Mindful Discoveries
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Schizophrenia and the Synapse
Genetic evidence suggests that overactive synaptic pruning drives development of schizophrenia.
3.2.4 Mindful Discoveries, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Compared to the brains of healthy individuals, those of people with schizophrenia have higher expression of a gene called C4, according to a paper published inNature today (January 27). The gene encodes an immune protein that moonlights in the brain as an eradicator of unwanted neural connections (synapses). The findings, which suggest increased synaptic pruning is a feature of the disease, are a direct extension of genome-wide association studies (GWASs) that pointed to the major histocompatibility (MHC) locus as a key region associated with schizophrenia risk.
“The MHC [locus] is the first and the strongest genetic association for schizophrenia, but many people have said this finding is not useful,” said psychiatric geneticist Patrick Sullivan of the University of North Carolina School of Medicine who was not involved in the study. “The value of [the present study is] to show that not only is it useful, but it opens up new and extremely interesting ideas about the biology and therapeutics of schizophrenia.”
Schizophrenia has a strong genetic component—it runs in families—yet, because of the complex nature of the condition, no specific genes or mutations have been identified. The pathological processes driving the disease remain a mystery.
Researchers have turned to GWASs in the hope of finding specific genetic variations associated with schizophrenia, but even these have not provided clear candidates.
“There are some instances where genome-wide association will literally hit one base [in the DNA],” explained Sullivan. While a 2014 schizophrenia GWAS highlighted the MHC locus on chromosome 6 as a strong risk area, the association spanned hundreds of possible genes and did not reveal specific nucleotide changes. In short, any hope of pinpointing the MHC association was going to be “really challenging,” said geneticist Steve McCarroll of Harvard who led the new study.
Nevertheless, McCarroll and colleagues zeroed in on the particular region of the MHC with the highest GWAS score—the C4 gene—and set about examining how the area’s structural architecture varied in patients and healthy people.
The C4gene can exist in multiple copies (from one to four) on each copy of chromosome 6, and has four different forms: C4A-short, C4B-short, C4A-long, and C4B-long. The researchers first examined the “structural alleles” of the C4 locus—that is, the combinations and copy numbers of the different C4 forms—in healthy individuals. They then examined how these structural alleles related to expression of both C4Aand C4B messenger RNAs (mRNAs) in postmortem brain tissues.From this the researchers had a clear picture of how the architecture of the C4 locus affected expression ofC4A and C4B. Next, they compared DNA from roughly 30,000 schizophrenia patients with that from 35,000 healthy controls, and a correlation emerged: the alleles most strongly associated with schizophrenia were also those that were associated with the highest C4A expression. Measuring C4A mRNA levels in the brains of 35 schizophrenia patients and 70 controls then revealed that, on average, C4A levels in the patients’ brains were 1.4-fold higher.C4 is an immune system “complement” factor—a small secreted protein that assists immune cells in the targeting and removal of pathogens. The discovery of C4’s association to schizophrenia, said McCarroll, “would have seemed random and puzzling if it wasn’t for work . . . showing that other complement components regulate brain wiring.” Indeed, complement protein C3 locates at synapses that are going to be eliminated in the brain, explained McCarroll, “and C4 was known to interact with C3 . . . so we thought well, actually, this might make sense.”McCarroll’s team went on to perform studies in mice that revealed C4 is necessary for C3 to be deposited at synapses. They also showed that the more copies of the C4 gene present in a mouse, the more the animal’s neurons were pruned.Synaptic pruning is a normal part of development and is thought to reflect the process of learning, where the brain strengthens some connections and eradicates others. Interestingly, the brains of deceased schizophrenia patients exhibit reduced neuron density. The new results, therefore, “make a lot of sense,” said Cardiff University’s Andrew Pocklington who did not participate in the work. They also make sense “in terms of the time period when synaptic pruning is occurring, which sort of overlaps with the period of onset for schizophrenia: around adolescence and early adulthood,” he added.
“[C4] has not been on anybody’s radar for having anything to do with schizophrenia, and now it is and there’s a whole bunch of really neat stuff that could happen,” said Sullivan. For one, he suggested, “this molecule could be something that is amenable to therapeutics.”
A. Sekar et al., “Schizophrenia risk from complexvariation of complement component 4,”Nature, http://dx.doi.com:/10.1038/nature16549, 2016.
Tags schizophrenia, neuroscience, gwas, genetics & genomics, disease/medicine and cell & molecular biology
Schizophrenia: From genetics to physiology at last
Ryan S. Dhindsa, & David B. Goldstein
Nature (2016) http://dx.doi.org://10.1038/nature16874
The identification of a set of genetic variations that are strongly associated with the risk of developing schizophrenia provides insights into the neurobiology of this destructive disease.
http://www.nytimes.com/2016/01/28/health/schizophrenia-cause-synaptic-pruning-brain-psychiatry.html
Genetic study provides first-ever insight into biological origin of schizophrenia
Finding explains clinical observations, opens new therapeutic avenues
BROAD INSTITUTE OF MIT AND HARVARD
A landmark study, based on genetic analysis of nearly 65,000 people, has revealed that a person’s risk of schizophrenia is increased if they inherit specific variants in a gene related to “synaptic pruning” — the elimination of connections between neurons. The findings represent the first time that the origin of this devastating psychiatric disease has been causally linked to specific gene variants and a biological process. They also help explain decades-old observations: synaptic pruning is particularly active during adolescence, which is the typical period of onset for schizophrenia symptoms, and brains of schizophrenic patients tend to show fewer connections between neurons. The gene, called complement component 4 (C4), plays a well-known role in the immune system but has now been shown to also play a key role in brain development and schizophrenia risk. The insight may allow future therapeutic strategies to be directed at the disorder’s roots, rather than just its symptoms.
The study, which appears online in the Jan. 27, 2016 issue of Nature, was led by researchers from the Broad Institute’s Stanley Center for Psychiatric Research, Harvard Medical School, and Boston Children’s Hospital. They include senior author Steven McCarroll, director of genetics for the Stanley Center and an associate professor of genetics at Harvard Medical School; Beth Stevens, a neuroscientist and assistant professor of neurology at Boston Children’s Hospital and institute member at the Broad; Michael Carroll, a professor at Harvard Medical School and researcher at Children’s Hospital; and first author Aswin Sekar, an M.D./Ph.D. student at Harvard Medical School.
The study has the potential to reinvigorate translational research on a debilitating disease. Schizophrenia is a devastating psychiatric disorder that afflicts approximately one percent of the population and is characterized by hallucinations, emotional withdrawal, and a decline in cognitive function. These symptoms most frequently begin in patients when they are teenagers or young adults. First described more than 130 years ago, schizophrenia lacks highly effective treatments and has seen few biological or medical breakthroughs over the past half-century. In summer 2014, an international consortium, led by researchers at the Broad Institute’s Stanley Center, identified more than 100 regions in the human genome that carry risk factors for schizophrenia. The newly published study now reports the discovery of the specific gene underlying the strongest of these risk factors and links it to a specific biological process in the brain.
“Since schizophrenia was first described over a century ago, its underlying biology has been a black box, in part because it has been virtually impossible to model the disorder in cells or animals,” said McCarroll. “The human genome is providing a powerful new way in to this disease. Understanding these genetic effects on risk is a way of prying open that black box, peering inside, and starting to see actual biological mechanisms.”
“This study marks a crucial turning point in the fight against mental illness,” said Bruce Cuthbert, acting director of the National Institute of Mental Health. “Because the molecular origins of psychiatric diseases are little-understood, efforts by pharmaceutical companies to pursue new therapeutics are few and far between. This study changes the game. Thanks to this genetic breakthrough we can finally see the potential for clinical tests, early detection, new treatments, and even prevention.”
The path to discovery
The remarkable story of discovery involved the collection of DNA from more than 100,000 people, detailed analysis of complex genetic variation in more than 65,000 human genomes, development of an innovative analytical strategy, examination of postmortem brain samples from hundreds of people, and the use of animal models to show that a protein from the immune system also plays a previously unsuspected role in the brain.
Worldwide search for data finds a clue; new research solves the mystery
Over the past five years, geneticists led by the Broad Institute’s Stanley Center for Psychiatric Research and its collaborators around the world collected more than 100,000 human DNA samples from 30 different countries to locate regions of the human genome harboring genetic variants that increase the risk of schizophrenia. The strongest signal by far was on chromosome 6, in a region of DNA long associated with infectious disease, causing some observers to suggest that schizophrenia might be triggered by an infectious agent. But researchers had no idea which of the hundreds of genes in the region was actually responsible or how it acted.
Based on analyses of the genetic data, McCarroll and Sekar focused on a region containing an unusual gene called complement component 4 (C4). Unlike most genes, C4 has a high degree of structural variability: different people have different numbers of copies and different types of the gene. McCarroll and Sekar developed a new molecular technique to characterize the C4 gene structure in human DNA samples. They also measured C4 gene activity in nearly 700 post-mortem brain samples. They found that the C4 gene structure (DNA) could predict the C4 gene activity (RNA) in each person’s brain – and used this information to infer C4 gene activity from genome data for 65,000 people with and without schizophrenia. These data revealed a striking correlation: patients who had particular structural forms of the C4 gene showed higher expression of that gene and, in turn, had a higher risk of developing schizophrenia.
Connecting cause and effect through neuroscience
But how exactly does C4 — a protein known to mark infectious microbes for destruction by immune cells — affect the risk of schizophrenia?
Answering this question required synthesizing genetics and neurobiology. Beth Stevens, recent recipient of the MacArthur “Genius Grant,” had found that other complement proteins in the immune system also played a role in brain development by studying an experimental model of synaptic pruning in the mouse visual system. Michael Carroll had long studied C4 for its role in immune disease, and developed mice with different numbers of copies of C4. The three labs set out to study the role of C4 in the brain.
They found that C4 played a key role in pruning synapses during maturation of the brain. In particular, they found that C4 was necessary for another protein (a complement component called C3) to be deposited onto synapses, as a signal that the synapses should be pruned. The data also suggested that the more C4 activity an animal had, the more synapses were eliminated in its brain at a key time in development.
The findings may help explain the longstanding mystery of why brains from people with schizophrenia tend to have a thinner cerebral cortex with fewer synapses than unaffected individuals do. The work may also help to explain why the onset of schizophrenia symptoms tends to occur in late adolescence: the human brain normally undergoes widespread synapse pruning during adolescence, especially in the cerebral cortex (the brain’s outer layer, responsible for many aspects of cognition). Excessive synaptic pruning during adolescence and early adulthood, due to increased complement (C4) activity, could lead to the cognitive symptoms seen in schizophrenia.
“Once we had the genetic findings in front of us we started thinking about the possibility that complement molecules are excessively tagging synapses in the developing brain,” Stevens said. “This discovery enriches our understanding of the complement system in brain development and in disease, and we could not have made that leap without the genetics. We’re far from having a treatment based on this, but it’s exciting to think that one day, we might be able to turn down the pruning process in some individuals and decrease their risk.”
Opening a path toward early detection and potential therapies
Beyond providing the first insights into the biological origins of schizophrenia, the work raises the possibility that therapies might someday be developed that could “turn down” the level of synaptic pruning in individuals who show early symptoms of schizophrenia. This would be a dramatically different approach from current medical therapies, which address only a specific symptom of schizophrenia (psychosis) rather than the disorder’s root causes, and which do not stop cognitive decline or other symptoms of the illness. The researchers emphasize that therapies based on these findings are still years down the road. Still, the fact that much is already known about the role of complement proteins in the immune system means that researchers can tap into a wealth of existing knowledge to identify possible therapeutic approaches. For example, anti-complement drugs are already under development for treating other diseases.
“For the first time, the origin of schizophrenia is no longer a complete black box,” said Eric Lander, director of the Broad Institute. “While it’s still early days, we’ve seen the power of understanding the biological mechanism of disease in other settings. Early discoveries about the biological mechanisms of cancer have led to many new treatments and hundreds of additional drug candidates in development. Understanding schizophrenia will similarly accelerate progress against this devastating disease that strikes young people.”
The success of this effort was enabled by catalytic funding from the Stanley Center for Psychiatric Research at the Broad Institute and this paper has been dedicated to the late Ted Stanley. “Through philanthropy, we have been able to take bets on risky science with potentially transformative results,” said Stanley Center Director Steven Hyman. “With support from the late Ted and Vada Stanley, Broad scientists have the freedom to bring together people, capabilities, and resources in innovative ways, at an unprecedented pace.”
“In this area of science, our dream has been to find disease mechanisms that lead to new kinds of treatments,” said McCarroll. “These results show that it is possible to go from genetic data to a new way of thinking about how a disease develops — something that has been greatly needed.”
