Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Cancer Surgery Rethought: Immunotherapy Takes the Lead
Curator: Dr. Sudipta Saha, Ph.D.
In a recent phase 2 study published in The New England Journal of Medicine, the efficacy of nonoperative management was assessed in patients with mismatch repair–deficient (dMMR) solid tumors. Instead of undergoing curative-intent surgery, patients with stage I to III dMMR tumors were administered immune checkpoint inhibitors.
The study was conducted across two cohorts involving 117 patients. After two years of follow-up, a recurrence-free survival rate of 92% (95% CI, 86 to 99) was achieved. It was found that complete clinical responses could be maintained without surgical intervention, and substantial preservation of organ function was observed.
The avoidance of surgery was associated with fewer treatment-related complications and a significant improvement in patients’ quality of life. It has been emphasized that dMMR tumors, being highly immunogenic, respond exceptionally well to immune checkpoint blockade, thereby offering a viable alternative to conventional surgery-based treatment plans.
While the study’s findings have been considered ground breaking, long-term data have been recommended to fully validate this approach. Future studies are expected to refine patient selection criteria and monitoring strategies to ensure sustained outcomes.
Overall, a potential shift in the standard of care for patients with early-stage dMMR tumors has been proposed, highlighting how personalized immunotherapy can redefine oncological practice.
A large clinical trial has shown that pembrolizumab (Keytruda), an immunotherapy drug, nearly doubles the cancer-free survival time for patients with high-risk, muscle-invasive bladder cancer following surgery. The study, published on September 15, 2024, in The New England Journal of Medicine, was led by NIH researchers and demonstrated that pembrolizumab outperforms traditional observation methods post-surgery. Patients receiving pembrolizumab had a median cancer-free survival of 29.6 months, compared to 14.2 months for the observation group.
The trial enrolled 702 participants, some of whom had previously undergone cisplatin-based chemotherapy (neoadjuvant therapy). Pembrolizumab was administered every three weeks for a year. The drug was well tolerated, with common side effects including fatigue, itching, diarrhea, and thyroid issues.
Interestingly, the benefit of pembrolizumab was seen regardless of the tumor’s PD-L1 status.
Patients with PD-L1-positive tumors had a median cancer-free survival of 36.9 months.
Patients with PD-L1-negative tumors experienced 17.3 months cancer-free.
These results suggest that PD-L1 status should not be the sole factor in selecting patients for this therapy.
While overall survival rates were similar between the pembrolizumab and observation groups, many patients in the observation group began taking nivolumab once it was approved, complicating survival comparisons. Researchers are continuing to explore other treatment combinations and biomarkers to better personalize adjuvant therapy for bladder cancer patients.
Armored CD7-CAR T Cells: A Fratricide-Resistant Solution for T-ALL Therapy
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
This research reported in Nature Medicine addresses the challenge of treating T-cell acute lymphoblastic leukemia (T-ALL) with CAR T-cell therapy, particularly focusing on CD7, a surface marker highly expressed on T-ALL cells. The authors develop a novel CAR T-cell therapy that targets CD7, but with a crucial innovation which is fratricide resistance.
Fratricide, a phenomenon where CAR T cells kill each other (killing sister cells) due to shared CD7 expression, has been a significant problem in using CD7-directed therapies. To overcome this, the researchers made CD7-negative CAR T cells (CD7-CAR T cells) by knocking out CD7 from the CAR T cells themselves, preventing them from attacking one another.
Their preclinical results show that these CD7-CAR T cells exhibit strong anti-leukemic activity in T-ALL models, both in vitro and in vivo.
The fratricide-resistant T cells not only maintain their potency but also display enhanced proliferation and persistence, crucial for sustained therapeutic effects. Additionally,
the study highlights the feasibility and safety of this approach by demonstrating no adverse off-target effects or side effects, making it a potentially promising treatment for T-ALL patients who have limited options.
The research presents a significant advancement in CAR T-cell therapy by addressing the challenge of fratricide, offering a new, effective, and safe therapeutic option for T-ALL patients. The development of fratricide-resistant CD7-CAR T cells could lead to more successful outcomes in clinical applications, revolutionizing the treatment for T-ALL patients.
Inhibitory CD161 receptor recognized as a potential immunotherapy target in glioma-infiltrating T cells by single-cell analysis
Reporter: Dr. Premalata Pati, Ph.D., Postdoc
Brain tumors, especially the diffused Gliomas are of the most devastating forms of cancer and have so-far been resistant to immunotherapy. It is comprehended that T cells can penetrate the glioma cells, but it still remains unknown why infiltrating cells miscarry to mount a resistant reaction or stop the tumor development.
Gliomas are brain tumors that begin from neuroglial begetter cells. The conventional therapeutic methods including, surgery, chemotherapy, and radiotherapy, have accomplished restricted changes inside glioma patients. Immunotherapy, a compliance in cancer treatment, has introduced a promising strategy with the capacity to penetrate the blood-brain barrier. This has been recognized since the spearheading revelation of lymphatics within the central nervous system. Glioma is not generally carcinogenic. As observed in a number of cases, the tumor cells viably reproduce and assault the adjoining tissues, by and large, gliomas are malignant in nature and tend to metastasize. There are four grades in glioma, and each grade has distinctive cell features and different treatment strategies. Glioblastoma is a grade IV glioma, which is the crucial aggravated form. This infers that all glioblastomas are gliomas, however, not all gliomas are glioblastomas.
Decades of investigations on infiltrating gliomas still take off vital questions with respect to the etiology, cellular lineage, and function of various cell types inside glial malignancies. In spite of the available treatment options such as surgical resection, radiotherapy, and chemotherapy, the average survival rate for high-grade glioma patients remains 1–3 years (1).
