Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
RNA from the SARS-CoV-2 virus taking over the cells it infects: Virulence – Pathogen’s ability to infect a Resistant Host: The Imbalance between Controlling Virus Replication versus Activation of the Adaptive Immune Response
Curator: Aviva Lev-Ari, PhD, RN – I added colors and bold face
UPDATED on 9/8/2020
What bats can teach us about developing immunity to Covid-19 | Free to read
Another duality and paradox in the Treatment of COVID-19 Patients in ICUs was expressed by Mike Yoffe, MD, PhD, David H. Koch Professor of Biology and Biological Engineering, Massachusetts Institute of Technology. Dr. Yaffe has a joint appointment in Acute Care Surgery, Trauma, and Surgical Critical Care, and in Surgical Oncology @BIDMC
on 6/29 at SOLUTIONS with/in/sight at Koch Institute @MIT
How Are Cancer Researchers Fighting COVID-19? (Part II)”Jun 29, 2020 11:30 AM EST
In COVID-19 patients: two life threatening conditions are seen in ICUs:
Blood Clotting – Hypercoagulability or Thrombophilia
Cytokine Storm – immuno-inflammatory response
The coexistence of 1 and 2 – HINDERS the ability to use effectively tPA as an anti-clotting agent while the cytokine storm is present.
Mike Yoffe’s related domain of expertise:
Signaling pathways and networks that control cytokine responses and inflammation
Misregulation of cytokine feedback loops, along with inappropriate activation of the blood clotting cascade causes dysregulation of cell signaling pathways in innate immune cells (neutrophils and macrophages), resulting in tissue damage and multiple organ failure following trauma or sepsis. Our research is focused on understanding the role of the p38-MK2 pathway in cytokine control and innate immune function, and on cross-talk between cytokines, clotting factors, and neutrophil NADPH oxidase-derived ROS in tissue damage, coagulopathy, and inflammation, using biochemistry, cell biology, and mouse knock-out/knock-in models. We recently discovered a particularly important link between abnormal blood clotting and the complement pathway cytokine C5a which causes excessive production of extracellular ROS and organ damage by neutrophils after traumatic injury.
SARS-CoV-2 infection induces low IFN-I and -III levels with a moderate ISG response
Strong chemokine expression is consistent across in vitro, ex vivo, and in vivo models
Low innate antiviral defenses and high pro-inflammatory cues contribute to COVID-19
Summary
Viral pandemics, such as the one caused by SARS-CoV-2, pose an imminent threat to humanity. Because of its recent emergence, there is a paucity of information regarding viral behavior and host response following SARS-CoV-2 infection. Here we offer an in-depth analysis of the transcriptional response to SARS-CoV-2 compared with other respiratory viruses. Cell and animal models of SARS-CoV-2 infection, in addition to transcriptional and serum profiling of COVID-19 patients, consistently revealed a unique and inappropriate inflammatory response. This response is defined by low levels of type I and III interferons juxtaposed to elevated chemokines and high expression of IL-6. We propose that reduced innate antiviral defenses coupled with exuberant inflammatory cytokine production are the defining and driving features of COVID-19.
Defining the Transcriptional Response to SARS-CoV-2 Relative to Other Respiratory Viruses
To compare the transcriptional response of SARS-CoV-2 with other respiratory viruses, including MERS-CoV, SARS-CoV-1, human parainfluenza virus 3 (HPIV3), respiratory syncytial virus (RSV), and IAV, we first chose to focus on infection in a variety of respiratory cell lines (Figure 1). To this end, we collected poly(A) RNA from infected cells and performed RNA sequencing (RNA-seq) to estimate viral load. These data show that virus infection levels ranged from 0.1% to more than 50% of total RNA reads (Figure 1A).
Discussion
In the present study, we focus on defining the host response to SARS-CoV-2 and other human respiratory viruses in cell lines, primary cell cultures, ferrets, and COVID-19 patients. In general, our data show that the overall transcriptional footprint of SARS-CoV-2 infection was distinct in comparison with other highly pathogenic coronaviruses and common respiratory viruses such as IAV, HPIV3, and RSV. It is noteworthy that, despite a reduced IFN-I and -III response to SARS-CoV-2, we observed a consistent chemokine signature. One exception to this observation is the response to high-MOI infection in A549-ACE2 and Calu-3 cells, where replication was robust and an IFN-I and -III signature could be observed. In both of these examples, cells were infected at a rate to theoretically deliver two functional virions per cell in addition to any defective interfering particles within the virus stock that were not accounted for by plaque assays. Under these conditions, the threshold for PAMP may be achieved prior to the ability of the virus to evade detection through production of a viral antagonist. Alternatively, addition of multiple genomes to a single cell may disrupt the stoichiometry of viral components, which, in turn, may itself generate PAMPs that would not form otherwise. These ideas are supported by the fact that, at a low-MOI infection in A549-ACE2 cells, high levels of replication could also be achieved, but in the absence of IFN-I and -III induction. Taken together, these data suggest that, at low MOIs, the virus is not a strong inducer of the IFN-I and -III system, as opposed to conditions where the MOI is high.
Taken together, the data presented here suggest that the response to SARS-CoV-2 is imbalanced with regard to controlling virus replication versus activation of the adaptive immune response. Given this dynamic, treatments for COVID-19 have less to do with the IFN response and more to do with controlling inflammation. Because our data suggest that numerous chemokines and ILs are elevated in COVID-19 patients, future efforts should focus on U.S. Food and Drug Administration (FDA)-approved drugs that can be rapidly deployed and have immunomodulating properties.
One of the features distinguishing SARS-CoV-2 from its more pathogenic counterpart SARS-CoV is the presence of premature stop codons in its ORF3b gene. Here, we show that SARS-CoV-2 ORF3b is a potent interferon antagonist, suppressing the induction of type I interferon more efficiently than its SARS-CoV ortholog. Phylogenetic analyses and functional assays revealed that SARS-CoV-2-related viruses from bats and pangolins also encode truncated ORF3b gene products with strong anti-interferon activity. Furthermore, analyses of more than 15,000 SARS-CoV-2 sequences identified a natural variant, in which a longer ORF3b reading frame was reconstituted. This variant was isolated from two patients with severe disease and further increased theability of ORF3b to suppress interferon induction. Thus, our findings not only help to explain the poor interferon response in COVID-19 patients, but also describe a possibility of the emergence of natural SARS-CoV-2 quasi-species with extended ORF3b that may exacerbate COVID-19 symptoms.
Highlights
ORF3b of SARS-CoV-2 and related bat and pangolin viruses is a potent IFN antagonist
SARS-CoV-2 ORF3b suppresses IFN induction more efficiently than SARS-CoV ortholog
The anti-IFN activity of ORF3b depends on the length of its C-terminus
An ORF3b with increased IFN antagonism was isolated from two severe COVID-19 cases
RNA (in green) from the SARS-CoV-2 virus is shown taking over the cells it infects.ICAHN SCHOOL OF MEDICINE AT MOUNT SINAI
A deep dive into how the new coronavirus infects cells has found that it orchestrates a hostile takeover of their genes unlike any other known viruses do, producing what one leading scientist calls “unique” and “aberrant” changes.Recent studies show that in seizing control of genes in the human cells it invades, the virus changes how segments of DNA are read, doing so in a way that might explain why the elderly are more likely to die of Covid-19 and why antiviral drugs might not only save sick patients’ lives but also prevent severe disease if taken before infection.“It’s something I have never seen in my 20 years of” studying viruses, said virologist Benjamin tenOever of the Icahn School of Medicine at Mount Sinai, referring to how SARS-CoV-2, the virus that causes Covid-19, hijacks cells’ genomes.The “something” he and his colleagues saw is how SARS-CoV-2 blocks one virus-fighting set of genes but allows another set to launch, a pattern never seen with other viruses. Influenza and the original SARS virus (in the early 2000s), for instance, interfere with both arms of the body’s immune response — what tenOever dubs “call to arms” genes and “call for reinforcement” genes.The first group of genes produces interferons. These proteins, which infected cells release, are biological semaphores, signaling to neighboring cells to activate some 500 of their own genes that will slow down the virus’ ability to make millions of copies of itself if it invades them. This lasts seven to 10 days, tenOever said, controlling virus replication and thereby buying time for the second group of genes to act.This second set of genes produce their own secreted proteins, called chemokines, that emit a biochemical “come here!” alarm. When far-flung antibody-making B cells and virus-killing T cells sense the alarm, they race to its source. If all goes well, the first set of genes holds the virus at bay long enough for the lethal professional killers to arrive and start eradicating viruses.
“Most other viruses interfere with some aspect of both the call to arms and the call for reinforcements,” tenOever said. “If they didn’t, no one would ever get a viral illness”: The one-two punch would pummel any incipient infection into submission.
SARS-CoV-2, however, uniquely blocks one cellular defense but activates the other, he and his colleagues reported in a study published last week in Cell. They studied healthy human lung cells growing in lab dishes, ferrets (which the virus infects easily), and lung cells from Covid-19 patients. In all three, they found that within three days of infection, the virus induces cells’ call-for-reinforcement genes to produce cytokines. But it blocks their call-to-arms genes — the interferons that dampen the virus’ replication.
The result is essentially no brakes on the virus’s replication, but a storm of inflammatory molecules in the lungs, which is what tenOever calls an “unique” and “aberrant” consequence of how SARS-CoV-2 manipulates the genome of its target.
In another new study, scientists in Japan last week identified how SARS-CoV-2 accomplishes that genetic manipulation. Its ORF3b gene produces a protein called a transcription factor that has “strong anti-interferon activity,” Kei Sato of the University of Tokyo and colleagues found — stronger than the original SARS virus or influenza viruses. The protein basically blocks the cell from recognizing that a virus is present, in a way that prevents interferon genes from being expressed.
In fact, the Icahn School team found no interferons in the lung cells of Covid-19 patients. Without interferons, tenOever said, “there is nothing to stop the virus from replicating and festering in the lungs forever.”
That causes lung cells to emit even more “call-for-reinforcement” genes, summoning more and more immune cells. Now the lungs have macrophages and neutrophils and other immune cells “everywhere,” tenOever said, causing such runaway inflammation “that you start having inflammation that induces more inflammation.”
At the same time, unchecked viral replication kills lung cells involved in oxygen exchange. “And suddenly you’re in the hospital in severe respiratory distress,” he said.
