Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Lesson 4 Cell Signaling And Motility: G Proteins, Signal Transduction: Curations and Articles of reference as supplemental information: #TUBiol3373
Curator: Stephen J. Williams, Ph.D.
Updated 7/15/2019
Below please find the link to the Powerpoint presentation for lesson #4 for #TUBiol3373. The lesson first competes the discussion on G Protein Coupled Receptors, including how cells terminate cell signals. Included are mechanisms of receptor desensitization. Please NOTE that desensitization mechanisms like B arrestin decoupling of G proteins and receptor endocytosis occur after REPEATED and HIGH exposures to agonist. Hydrolysis of GTP of the alpha subunit of G proteins, removal of agonist, and the action of phosphodiesterase on the second messenger (cAMP or cGMP) is what results in the downslope of the effect curve, the termination of the signal after agonist-receptor interaction.
Stroke is a leading cause of death worldwide and the most common cause of long-term disability amongst adults, more particularly in patients with diabetes mellitus and arterial hypertension. Increasing evidence suggests that disordered physiological variables following acute ischaemic stroke, especially hyperglycaemia, adversely affect outcomes.
Post-stroke hyperglycaemia is common (up to 50% of patients) and may be rather prolonged, regardless of diabetes status. A substantial body of evidence has demonstrated that hyperglycaemia has a deleterious effect upon clinical and morphological stroke outcomes. Therefore, hyperglycaemia represents an attractive physiological target for acute stroke therapies.
However, whether intensive glycaemic manipulation positively influences the fate of ischaemic tissue remains unknown. One major adverse event of management of hyperglycaemia with insulin (either glucose-potassium-insulin infusions or intensive insulin therapy) is the occurrence of hypoglycaemia, which can also induce cerebral damage.
Doctors all over the world have debated whether intensive glucose management, which requires the use of IV insulin to bring blood sugar levels down to 80-130 mg/dL, or standard glucose control using insulin shots, which aims to get glucose below 180 mg/dL, lead to better outcomes after stroke.
A period of hyperglycemia is common, with elevated blood glucose in the periinfarct period consistently linked with poor outcome in patients with and without diabetes. The mechanisms that underlie this deleterious effect of dysglycemia on ischemic neuronal tissue remain to be established, although in vitro research, functional imaging, and animal work have provided clues.
While prompt correction of hyperglycemia can be achieved, trials of acute insulin administration in stroke and other critical care populations have been equivocal. Diabetes mellitus and hyperglycemia per se are associated with poor cerebrovascular health, both in terms of stroke risk and outcome thereafter.
Interventions to control blood sugar are available but evidence of cerebrovascular efficacy are lacking. In diabetes, glycemic control should be part of a global approach to vascular risk while in acute stroke, theoretical data suggest intervention to lower markedly elevated blood glucose may be of benefit, especially if thrombolysis is administered.
Both hypoglycaemia and hyperglycaemia may lead to further brain injury and clinical deterioration; that is the reason these conditions should be avoided after stroke. Yet, when correcting hyperglycaemia, great care should be taken not to switch the patient into hypoglycaemia, and subsequently aggressive insulin administration treatment should be avoided.
Early identification and prompt management of hyperglycaemia, especially in acute ischaemic stroke, is recommended. Although the appropriate level of blood glucose during acute stroke is still debated, a reasonable approach is to keep the patient in a mildly hyperglycaemic state, rather than risking hypoglycaemia, using continuous glucose monitoring.
The primary results from the Stroke Hyperglycemia Insulin Network Effort (SHINE) study, a large, multisite clinical study showed that intensive glucose management did not improve functional outcomes at 90 days after stroke compared to standard glucose therapy. In addition, intense glucose therapy increased the risk of very low blood glucose (hypoglycemia) and required a higher level of care such as increased supervision from nursing staff, compared to standard treatment.
Today’s lesson 3 explains how extracellular signals are transduced (transmitted) into the cell through receptors to produce an agonist-driven event (effect). This lesson focused on signal transduction from agonist through G proteins (GTPases), and eventually to the effectors of the signal transduction process. Agonists such as small molecules like neurotransmitters, hormones, nitric oxide were discussed however later lectures will discuss more in detail the large growth factor signalings which occur through receptor tyrosine kinases and the Ras family of G proteins as well as mechanosignaling through Rho and Rac family of G proteins.
Transducers: The Heterotrimeric G Proteins (GTPases)
An excellent review of heterotrimeric G Proteins found in the brain is given by
Cyclic AMP is an important second messenger. It forms, as shown, when the membrane enzyme adenylyl cyclase is activated (as indicated, by the alpha subunit of a G protein).
The cyclic AMP then goes on the activate specific proteins. Some ion channels, for example, are gated by cyclic AMP. But an especially important protein activated by cyclic AMP is protein kinase A, which goes on the phosphorylate certain cellular proteins. The scheme below shows how cyclic AMP activates protein kinase A.
Updated 7/15/2019
Additional New Studies on Regulation of the Beta 2 Adrenergic Receptor
We had discussed regulation of the G protein coupled beta 2 adrenergic receptor by the B-AR receptor kinase (BARK)/B arrestin system which uncouples and desensitizes the receptor from its G protein system. In an article by Xiangyu Liu in Science in 2019, the authors describe another type of allosteric modulation (this time a POSITIVE allosteric modulation) in the intracellular loop 2. See below:
Mechanism of β2AR regulation by an intracellular positive allosteric modulator
Xiangyu Liu1,*, Ali Masoudi2,*, Alem W. Kahsai2,*, Li-Yin Huang2, Biswaranjan Pani2, Dean P. Staus2, Paul J. Shim2, Kunio Hirata3,4, Rishabh K. Simhal2, Allison M. Schwalb2, Paula K. Rambarat2, Seungkirl Ahn2, Robert J. Lefkowitz2,5,6,†, Brian Kobilka1
Positive reinforcement in a GPCR
Many drug discovery efforts focus on G protein–coupled receptors (GPCRs), a class of receptors that regulate many physiological processes. An exemplar is the β2-adrenergic receptor (β2AR), which is targeted by both blockers and agonists to treat cardiovascular and respiratory diseases. Most GPCR drugs target the primary (orthosteric) ligand binding site, but binding at allosteric sites can modulate activation. Because such allosteric sites are less conserved, they could possibly be targeted more specifically. Liu et al. report the crystal structure of β2AR bound to both an orthosteric agonist and a positive allosteric modulator that increases receptor activity. The structure suggests why the modulator compound is selective for β2AR over the closely related β1AR. Furthermore, the structure reveals that the modulator acts by enhancing orthosteric agonist binding and stabilizing the active conformation of the receptor.
Abstract
Drugs targeting the orthosteric, primary binding site of G protein–coupled receptors are the most common therapeutics. Allosteric binding sites, elsewhere on the receptors, are less well-defined, and so less exploited clinically. We report the crystal structure of the prototypic β2-adrenergic receptor in complex with an orthosteric agonist and compound-6FA, a positive allosteric modulator of this receptor. It binds on the receptor’s inner surface in a pocket created by intracellular loop 2 and transmembrane segments 3 and 4, stabilizing the loop in an α-helical conformation required to engage the G protein. Structural comparison explains the selectivity of the compound for β2– over the β1-adrenergic receptor. Diversity in location, mechanism, and selectivity of allosteric ligands provides potential to expand the range of receptor drugs.
Recent structures of GPCRs bound to allosteric modulators have revealed that receptor surfaces are decorated with diverse cavities and crevices that may serve as allosteric modulatory sites (1). This substantiates the notion that GPCRs are structurally plastic and can be modulated by a variety of allosteric ligands through distinct mechanisms (2-7). Most of these structures have been solved with negative allosteric modulators (NAMs), which stabilize receptors in their inactive states (1). To date, only a single structure of an active GPCR bound to a small-molecule positive allosteric modulator (PAM) has been reported, namely, the M2 muscarinic acetylcholine receptor with LY2119620 (8). Thus, mechanisms of PAMs and their potential binding sites remain largely unexplored.
Fig 1. Structure of the active state T4L-B2AR in complex with the orthosteric agonist BI-167107, nanobody 689, and compound 6FA. (A) The chemical structure of compound-6FA (Cmpd-6FA). (B) Isoproterenol (ISO) competition binding with 125I-cyanopindolol (CYP) to the β2AR reconstituted in nanodisks in the presence of vehicle (0.32% dimethylsulfoxide; DMSO), Cmpd-6, or Cmpd-6FA at 32 μM. Values were normalized to percentages of the maximal 125I-CYP binding level obtained from a one-site competition binding–log IC50 (median inhibitory concentration) curve fit. Binding curves were generated by GraphPad Prism. Points on curves represent mean ± SEM obtained from five independent experiments performed in duplicate. (C) Analysis of Cmpd-6FA interaction with the BI-167107–bound β2AR by ITC. Representative thermogram (inset) and binding isotherm, of three independent experiments, with the best titration curve fit are shown. Summary of thermodynamic parameters obtained by ITC: binding affinity (KD = 1.2 ± 0.1 μM), stoichiometry (N = 0.9 ± 0.1 sites), enthalpy (ΔH = 5.0 ± 1.2 kcal mol−1), and entropy (ΔS =13 ± 2.0 cal mol−1 deg−1). (D) Side view of T4L-β2AR bound to the orthosteric agonist BI-167107, nanobody 6B9 (Nb6B9), and Cmpd-6FA. The gray box indicates the membrane layer as defined by the OPM database. (E) Close-up view of Cmpd-6FA binding site. Covering Cmpd-6FA is 2Fo– Fc electron density contoured at 1.0 σ (green mesh).From Science 28 Jun 2019:
Vol. 364, Issue 6447, pp. 1283-1287
Fig 3. Fig. 3Mechanism of allosteric activation of the β2AR by Cmpd-6FA.
(A) Superposition of the inactive β2AR bound to the antagonist carazolol (PDB code: 2RH1) and the active β2AR bound to the agonist BI-167107, Cmpd-6FA, and Nb6B9. Close-up view of the Cmpd-6FA binding site is shown. The residues of the inactive (yellow) and active (blue) β2AR are depicted, and the hydrogen bond formed between Asp1303.49and Tyr141ICL2 in the active state is indicated by a black dashed line. (B) Topography of Cmpd-6FA binding surface on the active β2AR (left, blue) and the corresponding surface of the inactive β2AR (right, yellow) with Cmpd-6FA (orange sticks) docked on top. Molecular surfaces are of only those residues involved in interaction with Cmpd-6FA. Steric clash between Cmpd-6FA and the surface of inactive β2AR is represented by a purple asterisk. (C) Overlay of the β2AR bound to BI-167107, Nb6B9, and Cmpd-6FA with the β2AR–Gscomplex (PDB code: 3SN6). The inset shows the position of Phe139ICL2 relative to the α subunit of Gs. (D) Superposition of the active β2AR bound to the agonist BI-167107, Nb6B9, and Cmpd-6FA (blue) with the inactive β2AR bound to carazolol (yellow) (PDB code: 2RH1) as viewed from the cytoplasm. For clarity, Nb6B9 and the orthosteric ligands are omitted. The arrows indicate shifts in the intracellular ends of the TM helices 3, 5, and 6 upon activation and their relative distances.
Allosteric sites may not face the same evolutionary pressure as do orthosteric sites, and thus are more divergent across subtypes within a receptor family (24–26). Therefore, allosteric sites may provide a greater source of specificity for targeting GPCRs.
