Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
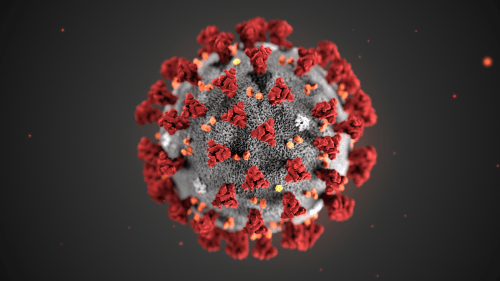
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
$The authors would like to note that the first eight authors are listed alphabetically.
Abstract
During its first month, the recently emerged 2019 Wuhan novel coronavirus (2019-nCoV) has already infected many thousands of people in mainland China and worldwide and took hundreds of lives. However, the swiftly spreading virus also caused an unprecedentedly rapid response from the research community facing the unknown health challenge of potentially enormous proportions. Unfortunately, the experimental research to understand the molecular mechanisms behind the viral infection and to design a vaccine or antivirals is costly and takes months to develop. To expedite the advancement of our knowledge we leverage the data about the related coronaviruses that is readily available in public databases, and integrate these data into a single computational pipeline. As a result, we provide a comprehensive structural genomics and interactomics road-maps of 2019-nCoV and use these information to infer the possible functional differences and similarities with the related SARS coronavirus. All data are made publicly available to the research community at http://korkinlab.org/wuhan
Figure 2. Structurally characterized non-structural proteins of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. The structurally resolved part of wNsp7 is sequentially identical to its homolog.
Figure 3. Structurally characterized structural proteins and an ORF of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. Highlighted in yellow are novel protein inserts found in wS.
Figure 4. Structurally characterized intra-viral and host-viral protein-protein interaction complexes of 2019-nCoV. Human proteins (colored in orange) are identified through their gene names. For each intra-viral structure, the number of subunits involved in the interaction is specified.
Figure 5. Evolutionary conservation of functional sites in 2019-nCoV proteins. A. Fully conserved protein binding sites (PBS, light orange) of wNsp12 in its interaction with wNsp7 and wNsp8 while other parts of the protein surface shows mutations (magenta); B. Both major monoclonal antibody binding site (light orange) and ACE2 receptor binding site (dark green) of wS are heavily mutated (binding site mutations are shown in red) compared to the same binding sites in other coronaviruses; mutations not located on the two binding sites are shown in magenta; C. Nearly intact protein binding site (light orange) of wNsp (papain-like protease PLpro domain) for its putative interaction with human ubiquitin-aldehyde (binding site mutations for the only two residues are shown in red, non-binding site mutations are shown in magenta); D. Fully conserved inhibitor ligand binding site (LBS, green) for wNsp5; non-binding site mutations are shown in magenta.
According to the World Health Organization, coronaviruses make up a large family of viruses named for the crown-like spikes found on their surface (Figure 1). They carry their genetic material in single strands of RNA and cause respiratory problems and fever. Like HIV, coronaviruses can be transmitted between animals and humans. Coronaviruses have been responsible for the Severe Acute Respiratory Syndrome (SARS) pandemic in the early 2000s and the Middle East Respiratory Syndrome (MERS) outbreak in South Korea in 2015. While the most recent coronavirus, COVID-19, has caused international concern, accessible and inexpensive sequencing is helping us understand COVID-19 and respond to the outbreak quickly.
Figure 1. Coronaviruses with the characteristic spikes as seen under a microscope.
First studies that explore genetic susceptibility to COVID-19 are now being published. The first results indicate that COVID-19 infects cells using the ACE2 cell-surface receptor. Genetic variants in the ACE2 receptor gene are thus likely to influence how effectively COVID-19 can enter the cells in our bodies. Researchers hope to discover genetic variants that confer resistance to a COVID-19 infection, similar to how some variants in the CCR5 receptor gene make people immune to HIV. At Nebula Genomics, we are monitoring the latest COVID-19 research and will add any relevant discoveries to the Nebula Research Library in a timely manner.
