
Perspectives on Nitric Oxide in Disease Mechanisms
available on Kindle Store @ Amazon.com
http://www.amazon.com/dp/B00DINFFYC
PRLog site Press Release

Nitric Oxide Synthase (Photo credit: Wikipedia)
Perspectives on Nitric Oxide in Disease Mechanisms
The Nitric Oxide Discovery, Function, and Targeted Therapy Opportunities
From Discovery to Innovation
From Innovation to Therapeutic Targets
From Therapeutic Targets to Clinical Applications
Leaders in Pharmaceutical Business Intelligence, Scotland
aviralvatsa@gmail.com
and
Triplex Medical Science, Trumbull, CT
Larry.bernstein@gmail.com
Leaders in Pharmaceutical Business Intelligence
avivalev-ari@alum.berkeley.edu
Director and Founder
Editor-in-Chief
Other e-Books in the BioMedicine e-Series
Series A: e-Books on Cardiovascular Diseases
Content Consultant: Justin D Pearlman, MD, PhD, FACC
Volume One: Perspectives on Nitric Oxide
Sr. Editor: Larry Bernstein, MD, FCAP, Editor: Aviral Vatsa, PhD and Content Consultant: Stephen J Williams, PhD
available on Kindle Store @ Amazon.com
http://www.amazon.com/dp/B00DINFFYC
Volume Two: Cardiovascular Original Research: Cases in Methodology Design for Content Co-Curation
Curators: Justin D Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP, Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Three: Etiologies of CVD: Epigenetics, Genetics & Genomics
Curators: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Four: Therapeutic Promise: CVD, Regenerative & Translational Medicine
Curators: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Five: Pharmaco-Therapies for CVD
Volume Curators: Justin D Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Six: Interventional Cardiology and Cardiac Surgery for Disease Diagnosis and Guidance of Treatment
Volume Curators: Justin D Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Seven: Cardiac Imaging
Volume Co-Editors: Justin D Pearlman, MD, PhD, FACC and Adan Sonnenberg, BSc
- Causes
- Risks and Biomarkers
- Therapeutic Implications
In addition to the Seven Volumes of SERIES A: Cardiovascular Diseases, Not included in SERIES A is a Three Volume Series by Dr. Pearlman, Editor, on Cardiovascular Diseases, positioned as Academic Textbooks for Training Residents in Cardiology and Texts for CEU Courses in Cardiology [Hardcover, Softcover, e-Books].
- CVD 1: Causes of Cardiovascular Diseases
- CVD 2: Risk Assessment of Cardiovascular Diseases
- CVD 3: Management of Cardiovascular Diseases
Series B: e-Books on Genomics & Medicine
Content Consultant: Larry H Bernstein, MD, FCAP
Volume 1: Genomics and Individualized Medicine
Sr. Editor: Stephen J Williams, PhD
Editors: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Volume 2: Methodological Breakthroughs in NGS
Editor: Marcus Feldman, PhD, Prof. of Genetics, Stanford University
Volume 3: Institutional Leadership in Genomics
Editors: Marcus Feldman, PhD and Aviva Lev-Ari, PhD, RN
Series C: e-Books on Cancer & Oncology
Content Consultant: Larry H Bernstein, MD, FCAP
Volume 1: Cancer and Genomics
Sr. Editor: Stephen J Williams, PhD
Editors: Ritu Saxena, PhD, Tilda Barliya, PhD
Volume 2: Cancer Therapies: Metabolic, Genomics, Interventional, Immunotherapy and Nanotechnology in Therapy Delivery
Author, Curator and Editor: Larry H Bernstein, MD, FCAP
Guest Authors: Stephen J Williams, PhD, Dror Nir, PhD and Tilda Barliya, PhD, Demet Sag, PhD, Raphael Nir, PhD, Michael Briggs, PhD
Volume 3: Cancer Patients’ Resources on Therapies
Sr. Editor: TBA
Series D: e-Books on BioMedicine
Volume 1: Metabolic Genomics & Pharmaceutics
Author, Curator and Editor: Larry H Bernstein, MD, FCAP
Volume 2: Infectious Diseases
Editor: TBA
Volume 3: Immunology and Therapeutics
Editor: TBA
Series E: Titles in the Strategic Plan for 2015
Volume 1: The Patient’s Voice: Personal Experience with Invasive Medical Procedures
Editor: TBA
Volume 2: Interviews with Scientific Leaders
Editor: TBA
Volume 3: Milestones in Physiology & Discoveries in Medicine and Genomics
Author, Curator and Editor: Larry H Bernstein, MD, FCAP
This book is a comprehensive review of Nitric Oxide, its discovery, function, and related opportunities for Targeted Therapy written by Experts, Authors, Writers. The e-Book’s articles have been published on the Open Access Online Scientific Journal, since April 2012. All new articles on this subject, will continue to be incorporated, as published, in real time in the e-Book which is a live book.
http://www.pharmaceuticalIntelligence.com
The Journal is a scientific, medical and business, multi-expert authoring environment for information syndication in several domains of Life Sciences, Medicine, Pharmaceutical and Healthcare Industries, BioMedicine, Medical Technologies & Devices. Scientific critical interpretations and original articles are written by PhDs, MDs, MD/PhDs, PharmDs, Technical MBAs as Experts, Authors, Writers (EAWs) on an Equity Sharing basis.
List of Contributors
2.1.3
3.1.3
1.4.2, 1.5.1, 1.6.1, 2.2.1, 2.2.2, 3.4.1, 3.4.2, 3.5.1, 7.3, 8.1, 8.2, 8.3, 8.4, 8.5
1.1.2, 1.6.2, 2.2.3, 3.1.1, 3.2.1, 3.2.2, 3.2.3, 3.2.4, 3.3.1, 7.4
2.1.2, 7.2
2.1.1, 7.1, 9.1
1.1.1, 1.2.1, 1.2.2, 1.3.1, 1.4.1, 5.1, 6.1, 6.2, 6.3
3.1.2, 4.1
Introduction
Nitric oxide is a gas produced by cells in the body to communicate requests for changes in neighboring tissue. One such role communicates from cells short of nutrients the need for increased blood delivery, achieved by triggering relaxation of muscles that control nearby blood vessel diameters. If the demand for increased blood vessel diameter is wide spread that can result in a drop in blood pressure. Evolution has yielded additional roles of nitric oxide. The distress signal of nitric oxide indicating inadequate local nutrition flags not only an increase in local blood delivery mediated by vessel dilation, it also promotes the breakdown of any clot fibers that may have contributed to impaired blood delivery, and it contributes to regulation of the amount of muscle in the blood vessel walls.
In the cardiovascular system disruption of NITRIC OXIDE pathways or alterations in NITRIC OXIDE production can result in predisposition to hypertension, hypercholesterolemia, diabetes mellitus, atherosclerosis and thrombosis.
There are three enzyme isoforms of NITRIC OXIDE synthase family that are responsible for generating NITRIC OXIDE in different tissues under various circumstances. The isoforms are:
- endothelial NOS (eNOS) expressed in endothelial cells,
- inducible NOS (iNOS) expressed in macrophages, and
- neuronal NOS (nNOS) expressed in certain neurons and skeletal muscle.
The basic mechanism of action for NITRIC OXIDE production is the same for all three NOS isoforms, and there is a pleiotrophic effect expressed by the eNOS. That is, low levels are anti-inflammatory, and high levels are proinflammatory. Deficiencies of each one of them manifest differently or with varying severity in the body e.g. eNOS deficiency (secondary to high oxidative stress) might lead to hypertension, more severe form of vascular injury to cerebral ischaemia and more severe form of atherosclerosis induced by hypercholesterolemic diet, but high levels are also toxic. So, there is a range of activity that is optimal, but that range is not known.
The activity of eNOS has to be kept in homeostatic balance with iNOS, which itself can be toxic. Steady-state NITRIC OXIDE concentration is a key determinant of its role in stress regulation as precise cellular responses are differentially regulated by specific NITRIC OXIDE concentrations. As indicated to maintain lower NITRIC OXIDE concentrations is favorable for cell survival and proliferation.
The toxicity of high concentrations of NITRIC OXIDE results in mitochondrial damage, cell proliferation with increased aerobic glycolysis, apoptosis, and/or senescence. Free radicals are closely tied to NITRIC OXIDE signaling, decreasing NITRIC OXIDE bioavailability. This is inherently related to endothelial damage and the development of atheromatous plaque, and cardiovascular disease genesis.
NITRIC OXIDES deficiency might show a less severe form of vascular injury to cerebral ischemia and absence of iNOS might lead to reduced hypotension in septic shock (with eNOS unbalanced and toxic).