A recent in vitro study performed by the researchers of Dana-Farber Cancer Institute, Massachusetts General Hospital, and the Broad Institute of MIT and Harvard, USA, has recognized that CD161 is identified as a potential new target for immunotherapy of malignant brain tumors. The scientific team depicted their work in the Cell Journal, in a paper entitled, “Inhibitory CD161 receptor recognized in glioma-infiltrating T cells by single-cell analysis.” on 15th February 2021.
To further expand their research and findings, Dr. Kai Wucherpfennig, MD, PhD, Chief of the Center for Cancer Immunotherapy, at Dana-Farber stated that their research is additionally important in a number of other major human cancer types such as
melanoma,
lung,
colon, and
liver cancer.
Dr. Wucherpfennig has praised the other authors of the report Mario Suva, MD, PhD, of Massachusetts Common Clinic; Aviv Regev, PhD, of the Klarman Cell Observatory at Broad Institute of MIT and Harvard, and David Reardon, MD, clinical executive of the Center for Neuro-Oncology at Dana-Farber.
Hence, this new study elaborates the effectiveness of the potential effectors of anti-tumor immunity in subsets of T cells that co-express cytotoxic programs and several natural killer (NK) cell genes.
The Study-
IMAGE SOURCE: Experimental Strategy (Mathewson et al., 2021)
The group utilized single-cell RNA sequencing (RNA-seq) to mull over gene expression and the clonal picture of tumor-infiltrating T cells. It involved the participation of 31 patients suffering from diffused gliomas and glioblastoma. Their work illustrated that the ligand molecule CLEC2D activates CD161, which is an immune cell surface receptor that restrains the development of cancer combating activity of immune T cells and tumor cells in the brain. The study reveals that the activation of CD161 weakens the T cell response against tumor cells.
Based on the study, the facts suggest that the analysis of clonally expanded tumor-infiltrating T cells further identifies the NK gene KLRB1 that codes for CD161 as a candidate inhibitory receptor. This was followed by the use of
CRISPR/Cas9 gene-editing technology to inactivate the KLRB1 gene in T cells and showed that CD161 inhibits the tumor cell-killing function of T cells. Accordingly,
genetic inactivation of KLRB1 or
antibody-mediated CD161 blockade
enhances T cell-mediated killing of glioma cells in vitro and their anti-tumor function in vivo. KLRB1 and its associated transcriptional program are also expressed by substantial T cell populations in other forms of human cancers. The work provides an atlas of T cells in gliomas and highlights CD161 and other NK cell receptors as immune checkpoint targets.
Further, it has been identified that many cancer patients are being treated with immunotherapy drugs that disable their “immune checkpoints” and their molecular brakes are exploited by the cancer cells to suppress the body’s defensive response induced by T cells against tumors. Disabling these checkpoints lead the immune system to attack the cancer cells. One of the most frequently targeted checkpoints is PD-1. However, recent trials of drugs that target PD-1 in glioblastomas have failed to benefit the patients.
In the current study, the researchers found that fewer T cells from gliomas contained PD-1 than CD161. As a result, they said, “CD161 may represent an attractive target, as it is a cell surface molecule expressed by both CD8 and CD4 T cell subsets [the two types of T cells engaged in response against tumor cells] and a larger fraction of T cells express CD161 than the PD-1 protein.”
However, potential side effects of antibody-mediated blockade of the CLEC2D-CD161 pathway remain unknown and will need to be examined in a non-human primate model. The group hopes to use this finding in their future work by
utilizing their outline by expression of KLRB1 gene in tumor-infiltrating T cells in diffuse gliomas to make a remarkable contribution in therapeutics related to immunosuppression in brain tumors along with four other common human cancers ( Viz. melanoma, non-small cell lung cancer (NSCLC), hepatocellular carcinoma, and colorectal cancer) and how this may be manipulated for prevalent survival of the patients.
References
(1) Anders I. Persson, QiWen Fan, Joanna J. Phillips, William A. Weiss, 39 – Glioma, Editor(s): Sid Gilman, Neurobiology of Disease, Academic Press, 2007, Pages 433-444, ISBN 9780120885923, https://doi.org/10.1016/B978-012088592-3/50041-4.
