tumors were hypermutant, harboring more than 100 exonic mutations (mean, 1,589 ± SD, 1,043; Fig 2A). Lower-grade bMMRD tumors (n = 5) did not exhibit hypermutation (mean, 40 ± SD, 18). Importantly, bMMRD GBMs had a significantly higher mutational load than sporadic pediatric and adult gliomas and all other brain tumors (P < .001; Fig 2A). To test the extent to which hypermutation translates to a strong neoantigen signature, a current predictor of response to immune checkpoint inhibition, we performed genome-wide somatic neoepitope analysis using similar algorithms previously used for melanoma, lung, and colon cancers.9,14,15 For each study, we compared our cohort of tumors with other tumors that were reported to respond to immune checkpoint inhibitors (Fig 2B). Strikingly, bMMRD GBMs had a significantly higher number of predicted neoantigens, whereas other tumors responded with a fraction of the neoantigens found in our patients (P < .001; Fig 2B). The mean neoantigen load was seven to 16 times higher than those of immunoresponsive melanomas, lung cancers, and microsatellite-unstable GI cancers.
Fig 2. Tumor mutation and neoantigen analysis. (A) Boxplot comparing the number of mutations per tumor exome in several biallelic mismatch repair deficiency (bMMRD) cancer types with pediatric and adult brain tumors. (B) Ratio of the number of neoantigens found in immunoresponsive tumors from melanoma (n = 27), lung cancer (n = 14), and colon cancer (n = 7) data sets compared with median number of neoantigens in bMMRD glioblastoma multiforme (GBM; n = 13). ATRT, atypical teratoid rhabdoid tumor; DIPG, diffuse intrinsic pontine glioma; L/L, leukemia/lymphoma; LGG, low-grade glioma; MB, medulloblastoma; PA, pilocytic astrocytoma; PNET, primitive neuroectodermal tumor.
We describe two pediatric patients with recurrent multifocal GBM refractory to current standard therapies who exhibited remarkable and durable responses to immune checkpoint inhibition with single-agent nivolumab. This observation is especially encouraging because these children are still clinically stable, whereas most relapsed pediatric GBMs will progress within 1 to 2 months19 despite salvage treatment, and survival is usually 3 to 6 months postrecurrence.20 Furthermore, bMMRD GBMs have outcomes similar to those of sporadic childhood GBMs,21 and data gathered from the consortium reveal a mean time from relapse to death of 2.6 months in bMMRD GBM. To our knowledge, this is the first report of such a response in childhood or adult GBM. It also highlights the utility of germline predisposition in guiding novel treatment options—in this case, immunotherapy—for cancer treatment. ….
sjwilliamspa
Not sure if the link between PD-L1 response and MMR status is causal in this ase. there are many tumors with MMR and especially all tumors had high degree of MMR. Perhaps they need to look at tumors that have a more stable genome like certain hepatocarcinomas.
hysical chemists have devised a rolling DNA-based motor that’s 1,000 times faster than any other synthetic DNA motor, giving it potential for real-world applications, such as disease diagnostics. Nature Nanotechnology is publishing the finding.
“Unlike other synthetic DNA-based motors, which use legs to ‘walk’ like tiny robots, ours is the first rolling DNA motor, making it far faster and more robust,” says Khalid Salaita, the Emory University chemist who led the research. “It’s like the biological equivalent of the invention of the wheel for the field of DNA machines.”
The speed of the new DNA-based motor, which is powered by ribonuclease H, means a simple smart phone microscope can capture its motion through video. The researchers have filed an invention disclosure patent for the concept of using the particle motion of their rolling molecular motor as a sensor for everything from a single DNA mutation in a biological sample to heavy metals in water.
“Our method offers a way of doing low-cost, low-tech diagnostics in settings with limited resources,” Salaita says.
The field of synthetic DNA-based motors, also known as nano-walkers, is about 15 years old. Researchers are striving to duplicate the action of nature’s nano-walkers. Myosin, for example, are tiny biological mechanisms that “walk” on filaments to carry nutrients throughout the human body.
“It’s the ultimate in science fiction,” Salaita says of the quest to create tiny robots, or nano-bots, that could be programmed to do your bidding. “People have dreamed of sending in nano-bots to deliver drugs or to repair problems in the human body.”
So far, however, mankind’s efforts have fallen far short of nature’s myosin, which speeds effortlessly about its biological errands. “The ability of myosin to convert chemical energy into mechanical energy is astounding,” Salaita says. “They are the most efficient motors we know of today.”
Some synthetic nano-walkers move on two legs. They are essentially enzymes made of DNA, powered by the catalyst RNA. These nano-walkers tend to be extremely unstable, due to the high levels of Brownian motion at the nano-scale. Other versions with four, and even six, legs have proved more stable, but much slower. In fact, their pace is glacial: A four-legged DNA-based motor would need about 20 years to move one centimeter.
Kevin Yehl, a post-doctoral fellow in the Salaita lab, had the idea of constructing a DNA-based motor using a micron-sized glass sphere. Hundreds of DNA strands, or “legs,” are allowed to bind to the sphere. These DNA legs are placed on a glass slide coated with the reactant: RNA.
The DNA legs are drawn to the RNA, but as soon as they set foot on it they destroy it through the activity of an enzyme called RNase H. As the legs bind and then release from the substrate, they guide the sphere along, allowing more of the DNA legs to keep binding and pulling.
“It’s called a burnt-bridge mechanism,” Salaita explains. “Wherever the DNA legs step, they trample and destroy the reactant. They have to keep moving and step where they haven’t stepped in order to find more reactant.”
The combination of the rolling motion, and the speed of the RNase H enzyme on a substrate, gives the new DNA motor its stability and speed.
“Our DNA-based motor can travel one centimeter in seven days, instead of 20 years, making it 1,000 times faster than the older versions,” Salaita says. “In fact, nature’s myosin motors are only 10 times faster than ours, and it took them billions of years to evolve.”
Emory post-doctoral fellow Kevin Yehl sets up a smart-phone microscope to get a readout for the particle motion of the rolling DNA-based motor.
The researchers demonstrated that their rolling motors can be used to detect a single DNA mutation by measuring particle displacement. They simply glued lenses from two inexpensive laser pointers to the camera of a smart phone to turn the phone into a microscope and capture videos of the particle motion.
“Using a smart phone, we can get a readout for anything that’s interfering with the enzyme-substrate reaction, because that will change the speed of the particle,” Salaita says. “For instance, we can detect a single mutation in a DNA strand.”
This simple, low-tech method could come in handy for doing diagnostic sensing of biological samples in the field, or anywhere with limited resources.
The proof that the motors roll came by accident, Salaita adds. During their experiments, two of the glass spheres occasionally became stuck together, or dimerized. Instead of making a wandering trail, they left a pair of straight, parallel tracks across the substrate, like a lawn mower cutting grass. “It’s the first example of a synthetic molecular motor that goes in a straight line without a track or a magnetic field to guide it,” Salaita says.
In addition to Salaita and Yehl, the co-authors on the Nature Nanotechnology paper include Emory researchers Skanda Vivek, Yang Liu, Yun Zhang, Megzhen Fan, Eric Weeks and Andrew Mugler (who is now at Purdue University).
DNA-based machines that walk by converting chemical energy into controlled motion could be of use in applications such as next-generation sensors, drug-delivery platforms and biological computing. Despite their exquisite programmability, DNA-based walkers are challenging to work with because of their low fidelity and slow rates (∼1 nm min–1). Here we report DNA-based machines that roll rather than walk, and consequently have a maximum speed and processivity that is three orders of magnitude greater than the maximum for conventional DNA motors. The motors are made from DNA-coated spherical particles that hybridize to a surface modified with complementary RNA; the motion is achieved through the addition of RNase H, which selectively hydrolyses the hybridized RNA. The spherical motors can move in a self-avoiding manner, and anisotropic particles, such as dimerized or rod-shaped particles, can travel linearly without a track or external force. We also show that the motors can be used to detect single nucleotide polymorphism by measuring particle displacement using a smartphone camera.
A 3-D rendering of a fluorescence image mapping the piconewton forces applied by T cells. The height and color indicates the magnitude of the applied force. (Microscopy image by Yang Liu.)
By Carol Clark
T cells, the security guards of the immune system, use a kind of mechanical “handshake” to test whether a cell they encounter is a friend or foe, a new study finds.
“We’ve provided the first direct evidence that a T cell gives precise mechanical tugs to other cells,” Salaita says. “And we’ve shown that these tugs are central to a T cell’s process of deciding whether to mount an immune response. A tug that releases easily, similar to a casual handshake, signals a friend. A stronger grip indicates a foe.”
T cells continuously patrol through the body in search of foreign invaders. They have molecules known as T-cell receptors (TCR) that can recognize specific antigenic peptides on the surface of a pathogenic or cancerous cell. When a T cell detects an antigen-presenting cell (APC), its TCR connects to a ligand, or binding molecule, of the APC. If the T cell determines the ligand is foreign, it becomes activated and starts pumping calcium. The calcium is part of a signaling chain that recruits other cells to come and help mount an immune response.
Scientists have known about this process for decades, but they have not fully understood how the T cell distinguishes small modifications to the antigenic ligand and how it decides to respond to it. “If you view this T cell response purely as a chemical process, it does not fully explain the remarkable specifity of the binding,” Salaita says. “When you take the two components – the TCR and the ligand on the surface of cells – and just let them chemically bind in a solution, for example, you can’t predict what will trigger a strong or a weak immune response.”
The researchers hypothesized that mechanical strain might also play a role in a T cell response, since the T cell continues to move even as it locks into a bind with an antigenic ligand.
To test this idea, the Salaita lab developed DNA-based gold nanoparticle tension sensors that light up, or fluoresce, in response to a miniscule mechanical force of a piconewton – about one million-millionth the weight of an apple.
The researchers designed experiments using T cells from a mouse and allowed them to test ligands containing eight amino acid peptides that had slight mutations.
“We swapped out the fourth amino acid position to create really subtle chemical changes in the ligand that would be very difficult to distinguish without a mechanical component,” Salaita says.
Some of the mutated ligands were given a firmer anchor to give them a tighter “grip” to the moving TCR.
Through the experiments, captured on microscopy video, the researchers were able to see, record and measure the responses of the T cells as they moved across the ligands.
“As a T cell moves across a cell’s surface and encounters a ligand, it pulls on it,” Salaita explains. “It doesn’t pull very hard, it’s a very precise and tiny tug that is not sustained. The T cell pulls and stops, pulls and stops, all across the surface. It’s like the T cell is doing a mechanical test of the ligand.”
During the experiments, the T cells did not activate fully when they encountered ligands with weak anchors. In contrast, when a T cell encountered a ligand with a firm anchor, the T cell became activated, showing that it experienced a piconewton level of resistance.
The amount of force that was applied by the T cell was mapped by using tension probes of different stiffness. Probes that responded to 19 piconewtons did not fluoresce, while softer, 12-piconewton probes produced high signal.
Following the fluorescence of the probe, the T cells switched on their calcium pumps and increased the calcium concentration within the cell, indicating that the T cell is mounting an immune response.
“We were able to map out the order of the cascade of chemical and mechanical reactions,” Salaita says. “First, the T cell uses a very specific and finely tuned mechanical tug to distinguish friend from foe. And when it senses a precise, piconewton level of force in response to that tug, the T cell realizes that it has encountered a foreign body and gives the signal for attack.”
The discovery could help in the search for treatments of auto-immune diseases and the development of immune therapies for cancer.
“Cancer cells have an extra molecule that can make T cell security guards ‘drunk’ or ‘sleepy’ so that they are not able to function properly,” Salaita says. “Learning more about the mechanical forces involved in an effective immune response may help us develop ways to evade this defense system of cancer cells.”
Co-authors on the study include Yang Liu, Victor Pui-Yan Ma, Kornelia Galior and Zheng Liu (from the Salaita lab); and Lori Blanchfield and Rakieb Andargachew (from the Evavold lab).
T cells protect the body against pathogens and cancer by recognizing specific foreign peptides on the cell surface. Because antigen recognition occurs at the junction between a migrating T cell and an antigen-presenting cell (APC), it is likely that cellular forces are generated and transmitted through T-cell receptor (TCR)-ligand bonds. Here we develop a DNA-based nanoparticle tension sensor producing the first molecular maps of TCR-ligand forces during T cell activation. We find that TCR forces are orchestrated in space and time, requiring the participation of CD8 coreceptor and adhesion molecules. Loss or damping of TCR forces results in weakened antigen discrimination, showing that T cells harness mechanics to optimize the specificity of response to ligand.
T cells are triggered when the T-cell receptor (TCR) encounters its antigenic ligand, the peptide-major histocompatibility complex (pMHC), on the surface of antigen presenting cells (APCs). Because T cells are highly migratory and antigen recognition occurs at an intermembrane junction where the T cell physically contacts the APC, there are long-standing questions of whether T cells transmit defined forces to their TCR complex and whether chemomechanical coupling influences immune function. Here we develop DNA-based gold nanoparticle tension sensors to provide, to our knowledge, the first pN tension maps of individual TCR-pMHC complexes during T-cell activation. We show that naïve T cells harness cytoskeletal coupling to transmit 12–19 pN of force to their TCRs within seconds of ligand binding and preceding initial calcium signaling. CD8 coreceptor binding and lymphocyte-specific kinase signaling are required for antigen-mediated cell spreading and force generation. Lymphocyte function-associated antigen 1 (LFA-1) mediated adhesion modulates TCR-pMHC tension by intensifying its magnitude to values >19 pN and spatially reorganizes the location of TCR forces to the kinapse, the zone located at the trailing edge of migrating T cells, thus demonstrating chemomechanical crosstalk between TCR and LFA-1 receptor signaling. Finally, T cells display a dampened and poorly specific response to antigen agonists when TCR forces are chemically abolished or physically “filtered” to a level below ∼12 pN using mechanically labile DNA tethers. Therefore, we conclude that T cells tune TCR mechanics with pN resolution to create a checkpoint of agonist quality necessary for specific immune response.
Andrej Kosmrlj et al., Proc Natl Acad Sci U S A, 2008
Larry H. Bernstein, MD, FCAP, Curator
LPBI
The two articles above are connected in an interesting way by the fact that cellular forces are generated and transmitted through T-cell receptor (TCR)-ligand bonds. The T-cell receptor (TCR) encounters its antigenic ligand, the peptide-major histocompatibility complex (pMHC), on the surface of antigen presenting cells (APCs). The movement detected by the fluorescent sensor may be based on only a single amino acid at the cell surface ligand. The result is chemomechanical crosstalk between TCR and LFA-1 receptor signaling
Generalized gene regulation mechanisms of miRNAs. [NIH]
Increasingly, cancer researchers are discovering novel biological pathways that regulate the expression of various genes that are often strongly associated with tumorigenesis. These new molecular mechanisms represent important potential therapeutic targets for aggressive and difficult-to-treat cancers. In particular, microRNAs (miRNAs)—small, noncoding genetic material that regulates gene expression—have steadily become implicated in the progression of some cancers.
Now, researchers at the University of Cincinnati (UC) have found a particular signaling route for a microRNA, miR-22, that they believe leads to targets for acute myeloid leukemia (AML), the most common type of fast-growing cancer of the blood and bone marrow.
The findings from this study were published recently in Nature Communications in an article entitled “miR-22 Has a Potent Anti-Tumour Role with Therapeutic Potential in Acute Myeloid Leukaemia.”
Structure of mi-22 miccroRNA. [Ppgardne at el., via Wikimedia Commons]
Increasingly, cancer researchers are discovering novel biological pathways that regulate the expression of various genes that are often strongly associated with tumorigenesis. These new molecular mechanisms represent important potential therapeutic targets for aggressive and difficult-to-treat cancers. In particular, microRNAs (miRNAs)—small, noncoding genetic material that regulates gene expression—have steadily become implicated in the progression of some cancers.
Now, researchers at the University of Cincinnati (UC) have found a particular signaling route for a microRNA, miR-22, that they believe leads to targets for acute myeloid leukemia (AML), the most common type of fast-growing cancer of the blood and bone marrow.
The findings from this study were published recently in Nature Communications in an article entitled “miR-22 Has a Potent Anti-Tumour Role with Therapeutic Potential in Acute Myeloid Leukaemia.”
“MicroRNAs make up a class of small, noncoding internal RNAs that control a gene’s job, or expression, by directing their target messaging RNAs, or mRNAs, to inhibit or stop. Cellular organisms use mRNA to convey genetic information,” explained senior study author Jianjun Chen, Ph.D., associate professor in the department of cancer biology at the UC College of Medicine. “Previous research has shown that microRNA miR-22 is linked to breast cancer and other blood disorders which sometimes turn into AML, but we found in this study that it could be an essential anti-tumor gatekeeper in AML when it is down-regulated, meaning its function is minimized.”
AML—most common type of acute leukemia—arises when the bone marrow begins to make blasts, cells that have not yet completely matured. These blast cells typically develop into white blood cells; however, in AML the cells do not develop and are unable to aid in warding off infections. In the current study, the UC team describes how altering the expression of miR-22 affected AML pathogenesis.
“When we forced miR-22 expression, we saw difficulty in leukemia cells developing, growing, and thriving. miR-22 targets multiple cancer-causing genes (CRTC1, FLT3, and MYCBP) and blocks certain pathways (CREB and MYC),” Dr. Chen noted. “The downregulation, or decreased output, of miR-22 in AML, is caused by the loss of the number of DNA being copied and/or stopping their expression through a pathway called TET1/GFI1/EZH2/SIN3A. Also, nanoparticles carrying miR-22 DNA oligonucleotides (short nucleic acid molecules) prevented leukemia advancement.”
The investigators conducted the study using bone marrow transplant samples and animal models. The researchers showed that the ten-eleven translocation proteins (TET1/2/3) in mammals helped to control genetic expression in normal developmental processes. This was in sharp contrast to mutations that cause function loss and tumor-slowing with TET2, which has been observed previously in blood and stem cell cancers.