The Role of Genomics in Responding to COVID-19
Scientists in China sequenced COVID-19’s genome just a few weeks after the first case was reported in Wuhan. This stands in contrast to SARS, which was discovered in late 2002 but was not sequenced until April of 2003. It is through inexpensive genome-sequencing that many scientists across the globe are learning and sharing information about COVID-19, allowing us to track the evolution of COVID-19 in real-time. Ultimately, sequencing can help remove the fear of the unknown and allow scientists and health professionals to prepare to combat the spread of COVID-19.
Next-generation DNA sequencing technology has enabled us to understand COVID-19 is ~30,000 bases long. Moreover, researchers in China determined that COVID-19 is also almost identical to a coronavirus found in bats and is very similar to SARS. These insights have been critical in aiding in the development of diagnostics and vaccines. For example, the Centers for Disease Control and Prevention developed a diagnostic test to detect COVID-19 RNA from nose or mouth swabs.
Moreover, a number of different government agencies and pharmaceutical companies are in the process of developing COVID-19 vaccines to stop the COVID-19 from infecting more people. To protect humans from infection inactivated virus particles or parts of the virus (e.g. viral proteins) can be injected into humans. The immune system will recognize the inactivated virus as foreign, priming the body to build immunity against possible future infection. Of note, Moderna Inc., the National Institute of Allergy and Infectious Diseases, and Coalition for Epidemic Preparedness Innovations identified a COVID-19 vaccine candidate in a record 42 days. This vaccine will be tested in human clinical trials starting in April.
For more information about COVID-19, please refer to the World Health Organization website.
The problem w/ visionaries is that we don’t recognize them in a timely manner (too late) Ralph Baric @UNCpublichealth and Vineet Menachery deserve recognition for being 5 yrs ahead of #COVID19https://nature.com/articles/nm.3985…@NatureMedicinehttps://pnas.org/content/113/11/3048…@PNASNews via @hoondy
Senior, A.W., Evans, R., Jumper, J. et al.Improved protein structure prediction using potentials from deep learning. Nature577, 706–710 (2020). https://doi.org/10.1038/s41586-019-1923-7
Abstract
Protein structure prediction can be used to determine the three-dimensional shape of a protein from its amino acid sequence1. This problem is of fundamental importance as the structure of a protein largely determines its function2; however, protein structures can be difficult to determine experimentally. Considerable progress has recently been made by leveraging genetic information. It is possible to infer which amino acid residues are in contact by analysing covariation in homologous sequences, which aids in the prediction of protein structures3. Here we show that we can train a neural network to make accurate predictions of the distances between pairs of residues, which convey more information about the structure than contact predictions. Using this information, we construct a potential of mean force4 that can accurately describe the shape of a protein. We find that the resulting potential can be optimized by a simple gradient descent algorithm to generate structures without complex sampling procedures. The resulting system, named AlphaFold, achieves high accuracy, even for sequences with fewer homologous sequences. In the recent Critical Assessment of Protein Structure Prediction5 (CASP13)—a blind assessment of the state of the field—AlphaFold created high-accuracy structures (with template modelling (TM) scores6 of 0.7 or higher) for 24 out of 43 free modelling domains, whereas the next best method, which used sampling and contact information, achieved such accuracy for only 14 out of 43 domains. AlphaFold represents a considerable advance in protein-structure prediction. We expect this increased accuracy to enable insights into the function and malfunction of proteins, especially in cases for which no structures for homologous proteins have been experimentally determined7. https://doi.org/10.1038/s41586-019-1923-7
The scientific community has galvanised in response to the recent COVID-19 outbreak, building on decades of basic research characterising this virus family. Labs at the forefront of the outbreak response shared genomes of the virus in open access databases, which enabled researchers to rapidly develop tests for this novel pathogen. Other labs have shared experimentally-determined and computationally-predicted structures of some of the viral proteins, and still others have shared epidemiological data. We hope to contribute to the scientific effort using the latest version of our AlphaFold system by releasing structure predictions of several under-studied proteins associated with SARS-CoV-2, the virus that causes COVID-19. We emphasise that these structure predictions have not been experimentally verified, but hope they may contribute to the scientific community’s interrogation of how the virus functions, and serve as a hypothesis generation platform for future experimental work in developing therapeutics. We’re indebted to the work of many other labs: this work wouldn’t be possible without the efforts of researchers across the globe who have responded to the COVID-19 outbreak with incredible agility.