4.1.3 Single-cell Genomics: Directions in Computational and Systems Biology – Contributions of Prof. Aviv Regev @Broad Institute of MIT and Harvard, Cochair, the Human Cell Atlas Organizing Committee with Sarah Teichmann of the Wellcome Trust Sanger Institute
4.1.7 Norwich Single-Cell Symposium 2019, Earlham Institute, single-cell genomics technologies and their application in microbial, plant, animal and human health and disease, October 16-17, 2019, 10AM-5PM
Positron Emission Tomography (PET) and Near-Infrared Fluorescence Imaging: Noninvasive Imaging of Cancer Stem Cells (CSCs) monitoring of AC133+ glioblastoma in subcutaneous and intracerebral xenograft tumors
Structure-guided Drug Discovery: (1) The Coronavirus 3CL hydrolase (Mpro) enzyme (main protease) essential for proteolytic maturation of the virus and (2) viral protease, the RNA polymerase, the viral spike protein, a viral RNA as promising two targets for discovery of cleavage inhibitors of the viral spike polyprotein preventing the Coronavirus Virion the spread of infection
Curators and Reporters: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Therapeutical options to coronavirus (2019-nCoV) include consideration of the following:
(a) Monoclonal and polyclonal antibodies
(b) Vaccines
(c) Small molecule treatments (e.g., chloroquinolone and derivatives), including compounds already approved for other indications
(d) Immuno-therapies derived from human or other sources
Structure of the nCoV trimeric spike
The World Health Organization has declared the outbreak of a novel coronavirus (2019-nCoV) to be a public health emergency of international concern. The virus binds to host cells through its trimeric spike glycoprotein, making this protein a key target for potential therapies and diagnostics. Wrapp et al. determined a 3.5-angstrom-resolution structure of the 2019-nCoV trimeric spike protein by cryo–electron microscopy. Using biophysical assays, the authors show that this protein binds at least 10 times more tightly than the corresponding spike protein of severe acute respiratory syndrome (SARS)–CoV to their common host cell receptor. They also tested three antibodies known to bind to the SARS-CoV spike protein but did not detect binding to the 2019-nCoV spike protein. These studies provide valuable information to guide the development of medical counter-measures for 2019-nCoV. [Bold Face Added by ALA]
The outbreak of a novel coronavirus (2019-nCoV) represents a pandemic threat that has been declared a public health emergency of international concern. The CoV spike (S) glycoprotein is a key target for vaccines, therapeutic antibodies, and diagnostics. To facilitate medical countermeasure development, we determined a 3.5-angstrom-resolution cryo–electron microscopy structure of the 2019-nCoV S trimer in the prefusion conformation. The predominant state of the trimer has one of the three receptor-binding domains (RBDs) rotated up in a receptor-accessible conformation. We also provide biophysical and structural evidence that the 2019-nCoV S protein binds angiotensin-converting enzyme 2 (ACE2) with higher affinity than does severe acute respiratory syndrome (SARS)-CoV S. Additionally, we tested several published SARS-CoV RBD-specific monoclonal antibodies and found that they do not have appreciable binding to 2019-nCoV S, suggesting that antibody cross-reactivity may be limited between the two RBDs. The structure of 2019-nCoV S should enable the rapid development and evaluation of medical countermeasures to address the ongoing public health crisis.
SOURCE
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Recent emergence of the COVID-19 coronavirus has resulted in a WHO-declared public health emergency of international concern. Research efforts around the world are working towards establishing a greater understanding of this particular virus and developing treatments and vaccines to prevent further spread.
While PDB entry 6lu7 is currently the only public-domain 3D structure from this specific coronavirus, the PDB contains structures of the corresponding enzyme from other coronaviruses. The 2003 outbreak of the closely-related Severe Acute Respiratory Syndrome-related coronavirus (SARS) led to the first 3D structures, and today there are more than 200 PDB structures of SARS proteins. Structural information from these related proteins could be vital in furthering our understanding of coronaviruses and in discovery and development of new treatments and vaccines to contain the current outbreak.
The coronavirus 3CL hydrolase (Mpro) enzyme, also known as the main protease, is essential for proteolytic maturation of the virus. It is thought to be a promising target for discovery of small-molecule drugs that would inhibit cleavage of the viral polyprotein and prevent spread of the infection.
Comparison of the protein sequence of the COVID-19 coronavirus 3CL hydrolase (Mpro) against the PDB archive identified 95 PDB proteins with at least 90% sequence identity. Furthermore, these related protein structures contain approximately 30 distinct small molecule inhibitors, which could guide discovery of new drugs. Of particular significance for drug discovery is the very high amino acid sequence identity (96%) between the COVID-19 coronavirus 3CL hydrolase (Mpro) and the SARS virus main protease (PDB 1q2w). Summary data about these closely-related PDB structures are available (CSV) to help researchers more easily find this information. In addition, the PDB houses 3D structure data for more than 20 unique SARS proteins represented in more than 200 PDB structures, including a second viral protease, the RNA polymerase, the viral spike protein, a viral RNA, and other proteins (CSV).
Public release of the COVID-19 coronavirus 3CL hydrolase (Mpro), at a time when this information can prove most vital and valuable, highlights the importance of open and timely availability of scientific data. The wwPDB strives to ensure that 3D biological structure data remain freely accessible for all, while maintaining as comprehensive and accurate an archive as possible. We hope that this new structure, and those from related viruses, will help researchers and clinicians address the COVID-19 coronavirus global public health emergency.
Update: Released COVID-19-related PDB structures include
PDB structure 6lu7 (X. Liu, B. Zhang, Z. Jin, H. Yang, Z. Rao Crystal structure of COVID-19 main protease in complex with an inhibitor N3 doi: 10.2210/pdb6lu7/pdb) Released 2020-02-05
PDB structure 6vsb (D. Wrapp, N. Wang, K.S. Corbett, J.A. Goldsmith, C.-L. Hsieh, O. Abiona, B.S. Graham, J.S. McLellan (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation Science doi: 10.1126/science.abb2507) Released 2020-02-26
PDB structure 6lxt (Y. Zhu, F. Sun Structure of post fusion core of 2019-nCoV S2 subunit doi: 10.2210/pdb6lxt/pdb) Released 2020-02-26
PDB structure 6lvn (Y. Zhu, F. Sun Structure of the 2019-nCoV HR2 Domain doi: 10.2210/pdb6lvn/pdb) Released 2020-02-26
PDB structure 6vw1
J. Shang, G. Ye, K. Shi, Y.S. Wan, H. Aihara, F. Li Structural basis for receptor recognition by the novel coronavirus from Wuhan doi: 10.2210/pdb6vw1/pdb
Released 2020-03-04
PDB structure 6vww
Y. Kim, R. Jedrzejczak, N. Maltseva, M. Endres, A. Godzik, K. Michalska, A. Joachimiak, Center for Structural Genomics of Infectious Diseases Crystal Structure of NSP15 Endoribonuclease from SARS CoV-2 doi: 10.2210/pdb6vww/pdb
Released 2020-03-04
PDB structure 6y2e
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure of the free enzyme of the SARS-CoV-2 (2019-nCoV) main protease doi: 10.2210/pdb6y2e/pdb
Released 2020-03-04
PDB structure 6y2f
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (monoclinic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2f/pdb
Released 2020-03-04
PDB structure 6y2g
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (orthorhombic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2g/pdb
Released 2020-03-04
Coronavirus disease 2019 (COVID-19) is a global pandemic impacting nearly 170 countries/regions and more than 285,000 patients worldwide. COVID-19 is caused by the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), which invades cells through the angiotensin converting enzyme 2 (ACE2) receptor. Among those with COVID-19, there is a higher prevalence of cardiovascular disease and more than 7% of patients suffer myocardial injury from the infection (22% of the critically ill). Despite ACE2 serving as the portal for infection, the role of ACE inhibitors or angiotensin receptor blockers requires further investigation. COVID-19 poses a challenge for heart transplantation, impacting donor selection, immunosuppression, and post-transplant management. Thankfully there are a number of promising therapies under active investigation to both treat and prevent COVID-19. Key Words: COVID-19; myocardial injury; pandemic; heart transplant
Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW (Apr 2004). “ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis”. The Journal of Biological Chemistry. 279 (17): 17996–8007. doi:10.1074/jbc.M311191200. PMID14754895.
