Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Structure-guided Drug Discovery: (1) The Coronavirus 3CL hydrolase (Mpro) enzyme (main protease) essential for proteolytic maturation of the virus and (2) viral protease, the RNA polymerase, the viral spike protein, a viral RNA as promising two targets for discovery of cleavage inhibitors of the viral spike polyprotein preventing the Coronavirus Virion the spread of infection
Curators and Reporters: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Therapeutical options to coronavirus (2019-nCoV) include consideration of the following:
(a) Monoclonal and polyclonal antibodies
(b) Vaccines
(c) Small molecule treatments (e.g., chloroquinolone and derivatives), including compounds already approved for other indications
(d) Immuno-therapies derived from human or other sources
Structure of the nCoV trimeric spike
The World Health Organization has declared the outbreak of a novel coronavirus (2019-nCoV) to be a public health emergency of international concern. The virus binds to host cells through its trimeric spike glycoprotein, making this protein a key target for potential therapies and diagnostics. Wrapp et al. determined a 3.5-angstrom-resolution structure of the 2019-nCoV trimeric spike protein by cryo–electron microscopy. Using biophysical assays, the authors show that this protein binds at least 10 times more tightly than the corresponding spike protein of severe acute respiratory syndrome (SARS)–CoV to their common host cell receptor. They also tested three antibodies known to bind to the SARS-CoV spike protein but did not detect binding to the 2019-nCoV spike protein. These studies provide valuable information to guide the development of medical counter-measures for 2019-nCoV. [Bold Face Added by ALA]
The outbreak of a novel coronavirus (2019-nCoV) represents a pandemic threat that has been declared a public health emergency of international concern. The CoV spike (S) glycoprotein is a key target for vaccines, therapeutic antibodies, and diagnostics. To facilitate medical countermeasure development, we determined a 3.5-angstrom-resolution cryo–electron microscopy structure of the 2019-nCoV S trimer in the prefusion conformation. The predominant state of the trimer has one of the three receptor-binding domains (RBDs) rotated up in a receptor-accessible conformation. We also provide biophysical and structural evidence that the 2019-nCoV S protein binds angiotensin-converting enzyme 2 (ACE2) with higher affinity than does severe acute respiratory syndrome (SARS)-CoV S. Additionally, we tested several published SARS-CoV RBD-specific monoclonal antibodies and found that they do not have appreciable binding to 2019-nCoV S, suggesting that antibody cross-reactivity may be limited between the two RBDs. The structure of 2019-nCoV S should enable the rapid development and evaluation of medical countermeasures to address the ongoing public health crisis.
SOURCE
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Recent emergence of the COVID-19 coronavirus has resulted in a WHO-declared public health emergency of international concern. Research efforts around the world are working towards establishing a greater understanding of this particular virus and developing treatments and vaccines to prevent further spread.
While PDB entry 6lu7 is currently the only public-domain 3D structure from this specific coronavirus, the PDB contains structures of the corresponding enzyme from other coronaviruses. The 2003 outbreak of the closely-related Severe Acute Respiratory Syndrome-related coronavirus (SARS) led to the first 3D structures, and today there are more than 200 PDB structures of SARS proteins. Structural information from these related proteins could be vital in furthering our understanding of coronaviruses and in discovery and development of new treatments and vaccines to contain the current outbreak.
The coronavirus 3CL hydrolase (Mpro) enzyme, also known as the main protease, is essential for proteolytic maturation of the virus. It is thought to be a promising target for discovery of small-molecule drugs that would inhibit cleavage of the viral polyprotein and prevent spread of the infection.
Comparison of the protein sequence of the COVID-19 coronavirus 3CL hydrolase (Mpro) against the PDB archive identified 95 PDB proteins with at least 90% sequence identity. Furthermore, these related protein structures contain approximately 30 distinct small molecule inhibitors, which could guide discovery of new drugs. Of particular significance for drug discovery is the very high amino acid sequence identity (96%) between the COVID-19 coronavirus 3CL hydrolase (Mpro) and the SARS virus main protease (PDB 1q2w). Summary data about these closely-related PDB structures are available (CSV) to help researchers more easily find this information. In addition, the PDB houses 3D structure data for more than 20 unique SARS proteins represented in more than 200 PDB structures, including a second viral protease, the RNA polymerase, the viral spike protein, a viral RNA, and other proteins (CSV).
Public release of the COVID-19 coronavirus 3CL hydrolase (Mpro), at a time when this information can prove most vital and valuable, highlights the importance of open and timely availability of scientific data. The wwPDB strives to ensure that 3D biological structure data remain freely accessible for all, while maintaining as comprehensive and accurate an archive as possible. We hope that this new structure, and those from related viruses, will help researchers and clinicians address the COVID-19 coronavirus global public health emergency.