“We recently reported that TET1 plays an essential cancer generating role in certain AML where it activates expression of homeobox genes, which are a large family of similar genes that direct the formation of many body structures during early embryonic development,” remarked Dr. Chen. “However, it is unknown whether TET1 can also function as a repressor for cellular function in cancer, and its role in microRNA expression has rarely been studied.”
Dr. Chen added that these findings are important in targeting a cancer that is both common and fatal, stating that “the majority of patients with ALM usually don’t survive longer than 5 years, even with chemotherapy, which is why the development of new effective therapies based on the underlying mechanisms of the disease is so important.”
“Our study uncovers a previously unappreciated signaling pathway (TET1/GFI1/EZH2/SIN3A/miR-22/CREB-MYC) and provides new insights into genetic mechanisms causing and progressing AML and also highlights the clinical potential of miR-22-based AML therapy. More research on this pathway and ways to target it are necessary,” Dr. Chen concluded.
miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia
MicroRNAs are subject to precise regulation and have key roles in tumorigenesis. In contrast to the oncogenic role of miR-22 reported in myelodysplastic syndrome (MDS) and breast cancer, here we show that miR-22 is an essential anti-tumour gatekeeper in de novo acute myeloid leukaemia (AML) where it is significantly downregulated. Forced expression of miR-22 significantly suppresses leukaemic cell viability and growth in vitro, and substantially inhibits leukaemia development and maintenance in vivo. Mechanistically, miR-22 targets multiple oncogenes, including CRTC1, FLT3 and MYCBP, and thus represses the CREB and MYC pathways. The downregulation of miR-22 in AML is caused by TET1/GFI1/EZH2/SIN3A-mediated epigenetic repression and/or DNA copy-number loss. Furthermore, nanoparticles carrying miR-22 oligos significantly inhibit leukaemia progression in vivo. Together, our study uncovers a TET1/GFI1/EZH2/SIN3A/miR-22/CREB-MYC signalling circuit and thereby provides insights into epigenetic/genetic mechanisms underlying the pathogenesis of AML, and also highlights the clinical potential of miR-22-based AML therapy.
As one of the most common and fatal forms of hematopoietic malignancies, acute myeloid leukaemia (AML) is frequently associated with diverse chromosome translocations (for example t(11q23)/MLL-rearrangements, t(15;17)/PML-RARA and t(8;21)/AML1-ETO) and molecular abnormalities (for example, internal tandem duplications of FLT3 (FLT3-ITD) and mutations in nucleophosmin (NPM1c+))1. Despite intensive chemotherapies, the majority of patients with AML fail to survive longer than 5 years2, 3. Thus, development of effective therapeutic strategies based on a better understanding of the molecular mechanisms underlying the pathogenesis of AML is urgently needed.
MicroRNAs (miRNAs) are a class of small, non-coding RNAs that post-transcriptionally regulate gene expression4. Individual miRNAs may play distinct roles in cancers originating from different tissues or even from different lineages of hematopoietic cells4. It is unclear whether a single miRNA can play distinct roles between malignancies originating from the same hematopoietic lineage, such as de novo AML and myelodysplastic syndrome (MDS). Although around 30% of MDS cases transform to AML, the genetic and epigenetic landscapes of MDS or MDS-derived AML are largely different from those of de novo AML5, 6. MDS and MDS-derived AML are more responsive to hypomethylating agents than de novo AML7. The molecular mechanisms underlying the distinct pathogenesis and drug response between MDS (or MDS-derived AML) and de novo AML remain unclear.
The ten-eleven translocation (Tet1/2/3) proteins play critical transcriptional regulatory roles in normal developmental processes as activators or repressors8, 9, 10. In contrast to the frequent loss-of-function mutations and tumour-suppressor role of TET2 observed in hematopoietic malignancies11, 12, 13, we recently reported that TET1 plays an essential oncogenic role in MLL-rearranged AML where it activates expression of homeobox genes14. However, it is unknown whether TET1 can also function as a transcriptional repressor in cancer. Moreover, Tet1-mediated regulation of miRNA expression has rarely been studied10.
In the present study, we demonstrate that miR-22, an oncogenic miRNA reported in breast cancer and MDS15, 16, is significantly downregulated in most cases of de novo AML due to TET1/GFI1/EZH2/SIN3A-mediated epigenetic repression and/or DNA copy-number loss. miR-22 functions as an essential anti-tumour gatekeeper in various AML and holds great therapeutic potential to treat AML.
The downregulation of miR-22 in de novo AML
Through Exiqon miRNA array profiling, we previously identified a set of miRNAs, such as miR-150, miR-148a, miR-29a, miR-29b, miR-184, miR-342, miR-423 and miR-22, which are significantly downregulated in AML compared with normal controls17. Here we showed that among all the above miRNAs, miR-150 and especially miR-22 exhibited the most significant and consistent inhibitory effect on MLL-AF9-induced cell immortalization in colony-forming/replating assays (CFA) (Supplementary Fig. 1a). In contrast to the reported upregulation of miR-22 in MDS16, our original microarray data17 (Fig. 1a,b) and new quantitative PCR-independent validation data (Supplementary Fig. 1b) demonstrated a significant and global downregulation of miR-22 in de novo AML relative to normal controls. Notably, miR-22 is significantly downregulated in AML samples (P<0.05) compared with all three sub-populations of normal control cells, that is, normal CD34+ hematopoietic stem/progenitor cells (HSPCs), CD33+ myeloid progenitor cells, or mononuclear cells (MNCs) (Fig. 1a). Expression of miR-22 is significantly downregulated in all or the majority of individual subsets of AML samples than in the normal CD33+ or CD34+ cell samples (Fig. 1b).
Figure 1: miR-22 inhibits AML cell transformation and leukemogenesis.
(a,b) Exiqon microRNA profiling assay showed that miR-22 is significantly (P<0.05) downregulated in the entire set of AML set (n=85) (a) or each individual subset (b), relative to normal controls. The expression data were log(2) transformed and mean-centred. Mean±s.e.m. values were shown. (c) Comparison of effects of in-house miR-22, miR-22_Song16 and miR-22 mutant (miR-22mut; see the mutation sequence at the top) on MLL-AF9-induced colony forming. CFAs were performed using mouse BM progenitor (Lin–) cells transduced with MSCV-neo+MSCV-PIG (Ctrl), MSCV-neo-MLL-AF9+MSCV-PIG (MLL-AF9), or MSCV-neo-MLL-AF9+MSCV-PIG-miR-22/miR-22_Song/miR-22mut. (d) Effects of miR-22 on the colony forming induced by multiple fusion genes. CFA was performed using wild-type BM progenitor cells co-transduced with MSCV-neo-MLL-AF9 (MA9), -MLL-AF10 (MA10), -PML-RARA (PR) or –AML1-ETO9a(AE9a)19, together with MSCV-PIG (Ctrl) or MSCV-PIG-miR-22 (+miR-22), as well as miR-22−/− BM progenitors co-transduced with individual fusion genes and MSCV-PIG. Colony counts (mean±s.d.) of the second round of plating are shown. *P<0.05; **P<0.01. (e,f) Effect of miR-22 on MLL-AF9-induced primary leukemogenesis. Kaplan–Meier curves are shown for six cohorts of transplanted mice including MSCVneo+MSCV-PIG (Ctrl; n=5), MSCVneo+MSCV-PIG-miR-22 (miR-22; n=5), MSCVneo-MLL-AF9+MSCV-PIG (MA9; n=8), MSCVneo-MLL-AF9+MSCV-PIG-miR-150 (MA9+miR-150, n=6), MSCVneo-MLL-AF9+MSCV-PIG-miR-22 (MA9+miR-22; n=10) and MSCVneo-MLL-AF9+MSCV-PIG-miR-22mutant (MA9+miR-22mut; n=5) (e); Wright–Giemsa stained PB and bone marrow (BM), and hematoxylin and eosin (H&E) stained spleen and liver of the primary BMT recipient mice at the end point are shown (f). (g) Effect of miR-22 on MLL-AF10-induced primary leukemogenesis. Kaplan–Meier curves are shown for two cohorts of transplanted mice including MSCVneo-MLL-AF10+MSCV-PIG (MA10; n=5) and MSCVneo-MLL-AF10+MSCV-PIG-miR-22 (MA10+miR-22; n=5). (h) miR-22 knockout promotes AE9a-induced leukemogenesis. Kaplan–Meier curves are shown for mice transplanted with wild-type or miR-22−/− BM progenitor cells transduced MSCV-PIG-AE9a (n=5 for each group). The P values were generated by t-test (a–d) or log-rank test (e,g,h).
To rule out the possibility that the inhibitory effect of miR-22 shown in Supplementary Fig. 1a was due to a non-specific effect of our miR-22 construct, we included the MSCV-PIG-miR-22 construct from Song et al.16 in a repeated CFA. Both miR-22 constructs dramatically inhibited MLL-AF9-induced colony formation (Fig. 1c). As the ‘seed’ sequences at the 5′ end of individual miRNAs are essential for the miRNA-target binding18, we also mutated the 6-bases ‘seed’ sequence of miR-22 and found that the miR-22 mutant did not inhibit colony formation anymore (Fig. 1c). In human AML cells, forced expression of miR-22, but not miR-22 mutant, significantly inhibited cell viability and growth/proliferation, while promoting apoptosis (Supplementary Fig. 1c,d).
Furthermore, as miR-22 is globally downregulated in all major types of AML (Fig. 1b), we also investigated the role of miR-22 in colony formation induced by other oncogenic fusion genes, including MLL-AF10/t(10;11), PML-RARA/t(15;17) and AML1-ETO9a/t(8;21) (ref. 19). As expected, forced expression of miR-22 significantly inhibited colony formation induced by all individual oncogenic fusions; conversely, miR-22 knockout20 significantly enhanced colony forming (Fig. 1d). These results suggest that miR-22 likely plays a broad anti-tumour role in AML.
In accordance with the potential anti-tumour function of miR-22 in AML, miR-22 was expressed at a significantly higher level (P<0.05) in human normal CD33+ myeloid progenitor cells than in more immature CD34+ HSPCs or MNC cells (a mixed population containing both primitive progenitors and committed cells) (Fig. 1a,b), implying that miR-22 is upregulated during normal myelopoiesis. Similarly, we showed that miR-22 was also expressed at a significantly higher level in mouse normal bone marrow (BM) myeloid (Gr-1+/Mac-1+) cells, relative to lineage negative (Lin−) progenitor cells, long-term hematopoietic stem cells (LT-HSCs), short-term HSCs (ST-HSCs), and committed progenitors (CPs) (Supplementary Fig. 1e), further suggesting that miR-22 is upregulated in normal myelopoiesis.
The anti-tumour effect of miR-22 in the pathogenesis of AML
Through bone marrow transplantation (BMT) assays, we showed that forced expression of miR-22 (but not miR-22 mutant) dramatically blocked MLL-AF9 (MA9)-mediated leukemogenesis in primary BMT recipient mice, with a more potent inhibitory effect than miR-150 (Fig. 1e;Supplementary Fig. 2a). All MA9+miR-22 mice exhibited normal morphologies in peripheral blood (PB), BM, spleen and liver tissues (Fig. 1f), with a substantially reduced c-Kit+ blast cell population in BM (Supplementary Fig. 2b). Forced expression of miR-22 also almost completely inhibited leukemogenesis induced by MLL-AF10 (Fig. 1g; Supplementary Fig. 2a). Conversely, miR-22 knockout significantly promoted AML1-ETO9a (AE9a)-induced AML (Fig. 1h). Thus, the repression of miR-22 is critical for the development of primary AML. Notably, forced expression of miR-22 inMLL-AF9 and MLL-AF10 leukaemia mouse models caused only a 2–3-fold increase in miR-22 expression level (Supplementary Fig. 2a), in a degree comparable to the difference in miR-22 expression levels between human AML samples and normal controls (Fig. 1a), suggesting that a 2–3-fold change in miR-22 expression level appears to be able to exert significant physiological or pathological effects.
To examine whether the maintenance of AML is also dependent on the repression of miR-22, we performed secondary BMT assays. Forced expression of miR-22 remarkably inhibited progression of MLL-AF9-, AE9a– or FLT3-ITD/NPM1c+-induced AML in secondary recipient mice (Fig. 2a–d), resulting in largely normal morphologies in PB, BM, spleen and liver tissues (Fig. 2b;Supplementary Fig. 2c). Collectively, our findings demonstrate that miR-22 is a pivotal anti-tumour gatekeeper in both development and maintenance of various AML.
Figure 2: Effect of miR-22 on the maintenance of AMLin vivo.
(a,b) Effect of miR-22 on the maintenance of MLL-AF9-induced AML in secondary BMT recipient mice. The secondary BMT recipients were transplanted with BM blast cells from the primary MLL-AF9 AML mice retrovirally transduced with MSCV-PIG+MSCVneo (MA9-AML+Ctrl; n=7) or MSCV-PIG+MSCVneo-miR-22 (MA9-AML+miR-22; n=10). Kaplan–Meier curves (a) and Wright–Giemsa or H&E-stained PB, BM, spleen and liver (b) of the secondary leukaemic mice are shown. (c,d) Effect of miR-22 on the maintenance/progression of AML1-ETO9a (AE9a)-induced AML (c) or FLT3-ITD/NPM1c+-induced AML (d) in secondary BMT recipient mice (n=5 for each group). Kaplan–Meier curves and P values (log-rank test) are shown.
Identification of critical target genes of miR-22 in AML
To identify potential targets of miR-22 in AML, we performed a series of data analysis. Analysis of In-house_81S (ref. 21) and TCGA_177S (ref. 22) data sets revealed a total of 999 genes exhibiting significant inverse correlations with miR-22 in expression. Of them, 137 genes, including 21 potential targets of miR-22 as predicted by TargetScan18 (Supplementary Table 1), were significantly upregulated in both human and mouse AML compared with normal controls as detected in two additional in-house data sets14, 23. Among the 21 potential targets, CRTC1, ETV6and FLT3 are known oncogenes24, 25, 26, 27, 28, 29. We then focused on these three genes, along with MYCBP that encodes the MYC-binding protein and is an experimentally validated target of miR-22 (ref. 30) although due to a technical issue it was not shown in the 21-gene list (Supplementary Table 1), for further studies.
As expected, all four genes were significantly downregulated in expression by ectopic expression of miR-22 in human MONOMAC-6/t(9;11) cells (Fig. 3a). The coincidence of downregulation of those genes and upregulation of miR-22 was also observed in mouse MLL-ENL-ERtm cells, a leukaemic cell line with an inducible MLL-ENL derivative31, when MLL-ENL was depleted by 4-hydroxy-tamoxifen (4-OHT) withdrawal (Fig. 3b; Supplementary Fig. 3a). While MLL-AF9 remarkably promoted expression of those four genes in mouse BM progenitor cells, co-expressed miR-22 reversed the upregulation (Fig. 3c). In leukaemia BM blast cells of mice with MLL-AF9-induced AML, the expression of Crtc1, Flt3 and Mycbp, but not Etv6, was significantly downregulated by co-expressed miR-22 (but not by miR-22 mutant) (Fig. 3d). Because miR-22-mediated downregulation of Etv6 could be observed only in the in vitro models (Fig. 3a–c), but not in the in vivo model (Fig. 3d), which was probably due to the difference between in vitro and in vivo microenvironments, we decided to focus on the three target genes (that is, Crtc1, Flt3 and Mycbp) that showed consistent patterns between in vitro and in vivo for further studies. The repression of Crtc1, Flt3 and Mycbpwas also found in leukaemia BM cells of mice with AE9a or FLT3-ITD/NPM1c+-induced AML (Fig. 3e,f). As Mycbp is already a known target of miR-22 (ref. 30), here we further confirmed that FLT3and CRTC1 are also direct targets of miR-22 (Fig. 3g,h). The downregulation of CRTC1, FLT3 and MYCBP by miR-22 at the protein level was confirmed in both human and mouse leukaemic cells (Supplementary Fig. 3b,c). Overexpression of miR-22 had no significant influence on the level of leukaemia fusion genes (Supplementary Fig. 3d).
Figure 3: miR-22 targets multiple oncogenes.
(a) Downregulation of CRTC1, FLT3, MYCBP and ETV6 by forced expression of miR-22 in MONOMAC-6 cells. Expression of these genes was detected 48h post transfection of MSCV-PIG (Ctrl) or MSCV-PIG-miR-22 (miR-22). (b) Crtc1, Flt3, Mycbp and Etv6 levels in MLL-ENL-ERtm cells after withdrawal of 4-OHT for 0, 7 or 10 days. (c) Expression levels of Crtc1, Flt3, Mycbp and Etv6 in mouse BM progenitor cells retrovirally transduced with MSCV-PIG+MSCV-neo (Ctrl), MSCV-PIG-miR-22+MSCV-neo (miR-22), MSCV-PIG+MSCV-neo-MLL-AF9 (MLL-AF9) or MSCV-PIG-miR-22+MSCV-neo-MLL-AF9 (MLL-AF9+miR-22). (d) Expression levels of Crtc1, Flt3, Mycbp and Etv6 in BM blast cells of leukaemic mice transplanted with MLL-AF9, MLL-AF9+miR-22 or MLL-AF9+miR-22mut primary leukaemic cells. (e,f) Expression levels of Crtc1, Flt3 and Mycbp in BM blast cells of leukaemic mice transplanted with MSCV-PIG or MSCV-PIG-miR-22-retrovirally transduced AE9a (e) or FLT3-ITD/NPM1c+ (f) primary leukaemic cells. (g) Putative miR-22 target sites and mutants in the 3′UTRs of CRTC1 (upper panel) and FLT3(lower panel). (h) Effects of miR-22 on luciferase activity of the reporter gene bearing wild type or mutant 3′UTRs of CRTC1 or FLT3 in HEK293T cells. The mean±s.d. values from three replicates are shown.*P<0.05, t-test.