Turner AJ, Tipnis SR, Guy JL, Rice G, Hooper NM (Apr 2002). “ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors”. Canadian Journal of Physiology and Pharmacology. 80 (4): 346–53. doi:10.1139/y02-021. PMID12025971.
Zhang, Haibo; Penninger, Josef M.; Li, Yimin; Zhong, Nanshan; Slutsky, Arthur S. (3 March 2020). “Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target”. Intensive Care Medicine. Springer Science and Business Media LLC. doi:10.1007/s00134-020-05985-9. ISSN0342-4642. PMID32125455.
^Gurwitz, David (2020). “Angiotensin receptor blockers as tentative SARS‐CoV‐2 therapeutics”. Drug Development Research. doi:10.1002/ddr.21656. PMID32129518.
ACE2 receptors have been shown to be the entry point into human cells for some coronaviruses, including the SARSvirus.[10] A number of studies have identified that the entry point is the same for SARS-CoV-2,[11] the virus that causes COVID-19.[12][13][14][15]
Some have suggested that a decrease in ACE2 could be protective against Covid-19 disease[16], but others have suggested the opposite, that Angiotensin II receptor blocker drugs could be protective against Covid-19 disease via increasing ACE2, and that these hypotheses need to be tested by datamining of clinical patient records.[17]
We need your help! Folding@home is joining researchers around the world working to better understand the 2019 Coronavirus (2019-nCoV) to accelerate the open science effort to develop new life-saving therapies. By downloading Folding@Home, you can donate your unused computational resources to the Folding@home Consortium, where researchers working to advance our understanding of the structures of potential drug targets for 2019-nCoV that could aid in the design of new therapies. The data you help us generate will be quickly and openly disseminated as part of an open science collaboration of multiple laboratories around the world, giving researchers new tools that may unlock new opportunities for developing lifesaving drugs.
2019-nCoV is a close cousin to SARS coronavirus (SARS-CoV), and acts in a similar way. For both coronaviruses, the first step of infection occurs in the lungs, when a protein on the surface of the virus binds to a receptor protein on a lung cell. This viral protein is called the spike protein, depicted in red in the image below, and the receptor is known as ACE2. A therapeutic antibody is a type of protein that can block the viral protein from binding to its receptor, therefore preventing the virus from infecting the lung cell. A therapeutic antibody has already been developed for SARS-CoV, but to develop therapeutic antibodies or small molecules for 2019-nCoV, scientists need to better understand the structure of the viral spike protein and how it binds to the human ACE2 receptor required for viral entry into human cells.
Proteins are not stagnant—they wiggle and fold and unfold to take on numerous shapes. We need to study not only one shape of the viral spike protein, but all the ways the protein wiggles and folds into alternative shapes in order to best understand how it interacts with the ACE2 receptor, so that an antibody can be designed. Low-resolution structures of the SARS-CoV spike protein exist and we know the mutations that differ between SARS-CoV and 2019-nCoV. Given this information, we are uniquely positioned to help model the structure of the 2019-nCoV spike protein and identify sites that can be targeted by a therapeutic antibody. We can build computational models that accomplish this goal, but it takes a lot of computing power.
This is where you come in! With many computers working towards the same goal, we aim to help develop a therapeutic remedy as quickly as possible. By downloading Folding@home here [LINK] and selecting to contribute to “Any Disease”, you can help provide us with the computational power required to tackle this problem. One protein from 2019-nCoV, a protease encoded by the viral RNA, has already been crystallized. Although the 2019-nCoV spike protein of interest has not yet been resolved bound to ACE2, our objective is to use the homologous structure of the SARS-CoV spike protein to identify therapeutic antibody targets.