Update: Released COVID-19-related PDB structures include
PDB structure 6lu7 (X. Liu, B. Zhang, Z. Jin, H. Yang, Z. Rao Crystal structure of COVID-19 main protease in complex with an inhibitor N3 doi: 10.2210/pdb6lu7/pdb) Released 2020-02-05
PDB structure 6vsb (D. Wrapp, N. Wang, K.S. Corbett, J.A. Goldsmith, C.-L. Hsieh, O. Abiona, B.S. Graham, J.S. McLellan (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation Science doi: 10.1126/science.abb2507) Released 2020-02-26
PDB structure 6lxt (Y. Zhu, F. Sun Structure of post fusion core of 2019-nCoV S2 subunit doi: 10.2210/pdb6lxt/pdb) Released 2020-02-26
PDB structure 6lvn (Y. Zhu, F. Sun Structure of the 2019-nCoV HR2 Domain doi: 10.2210/pdb6lvn/pdb) Released 2020-02-26
PDB structure 6vw1
J. Shang, G. Ye, K. Shi, Y.S. Wan, H. Aihara, F. Li Structural basis for receptor recognition by the novel coronavirus from Wuhan doi: 10.2210/pdb6vw1/pdb
Released 2020-03-04
PDB structure 6vww
Y. Kim, R. Jedrzejczak, N. Maltseva, M. Endres, A. Godzik, K. Michalska, A. Joachimiak, Center for Structural Genomics of Infectious Diseases Crystal Structure of NSP15 Endoribonuclease from SARS CoV-2 doi: 10.2210/pdb6vww/pdb
Released 2020-03-04
PDB structure 6y2e
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure of the free enzyme of the SARS-CoV-2 (2019-nCoV) main protease doi: 10.2210/pdb6y2e/pdb
Released 2020-03-04
PDB structure 6y2f
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (monoclinic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2f/pdb
Released 2020-03-04
PDB structure 6y2g
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (orthorhombic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2g/pdb
Released 2020-03-04
Coronavirus disease 2019 (COVID-19) is a global pandemic impacting nearly 170 countries/regions and more than 285,000 patients worldwide. COVID-19 is caused by the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), which invades cells through the angiotensin converting enzyme 2 (ACE2) receptor. Among those with COVID-19, there is a higher prevalence of cardiovascular disease and more than 7% of patients suffer myocardial injury from the infection (22% of the critically ill). Despite ACE2 serving as the portal for infection, the role of ACE inhibitors or angiotensin receptor blockers requires further investigation. COVID-19 poses a challenge for heart transplantation, impacting donor selection, immunosuppression, and post-transplant management. Thankfully there are a number of promising therapies under active investigation to both treat and prevent COVID-19. Key Words: COVID-19; myocardial injury; pandemic; heart transplant
Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW (Apr 2004). “ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis”. The Journal of Biological Chemistry. 279 (17): 17996–8007. doi:10.1074/jbc.M311191200. PMID14754895.
Turner AJ, Tipnis SR, Guy JL, Rice G, Hooper NM (Apr 2002). “ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors”. Canadian Journal of Physiology and Pharmacology. 80 (4): 346–53. doi:10.1139/y02-021. PMID12025971.
Zhang, Haibo; Penninger, Josef M.; Li, Yimin; Zhong, Nanshan; Slutsky, Arthur S. (3 March 2020). “Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target”. Intensive Care Medicine. Springer Science and Business Media LLC. doi:10.1007/s00134-020-05985-9. ISSN0342-4642. PMID32125455.
^Gurwitz, David (2020). “Angiotensin receptor blockers as tentative SARS‐CoV‐2 therapeutics”. Drug Development Research. doi:10.1002/ddr.21656. PMID32129518.
ACE2 receptors have been shown to be the entry point into human cells for some coronaviruses, including the SARSvirus.[10] A number of studies have identified that the entry point is the same for SARS-CoV-2,[11] the virus that causes COVID-19.[12][13][14][15]
Some have suggested that a decrease in ACE2 could be protective against Covid-19 disease[16], but others have suggested the opposite, that Angiotensin II receptor blocker drugs could be protective against Covid-19 disease via increasing ACE2, and that these hypotheses need to be tested by datamining of clinical patient records.[17]
We need your help! Folding@home is joining researchers around the world working to better understand the 2019 Coronavirus (2019-nCoV) to accelerate the open science effort to develop new life-saving therapies. By downloading Folding@Home, you can donate your unused computational resources to the Folding@home Consortium, where researchers working to advance our understanding of the structures of potential drug targets for 2019-nCoV that could aid in the design of new therapies. The data you help us generate will be quickly and openly disseminated as part of an open science collaboration of multiple laboratories around the world, giving researchers new tools that may unlock new opportunities for developing lifesaving drugs.
2019-nCoV is a close cousin to SARS coronavirus (SARS-CoV), and acts in a similar way. For both coronaviruses, the first step of infection occurs in the lungs, when a protein on the surface of the virus binds to a receptor protein on a lung cell. This viral protein is called the spike protein, depicted in red in the image below, and the receptor is known as ACE2. A therapeutic antibody is a type of protein that can block the viral protein from binding to its receptor, therefore preventing the virus from infecting the lung cell. A therapeutic antibody has already been developed for SARS-CoV, but to develop therapeutic antibodies or small molecules for 2019-nCoV, scientists need to better understand the structure of the viral spike protein and how it binds to the human ACE2 receptor required for viral entry into human cells.
