MedChemComm articles -3rd Q 2015
Larry H. Bernstein, MD, FCAP, Curator
LPBI
MedChemComm articles in July, August and September 2015.
MedChemComm medchemcomm-rsc@rsc.org
|
|
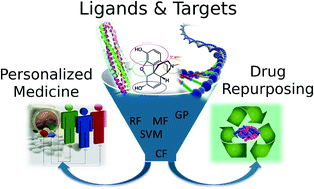
Polypharmacology modelling using proteochemometrics (PCM): recent methodological developments, applications to target families, and future prospects
Isidro Cortés-Ciriano, Qurrat Ul Ain, Vigneshwari Subramanian, Eelke B. Lenselink, Oscar Méndez-Lucio, Adriaan P. IJzerman, Gerd Wohlfahrt, Peteris Prusis, Thérèse E. Malliavin, Gerard J. P. van Westen and Andreas Bender
DOI: 10.1039/C4MD00216D, Review Article
|
|
Transition metal diamine complexes with antimicrobial activity againstStaphylococcus aureus and methicillin-resistant S. aureus (MRSA)
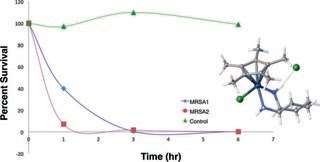
Pentaalkylcyclopentadienyl (Cp*R) iridium (Ir) and cobalt (Co) 1,2-diamine complexes were synthesized. Susceptibility of Staphylococcus aureus and recent patient methicillin-resistant S. aureus (MRSA) isolates to the transition metal–diamine complexes were measured by broth microdilution and reported as the MIC and MBC. Hemolytic activities of the transition metal-complexes as well as toxicity toward Vero cells were also measured. The transition metal complex of Cp*RIr with cis-1,2-diaminocyclohexane, had strong antibiotic activity against S. aureus and MRSA (MIC = 4 μg mL−1, MBC = 8 μg mL−1) strains and killed 99% of S. aureus cells in 6 hours. Stronger antibiotic activity was associated with the presence of octyl linked to the cyclopentadienyl group and cyclohexane as the diamine backbone. Activity was greatly diminished by tri- or tetramethylation of the nitrogen of the diamine. A cyclopentadienylcobalt complex of cis-1,2-diaminocyclohexane also showed significant anti-microbial activity against both S. aureus and MRSA strains. The absence of hemolytic activity, Vero cell cytotoxicity and the significant anti-microbial activity of several members of the family of compounds reported suggest this is an area worth further development.

Daniel Sykes, Ahmet J. Cankut, Noorshida Mohd Ali, Andrew Stephenson, Steven J. P. Spall, Simon C. Parker, Julia A. Weinstein and Michael D. Ward
Dalton Trans., 2014,43, 6414-6428
Abstract
In Ir(III)/Eu(III) and Ir(III)/Tb(III) dyads, sensitization of lanthanide luminescence occurs via both energy-transfer and electron-transfer pathways on similar timescales.
Synthesis and properties of a dendritic FRET donor–acceptor system with cationic iridium(III) complex core and carbazolyl periphery
Junpei Kuwabara, Tomomi Namekawa, Masa-aki Haga and Takaki Kanbara
Dalton Trans., 2012,41, 44-46
Abstract
Ir(III) complexes that contain benzothiazole-based tridentate ligands were synthesized and their crystal structures and luminescent properties were examined. A neutral complex had a high quantum yield (89%) and performed well as an emissive material for organic light-emitting diodes.
Multivalent glycoconjugates as vaccines and potential drug candidates
Pathogens adhere to the host cells during the first steps of infection through multivalent interactions which involve protein–glycan recognition. Multivalent interactions are also involved at different stages of immune response. Insights into these multivalent interactions generate a way to use suitable carbohydrate ligands that are attached to a basic scaffold consisting of e.g., dendrimer, polymer, nanoparticle, etc., with a suitable linker. Thus a multivalent architecture can be obtained with controllable spatial and topology parameters which can interfere with pathogen adhesion. Multivalent glycoconjugates bearing natural or unnatural carbohydrate antigen epitopes have also been used as carbohydrate based vaccines to stimulate an innate and adaptive immune response. Designing and synthesizing an efficient multivalent architecture with optimal ligand density and a suitable linker is a challenging task. This review presents a concise report on the endeavors to potentially use multi- and polyvalent glycoconjugates as vaccines as well as anti-infectious and anti-inflammatory drug candidates.
http://pubs.rsc.org/services/images/RSCpubs.ePlatform.Service.FreeContent.ImageService.svc/ImageService/image/GA?id=C4MD00143E
Polypharmacology modelling using proteochemometrics (PCM): recent methodological developments, applications to target families, and future prospects
Proteochemometric (PCM) modelling is a computational method to model the bioactivity of multiple ligands against multiple related protein targets simultaneously. Hence it has been found to be particularly useful when exploring the selectivity and promiscuity of ligands on different proteins. In this review, we will firstly provide a brief introduction to the main concepts of PCM for readers new to the field. The next part focuses on recent technical advances, including the application of support vector machines (SVMs) using different kernel functions, random forests, Gaussian processes and collaborative filtering. The subsequent section will then describe some novel practical applications of PCM in the medicinal chemistry field, including studies on GPCRs, kinases, viral proteins (e.g. from HIV) and epigenetic targets such as histone deacetylases. Finally, we will conclude by summarizing novel developments in PCM, which we expect to gain further importance in the future. These developments include adding three-dimensional protein target information, application of PCM to the prediction of binding energies, and application of the concept in the fields of pharmacogenomics and toxicogenomics. This review is an update to a related publication in 2011 and it mainly focuses on developments in the field since then.
http://pubs.rsc.org/services/images/RSCpubs.ePlatform.Service.FreeContent.ImageService.svc/ImageService/image/GA?id=C4MD00216D
Related articles
Experimental and computational studies of fatty acid distribution networks
Yong Liu, Germán Buendía-Rodríguez, Claudia Giovanna Peñuelas-Rívas, Zhiliang Tan, María Rívas-Guevara, Esvieta Tenorio-Borroto, Cristian R. Munteanu, Alejandro Pazos and Humberto González-Díaz
Mol. BioSyst., 2015,11, 2964-2977
Abstract
A new PT-LFER model is useful for predicting a distribution network in terms of specific fatty acid distribution.
Kinetics and non-exponential binding of DNA-coated colloids
W. Benjamin Rogers, Talid Sinno and John C. Crocker Soft Matter, 2013,9, 6412-6417
Abstract
Dynamic binding of DNA-grafted microspheres is found to be diffusion-limited and provides evidence of anomalous kinetics at the single-molecule scale.
Configuration of nonspherical amphiphilic particles at a fluid–fluid interface
Bum Jun Park and Daeyeon Lee Soft Matter, 2012,8, 7690-7698
Abstract
The equilibrium configuration of amphiphilic ellipsoids and dumbbells at the oil–water interface strongly depends on the particle characteristics, such as their shape and wettability.
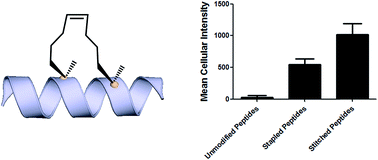
Towards understanding cell penetration by stapled peptides
Hydrocarbon-stapled α-helical peptides are a new class of targeting molecules capable of penetrating cells and engaging intracellular targets formerly considered intractable. This technology has been applied to the development of cell-permeable ligands targeting key intracellular protein–protein interactions. However, the properties governing cell penetration of hydrocarbon-stapled peptides have not yet been rigorously investigated. Herein we report our studies to systematically probe cellular uptake of stapled peptides. We developed a high-throughput epifluorescence microscopy assay to quantitatively measure stapled peptide intracellular accumulation and demonstrated that this assay yielded highly reproducible results. Using this assay, we analyzed more than 200 peptides with various sequences, staple positions and types, and found that cell penetration ability is strongly related to staple type and formal charge, whereas other physicochemical parameters do not appear to have a significant effect. We next investigated the mechanism(s) involved in stapled peptide internalization and have demonstrated that stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that involves, in part, sulfated cell surface proteoglycans, but that also seems to exploit a novel, uncharacterized pathway. Taken together, staple type and charge are the key physical properties in determining the cell penetration ability of stapled peptides, and anionic cell surface proteoglycans might serve as receptors to mediate stapled peptide internalization. These findings improve our understanding of stapled peptides as chemical probes and potential targeted therapeutics, and provide useful guidelines for the design of next-generation stapled peptides with enhanced cell permeability.
http://pubs.rsc.org/services/images/RSCpubs.ePlatform.Service.FreeContent.ImageService.svc/ImageService/image/GA?id=C4MD00131A
Introduction Hydrocarbon stapled a-helical peptides are an exciting new class of investigational agents capable of targeting and interfering with intracellular protein–protein interactions.1,2 (For reviews on hydrocarbon stapled peptides, see ref. 3 and 4, and for reviews on synthetic a-helix stabilization in general, see ref. 5 and 6.) These peptides contain a synthetic brace, referred to as a staple, introduced across one face of an a-helix (Fig. 1), that in favorable cases can increase a-helical content and protease resistance, enhance target binding affinity, promote cell membrane penetration, and suppress clearance in vivo. 7–10
Fig. 1 All-hydrocarbon stapled peptide technology. (a) Schematic illustration of peptide stapling. Two alpha-methylated, alkenyl-bearing non-natural amino acids are incorporated at two or more positions in the peptide chain and then cross-linked by ruthenium-catalyzed ringclosing olefin metathesis. (b) Different types of alkenyl-containing non-natural amino acids with distinct stereochemistry at the a-carbon and varied lengths of alkenyl side chains. (c) Three types of stapled peptides used in this study with optimized combinations of nonnatural amino acids.
