Summary of Transcription, Translation ond Transcription Factors
Author and Curator: Larry H. Bernstein, MD, FCAP

Article ID #158: Summary of Transcription, Translation and Transcription Factors. Published on 11/5/2014
WordCloud Image Produced by Adam Tubman
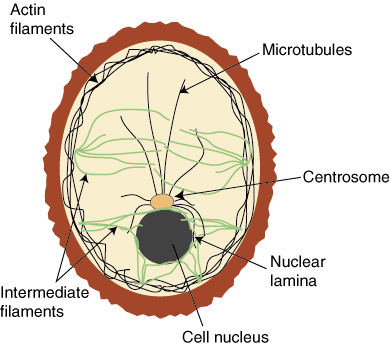
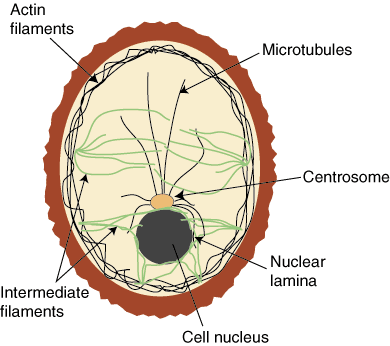
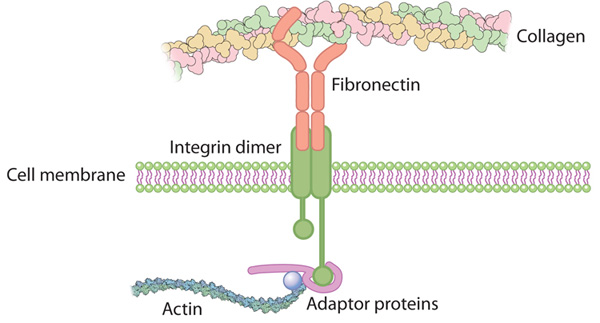
Proteins are integral to the composition of the cytoskeleton, and also to the extracellular matrix. Many proteins are actually enzymes, carrying out the transformation of some substrate, a derivative of the food we ingest. They have a catalytic site, and they function with a cofactor – either a multivalent metal or a nucleotide. Proteins also are critically involved in the regulation of cell metabolism, and they are involved in translation of the DNA code, as they make up transcription factors (TFs). There are 20 essential amino acids that go into protein synthesis that are derived from animal or plant protein. Protein synthesis is carried out by the transport of mRNA out of the nucleus to the ribosome, where tRNA is paired with a matching amino acid, and the primary sequence of a protein is constructed as a linear string of amino acids.
This is illustrated in the following three pictures:

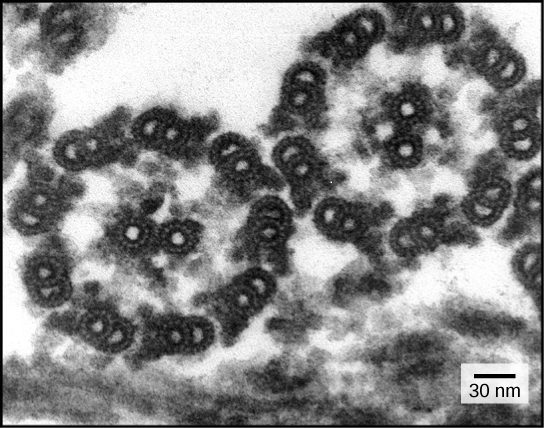
protein synthesis

mcell-transcription-translation

transcription_translation
Proteins synthesized at distal locations frequently contain intrinsically disordered segments. These regions are generally rich in assembly-promoting modules and are often regulated by post-translational modifications. Such proteins are tightly regulated but display distinct temporal dynamics upon stimulation with growth factors. Thus, proteins synthesized on-site may rapidly alter proteome composition and act as dynamically regulated scaffolds to promote the formation of reversible cellular assemblies.