Proteins are not stagnant—they wiggle and fold and unfold to take on numerous shapes. We need to study not only one shape of the viral spike protein, but all the ways the protein wiggles and folds into alternative shapes in order to best understand how it interacts with the ACE2 receptor, so that an antibody can be designed. Low-resolution structures of the SARS-CoV spike protein exist and we know the mutations that differ between SARS-CoV and 2019-nCoV. Given this information, we are uniquely positioned to help model the structure of the 2019-nCoV spike protein and identify sites that can be targeted by a therapeutic antibody. We can build computational models that accomplish this goal, but it takes a lot of computing power.
This is where you come in! With many computers working towards the same goal, we aim to help develop a therapeutic remedy as quickly as possible. By downloading Folding@home here [LINK] and selecting to contribute to “Any Disease”, you can help provide us with the computational power required to tackle this problem. One protein from 2019-nCoV, a protease encoded by the viral RNA, has already been crystallized. Although the 2019-nCoV spike protein of interest has not yet been resolved bound to ACE2, our objective is to use the homologous structure of the SARS-CoV spike protein to identify therapeutic antibody targets.
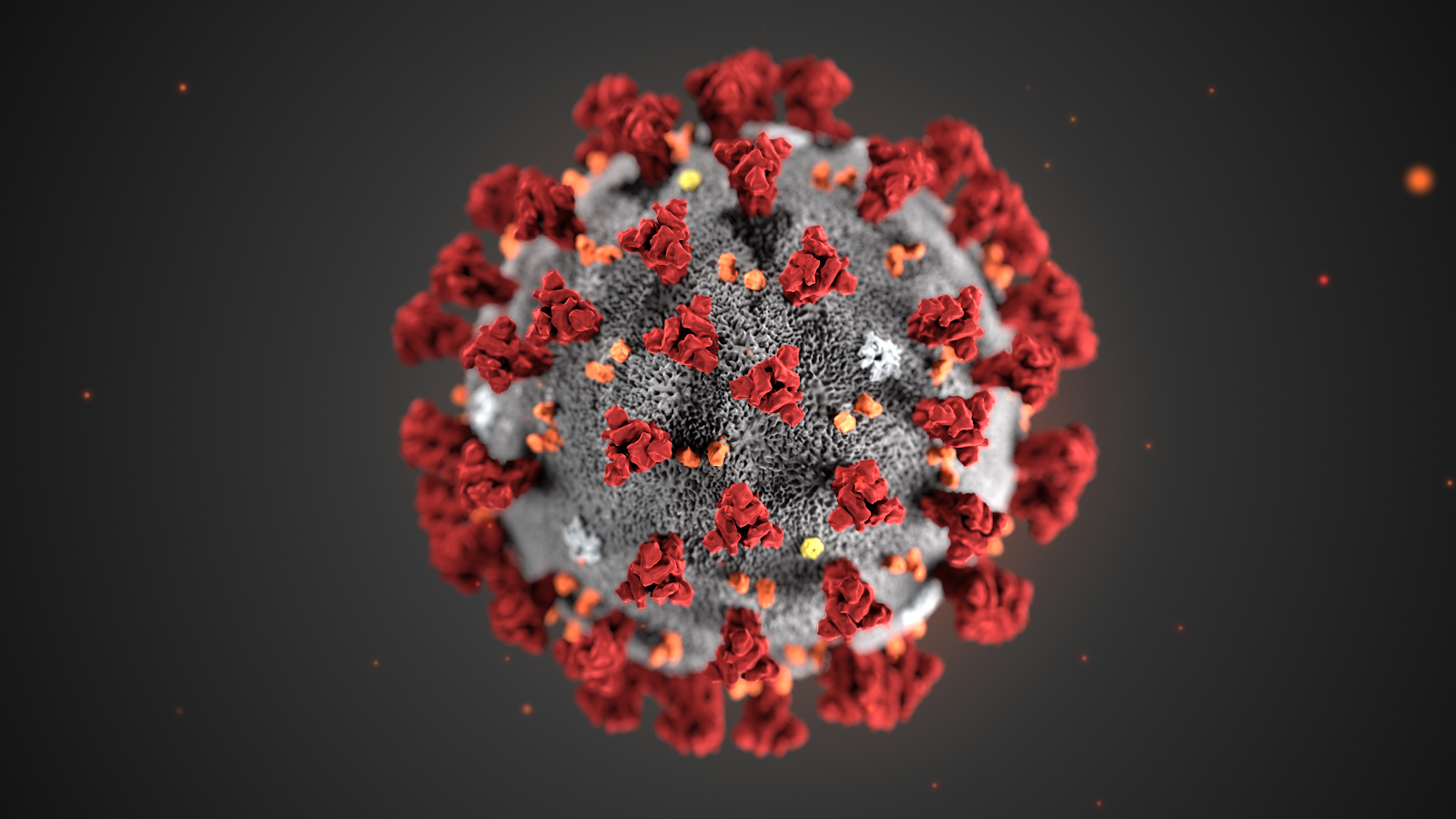
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
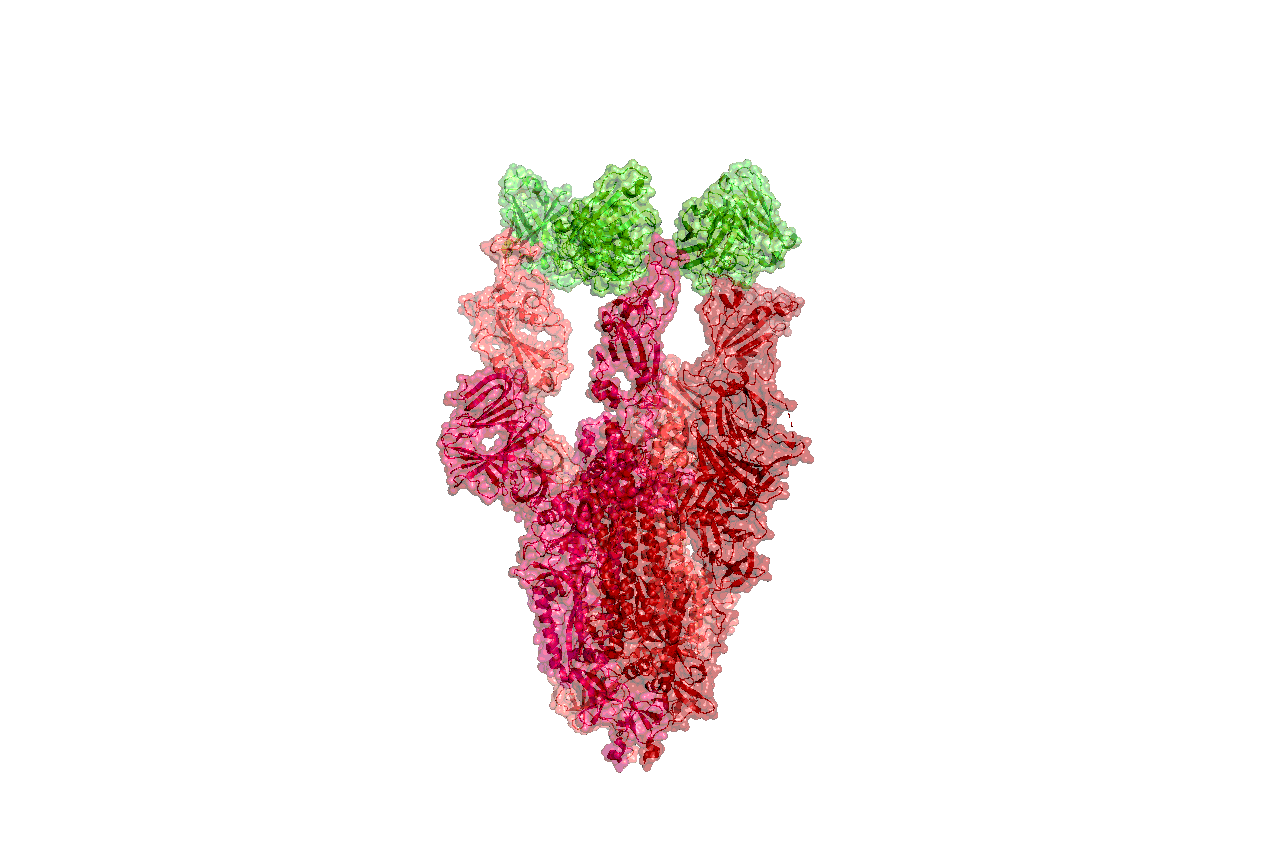
Structures of the closely related SARS-CoV spike protein bound by therapeutic antibodies may help rapidly design better therapies. The three monomers of the SARS-CoV spike protein are shown in different shades of red; the antibody is depicted in green. [PDB: 6NB7 https://www.rcsb.org/structure/6nb7]