Stapled peptides are synthesized via incorporation of two amethyl, a-alkenyl amino acids at defined positions in a synthetic peptide, followed by ring-closing olefin metathesis to close the helix-spanning hydrocarbon bridge (Fig. 1a).11,12 The two components of the staple, namely the hydrocarbon bridge and terminal methyl groups, are both important to obtain maximal effectiveness of the conformationally constrained peptide products. This technology has been successfully utilized to target several classes of proteins formerly considered intractable, including multi-component transcription factor complexes and protein–protein interactions having extended interfaces, such as the NOTCH transcription factor complex,13 the b-catenin–TCF interaction in the oncogenic Wnt signaling pathway,14 and the epigenetic modulator PRC2 complex.15 Given the difficulties of developing traditional small molecule drugs that can successfully target intracellular protein–protein interactions, hydrocarbon stapling technology is widely considered to represent a promising avenue of research for the development of chemical probes and potential targeted therapeutics.
Multiple types of hydrocarbon staples have been obtained by varying the relative placement of the cross-linking a,a-disubstituted amino acids, as well as the stereochemistry at the acarbon and the lengths of the alkenyl substituents (Fig. 1b).16,17 These staple types were optimized to provide robust a-helical stabilization and confer the potential for in vitro and in vivo activity. As a result of the combinatorial search process used to identify helix-stabilizing hydrocarbon staples, the diversity of the resulting macrocyclic bridges has revealed stapled peptides with different physicochemical properties. Recently, a new hyperstable version of stapled peptide with tandem crosslinks, referred to as a stitched peptide, was generated by introduction of S5 at the i position, B5 at the i + 4 position, and S8 at the i + 11 position (Fig. 1c) (Y.-W. Kim and G. L. Verdine, to be published).
Of the physicochemical properties demonstrated by peptide bearing hydrocarbon staples, the capacity to promote cellular membrane penetration is perhaps the most signicant and yet remains the most poorly understood. Independent of hydrocarbon-stapled peptides, several classes of cell penetrating peptides (CPPs) have been discovered, including naturally occuring transcription factor domains such as pennetratin18 and HIV-Tat19 and synthetic cationic peptides such as polyArginine peptides.20 Notably, despite extensive exploration during the past two decades, the mechanism(s) by which CPPs enter cells remain unclear.21–23 In contrast to CPPs, in which cell penetration appears to be sequence-dependent, numerous cell permeable stapled peptides have been discovered for peptide scaffolds with little sequence homology. These divergent observations regarding cell penetration is proposed to result from several features of stapled peptides that differentiate them from typical CPPs. For example, the introduction of an allhydrocarbon cross-link results in a constrained a-helical conformation, which embeds the hydrophilic amide backbone in the core of the folded structure. Furthermore, the hydrocarbon brace itself introduces a significantly hydrophobic patch to one face of the peptide. The exposure of the hydrophobic moiety as well as the masking of the hydrophilic peptide backbone may facilitate the interaction of stapled peptides with the hydrophobic interior of the cell membrane and thereby enhance the cellular uptake. As cell penetration is a critical property of stapled peptides, we sought to develop quantitative methods to correlate a battery of stapled peptide properties with the capacity for cellular uptake. A direct comparison with several well-known CPPs has revealed that stapled peptides, including some stapled versions of the CPPs, exhibit more robust cell penetration. Lastly, we have demonstrated that stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that involves, in part, sulfated cell surface proteoglycans. These findings significantly expand our current understanding of cell penetration by stapled peptides and provide useful information for the future rational design of cell penetrating stapled peptides with novel applications.
Results and discussion
Development of a high-throughput assay to quantitatively measure cellular uptake of peptides
Understanding the internalization process of cell penetrating peptides (CPPs), especially stapled peptides, has been a subject of great interest. The majority of previous studies have been performed by either using high-resolution microscopy to show the existence of fluorophore-labeled CPPs inside cells, or by quantitatively measuring intracellular fluorescence by flow cytometry.24,25 Although these two methods can provide important information regarding cell penetration, their respective limitations prompted us to adopt an assay that combines high-resolution imaging with reliable quantitation of intracellular accumulation to better analyze and understand the cell penetration of stapled peptides. In recent years, highthroughput cell-based imaging platforms have become increasingly popular to screen for small molecule modulators of various biological processes.26,27 Taking advantage of one of these platforms, high-content epifluorescence microscopy, we developed a high-throughput quantitative assay to measure stapled peptide intracellular access.
Proof-of-principle experiments were performed to determine whether epifluorescence microscopy could be used to quantitatively compare stapled peptide intracellular access. Human U2OS osteosarcoma cells were seeded in black, clear-bottom 384-well plates and then incubated in serum-containing media supplemented with fluorescein-labeled peptides or DMSO vehicle for 12 hours. After the treatment, cells were washed thoroughly with PBS to remove excess peptide, fixed with 4% formaldehyde, and stained with Hoechst dye to visualize nuclei. Once prepared, the plates were imaged and quantified by epi- fluorescence microscopy according to a protocol developed and discussed in detail in Experimental methods. An initial z-scan was performed using the Hoechst channel to locate the cells, and the microscope parameters were subsequently adjusted to optimize the cell size and fluorescence intensity. The parameters from this acquisition were then applied to the FITC channel, and the microscope scanned and recorded images of the FITC-labeled peptides within the z-plane of the cell. This assay was performed in a high-throughput manner, resulting in a panel of Hoechst/FITC images from individual wells (Fig. S1†). The raw image data was then analyzed using MetaXpress® software (Fig. 2a). Cells were identified based on the Hoechst stain of nuclei, with the requirement that they were a contiguous fluorescent region having a specific intensity above local background as well as having a diameter between defined minimum and maximum to be designated as “positive” cells. The cytoplasm of each cell was then identied according to the spatial location of FITC signal in relation to the nuclei as well as empiric parameters (details in Experimental methods). The FITC intensities in the cytoplasm and nuclei were then quantified separately, and the sum of these two values yielded the FITC signal for the whole cell, which can be considered the relative intracellular peptide intensity. In addition, FITC negative cells were identified on the basis of a positive Hoechst stain, which was accompanied by an absence of appreciable signal in the FITC channel.
Fig. 2 Quantitative measurement of cellular peptide intensity. (a) Hoechst channel (left) showing the location and size of nuclei, FITC channel (middle) showing the fluorescence intensity of the same cells. Information about cell size and fluorescence intensity was integrated to identify the FITC positive (green mask) and negative (red mask) cell (right). For positive cells, additional parameters allowed determination of the fluorescence intensity in the nucleus (inner intense green) and the cytoplasm (outer dim green). (b) The background fluorescence the DMSO vehicle was almost identical among different experiments. (c) Four stapled peptides from different batches of synthesis generated similar intracellular fluorescence intensity in different tests. Error bars represent the S.D. of two measurements.
We found that this system generated highly reproducible and reliable results from assay-to-assay and with different stocks of the same stapled peptides. As shown in Fig. 2b, there were negligible fluorescence differences among experiments for cells treated with DMSO vehicle, which could be used as a fluorescence background for all subsequent experiments. In addition, the same stapled peptides from different batches of synthesis and stocks featured almost identical intracellular fluorescence signals in different tests (Fig. 2c), indicating that the assay developed in this study produces repeatable and reliable results that could be directly combined and compared from a large set of experiments. Furthermore, to determine how this assay performs as a screening tool, we have calculated Z0 factor of 0.54 by using the most penetrant A6 peptide as a positive control and DMSO background as negative control, which also indicates a statistically good assay quality.