In elderly people, as well as those with diabetes, heart disease, and other underlying conditions, the call-to-arms part of the immune system is weaker than in younger, healthier people, even before the coronavirus arrives. That reduces even further the cells’ ability to knock down virus replication with interferons, and imbalances the immune system toward the dangerous inflammatory response.
The discovery that SARS-CoV-2 strongly suppresses infected cells’ production of interferons has raised an intriguing possibility: that taking interferons might prevent severe Covid-19 or even prevent it in the first place, said Vineet Menachery of the University of Texas Medical Branch.
In a study of human cells growing in lab dishes, described in a preprint (not peer-reviewed or published in a journal yet), he and his colleagues also found that SARS-CoV-2 “prevents the vast amount” of interferon genes from turning on. But when cells growing in lab dishes received the interferon IFN-1 before exposure to the coronavirus, “the virus has a difficult time replicating.”
After a few days, the amount of virus in infected but interferon-treated cells was 1,000- to 10,000-fold lower than in infected cells not pre-treated with interferon. (The original SARS virus, in contrast, is insensitive to interferon.)
Ending the pandemic and preventing its return is assumed to require an effective vaccine to prevent infectionand antiviral drugs such as remdesivir to treat the very sick, but the genetic studies suggest a third strategy: preventive drugs.
It’s possible that treatment with so-called type-1 interferon “could stop the virus before it could get established,” Menachery said.
Giving drugs to healthy people is always a dicey proposition, since all drugs have side effects — something considered less acceptable than when a drug is used to treat an illness. “Interferon treatment is rife with complications,” Menachery warned. The various interferons, which are prescribed for hepatitis, cancers, and many other diseases, can cause flu-like symptoms.
But the risk-benefit equation might shift, both for individuals and for society, if interferons or antivirals or other medications are shown to reduce the risk of developing serious Covid-19 or even make any infection nearly asymptomatic.
Interferon “would be warning the cells the virus is coming,” Menachery said, so such pretreatment might “allow treated cells to fend off the virus better and limit its spread.” Determining that will of course require clinical trials, which are underway.
Other related articles in this Open Access Online Scientific Journal include the following:
Structure-guided Drug Discovery: (1) The Coronavirus 3CL hydrolase (Mpro) enzyme (main protease) essential for proteolytic maturation of the virus and (2) viral protease, the RNA polymerase, the viral spike protein, a viral RNA as promising two targets for discovery of cleavage inhibitors of the viral spike polyprotein preventing the Coronavirus Virion the spread of infection
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Glycobiology vs Proteomics: Glycobiologists Prespective in the effort to explain the origin, etiology and potential therapeutics for the Coronavirus Pandemic (COVID-19).
Actemra, immunosuppressive which was designed to treat rheumatoid arthritis but also approved in 2017 to treat cytokine storms in cancer patients SAVED the sickest of all COVID-19 patients
The Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) Partnership on May 18, 2020: Leadership of AbbVie, Amgen, AstraZeneca, Bristol Myers Squibb, Eisai, Eli Lilly, Evotec, Gilead, GlaxoSmithKline, Johnson & Johnson, KSQ Therapeutics, Merck, Novartis, Pfizer, Roche, Sanofi, Takeda, and Vir. We also thank multiple NIH institutes (especially NIAID), the FDA, BARDA, CDC, the European Medicines Agency, the Department of Defense, the VA, and the Foundation for NIH
Tweets & Retweets 2020 World Medical Innovation Forum – COVID-19, AI and the Future of Medicine, Featuring Harvard and Industry Leader Insights – MGH & BWH, Virtual Event: Monday, May 11, 8:15 a.m. – 5:15 p.m. ET
Actemra, immunosuppressive which was designed to treat rheumatoid arthritis but also approved in 2017 to treat cytokine storms in cancer patients SAVED the sickest of all COVID-19 patients
Reporter: Aviva Lev-Ari, PhD, RN
Emergency room doctor, near death with coronavirus, saved with experimental treatment
Soon after being admitted to his own hospital with a fever, cough and difficulty breathing, he was placed on a ventilator. Five days after that, his lungs and kidneys were failing, his heart was in trouble, and doctors figured he had a day or so to live.
He owes his survival to an elite team of doctors who tried an experimental treatment pioneered in China and used on the sickest of all COVID-19 patients.
Lessons from his dramatic recovery could help doctors worldwide treat other extremely ill COVID-19 patients.
Based on the astronomical level of inflammation in his body and reports written by Chinese and Italian physicians who had treated the sickest COVID-19 patients, the doctors came to believe that it was not the disease itself killing him but his own immune system.
It had gone haywire and began to attack itself — a syndrome known as a “cytokine storm.”
The immune system normally uses proteins called cytokines as weapons in fighting a disease. For unknown reasons in some COVID-19 patients, the immune system first fails to respond quickly enough and then floods the body with cytokines, destroying blood vessels and filling the lungs with fluid.
Dr. Matt Hartman, a cardiologist, said that after four days on the immunosuppressive drug, supplemented by high-dose vitamin C and other therapies, the level of oxygen in Padgett’s blood improved dramatically. On March 23, doctors were able to take him off life support.
Four days later, they removed his breathing tube. He slowly came out of his sedated coma, at first imagining that he was in the top floor of the Space Needle converted to a COVID ward.
Structure-guided Drug Discovery: (1) The Coronavirus 3CL hydrolase (Mpro) enzyme (main protease) essential for proteolytic maturation of the virus and (2) viral protease, the RNA polymerase, the viral spike protein, a viral RNA as promising two targets for discovery of cleavage inhibitors of the viral spike polyprotein preventing the Coronavirus Virion the spread of infection
Curators and Reporters: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Therapeutical options to coronavirus (2019-nCoV) include consideration of the following:
(a) Monoclonal and polyclonal antibodies
(b) Vaccines
(c) Small molecule treatments (e.g., chloroquinolone and derivatives), including compounds already approved for other indications
(d) Immuno-therapies derived from human or other sources
Structure of the nCoV trimeric spike
The World Health Organization has declared the outbreak of a novel coronavirus (2019-nCoV) to be a public health emergency of international concern. The virus binds to host cells through its trimeric spike glycoprotein, making this protein a key target for potential therapies and diagnostics. Wrapp et al. determined a 3.5-angstrom-resolution structure of the 2019-nCoV trimeric spike protein by cryo–electron microscopy. Using biophysical assays, the authors show that this protein binds at least 10 times more tightly than the corresponding spike protein of severe acute respiratory syndrome (SARS)–CoV to their common host cell receptor. They also tested three antibodies known to bind to the SARS-CoV spike protein but did not detect binding to the 2019-nCoV spike protein. These studies provide valuable information to guide the development of medical counter-measures for 2019-nCoV. [Bold Face Added by ALA]
The outbreak of a novel coronavirus (2019-nCoV) represents a pandemic threat that has been declared a public health emergency of international concern. The CoV spike (S) glycoprotein is a key target for vaccines, therapeutic antibodies, and diagnostics. To facilitate medical countermeasure development, we determined a 3.5-angstrom-resolution cryo–electron microscopy structure of the 2019-nCoV S trimer in the prefusion conformation. The predominant state of the trimer has one of the three receptor-binding domains (RBDs) rotated up in a receptor-accessible conformation. We also provide biophysical and structural evidence that the 2019-nCoV S protein binds angiotensin-converting enzyme 2 (ACE2) with higher affinity than does severe acute respiratory syndrome (SARS)-CoV S. Additionally, we tested several published SARS-CoV RBD-specific monoclonal antibodies and found that they do not have appreciable binding to 2019-nCoV S, suggesting that antibody cross-reactivity may be limited between the two RBDs. The structure of 2019-nCoV S should enable the rapid development and evaluation of medical countermeasures to address the ongoing public health crisis.
SOURCE
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Recent emergence of the COVID-19 coronavirus has resulted in a WHO-declared public health emergency of international concern. Research efforts around the world are working towards establishing a greater understanding of this particular virus and developing treatments and vaccines to prevent further spread.
While PDB entry 6lu7 is currently the only public-domain 3D structure from this specific coronavirus, the PDB contains structures of the corresponding enzyme from other coronaviruses. The 2003 outbreak of the closely-related Severe Acute Respiratory Syndrome-related coronavirus (SARS) led to the first 3D structures, and today there are more than 200 PDB structures of SARS proteins. Structural information from these related proteins could be vital in furthering our understanding of coronaviruses and in discovery and development of new treatments and vaccines to contain the current outbreak.
The coronavirus 3CL hydrolase (Mpro) enzyme, also known as the main protease, is essential for proteolytic maturation of the virus. It is thought to be a promising target for discovery of small-molecule drugs that would inhibit cleavage of the viral polyprotein and prevent spread of the infection.
Comparison of the protein sequence of the COVID-19 coronavirus 3CL hydrolase (Mpro) against the PDB archive identified 95 PDB proteins with at least 90% sequence identity. Furthermore, these related protein structures contain approximately 30 distinct small molecule inhibitors, which could guide discovery of new drugs. Of particular significance for drug discovery is the very high amino acid sequence identity (96%) between the COVID-19 coronavirus 3CL hydrolase (Mpro) and the SARS virus main protease (PDB 1q2w). Summary data about these closely-related PDB structures are available (CSV) to help researchers more easily find this information. In addition, the PDB houses 3D structure data for more than 20 unique SARS proteins represented in more than 200 PDB structures, including a second viral protease, the RNA polymerase, the viral spike protein, a viral RNA, and other proteins (CSV).
Public release of the COVID-19 coronavirus 3CL hydrolase (Mpro), at a time when this information can prove most vital and valuable, highlights the importance of open and timely availability of scientific data. The wwPDB strives to ensure that 3D biological structure data remain freely accessible for all, while maintaining as comprehensive and accurate an archive as possible. We hope that this new structure, and those from related viruses, will help researchers and clinicians address the COVID-19 coronavirus global public health emergency.
