Notable Papers in Neurosciences
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Notable Papers in Neurosciences
NIH researchers’ new mouse model points to a gene therapy for eye disease
Oliver Worsley
A mouse model has been established for Leber hereditary optic neuropathy (LHON), a vision disorder caused by mutations within genes in the “battery packs” of our cells–the mitochondria. And investigators at the NIH say they were able to develop a gene therapy that could be used to treat it.
Within the mitochondrion are mitochondrial DNA (mtDNA) which carries the instructions for important metabolic processes required to keep the cell topped up with energy. Mutations in genes found in mtDNA can lead to various diseases; one of these is LHON which affects around 1 in 30,000 in England.
“[Until now] there was no efficient way to get DNA into mitochondria,” said John Guy, who is a professor of ophthalmology and is lead author of this study. Their work has been published in the Proceedings of the National Academy of Sciences.
Early symptoms of the disease include blurred vision and eyesight will eventually deteriorate over time. A loss of retinal ganglion cells is at the crux of the pathology and these cells are crucial for carrying visual signals from the retina to the brain, via the optic nerve.
The most prevalent mutation responsible for LHON is in a mitochondrial gene called ND4. Dr. Guy and his lab have been attempting to develop a gene therapy approach to correct this mutation for 15 years now. But one issue with adopting the widely used viral vector is that despite its efficacy in integrating into nuclear DNA–viruses have a harder time penetrating the mitochondria.
In developing his mouse model, Dr. Guy found a way around this. He fixed a virus with the same mutation in ND4 seen in 70% of LHON patients–adding a protein that mitochondria require from outside the organelle, as they cannot produce it on their own.
In the hijacked virus they included a fluorescent tag so they could confirm the future progeny of mice which had the defective gene. The mouse model does what is seen in patients with the same disease and optic nerve atrophy, loss of retinal ganglion cells and a decline in visual response is consistently observed.
The next step was providing a gene therapy to reverse it. The researchers packaged a normal ND4 gene into the same type of virus and injected it directly into the eye–leading to marked visual improvements without any side effects from the virus itself.
Related Articles:
Study: Eyes may signal brain pathology in schizophrenia
Stem cell therapy protects vision in preclinical retinal disease study
Retinas made from embryonic stem cells implanted into mice for the first time
GEN News Highlights
Oct 7, 2015
Stem Cell Advance Brings Vision Repair in Sight
http://www.genengnews.com/gen-news-highlights/stem-cell-advance-brings-vision-repair-in-sight/81251832/
Transplantation of cones produced from stem cells could reverse macular degeneration. A new differentiation approach yields abundant cones from human embryonic stem cells. When allowed to grow to confluence, the cones spontaneously form sheets of organized retinal tissue. [G. Bernier, University of Montreal]
http://www.genengnews.com/Media/images/GENHighlight/thumb_Oct7_2015_UnivMontreal_StemCellVision1867674218.jpg
A dearth of cone cells means degraded vision, so perhaps cone cell numbers could be raised, if only there were a way to produce cone cells in abundance. Then, cone cells could be transplanted en masse, potentially reversing the vision losses due to age-related macular degeneration.
We are born with a fixed number of cone cells. Additional cone cells must be contrived if degradation of the retina, a condition that is accelerated in nearly one out of four people, is to be reversed. Although cone cells have been produced by means of stem cell differentiation, the output has been meager. Now, however, scientists at the University of Montreal report that they have developed an efficient technique for producing cone cells from human embryonic stem cells.
These scientists, led by Gilbert Bernier, Ph.D., essentially closed a number of signaling pathways in stem cells, leaving open a default pathway that led to photoreceptor genesis. The scientists detailed their work in the journal Development, in an article that appeared online October 1. The article—“Differentiation of human embryonic stem cells into cone photoreceptors through simultaneous inhibition of BMP, TGFβ, and Wnt signaling”—is the culmination of years of work.
Bernier has been interested in the genes that code and enable the induction of the retina during embryonic development since completing his doctorate in molecular biology in 1997. “During my post-doc at the Max-Planck Institute in Germany, I developed the idea that there was a natural molecule that must exist and be capable of forcing embryonic stem cells into becoming cones,” he said. Indeed, bioinformatic analysis led him to predict the existence of a mysterious protein: COCO, a “recombinational” human molecule that is normally expressed within photoreceptors during their development.