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
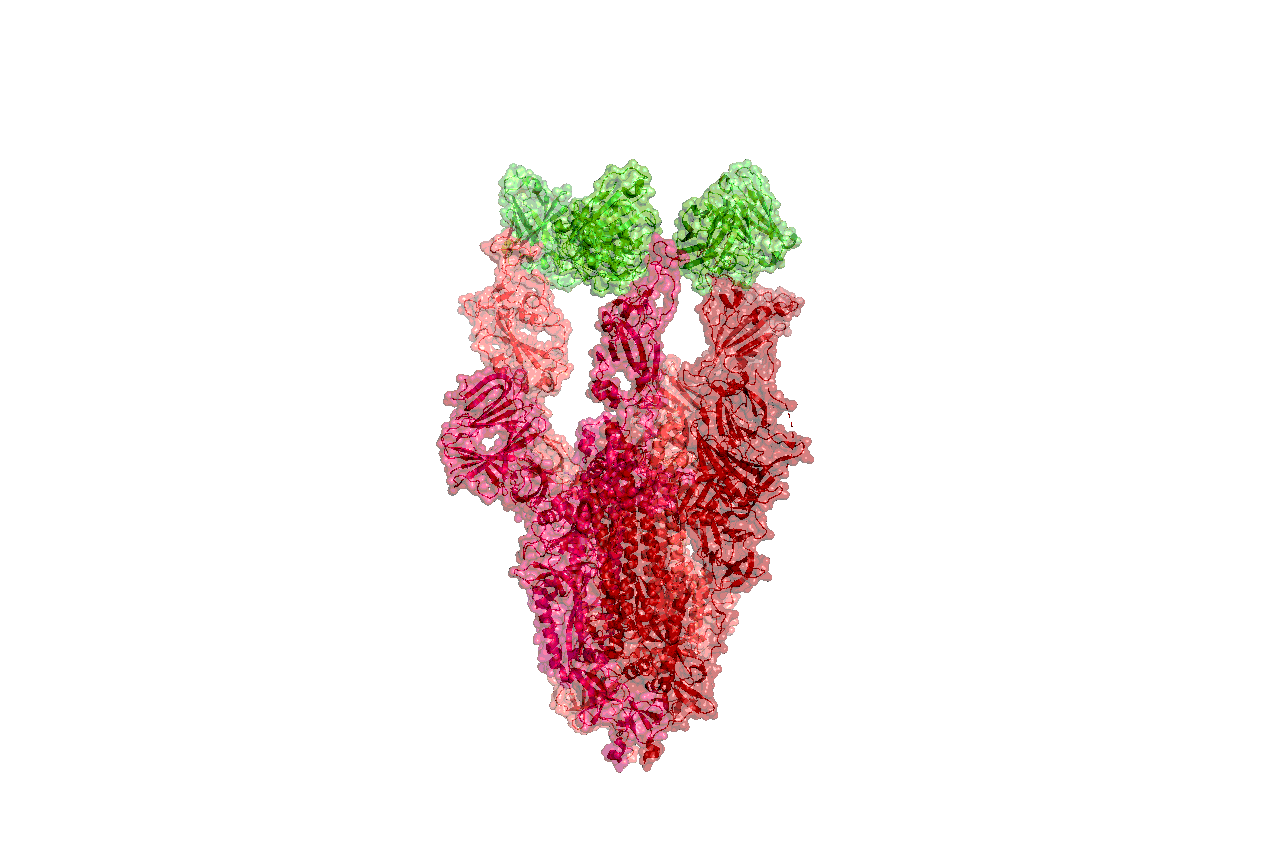
Structures of the closely related SARS-CoV spike protein bound by therapeutic antibodies may help rapidly design better therapies. The three monomers of the SARS-CoV spike protein are shown in different shades of red; the antibody is depicted in green. [PDB: 6NB7 https://www.rcsb.org/structure/6nb7]
I am reposting the following Science blog post from Derrick Lowe as is and ask people go browse through the comments on his Science blog In the Pipeline because, as Dr. Lowe states that in this current crisis it is important to disseminate good information as quickly as possible so wanted the readers here to have the ability to read his great posting on this matter of Covid-19. Also i would like to direct readers to the journal Science opinion letter concerning how important it is to rebuild the trust in good science and the scientific process. The full link for the following In the Pipeline post is: https://blogs.sciencemag.org/pipeline/archives/2020/03/06/covid-19-small-molecule-therapies-reviewed
A Summary of current potential repurposed therapeutics for COVID-19 Infection from In The Pipeline: A Science blog from Derick Lowe
Let’s take inventory on the therapies that are being developed for the coronavirus epidemic. Here is a very thorough list of at Biocentury, and I should note that (like Stat and several other organizations) they’re making all their Covid-19 content free to all readers during this crisis. I’d like to zoom in today on the potential small-molecule therapies, since some of these have the most immediate prospects for use in the real world.
The ones at the front of the line are repurposed drugs that are already approved for human use, for a lot of obvious reasons. The Biocentury list doesn’t cover these, but here’s an article at Nature Biotechnology that goes into detail. Clinical trials are a huge time sink – they sort of have to be, in most cases, if they’re going to be any good – and if you’ve already done all that stuff it’s a huge leg up, even if the drug itself is not exactly a perfect fit for the disease. So what do we have? The compound that is most advanced is probably remdesivir from Gilead, at right. This has been in development for a few years as an RNA virus therapy – it was originally developed for Ebola, and has been tried out against a whole list of single-strand RNA viruses. That includes the related coronaviruses SARS and MERS, so Covid-19 was an obvious fit.
The compound is a prodrug – that phosphoramide gets cleaved off completely, leaving the active 5-OH compound GS-44-1524. It mechanism of action is to get incorporated into viral RNA, since it’s taken up by RNA polymerase and it largely seems to evade proofreading. This causes RNA termination trouble later on, since that alpha-nitrile C-nucleoside is not exactly what the virus is expecting in its genome at that point, and thus viral replication is inhibited.
There are five clinical trials underway (here’s an overview at Biocentury). The NIH has an adaptive-design Phase II trial that has already started in Nebraska, with doses to be changed according to Bayesian readouts along the way. There are two Phase III trials underway at China-Japan Friendship Hospital in Hubei, double-blinded and placebo-controlled (since placebo is, as far as drug therapy goes, the current standard of care). And Gilead themselves are starting two open-label trials, one with no control arm and one with an (unblinded) standard-of-care comparison arm. Those might read out first, depending on when they get off the ground, but will be only rough readouts due to the fast-and-loose trial design. The two Hubei trials and the NIH one will add some rigor to the process, but I’m not sure when they’re going to report. My personal opinion is that I like the chances of this drug more than anything else on this list, but it’s still unlikely to be a game-changer.