Update: Released COVID-19-related PDB structures include
PDB structure 6lu7 (X. Liu, B. Zhang, Z. Jin, H. Yang, Z. Rao Crystal structure of COVID-19 main protease in complex with an inhibitor N3 doi: 10.2210/pdb6lu7/pdb) Released 2020-02-05
PDB structure 6vsb (D. Wrapp, N. Wang, K.S. Corbett, J.A. Goldsmith, C.-L. Hsieh, O. Abiona, B.S. Graham, J.S. McLellan (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation Science doi: 10.1126/science.abb2507) Released 2020-02-26
PDB structure 6lxt (Y. Zhu, F. Sun Structure of post fusion core of 2019-nCoV S2 subunit doi: 10.2210/pdb6lxt/pdb) Released 2020-02-26
PDB structure 6lvn (Y. Zhu, F. Sun Structure of the 2019-nCoV HR2 Domain doi: 10.2210/pdb6lvn/pdb) Released 2020-02-26
PDB structure 6vw1
J. Shang, G. Ye, K. Shi, Y.S. Wan, H. Aihara, F. Li Structural basis for receptor recognition by the novel coronavirus from Wuhan doi: 10.2210/pdb6vw1/pdb
Released 2020-03-04
PDB structure 6vww
Y. Kim, R. Jedrzejczak, N. Maltseva, M. Endres, A. Godzik, K. Michalska, A. Joachimiak, Center for Structural Genomics of Infectious Diseases Crystal Structure of NSP15 Endoribonuclease from SARS CoV-2 doi: 10.2210/pdb6vww/pdb
Released 2020-03-04
PDB structure 6y2e
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure of the free enzyme of the SARS-CoV-2 (2019-nCoV) main protease doi: 10.2210/pdb6y2e/pdb
Released 2020-03-04
PDB structure 6y2f
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (monoclinic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2f/pdb
Released 2020-03-04
PDB structure 6y2g
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (orthorhombic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2g/pdb
Released 2020-03-04
Coronavirus disease 2019 (COVID-19) is a global pandemic impacting nearly 170 countries/regions and more than 285,000 patients worldwide. COVID-19 is caused by the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), which invades cells through the angiotensin converting enzyme 2 (ACE2) receptor. Among those with COVID-19, there is a higher prevalence of cardiovascular disease and more than 7% of patients suffer myocardial injury from the infection (22% of the critically ill). Despite ACE2 serving as the portal for infection, the role of ACE inhibitors or angiotensin receptor blockers requires further investigation. COVID-19 poses a challenge for heart transplantation, impacting donor selection, immunosuppression, and post-transplant management. Thankfully there are a number of promising therapies under active investigation to both treat and prevent COVID-19. Key Words: COVID-19; myocardial injury; pandemic; heart transplant
Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW (Apr 2004). “ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis”. The Journal of Biological Chemistry. 279 (17): 17996–8007. doi:10.1074/jbc.M311191200. PMID14754895.
Turner AJ, Tipnis SR, Guy JL, Rice G, Hooper NM (Apr 2002). “ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors”. Canadian Journal of Physiology and Pharmacology. 80 (4): 346–53. doi:10.1139/y02-021. PMID12025971.
Zhang, Haibo; Penninger, Josef M.; Li, Yimin; Zhong, Nanshan; Slutsky, Arthur S. (3 March 2020). “Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target”. Intensive Care Medicine. Springer Science and Business Media LLC. doi:10.1007/s00134-020-05985-9. ISSN0342-4642. PMID32125455.
^Gurwitz, David (2020). “Angiotensin receptor blockers as tentative SARS‐CoV‐2 therapeutics”. Drug Development Research. doi:10.1002/ddr.21656. PMID32129518.
ACE2 receptors have been shown to be the entry point into human cells for some coronaviruses, including the SARSvirus.[10] A number of studies have identified that the entry point is the same for SARS-CoV-2,[11] the virus that causes COVID-19.[12][13][14][15]
Some have suggested that a decrease in ACE2 could be protective against Covid-19 disease[16], but others have suggested the opposite, that Angiotensin II receptor blocker drugs could be protective against Covid-19 disease via increasing ACE2, and that these hypotheses need to be tested by datamining of clinical patient records.[17]
We need your help! Folding@home is joining researchers around the world working to better understand the 2019 Coronavirus (2019-nCoV) to accelerate the open science effort to develop new life-saving therapies. By downloading Folding@Home, you can donate your unused computational resources to the Folding@home Consortium, where researchers working to advance our understanding of the structures of potential drug targets for 2019-nCoV that could aid in the design of new therapies. The data you help us generate will be quickly and openly disseminated as part of an open science collaboration of multiple laboratories around the world, giving researchers new tools that may unlock new opportunities for developing lifesaving drugs.
2019-nCoV is a close cousin to SARS coronavirus (SARS-CoV), and acts in a similar way. For both coronaviruses, the first step of infection occurs in the lungs, when a protein on the surface of the virus binds to a receptor protein on a lung cell. This viral protein is called the spike protein, depicted in red in the image below, and the receptor is known as ACE2. A therapeutic antibody is a type of protein that can block the viral protein from binding to its receptor, therefore preventing the virus from infecting the lung cell. A therapeutic antibody has already been developed for SARS-CoV, but to develop therapeutic antibodies or small molecules for 2019-nCoV, scientists need to better understand the structure of the viral spike protein and how it binds to the human ACE2 receptor required for viral entry into human cells.
Proteins are not stagnant—they wiggle and fold and unfold to take on numerous shapes. We need to study not only one shape of the viral spike protein, but all the ways the protein wiggles and folds into alternative shapes in order to best understand how it interacts with the ACE2 receptor, so that an antibody can be designed. Low-resolution structures of the SARS-CoV spike protein exist and we know the mutations that differ between SARS-CoV and 2019-nCoV. Given this information, we are uniquely positioned to help model the structure of the 2019-nCoV spike protein and identify sites that can be targeted by a therapeutic antibody. We can build computational models that accomplish this goal, but it takes a lot of computing power.
This is where you come in! With many computers working towards the same goal, we aim to help develop a therapeutic remedy as quickly as possible. By downloading Folding@home here [LINK] and selecting to contribute to “Any Disease”, you can help provide us with the computational power required to tackle this problem. One protein from 2019-nCoV, a protease encoded by the viral RNA, has already been crystallized. Although the 2019-nCoV spike protein of interest has not yet been resolved bound to ACE2, our objective is to use the homologous structure of the SARS-CoV spike protein to identify therapeutic antibody targets.
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
Structures of the closely related SARS-CoV spike protein bound by therapeutic antibodies may help rapidly design better therapies. The three monomers of the SARS-CoV spike protein are shown in different shades of red; the antibody is depicted in green. [PDB: 6NB7 https://www.rcsb.org/structure/6nb7]
I am reposting the following Science blog post from Derrick Lowe as is and ask people go browse through the comments on his Science blog In the Pipeline because, as Dr. Lowe states that in this current crisis it is important to disseminate good information as quickly as possible so wanted the readers here to have the ability to read his great posting on this matter of Covid-19. Also i would like to direct readers to the journal Science opinion letter concerning how important it is to rebuild the trust in good science and the scientific process. The full link for the following In the Pipeline post is: https://blogs.sciencemag.org/pipeline/archives/2020/03/06/covid-19-small-molecule-therapies-reviewed
A Summary of current potential repurposed therapeutics for COVID-19 Infection from In The Pipeline: A Science blog from Derick Lowe
Let’s take inventory on the therapies that are being developed for the coronavirus epidemic. Here is a very thorough list of at Biocentury, and I should note that (like Stat and several other organizations) they’re making all their Covid-19 content free to all readers during this crisis. I’d like to zoom in today on the potential small-molecule therapies, since some of these have the most immediate prospects for use in the real world.
The ones at the front of the line are repurposed drugs that are already approved for human use, for a lot of obvious reasons. The Biocentury list doesn’t cover these, but here’s an article at Nature Biotechnology that goes into detail. Clinical trials are a huge time sink – they sort of have to be, in most cases, if they’re going to be any good – and if you’ve already done all that stuff it’s a huge leg up, even if the drug itself is not exactly a perfect fit for the disease. So what do we have? The compound that is most advanced is probably remdesivir from Gilead, at right. This has been in development for a few years as an RNA virus therapy – it was originally developed for Ebola, and has been tried out against a whole list of single-strand RNA viruses. That includes the related coronaviruses SARS and MERS, so Covid-19 was an obvious fit.
The compound is a prodrug – that phosphoramide gets cleaved off completely, leaving the active 5-OH compound GS-44-1524. It mechanism of action is to get incorporated into viral RNA, since it’s taken up by RNA polymerase and it largely seems to evade proofreading. This causes RNA termination trouble later on, since that alpha-nitrile C-nucleoside is not exactly what the virus is expecting in its genome at that point, and thus viral replication is inhibited.
There are five clinical trials underway (here’s an overview at Biocentury). The NIH has an adaptive-design Phase II trial that has already started in Nebraska, with doses to be changed according to Bayesian readouts along the way. There are two Phase III trials underway at China-Japan Friendship Hospital in Hubei, double-blinded and placebo-controlled (since placebo is, as far as drug therapy goes, the current standard of care). And Gilead themselves are starting two open-label trials, one with no control arm and one with an (unblinded) standard-of-care comparison arm. Those might read out first, depending on when they get off the ground, but will be only rough readouts due to the fast-and-loose trial design. The two Hubei trials and the NIH one will add some rigor to the process, but I’m not sure when they’re going to report. My personal opinion is that I like the chances of this drug more than anything else on this list, but it’s still unlikely to be a game-changer.
There’s an RNA polymerase inhibitor (favipiravir) from Toyama, at right, that’s in a trial in China. It’s a thought – a broad-spectrum agent of this sort would be the sort of thing to try. But unfortunately, from what I can see, it has already turned up as ineffective in in vitro tests. The human trial that’s underway is honestly the sort of thing that would only happen under circumstances like the present: a developing epidemic with a new pathogen and no real standard of care. I hold out little hope for this one, but given that there’s nothing else at present, it probably should be tried. As you’ll see, this is far from the only situation like this.