RJ Weatheritt, et al. Nature Structural & Molecular Biology 24 Aug, 2014; 21: 833–839 http://dx.do.orgi:/10.1038/nsmb.2876

An overview of the potential advantages conferred by distal-site protein synthesis
Turquoise and red filled circle represents off-target and correct interaction partners, respectively. Wavy lines represent a disordered region within a distal site synthesis protein. Grey and red line in graphs represents profiles of t… http://www.nature.com/nsmb/journal/v21/n9/carousel/nsmb.2876-F5.jpg
In the the transcription process an RNA sequence is read. This is essential for protein synthesis through the ordering of the amino acids in the primary structure. However, there are microRNAs and noncoding RNAs, and there are transcription factors. The transcription factors bind to chromatin, and the RNAs also have some role in regulating the transcription process. (see picture above)
Transcription factors (TFs) interact dynamically in vivo with chromatin binding sites. Four different techniques are currently used to measure their kinetics in live cells,
- fluorescence recovery after photobleaching (FRAP),
- fluorescence correlation spectroscopy (FCS),
- single molecule tracking (SMT) and
- competition ChIP (CC).
A comparison of data from each of these techniques raises an important question:
- do measured transcription kinetics reflect biologically functional interactions at specific sites (i.e. working TFs) or
- do they reflect non-specific interactions (i.e. playing TFs)?
There are five key unresolved biological questions related to
- the functionality of transient and prolonged binding events at both
- specific promoter response elements as well as non-specific sites.
In support of functionality,
- there are data suggesting that TF residence times are tightly regulated, and
- that this regulation modulates transcriptional output at single genes.
In addition to this site-specific regulatory role, TF residence times
- also determine the fraction of promoter targets occupied within a cell
- thereby impacting the functional status of cellular gene networks.
- TF residence times, then, are key parameters that could influence transcription in multiple ways.
Quantifying transcription factor kinetics: At work or at play? Mueller F., et al. http://dx.doi.org:/10.3109/10409238.2013.833891
Dr. Virginie Mattot works in the team “Angiogenesis, endothelium activation and Cancer” directed by Dr. Fabrice Soncin at the Institut de Biologie de Lille in France where she studies the roles played by microRNAs in endothelial cells during physiological and pathological processes such as angiogenesis or endothelium activation. She has been using Target Site Blockers to investigate the role of microRNAs on putative targets.
A few years ago, the team identified
- an endothelial cell-specific gene which
- harbors a microRNA in its intronic sequence.
They have since been working on understanding the functions of
- both this new gene and its intronic microRNA in endothelial cells.
While they were searching for the functions of the intronic microRNA,
- theye identified an unknown gene as a putative target.
The aim of my project was to investigate if this unknown gene was actually a genuine target and
- if regulation of this gene by the microRNA was involved in endothelial cell function.
They had already shown the endothelial cell phenotype is associated with the inhibition of the intronic microRNA.
They then used miRCURY LNA™ Target Site Blockers to demonstrate
- the expression of this unknown gene is actually controlled by this microRNA.
- the microRNA regulates specific endothelial cell properties through regulation of this unknown gene.
MicroRNA function in endothelial cells – Solving the mystery of an unknown target gene using Target Site Blockers to investigate the role of microRNAs on putative targets
We first verified that this TSB was functional by analyzing
- the expression of the miRNA target against which the TSB was directed
- we then showed the TSB induced similar phenotypes as those when we inhibited the microRNA in the same cells.
Target Site Blockers were shown to be efficient tools to demonstrate the specific involvement of
- putative microRNA targets
- in the function played by this microRNA.
Some genes are known to have several different alternatively spliced protein variants, but the Scripps Research Institute’s Paul Schimmel and his colleagues have uncovered almost 250 protein splice variants of an essential, evolutionarily conserved family of human genes. The results were published July 17 in Science.
Focusing on the 20-gene family of aminoacyl tRNA synthetases (AARSs),
- the team captured AARS transcripts from human tissues—some fetal, some adult—and showed that
- many of these messenger RNAs (mRNAs) were translated into proteins.
Previous studies have identified several splice variants of these enzymes that have novel functions, but uncovering so many more variants was unexpected, Schimmel said. Most of these new protein products
- lack the catalytic domain but retain other AARS non-catalytic functional domains.