###
Paper cited: Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. DOI: 10.1038/nature16549
Schizophrenia’s strongest known genetic risk deconstructed
Suspect gene may trigger runaway synaptic pruning during adolescence — NIH-funded study
NIH/NATIONAL INSTITUTE OF MENTAL HEALTH

http://media.eurekalert.org/multimedia_prod/pub/web/107629_web.jpg
The site in Chromosome 6 harboring the gene C4 towers far above other risk-associated areas on schizophrenia’s genomic “skyline,” marking its strongest known genetic influence. The new study is the first to explain how specific gene versions work biologically to confer schizophrenia risk. CREDIT Psychiatric Genomics Consortium
Versions of a gene linked to schizophrenia may trigger runaway pruning of the teenage brain’s still-maturing communications infrastructure, NIH-funded researchers have discovered. People with the illness show fewer such connections between neurons, or synapses. The gene switched on more in people with the suspect versions, who faced a higher risk of developing the disorder, characterized by hallucinations, delusions and impaired thinking and emotions.
“Normally, pruning gets rid of excess connections we no longer need, streamlining our brain for optimal performance, but too much pruning can impair mental function,” explained Thomas Lehner, Ph.D., director of the Office of Genomics Research Coordination of the NIH’s National Institute of Mental Health (NIMH), which co-funded the study along with the Stanley Center for Psychiatric Research at the Broad Institute and other NIH components. “It could help explain schizophrenia’s delayed age-of-onset of symptoms in late adolescence/early adulthood and shrinkage of the brain’s working tissue. Interventions that put the brakes on this pruning process-gone-awry could prove transformative.”
The gene, called C4 (complement component 4), sits in by far the tallest tower on schizophrenia’s genomic “skyline” (see graph below) of more than 100 chromosomal sites harboring known genetic risk for the disorder. Affecting about 1 percent of the population, schizophrenia is known to be as much as 90 percent heritable, yet discovering how specific genes work to confer risk has proven elusive, until now.
A team of scientists led by Steve McCarroll, Ph.D., of the Broad Institute and Harvard Medical School, Boston, leveraged the statistical power conferred by analyzing the genomes of 65,000 people, 700 postmortem brains, and the precision of mouse genetic engineering to discover the secrets of schizophrenia’s strongest known genetic risk. C4’s role represents the most compelling evidence, to date, linking specific gene versions to a biological process that could cause at least some cases of the illness.
“Since schizophrenia was first described over a century ago, its underlying biology has been a black box, in part because it has been virtually impossible to model the disorder in cells or animals,” said McCarroll. “The human genome is providing a powerful new way in to this disease. Understanding these genetic effects on risk is a way of prying open that block box, peering inside and starting to see actual biological mechanisms.”
McCarroll’s team, including Harvard colleagues Beth Stevens, Ph.D., Michael Carroll, Ph.D., and Aswin Sekar, report on their findings online Jan. 27, 2016 in the journal Nature.
A swath of chromosome 6 encompassing several genes known to be involved in immune function emerged as the strongest signal associated with schizophrenia risk in genome-wide analyses by the NIMH-funded Psychiatric Genomics Consortium over the past several years. Yet conventional genetics failed to turn up any specific gene versions there linked to schizophrenia.
To discover how the immune-related site confers risk for the mental disorder, McCarroll’s team mounted a search for “cryptic genetic influences” that might generate “unconventional signals.” C4, a gene with known roles in immunity, emerged as a prime suspect because it is unusually variable across individuals. It is not unusual for people to have different numbers of copies of the gene and distinct DNA sequences that result in the gene working differently.
The researchers dug deeply into the complexities of how such structural variation relates to the gene’s level of expression and how that, in turn, might relate to schizophrenia. They discovered structurally distinct versions that affect expression of two main forms of the gene in the brain. The more a version resulted in expression of one of the forms, called C4A, the more it was associated with schizophrenia. The more a person had the suspect versions, the more C4 switched on and the higher their risk of developing schizophrenia. Moreover, in the human brain, the C4 protein turned out to be most prevalent in the cellular machinery that supports connections between neurons.
Adapting mouse molecular genetics techniques for studying synaptic pruning and C4’s role in immune function, the researchers also discovered a previously unknown role for C4 in brain development. During critical periods of postnatal brain maturation, C4 tags a synapse for pruning by depositing a sister protein in it called C3. Again, the more C4 got switched on, the more synapses got eliminated.
In humans, such streamlining/pruning occurs as the brain develops to full maturity in the late teens/early adulthood – conspicuously corresponding to the age-of-onset of schizophrenia symptoms.
Future treatments designed to suppress excessive levels of pruning by counteracting runaway C4 in at risk individuals might nip in the bud a process that could otherwise develop into psychotic illness, suggest the researchers. And thanks to the head start gained in understanding the role of such complement proteins in immune function, such agents are already in development, they note.
“This study marks a crucial turning point in the fight against mental illness. It changes the game,” added acting NIMH director Bruce Cuthbert, Ph.D. “Thanks to this genetic breakthrough, we can finally see the potential for clinical tests, early detection, new treatments and even prevention.”
###
VIDEO: Opening Schizophrenia’s Black Box https://youtu.be/s0y4equOTLg
Reference: Sekar A, Biala AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K Presumey J Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Daly MJ, Carroll MC, Stevens B, McCarroll SA. Schizophrenia risk from complex variation of complement component 4.Nature. Jan 27, 2016. DOI: 10.1038/nature16549.
Schizophrenia risk from complex variation of complement component 4
Aswin Sekar, Allison R. Bialas, Heather de Rivera, Avery Davis, Timothy R. Hammond, …., Michael C. Carroll, Beth Stevens & Steven A. McCarroll
Nature(2016) http://dx.doi.org:/10.1038/nature16549
a, Location of the C4 genes within the major histocompatibility complex (MHC) locus on human chromosome 6. b, Human C4 exists as two paralogous genes (isotypes), C4A and C4B; the encoded proteins are distinguished at a key site
Scientists have pinpointed the cells that are likely to trigger common brain disorders, including Alzheimer’s disease, Multiple Sclerosis and intellectual disabilities.
It is the first time researchers have been able to identify the particular cell types that malfunction in a wide range of brain diseases.
Scientists say the findings offer a roadmap for the development of new therapies to target the conditions.
The researchers from the University of Edinburgh’s Centre for Clinical Brain Sciences used advanced gene analysis techniques to investigate which genes were switched on in specific types of brain cells.
They then compared this information with genes that are known to be linked to each of the most common brain conditions — Alzheimer’s disease, anxiety disorders, autism, intellectual disability, multiple sclerosis, schizophrenia and epilepsy.
Their findings reveal that for some conditions, the support cells rather than the neurons that transmit messages in the brain are most likely to be the first affected.
Alzheimer’s disease, for example, is characterised by damage to the neurons. Previous efforts to treat the condition have focused on trying to repair this damage.
The study found that a different cell type — called microglial cells — are responsible for triggering Alzheimer’s and that damage to the neurons is a secondary symptom of disease progression.
Researchers say that developing medicines that target microglial cells could offer hope for treating the illness.
The approach could also be used to find new treatment targets for other diseases that have a genetic basis, the researchers say.
Dr Nathan Skene, who carried out the study with Professor Seth Grant, said: “The brain is the most complex organ made up from a tangle of many cell types and sorting out which of these cells go wrong in disease is of critical importance to developing new medicines.”
Professor Seth Grant said: “We are in the midst of scientific revolution where advanced molecular methods are disentangling the Gordian Knot of the brain and completely unexpected new pathways to solving diseases are emerging. There is a pressing need to exploit the remarkable insights from the study.”
Quantitative multimodal multiparametric imaging in Alzheimer’s disease
Qian Zhao, Xueqi Chen, Yun Zhou Brain Informatics http://link.springer.com/article/10.1007/s40708-015-0028-9
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, causing changes in memory, thinking, and other dysfunction of brain functions. More and more people are suffering from the disease. Early neuroimaging techniques of AD are needed to develop. This review provides a preliminary summary of the various neuroimaging techniques that have been explored for in vivo imaging of AD. Recent advances in magnetic resonance (MR) techniques, such as functional MR imaging (fMRI) and diffusion MRI, give opportunities to display not only anatomy and atrophy of the medial temporal lobe, but also at microstructural alterations or perfusion disturbance within the AD lesions. Positron emission tomography (PET) imaging has become the subject of intense research for the diagnosis and facilitation of drug development of AD in both animal models and human trials due to its non-invasive and translational characteristic. Fluorodeoxyglucose (FDG) PET and amyloid PET are applied in clinics and research departments. Amyloid beta (Aβ) imaging using PET has been recognized as one of the most important methods for the early diagnosis of AD, and numerous candidate compounds have been tested for Aβ imaging. Besides in vivo imaging method, a lot of ex vivo modalities are being used in the AD researches. Multiphoton laser scanning microscopy, neuroimaging of metals, and several metal bioimaging methods are also mentioned here. More and more multimodality and multiparametric neuroimaging techniques should improve our understanding of brain function and open new insights into the pathophysiology of AD. We expect exciting results will emerge from new neuroimaging applications that will provide scientific and medical benefits.
Keywords – Alzheimer’s disease Neuroimaging PET MRI Amyloid beta Multimodal
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that gradually destroys brain cells, causing changes in memory, thinking, and other dysfunction of brain functions [1]. AD is considered to a prolonged preclinical stage where neuropathological changes precede the clinical symptoms [2]. An estimation of 35 million people worldwide is living with this disease. If effective treatments are not discovered in a timely fashion, the number of AD cases is anticipated to rise to 113 million by 2050 [3].
Amyloid beta (Aβ) and tau are two of the major biomarkers of AD, and have important and different roles in association with the progression of AD pathophysiology. Jack et al. established hypothetical models of the major biomarkers of AD. By renewing and modifying the models, they found that the two major proteinopathies underlying AD biomarker changes, Aβ and tau, may be initiated independently in late onset AD where they hypothesize that an incident Aβ pathophysiology can accelerate an antecedent limbic and brainstem tauopathy [4]. MRI technique was used in the article, which revealed that the level of Aβ load was associated with a shorter time-to-progression of AD [5]. This warrants an urgent need to develop early neuroimaging techniques of AD neuropathology that can detect and predict the disease before the onset of dementia, monitor therapeutic efficacy in halting and slowing down progression in the earlier stage of the disease.
There have been various reports on the imaging assessments of AD. Some measurements reflect the pathology of AD directly, including positron emission tomography (PET) amyloid imaging and cerebrospinal fluid (CSF) beta-amyloid 42 (Aβ42), while others reflect neuronal injury associated with AD indirectly, including CSF tau (total and phosphorylated tau), fluorodeoxy-d-glucose (FDG)-PET, and MRI. AD Neuroimaging Initiative (ADNI) has been to establish the optimal panel of clinical assessments, MRI and PET imaging measures, as well as other biomarkers from blood and CSF, to inform clinical trial design for AD therapeutic development. At the same time, it has been highly productive in generating a wealth of data for elucidating disease mechanisms occurring during early stages of preclinical and prodromal AD [6].
Single neuroimaging often reflects limit information of AD. As a result, multimodal neuroimaging is widely used in neuroscience researches, as it overcomes the limitations of individual modalities. Multimodal multiparametric imaging mean the combination of different imaging techniques, such as PET, MRI, simultaneously or separately. The multimodal multiparametric imaging enables the visualization and quantitative analysis of the alterations in brain structure and function, such as PET/CT, and PET/MRI. [7]. In this review article, we summarize and discuss the main applications, findings, perspectives as well as advantages and challenges of different neuroimaging in AD, especially MRI and PET imaging.
2 Magnetic resonance imaging
MRI demonstrates specific volume loss or cortical atrophy patterns with disease progression in AD patients [8–10]. There are several MRI techniques and analysis methods used in clinical and scientific research of AD. Recent advances in MR techniques, such as functional MRI (fMRI) and diffusion MRI, depict not only anatomy and atrophy of the medial temporal lobe (MTL), but also microstructural alterations or perfusion disturbance within this region.
2.1 Functional MRI
Because of the cognitive reserve (CR), the relationship between severity of AD patients’ brain damage and corresponding clinical symptoms is not always paralleled [11, 12]. Recently, resting-state fMRI (RS-fMRI) is popular for its ability to map brain functional connectivity non-invasively [13]. By using RS-fMRI, Bozzali et al. reported that the CR played a role in modulating the effect of AD pathology on default mode network functional connectivity, which account for the variable clinical symptoms of AD [14]. Moreover, AD patients with higher educated experience were able to recruit compensatory neural mechanisms, which can be measured using RS-fMRI. Arterial spin-labeled (ASL) MRI is another functional brain imaging modality, which measures cerebral blood flow (CBF) by magnetically labeled arterial blood water following through the carotid and vertebral arteries as an endogenous contrast medium. Several studies have concluded the characteristics of CBF changes in AD patients using ASL-MRI [15–17].
