
Genomics and epigenetics link to DNA structure
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Sequence and Epigenetic Factors Determine Overall DNA Structure

Atomic-level simulations show electrostatic forces between each atom. [Alek Aksimentiev, University of Illinois at Urbana-Champaign]
The traditionally held hypothesis about the highly ordered organization of DNA describes the interaction of various proteins with DNA sequences to mediate the dynamic structure of the molecule. However, recent evidence has emerged that stretches of homologous DNA sequences can associate preferentially with one another, even in the absence of proteins.
Researchers at the University of Illinois Center for the Physics of Living Cells, Johns Hopkins University, and Ulsan National Institute of Science and Technology (UNIST) in South Korea found that DNA molecules interact directly with one another in ways that are dependent on the sequence of the DNA and epigenetic factors, such as methylation.
The researchers described evidence they found for sequence-dependent attractive interactions between double-stranded DNA molecules that neither involve intermolecular strand exchange nor are mediated by DNA-binding proteins.
“DNA molecules tend to repel each other in water, but in the presence of special types of cations, they can attract each other just like nuclei pulling each other by sharing electrons in between,” explained lead study author Hajin Kim, Ph.D., assistant professor of biophysics at UNIST. “Our study suggests that the attractive force strongly depends on the nucleic acid sequence and also the epigenetic modifications.”
The investigators used atomic-level supercomputer simulations to measure the forces between a pair of double-stranded DNA helices and proposed that the distribution of methyl groups on the DNA was the key to regulating this sequence-dependent attraction. To verify their findings experimentally, the scientists were able to observe a single pair of DNA molecules within nanoscale bubbles.
“Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation,” the authors wrote. “We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine act as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction.”
The findings from this study were published recently in Nature Communications in an article entitled “Direct Evidence for Sequence-Dependent Attraction Between Double-Stranded DNA Controlled by Methylation.”
After conducting numerous further simulations, the research team concluded that direct DNA–DNA interactions could play a central role in how chromosomes are organized in the cell and which ones are expanded or folded up compactly, ultimately determining functions of different cell types or regulating the cell cycle.
“Biophysics is a fascinating subject that explores the fundamental principles behind a variety of biological processes and life phenomena,” Dr. Kim noted. “Our study requires cross-disciplinary efforts from physicists, biologists, chemists, and engineering scientists and we pursue the diversity of scientific disciplines within the group.”
Dr. Kim concluded by stating that “in our lab, we try to unravel the mysteries within human cells based on the principles of physics and the mechanisms of biology. In the long run, we are seeking for ways to prevent chronic illnesses and diseases associated with aging.”
Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation
Jejoong Yoo, Hajin Kim, Aleksei Aksimentiev, and Taekjip Ha
Nature Communications 7 11045 (2016) DOI:10.1038/ncomms11045BibTex