There’s an RNA polymerase inhibitor (favipiravir) from Toyama, at right, that’s in a trial in China. It’s a thought – a broad-spectrum agent of this sort would be the sort of thing to try. But unfortunately, from what I can see, it has already turned up as ineffective in in vitro tests. The human trial that’s underway is honestly the sort of thing that would only happen under circumstances like the present: a developing epidemic with a new pathogen and no real standard of care. I hold out little hope for this one, but given that there’s nothing else at present, it probably should be tried. As you’ll see, this is far from the only situation like this.
One of the screens of known drugs in China that also flagged remdesivir noted that the old antimalarial drug chloroquine seemed to be effective in vitro. It had been reported some years back as a possible antiviral, working through more than one mechanism, probably both at viral entry and intracellularly thereafter. That part shouldn’t be surprising – chloroquine’s actual mode(s) of action against malaria parasites are still not completely worked out, either, and some of what people thought they knew about it has turned out to be wrong. There are several trials underway with it at Chinese facilities, some in combination with other agents like remdesivir. Chloroquine has of course been taken for many decades as an antimalarial, but it has a number of liabilities, including seizures, hearing damage, retinopathy and sudden effects on blood glucose. So it’s going to be important to establish just how effective it is and what doses will be needed. Just as with vaccine candidates, it’s possible to do more harm with a rushed treatment than the disease is doing itself
There are several other known antiviral drugs are being tried in China, but I don’t have too much hope for those, either. The neuraminidase inhibitors such as oseltamivir (better known as Tamiflu) were tried against SARS and were ineffective; there is no reason to expect anything versus Covid-19 although these drugs are a component of some drug cocktail trials. The HIV protease therapies such as darunavir and the combination therapy Kaletra are in trials, but that’s also a rather desperate long shot, since there’s no particular reason to think that they will have any such protease inhibition against what this new virus has to offer (and indeed, such agents weren’t much help against SARS in the end, either). The classic interferon/ribavirin combination seems to have had some activity against SARS and MERS, and is in two trials from what I can see. That’s not an awful idea by any means, but it’s not a great one, either: if your viral disease has interferon/ribavirin as a front line therapy, it generally means that there’s nothing really good available. No, unless we get really lucky none of these ideas are going to slow the disease down much.
There are a few other repurposed-protease-inhibitors ideas out there, such as this one. (Edit: I had seen this paper but couldn’t track it down, so thanks to those who sent it along). This paper suggests that the TMPRSS2 protease is important for viral entry on the human-cell-side of the process, a pathway that has been noted for other coronaviruses. And it points out that there is a an approved inhibitor (in Japan) for this enzyme (camostat), so that would definitely seem to be worth a trial, probably in combination with remdesivir.
That’s about it for the existing small molecules, from what I can see. What about new ones? Don’t hold your breath, is all I can say. A drug discovery program from scratch against a new pathogen is, as many readers here well know, not a trivial exercise. As this Bloomberg article details, many such efforts in the past (small molecules and vaccines alike) have come to grief because by the time they had anything to deliver the epidemic itself had passed. Indeed, Gilead’s remdesivir had already been dropped as a potential Ebola therapy.
You will either need to have a target in mind up front or go phenotypic. For the former, what you’d see are better characterizations of the viral protease and more extensive screens against it. Two other big target areas are viral entry (which involves the “spike” proteins on the virus surface and the ACE2 protein on human cells) and viral replication. To the former, it’s worth quickly noting that ACE2 is so much unlike the more familiar ACE protein that none of the cardiovascular ACE inhibitors do anything to it at all. And targeting the latter mechanisms is how remdesivir was developed as a possible Ebola agent, but as you can see, that took time, too. Phenotypic screens are perfectly reasonable against viral pathogens as well, but you’ll need to put time and effort into that assay up front, just as with any phenotypic effort, because as anyone who does that sort of work will tell you, a bad phenotypic screen is a complete waste of everyone’s time.
One of the key steps for either route is identifying an animal model. While animal models of infectious disease can be extremely well translated to human therapy, that doesn’t happen by accident: you need to choose the right animal. Viruses in general (and coronaviruses are no exception) vary widely in their effects in different species, and not just across the gaps of bird/reptile/human and the like. No, you’ll run into things where even the usual set of small mammals are acting differently from each other, with some of them not even getting sick at all. This current virus may well have gone through a couple of other mammalian species before landing on us, but you’ll note that dogs (to pick one) don’t seem to have any problem with it.
All this means that any new-target new-chemical-matter effort against Covid-19 (or any new pathogen) is going to take years, and there is just no way around that. Update: see here for just such an effort to start finding fragment hits for the viral protease. This puts small molecules in a very bimodal distribution: you have the existing drugs that might be repurposed, and are presumably available right now. Nothing else is! At the other end, for completely new therapies you have the usual prospects of drug discovery: years from now, lots of money, low success rate, good luck to all of us. The gap between these two could in theory be filled by vaccines and antibody therapies (if everything goes really, really well) but those are very much their own area and will be dealt with in a separate post.
Either way, the odds are that we (and I mean “we as a species” here) are going to be fighting this epidemic without any particularly amazing pharmacological weapons. Eventually we’ll have some, but I would advise people, pundits, and politicians not to get all excited about the prospects for some new therapies to come riding up over the hill to help us out. The odds of that happening in time to do anything about the current outbreak are very small. We will be going for months, years, with the therapeutic options we have right now. Look around you: what we have today is what we have to work with.