At some point in time, sufficient brain damage accumulates to result in cognitive symptoms and impairment. Mild cognitive impairment (MCI) is a condition in which subjects are usually only mildly impaired in memory with relative preservation of other cognitive domains and functional activities and do not meet the criteria for dementia [18], or as the prodromal state AD [19]. MCI patients are at a higher risk of developing AD and up to 15 % convert to AD per year [18]. Binnewijzend et al. have reported the pseudocontinuous ASL could distinguish both MCI and AD from healthy controls, and be used in the early diagnosis of AD [20]. In their continuous study, they used quantitative whole brain pseudocontinuous ASL to compare regional CBF (rCBF) distribution patterns in different types of dementia, and concluded that ASL-MRI could be a non-invasive and easily accessible alternative to FDG-PET imaging in the assessment of CBF of AD patients [21].
2.2 Structure MRI
Structural MRI (sMRI) has already been a reliable imaging method in the clinical diagnosis of AD, characterized as gray matter reduction and ventricular enlargement in standard T1-weighted sequences [9]. Locus coeruleus (LC) and substantia nigra (SN) degeneration was seen in AD. By using new quantitative calculating method, Chen et al. presented a new quantitative neuromelanin MRI approach for simultaneous measurement of locus LC and SN of brainstem in living human subjects [22]. The approach they used demonstrated advantages in image acquisition, pre-processing, and quantitative analysis. Numerous transgenic animal models of amyloidosis are available, which can manipulate a lot of neuropathological features of AD progression from the deposition of β-amyloid [23]. Braakman et al. demonstrated the dynamics of amyloid plaque formation and development in a serial MRI study in a transgenic mouse model [24]. Increased iron accumulation in gray matter is frequently observed in AD. Because of the paramagnetic nature of iron, MRI shows nice potential in the investigating iron levels in AD [25]. Quantitative MRI was shown high sensitivity and specificity in mapping cerebral iron deposition, and helped in the research on AD diagnosis [26].
The imaging patterns are always associated with the pathologic changes, such as specific protein markers. Spencer et al. manifested the relationship between quantitative T1 and T2 relaxation time changes and three immunohistochemical markers: β-amyloid, neuron-specific nuclear protein (a marker of neuronal cell load), and myelin basic protein (a marker of myelin load) in AD transgenic mice [27].
High-field MRI has been successfully applied to imaging plaques in transgenic mice for over a decade without contrast agents [24, 28–30]. Sillerud et al. devised a method using blood–brain barrier penetrating, amyloid-targeted, superparamagnetic iron oxide nanoparticles (SPIONs) for better imaging of amyloid plaque [31]. Then, they successfully used this SPION-MRI to assess the drug efficacy on the 3D distribution of Aβ plaques in transgenic AD mouse [32].
2.3 Diffusion MRI
Diffusion-weighted imaging (DWI) is a sensitive tool that allows quantifying of physiologic alterations in water diffusion, which result from microscopic structural changes.
Diffusion tensor imaging (DTI) is a well-established and commonly employed diffusion MRI technique in clinical and research on neuroimaging studies, which is based on a Gaussian model of diffusion processes [33]. In general, AD is associated with widespread reduced fractional anisotropy (FA) and increased mean diffusivity (MD) in several regions, most prominently in the frontal and temporal lobes, and along the cingulum, corpus callosum, uncinate fasciculus, superior longitudinal fasciculus, and MTL-associated tracts than healthy controls [34–37]. Acosta-Cabronero et al. reported increased axial diffusivity and MD in the splenium, which were the earliest abnormalities in AD [38]. FA and radial diffusivity (DR) differences in the corpus callosum, cingulum, and fornix were found to separate individuals with MCI who converted to AD from non-converters [39]. DTI was also found to be a better predictor of AD-specific MTL atrophy when compared to CSF biomarkers [40]. These findings suggested the potential clinical utility of DTI as early biomarkers of AD and its progression. However, an increase in MD and DR and a decrease in FA with advancing age in selective brain regions have been previously reported [41, 42]. Diffusion MRI can be also used in the classifying of various stages of AD. Multimodal classification method, which combined fMRI and DTI, separated more MCI from healthy controls than single approaches [43].
In recent years, tau has emerged as a potential target for therapeutic intervention. Tau plays a critical role in the neurodegenerative process forming neurofibrillary tangles, which is a major hallmark of AD and correlates with clinical disease progression. Wells et al. applied multiparametric MRI, containing high-resolution structure MRI (sMRI), a novel chemical exchange saturation transfer (CEST) MRI, DTI, and ASL, and glucose CEST to measure changes of tau pathology in AD transgenic mouse [44].
Besides DWI MRI, perfusion-weighted imaging (PWI) is another advanced MR technique, which could measure the cerebral hemodynamics at the capillary level. Zimny et al. evaluated the correlation of MTL with both DWI and PWI in AD and MCI patients [45].
3 Positron emission tomography
PET is a specific imaging technique applying in researches of brain function and neurochemistry of small animals, medium-sized animals, and human subjects [46–48]. As a particular brain imaging technique, PET imaging has become the subject of intense research for the diagnosis and facilitation of drug development of AD in both animal models and human trials due to its non-invasive and translational characteristic. PET with various radiotracers is considered as a standard non-invasive quantitative imaging technique to measure CBF, glucose metabolism, and β-amyloid and tau deposition.
3.1 FDG-PET
To date, 18F-FDG is one of the best and widely used neuroimaging tracers of PET, which employed for research and clinical assessment of AD [49]. Typical lower FDG metabolism was shown in the precuneus, posterior cingulate, and temporal and parietal cortex with progression to whole brain reductions with increasing disease progress in AD brains [50, 51]. FDG-PET imaging reflects the cerebral glucose metabolism, neuronal injury, which provides indirect evidence on cognitive function and progression that cannot be provided by amyloid PET imaging.
Schraml et al. [52] identified a significant association between hypometabolic convergence index and phenotypes using ADNI data. Some researchers also used 18F-FDG-PET to analyze genetic information with multiple biomarkers to classify AD status, predicting cognitive decline or MCI to AD conversion [53–55]. Trzepacz et al. [56] reported multimodal AD neuroimaging study, using MRI, 11C-PiB PET, and 18F-FDG-PET imaging to predict MCI conversion to AD along with APOE genotype. Zhang et al. [57] compared the genetic modality single-nucleotide polymorphism (SNP) with sMRI, 18F-FDG-PET, and CSF biomarkers, which were used to differentiate healthy control, MCI, and AD. They found FDG-PET is the best modality in terms of accuracy.
3.2 Amyloid beta PET
Aβ, the primary constituent of senile plaques, and tau tangles are hypothesized to play a primary role in the pathogenesis of AD, but it is still hard to identify the fundamental mechanisms [58–60]. Aβ plaque in brain is one of the pathological hallmarks of AD [61,62]. Accumulation of Aβ peptide in the cerebral cortex is considered one cause of dementia in AD [63]. Numerous studies have involved in vivo PET imaging assessing cortical β-amyloid burden [64–66].
Aβ imaging using PET has been recognized as one of the most important methods for the early diagnosis of AD [67]. Numerous candidate compounds have been tested for Aβ imaging, such as 11C-PiB [68], 18F-FDDNP [69], 11C-SB-13 [70], 18F-BAY94-9172 [71], 18F-AV-45 [72], 18F-flutemetamol [73, 74], 11C-AZD2184 [75], and 18F-ADZ4694 [76], 11C-BF227 and 18F-FACT [77].
Several amyloid PET studies examined genotypes, phenotypes, or gene–gene interactions. Ramanan et al. [78] reported the GWAS results with 18F-AV-45 reflecting the cerebral amyloid metabolism in AD for the first time. Swaminathan et al. [79] revealed the association between plasma Aβ from peripheral blood and cortical amyloid deposition on 11C-PiB. Hohman et al. [80] reported the relationship between SNPs involved in amyloid and tau pathophysiology with 18F-AV-45 PET.
Among the PET tracers, 11C-PiB, which has a high affinity for fibrillar Aβ, is a reliable biomarker of underlying AD pathology [68, 81]. It shows cortical uptake well paralleled with AD pathology [82, 83], has recently been approved for use by the Food and Drug Administration (FDA, April 2012) and the European Medicines Agency (January 2013). 18F-GE-067 (flutemetamol) and 18F-BAY94-9172 (florbetaben) have also been approved by the US FDA in the last 2 years [84, 85].
18F-Florbetapir (also known as 18F-AV-45) exhibits high affinity specific binding to amyloid plaques. 18F-AV-45 labels Aβ plaques in sections from patients with pathologically confirmed AD [72].
It was reported in several research groups that 18F-AV-45 PET imaging showed a reliability of both qualitative and quantitative assessments in AD patients, and Aβ+ increased with diagnostic category (healthy control < MCI < AD) [82, 86, 87]. Johnson et al. used 18F-AV-45 PET imaging to evaluate the amyloid deposition in both MCI and AD patients qualitatively and quantitatively, and found that amyloid burden increased with diagnostic category (MCI < AD), age, and APOEε4 carrier status [88]. Payoux et al. reported the equivocal amyloid PET scans using 18F-AV-45 associated with a specific pattern of clinical signs in a large population of non-demented older adults more than 70 years old [89].
More and more researchers consider combination and comparison of multiple PET tracers targeting amyloid plaque imaging together. Bruck et al. compared the prognostic ability of 11C-PiB PET, 18F-FDG-PET, and quantitative hippocampal volumes measured with MR imaging in predicting MCI to AD conversion. They found that the FDG-PET and 11C-PiB PET imaging are better in predicting MCI to AD conversion [90]. Hatashita et al. used 11C-PiB and FDG-PET imaging to identify MCI due to AD, 11C-PiB showed a higher sensitivity of 96.6 %, and FDG-PET added diagnostic value in predicting AD over a short period [91].
Besides, new Aβ imaging agents were radiosynthesized. Yousefi et al. radiosynthesized a new Aβ imaging agent 18F-FIBT, and compared the three different Aβ-targeted radiopharmaceuticals for PET imaging, including 18F-FIBT, 18F-florbetaben, and 11C-PiB [92]. 11C-AZD2184 is another new PET tracer developed for amyloid senile plaque imaging, and the kinetic behavior of 11C-AZD2184 is suitable for quantitative analysis and can be used in clinical examination without input function [75,93, 94].
4 Multimodality imaging: PET/MRI
Several diagnostic techniques, including MRI and PET, are employed for the diagnosis and monitoring of AD [95]. Multimodal imaging could provide more information in the formation and key molecular event of AD than single method. It drives the progression of neuroimaging research due to the recognition of the clinical benefits of multimodal data [96], and the better access to hybrid devices, such as PET/MRI [97].
Maier et al. evaluated the dynamics of 11C-PiB PET, 15O-H2O-PET, and ASL-MRI in transgenic AD mice and concluded that the AD-related decline of rCBF was caused by the cerebral Aβ angiopathy [98]. Edison et al. systematically compared 11C-PiB PET and MRI in AD, MCI patients, and controls. They thought that 11C-PiB PET was adequate for clinical diagnostic purpose, while MRI remained more appropriate for clinical research [99]. Zhou et al. investigated the interactions between multimodal PET/MRI in elder patients with MCI, AD, and healthy controls, and confirmed the invaluable application of amyloid PET and MRI in early diagnosis of AD [100]. Kim et al. reported that Aβ-weighted cortical thickness, which incorporates data from both MRI and amyloid PET imaging, is a consistent and objective imaging biomarker in AD [101].
5 Other imaging modalities
Multiphoton non-linear optical microscope imaging systems using ultrafast lasers have powerful advantages such as label-free detection, deep penetration of thick samples, high sensitivity, subcellular spatial resolution, 3D optical sectioning, chemical specificity, and minimum sample destruction [102, 103]. Coherent anti-Stokes–Raman scattering (CARS), two-photon excited fluorescence (TPEF), and second-harmonic generation (SHG) microscopy are the most widely used biomedical imaging techniques [104–106].
Quantitative electroencephalographic and neuropsychological investigation of an alternative measure of frontal lobe executive functions: the Figure Trail Making Test
Paul S. Foster,
Valeria Drago,
Brad J. Ferguson,
Patti Kelly Harrison,
David W. Harrison Brain Informatis http://dx.doi.org:/10.1007/s40708-015-0025-z http://link.springer.com/article/10.1007/s40708-015-0025-z/fulltext.html
The most frequently used measures of executive functioning are either sensitive to left frontal lobe functioning or bilateral frontal functioning. Relatively little is known about right frontal lobe contributions to executive functioning given the paucity of measures sensitive to right frontal functioning. The present investigation reports the development and initial validation of a new measure designed to be sensitive to right frontal lobe functioning, the Figure Trail Making Test (FTMT). The FTMT, the classic Trial Making Test, and the Ruff Figural Fluency Test (RFFT) were administered to 42 right-handed men. The results indicated a significant relationship between the FTMT and both the TMT and the RFFT. Performance on the FTMT was also related to high beta EEG over the right frontal lobe. Thus, the FTMT appears to be an equivalent measure of executive functioning that may be sensitive to right frontal lobe functioning. Applications for use in frontotemporal dementia, Alzheimer’s disease, and other patient populations are discussed.