Co-expression of the coding region (CDS) of each of the three target genes (that is, CRTC1, FLT3and MYCBP) largely reversed the effects of miR-22 on cell viability, apoptosis and proliferation (Fig. 4a–e). More importantly, in vivo BMT assays showed that co-expressing CRTC1, FLT3 orMYCBP largely rescued the inhibitory effect of miR-22 on leukemogenesis (Fig. 4f,g;Supplementary Fig. 3e). Our data thus suggest that CRTC1, FLT3 and MYCBP are functionally important targets of miR-22 in AML.
Figure 4: Multiple onocgenes are functionally important targets of miR-22 in AML.
(a,b) Relative viability (a) and apoptosis (b) levels of MONOMAC-6 cells transfected with MSCV-PIG-CRTC1, -FLT3 or –MYCBP alone, or together with MSCVneo-miR-22. Values were detected 48h post transfection. (c–e) Rescue effects of CRTC1 (c), FLT3 (d) and MYCBP (e) on the inhibition of MONOMAC-6 growth mediated by miR-22. Cell counts at the indicated time points are shown. Mean±s.d. values are shown. *P<0.05, t-test. (f) In vivo rescue effects of CRTC1, FLT3 and MYCBP on the inhibition of MLL-AF9-induced leukemogenesis mediated by miR-22. The secondary recipients were transplanted with BM blast cells of the primary MLL-AF9 leukaemic mice retrovirally transduced with MSCVneo+MSCV-PIG (MA9-AML+Ctrl; n=7), MSCVneo-miR-22+MSCV-PIG (MA9-AML+miR-22; n=10), MSCVneo-miR-22+MSCV-PIG-CRTC1 (MA9-AML+miR-22+CRTC1; n=5), MSCVneo-miR-22+MSCV-PIG-FLT3 (MA9-AML+miR-22+FLT3; n=6) or MSCVneo-miR-22+MSCV-PIG-MYCBP (MA9-AML+miR-22+MYCBP; n=6). Kaplan–Meier curves for all the five groups of transplanted mice are shown. MA9-AML+Ctrl versus MA9-AML+miR-22, P<0.001 (log-rank test); MA9-AML+Ctrl versus any other groups,P>0.05 (log-rank test). (g) Wright–Giemsa stained PB and BM, and H&E stained spleen and liver of the secondary leukaemic mice.
(a) Correlation between the expression levels of miR-22 and TET1 in three independent AML patient databases. All expression data were log(2) transformed; the data in In-house_81S were also mean-centred. The correlation coefficient (r) and P values were detected by ‘Pearson Correlation’, and the correlation regression lines were drawn with the ‘linear regression’ algorithm. (b) Expression of pri-, pre- and mature miR-22, and Tet1/2/3 in colony-forming cells of wild-type mouse BM progenitors retrovirally transduced with MSCVneo (Ctrl), MSCVneo-MLL-AF9 (MLL-AF9), MSCVneo-MLL-AF10 (MLL-AF10) or MSCVneo-AE9a (AE9a), or of FLT3-ITD/NPM1c+ mouse BM progenitors transduced with MSCVneo (FLT3-ITD+/NPM1c+). (c) Expression of miR-22 and Tet1/2/3 in MLL-ENL-ERtm cells. Expression levels were detected at the indicated time points post 4-OHT withdrawal. (d) Effect of miR-22 overexpression onTet1 expression in colony-forming cells with MLL-AF9, AE9a or FLT3-ITD/NPM1c+. (e) Expression ofTet1 in BM progenitor cells of 6-weeks old miR-22−/− or wild-type mice. (f) Effect of miR-22 overexpression on TET1 expression in THP-1 and KOCL-48 AML cells 48h post transfection. (g) Expression of pri-, pre- and mature miR-22 in BM progenitor cells of 6-weeks old Tet1−/− or wild-type mice. Mean±s.d. values are shown. *P<0.05, t-test.
(a) Tet1 targets miR-22 promoter region (−1,100/+55bp), as detected by luciferase reporter assay 48h post transfection in HEK293T cells. (b) Expression of TET1/2/3, EZH2, SIN3A, GFI1 and miR-22 in THP-1 cells 72h post treatment with 1μM ATRA or DMSO control. (c) Co-immunoprecipitation assay showing the binding of endogenous GFI1 and TET1 in THP1 cells. (d) ChIP-qPCR analyses of the promoter region of miR-22 in THP-1 cells 72h post treatment with 1μM ATRA or DMSO. Upper panel: PCR site on the CpG-enriched region of miR-22 gene locus. Note: miR-22 is coded within the second exon of a long non-coding RNA (MIR22HG), which represents the primary transcript of miR-22. Lower panels: enrichment of MLL-N terminal (for both wild-type MLL and MLL-fusion proteins), MLL-C terminal (for wild-type MLL), TET1, EZH2, SIN3A, GFI1, H3K27me3, H3K4me3 or RNA pol II at miR-22 promoter region. (e) Expression levels of TET1, EZH2, SIN3A and miR-22 in GFI1 knockdown cells. (f) ChIP-qPCR analyses of the promoter region of miR-22 in THP-1 cells transduced with GFI1 shRNA or control shRNA. Enrichment of GFI1, TET1, EZH2 and SIN3A are shown. (g) Effects of knockdown of TET1, EZH2 and/orSIN3A on miR-22 expression. The expression level of miR-22 was detected in THP-1 cells 72h post transfection with siRNAs targeting TET1, EZH2 and/or SIN3A. Mean±s.d. values are shown. *P<0.05;**P<0.01 (t-test). (h) Schematic model of the regulatory pathway involving miR-22 in AML and ATRA treatment.
The miR-22-associated regulatory circuit in AML
Restoration of miR-22 expression and function to treat AML
Figure 7: Therapeutic effect of miR-22-nanoparticles in treating AML.
(a,b) Primary leukaemia BM cells bearing MLL-AF9 (a) or AE9a (b) were transplanted into sublethally irradiated secondary recipient mice. After the onset of secondary AML (usually 10 days post transplantation), the recipient mice were treated with PBS control, or 0.5mgkg−1 miR-22 or miR-22 mutant RNA oligos formulated with G7 PAMAM dendrimer nanoparticles, i.v., every other day, until the PBS-treated control group all died of leukaemia. (c) NSGS mice49 were transplanted with MV4;11/t(4;11) AML cells. Five days post transplantation, these mice started to be treated with PBS control, miR-22 or miR-22 mutant nanoparticles at the same dose as described above. Kaplan–Meier curves are shown; the drug administration period and frequency were indicated with yellow arrows. The P values were detected by log-rank test. (d) Wright–Giemsa stained PB and BM, and H&E stained spleen and liver of the MLL-AF9-secondary leukaemic mice treated with PBS control, miR-22 or miR-22 mutant nanoparticles.
We then tested the miR-22 nanoparticles in a xeno-transplantation model49. Similarly, the nanoparticles carrying miR-22 oligos, but not miR-22 mutant, significantly delayed AML progression induced by human MV4;11/t(4;11) cells (Fig. 7c). The miR-22-nanoparticle administration also resulted in less aggressive leukaemic pathological phenotypes in the recipient mice (Supplementary Fig. 6e). Thus, our studies demonstrated the therapeutic potential of using miR-22-based nanoparticles to treat AML.
It remains poorly understood how TET proteins mediate gene regulation in cancer. Here we show that in de novo AML, it is TET1, but not TET2 (a reported direct target of miR-22 in MDS and breast cancer15, 16), that inversely correlates with miR-22 in expression and negatively regulates miR-22 at the transcriptional level. Likely together with GFI1, TET1 recruits polycomb cofactors (for example, EZH2/SIN3A) to the miR-22 promoter, leading to a significant increase in H3K27me3 occupancy and decrease in RNA pol II occupancy at that region, and thereby resulting in miR-22 repression in AML cells; such a repression can be abrogated by ATRA treatment. Thus, our study uncovers a novel epigenetic regulation mechanism in leukaemia involving the cooperation between TET1/GFI1 and polycomb factors.
Besides GFI1, it was reported that LSD1 is also a binding partner of TET1 (ref. 50). Interestingly, LSD1 is known as a common binding partner shared by TET1 and GFI1, and mediates the effect of GFI1 on hematopoietic differentiation51, 52. Thus, it is possible that LSD1 might also participate in the transcriptional repression of miR-22 as a component of the GFI1/TET1 repression complex.
We previously reported that TET1 cooperates with MLL fusions in positively regulating their oncogenic co-targets in MLL-rearranged AML14. Here we show that TET1 can also function as a transcriptional repressor (of a miRNA) in cancer. The requirement of TET1-mediated regulation on expression of its positive (for example, HOXA/MEIS1/PBX3)14 or negative (for example, miR-22) downstream effectors in leukemogenesis likely explains the rareness of TET1 mutations in AML53, and highlights its potent oncogenic role in leukaemia.
The aberrant activation of both CREB and MYC signalling pathways has been shown in AML24, 25,26, 54, 55, but the underlying molecular mechanisms remain elusive. Our data suggest that the activation of these two signalling pathways in AML can be attributed, at least in part, to the repression of miR-22, which in turn, results in the de-repression of CRTC1 (CREB pathway), FLT3and MYCBP (MYC pathway), and leads to the upregulation of oncogenic downstream targets (for example, CDK6, HOXA7, BMI1, FASN and HMGA1) and downregulation of tumour-suppressor downstream targets (for example, RGS2).
In summary, we uncover a TET1/GFI1/EZH2/SIN3A⊣miR-22⊣CREB-MYC signalling circuit in de novo AML, in which miR-22 functions as a pivotal anti-tumour gate-keeper, distinct from its oncogenic role reported in MDS or MDS-derived AML16. Thus, our study together with the study of Song et al.16 highlight the complexity and functional importance of miR-22-associated gene regulation and signalling pathways in hematopoietic malignancies, and may provide novel insights into the genetic/epigenetic differences between de novo AML and MDS.
Our findings also highlight the possibility of using miR-22-based therapy to treat AML patients. Our proof-of-concept studies demonstrate that the nanoparticles carrying miR-22 oligos significantly inhibit AML progression and prolong survival of leukaemic mice in both BMT and xeno-transplantation models. Notably, miRNA-based nanoparticles have already entered clinical trials56. It would be important, in the future, to further test the combination of miR-22-carrying nanoparticles (or small-molecule compounds that can induce endogenous expression of miR-22) with standard chemotherapy agents (cytosine arabinoside and anthracycline), or with the emerging small molecule inhibitors against MYC and/or CREB pathway effectors, to achieve optimal anti-leukaemia effect with minimal side effects. Overall, our results suggest that restoration of miR-22 expression/function (for example, using miR-22-carrying nanoparticles or small-molecule compounds) holds great therapeutic potential to treat AML, especially those resistant to current therapies.
MicroRNAs: A Gene Silencing Mechanism with Therapeutic Implications
MicroRNAs (miRNAs) are single-stranded RNAs about 22 nucleotides in length that repress the expression of specific proteins by annealing to complementary sequences in the 3′ untranslated regions (UTRs) of target mRNAs. Apart from their posttranscriptional expression, or silencing, miRNAs may also direct mRNA destabilization and cleavage. Moreover, rather than targeting a single disease-associated protein target as many small molecule drugs and antibodies do, each miRNA may serve to repress the expression of numerous proteins involved in the pathogenesis and progression of various diseases and could therefore potentially interfere with multiple disease-promoting signal transduction pathways. Because aberrant expression of miRNAs has been implicated in numerous disease states, miRNA-based therapies have sparked much interest for the treatment of a variety of diseases. The objective of this symposium is to bring together investigators who have led the field in describing what miRNAs do and their potential in treating diseases, as well as those who are translating these findings into promising drug candidates, some of which have already advanced into early stage clinical trials.
Call for Poster Abstracts
Abstract submissions are invited for a poster session. For complete submission instructions, please send an email to miRNA@nyas.org with the words “Abstract Information” in the subject line. The deadline for abstract submission is May 13, 2016.
Chemotherapy Benefit in Early Breast Cancer Patients
Larry H Bernstein, MD, FCAP, Curator
LPBI
Agendia’s MammaPrint® First and Only Genomic Assay to Receive Level 1A Clinical Utility Evidence for Chemotherapy Benefit in Early Breast Cancer Patients
Clinical high-risk patients with a low-risk MammaPrint® result, including 48 percent node-positive, had five-year distant metastasis-free survival rate in excess of 94 percent, whether randomized to receive adjuvant chemotherapy or not
MammaPrint could change clinical practice by substantially de-escalating the use of adjuvant chemotherapy and sparing many patients an aggressive treatment they will not benefit from
Forty-six percent overall reduction in chemotherapy prescription among clinically high-risk patients
April 19, 2016 / B3C newswire / —Agendia, Inc., together with the European Organisation for Research and Treatment of Cancer (EORTC) and Breast International Group (BIG), announced results from the initial analysis of the primary objective of the Microarray In Node-negative (and 1 to 3 positive lymph node) Disease may Avoid ChemoTherapy (MINDACT) study at the American Association for Cancer Research Annual Meeting 2016 in New Orleans, LA.
Using the company’s MammaPrint® assay, patients with early-stage breast cancer who were considered at high risk for disease recurrence based on clinical and biological criteria had a distant metastasis-free survival at five years in excess of 94 percent.The MammaPrint test—the first and only genomic assay with FDA 510(k) clearance for use in risk assessment for women of all ages with early stage breast cancer—identified a large group of patients for whom five-year distant metastasis–free survival was equally good whether or not they received adjuvant chemotherapy (chemotherapy given post-surgery).
“The MINDACT trial design is the optimal way to prove clinical utility of a genomic assay,” said Prof. Laura van ’t Veer, CRO at Agendia, Leader, Breast Oncology Program, and Director, Applied Genomics at UCSF Helen Diller Family Comprehensive Cancer Center. “It gives the level 1A clinical evidence (prospective, randomized and controlled) that empowers physicians to clearly and confidently know when chemotherapy is part of optimal early-stage breast cancer therapy. In this trial, MammaPrint (70-gene assay) was compared to the standard of care physicians use today, to decide what is the best treatment option for an early-stage breast cancer patient.”
The MINDACT trial is the first prospective randomized controlled clinical trial of a breast cancer recurrence genomic assay with level 1A clinical evidence and the first prospective translational research study of this magnitude in breast cancer to report the results of its primary objective.
Among the 3,356 patients enrolled in the MINDACT trial, who were categorized as having a high risk of breast cancer recurrence based on common clinical and pathological criteria (C-high), the MammaPrint assay reduced the chemotherapy treatment prescription by 46 percent.Using the 70-gene assay, MammaPrint, 48 percent of lymph-node positive breast cancer patients considered clinically high-risk (Clinical-high) and genomic low-risk (MammaPrint-low) had an excellent distant metastasis-free survival at five years in excess of 94 percent.
“Traditionally, physicians have relied on clinical-pathological factors such as age, tumor size, tumor grade, lymph node involvement, and hormone receptor status to make breast cancer treatment decisions,” said Massimo Cristofanilli, MD, Associate Director of Translational Research and Precision Medicine at the Robert H. Lurie Comprehensive Cancer Center, Northwestern University in Chicago. “These findings provide level 1A clinical utility evidence by demonstrating that the detection of low-risk of distant recurrence reported by the MammaPrint test can be safely used in the management of thousands of women by identifying those who can be spared from a toxic and unnecessary treatment.”
MINDACT is a randomized phase III trial that investigates the clinical utility of MammaPrint, when compared (or – “used in conjunction with”) to the standard clinical pathological criteria, for the selection of patients unlikely to benefit from adjuvant chemotherapy. From 2007 to 2011, 6,693 women who had undergone surgery for early-stage breast cancer enrolled in the trial (111 centers in nine countries). Participants were categorized as low or high risk for tumor recurrence in two ways: first, through analysis of tumor tissue using MammaPrint at a central location in Amsterdam; and second, using Adjuvant! Online, a tool that calculates risk of breast cancer recurrence based on common clinical and biological criteria.
Patients characterized in both clinical and genomic assessments as “low- risk” are spared chemotherapy, while patients characterized as “high- risk” are advised chemotherapy. Those with conflicting results are randomized to use either clinical or genomic risk (MammaPrint) evaluation to decide on chemotherapy treatment.
The MINDACT trial is managed and sponsored by the EORTC as part of an extensive and complex partnership in collaboration with Agendia and BIG, and many other academic and commercial partners, as well as patient advocates.
“These MINDACT trial results are a testament that the science of the MammaPrint test is the most robust in the genomic breast recurrence assay market. Agendia will continue to collaborate with pharmaceutical companies, leading cancer centers and academic groups on additional clinical research and in the pursuit of bringing more effective, individualized treatments within reach of cancer patients,” said Mark Straley, Chief Executive Officer at Agendia. “We value the partnership with the EORTC and BIG and it’s a great honor to share this critical milestone.”
Breast cancer is the most frequently diagnosed cancer in women worldwide(1). In 2012, there were nearly 1.7 million new breast cancer cases among women worldwide, accounting for 25 percent of all new cancer cases in women(2).
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….
In 2013, two independent teams of scientists, one in Maryland and one in France, made a surprising observation: both germ-free mice and mice treated with a heavy dose of antibiotics responded poorly to a variety of cancer therapies typically effective in rodents. The Maryland team, led by Romina Goldszmidand Giorgio Trinchieri of the National Cancer Institute, showed that both an investigational immunotherapy and an approved platinum chemotherapy shrank a variety of implanted tumor types and improved survival to a far greater extent in mice with intact microbiomes.1 The French group, led by INSERM’s Laurence Zitvogel, got similar results when testing the long-standing chemotherapeutic agent cyclophosphamide in cancer-implanted mice, as well as in mice genetically engineered to develop tumors of the lung.2
The findings incited a flurry of research and speculation about how gut microbes contribute to cancer cell death, even in tumors far from the gastrointestinal tract. The most logical link between the microbiome and cancer is the immune system. Resident microbes can either dial up inflammation or tamp it down, and can modulate immune cells’ vigilance for invaders. Not only does the immune system appear to be at the root of how the microbiome interacts with cancer therapies, it also appears to mediate how our bacteria, fungi, and viruses influence cancer development in the first place.