One of the screens of known drugs in China that also flagged remdesivir noted that the old antimalarial drug chloroquine seemed to be effective in vitro. It had been reported some years back as a possible antiviral, working through more than one mechanism, probably both at viral entry and intracellularly thereafter. That part shouldn’t be surprising – chloroquine’s actual mode(s) of action against malaria parasites are still not completely worked out, either, and some of what people thought they knew about it has turned out to be wrong. There are several trials underway with it at Chinese facilities, some in combination with other agents like remdesivir. Chloroquine has of course been taken for many decades as an antimalarial, but it has a number of liabilities, including seizures, hearing damage, retinopathy and sudden effects on blood glucose. So it’s going to be important to establish just how effective it is and what doses will be needed. Just as with vaccine candidates, it’s possible to do more harm with a rushed treatment than the disease is doing itself
There are several other known antiviral drugs are being tried in China, but I don’t have too much hope for those, either. The neuraminidase inhibitors such as oseltamivir (better known as Tamiflu) were tried against SARS and were ineffective; there is no reason to expect anything versus Covid-19 although these drugs are a component of some drug cocktail trials. The HIV protease therapies such as darunavir and the combination therapy Kaletra are in trials, but that’s also a rather desperate long shot, since there’s no particular reason to think that they will have any such protease inhibition against what this new virus has to offer (and indeed, such agents weren’t much help against SARS in the end, either). The classic interferon/ribavirin combination seems to have had some activity against SARS and MERS, and is in two trials from what I can see. That’s not an awful idea by any means, but it’s not a great one, either: if your viral disease has interferon/ribavirin as a front line therapy, it generally means that there’s nothing really good available. No, unless we get really lucky none of these ideas are going to slow the disease down much.
There are a few other repurposed-protease-inhibitors ideas out there, such as this one. (Edit: I had seen this paper but couldn’t track it down, so thanks to those who sent it along). This paper suggests that the TMPRSS2 protease is important for viral entry on the human-cell-side of the process, a pathway that has been noted for other coronaviruses. And it points out that there is a an approved inhibitor (in Japan) for this enzyme (camostat), so that would definitely seem to be worth a trial, probably in combination with remdesivir.
That’s about it for the existing small molecules, from what I can see. What about new ones? Don’t hold your breath, is all I can say. A drug discovery program from scratch against a new pathogen is, as many readers here well know, not a trivial exercise. As this Bloomberg article details, many such efforts in the past (small molecules and vaccines alike) have come to grief because by the time they had anything to deliver the epidemic itself had passed. Indeed, Gilead’s remdesivir had already been dropped as a potential Ebola therapy.
You will either need to have a target in mind up front or go phenotypic. For the former, what you’d see are better characterizations of the viral protease and more extensive screens against it. Two other big target areas are viral entry (which involves the “spike” proteins on the virus surface and the ACE2 protein on human cells) and viral replication. To the former, it’s worth quickly noting that ACE2 is so much unlike the more familiar ACE protein that none of the cardiovascular ACE inhibitors do anything to it at all. And targeting the latter mechanisms is how remdesivir was developed as a possible Ebola agent, but as you can see, that took time, too. Phenotypic screens are perfectly reasonable against viral pathogens as well, but you’ll need to put time and effort into that assay up front, just as with any phenotypic effort, because as anyone who does that sort of work will tell you, a bad phenotypic screen is a complete waste of everyone’s time.
One of the key steps for either route is identifying an animal model. While animal models of infectious disease can be extremely well translated to human therapy, that doesn’t happen by accident: you need to choose the right animal. Viruses in general (and coronaviruses are no exception) vary widely in their effects in different species, and not just across the gaps of bird/reptile/human and the like. No, you’ll run into things where even the usual set of small mammals are acting differently from each other, with some of them not even getting sick at all. This current virus may well have gone through a couple of other mammalian species before landing on us, but you’ll note that dogs (to pick one) don’t seem to have any problem with it.
All this means that any new-target new-chemical-matter effort against Covid-19 (or any new pathogen) is going to take years, and there is just no way around that. Update: see here for just such an effort to start finding fragment hits for the viral protease. This puts small molecules in a very bimodal distribution: you have the existing drugs that might be repurposed, and are presumably available right now. Nothing else is! At the other end, for completely new therapies you have the usual prospects of drug discovery: years from now, lots of money, low success rate, good luck to all of us. The gap between these two could in theory be filled by vaccines and antibody therapies (if everything goes really, really well) but those are very much their own area and will be dealt with in a separate post.
Either way, the odds are that we (and I mean “we as a species” here) are going to be fighting this epidemic without any particularly amazing pharmacological weapons. Eventually we’ll have some, but I would advise people, pundits, and politicians not to get all excited about the prospects for some new therapies to come riding up over the hill to help us out. The odds of that happening in time to do anything about the current outbreak are very small. We will be going for months, years, with the therapeutic options we have right now. Look around you: what we have today is what we have to work with.
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Record Innovations in Drug Discovery by Koch Institute @MIT Members and Affiliates
Reporter: Aviva Lev-Ari, PhD, RN
In Good Company
Trovagene announced a new patent for the use of the drug onvansertib in combination with other anti-androgen drugs for the treatment of prostate cancer. Last fall, Trovagene secured exclusive rights to develop combination therapies and clinical biomarkers for prostate cancer based in part on Bridge Project-funded research. Read more.
Lyndra Therapeutics, co-founded by KI member Bob Langer, raised $55 million in its Series B round, with new investors including the Bill and Melinda Gates Foundation and Gilead Sciences. Phase 2 trials for its ultra long-acting drug delivery capsule are expected to begin next year. Read more.
Dragonfly Therapeutics, co-founded by KI director Tyler Jacks, has committed $10 million to launch the first clinical studies of its TriNKETs (Tri-specific, NK cell Engager Therapies) platform for both solid tumor and hematological cancers. Read more.
KI member Bob Langer and collaborator Omid Farokhzad co-founded Seer— combining nanotechnology, protein chemistry, and machine learning—to develop liquid biopsy tests for the early detection of cancer and other diseases. Read more.
Epizyme, co-founded by KI member Bob Horvitz, is submitting a New Drug Application to gain accelerated approval of tazemetostat for patients with relapsed or refractory follicular lymphoma. Read more.
Ribon Therapeutics, founded by former KI member PaulChang, launched with $65 million in a Series B funding round with Victoria Richon, a veteran of Sanofi and Epizyme, at the helm. Ribon focuses on developing PARP7 inhibitors for cancer treatment. Read more.
TWEETS by @pharma_BI and @AVIVA1950 at #IESYMPOSIUM – @kochinstitute 2019 #Immune #Engineering #Symposium, 1/28/2019 – 1/29/2019
Real Time Press Coverage: Aviva Lev-Ari, PhD, RN
2.1.3.4 TWEETS by @pharma_BI and @AVIVA1950 at #IESYMPOSIUM – @kochinstitute 2019 #Immune #Engineering #Symposium, 1/28/2019 – 1/29/2019, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
eProceedings for Day 1 and Day 2
LIVE Day One – Koch Institute 2019 Immune Engineering Symposium, January 28, 2019, Kresge Auditorium, MIT
#IESYMPOSIUM@pharma_BI@AVIVA1950 Aviv Regev @kochinstitute Melanoma: malignant cells with resistance in cold niches in situ cells express the resistance program pre-treatment: resistance UP – cold Predict checkpoint immunotherapy outcomes CDK4/6 abemaciclib in cell lines
#IESYMPOSIUM@pharma_BI@AVIVA1950 Diane Mathis @HMS Age-dependent Treg and mSC changes – Linear with increase in age Sex-dependent Treg and mSC changes – Female Treg loss in cases of Obesity leading to fibrosis Treg keep IL-33-Producing mSCs under rein Lean tissue/Obese tissue
#IESYMPOSIUM@pharma_BI@AVIVA1950 Martin LaFleur @HMS Loss of Ptpn2 enhances CD8+ T cell responses to LCMV and Tumors PTpn2 deletion in the immune system enhanced tumor immunity CHIME enables in vivo screening
#IESYMPOSIUM@pharma_BI@AVIVA1950 Alex Shalek @MIT@kochinstitute Identifying and rationally modulating cellular drivers of enhanced immunity T Cells, Clusters Expression of Peak and Memory Immunotherapy- Identifying Dendritic cells enhanced in HIV-1 Elite Controllers
#IESYMPOSIUM@pharma_BI@AVIVA1950 Glenn Dranoff @Novartis Adenosine level in blood or tissue very difficult to measure in blood even more than in tissue – NIR178 + PDR 001 Monotherapy (NIR178) combine with PD receptor blockage (PDR) show benefit A alone vs A+B in Clinical trial
#IESYMPOSIUM@pharma_BI@AVIVA1950 Glenn Dranoff @Novartis PD-L1 blockade elicits responses in some patients: soft part sarcoma LAG-3 combined with PD-1 – human peripheral blood tumor TIM-3 key regulator of T cell and Myeloid cell function: correlates in the TCGA DB myeloid