In 2001, Bernier launched his laboratory in Montreal and immediately isolated the molecule. But it took several years of research to demystify the molecular pathways involved in the photoreceptors development mechanism. The Bernier laboratory’s current work has established that Coco (Dand5), a member of the Cerberus gene family, is expressed in the developing and adult mouse retina.
“Upon exposure to recombinant COCO, human embryonic stem cells (hESCs) differentiated into S-cone photoreceptors, developed an inner segment-like protrusion, and could degrade cGMP when exposed to light,” Bernier and colleagues wrote in the Development article. “Addition of thyroid hormone resulted in a transition from a unique S-cone population toward a mixed M/S-cone population.”
In addition, when the COCO-exposed hESCs were cultured at confluence for a prolonged period of time, they spontaneously developed into a cellular sheet composed of polarized cone photoreceptors. “Within 45 days, the cones that we allowed to grow toward confluence spontaneously formed organized retinal tissue that was 150 microns thick,” Dr. Bernier noted. “This has never been achieved before.”
In order to verify the technique, Dr. Bernier injected clusters of retinal cells into the eyes of healthy mice. The transplanted photoreceptors migrated naturally within the retina of their host.
Although Dr. Bernier acknowledged that the transplantation of photoreceptors in clinical trials was years away, he expressed optimism that his laboratory had made a significant advance, one that could, ultimately, benefit countless patients. “Our method has the capacity to differentiate 80% of the stem cells into pure cones,” Dr. Gilbert explained. “Thanks to our simple and effective approach, any laboratory in the world will now be able to create masses of photoreceptors.”
Beyond the clinical applications, Dr. Bernier’s findings could enable the modeling of human retinal degenerative diseases through the use of induced pluripotent stem cells, offering the possibility of directly testing potential avenues for therapy on the patient’s own tissues. “Our work,” the Development article concluded, “provides a unique platform to produce human cones for developmental, biochemical, and therapeutic studies.”
Neurogenesis in the Mammalian Brain
Neuron nurseries in the adult brains of rodents and humans appear to influence cognitive function.
By Jef Akst | October 1, 2015
http://www.the-scientist.com//?articles.view/articleNo/44047/title/Neurogenesis-in-the-Mammalian-Brain/
In rodents, there are two populations of neural stem cells in the adult brain. The majority of new neurons are born in the subventricular zone along the lateral ventricle wall and migrate through the rostral migratory stream (RMS) to the olfactory bulb. About one-tenth as many new neurons are produced in the subgranular zone of the dentate gyrus (white) of the hippocampus.
In the rodent dentate gyrus, neural stem cells differentiate into neuroblasts before maturing and integrating with hippocampal circuits important in learning and memory.
In the rodent subventricular zone, neural stem cells differentiate into neuroblasts, which make their way to the olfactory bulb, where they complete their development.
Researchers have also demonstrated that neurogenesis occurs in the adult human brain, though the locations and degree of cell proliferation appear to differ somewhat from rodents. Strong evidence now exists that new neurons are born in the dentate gyrus of the hippocampus, where they integrate into existing circuits. But so far, there is no definitive support for the migration of new neurons migrating from the subventricular zone (SVZ) of the lateral ventricle to the olfactory bulb, which is atrophied relative to the olfactory bulb of rodents and other mammals that rely more heavily on smell. However, one study did report signs of neurogenesis in an area next to the SVZ, the striatum, which is important for cognitive function and motor control.
Brain Gain
Young neurons in the adult human brain are likely critical to its function.
By Jef Akst | October 1, 2015
http://www.the-scientist.com/?articles.view/articleNo/44097/title/Brain-Gain/
How the Brain Builds New Thoughts
10/06/2015 Harvard University
http://www.biosciencetechnology.com/news/2015/10/how-brain-builds-new-thoughts?