Although proteins mediate highly ordered DNA organization in vivo, theoretical studies suggest that homologous DNA duplexes can preferentially associate with one another even in the absence of proteins. Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation. We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine acts as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction. Indeed, methylation of cytosines makes attraction between GC-rich DNA as strong as that between AT-rich DNA. Recent genome-wide chromosome organization studies showed that remote contact frequencies are higher for AT-rich and methylated DNA, suggesting that direct DNA–DNA interactions that we report here may play a role in the chromosome organization and gene regulation.
Formation of a DNA double helix occurs through Watson–Crick pairing mediated by the complementary hydrogen bond patterns of the two DNA strands and base stacking. Interactions between double-stranded (ds)DNA molecules in typical experimental conditions containing mono- and divalent cations are repulsive1, but can turn attractive in the presence of high-valence cations2. Theoretical studies have identified the ion–ion correlation effect as a possible microscopic mechanism of the DNA condensation phenomena3, 4, 5. Theoretical investigations have also suggested that sequence-specific attractive forces might exist between two homologous fragments of dsDNA6, and this ‘homology recognition’ hypothesis was supported by in vitro atomic force microscopy7 and in vivo point mutation assays8. However, the systems used in these measurements were too complex to rule out other possible causes such as Watson–Crick strand exchange between partially melted DNA or protein-mediated association of DNA.
Here we present direct evidence for sequence-dependent attractive interactions between dsDNA molecules that neither involve intermolecular strand exchange nor are mediated by proteins. Further, we find that the sequence-dependent attraction is controlled not by homology—contradictory to the ‘homology recognition’ hypothesis6—but by a methylation pattern. Unlike the previous in vitro study that used monovalent (Na+) or divalent (Mg2+) cations7, we presumed that for the sequence-dependent attractive interactions to operate polyamines would have to be present. Polyamine is a biological polycation present at a millimolar concentration in most eukaryotic cells and essential for cell growth and proliferation9, 10. Polyamines are also known to condense DNA in a concentration-dependent manner2, 11. In this study, we use spermine4+(Sm4+) that contains four positively charged amine groups per molecule.
Sequence dependence of DNA–DNA forces
To characterize the molecular mechanisms of DNA–DNA attraction mediated by polyamines, we performed molecular dynamics (MD) simulations where two effectively infinite parallel dsDNA molecules, 20 base pairs (bp) each in a periodic unit cell, were restrained to maintain a prescribed inter-DNA distance; the DNA molecules were free to rotate about their axes. The two DNA molecules were submerged in 100 mM aqueous solution of NaCl that also contained 20 Sm4+molecules; thus, the total charge of Sm4+, 80 e, was equal in magnitude to the total charge of DNA (2 × 2 × 20 e, two unit charges per base pair; Fig. 1a). Repeating such simulations at various inter-DNA distances and applying weighted histogram analysis12 yielded the change in the interaction free energy (ΔG) as a function of the DNA–DNA distance (Fig. 1b,c). In a broad agreement with previous experimental findings13, ΔG had a minimum, ΔGmin, at the inter-DNA distance of 25−30 Å for all sequences examined, indeed showing that two duplex DNA molecules can attract each other. The free energy of inter-duplex attraction was at least an order of magnitude smaller than the Watson–Crick interaction free energy of the same length DNA duplex. A minimum of ΔG was not observed in the absence of polyamines, for example, when divalent or monovalent ions were used instead14, 15.