D. M. Thal, A. Glukhova, P. M. Sexton, A. Christopoulos, Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53 (2018). doi:10.1038/s41586-018-0259-zpmid:29973731CrossRefPubMedGoogle Scholar
D. Wacker, R. C. Stevens, B. L. Roth, How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427 (2017).
D. P. Staus, R. T. Strachan, A. Manglik, B. Pani, A. W. Kahsai, T. H. Kim, L. M. Wingler, S. Ahn, A. Chatterjee, A. Masoudi, A. C. Kruse, E. Pardon, J. Steyaert, W. I. Weis, R. S. Prosser, B. K. Kobilka, T. Costa, R. J. Lefkowitz, Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–452 (2016). doi:10.1038/nature18636pmid:27409812CrossRefPubMedGoogle Scholar
A. Manglik, T. H. Kim, M. Masureel, C. Altenbach, Z. Yang, D. Hilger, M. T. Lerch, T. S. Kobilka, F. S. Thian, W. L. Hubbell, R. S. Prosser, B. K. Kobilka, Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell 161, 1101–1111 (2015). doi:10.1016/j.cell.2015.04.043pmid:25981665CrossRefPubMedGoogle Scholar
5, L. Ye, N. Van Eps, M. Zimmer, O. P. Ernst, R. S. Prosser, Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268 (2016). doi:10.1038/nature17668pmid:27144352CrossRefPubMedGoogle Scholar
N. Van Eps, L. N. Caro, T. Morizumi, A. K. Kusnetzow, M. Szczepek, K. P. Hofmann, T. H. Bayburt, S. G. Sligar, O. P. Ernst, W. L. Hubbell, Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc. Natl. Acad. Sci. U.S.A. 114, E3268–E3275 (2017). doi:10.1073/pnas.1620405114pmid:28373559Abstract/FREE Full TextGoogle Scholar
R. O. Dror, H. F. Green, C. Valant, D. W. Borhani, J. R. Valcourt, A. C. Pan, D. H. Arlow, M. Canals, J. R. Lane, R. Rahmani, J. B. Baell, P. M. Sexton, A. Christopoulos, D. E. Shaw, Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 (2013). doi:10.1038/nature12595pmid:24121438CrossRefPubMedWeb of ScienceGoogle Scholar
A. C. Kruse, A. M. Ring, A. Manglik, J. Hu, K. Hu, K. Eitel, H. Hübner, E. Pardon, C. Valant, P. M. Sexton, A. Christopoulos, C. C. Felder, P. Gmeiner, J. Steyaert, W. I. Weis, K. C. Garcia, J. Wess, B. K. Kobilka, Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013). doi:10.1038/nature12735pmid:24256733
Additional information on Nitric Oxide as a Cellular Signal
Nitric oxide is actually a free radical and can react with other free radicals, resulting in a very short half life (only a few seconds) and so in the body is produced locally to its site of action (i.e. in endothelial cells surrounding the vascular smooth muscle, in nerve cells). In the late 1970s, Dr. Robert Furchgott observed that acetylcholine released a substance that produced vascular relaxation, but only when the endothelium was intact. This observation opened this field of research and eventually led to his receiving a Nobel prize. Initially, Furchgott called this substance endothelium-derived relaxing factor (EDRF), but by the mid-1980s he and others identified this substance as being NO.
Nitric oxide is implicated in many pathologic processes as well. Nitric oxide post translational modifications have been attributed to nitric oxide’s role in pathology however, although the general mechanism by which nitric oxide exerts its physiological effects is by stimulation of soluble guanylate cyclase to produce cGMP, these post translational modifications can act as a cellular signal as well. For more information of NO pathologic effects and how NO induced post translational modifications can act as a cellular signal see the following:
UPDATED on 12/26/2020 – CABG: a Superior Revascularization Modality to PCI in Patients with poor LVF, Multivessel disease and Diabetes, Similar Risk of Stroke between 31 days and 5 years, post intervention
Reporter: Aviva Lev-Ari, PhD, RN
UPDATED ON 1/16/2025
Surgery outperforms PCI in NSTEMI patients with multivessel CAD
Bypass surgery is associated with better long-term outcomes than percutaneous coronary intervention (PCI) when treating patients who present with non-ST-segment elevation myocardial infarction (NSTEMI) and multivessel disease, according to new research published in European Heart Journal.[1]
Researchers tracked more than 57,000 patients with NSTEMI and multivessel coronary artery disease (CAD) who underwent treatment from 2005 to 2022. The mean patient age was 68.7 years old, 75.8% were men and the median follow-up period was 7.1 years. All data came from the SWEDEHEART registry, which monitors heart patients treated in Sweden.
“Our findings indicate that CABG is associated with lower risks of all-cause mortality and myocardial infarction compared with PCI,” wrote first author Elmir Omerovic, a cardiologist and professor of cardiology with the University of Gothenburg in Sweden, and colleagues. “Specifically, the long-term risk of all-cause mortality was 41% lower in the CABG group, and the risk of myocardial infarction was 34% lower. The mortality benefit of CABG over PCI was evident at each yearly follow-up interval.”
Additionally, exploring the role of hybrid revascularization approaches and personalized medicine strategies could provide valuable insights into optimizing treatment for this complex patient population.”
Evidence suggests that a high-dose statin loading before a percutaneous coronary revascularization improves outcomes in patients receiving long-term statins. This study aimed to analyse the effects of such an additional statin therapy before surgical revascularization.
Additional statin loading before CABG failed to reduce the rate of MACCE occuring within 30 days of surgery.
Long-term outcomes after percutaneous coronary intervention (PCI) with contemporary drug-eluting stents, as compared with coronary-artery bypass grafting (CABG), in patients with left main coronary artery disease are not clearly established.
METHODS
We randomly assigned 1905 patients with left main coronary artery disease of low or intermediate anatomical complexity (according to assessment at the participating centers) to undergo either PCI with fluoropolymer-based cobalt–chromium everolimus-eluting stents (PCI group, 948 patients) or CABG (CABG group, 957 patients). The primary outcome was a composite of death, stroke, or myocardial infarction.
RESULTS
At 5 years, a primary outcome event had occurred in 22.0% of the patients in the PCI group and in 19.2% of the patients in the CABG group (difference, 2.8 percentage points; 95% confidence interval [CI], −0.9 to 6.5; P=0.13). Death from any cause occurred more frequently in the PCI group than in the CABG group (in 13.0% vs. 9.9%; difference, 3.1 percentage points; 95% CI, 0.2 to 6.1). In the PCI and CABG groups, the incidences of definite cardiovascular death (5.0% and 4.5%, respectively; difference, 0.5 percentage points; 95% CI, −1.4 to 2.5) and myocardial infarction (10.6% and 9.1%; difference, 1.4 percentage points; 95% CI, −1.3 to 4.2) were not significantly different. All cerebrovascular events were less frequent after PCI than after CABG (3.3% vs. 5.2%; difference, −1.9 percentage points; 95% CI, −3.8 to 0), although the incidence of stroke was not significantly different between the two groups (2.9% and 3.7%; difference, −0.8 percentage points; 95% CI, −2.4 to 0.9). Ischemia-driven revascularization was more frequent after PCI than after CABG (16.9% vs. 10.0%; difference, 6.9 percentage points; 95% CI, 3.7 to 10.0).
CONCLUSIONS
In patients with left main coronary artery disease of low or intermediate anatomical complexity, there was no significant difference between PCI and CABG with respect to the rate of the composite outcome of death, stroke, or myocardial infarction at 5 years. (Funded by Abbott Vascular; EXCEL ClinicalTrials.gov number, NCT01205776. opens in new tab.)
Is the Tide Turning on the ‘Grubby’ Affair of EXCEL and the European Guidelines?
Taggart was chair of the surgical committee for the Abbott-sponsored EXCEL trial, which compared two procedures for patients who had blockages in their left main coronary artery: percutaneous coronary intervention (PCI) using coronary stents, and coronary artery bypass graft surgery (CABG). The investigators designed the trial to compare outcomes for the two treatments using a composite endpoint of death, stroke, and myocardial infarction (MI). The 3-year follow-up data had been published in NEJM without controversy — or, at least, without public controversy.
But when it came time to publish the 5-year follow-up, there was a significantly higher rate of death in the stent group, and both Taggart and the journal editors were concerned that this finding was being downplayed in the manuscript.
In their comments to the authors, the journal editors had recommended including the mortality difference (unless clearly trivial) ‘”in the concluding statement in the final paragraph.” Yet, the concluding statement of the published paper read that there “was no significant difference between PCI and CABG.”
Over a year after the BBC received the leaked data, the EXCEL investigators published an analysis of the primary outcome using the universal definition of MI data in the Journal of the American College of Cardiology.
It shows 141 events in the PCI arm compared to 102 in the CABG arm. The investigators acknowledge that the rates of procedural MI differ depending on the definition used. According to their analysis, the protocol definition was predictive of mortality after both treatments, whereas the universal definition of procedural MI was predictive of mortality only after CABG. Not everyone agrees with this interpretation, and an accompanying editorial questioned these conclusions.
As for the guidelines, the tide may be turning.
In a joint statement with EACTS on October 6, 2020, the ESC agreed to review its guidelines for left main disease in the light of emerging, longer-term outcome data from the trials of CABG vs PCI.
SYNTAX at 10 Years: Bypass vs PCI Still a Toss-Up Overall
But CABG beats stenting for important subgroups
SYNTAX was funded by Boston Scientific.
Thuijs, Stone, and Taggart disclosed no conflicts.
Baumbach reported receiving institutional research support from Abbott Vascular; and consultation and speaker fees from AstraZeneca, Sinomed, Microport, Abbott Vascular, Cardinal Health, KSH, Medtronic.
Pagano is the Secretary General of the European Association for Cardiothoracic Surgery.
LAST UPDATED
Overall, mortality curves did not diverge over time for SYNTAX participants now included in the trial’s extension study SYNTAXES: by 1o years, all-cause death reached 27.0% among those randomized to percutaneous coronary intervention (PCI) and 23.5% of the CABG arm (HR 1.17, 95% CI 0.97-1.41), according to Daniel Thuijs, MD, of Erasmus University Medical Centre in Rotterdam, the Netherlands.
However, people with three-vessel disease had particularly high mortality when assigned to PCI in lieu of CABG (27.7% vs 20.6%, HR 1.41, 95% CI 1.10-1.80), whereas peers with left main disease were no worse off after either procedure (26.1% vs 26.7%, HR 0.90, 95% CI 0.68-1.52),Thuijs told the audience during a late-breaking trial session at the European Society of Cardiology’s annual congress. The study was also published online in The Lancet.
CABG had a mortality benefit over PCI in patients with multivessel disease, particularly those with diabetes and higher coronary complexity. No benefit for CABG over PCI was seen in patients with left main disease. Longer follow-up is needed to better define mortality differences between the revascularisation strategies.
JACC Study, 7/2018
CONCLUSIONS
This individual patient-data pooled analysis demonstrates that 5-year stroke rates are significantly lower after PCI compared with CABG, driven by a reduced risk of stroke in the 30-day post-procedural period but a similar risk of stroke between 31 days and 5 years. The greater risk of stroke after CABG compared with PCI was confined to patients with multivessel disease and diabetes. Five-year mortality was markedly higher for patients experiencing a stroke within 30 days after revascularization.
European Journal of Cardiothoracic Surgery Study, 6/2018
CONCLUSIONS
Despite a longer length of hospital stay, patients with impaired LVF requiring intervention for coronary artery disease experienced a greater post-procedural survival benefit if they received CABG compared to PCI. We have demonstrated this at 30 days, 90 days, 1 year, 3 years, 5 years and 8 years following revascularization. At present, CABG remains a superior revascularization modality to PCI in patients with poor LVF.
New Studies on Clinical Outcomes from two Revascularization Strategies: CABG and PCI
J Am Coll Cardiol. 2018 Jul 24;72(4):386-398. doi: 10.1016/j.jacc.2018.04.071.
Stroke Rates Following Surgical Versus Percutaneous Coronary Revascularization.
Coronary artery bypass grafting (CABG) and percutaneous coronary intervention (PCI) are used for coronary revascularization in patients with multivessel and left main coronary artery disease. Stroke is among the most feared complications of revascularization. Due to its infrequency, studies with large numbers of patients are required to detect differences in stroke rates between CABG and PCI.