“One of the big mysteries of human cognition is how the brain takes ideas and puts them together in new ways to form new thoughts,” said postdoctoral fellow Steven Frankland. (Kris Snibbe/Harvard Staff Photographer)Let’s start with a simple sentence: Last week Joe Biden beat Vladimir Putin in a game of Scrabble.
http://www.biosciencetechnology.com/sites/biosciencetechnology.com/files/bt1510_harvard_snibbe.jpg
It’s a strange notion to entertain, certainly, but one humans can easily make sense of, researchers say, thanks to the way the brain constructs new thoughts.
A new study, co-authored by postdoctoral fellow Steven Frankland and Professor of Psychology Joshua Greene, suggests that two adjacent brain regions allow humans to build new thoughts using a sort of conceptual algebra, mimicking the operations of silicon computers that represent variables and their changing values. The study is described in a Sept. 17 paper in the Proceedings of the National Academy of Sciences.
“One of the big mysteries of human cognition is how the brain takes ideas and puts them together in new ways to form new thoughts,” said Frankland, the lead author of the study. “Most people can understand ‘Joe Biden beat Vladimir Putin at Scrabble’ even though they’ve never thought about that situation, because, as long as you know who Putin is, who Biden is, what Scrabble is, and what it means to win, you’re able to put these concepts together to understand the meaning of the sentence. That’s a basic, but remarkable, cognitive ability.”
But how are such thoughts constructed? According to one theory, the brain does it by representing conceptual variables, answers to recurring questions of meaning such as “What was done?” and “Who did it?” and “To whom was it done?” A new thought such as “Biden beats Putin” can then be built by making “beating” the value of the action variable, “Biden” the value of the “agent” variable (“Who did it?”), and “Putin” the value of the “patient” variable (“To whom was it done?”). Frankland and Greene are the first to point to specific regions of the brain that encode such mental syntax.
“This has been a central theoretical discussion in cognitive science for a long time, and although it has seemed like a pretty good bet that the brain works this way, there’s been little direct empirical evidence for it,” Frankland said.
To identify the regions, Frankland and Greene used functional magnetic resonance imaging (fMRI) to scan students’ brains as they read a series of simple sentences such as “The dog chased the man” and “The man chased the dog.”
Equipped with that data, they then turned to algorithms to identify patterns of brain activity that corresponded with “dog” and “boy.”
“What we found is there are two regions in the left superior temporal lobe, one which is situated more toward the center of the head, that carries information about the agent, the one doing an action,” Frankland said. “An immediately adjacent region, located closer to the ear, carries information about the patient, or who the action was done to.”
Importantly, Frankland added, the brain appears to reuse the same patterns across multiple sentences, implying that these patterns function like symbols.
“So we might say ‘the dog chased the boy,’ or ‘the dog scratched the boy,’ but if we use some new verb the algorithms can still recognize the ‘dog’ pattern as the agent,” Frankland said. “That’s important because it suggests these symbols are used over and over again to compose new thoughts. And, moreover, we find that the structure of the thought is mapped onto the structure of the brain in a systematic way.”
That ability to use a series of repeatable concepts to formulate new thoughts may be part of what makes human thought unique ― and uniquely powerful.
“This paper is about language,” Greene said. “But we think it’s about more than that. There’s a more general mystery about how human thinking works.
“What makes human thinking so powerful is that we have this library of concepts that we can use to formulate an effectively infinite number of thoughts,” he continued. “Humans can engage in complicated behaviors that, for any other creature on Earth, would require an enormous amount of training. Humans can read or hear a string of concepts and immediately put those concepts together to form some new idea.”
Unlike models of perception, which put more complex representations at the top of a processing hierarchy, Frankland and Greene’s study supports a model of higher cognition that relies on the dynamic combination of conceptual building blocks to formulate thoughts.
“You can’t have a set of neurons that are there just waiting for someone to say ‘Joe Biden beat Vladimir Putin at Scrabble,’ ” Greene said. “That means there has to be some other system for forming meanings on the fly, and it has to be incredibly flexible, incredibly quick and incredibly precise.” He added, “This is an essential feature of human intelligence that we’re just beginning to understand.”
Source: Harvard Gazette
Predicting Change in the Alzheimer’s Brain
Tue, 10/06/2015 – 9:14am
Larry Hardesty, MIT News Office
http://www.biosciencetechnology.com/news/2015/10/predicting-change-alzheimers-brain?