This study fundamentally effects how we view protein-synthesis, according to Michael Ibba (who was not involved in the work), The Scientist reported. “The unexpected and potentially vast expanded functional networks that emerge from this study have the potential to influence virtually any aspect of cell growth.”
The team—comprehensively captured and sequenced the AARS mRNAs from six human tissue types using high-throughput deep sequencing. They next showed that a proportion of these transcripts, including those missing the catalytic domain, indeed resulted in stable protein products:
- 48 of these splice variants associated with polysomes.
In vitro translation assays and the expression of more than 100 of these variants in cells confirmed that
- many of these variants could be made into stable protein products.
The AARS enzymes—of which there’s one for each of the 20 amino acids—bring together an amino acid with its appropriate transfer RNA (tRNA) molecule. This reaction allows a ribosome to add the amino acid to a growing peptide chain during protein translation. AARS enzymes can be found in all living organisms and are thought to be among the first proteins to have originated on Earth.
One goal of human genetics is to understand how the information for precise and dynamic gene expression programs is encoded in the genome. The interactions of transcription factors (TFs) with DNA regulatory elements clearly
- play an important role in determining gene expression outputs, yet
- the regulatory logic underlying functional transcription factor binding is poorly understood.
An important question in genomics is to understand how a class of proteins called ‘‘transcription factors’’ controls the expression level of other genes in the genome in a cell type-specific manner – a process that is essential to human development. One major approach to this problem is to study where these transcription factors bind in the genome, but this does not tell us about the effect of that binding on gene expression levels and
- it is generally accepted that much of the binding does not strongly influence gene expression.
DA Cusanovich et al. PLoS Genet 2014;10(3):e1004226. http://dx.doi.org:/10.1371/journal.pgen.1004226
We knocked down 59 TFs and chromatin modifiers in one HapMap lymphoblastoid cell line
- to evaluate the context of functional TF binding.
We then identified genes whose expression was affected by the knockdowns
- by intersecting the gene expression data with transcription factor binding data
(based on ChIP-seq and DNase-seq) - within 10 kb of the transcription start sites of expressed genes.
This combination of data allowed us to infer functional TF binding.
Only a small subset of genes bound by a factor were
- differentially expressed following the knockdown of that factor,
- suggesting that most interactions between TF and chromatin
- do not result in measurable changes in gene expression levels
- of putative target genes.
We found that functional TF binding is enriched
- in regulatory elements that harbor a large number of TF binding sites,
- at sites with predicted higher binding affinity, and
- at sites that are enriched in genomic regions annotated as ‘‘active enhancers.’’
We aim to be able to predict the expression pattern of a gene based on its regulatory
sequence alone.
Combining a TF knockdown approach with TF binding data can help us to
- distinguish functional binding from non-functional binding
This approach has previously been applied to the study of human TFs, although for the most part studies have only focused on
- the regulatory relationship of a single factor with its downstream targets.
The FANTOM consortium knocked down 52 different transcription factors in
- the THP-1 cell line, an acute monocytic leukemia-derived cell line, and
- used a subset of these to validate certain regulatory predictions based on binding motif enrichments.
We and others previously studied the regulatory architecture of gene expression in
- the model system of HapMap lymphoblastoid cell lines (LCLs) using both
- binding map strategies and QTL mapping strategies.
We now sought to use knockdown experiments targeting transcription factors in a HapMap LCL
- to refine our understanding of the gene regulatory circuitry of the human genome.
Therefore, We integrated the results of the knockdown experiments with previous data on TF binding to
- better characterize the regulatory targets of 59 different factors and
- to learn when a disruption in transcription factor binding
- is most likely to be associated with variation in the expression level of a nearby gene.
Gene expression levels following the knockdown were compared to
- expression data collected from six samples that were transfected with negative control siRNA.
Depending on the factor targeted, the knockdowns resulted in
- between 39 and 3,892 differentially expressed genes at an FDR of 5%
(Figure 1B; see Table S3 for a summary of the results).