Keywords – Frontal lobes, Executive functioning, Trail making test, Sequencing, Behavioral speed, Designs, Nonverbal, Neuropsychological assessment, Regulatory control, Effortful control
A recent survey indicated that the vast majority of neuropsychologists frequently assess executive functioning as part of their neuropsychological evaluations [1]. Surveys of neuropsychologists have indicated that the Trail Making Test (TMT), Controlled Oral Word Association Test (COWAT), Wisconsin Card Sorting Test (WCST), and the Stroop Color-Word Test (SCWT) are among the most commonly used instruments [1,2]. Further, the Rabin et al. [1] survey indicated that these same tests are among the most frequently used by neuropsychologists when specifically assessing executive or frontal lobe functioning. The frequent use of the TMT, WCST, and the SCWT, as well as the assumption that they are measures of executive functioning, led Demakis (2003–2004) to conduct a series of meta-analyses to determine the sensitivity of these test to detect frontal lobe dysfunction, particularly lateralized frontal lobe dysfunction. The findings indicated that the SCWT and Part A of the TMT [3], as well as the WCST [4], were all sensitive to frontal lobe dysfunction. However, only the SCWT differentiated between left and right frontal lobe dysfunction, with the worst performance among those with left frontal lobe dysfunction [3].
The finding of the Demakis [4] meta-analysis, that the WCST was not sensitive to lateralized frontal lobe dysfunction, is not surprising given the equivocal findings that have been reported. Whereas performance on the WCST is sensitive to frontal lobe dysfunction [5, 6], demonstration of lateralized frontal dysfunction has been quite problematic. Unilateral left or right dorsolateral frontal dysfunction has been associated with impaired performance on the WCST [6]. Fallgatter and Strik [7] found bilateral frontal lobe activation during performance of the WCST. However, other imaging studies have found right lateralized frontal lobe activation [8] and left lateralized frontal activation [9] in response to performance on the WCST. Further, left frontal lobe alpha power is negatively correlated with performance on the WCST [10]. Finally, patients with left frontal lobe tumors exhibit more impaired performance on the WCST than those with right frontal tumors [11].
Unlike the data for the WCST, more consistent findings have been reported regarding lateralized frontal lobe functioning for the other commonly used measures of executive functioning. For instance, as with the Demakis [3] study, many investigations have found the SCWT to be sensitive to left frontal lobe functioning, although the precise localization within the left frontal lobe has varied. Impaired performance on the SCWT results from left frontal lesions [12] and specifically from lesions localized to the left dorsolateral frontal lobe [13, 14], though bilateral frontal lesions have also yielded impaired performance [13, 14]. Further, studies using neuroimaging to investigate the neural basis of performance on the SCWT have indicated involvement of the left anterior cingulated cortex [15], left lateral prefrontal cortex [16], left inferior precentral sulcus [17], and the left dorsolateral frontal lobe [18].
Wide agreement exists among investigations of the frontal lateralization of verbal or lexical fluency to confrontation. Specifically, patients with left frontal lobe lesions are known to exhibit impaired performance on lexical fluency to confrontation tasks, relative to either patients with right frontal lesions [12, 19, 20] or controls [21]. A recent meta-analysis also indicated that the largest deficits in performance on measures of lexical fluency are associated with left frontal lobe lesions [22]. Troster et al. [23] found that, relative to patients with right pallidotomy, patients with left pallidotomy exhibited more impaired lexical fluency. Several neuroimaging investigations have further supported the role of the left frontal lobe in lexical fluency tasks [15, 24–27]. Performance on lexical fluency tasks also varies as a function of lateral frontal lobe asymmetry, as assessed by electroencephalography [28].
The Trail Making Test is certainly among the most widely used tests [1] and perhaps the most widely researched. Various norms exist for the TMT (see [29]), with Tombaugh [30] providing the most recent comprehensive set of normative data. Different methods of analyzing and interpreting the data have also been proposed and used, including error analysis [13, 14, 31–33], subtraction scores [13, 14, 34], and ratio scores [13, 14, 35].
Several different language versions of the test have been developed and reported, including Arabic [36], Chinese [37, 38], Greek [39], and Hebrew [40]. Numerous alternative versions of the TMT have been developed to address perceived shortcomings of the original TMT. For instance, the Symbol Trail Making Test [41] was developed to reduce the cultural confounds associated with the use of the Arabic numeral system and English alphabet in the original TMT. The Color Trails Test (CTT; [42]) was also developed to control for cultural confounds, although mixed results have been reported regarding whether the CTT is indeed analogous to the TMT [43–45]. A version of the TMT for preschool children, the TRAILS-P, has also been reported [46].
Additionally, the Comprehensive Trail Making Test [47] was developed to control for perceived psychometric shortcomings of the original TMT (for a review see [48] and the Oral Trail Making Test (OTMT; [49]) was developed to reduce confounds associated with motor speed and visual search abilities, with research supporting the OTMT as an equivalent measure [50, 51]. Alternate forms of the TMT have also been developed to permit successive administrations [32, 52] and to assess the relative contributions of the requisite cognitive skills [53].
Delis et al. [54] stated that the continued development of new instrumentation for improving diagnosis and treatment is a critical undertaking in all health-related fields. Further, in their view, the field of neuropsychology has recognized the importance of continually striving to develop new clinical measures. Delis and colleagues developed the extensive Delis-Kaplan Executive Functioning System (D-KEFS; [55]) in the spirit of advancing the instrumentation of neuropsychology. The D-KEFS includes a Trail Making Test consisting of five separate conditions. The Number-Letter Switching condition involves a sequencing procedure similar to that of the classic TMT. The other four conditions are designed to assess the component processes involved in completing the Number-Letter Switching condition so that a precise analysis of the nature of any underlying dysfunction may be accomplished. Specifically, these additional components include Visual Scanning, Number Sequencing, Letter Sequencing, and Motor Speed.
Given that the TMT comprises numbers and letters and is a measure of executive functioning, it may preferentially involve the left frontal lobe. Although the literature is somewhat controversial, neuropsychological and neuroimaging studies seem to provide support for the sensitivity of the TMT to detect left frontal dysfunction [56]. Recent clinically oriented studies investigating frontal lobe involvement of the TMT using transcranial magnetic stimulation (TMS) and near-infrared spectroscopy (NIRS) also support this localization [57]. Performance on Part B of the TMT improved following repetitive TMS applied to the left dorsolateral frontal lobe [57].
With 9–13-year-old boys performing TMT Part B, Weber et al. [58] found a left lateralized increase in the prefrontal cortex in deoxygenated hemoglobin, an indicator of increased oxygen consumption. Moll et al. [59] demonstrated increased activation specific to the prefrontal cortex, especially the left prefrontal region, in healthy controls performing Part B of the TMT. Foster et al. [60] found a significant positive correlation between performance on Part A of the TMT and low beta (13–21 Hz) magnitude (μV) at the left lateral frontal lobe, but not at the right lateral frontal lobe. Finally, Stuss et al. [13, 14] found that patients with left dorsolateral frontal dysfunction evidenced more errors than patients with lesions in other areas of the frontal lobes and those patients with left frontal lesions were the slowest to complete the test.
Taken together, the possibility exists that the aforementioned tests are largely associated with left frontal lobe activity and the TMT, in particular, provides information concerning mental processing speed as well as cognitive flexibility and set-shifting. While some studies have found that deficits in visuomotor set-shifting are specific to the frontal lobe damage [61], others investigators have reported such impairment in patients with posterior brain lesions and widespread cerebral dysfunctions, including cerebellar damage [62] and Alzheimer disease [63]. Thus, it remains unclear whether impairments in visuomotor set-shifting are specific to frontal lobe dysfunction or whether they are non-specific and can result from more posterior or widespread brain dysfunction.
Compared to the collective knowledge we have regarding the cognitive roles of the left frontal lobe, relatively little is known about right frontal lobe contributions to executive functioning. This is likely a result of the dearth of tests that are associated with right frontal activity. The Ruff Figural Fluency Test (RFFT; [64]) is among the few standardized tests of right frontal lobe functioning and was listed as the 14th most commonly used instrument to assess executive functioning in the Rabin et al. [1] survey. The RFFT is known to be sensitive to right frontal lobe functioning [65, 66]; see also [67] pp. 297–298), as is a measure based on the RFFT [19].
The present investigation, with the same intent and spirit as that reported by Delis et al. [54], sought to develop and initially validate a measure of right frontal lobe functioning in an effort to attain a greater understanding of right frontal contributions to executive functioning and to advance the instrumentation of neuropsychology. To meet this objective, a version of the Trail Making Test comprising figures, as opposed to numbers and letters, was developed. The TMT was used as a model for the new test, referred to as the Figure Trail Making Test (FTMT), due to the high frequency of use, the volume of research conducted, and the ease of administration of the TMT. Given that the TMT and the FTMT are both measuring executive functioning, we felt that a moderate correlation would exist between these two measures. Specifically, we hypothesized that performance on the FTMT would be positively correlated with performance on the TMT, in terms of the total time required to complete each part of the tests, an additive and subtractive score, and a ratio score. The total time required to complete each part of the FTMT was also hypothesized to be negatively correlated with the total number of unique designs produced on the RFFT and positively correlated with the number of perseverative errors committed on the RFFT and the perseverative error ratio. We also sought to determine whether the TMT and the FTMT were measuring different constructs by conducting a factor analysis, anticipating that the two tests would load on separate factors.
Additionally, we sought to obtain neurophysiological evidence that the FTMT is sensitive to right frontal lobe functioning. Specifically, we used quantitative electroencephalography (QEEG) to measure electrical activity over the left and right frontal lobes. A previous investigation we conducted found that performance on Part A of the TMT was related to left frontal lobe (F7) low beta magnitude [60]. For the present investigation, we predicted that significant negative correlations would exist between performance on Parts A and B of the TMT and both low and high beta magnitude at the F7 electrode site. We further predicted that significant negative correlations would exist between performance on Parts C and D of the FTMT and both low and high beta magnitude at the F8 electrode site.
3 Discussion
The need for additional measures of executive functions and especially instruments which may provide implications relevant to cerebral laterality is clear. There remains especially a void for neuropsychological instruments using a TMT format, which may provide information pertaining to the functional integrity of the right frontal region. Consistent with the hypotheses forwarded, significant correlations were found between performance on the TMT and the FTMT, in terms of the raw time required to complete each respective part of the tests as well as the additive and subtraction scores. The fact that the ratio scores were not significantly correlated is not surprising given that research has generally indicated a lack of clinical utility for this score [13, 14, 35]. Given the present findings, the TMT and the FTMT appear to be equivalent measures of executive functioning. Further, the present findings not only suggest that the FTMT may be a measure of executive functioning but also extend the realm of executive functioning to the sequencing and set-shifting of nonverbal stimuli.
However, the finding of significant correlations between the TMT and the FTMT represents somewhat of a caveat in that the TMT has been found to be sensitive to left frontal lobe functioning [13, 14, 57, 59]. This would seem to suggest the possibility that the FTMT is also sensitive to left frontal lobe functioning. The possibility that FTMT is related to left frontal lobe functioning is tempered, though, by the fact that the many of the hypothesized correlations between performance on the RFFT and the FTMT were also significant. Performance on the RFFT is related to right frontal lobe functioning [65,66]. Thus, the significant correlations between the RFFT and the FTMT suggest that the FTMT may also be sensitive to right frontal lobe functioning. Additionally, it should also be noted that the TMT was not significantly correlated with performance on the RFFT, with the exception of the significant correlation between performance on the TMT Part A and the total number of unique designs produced on the RFFT. Taken together, the results suggest that the FTMT may be a measure of right frontal executive functioning.
Additional support for the sensitivity of the FTMT to right frontal lobe functioning is provided by the finding of a significant negative correlation between performance on Part D of the FTMT and high beta magnitude. We have previously used QEEG to provide neurophysiological validation of the RFFT [65] and the Rey Auditory Verbal Learning Test [70] and the present findings provide further support for the use of QEEG in validating neuropsychological tests. The lack of significant correlations between the TMT and either low or high beta magnitude may be related to a restricted range of scores on the TMT. As a whole, performance on the FTMT was more variable than performance on the TMT and this relatively restricted range for the TMT may have impacted the obtained correlations. Given the present findings, together with those of the Foster et al. [65, 70] investigations, further support is also provided for the use of EEG in establishing neurophysiological validation for neuropsychological tests.