MIT researchers are developing a computer system that uses genetic, demographic, and clinical data to help predict the effects of disease on brain anatomy.
http://www.biosciencetechnology.com/sites/biosciencetechnology.com/files/bt1510_MIT_Predicting.jpg
In experiments, they trained a machine-learning system on MRI data from patients with neurodegenerative diseases and found that supplementing that training with other patient information improved the system’s predictions. In the cases of patients with drastic changes in brain anatomy, the additional data cut the predictions’ error rate in half, from 20 percent to 10 percent.
“This is the first paper that we’ve ever written on this,” said Polina Golland, a professor of electrical engineering and computer science at MIT and the senior author on the new paper. “Our goal is not to prove that our model is the best model to do this kind of thing; it’s to prove that the information is actually in the data. So what we’ve done is, we take our model, and we turn off the genetic information and the demographic and clinical information, and we see that with combined information, we can predict anatomical changes better.”
First author on the paper is Adrian Dalca, an MIT graduate student in electrical engineering and computer science and a member of Golland’s group at MIT’s Computer Science and Artificial Intelligence Laboratory. They’re joined by Ramesh Sridharan, another Ph.D. student in Golland’s group, and by Mert Sabuncu, an assistant professor of radiology at Massachusetts General Hospital, who was a postdoc in Golland’s group.
The researchers are presenting the paper at the International Conference on Medical Image Computing and Computer Assisted Intervention this week. The work is a project of the Neuroimage Analysis Center, which is based at Brigham and Women’s Hospital in Boston and funded by the National Institutes of Health.
Common denominator
In their experiments, the researchers used data from the Alzheimer’s Disease Neuroimaging Initiative, a longitudinal study on neurodegenerative disease that includes MRI scans of the same subjects taken months and years apart.
Each scan is represented as a three-dimensional model consisting of millions of tiny cubes, or “voxels,” the 3-D equivalent of image pixels.
The researchers’ first step is to produce a generic brain template by averaging the voxel values of hundreds of randomly selected MRI scans. They then characterize each scan in the training set for their machine-learning algorithm as a deformation of the template. Each subject in the training set is represented by two scans, taken between six months and seven years apart.
The researchers conducted two experiments: one in which they trained their system on scans of both healthy subjects and those displaying evidence of either Alzheimer’s disease or mild cognitive impairment, and one in which they trained it only on data from healthy subjects.
In the first experiment, they trained the system twice, once using just the MRI scans and the second time supplementing them with additional information. This included data on genetic markers known as single-nucleotide polymorphisms; demographic data, such as subject age, gender, marital status, and education level; and rudimentary clinical data, such as patients’ scores on various cognitive tests.
The brains of healthy subjects and subjects in the early stages of neurodegenerative disease change little over time, and indeed, in cases where the differences between a subject’s scans were slight, the system trained only on MRI data fared well. In cases where the changes were more marked, however, the addition of the supplementary data made a significant difference.
Counterfactuals
In the second experiment, the researchers trained the system just once, on both the MRI data and the supplementary data of healthy subjects. But they instead used it to predict what the brains of Alzheimer’s patients would have looked like had they not been disfigured by disease.
In this case, there are no clinical data that could validate the system’s predictions. But the researchers believe that exploring this sort of counterfactual could be scientifically useful.
“It would illuminate how changes in individual subjects — for example, with mild cognitive impairment, which is a precursor to Alzheimer’s — evolve along this trajectory of degeneration, as compared to what normal degeneration would be,” Golland said. “We think that there are very interesting research applications of this. But I have to be honest and say that the original motivation was curiosity about how much of anatomy we could predict from genetics and other non-image data.”
“It’s not surprising that clinical and genetic data would help,” said Bruce Rosen, a professor of radiology at Harvard Medical School and director of the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital. “But the fact that it did as well as it did is encouraging.”
“There are lots of ways these tools could be beneficial to the research community,” Rosen adds. “To my mind, the more challenging question is whether they could be useful clinically.”
Some promising experimental Alzheimer’s drugs require early determination of how the disease is likely to progress, Rosen said. Currently, he said, that determination relies on a combination of MRI and PET scan data. “People think MRI is expensive, but it’s only a fraction of what PET scans cost,” Rosen said. “If machine-learning tools can help avoid the need for PET scans in evaluating patients early in the disease course, that will be very impactful.”