OBJECTIVES:
This study sought to compare rates of stroke after CABG and PCI and the impact of procedural stroke on long-term mortality.
METHODS:
We performed a collaborative individual patient-data pooled analysis of 11 randomized clinical trials comparing CABG with PCI using stents; ERACI II (Argentine Randomized Study: Coronary Angioplasty With Stenting Versus Coronary Bypass Surgery in Patients With Multiple Vessel Disease) (n = 450), ARTS (Arterial Revascularization Therapy Study) (n = 1,205), MASS II (Medicine, Angioplasty, or Surgery Study) (n = 408), SoS (Stent or Surgery) trial (n = 988), SYNTAX (Synergy Between Percutaneous Coronary Intervention With Taxus and Cardiac Surgery) trial (n = 1,800), PRECOMBAT (Bypass Surgery Versus Angioplasty Using Sirolimus-Eluting Stent in Patients With Left Main Coronary Artery Disease) trial (n = 600), FREEDOM (Comparison of Two Treatments for Multivessel Coronary Artery Disease in Individuals With Diabetes) trial (n = 1,900), VA CARDS (Coronary Artery Revascularization in Diabetes) (n = 198), BEST (Bypass Surgery Versus Everolimus-Eluting Stent Implantation for Multivessel Coronary Artery Disease) (n = 880), NOBLE (Percutaneous Coronary Angioplasty Versus Coronary Artery Bypass Grafting in Treatment of Unprotected Left Main Stenosis) trial (n = 1,184), and EXCEL (Evaluation of Xience Versus Coronary Artery Bypass Surgery for Effectiveness of Left Main Revascularization) trial (n = 1,905). The 30-day and 5-year stroke rates were compared between CABG and PCI using a random effects Cox proportional hazards model, stratified by trial. The impact of stroke on 5-year mortality was explored.
RESULTS:
The analysis included 11,518 patients randomly assigned to PCI (n = 5,753) or CABG (n = 5,765) with a mean follow-up of 3.8 ± 1.4 years during which a total of 293 strokes occurred. At 30 days, the rate of stroke was 0.4% after PCI and 1.1% after CABG (hazard ratio [HR]: 0.33; 95% confidence interval [CI]: 0.20 to 0.53; p < 0.001). At 5-year follow-up, stroke remained significantly lower after PCI than after CABG (2.6% vs. 3.2%; HR: 0.77; 95% CI: 0.61 to 0.97; p = 0.027). Rates of stroke between 31 days and 5 years were comparable: 2.2% after PCI versus 2.1% after CABG (HR: 1.05; 95% CI: 0.80 to 1.38; p = 0.72). No significant interactions between treatment and baseline clinical or angiographic variables for the 5-year rate of stroke were present, except for diabetic patients (PCI: 2.6% vs. CABG: 4.9%) and nondiabetic patients (PCI: 2.6% vs. CABG: 2.4%) (p for interaction = 0.004). Patients who experienced a stroke within 30 days of the procedure had significantly higher 5-year mortality versus those without a stroke, both after PCI (45.7% vs. 11.1%, p < 0.001) and CABG (41.5% vs. 8.9%, p < 0.001).
CONCLUSIONS:
This individual patient-data pooled analysis demonstrates that 5-year stroke rates are significantly lower after PCI compared with CABG, driven by a reduced risk of stroke in the 30-day post-procedural period but a similar risk of stroke between 31 days and 5 years. The greater risk of stroke after CABG compared with PCI was confined to patients with multivessel disease and diabetes. Five-year mortality was markedly higher for patients experiencing a stroke within 30 days after revascularization.
Head SJ, Milojevic M, Daemen J, Ahn JM, Boersma E, Christiansen EH, Domanski MJ, Farkouh ME, Flather M, Fuster V, Hlatky MA, Holm NR, Hueb WA, Kamalesh M, Kim YH, Mäkikallio T, Mohr FW, Papageorgiou G, Park SJ, Rodriguez AE, Sabik JF, Stables RH, Stone GW, Serruys PW, Kappetein AP. Mortality after coronary artery bypass grafting versus percutaneous coronary intervention with stenting for coronary artery disease: a pooled analysis of individual patient data. Lancet. 2018 Feb 22 [Epub ahead of print]. doi: 10.1016/S0140-6736(18)30423-9. PMID: 29478841
Numerous randomised trials have compared coronary artery bypass grafting (CABG) with percutaneous coronary intervention (PCI) for patients with coronary artery disease. However, no studies have been powered to detect a difference in mortality between the revascularisation strategies.
Methods
We did a systematic review up to July 19, 2017, to identify randomised clinical trials comparing CABG with PCI using stents. Eligible studies included patients with multivessel or left main coronary artery disease who did not present with acute myocardial infarction, did PCI with stents (bare-metal or drug-eluting), and had more than 1 year of follow-up for all-cause mortality. In a collaborative, pooled analysis of individual patient data from the identified trials, we estimated all-cause mortality up to 5 years using Kaplan-Meier analyses and compared PCI with CABG using a random-effects Cox proportional-hazards model stratified by trial. Consistency of treatment effect was explored in subgroup analyses, with subgroups defined according to baseline clinical and anatomical characteristics.
Findings
We included 11 randomised trials involving 11 518 patients selected by heart teams who were assigned to PCI (n=5753) or to CABG (n=5765). 976 patients died over a mean follow-up of 3·8 years (SD 1·4). Mean Synergy between PCI with Taxus and Cardiac Surgery (SYNTAX) score was 26·0 (SD 9·5), with 1798 (22·1%) of 8138 patients having a SYNTAX score of 33 or higher. 5 year all-cause mortality was 11·2% after PCI and 9·2% after CABG (hazard ratio [HR] 1·20, 95% CI 1·06–1·37; p=0·0038). 5 year all-cause mortality was significantly different between the interventions in patients with multivessel disease (11·5% after PCI vs 8·9% after CABG; HR 1·28, 95% CI 1·09–1·49; p=0·0019), including in those with diabetes (15·5% vs 10·0%; 1·48, 1·19–1·84; p=0·0004), but not in those without diabetes (8·7% vs 8·0%; 1·08, 0·86–1·36; p=0·49). SYNTAX score had a significant effect on the difference between the interventions in multivessel disease. 5 year all-cause mortality was similar between the interventions in patients with left main disease (10·7% after PCI vs 10·5% after CABG; 1·07, 0·87–1·33; p=0·52), regardless of diabetes status and SYNTAX score.
Interpretation
CABG had a mortality benefit over PCI in patients with multivessel disease, particularly those with diabetes and higher coronary complexity. No benefit for CABG over PCI was seen in patients with left main disease. Longer follow-up is needed to better define mortality differences between the revascularisation strategies.
Comparison of the survival between coronary artery bypass graft surgery versus percutaneous coronary intervention in patients with poor left ventricular function (ejection fraction <30%): a propensity-matched analysis.
Existing evidence comparing the outcomes of coronary artery bypass graft (CABG) surgery versus percutaneous coronary intervention (PCI) in patients with poor left ventricular function (LVF) is sparse and flawed. This is largely due to patients with poor LVF being underrepresented in major research trials and the outdated nature of some studies that do not consider drug-eluting stent PCI.
METHODS:
Following strict inclusion criteria, 717 patients who underwent revascularization by CABG or PCI between 2002 and 2015 were enrolled. All patients had poor LVF (defined by ejection fraction <30%). By employing a propensity score analysis, 134 suitable matches (67 CABG and 67 PCI) were identified. Several outcomes were evaluated, in the matched population, using data extracted from national registry databases.
RESULTS:
CABG patients required a longer length of hospital stay post-revascularization compared to PCI in the propensity-matched population, 7 days (lower-upper quartile; 6-12) and 2 days (lower-upper quartile; 1-6), respectively (Mood’s median test, P = 0.001). Stratified Cox-regression proportional-hazards analysis of the propensity-matched population found that PCI patients experienced a higher adjusted 8-year mortality rate (hazard ratio 3.291, 95% confidence interval 1.776-6.101; P < 0.001). This trend was consistent amongst urgent cases of revascularization: patients with 3 or more vessels with coronary artery disease and patients where complete revascularization was achieved. Although sub-analyses found no difference between survival distributions of on-pump versus off-pump CABG (log-rank P = 0.726), both modes of CABG were superior to PCI (stratified log-rank P = 0.002).
CONCLUSIONS:
Despite a longer length of hospital stay, patients with impaired LVF requiring intervention for coronary artery disease experienced a greater post-procedural survival benefit if they received CABG compared to PCI. We have demonstrated this at 30 days, 90 days, 1 year, 3 years, 5 years and 8 years following revascularization. At present, CABG remains a superior revascularization modality to PCI in patients with poor LVF.
Using “Cerebral Organoids” to Trace the Elemental Composition of a Developing Brain
Curator: Marzan Khan, B.Sc
A research focused on the detection of micronutrient accumulation in the developing brain has been conducted recently by a team of scientific researchers in Brazil(1). Their study was comprised of a cutting-edge technology – human cerebral organoids, which are a close equivalent of the embryonic brain, in in-vitro models to identify some of the minerals essential during brain development using synchroton radiation(1). Since the majority of studies done on this matter have relied on samples from animal models, the adult brain or post-mortem tissue, this technique has been dubbed the “closest and most complete study system to date for understanding human neural development and its pathological manifestations”(2).
Cerebral organoids are three-dimensional miniature structures derived from human pluripotent stem cells that further differentiate into structures closely resembling the developing brain(2). Concentrating on two different time points during the developmental progression, the researchers illustrated the micronutrient content during an interval of high cell division marked on day 30 as well as day 40 when the organoids were starting to become mature neurons that secrete neurotransmitters, arranging into layers and forming synapses(2).
Synchrotron radiation X-ray fluorescence (SR-XRF) spectroscopy was used to discern each type of element present(2). After an incident beam of X-ray was directed at the sample, each atom emitted a distinct photon signature(2). Phosphorus (P), Potassium (P), Sulphur (S), Calcium (Ca), Iron (Fe), and Zinc (Zn) were found to be present in the samples in significant concentrations(2). Manganese (Mn), Nickel (Ni) and Copper (Cu) were also detected, but in negligible amounts, and therefore tagged as “ultratrace” elements(2). The distribution of these minerals, their concentration as well as their occurrence in pairs were examined at each interval(2).
Phosphorus was discovered to be the most abundant element in the cerebral organoid samples(3). Between the two time points at 30 days (cell proliferation) and 45 days (neuronal maturation) there was a marked decrease in P content(2). Since phosphorus is a major component of nucleotides and phospholipids, this reduction was clarified as a shift from a stage of cell division that requires the production of DNA and phospholipids, to a migratory and differentiation phase(2). Potassium levels were maintained during both phases, substantiating its role in mitotic cell division as well as cell migration over long distances(2). Sulfur levels were reportedly high at 30 days and 45 days(2). It was hypothesized that this element was responsible for the patterning of the organoids(2). Calcium, known to control transcription factors involved in neuronal differentiation and survival were detected in the micromolar range, along with zinc and iron(2). Zinc commits the differentiation of pluripotent stem cells into neuronal cells and iron is necessary for neuronal tissue expansion(2).
The cells in an embryo start to differentiate very early on- the neural plate is formed on the 16th day of contraception, which further folds and bulges out to become the nervous system (containing the brain and spinal cord regions)(3). Nutrients obtained from the mother are the primary sources of diet and energy for a developing embryo to fully differentiate and specialize into different organs(2). Lack of proper nutrition in pregnant mothers has been linked to many neurodegenerative diseases occurring in their progeny(2). Spina bifida which is characterized by the incomplete development of the brain and spinal cord, is a classic example of maternal malnutrition(2,4). Paucity of minerals in the diet of pregnant women are known to hamper learning and memory in children(2). Even Schizophrenia, Parkinson’s and Huntington’s disease have been associated to malnourishment(2). By showing the different types of elements present in statistically significant concentrations in cerebral organoids, the results of this study underscore the necessity of a healthy nourishment available to mothers during pregnancy for optimal development of the fetal brain(2).