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Engineered Bacteria used as Trojan Horse for Cancer Immunotherapy
Reporter: Irina Robu, PhD
Researchers are using synthetic biology— design and construction of new biological entities such as enzymes, genetic circuits, and cells or the redesign of existing biological systems—is changing medicine leading to innovative solution in molecular-based therapeutics. To address the issue of designing therapies that can induce a potent, anti-tumor immune response researchers at Columbia Engineering and Columbia Irving Medical Center engineered a strain of non-pathogenic bacteria that can colonize tumors in mice. The non-pathogenic bacteria act as Trojan Horse that can lead to complete tumor regression in a mouse model of lymphoma. Their results are currently published in Nature Medicine.
The scientists led by Nicholas Arpaia, used their expertise in synthetic biology and immunology to engineer a strain of bacteria able to grow and multiply in the necrotic core of tumors. The non-pathogenic E. coli are programmed to self-destruct when the bacteria numbers reach a critical threshold, allowing for actual release of therapeutics and averting them from causing havoc somewhere else in the body. Afterward, a small portion of bacteria survive lysis and repopulate the population which allows repeated rounds of drug delivery inside treated tumors.
In the present study, the scientists release a nanobody that targets CD47 protein, which defends cancer cells from being eaten by distinctive immune cells. The mutual effects of bacteria, induced local inflammation within the tumor and the blockage of the CD47 leads to better ingestion and activation of T-cells within the treated tumors. The team deduced that the treatment with their engineered bacteria not only cleared the treated tumors but also reduced the incidence of tumor metastasis.
Before moving to clinical trials, the team is performing proof-of-concept tests, safety and toxicology studies of their immunotherapeutic bacteria in a rand of advanced solid tumor settings in mouse models. They have currently collaborated with Gary Schwartz, deputy director of the Herbert Irving Comprehensive Cancer and have underway a company to translate their promising technology to patients.
Nanoparticles Could Boost Effectiveness of Allergy Shots
Reporter : Irina Robu, PhD
Immunotherapy is a preventive treatment for allergic reactions to substances such as grass pollens, house dust mites and bee venom. The only existing therapy that treats their causes is allergen-specific immunotherapy or allergy shots which can cause severe side effects. For many people, allergies are a seasonal annoyance. But for others, exposure to a particular allergen can cause antagonistic reactions such as itching, breathing problems or even death. Allergy shots can diminish sensitivity by gradually ramping up exposure to the offending substance. Each allergy shot contains a tiny amount of the specific substance or substances that trigger your allergic reactions.
Holger Frey and colleagues report in Biomacromolecules the development of a potentially better allergy shot that uses nanocarriers to address these unwanted issues. In order to develop a safer, cause-based therapy scientist have developed nanoparticles that enclose an allergen and deliver it to specific cells. However, these nanocarriers degrade slowly, hindering the efficiency of the treatment.
Nanocarriers offer the following potential advantages: site-specific delivery of drugs, peptides, and genes, improved in-vitro and in-vivo stability and reduced side effect profile. However, nanoparticles are usually first picked up by the phagocytic cells of the immune system which may promote inflammatory disorders. In order to overcome the limitations, the researchers designed a novel type of nanocarrier created on the biocompatible molecule poly (ethylene glycol) that releases its cargo only in targeted immune cells.
This approach could be used not only for allergies but also can be used for other immunotherapies such as cancer and AIDS.
Immunotherapy may help in glioblastoma survival, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
Glioblastoma is the most common primary malignant brain tumor in adults and is associated with poor survival. But, in a glimmer of hope, a recent study found that a drug designed to unleash the immune system helped some patients live longer. Glioblastoma powerfully suppresses the immune system, both at the site of the cancer and throughout the body, which has made it difficult to find effective treatments. Such tumors are complex and differ widely in their behavior and characteristics.
A small randomized, multi-institution clinical trial was conducted and led by researchers at the University of California at Los Angeles involved patients who had a recurrence of glioblastoma, the most common central nervous system cancer. The aim was to evaluate immune responses and survival following neoadjuvant and/or adjuvant therapy with pembrolizumab (checkpoint inhibitor) in 35 patients with recurrent, surgically resectable glioblastoma. Patients who were randomized to receive neoadjuvant pembrolizumab, with continued adjuvant therapy following surgery, had significantly extended overall survival compared to patients that were randomized to receive adjuvant, post-surgical programmed cell death protein 1 (PD-1) blockade alone.
Neoadjuvant PD-1 blockade was associated with upregulation of T cell– and interferon-γ-related gene expression, but downregulation of cell-cycle-related gene expression within the tumor, which was not seen in patients that received adjuvant therapy alone. Focal induction of programmed death-ligand 1 in the tumor microenvironment, enhanced clonal expansion of T cells, decreased PD-1 expression on peripheral blood T cells and a decreasing monocytic population was observed more frequently in the neoadjuvant group than in patients treated only in the adjuvant setting. These findings suggest that the neoadjuvant administration of PD-1 blockade enhanced both the local and systemic antitumor immune response and may represent a more efficacious approach to the treatment of this uniformly lethal brain tumor.
Immunotherapy has not proved to be effective against glioblastoma. This small clinical trial explored the effect of PD-1 blockade on recurrent glioblastoma in relation to the timing of administration. A total of 35 patients undergoing resection of recurrent disease were randomized to either neoadjuvant or adjuvant pembrolizumab, and surgical specimens were compared between the two groups. Interestingly, the tumoral gene expression signature varied between the two groups, such that those who received neoadjuvant pembrolizumab displayed an INF-γ gene signature suggestive of T-cell activation as well as suppression of cell-cycle signaling, possibly consistent with growth arrest. Although the study was not powered for efficacy, the group found an increase in overall survival in patients receiving neoadjuvant pembrolizumab compared with adjuvant pembrolizumab of 13.7 months versus 7.5 months, respectively.