Source: Massachusetts Institute of Technology
Alzheimer’s: Investigators spotlight a pathway for amyloid beta clearance
There are a variety of theories as to why people develop Alzheimer’s. And one of the best known is that toxic clusters of amyloid beta in the brain wipe out memories and trigger dementia in the elderly.
Now researchers at Indiana University say that they have determined that the IL1RAP immune pathway could provide a promising avenue for drug developers. And they’re quick to add that some experimental therapies that already hit this target could offer a quick way to help determine their utility against Alzheimer’s.
The team confirmed an observation that has been made before: the APOE e4 allele is associated with a significant accumulation of amyloid beta. But they were surprised to find that the IL1RAP gene–which they note codes for the immune signaling factor interleukin-1 receptor accessory protein–“showed an independent and even stronger influence on amyloid accumulation.”
They also determined that the gene was linked to a lower level of microglial activity as measured by PET scans; increased atrophy of the temporal cortex; swift cognitive decline and a “greater likelihood among study participants of progression from mild cognitive impairment to Alzheimer’s disease.”
“This was an intriguing finding because IL1RAP is known to play a central role in the activity of microglia, the immune system cells that act as the brain’s “garbage disposal system” and the focus of heavy investigation in a variety of neurodegenerative diseases,” said Dr. Vijay Ramanan, postdoctoral researcher at the IU School of Medicine.
There are already experimental anti-inflammatories and antibodies that are designed to hit this target, offering a shortcut in determining the impact on patients.
“These findings suggest that targeting the IL1RAP immune pathway may be a viable approach for promoting the clearance of amyloid deposits and fighting an important cause of progression in Alzheimer’s disease,” said Andrew Saykin, director of the Indiana Alzheimer Disease Center and the national Alzheimer’s Disease Neuroimaging Initiative Genetics Core.
It’s also useful to note that while many researchers believe that amyloid beta causes Alzheimer’s, there’s no consensus at the FDA on that point. And while many programs have been put in place to treat the disease, the vast majority have failed in the clinic, including drugs that aim at amyloid beta clearance.
Related Articles:
Mayo Clinic team renews Alzheimer’s feud, fingers tau over amyloid
Alzheimer’s study finds a molecule that might stymie critical stage of the disease
Neuroscience project tries to put the immune system to work against Alzheimer’s
An Accessible Approach to Making a Mini-brain
10/05/2015 – Brown University
http://www.biosciencetechnology.com/news/2015/10/accessible-approach-making-mini-brain
A bioengineering team at Brown University can grow “mini-brains” of neurons and supporting cells that form networks and are electrically active. (Image: Hoffman-Kim lab/Brown University)
http://www.biosciencetechnology.com/sites/biosciencetechnology.com/files/bt1510_brown_minibrain.jpg
If you need a working miniature brain — say for drug testing, to test neural tissue transplants, or to experiment with how stem cells work — a new paper describes how to build one with what the Brown University authors say is relative ease and low expense. The little balls of brain aren’t performing any cogitation, but they produce electrical signals and form their own neural connections — synapses — making them readily producible testbeds for neuroscience research, the authors said.
“We think of this as a way to have a better in vitro [lab] model that can maybe reduce animal use,” said graduate student Molly Boutin, co-lead author of the new paper in the journal Tissue Engineering: Part C. “A lot of the work that’s done right now is in two-dimensional culture, but this is an alternative that is much more relevant to the in vivo [living] scenario.”
Just a small sample of living tissue from a single rodent can make thousands of mini-brains, the researchers said. The recipe involves isolating and concentrating the desired cells with some centrifuge steps and using that refined sample to seed the cell culture in medium in an agarose spherical mold.
The mini-brains, about a third of a millimeter in diameter, are not the first or the most sophisticated working cell cultures of a central nervous system, the researchers acknowledged, but they require fewer steps to make and they use more readily available materials.
“The materials are easy to get and the mini-brains are simple to make,” said co-lead author Yu-Ting Dingle, who earned her Ph.D. at Brown in May 2015. She compared them to retail 3-D printers which have proliferated in recent years, bringing that once-rare technology to more of a mass market. “We could allow all kinds of labs to do this research.”