2.Sartore RC, Cardoso SC, Lages YVM, Paraguassu JM, Stelling MP, Madeiro da Costa RF, Guimaraes MZ, Pérez CA, Rehen SK.(2017)Trace elements during primordial plexiform network formation in human cerebral organoids. PeerJ 5:e2927https://doi.org/10.7717/peerj.292
Alpha-linolenic acid given as enteral or parenteral nutritional intervention against sensorimotor and cognitive deficits in a mouse model of ischemic stroke
Stroke is a leading cause of disability and death worldwide. Numerous therapeutics applied acutely after stroke have failed to improve long-term clinical outcomes. An emerging direction is nutritional intervention with omega-3 polyunsaturated fatty acids acting as disease-modifying factors and targeting post-stroke disabilities. Our previous studies demonstrated that the omega-3 precursor, alpha-linolenic acid (ALA) administrated by injections or dietary supplementation reduces stroke damage by direct neuroprotection, and triggering brain artery vasodilatation and neuroplasticity. Successful translation of putative therapies will depend on demonstration of robust efficacy on common deficits resulting from stroke like loss of motor control and memory/learning. This study evaluated the value of ALA as adjunctive therapy for stroke recovery by comparing whether oral or intravenous supplementation of ALA best support recovery from ischemia. Motor and cognitive deficits were assessed using rotarod, pole and Morris water maze tests. ALA supplementation in diet was better than intravenous treatment in improving motor coordination, but this improvement was not due to a neuroprotective effect since infarct size was not reduced. Both types of ALA supplementation improved spatial learning and memory after stroke. This cognitive improvement correlated with higher survival of hippocampal neurons. These results support clinical investigation establishing therapeutic plans using ALA supplementation
A research team led by scientists at SUNY Downstate Medical Center has identified a brain receptor that appears to initiate adolescent synaptic pruning, a process believed necessary for learning, but one that appears to go awry in both autism and schizophrenia.
Sheryl Smith, Ph.D., professor of physiology and pharmacology at SUNY Downstate, explained that “Memories are formed at structures in the brain known as dendritic spines that communicate with other brain cells through synapses. The number of brain connections decreases by half after puberty, a finding shown in many brain areas and for many species, including humans and rodents.”
This process is referred to as adolescent “synaptic pruning” and is thought to be important for normal learning in adulthood. Synaptic pruning is believed to remove unnecessary synaptic connections to make room for relevant new memories, but because it is disrupted in diseases such as autism and schizophrenia, there has recently been widespread interest in the subject.
“Our report is the first to identify the process which initiates synaptic pruning at puberty. Previous studies have shown that scavenging by the immune system cleans up the debris from these pruned connections, likely the final step in the pruning process,” added Dr. Smith. “Working with a mouse model we have shown that, at puberty, there is an increase in inhibitory GABA [gamma-aminobutyric acid] receptors, which are targets for brain chemicals that quiet down nerve cells. We now report that these GABA receptors trigger synaptic pruning at puberty in the mouse hippocampus, a brain area involved in learning and memory.”
The study (“Synaptic Pruning in the Female Hippocampus Is Triggered at Puberty by Extrasynaptic GABAA Receptors on Dendritic Spines”) is published online in eLife.
Dr. Smith noted that by reducing brain activity, these GABA receptors also reduce levels of a protein in the dendritic spine, kalirin-7, which stabilizes the scaffolding in the spine to maintain its structure. Mice that do not have these receptors maintain the same high level of brain connections throughout adolescence.
Dr. Smith pointed out that the mice with too many brain connections, which do not undergo synaptic pruning, are able to learn spatial locations, but are unable to relearn new locations after the initial learning, suggesting that too many brain connections may limit learning potential.
These findings may suggest new treatments targeting GABA receptors for “normalizing” synaptic pruning in diseases such as autism and schizophrenia, where synaptic pruning is abnormal. Research has suggested that children with autism may have an over-abundance of synapses in some parts of the brain. Other research suggests that prefrontal brain areas in persons with schizophrenia have fewer neural connections than the brains of those who do not have the condition.
Synaptic pruning in the female hippocampus is triggered at puberty by extrasynaptic GABAAreceptors on dendritic spines
JulieParato
Department of Physiology and Pharmacology, SUNY Downstate Medical Center, Brooklyn, United States
No competing interests declared
</div>”>Sonia Afroz, JulieParato,
HuiShen
School of Biomedical Engineering, Tianjin Medical University, Tianjin, China
No competing interests declared
</div>”>HuiShen,
Sheryl SueSmith
Department of Physiology and Pharmacology, SUNY Downstate Medical Center, Brooklyn, United Statessheryl.smith@downstate.edu
Adolescent synaptic pruning is thought to enable optimal cognition because it is disrupted in certain neuropathologies, yet the initiator of this process is unknown. One factor not yet considered is the α4βδ GABAA receptor (GABAR), an extrasynaptic inhibitory receptor which first emerges on dendritic spines at puberty in female mice. Here we show that α4βδ GABARs trigger adolescent pruning. Spine density of CA1 hippocampal pyramidal cells decreased by half post-pubertally in female wild-type but not α4 KO mice. This effect was associated with decreased expression of kalirin-7 (Kal7), a spine protein which controls actin cytoskeleton remodeling. Kal7 decreased at puberty as a result of reduced NMDAR activation due to α4βδ-mediated inhibition. In the absence of this inhibition, Kal7 expression was unchanged at puberty. In the unpruned condition, spatial re-learning was impaired. These data suggest that pubertal pruning requires α4βδ GABARs. In their absence, pruning is prevented and cognition is not optimal.
Searches Related to Synaptic Pruning in the Female Hippocampus Is Triggered at Puberty by Extrasynaptic GABAA Receptors on Dendritic Spines
Researchers at Ruhr University Bochum (RUB; Germany) coupled nerve cell receptors to light-sensitive retinal pigments to understand how the serotonin neurotransmitter works and, therefore, learn what causes anxiety anddepression.
Prof. Dr. Olivia Masseck, who led the work, researches the causes of anxiety and depression. For more than 60 years, researchers have been hypothesising that the diseases are caused by, among other factors, changes to the level of serotonin. But understanding how the serotonin system works is quite difficult, says Masseck, who became junior professor for Super-Resolution Fluorescence Microscopy at RUB in April 2016.
With a method called optogenetics, Olivia Masseck (right) creates nerve cell receptors that are controllable with light. (Copyright: RUB, Damian Gorczany)
The number of receptors for serotonin in the brain amounts to 14, occurring in different cell types. Consequently, determining the functions that different receptors fulfill in the individual cell types is a complicated task. If, however, the proteins are coupled to light-sensitive pigments, they can be switched on and off with light of a specific color at high spatial and temporal precision. Masseck used this method, known as optogenetics, to characterize, for example, the properties of different light-sensitive proteins and identified the ones that are best suited as optogenetic tools. She has analyzed several light-sensitive varieties of the serotonin receptors 5-HT1A and 5-HT2C in great detail. Together with her collaborators, she has demonstrated in several studies that both receptors can control the anxiety behavior of mice.
To investigate the serotonin system more closely, Masseck and her research team is currently developing a sensor that is going to indicate the neurotransmitter in real time. One potential approach involves the integration of a modified form of a green fluorescent protein into a serotonin receptor.
In a brain slice, Olivia Masseck measures the activity of nerve cells in which she switches on their receptors using light stimulation. Via the pipette a red dye diffuses into the cell, rendering them visible in the brain slice. (Copyright: RUB, Damian Gorczany)
This protein produces green light only if it is embedded in a specific spatial structure. If a serotonin molecule binds to a receptor, the receptor changes its three-dimensional conformation. The objective is to integrate the fluorescent protein in the receptor so that its spatial structure changes together with that of the receptor when it binds a serotonin molecule, in such a way that the protein begins to glow.
New optogenetic tools by Julia Weiler
Anxiety and depression are two of the most frequently occurring mental disorders worldwide. Light-activated nerve cells may indicate how they are formed.
Statistically, every fifth individual suffers from depression or anxiety in the course of his or her life. The mechanisms that trigger these disorders are not yet fully understood, despite the fact that researchers have been studying the hypothesis that one of the underlying cause are changes to the level of the neurotransmitter serotonin for 60 years.
“Unfortunately, it is very difficult to understand how the serotonin system works,” says Prof Dr Olivia Masseck, who is junior professor for Super-Resolution Fluorescence Microscopy since the end of April 2016. She intends to fathom the mysteries of the complex system. The number of receptors for the neurotransmitter in the brain amounts to 14 in total, and they occur in different cell types. Consequently, determining the functions that different receptors fulfil in the individual cell types is a complicated task.
In order to fathom the purpose of such receptors, researchers used to observe which functions were inhibited after they had been activated or blocked with the aid of pharmaceutical drugs. However, many substances affect not just one receptor, but several at the same time. Moreover, researchers cannot tell receptors in the individual cell types apart when pharmaceutical drugs have been applied. “It had been impossible to study serotonin signalling pathways at high spatial and temporal resolution,” adds Masseck. Until the development of optogenetics.
“This method has revolutionised neuroscience,” says Olivia Masseck, whose collaborator Prof Dr Stefan Herlitze was one of the pioneers in this field. Optogenetics allows precise control over the activity of specific nerve cells or receptors with light. What sounds like science fiction, is routine at RUB’s Neuroscience Research Department. Masseck: “Until now, we had been passive observers, and monitoring cell activity was all we could do; now, we are able to manipulate it precisely.”
The researcher from Bochum is mainly interested in the 5-HT1A and 5-HT1B receptors, the so-called autoreceptors of the serotonin system. They occur in serotonin-producing cells, where they regulate the amount of released neurotransmitters; that means they determine the serotonin level in the brain.
Normally, 5-HT1A and 5-HT1B are activated when a serotonin molecule bonds to the receptor. The docking triggers a chain reaction in the cell. The effects of this signalling cascade include a reduced activity of the neural cell, which releases less neurotransmitter.
By modifying certain brain cells in the brains of mice, Olivia Masseck successfully activated the 5-HT1Areceptor without the aid of serotonin. She combined it with a visual pigment – so-called opsin. More specifically, she utilised blue or red visual pigments from the cones responsible for colour vision. This is how she generated a serotonin receptor that she could switch on with red or blue light. This method enables the RUB researcher to identify the role the 5-HT1A receptor plays in anxiety and depression.
To this end, she delivered the combined protein made up of light-sensitive opsin and serotonin receptor into the brain of mice using a virus that had been rendered harmless. Like a shuttle, it transports genetic information which contains the blueprint for the combined protein. Once injected into brain tissue, the virus implants the gene for the light-activated receptor in specific nerve cells. There, it is read, and the light-activated receptor is incorporated into the cell membrane.
The researcher was now able to switch the receptors on and off in a living mouse using light. She analysed in what way this manipulation affected the animals’ behaviour in an anxiety test, i.e. Open Field Test. For the purpose of the experiment, she placed individual mice in a large, empty Plexiglas box.
Under normal circumstances, the animals avoid the centre of the brightly-lit box, because it doesn’t offer any cover. Most of the time, they stay close to the walls. When Olivia Masseck switched on the 5-HT1Areceptor using light, the behaviour of the mice changed. They were less anxious and spent more time in the middle of the Plexiglas box.
These results were confirmed in a further test. Olivia Masseck stopped the time it took the mice to eat a food pellet in the middle of a large Plexiglas box. Normal animals waited between six and seven minutes before they ventured into the centre to feed. However, mice whose serotonin receptor was switched on started to feed after one or two minutes. “This is important evidence indicating that the 5-HT1A receptor signalling pathway in the serotonin system is linked to anxiety,” concludes Masseck.