The knockdown efficiency for the 59 factors ranged
- from 50% to 90% (based on qPCR; Table S1).
The qPCR measurements of the knockdown level were significantly
- correlated with estimates of the TF expression levels
- based on the microarray data (P =0.001; Figure 1C).
Did the factors tended to have a consistent effect (either up- or down-regulation)
- on the expression levels of genes they purportedly regulated?
All factors we tested are associated with both up- and down-regulation of downstream targets (Figure 6).
While there is compelling evidence for our inferences, the current chromatin functional annotations
- do not fully explain the regulatory effects of the knockdown experiments.
For example, the enrichments for binding in ‘‘strong enhancer’’ regions of the genome range from 7.2% to 50.1% (median = 19.2%),
- much beyond what is expected by chance alone, but far from accounting for all functional binding.
A slight majority of downstream target genes were expressed at higher levels
- following the knockdown for 15 of the 29 factors for which we had binding information (Figure 6B).
The factor that is associated with the largest fraction (68.8%) of up-regulated target genes following the knockdown is EZH2,
- the enzymatic component of the Polycomb group complex.
On the other end of the spectrum was JUND, a member of the AP-1 complex, for which
- 66.7% of differentially expressed targets were down-regulated following the knockdown.
Our results, combined with the previous work from our group and others make for a complicated view
- of the role of transcription factors in gene regulation as
- it seems difficult to reconcile the inference from previous work that
- many transcription factors should primarily act as activators with the results presented here.
One somewhat complicated hypothesis, which nevertheless can resolve the apparent discrepancy, is that
- the ‘‘repressive’’ effects we observe for known activators may be
- at sites in which the activator is acting as a weak enhancer of transcription and
- that reducing the cellular concentration of the factor
- releases the regulatory region to binding by an alternative, stronger activator.
Integrative study of Arabidopsis thaliana metabolomic and transcriptomic data
with the interactiveMarVis-Graph software
M Landesfeind, A Kaever, K Feussner, C Thurow, C Gatz, I Feussner and P Meinicke
PeerJ 2:e239; http://dx.doi.org /10.7717/peerj.239
High-throughput technologies notoriously generate large datasets often including data from different omics platforms. Each dataset contains data for several thousand experimental markers, e.g., mass-to-charge ratios in mass spectrometry or spots in DNA microarray analysis. An experimental marker is associated with an intensity profile which may include several measurements according to different experimental conditions (Dettmer, Aronov & Hammock, 2007).
The combined analysis and visualization of data from different high-throughput technologies remains a key challenge in bioinformatics.We present here theMarVis-Graph software for integrative analysis of metabolic and transcriptomic data. All experimental data is investigated in terms of the full metabolic network obtained from a reference database. The reactions of the network are scored based on the associated data, and
- sub-networks, according to connected high-scoring reactions, are identified.
Finally, MarVis-Graph scores the detected sub-networks,
- evaluates them by means of a random permutation test and
- presents them as a ranked list.
Furthermore, MarVis-Graph features an interactive network visualization that provides researchers with a convenient view on the results.
The key advantage ofMarVis-Graph is the analysis of reactions detached from their pathways so that
- it is possible to identify new pathways or
- to connect known pathways by previously unrelated reactions.
TheMarVis-Graph software is freely available for academic use and can be downloaded at: http://marvis.gobics.de/marvis-graph.
Significant differences or clusters may be explained by associated annotations, e.g., in terms of metabolic pathways or biological functions. During recent years, numerous specialized tools have been developed to aid biological researchers in automating all these steps (e.g., Medina et al., 2010; Kaever et al., 2009; Waegele et al., 2012). Comprehensive studies can be performed by combining technologies from different omics fields. The combination of transcriptomic and proteomic data sets revealed a strong
correlation between both kinds of data (Nie et al., 2007) and supported the detection of complex interactions, e.g., in RNA silencing (Haq et al., 2010). Moreover, correlations
were detected between RNA expression levels and metabolite abundances (Gibon et al., 2006). Therefore, tools that integrate, analyze and visualize experimental markers from different platforms are needed. To cope with the complexity of genome-wide studies, pathway models are utilized extensively as a simple abstraction of the underlying complex mechanisms. Set Enrichment Analysis (Subramanian et al., 2005) and Over-Representation Analysis (Huang, Sherman & Lempicki, 2009) have become state-of-the-art tools for analyzing large-scale datasets: both methods evaluate predefined sets of entities, e.g., the accumulation of differentially expressed genes in a pathway.