In this small pilot study, neoadjuvant PD-1 blockade followed by surgical resection was associated with intratumoral T-cell activation and inhibition of tumor growth as well as longer survival. How the drug works in glioblastoma has not been totally established. The researchers speculated that giving the drug before surgery prompted T-cells within the tumor, which had been impaired, to attack the cancer and extend lives. The drug didn’t spur such anti-cancer activity after the surgery because those T-cells were removed along with the tumor. The results are very important and very promising but would need to be validated in much larger trials.
The human body is often described as being ‘at war’. By this, it is meant that the body is constantly under attack from things that are trying to do it harm. These include toxins, bacteria, fungi, parasites and viruses. The human immune system is one of the most effective defense mechanisms known to nature and can sometimes can be overwhelmed by disease. Yet, on occasions our immune systems turn on our own tissue and attack it which can trigger conditions such as type I diabetes, rheumatoid arthritis and lupus.
In the case of rheumatoid arthritis, immune cells start to attack tissues in the joins which causes them to become painful, stiff and swollen. It is known that one third of those who develop rheumatoid arthritis, feel the horrible effects of the disease within two years of its onset. Immunologist Adrian Hayday, which is a researcher at Francis Crick Institute of London says that the current treatment for rheumatoid arthritis require patients to take the drugs for the rest of their lives. But, researchers such as Hayday found an unexpected ally in the battle against autoimmune disease, cancer.
However, there is a positive consequence to the discovery that cancer immunotherapies have the effect of triggering autoimmune diseases and for the first-time rheumatoid arthritis can be detected at the earliest stages. At present, people are not diagnosed with the condition until symptoms have already made their lives so unpleasant, they have gone to see their doctors. As a result, research backed by Cancer Research UK and Arthritis Research UK, has been launched with the aim of uncovering the roots of autoimmune disease from research on cancer patients.
The scientists mentioned stress that their work is only now start and warn that it will still take several years of research to get substantial results. Nevertheless, uncovering the first stages of an autoimmune disease emerging in a person’s body should give researchers a vital lead in ultimately developing treatments that will prevent or halt a range of conditions that currently cause a great deal of misery and require constant medication.
Our immune defenses consist of a range of cells and proteins that notice invading micro-organisms and attack them. The first line of defense, yet, consists of simple physical barriers similar to skin, which blocks invaders from entering your body. When this defense is penetrated, they are attacked by a number of agents. The key cells, leukocytes seek out and destroy disease-causing organisms. Neutrophils rush to the site of an infection and attack invading bacteria. Helper T-cells give instructions to other cells while killer T-cells punch holes in infected cells so that their contents ooze out. After these macrophages clean up the mess left behind.
Another significant agent is the B-cell, which produces antibodies that lock on to sites on the surface of bacteria or viruses and immobilize them until macrophages consume them. These cells can live a long time and can answer quickly following a second exposure to the same infections. In conclusion, suppressor T-cells act when an infection has been distributed with and the immune system needs to be reassured, the killer cells may keep on attacking, as they do in autoimmune diseases. By slowing down the immune system, regulatory T-cells prevent damage to “good” cells.
Two research groups from Harvard Medical School based at Dana Faber Cancer Institute have discovered a genetic mechanism in a cancer cells that influence whether they respond or resist to immunotherapy drugs, otherwise called as checkpoint inhibitors. The results are published in Science as part of two articles. One article is focused on clinical trial patients with advanced kidney cancer treated with checkpoint inhibitors comes from Eliezer van Allen’s group at Dana Farber Cancer Institute and Toni Choueiri group at Lank Center for Genitourinary Oncology at Dana Farber. The second articles is focused on identifying the immunotherapy resistance mechanism in melanoma cells comes from Kai Wucherpfennig at Dana-Farber and Shirley Liu at Dana -Farber. The two groups joined on that the resistance to immune checkpoint blockade is critically controlled by changes in a group of proteins that regulate how DNA is packaged in cells. The assortment of proteins, called a chromatin remodeling complex, is known as SWI/SNF. Its components are encoded by different genes, among them ARID2, PBRM1 and BRD7. SWI/SNF’s job is to open up stretches of tightly wound DNA so that its blueprints can be read by the cell to activate certain genes to make proteins.
Scientists led by Van Allen and Choueiri wanted a clarification for why some patients with a form of metastatic kidney cancer, clear cell renal carcinoma (ccRCC) gain clinical benefit from treatment with immune checkpoint inhibitors that block the PD-1 checkpoint while others patients don’t. The researchers use whole exome DNA sequencing to analyze tumor samples from 35 patients treated in a clinical trial with Opdivo, a checkpoint blocker nivolumab to search for other characteristics of ccRCC tumors that influence immunotherapy response and/or resistance. The scientist discovered that patients from the trial benefited from the immunotherapy treatment with longer survival and progression free survival were those whose tumors lacked a functioning PRBM1 gene. Loss of PRBM1 gene function caused cancer cells to have increased expression of other genes including those in the gene pathway known as IL6/JAK-STAT3, which is involved in immune system stimulation.
When the PBRM1 gene was knocked out in experiments, the melanoma cells became more sensitive to interferon gamma produced by T cells and, in response, produced signaling molecules that recruited more tumor-fighting T cells into the tumor. The two other genes in the PBAF complex—ARID2 and BRD7—are also found mutated in some cancers, according to the researchers, and those cancers, like the melanoma lacking ARID2 function, may also respond better to checkpoint blockade. According to the researchers, finding ways to alter those target molecules “will be important to extend the benefit of immunotherapy to larger patient populations, including cancers that thus far are refractory to immunotherapy.”