In the next step, the researcher intends to find out in what way depressive behaviour is affected by the activation of the 5-HT1A receptor. “If the animals are exposed to chronic stress, they develop symptoms similar to those in humans with depression,” describes Masseck. “They might, for example, withdraw from social interactions.”
However, just like in humans, this applies to only a certain percentage of the mice. “Not every individual who suffers from chronic stress or experiences negative situations develops depression,” points out Masseck. What happens in the serotonin system of animals that are susceptible to depression, as opposed to that of animals that do not present any depressive symptoms? This is what the researcher intends to find out by deploying the optogenetic methods described above; in addition, she is currently developing a custom-built serotonin sensor.
Olivia Masseck’s assumption is that her findings regarding the neuronal circuits and molecular mechanisms of anxiety and depression are applicable to humans. Mice have similar cell functions, and their nervous system has a similar structure. The neuroscientist expects that optogenetics will one day be deployed in human applications.
“Genetic manipulation of cells for the purpose of controlling them with light might sound like science fiction,” she says, “but I am convinced that optogenetics will be used in human applications in the next decades.” It could, for example, be utilised for deep brain stimulation in Parkinson’s patients, because it facilitates precise activation of the required signalling pathways, with fewer side effects, at that.
“In the first step, optogenetics will be used in therapy of retinal diseases,” believes Olivia Masseck. Researchers are currently conducting experiments aiming at restoring the visual function in blind mice.
Olivia Masseck is aware that her research raises ethical questions. “We have to discuss in which applications we want or don’t want to use these techniques,” she says. Her research demonstrates how easily the lines between science-fiction films and scientific research can blur.
Detect cancer hallmarks with targeted fluorescent probes.
The smart iABP™ targeted imaging probes are based on cysteine cathepsin activity, which are highly expressed in tumor and tumor-associated cells of numerous cancers. Additionally, cathepsins are consistently expressed across multiple tumor types compared to other affinity based probes such as integrin or MMPs, which are inconsistently expressed.
The small molecule iABP™ probes are based on smart, activatable technology:
Penetrates tumor tissues quickly
Gives you superior target localization and retention at the proteolytic site
only fluorescences once it’s bound to the active target giving extremely low background fluorescence and high signal to noise ratio
Eliminates any need to wash prior to staining cells or tissue in ex vivo analysis.
The probes are suitable for use in non-invasive small animal imaging studies, live cell imaging, fluorescence microscopy, flow cytometry and SDS-PAGE applications.
Pinchas Cohen led a team that identified tiny proteins that appear to play a role in controlling how the body ages. (Photo/Beth Newcomb)
A group of six newly discovered proteins may help to divulge secrets of how we age, potentially unlocking insights into diabetes, Alzheimer’s, cancer and other aging-related diseases.
The tiny proteins appear to play several big roles in our bodies’ cells, from decreasing the amount of damaging free radicals and controlling the rate at which cells die to boosting metabolism and helping tissues throughout the body respond better to insulin. The naturally occurring amounts of each protein decrease with age, leading researchers to believe that they play an important role in the aging process and the onset of diseases linked to older age.
The research team led by Pinchas Cohen, dean of the USC Davis School of Gerontology, identified the tiny proteins for the first time and observed their surprising origin from organelles in the cell called mitochondria and their game-changing roles in metabolism and cell survival. This latest finding builds upon prior research by Cohen and his team that uncovered two significant proteins, humanin and MOTS-c, hormones that appear to have significant roles in metabolism and diseases of aging.
Unlike most other proteins, humanin and MOTS-c are encoded in mitochondria, the structure within cells that produces energy from food, instead of in the cell’s nucleus where most genes are contained.
Key functions
Mitochondria have their own small collection of genes, which were once thought to play only minor roles within cells but now appear to have important functions throughout the body. Cohen’s team used computer analysis to see if the part of the mitochondrial genome that provides the code for humanin was coding for other proteins as well. The analysis uncovered the genes for six new proteins, which were dubbed small humanin-like peptides, or SHLPs, 1 through 6 (the name of this hardworking group of proteins is appropriately pronounced “schlep”).
After identifying the six SHLPs and successfully developing antibodies to test for several of them, the team examined both mouse tissues and human cells to determine their abundance in different organs as well as their functions. The proteins were distributed quite differently among organs, which suggests that the proteins have varying functions based on where they are in the body.
Of particular interest is SHLP 2, Cohen said. The protein appears to have profound insulin-sensitizing, anti-diabetic effects as well as potent neuro-protective activity that may emerge as a strategy to combat Alzheimer’s disease. He added that SHLP 6 is also intriguing, with a unique ability to promote cancer cell death and thus potentially target malignant diseases.
“Together with the previously identified mitochondrial peptides, the newly recognized SHLP family expands the understanding of the mitochondria as an intracellular signaling organelle that communicates with the rest of the body to regulate metabolism and cell fate,” Cohen said. “The findings are an important advance that will be ripe for rapid translation into drug development for diseases of aging.”
The study first appeared online in the journal Aging on April 10. Cohen’s research team included collaborators from the Albert Einstein College of Medicine; the findings have been licensed to the biotechnology company CohBar for possible drug development.
The research was supported by a Glenn Foundation Award and National Institutes of Health grants to Cohen (1P01AG034906, 1R01AG 034430, 1R01GM 090311, 1R01ES 020812) and an Ellison/AFAR postdoctoral fellowship to Kelvin Yen. Study authors Laura Cobb, Changhan Lee, Nir Barzilai and Pinchas Cohen are consultants and stockholders of CohBar Inc.
On a blazingly hot morning this past June, a half-dozen scientists convened in a hotel conference room in suburban Maryland for the dress rehearsal of what they saw as a landmark event in the history of aging research. In a few hours, the group would meet with officials at the U.S. Food and Drug Administration (FDA), a few kilometers away, to pitch an unprecedented clinical trial—nothing less than the first test of a drug to specifically target the process of human aging.
“We think this is a groundbreaking, perhaps paradigm-shifting trial,” said Steven Austad, chairman of biology at the University of Alabama, Birmingham, and scientific director of the American Federation for Aging Research (AFAR). After Austad’s brief introductory remarks, a scientist named Nir Barzilai tuned up his PowerPoint and launched into a practice run of the main presentation.
Barzilai is a former Israeli army medical officer and head of a well-known study of centenarians based at the Albert Einstein College of Medicine in the Bronx, New York. To anyone who has seen the ebullient scientist in his natural laboratory habitat, often in a short-sleeved shirt and always cracking jokes, he looked uncharacteristically kempt in a blue blazer and dress khakis. But his practice run kept hitting a historical speed bump. He had barely begun to explain the rationale for the trial when he mentioned, in passing, “lots of unproven, untested treatments under the category of anti-aging.” His colleagues pounced.
“Nir,” interrupted S. Jay Olshansky, a biodemographer of aging from the University of Illinois, Chicago. The phrase “anti-aging … has an association that is negative.”
“I wouldn’t dignify them by calling them ‘treatments,’” added Michael Pollak, director of cancer prevention at McGill University in Montreal, Canada. “They’re products.”
Barzilai, a 59-year-old with a boyish mop of gray hair, wore a contrite grin. “We know the FDA is concerned about this,” he conceded, and deleted the offensive phrase.
Then he proceeded to lay out the details of an ambitious clinical trial. The group—academics all—wanted to conduct a double-blind study of roughly 3000 elderly people; half would get a placebo and half would get an old (indeed, ancient) drug for type 2 diabetes called metformin, which has been shown to modify aging in some animal studies. Because there is still no accepted biomarker for aging, the drug’s success would be judged by an unusual standard—whether it could delay the development of several diseases whose incidence increases dramatically with age: cardiovascular disease, cancer, and cognitive decline, along with mortality. When it comes to these diseases, Barzilai is fond of saying, “aging is a bigger risk factor than all of the other factors combined.”
But the phrase “anti-aging” kept creeping into the rehearsal, and critics kept jumping in. “Okay,” Barzilai said with a laugh when it came up again. “Third time, the death penalty.”
The group’s paranoia about the term “anti-aging” captured both the audacity of the proposed trial and the cultural challenge of venturing into medical territory historically associated with charlatans and quacks. The metformin initiative, which Barzilai is generally credited with spearheading, is unusual by almost any standard of drug development. The people pushing for the trial are all academics, none from industry (although Barzilai is co-founder of a biotech company, CohBar Inc., that is working to develop drugs targeting age-related diseases). The trial would be sponsored by the nonprofit AFAR, not a pharmaceutical company. No one stood to make money if the drug worked, the scientists all claimed; indeed, metformin is not only generic, costing just a few cents a dose, but belongs to a class of drugs that has been part of the human apothecary for 500 years. Patient safety was unlikely to be an issue; millions of diabetics have taken metformin since the 1960s, and its generally mild side effects are well-known.
Finally, the metformin group insisted they didn’t need a cent of federal money to proceed (although they do intend to ask for some). Nor did they need formal approval from FDA to proceed. But they very much wanted the agency’s blessing. By recognizing the merit of such a trial, Barzilai believes, FDA would make aging itself a legitimate target for drug development.
By the time the scientists were done, the rehearsal—which was being filmed for a television documentary—had the feel of a pep rally. They spoke with unguarded optimism. “What we’re talking about here,” Olshansky said, “is a fundamental sea change in how we look at aging and disease.” To Austad, it is “the key, potentially, to saving the health care system.”
As the group piled into a van for the drive to FDA headquarters, there was more talk about setting precedents and opening doors. So it was a little disconcerting when Austad led the delegation up to the main entrance of FDA—and couldn’t get the door open. ……
Mitochondrial Peptides Found in a Preclinical Study Seen to Control Cell Metabolism
CohBar, a developer of mitochondria-based therapeutics, announced that preclinical research by its academic collaborators has found small humanin-like peptides (SHLPs) that can control metabolism and cell survival. The findings have implications for age-related diseases such as Alzheimer’s and cancer.
Researchers discovered the SHLPs by examining the genome of mitochondria with the help of a bioinformatics approach, which identified six peptides. The team then verified the presence of the factors and explored their function in laboratory animals.
CohBar, who have the exclusive license to develop SHLPs into therapeutics, works closely with its academic partners to explore the peptides in preclinical models.
While it was previously believed that mitochondria only have 37 genes, research has revealed that the mitochondrial genome is far more versatile, potentially harboring a multitude of new genes, which can encode peptides acting as cellular signaling factors. The peptides, it has turned out, have shown neuroprotective and anti-inflammatory effects, and act to protect cells in disease-modifying ways in preclinical models of aging.
CohBar’s goal is to bring these peptides to the market as therapies for age-related diseases, such as obesity, type 2 diabetes, cancer, atherosclerosis and neurodegenerative disorders.
“Together with the previously described mitochondrial-derived peptides humanin and MOTS-c, the SHLP family expands our understanding of the role that these peptides play in intracellular signaling throughout the body to regulate both metabolism and cell survival,” Pinchas Cohen, dean of the USC Leonard Davis School of Gerontology, founder and director of CohBar, and the study’s senior author, said in a press release. “These findings further illustrate the enormous potential that mitochondria-based therapeutics could have on treating age-associated diseases like Alzheimer’s and cancer.”
“The pre-clinical evidence continues to confirm that these peptides represent a new class of naturally occurring metabolic regulators,” added Simon Allen, CohBar’s CEO. “They form the foundation of our pipeline of first-in-class treatments for age-related diseases, and we are committed to rapidly advancing them through pre-clinical and clinical activities as we move forward.”
Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers
Laura J. Cobb1,5, Changhan Lee2, Jialin Xiao2, Kelvin Yen2, Richard G. Wong2, Hiromi K. Nakamura1, ….., Derek M. Huffman4, Junxiang Wan2, Radhika Muzumdar3, Nir Barzilai4 , and Pinchas Cohen2 http://www.impactaging.com/papers/v8/n4/full/100943.html
Mitochondria are key players in aging and in the pathogenesis of age-related diseases. Recent mitochondrial transcriptome analyses revealed the existence of multiple small mRNAs transcribed from mitochondrial DNA (mtDNA). Humanin (HN), a peptide encoded in the mtDNA 16S ribosomal RNA region, is a neuroprotective factor. An in silico search revealed six additional peptides in the same region of mtDNA as humanin; we named these peptides small humanin-like peptides (SHLPs). We identified the functional roles for these peptides and the potential mechanisms of action. The SHLPs differed in their ability to regulate cell viability in vitro. We focused on SHLP2 and SHLP3 because they shared similar protective effects with HN. Specifically, they significantly reduced apoptosis and the generation of reactive oxygen species, and improved mitochondrial metabolism in vitro. SHLP2 and SHLP3 also enhanced 3T3-L1 pre-adipocyte differentiation. Systemic hyperinsulinemic-euglycemic clamp studies showed that intracerebrally infused SHLP2 increased glucose uptake and suppressed hepatic glucose production, suggesting that it functions as an insulin sensitizer both peripherally and centrally. Similar to HN, the levels of circulating SHLP2 were found to decrease with age. These results suggest that mitochondria play critical roles in metabolism and survival through the synthesis of mitochondrial peptides, and provide new insights into mitochondrial biology with relevance to aging and human biology.
Human mitochondrial DNA (mtDNA) is a double-stranded, circular molecule of 16,569 bp and contains 37 genes encoding 13 proteins, 22 tRNAs, and 2 rRNAs. Recent mitochondrial transcriptome analyses revealed the existence of small RNAs derived from mtDNA [1]. In 2001, Nishimoto and colleagues identified humanin (HN), a 24-amino-acid peptide encoded from the 16S ribosomal RNA (rRNA) region of mtDNA. HN is a potent neuroprotective factor capable of antagonizing Alzheimer’s disease (AD)-related cellular insults [2]. HN is a component of a novel retrograde signaling pathway from the mitochondria to the nucleus, which is distinct from mitochondrial signaling pathways, such as the SIRT4-AMPK pathway [3]. HN-dependent cellular protection is mediated in part by interacting with and antagonizing pro-apoptotic Bax-related peptides [4] and IGFBP-3 (IGF binding protein 3) [5].
Because of their involvement in energy production and free radical generation, mitochondria likely play a major role in aging and age-related diseases [6–8]. In fact, improvement of mitochondrial function has been shown to ameliorate age-related memory loss in aged mice [9]. Recent studies have shown that HN levels decrease with age, suggesting that HN could play a role in aging and age-related diseases, such as Alzheimer’s disease (AD), atherosclerosis, and diabetes. Along with lower HN levels in the hypothalamus, skeletal muscle, and cortex of older rodents, the circulating levels of HN were found to decline with age in both humans and mice [10]. Notably, circulating HN levels were found to be (i) significantly higher in long-lived Ames dwarf mice but lower in short-lived growth hormone (GH) transgenic mice, (ii) significantly higher in a GH-deficient cohort of patients with Laron syndrome, and (iii) reduced in mice and humans treated with GH or IGF-1 (insulin-like growth factor 1) [11]. Age-dependent declines in the circulating HN levels may be due to higher levels of reactive oxygen species (ROS) that contribute to atherosclerosis development. Using mouse models of atherosclerosis, it was found that HN-treated mice had a reduced disease burden and significant health improvements [12,13]. In addition, HN improved insulin sensitivity, suggesting clinical potential for mitochondrial peptides in diseases of aging [10]. The discovery of HN represents a unique addition to the spectrum of roles that mitochondria play in the cell [14,15]. A second mitochondrial-derived peptide (MDP), MOTS-c (mitochondrial open reading frame of the 12S rRNA-c), has also been shown to have metabolic effects on muscle and may also play a role in aging [16].
We further investigated mtDNA for the presence of other MDPs. Recent technological advances have led to the identification of small open reading frames (sORFs) in the nuclear genomes ofDrosophila[17,18] and mammals [19,20]. Therefore, we attempted to identify novel sORFs using the following approaches: 1) in silico identification of potential sORFs; 2) determination of mRNA expression levels; 3) development of specific antibodies against these novel peptides to allow for peptide detection in cells, organs, and plasma; 4) elucidating the actions of these peptides by performing cell-based assays for mitochondrial function, signaling, viability, and differentiation; and 5) delivering these peptides in vivo to determine their systemic metabolic effects. Focusing on the 16S rRNA region of the mtDNA where the humanin gene is located, we identified six sORFs and named them small humanin-like peptides (SHLPs) 1-6. While surveying the biological effects of SHLPs, we found that SHLP2 and SHLP3 were cytoprotective; therefore, we investigated their effects on apoptosis and metabolism in greater detail. Further, we showed that circulating SHLP2 levels declined with age, similar to HN, suggesting that SHLP2 is involved in aging and age-related disease progression.
SHLP2 and SHLP3 regulate the expression of metabolic and inflammatory markers
Epidemiological studies have demonstrated that increased levels of mediators of inflammation and acute-phase reactants, such as fibrinogen, C-reactive protein (CRP), and IL-6, correlate with the incidence of type 2 diabetes mellitus (T2DM) [34–36]. In humans, anti-inflammatory drugs, such as aspirin and sodium salicylate, reduce fasting plasma glucose levels and ameliorate the symptoms of T2DM. In addition, anti-diabetic drugs, such as fibrates [37] and thiazolidinediones [38], have been found to lower some markers of inflammation. SHLP2 increased the levels of leptin, which is known to improve insulin sensitivity, but had no effect on the levels of the pro-inflammatory cytokines IL-6 and MCP-1. SHLP3 significantly increased the leptin levels, but also elevated IL-6 and MCP-1 levels, which could explain the lack of an in vivo insulin-sensitizing effect of SHLP3. The mechanism by which SHLPs regulate the expression of metabolic and inflammatory markers remains unclear and needs to be further investigated. Furthermore, SHLPs have different effects on inflammatory marker expression, suggesting differential regulation and function of individual SHLPs.
SHLP2 in aging
Mitochondria have been implicated in increased lifespan in several life-extending treatments [39,40]; however, it is not known whether the relationship is correlative or causative [40]. Additionally, it is well known that hormone levels change with aging. For example, levels of aldosterone, calcitonin, growth hormone, and IGF-I decrease with age. Circulating HN levels decline with age in humans and rodents, specifically in the hypothalamus and skeletal muscle of older rats. These changes parallel increases in the incidence of age-associated diseases such as AD and T2DM. The decline in circulating SHLP2 levels with age (Fig. 6), the anti-oxidative stress function of SHLP2 (Fig. 3C), and its neuroprotective effect (Fig. 6B) indicate that SHLP2 has a role in the regulation of aging and age-related diseases.
Conclusion
By analyzing the mitochondrial transcriptome, we found that sORFs from mitochondrial DNA encode functional peptides. We identified many mRNA transcripts within 13 protein-coding mitochondrial genes [1]. Such previously underappreciated sORFs have also been described in the nuclear genome [41]. The MDPs we describe here may represent retrograde communication signals from the mitochondria to the nucleus and may explain important aspects of mitochondrial biology that are implicated in health and longevity.
Larry, John Walker is working on mt proteins dynamics. His rotor – stator mechanism in ATPase synthase, a ‘complex’ that biologist accepted as energy generator is likely wrong. I was suppose to have met him in Germany few years ago. Energy in biological systems has nothing to do with heat. Heat is an outcome of a reaction, meaning that IR spectra accordingly to wave theory is a source of information memorized in water interference with carbon open systems within protein and glyo-proteins complexes as well as genome space-time outcomes. Physically speaking from a pure perspective of science ATP is highly unstable form of phosphate ‘chains’. It cannot hold energy, it is actually in contrary, it is like a resonator, trapping negativity, thus functioning as space propeller by expanding carbon skeleton of protein ‘machines’ Now, we don’t know what is ‘aging’ in a pure physical sense, except that we observe structural changes in what we call complexes. We we know is that proteins are not stationary structures, but highly dynamic forms of matter, seemingly occupying discrete and relative spaces. A piece of mt ATP ase could be discovered in the nucleus as transcription factor. Our notion of operational space in terms of electro dynamics from a motor – stator perspective is now translated toward defining semi conducting and supracoductive strings. The reality of which is so much more fascinating and beautiful as time progresses overally. There are spaces where time does not change, and there are spaces where time walks, and there are spaces, where time flies, and there are spaces where time runs. Amazing, indeed! The story of aging gets a lot deeper that science could even imagine, probably to roots of immortal energy- spaces. We know that matter is transient, that is nearly all living matter, replenishes of about 3 to 7 weeks.
Take a glass full of some kind of liquid, you know the mass of the glass and the mass of the liquid (say wine, beer, water, or milk) You also know to an approximate reality the composition of both. Now lift the glass full of liquid and let it break on a surface of your choice. Depending on the surface pieces of the glass would travel differential from a center projected by the vertical axis of your hand. What technology does today is recollecting those pieces and modelling them to fit in a form again that would resemble a holding device, a glass. The liquid we don’t know exactly how it spilled due the nature of its absorbancy of both surface physics and physical ‘state’ properties. Thus we can say how much approximate energy we have held thinking of m/z as time flight objectives. Each technology can read 1D and approximate the 2D, absolutely lacking computational methodology for 3D dynamic reality. Many scientists confuse space and volume. Volume is a one dimensional characteristic! So is crystalography! BY taking quantum chemical method computing principles following imaginative rules we could approach 2D, however , that is not enough to define 3D. Time we use as a reference frame of clocks we have invented in order to keep track of a sense to observable ‘change’ . But remember, time is absolute and parallel in continuity while energy is discrete , coming in quantum packages, realization of accumulated information. Information is highly redundant we see, so annotating information is an objective to modern days simulations that could predict outcomes of possible parallel realities we call worlds. One could ‘jump’ from one reality to another through guidance of light and water, but what remains unsolved is why people make mistakes, constantly by accusing in name of greed and power , or disobedience of commandments of the Lord!
On Thu, Apr 21, 2016 at 3:41 AM, Leaders in Pharmaceutical Business Intelligence (LPBI) Group wrote:
> larryhbern posted: “New Insights into mtDNA, mitochondrial proteins, > aging, and metabolic control Larry H. Bernstein, MD, FCAP, Curator LPBI > Newly discovered proteins may protect against age-related illnesses The > proteins could play a key role in the ” >
Metabolic features of the cell danger response
– Mitochondria in Health and Disease
The Cell Danger Response (CDR) is defined in terms of an ancient metabolic response to threat.
The CDR encompasses inflammation, innate immunity, oxidative stress, and the ER stress response.
The CDR is maintained by extracellular nucleotide (purinergic) signaling.
Abnormal persistence of the CDR lies at the heart of many chronic diseases.