“We clearly see shifts in the [microbial] community that precede development of tumors,” says microbiologist and immunologist Patrick Schloss, who studies the influence of the microbiome on colon cancer at the University of Michigan.
But the relationship between the microbiome and cancer is complex: while some microbes promote cell proliferation, others appear to protect us against cancerous growth. And in some cases, the conditions that spur one cancer may have the opposite effect in another. “It’s become pretty obvious that the commensal microbiota affect inflammation and, through that or through other mechanisms, affect carcinogenesis,” says Trinchieri. “What we really need is to have a much better understanding of which species, which type of bug, is doing what and try to change the balance.”
Gut feeling
In the late 1970s, pathologist J. Robin Warren of Royal Perth Hospital in Western Australia began to notice that curved bacteria often appeared in stomach tissue biopsies taken from patients with chronic gastritis, an inflammation of the stomach lining that often precedes the development of stomach cancer. He and Barry J. Marshall, a trainee in internal medicine at the hospital, speculated that the bacterium, now called Helicobacter pylori, was somehow causing the gastritis.3 So committed was Marshall to demonstrating the microbe’s causal relationship to the inflammatory condition that he had his own stomach biopsied to show that it contained no H. pylori, then infected himself with the bacterium and documented his subsequent experience of gastritis.4 Scientists now accept that H. pylori, a common gut microbe that is present in about 50 percent of the world’s population, is responsible for many cases of gastritis and most stomach ulcers, and is a strong risk factor for stomach cancer.5 Marshall and Warren earned the 2005 Nobel Prize in Physiology or Medicine for their work.
H. pylori may be the most clear-cut example of a gut bacterium that influences cancer development, but it is likely not the only one. Researchers who study cancer in mice have long had anecdotal evidence that shifts in the microbiome influence the development of diverse tumor types. “You have a mouse model of carcinogenesis. It works beautifully,” says Trinchieri. “You move to another institution. It works completely differently,” likely because the animals’ microbiomes vary with environment.
Around the turn of the 21st century, cancer researchers began to systematically experiment with the rodent microbiome, and soon had several lines of evidence linking certain gut microbes with a mouse’s risk of colon cancer. In 2001, for example, Shoichi Kado of the Yakult Central Institute for Microbiological Research in Japan and colleagues found that a strain of immunocompromised mice rapidly developed colon tumors, but that germ-free versions of these mice did not.6 That same year, an MIT-based group led by the late David Schauer demonstrated that infecting mice with the bacterium Citrobacter rodentium spurred colon tumor development.7 And in 2003, MIT’s Susan Erdman and her colleagues found that they could induce colon cancer in immunocompromised mice by infecting them with Helicobacter hepaticus, a relative of? H. pylori that commonly exists within the murine gut microbiome.8
More recent work has documented a similar link between colon cancer and the gut microbiome in humans. In 2014, a team led by Schloss sequenced 16S rRNA genes isolated from the stool of 90 people, some with colon cancer, some with precancerous adenomas, and still others with no disease.9 The researchers found that the feces of people with cancer tended to have an altered composition of bacteria, with an excess of the common mouth microbes Fusobacterium or Porphyromonas. A few months later, Peer Bork of the European Molecular Biology Laboratory performed metagenomic sequencing of stool samples from 156 people with or without colorectal cancer. Bork and his colleagues found they could predict the presence or absence of cancer using the relative abundance of 22 bacterial species, including Porphyromonas andFusobacterium.10 They could also use the method to predict colorectal cancer with about the same accuracy as a blood test, correctly identifying about 50 percent of cancers while yielding false positives less than 10 percent of the time. When the two tests were combined, they caught more than 70 percent of cancers.
Whether changes in the microbiota in colon cancer patients are harbingers of the disease or a consequence of tumor development remained unclear. “What comes first, the change in the microbiome or tumor development?” asks Schloss. To investigate this question, he and his colleagues treated mice with microbiome-altering antibiotics before administering a carcinogen and an inflammatory agent, then compared the outcomes in those animals and in mice that had received only the carcinogenic and inflammatory treatments, no antibiotics. The antibiotic-treated animals had significantly fewer and smaller colon tumors than the animals with an undisturbed microbiome, suggesting that resident bacteria were in some way promoting cancer development. And when the researchers transferred microbiota from healthy mice to antibiotic-treated or germ-free mice, the animals developed more tumors following carcinogen exposure. Sterile mice that received microbiota from mice already bearing malignancies developed the most tumors of all.11
Most recently, Schloss and his colleagues showed that treating mice with seven unique combinations of antibiotics prior to exposing them to carcinogens yielded variable but predictable levels of tumor formation. The researchers determined that the number of tumors corresponded to the unique ways that each antibiotic cocktail modulated the microbiome.12
“We’ve kind of proven to ourselves, at least, that the microbiome is involved in colon cancer,” says Schloss, who hypothesizes that gut bacteria–driven inflammation is to blame for creating an environment that is hospitable to tumor development and growth. Gain or loss of certain components of the resident bacterial community could lead to the release of reactive oxygen species, damaging cells and their genetic material. Inflammation also involves increased release of growth factors and blood vessel proliferation, potentially supporting the growth of tumors. (See illustration above.)
Recent research has also yielded evidence that the gut microbiota impact the development of cancer in sites far removed from the intestinal tract, likely through similar immune-modulating mechanisms.
Systemic effects
In the mid-2000s, MIT’s Erdman began infecting a strain of mice predisposed to intestinal tumors withH. hepaticus and observing the subsequent development of colon cancer in some of the animals. To her surprise, one of the mice developed a mammary tumor. Then, more of the mice went on to develop mammary tumors. “This told us that something really interesting was going on,” Erdman recalls. Sure enough, she and her colleagues found that mice infected with H. hepaticus were more likely to develop mammary tumors than mice not exposed to the bacterium.13The researchers showed that systemic immune activation and inflammation could contribute to mammary tumors in other, less cancer-prone mouse models, as well as to the development of prostate cancer.
At the University of Chicago, Thomas Gajewski and his colleagues have taken a slightly different approach to studying the role of the microbiome in cancer development. By comparing Black 6 mice coming from different vendors—Taconic Biosciences (formerly Taconic Farms) and the Jackson Laboratory—Gajewski takes advantage of the fact that the animals’ different origins result in different gut microbiomes. “We deliberately stayed away from antibiotics, because we had a desire to model how intersubject heterogeneity [in cancer development] might be impacted by the commensals they happen to be colonized with,” says Gajewski in an email to The Scientist.
Last year, the researchers published the results of a study comparing the progression of melanoma tumors implanted under the mice’s skin, finding that tumors in the Taconic mice grew more aggressively than those in the Jackson mice. When the researchers housed the different types of mice together before their tumors were implanted, however, these differences disappeared. And transferring fecal material from the Jackson mice into the Taconic mice altered the latter’s tumor progression.14
Instead of promoting cancer, in these experiments the gut microbiome appeared to slow tumor growth. Specifically, the reduced tumor growth in the Jackson mice correlated with the presence of Bifidobacterium, which led to the greater buildup of T?cells in the Jackson mice’s tumors. Bifidobacteriaactivate dendritic cells, which present antigens from bacteria or cancer cells to T?cells, training them to hunt down and kill these invaders. Feeding Taconic mice bifidobacteria improved their response to the implanted melanoma cells.
“One hypothesis going into the experiments was that we might identify immune-suppressive bacteria, or commensals that shift the immune response towards a character that was unfavorable for tumor control,” says Gajewski. “But in fact, we found that even a single type of bacteria could boost the antitumor immune response.”
Ideally, the immune system should recognize cancer as invasive and nip tumor growth in the bud. But cancer cells display “self” molecules that can inhibit immune attack. A new type of immunotherapy, dubbed checkpoint inhibition or blockade, spurs the immune system to attack cancer by blocking either the tumor cells’ surface molecules or the receptors on T?cells that bind to them.
CANCER THERAPY AND THE MICROBIOME
In addition to influencing the development and progression of cancer by regulating inflammation and other immune pathways, resident gut bacteria appear to influence the effectiveness of many cancer therapies that are intended to work in concert with host immunity to eliminate tumors.
Some cancer drugs, such as oxaliplatin chemotherapy and CpG-oligonucleotide immunotherapy, work by boosting inflammation. If the microbiome is altered in such a way that inflammation is reduced, these therapeutic agents are less effective.
Cancer-cell surface proteins bind to receptors on T cells to prevent them from killing cancer cells. Checkpoint inhibitors that block this binding of activated T cells to cancer cells are influenced by members of the microbiota that mediate these same cell interactions.
Cyclophosphamide chemotherapy disrupts the gut epithelial barrier, causing the gut to leak certain bacteria. Bacteria gather in lymphoid tissue just outside the gut and spur generation of T helper 1 and T helper 17 cells that migrate to the tumor and kill it.
As part of their comparison of Jackson and Taconic mice, Gajewski and his colleagues decided to test a type of investigational checkpoint inhibitor that targets PD-L1, a ligand found in high quantities on the surface of multiple types of cancer cells. Monoclonal antibodies that bind to PD-L1 block the PD-1 receptors on T?cells from doing so, allowing an immune response to proceed against the tumor cells. While treating Taconic mice with PD-L1–targeting antibodies did improve their tumor responses, they did even better when that treatment was combined with fecal transfers from Jackson mice, indicating that the microbiome and the immunotherapy can work together to take down cancer. And when the researchers combined the anti-PD-L1 therapy with a bifidobacteria-enriched diet, the mice’s tumors virtually disappeared.14
Gajewski’s group is now surveying the gut microbiota in humans undergoing therapy with checkpoint inhibitors to better understand which bacterial species are linked to positive outcomes. The researchers are also devising a clinical trial in which they will give Bifidobacterium supplements to cancer patients being treated with the approved anti-PD-1 therapy pembrolizumab (Keytruda), which targets the immune receptor PD-1 on T?cells, instead of the cancer-cell ligand PD-L1.
Meanwhile, Zitvogel’s group at INSERM is investigating interactions between the microbiome and another class of checkpoint inhibitors called CTLA-4 inhibitors, which includes the breakthrough melanoma treatment ipilimumab (Yervoy). The researchers found that tumors in antibiotic-treated and germ-free mice had poorer responses to a CTLA-4–targeting antibody compared with mice harboring unaltered microbiomes.15 Particular Bacteroides species were associated with T-cell infiltration of tumors, and feedingBacteroides fragilis to antibiotic-treated or germ-free mice improved the animals’ responses to the immunotherapy. As an added bonus, treatment with these “immunogenic” Bacteroides species decreased signs of colitis, an intestinal inflammatory condition that is a dangerous side effect in patients using checkpoint inhibitors. Moreover, Zitvogel and her colleagues showed that human metastatic melanoma patients treated with ipilimumab tended to have elevated levels of B. fragilis in their microbiomes. Mice transplanted with feces from patients who showed particularly strong B. fragilis gains did better on anti-CTLA-4 treatment than did mice transplanted with feces from patients with normal levels of B. fragilis.
“There are bugs that allow the therapy to work, and at the same time, they protect against colitis,” says Trinchieri. “That is very exciting, because not only [can] we do something to improve the therapy, but we can also, at the same time, try to reduce the side effect.”
And these checkpoint inhibitors aren’t the only cancer therapies whose effects are modulated by the microbiome. Trinchieri has also found that an immunotherapy that combines antibodies against interleukin-10 receptors with CpG oligonucleotides is more effective in mice with unaltered microbiomes.1He and his NCI colleague Goldszmid further found that the platinum chemotherapy oxaliplatin (Eloxatin) was more effective in mice with intact microbiomes, and Zitvogel’s group has shown that the chemotherapeutic agent cyclophosphamide is dependent on the microbiota for its proper function.
Although the mechanisms by which the microbiome influences the effectiveness of such therapies remains incompletely understood, researchers once again speculate that the immune system is the key link. Cyclophosphamide, for example, spurs the body to generate two types of T?helper cells, T?helper 1 cells and a subtype of T?helper 17 cells referred to as “pathogenic,” both of which destroy tumor cells. Zitvogel and her colleagues found that, in mice with unaltered microbiomes, treatment with cyclophosphamide works by disrupting the intestinal mucosa, allowing bacteria to escape into the lymphoid tissues just outside the gut. There, the bacteria spur the body to generate T?helper 1 and T?helper 17 cells, which translocate to the tumor. When the researchers transferred the “pathogenic” T?helper 17 cells into antibiotic-treated mice, the mice’s response to chemotherapy was partly restored.
Microbiome modification
As the link between the microbiome and cancer becomes clearer, researchers are thinking about how they can manipulate a patient’s resident microbial communities to improve their prognosis and treatment outcomes. “Once you figure out exactly what is happening at the molecular level, if there is something promising there, I would be shocked if people don’t then go in and try to modulate the microbiome, either by using pharmaceuticals or using probiotics,” says Michael Burns, a postdoc in the lab of University of Minnesota genomicist Ran Blekhman.
Even if researchers succeed in identifying specific, beneficial alterations to the microbiome, however, molding the microbiome is not simple. “It’s a messy, complicated system that we don’t understand,” says Schloss.
So far, studies of the gut microbiome and colon cancer have turned up few consistent differences between cancer patients and healthy controls. And the few bacterial groups that have repeatedly shown up are not present in every cancer patient. “We should move away from saying, ‘This is a causal species of bacteria,’” says Blekhman. “It’s more the function of a community instead of just a single bacterium.”
But the study of the microbiome in cancer is young. If simply adding one type of microbe into a person’s gut is not enough, researchers may learn how to dose people with patient-specific combinations of microbes or antibiotics. In February 2016, a team based in Finland and China showed that a probiotic mixture dubbed Prohep could reduce liver tumor size by 40 percent in mice, likely by promoting an anti-inflammatory environment in the gut.16
“If it is true that, in humans, we can alter the course of the disease by modulating the composition of the microbiota,” says José Conejo-Garcia of the Wistar Institute in Philadelphia, “that’s going to be very impactful.”
Kate Yandell has been a freelance writer living Philadelphia, Pennsylvania. In February she became an associate editor at Cancer Today.
GENETIC CONNECTION
The microbiome doesn’t act in isolation; a patient’s genetic background can also greatly influence response to therapy. Last year, for example, the Wistar Institute’s José Garcia-Conejo and Melanie Rutkowski, now an assistant professor at the University of Virginia, showed that a dominant polymorphism of the gene for the innate immune protein toll-like receptor 5 (TLR5) influences clinical outcomes in cancer patients by changing how the patients’ immune cells interact with their gut microbes (Cancer Cell, 27:27-40, 2015).
More than 7 percent of people carry a specific mutation in TLR5 that prevents them from mounting a full immune response when exposed to bacterial flagellin. Analyzing both genetic and survival data from the Cancer Genome Atlas, Conejo-Garcia, Rutkowski, and their colleagues found that estrogen receptor–positive breast cancer patients who carry the TLR5 mutation, called the R392X polymorphism, have worse outcomes than patients without the mutation. Among patients with ovarian cancer, on the other hand, those with the TLR5 mutation were more likely to live at least six years after diagnosis than patients who don’t carry the mutation.
Investigating the mutation’s contradictory effects, the researchers found that mice with normal TLR5produce higher levels of the cytokine interleukin 6 (IL-6) than those carrying the mutant version, which have higher levels of a different cytokine called interleukin 17 (IL-17). But when the researchers knocked out the animals’ microbiomes, these differences in cytokine production disappeared, as did the differences in cancer progression between mutant and wild-type animals.
“The effectiveness of depleting specific populations or modulating the composition of the microbiome is going to affect very differently people who are TLR5-positive or TLR5-negative,” says Conejo-Garcia. And Rutkowski speculates that many more polymorphisms linked to cancer prognosis may act via microbiome–immune system interactions. “I think that our paper is just the tip of the iceberg.”
N. Iida et al., “Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment,” Science, 342:967-70, 2013.
S. Viaud et al., “The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide,” Science, 342:971-76, 2013.
J.R. Warren, B. Marshall, “Unidentified curved bacilli on gastric epithelium in active chronic gastritis,”Lancet, 321:1273-75, 1983.
B.J. Marshall et al., “Attempt to fulfil Koch’s postulates for pyloric Campylobacter,” Med J Aust, 142:436-39, 1985.
J. Parsonnet et al., “Helicobacter pylori infection and the risk of gastric carcinoma,” N Engl J Med, 325:1127-31, 1991.
S. Kado et al., “Intestinal microflora are necessary for development of spontaneous adenocarcinoma of the large intestine in T-cell receptor β chain and p53 double-knockout mice,” Cancer Res, 61:2395-98, 2001.
J.V. Newman et al., “Bacterial infection promotes colon tumorigenesis in ApcMin/+ mice,” J Infect Dis, 184:227-30, 2001.
S.E. Erdman et al., “CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice,” Am J Pathol, 162:691-702, 2003.
J.P. Zackular et al., “The human gut microbiome as a screening tool for colorectal cancer,” Cancer Prev Res, 7:1112-21, 2014.
G. Zeller et al., “Potential of fecal microbiota for early-stage detection of colorectal cancer,” Mol Syst Biol, 10:766, 2014.
J.P. Zackular et al., “The gut microbiome modulates colon tumorigenesis,” mBio, 4:e00692-13, 2013.
J.P. Zackular et al., “Manipulation of the gut microbiota reveals role in colon tumorigenesis,”mSphere, doi:10.1128/mSphere.00001-15, 2015.
V.P. Rao et al., “Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice,” Cancer Res, 66:7395, 2006.