The spheres of brain tissue begin to form within a day after the cultures are seeded and have formed complex 3-D neural networks within two to three weeks, the paper shows.
25-cent mini-brains
There are fixed costs, of course, but an approximate cost for each new mini-brain is on the order of $0.25, said study senior author Diane Hoffman-Kim, associate professor of molecular pharmacology, physiology and biotechnology and associate professor of engineering at Brown.
“We knew it was a relatively high-throughput system, but even we were surprised at the low cost per mini-brain when we computed it,” Hoffman-Kim said.
Hoffman-Kim’s lab collaborated with fellow biologists and bioengineers at Brown — faculty colleagues Julie Kauer, Jeffrey Morgan, and Eric Darling are all co-authors — to build the mini-brains. She wanted to develop a testbed for her lab’s basic biomedical research. She was interested, for example, in developing a model to test aspects of neural cell transplantation, as has been proposed to treat Parkinson’s disease. Boutin was interested in building working 3-D cell cultures to study how adult neural stem cells develop.
Morgan’s Providence startup company, MicroTissues Inc., makes the 3-D tissue engineering molds used in the study.
The method they developed yields mini-brains with several important properties:
- Diverse cell types: The cultures contain both inhibitory and excitatory neurons and several varieties of essential neural support cells called glia.
- Electrically active: the neurons fire and spike and form synaptic connections, producing complex networks.
- 3-D: Cells connect and communicate within a realistic geometry, rather than merely across a flat plane as in a 2-D culture.
- Natural density: Experiments showed that the mini-brains have a density of a few hundred thousand cells per cubic millimeter, which is similar to a natural rodent brain.
- Physical structure: Cells in the mini-brain produce their own extracellular matrix, producing a tissue with the same mechanical properties (squishiness) as natural tissue. The cultures also don’t rely on foreign materials such as scaffolds of collagen.
- Longevity: In testing, cultured tissues live for at least a month.
Hoffman-Kim, who is affiliated with the Brown Institute for Brain Science and the Center for Biomedical Engineering, said she hopes the mini-brains might proliferate to many different labs, including those of researchers who have questions about neural tissue but not necessarily the degree of neuroscience and cell culture equipment required of other methods.
“If you are that person in that lab, we think you shouldn’t have to equip yourself with a microelectronics facility, and you shouldn’t have to do embryonic dissections in order to generate an in vitro model of the brain,” Hoffman-Kim said.
The National Science Foundation, the National Institutes of Health, the Brown Institute for Brain Science, and the U.S. Department of Education funded the research.
Source: Brown University
| Rat Brain Simulation Runs Neocortical Maze
http://www.genengnews.com/gen-news-highlights/rat-brain-simulation-runs-neocortical-maze/81251842/
http://www.genengnews.com/Media/images/GENHighlight/thumb_Oct9_2015_BBPEPFL2015_VirtualRatBrain9128157182.jpg
In this depiction of in silico retrograde staining, a digital reconstruction of neocortical microcircuitry, the presynaptic neurons of a layer 2/3 nest basket cell (red) are stained in blue. Only immediate neighboring presynaptic neurons are shown. [© BBP/EPFL 2015]
It’s a piece of rat brain containing about 30,000 neurons and 40 million synaptic connections, and there’s nothing remarkable about it, except that it isn’t real. It’s a digital reconstruction—a representation of a one-third cubic millimeter of rat neocortex—and it seems to work like the real thing.
Needless to say, its many creators are proud. They include 82 scientists and engineers from around the world, collaborators who are aware that their reconstruction represents the culmination of 20 years of biological experimentation and 10 years of computational science work. They are also aware that their work is controversial. It was criticized last year in an open letter. Signed by hundreds of neuroscientists, the letter argued that attempts to digitally reconstruct brain tissue were premature and represented an “overly narrow” approach that risked a misallocation of resources.