This book is a series of articles delineating the basic functioning of the NOS isoforms, their production widely by endothelial cells, and the effect of NITRIC OXIDE production by endothelial cells, by neutrophils and macrophages. The effect on intercellular adhesion, and the effect of circulatory shear and turbulence on NITRIC OXIDE production. The essential role of NITRIC OXIDE is seen widely in organ function and in disease development. There is considerable coverage of NITRIC OXIDE and the cardiovascular system, in sepsis and septic shock, in gastrointestinal disease, in renal disease, and on neurological disorders. The final chapter is the essential role of NITRIC OXIDE in carcinogenesis.
The higher solubility of NITRIC OXIDE in membranes than in water has consequences. The reactions of nitric oxide and reactive nitrogen species involving protein as well as lipid oxidation and nitration are key agents in the systemic injury processes. The final protective versus toxic effect of lipid and protein nitration processes during inflammation depends on a delicate balance in cells. The role of protein modification is seen in peroxynitrite reactions with proteins that promote protein aggregation, turnover, and signaling. Soluble guanine cyclase (sGC) is an exquisite sensor of changes in cellular redox status and can adapt accordingly to preserve cytoprotective cGMP-dependent signaling. sGC is a principal receptor for the ubiquitous signaling molecule, nitric oxide (NO), and thereby plays a pivotal role in regulating cellular function in most organ systems. A treatment of this will be developed elsewhere. There is ample discussion of therapeutic targets.
A brief overview of the coverage would include:
- NITRIC OXIDE production from arginine.
2. NITRIC OXIDE chemistry.
3. NITRIC OXIDE signaling in vascular endothelium.
4. The endothelins, and the relationship of endothelin-1 to NITRIC OXIDE.
5. S-nitrosation of NITRIC OXIDE.
6. NITRIC OXIDE, peroxynitrite, and oxidative stress.
7. Inhaled NITRIC OXIDE therapy.
8. NITRIC OXIDE and immunity related diseases.
9. NITRIC OXIDE in cancer.
10. NITRIC OXIDE in coagulation.
11. Vascular damage and cardiovascular disease
12. Regulation of signaling pathways and NITRIC OXIDE
Table of Contents
Chapter 1:
Nitric Oxide Basic Research
1.1 Discovery of Nitric Oxide
1.1.1 Discovery of Nitric Oxide and its Role in Vascular Biology
Aviral Vatsa, PhD, MBBS
1.1.2 Nitric Oxide: The Nobel Prize in Physiology or Medicine
Aviva Lev-Ari, PhD, RN
1.2 Nitric Oxide Synthase(s)
1.2.1 Nitric Oxide: A Short Historic Perspective
Aviral Vatsa, PhD, MBBS
1.2.2 Nitric Oxide: Role in Cardiovascular Health and Disease
Aviral Vatsa, PhD, MBBS
1.3 Endothelial Blood Cell Interactions: Platelet, Leukocyte and Monocyte
1.3.1 Nitric Oxide: Chemistry and Function
Aviral Vatsa, PhD, MBBS
1.4 Signaling Pathways
1.4.1 Nitric Oxide Signaling Pathways
Aviral Vatsa, PhD, MBBS
1.4.2 Nitric Oxide has a Ubiquitous Role in the Regulation of Glycolysis – with a Concomitant Influence on Mitochondrial Function
Larry H. Bernstein, MD, FCAP
1.5 Oxidative Stress
1.5.1 Mitochondrial Damage and Repair under Oxidative Stress
Larry H. Bernstein, MD, FCAP
1.6 Oxygen and Nitrogen Reactive Species
1.6.1 Interaction of Nitric Oxide and Prostacyclin in Vascular Endothelium
Larry H Bernstein, MD, FCAP
1.6.2 Prostacyclin and Nitric Oxide: Adventures in vascular biology – a tale of two mediators
Aviva Lev-Ari, PhD, RN
Chapter 2:
Nitric Oxide and Circulatory Diseases
2.1 Endothelial Dysruption and Denudation
2.1.1 Blood-vessels-generating Stem Cells Discovered
Ritu Saxena, PhD
2.1.2 Differential Distribution of Nitric Oxide – A 3-D Mathematical Model
Anamika Sarkar, PhD
2.1.3 Nitric Oxide Nutritional Remedies for Hypertension and Atherosclerosis. It’s 12AM: Do you know where your electrons are?
Meg Baker, PhD
2.2 Endothelin and ET Receptors
2.2.1 Statins’ Nonlipid Effects on Vascular Endothelium through eNOS Activation
Larry H Bernstein, MD, FCAP
2.2.2 Endothelial Function and Cardiovascular Disease
Larry H Bernstein, MD, FCAP
2.2.3 Endothelin Receptors in Cardiovascular Diseases: The Role of eNOS Stimulation: Observations on Intellectual Property Development for an Unrecognized Future Fast Acting Therapy for Patients at High Risk for Macrovascular Events
Aviva Lev-Ari, PhD, RN
Chapter 3:
Therapeutic Cardiovascular Targets
3.1 Nitric oxide and therapeutic Targets
3.1.1 Cardiovascular Disease (CVD) and the Role of Agent Alternatives in Endothelial Nitric Oxide Synthase (eNOS) Activation and Nitric Oxide Production
Aviva Lev-Ari, PhD, RN
3.1.2 Telling NO to Cardiac Risk
Stephen J Williams, PhD
3.1.3 Nitric Oxide and its Impact on Cardiothoracic Surgery
Tilda Barliya PhD
3.2 Therapeutic opportunities for Endothelial Progenitor Cells
3.2.1 Inhibition of ET-1, ETA and ETA-ETB, Induction of Nitric Oxide production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): eEPCs Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
Aviva Lev-Ari, PhD, RN
3.2.2 Bystolic’s generic Nebivolol – Positive Effect on circulating Endothelial Progenitor Cells Endogenous Augmentation
Aviva Lev-Ari, PhD, RN
3.2.3 Positioning a Therapeutic Concept for Endogenous Augmentation of cEPCs — Therapeutic Indications for Macrovascular Disease: Coronary, Cerebrovascular and Peripheral
Aviva Lev-Ari, PhD, RN
3.2.4 Endothelial Dysfunction, Diminished Availability of cEPCs, Increasing CVD Risk for Macrovascular Disease – Therapeutic Potential of cEPCs
Aviva Lev-Ari, PhD, RN
3.3 Hypertension, Congestive Heart Failure and Endothelin Biomarker
3.3.1 Clinical Trials Results for Endothelin System: Pathophysiological Role in Chronic Heart Failure, Acute Coronary Syndromes and MI – Markers of Disease Severity or Genetic Determination?
Aviva Lev-Ari, PhD, RN
3.4 Hypotension and Shock: Cardiovascular Collapse
3.4.1 Nitric Oxide and Sepsis, Hemodynamic Collapse and the Search for Therapeutic Options
Larry H Bernstein, MD, FCAP
3.4.2 Sepsis, Multi-organ Dysfunction Syndrome, and Septic Shock: A Conundrum of Signaling Pathways Cascading Out of Control
Larry H Bernstein, MD, FCAP
3.5 Hemorrhagic and Thrombo-embolic Events
3.5.1 Nitric Oxide Function in Coagulation
Larry H Bernstein, MD, FCAP
Chapter 4:
Nitric Oxide and Neurodegenerative Diseases
4.1 Nitric Oxide Covalent Modifications: A Putative Therapeutic Target?
Stephen J. Williams, PhD
Chapter 5:
Bone Metabolism
5.1 Nitric Oxide in Bone Metabolism
Aviral Vatsa, PhD, MBBS
Chapter 6:
Nitric Oxide and Systemic Inflammatory Disease
6.1 Nitric Oxide and Immune Responses: Part 1
Aviral Vatsa, PhD, MBBS
6.2 Nitric Oxide and Immune Responses: Part 2
Aviral Vatsa, PhD, MBBS
6.3 Nitric Oxide Production in Systemic Sclerosis
Aviral Vatsa, PhD. MBBS
Chapter 7:
Nitric Oxide: Lung and Alveolar Gas Exchange
7.1 ’Lung on a Chip’
Ritu Saxena, Ph.D.
7.2 Low Bioavailability of Nitric Oxide due to Misbalance in Cell Free Hemoglobin in Sickle Cell Disease – A Computational Model
Anamika Sarkar, Ph.D.
7.3 The Rationale and Use of Inhaled Nitric Oxide in Pulmonary Artery Hypertension and Right Sided Heart Failure
Larry H Bernstein, MD, FCAP
7.4 Transposon-mediated Gene Therapy improves Pulmonary Hemodynamics and attenuates Right Ventricular Hypertrophy: eNOS gene therapy reduces Pulmonary vascular remodeling and Arterial wall hyperplasia
Aviva Lev-Ari, PhD, RN
Chapter 8:
Nitric Oxide and Kidney Dysfunction
8.1 Part I: The Amazing Structure and Adaptive Functioning of the Kidneys: Nitric Oxide
Larry H. Bernstein, MD, FCAP
8.2 Part II: Nitric Oxide and iNOS have Key Roles in Kidney Diseases
Larry H. Bernstein, MD, FCAP
8.3 Part III: The Molecular Biology of Renal Disorders: Nitric Oxide
Larry H. Bernstein, MD, FCAP
8.4 Part IV: New Insights on Nitric Oxide Donors
Larry H. Bernstein, MD, FCAP
8.5 The Essential Role of Nitric Oxide and Therapeutic Nitric Oxide Donor Targets in Renal Pharmacotherapy
Larry H. Bernstein, MD, FCAP
Chapter 9:
Nitric Oxide and Cancer
9.1 Crucial role of Nitric Oxide in Cancer
Ritu Saxena, Ph.D.