Analysis of cell penetration by stapled peptides The development of this quantitative high-throughput assay enabled a broad investigation of the physicochemical properties governing the cell uptake of a diverse set of hydrocarbonstapled peptides synthesized in our laboratory. We postulated that any correlation between cellular uptake and physicochemical properties would illuminate characteristics associated with productive cellular uptake and inform the future design of stapled peptides with improved cell penetration.28 To this end, we screened and analyzed more than 200 discrete FITC-labeled peptides belonging to three different classes: wild-type (unmodified), stapled and stitched peptides. All peptides were converted to two-dimensional structures and analyzed for theoretical physicochemical properties with the publicly available Marvin View software package from ChemAxon. Properties including the molecular weight, theoretical pI, calculated 2D polar surface area (PSA), theoretical log P and formal charge at pH 7.5 were calculated for each peptide (Table S1†). In general, the unmodified, stapled and stitched peptide libraries present in this screen had relatively similar physicochemical characteristics (Fig. S2†). The mean molecular weight and calculated PSA values were nearly identical among the three peptide classes. A notable difference was observed among theoretical log P values, which were significantly higher for the stapled and stitched peptides relative to the unmodified peptides, which is not surprising as these modified peptides contain a solvent exposed hydrocarbon crosslink. Additionally, the stapled peptide class had a mean formal charge of approximately zero while the stitched and unmodified peptide classes exhibited a positive mean charge. Overall, the calculated physicochemical properties indicated that the peptide classes were quite similar in terms of their mean properties, which is useful when making comparisons among their cell penetration properties.
We next performed an intracellular access screen by treating U2OS cells with 1 mM of FITC-labeled peptide for 12 hours in duplicate. All assays contained control DMSO wells and positive control peptides, which were compared among assays to ensure plate-to-plate reproducibility (Fig. 2b and c). The primary readout of the screen was mean cellular fluorescence intensity. As the DMSO background was highly consistent between wells and experiments, a mean background value was subtracted from all data. The results of the screen were used to generate plots comparing cell penetration with peptide physicochemical parameters. Interestingly, as a class, stapled and stitched peptides exhibited significantly higher cell penetration compared with wild-type unmodified peptides, which contained several established cell penetrating peptides (CPPs; Fig. 3a). Given that all three peptide classes have similar physicochemical properties in general, the benefit in cell penetration can be largely attributed to the synthetic stabilization of the a-helical peptides with all-hydrocarbon peptide stapling technology. Furthermore, we found that peptide charge near physiologic pH exhibited a strong correlation with intracellular access and could be fitted into a Gaussian distribution with a population centroid at a formal charge of +4 (Fig. 3b). In particular, peptides exhibiting a net negative charge (7 to 1) exhibited little cellular uptake, whereas peptides of approximately neutral charge (1 to +1) displayed moderate cell penetration above background. Interestingly, peptides with a net positive charge (+1 to +7) showed significantly higher cell penetration as a group. Cellular uptake did not appear to increase linearly with charge, as the cell penetration decreases dramatically for the peptides in this study with charge greater than +7. The same trend between formal charge and cellular uptake were observed for individual stapled and stitched peptide classes as well (Fig. S3†). This observation is not consistent with previously reported models that indicate that peptides/mini-proteins with more positive charge have better penetration properties due to tighter electrostatic interactions with the negatively charged phospholipid membrane.29,30 The lower penetration for highly charged peptides in this study could result from any one of many factors including, for example, peptide aggregation in solution, the disruption of peptide packing during internalization or difficulty in dissociation from cell membrane. Additional tests with a larger number of peptides could further our understanding of this phenomenon. In addition, there was no discernible correlation between cell penetration and peptide molecular weight, log P, pI value or PSA (Fig. S4†). Taken together, these data demonstrated that the staple type and peptide charge are key physical properties correlated with peptide cell penetration ability, whereas the other parameters do not appear to be significantly associated.
In order to further investigate the cell penetration properties for stapled peptides and to systematically analyze the similarities and differences in cellular uptake between stapled peptides and other wild-type cell penetrating peptides, we compared cell penetration of several stapled peptides to that of three well known wild-type CPPs: Tat (48–60), penetratin (Antennapedia 43–58) and poly-Arg8 (Table S2†). First, we investigated the cellular uptake at varied peptide concentrations. As shown in Fig. 4a and b, both wild-type CPPs and stapled peptides showed dose-dependent increases in cell penetration. Strong intracellular fluorescence was detected in the low micromolar range, and although the levels of accumulation were different for distinct peptides, stapled peptides featured more robust dosedependent cell penetration at lower concentrations relative to wild-type CPPs, in general. It is also interesting to note that while significant increases in intracellular fluorescence were mostly evident in the 1–10 mM range for stapled peptides, distinct profiles were observed for specific peptides. For example, TNG147 showed little cell penetration at 1 mM but showed a dramatic increase at 5 mM, which might suggest that concentration-dependent peptide packing or a receptor-mediated mechanism may facilitate the cell penetration process, and these processes may be triggered at different concentrations for distinct peptides. Furthermore, it is worth noting that the stapled peptides studied here were more cell permeable than wild-type CPPs at most concentrations tested, exhibiting nearly an order of magnitude higher intracellular fluorescence at the same treatment concentrations.
Fig. 4 Effects of peptide concentration and incubation time on cellular uptake of stapled and wild-type peptides. (a) Wild-type and (b) stapled peptides showed a dose-dependent increase in cell internalization. Cellular uptake for (c) penetratin and (d) SAHM1 peptides over time at concentrations of 5 and 10 mM. (e) A pulse-chase penetration assay for SAHM1 peptide in which fresh medium containing either a new batch of peptide or DMSO vehicle were exchanged at 12 hours after initial treatment. Error bars represent the S.D. of triplicate samples.
We next performed a time-course penetration assay to better understand the kinetics of peptide internalization using a representative CPP and stapled peptide. Penetratin and SAHM1 showed distinct kinetics of uptake and stabilization throughout a 24 hour time course. 5 mM and 10 mM penetratin peptide exhibited similar intracellular cellular fluorescence after 2 hours, which then decreased until approximately 8 hours and finally stabilized at different intracellular levels until 24 hours (Fig. 4c). On the other hand, the stapled peptide SAHM1 showed time- and dose-dependent cellular uptake, which stabilized after approximately 8 hours (Fig. 4d). Compared to the wild-type penetratin, the SAHM1 profile was unique in that dose-dependent accumulation was evident at all time points and no loss of signal was observed. One explanation for the loss of signal observed with penetratin could be attributed to an equilibrium between cell penetration and subsequent intracellular proteolysis followed by export of the fluorophore. The presence of the all-hydrocarbon crosslink in its peptide sequence and lower net charge of SAHM1 relative to penetratin, could contribute to enhanced cellular uptake and reduced intracellular proteolysis, leading to continuous accumulation in cells. To further explore the equilibrium observed for stapled peptides, we performed a pulse-chase experiment using SAHM1. After 12 hours of incubation with SAHM1, cell culture medium was aspirated and the cells were extensively washed with PBS to completely remove excess peptide. Then fresh medium containing either a new batch of 1 mM peptide or DMSO vehicle was added to cells and incubated for the indicated time points (Fig. 4e). As expected, the cellular uptake increased for the first 12 hours incubation. After medium exchange, cells incubated with fresh medium containing DMSO vehicle retained the intracellular fluorescence intensity. Interestingly, the signal for cells treated with a new batch of staple peptide continued to increase up to 24 hours (Fig. 4e). This observation indicates that despite incubation over a time course previously shown to reach equilibrium, the mechanism(s) responsible for cellular uptake are not saturated, as evidenced by further uptake upon replacement with fresh stapled peptide. Taken together, these data indicate that the mechanism(s) underlying cellular uptake by both CPPs and stapled peptides exhibit time- and dose-dependency that is not saturable at early time points or low micromolar doses and, importantly, appears to be more robustly utilized by stapled peptides.15,31
Given that stapled peptides exhibit better cell penetration properties in general than parent unmodified peptides, we wondered whether the peptide stapling strategy could be applied generally to improve cellular uptake of parent unmodified peptides. To test this hypothesis, we designed a panel of stapled peptides based on Tat (48–60), penetratin and poly-Arg8 (Fig. 5a). These stapled peptides and their parent unmodified peptides were incubated in U2OS cells for 12 hours with a concentration range from 10 nM to 20 mM, mirroring the dosedependent uptake studies shown in Fig. 4. As expected, all peptides showed dose-dependent cell penetration (Fig. 5b–d). Interestingly, stapled peptides derived from penetratin and poly-Arg8 showed improved cell permeability at concentrations starting from 1 mM for stapled penetratin and 5 mM for stapled poly-Arg8. It is noteworthy that the staple position also affected the cellular uptake as the two stapled penetratin peptides with different crosslink positions exhibited varied cell penetration, though both were superior to wild-type penetratin. In contrast, reduced cellular uptake was observed for both stapled peptide variants derived from the Tat sequence (Fig. 5b). This could result from several possible effects, including disruption of peptide secondary structure, masking of residues essential for surface recognition or altering peptide packing interactions involved in cell penetration. Further focused study of these variants is warranted to elucidate the source of altered cellular uptake, however these data clearly demonstrate that peptide stapling may be a general method to further improve the cell permeability of CPPs, which could serve as more efficient transduction domains for molecular cargoes. In addition, while increasing the helical content of stabilized peptides has been stated to be a guiding principle in the successful design of biologically active stapled peptides, it has not been shown to be generally correlated with cell penetration. To specifically address whether increasing the helical content of a peptide is correlated with augmented cell penetration, we have measured the relative helicity of hydrocarbon stapled variants of Tat, penetratin and poly-Arg8 (Fig. S5†). Notably, we did not observe a general correlation between increased helical character and cell penetration of these peptides. Peptide stapling increased the helical content of both Tat and poly-Arg8 peptide sequences, which were largely unstructured when unmodified. In contrast, the unmodified penetratin peptide had signicant helical content (>50%), and the hydrocarbon stapled variants of this sequence largely retained their helicity, albeit lower overall helicity. Intriguingly, these species demonstrated the differing effect of hydrocarbon stapling and increased helical content on cell penetration since introduction of the hydrocarbon staple increased the cellular uptake of both penetratin and poly-Arg8 sequences, while it decreased uptake for Tat peptides. Therefore, we cannot conclusively state, a priori, that the incorporation of a hydrocarbon staple or increased a-helicity will lead to more productive cellular penetration, although in general stapling can increase the uptake of specific sequences (Fig. 5) and as a class stapled and stitched peptides are more cell penetrant (Fig. 3a). A more comprehensive follow-up study with CD analyses on a larger peptide library is needed to better address this question.