#IESYMPOSIUM@pharma_BI@AVIVA1950 Yvonne Chen @UCLA Activation of t Cell use CAR t Engineer CAR-T to respond to soluble form of antigens: CD19 CAR Responds to soluble CD19 GFP MCAR responds to Dimeric GFP “Tumor microenvironment is a scary place”
#IESYMPOSIUM@pharma_BI@AVIVA1950 Yvonne Chen @UCLA “Engineering smarter and stronger T cells for cancer immunotherapy” OR-Gate cause no relapse – Probing limits of modularity in CAR Design Bispecific CARs are superior to DualCAR: One vs DualCAR (some remained single CAR)
Ending the 1st session is Cathy Wu of @DanaFarber detailing some amazing work on vaccination strategies for melanoma and glioblastoma patients. They use long peptides engineered from tumor sequencing data. #iesymposium
Some fancy imaging: Duggan gives a nice demo of how dSTORM imaging works using a micropatterend image of Kennedy Institute for Rheumatology! yay! #iesymposium
Lots of interesting talks in the second session of the #iesymposium – effects of lymphoangiogenesis on anti-tumor immune responses, nanoparticle based strategies to improve bNAbs titers/affinity for HIV therapy, and IAPi cancer immunotherapy
Looking forward to another day of the #iesymposium. One more highlight from yesterday – @nm0min from our own lab showcased her work developing cytokine fusions that bind to collagen, boosting efficacy while drastically reducing toxicities
#IESYMPOSIUM@pharma_BI@AVIVA1950 Preeti Sharma, U Illinois T cell receptor and CAR-T engineering TCR engineering for Targeting glycosylated cancer antigens Nornal glycosylation vs Aberrant Engineering 237-CARs libraries with conjugated (Tn-OTS8) against Tn-antigend In vitro
#IESYMPOSIUM@pharma_BI@AVIVA1950 Bryan Bryson @MIT Loss of polarization potential: scRNAseq reveals transcriptional differences Thioredoxin facilitates immune response to Mtb is a marker of an inflammatory macrophage state functional spectrum of human microphages
#IESYMPOSIUM@pharma_BI@AVIVA1950 Bryan Bryson @MIT macrophage axis in Mycobacterium tuberculosis Building “libraries” – surface marker analysis of Microphages Polarized macrophages are functionally different quant and qual differences History of GM-CSF suppresses IL-10
#IESYMPOSIUM@pharma_BI@AVIVA1950 Jamie Spangler John Hopkins University “Reprogramming anti-cancer immunity RESPONSE through molecular engineering” De novo IL-2 potetiator in therapeutic superior to the natural cytokine by molecular engineering mimicking other cytokines
#IESYMPOSIUM@pharma_BI@AVIVA1950 Michael Dustin @UniofOxford ESCRT pathway associated with synaptic ectosomes Locatization, Microscopy Cytotoxic T cell granules CTLs release extracellular vescicles similar to T Helper with perforin and granzyme – CTL vesicles kill targets
#IESYMPOSIUM@pharma_BI@AVIVA1950 Michael Dustin @Oxford Delivery of T cell Effector function through extracellular vesicles Synaptic ectosome biogenisis Model: T cells: DOpamine cascade in germinal cell delivered to synaptic cleft – Effector CD40 – Transfer is cooperative
#IESYMPOSIUM@pharma_BI@AVIVA1950 Michael Dustin @Oxford Delivery of T cell Effector function through extracellular vesicles Laterally mobile ligands track receptor interaction ICAM-1 Signaling of synapse – Sustain signaling by transient in microclusters TCR related Invadipodia
#IESYMPOSIUM@pharma_BI@AVIVA1950 Mikael Pittet @MGH Myeloid Cells in Cancer Indirect mechanism AFTER a-PD-1 Treatment IFN-gamma Sensing Fosters IL-12 & therapeutic Responses aPD-1-Mediated Activation of Tumor Immunity – Direct activation and the ‘Licensing’ Model
#IESYMPOSIUM@pharma_BI@AVIVA1950 Stefani Spranger @MIT KI Response to checkpoint blockade Non-T cell-inflamed – is LACK OF T CELL INFILTRATION Tumor CD103 dendritic cells – Tumor-residing Batf3-drivenCD103 Tumor-intrinsic Beta-catenin mediates lack of T cell infiltration
#IESYMPOSIUM@pharma_BI@AVIVA1950 Max Krummel @UCSF Gene expression association between two genes: #NK and #cDC1 numbers are tightly linked to response to checkpoint blockage IMMUNE “ACCOMODATION” ARCHYTYPES: MYELOID TUNING OF ARCHITYPES Myeloid function and composition
#IESYMPOSIUM@pharma_BI@AVIVA1950 Noor Momin, MIT Lumican-cytokines improve control of distant lesions – Lumican-fusion potentiates systemic anti-tumor immunity
#IESYMPOSIUM@pharma_BI@AVIVA1950 Noor Momin, MIT Lumican fusion to IL-2 improves treatment efficacy reduce toxicity – Anti-TAA mAb – TA99 vs IL-2 Best efficacy and least toxicity in Lumican-MSA-IL-2 vs MSA-IL2 Lumican synergy with CAR-T
excited to attend the @kochinstitute@MIT immune engineering symposium #iesymposium this week! find me there to chat about @CellCellPress and whether your paper could be a good fit for us!
April Pawluk added,
Koch Institute at MITVerified account@kochinstitute
Join leading immunology researchers at our Immune Engineering Symposium on Jan. 28 & 29. Register now: http://bit.ly/2AOUWH6#iesymposium
Bob Schreiber and Tyler Jacks kicked off the #iesymposium with 2 great talks on the role of Class I and Class II neo-Ag in tumor immunogenicity and how the tumor microenvironment alters T cell responsiveness to tumors in vivo
Scott Wilson from @UChicago gave a fantastic talk on glycopolymer conjugation to antigens to improve trafficking to HAPCs and enhanced tolerization in autoimmunity models. Excited to learn more about his work at his @MITChemE faculty talk! #iesymposium
Spending the (literal) first day of my fellowship at the @kochinstitute#iesymposium! @DanaFarber Cathy Wu talking about the use of neoantigen targeting cancer vaccines for the treatment of ‘cold’ glioblastoma tumors in pts
Tyler Jacks talk was outstanding, Needs be delivered A@TED TALKs, needs become contents in the curriculum of Cell Biology graduate seminar as an Online class. BRAVO @pharma_BI@AVIVA1950
Aviva Lev-Ari added,
Anne E Deconinck@AEDeconinck
My boss, @kochinstitute director Tyler Jacks, presenting beautiful, unpublished work at our 3rd #iesymposium.
#IESYMPOSIUM@pharma_BI@AVIVA1950 Stephanie Dougan (Dana-Farber Cancer Institute) Dept. Virology IAPi outperforms checkpoint blockade in T cell cold tumors reduction of tumor burden gencitabine cross-presenting DCs and CD8 T cells – T cell low 6694c2
#IESYMPOSIUM@pharma_BI@AVIVA1950 Melody Swartz (University of Chicago) Lymphangiogenesis attractive to Native T cells, in VEGF-C tumors T cell homing inhibitors vs block T cell egress inhibitors – Immunotherapy induces T cell killing
#IESYMPOSIUM@pharma_BI@AVIVA1950 Cathy Wu @MGH breakthrough for Brain Tumor #vaccine based neoantigen-specific T cell at intracranial site Single cells brain tissue vs single cells from neoantigen specific T cells – intratumoral neoantigen-specific T cells: mutARGAP35-spacific
#IESYMPOSIUM@pharma_BI@AVIVA1950 Cathy Wu (Massachusetts General Hospital) – CoFounder of NEON Enduring complete radiographic responses after #Neovax + alpha-PD-1 treatment (anti-PD-1) NeoVax vs IVAC Mutanome for melanoma and Glioblastoma clinical trials
#IESYMPOSIUM@pharma_BI@AVIVA1950@TylerJacks@MIT Interrogating markers of T cell dysfunction – chance biology of cells by CRISPR – EGR2 at 2 weeks dysfuntioning is reduced presence of EDR2 mutant class plays role in cell metabolism cell becomes functional regulator CD8 T cell
MISSION The mission of the Koch Institute (KI) is to apply the tools of science and technology to improve the way cancer is detected, monitored, treated and prevented.
APPROACH We bring together scientists and engineers – in collaboration with clinicians and industry partners – to solve the most intractable problems in cancer. Leveraging MIT’s strengths in technology, the life sciences and interdisciplinary research, the KI is pursuing scientific excellence while also directly promoting innovative ways to diagnose, monitor, and treat cancer through advanced technology.
HISTORY The Koch Institute facility was made possible through a $100 million gift from MIT alumnus David H. Koch. Our new building opened in March 2011, coinciding with MIT’s 150th anniversary. Our community has grown out of the MIT Center for Cancer Research (CCR), which was founded in 1974 by Nobel Laureate and MIT Professor Salvador Luria, and is one of seven National Cancer Institute-designated basic (non-clinical) research centers in the U.S.
Biological, chemical, and materials engineers are engaged at the forefront of immunology research. At their disposal is an analytical toolkit honed to solve problems in the petrochemical and materials industries, which share the presence of complex reaction networks, and convective and diffusive molecular transport. Powerful synthetic capabilities have also been crafted: binding proteins can be engineered with effectively arbitrary specificity and affinity, and multifunctional nanoparticles and gels have been designed to interact in highly specific fashions with cells and tissues. Fearless pursuit of knowledge and solutions across disciplinary boundaries characterizes this nascent discipline of immune engineering, synergizing with immunologists and clinicians to put immunotherapy into practice.
The 2019 symposium will include two poster sessions and four abstract-selected talks. Abstracts should be uploaded on the registration page. Abstract submission deadline is November 15, 2018. Registration closes December 14.
Featuring on Day 2, 1/29, 2019:
Session IV
Moderator: Michael Birnbaum, Koch Institute, MIT
Jamie Spangler (John Hopkins University)
“Reprogramming anti-cancer immunity through molecular engineering”
Reprogramming anti-cancer immunity response through molecular engineering”
Cytokines induce receptor dimerization
Clinical Use of cytokines: Pleiotropy, expression and stability isssues
poor pharmacological properties
cytokine therapy: New de novo protein using computational methods
IL-2 signals through a dimeric nad a trimeric receptor complex
IL-2 pleiotropy hinders its therapeutic efficacy
IL-2 activate immunosuppression
potentiation of cytokine activity by anti-IL-2 antibody selectivity
Cytokine binding – Antibodies compete with IL-2 receptor subunits
IL-2Ralpha, IL-2 Rbeta: S4B6 mimickry of alpha allosterically enhances beta
Affinity – molecular eng De Novo design of a hyper-stable, effector biased IL-2
De novo IL-2 poteniator in therapeutic superior to the natural cytokine by molecular engineering
Bryan Bryson (MIT, Department of Biological Engineering)
“Exploiting the macrophage axis in Mycobacterium tuberculosis (Mtb) infection”
TB – who develop Active and why?