Summary
Nitric oxide and its role in vascular biology
Signal transmission by a gas that is produced by one cell, penetrates through membranes and regulates the function of another cell represents an entirely new principle for signaling in biological systems. All compounds that inhibit endothelium-derived relaxation-factor (EDRF) have one property in common, redox activity, which accounts for their inhibitory action on EDRF. One exception is hemoglobin, which inactivates EDRF by binding to it. Furchgott, Ignarro and Murad received the Nobel Prize in Physiology and Medicine for discovery of EDRF in 1998 and demonstrating that it might be nitric oxide (NO) based on a study of the transient relaxations of endothelium-denuded rings of rabbit aorta. These investigators working independently demonstrated that NO is indeed produced by mammalian cells and that NO has specific biological roles in the human body. These studies highlighted the role of NO in cardiovascular, nervous and immune systems. In cardiovascular system NO was shown to cause relaxation of vascular smooth muscle cells causing vasodilatation, in nervous system NO acts as a signaling molecule and in immune system it is used against pathogens by the phagocytosis cells. These pioneering studies opened the path of investigation of role of NO in biology.
NO modulates vascular tone, fibrinolysis, blood pressure and proliferation of vascular smooth muscles. In cardiovascular system disruption of NO pathways or alterations in NO production can result in preponderance to hypertension, hypercholesterolemia, diabetes mellitus, atherosclerosis and thrombosis. The three enzyme isoforms of NO synthase family are responsible for generating NO in different tissues under various circumstances.
Reduction in NO production is implicated as one of the initial factors in initiating endothelial dysfunction. This reduction could be due to
- reduction in eNOS production
- reduction in eNOS enzymatic activity
- reduced bioavailability of NO
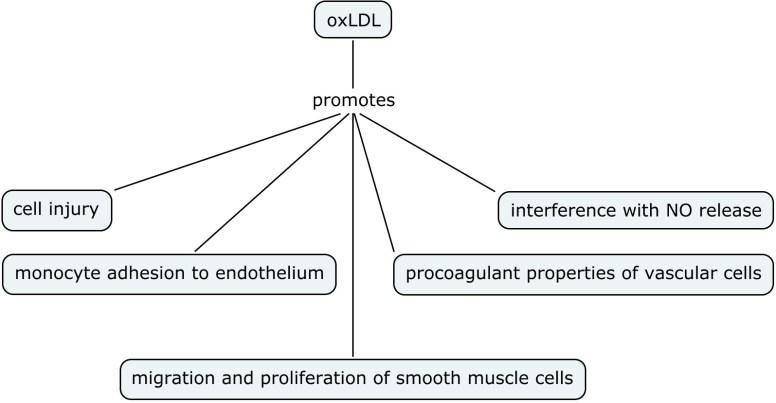
Nitric oxide is one of the smallest molecules involved in physiological functions in the body. It seeks formation of chemical bonds with its targets. Nitric oxide can exert its effects principally by two ways:
- Direct
Direct actions, as the name suggests, result from direct chemical interaction of NO with its targets e.g. with metal complexes, radical species. These actions occur at relatively low NO concentrations (<200 nM)
- Indirect
Indirect actions result from the effects of reactive nitrogen species (RNS) such as NO2 and N2O3. These reactive species are formed by the interaction of NO with superoxide or molecular oxygen. RNS are generally formed at relatively high NO concentrations (>400 nM)
Although it can be tempting for scientists to believe that RNS will always have deleterious effects and NO will have anabolic effects, this is not entirely true as certain RNS mediated actions mediate important signaling steps e.g. thiol oxidation and nitrosation of proteins mediate cell proliferation and survival, and apoptosis respectively.
- Cells subjected to NO concentration between 10-30 nM were associated with cGMP dependent phosphorylation of ERK
- Cells subjected to NO concentration between 30-60 nM were associated with Akt phosphorylation
- Concentration nearing 100 nM resulted in stabilization of hypoxia inducible factor-1
- At nearly 400 nM NO, p53 can be modulated
- >1μM NO, it inhibits mitochondrial respiration
Nitric oxide signaling, oxidative stress, mitochondria, cell damage
Recent data suggests that other NO containing compounds such as S- or N-nitrosoproteins and iron-nitrosyl complexes can be reduced back to produce NO. These NO containing compounds can serve as storage and can reach distant tissues via blood circulation, remote from their place of origin. Hence NO can have both paracrine and ‘endocrine’ effects.
Intracellularly the oxidants present in the cytosol determine the amount of bio-acitivity that NO performs. NO can travel roughly 100 microns from NOS enzymes where it is produced.
NO itself in low concentrations have protective action on mitochondrial signaling of cell death.
The aerobic cell was an advance in evolutionary development, but despite the energetic advantage of using oxygen, the associated toxicity of oxygen abundance required adaptive changes.
Oxidation-reduction reactions that are necessary for catabolic and synthetic reactions, can cumulatively damage the organism associated with cancer, cardiovascular disease, neurodegerative disease, and inflammatory overload. The normal balance between production of pro-oxidant species and destruction by the antioxidant defenses is upset in favor of overproduction of the toxic species, which leads to oxidative stress and disease.
We reviewed the complex interactions and underlying regulatory balances/imbalances between the mechanism of vasorelaxation and vasoconstriction of vascular endothelium by way of nitric oxide (NO), prostacyclin, in response to oxidative stress and intimal injury.
Nitric oxide has a ubiquitous role in the regulation of glycolysis with a concomitant influence on mitochondrial function. The influence on mitochondrial function that is active in endothelium, platelets, vascular smooth muscle and neural cells and the resulting balance has a role in chronic inflammation, asthma, hypertension, sepsis and cancer.
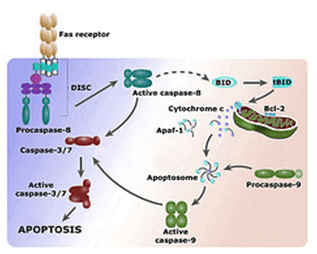
Potential cytotoxic mediators of endothelial cell (EC) apoptosis include increased formation of reactive oxygen and nitrogen species (ROSRNS) during the atherosclerotic process. Nitric oxide (NO) has a biphasic action on oxidative cell killing with low concentrations protecting against cell death, whereas higher concentrations are cytotoxic.
ROS induces mitochondrial DNA damage in ECs, and this damage is accompanied by a decrease in mitochondrial RNA (mtRNA) transcripts, mitochondrial protein synthesis, and cellular ATP levels.
NO and circulatory diseases
Blood vessels arise from endothelial precursors that are thin, flat cells lining the inside of blood vessels forming a monolayer throughout the circulatory system. ECs are defined by specific cell surface markers that characterize their phenotype.
Scientists at the University of Helsinki, Finland, wanted to find out if there exists a rare vascular endothelial stem cell (VESC) population that is capable of producing very high numbers of endothelial daughter cells, and can lead to neovascular growth in adults.
VESCs discovered that reside at the blood vessel wall endothelium are a small population of CD117+ ECs capable of self-renewal. These cells are capable of undergoing clonal expansion unlike the surrounding ECs that bear limited proliferating potential. A single VESC cell isolated from the endothelial population was able to generate functional blood vessels.
Among many important roles of Nitric oxide (NO), one of the key actions is to act as a vasodilator and maintain cardiovascular health. Induction of NO is regulated by signals in tissue as well as endothelium.
Chen et. al. (Med. Biol. Eng. Comp., 2011) developed a 3-D model consisting of two branched arterioles and nine capillaries surrounding the vessels. Their model not only takes into account of the 3-D volume, but also branching effects on blood flow.
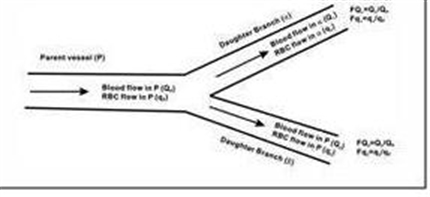
The model indicates that wall shear stress changes depending upon the distribution of RBC in the micro-circulations of blood vessels, lead to differential production of NO along the vascular network.