Fig. 5 Effects of all-hydrocarbon staples on cell penetration by wild-type cell penetrating peptides. (a) List of wild-type cell penetrating peptides and their stapled derivatives investigated in this study. (b–d) Dose-dependent cell penetration assays showed that stapling strategy greatly improves the cellular uptake of penetratin and poly-Arg8 peptides. Experiments were performed in triplicate, and error bars represent S.D. of three measurements.
Mechanistic studies of cell penetration by stapled peptides The aforementioned studies indicate that stapled peptides exhibit better cellular uptake properties than wild-type peptides in general, and that internalization correlated primarily with hydrocarbon staple type and formal peptide charge. However, the mechanism(s) utilized by peptides to translocate across the cell membrane are still unclear. Therefore, we sought to investigate the uptake mechanism(s) for stapled peptides. The uptake mechanism(s) of wild-type CPPs have been extensively studied. Some evidence indicates that they enter cells via energy-dependent endocytosis, which is an active transport process, however data suggesting passive diffusion for CPPs have also been reported; hence, the mechanism(s) of cell uptake by CPPs remains ambiguous.32–34 We first sought to determine whether cell penetration by stapled peptides and wild-type CPPs occurs via ATP-dependent endocytosis.2 Cells were pre-treated with NaN3 and 2-deoxyglucose (2-DG) to reduce cellular ATP levels, and then incubated with FITC-labeled peptides (wildtype and stapled) for 4 hours and compared to normal cells for intracellular fluorescence. Cellular ATP levels were confirmed to be decreased by approximately 90% after NaN3 and 2-DG treatment (Fig. S6†), but Tat and poly-Arg8 exhibited almost identical cellular uptake in ATP-depleted and normal cells, supporting the model that they utilize passive diffusion to translocate across the cell membrane. However, penetratin and all stapled peptides showed 20–50% lower accumulation in ATP-depleted cells, indicating an active trans-membrane process requiring cellular ATP (Fig. 6a). These data indicate that there may be more than one uptake mechanism for CPPs and stapled peptides, but that for the most robust cell penetrating peptides (penetratin and stapled peptides studied here), the internalization mechanism(s) involves ATP-dependent endocytosis.
Fig. 6 Mechanistic study of cell penetration by stapled peptides and wild-type cell penetrating peptides. (a) Cellular uptake in normal and ATP-depleted cells indicated that stapled peptides penetrate cells via an ATP-dependent endocytosis. (b) Impaired uptake was observed in NaClO3 treated cells, which inhibit proteoglycan biosynthesis. (c) Cell penetration of wild-type and stapled peptides in wild-type CHO and proteoglycan-deficient CHO cells. Experiments were performed in triplicate, and error bars represent S.D. of three measurements.*P < 0.05, **P < 0.01, ***P < 0.001.
Next, we sought to identify the specific pathway(s) utilized for cellular uptake, since energy-dependent endocytosis can be accomplished by several different pathways including caveolinand clathrin-mediated endocytosis. We repeated the cell penetration experiments under a variety of conditions that each blocked a different endocytosis pathway (Table S3†).35–37 We found that uptake was partially blocked in cells treated with sodium chlorate (Fig. 6b), which aborts the decoration of cells with sulfated proteoglycans, but was unaffected by inhibitors of other endocytic pathways (Fig. S7†). It thus appears that interaction with sulfated proteoglycans is responsible for some, but not all, endocytic uptake of stapled peptides and wild-type CPPs. It is reasonable to connect this result with the previous discovery that peptide charge is a key factor determining cell penetration. Proteoglycans are negatively charged under physiologic conditions due to the occurrence of sulfate groups, and these might form electrostatic pairs with positively charged peptides to facilitate anchoring on the cell membrane.38–40 To further confirm that sulfated proteoglycans are important to mediate cellular uptake for peptides, we performed a secondary assay using wild-type CHO cells (CHO-K1) and proteoglycan deficient CHO cells (pgsA-745) which harbor a defect in xylosyltransferase, thereby preventing glycosaminoglycan biosynthesis. All peptides showed similar penetration properties in wild-type CHO cells, but uptake was decreased by approximately 50% in proteoglycan-deficient CHO cells, consistent with the experiment using a small molecule inhibitor (Fig. 6c). Taken together, our data suggest that CPPs and stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that is in part mediated by interaction with anionic cell surface proteoglycans. This result is very similar to the previous reports on the mechanism of cellular uptake for supercharged GFP (scGFP), which likewise does not utilize clathrin- or caveolin-mediated endocytosis.41 Notably, scGFP internalization requires actin polymerization, which may not be required for peptide penetration (Fig. S7c†) types, and distinct physicochemical properties. As a result, we found that stapled peptides penetrate cells more efficiently than unmodified peptides, including well-characterized cell penetrating peptides. For the panel of peptides used in this study, only staple type and formal charge were significantly correlated with cell penetration potential, whereas the other physical parameters did not appear to have a signicant effect. We further studied the relationships between cellular uptake and
In conclusion, we sought to investigate the cell penetration properties of stapled peptides, which is one of the most significant yet poorly understood aspects of peptide stapling technology and cellular transduction technologies in general. In order to address this problem, we developed a high-throughput assay to quantitatively measure stapled peptide intracellular accumulation. Using this assay, we analyzed more than 200 discrete peptides with various sequences, staple positions and peptide concentration or incubation time, revealing that stapled peptides accumulate in cells in a dose-dependent fashion and reach steady intracellular levels over a course of a few hours. These studies revealed similar time- and dosedependent behavior for CPPs and stapled peptides, but stapled peptides, including stapled versions of CPPs, were shown to be 10- to 20-fold more penetrant, measured by intracellular fluorescence level at a given dose, than the most potent CPP. We also propose that the specific intracellular accumulation and stabilization kinetics of stapled peptides or unmodified CPPs may be a consequence of equilibria between peptide penetration, cellular proteolysis and/or retrograde transport of the species. Finally, we investigated the mechanism(s) involved in the internalization of stapled peptide and unmodified CPPs and demonstrated that cell penetration occurs through a clathrinand caveolin-independent, energy-dependent endocytosis pathway that utilizes, in part, sulfated cell surface proteoglycans. This dataset provides significant insight into the physicochemical properties correlated with productive cellular penetration as well as a more detailed understanding of the mechanism(s) utilized by stapled peptides to access intracellular compartments, which together should aid in the design of and characterization of novel stapled peptides in the future.
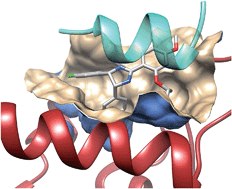
Rational design of protein–protein interaction inhibitors
Protein–protein interactions are at the heart of most physiopathological processes. As such, they have attracted considerable attention for designing drugs of the future. Although initially considered as high-value but difficult to identify, low molecular weight compounds able to selectively and potently modulate protein–protein interactions have recently reached clinical trials. Along with high-throughput screening of compound libraries, combining structural and computational approaches has boosted this formerly minor area of research into a currently tremendously active field. This review highlights the very recent developments in the rational design of protein–protein interaction inhibitors.