A. Sivan et al., “Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy,” Science, 350:1084-89, 2015.
M. Vétizou et al., “Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota,”Science, 350:1079-84, 2015.
ABSTRACT: Intestinal Th17 cells are induced and accumulate in response to colonization with a subgroup of intestinal microbes such as segmented filamentous bacteria (SFB) and certain extracellular pathogens. Here, we show that adhesion of microbes to intestinal epithelial cells (ECs) is a critical cue for Th17 induction. Upon monocolonization of germ-free mice or rats with SFB indigenous to mice (M-SFB) or rats (R-SFB), M-SFB and R-SFB showed host-specific adhesion to small intestinal ECs, accompanied by host-specific induction of Th17 cells. Citrobacter rodentium and Escherichia coli O157 triggered similar Th17 responses, whereas adhesion-defective mutants of these microbes failed to do so. Moreover, a mixture of 20 bacterial strains, which were selected and isolated from fecal samples of a patient with ulcerative colitis on the basis of their ability to cause a robust induction of Th17 cells in the mouse colon, also exhibited EC-adhesive characteristics….. · Sep 2015 · Cell 163(2) https://www.researchgate.net/profile/Shoichi_Kadohttp://dx.doi.org:/10.1016/j.cell.2015.08.058
MIT Faculty Newsletter Vol. XXII No. 2 Nov / Dec 2009 Memorial Resolution for David B. Schauer
We regretfully acknowledge the sudden death of our friend and colleague, Dr. David Schauer, on June 7, 2009 – one day after his 48th birthday. David Schauer was known not only for his keen scientific mind, but also as a friend whose empathy and compassion touched countless individuals.
David’s research was supported on a continuous basis by the National Institutes of Health throughout his career at MIT. His studies on microbial pathogenesis of gastrointestinal pathogenic bacteria, particularly Citrobacter rodentium, a murine model of enteropathogenic Escherichia coli, and enterohepatic helicobacter are widely known and respected.
David’s research provided important insights into the molecular mechanisms evoked by enteric pathogens and how the infections caused by these bacteria perturb the gastrointestinal barrier, elicit inflammation, and produce clinically relevant disease.
Susan Erdman is a Principal Research Scientist and Assistant Director in the Division of Comparative Medicine at MIT.
Invited Speaker. National Cancer Institute (NCI) Mucosal, Immunosurveillance, Inflammation and Cancer Workshop. April, 2005.
Invited reviewer. National Cancer Institute (NCI) RFA-CA-06-014 Tumor Microenvironment Network. August, 2006.
Invited speaker. Keystone Symposium on Mechanisms Linking Inflammation and Cancer, February, 2007
Alumni Fellow, College of Veterinary Medicine, Mississippi State University, 2007
Invited Speaker. National Cancer Institute (NCI) Changing Concepts in Cancer Etiology: The Role of the Human Microbiome. November, 2009.
Invited speaker. Keystone Symposium on Role of Inflammation in Oncogenesis, February, 2010
Invited reviewer. National Cancer Institute (NCI) Tumor Microenvironment Network. June, 2011
Invited Speaker, National Institute of Health (NIH) Human Microbiome Science Conference: Vision for the Future. Wound healing to longevity: Microbe-induced immune proficiency for human health. July, 2013.
Invited speaker, Wellcome Trust, Human Host-Microbiome Interactions Conference, April 2014.
Invited speaker, Van Andel Institute, Origins of Cancer Conference, July 2014
Peer Bork
Current position: senior group leader (bioinformatics) at the EMBL (Heidelberg);
joint coordinator of the EMBL Structural and Computational Biology unit;
visiting group leader at the MDC (Berlin-Buch)
Scientific interests: prediction of protein function at different spatial scales,
protein and genome evolution (iTOL), protein domain analysis,
comparative genome and proteome analysis, bridging genotype and phenotype
Some Projects: genome annotation,
comparative analysis of protein modules (SMART),
biochemical pathways and networks (STRING),
networks of chemicals and proteins (STITCH)
bioinformatics applications in molecular medicine
comparative metagenomics
•TLR5-dependent IL-6 mobilizes MDSCs that drive galectin-1 production by γδ T cells
•IL-17 drives malignant progression in IL-6-unresponsive tumors
•TLR5-dependent differences in tumor growth are abrogated upon microbiota depletion
•A common dominant TLR5 polymorphism influences the outcome of human cancers
The dominant TLR5R392X polymorphism abrogates flagellin responses in >7% of humans. We report that TLR5-dependent commensal bacteria drive malignant progression at extramucosal locations by increasing systemic IL-6, which drives mobilization of myeloid-derived suppressor cells (MDSCs). Mechanistically, expanded granulocytic MDSCs cause γδ lymphocytes in TLR5-responsive tumors to secrete galectin-1, dampening antitumor immunity and accelerating malignant progression. In contrast, IL-17 is consistently upregulated in TLR5-unresponsive tumor-bearing mice but only accelerates malignant progression in IL-6-unresponsive tumors. Importantly, depletion of commensal bacteria abrogates TLR5-dependent differences in tumor growth. Contrasting differences in inflammatory cytokines and malignant evolution are recapitulated in TLR5-responsive/unresponsive ovarian and breast cancer patients. Therefore, inflammation, antitumor immunity, and the clinical outcome of cancer patients are influenced by a common TLR5 polymorphism.
see also… Immune Influence
In recent years, research has demonstrated that microbes living in and on the mammalian body can affect cancer risk, as well as responses to cancer treatment.
Although the details of this microbe-cancer link remain unclear, investigators suspect that the microbiome’s ability to modulate inflammation and train immune cells to react to tumors is to blame. Here are some of the hypotheses that have come out of recent research in rodents for how gut bacteria shape immunity and influence cancer.
HOW THE MICROBIOME PROMOTES CANCER
Gut bacteria can dial up inflammation locally in the colon, as well as in other parts of the body, leading to the release of reactive oxygen species, which damage cells and DNA, and of growth factors that spur tumor growth and blood vessel formation.
Helicobacter pylori can cause inflammation and high cell turnover in the stomach wall, which may lead to cancerous growth.
HOW THE MICROBIOME STEMS CANCER
Gut bacteria can also produce factors that lower inflammation and slow tumor growth.
Some gut bacteria (e.g., Bifidobacterium)
appear to activate dendritic cells,
which present cancer-cell antigens to T cells that in turn kill the cancer cells.
Digital PCR (dPCR) has generated intense interest because it is showing potential as a clinical diagnostics tool. It has already proven to be a useful technique for any application where extreme sensitivity or precise quantification is essential, such as identifying mutations or copy number variations in tumor cells, or examining gene expression at the single-cell level.
GEN interviewed several dPCR experts to find out specifically why the technique is increasing in popularity. GEN also asked the experts to envision dPCR’s future capabilities.
GEN: What makes dPCR technology such a superior tool for discovery and diagnostic applications?
Dr. Shelton The high levels of sensitivity, precision, and reproducibility in DNA and quantification are the major strengths of dPCR. The technology is robust where differences in primer efficiency or the presence of sample-specific PCR inhibitors are trivial to the final quantification through an end-point amplification reaction.
This provides value to discovery as a trusted tool for validating potential biomarkers and hypotheses generated by broad profiling techniques such as microarrays or next-generation sequencing (NGS). In diagnostics applications, the reproducibility and rapid results of dPCR are critical for labs around the world to quickly compare and share data, especially for ultra-low detection of DNA where variability is high.
Dr. Garner Digital PCR provides a precise direct counting approach for single molecule detection, thereby providing a straightforward process for the absolute quantification of nucleic acids in samples. One of the biggest advantages of using a system such as ours is its ability to do real-time reads on digital samples. When samples go through PCR, their results are recorded after each cycle.
These results build a curve, and customers can analyze the data if something went wrong. If it isn’t a clean read—from either a contamination issue, primer-dimer issue, or off-target issue—the curve isn’t the classic PCR curve.
Dr. Menezes Digital PCR allows absolute quantification of target concentration in samples without the need for standard curves. Obtaining consistent, precise, and absolute quantification with regular qPCR is dependent on standard curve generation and amplification efficiency calculations, which can introduce errors.
Ms. Hibbs At MilliporeSigma Cell Design Studio, the implementation of dPCR has improved and accelerated the custom cell engineering workflow. After the application of zinc finger nuclease or CRISPR/Cas to create precise genetic modifications in mammalian cell lines, dPCR is used to characterize the expected frequency of homologous recombination and develop a screening strategy based on this expected frequency.
In some cell lines, homologous recombination occurs at a low frequency. In such cases, dPCR is used to screen cell pools and subsequently identify rare clones having the desired mutation. Digital PCR is also used to accurately and expeditiously measure target gene copy number. It is used this way, for example, in polyploid cell lines.
Dr. Price The ability to partition genomic samples to a level that enables robust detection of single target molecules is what sets dPCR apart as an innovative tool. Each partition (droplet in the case of the RainDrop System) operates as an individual PCR reaction, allowing for sensitive, reproducible, and precise quantification of nucleic acid molecules without the need for reference standards or endogenous controls.
Partitioning also provides greater tolerance to PCR inhibitors compared to quantitative PCR (qPCR). In doing so, dPCR can remedy many shortcomings of qPCR by transforming the analog, exponential nature of PCR into a digital signal.
Mr. Wakida Digital PCR is an ideal technology for detecting rare targets at concentrations of 0.1% or lower. By partitioning samples prior to PCR, exceptionally rare targets can be isolated into individual partitions and amplified.
Digital PCR produces absolute quantitative results, so in some respects, it is easier than qPCR because it doesn’t require a standard curve, with the added advantages of being highly tolerant of inhibitors and being able to detect more minute fold changes. Absolute quantification is useful for generating reference standards, detecting viral load, and preparing NGS libraries.
GEN: In what field do you think dPCR will have the greatest impact in the future?
Dr. Shelton dPCR will have a great impact on precision medicine, especially in liquid biopsy analysis. Cell-free DNA from bodily fluids such as urine or blood plasma can be analyzed quickly and cost-effectively using dPCR. For example, a rapid dPCR test can be performed to determine mutations present in a patient’s tumor and help drive treatment decisions.
Iterative monitoring of disease states can also be achieved due to the relatively low cost of dPCR, providing faster response times when medications are failing. Gene editing will also be greatly impacted by dPCR. Digital PCR enables refinement and optimization of gene-editing tools and conditions. Digital PCR also serves as quality control of therapeutically modified cells and viral transfer vectors used in gene-therapy efforts.
Dr. Garner The BioMark™ HD system combines dPCR with simultaneous real-time data for counting and validation. This capability is important for applications such as rare mutation detection, GMO quantitation, and aneuploidy detection—where false positives are intolerable and precision is paramount.
Any field that requires precision and the ability to detect false positives is a likely target for Fluidigm’s dPCR. Suitable applications include detecting and quantifying cancer-causing genes in patients’ cells, viral RNA that infects bacteria, or fetal DNA in an expectant mother’s plasma.
Dr. Menezes This technology is particularly useful for samples with low frequency sequences as, for example, those containing rare alleles, low levels of pathogen, or low levels of target gene expression. Teasing out fine differences in copy number variants is another area where this technology delivers more precise data.
Ms. Hibbs Digital PCR overcomes limitations associated with low-abundance template material and quantification of rare mutations in a high background of wild-type DNA sequence. For this reason, dPCR is poised to have significant impacts in diverse clinical applications such as detection and quantification of rare mutations in liquid biopsies, detection of viral pathogens, and detection of copy number variation and mosaicism.
Dr. Price Due to its high sensitivity, precision, and absolute quantification, the RainDrop dPCR has the potential to extend the range of nucleic acid analysis beyond the reach of other methods in a number of applications that could lend themselves to diagnostic, prognostic, and predictive applications. The precision of dPCR can be extremely useful in applications that require finer measures of fold change and rare variant detection.
Digital PCR is suitable for addressing varied research and clinical challenges. These include the early detection of cancer, pathogen/viral detection and quantitation, copy number variation, rare mutation detection, fetal genetic screening, and predicting transplant rejection. Additional applications include gene expression analysis, microRNA analysis, and NGS library quantification.
Mr. Wakida Digital PCR will have an impact on applications for detecting rare targets by enabling investigators to complement and extend their capabilities beyond traditionally employed methods. One such application is using dPCR to monitor rare targets in peripheral blood, as in liquid biopsies.
The monitoring of peripheral blood by means of dPCR has been described in several peer-reviewed articles. In one such article, investigators considered the clinical value of Thermo’s QuantStudio™ 3D Digital PCR system for the detection of circulating DNA in metastatic colorectal cancer (Dig Liver Dis. 2015 Oct; 47(10): 884–90).
GEN: Is there a new technology on the horizon that will increase the speed and/or efficiency of dPCR?
Dr. Shelton High-throughput sample analysis can be an issue with some dPCR systems. However, Bio-Rad’s Automated Droplet Generator allows labs to process 96 samples simultaneously, a capability that eliminates user-to-user variability and minimizes hands-on time.
We also want users to get the most information from one sample. Therefore, we are focused on expanding the multiplexing capabilities of our system. In development at Bio-Rad are new technologies that increase the multiplexing capabilities without loss of specificity or accuracy in the downstream workflow.
Dr. Garner Much of the industry direction seems to be in offering ever-higher resolution, or the ability to run more samples at the same resolution. Thus far, however, customers haven’t found commercial uses for these tools. Also, with increasing resolution and the search for even rarer mutations, the challenge of detecting false positives becomes an even bigger issue.
Dr. Menezes Use of ZEN™ Double-Quenched Probes by IDT in digital PCR provides increased sensitivity and a lower limit of detection. Due to the second quencher, ZEN probes provide even lower background than traditional single-quenched probes. And this lower background enables increased sensitivity when analyzing samples with low copy number targets, where every droplet matters.
Ms. Hibbs Quantification relies upon counting the number of positive partitions at the end point of the reaction. Accordingly, precision and resolution can be increased by increasing the number of partitions. We are now capable of analyzing on the order of millions of partitions per run, further extending the lower limit of detection. Additionally, the workflow is amenable to the integration of automation in order to increase throughput and standardize reaction set up.
Dr. Price Although dPCR is still an emerging technology, there is tremendous interest in its potential clinical diagnostics applications. Enabling adoption of dPCR in the clinical lab requires addressing current gaps in workflow, cost, throughput, and turnaround time.
Digital PCR technology has the potential for being improved significantly in two dimensions. First, one can address the problem of serially detecting positive versus negative partitions by leveraging lower-cost imaging detection technologies. Alternatively, one may capitalize on the small partition volumes to dramatically reduce the time to perform PCR. Ideally, the future will bring both capabilities to bear.
Mr. Wakida Compared to qPCR, dPCR currently requires more hands-on time to set up experiments. We are investigating methods to address this.
PCR Shows Off Its Clinical Chops
Thanks to Advances in Genomics, PCR Is Becoming More Common in Clinical Applications
Last May, Roche Molecular Systems announced that its cobas Liat Strep A assay received a CLIA waiver. This clinic-ready assay can detect Streptococcus pyogenes (group A ß-hemolytic streptococcus) DNA in throat swabs by targeting a segment of the S. pyogenes genome.
Since its invention by Kary B. Mullis in 1985, the polymerase chain reaction (PCR) has become well established, even routine, in research laboratories. And now PCR is becoming more common in clinical applications, thanks to advances in genomics and the evolution of more sensitive quantitative PCR methodologies.
Examples of clinical applications of PCR include point-of-care (POC) molecular tests for bacterial and viral detection, as well as mutation detection in liquid or tumor biopsies for patient stratification and treatment monitoring.
Industry leaders recently participated in a CHI conference that was held in San Francisco. This conference—PCR for Molecular Medicine—encompassed research and clinical perspectives and emphasized advanced techniques and tools for effective disease diagnosis.
To kick off the event, speakers shared their views on POC molecular tests. These tests, the speakers insisted, can provide significant value to healthcare only if they support timely decision making.
Clinic-ready PCR platforms need to combine speed, ease of use, and accuracy. One such platform, the cobas Liat (“laboratory in a tube”), is manufactured by Roche Molecular Systems. The system employs nucleic acid purification and state-of-art PCR-based assay chemistry to enable POC sites to rapidly provide lab-quality results.
The cobas Liat Strep A Assay detects Streptococcus pyogenes (group A β-hemolytic streptococcus) DNA by targeting a segment of the S. pyogenes genome. The operator transfers an aliquot of a throat swab sample in Amies medium into a cobas Liat Strep A Assay tube, scans the relevant tube and sample identification barcodes, and then inserts the tube into the analyzer for automated processing and result interpretation. No other operator intervention or interpretation is required. Results are ready in approximately 15 minutes.
According to Shuqi Chen, Ph.D., vp of Point-of-Care R&D at Roche Molecular Systems, clinical studies of the cobas Liat Strep A Assay demonstrated 97.7% sensitivity when the test was used at CLIA-waived, intended-use sites, such as physicians’ offices. In comparison, rapid antigen tests and diagnostic culture have sensitivities of 70% and 81%, respectively (according to a 2009 study Tanz et al. in Pediatrics).
The cobas Liat assay preserved the same ease-of-use and rapid turnaround as the rapid antigen tests. It addition, it provided significantly faster turnaround than the lab-based culture test, which can take 24–48 hours.
A CLIA waiver was announced for the cobas Liat Strep A assay in May 2015. CLIA wavers have been submitted for cobas Liat flu assays, and Roche intends to extend the assay menu.
POC tests are also moving into field applications. Coyote Bioscience has developed a novel method for one-step gene testing without nucleic acid extraction that can be as fast as 10 minutes from blood sample to result. Their portable devices for molecular diagnostics can be used as genetic biosensors to bring complex clinical testing directly to the patient.
“Instead of sequential steps, reactions happen in parallel, significantly reducing analysis time. Buffer, enzyme, and temperature profiles are optimized to maximize sensitivity,” explained Sabrina Li, CEO, Coyote Bioscience. “Both RNA and DNA can be analyzed simultaneously from a drop of blood in the same reaction.”
The first-generation Mini-8 system was used for Ebola detection in Africa where close to 600 samples were tested with 98.8% sensitivity. Recently in China, the Mini-8 system was applied in hospitals and small community clinics for hepatitis B and C and Bunia virus detection. The second-generation InstantGene system is currently being tested internally with clinical samples.