Endothelial dysfunction, the hallmark of which is reduced activity of endothelial cell derived nitric oxide (NO), is a key factor in developing atherosclerosis and cardiovascular disease. Vascular endothelial cells play a pivotal role in modulation of leukocyte and platelet adherence, thrombogenicity, anticoagulation, and vessel wall contraction and relaxation, so that endothelial dysfunction has become almost a synonym for vascular disease. A single layer of endothelial cells is the only constituent of capillaries, which differ from other vessels, which contain smooth muscle cells and adventitia. Capillaries directly mediate nutritional supply as well as gas exchange within all organs. The failure of the microcirculation leads to tissue apoptosis/necrosis.
Endothelial Function and Cardiovascular Disease
Nitric Oxide, vascular relaxation, vascular integrity, and systemic organ dysfunctions are related to inflammatory and circulatory disorders. In some of these, the relationships are more clear than others, and in other cases the vascular disorders are aligned with serious metabolic disturbances. We focus on the regulation of NO production, NO synthase, and elaborate on the assymetrical dimethylarginine (ADMA) inhibition and cardiovascular disease, including:
- Acute myocardial infarction
- Congestive heart failure
- Chronic renal disease and dialysis
- Hypertension
The endothelium plays a crucial role in the maintenance of vascular tone and structure by means of eNOS, producing the endothelium-derived vasoactive mediator nitric oxide (NO), an endogenous messenger molecule formed in healthy vascular endothelium from the amino acid precursor L-arginine. Nitric oxide synthases (NOS) are the enzymes responsible for nitric oxide (NO) generation. Increased ADMA levels are associated with reduced NO synthesis as assessed by impaired endothelium-dependent vasodilation.
The physiological regulation of nitric oxide (NO) is tied to an endogenous NO synthase inhibitor asymmetrical dimethylarginine (ADMA) and ADMA metabolism by the enzyme DDAH. Plasma ADMA levels are elevated in patients with cardiovascular disease and several large studies have shown that plasma ADMA is an independent biomarker for cardiovascular-related morbidity and mortality.
Several animal models genetically lacking ET-1 and ET receptors have also been used as a tool for determining the physiological and pathophysiological roles of ET-1 and ET receptors in CVD. Further, The discovery of endothelin-1 (ET-1) almost 20 years ago (Yanagisawa et al., 1988) was rapidly followed by prospects that pharmacological manipulation of the ET-1 system might provide powerful new treatments for many clinically significant cardiovascular conditions.
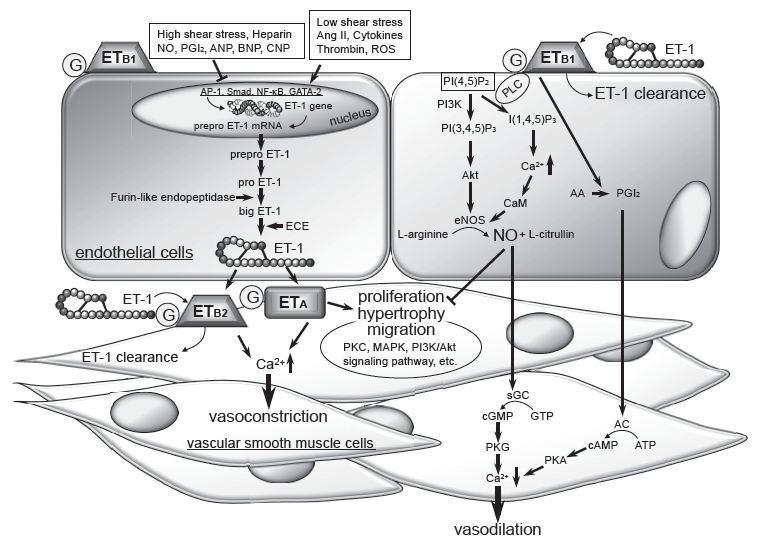
Cardiovascular Disease (CVD) and the Role of agent alternatives in endothelial Nitric Oxide Synthase (eNOS) activation
Sufficient evidence now supports a hypothesis that cholesterol-independent or “pleiotropic” effects of statins improve endothelial dysfunction, effects on angiogenesis, and reduce vascular inflammation. The statins’ cholesterol-independent vascular effects appear to directly restore or improve endothelial function by increasing NO production, promote endothelial repair after arterial injury, and decrease vascular inflammation.
Enhancing the presence of L-Arg or the one or more of the NOS enzymes are obviously essential for NO production. However, NOS enzymes are covalently bound to heme (heme, iron), and flavin co-factors (Vit B2), and require soluble cofactors NADPH (triphosphopyridine nucleotide [nicotinamide adenine diphosphate] containing niacin , Vitamin B3), and BH4 (from Vit B9).
Foods high in Arginine and Citrulline include melons and cucumber, peanuts, salmon, and soy. Arginine is found in varying degrees (3-15% by weight) in all animal proteins.
- Niacin (Vitamin B3), Fibrates and Genistein
- 5-hydroxytryptamine evokes endothelial nitric oxide synthase activation
- 3 orpholinosyndnonimine inhibits 5-hydroxytryptamine induced phosphorylation of nitric oxide synthase in endothelial cells.
- Nebivolol
- time concentration dependence on eNOS reuptake
- dose concentration dependence on NO production
Bystolic’s generic Nebivolol – FDA approved for Treatment of Hypertension since 2008
- Pharmacological agent hypothesized to have positive effect on circulating Endothelial Progenitor Cells (cEPCs)
- endogenous augmentation: Low number of cEPCs found to be associated with high Macrovascular Risk Events
Circulating Endothelial Progenitor Cells (cEPCs)
Cardinal to the study of reendothelialization and neovascularization is the mechanism of action (MOA) of EPCs. It requires exact biological phenotype of the true EPC and its MOA on the endothelium. Is the EPC autocrine or paracrine in its functional role? It is critical to understand this biological unknown for planning therapeutic approaches. Patients with unstable angina and no evidence of cardiac necrosis exhibited increased cEPCs. Systemic inflammation and recognized growth factors may play a role in peripheral mobilization of EPCs in patients with unstable anginal syndromes.
In normal conditions, the vascular endothelium produces and secretes substances that modulate vascular tone and protect the vessel wall from inflammatory cell infiltration, thrombus formation, and vascular smooth muscle cell proliferation (Rubanyi, 1993). Pathologic conditions such as hyperlipidemia, hyperglycemia, and hypertension impair the ability of the vascular endothelium to produce vasodilatory and anti-adhesion moieties and increase the production of vasoconstrictor, pro-adhesion, and pro-thrombotic molecules, leading to elevated vascular tone, enhanced cell adhesion, proliferation of media smooth muscle cells, and propensity toward thrombosis (Drexler & Hornig, 1999),(Endemann & Schiffrin, 2004). Endothelial cell loss and turnover are accelerated in the presence of hemodynamic and biochemical alterations and are a prominent feature of vascular injury resulting from percutaneous coronary intervention.
Since the discovery of NO and its role in vascular biology, a main focus in vascular research has been to create novel mechanisms to use NO to combat neointimal hyperplasia. To date, numerous animal studies have restored NO production to the vasculature and have shown that this inhibits neointimal hyperplasia, improves patency rates, and is safe to the animal.
Nitric Oxide, sepsis and hemodynamic collapse
This document explored the current understanding of sepsis as a cascade of events that involves the microcirculation unevenly because of a differential effect on the large and contiguous intestinal epithelium, secondary effects on cardiopulmonary blood flows and cardiac output, and the role of Nitric Oxide in the emergence of beneficial and potentially deleterious effects. This leads to a substantial body of work on therapeutic targets, either aimed at total inhibition or selective inhibition of NO synthase, and the special role of iNOS.
It is basic to understanding the metabolic and regulatory role of NO in health and disease.
The systemic inflammatory response syndrome (SIRS) is the massive inflammatory reaction resulting from systemic mediator release that may lead to multiple organ dysfunction. I introduce an analysis of the roles of cytokines, cytokine production, and the relationship of cytokine production to the development of SIRS. The article postulates a three-stage development of SIRS, in which stage 1 is a local production of cytokines in response to an injury or infection. Stage 2 is the protective release of a small amount of cytokines into the body’s circulation. Stage 3 is the massive systemic reaction where cytokines turn destructive by compromising the integrity of the capillary walls and flooding end organs.
Sepsis is accompanied by intravascular coagulation with a drop in blood pressure, decreasing platelet count and dissemination of fibrin clots (DIC). The stage is set with a hyper-coagulable state from the pro-thrombotic milieu. There are diverse effects of NO on platelets, the coagulation cascade, and protein-membrane interactions with low flow states, local and systemic inflammatory disease, oxidative stress, and hematologic disorders. It is highly complex as the distinction between the intrinsic and extrinsic pathways become blurred as a result of endothelial shear stress, distinctly different than penetrating or traumatic injury. In addition, there are other factors that come into play.
NO and neurodegenerative disease
The finding that a substance, derived from vascular endothelium, that could control vascular tone and induce smooth muscle relaxation, led to the discovery of nitric oxide (NO) as a major physiological mediator (1) in many cell types and processes.