I am reposting the following Science blog post from Derrick Lowe as is and ask people go browse through the comments on his Science blog In the Pipeline because, as Dr. Lowe states that in this current crisis it is important to disseminate good information as quickly as possible so wanted the readers here to have the ability to read his great posting on this matter of Covid-19. Also i would like to direct readers to the journal Science opinion letter concerning how important it is to rebuild the trust in good science and the scientific process. The full link for the following In the Pipeline post is: https://blogs.sciencemag.org/pipeline/archives/2020/03/06/covid-19-small-molecule-therapies-reviewed
A Summary of current potential repurposed therapeutics for COVID-19 Infection from In The Pipeline: A Science blog from Derick Lowe
Let’s take inventory on the therapies that are being developed for the coronavirus epidemic. Here is a very thorough list of at Biocentury, and I should note that (like Stat and several other organizations) they’re making all their Covid-19 content free to all readers during this crisis. I’d like to zoom in today on the potential small-molecule therapies, since some of these have the most immediate prospects for use in the real world.
The ones at the front of the line are repurposed drugs that are already approved for human use, for a lot of obvious reasons. The Biocentury list doesn’t cover these, but here’s an article at Nature Biotechnology that goes into detail. Clinical trials are a huge time sink – they sort of have to be, in most cases, if they’re going to be any good – and if you’ve already done all that stuff it’s a huge leg up, even if the drug itself is not exactly a perfect fit for the disease. So what do we have? The compound that is most advanced is probably remdesivir from Gilead, at right. This has been in development for a few years as an RNA virus therapy – it was originally developed for Ebola, and has been tried out against a whole list of single-strand RNA viruses. That includes the related coronaviruses SARS and MERS, so Covid-19 was an obvious fit.