While manually curated pathways are convenient and easy to interpret, experimental studies have shown that all metabolic and signaling pathways are heavily interconnected (Kunkel & Brooks, 2002; Laule et al., 2003). Data from biomolecular databases support these studies: the metabolic network of Arabidopsis thaliana in the KEGG database (Kanehisa et al., 2012; Kanehisa & Goto, 2000) contains 1606 reactions from which 1464 are connected in a single sub-network (>91%), i.e., they
share a metabolite as product or substrate. In the AraCyc 10.0 database (Mueller, Zhang & Rhee, 2003; Rhee et al., 2006), more than 89% of the reactions are counted in a single sub-network. In both databases, most other reactions are completely disconnected. Additionally, Set Enrichment Analyses can not identify links between the predefined sets easily. This becomes even more important when analyzing smaller pathways as provided by the MetaCyc (Caspi et al., 2008; Caspi et al., 2012) database. Moreover, methods that utilize pathways as predefined sets ignore reactions and related biomolecular entities (e.g., metabolites, genes) which are not associated with a single pathway. For example, this affects 4000 reactions in MetaCyc and 2500 in KEGG, respectively (Altman et al., 2013). Therefore, it is desirable to develop additional methods
- that do not require predefined sets but may detect enriched sub-networks in the full metabolic network.
While several tools support the statistical analysis of experimental markers from one or more omics technologies and then utilize variants of Set Enrichment Analysis (Xia et al., 2012; Chen et al., 2013; Howe et al., 2011),
- no tool is able to explicitly search for connected reactions that include
- most of the metabolites, genes, and enyzmes with experimental evidence.
However, the automatic identification of sub-networks has been proven useful in other contexts, e.g., in the analysis of protein–protein-interaction networks (Alcaraz et al., 2012; Baumbach et al., 2012; Maeyer et al., 2013).
MarVis-Graph imports experimental markers from different high-throughput experiments and
- analyses them in the context of reaction-chains in full metabolic networks.
Then, MarVis-Graph scores the reactions in the metabolic network
- according to the number of associated experimental markers and
- identifies sub-networks consisting of subsequent, high-scoring reactions.
The resulting sub-networks are
- ranked according to a scoring method and visualized interactively.
Hereby, sub-networks consisting of reactions from different pathways may be identified to be important
- whereas the single pathways may not be found to be significantly enriched.
MarVis-Graph may also connect reactions without an assigned pathway
- to reactions within a particular pathway.
TheMarVis-Graph tool was applied in a case-study investigating the wound response in Arabidopsis thaliana to analyze combined metabolomic and transcriptomic high-throughput data.
Figure 1 Schema of the metabolic network representation in MarVis-Graph. Metabolite markers are shown in gray, metabolites in red, reactions in blue, enzymes in green, genes in yellow, transcript markers in pink, and pathways in turquoise color. The edges are shown in black with labels that comply with the biological meaning. The orange arrows depict the flow of score for the initial scoring (described in section “Initial Scoring”). (not shown)
In MarVis-Graph, metabolite markers obtained from mass-spectrometry experiments additionally contain the experimental mass. The experimental mass has to be
calculated based on the mass-to-charge ratio (m/z-value) and specific isotope- or adduct-corrections (Draper et al., 2009) by means of specialized tools, e.g.,MarVis-Filter
(Kaever et al., 2012).
For each transcript marker the corresponding annotation has to be given. In DNA microarray experiments, each spot (transcript marker) is specific for a gene and can
therefore be used for annotation. For other technologies an annotation has to be provided by external tools.