The results from the factor analysis provide support for the contention that the FMT may be a measure of right frontal lobe activity and also provide initial discriminant validity for the FTMT. Specifically, Parts C and D of the FTMT were found to load on the same factor as the number of designs generated on the RFFT, although the time required to complete Part A of the TMT is also included. Additionally, the number of errors committed on Parts C and D of the FTMT comprises a single factor, separate from either the TMT or the RFFT. Although these results support the FTMT as a measure of nonverbal executive functioning, it would be helpful to conduct an additional factor analysis including additional measures of right frontal functioning, and perhaps other measures of right hemisphere functioning as marker variables.
We sought to develop a measure sensitive to right frontal lobe functioning due to the paucity of such tests and the potentially important uses that right frontal lobe tests may have clinically. Tests of right frontal lobe functioning may, for instance, be useful in identifying and distinguishing left versus right frontotemporal dementia (FTD). Research has indicated that FTD is associated with cerebral atrophy at the right dorsolateral frontal and left premotor cortices [71]. Fukui and Kertesz [72] found right frontal lobe volume reduction in FTD relative to Alzheimer’s disease and progressive nonfluent aphasia. Some have suggested that FTD should not be considered as a unitary disorder and that neuropsychological testing may aid in differentially diagnosing left versus right FTD [73].
Whereas right FTD has been associated with more errors and perseverative responses on the Wisconsin Card Sorting Test (WCST), left FTD has been associated with significantly worse performance on the Boston Naming Test (BNT) and the Stroop Color-Word test [73]. Razani et al. [74] also distinguished between left and right FTD in finding that left FTD performed worse on the BNT and the right FTD patients performed worse on the WCST. However, as noted earlier, the WCST has been associated with left frontal activity [9], right frontal activation [8], and bilateral frontal activation [7]. Further, patients with left frontal tumors perform worse than those with right frontal tumors [11].
Patients with FTD that predominantly involves the right frontotemporal region have behavioral and emotional abnormalities and those with predominantly left frontotemporal region damage have a loss of lexical semantic knowledge. Patients, in whom neural degeneration begins on the left side, often present to the clinicians at an early stage of the disease due to the presence of language abnormalities, but maintain their emotion processing abilities, being preserved the right anterior temporal lobe. However, as this disease advances, the disease may progress to the right frontotemporal regions. Tests sensitive to right frontal lobe functioning may be useful tools to identify in advance the course of the disease, providing immediate and specific treatments and informing the caregivers on the possible prospective frame of the disease.
A potentially more important use of tests sensitive to right frontal lobe functioning, though, may be in predicting dementia patients that will develop significant and disruptive behavioral deficits. Research has found that approximately 92 % of right-sided FTD patients exhibit socially undesirable behaviors as their initial symptom, as compared to only 11 % of left-sided FTD patients [75]. Behavioral deficits in FTD are associated with gray matter loss at the dorsomedial frontal region, particularly on the right [76].
Alzheimer’s disease (AD) is also often associated with significant behavioral disturbances. Even AD patients with mild dementia are noted to exhibit behavioral deficits such as delusions, hallucinations, agitation, dysphoria, anxiety, apathy, and irritability [77]. Indeed, Shimabukuro et al. [77] found that regardless of dementia severity, over half of all AD patients exhibited apathy, delusions, irritability, dysphoria, and anxiety. Delusions in AD patients are associated with relative right frontal hypoperfusion as indicated by SPECT imaging [78, 79]. Further, positron emission tomography (PET) has indicated that AD patients exhibiting delusions exhibit hypometabolism at the right superior dorsolateral frontal and right inferior frontal pole [80].
Although research clearly implicates right frontal lobe dysfunction in the expression of behavioral deficits, data from neuropsychological testing are not as clear. Negative symptoms in patients with AD and FTD have been related to measures of nonverbal and verbal executive functioning as well as verbal memory [81]. Positive symptoms, in contrast, were related to constructional skills and attention. However, Staff et al. [78] failed to dissociate patients with delusions from those without delusions based on neuropsychological test performance, despite significant differences existing in right frontal and limbic functioning as revealed by functional imaging. The inclusion of other measures of right frontal lobe functioning may result in improved neuropsychological differentiation of dementia patients with and without significant behavioral disturbances. Further, it may be possible to predict early in the disease process those patients that will ultimately develop behavioral disturbances with improved measures of right frontal functioning. Predicting those that may develop behavioral problems will permit earlier treatment and will provide the family with more time to prepare for the potential emergence of such difficulties. Certainly, future research needs to be conducted that incorporates measures of right and left frontal lobe functioning in regression analyses to determine the plausibility of such prediction.
Tests sensitive to right frontal lobe functioning may also be useful in identifying more subtle right frontal lobe dysfunction and the cognitive and behavioral changes that follow. The right frontal lobe mediates language melody or prosody and forms a cohesive discourse, interprets abstract communication in spoken and written languages, and interprets the inferred relationships involved in communications. Subtle difficulties in interpreting abstract meaning in communication, comprehending metaphors, and even understanding jokes that are often seen in right frontal lobe stroke patients may not be detected by the family and may also be under diagnosed by clinicians [82]. Further, patients with right frontal lobe lesions are generally more euphoric and unconcerned, often minimizing their symptoms [82] or denying the illness, which may delay referral to a clinician and diagnosis.
Attention deficit hyperactivity disorder (ADHD) is a neurological disease characterized by motor inhibition deficit, problems with cognitive flexibility, social disruption, and emotional disinhibition [83, 84]. Functional MRI studies reveal reduced right prefrontal activation during “frontal tasks,” such as go/no go [85], Stroop [86], and attention task performance [87]. The right frontal lobe deficit hypothesis is further supported by structural studies [88, 89]. Tests of right frontal lobe functioning may be useful in further characterizing the nature of this deficit and in specifying the likely hemispheric locus of dysfunction.
To summarize, we feel that right frontal lobe functioning has been relatively neglected in neuropsychological assessment and that many uses for such tests exist. Our intent was to develop a test purportedly sensitive to right frontal functioning that would be easy and quick to administer in a clinical setting. However, we are certainly not meaning to assert that our FTMT would be applicable in all the aforementioned conditions. Additional research should be conducted to determine the precise clinical utility of the FTMT.
Further validation of the FTMT should also be undertaken. Establishing convergent validation may involve correlating tests measuring the same domain, such as executive functioning. This was initially accomplished in the present investigation through the significant correlations between the TMT and the FTMT. Additionally, convergent validation may also involve correlating tests that purportedly measure the same region of the brain. This was also initially accomplished in the present investigation through the significant correlations between the FTMT and the RFFT. However, additional convergent validation certainly needs to be obtained, as well as validation using patient populations and neurophysiological validation.
We are currently collecting data that hopefully will provide neurophysiological validation of the FTMT. Certainly, though, it is hoped that the present investigation will not only stimulate further research seeking to validate the FTMT and provide more comprehensive normative data, but also stimulate research investigating whether the FTMT or other measures of right frontal lobe functioning may be used to predict patients that will develop behavioral disturbances.
http://www.scientificcomputing.com/news/2016/01/worlds-greatest-literature-reveals-multifractals-cascades-consciousness
http://www.scientificcomputing.com/sites/scientificcomputing.com/files/Worlds_Greatest_Literature_Reveals_Multifractals_Cascades_of_Consciousness_440.jpg
Multifractal analysis of Finnegan’s Wake by James Joyce. The ideal shape of the graph is virtually indistinguishable from the results for purely mathematical multifractals. The horizontal axis represents the degree of singularity, and the vertical axis shows the spectrum of singularity. Courtesy of IFJ PAN
Arthur Conan Doyle, Charles Dickens, James Joyce, William Shakespeare and JRR Tolkien. Regardless of the language they were working in, some of the world’s greatest writers appear to be, in some respects, constructing fractals. Statistical analysis, however, revealed something even more intriguing. The composition of works from within a particular genre was characterized by the exceptional dynamics of a cascading (avalanche) narrative structure. This type of narrative turns out to be multifractal. That is, fractals of fractals are created.
As far as many bookworms are concerned, advanced equations and graphs are the last things which would hold their interest, but there’s no escape from the math. Physicists from the Institute of Nuclear Physics of the Polish Academy of Sciences (IFJ) in Cracow, Poland, performed a detailed statistical analysis of more than one hundred famous works of world literature, written in several languages and representing various literary genres. The books, tested for revealing correlations in variations of sentence length, proved to be governed by the dynamics of a cascade. This means that the construction of these books is, in fact, a fractal. In the case of several works, their mathematical complexity proved to be exceptional, comparable to the structure of complex mathematical objects considered to be multifractal. Interestingly, in the analyzed pool of all the works, one genre turned out to be exceptionally multifractal in nature.
Fractals are self-similar mathematical objects: when we begin to expand one fragment or another, what eventually emerges is a structure that resembles the original object. Typical fractals, especially those widely known as the Sierpinski triangle and the Mandelbrot set, are monofractals, meaning that the pace of enlargement in any place of a fractal is the same, linear: if they at some point were rescaled x number of times to reveal a structure similar to the original, the same increase in another place would also reveal a similar structure.
Multifractals are more highly advanced mathematical structures: fractals of fractals. They arise from fractals ‘interwoven’ with each other in an appropriate manner and in appropriate proportions. Multifractals are not simply the sum of fractals and cannot be divided to return back to their original components, because the way they weave is fractal in nature. The result is that, in order to see a structure similar to the original, different portions of a multifractal need to expand at different rates. A multifractal is, therefore, non-linear in nature.
“Analyses on multiple scales, carried out using fractals, allow us to neatly grasp information on correlations among data at various levels of complexity of tested systems. As a result, they point to the hierarchical organization of phenomena and structures found in nature. So, we can expect natural language, which represents a major evolutionary leap of the natural world, to show such correlations as well. Their existence in literary works, however, had not yet been convincingly documented. Meanwhile, it turned out that, when you look at these works from the proper perspective, these correlations appear to be not only common, but in some works they take on a particularly sophisticated mathematical complexity,” says Professor Stanislaw Drozdz, IFJ PAN, Cracow University of Technology.
The study involved 113 literary works written in English, French, German, Italian, Polish, Russian and Spanish by such famous figures as Honore de Balzac, Arthur Conan Doyle, Julio Cortazar, Charles Dickens, Fyodor Dostoevsky, Alexandre Dumas, Umberto Eco, George Elliot, Victor Hugo, James Joyce, Thomas Mann, Marcel Proust, Wladyslaw Reymont, William Shakespeare, Henryk Sienkiewicz, JRR Tolkien, Leo Tolstoy and Virginia Woolf, among others. The selected works were no less than 5,000 sentences long, in order to ensure statistical reliability.
To convert the texts to numerical sequences, sentence length was measured by the number of words (an alternative method of counting characters in the sentence turned out to have no major impact on the conclusions). The dependences were then searched for in the data — beginning with the simplest, i.e. linear. This is the posited question: if a sentence of a given length is x times longer than the sentences of different lengths, is the same aspect ratio preserved when looking at sentences respectively longer or shorter?
“All of the examined works showed self-similarity in terms of organization of the lengths of sentences. Some were more expressive — here The Ambassadors by Henry James stood out — while others to far less of an extreme, as in the case of the French seventeenth-century romance Artamene ou le Grand Cyrus. However, correlations were evident and, therefore, these texts were the construction of a fractal,” comments Dr. Pawel Oswiecimka (IFJ PAN), who also noted that fractality of a literary text will, in practice, never be as perfect as in the world of mathematics. It is possible to magnify mathematical fractals up to infinity, while the number of sentences in each book is finite and, at a certain stage of scaling, there will always be a cut-off in the form of the end of the dataset.
Things took a particularly interesting turn when physicists from IFJ PAN began tracking non-linear dependence, which in most of the studied works was present to a slight or moderate degree. However, more than a dozen works revealed a very clear multifractal structure, and almost all of these proved to be representative of one genre, that of stream of consciousness. The only exception was the Bible, specifically the Old Testament, which has, so far, never been associated with this literary genre.
“The absolute record in terms of multifractality turned out to be Finnegan’s Wakeby James Joyce. The results of our analysis of this text are virtually indistinguishable from ideal, purely mathematical multifractals,” says Drozdz.
The most multifractal works also included A Heartbreaking Work of Staggering Genius by Dave Eggers, Rayuela by Julio Cortazar, The US Trilogy by John Dos Passos, The Waves by Virginia Woolf, 2666 by Roberto Bolano, and Joyce’sUlysses. At the same time, a lot of works usually regarded as stream of consciousness turned out to show little correlation to multifractality, as it was hardly noticeable in books such as Atlas Shrugged by Ayn Rand and A la recherche du temps perdu by Marcel Proust.