Immunological life cycle of Mtb
Global disease Mtb infection outcome varies within individual host
lesion are found by single bacteria
What are the cellular players in immune success
MACROPHAGES – molecular signals enhancing Mtb control of macrophages
modeling the host- macrophages are plastic and polarize
Building “libraries” – surface marker analysis of Microphages
Polarized macrophages are functionally different
quant and qual differences
History of GM-CSF suppresses IL-10
Loss of polarization potential: scRNAseq reveals transcriptional differences Thioredoxin facilitates immune response to Mtb is a marker of an inflammatory macrophage state
functional spectrum of human microphages
Facundo Batista (Ragon Institute (HIV Research) @MGH, MIT and Harvard)
“Vaccine evaluation in rapidly produced custom humanized mouse models”
Effective B cell activation requires 2 signals Antigen and binding to T cell
VDJ UCA (Unmutated common Ancestor)
B Cell Receptor (BCR) co-receptors and cytoskeleton
44% in Women age 24-44
Prototype HIV broadly neutralizing Antibodies (bnAb) do not bind to Env protein – Immunogen design and validation
Human Ig Knock-ins [Light variable 5′ chain length vs 7′ length] decisive to inform immunogenicity – One-Step CRISPR approach does not require ES cell work
Proof of principle with BG18 Germline Heavy Chain (BG18-gH) High-mannose patch – mice exhibit normal B cell development
B cells from naive human germline BG18-gH bind to GT2 immunogen
Interrogate immune response for HIV, Malaria, Zika, Flu
Session V
Moderator: Dane Wittrup, Koch Institute, MIT
Yvonne Chen (University of California, Los Angeles)
“Engineering smarter and stronger T cells for cancer immunotherapy”
Adoptive T-Cell Therapy
Tx for Leukemia – Tumor Antigen escape fro CAR T-cell therapy, CD19/CD20 OR-Gate CARs for prevention of antigen escape – 15 month of development
reduce probability of antigen escape due to two antigen CD19/CD20: Probing limits of modularity in CAR design
In vivo model: 75% wild type & 25% CD19 – relapse occur in the long term, early vs late vs no relapse: Tx with CAR t had no relapse
OR-Gate cause no relapse – Probing limits of modularity in CAR Design
Bispecific CARs are superior to DualCAR: One vs DualCAR (some remained single CAR)
Bispecific CARs exhibit superior antigen-stimulation capacity – OR-Gate CAR Outperforms Single-Input CARs
Lymphoma and Leukemia are 10% of all Cancers
TGF-gamma Rewiring T Cell Response
Activation of t Cell use CAR t
Engineer CAR-T to respond to soluble form of antigens: CD19 CAR Responds to soluble CD19
GFP MCAR responds to Dimeric GFP
“Tumor microenvironment is a scary place”
Michael Birnbaum, MIT, Koch Institute
“A repertoire of protective tumor immunity”
Decoding T and NK cell recognition – understanding immune recognition and signaling function for reprogramming the Immune system – Neoantigen vaccine pipeline
Personal neoantigen vax improve immunotherapy
CLASS I and CLASS II epitomes: MHC prediction performance – more accurate for CLASS I HLA polymorphisms
Immune Epitope DB and Analysis Resources 448,630 Peptide Epitomes
PD-L1 blockade elicits responses in some patients: soft part sarcoma
LAG-3 combined with PD-1 – human peripheral blood tumor
TIM-3 key regulator of T cell and Myeloid cell function: correlates in the TCGA DB with myeloid
Adenosine level in blood or tissue very difficult to measure in blood even more than in tissue – NIR178 + PDR 001 Mono-therapy (NIR178) combine with PD receptor blockage (PDR) – shows benefit
A alone vs A+B in Clinical trial
Session VI
Moderator: Stefani Spranger, Koch Institute, MIT
Tim Springer, Boston Children’s Hospital, HMS
The Milieu Model for TGF-Betta Activation”
Protein Science – Genomics with Protein
Antibody Initiative – new type of antibodies not a monoclonal antibody – a different type
Pro TGF-beta
TGF-beta – not a typical cytokine it is a prodamine for Mature growth factor — 33 genes mono and heterogeneous dimers
Latent TGF-Beta1 crystal structure: prodomaine shields the Growth Factor
Mechanism od activation of pro-TGF-beta – integrin alphaVBeta 6: pro-beta1:2
Simulation in vivo: actin cytoskeleton cytoplasmic domain
blocking antibodies LRRC33 mitigate toxicity on PD-L1 treatment
Alex Shalek, MIT, Department of Chemistry, Koch Institute
“Identifying and rationally modulating cellular drivers of enhanced immunity”
Balance in the Immune system
Profiling Granulomas using Seq-Well 2.0
lung tissue in South Africa of TB patients
Granulomas, linking cell type abundance with burden
Exploring T cells Phenotypes
Cytotoxic & Effector ST@+ Regulatory
Vaccine against TB – 19% effective, only 0 IV BCG vaccination can elicit sterilizing Immunity
Profiling cellular response to vaccination
T cell gene modules across vaccine routes
T Cells, Clusters
Expression of Peak and Memory
Immunotherapy- Identifying Dendritic cells enhanced in HIV-1 Elite Controllers
moving from Observing to Engineering
Cellular signature: NK-kB Signaling
Identifying and testing Cellular Correlates of TB Protection
Beyond Biology: Translation research: Data sets: dosen
Session VII
Moderator: Stefani Spranger, Koch Institute, MIT
Diane Mathis, Harvard Medical School
“Tissue T-regs”
T reg populations in Lymphoid Non–lymphoid Tissues
2009 – Treg tissue homeostasis status – sensitivity to insulin, 5-15% CD4+ T compartment
transcriptome
expanded repertoires TCRs
viceral adipose tissue (VAT) – Insulin
Dependencies: Taget IL-33 its I/1r/1 – encoded Receptor ST2
VAT up-regulate I/1r/1:ST2 Signaling
IL-33 – CD45 negative CD31 negative
mSC Production of IL-33 is Important to Treg
The mesenchyme develops into the tissues of the lymphatic and circulatory systems, as well as the musculoskeletal system. This latter system is characterized as connective tissues throughout the body, such as bone, muscle and cartilage. A malignant cancer of mesenchymal cells is a type of sarcoma.
Age-dependent Treg and mSC changes – Linear with increase in age
Sex-dependent Treg and mSC changes – Female
Treg loss in cases of Obesity leading to fibrosis
Treg keep IL-33-Producing mSCs under rein
Lean tissue vs Obese tissue
Aged mice show poor skeletal muscle repair – it is reverses by IL-33 Injection
Immuno-response: target tissues systemic T reg
Treg and mSC
Aviv Regev, Broad Institute; Koch Institute
“Cell atlases as roadmaps to understand Cancer”
Colon disease UC – genetic underlining risk, – A single cell atlas of healthy and UC colonic mucosa inflammed and non-inflammed: Epithelial, stromal, Immune – fibroblast not observed in UC colon IAFs; IL13RA2 + IL11
Anti TNF responders – epithelial cells
Anti TNF non-responders – inflammatory monocytes fibroblasts
RESISTANCE to anti-cancer therapy: OSM (Inflammatory monocytes-OSMR (IAF)
cell-cell interactions from variations across individuals
Most UC-risk genes are cell type specific
Variation within a cell type helps predict GWAS gene functions – epithelial cell signature – organize US GWAS into cell type specific – genes in associated regions: UC and IBD
Melanoma
malignant cells with resistance in cold niches in situ
cells express the resistance program pre-treatment: resistance UP – cold
Predict checkpoint immunotherapy outcomes
CDK4/6 – computational search predict as program regulators: abemaciclib in cell lines
Poster Presenters
Preeti Sharma, University of Illinois
T cell receptor and CAR-T engineering – T cell therapy
TCR Complex: Vbeta Cbeta P2A Valpha Calpha
CAR-T Aga2 HA scTCR/scFv c-myc
Directed elovution to isolate optimal TCR or CAR
Eng TCR and CARt cell therapy
Use of TCRs against pep/MHC allows targeting a n array of cancer antigens
TCRs are isolated from T cell clones
Conventional TCR identification method vs In Vitro TCR Eng directed evolution
T1 and RD1 TCRs drive activity against MART-1 in CD4+ T cells
CD8+
TCR engineering for Targeting glycosylated cancer antigens
Normal glycosylation vs Aberrant glycosylation
Engineering 237-CARs libraries with conjugated (Tn-OTS8) against multiple human Tn-antigend
In vitro engineering: broaden specificity to multiple peptide backbone
CAR engineering collaborations with U Chicago, U Wash, UPenn, Copenhagen, Germany
Martin LaFleur, HMS
CRISPR- Cas9 Bone marrow stem cells for Cancer Immunotherapy
CHIME: CHimeric IMmune Editing system
sgRNA-Vex
CHIME can be used to KO genes in multiple immune lineages
identify T cell intrinsic effects in the LCMV model Spleen-depleted, Spleen enhanced
Loss of Ptpn2 enhances CD8+ T cell responses to LCMV and Tumors
Ptpn2 deletion in the immune system enhanced tumor immunity
MISSION The mission of the Koch Institute (KI) is to apply the tools of science and technology to improve the way cancer is detected, monitored, treated and prevented.
APPROACH We bring together scientists and engineers – in collaboration with clinicians and industry partners – to solve the most intractable problems in cancer. Leveraging MIT’s strengths in technology, the life sciences and interdisciplinary research, the KI is pursuing scientific excellence while also directly promoting innovative ways to diagnose, monitor, and treat cancer through advanced technology.
HISTORY The Koch Institute facility was made possible through a $100 million gift from MIT alumnus David H. Koch. Our new building opened in March 2011, coinciding with MIT’s 150th anniversary. Our community has grown out of the MIT Center for Cancer Research (CCR), which was founded in 1974 by Nobel Laureate and MIT Professor Salvador Luria, and is one of seven National Cancer Institute-designated basic (non-clinical) research centers in the U.S.
Biological, chemical, and materials engineers are engaged at the forefront of immunology research. At their disposal is an analytical toolkit honed to solve problems in the petrochemical and materials industries, which share the presence of complex reaction networks, and convective and diffusive molecular transport. Powerful synthetic capabilities have also been crafted: binding proteins can be engineered with effectively arbitrary specificity and affinity, and multifunctional nanoparticles and gels have been designed to interact in highly specific fashions with cells and tissues. Fearless pursuit of knowledge and solutions across disciplinary boundaries characterizes this nascent discipline of immune engineering, synergizing with immunologists and clinicians to put immunotherapy into practice.
The 2019 symposium will include two poster sessions and four abstract-selected talks. Abstracts should be uploaded on the registration page. Abstract submission deadline is November 15, 2018. Registration closes December 14.