Knowing a protein’s structure provides an important resource for understanding how it functions, but experiments to determine the structure can take months or longer, and some prove to be intractable. For this reason, researchers have been developing computational methods to predict protein structure from the amino acid sequence. In cases where the structure of a similar protein has already been experimentally determined, algorithms based on “template modelling” are able to provide accurate predictions of the protein structure. AlphaFold, our recently published deep learning system, focuses on predicting protein structure accurately when no structures of similar proteins are available, called “free modelling”. We’ve continued to improve these methods since that publication and want to provide the most useful predictions, so we’re sharing predicted structures for some of the proteins in SARS-CoV-2 generated using our newly-developed methods.
It’s important to note that our structure prediction system is still in development and we can’t be certain of the accuracy of the structures we are providing, although we are confident that the system is more accurate than our earlier CASP13 system. We confirmed that our system provided an accurate prediction for the experimentally determined SARS-CoV-2 spike protein structure shared in the Protein Data Bank, and this gave us confidence that our model predictions on other proteins may be useful. We recently shared our results with several colleagues at the Francis Crick Institute in the UK, including structural biologists and virologists, who encouraged us to release our structures to the general scientific community now. Our models include per-residue confidence scores to help indicate which parts of the structure are more likely to be correct. We have only provided predictions for proteins which lack suitable templates or are otherwise difficult for template modeling. While these understudied proteins are not the main focus of current therapeutic efforts, they may add to researchers’ understanding of SARS-CoV-2.
Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them.
Interested researchers can download the structures here, and can read more technical details about these predictions in a document included with the data. The protein structure predictions we’re releasing are for SARS-CoV-2 membrane protein, protein 3a, Nsp2, Nsp4, Nsp6, and Papain-like proteinase (C terminal domain). To emphasise, these are predicted structures which have not been experimentally verified. Work on the system continues for us, and we hope to share more about it in due course.
DeepMind has shared its results with researchers at the Francis Crick Institute, a biomedical research lab in the UK, as well as offering it for download from its website.
“Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them,” it said. [ALA added bold face]
There are 93,090 cases of COVID-19, and 3,198 deaths, spread across 76 countries, according to the latest report from the World Health Organization at time of writing. ®
MHC content – The spike protein is thought to be the key to binding to cells via the angiotensin II receptor, the major mechanism the immune system uses to distinguish self from non-self
Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies
Syed Faraz Ahmed 1,† , Ahmed A. Quadeer 1, *,† and Matthew R. McKay 1,2, *
1 Department of Electronic and Computer Engineering, The Hong Kong University of Science and
Technology, Hong Kong, China; sfahmed@connect.ust.hk
2 Department of Chemical and Biological Engineering, The Hong Kong University of Science and
Received: 9 February 2020; Accepted: 24 February 2020; Published: 25 February 2020
Abstract:
The beginning of 2020 has seen the emergence of COVID-19 outbreak caused by a novel coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). There is an imminent need to better understand this new virus and to develop ways to control its spread. In this study, we sought to gain insights for vaccine design against SARS-CoV-2 by considering the high genetic similarity between SARS-CoV-2 and SARS-CoV, which caused the outbreak in 2003, and leveraging existing immunological studies of SARS-CoV. By screening the experimentally determined SARS-CoV-derived B cell and T cell epitopes in the immunogenic structural proteins of SARS-CoV, we identified a set of B cell and T cell epitopes derived from the spike (S) and nucleocapsid (N) proteins that map identically to SARS-CoV-2 proteins. As no mutation has been observed in these identified epitopes among the 120 available SARS-CoV-2 sequences (as of 21 February 2020), immune targeting of these epitopes may potentially offer protection against this novel virus. For the T cell epitopes, we performed a population coverage analysis of the associated MHC alleles and proposed a set of epitopes that is estimated to provide broad coverage globally, as well as in China. Our findings provide a screened set of epitopes that can help guide experimental efforts towards the development of vaccines against SARS-CoV-2.