Tania Gamberi, Francesca Magherini, Tania Fiaschi, Ida Landini, Lara Massai, Elisa Valocchia, Laura Bianchi, Luca Bini, Chiara Gabbiani, Stefania Nobili, Enrico Mini, Luigi Messori and Alessandra Modesti
Mol. BioSyst., 2015,11, 1653-1667
Abstract
The cytotoxic mechanisms of the organogold(III) Aubipyc.
Interactions of arene ruthenium metallaprisms with human proteins
Lydia E. H. Paul, Bruno Therrien and Julien Furrer
Org. Biomol. Chem., 2015,13, 946-953
Abstract
Interactions between three hexacationic arene ruthenium metallaprisms and human proteins have been studied using NMR spectroscopy, mass spectrometry and circular dichroism spectroscopy, showing that proteins are potential biological targets for these metallaprisms.
Didier Rognan heads the Laboratory of Structural Chemogenomics at the Faculty of Pharmacy of Strasbourg (France). He studied Pharmacy at the University of Rennes (France) and did a Ph.D. in Medicinal Chemistry in Strasbourg (France) under the supervision of Prof. C.G. Wermuth. Aer a postdoctoral fellowship at the University of Tubingen (Ger- ¨ many), he moved as an Assistant Professor to the Swiss Federal Institute of Technology (ETH) until October 2000. He was then appointed Research Director at the CNRS to build a new group in Strasbourg. He is mainly interested in all aspects (method development and applications) of structurebased drug design, notably on G protein-coupled receptor ligands and protein–protein interaction inhibitors.
Introduction Drug discovery is a long, costly, multi-step endeavour which aims at reducing all possible risks to deliver a novel therapeutic solution to previously unmet clinical needs. To reduce chemical risks, empirical rules are used to filter the chemical space and retain drug-like low molecular weight compounds. Reduction of the biological risk is addressed by considering privileged target families (e.g., G protein-coupled receptors and kinases) whose activation/inhibition by drug-like compounds is likely to correct or reverse pathological states. Until recently, mostly single macromolecules (proteins and nucleic acids) have been considered as potential drug targets. Out of 68 000 proteins currently annotated in UniProt for the human proteome,1 only about 300 targets2 have been addressed by current drugs, and the large majority of single targets is still awaiting first-in class drugs.
Besides single targets, large scale genomics and proteomics3 have identified complex networks of targets and pathways regulating physiopathological processes in a coordinated manner. The current human protein–protein interactome has been estimated between 130 000 (ref. 4) and 650 000 (ref. 5) complexes, out of which only a tiny amount is known, and only a very few6–8 have been the object of a drug discovery initiative. Protein–protein interactions (PPIs) therefore describe a totally new biological space that attracts more and more attention, with 26PPI inhibitors9,10 already under clinical development, notably in the oncology field.11 Despite PPIs may adopt quite different sizes, shapes and electrostatics,12 identifying highaffinity PPI inhibitors is a considerable challenge for many reasons: (i) in contrast to conventional targets, a medicinal chemist cannot start inhibitor design from the structure of endogenous ligands, (i) PPIs often involve flat surfaces delocalized over multiple epitopes, usually lack well-defined buried cavities13 typical of conventional targets, and are significantly larger (ca. 1000–3000 A˚2 ) than enzyme/receptor pockets (300– 1000 A˚2 ), (iii) high-throughput screening of traditional compound libraries often returns no viable hits14 for the main reason that PPI inhibitor chemical space is quite different from that described by traditional drug-like compounds.10 Nonetheless, thanks to bioinformatics and proteomics-guided prioritization of therapeutically relevant protein–protein complexes, more and more PPI inhibitors are currently reported. Several excellent reviews6,7,9,11,15–18 have already been published on experimental methods (high throughput screening, biochemical and cellular assays, and fragment-based approaches) suitable to discover PPI inhibitors. The present report will only cover computer-aided approaches, with a major emphasis on structure-based methods and recent discoveries (2012–2014).
Databases Preliminary access to experimentally validated data is key to launch a drug discovery program on PPI modulators. A multitude of databases storing genomics, proteomics and structural data are currently available to help the medicinal chemist. We will here briefly review these archives, focusing mostly on easily interpretable structural data.
PPI databases Many experimental methods with different throughputs (from low to high) have been developed to characterize binary interactomes in various species, among which the most prominent has been the yeast two-hybrid (Y2H) assays, and mass-spectrometry (MS) coupled with co-immunoprecipitation or coaffinity purification.19 These experimental data are stored in many primary databases (Table 1) that are difficult to mine due to their large heterogeneity. Metadatabases have been derived thereof to facilitate their analysis, among which the most popular are APID and PRIMOS (Table 1). These metadatabases cover a wide range of organisms and notably offer the possibility to mine experimental PPI data according to disease relevance or inter-organism crosstalk, and provide graphic tools to visualize complex networks of interacting proteins and identifying important protein nodes (hubs). It is however very difficult, from this large amount of data, to clearly prioritize PPIs for a drug discovery program. Attempts to classify the PPIs by structural druggability25 (although ligandability26 is probably a better term) are worth mentioning but should be taken with care due to the still insufficient number of existing PPI three-dimensional (3D) structures.
Table 1 Protein–protein interaction databases
Database Interactions Website References
BIND 32 211 http://bond.unleashedinformatics.com 20
DIP 78 191 http://dip.doe-mbi.ucla.edu/dip/Main.cgi 21
HPRD 41 327 http://www.hprd.org/ 22
IntAct 448 986 http://www.ebi.ac.uk/intact/ 17
MIPS 9 835 http://mips.helmholtz-muenchen.de/proj/ppi/ 23
APID 196 700 http://bioinfow.dep.usal.es/apid/index.htm 24
PRIMOS 384 127 http://primos.fh-hagenberg.at/ 19
Table 2 Database of low molecular-weight PPI inhibitors
Database Ligands Website References
2P2I 71 http://2p2idb.cnrs-mrs.fr/ 12
iPPI-DB 1650 http://www.ippidb.cdithem.fr/ 10
TIMBAL 6896 http://mordred.bioc.cam.ac.uk/ timbal 29
Ligand databases Initially limited to a limited subset of inhibitors able to disrupt few PPIs (e.g. p53/MDM2, Bcl-Xl/Bak, and IL-2/IL-2Ra),7,27 the repertoire of PPI inhibitors rises constantly thanks to exciting developments in biophysical fragment screening.15,28 Three publicly available databases storing information on PPIs and their inhibitors (Table 2) may be used to better describe the structural properties of druggable PPIs and the chemical space associated with their disruptors.
The 2P2Idb database12 is a hand-curated repository of protein–protein complexes of known X-ray structures (X-ray diffraction and nuclear magnetic resonance spectroscopy) for which at least one low molecular weight orthosteric inhibitor has been co-crystallized with one of the two protein partners. It currently describes 71 inhibitors for 14 PPIs, clustered in two groups (Fig. 1) with respect to the nature of the interface (protein–peptide and protein–protein). Companion tools (2P2I inspector,30 2P2I score,30 and 2P2I hunter31) are provided to analyse PPIs at a structural level, predict their structural druggability and design PPI focussed libraries, respectively.
Fig. 1 Prototypical examples of class I (left panel) and class II PPIs (right panel), exemplified by the Bcl-Xl/Bak (PDB id 1BXL) and integrase/LEDGF (PDB id 2B4J) complexes, respectively. Class I PPIs involve the interaction of a globular protein with a peptide or a single secondary structure (a-helix and b-strand) of a second protein partner. Class II PPIs are characterized by the interaction of two globular proteins.
The iPPI-DB10 is another manually curated database from world patents and the medicinal chemistry literature, focussing on low molecular weight orthosteric inhibitors, disease-related protein–protein interfaces and a clear biochemical readout (e.g. fluorescence polarisation and enzyme-linked immunosorbent assay). The database archives 1650 PPI inhibitors targeting 13 families of homologous PPI targets mainly involved in cancer, immune disorders and infectious diseases.
Finally, the TIMBAL database29 reports ca. 7000 inhibitors for 50 known PPIs. In contrast to the two other databases, TIMBAL is maintained through a predefined list of PPIs and automated searches in ChEMBL32 and the Protein Data Bank.33 In contrast to the other databases, TIMBAL also registers short peptides with an upper molecular weight limit of 1200 Da. It should be pointed that most of the 15 000 uncurated biological data present in TIMBAL arise from a single target family (integrins) and should be considered with care.