Digital PCR
Conventional real-time PCR technology, while suited to the analysis of high-quality clinical samples, may effectively conceal amplification efficiency changes when sample quality is inconsistent. A more effective alternative, Bio-Rad suggests, is its droplet-digital PCR (ddPCR) technology, which can provide absolute quantification of target DNA or RNA, a critical advantage when samples are limited, degraded, or contain PCR inhibitors. The company says that of the half-dozen clinical trials that are using digital PCR, half rely on the Bio-Rad QX200 ddPCR system.
Personalized cancer care requires ultra-sensitive detection and monitoring of actionable mutations from patient samples. The high sensitivity and precision of droplet-digital PCR (ddPCR) from Bio-Rad Laboratories offers critical advantages when clinical samples are limited, degraded, or contain PCR inhibitors.
Typically, formalin-fixed and paraffin-embedded (FFPE) tissue samples are processed. FFPE samples work well for immunohistochemistry and protein analysis; however, the formalin fixation can damage nucleic acids and inhibit the PCR reaction. Samples may yield 100 ng of purified nucleic acid, but the actual amplifiable material is less than 1%, or 1 ng, in most cases.
“Current qPCR technology depends on real-time fluorescence accumulation as the PCR is occurring, which can be an effective means of detecting and quantifying DNA targets in nondegraded samples,” commented Dawne Shelton, Ph.D., staff scientist, Digital Biology Center, Applications Development Group, Bio-Rad Laboratories. “Amplification efficiency is critical; if that amplification efficiency changes because of sample quality it is hidden in the qPCR methodology.”
“In ddPCR, that is a big red flag,” Dr. Shelton continued. “It changes the format of how the data look immediately so you know the amount of inhibition and which samples are too inhibited to use.”
Tissue types vary and contain different degrees of fat or other content that can also act as PCR inhibitors. In blood monitoring, the small circulating fragments of DNA are extremely degraded; in addition, food, supplements, or other compounds ingested by the patient may have an inhibitory effect.
Clinical labs test for these variabilities and clean the blood, but remnant PCR inhibitors can remain. In ddPCR, a single template is partitioned into a droplet. If the droplet contains a good template, it produces a signal; otherwise, it does not—a simple yes or no answer.
“Even if there is no PCR inhibition, most clinical samples yield very small amounts of nucleic acid,” Dr. Shelton added. “To make a secure decision using qPCR is difficult because you are in a gray zone at the very end of its linear range. ddPCR operates best with small sample amounts and provides good statistics for confidence in your results.”
Currently, at least a half dozen clinical trials worldwide are using digital PCR, half of them are using the Bio-Rad QX200 Droplet Digital PCR system. Examples of studies include examining BCR-ABL monitoring in patients with chronic myelogenous leukemia (CML); identifying activating mutations in epidermal growth factor receptor (EGFR) for first-line therapy of new drugs in patients with lung cancer; and the monitoring of resistance mutations such as EFGR T790M in patients with non-small cell lung cancer (NSCLC).
Clovis Oncology used a technology called BEAMing (Beads, Emulsions, Amplification, and Magnetics), a type of digital PCR for blood-based molecular testing, to perform EGFR testing on almost 250 patients in clinical trials. In BEAMing, individual EGFR gene copies from plasma are separated into individual water droplets in a water-in-oil emulsion. The gene copies are then amplified by PCR on magnetic beads.
The beads are counted by flow cytometry using fluorescently labeled probes to distinguish mutant beads from wild-type. Because each bead can be traced to an individual EGFR molecule in the patient’s plasma, the method is highly quantitative.
“BEAMing is particularly well-suited for the detection of known mutations in circulating tumor DNA. In this circumstance, the mutation of interest often occurs at low levels, perhaps only 1–2 copies per milliliter or even less, and in a high background of wild-type DNA that comes from normal tissue. BEAMing can detect one mutant molecule in a background of 5,000 wild-type molecules in clinical samples,” stated Andrew Allen, MRCP, Ph.D., chief medical officer, Clovis Oncology.
In the studies, the EGFR-resistance mutation T790M could be identified in plasma 81% of the time that it was seen in the matched patient tumor biopsy. Additionally, about 10% of patients in the study had a T790M mutation in plasma that was not identified in tissue, presumably because of tumor heterogeneity. Another 5–10% of the patients did not provide an EGFR result, usually because the tissue biopsy had no tumor cells.
In aggregate, these results suggest that plasma EGFR testing can be a valuable complement to tumor testing in the clinical management of NSCLC patients, and can provide an alternative when a biopsy is not available. Tumor biopsies may provide only limited tissue, if in fact any tissue is available, for molecular analysis. Also, mutations may be missed due to tumor heterogeneity. These mutations may be captured by sampling the blood, which acts as a reservoir for mutations from all parts of a patient’s tumor burden.
In the last few years, a panoply of clinically actionable driver mutations have been identified for NSCLC, including mutations in EGFR, BRAF, and HER2, as well as ALK, ROS, and RET rearrangements. These driver mutations will migrate NSCLC molecular diagnostic testing in the next few years toward panel testing of relevant cancer genes using various digital technologies, including next-generation sequencing.
PCR is a fast and inexpensive technique used to amplify segments of DNA that continues to adapt and evolve for the demanding needs of molecular biology researchers. This diagram shows the basic principles of PCR amplification. [NHGRI]
The influence that the polymerase chain reaction (PCR) has had on modern molecular biology is nothing short of remarkable. This technique, which is akin to molecular photocopying, has been the centerpiece of everything from the OJ Simpson Trial to the completion of the Human Genome Project. Clinical laboratories use this DNA amplification method for infectious disease testing and tissue typing in organ transplantation. Most recently, with the explosion of the molecular diagnostics field and meteoric rise in the use of next-generation sequencing platforms, PCR has enhanced its standing as an essential pillar of genomic science.
Let’s open the door to the past and take a look back around 35 years ago when GEN started reporting on the relatively new disciplines of genetic engineering and molecular biology. At that time, GEN was among the first to hear the buzz surrounding a new method to synthesize and amplify DNA in the laboratory. In reviewing the fascinating history of PCR, we will see how the molecular diagnostics field took shape and where it could be headed in the future.
Some Like It Hot
The biological sciences rarely advance within a vacuum—rather they rely on previous discoveries to promote directly or indirectly our understanding. The contributions made by scientists in the field of molecular biology that contributed to the functional pieces of PCR were numerous and spread out over more than two decades.
It began with H. Gobind Khorana’s advances in understanding the genetic code, leading to the use of synthetic DNA oligonucleotides, continued through Kjell Kleepe’s 1971 vision of a two-primer system for replicating DNA segments, to Fredrick Sanger’s method of DNA sequencing—a process that would win him the Nobel prize in 1980—which utilized DNA oligo primers, nucleotide precursors, and a DNA synthesis enzyme.
All of the previous discoveries were essential to PCR’s birth, yet it would be an egregious mistake to begin a retrospective on PCR and not discuss the enzyme upon which the entire reaction hinges upon—DNA polymerase. In 1956, Nobel laureate Arthur Kornberg and his colleagues discovered DNA polymerase (Pol I), in Escherichia coli. Moreover, the researchers described the fundamental process by which the polymerase enzyme copies the base sequence of a DNA template strand. However, it would take biologists another 20 years to discover a version of DNA polymerase that was stable enough for use for any meaningful laboratory purposes.
That discovery came in 1976 when a team of researchers from the University of Cincinnati described the activity of a DNA polymerase (Taq) they isolated from the extreme thermophile bacteria, Thermus aquaticus, which lives in hot springs and hydrothermal vents. The fact that this enzyme could withstand typical protein-denaturing temperatures and function optimally around 75–80°C fortuitously set the stage for the development of PCR.
By 1983, all of the ingredients to bake the molecular cake were sitting in the biological cupboard waiting to be assembled in the proper order. At that time, Nobel laureate Kary Mullis was working as a scientist for the Cetus Corporation trying to perfect oligonucleotide synthesis. Mullis stumbled upon the idea of amplifying segments of DNA using multiple rounds of replication and the two primer system—essentially modifying and expanding upon Sanger’s sequencing reaction. Mullis discovered that the temperatures for each step (melting, annealing, and extension) in the reaction would need to be painstakingly controlled by hand. In addition, he realized that since the reactions were using a non-thermostable DNA polymerase, fresh enzyme would need to be “spiked in” after each successive cycle.
Mullis’ hard work and persistence paid off as the reaction was successful at amplifying a particular segment of DNA that was flanked by two opposing nucleotide primer molecules. Two years later, the Cetus team presented their work at the annual meeting of the American Society for Human Genetics, and the first mention of the method was published in Science that same year; however, that article did not go into detail about the specifics of the newly developed PCR method—a paper that would be rejected by roughly 15 journals and would not be published until 1987.
Although scientists were a bit slow on the uptake for the new method, the researchers at Cetus were developing ways to improve upon the original assay. In 1986, the scientists substituted the original heat-liable DNA polymerase for the temperature-resistant Taq polymerase, removing the need to spike in enzyme and dramatically reducing errors while increasing sensitivity. A year later, PerkinElmer launched their creation of a thermal cycler, allowing scientists to regulate the heating and cooling parts of the PCR reaction with greater efficiency.
Extremely soon after the introduction of Taq and the launch of the thermal cycler, the PCR reaction exploded exponentially among research laboratories and not only vaulted molecular biology to the pinnacle of researcher interests, it also launched a molecular diagnostics revolution that continues today and shows no signs of slowing down.
Molecular Workhorse
In the years since PCR first burst onto the scene, there have been a number of significant advancements to the technique that have widely improved the overall method. For example, in 1991, a new DNA polymerase from the hyperthermophilic bacteria Pyrococcus furiosus, or Pfu, was introduced as a high-fidelity alternative enzyme to Taq. Unlike Taq polymerase, Pfu has built in 3′ to 5′ exonuclease proofreading activity, which allows the enzyme to correct nucleotide incorporation errors on the fly—dramatically increasing base specificity, albeit at a reduced rate of amplification versusTaq.
In 1995, two advancements were introduced to PCR users. The first, called antibody “hot-start” PCR, utilized an immunoglobulin molecule that is directed to the DNA polymerase and inhibits its activity until the first 95°C melt stage, denaturing the antibody and allowing the polymerase to become active. Although this process was effective in increasing the specificity of the PCR reaction, many researchers found that the technique was time consuming and often caused cross-contamination of samples.
The second innovation introduced that year began another revolution for molecular biology and the PCR method. Real-time PCR, or quantitative PCR (qPCR), allowed researchers to quantitatively create DNA templates for PCR amplification from RNA transcripts through the use of the reverse-transcriptase enzyme and specifically incorporated fluorescent reporter dyes. The technique is still widely used by researchers to monitor gene expression extremely accurately. Over the past 20 years, many companies have spent many R&D dollars to create more accurate, higher throughput, and simple qPCR machines to meet researcher demands.
With the advent of next-generation sequencing techniques—and the rise of techniques that started commanding the attention of more and more researchers—PCR machines and methods needed to evolve and modernize to keep pace. PCR remained the lynchpin in almost all the next-generation sequencing reactions that came along, but the traditional technique wasn’t nearly as precise as required.
Digital PCR (dPCR) was introduced as a refinement of the conventional method, with the first real commercial system emerging around 2006. dPCR can be used to quantify directly and clonally amplify DNA or RNA.
The apparatus carries out a single reaction within a sample. The sample, however, is separated into a large number of partitions. Moreover, the reaction is performed in each partition individually—allowing a more reliable measurement of nucleic acid content. Researchers often use this method for studying gene-sequence variations, such as copy number variants (CNV), point mutations, rare-sequence detection, and microRNA analysis, as well as for routine amplification of next-generation sequencing samples.
Future of PCR: Better, Faster, Stronger!
It is almost impossible to envision a future laboratory setting that wouldn’t utilize PCR in some fashion, especially due to the heavy reliance of next-generation sequencing techniques for accurate PCR samples and at the very least using the method as a simple amplification tool for creating DNA fragments of interest.
Yet there is at least one new next-generation sequencing technique that can identify native DNA sequences without an amplification step—nanopore sequencing. Although this technique has performed well in many preliminary trials, it is in its relative infancy. It will probably undergo additional development lasting several years before it approaches large-scale adoption by researchers. Even then, PCR has become so engrained into daily laboratory life that to try to phase out the technique would be like asking molecular biologists to give up their pipettes or restriction enzymes.
Most PCR equipment manufacturers continue to seek ways to improve the speed and sensitivity of their thermal cyclers, while biologists continue to look toward ways to genetically engineer better DNA polymerase molecules with even greater fidelity than their naturally occurring cousins. Whatever the new advancements are, and wherever they lead the life sciences field, you can count on us at GEN to continue to provide our readers with detailed information for another 35 years … at least!
The late Cambridge Mayor Alfred Vellucci welcomed Life Sciences Labs to Cambridge, MA – June 1976
Reporter: Aviva Lev-Ari, PhD, RN
How Cambridge became the Life Sciences Capital
Worth watching is the video below, which captures the initial Cambridge City Council hearing on recombinant DNA research from June 1976. The first speaker is the late Cambridge mayor Alfred Vellucci.
Vellucci hoped to pass a two-year moratorium on gene splicing in Cambridge. Instead, the council passed a three-month moratorium, and created a board of nine Cambridge citizens — including a nun and a nurse — to explore whether the work should be allowed, and if so, what safeguards would be necessary. A few days after the board was created, the pro and con tables showed up at the Kendall Square marketplace.
At the time, says Phillip Sharp, an MIT professor, Cambridge felt like a manufacturing town that had seen better days. He recalls being surrounded by candy, textile, and leather factories. Sharp hosted the citizens review committee at MIT, explaining what the research scientists there planned to do. “I think we built a relationship,” he says.
By early 1977, the citizens committee had proposed a framework to ensure that any DNA-related experiments were done under fairly stringent safety controls, and Cambridge became the first city in the world to regulate research using genetic material.
Triglycerides: Is it a Risk Factor or a Risk Marker for Atherosclerosis and Cardiovascular Disease ? The Impact of Genetic Mutations on (ANGPTL4) Gene, encoder of (angiopoietin-like 4) Protein, inhibitor of Lipoprotein Lipase
Reporters, Curators and Authors: Aviva Lev-Ari, PhD, RN and Larry H. Bernstein, MD, FCAP
Introduction
The role for triglycerides as a risk factor for cardiovascular disease is not new, going back to Donald Frederickson’s classification of hyperlipidemias, at least with respect to Type I and Type IIb. Whether there was a mechanism beyond the observations was yet an open question. The paper that follows addresses such a question.
Large Genetic Studies Support Role For Triglycerides In Cardiovascular Disease
Two papers published in the New England Journal of Medicine offer new genetic evidence to support the increasingly accepted though still controversial view that triglycerides play an important causal role in cardiovascular disease. If fully validated the new findings could lead to new drugs to prevent and treat cardiovascular disease, though others caution that there is still a long way to go before this could happen.
Both studies describe the impact of genetic mutations on a gene (ANGPTL4) which encodes for a protein (angiopoietin-like 4) that inhibits lipoprotein lipase, an enzyme that plays a key role in breaking down and removing triglycerides from the blood. The large studies found that people with mutations that inactivate ANGPTL4 have lower levels of triglycerides, higher levels of HDL cholesterol, and decreased risk for cardiovascular disease.
The findings, writes Sander Kersten (Wageningen University, the Netherlands) in an accompanying editorial, “suggest that lowering plasma triglyceride levels is a viable approach to reducing the risk of coronary artery disease.”
Two groups of investigators now describe in the Journal important genetic evidence showing a causal role of plasma triglycerides in coronary heart disease. Stitziel and colleagues2 tested 54,003 coding-sequence variants covering 13,715 human genes in more than 72,000 patients with coronary artery disease and 120,000 controls. Dewey and colleagues3 sequenced the exons of the gene encoding angiopoietin-like 4 (ANGPTL4) in samples obtained from nearly 43,000 participants in the DiscovEHR human genetics study. The two groups found a significant association between an inactivating mutation (E40K) in ANGPTL4 and both low plasma triglyceride levels and high levels of HDL cholesterol. ANGPTL4 is an inhibitor of lipoprotein lipase, the enzyme that breaks down plasma triglycerides along the capillaries in heart, muscle, and fat.4 Extensive research has shown that ANGPTL4 orchestrates the processing of triglyceride-rich lipoproteins during physiologic conditions such as fasting, exercise, and cold exposure.4 The E40K mutation in ANGPTL4 was previously shown to nearly eliminate the ability of ANGPTL4 to inhibit lipoprotein lipase, a mechanism that may result in part from the destabilization of ANGPTL4.5
The key finding in each study was that carriers of the E40K mutation and other rare mutations in ANGPTL4 had a lower risk of coronary artery disease than did noncarriers, a result that is consistent with the lower triglyceride levels and higher HDL cholesterol levels among mutation carriers. These findings confirm previous data6 and provide convincing genetic evidence that an elevated plasma triglyceride level increases the risk of coronary heart disease. In combination with extensive recent data on other genetic variants that modulate plasma triglyceride levels, the studies suggest that lowering plasma triglyceride levels is a viable approach to reducing the risk of coronary artery disease.
However, as a cautionary note, Talmud and colleagues7 previously found that the presence of the E40K variant was associated with an increased risk of coronary heart disease after adjustment for the altered plasma lipids. Consistent with this hypothesis, the overexpression of Angptl4 in mice was found to protect against atherosclerosis independent of plasma lipids.8
The studies also “implicate targeted inactivation of ANGPTL4 as a potential weapon in the war on heart disease,” though he also points to a previous study that did not support this hypothesis. Sekar Kathiresan (Broad Institute), senior author of one of the NEJM studies, told me that the previous study was small and “basically got the result wrong. Between, the two papers in this NEJM issue, we are looking at 10X more data.”
Recent large genetic studies have resulted in an important change in the field. Many researchers now believe that HDL, which was once thought to play an important protective role in atherosclerosis, is only a marker of disease. In contrast, triglycerides are now thought by many to play an important functional role.