The compound is a prodrug – that phosphoramide gets cleaved off completely, leaving the active 5-OH compound GS-44-1524. It mechanism of action is to get incorporated into viral RNA, since it’s taken up by RNA polymerase and it largely seems to evade proofreading. This causes RNA termination trouble later on, since that alpha-nitrile C-nucleoside is not exactly what the virus is expecting in its genome at that point, and thus viral replication is inhibited.
There are five clinical trials underway (here’s an overview at Biocentury). The NIH has an adaptive-design Phase II trial that has already started in Nebraska, with doses to be changed according to Bayesian readouts along the way. There are two Phase III trials underway at China-Japan Friendship Hospital in Hubei, double-blinded and placebo-controlled (since placebo is, as far as drug therapy goes, the current standard of care). And Gilead themselves are starting two open-label trials, one with no control arm and one with an (unblinded) standard-of-care comparison arm. Those might read out first, depending on when they get off the ground, but will be only rough readouts due to the fast-and-loose trial design. The two Hubei trials and the NIH one will add some rigor to the process, but I’m not sure when they’re going to report. My personal opinion is that I like the chances of this drug more than anything else on this list, but it’s still unlikely to be a game-changer.
There’s an RNA polymerase inhibitor (favipiravir) from Toyama, at right, that’s in a trial in China. It’s a thought – a broad-spectrum agent of this sort would be the sort of thing to try. But unfortunately, from what I can see, it has already turned up as ineffective in in vitro tests. The human trial that’s underway is honestly the sort of thing that would only happen under circumstances like the present: a developing epidemic with a new pathogen and no real standard of care. I hold out little hope for this one, but given that there’s nothing else at present, it probably should be tried. As you’ll see, this is far from the only situation like this.
One of the screens of known drugs in China that also flagged remdesivir noted that the old antimalarial drug chloroquine seemed to be effective in vitro. It had been reported some years back as a possible antiviral, working through more than one mechanism, probably both at viral entry and intracellularly thereafter. That part shouldn’t be surprising – chloroquine’s actual mode(s) of action against malaria parasites are still not completely worked out, either, and some of what people thought they knew about it has turned out to be wrong. There are several trials underway with it at Chinese facilities, some in combination with other agents like remdesivir. Chloroquine has of course been taken for many decades as an antimalarial, but it has a number of liabilities, including seizures, hearing damage, retinopathy and sudden effects on blood glucose. So it’s going to be important to establish just how effective it is and what doses will be needed. Just as with vaccine candidates, it’s possible to do more harm with a rushed treatment than the disease is doing itself
There are several other known antiviral drugs are being tried in China, but I don’t have too much hope for those, either. The neuraminidase inhibitors such as oseltamivir (better known as Tamiflu) were tried against SARS and were ineffective; there is no reason to expect anything versus Covid-19 although these drugs are a component of some drug cocktail trials. The HIV protease therapies such as darunavir and the combination therapy Kaletra are in trials, but that’s also a rather desperate long shot, since there’s no particular reason to think that they will have any such protease inhibition against what this new virus has to offer (and indeed, such agents weren’t much help against SARS in the end, either). The classic interferon/ribavirin combination seems to have had some activity against SARS and MERS, and is in two trials from what I can see. That’s not an awful idea by any means, but it’s not a great one, either: if your viral disease has interferon/ribavirin as a front line therapy, it generally means that there’s nothing really good available. No, unless we get really lucky none of these ideas are going to slow the disease down much.
There are a few other repurposed-protease-inhibitors ideas out there, such as this one. (Edit: I had seen this paper but couldn’t track it down, so thanks to those who sent it along). This paper suggests that the TMPRSS2 protease is important for viral entry on the human-cell-side of the process, a pathway that has been noted for other coronaviruses. And it points out that there is a an approved inhibitor (in Japan) for this enzyme (camostat), so that would definitely seem to be worth a trial, probably in combination with remdesivir.
That’s about it for the existing small molecules, from what I can see. What about new ones? Don’t hold your breath, is all I can say. A drug discovery program from scratch against a new pathogen is, as many readers here well know, not a trivial exercise. As this Bloomberg article details, many such efforts in the past (small molecules and vaccines alike) have come to grief because by the time they had anything to deliver the epidemic itself had passed. Indeed, Gilead’s remdesivir had already been dropped as a potential Ebola therapy.
You will either need to have a target in mind up front or go phenotypic. For the former, what you’d see are better characterizations of the viral protease and more extensive screens against it. Two other big target areas are viral entry (which involves the “spike” proteins on the virus surface and the ACE2 protein on human cells) and viral replication. To the former, it’s worth quickly noting that ACE2 is so much unlike the more familiar ACE protein that none of the cardiovascular ACE inhibitors do anything to it at all. And targeting the latter mechanisms is how remdesivir was developed as a possible Ebola agent, but as you can see, that took time, too. Phenotypic screens are perfectly reasonable against viral pathogens as well, but you’ll need to put time and effort into that assay up front, just as with any phenotypic effort, because as anyone who does that sort of work will tell you, a bad phenotypic screen is a complete waste of everyone’s time.
One of the key steps for either route is identifying an animal model. While animal models of infectious disease can be extremely well translated to human therapy, that doesn’t happen by accident: you need to choose the right animal. Viruses in general (and coronaviruses are no exception) vary widely in their effects in different species, and not just across the gaps of bird/reptile/human and the like. No, you’ll run into things where even the usual set of small mammals are acting differently from each other, with some of them not even getting sick at all. This current virus may well have gone through a couple of other mammalian species before landing on us, but you’ll note that dogs (to pick one) don’t seem to have any problem with it.