Analysing the content of these databases enables a first comparison of PPI inhibitors versus drugs, as well as PPIs amenable to disruption versus standard heterodimers. PPI surfaces disrupted by inhibitors tend to be smaller, more hydrophobic and accessible than standard heterodimers.12 As a consequence, low molecular weight PPI inhibitors tend to be larger, more hydrophobic and more aromatic-rich than standard drugs. Interestingly, many of them (ca. 60%) still comply with Lipinski’s rule-of-five, 10 revealing some hopes in the developability of such compounds.
However, it should be stated that the set of empirical rules designed to discriminate druggable from non-druggable PPIs, as well as to distinguish PPI inhibitors from conventional druglike compounds still rely on a very limited set of highly homologous data (PPIs, inhibitors), and should therefore be regarded with caution. Increasing coverage of the PPI repertoire by future experimental screens will undoubtedly lead to a better denition of PPI biological and chemical spaces. We therefore expect in the future the above-mentioned rules to be rened and be more descriptive of the true world of PPI inhibitors, notably with respect to rational design of PPI focussed libraries.
Rational design of PPI modulators
Sequence-based approaches Whatever the nature of the PPI (type I or type II, see the definition above), PPI interfaces are often characterized by the presence of hotspots,34 in other words anchor residues that contribute the most to the binding free energy of the protein– protein complex. The interaction of a single modified amino acid with a single anchor residue might be sufficient to disrupt a PPI as elegantly demonstrated by Lin et al. in a recent study.35 Capitalizing on the presence of a reactive cysteine (C246) at the interface of the complex between caspase-7 (CASP7) and the Xlinked inhibitor of apoptosis protein (XIAP), they designed the N-iodoacetyl-lysine amino acid derivative 1 (Fig. 2) that covalently traps C246 and further disrupts the XIAP–CASP7 complex, therefore triggering CASP7-dependent apoptosis and killing MCF-7 breast cancer cells (EC50 ¼ 0.64 mM) previously resistant to chemotherapy.
The easiest way to inhibit a PPI is to start with the amino acid sequence of one interacting epitope, notably if the latter is part of regular secondary structures (a-helix, b-strand, and b-turn). For example, a-helical peptides mimicking the sequence of protein transmembrane domains may disrupt PPIs quite efficiently.36,37
Fig. 2 Peptidomimetics as PPI disruptors
Due to poor pharmacokinetic profiles, linear peptides are good in vitro tools but usually not efficient clinical candidates. Chemical modifications are required to stabilize their secondary structures in physiological media and prevent early degradation. Among the most exciting developments in this area38,39 is the design of stapled peptides.40,41 Stapled peptides are synthetic analogues of a-helical protein epitopes involved in a PPI, and in which a covalent hydrocarbon linkage connects adjacent turns of the helix. Stapling is known to significantly increase the in vivo half-life of the natural peptide (increasing proteolytic stability), decrease the entropic cost of binding, and even enable cellular uptake.42 Many stapled peptides with potent in vivo activities have already been reported.39 One of these stapled peptides (ATSP-7041, compound 2, Fig. 2) just entered clinical development as a dual nM MDM2/MDMX inhibitor for p53-dependent cancer therapy.43
Heterocyclic scaffolds mimicking secondary structures can also be obtained by solution-phase synthesis to afford peptidomimetic libraries amenable to PPI inhibition. Whitby et al. notably reported the design of 8000 member 4-acetamido-3- alkoxy-benzamide focused library featuring weak p53/MDM2 inhibitors and potent HIV-1/gp41 inhibition (compound 3, Fig. 2).44 When the peptide epitope is not structured, developing macrocyclic analogues is more difficult but still feasible as recently demonstrated by Glas et al.38 who successfully improved 14-3-3 binding of a 11-mer peptide from a bacterial ExoS virulence factor by cross-linking binding amino acids with polymethylene linkers, up to an in vitro 40 nM disruptor of the ExoS/14-3-3 interaction (compound 4, Fig. 2). Interestingly, the cross-linker was not only chosen to rigidify the natural ExoS peptide structure but also to directly provide additional hydrophobic interactions to the 14-3-3 binding site.38 Only in exceptional cases the natural unmodified peptide is directly usable as a PPI inhibitor. One recent example is the 28 amino acid cell-penetrating peptide (p28) from a bacterial azurin redox protein, that binds to the DNA-binding domain of the p53 tumor suppressor and inhibits p53 degradation by interfering with the Cop1-mediated ubiquitination,45 thereby enhancing p53 levels in cancer cells and exhibiting antitumoral efficacy in patients with advanced solid tumors.46
Pharmacophore-based approaches As defined by the IUPAC,47 a pharmacophore is “an ensemble of steric and electronic features that are necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response.” Although pharmacophores are mainly used to align and compare ligands sharing the same target,48 the same concept can be easily transferred to PPIs in which one partner is the “receptor” and the second one the “ligand”. Pharmacophore features (hydrophobic, aromatic, H-bond donor and H-bond acceptor, positively and negatively ionisable) can therefore be manually or automatically mapped to atoms of the ligand in direct interactions with the receptor. The resulting pharmacophore can then be used to identify a compound library for hits fulfilling the defined query. Several tools (e.g. LigandScout,49 Discovery Studio,50 and Pocket Query51) can be directly used to map PPI pharmacophores onto protein–protein X-ray structures (Fig. 3).
Fig. 3 Example of a PPI pharmacophore mapped onto interacting atoms of human LEDGF (yellow ribbons) bound to HIV-1 integrase (red ribbons, PDB ID 2B4J). The PPI pharmacophore is composed of 2 Hbond donors (magenta balls), two H-bond acceptors (green balls), one hydrophobic feature (cyan ball) and 6 exclusion volumes (gray balls).
Using a manual PPI pharmacophore defined from the X-ray structure of the Annexin A2/S100A10 complex, a pro-angiogenic complex, Reddy et al.52 derived a simple pharmacophore (2 hydrophobes, 2 H-bond donors, and 2 H-bond acceptors) using the Unity program,53 and screened a library of 700 000 compounds to select 586 hits which were further docked to the Annexin A2 binding site to retain only 190 candidates with both a good docking and pharmacophore fitness score (Table 3). Out of 190 tested compounds, 7 hits blocked the interaction between S100A10 and the Annexin A2 N-terminus in a competitive fluorescent binding assay, with the most potent PPI inhibitor (compound 5, Fig. 4) exhibiting an IC50 of 24 mM.52 Geppert et al.54 reported the rational discovery of a low molecular weight inhibitor of the complex between interferon-a (IFN-a) and its receptor (IFNAR2). Fortunately, the PPI interface was small enough (ca. 800 A˚2 ) to be targeted by a small heterocyclic compound. After identifying major hotspots at the IFN-a surface, a fuzzy receptor-based pharmacophore was determined using the VirtualLigand approach,55 which assigns pharmacophoric features to Gaussian densities. Screening a collection of 556 000 commercially available compounds retained six virtual hits, out of which two were weak IFN-a inhibitors, but one (compound 6, Fig. 4) was confirmed by NMR and surface plasmon resonance (SPR) to bind to IFN-a with a dissociation constant (Kd) of 4 mM and to inhibit IFN-a responses in various cell assays. The novel inhibitor may be useful to reduce IFN-a titers in autoimmune disorders.
Table 3 Protein–protein pharmacophore searches to identify PPI inhibitors
Target Library size Tested Hits Ref.
Annexin A2/S100A 10 700 000 190 7 52
INFAR2/IFN-a 556 000 6 3 54
p53/MDM2 21 287 15 6 56
Nrf2/Keap1 21 199 17 1 57
PKC3/RACK2 330 000 19 1 58
Due to the inherent complexity of PPI pharmacophores (many features covering a large surface), combining several pharmacophores into a consensus model may help to retrieve essential features and simplify pharmacophore queries. Xue et al. applied this approach to the identification of p53–MDM2 inhibitors.56 The p53–MDM2 complex has become a prototypical PPI for its biological background (this interaction plays an important role in regulating the transcriptional activity of tumour cells) and many high affinity low molecular-weight inhibitors of this PPI identified by various screening approaches.59 Starting from a set of 15 MDM2-peptide X-ray structures, a common feature structure-based pharmacophore (2 H-bond donors, one H-bond acceptor, 2 aromatic rings, and one hydrophobe) was first identified. In addition, a receptorligand pharmacophore (five hydrophobes, one aromatic, and one H-bond donor) was generated from a separate set of 10 MDM2-non peptide complexes. Merging both pharmacophores and retaining the most common features led to an ensemble pharmacophore definition (two aromatic rings, two hydrophobes, and one H-bond donor) taking into account both peptide and non-peptide binding. This pharmacophore was used to screen a collection of 21 287 commercially available compounds, and led to a hit list of 15 compounds out of which 6 were confirmed as p53–MDM2 inhibitors using an in vitro uorescence polarization assay.56 The most potent inhibitor (compound 7, Fig. 4) is a 180 nM MDM2 inhibitor. Despite a good selectivity in a MTT tumour cell proliferation assay (p53+/+ vs. p53/ cells), compound 7 was a weak inhibitor (IC50 ¼ 85 mM) of tumour cell growth, because of poor pharmacokinetic properties.