In MarVis-Graph, each reaction is scored initially based on the associated experimental data (see “Initial scoring”). This initial scoring is refined (see “Refining the scoring”) and afterwards reactions with a score below a user-defined threshold are removed. The network is
- decomposed into subsequent high-scoring reactions that constitute the sub-networks.
The weight of each experimental marker (see “Experimental markers”) is equally distributed over all metabolites and genes associated with the metabolite marker or
transcript marker, respectively. For all vertices, this is repeated as illustrated in Fig. 1 until the weights are accumulated by the reactions.
The initial reaction scores are used as input scoring for the random walk algorithm. The algorithm is performed as described by Glaab et al. (2012) with a user-defined
restart-probability r (default value 0.8). After convergence of the algorithm, reactions with a score lower than the user-defined threshold t (default value t = 1−r) are removed from the reaction network. During the removal process,
- the network is decomposed into pairwise disconnected sub-networks containing only high-scoring reactions.
In the following, a resulting sub-network is denoted by a prime: G′ = (V′,L′) with V′ = M′ ∪C′ ∪R′ ∪E′ ∪G′ ∪T′ ∪P′.
The scores of the identified sub-networks can be assessed using a random permutation test, evaluating the marker annotations under the null hypothesis of being connected
randomly. Here, the assignments
- from metabolite markers to metabolites and from transcript markers to genes are randomized.
For each association between a metabolite marker and a metabolite,
- this connection is replaced by a connection between a randomly chosen metabolite marker and a randomly chosen metabolite.
The random metabolite marker is chosen from the pool of formerly connected metabolite markers. Each connected transcript marker
- is associated with a randomly chosen gene.
Choosing from the list of already connected experimental markers ensures that
- the sum of weights from the original and the permuted network are equal.
This method differs from the commonly utilized XSwap permutation (Hanhij¨arvi, Garriga & Puolam¨aki, 2009) that is based on swapping endpoints of two random edges. The main difference of our permutation method is that it results in a network with different topological structure, i.e., different degree of the metabolite and gene nodes.
Finally, the sub-networks are detected and scored with the same parameters applied for the original network. Based on the scores of the networks identified in the random
permutations, the family-wise-error-rate (FWER) and false-discovery-rate (FDR) are calculated for each originally identified sub-network.
MarVis-Graph was applied in a case study investigating the A. thaliana wound response. Data from a metabolite fingerprinting (Meinicke et al., 2008) and a DNA microarray
experiment (Yan et al., 2007) were imported into a metabolic network specific for A. thaliana created from the AraCyc 10.0 database (Lamesch et al., 2011). The metabolome
and transcriptome have been measured before wounding as control and at specific time points after wounding in wild-type and in the allene oxide synthase (AOS) knock-out
mutant dde-2-2 (Park et al., 2002) of A. thaliana Columbia (see Table 1). The AOS mutant was chosen, because AOS catalyzes the first specific step in the biosynthesis of the hormone jasmonic acid, which is the key regulator in wound response of plants (Wasternack & Hause, 2013).
Both datasets have been preprocessed with theMarVis-Filter tool (Kaever et al., 2012) utilizing the Kruskal–Wallis p-value calculation on the intensity profiles. Based on the ranking of ascending p-values,
- the first 25% of the metabolite markers and 10% of the transcript markers have been selected for further investigation (Data S2).
The filtered metabolite and transcript markers were imported into the metabolic network. For metabolite markers, metabolites were associated
- if the metabolite marker’s detected mass differs from the metabolites monoisotopic mass by a maximum of 0.005u.
Transcript markers were linked to the genes whose ID equaled the ID given in the CATMA database (Sclep et al., 2007) for that transcript marker.