One of the NEJM papers showed that a human monoclonal antibody to ANGPTL4 lowered triglyceride levels in animals. The study was funded by Regeneron and was performed by researchers at Regeneron and Geisinger, as part of an ongoing collaboration using deidentified genetic data from Geisinger patients. In their NEJM paper the researchers reported inflammation and other side effects in the animals treated with the antibody, but they said that no such problem has been observed in humans who have mutations that have the same functional effect as the antibody.
Coding Variation in ANGPTL4, LPL, and SVEP1and the Risk of Coronary Disease Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators
Although genomewide association studies have identified more than 56 loci associated with the risk of coronary artery disease,1-3 the disease-associated variants are typically common (minor-allele frequency >5%) and located in noncoding sequences; this has made it difficult to pinpoint causal genes and affected pathways. This lack of a causal mechanism has in part hindered the immediate translation of the findings of genomewide association studies into new therapeutic targets. However, the discovery of rare or low-frequency coding-sequence variants that affect the risk of coronary artery disease has facilitated advances in the prevention and treatment of disease. The most recent example of such advances is the development of a new class of therapeutic agents that is based on the discovery of the gene encoding proprotein convertase subtilisin/kexin type 9 (PCSK9) as a regulator of low-density lipoprotein (LDL) cholesterol4 and the discovery that low-frequency and loss-of-function variants in this gene protect against coronary artery disease.5,6
Recently, low-frequency coding variation across the genome was systematically tabulated with the use of next-generation exome and whole-genome sequencing data from more than 12,000 persons of various ancestries (including a major contribution from the National Heart, Lung, and Blood Institute Exome Sequencing Project). Protein-altering variants (i.e., nonsynonymous, splice-site, and nonsense single-nucleotide substitutions) that were observed at least twice among these 12,000 persons were included in a genotyping array (hereafter referred to as the exome array). In addition, the exome array contains previously described variants from genomewide association studies, a sparse genomewide grid of common markers, markers that are informative with regard to ancestry (i.e., African American, Native American, and European), and some additional content. Additional information on the design of the exome array is provided at http://genome.sph.umich.edu/wiki/Exome_Chip_Design. In this study, we focused on the 220,231 autosomal variants that were present on the array and were expected to alter protein sequence (i.e., missense, nonsense, splice-site, and frameshift variants) and used these to test the contribution of low-frequency coding variation to the risk of coronary artery disease.
Low-Frequency Coding Variants Associated with Coronary Artery Disease
The discovery cohort comprised 120,575 persons (42,335 patients and 78,240 controls) (Table S1 in the Supplementary Appendix). In the discovery cohort, we found significant associations between low-frequency coding variants in theLPA and PCSK9 genes and coronary artery disease (Table 1
TABLE 1
Low-Frequency Coding Variations Previously Associated with Coronary Artery Disease.). Both gene loci also harbor common noncoding variants associated with coronary artery disease that had previously been discovered through genomewide association studies. These variants were also present on the exome array and had significant associations with coronary artery disease in our study (Table 1). In a conditional analysis, the associations between coronary artery disease and the low-frequency coding variants in both LPA and PCSK9 were found to be independent of the associations between coronary artery disease and the more common variants (Table 1). ….
We found a significant association between SVEP1 p.D2702G and blood pressure (Table 3TABLE 3Association between Low-Frequency Variants and Traditional Risk Factors., and Table S7 in the Supplementary Appendix). The allele associated with an increased risk of coronary artery disease was also associated with higher systolic blood pressure (0.94 mm Hg higher for each copy of the allele among allele carriers, P=3.0×10−7) and higher diastolic blood pressure (0.57 mm Hg higher for each copy of the allele among allele carriers, P=4.4×10−7). We did not find an association between SVEP1 p.D2702G and any plasma lipid trait. In contrast, ANGPTL4 p.E40K was not associated with blood pressure but instead was found to be associated with significantly lower levels of triglycerides (approximately 0.3 standard deviation units lower for each copy of the allele among allele carriers, P=1.6×10−13) (Table 3) and with higher levels of high-density lipoprotein (HDL) cholesterol (approximately 0.3 standard deviation units higher for each copy of the allele among allele carriers, P=8.2×10−11) (Table 3). In a conditional analysis, these effects appeared to be at least partially independent of each other (Table S8 in the Supplementary Appendix). We did not observe any significant association between ANGPTL4 p.E40K and LDL cholesterol level (Table 3). Both SVEP1 p.D2702G and ANGPTL4 p.E40K were nominally associated with type 2 diabetes in a direction concordant with the associated risk of coronary artery disease.
ANGPTL4 Loss-of-Function Mutations, Plasma Lipid Levels, and Coronary Artery Disease
The finding that a missense allele in ANGPTL4 reduced the risk of coronary artery disease, potentially by reducing triglyceride levels, raised the possibility that complete loss-of-function variants in ANGPTL4 may have an even more dramatic effect on triglyceride concentrations and the risk of coronary artery disease. We therefore examined sequence data for the seven protein-coding exons of ANGPTL4 in 6924 patients with early-onset myocardial infarction and 6834 controls free from coronary artery disease (details of the patients and controls are provided in Table S3 in the Supplementary Appendix). We discovered a total of 10 variants that were predicted to lead to loss of gene function (Figure 1AFIGURE 1
Loss-of-Function Alleles in ANGPTL4 and Plasma Lipid Levels., and Table S9 in the Supplementary Appendix), carried by 28 heterozygous persons; no homozygous or compound heterozygous persons were discovered. Carriers of loss-of-function alleles had significantly lower levels of triglycerides than did noncarriers (a mean of 35% lower among carriers, P=0.003) (Figure 1B, and Table S10 in the Supplementary Appendix), with no significant difference in LDL or HDL cholesterol levels. Moreover, we found a lower risk of coronary artery disease among carriers of loss-of-function alleles (9 carriers among 6924 patients vs. 19 carriers among 6834 controls; odds ratio for disease, 0.47; P=0.04) (Table S11 in the Supplementary Appendix). A similar investigation was performed for the 48 protein-coding exons of SVEP1; however, only 3 loss-of-function allele carriers were discovered (2 carriers among 6924 patients vs. 1 carrier among 6834 controls).
Coding Variation in LPL and the Risk of Coronary Artery Disease
On the basis of the fact that a loss of ANGPTL4 function was associated with reduced risk of coronary artery disease and that ANGPTL4 inhibits lipoprotein lipase (LPL), one would expect a gain of LPL function to also be associated with a lower risk of coronary artery disease, whereas a loss of LPL function would be expected to be associated with a higher risk. In observations consistent with these expectations, we found a low-frequency missense variant in LPL on the exome array that was associated with an increased risk of coronary artery disease (p.D36N; minor-allele frequency, 1.9%; odds ratio for disease, 1.13; P=2.0×10−4) (Table S12 in the Supplementary Appendix); previous studies have shown that this allele (also known as p.D9N) is associated with LPL activity that is 20% lower in allele carriers than in noncarriers.8 We also identified a nonsense mutation in LPL on the exome array that was significantly associated with a reduced risk of coronary artery disease (p.S447*; minor-allele frequency, 9.9%; odds ratio, 0.94; P=2.5×10−7) (Table S12 in the Supplementary Appendix). Contrary to most instances in which the premature introduction of a stop codon leads to loss of gene function, this nonsense mutation, which occurs in the penultimate codon of the gene, paradoxically induces a gain of LPL function.9 …..
Through large-scale exomewide screening, we identified a low-frequency coding variant in ANGPTL4 that was associated with protection against coronary artery disease and a low-frequency coding variant in SVEP1 that was associated with an increased risk of the disease. Moreover, our results highlight LPL as a significant contributor to the risk of coronary artery disease and support the hypothesis that a gain of LPL function or loss of ANGPTL4 inhibition protects against the disease.
ANGPTL4 has previously been found to be involved in cancer pathogenesis and wound healing.10 Previous functional studies also revealed that ANGPTL4 regulates plasma triglyceride concentration by inhibiting LPL.11 The minor allele at p.E40K has previously been associated with lower levels of triglycerides and higher levels of HDL cholesterol.12 We now provide independent confirmation of these lipid effects. In vitro and in vivo experimental evidence suggests that the lysine allele at p.E40K results in destabilization of ANGPTL4 after its secretion from the cell in which it was synthesized. It may be that the p.E40K variant leads to increases in the enzymatic activity of LPL because of this destabilization.13 Previous, smaller studies produced conflicting results regarding p.E40K and the risk of coronary artery disease14,15; we now provide robust support for an association between p.E40K and a reduced risk of coronary artery disease.
An important caveat to this research is that it is still very early. Most promising therapeutic targets do not work out. James Stein (University of Wisconsin) praised the papers but also offered a word of caution. “This is great science and important research that sheds light on the genetic regulation of TG-rich lipoproteins, serum TG levels, and CVD risk,” he said. “Since it is hard, if not impossible, to disconnect TG-rich lipoproteins from LDL, we should be humble in extrapolating these findings to clinical medicine in an era of low LDL due to statins and PCSK9 inhibitors. I hope this research identifies new targets for drug therapy and better understanding of CVD risk prediction– only time will tell.”
Previous studies with fibrates and other drugs have failed to convincingly show that lowering triglycerides is beneficial. Kathiresan said that what really seems to matter is “how you alter the plasma triglyceride-rich lipoproteins (TRLs).” Some genes that alter TRLs have other metabolic effects. As an example he cited a gene that lowers TRLs but increases the risk for type 2 diabetes. The NEJM papers, by observing the effect of specific mutations, therefore point the way to targets that may be clinically significant.
Conclusions: The work that has been presented puts a new light on the possible role of triglycerides in the development of congenitally predetermined cardiovascular disease. It does not necessarily establish a general link to mechanism of cardiovascular disease, but it opens up new pathways to our understanding.
“Are triglycerides a CHD risk factor? The answer is still maybe. Triglyceride-rich lipoproteins are inextricably linked to LDL metabolism and LDL particle number (and apo B). Still these are important new data and targets for novel therapeutics.”
Risk of Dis-lipids Syndrome in Modern Society
Aurelian Udristioiuᶪ, Manole Cojocaru²
¹Department of Biochemistry, Clinical Laboratory, Emergency County Hospital Targu Jiu & Titu Maiorescu University, Bucharest, Romania,
Department of Physiology, Faculty of Medicine, Titu Maiorescu University, Bucharest, Romania
Abstract
Aim of this work was to emphasis the preclinical evaluation of dis-lipids syndromes types at the patients which were presented to a routine control for checking health status, in the hospital ambulatory.
Material and Method:
Were analyzed 60 patients, registered in Clinical Laboratory, assessing by running on the Hitachi 912 Analyzer, the principal biochemical parameters of lipid metabolism: Cholesterol, Triglycerides and fractions of Cholesterol, HDL and LDL. From the total of 60 patients 35 were females and 25 males.
Results
The persons with an alarm signal of atherosclerotic process were in 28 % and persons with low HDL was in 17%. The cases with atherosclerotic index, report-LDL/HDL>3.5 for men and 2.5 for women were in 14 % , the cases with predictive value with coronary risk, report-CO/HDL>5 were presented in 5 % and the cases with dis-lipid syndrome type 2- 4, with high Cholesterol and Triglycerides, were presented in 30% percent.
Conclusions
Lipids controls, and its fractions, are necessary to be prevented atherosclerotic process in the incipient status of ill.
March 2, 2016 Regeneron Genetics Center Publication in New England Journal of Medicine Links ANGPTL4 Inhibition and Risk of Coronary Artery Disease Demonstrates power of large-scale Precision Medicine initiatives
Cardiovascular Diseases, Volume Four: Regenerative and Translational Medicine: The Therapeutics Promise for Cardiovascular Diseases, on Amazon since 12/26/2015
Researchers at the University of Illinois at Chicago have discovered a molecular regulator that allows salmonella bacteria to switch from actively causing disease to lurking in a chronic but asymptomatic state called a biofilm.
Their findings are published in the online journal, eLife.
Biofilms cling to surfaces in the body, such as the bronchial tubes or artificial joints, often without causing illness. But they can be a reservoir of bacteria that detach and cause disease or infect new hosts. The biofilms are resistant to host defenses and antibiotics because their tightly-packed structure exposes little surface area for drugs to reach. Many pathogenic bacteria are able to switch from an infectious to a dormant state as a strategy for survival inside their hosts.
Linda Kenney, professor of microbiology and immunology at the UIC College of Medicine and lead author of the study, had been studying how salmonella survive inside immune system cells called macrophages. These white blood cells patrol the body and engulf viruses and bacteria they encounter. They encase their prey in a bubble called a vacuole that protects them from the invader until it can be destroyed.
Macrophages digest their quarry when the acidity inside the vacuole drops in response to the captive. But the bacteria have evolved a unique defense, enabling them to survive inside the vacuole and use the macrophage as a Trojan horse to travel elsewhere in the body undetected by other immune cells.
Kenney knew that a type of salmonella that causes typhoid fever in humans, called Salmonella typhi, and its mouse counterpart, Salmonella typhimurium, were able to survive inside macrophage vacuoles. She noticed that these bacteria did two things: inside the vacuole, they formed a kind of syringe – a long, hollow filament to inject the vacuole with a host of proteins that altered it. They also quickly assumed the same acidity of the vacuole.
“These two defenses, together, allow salmonella to survive and replicate in the harsh conditions of the vacuole,” Kenney said.
Further experiments revealed that sensing and mirroring the acidity, or pH, of the vacuole is what triggers salmonella to form the syringe.
“The syringe-forming and pH-adjusting genes are signaled to turn on by the lower pH inside the vacuole,” Kenney said. But these same salmonella, equipped to survive the hostile environment inside a macrophage vacuole, were also able to exist free in the body of the host—as biofilms.
“I wanted to know how Salmonella ‘decide’ between these two very different lifestyles,” Kenney said.
Studying S. typhimurium, Kenney discovered that the molecular switch is a bacterial molecule called SsrB. As the macrophage vacuole starts to acidify, SsrB is activated and it turns on the genes needed to form the syringe and adjust the pH. When salmonella lives outside the vacuole, where pH levels are neutral, SsrB instead turns on genes for sticky proteins in the membrane that help bacteria bind to one another to form biofilms.
Kenney said that many disease-causing salmonella evolved from harmless strains partly by acquiring new genes from other germs in a process called horizontal gene transfer.
“Salmonella acquired their pH-adjusting and syringe-forming genes in this way, as well as the switch that turns them on and off – SsrB,” she said. “The default mode, or its ancestral program, dictates that it make biofilms, cause no illness, and survive long enough to infect new hosts when the opportunity arises. The new genes allow it to survive the host’s main defense—the acidifying macrophage vacuole.”
Understanding how bacteria switch from the disease-causing state to the biofilm state could help scientists develop anticancer drugs that encourage the formation of biofilms on tumors, Kenney said.
“When salmonella forms biofilms on tumors, it releases TNF-alpha, a powerful anti-tumor molecule,” she said. “If we can better control the formation of biofilms, we can target them to tumors for cancer therapy.”
The horizontally-acquired response regulator SsrB drives a Salmonella lifestyle switch by relieving biofilm silencing
Stuti KDesai,
Ricksen SWinardhi
Mechanobiology Institute, National University of Singapore, Singapore, Singapore; Department of Physics, National University of Singapore, Singapore, Singapore
Contribution: Acquisition of data, Analysis and interpretation of data, Drafting or revising the article
No competing interests declared
</div>”>Ricksen SWinardhi,
SaravananPeriasamy
Singapore Centre on Environmental Life Sciences Engineering, Nanyang Technological University, Singapore, Singapore
Contribution: Conception and design, Acquisition of data
No competing interests declared
</div>”>SaravananPeriasamy,
Michal MDykas
Nanoscience and Nanotechnology Institute, National University of Singapore, Singapore, Singapore; Graduate School for Integrative Sciences and Engineering, National University of Singapore, Singapore, Singapore
Contribution: Acquisition of data, Drafting or revising the article
Mechanobiology Institute, National University of Singapore, Singapore, Singapore; Department of Physics, National University of Singapore, Singapore, Singapore
Contribution: Analysis and interpretation of data, Drafting or revising the article
No competing interests declared
</div>”>YanJie,
Linda JKenney
Mechanobiology Institute, National University of Singapore, Singapore, Singapore; Jesse Brown Veterans Affairs Medical Center, University of Illinois-Chicago, Chicago, United States; Department of Microbiology and Immunology, University of Illinois-Chicago, Chicago, United States
Contribution: Conception and design, Analysis and interpretation of data, Drafting or revising the article
A common strategy by which bacterial pathogens reside in humans is by shifting from a virulent lifestyle, (systemic infection), to a dormant carrier state. Two major serovars of Salmonella enterica, Typhi and Typhimurium, have evolved a two-component regulatory system to exist insideSalmonella-containing vacuoles in the macrophage, as well as to persist as asymptomatic biofilms in the gallbladder. Here we present evidence that SsrB, a transcriptional regulator encoded on the SPI-2 pathogenicity-island, determines the switch between these two lifestyles by controlling ancestral and horizontally-acquired genes. In the acidic macrophage vacuole, the kinase SsrA phosphorylates SsrB, and SsrB~P relieves silencing of virulence genes and activates their transcription. In the absence of SsrA, unphosphorylated SsrB directs transcription of factors required for biofilm formation specifically by activating csgD (agfD), the master biofilm regulator by disrupting the silenced, H-NS-bound promoter. Anti-silencing mechanisms thus control the switch between opposing lifestyles.
Introduction
Salmonella enterica serovar Typhimurium is a rod-shaped enteric bacterium which easily infects diverse hosts such as humans, cattle, poultry and reptiles through contaminated food or water, causing gastroenteritis. A human-restricted serovar of Salmonella enterica, serovar Typhi, causes typhoid fever and continues to be a dangerous pathogen throughout the world. Salmonella lives as a facultative pathogen in various natural and artificial environments as independent planktonic cells, cooperative swarms (Harshey and Matsuyama, 1994) or as multi-cellular communities called biofilms (see Steenackers et al., 2012 for a review). Upon successful invasion of host cells, Salmonella is phagocytosed by macrophages, where it resides in a modified vacuole in a self-nourishing niche called a Salmonella-Containing Vacuole (SCV). This intracellular lifestyle eventually adversely affects the host. Salmonella also resides as multi-cellular communities on intestinal epithelial cells (Boddicker et al., 2002), gallstones (Prouty et al., 2002) and tumors (Crull et al., 2011). It is believed that biofilms in the gall bladder are important for maintaining the carrier state, allowing Salmonella to persist (Crawford et al., 2010).