In addition nitric oxide is involved in macrophage-mediated cytotoxicity, (5)based on the observation the cytotoxic action of macrophages required external arginine, which summarily was converted to citrulline, releasing the nitric oxide involved in the cell-killing process.
However nitric oxide also produces some physiologically, pharmacologically, and pathologically relevant changes, lasting longer time periods, which is the main focus of this article. For example, nitric oxide is important in the development of long term potentiation (a model of learning and memory), neural plasticity, and neurite outgrowth, revealing nitric oxide can induce more permanent changes in cellular and tissue reorganization.
Other pathologic and toxicological responses to nitric oxide include cell death from excitotoxic amino acids (glutamate, kainite), oxidative stress, DNA and protein damage, and disease progression in Alzheimer’s disease, epilepsy, aging, apoptosis and Huntington’s chorea (10-12). These effects persist over longer time frames than the effects which most second messenger systems occur.
There is ample evidence that alterations of GAPDH structure/function exist in these neurodegenerative diseases and evidence that this type of modification may be important in the etiology of neurodegenerative diseases. Further, this modification is unique for GAPDH and would offer a disease-specific target. Finally, the multi-functionality of GAPDH and its modification has the possibility for affecting many processes involved in the disease progression.
Nitric Oxide in bone metabolism
In bone, NO plays a vital role in mechanosensation and mechanotransduction. Osteocytes are widely accepted as the ‘professional’ mechanosensors in bone. They sense external mechanical loads on bone and produce chemical signals such as NO and prostaglandins. NO in turn has been shown to modulate the activity of both bone forming osteoblasts and bone resorbing osteoclasts. NO is essential for load-induced bone formation in vivo. Studies using single gene deletions have shown that NO is an important cog in the wheel for bone metabolism and bone remodeling. Although eNOS isotype is widely implicated in NO production in bone, but recent studies indicate that iNOS isotype might also be involved in NO production in bone in response to mechanical loading.
NO and immune mediated disease
Nitric oxide (NO), reactive nitrogen species (RNS) and reactive oxygen species (ROS) perform dual roles as immuno-toxins and immuno-modulators. An incoming immune signal initiates NO and ROS production both for tackling the pathogens and modulating the downstream immune response via complex signaling pathways. The complexity of these interactions is a reflection of involvement of redox chemistry in biological setting.
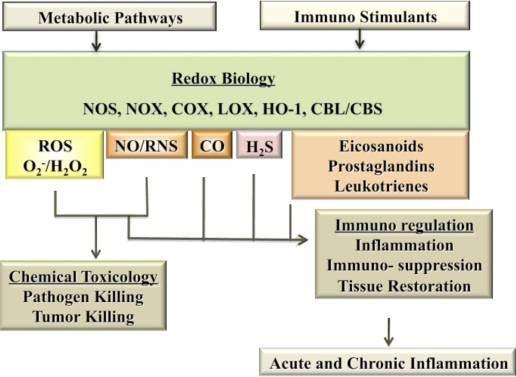
In 1980s a number of authors showed the direct evidence that macrophages made nitrite, nitrates and nitrosamines. It was also shown that NO generated by macrophages could kill leukemia cells (Stuehr and Nathan, 1989). Collectively these studies along with others demonstrated the important role NO plays in immunity and lay the path for further research in understanding the role of redox molecules in immunity.
In addition to eradicating pathogens, NO/RNS and ROS and their chemical interactions act as effective immuno-modulators that regulate many cellular metabolic pathways and tissue repair and proinflammatory pathways.
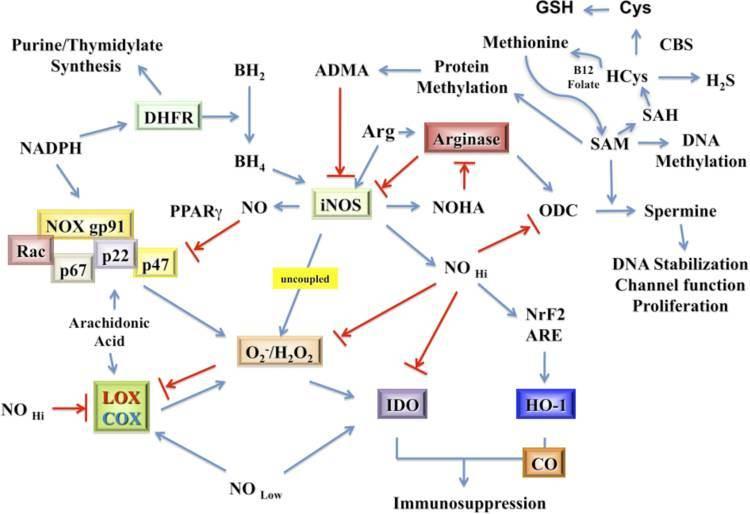
Regulation of iNOS enzyme activity is critical to NO production. Factors such as the availability of arginine, BH4, NADPH, and superoxide affect iNOS activity and thus NO production. In the absence of arginine and BH4 iNOS becomes a O2_/H2O2 generator (Vásquez-Vivar et al., 1999). Further, NO/RNS and ROS actively control innate and adaptive immune signaling by participating in induction, maintenance, and/or termination of proinflammatory and anti-inflammatory signaling.
Systemic sclerosis (SSc) is a type of autoimmune disease when the body’s immune system attacks and destroys body’s healthy tissue. It is characterized by lesions in the vessels and accumulation of collagen in the tissues. The pathogenesis of this disease is not clear, but one of the suggestions is that the endothelium fails to produce NO upon cold stimulation. Physiologically, NO acts as a vasodilator and its deficiency has been implicated in diseases such as hypertension and atherosclerosis.
In the body NO is generated when L-arginine is converted to L-citruline in the presence of NO synthase (NOS) enzyme, molecular oxygen, NADPH, and other cofactors. Principally, three isoenzymes of NOS are present in the body to catalyze the production of NO in various anatomic locations and under various physiological conditions. Three distinct genes encode for the three types of NOS i.e. endothelial (eNOS or NOS-3), neuronal (nNOS or NOS-1), and inducible (iNOS or NOS-2) NOS.
The authors of this study investigated NO metabolites in plasma and PBMC supernatants, and iNOS synthesis in PBMC to see if the level of NO production by peripheral blood mononuclear cells (PBMC) was low in SSc, as this might contribute to the vasodilatory abnormalities observed in this disease.
Nitric Oxide and pulmonary function modeling
Researchers at the Wyss Institute, Harvard, have developed lung-on-a-microfluid chip and shown that it mimics human lung function in response to Interleukin-2 (IL-2) and mechanical strain. The “lung-on-a-chip” reconstitutes the alveolar-capillary interface of the human lung and exposes it to physiological mechanical deformation and flow; in other words, it breathes like a living lung.
Decrease in availability of NO can lead to many complications like pulmonary hypertension. Some of the causes of decrease in NO have been identified as clinical hypertension, right ventricular overload which can lead to cardiac heart failure, low levels of zinc and high levels of cardiac necrosis
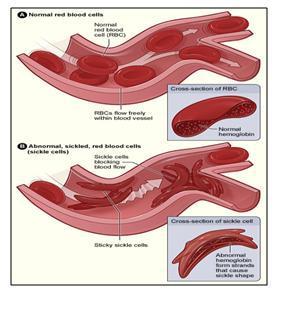
The question to be answered is “what is the quantitative relationship between cell free Hb (released by hemolysis) and depletion of NO”. Deonikar and Kavdia (J. Appl. Physiol., 2012) addressed this question by developing a 2 dimensional Mathematical Model of a single idealized arteriole, with different layers of blood vessels diffusing nutrients to tissue layers.
Inhaled nitric oxide (NO), a mediator of vascular tone produces pulmonary vasodilatation with low pulmonary vascular resistance. The route of administration delivers NO selectively improving oxygenation. Developments in our understanding of the cellular and molecular actions of NO may help to explain the results of randomized controlled trials of inhaled NO.
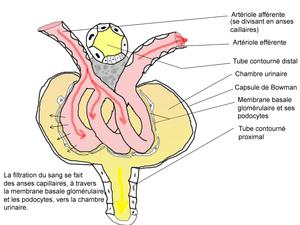
NO and Kidney Dysfunction
Nitric oxide (NO) and its metabolite, peroxynitrite (ONOO-), are involved in renal tubular cell injury. However, while eNOS generated NO is beneficial, inducible iNOS generated NO is a large player in ATM. We learn that both forms have an important counter-balancing effect. NO/ONOO- has an effect of reducing cell adhesion to the basement membrane. The converted ONOO- peroxynitrite has the most damaging effect. The damaged cells in the tubule are a precursor of Tamm Horsfall protein (TFP), which obstructs flow. The exposure to NO donor SIN-1 caused a dose-dependent impairment in cell-matrix adhesions. The .ONOO- generated in the tubular epithelium during ischemia/reperfusion has the potential to impair the adhesion properties of tubular cells, which then may contribute to the tubular obstruction in ARF.