Re: Protein structure prediction has been done for ages…
Not quite, Natural Selection does not measure methods, it measures outputs, usually at the organism level.
Sure correct folding is necessary for much protein function and we have prions and chaperone proteins to get it wrong and right.
The only way NS measures methods and mechanisms is if they are very energetically wasteful. But there are some very wasteful ones out there. Beta-Catenin at the end of point of Wnt signalling comes particularly to mind.
“Determining the structure of the virus proteins might also help in developing a molecule that disrupts the operation of just those proteins, and not anything else in the human body.”
Well it might, but predicting whether a ‘drug’ will NOT interact with any other of the 20000+ protein in complex organisms is well beyond current science. If we could do that we could predict/avoid toxicity and other non-mechanism related side-effects & mostly we can’t.
There are 480 structures on PDBe resulting from a search on ‘coronavirus,’ the top hits from MERS and SARS. PR stunt or not, they did win the most recent CASP ‘competition’, so arguably it’s probably our best shot right now – and I am certainly not satisfied that they have been sufficiently open in explaining their algorithms though I have not checked in the last few months. No one is betting anyone’s health on this, and it is not like making one wrong turn in a series of car directions. Latest prediction algorithms incorporate contact map predictions, so it’s not like a wrong dihedral angle sends the chain off in the wrong direction. A decent model would give something to run docking algorithms against with a series of already approved drugs, then we take that shortlist into the lab. A confirmed hit could be an instantly available treatment, no two year wait as currently estimated. [ALA added bold face]
Re: these structure predictions have not been experimentally verified
Naaaah. Can’t possibly be a stupid marketing stunt.
Well yes, a good possibility. But it can also be trying to build on the open-source model of putting it out there for others to build and improve upon. Essentially opening that “peer review” to a larger audience quicker. [ALA added bold face]
What bothers me, besides the obvious PR stunt, is that they say this prediction is licensed. How can a prediction from software be protected by, I presume, patents? And if this can be protected without even verifying which predictions actually work, what’s to stop someone spitting out millions of random, untested predictions just in case they can claim ownership later when one of them is proven to work? [ALA added bold face]
AI-predicted protein structures could unlock vaccine for Wuhan coronavirus… if correct… after clinical trials It’s not quite DeepMind’s ‘Come with me if you want to live’ moment, but it’s close, maybe
Experimentally derived by a group of scientists at the University of Texas at Austin and the National Institute of Allergy and Infectious Diseases, an agency under the US National Institute of Health. They both feature a “Spike protein structure.”
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN







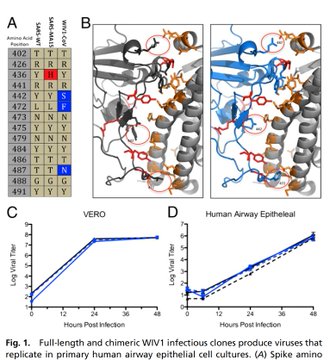

What bothers me, besides the obvious PR stunt, is that they say this prediction is licensed. How can a prediction from software be protected by, I presume, patents? And if this can be protected without even verifying which predictions actually work, what’s to stop someone spitting out millions of random, untested predictions just in case they can claim ownership later when one of them is proven to work? [ALA added bold face]
Paper in collection COVID-19 SARS-CoV-2 preprints from medRxiv and bioRxiv