“It is not entirely clear whether stream of consciousness writing actually reveals the deeper qualities of our consciousness, or rather the imagination of the writers. It is hardly surprising that ascribing a work to a particular genre is, for whatever reason, sometimes subjective. We see, moreover, the possibility of an interesting application of our methodology: it may someday help in a more objective assignment of books to one genre or another,” notes Drozdz.
Multifractal analyses of literary texts carried out by the IFJ PAN have been published in Information Sciences, the journal of computer science. The publication has undergone rigorous verification: given the interdisciplinary nature of the subject, editors immediately appointed up to six reviewers.
Citation: “Quantifying origin and character of long-range correlations in narrative texts” S. Drożdż, P. Oświęcimka, A. Kulig, J. Kwapień, K. Bazarnik, I. Grabska-Gradzińska, J. Rybicki, M. Stanuszek; Information Sciences, vol. 331, 32–44, 20 February 2016; DOI: 10.1016/j.ins.2015.10.023
New Quantum Approach to Big Data could make Impossibly Complex Problems Solvable
David L. Chandler, MIT
http://www.scientificcomputing.com/news/2016/01/new-quantum-approach-big-data-could-make-impossibly-complex-problems-solvable
http://www.scientificcomputing.com/sites/scientificcomputing.com/files/New_Quantum_Approach_to_Big_Data_could_make_Impossibly_Complex_Problems_Solvable_440.jpg
This diagram demonstrates the simplified results that can be obtained by using quantum analysis on enormous, complex sets of data. Shown here are the connections between different regions of the brain in a control subject (left) and a subject under the influence of the psychedelic compound psilocybin (right). This demonstrates a dramatic increase in connectivity, which explains some of the drug’s effects (such as “hearing” colors or “seeing” smells). Such an analysis, involving billions of brain cells, would be too complex for conventional techniques, but could be handled easily by the new quantum approach, the researchers say. Courtesy of the researchers
From gene mapping to space exploration, humanity continues to generate ever-larger sets of data — far more information than people can actually process, manage or understand.
Machine learning systems can help researchers deal with this ever-growing flood of information. Some of the most powerful of these analytical tools are based on a strange branch of geometry called topology, which deals with properties that stay the same even when something is bent and stretched every which way.
Such topological systems are especially useful for analyzing the connections in complex networks, such as the internal wiring of the brain, the U.S. power grid, or the global interconnections of the Internet. But even with the most powerful modern supercomputers, such problems remain daunting and impractical to solve. Now, a new approach that would use quantum computers to streamline these problems has been developed by researchers at MIT, the University of Waterloo, and the University of Southern California.
The team describes their theoretical proposal this week in the journal Nature Communications. Seth Lloyd, the paper’s lead author and the Nam P. Suh Professor of Mechanical Engineering, explains that algebraic topology is key to the new method. This approach, he says, helps to reduce the impact of the inevitable distortions that arise every time someone collects data about the real world.
In a topological description, basic features of the data (How many holes does it have? How are the different parts connected?) are considered the same no matter how much they are stretched, compressed, or distorted. Lloyd explains that it is often these fundamental topological attributes “that are important in trying to reconstruct the underlying patterns in the real world that the data are supposed to represent.”
It doesn’t matter what kind of dataset is being analyzed, he says. The topological approach to looking for connections and holes “works whether it’s an actual physical hole, or the data represents a logical argument and there’s a hole in the argument. This will find both kinds of holes.”
Using conventional computers, that approach is too demanding for all but the simplest situations. Topological analysis “represents a crucial way of getting at the significant features of the data, but it’s computationally very expensive,” Lloyd says. “This is where quantum mechanics kicks in.” The new quantum-based approach, he says, could exponentially speed up such calculations.
Lloyd offers an example to illustrate that potential speedup: If you have a dataset with 300 points, a conventional approach to analyzing all the topological features in that system would require “a computer the size of the universe,” he says. That is, it would take 2300 (two to the 300th power) processing units — approximately the number of all the particles in the universe. In other words, the problem is simply not solvable in that way.
“That’s where our algorithm kicks in,” he says. Solving the same problem with the new system, using a quantum computer, would require just 300 quantum bits — and a device this size may be achieved in the next few years, according to Lloyd.
“Our algorithm shows that you don’t need a big quantum computer to kick some serious topological butt,” he says.
There are many important kinds of huge datasets where the quantum-topological approach could be useful, Lloyd says, for example understanding interconnections in the brain. “By applying topological analysis to datasets gleaned by electroencephalography or functional MRI, you can reveal the complex connectivity and topology of the sequences of firing neurons that underlie our thought processes,” he says.
The same approach could be used for analyzing many other kinds of information. “You could apply it to the world’s economy, or to social networks, or almost any system that involves long-range transport of goods or information,” Lloyd says. But the limits of classical computation have prevented such approaches from being applied before.
While this work is theoretical, “experimentalists have already contacted us about trying prototypes,” he says. “You could find the topology of simple structures on a very simple quantum computer. People are trying proof-of-concept experiments.”
Ignacio Cirac, a professor at the Max Planck Institute of Quantum Optics in Munich, Germany, who was not involved in this research, calls it “a very original idea, and I think that it has a great potential.” He adds “I guess that it has to be further developed and adapted to particular problems. In any case, I think that this is top-quality research.”
The team also included Silvano Garnerone of the University of Waterloo in Ontario, Canada, and Paolo Zanardi of the Center for Quantum Information Science and Technology at the University of Southern California. The work was supported by the Army Research Office, Air Force Office of Scientific Research, Defense Advanced Research Projects Agency, Multidisciplinary University Research Initiative of the Office of Naval Research, and the National Science Foundation.
Beyond Chess: Computer Beats Human in Ancient Chinese Game
http://www.rdmag.com/news/2016/01/beyond-chess-computer-beats-human-ancient-chinese-game
http://www.rdmag.com/sites/rdmag.com/files/rd1601_chess.jpg
A player places a black stone while his opponent waits to place a white one as they play Go, a game of strategy, in the Seattle Go Center, Tuesday, April 30, 2002. The game, which originated in China more than 2,500 years ago, involves two players who take turns putting markers on a grid. The object is to surround more area on the board with the markers than one’s opponent, as well as capturing the opponent’s pieces by surrounding them. A paper released Wednesday, Jan. 27, 2016 describes how a computer program has beaten a human master at the complex board game, marking significant advance for development of artificial intelligence. (AP Photo/Cheryl Hatch)
A computer program has beaten a human champion at the ancient Chinese board game Go, marking a significant advance for development of artificial intelligence.
The program had taught itself how to win, and its developers say its learning strategy may someday let computers help solve real-world problems like making medical diagnoses and pursuing scientific research.
The program and its victory are described in a paper released Wednesday by the journal Nature.
Computers previously have surpassed humans for other games, including chess, checkers and backgammon. But among classic games, Go has long been viewed as the most challenging for artificial intelligence to master.
Go, which originated in China more than 2,500 years ago, involves two players who take turns putting markers on a checkerboard-like grid. The object is to surround more area on the board with the markers than one’s opponent, as well as capturing the opponent’s pieces by surrounding them.
While the rules are simple, playing it well is not. It’s “probably the most complex game ever devised by humans,” Dennis Hassabis of Google DeepMind in London, one of the study authors, told reporters Tuesday.
The new program, AlphaGo, defeated the European champion in all five games of a match in October, the Nature paper reports.
In March, AlphaGo will face legendary player Lee Sedol in Seoul, South Korea, for a $1 million prize, Hassabis said.
Martin Mueller, a computing science professor at the University of Alberta in Canada who has worked on Go programs for 30 years but didn’t participate in AlphaGo, said the new program “is really a big step up from everything else we’ve seen…. It’s a very, very impressive piece of work.”
Biological Origin of Schizophrenia
Excessive ‘pruning’ of connections between neurons in brain predisposes to disease
http://hms.harvard.edu/sites/default/files/uploads/news/McCarroll_C4_600x400.jpg
Imaging studies showed C4 (in green) located at the synapses of primary human neurons. Image: Heather de Rivera, McCarroll lab
PAUL GOLDSMITH http://hms.harvard.edu/news/biological-origin-schizophrenia
The risk of schizophrenia increases if a person inherits specific variants in a gene related to “synaptic pruning”—the elimination of connections between neurons—according to a study from Harvard Medical School, the Broad Institute and Boston Children’s Hospital. The findings were based on genetic analysis of nearly 65,000 people.
The study represents the first time that the origin of this psychiatric disease has been causally linked to specific gene variants and a biological process.
Get more HMS news here
It also helps explain two decades-old observations: synaptic pruning is particularly active during adolescence, which is the typical period of onset for symptoms of schizophrenia, and the brains of schizophrenic patients tend to show fewer connections between neurons.
The gene, complement component 4 (C4), plays a well-known role in the immune system. It has now been shown to also play a key role in brain development and schizophrenia risk. The insight may allow future therapeutic strategies to be directed at the disorder’s roots, rather than just its symptoms.
The study, which appears online Jan. 27 in Nature, was led by HMS researchers at the Broad Institute’s Stanley Center for Psychiatric Research and Boston Children’s. They include senior author Steven McCarroll, HMS associate professor of genetics and director of genetics for the Stanley Center; Beth Stevens, HMS assistant professor of neurology at Boston Children’s and institute member at the Broad; Michael Carroll, HMS professor of pediatrics at Boston Children’s; and first author Aswin Sekar, an MD-PhD student at HMS.
The study has the potential to reinvigorate translational research on a debilitating disease. Schizophrenia afflicts approximately 1 percent people worldwide and is characterized by hallucinations, emotional withdrawal and a decline in cognitive function. These symptoms most frequently begin in patients when they are teenagers or young adults.
“These results show that it is possible to go from genetic data to a new way of thinking about how a disease develops—something that has been greatly needed.”
First described more than 130 years ago, schizophrenia lacks highly effective treatments and has seen few biological or medical breakthroughs over the past half-century.
In the summer of 2014, an international consortium led by researchers at the Stanley Center identified more than 100 regions in the human genome that carry risk factors for schizophrenia.
The newly published study now reports the discovery of the specific gene underlying the strongest of these risk factors and links it to a specific biological process in the brain.
“Since schizophrenia was first described over a century ago, its underlying biology has been a black box, in part because it has been virtually impossible to model the disorder in cells or animals,” said McCarroll. “The human genome is providing a powerful new way in to this disease. Understanding these genetic effects on risk is a way of prying open that black box, peering inside and starting to see actual biological mechanisms.”
“This study marks a crucial turning point in the fight against mental illness,” said Bruce Cuthbert, acting director of the National Institute of Mental Health. “Because the molecular origins of psychiatric diseases are little-understood, efforts by pharmaceutical companies to pursue new therapeutics are few and far between. This study changes the game. Thanks to this genetic breakthrough we can finally see the potential for clinical tests, early detection, new treatments and even prevention.”
The path to discovery
The discovery involved the collection of DNA from more than 100,000 people, detailed analysis of complex genetic variation in more than 65,000 human genomes, development of an innovative analytical strategy, examination of postmortem brain samples from hundreds of people and the use of animal models to show that a protein from the immune system also plays a previously unsuspected role in the brain.
Over the past five years, Stanley Center geneticists and collaborators around the world collected more than 100,000 human DNA samples from 30 different countries to locate regions of the human genome harboring genetic variants that increase the risk of schizophrenia. The strongest signal by far was on chromosome 6, in a region of DNA long associated with infectious disease. This caused some observers to suggest that schizophrenia might be triggered by an infectious agent. But researchers had no idea which of the hundreds of genes in the region was actually responsible or how it acted.
Based on analyses of the genetic data, McCarroll and Sekar focused on a region containing the C4 gene. Unlike most genes, C4 has a high degree of structural variability. Different people have different numbers of copies and different types of the gene.
McCarroll and Sekar developed a new molecular technique to characterize the C4 gene structure in human DNA samples. They also measured C4 gene activity in nearly 700 post-mortem brain samples.
They found that the C4 gene structure (DNA) could predict the C4 gene activity (RNA) in each person’s brain. They then used this information to infer C4 gene activity from genome data from 65,000 people with and without schizophrenia.
These data revealed a striking correlation. People who had particular structural forms of the C4 gene showed higher expression of that gene and, in turn, had a higher risk of developing schizophrenia.
Connecting cause and effect through neuroscience
But how exactly does C4—a protein known to mark infectious microbes for destruction by immune cells—affect the risk of schizophrenia?
Answering this question required synthesizing genetics and neurobiology.
Stevens, a recent recipient of a MacArthur Foundation “genius grant,” had found that other complement proteins in the immune system also played a role in brain development. These results came from studying an experimental model of synaptic pruning in the mouse visual system.