Featuring on Day 1, 1/28, 2019:
Dane Wittrup,, Koch Institute, MIT
IMMUNE BIOLOGY,
7 — Stephanie Dougan (Dana-Farber Cancer Institute) HMS, Department of Virology
Shared antigens may be the only option for many patients
T cell affinity low or high TCRs – Augment priming
Radiation plus anti-CD40 induces vigorous T cell priming
TNF family co-stimulatory receptor signaling can be mimicked by IAP antagonists
SMACK – c-IAP12 – IAPi enhances function of many immune cells: B Cells, Dendritic cells,
Pancreatic cancer cell immunologic memory : Primary challenge, re-challenge
IAPi outperforms checkpoint blockade in T cell cold tumors
reduction of tumor burden gencitabine cross-presenting DCs and CD8 T cells – T cell low 6694c2
IAPi is a T cell-dependent immunotherapy in pancreatic cancer: MHC class I and IFN gemma sensing by tumor cells are critical for endogenous anti-tumor immunity and response to checkpoint blockade
T cells are catalytic, they can kill some tumors not all – Genes deleted in tumor cells
Intratumoral phagocytes are critical for endogenous: IAP antagonism increases phagocytosis in vivo
Model: T cells provide antigen specificity for sustained innate immune response
Antigen and adjuvants
12 — Michael Dustin (University of Oxford)
Delivery of T cell Effector function through extracellular vesicles
Laterally mobile ligands track receptor interaction
ICAM-1
Signaling of synapse – Sustain signaling by transient in microclusters TCR related to Invadipodia
Synaptic ectosome biogenisis Model: T cells: DOpamine cascade in germinal cell delivered to synaptic cleft – Effector CD40 – Transfer is cooperative
Synaptic ectosome composition
ESCRT pathway associated with synaptic ectosomes
Locatization, Microscopy (STORM, PALM, GSD)
Updated Model T cells Exosome transport Cytotoxic T cell granules CTLs release extracellular vescicles similar to T Helper with perforin and granzyme – CTL vesicles kill targets
6 — Darrell Irvine (MIT, Koch Institute; HHMI)
Innate immune recognition of glycosylation in nano particle vaccines
HIV Vaccines: Why is it such a challenge
HIV vaccine – Immunogen design – CD4 binding site-targeting
rational for nanoparticles forms of env immunogens
Exploring tumor-immune interactions with genetically engineered Cancer Models – A case of Lung Cancer
Factors controlling tumor progression – genetically-engineered model of lung adenocarcinoma, metastasis causing death
Infiltration of cells: SEQUENCE EXOME – NO TUMOR BURDEN,
Exome sequencing reveals few mutations in KP model
Programmed neoantogen expression in the KP model: Kras, p53 – both are well researched in Lung cancer – immune cell dependent – tumors escape immune response due to immunosuppression – regulatory T cells most important in this model system
tissue specific responses to antigens
Lung Cancer – late stage — Programmed neo-antigen expression
Single cell mRNA sequencing of CD* T cell over time – sort cells, 8 weeks, 12 weeks, 20 weeks – progression of single cell similarity lymph cells vs lungs cells – cell identities – transcription activation of dysfunction in cells
SIIN+ CD8 T cells show markers of dysfunction over time – up regulated signs of exhaustion,
T cells becomes exhausted, checkpoint inhibitors beyond a certain point – has no capacity –
Interrogating markers of T cell dysfunction – chance biology of cells by CRISPR Cas9 – EGR2 at 2 weeks dysfunctioning is reduced – presence of EDR2 mutant class plays a role in cell metabolism – cell becomes more functional by modification protocols
Effects of CRISPR-mediated vs Combinatorial effects of CRISPR-mediated mutation of inhibitory models
8 — Max Krummel (University of California, San Francisco)
Dynamic Emergent behavior in Immune Systems
T cells are captured on tumor margins (without desired cytotoxicity)
Myeloid cells Underlie Intratumoral T cell capture
Anti tumor (CD4 CD8) vs Pro-tumor (CD9)
If many cells predicting Outcome more favorable – cellular abundance
Alternative T Cell reactions in Tissue: T-Helper 1, T-Helper 2
Gene expression association between two genes:
NK and cDC1 numbers are tightly linked and correlated with response to checkpoint blockage
A CD4-Enhaced Class of Melanoma Patients Also can be Checkpoint
CD4 T cells in Cancer – control tumors on their on
If high ICOS and CD4
Stimulate CD4: pull out of lymph nodes cells mCD301B
CD4 T cell proliferation but they don’t make PD1 ICOS CD4T
CD4 – required: Regulatory T Cells control CS4-dependent Tumor control via Lymph Node depletion (dLN)
If CD4 depleted, Lymph Node (LN) connected
Regulatory of PD1 ICOS CD4T
CD8 CD4 Tumor Affinity
Melanoma – T-reg hi or low – Responders are T-reg hi they have CD8
Existing Paired presence of T-reg, together with cDC2 number classifies Pt with better CD4
In Head and Neck: DC needed to stimulate immune response by CD4
Architypes of Immune systems in Tumors – Generally
CLASS I, II, III, IV – phynotypic
IMMUNE “ACCOMODATION” ARCHYTYPES: MYELOID TUNING OF ARCHITYPES
Myeloid function and composition
11 — Mikael Pittet (Massachusetts General Hospital)
Myeloid Cells in Cancer
complexity of Myeloid
Myeloid cells for cancer therapy: Outcomes good and bad: Tumor suppressing vs Tumor Promoting
Myeloid and immunotherapy
aPD-1 mAbs do not bind IL-12+DCs (scRNAseq): DC Classical and PlasmaCytoid (Allon Klein)
Cross-presenting cDC1 are essential for effector T cells
How can we raise the curve and increase the number of long-term survivors
Understanding the role of tumor-resident DC
Accumulation of CD103 DC independent of T cells
Regression tumor mount T cell response independent of DC1 DC
Induction of anti-tumor immunity is independent of the canonical
Single cell RNA-Seq reveal new subset to regressiong tumors and stimulate T cells via non-conventional
Working hypothesis: productive anti-tumor immunity depends on multiple tumor-resident DC subsets
5 — Melody Swartz (University of Chicago)
Lymphangiogenesis and immunomodulation
Lymphangiogenesisfor in Inflammation
Immunosuppression drives metastasis
promotion of resolution in disease progression
Tumors uses lymphatic system vessels
Tumor VEGF-C enhances immune cell interactions with lymphatic system
Lymphangiogenesis promore immune suppression in the tumor microenvironment
Recruitment of immune cells system: Dendritic Cells,
Lymphangiogenesis melanomas – highly responsive to immunotherapy : Vaccination
Lymphangiogenesis promote antigen spreading
Lymphangiogenesis potentiation: CCL21, CCR7
Lymphangiogenesis attractive to Native T cells, in VEGF-C tumors
T cell homing inhibitors vs block T cell egress inhibitors – Immunotherapy induces T cell killing
Allergic airway inflammation is driven lung and lymph node Lymphangiogenesis
Innate Immune cell infiltration reduced
Memory recall responses reflect adaptive immunity
pathology exacerbated with VEGFR-3 blockade response of memory recall cell is enhanced
VEGFR-3 signaling shifts T call balance, and CCL@1, from Lymph nodes to Lung
Differential changes in T cell balance between lung vs adaptive immune response to allergic airway inflammation
Lymphangiogenesis in the lung, competition with adaptive immune response to allergic airway inflammation in the lung
4 — Cathy Wu, Dana Farber Cancer Institute, HMS – CoFounder of NEON
Building better personal cancer vaccines
Vaccine: up to 20 personalized neoantigens as SLPs with adjuvant (polyICLC)
high risk melanoma – RESULTS: new immune responses – new responses mutiple immune responses CD4 & CD8: mutated vs Wild type differences
Enduring complete radiographic responses after Neovax + alpha-PD-1 treatment (anti-PD-1)
NeoVax vs IVAC MutaNOME
Ex vivo responses to assay peptide pools – immune response identified
NeoVax: ‘warming’ a cold tumor
immune cell infiltration – not studied in Glioblastoma which is a pooled tumor: TCR repertoire and MHC. Available materials: PBMC vs Fresh frozen and FFPE tumor material: Blood va FF brain tissue sequencing
Pt 8 neoantigen-specific clonotypesID’s – reactive T cells track to the brain after vaccination
Single cells from brain tissue vs single cells from neoantigen specific T cells – intratumoral neoantigen-specific T cells: mutARGAP35-specific T cell identified at site of disease – breakthrough for Brain Tumor #vaccine based neoantigen-specific T cell at intracranial site
VAX steering the Immune system
commission at Dana Farber – Prediction algorithms of denovo neoantigen targets: Newly profiled peptides to train a model vs peptide in the DB – Single vs Multi-allele HLA peptide sequencing by MassSpectroscopy
Mono-allelic MS data reveals novel motifs and sub-motifs
Endogenous signals contribution to predictive power
NeuroNets Algoriths : Integrative models identify tumor-presented epitopes more accurately than models without training like NeuroNets
5778 class I peptides from 4 cancers class I allele
The Immune System, Stress Signaling, Infectious Diseases and Therapeutic Implications: VOLUME 2: Infectious Diseases and Therapeutics and VOLUME 3: The Immune System and Therapeutics (Series D: BioMedicine & Immunology) Kindle Edition – on Amazon.com since September 4, 2017
8:30 – 9:45 Session V Moderator: Stefani Spranger | MIT, Koch Institute
K. Christopher Garcia – Stanford University Exploiting T Cell and Cytokine Receptor Structure and Mechanism to Develop New Immunotherapeutic Strategies
T Cell Receptor, peptide-MHC, 10 to the power of 10 is combinatorics – Library for selection to determine enrichment possibilities
Ligand identification for orphan TCRs
Industrializing process
use pMHC
IL-2 – Receptor Signaling Complex
Effector cells (NK, T)
Engineered T Cell – Tunable expansion, ligand-Receptor interface
Randomize IL-2RBeta interface: Orthogonal receptor vs wild type
In Vivo adoptive transfer model: to quantify orthogonality ratio
CD4, CD8, Treg,C57BL/6J
Ligand discovery
Orthogonal IL-2
Stefani Spranger – MIT, Koch Institute Batf3-DC as Mediators of the T Cell-Inflamed Tumor Microenvironment
Melanoma – solid cancer and other types, Immune inhibitory regulatory pathway patient with Immune response present
T cell-inflamed Tumor vs Non-T cell-inflamed Tumor
identify oncogenic pathways differentially activated between T cell-inflamed and non-Tcell-inflamed infiltration
If on Tumor:
Braf/PTEN
Braf/CAT
Braf/PTEN/CAT
The role of T cell priming – lack of initial
Beta-catenin-expressing tumors fail to prime 2C TCR-transgenic T cells
Deficiency in number of CD8+ and CD103+ dendritic cells
CD103+ DC are essential for T cell Priming and T cell-inflammation #StefaniSpranger
Adoptive transfer of effector 2C T cells fails to control Beta-catenin+ tumors
Vaccination induced anti-gen specific T cell memory fails to control Beta-catenin+ tumors
What cell type in tumor microenvironment effect monilization of T cell
CD103+ Dendritic cellsare source chymokine
Recruitment of effector T cells: Reconstitution od Beta-catenin-expressing SIY+
Are Batf3-DC within the tumor required for the recruitment of effector T cells?