Antipurinergic therapy (APT) has proven effective in many chronic disorders in animal models
The cell danger response (CDR) is the evolutionarily conserved metabolic response that protects cells and hosts from harm. It is triggered by encounters with chemical, physical, or biological threats that exceed the cellular capacity for homeostasis. The resulting metabolic mismatch between available resources and functional capacity produces a cascade of changes in cellular electron flow, oxygen consumption, redox, membrane fluidity, lipid dynamics, bioenergetics, carbon and sulfur resource allocation, protein folding and aggregation, vitamin availability, metal homeostasis, indole, pterin, 1-carbon and polyamine metabolism, and polymer formation. The first wave of danger signals consists of the release of metabolic intermediates like ATP and ADP, Krebs cycle intermediates, oxygen, and reactive oxygen species (ROS), and is sustained by purinergic signaling. After the danger has been eliminated or neutralized, a choreographed sequence of anti-inflammatory and regenerative pathways is activated to reverse the CDR and to heal. When the CDR persists abnormally, whole body metabolism and the gut microbiome are disturbed, the collective performance of multiple organ systems is impaired, behavior is changed, and chronic disease results. Metabolic memory of past stress encounters is stored in the form of altered mitochondrial and cellular macromolecule content, resulting in an increase in functional reserve capacity through a process known as mitocellular hormesis. The systemic form of the CDR, and its magnified form, the purinergic life-threat response (PLTR), are under direct control by ancient pathways in the brain that are ultimately coordinated by centers in the brainstem. Chemosensory integration of whole body metabolism occurs in the brainstem and is a prerequisite for normal brain, motor, vestibular, sensory, social, and speech development. An understanding of the CDR permits us to reframe old concepts of pathogenesis for a broad array of chronic, developmental, autoimmune, and degenerative disorders. These disorders include autism spectrum disorders (ASD), attention deficit hyperactivity disorder (ADHD), asthma, atopy, gluten and many other food and chemical sensitivity syndromes, emphysema, Tourette’s syndrome, bipolar disorder, schizophrenia, post-traumatic stress disorder (PTSD), chronic traumatic encephalopathy (CTE), traumatic brain injury (TBI), epilepsy, suicidal ideation, organ transplant biology, diabetes, kidney, liver, and heart disease, cancer, Alzheimer and Parkinson disease, and autoimmune disorders like lupus, rheumatoid arthritis, multiple sclerosis, and primary sclerosing cholangitis.
This is a confocal microscopy image of human fibroblasts derived from embryonic stem cells. The nuclei appear in blue, while smaller and more numerous mitochondria appear in red. [Shoukhrat Mitalipov]
Mutations in our mitochondrial DNA tend to be inconspicuous, but they can become more prevalent as we age. They can even vary in frequency from cell to cell. Naturally, some cells will be relatively compromised because they happen to have a higher percentage of mutated mitochondrial DNA. Such cells make a poor basis for stem cell lines. They should be excluded. But how?
To answer this question, a team of scientists scrutinized skin fibroblasts, blood cells, and induced pluripotent stem cells (iPSCs) for mitochondrial genome integrity. When the scientists tested the samples for mitochondrial DNA mutations, the levels of mutations appeared low. But when the scientists sequenced the iPS cell lines, they found higher numbers of mitochondrial DNA mutations, particularly in cells from patients over 60.
The scientists were led by Shoukhrat Mitalipov, Ph.D., director of the Center for Embryonic Cell and Gene Therapy at Oregon Health & Science University, and Taosheng Huang, M.D., a medical geneticist and director of the Mitochondrial Medicine Program at Cincinnati Children’s Hospital. The Mitalipov/Huang-led team also found higher percentages of mitochondria containing mutations within a cell. The higher the load of mutated mitochondrial DNA in a cell, the more compromised the cell’s function.
Since each iPSC line is created from a different cell, each line may contain different types of mitochondrial DNA mutations and mutation loads. To choose the least damaged line, the authors recommend screening multiple lines per patient. “It’s a good idea to check the iPS clones for mitochondrial DNA mutations and make sure you pick a good cell line,” said Dr. Huang.
This recommendation appeared April 14 in the journal Cell Stem Cell, in an article entitled, “Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs.” This article holds that mitochondrial genome integrity is a vital readout in assessing the proficiency of patient-derived regenerative products destined for clinical applications.
“We found that pooled skin and blood mtDNA contained low heteroplasmic point mutations, but a panel of ten individual iPSC lines from each tissue or clonally expanded fibroblasts carried an elevated load of heteroplasmic or homoplasmic mutations, suggesting that somatic mutations randomly arise within individual cells but are not detectable in whole tissues,” wrote the article’s authors. “The frequency of mtDNA defects in iPSCs increased with age, and many mutations were nonsynonymous or resided in RNA coding genes and thus can lead to respiratory defects.”
Potential therapies using stem cells hold tremendous promise for treating human disease. However, defects in the mitochondria could undermine the iPS cells’ ability to repair damaged tissue or organs.
“If you want to use iPS cells in a human, you must check for mutations in the mitochondrial genome,” declared Dr. Huang. “Every single cell can be different. Two cells next to each other could have different mutations or different percentages of mutations.”
Prior to the creation of a therapeutic iPS cell line, a collection of cells is taken from the patient. These cells will be tested for mutations. If the tester uses Sanger sequencing, older technology that is not as sensitive as newer next-generation sequencing, any mutation that occurs in less than 20% of the sample will go undetected. But mitochondrial DNA mutations might occur in less than 20% of mitochondria in the pooled cells. As a result, mutation rates have not been well understood. “These mitochondrial mutations are actually hidden,” explained Dr. Mitalipov.
The mitochondrial genome is relatively small, containing just 37 genes, so screening should be feasible using next generation sequencing, Dr. Mitalipov added. “It should be relatively cheap and do-able.”
Dr. Mitalipov also commented on a more general point, the implications of the current study on illuminating the mechanisms of age-related disease: “Pathogenic mutations in our mitochondrial DNA have long been thought to be a driving force in aging and age-onset diseases, though clear evidence was missing. This foundational knowledge of how cells are damaged in the natural process of aging may help to illuminate the role of mutated mitochondria in degenerative disease.”
A new paperfrom Shoukhrat Mitalipov’s lab on stem cell mitochondria points to a pattern whereby induced pluripotent stem (IPS) cells tend to have more problems if they are from older patients.
What does this paper mean for the stem cell field and could it impact more specifically the clinical applications of IPS cells?
The new paper Kang, et al is entitled “Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs”.
This paper reminds us of the very important realities that mitochondria are key players in stem cell function and that mitochondria have their own genomes that impact that function. A lot of us don’t think about mitochondria and their genome as often as we should.
The paper came to three major scientific conclusions (this from the Highlights section of the paper and also see the graphical abstract for a visual sense of the results overall):
Human iPSC clones derived from elderly adults show accumulation of mtDNA mutations
Fewer mtDNA mutations are present in ESCs and iPSCs derived from younger adults
Accumulated mtDNA mutations can impact metabolic function in iPSCs
Importantly the team looked at IPS cells derived from both blood and skin cells and found that the former were less likely to have mitochondrial mutations.
This study suggests that those teams producing or working with human IPS cells (hIPSCs) should be screening the different lines for mitochondrial mutations. This excellent piece from Sara Reardon on the Mitalipov paper quotes IPS cell expert Jeanne Loring on this very point:
“It’s one of those things most of us don’t think about,” says Jeanne Loring, a stem-cell biologist at the Scripps Research Institute in La Jolla, California. Her lab is working towards using iPS cells to treat Parkinson’s disease, and Loring now plans to go back and examine the mitochondria in her cell lines. She suspects that it will be fairly easy for researchers to screen cells for use in therapies.”
Mitalipov goes further and suggests that his team’s new findings could support the use of human embryonic stem cells (hESC) derived by somatic cell nuclear transfer (SCNT) which would be expected to have mitochondria with fewer mutations. However, as Loring points out in the Reardon article, SCNT is really difficult to successfully perform and only a few labs in the world can do it at present. In that context, working with hIPSC and adding on the additional layer of mitochondrial DNA mutation screening could be more practical.
New York stem cell researcher Dieter Egli, however, is quoted that hIPSC have other differences with hESC as well such as epigenetic differences and he’s quoted in the Reardon piece, “It’s going to be very hard to find a cell line that’s perfect.”
One might reasonably ask both Egli and oneself, “What is a perfect cell line”?
In the end the best approach for use of human pluripotent stem cells of any kind is going to involve a balance between practicality of production and the potentially positive or negative traits of those cells as determined by rigorous validation screening.
With this new paper we’ve just learned more about another layer of screening that is needed. An interesting question is whether adult stem cells such as mesenchymal stromal/stem cells (MSC) also should be screened for mitochondrial mutations. They are often produced from patients who are getting up there in years. I hope that someone will publish on that too.
As to pluripotent cells, I expect that sometimes the best lines, meaning those most perfect for a given clinical application, will be hIPSC (autologous or allogeneic in some instances) and in other cases they may be hESC made from leftover IVF embryos. If SCNT-derived hESC can be more widely produced in an affordable manner and they pass validation as well then those (sometimes called NT-hESC) may also come into play clinically. So far that hasn’t happened for the SCNT cells, but it may over time. …..
Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs
In Brief Mitalipov, Huang, and colleagues show that human iPSCs derived from older adults carry more mitochondrial DNA mutations than those derived from younger individuals. Defects in metabolic function caused by mtDNA mutations suggest careful screening of hiPSC clones for mutational load before clinical application.
Highlights
Human iPSC clones derived from elderly adults show accumulation of mtDNA mutations
Fewer mtDNA mutations are present in ESCs and iPSCs derived from younger adults
Accumulated mtDNA mutations can impact metabolic function in iPSCs
The genetic integrity of iPSCs is an important consideration for therapeutic application. In this study, we examine the accumulation of somatic mitochondrial genome (mtDNA) mutations in skin fibroblasts, blood, and iPSCs derived from young and elderly subjects (24–72 years). We found that pooled skin and blood mtDNA contained low heteroplasmic point mutations, but a panel of ten individual iPSC lines from each tissue or clonally expanded fibroblasts carried an elevated load of heteroplasmic or homoplasmic mutations, suggesting that somatic mutations randomly arise within individual cells but are not detectable in whole tissues. The frequency of mtDNA defects in iPSCs increased with age, and many mutations were non-synonymous or resided in RNA coding genes and thus can lead to respiratory defects. Our results highlight a need to monitor mtDNA mutations in iPSCs, especially those generated from older patients, and to examine the metabolic status of iPSCs destined for clinical applications.
Induced pluripotent stem cells (iPSCs) offer an unlimited source for autologous cell replacement therapies to treat age-associated degenerative diseases. Aging is generally characterized by increased DNA damage and genomic instability (Garinis et al., 2008; Lombard et al., 2005); thus, iPSCs derived from elderly subjects may harbor point mutations and larger genomic rearrangements. Indeed, iPSCs display increased chromosome aberrations (Mayshar et al., 2010), subchromosomal copy number variations (CNVs) (Abyzov et al., 2012; Laurent et al., 2011), and exome mutations (Johannesson et al., 2014), compared to natural embryonic stem cell (ESC) counterparts (Ma et al., 2014). The rate of mtDNA mutations is believed to be at least 10- to 20-fold higher than that observed in the nuclear genome (Wallace, 1994), and often both mutated and wild-type mtDNA (heteroplasmy) can coexist in the same cell (Rossignol et al., 2003). Large deletions are most frequently observed mtDNA abnormalities in aged post-mitotic tissues such as brain, heart, and muscle (Bender et al., 2006; Bua et al., 2006; Corral-Debrinski et al., 1992; Cortopassi et al., 1992; Mohamed et al., 2006) and have been implicated in aging and diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and diabetes (Larsson, 2010; Lin and Beal, 2006; Petersen et al., 2003; Wallace, 2005). In addition, mtDNA point mutations were reported in some tumors and replicating tissues (Chatterjee et al., 2006; Ju et al., 2014; Michikawa et al., 1999; Taylor et al., 2003). However, the extent of mtDNA defects in proliferating peripheral tissues commonly used for iPSC induction, such as skin and blood, is thought to be low and limited to common non-coding variants (Schon et al., 2012; Yao et al., 2015). Accumulation of mtDNA variants in these tissues with age was insignificant (Greaves et al., 2010; Hashizume et al., 2015). Several point mutations were identified in iPSCs generated from the newborn foreskin fibroblasts, although most of these variants were non-coding, common for the general population, and did not affect their metabolic activity (Prigione et al., 2011). Somatic mtDNA mutations may be under-reported secondary to the level of sample interrogation. …..