“This discovery enriches our understanding of the complement system in brain development and in disease, and we could not have made that leap without the genetics.”
Carroll had long studied C4 for its role in immune disease, and developed mice with different numbers of copies of C4.
The three labs set out to study the role of C4 in the brain.
They found that C4 played a key role in pruning synapses during maturation of the brain. In particular, they found that C4 was necessary for another protein—a complement component called C3—to be deposited onto synapses as a signal that the synapses should be pruned. The data also suggested that the more C4 activity an animal had, the more synapses were eliminated in its brain at a key time in development.
The findings may help explain the longstanding mystery of why the brains of people with schizophrenia tend to have a thinner cerebral cortex (the brain’s outer layer, responsible for many aspects of cognition) with fewer synapses than do brains of unaffected individuals. The work may also help explain why the onset of schizophrenia symptoms tends to occur in late adolescence.
The human brain normally undergoes widespread synapse pruning during adolescence, especially in the cerebral cortex. Excessive synaptic pruning during adolescence and early adulthood, due to increased complement (C4) activity, could lead to the cognitive symptoms seen in schizophrenia.
“Once we had the genetic findings in front of us we started thinking about the possibility that complement molecules are excessively tagging synapses in the developing brain,” Stevens said.
“This discovery enriches our understanding of the complement system in brain development and in disease, and we could not have made that leap without the genetics,” she said. “We’re far from having a treatment based on this, but it’s exciting to think that one day we might be able to turn down the pruning process in some individuals and decrease their risk.”
Opening a path toward early detection and potential therapies
Beyond providing the first insights into the biological origins of schizophrenia, the work raises the possibility that therapies might someday be developed that could turn down the level of synaptic pruning in people who show early symptoms of schizophrenia.
This would be a dramatically different approach from current medical therapies, which address only a specific symptom of schizophrenia—psychosis—rather than the disorder’s root causes, and which do not stop cognitive decline or other symptoms of the illness.
The researchers emphasize that therapies based on these findings are still years down the road. Still, the fact that much is already known about the role of complement proteins in the immune system means that researchers can tap into a wealth of existing knowledge to identify possible therapeutic approaches. For example, anticomplement drugs are already under development for treating other diseases.
“In this area of science, our dream has been to find disease mechanisms that lead to new kinds of treatments,” said McCarroll. “These results show that it is possible to go from genetic data to a new way of thinking about how a disease develops—something that has been greatly needed.”
This work was supported by the Broad Institute’s Stanley Center for Psychiatric Research and by the National Institutes of Health (grants U01MH105641, R01MH077139 and T32GM007753).
Adapted from a Broad Institute news release.
Scientists open the ‘black box’ of schizophrenia with dramatic genetic discovery
Amy Ellis Nutt https://www.washingtonpost.com/news/speaking-of-science/wp/2016/01/27/scientists-open-the-black-box-of-schizophrenia-with-dramatic-genetic-finding/
Scientists Prune Away Schizophrenia’s Hidden Genetic Mechanisms
http://www.genengnews.com/gen-news-highlights/scientists-prune-away-schizophrenia-s-hidden-genetic-mechanisms/81252297/
https://youtu.be/s0y4equOTLg
A landmark study has revealed that a person’s risk of schizophrenia is increased if they inherit specific variants in a gene related to “synaptic pruning”—the elimination of connections between neurons. The findings represent the first time that the origin of this devastating psychiatric disease has been causally linked to specific gene variants and a biological process.
http://www.genengnews.com/Media/images/GENHighlight/thumb_107629_web2209513618.jpg
The site in Chromosome 6 harboring the gene C4 towers far above other risk-associated areas on schizophrenia’s genomic “skyline,” marking its strongest known genetic influence. The new study is the first to explain how specific gene versions work biologically to confer schizophrenia risk. [Psychiatric Genomics Consortium]
- A new study by researchers at the Broad Institute’s Stanley Center for Psychiatric Research, Harvard Medical School, and Boston Children’s Hospital genetically analyzed nearly 65,000 people and revealed that an individual’s risk of schizophrenia is increased if they inherited distinct variants in a gene related to “synaptic pruning”—the elimination of connections between neurons. This new data represents the first time that the origin of this psychiatric disease has been causally linked to particular gene variants and a biological process.
The investigators discovered that versions of a gene commonly thought to be involved in immune function might trigger a runaway pruning of an adolescent brain’s still-maturing communications infrastructure. The researchers described a scenario where patients with schizophrenia show fewer such connections between neurons or synapses.
“Normally, pruning gets rid of excess connections we no longer need, streamlining our brain for optimal performance, but too much pruning can impair mental function,” explained Thomas Lehner, Ph.D., director of the Office of Genomics Research Coordination at the NIH’s National Institute of Mental Health (NIMH), which co-funded the study along with the Stanley Center for Psychiatric Research at the Broad Institute and other NIH components. “It could help explain schizophrenia’s delayed age-of-onset of symptoms in late adolescence and early adulthood and shrinkage of the brain’s working tissue. Interventions that put the brakes on this pruning process-gone-awry could prove transformative.”
The gene the research team called into question, dubbed C4 (complement component 4), was associated with the largest risk for the disorder. C4’s role represents some of the most compelling evidence, to date, linking specific gene versions to a biological process that could cause at least some cases of the illness.
The findings from this study were published recently in Nature through an article entitled “Schizophrenia risk from complex variation of complement component 4.”
“Since schizophrenia was first described over a century ago, its underlying biology has been a black box, in part because it has been virtually impossible to model the disorder in cells or animals,” noted senior study author Steven McCarroll, Ph.D., director of genetics for the Stanley Center and an associate professor of genetics at Harvard Medical School. “The human genome is providing a powerful new way into this disease. Understanding these genetic effects on risk is a way of prying open that block box, peering inside and starting to see actual biological mechanisms.”
Dr. McCarroll and his colleagues found that a stretch of chromosome 6 encompassing several genes known to be involved in immune function emerged as the strongest signal associated with schizophrenia risk in genome-wide analyses. Yet conventional genetics failed to turn up any specific gene versions there that were linked to schizophrenia.
In order to uncover how the immune-related site confers risk for the mental disorder, the scientists mounted a search for cryptic genetic influences that might generate unconventional signals. C4, a gene with known roles in immunity, emerged as a prime suspect because it is unusually variable across individuals.
Upon further investigation into the complexities of how such structural variation relates to the gene’s level of expression and how that, in turn, might link to schizophrenia, the team discovered structurally distinct versions that affect expression of two main forms of the gene within the brain. The more a version resulted in expression of one of the forms, called C4A, the more it was associated with schizophrenia. The greater number of copies an individual had of the suspect versions, the more C4 switched on and the higher their risk of developing schizophrenia. Furthermore, the C4 protein turned out to be most prevalent within the cellular machinery that supports connections between neurons.
“Once we had the genetic findings in front of us we started thinking about the possibility that complement molecules are excessively tagging synapses in the developing brain,” remarked co-author Beth Stevens, Ph.D. a neuroscientist and assistant professor of neurology at Boston Children’s Hospital and institute member at the Broad. “This discovery enriches our understanding of the complement system in brain development and disease, and we could not have made that leap without the genetics. We’re far from having a treatment based on this, but it’s exciting to think that one day we might be able to turn down the pruning process in some individuals and decrease their risk.”
“This study marks a crucial turning point in the fight against mental illness. It changes the game,” added acting NIMH director Bruce Cuthbert, Ph.D. “Because the molecular origins of psychiatric diseases are little-understood, efforts by pharmaceutical companies to pursue new therapeutics are few and far between. This study changes the game. Thanks to this genetic breakthrough, we can finally see the potential for clinical tests, early detection, new treatments, and even prevention.”
Connecting cause and effect through neuroscience
But how exactly does C4—a protein known to mark infectious microbes for destruction by immune cells—affect the risk of schizophrenia?
Answering this question required synthesizing genetics and neurobiology.
Stevens, a recent recipient of a MacArthur Foundation “genius grant,” had found that other complement proteins in the immune system also played a role in brain development. These results came from studying an experimental model of synaptic pruning in the mouse visual system.
“This discovery enriches our understanding of the complement system in brain development and in disease, and we could not have made that leap without the genetics.”
Carroll had long studied C4 for its role in immune disease, and developed mice with different numbers of copies of C4.
The three labs set out to study the role of C4 in the brain.
They found that C4 played a key role in pruning synapses during maturation of the brain. In particular, they found that C4 was necessary for another protein—a complement component called C3—to be deposited onto synapses as a signal that the synapses should be pruned. The data also suggested that the more C4 activity an animal had, the more synapses were eliminated in its brain at a key time in development.
The findings may help explain the longstanding mystery of why the brains of people with schizophrenia tend to have a thinner cerebral cortex (the brain’s outer layer, responsible for many aspects of cognition) with fewer synapses than do brains of unaffected individuals. The work may also help explain why the onset of schizophrenia symptoms tends to occur in late adolescence.
The human brain normally undergoes widespread synapse pruning during adolescence, especially in the cerebral cortex. Excessive synaptic pruning during adolescence and early adulthood, due to increased complement (C4) activity, could lead to the cognitive symptoms seen in schizophrenia.
“Once we had the genetic findings in front of us we started thinking about the possibility that complement molecules are excessively tagging synapses in the developing brain,” Stevens said.
“This discovery enriches our understanding of the complement system in brain development and in disease, and we could not have made that leap without the genetics,” she said. “We’re far from having a treatment based on this, but it’s exciting to think that one day we might be able to turn down the pruning process in some individuals and decrease their risk.”
Opening a path toward early detection and potential therapies
Beyond providing the first insights into the biological origins of schizophrenia, the work raises the possibility that therapies might someday be developed that could turn down the level of synaptic pruning in people who show early symptoms of schizophrenia.
This would be a dramatically different approach from current medical therapies, which address only a specific symptom of schizophrenia—psychosis—rather than the disorder’s root causes, and which do not stop cognitive decline or other symptoms of the illness.
The researchers emphasize that therapies based on these findings are still years down the road. Still, the fact that much is already known about the role of complement proteins in the immune system means that researchers can tap into a wealth of existing knowledge to identify possible therapeutic approaches. For example, anticomplement drugs are already under development for treating other diseases.
“In this area of science, our dream has been to find disease mechanisms that lead to new kinds of treatments,” said McCarroll. “These results show that it is possible to go from genetic data to a new way of thinking about how a disease develops—something that has been greatly needed.”
This work was supported by the Broad Institute’s Stanley Center for Psychiatric Research and by the National Institutes of Health (grants U01MH105641, R01MH077139 and T32GM007753).
Adapted from a Broad Institute news release.
https://img.washingtonpost.com/wp-apps/imrs.php?src=https://img.washingtonpost.com/rf/image_908w/2010-2019/WashingtonPost/2011/09/27/Production/Sunday/SunBiz/Images/mental2b.jpg&w=1484
This post has been updated.
For the first time, scientists have pinned down a molecular process in the brain that helps to trigger schizophrenia. The researchers involved in the landmark study, which was published Wednesday in the journal Nature, say the discovery of this new genetic pathway probably reveals what goes wrong neurologically in a young person diagnosed with the devastating disorder.
The study marks a watershed moment, with the potential for early detection and new treatments that were unthinkable just a year ago, according to Steven Hyman, director of the Stanley Center for Psychiatric Research at the Broad Institute at MIT. Hyman, a former director of the National Institute of Mental Health, calls it “the most significant mechanistic study about schizophrenia ever.”
“I’m a crusty, old, curmudgeonly skeptic,” he said. “But I’m almost giddy about these findings.”
The researchers, chiefly from the Broad Institute, Harvard Medical School and Boston Children’s Hospital, found that a person’s risk of schizophrenia is dramatically increased if they inherit variants of a gene important to “synaptic pruning” — the healthy reduction during adolescence of brain cell connections that are no longer needed.
[Schizophrenic patients have different oral bacteria than non-mentally ill individuals]
In patients with schizophrenia, a variation in a single position in the DNA sequence marks too many synapses for removal and that pruning goes out of control. The result is an abnormal loss of gray matter.
The genes involved coat the neurons with “eat-me signals,” said study co-author Beth Stevens, a neuroscientist at Children’s Hospital and Broad. “They are tagging too many synapses. And they’re gobbled up.
The Institute’s founding director, Eric Lander, believes the research represents an astonishing breakthrough. “It’s taking what has been a black box…and letting us peek inside for the first time. And that is amazingly consequential,” he said.
The timeline for this discovery has been relatively fast. In July 2014, Broad researchers published the results of the largest genomic study on the disorder and found more than 100 genetic locations linked to schizophrenia. Based on that research, Harvard and Broad geneticist Steven McCarroll analyzed data from about 29,000 schizophrenia cases, 36,000 controls and 700 post mortem brains. The information was drawn from dozens of studies performed in 22 countries, all of which contribute to the worldwide database called the Psychiatric Genomics Consortium.