Tumor-residing Batf3-drive CD103+ DC are required for the recruitment of effector T cells
Gene spore for correlation with recturment of effector cells
T cell Priming – CD103+ DC are essential for effector T cells
George Georgiou – University of Texas at Austin The Human Circulating Antibody Repertoire in Infection, Vaccination or Cancer
Serological Antibody Repertoire: in blood or in secretions
Antibody in serum – is difficult sequence identity
Serum IgG – 7-17 mg/ml if less immune deficient if more hyper globular
antibodies produced in long lived plasma cells in the bone marrow — experimentally inaccessible
Discovery of antibodies from the serological repertoire – not B cells
BM-PCs
Serum antibodies function via Fc effector mechanism – complement activation
Ig-SEQ – BCR-SEQ
Repertoire-wide computational modelling of antibody structures
En masse analysis & Mining of the Human Native Antibody Repertoire
hypervariable – High-Throughput Single B Cell VH:VL (or TCRalpha, beta) sequencing
HuNoV causes 800 death in the US per year of immune deficient
Influenza Trivalent Vaccine: Antibodies to hemaggiutinin: H1, H3, and B COmponenet
Abundant H1 +H3 Serum IgGs do not neutralize but confer Protection toInfluenza challenge with Live Virus #GeorgeGeorgiou
Non-Neutralizing Antibodies: The role of Complement in Protection
9:45 – 10:15 Break
10:15 – 11:30 Session VI Moderator: K. Dane Wittrup | MIT, Koch Institute
Harvey Lodish – Whitehead Institute and Koch Institute Engineered Erythrocytes Covalently Linked to Antigenic Peptides Can Protect Against Autoimmune Disease
Modified Red blood cells are microparticles for introducing therapeutics & diagnostics into the human body
Bool transfusion is widely used therapeutics
Covalently linking unique functional modalities to mouse or human red cells produced in cell culture:
PRODUCTION OF HUMAN RED BLOD CELLS EXPRESSING A FOREIN PROTEIN: CD34+ stem/progenitor cells that generates normal enucleated RBC.
PPAR-alpha and glucocorticoticoid receptor
Norman morphology: Sortase A is a bactrial transpeptidase that covalently links a “donor”
Engineering Normal Human RBC biotin-LPETG
Covelantely – Glycophorin A with camelid VHHs specific for Botulinum toxin A or B
HLA-DR4 library design and selection to enrich HLA-DM: Amino Acid vs Peptide position: Depleted vs Enriched – relative to expected for NNK codon
6852 _ predicted to bind vs 220 Non-binding peptides
HLA polymorphism: repertoire differences caused by
Antigen – T cell-driven antigen discovery: engaging Innate and Adaptive Immune response
Sorting TIL and select: FOcus of T cell-driven antigen discovery
T cell-driven antigen discovery: TCR
Jennifer R. Cochran – Stanford University Innate and Adaptive Integrin-targeted Combination Immunotherapy
alpa-TAA
TargetingIntegrin = universal targetinvolved in binding to several receptors: brest, lung, pancreatic, brain tumors arising by mutations – used as a handle for binding to agents
NOD201 Peptide-Fc Fusion: A Psudo Ab
Handle the therapeutics: NOD201 + alphaPD1
NOD201 effectively combines with alphaPD-L1, alphaCTLA-4, and alpha4-1BB/CD137
Corresponding monotherapies vs ComboTherapy invoking Innate and Adaptive Immune System
Microphages, CD8+ are critical vs CD4+ Neutrophils, NK cells, B cells #JenniferR. Cochran
Macrophages activation is critical – Day 4, 4 and 5
NOD201 + alphaPD1 combo increases M1 macrophages
Who are the best responders to PD1 – genes that are differentially expressed
NOD201 deives T cells reaponses through a “vaccinal” effect
CAncer Immune CYcle
Integrin – localization
Prelim NOD201 toxicity studies: no significant effects
Targeting multiple integrins vs antibodies RJ9 – minimal effect
NOD201 – manufacturability – NEW AGENT in Preclinical stage
2:15 – 2:45 Break
2:45 – 3:35 Session VIII Moderator: Jianzhu Chen | MIT, Koch Institute
Jennifer Wargo – MD Anderson Cancer Center Understanding Responses to Cancer Therapy: The Tissue is the Issue, but the Scoop is in the Poop
Optimize Targeted Treatment response
Translational research in patients on targeted therapy revealed molecular and immune mechanisms of response and resistance
Molecular mechanisms – T cell infiltrate after one week of therapy
Role of tumor stroma in mediating resistance to targeted therapy
Tumor microenvironment
Intra-tumoral bacteria identified in patients with Pancreatic Cancer
Translational research in patients on immune checkpoint blockade revealed molecualr and immune mechanism of response and resistance
Biomarkers not found
SYstemic Immunity and environment (temperature) on response to checkpoint blockade – what is the role?
Role of mIcrobiome in shaping response to checkpoint blockade in Melanoma
Microbime and GI Cancer
Diversity of the gut microbiome is associated with differential outcomes in the setting of stem cell transplant in AML
Oral and gut fecal microbiome in large cohort patient with metastatic melanoma undergoing systemic therapy
Repeat oral & gut AFTER chemo
WGSeq – Diversity of microbiome and response (responders vs non-responders to anti PD-1 – High diversity of microbiome have prolonged survival to PD-1 blockade
Anti tumor Immunity and composition of gut microbiome in patient on anti-PD-1 favorable AND higher survival #JenniferWargo
Enhance therapeutic responses in lang and renal carcinoma: If on antibiotic – poorer survival
sharing data important across institutions
Jianzhu Chen – MIT, Koch Institute Modulating Macrophages in Cancer Immunotherapy
Humanized mouth vs de novo human cancer
B cell hyperplasia
double hit lymphoma
AML
Overexpression of Bcl-2 & Myc in B cells leads to double-hit lymphoma
antiCD52 – CLL
Spleen, Bone marrow, Brain
Microphages are required to kill Ab-bound lymphoma cells in vivo #JianzhuChen
COmbinatorial chemo-Immunotherapy works for solid tumors: treating breast cancer in humanized mice
Infiltration of monocytic cells in the bone marrow
Cyclophosphophamide-antibody synergy extending to solid tumor and different antibodies #JianzhuChen
Polarization of macrophages it is dosage-dependent M1 and M2
Antibiotic induces expression of M1 polarizing supresses development and function of tumor-associated macrophages (TAM)
Antibiotic inhibits melanoma growth by activating macrophages in vivo #JianzhuChen





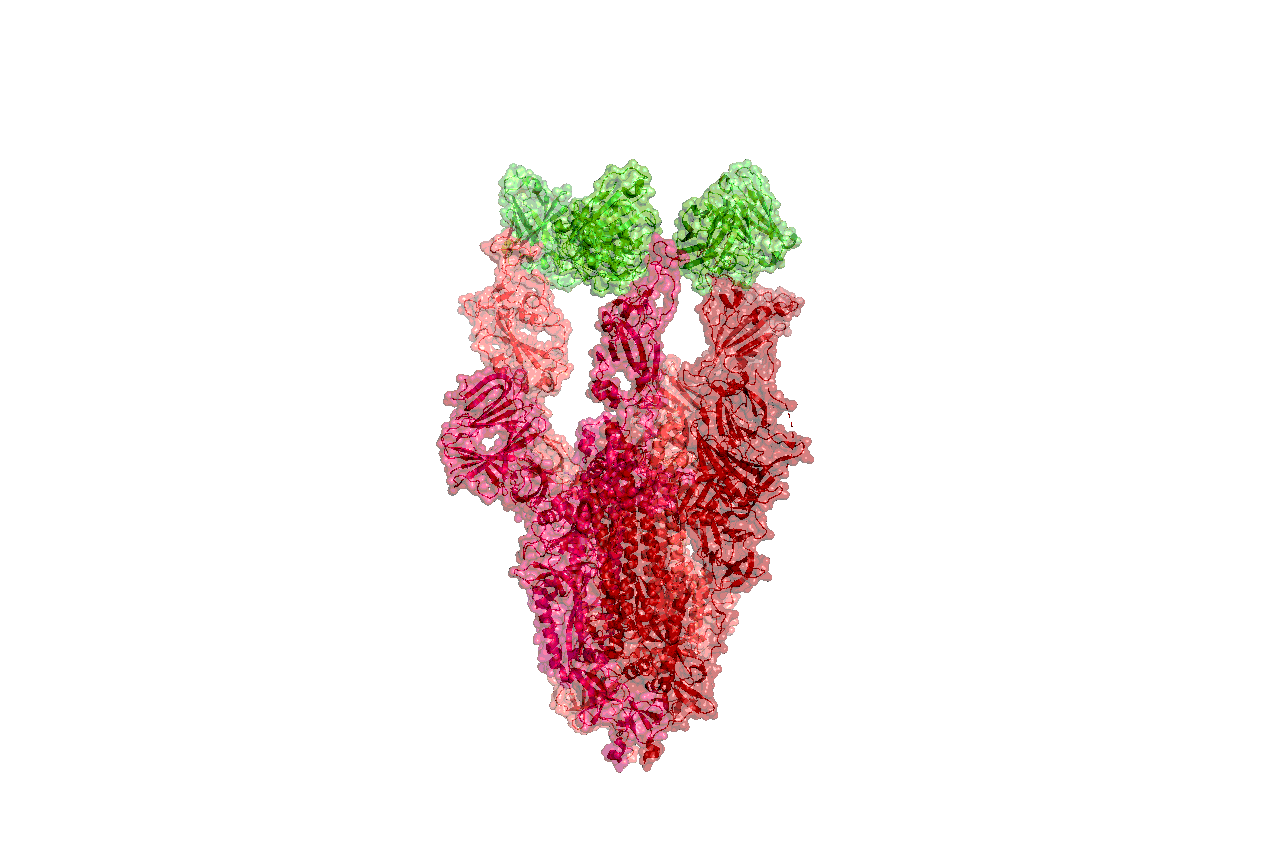




































30489-X&id=fx1.jpg){kind=link}