In addition, inflammatory cytokines, released early in sepsis, cause Proximal Tubular Epithelial Cells (PTEC) cytoskeletal damage and alter integrin-dependent cell-matrix adhesion. After exposure of human PTEC to tumor necrosis factor-α, interleukin-1α, and interferon-γ, the actin cytoskeleton is disrupted. The cytokines induce shedding of PTEC, dependent on NO synthesized by inducible NO synthase (iNOS) produced as a result of cytokine actions on PTEC. The major ligand involved in cell anchorage was laminin, probably through interactions with the integrin α3β1, which was down-regulated by the cytokines. The apoptotic effects of pro-inflammatory stimuli are due to the expression of inducible NO synthase. Other experiments indicated:
- Exposure to SIN-1, which generated peroxynitrite (.ONOO) produced a concentration- and time-dependent delayed cell death
- a critical threshold concentration (>440 nM/min) was required for . NO to produce significant cell injury.
- N acetyl cysteine (NAC) given within the first 3 h posttreatment further delayed cell death and increased the intracellular thiol level in SIN-1 but not . NO-exposed cells
- but cell injury from . NO was independent of cGMP, caspases, and superoxide or peroxynitrite formation.
- Exposure of non-.NO-producing cells to .NO or peroxynitrite results in delayed cell death, which,
- although occurring by different mechanisms, is mediated by the loss of intracellular redox balance.
The mechanisms of ARF involve both vascular and tubular factors. An ischemic insult to the kidney will in general be the cause of the ARF. A relative increase in oxygen demand by the tubule is also a factor in renal ischemia. Abnormalities occur in the proximal tubule cytoskeleton that are associated with translocation of Na+/K+-ATPase from the basolateral to the apical membrane., which may explain the decrease in tubular sodium reabsorption in ARF.
Calpain-mediated breakdown products of the actin-binding protein spectrin occur with renal ischemia. Calpain activity increased during hypoxia in isolated proximal tubules. The existence of proteolytic pathways involving cysteine proteases, namely calpain and caspases, may explain the decrease in proximal tubule sodium reabsorption and proteolytic uncoupling of Na+/K+-ATPase from its basolateral membrane anchoring proteins. An antisense oligonucleotide was shown to block the upregulation of iNOS and afford functional protection against acute renal ischemia.
What about the NO donors as therapeutic targets? IFN-α produced dose-dependent and time-dependent decrease in transepithelial resistance (TER) ameliorated by tyrphostin, an inhibitor of phosphotyrosine kinase with increased expression of occludin and E-cadherin. Therefore, IFN-α can directly affect barrier function in renal epithelial cells via overexpression or missorting of the junctional proteins occludin and E-cadherin.
There is agreement that oxygen-derived reactive species are important in renal ischemia-reperfusion (I-R) injury. Treatment with oxygen radical scavengers, antioxidants, and iron chelators such as superoxide dismutase, dimethylthiourea, allopurinol, and deferoxamine are protective. They all directly scavenge or inhibit the formation of peroxynitrite (ONOO−), a highly toxic species derived from nitric oxide (NO) and superoxide. Thus, the protective effects seen with these inhibitors may be due in part to their ability to inhibit ONOO− formation. L-NIL administered to animals subjected to I-R significantly decreased plasma creatinine levels to 1.2 ± 0.10 mg/dl and reduced tubular damage. Selective inhibition of iNOS by L-NIL decreased injury, improved renal function, and decreased apparent ONOO− formation. Therefore, reactive nitrogen species should be considered potential therapeutic targets in the prevention and treatment of renal I-R injury.
NO functions to promote natriuresis and diuresis, contributing to adaptation to variations of dietary salt intake and maintenance of normal blood pressure. A pretreatment with nitric oxide donors or L-arginine may prevent the ischemic acute renal injury. In chronic kidney diseases, the systolic blood pressure is correlated with the plasma level of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase. A reduced production and biological action of nitric oxide is associated with an elevation of arterial pressure, and conversely, an exaggerated activity may represent a compensatory mechanism to mitigate the hypertension.
Adequate medullary tissue oxygenation, in terms of balanced oxygen supply and demand, is dependent on the maintenance of medullary perfusion by adequate cortical perfusion and also on the high rate of O2 consumption required for active electrolyte transport. The sensitivity of the medulla to hypoxic conditions results from high O2 consumption. Renal sodium transport is the main O2-consuming function of the kidney and is closely linked to renal blood flow for sodium transport, particularly in the thick ascending limbs of the loop of Henle and the S3 segments of the proximal tubules. The medulla has been found to be the main site of production of NO in the kidney. In addition to the actions described above, NO appears to be a key regulator of renal tubule cell metabolism by inhibiting the activity of the Na+-K+-2Cl- co-transporter and reducing Na+/H+ exchange.
The kidney is not only a major source of arginine and nitric oxide but NO plays an important role in the
- water and electrolyte balance and
- acid-base physiology and
- many other homeostatic functions in the kidney.
We know that there is an unquestionable role of NO, and a competing balance to be achieved between eNOS, iNOS, an effect on tubular water and cation reabsorption, a role of TNF-α, and consequently an important role in essential/malignant hypertension, with the size of the effect related to the stage of disorder, the amount of interstitial fibrosis, the remaining nephron population, the hypertonicity of the medulla, the vasodilation of the medullary circulation, and the renin-angiotensin-aldosterone system.
Crucial role of Nitric Oxide in Cancer
Nitric oxide is a molecule that acts as a physiological messenger and has been known to regulate a variety of important physiological responses including vasodilation, respiration, cell migration, immune response and apoptosis.
The intracellular localization is relevant for the activity of NOS. In fact, NOSs are subject to specific targeting to sub-cellular compartments (plasma membrane, Golgi, cytosol, nucleus and mitochondria). This is crucial for NO production and specific post-translational modifications of target proteins.
The expression of iNOS induced by inflammatory stimuli coupled with the constitutive expression of nNOS and eNOS may contribute to increased cancer risk. NO can have varied roles in the tumor environment influencing DNA repair, cell cycle, and apoptosis.
Post-translational modifications of proteins by nitration, nitrosation, phosphorylation, acetylation or polyADP-ribosylation could lead to an increase in the cancer risk driving carcinogenesis by altering targets and pathways that are crucial for cancer progression
PUT IT IN CONTEXT OF CANCER CELL MOVEMENT
The contraction of skeletal muscle is triggered by nerve impulses, which stimulate the release of Ca2+ from the sarcoplasmic reticuluma specialized network of internal membranes, similar to the endoplasmic reticulum, that stores high concentrations of Ca2+ ions. The release of Ca2+ from the sarcoplasmic reticulum increases the concentration of Ca2+ in the cytosol from approximately 10-7 to 10-5 M. The increased Ca2+ concentration signals muscle contraction via the action of two accessory proteins bound to the actin filaments: tropomyosin and troponin (Figure 11.25). Tropomyosin is a fibrous protein that binds lengthwise along the groove of actin filaments. In striated muscle, each tropomyosin molecule is bound to troponin, which is a complex of three polypeptides: troponin C (Ca2+-binding), troponin I (inhibitory), and troponin T (tropomyosin-binding). When the concentration of Ca2+ is low, the complex of the troponins with tropomyosin blocks the interaction of actin and myosin, so the muscle does not contract. At high concentrations, Ca2+ binding to troponin C shifts the position of the complex, relieving this inhibition and allowing contraction to proceed.
Figure 11.25
Association of tropomyosin and troponins with actin filaments. (A) Tropomyosin binds lengthwise along actin filaments and, in striated muscle, is associated with a complex of three troponins: troponin I (TnI), troponin C (TnC), and troponin T (TnT). In (more ) Contractile Assemblies of Actin and Myosin in Nonmuscle Cells
Contractile assemblies of actin and myosin, resembling small-scale versions of muscle fibers, are present also in nonmuscle cells. As in muscle, the actin filaments in these contractile assemblies are interdigitated with bipolar filaments of myosin II, consisting of 15 to 20 myosin II molecules, which produce contraction by sliding the actin filaments relative to one another (Figure 11.26). The actin filaments in contractile bundles in nonmuscle cells are also associated with tropomyosin, which facilitates their interaction with myosin II, probably by competing with filamin for binding sites on actin.
Figure 11.26
Contractile assemblies in nonmuscle cells. Bipolar filaments of myosin II produce contraction by sliding actin filaments in opposite directions. Two examples of contractile assemblies in nonmuscle cells, stress fibers and adhesion belts, were discussed earlier with respect to attachment of the actin cytoskeleton to regions of cell-substrate and cell-cell contacts (see Figures 11.13 and 11.14). The contraction of stress fibers produces tension across the cell, allowing the cell to pull on a substrate (e.g., the extracellular matrix) to which it is anchored. The contraction of adhesion belts alters the shape of epithelial cell sheets: a process that is particularly important during embryonic development, when sheets of epithelial cells fold into structures such as tubes.