Each of these lifestyles of Salmonella are regulated by two-component regulatory systems (TCRS). TCRSs are comprised of a membrane-bound sensor histidine kinase and a cytoplasmic response regulator. The virulence genes of Salmonella are encoded on horizontally acquired AT-rich segments of the genome called Salmonella Pathogenecity Islands (SPIs), and are also tightly regulated by TCRSs. For example, the SsrA/B TCRS is essential for the activation of the SPI-2 regulon genes encoding a type-three secretory needle and effectors that are involved in formation of the SCV (Cirillo et al., 1998). Interestingly, the SsrA/B system itself is regulated by upstream two-component systems such as EnvZ/OmpR and PhoP/Q, which regulate gene expression in response to changes in osmolality, pH and the presence of anti-microbial peptides (Fields et al., 1989; Miller et al., 1989;Lee et al., 2000; Feng et al., 2003). The ssrA and ssrB genes are present on the SPI-2 pathogenecity island adjacent to each other and are regulated by a set of divergent promoters (Feng et al., 2003; Ochman et al., 1996). Under acidic pH and low osmolality, the ssrA and ssrB genes are transcriptionally activated by the binding of OmpR~P and PhoP~P to their promoters (Feng et al., 2003; Bijlsma and Groisman, 2005; Walthers and Kenney unpublished) whose levels are in turn regulated by the respective sensor kinases, EnvZ and PhoQ. SsrA is a tripartite membrane-bound histidine sensor kinase that undergoes a series of intra-molecular phosphorylation reactions before it transfers the phosphoryl group to the N-terminal aspartate residue of the response regulator, SsrB.
SsrB belongs to the NarL/FixJ family of transcriptional regulators that require phosphorylation-dependent dimerization to bind DNA. The X-ray crystal structure of NarL revealed that the C-terminal DNA binding domain was occluded by the N-terminus (Baikalov et al., 1996), and phosphorylation was predicted to relieve this inhibition. Full-length SsrB is unstable in solution, but an isolated C-terminal domain of SsrB, SsrBc, is capable of binding to the regulatory regions of nine genes belonging to the SPI-2 regulon, including ssrA and ssrB (Feng et al., 2004; Walthers et al., 2007) and activating transcription. A role for SsrB~P was identified by its dual function as a direct transcriptional activator and as an anti-silencer of H-NS-mediated repression (Walthers et al., 2007). The Histone like Nucleoid Structuring protein H-NS is involved in silencing many of the SPI-2 regulon genes in accordance with its role in binding to xenogenic AT-rich sequences and repressing their expression (Walthers et al., 2007; Navarre et al., 2006). H-NS binding to DNA leads to the formation of a stiff nucleoprotein filament which is essential in gene silencing (Lim et al., 2012; Liu et al., 2010; Amit et al., 2003; Winardhi et al., 2015). Moreover, relief of repression occurs due to the binding of SsrBc to this rigid H-NS-DNA complex (Walthers et al., 2011).
Salmonella reservoirs in host and non-host environments produce a three-dimensional extracellular matrix which consists of curli fimbriae, cellulose, proteins and extracellular DNA, to encase clusters of bacteria and form a mature biofilm. CsgD (AgfD) is the master regulator of biofilm formation (Gerstel et al., 2003); it is a LuxR family transcriptional activator that activates the expression of curli fimbriae encoded by csgDEFG/csgBAC operons (Collinson et al., 1996; Romling et al., 1998). CsgD also activates expression of adrA, increasing intracellular c-di-GMP levels, and activating the cellulose biosynthetic operon bcsABZC (Zogaj et al., 2001). Two other biofilm matrix components are also positively regulated by CsgD: BapA and the O-antigen capsule (Latasa et al., 2005; Gibson et al., 2006).
Transcriptional profiling of biofilms formed by S. Typhimurium SL1344 showed that many SPI-2 genes were down-regulated, yet SsrA was required for biofilms (Hamilton et al., 2009). This apparent paradox drove us to explore the underlying mechanism of biofilm formation. The role of SsrA/B in this process was of particular interest, since our previous comparison of SsrA and SsrB levels at neutral and acidic pH had shown that the expression of ssrA and ssrB was uncoupled (Feng et al., 2004).
We examined the ability of the wild type S. Typhimurium strain 14028s to form biofilms in the absence of ssrA and ssrB and found it to be dependent only on the expression of ssrB. We further showed that H-NS was a negative regulator of csgD. Surprisingly, the SsrB response regulator positively regulated the formation of biofilms by activating csgD expression in the absence of any phospho-donors. Moreover, AFM imaging revealed that unphosphorylated SsrB was able to bind to the csgD regulatory region and binding was sufficient to relieve H-NS-mediated repression and favor formation of S. Typhimurium biofilms.
As a result of these studies, we propose that SsrB, a pathogenicity island-2-encoded response regulator, sits at a pivotal position in governing Salmonella lifestyle fate: to either exist inside the host (in the SCV) as a promoter of virulence; or as a surface-attached multicellular biofilm, maintaining the carrier state. This switch is achieved merely by the ability of unphosphorylated SsrB to function as an anti-repressor of H-NS and the additional role of SsrB~P in activating SPI-2 transcription (Walthers et al., 2011).
eLife digest
Salmonella bacteria can infect a range of hosts, including humans and poultry, and cause sickness and diseases such as typhoid fever. Disease-causing Salmonella evolved from harmless bacteria in part by acquiring new genes from other organisms through a process called horizontal gene transfer. However, some strains of disease-causing Salmonella can also survive inside hosts as communities called biofilms without causing any illness to their hosts, who act as carriers of the disease and are able to pass their infection on to others.
So how do Salmonella bacteria ‘decide’ between these two lifestyles? Previous studies have uncovered a regulatory system that controls the decision in Salmonella, which is made up of two proteins called SsrA and SsrB. To trigger the disease-causing lifestyle, SsrA is activated and adds a phosphate group onto SsrB. This in turn causes SsrB to bind to and switch on disease-associated genes in the bacterium. However, it was less clear how the biofilm lifestyle was triggered.
Desai et al. now reveal that the phosphate-free form of SsrB – which was considered to be the inactive form of this protein – plays an important role in the formation of biofilms. Experiments involving an approach called atomic force microscopy showed that the unmodified SsrB acts to stop a major gene that controls biofilm formation from being switched off by a so-called repressor protein.
Salmonella acquired SsrB through horizontal gene transfer, and these findings show how this protein now acts as a molecular switch between disease-causing and biofilm-based lifestyles. SsrB protein is also involved in the decision to switch between these states, but how it does so remains a question for future work.
(A) (i) AFM imaging in the presence of 600 nM H-NS shows a straight and rigid filament on csgD755. (ii) Addition of 600 nM SsrB to the H-NS bound csgD DNA resulted in areas of condensation (pink arrows; an ‘SsrB signature’) along with a few areas where the straight H-NS bound conformation persisted (yellow line; an ‘H-NS signature’); Scale bar = 200 nm as in Figure 5A. (B) A model for the mechanism of anti-silencing by SsrB at csgD wherein SsrB likely displaces H-NS from the ends of a stiffened nucleoprotein filament and relieves the blockade on the promoter for RNA polymerase to activate transcription. For details refer to (Winardhi et al., 2015).
Discussion
Pathogenic microbes constantly evolve novel means to counter the multitude of challenges posed by complex eukaryotic hosts. Successful acquisition and integeration of laterally acquired genes into the native genome of pathogens leads to novel capabilities enabling their survival in a wide range of environmental stresses. The present work demonstrates how the presence or absence of the horizontally acquired SsrA kinase controls post-translational modification of the transcription factor SsrB (i.e. phosphorylation at aspartate-56). This event controls the fate of Salmonella Typhimurium, resulting in either acute or chronic, but asymptomatic infection. A variation on two-component signaling in a similar lifestyle fate in Pseudomonas aeruginosa involved the presence or absence of the hybrid kinase RetS (Goodman et al., 2004).
SsrB sits at a pivotal decision point that determines Salmonella lifestyles
When the SsrA kinase is present and activated by acid stress, SsrB is phosphorylated and SsrB~P de-represses H-NS and activates transcription at SPI-2 and SPI-2 co-regulated genes, including: sifA(Walthers et al., 2011), ssaB, ssaM, sseA and ssaG (Walthers et al., 2007). In the absence of the SsrA kinase, SsrB is not phosphorylated, but it can counter H-NS silencing at csgD (Figure 4A–D andFigure 6A). SsrB binding and bending at the csgD promoter causes a sufficient change in the DNA secondary structure (Figure 5B,C) that likely enables access for RNA polymerase, stimulating csgDtranscription. It is interesting to note that SsrB is located on the SPI-2 pathogenicity island, and thus was acquired as Salmonella enterica diverged from Salmonella bongori. However, the capability to form biofilms is an ancestral trait, as phylogeny studies have shown that most of the natural or clinical isolates of Salmonella belonging to all the five sub-groups form rdar colonies (White and Surette, 2006). The SsrB response regulator can control two distinct lifestyle choices: the ability to assemble a type three secretory system and survive in the macrophage vacuole or the ability to form biofilms on gallstones in the gall bladder to establish the carrier state.
What then controls the presence or activation of the kinase SsrA? Our early experiments indicated that SsrA and SsrB were uncoupled from one another (i.e., SsrB was present in the absence of SsrA) and ssrA transcription was completely dependent on OmpR (Feng et al., 2004). The EnvZ/OmpR system is stimulated by a decrease in cytoplasmic pH when Salmonella enters the macrophage vacuole (Chakraborty et al., 2015). This may also be the stimulus for activating SsrA, since theSalmonella cytoplasm acidifies to pH 5.6 during infection and the cytoplasmic domain of EnvZ (EnvZc) was sufficient for signal transduction (Wang et al., 2012; Chakraborty et al., 2015). Previous reports also identified a role for PhoP in ssrA translation (Bijlsma and Groisman, 2005), which would further add to fluctuating SsrA levels. The present work describes a novel role for the unphosphorylated response regulator SsrB in de-repressing H-NS (Figure 6B). We show that under biofilm-inducing conditions, unphosphorylated SsrB is sufficient to activate the expression of csgD. There are only a few such examples of unphosphorylated response regulators playing a role in transcription such as DegU (Dahl et al., 1992) in Bacillus subtilis and RcsB (Latasa et al., 2012) in S.Typhimurium.
The importance of anti-silencing in gene regulation
In recent years, it has become apparent that H-NS silences pathogenicity island genes in Salmonella(Lucchini et al., 2006; Navarre et al., 2006; Walthers et al., 2007; 2011). Understanding how H-NS silences genes and how this silencing is relieved is an active area of research (Will et al., 2015;Winardhi et al., 2015). Because the anti-silencing style of gene regulation is indirect and does not rely on specific DNA interactions, searching for SsrB binding sites has not been informative in uncovering this type of regulation (Tomljenovic-Berube et al., 2010; Worley et al., 2000; Shea et al., 1996). Even a recent report in which the proteomes of wild type, hilA null (a transcriptional regulator of SPI-1 genes) and ssrB null were analyzed by SILAC and compared with an existing CHIP dataset failed to identify csgD as an SsrB-regulated locus (Brown et al., 2014), as sequence gazing alone does not help in identifying mechanisms of transcriptional regulation.
SsrB is well suited to this style of regulation, because it does not recognize a well-defined binding site (Feng et al., 2004; Walthers et al., 2007; Tomljenovic-Berube et al., 2010), it has a high non-specific binding component (Carroll et al., 2009) and it bends DNA upon binding (Carroll et al., 2009; Figure 6B, this work). Furthermore, previous microarray studies disrupted both ssrA and ssrB, which would not uncover a distinct role for SsrB in gene regulation under non SPI-2-inducing conditions in the absence of the SsrA kinase. It is worth mentioning here that in our AFM images, it was apparent that H-NS was still bound to some regions of the csgD promoter when SsrB condensed the DNA (Figure 6A(ii)). Thus, H-NS does not have to be completely stripped off the DNA for de-repression to occur, a finding that was also evident in our previous studies (Liu et al., 2010) and others (Will et al., 2014).
SsrB binds and bends DNA, resulting in highly curved DNA conformations. This DNA binding property of SsrB is distinct from H-NS, which forms rigid nucleoprotein filaments and thus straight DNA conformations (Figure 6A(i)). Bent DNA is therefore an energetically unfavorable substrate for H-NS binding, and a likely mechanism of SsrB-mediated anti-silencing of H-NS repressed genes. SsrB-dependent displacement of H-NS is more energetically favored to occur predominantly at the ends of H-NS-bound filaments, which requires disruption of fewer H-NS protein-protein interactions (Winardhi et al., 2015 and Figure 6B). In an equal mixture of H-NS and SsrB (Figure 6A(ii)), we do not see evidence of sharply bent filaments. This is expected because H-NS dissociation is likely restricted to the filament ends. Such events occur due to the cooperative nature of H-NS binding that results in a chain of linked H-NS proteins. Hence, H-NS displacement by SsrB likely occurs progressively from the filament end. This behavior has been observed in our single-molecule stretching experiments with H-NS filaments in the presence of SsrB. This ability of H-NS to re-orient on the DNA without being released would also promote its re-binding and silencing when SsrB or other anti-silencers are released (Figure 6B).
Structural homology does not indicate functional homology
Response regulators are grouped into subfamilies on the basis of the structures of their DNA binding domains. SsrB is in the NarL/FixJ subfamily, which possess a helix-turn-helix (HTH) motif in the C-terminus (Baikalov et al., 1996). NarL was the first full-length structure of a response regulator and it showed that the N-terminal phosphorylation domain physically blocked the recognition helix in the HTH motif (Maris et al., 2002). Thus, phosphorylation is required to relieve the inhibition of the N-terminus. In the results presented herein, it is apparent that SsrB has adapted to relieving H-NS-silencing and that phosphorylation is not required for this behavior, nor is it required for DNA binding (Figure 5B).
In summary, we showed that the response regulator SsrB is required for biofilm formation because it can de-repress H-NS at the csgD promoter (Figure 6B). This leads to the production of CsgD, the master regulator of biofilms. It is noteworthy that a laterally acquired gene product, SsrB, has evolved the job of regulating the levels of csgD, a transcriptional regulator encoded by the core genome. For this activity, phosphorylation of SsrB was not required, which is rare amongst response regulators. Furthermore, we identify H-NS as a repressor of csgD in Salmonella, instead of an activator (Gerstel et al., 2003). This unifies the regulation of CsgD by H-NS in E. coli (Ogasawara et al., 2010) and Salmonella. This work places SsrB at a unique decision point in the choice between lifestyles bySalmonella and makes it crucial for the entire gamut of pathogenesis, i.e., biofilms and virulence.


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
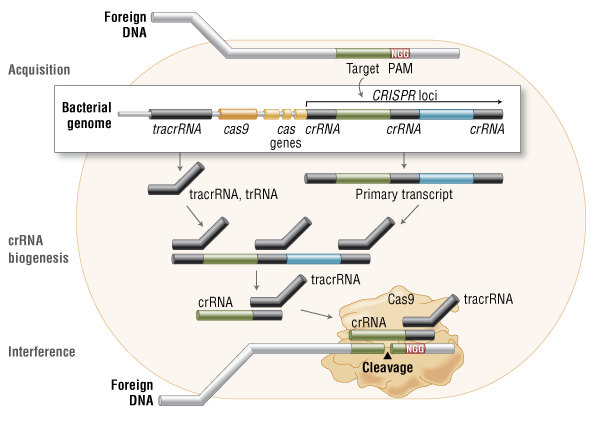{kind=link}
{kind=link}
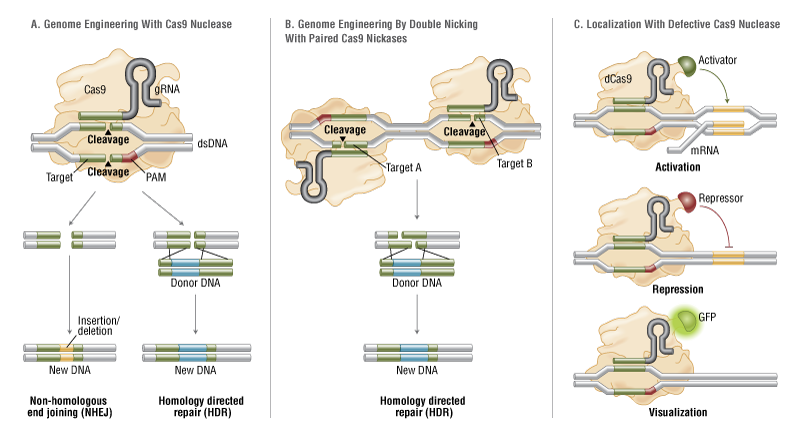{kind=link}
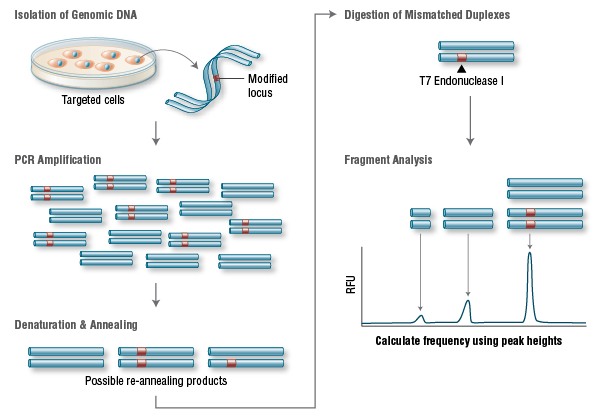{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}