Table 2 Vertices in the A. thaliana specific metabolic network after import of experimental markers. Number of objects in the metabolic network
in absolute counts and relative abundances. For experimental markers, the with annotation column gives the number of metabolite markers and
transcript markers that were annotated with a metabolite or gene, respectively. The direct evidence column contains the number of metabolites
and genes, that are associated with a metabolite marker or transcript marker. For enzymes, this is the number of enzymes encoded by a gene with
direct evidence. The number of vertices with an association to a reaction is given in the with reaction column. In the last column, this is given for
associations to metabolic pathways. (not shown)
MarVis-Graph detected a total of 133 sub-networks. The sub-networks were ranked according to size Ss, diameter Sd, and sum-of-weights Ssow
scores (Table S4). Interestingly, the different rankings show a high correlation with all pairwise correlations higher than 0.75 (Pearson correlation
coefficient) and 0.6 (Spearman rank correlation).
Allene-oxide cyclase sub-network
In all rankings, the sub-network allene-oxide cyclase (named after the reaction with the highest score in this sub-network) appeared as top candidate.
This sub-network is constituted of reactions from different pathways related to fatty acids. Figure 2 shows a visualization of the sub-network.
Jasmonic acid biosynthesis. The main part of the sub-network is formed by reactions from the “jasmonic acid biosynthesis” (PlantMetabolic Network, 2013)
resulting in jasmonic acid (jasmonate). The presence of this pathway is very well established because of its central role in mediating the plants wound response
(Reymond & Farmer, 1998; Creelman, Tierney & Mullet, 1992). Additionally, metabolites and transcripts from this pathway were expected to show prominent
expression profiles because AOS, a key enzyme in this pathway, is knocked-out in themutant plant. Jasmonic acid derivatives and hormones.
Jasmonic acid derivatives and hormones. Jasmonate is a precursor for a broad variety of plant hormones (Wasternack & Hause, 2013), e.g., the derivative (-)-
jasmonic acid methyl ester (also Methyl Jasmonic Acid; MeJA) is a volatile, airborne signal mediating wound response between plants (Farmer&Ryan, 1990).
Reactions from the jasmonoyl-amino acid conjugates biosynthesis I (PMN, 2013a) pathway connect jasmonate to different amino acids, including L-valine,
L-leucine, and L-isoleucine. Via these amino acids, this sub-network is connected to the indole-3-acetylamino acid biosynthesis (PMN, 2013b) (IAA biosynthesis).
Again, this pathway produces a well known plant hormone: Auxine (Woodward & Bartel, 2005). Even though, jasmonate and auxin are both plant hormones, their
connection in this subnetwork is of minor relevance because amino acid conjugates are often utilized as active or storage forms of signaling molecules.While
jasmonoyl-amino acid conjugates represent the active signaling form of jasmonates, IAA amino acid conjugates are the storage form of this hormone (Staswick et al.,
2005).

polyhydroxy fatty acids synthesis
Figure 2 Schema of the allene-oxide cyclase sub-network. Metabolites are shown in red, reactions in blue, and enzymes in green color. Metabolites and reactions without direct experimental evidence are marked by a dashed outline and a brighter color while enzymes without experimental evidence are hidden. The metabolic pathways described in section “Resulting sub-networks” are highlighted with different colors. The orange and green parts indicate the reaction chains required to build jasmonate and its amino acid conjugates. The coloring of pathways was done manually after export from MarVis-Graph.
The ω-3-fatty acid desaturase should catalyze a reaction from linoleate to α-linolenate. Metabolite markers that match the mass of crepenynic acid do also match α-linolenate
because both molecules have the same sum-formula and monoisotopic mass. As mentioned above, MarVis-Graph compiled the metabolic network for this study
from the AraCyc database version 10.0. On June 4th, a curator changed the database to remove theΔ12-fatty acid dehydrogenase prior to the release of AraCyc version 11.0.
The presented new software tool MarVis-Graph supports the investigation and visualization of omics data from different fields of study. The introduced algorithm for
identification of sub-networks is able to identify reaction-chains across different pathways and includes reactions that are not associated with a single pathway. The application of MarVis-Graph in the case study on A. thaliana wound response resulted in a convenient graphical representation of high-throughput data which allows the analysis of the complex dynamics in a metabolic network.


































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}