Figure 2. mtDNA Mutations in Skin Fibroblasts, Blood, and the iPSCs of a 72-YearOld B Subject (A) Sixteen mutations at low heteroplasmy levels were detected in the DNA of PF, while a panel of ten FiPSC lines carried nine mutations, including four that were homoplasmic. Gray rectangles define the mutations shared between PF and FiPSCs. (B) Venn diagram showing only one mutation in FiPSCs shared with PF. (C) All ten FiPSC lines carried between one and five high-heteroplasmy (>15%) mutations. (D) Mutation distribution in whole blood and BiPSCs was similar to that in PF and FiPSCs. Six mutations at low-heteroplasmy levels were observed in blood, while BiPSC lines displayed 21 mutations, including four over the 80% heteroplasmy level. (E) Venn diagram showing four mutations in BiPSCs shared with whole blood and the 17 novel variants. (F) Distribution of mutations in individual BiPSC lines. See also Figures S2 and S3; Table S1; Table S3, sheet 2; and Table S4, sheet 1 ….
Figure 4. Transmission and Distribution of Somatic mtDNA Mutations to iPSCs (A) A total of 112 mtDNA mutations were discovered in parental cells (PF, CF, and blood) from 11 subjects. Of these, 39 variants (35%) were found in corresponding 130 iPSC lines. Among non-transmitted, transmitted, and novel mutations in iPSCs, comparable percentages of variants (68%, 69%, and 79%, respectively) were coding mutations in protein, rRNA, or tRNA genes. This suggests that most pathogenic mutations do not affect iPSC induction. However, certain coding mutations including in ND3, ND4L, and 14 tRNA genes were not detected in iPSCs, suggesting possible pathogenicity. n, the number of mtDNA mutations. Blue font genes were detected in parental cells. (B–D) A total of 80 high heteroplasmic (>15%) variants were detected in the present study in 130 FiPSC or BiPSC lines from 11 subjects. (B) The majority of these variants (76%) were non-synonymous or frame-shift mutations in protein-coding genes or affected rRNA and tRNA genes. (C) More than half of the mutations (56%) were never reported in a database containing whole mtDNA sequences from 26,850 healthy subjects representing the general human population (http://www.mitomap.org/MITOMAP). (D) Most mutations (90%) were never reported in a database containing sequences from healthy subjects with corresponding mtDNA haplotypes. freq., frequent. See also Figure S5 and Tables S3 and S4. ….
sjwilliamspa
Mutations will accumulate over age in mitochondrial DNA, however the current study has the difficulty that the authors could not use patient-age-matched controls, in essence they could only compare induced pluripotent stem cells derived from different patients. This could confound the results but the result with higher frequency of mutation in mtDNA in cells reprogrammed from younger patients is interesting but might limit the ability of autologous regenerative therapy in older patients. However reprogramming, although the method not mentioned here although I am assuming by transfection with lentivirus is a rough procedure, involving multiple dedifferentiation steps. Therefore it is very understandable that cells obtained from elderly patients would respond less favorably to such a rough reprogramming regimen, especially if it produced a higher degree of ROS, which has been shown to alter mtDNA. This is why I feel it is more advantageous to obtain a stem cell population from fat cells and forgo the Oct4, htert, reprogramming with lentiviral vectors.
Salk scientists show how immune receptors clear dead and dysfunctional brain cells and how they might be targets for treating neurodegenerative diseases
By adolescence, your brain already contains most of the neurons that you’ll have for the rest of your life. But a few regions continue to grow new nerve cells—and require the services of cellular sentinels, specialized immune cells that keep the brain safe by getting rid of dead or dysfunctional cells.
Now, Salk scientists have uncovered the surprising extent to which both dying and dead neurons are cleared away, and have identified specific cellular switches that are key to this process. The work was detailed in Nature on April 6, 2016.
“We discovered that receptors on immune cells in the brain are vital for both healthy and injured states,” says Greg Lemke, senior author of the work, a Salk professor of molecular neurobiology and the holder of the Françoise Gilot-Salk Chair. “These receptors could be potential therapeutic targets for neurodegenerative conditions or inflammation-related disorders, such as Parkinson’s disease.”
An accumulation of dead cells (green spots) is seen in the subventricular zone (SVZ)—a neurogenic region—of the brain in a mouse lacking the receptors Mer and Axl. (Blue staining marks all cells.) No green spots are seen in the SVZ from a normal mouse. IMAGE CREDIT: SALK INSTITUTE
Two decades ago, the Lemke lab discovered that immune cells express critical molecules called TAM receptors, which have since become a focus for autoimmune and cancer research in many laboratories. Two of the TAM receptors, dubbed Mer and Axl, help immune cells called macrophages act as garbage collectors, identifying and consuming the over 100 billion dead cells that are generated in a human body every day.
For the current study, the team asked if Mer and Axl did the same job in the brain. Specialized central nervous system macrophages called microglia make up about 10 percent of cells in the brain, where they detect, respond to and destroy pathogens. The researchers removed Axl and Mer in the microglia of otherwise healthy mice. To their surprise, they found that the absence of the two receptors resulted in a large pile-up of dead cells, but not everywhere in the brain. Cellular corpses were seen only in the small regions where the production of new neurons—neurogenesis—is observed.
Many cells die normally during adult neurogenesis, but they are immediately eaten by microglia. “It is very hard to detect even a single dead cell in a normal brain, because they are so efficiently recognized and cleared by microglia,” says Paqui G. Través, a co-first author on the paper and former Salk research associate. “But in the neurogenic regions of mice lacking Mer and Axl, we detected many such cells.”
When the researchers more closely examined this process by tagging the newly growing neurons in mice’s microglia missing Mer and Axl, they noticed something else interesting. New neurons that migrate to the olfactory bulb, or smell center, increased dramatically without Axl and Mer around. Mice lacking the TAM receptors had a 70 percent increase in newly generated cells in the olfactory bulb than normal mice.
How—and to what extent—this unchecked new neural growth affects a mouse’s sense of smell is not yet known, according to Lemke, though it is an area the lab will explore. But the fact that so many more living nerve cells were able to migrate into the olfactory bulb in the absence of the receptors suggests that Mer and Axl have another role aside from clearing dead cells—they may actually also target living, but functionally compromised, cells.
“It appears as though a significant fraction of cell death in neurogenic regions is not due to intrinsic death of the cells but rather is a result of the microglia themselves, which are killing a fraction of the cells by engulfment,” says Lemke. “In other words, some of these newborn neuron progenitors are actually being eaten alive.”
This isn’t necessarily a bad thing in the healthy brain, Lemke adds. The brain produces more neurons than it can use and then prunes back the cells that aren’t needed. However, in an inflamed or diseased brain, the destruction of living cells may backfire.
The Lemke lab did one more series of experiments to understand the role of TAM receptors in disease: they looked at the activity of Axl and Mer in a mouse model of Parkinson’s disease. This model produces a human protein present in an inherited form of the disease that results in a slow degeneration of the brain. The team saw that Axl was far more active in this setting, consistent with other studies showing that increased Axl is a reliable indicator of inflammation in tissues.
In the area of a brain lacking Mer and Axl a ‘trail of death’ is apparent from the migratory pathway from the neurogenic region to the olfactory bulb (smell center of the brain). Blue staining marks all cells, and green spots are dead cells. No green spots are seen in the same section from a normal mouse. IMAGE CREDIT: SALK INSTITUTE
“It seems that we can modify the course of the disease in an animal model by manipulating Axl and Mer,” says Lawrence Fourgeaud, a co-first author on the paper and former Salk research associate. The team cautions that more research needs to be done to determine if modulating the TAM receptors could be a viable therapy for neurodegenerative disease involving microglia.
Other researchers on the paper were Yusuf Tufail, Humberto Leal-Bailey, Erin D. Lew, Patrick G. Burrola, Perri Callaway, Anna Zagórska and Axel Nimmerjahn of the Salk Institute; and Carla V. Rothlin of the Yale University School of Medicine.
Microglia are damage sensors for the central nervous system (CNS), and the phagocytes responsible for routine non-inflammatory clearance of dead brain cells1. Here we show that the TAM receptor tyrosine kinases Mer and Axl2 regulate these microglial functions. We find that adult mice deficient in microglial Mer and Axl exhibit a marked accumulation of apoptotic cells specifically in neurogenic regions of the CNS, and that microglial phagocytosis of the apoptotic cells generated during adult neurogenesis3, 4 is normally driven by both TAM receptor ligands Gas6 and protein S5. Using live two-photon imaging, we demonstrate that the microglial response to brain damage is also TAM-regulated, as TAM-deficient microglia display reduced process motility and delayed convergence to sites of injury. Finally, we show that microglial expression of Axl is prominently upregulated in the inflammatory environment that develops in a mouse model of Parkinson’s disease6. Together, these results establish TAM receptors as both controllers of microglial physiology and potential targets for therapeutic intervention in CNS disease.
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….









 Article Info
Article Info





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
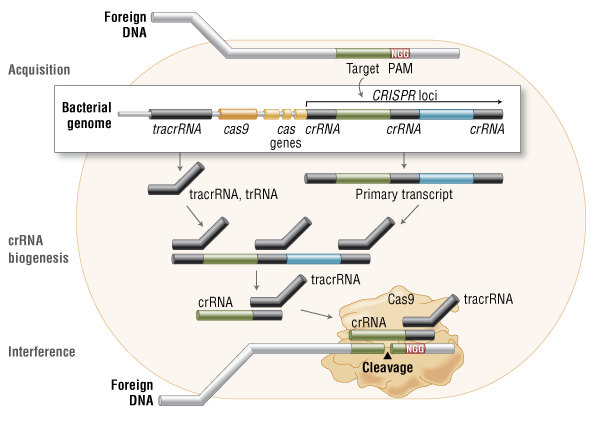{kind=link}
{kind=link}
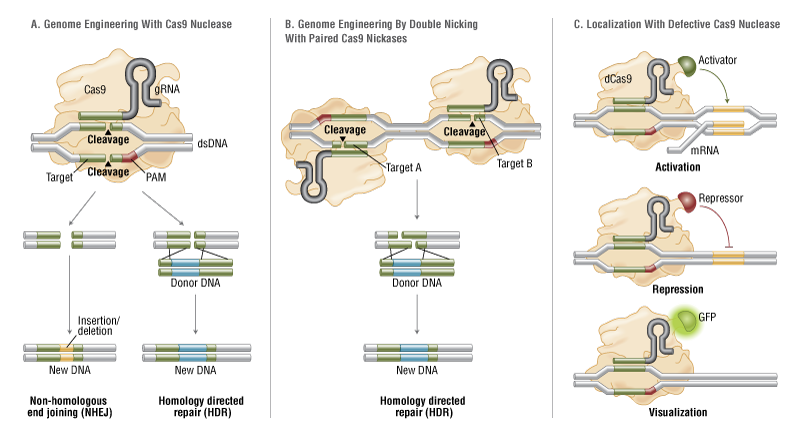{kind=link}
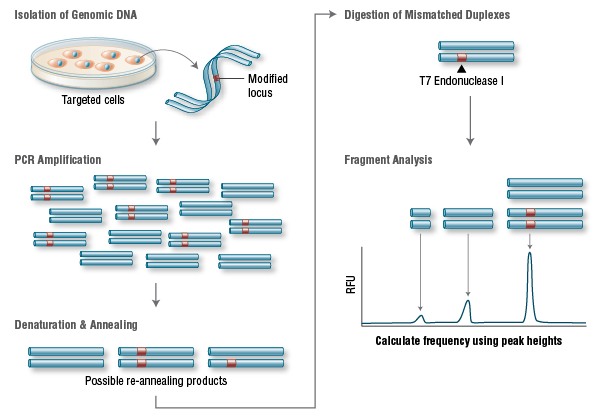{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}