(a) Set-up of MD simulations. A pair of parallel 20-bp dsDNA duplexes is surrounded by aqueous solution (semi-transparent surface) containing 20 Sm4+ molecules (which compensates exactly the charge of DNA) and 100 mM NaCl. Under periodic boundary conditions, the DNA molecules are effectively infinite. A harmonic potential (not shown) is applied to maintain the prescribed distance between the dsDNA molecules. (b,c) Interaction free energy of the two DNA helices as a function of the DNA–DNA distance for repeat-sequence DNA fragments (b) and DNA homopolymers (c). (d) Schematic of experimental design. A pair of 120-bp dsDNA labelled with a Cy3/Cy5 FRET pair was encapsulated in a ~200-nm diameter lipid vesicle; the vesicles were immobilized on a quartz slide through biotin–neutravidin binding. Sm4+ molecules added after immobilization penetrated into the porous vesicles. The fluorescence signals were measured using a total internal reflection microscope. (e) Typical fluorescence signals indicative of DNA–DNA binding. Brief jumps in the FRET signal indicate binding events. (f) The fraction of traces exhibiting binding events at different Sm4+ concentrations for AT-rich, GC-rich, AT nonhomologous and CpG-methylated DNA pairs. The sequence of the CpG-methylated DNA specifies the methylation sites (CG sequence, orange), restriction sites (BstUI, triangle) and primer region (underlined). The degree of attractive interaction for the AT nonhomologous and CpG-methylated DNA pairs was similar to that of the AT-rich pair. All measurements were done at [NaCl]=50 mM and T=25 °C. (g) Design of the hybrid DNA constructs: 40-bp AT-rich and 40-bp GC-rich regions were flanked by 20-bp common primers. The two labelling configurations permit distinguishing parallel from anti-parallel orientation of the DNA. (h) The fraction of traces exhibiting binding events as a function of NaCl concentration at fixed concentration of Sm4+ (1 mM). The fraction is significantly higher for parallel orientation of the DNA fragments.
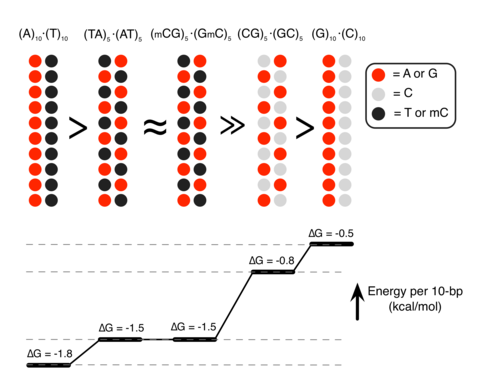
Unexpectedly, we found that DNA sequence has a profound impact on the strength of attractive interaction. The absolute value of ΔG at minimum relative to the value at maximum separation, |ΔGmin|, showed a clearly rank-ordered dependence on the DNA sequence: |ΔGmin| of (A)20>|ΔGmin| of (AT)10>|ΔGmin| of (GC)10>|ΔGmin| of (G)20. Two trends can be noted. First, AT-rich sequences attract each other more strongly than GC-rich sequences16. For example, |ΔGmin| of (AT)10 (1.5 kcal mol−1 per turn) is about twice |ΔGmin| of (GC)10 (0.8 kcal mol−1 per turn) (Fig. 1b). Second, duplexes having identical AT content but different partitioning of the nucleotides between the strands (that is, (A)20 versus (AT)10 or (G)20 versus (GC)10) exhibit statistically significant differences (~0.3 kcal mol−1 per turn) in the value of |ΔGmin|.
To validate the findings of MD simulations, we performed single-molecule fluorescence resonance energy transfer (smFRET)17 experiments of vesicle-encapsulated DNA molecules. Equimolar mixture of donor- and acceptor-labelled 120-bp dsDNA molecules was encapsulated in sub-micron size, porous lipid vesicles18 so that we could observe and quantitate rare binding events between a pair of dsDNA molecules without triggering large-scale DNA condensation2. Our DNA constructs were long enough to ensure dsDNA–dsDNA binding that is stable on the timescale of an smFRET measurement, but shorter than the DNA’s persistence length (~150 bp (ref. 19)) to avoid intramolecular condensation20. The vesicles were immobilized on a polymer-passivated surface, and fluorescence signals from individual vesicles containing one donor and one acceptor were selectively analysed (Fig. 1d). Binding of two dsDNA molecules brings their fluorescent labels in close proximity, increasing the FRET efficiency (Fig. 1e).
FRET signals from individual vesicles were diverse. Sporadic binding events were observed in some vesicles, while others exhibited stable binding; traces indicative of frequent conformational transitions were also observed (Supplementary Fig. 1A). Such diverse behaviours could be expected from non-specific interactions of two large biomolecules having structural degrees of freedom. No binding events were observed in the absence of Sm4+ (Supplementary Fig. 1B) or when no DNA molecules were present. To quantitatively assess the propensity of forming a bound state, we chose to use the fraction of single-molecule traces that showed any binding events within the observation time of 2 min (Methods). This binding fraction for the pair of AT-rich dsDNAs (AT1, 100% AT in the middle 80-bp section of the 120-bp construct) reached a maximum at ~2 mM Sm4+(Fig. 1f), which is consistent with the results of previous experimental studies2, 3. In accordance with the prediction of our MD simulations, GC-rich dsDNAs (GC1, 75% GC in the middle 80 bp) showed much lower binding fraction at all Sm4+ concentrations (Fig. 1b,c). Regardless of the DNA sequence, the binding fraction reduced back to zero at high Sm4+ concentrations, likely due to the resolubilization of now positively charged DNA–Sm4+ complexes2, 3, 13.
Because the donor and acceptor fluorophores were attached to the same sequence of DNA, it remained possible that the sequence homology between the donor-labelled DNA and the acceptor-labelled DNA was necessary for their interaction6. To test this possibility, we designed another AT-rich DNA construct AT2 by scrambling the central 80-bp section of AT1 to remove the sequence homology (Supplementary Table 1). The fraction of binding traces for this nonhomologous pair of donor-labelled AT1 and acceptor-labelled AT2 was comparable to that for the homologous AT-rich pair (donor-labelled AT1 and acceptor-labelled AT1) at all Sm4+ concentrations tested (Fig. 1f). Furthermore, this data set rules out the possibility that the higher binding fraction observed experimentally for the AT-rich constructs was caused by inter-duplex Watson–Crick base pairing of the partially melted constructs.
Next, we designed a DNA construct named ATGC, containing, in its middle section, a 40-bp AT-rich segment followed by a 40-bp GC-rich segment (Fig. 1g). By attaching the acceptor to the end of either the AT-rich or GC-rich segments, we could compare the likelihood of observing the parallel binding mode that brings the two AT-rich segments together and the anti-parallel binding mode. Measurements at 1 mM Sm4+ and 25 or 50 mM NaCl indicated a preference for the parallel binding mode by ~30% (Fig. 1h). Therefore, AT content can modulate DNA–DNA interactions even in a complex sequence context. Note that increasing the concentration of NaCl while keeping the concentration of Sm4+ constant enhances competition between Na+ and Sm4+ counterions, which reduces the concentration of Sm4+ near DNA and hence the frequency of dsDNA–dsDNA binding events (Supplementary Fig. 2).
Methylation determines the strength of DNA–DNA attraction
Analysis of the MD simulations revealed the molecular mechanism of the polyamine-mediated sequence-dependent attraction (Fig. 2). In the case of the AT-rich fragments, the bulky methyl group of thymine base blocks Sm4+ binding to the N7 nitrogen atom of adenine, which is the cation-binding hotspot21, 22. As a result, Sm4+ is not found in the major grooves of the AT-rich duplexes and resides mostly near the DNA backbone (Fig. 2a,d). Such relocated Sm4+ molecules bridge the two DNA duplexes better, accounting for the stronger attraction16, 23, 24, 25. In contrast, significant amount of Sm4+ is adsorbed to the major groove of the GC-rich helices that lacks cation-blocking methyl group (Fig. 2b,e).