The most dramatic example of actin-myosin contraction in nonmuscle cells, however, is provided by cytokinesisthe division of a cell into two following mitosis (Figure 11.27). Toward the end of mitosis in animal cells, a contractile ring consisting of actin filaments and myosin II assembles just underneath the plasma membrane. Its contraction pulls the plasma membrane progressively inward, constricting the center of the cell and pinching it in two. Interestingly, the thickness of the contractile ring remains constant as it contracts, implying that actin filaments disassemble as contraction proceeds. The ring then disperses completely following cell division.
Figure 11.27
Cytokinesis. Following completion of mitosis (nuclear division), a contractile ring consisting of actin filaments and myosin II divides the cell in two.
http://www.ncbi.nlm.nih.gov/books/NBK9961/
This is good. I don’t recall seeing it in the original comment. I am very aware of the actin myosin troponin connection in heart and in skeletal muscle, and I did know about the nonmuscle work. I won’t deal with it now, and I have been working with Aviral now online for 2 hours.
I have had a considerable background from way back in atomic orbital theory, physical chemistry, organic chemistry, and the equilibrium necessary for cations and anions. Despite the calcium role in contraction, I would not discount hypomagnesemia in having a disease role because of the intracellular-extracellular connection. The description you pasted reminds me also of a lecture given a few years ago by the Nobel Laureate that year on the mechanism of cell division.
PUT IT IN CONTEXT OF CANCER CELL MOVEMENT
The contraction of skeletal muscle is triggered by nerve impulses, which stimulate the release of Ca2+ from the sarcoplasmic reticuluma specialized network of internal membranes, similar to the endoplasmic reticulum, that stores high concentrations of Ca2+ ions. The release of Ca2+ from the sarcoplasmic reticulum increases the concentration of Ca2+ in the cytosol from approximately 10-7 to 10-5 M. The increased Ca2+ concentration signals muscle contraction via the action of two accessory proteins bound to the actin filaments: tropomyosin and troponin (Figure 11.25). Tropomyosin is a fibrous protein that binds lengthwise along the groove of actin filaments. In striated muscle, each tropomyosin molecule is bound to troponin, which is a complex of three polypeptides: troponin C (Ca2+-binding), troponin I (inhibitory), and troponin T (tropomyosin-binding). When the concentration of Ca2+ is low, the complex of the troponins with tropomyosin blocks the interaction of actin and myosin, so the muscle does not contract. At high concentrations, Ca2+ binding to troponin C shifts the position of the complex, relieving this inhibition and allowing contraction to proceed.
Figure 11.25
Association of tropomyosin and troponins with actin filaments. (A) Tropomyosin binds lengthwise along actin filaments and, in striated muscle, is associated with a complex of three troponins: troponin I (TnI), troponin C (TnC), and troponin T (TnT). In (more ) Contractile Assemblies of Actin and Myosin in Nonmuscle Cells
Contractile assemblies of actin and myosin, resembling small-scale versions of muscle fibers, are present also in nonmuscle cells. As in muscle, the actin filaments in these contractile assemblies are interdigitated with bipolar filaments of myosin II, consisting of 15 to 20 myosin II molecules, which produce contraction by sliding the actin filaments relative to one another (Figure 11.26). The actin filaments in contractile bundles in nonmuscle cells are also associated with tropomyosin, which facilitates their interaction with myosin II, probably by competing with filamin for binding sites on actin.
Figure 11.26
Contractile assemblies in nonmuscle cells. Bipolar filaments of myosin II produce contraction by sliding actin filaments in opposite directions. Two examples of contractile assemblies in nonmuscle cells, stress fibers and adhesion belts, were discussed earlier with respect to attachment of the actin cytoskeleton to regions of cell-substrate and cell-cell contacts (see Figures 11.13 and 11.14). The contraction of stress fibers produces tension across the cell, allowing the cell to pull on a substrate (e.g., the extracellular matrix) to which it is anchored. The contraction of adhesion belts alters the shape of epithelial cell sheets: a process that is particularly important during embryonic development, when sheets of epithelial cells fold into structures such as tubes.
The most dramatic example of actin-myosin contraction in nonmuscle cells, however, is provided by cytokinesisthe division of a cell into two following mitosis (Figure 11.27). Toward the end of mitosis in animal cells, a contractile ring consisting of actin filaments and myosin II assembles just underneath the plasma membrane. Its contraction pulls the plasma membrane progressively inward, constricting the center of the cell and pinching it in two. Interestingly, the thickness of the contractile ring remains constant as it contracts, implying that actin filaments disassemble as contraction proceeds. The ring then disperses completely following cell division.
Figure 11.27
Cytokinesis. Following completion of mitosis (nuclear division), a contractile ring consisting of actin filaments and myosin II divides the cell in two.
http://www.ncbi.nlm.nih.gov/books/NBK9961/
This is good. I don’t recall seeing it in the original comment. I am very aware of the actin myosin troponin connection in heart and in skeletal muscle, and I did know about the nonmuscle work. I won’t deal with it now, and I have been working with Aviral now online for 2 hours.
I have had a considerable background from way back in atomic orbital theory, physical chemistry, organic chemistry, and the equilibrium necessary for cations and anions. Despite the calcium role in contraction, I would not discount hypomagnesemia in having a disease role because of the intracellular-extracellular connection. The description you pasted reminds me also of a lecture given a few years ago by the Nobel Laureate that year on the mechanism of cell division.
PUT IT IN CONTEXT OF CANCER CELL MOVEMENT
The contraction of skeletal muscle is triggered by nerve impulses, which stimulate the release of Ca2+ from the sarcoplasmic reticuluma specialized network of internal membranes, similar to the endoplasmic reticulum, that stores high concentrations of Ca2+ ions. The release of Ca2+ from the sarcoplasmic reticulum increases the concentration of Ca2+ in the cytosol from approximately 10-7 to 10-5 M. The increased Ca2+ concentration signals muscle contraction via the action of two accessory proteins bound to the actin filaments: tropomyosin and troponin (Figure 11.25). Tropomyosin is a fibrous protein that binds lengthwise along the groove of actin filaments. In striated muscle, each tropomyosin molecule is bound to troponin, which is a complex of three polypeptides: troponin C (Ca2+-binding), troponin I (inhibitory), and troponin T (tropomyosin-binding). When the concentration of Ca2+ is low, the complex of the troponins with tropomyosin blocks the interaction of actin and myosin, so the muscle does not contract. At high concentrations, Ca2+ binding to troponin C shifts the position of the complex, relieving this inhibition and allowing contraction to proceed.
Figure 11.25
Association of tropomyosin and troponins with actin filaments. (A) Tropomyosin binds lengthwise along actin filaments and, in striated muscle, is associated with a complex of three troponins: troponin I (TnI), troponin C (TnC), and troponin T (TnT). In (more ) Contractile Assemblies of Actin and Myosin in Nonmuscle Cells
Contractile assemblies of actin and myosin, resembling small-scale versions of muscle fibers, are present also in nonmuscle cells. As in muscle, the actin filaments in these contractile assemblies are interdigitated with bipolar filaments of myosin II, consisting of 15 to 20 myosin II molecules, which produce contraction by sliding the actin filaments relative to one another (Figure 11.26). The actin filaments in contractile bundles in nonmuscle cells are also associated with tropomyosin, which facilitates their interaction with myosin II, probably by competing with filamin for binding sites on actin.
Figure 11.26
Contractile assemblies in nonmuscle cells. Bipolar filaments of myosin II produce contraction by sliding actin filaments in opposite directions. Two examples of contractile assemblies in nonmuscle cells, stress fibers and adhesion belts, were discussed earlier with respect to attachment of the actin cytoskeleton to regions of cell-substrate and cell-cell contacts (see Figures 11.13 and 11.14). The contraction of stress fibers produces tension across the cell, allowing the cell to pull on a substrate (e.g., the extracellular matrix) to which it is anchored. The contraction of adhesion belts alters the shape of epithelial cell sheets: a process that is particularly important during embryonic development, when sheets of epithelial cells fold into structures such as tubes.
The most dramatic example of actin-myosin contraction in nonmuscle cells, however, is provided by cytokinesisthe division of a cell into two following mitosis (Figure 11.27). Toward the end of mitosis in animal cells, a contractile ring consisting of actin filaments and myosin II assembles just underneath the plasma membrane. Its contraction pulls the plasma membrane progressively inward, constricting the center of the cell and pinching it in two. Interestingly, the thickness of the contractile ring remains constant as it contracts, implying that actin filaments disassemble as contraction proceeds. The ring then disperses completely following cell division.