[Influential government-appointed panel recommends depression screening for everyone]
One area in particular, when graphed, showed the strongest association. It was dubbed the “Manhattan plot” for its resemblance to New York City’s towering buildings. The highest peak was on chromosome 6, where McCarroll’s team discovered the gene variant. C4 was “a dark corner of the human genome,” he said, an area difficult to decipher because of its “astonishing level” of diversity.
C4 and numerous other genes reside in a region of chromosome 6 involved in the immune system, which clears out pathogens and similar cellular debris from the brain. The study’s researchers found that one of C4’s variants, C4A, was most associated with a risk for schizophrenia.
More than 25 million people around the globe are affected by schizophrenia, according to the World Health Organization, including 2 million to 3 million Americans. Highly hereditable, it is one of the most severe mental illnesses, with an annual economic burden in this country of tens of billions of dollars.
“This paper is really exciting,” said Jacqueline Feldman, associate medical director of the National Alliance on Mental Illness. “We as scientists and physicians have to temper our enthusiasm because we’ve gone down this path before. But this is profoundly interesting.”
There have been hundreds of theories about schizophrenia over the years, but one of the enduring mysteries has been how three prominent findings related to each other: the apparent involvement of immune molecules, the disorder’s typical onset in late adolescence and early adulthood, and the thinning of gray matter seen in autopsies of patients.
[A low-tech way to help treat young schizophrenic patients]
“The thing about this result,” said McCarroll, the lead author, ” it makes a lot of other things understandable. To have a result to connect to these observations and to have a molecule and strong level of genetic evidence from tens of thousands of research participants, I think that combination sets [this study] apart.”
The authors stressed that their findings, which combine basic science with large-scale analysis of genetic studies, depended on an unusual level of cooperation among experts in genetics, molecular biology, developmental neurobiology and immunology.
“This could not have been done five years ago,” said Hyman. “This required the ability to reference a very large dataset . …When I was [NIMH] director, people really resisted collaborating. They were still in the Pharaoh era. They wanted to be buried with their data.”
The study offers a new approach to schizophrenia research, which has been largely stagnant for decades. Most psychiatric drugs seek to interrupt psychotic thinking, but experts agree that psychosis is just a single symptom — and a late-occurring one at that. One of the chief difficulties for psychiatric researchers, setting them apart from most other medical investigators, is that they can’t cut schizophrenia out of the brain and look at it under a microscope. Nor are there any good animal models.
All that now has changed, according to Stevens. “We now have a strong molecular handle, a pathway and a gene, to develop better models,” he said.
Which isn’t to say a cure is right around the corner.
“This is the first exciting clue, maybe even the most important we’ll ever have, but it will be decades” before a true cure is found,” Hyman said. “Hope is a wonderful thing. False promise is not.”
Insight Pharma Report
Three neurodegenerative disorders that are heavily focused on in this report include: Alzheimer’s Disease/Mild Cognitive Impairment, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Part II of the report will include all three of these disorders, highlighting specifics including background, history, and development of the disease. Deeper into the chapters, the report will unfold biomarkers under investigation, genetic targets, and an analysis of multiple studies investigating these elements.
Experts interviewed in these chapters include:
- Dr. Jens Wendland, Head of Neuroscience Genetics, Precision Medicine, Clinical Research, Pfizer Worldwide R&D
- Dr. Howard J. Federoff, Executive Vice President for Health Sciences, Georgetown University
- Dr. Andrew West, Associate Professor of Neurology and Neurobiology and Co-Director, Center for Neurodegeneration and Experimental Therapeutics
- Dr. Merit Ester Cudkowicz, Chief of Neurology at Massachusetts General Hospital
Part III of the report makes a shift from neurobiomarkers to neurodiagnostics. This section highlights several diagnostics in play and in the making from a number of companies, identifying company strategies, research underway, hypotheses, and institution goals. Elite researchers and companies highlighted in this part include:
- Dr. Xuemei Huang, Professor and Vice Chair, Department of Neurology; Professor of Neurosurgery, Radiology, Pharmacology, and Kinesiology Director; Hershey Brain Analysis Research Laboratory for Neurodegenerative Disorders, Penn State University-Milton, S. Hershey Medical Center Department of Neurology
- Dr. Andreas Jeromin, CSO and President of Atlantic Biomarkers
- Julien Bradley, Senior Director, Sales & Marketing, Quanterix
- Dr. Scott Marshall, Head of Bioanalytics, and Dr. Jared Kohler, Head of Biomarker Statistics, BioStat Solutions, Inc.
Further analysis appears in Part IV. This section includes a survey exclusively conducted for this report. With over 30 figures and graphics and an in depth analysis, this part features insight into targets under investigation, challenges, advantages, and desired features of future diagnostic applications. Furthermore, the survey covers more than just the featured neurodegenerative disorders in this report, expanding to Multiple Sclerosis and Huntington’s Disease.
Finally, Insight Pharma Reports concludes this report with clinical trial and pipeline data featuring targets and products from over 300 companies working in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis.
Epigenome Tapped to Understand Rise of Subtype of Brain Medulloblastoma
http://www.genengnews.com/gen-news-highlights/epigenome-tapped-to-understand-rise-of-subtype-of-brain-medulloblastoma/81252294/
Scientists have identified the cells that likely give rise to the brain tumor subtype Group 4 medulloblastoma. [V. Yakobchuk/ Fotolia]
http://www.genengnews.com/Media/images/GENHighlight/thumb_Jan_28_2016_Fotolia_6761569_ColorfulBrain_4412824411.jpg
An international team of scientists say they have identified the cells that likely give rise to the brain tumor subtype Group 4 medulloblastoma. The believe their study (“Active medulloblastoma enhancers reveal subgroup-specific cellular origins”), published in Nature, removes a barrier to developing more effective targeted therapies against the brain tumor’s most common subtype.
Medulloblastoma occurs in infants, children, and adults, but it is the most common malignant pediatric brain tumor. The disease includes four biologically and clinically distinct subtypes, of which Group 4 is the most common. In children, about half of medulloblastoma patients are of the Group 4 subtype. Efforts to improve patient outcomes, particularly for those with high-risk Group 4 medulloblastoma, have been hampered by the lack of accurate animal models.
Evidence from this study suggests Group 4 tumors begin in neural stem cells that are born in a region of the developing cerebellum called the upper rhomic lip (uRL), according to the researchers.
“Pinpointing the cell(s) of origin for Group 4 medulloblastoma will help us to better understand normal cerebellar development and dramatically improve our chances of developing genetically faithful preclinical mouse models. These models are desperately needed for learning more about Group 4 medulloblastoma biology and evaluating rational, molecularly targeted therapies to improve patient outcomes,” said Paul Northcott, Ph.D., an assistant member of the St. Jude department of developmental neurobiology. Dr. Northcott, Stefan Pfister, M.D., of the German Cancer Research Center (DKFZ), and James Bradner, M.D., of Dana-Farber Cancer Institute, are the corresponding authors.
The discovery and other findings about the missteps fueling tumor growth came from studying the epigenome. Researchers used the analytic tool ChiP-seq to identify and track medulloblastoma subtype differences based on the activity of epigenetic regulators, which included proteins known as master regulator transcription factors. They bind to DNA enhancers and super-enhancers. The master regulator transcription factors and super-enhancers work together to regulate the expression of critical genes, such as those responsible for cell identity.
Those and other tools helped investigators identify more than 3,000 super-enhancers in 28 medulloblastoma tumors as well as evidence that the activity of super-enhancers varied by subtype. The super-enhancers switched on known cancer genes, including genes like ALK, MYC, SMO, and OTX2 that are associated with medulloblastoma, the researchers reported.
Knowledge of the subtype super-enhancers led to identification of the transcription factors that regulate their activity. Using computational methods, researchers applied that information to reconstruct the transcription factor networks responsible for medulloblastoma subtype diversity and identity, providing previously unknown insights into the regulatory landscape and transcriptional output of the different medulloblastoma subtypes.
The approach helped to discover and nominate Lmx1A as a master regulator transcription factor of Group 4 tumors, which led to the identification of the likely Group 4 tumor cells of origin. Lmx1A was known to play an important role in normal development of cells in the uRL and cerebellum. Additional studies performed in mice with and without Lmx1A in this study supported uRL cells as the likely source of Group 4 tumors.
“By studying the epigenome, we also identified new pathways and molecular dependencies not apparent in previous gene expression and mutational studies,” explained Dr. Northcott. “The findings open new therapeutic avenues, particularly for the Group 3 and 4 subtypes where patient outcomes are inferior for the majority of affected children.”
For example, researchers identified increased enhancer activity targeting the TGFbeta pathway. The finding adds to evidence that the pathway may drive Group 3 medulloblastoma, currently the subtype with the worst prognosis. The pathway regulates cell growth, cell death, and other functions that are often disrupted in cancer, but it’s role in medulloblastoma is poorly understood.
The analysis included samples from 28 medulloblastoma tumors representing the four subtypes. Researchers believe it is the largest epigenetic study yet for any single cancer type and, importantly, the first to use a large cohort of primary patient tumor tissues instead of cell lines grown in the laboratory. Previous studies have suggested that cell lines may be of limited use for studying the tumor epigenome. The three Group 3 medulloblastoma cell lines used in this study reinforced the observation, highlighting significant differences in epigenetic regulators at work in medulloblastoma cell lines versus tumor samples.
Read Full Post »

















 These conclusions do not mean that the brain does not remain malleable, even in old age. Any mentally effortful new experience, such as learning a language, acquiring a motor skill, navigating in a new environment, and, yes, playing commercially available computer games, will produce changes in those neural systems that support acquisition of the new skill. For example, there may be an increase in the number of synapses, the number of neurons and supporting cells, or a strengthening of the connections among them. This type of brain plasticity is possible throughout the life span, though younger brains seem to have an advantage over the older ones. It would be appropriate to conclude from such work that the potential to learn new skills remains intact throughout the life span. However at this point it is not appropriate to conclude that training-induced changes go significantly beyond the learned skills, that they affect broad abilities with real-world relevance, or that they generally promote “brain health”.
These conclusions do not mean that the brain does not remain malleable, even in old age. Any mentally effortful new experience, such as learning a language, acquiring a motor skill, navigating in a new environment, and, yes, playing commercially available computer games, will produce changes in those neural systems that support acquisition of the new skill. For example, there may be an increase in the number of synapses, the number of neurons and supporting cells, or a strengthening of the connections among them. This type of brain plasticity is possible throughout the life span, though younger brains seem to have an advantage over the older ones. It would be appropriate to conclude from such work that the potential to learn new skills remains intact throughout the life span. However at this point it is not appropriate to conclude that training-induced changes go significantly beyond the learned skills, that they affect broad abilities with real-world relevance, or that they generally promote “brain health”. In a balanced evaluation of brain games, we also need to keep in mind opportunity costs. Time spent playing the games is time not spent reading, socializing, gardening, exercising, or engaging in many other activities that may benefit cognitive and physical health of older adults. Given that the effects of playing the games tend to be task-specific, it may be advisable to train an activity that by itself comes with benefits for everyday life. Another drawback of publicizing computer games as a fix to deteriorating cognitive performance is that it diverts attention and resources from prevention efforts. The promise of a magic bullet detracts from the message that cognitive vigor in old age, to the extent that it can be influenced by the lives we live, reflects the long-term effects of a healthy and active lifestyle.
In a balanced evaluation of brain games, we also need to keep in mind opportunity costs. Time spent playing the games is time not spent reading, socializing, gardening, exercising, or engaging in many other activities that may benefit cognitive and physical health of older adults. Given that the effects of playing the games tend to be task-specific, it may be advisable to train an activity that by itself comes with benefits for everyday life. Another drawback of publicizing computer games as a fix to deteriorating cognitive performance is that it diverts attention and resources from prevention efforts. The promise of a magic bullet detracts from the message that cognitive vigor in old age, to the extent that it can be influenced by the lives we live, reflects the long-term effects of a healthy and active lifestyle.{kind=link}
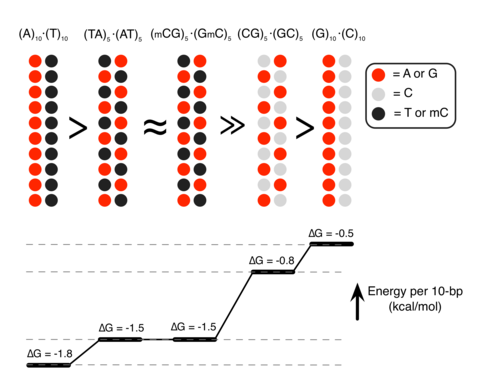{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}