Fig. 4 PPI inhibitors identified by pharmacophore-based virtual screening.
Along the same lines, two X-ray structures were used to derive inhibitors of the PPI between Keap1 and Nrf2, a complex involved in the response to oxidative stress.57 The two PPI pharmacophores were merged into a single query consisting of one H-bond donor, two H-bond acceptors and three negative ionisable centers. To afford some fuzziness in the search, up to two features were allowed to be missed by virtual hits. Since the Keap1-binding epitope of Nrf2 is composed of several acidic residues, only compounds bearing a negative charge were searched among a full commercial library of 251 774 compounds. The remaining 21 199 hit list was matched to the pharmacophore, and led after confirmation with docking and MM-PBSA scoring, to a list of 17 potential hits which were tested for Keap1–Nrf2 inhibition using an in vitro fluorescence polarization assay. A single compound (compound 9, Fig. 3) was confirmed in vitro as a moderately potent Keap1–Nrf2 inhibitor with an EC50 of 9.8 mM.57 Interestingly, the inhibitor activated the Nrf2 transcriptional activity .
When both protein partners involved in the PPI have not been co-crystallized, it is still possible to rationally discover PPI inhibitors, starting from the sole X-ray structure of one of the two proteins. This approach was followed by Rechfeld et al. in the discovery of PKC3–RACK2 inhibitors.58 Starting from the Xray structure of the PKC3 octameric epitope binding to RACK2 (a receptor for activated protein kinase C), a simple peptide-based pharmacophore model (3 H-bond donor/acceptor, one hydrophobe) was defined and used to screen a collection of 330 000 compounds. Out of 19 virtual hits, a thienoquinoline was found to disrupt the PPI in vitro and served as a query for a secondary screen for chemically similar analogues, which led to compound 8 (Fig. 4) as a micromolar potent PKC3-RACK2 inhibitor (IC50 ¼ 5.9 mM) which also inhibited PKC3 downstream signalling, HeLa cancer cell migration and invasion.58
Finally, pharmacophore searches may be used to prioritize privileged scaffolds for synthesizing PPI-focused libraries. For example, Fry et al. reported a rational approach to PPI library design targeting a-helical binding epitopes.60 Starting from the known X-ray structure of an a-helical p53 epitope binding to MDM2, a three point pharmacophore, featuring the three important hydrophobic side chains (Phe19, Trp23, and Leu26) of the p53 peptide, was designed and used to find heterocyclic scaffolds among the CSD database61 of small molecule X-ray structures. Several small-sized libraries (ca. 100 members) were synthesized from each hit and tested for general inhibition of PPIs involving an a-helical epitope (e.g. MDM2, BCL2, BCL-XL, and MCL1). Although no potent hit could be discovered, the average hit rate was far superior (4%) to what should be expected from a random screen. Moreover, many starting hits exhibited good ligand efficiencies,60 and are therefore interesting starting points for hit leading optimization.
Despite its apparent simplicity, PPI-based pharmacophore search is a fast, cost-effective and simple in silico approach to discover the very first inhibitors of a particular PPI. Of course, all successful examples mentioned above imply that the PPI is of manageable size and does not involve a too large and complex binding epitope. Beside the existence of a X-ray or NMR structure of the protein–protein (peptide) complex, it is therefore equally important to properly select PPIs amenable to pharmacophore-based searches, notably with respect to the complexity of the query (5–6 features) and its hydrophobic/ hydrophilic balance.
Docking-based approaches At the first sight, protein–ligand docking should be considered as the most intuitive and logical computational tool to predict likely ligands of any target of known 3D structures.62 Unfortunately, severe drawbacks associated with the scoring of protein– ligand interactions render that tool usually suitable for positioning a ligand into a binding site, but rarely to predict binding free energies or to precisely rank ligands by decreasing affinity.63 Moreover, the ability of docking algorithms to anchor ligands to flat PPI surfaces has long remained elusive. In a benchmark study, Kruger ¨ et al. used two popular docking tools (AutoDock and Glide) to reproduce the known X-ray structure of PPI inhibitors to their target.64 Surprisingly, the performance of these standard docking programs with respect to the positioning of the ligand (rmsd to the X-ray structure) was only moderately affected by switching from conventional targets to PPIs. Although PPI inhibitors with more than 10 rotatable bonds were found more difficult to properly dock, a good pose was generated in ca. 54% of the 80 PPI inhibitors considered. Docking to PPIs providing at least one charge residue was favoured over those purely hydrophobic.64 There are therefore no particular reasons to discard docking-based approaches from rational PPI inhibitor discovery scenarios. Many of the following success stories support this assumption.
We will not here review the many recent reports describing docking as a mean to predict the binding mode of a PPI inhibitor discovered by an experimental screening method.59,65–68 The next section will only focus on inhibitors discovered by a docking-based virtual screening campaign (Table 4).
Table 4 Protein–protein inhibitors discovered by docking-based screening
Target Library size Tested Hits Ref.
TLR4/MD- 2 50 000 14 3 69
uPA–uPAR 5 000 000 50 3 70
IL-6/gp130 9 2 2 71
Keap1–Nrf2 153 611 65 9 72
CRYAB/VEGF 139 735 40 4 73
NRP-1/VEGF- 429 623 1317 56 74
PPxY/Nedd4 4 800 000 20 1 75
p53/MDM2 87 430 295 1 76
Despite an apparent unsuitable large and concave cavity, the MD-2-binding site at the surface of the toll-like receptor 4 (TLR4) was selected for pharmacophore-constrained FlexX77 docking of a library of 49 600 compounds pre-filtered for 3D shape similarity to an existing TLR4 antagonist.69 40 virtual hits were selected for in vitro TLR4 binding and functional antagonism, and 3 of them could be confirmed experimentally. The most potent antagonist (compound 10, Fig. 5) blocked TLR4 in a gene receptor assay with an IC50 of 16.6 mM and inhibited proinflammatory cytokine release (e.g. TNF-a) from human peripheral blood mononuclear cells upon LPS activation. Due to unfavourable aqueous solubility, the compound could not be tested in vivo but represent a good starting hit for developing small molecule TLR4 antagonists for the treatment of neuropathic pain and sepsis.
Fig. 5 PPI inhibitors identified by docking-based virtual screening
To account for the conformational flexibility of proteins, Khanna et al. reported a cascade docking-based virtual screening for discovering inhibitors of the interaction between the urokinase-type plasminogen activator (uPA) and the urokinase receptor (uPAR).70 Two X-ray structures of the uPAR were first used for docking a collection of 5 million commercially available compounds using AutoDock4.78 10 000 top-ranked virtual hits were further docked, still with AutoDock, to 50 molecular dynamics snapshots of the uPAR structure, leading to 500 top-ranked compounds which, in a third step, were docked using a different program (Glide) on the 50 receptor conformers. After clustering the top 250 compounds by chemical similarity, the highest scoring compounds from each of the top 50 clusters were finally selected, purchased and evaluated in vitro in a fluorescence polarization assay. Among the three validated hits, the most potent inhibitor (compound 11, Fig. 5) binds to uPRA with a submicromolar affinity (Kd ¼ 310 nM) and inhibits the uPA–uPAR interaction with an IC50 of 10 mM.70 The hit blocked invasion of breast cancer cells but not their migration or adhesion. A close analogue of compound 11 was recently shown to be efficient in an in vivo breast cancer metastasis assay.79
Docking is not limited to the study of single protein–ligand interactions. In an elegant study, Li et al. reports a computational method enabling the simultaneous docking of multiple fragments to a single binding site, by analogy to experimental fragment screening.71 When applied to the PPI between IL-6 and gp130, simultaneous docking of two fragment pools (6 and 3 fragments, respectively) targeting two different hotspots at the PPI, two theoretical ligands could be reconstructed after tethering the best fragments at each hotspot. Searching for known drugs80 which are chemically similar to the two virtual hits suggested than two estrogen receptor modulators (raloxifene and bazedoxifene) may bind to the gp130/IL-6 PPI. Effective binding of both drugs to gp130 was confirmed experimentally, as well as inhibition of IL-6 induced STAT3 phosphorylation in various cancer cell lines defective in estrogen receptor expression. Bazedoxifene (compound 12, Fig. 5) was the most efficient (IC50 ¼ 25 mM) in inhibiting the ER-independent IL6-induced breast cancer cell proliferation, thereby offering some repositioning potential in the treatment of IL-6/gp130/STAT3 dependent tumours.71
The Nrf2–Keap1 complex, previously investigated using a pharmacophore-based approach (see the previous section), was also used for docking 300 000 commercially available compounds with the program Glide. Among the chemically diverse 65 top-ranking hits, 9 compounds were confirmed to be PPI inhibitors, the most potent disruptor (compound 13, Fig. 5) exhibiting a Kd of 2.9 mM in a fluorescence anisotropy-based assay.