Figure 11.27
Cytokinesis. Following completion of mitosis (nuclear division), a contractile ring consisting of actin filaments and myosin II divides the cell in two.
http://www.ncbi.nlm.nih.gov/books/NBK9961/
This is good. I don’t recall seeing it in the original comment. I am very aware of the actin myosin troponin connection in heart and in skeletal muscle, and I did know about the nonmuscle work. I won’t deal with it now, and I have been working with Aviral now online for 2 hours.
I have had a considerable background from way back in atomic orbital theory, physical chemistry, organic chemistry, and the equilibrium necessary for cations and anions. Despite the calcium role in contraction, I would not discount hypomagnesemia in having a disease role because of the intracellular-extracellular connection. The description you pasted reminds me also of a lecture given a few years ago by the Nobel Laureate that year on the mechanism of cell division.
PUT IT IN CONTEXT OF CANCER CELL MOVEMENT
The contraction of skeletal muscle is triggered by nerve impulses, which stimulate the release of Ca2+ from the sarcoplasmic reticuluma specialized network of internal membranes, similar to the endoplasmic reticulum, that stores high concentrations of Ca2+ ions. The release of Ca2+ from the sarcoplasmic reticulum increases the concentration of Ca2+ in the cytosol from approximately 10-7 to 10-5 M. The increased Ca2+ concentration signals muscle contraction via the action of two accessory proteins bound to the actin filaments: tropomyosin and troponin (Figure 11.25). Tropomyosin is a fibrous protein that binds lengthwise along the groove of actin filaments. In striated muscle, each tropomyosin molecule is bound to troponin, which is a complex of three polypeptides: troponin C (Ca2+-binding), troponin I (inhibitory), and troponin T (tropomyosin-binding). When the concentration of Ca2+ is low, the complex of the troponins with tropomyosin blocks the interaction of actin and myosin, so the muscle does not contract. At high concentrations, Ca2+ binding to troponin C shifts the position of the complex, relieving this inhibition and allowing contraction to proceed.
Figure 11.25
Association of tropomyosin and troponins with actin filaments. (A) Tropomyosin binds lengthwise along actin filaments and, in striated muscle, is associated with a complex of three troponins: troponin I (TnI), troponin C (TnC), and troponin T (TnT). In (more ) Contractile Assemblies of Actin and Myosin in Nonmuscle Cells
Contractile assemblies of actin and myosin, resembling small-scale versions of muscle fibers, are present also in nonmuscle cells. As in muscle, the actin filaments in these contractile assemblies are interdigitated with bipolar filaments of myosin II, consisting of 15 to 20 myosin II molecules, which produce contraction by sliding the actin filaments relative to one another (Figure 11.26). The actin filaments in contractile bundles in nonmuscle cells are also associated with tropomyosin, which facilitates their interaction with myosin II, probably by competing with filamin for binding sites on actin.
Figure 11.26
Contractile assemblies in nonmuscle cells. Bipolar filaments of myosin II produce contraction by sliding actin filaments in opposite directions. Two examples of contractile assemblies in nonmuscle cells, stress fibers and adhesion belts, were discussed earlier with respect to attachment of the actin cytoskeleton to regions of cell-substrate and cell-cell contacts (see Figures 11.13 and 11.14). The contraction of stress fibers produces tension across the cell, allowing the cell to pull on a substrate (e.g., the extracellular matrix) to which it is anchored. The contraction of adhesion belts alters the shape of epithelial cell sheets: a process that is particularly important during embryonic development, when sheets of epithelial cells fold into structures such as tubes.
The most dramatic example of actin-myosin contraction in nonmuscle cells, however, is provided by cytokinesisthe division of a cell into two following mitosis (Figure 11.27). Toward the end of mitosis in animal cells, a contractile ring consisting of actin filaments and myosin II assembles just underneath the plasma membrane. Its contraction pulls the plasma membrane progressively inward, constricting the center of the cell and pinching it in two. Interestingly, the thickness of the contractile ring remains constant as it contracts, implying that actin filaments disassemble as contraction proceeds. The ring then disperses completely following cell division.
Figure 11.27
Cytokinesis. Following completion of mitosis (nuclear division), a contractile ring consisting of actin filaments and myosin II divides the cell in two.
http://www.ncbi.nlm.nih.gov/books/NBK9961/
This is good. I don’t recall seeing it in the original comment. I am very aware of the actin myosin troponin connection in heart and in skeletal muscle, and I did know about the nonmuscle work. I won’t deal with it now, and I have been working with Aviral now online for 2 hours.
I have had a considerable background from way back in atomic orbital theory, physical chemistry, organic chemistry, and the equilibrium necessary for cations and anions. Despite the calcium role in contraction, I would not discount hypomagnesemia in having a disease role because of the intracellular-extracellular connection. The description you pasted reminds me also of a lecture given a few years ago by the Nobel Laureate that year on the mechanism of cell division.
PUT IT IN CONTEXT OF CANCER CELL MOVEMENT
The contraction of skeletal muscle is triggered by nerve impulses, which stimulate the release of Ca2+ from the sarcoplasmic reticuluma specialized network of internal membranes, similar to the endoplasmic reticulum, that stores high concentrations of Ca2+ ions. The release of Ca2+ from the sarcoplasmic reticulum increases the concentration of Ca2+ in the cytosol from approximately 10-7 to 10-5 M. The increased Ca2+ concentration signals muscle contraction via the action of two accessory proteins bound to the actin filaments: tropomyosin and troponin (Figure 11.25). Tropomyosin is a fibrous protein that binds lengthwise along the groove of actin filaments. In striated muscle, each tropomyosin molecule is bound to troponin, which is a complex of three polypeptides: troponin C (Ca2+-binding), troponin I (inhibitory), and troponin T (tropomyosin-binding). When the concentration of Ca2+ is low, the complex of the troponins with tropomyosin blocks the interaction of actin and myosin, so the muscle does not contract. At high concentrations, Ca2+ binding to troponin C shifts the position of the complex, relieving this inhibition and allowing contraction to proceed.
Figure 11.25
Association of tropomyosin and troponins with actin filaments. (A) Tropomyosin binds lengthwise along actin filaments and, in striated muscle, is associated with a complex of three troponins: troponin I (TnI), troponin C (TnC), and troponin T (TnT). In (more ) Contractile Assemblies of Actin and Myosin in Nonmuscle Cells
Contractile assemblies of actin and myosin, resembling small-scale versions of muscle fibers, are present also in nonmuscle cells. As in muscle, the actin filaments in these contractile assemblies are interdigitated with bipolar filaments of myosin II, consisting of 15 to 20 myosin II molecules, which produce contraction by sliding the actin filaments relative to one another (Figure 11.26). The actin filaments in contractile bundles in nonmuscle cells are also associated with tropomyosin, which facilitates their interaction with myosin II, probably by competing with filamin for binding sites on actin.
Figure 11.26
Contractile assemblies in nonmuscle cells. Bipolar filaments of myosin II produce contraction by sliding actin filaments in opposite directions. Two examples of contractile assemblies in nonmuscle cells, stress fibers and adhesion belts, were discussed earlier with respect to attachment of the actin cytoskeleton to regions of cell-substrate and cell-cell contacts (see Figures 11.13 and 11.14). The contraction of stress fibers produces tension across the cell, allowing the cell to pull on a substrate (e.g., the extracellular matrix) to which it is anchored. The contraction of adhesion belts alters the shape of epithelial cell sheets: a process that is particularly important during embryonic development, when sheets of epithelial cells fold into structures such as tubes.
The most dramatic example of actin-myosin contraction in nonmuscle cells, however, is provided by cytokinesisthe division of a cell into two following mitosis (Figure 11.27). Toward the end of mitosis in animal cells, a contractile ring consisting of actin filaments and myosin II assembles just underneath the plasma membrane. Its contraction pulls the plasma membrane progressively inward, constricting the center of the cell and pinching it in two. Interestingly, the thickness of the contractile ring remains constant as it contracts, implying that actin filaments disassemble as contraction proceeds. The ring then disperses completely following cell division.
Figure 11.27
Cytokinesis. Following completion of mitosis (nuclear division), a contractile ring consisting of actin filaments and myosin II divides the cell in two.
http://www.ncbi.nlm.nih.gov/books/NBK9961/
This is good. I don’t recall seeing it in the original comment. I am very aware of the actin myosin troponin connection in heart and in skeletal muscle, and I did know about the nonmuscle work. I won’t deal with it now, and I have been working with Aviral now online for 2 hours.
I have had a considerable background from way back in atomic orbital theory, physical chemistry, organic chemistry, and the equilibrium necessary for cations and anions. Despite the calcium role in contraction, I would not discount hypomagnesemia in having a disease role because of the intracellular-extracellular connection. The description you pasted reminds me also of a lecture given a few years ago by the Nobel Laureate that year on the mechanism of cell division.
I actually consider this amazing blog , âSAME SCIENTIFIC IMPACT: Scientific Publishing –
Open Journals vs. Subscription-based « Pharmaceutical Intelligenceâ, very compelling plus the blog post ended up being a good read.
Many thanks,Annette