(a–c) Representative configurations of Sm4+ molecules at the DNA–DNA distance of 28 Å for the (AT)10–(AT)10 (a), (GC)10–(GC)10 (b) and (GmC)10–(GmC)10 (c) DNA pairs. The backbone and bases of DNA are shown as ribbon and molecular bond, respectively; Sm4+ molecules are shown as molecular bonds. Spheres indicate the location of the N7 atoms and the methyl groups. (d–f) The average distributions of cations for the three sequence pairs featured in a–c. Top: density of Sm4+ nitrogen atoms (d=28 Å) averaged over the corresponding MD trajectory and the z axis. White circles (20 Å in diameter) indicate the location of the DNA helices. Bottom: the average density of Sm4+ nitrogen (blue), DNA phosphate (black) and sodium (red) atoms projected onto the DNA–DNA distance axis (x axis). The plot was obtained by averaging the corresponding heat map data over y=[−10, 10] Å. See Supplementary Figs 4 and 5 for the cation distributions at d=30, 32, 34 and 36 Å.
If indeed the extra methyl group in thymine, which is not found in cytosine, is responsible for stronger DNA–DNA interactions, we can predict that cytosine methylation, which occurs naturally in many eukaryotic organisms and is an essential epigenetic regulation mechanism26, would also increase the strength of DNA–DNA attraction. MD simulations showed that the GC-rich helices containing methylated cytosines (mC) lose the adsorbed Sm4+ (Fig. 2c,f) and that |ΔGmin| of (GC)10 increases on methylation of cytosines to become similar to |ΔGmin| of (AT)10 (Fig. 1b).
To experimentally assess the effect of cytosine methylation, we designed another GC-rich construct GC2 that had the same GC content as GC1 but a higher density of CpG sites (Supplementary Table 1). The CpG sites were then fully methylated using M. SssI methyltransferase (Supplementary Fig. 3; Methods). As predicted from the MD simulations, methylation of the GC-rich constructs increased the binding fraction to the level of the AT-rich constructs (Fig. 1f).
The sequence dependence of |ΔGmin| and its relation to the Sm4+ adsorption patterns can be rationalized by examining the number of Sm4+ molecules shared by the dsDNA molecules (Fig. 3a). An Sm4+ cation adsorbed to the major groove of one dsDNA is separated from the other dsDNA by at least 10 Å, contributing much less to the effective DNA–DNA attractive force than a cation positioned between the helices, that is, the ‘bridging’ Sm4+ (ref. 23). An adsorbed Sm4+ also repels other Sm4+ molecules due to like-charge repulsion, lowering the concentration of bridging Sm4+. To demonstrate that the concentration of bridging Sm4+ controls the strength of DNA–DNA attraction, we computed the number of bridging Sm4+ molecules, Nspm (Fig. 3b). Indeed, the number of bridging Sm4+ molecules ranks in the same order as |ΔGmin|: Nspm of (A)20>Nspm of (AT)10≈Nspm of (GmC)10>Nspm of (GC)10>Nspm of (G)20. Thus, the number density of nucleotides carrying a methyl group (T and mC) is the primary determinant of the strength of attractive interaction between two dsDNA molecules. At the same time, the spatial arrangement of the methyl group carrying nucleotides can affect the interaction strength as well (Fig. 3c). The number of methyl groups and their distribution in the (AT)10 and (GmC)10 duplex DNA are identical, and so are their interaction free energies, |ΔGmin| of (AT)10≈|ΔGmin| of (GmC)10. For AT-rich DNA sequences, clustering of the methyl groups repels Sm4+ from the major groove more efficiently than when the same number of methyl groups is distributed along the DNA (Fig. 3b). Hence, |ΔGmin| of (A)20>|ΔGmin| of (AT)10. For GC-rich DNA sequences, clustering of the cation-binding sites (N7 nitrogen) attracts more Sm4+ than when such sites are distributed along the DNA (Fig. 3b), hence |ΔGmin| is larger for (GC)10 than for (G)20.