All this means that any new-target new-chemical-matter effort against Covid-19 (or any new pathogen) is going to take years, and there is just no way around that. Update: see here for just such an effort to start finding fragment hits for the viral protease. This puts small molecules in a very bimodal distribution: you have the existing drugs that might be repurposed, and are presumably available right now. Nothing else is! At the other end, for completely new therapies you have the usual prospects of drug discovery: years from now, lots of money, low success rate, good luck to all of us. The gap between these two could in theory be filled by vaccines and antibody therapies (if everything goes really, really well) but those are very much their own area and will be dealt with in a separate post.
Either way, the odds are that we (and I mean “we as a species” here) are going to be fighting this epidemic without any particularly amazing pharmacological weapons. Eventually we’ll have some, but I would advise people, pundits, and politicians not to get all excited about the prospects for some new therapies to come riding up over the hill to help us out. The odds of that happening in time to do anything about the current outbreak are very small. We will be going for months, years, with the therapeutic options we have right now. Look around you: what we have today is what we have to work with.
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
$The authors would like to note that the first eight authors are listed alphabetically.
Abstract
During its first month, the recently emerged 2019 Wuhan novel coronavirus (2019-nCoV) has already infected many thousands of people in mainland China and worldwide and took hundreds of lives. However, the swiftly spreading virus also caused an unprecedentedly rapid response from the research community facing the unknown health challenge of potentially enormous proportions. Unfortunately, the experimental research to understand the molecular mechanisms behind the viral infection and to design a vaccine or antivirals is costly and takes months to develop. To expedite the advancement of our knowledge we leverage the data about the related coronaviruses that is readily available in public databases, and integrate these data into a single computational pipeline. As a result, we provide a comprehensive structural genomics and interactomics road-maps of 2019-nCoV and use these information to infer the possible functional differences and similarities with the related SARS coronavirus. All data are made publicly available to the research community at http://korkinlab.org/wuhan
Figure 2. Structurally characterized non-structural proteins of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. The structurally resolved part of wNsp7 is sequentially identical to its homolog.
Figure 3. Structurally characterized structural proteins and an ORF of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. Highlighted in yellow are novel protein inserts found in wS.
Figure 4. Structurally characterized intra-viral and host-viral protein-protein interaction complexes of 2019-nCoV. Human proteins (colored in orange) are identified through their gene names. For each intra-viral structure, the number of subunits involved in the interaction is specified.
Figure 5. Evolutionary conservation of functional sites in 2019-nCoV proteins. A. Fully conserved protein binding sites (PBS, light orange) of wNsp12 in its interaction with wNsp7 and wNsp8 while other parts of the protein surface shows mutations (magenta); B. Both major monoclonal antibody binding site (light orange) and ACE2 receptor binding site (dark green) of wS are heavily mutated (binding site mutations are shown in red) compared to the same binding sites in other coronaviruses; mutations not located on the two binding sites are shown in magenta; C. Nearly intact protein binding site (light orange) of wNsp (papain-like protease PLpro domain) for its putative interaction with human ubiquitin-aldehyde (binding site mutations for the only two residues are shown in red, non-binding site mutations are shown in magenta); D. Fully conserved inhibitor ligand binding site (LBS, green) for wNsp5; non-binding site mutations are shown in magenta.
According to the World Health Organization, coronaviruses make up a large family of viruses named for the crown-like spikes found on their surface (Figure 1). They carry their genetic material in single strands of RNA and cause respiratory problems and fever. Like HIV, coronaviruses can be transmitted between animals and humans. Coronaviruses have been responsible for the Severe Acute Respiratory Syndrome (SARS) pandemic in the early 2000s and the Middle East Respiratory Syndrome (MERS) outbreak in South Korea in 2015. While the most recent coronavirus, COVID-19, has caused international concern, accessible and inexpensive sequencing is helping us understand COVID-19 and respond to the outbreak quickly.
Figure 1. Coronaviruses with the characteristic spikes as seen under a microscope.
First studies that explore genetic susceptibility to COVID-19 are now being published. The first results indicate that COVID-19 infects cells using the ACE2 cell-surface receptor. Genetic variants in the ACE2 receptor gene are thus likely to influence how effectively COVID-19 can enter the cells in our bodies. Researchers hope to discover genetic variants that confer resistance to a COVID-19 infection, similar to how some variants in the CCR5 receptor gene make people immune to HIV. At Nebula Genomics, we are monitoring the latest COVID-19 research and will add any relevant discoveries to the Nebula Research Library in a timely manner.
The Role of Genomics in Responding to COVID-19
Scientists in China sequenced COVID-19’s genome just a few weeks after the first case was reported in Wuhan. This stands in contrast to SARS, which was discovered in late 2002 but was not sequenced until April of 2003. It is through inexpensive genome-sequencing that many scientists across the globe are learning and sharing information about COVID-19, allowing us to track the evolution of COVID-19 in real-time. Ultimately, sequencing can help remove the fear of the unknown and allow scientists and health professionals to prepare to combat the spread of COVID-19.