A major hurdle in PPI inhibitor development is the frequently objected high molecular weight and unfavourable pharmacokinetic properties. Chen et al. strikingly contradicted this dogma by reporting a very low molecular weight inhibitor of the aB-crystallin (CRYAB)/VEGF-A interaction.73 CRYAB is a protein overexpressed in triple-negative breast cancer cells that acts as a chaperone to several proteins including the proangiogenic vascular endothelial growth factor (VEGF). Disrupting the interaction between CRYAB and VEGF-A is therefore a potential approach to cancer cell proliferation and invasion. The VEGF-binding site on the surface of the CRYAB X-ray structure was therefore targeted by docking 140 000 compounds from the NCI database using the Dock6.5 program (UCSF). Despite a very modest molecular weight (161.16 Da), one compound (compound 14, Fig. 5) was identified as an in vitro disruptor of the CRYAB/VEGF-A interface with an IC50 of ca. 20 mM. Intraperitoneal injection of compound 14 (200 mg kg1 ) remarkably suppresses tumour growth in vivo in human breast cancer xenograph models. VEGF-A is an important angiogenic factor that interacts with many other partners, notably the family of neuropilin receptors (NRP-1, NRP-2) whose inhibition leads to cancer cell apoptosis. The PPI between the C-terminal end of VEGF-A165 and the tandem b1 and b2 domains of NRP-1 was targeted for docking 430 000 molecules with a consensus docking approach relying on two docking programs (SurflexDock81 and ICM82). A consensus list of 1317 top-scoring compounds was retained for their in vitro anti-proliferative activity and binding to NRP-1 using a chemiluminescent assay.74 56 molecules (hit rate of 4.2%) antagonized the NRP-1/ VEGF-A interaction by at least 30% at the concentration of 10 mM. The best hit (compound 15, Fig. 5) is the first non-peptide NRP-1/VEGF-A antagonist (IC50 ¼ 34 mM) and displays remarkable anti-proliferative effects (IC50 ¼ 0.2 mM) on breast cancer cells. Administered at the dose of 50 mg kg1 in NOG xenographed mice, compound 15 strongly inhibits tumour growth inhibition by inducing cell apoptosis, without any effect on pro-angiogenic kinases.
Although most of the above reported therapeutical indications remain in the oncology field, PPI inhibitors have clear potential in other areas, notably infectious diseases as recently demonstrated by Han et al.75 who reported the structure-based discovery of antiviral compounds inhibiting viral–host interactions. The PPI target is the complex between the conserved Ldomain PPxY sequence of several viral matrix proteins (e.g. Ebola, Marburg, Lassa fever, and VSV) and the ubiquitin ligase Nedd4 protein. Docking ca. 5 million compounds (ZINC database)83 on the Nedd4 X-ray structure with the AutoDock4 program, yielded to the evaluation of 20 compounds, out of which one molecule was confirmed as a true inhibitor of the PPI in a cellular assay. Acquiring close analogs of the initial hit led to two more potent inhibitors (compounds 16 and 17, Fig. 5) as submicromolar inhibitors of the PPxY–Nedd4 interaction in vitro. 75 Both compounds exhibit antibudding activity against Ebola, Lassa fever, Marburg and VSV viruses, thereby decreasing viral titers, without apparent cytotoxicity on HEK293T cells.
Natural compounds are also a major source of potentially interesting PPI inhibitors. By docking a library of commercially available compounds to the p53 binding site, Vogel et al. recently reported lithocholic acid (compound 18, Fig. 5), a secondary bile acid, as a weak binder (Kd of 15 mM) to MDM4 and MDM2 proteins with a slight preference for MDM4.76 The natural compound was further shown to inhibit p53–MDM4 interactions and promote apoptosis in a p53-dependent manner by inducting caspase3/7.
Conclusions
We should acknowledge that peptides usually remain a good starting point to derive PPI inhibitors. Given the increasing number of high resolution X-ray structures of biologically relevant protein–protein complexes, the number of potentially increasing PPIs is likely to significantly rise in the next years. Provided that molecular rules exist to prioritize the most interesting anchoring residues at the interface, continuous protein epitopes can be easily converted into linear peptides for quick experimental validation. Recent progress in peptide stabilisation by chemical stapling next opens an immense eld for deriving either pharmacological tools or drug candidates. Numerous successes in identifying non-peptide PPI inhibitors also exist. The present review has only considered inhibitors mostly discovered by a rational structure-based virtual screening approach. Despite the few cases described herein (15 in total), examples are pretty much indicative of results than can be reasonably achieved. Comparing the properties of PPIs (Fig. 6A and B) and their inhibitors (Fig. 6C) with previously reported larger PPI data,64 some trends could be verified. Considering success as the availability of low micromolar nonpeptide inhibitors, successfully targeted PPIs present a higher proportion of charged residues with respect to conventional targets (sc-PDB data).84 Unsurprisingly, PPI inhibitors bind to smaller cavities (200–350 A˚3 ) than that presented by conventional targets (450–800 A˚3 range). Consequently, PPI inhibitors present a high proportion of aromatic rings, amide moieties and charged groups (Fig. 4 and 5) that hamper their druggability potential, as estimated here by the QED metric85 (Fig. 6C). We notice a significant proportion of negatively charged compounds, suggesting that a strong electrostatic interaction with the target is often mandatory to reach detectable affinity to PPI-participating cavities.
Fig. 6 Properties of PPIs and their inhibitors: (A) cavity properties expressed in percentage according to the cavity detection VolSite program86 (Hydro, hydrophobic; Aro, aromatic; H-bond, H-bond accepting/donating properties; Neg: negatively charged; Pos, positively charged, Du: fully accessible); (B) cavity volumes targeted by PPI inhibitors (this review) and conventional ligands (sc-PDB data84). The box delimits the 25th and 75th percentiles, and the whiskers delimit the 5th and 95th percentiles. The median and mean values are indicated by a horizontal line and an empty square in the box; (C) quantitative estimate of druggability (QED)85 of the inhibitors. QED values for true drug-like compounds should be over 0.5 (red broken line).
However, the current survey also indicates that there is no absolute dogma with respect to PPI inhibitor identification. Very low molecular weight compounds (compounds 1, 6 and 14) have been successfully identified as PPI disruptors.
Beside interfacial inhibitors, there exist promising alternative ways of inhibiting PPIs. For example, PPI stabilizers87,88 (e.g. paclitaxel, rapmycine, and forskolin) bind to rim exposed pockets at or very close to the interface, and also lead to the functional inactivation of the protein–protein complex. Such stabilizers are frequent in the nature, and this area still has not been fully exploited until now. Likewise, the allosteric inhibition of PPIs, at pockets remote from the interface, clearly deserves some consideration. Such pockets have been shown to be frequent at the close vicinity of two protein chains in close interaction,89 and represent, at least for some of them, more ligandable pockets than those presented by PPIs.
Although dominated by a continent of flat and featureless interfaces, the PPI world is also populated by very different islands in terms of shape and electrostatics that should not been discarded. Many factors are likely to increase our knowledge of PPIs and their inhibitors among which: (i) the increasing number of biologically relevant and crystallized protein–protein complexes, (ii) the development of label-free experimental screening techniques, and (iii) the significant contribution of molecular simulations to detect transient interfaces. Medicinal chemistry will be a key factor to transform moderately potent PPI inhibitor hits into clinical candidates with desired pharmacokinetic properties.
References
1 http://www.uniprot.org/uniprot/
?query¼organism% 3A9606+AND+keyword:%22Complete+proteome+[KW- 0181]%22, (accessed 17/07/2014).
2 J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996.
3 P. Legrain and J. C. Rain, J. Proteomics, 2014, 107, 93–97.
4 K. Venkatesan, J. F. Rual, A. Vazquez, U. Stelzl, I. Lemmens, T. Hirozane-Kishikawa, T. Hao, M. Zenkner, X. Xin, K. I. Goh, M. A. Yildirim, N. Simonis, K. Heinzmann, ….A. L. Barabasi and M. Vidal, Nat. Methods, 2009, 6, 83–90.
5 M. P. Stumpf, T. Thorne, E. de Silva, R. Stewart, H. J. An, M. Lappe and C. Wiuf, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6959–6964
Read Full Post »


























{kind=link}
{kind=link}