(a) Typical spatial arrangement of Sm4+ molecules around a pair of DNA helices. The phosphates groups of DNA and the amine groups of Sm4+ are shown as red and blue spheres, respectively. ‘Bridging’ Sm4+molecules reside between the DNA helices. Orange rectangles illustrate the volume used for counting the number of bridging Sm4+ molecules. (b) The number of bridging amine groups as a function of the inter-DNA distance. The total number of Sm4+ nitrogen atoms was computed by averaging over the corresponding MD trajectory and the 10 Å (x axis) by 20 Å (y axis) rectangle prism volume (a) centred between the DNA molecules. (c) Schematic representation of the dependence of the interaction free energy of two DNA molecules on their nucleotide sequence. The number and spatial arrangement of nucleotides carrying a methyl group (T or mC) determine the interaction free energy of two dsDNA molecules.
Genome-wide investigations of chromosome conformations using the Hi–C technique revealed that AT-rich loci form tight clusters in human nucleus27, 28. Gene or chromosome inactivation is often accompanied by increased methylation of DNA29 and compaction of facultative heterochromatin regions30. The consistency between those phenomena and our findings suggest the possibility that the polyamine-mediated sequence-dependent DNA–DNA interaction might play a role in chromosome folding and epigenetic regulation of gene expression.
- , & Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: hydration forces between parallel DNA double helices. Proc. Natl Acad. Sci. USA 81, 2621–2625 (1984).
- , , & Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J. 74, 381–393 (1998).
- , & Charge inversion accompanies DNA condensation by multivalent ions. Nat. Phys. 3, 641–644 (2007).
- , , & Understanding nucleic acid-ion interactions.Annu. Rev. Biochem. 83, 813–841 (2014).
- , & The physics of charge inversion in chemical and biological systems. Rev. Mod. Phys. 74, 329–345 (2002).
- & Sequence recognition in the pairing of DNA duplexes. Phys. Rev. Lett. 86, 3666–3669 (2001).
- et al. Single molecule detection of direct, homologous, DNA/DNA pairing.Proc. Natl Acad. Sci. USA 106, 19824–19829 (2009).
- & Direct recognition of homology between double helices of DNA in Neurospora crassa. Nat. Commun. 5, 3509 (2014).
- & Polyamines. Annu. Rev. Biochem. 53, 749–790 (1984).
- & Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. 58, 244–258 (2001).










")



 Department of Health and Human Services
Department of Health and Human Services













 42875 genomic copy number arrays
42875 genomic copy number arrays 634 experimental series
634 experimental series 256 array platforms
256 array platforms 197 ICD-O cancer entities
197 ICD-O cancer entities 480 publications (Pubmed entries)
480 publications (Pubmed entries) The
The 
 The
The  The
The  The
The  The
The  The
The  The
The 





 The
The 


























")






{kind=link}
{kind=link}