Next-generation DNA sequencing technology has enabled us to understand COVID-19 is ~30,000 bases long. Moreover, researchers in China determined that COVID-19 is also almost identical to a coronavirus found in bats and is very similar to SARS. These insights have been critical in aiding in the development of diagnostics and vaccines. For example, the Centers for Disease Control and Prevention developed a diagnostic test to detect COVID-19 RNA from nose or mouth swabs.
Moreover, a number of different government agencies and pharmaceutical companies are in the process of developing COVID-19 vaccines to stop the COVID-19 from infecting more people. To protect humans from infection inactivated virus particles or parts of the virus (e.g. viral proteins) can be injected into humans. The immune system will recognize the inactivated virus as foreign, priming the body to build immunity against possible future infection. Of note, Moderna Inc., the National Institute of Allergy and Infectious Diseases, and Coalition for Epidemic Preparedness Innovations identified a COVID-19 vaccine candidate in a record 42 days. This vaccine will be tested in human clinical trials starting in April.
For more information about COVID-19, please refer to the World Health Organization website.
The problem w/ visionaries is that we don’t recognize them in a timely manner (too late) Ralph Baric @UNCpublichealth and Vineet Menachery deserve recognition for being 5 yrs ahead of #COVID19https://nature.com/articles/nm.3985…@NatureMedicinehttps://pnas.org/content/113/11/3048…@PNASNews via @hoondy
Senior, A.W., Evans, R., Jumper, J. et al.Improved protein structure prediction using potentials from deep learning. Nature577, 706–710 (2020). https://doi.org/10.1038/s41586-019-1923-7
Abstract
Protein structure prediction can be used to determine the three-dimensional shape of a protein from its amino acid sequence1. This problem is of fundamental importance as the structure of a protein largely determines its function2; however, protein structures can be difficult to determine experimentally. Considerable progress has recently been made by leveraging genetic information. It is possible to infer which amino acid residues are in contact by analysing covariation in homologous sequences, which aids in the prediction of protein structures3. Here we show that we can train a neural network to make accurate predictions of the distances between pairs of residues, which convey more information about the structure than contact predictions. Using this information, we construct a potential of mean force4 that can accurately describe the shape of a protein. We find that the resulting potential can be optimized by a simple gradient descent algorithm to generate structures without complex sampling procedures. The resulting system, named AlphaFold, achieves high accuracy, even for sequences with fewer homologous sequences. In the recent Critical Assessment of Protein Structure Prediction5 (CASP13)—a blind assessment of the state of the field—AlphaFold created high-accuracy structures (with template modelling (TM) scores6 of 0.7 or higher) for 24 out of 43 free modelling domains, whereas the next best method, which used sampling and contact information, achieved such accuracy for only 14 out of 43 domains. AlphaFold represents a considerable advance in protein-structure prediction. We expect this increased accuracy to enable insights into the function and malfunction of proteins, especially in cases for which no structures for homologous proteins have been experimentally determined7. https://doi.org/10.1038/s41586-019-1923-7
The scientific community has galvanised in response to the recent COVID-19 outbreak, building on decades of basic research characterising this virus family. Labs at the forefront of the outbreak response shared genomes of the virus in open access databases, which enabled researchers to rapidly develop tests for this novel pathogen. Other labs have shared experimentally-determined and computationally-predicted structures of some of the viral proteins, and still others have shared epidemiological data. We hope to contribute to the scientific effort using the latest version of our AlphaFold system by releasing structure predictions of several under-studied proteins associated with SARS-CoV-2, the virus that causes COVID-19. We emphasise that these structure predictions have not been experimentally verified, but hope they may contribute to the scientific community’s interrogation of how the virus functions, and serve as a hypothesis generation platform for future experimental work in developing therapeutics. We’re indebted to the work of many other labs: this work wouldn’t be possible without the efforts of researchers across the globe who have responded to the COVID-19 outbreak with incredible agility.
Knowing a protein’s structure provides an important resource for understanding how it functions, but experiments to determine the structure can take months or longer, and some prove to be intractable. For this reason, researchers have been developing computational methods to predict protein structure from the amino acid sequence. In cases where the structure of a similar protein has already been experimentally determined, algorithms based on “template modelling” are able to provide accurate predictions of the protein structure. AlphaFold, our recently published deep learning system, focuses on predicting protein structure accurately when no structures of similar proteins are available, called “free modelling”. We’ve continued to improve these methods since that publication and want to provide the most useful predictions, so we’re sharing predicted structures for some of the proteins in SARS-CoV-2 generated using our newly-developed methods.
It’s important to note that our structure prediction system is still in development and we can’t be certain of the accuracy of the structures we are providing, although we are confident that the system is more accurate than our earlier CASP13 system. We confirmed that our system provided an accurate prediction for the experimentally determined SARS-CoV-2 spike protein structure shared in the Protein Data Bank, and this gave us confidence that our model predictions on other proteins may be useful. We recently shared our results with several colleagues at the Francis Crick Institute in the UK, including structural biologists and virologists, who encouraged us to release our structures to the general scientific community now. Our models include per-residue confidence scores to help indicate which parts of the structure are more likely to be correct. We have only provided predictions for proteins which lack suitable templates or are otherwise difficult for template modeling. While these understudied proteins are not the main focus of current therapeutic efforts, they may add to researchers’ understanding of SARS-CoV-2.
Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them.
Interested researchers can download the structures here, and can read more technical details about these predictions in a document included with the data. The protein structure predictions we’re releasing are for SARS-CoV-2 membrane protein, protein 3a, Nsp2, Nsp4, Nsp6, and Papain-like proteinase (C terminal domain). To emphasise, these are predicted structures which have not been experimentally verified. Work on the system continues for us, and we hope to share more about it in due course.
DeepMind has shared its results with researchers at the Francis Crick Institute, a biomedical research lab in the UK, as well as offering it for download from its website.
“Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them,” it said. [ALA added bold face]
There are 93,090 cases of COVID-19, and 3,198 deaths, spread across 76 countries, according to the latest report from the World Health Organization at time of writing. ®
MHC content – The spike protein is thought to be the key to binding to cells via the angiotensin II receptor, the major mechanism the immune system uses to distinguish self from non-self
Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies
Syed Faraz Ahmed 1,† , Ahmed A. Quadeer 1, *,† and Matthew R. McKay 1,2, *
1 Department of Electronic and Computer Engineering, The Hong Kong University of Science and
Technology, Hong Kong, China; sfahmed@connect.ust.hk
2 Department of Chemical and Biological Engineering, The Hong Kong University of Science and
Received: 9 February 2020; Accepted: 24 February 2020; Published: 25 February 2020
Abstract:
The beginning of 2020 has seen the emergence of COVID-19 outbreak caused by a novel coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). There is an imminent need to better understand this new virus and to develop ways to control its spread. In this study, we sought to gain insights for vaccine design against SARS-CoV-2 by considering the high genetic similarity between SARS-CoV-2 and SARS-CoV, which caused the outbreak in 2003, and leveraging existing immunological studies of SARS-CoV. By screening the experimentally determined SARS-CoV-derived B cell and T cell epitopes in the immunogenic structural proteins of SARS-CoV, we identified a set of B cell and T cell epitopes derived from the spike (S) and nucleocapsid (N) proteins that map identically to SARS-CoV-2 proteins. As no mutation has been observed in these identified epitopes among the 120 available SARS-CoV-2 sequences (as of 21 February 2020), immune targeting of these epitopes may potentially offer protection against this novel virus. For the T cell epitopes, we performed a population coverage analysis of the associated MHC alleles and proposed a set of epitopes that is estimated to provide broad coverage globally, as well as in China. Our findings provide a screened set of epitopes that can help guide experimental efforts towards the development of vaccines against SARS-CoV-2.
Re: Protein structure prediction has been done for ages…
Not quite, Natural Selection does not measure methods, it measures outputs, usually at the organism level.
Sure correct folding is necessary for much protein function and we have prions and chaperone proteins to get it wrong and right.
The only way NS measures methods and mechanisms is if they are very energetically wasteful. But there are some very wasteful ones out there. Beta-Catenin at the end of point of Wnt signalling comes particularly to mind.
“Determining the structure of the virus proteins might also help in developing a molecule that disrupts the operation of just those proteins, and not anything else in the human body.”
Well it might, but predicting whether a ‘drug’ will NOT interact with any other of the 20000+ protein in complex organisms is well beyond current science. If we could do that we could predict/avoid toxicity and other non-mechanism related side-effects & mostly we can’t.
There are 480 structures on PDBe resulting from a search on ‘coronavirus,’ the top hits from MERS and SARS. PR stunt or not, they did win the most recent CASP ‘competition’, so arguably it’s probably our best shot right now – and I am certainly not satisfied that they have been sufficiently open in explaining their algorithms though I have not checked in the last few months. No one is betting anyone’s health on this, and it is not like making one wrong turn in a series of car directions. Latest prediction algorithms incorporate contact map predictions, so it’s not like a wrong dihedral angle sends the chain off in the wrong direction. A decent model would give something to run docking algorithms against with a series of already approved drugs, then we take that shortlist into the lab. A confirmed hit could be an instantly available treatment, no two year wait as currently estimated. [ALA added bold face]
Re: these structure predictions have not been experimentally verified
Naaaah. Can’t possibly be a stupid marketing stunt.
Well yes, a good possibility. But it can also be trying to build on the open-source model of putting it out there for others to build and improve upon. Essentially opening that “peer review” to a larger audience quicker. [ALA added bold face]
What bothers me, besides the obvious PR stunt, is that they say this prediction is licensed. How can a prediction from software be protected by, I presume, patents? And if this can be protected without even verifying which predictions actually work, what’s to stop someone spitting out millions of random, untested predictions just in case they can claim ownership later when one of them is proven to work? [ALA added bold face]
AI-predicted protein structures could unlock vaccine for Wuhan coronavirus… if correct… after clinical trials It’s not quite DeepMind’s ‘Come with me if you want to live’ moment, but it’s close, maybe
Experimentally derived by a group of scientists at the University of Texas at Austin and the National Institute of Allergy and Infectious Diseases, an agency under the US National Institute of Health. They both feature a “Spike protein structure.”
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN














Paper in collection COVID-19 SARS-CoV-2 preprints from medRxiv and bioRxiv