Undaunted, the investigators, led by scientists of the École Polytechnique Fédérale de Lausanne (EPFL), ran simulations on supercomputers to show that the electrical behavior of the virtual brain tissue matched the behavior of real rat neocortical tissue. Even though the digital reconstruction was not designed to reproduce any specific circuit phenomenon, a variety of experimental findings emerged. One such simulation examined how different types of neuron would respond if fibers coming into the neocortex were to convey signals encoding touch sensations. The researchers found that the responses of the different types of neurons in the digital reconstruction were very similar to those that had been previously observed in the laboratory.
These findings appeared October 8 in the journal Cell, in an article entitled, “Reconstruction and Simulation of Neocortical Microcircuitry.” This article also described how additional simulations revealed novel insights into the functioning of the neocortex.
“[We] find a spectrum of network states with a sharp transition from synchronous to asynchronous activity, modulated by physiological mechanisms,” wrote the authors. “The spectrum of network states, dynamically reconfigured around this transition, supports diverse information processing strategies.”
The authors even suggested that their work represents the first step toward the digital reconstruction and simulation of a whole brain. “They delivered what they promised,” said Patrick Aebischer, president of EPFL. This statement appeared in an EPFL press release that also indicated that the EPFL, together with the Swiss government, took the “bold step of funding the ambitious and controversial Blue Brain Project.”
The Blue Brain project is the simulation core of the Human Brain Project, a decade-long effort that is being allocated more than $1 billion.
“While a long way from the whole brain, the study demonstrates that it is feasible to digitally reconstruct and simulate brain tissue,” the release continued. “It is a first step and a significant contribution to Europe’s Human Brain Project, which Henry Markram founded, and where EPFL is the coordinating partner.”
Idan Segev, a senior author, sees the paper as building on the pioneering work of the Spanish anatomist, Ramon y Cajal from more than 100 years ago. “Ramon y Cajal began drawing every type of neuron in the brain by hand. He even drew in arrows to describe how he thought the information was flowing from one neuron to the next. Today, we are doing what Cajal would be doing with the tools of the day—building a digital representation of the neurons and synapses and simulating the flow of information between neurons on supercomputers. Furthermore, the digitization of the tissue allows the data to be preserved and reused for future generations.”
Now that the Blue Brain team has published the experimental results and the digital reconstruction, other scientists will be able to use the data and reconstruction to test other theories of brain function.
“The reconstruction is a first draft, it is not complete and it is not yet a perfect digital replica of the biological tissue,” explained Henry Markram. In fact, the current version explicitly leaves out many important aspects of the brain, such as glia, blood vessels, gap-junctions, plasticity, and neuromodulation. According to Sean Hill, a senior author: “The job of reconstructing and simulating the brain is a large-scale collaborative one, and the work has only just begun. The Human Brain Project represents the kind of collaboration that is required.”
Neuronal Waste Removal Gene Found to Prevent Parkinson’s
http://www.genengnews.com/gen-news-highlights/neuronal-waste-removal-gene-found-to-prevent-parkinson-s/81251836/
Researchers at the University of Copenhagen in Denmark say they have discovered that noninheritable Parkinson’s Disease (PD) may be caused by functional changes in the Interferon-beta (IFNβ) gene, which plays a vital role in keeping neurons healthy by regulating waste management. Treatment with IFNβ-gene therapy successfully prevented neuronal death and disease effects in an experimental model of PD.
The team’s study (“Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia”) was published in Cell.
“We found that IFNβ is essential for neurons ability to recycle waste proteins,” explained Patrick Ejlerskov, Ph.D., an assistant professor in the lab of Shohreh Issazadeh-Navikas, Ph.D., at the university’s Biotech Research and Innovation Center (BRIC) and first author on the paper. “Without this, the waste proteins accumulate in disease-associated structures called Lewy bodies and with time the neurons die.”
The scientists found that mice missing IFNβ developed Lewy bodies in parts of the brain, which control body movement and restoration of memory, and as a result they developed disease and clinical signs similar to patients with PD and dementia with Lewy bodies (DLB).
While hereditary gene mutations have long been known to play a role in familial PD, the study from BRIC offers one of the first models for so-called nonfamilial PD, which comprises the majority (90-95%) of patients suffering from PD. According to Dr. Issazadeh-Navikas, the new knowledge opens new therapeutic possibilities.
“This is one of the first genes found to cause pathology and clinical features of nonfamilial PD and DLB, through accumulation of disease-causing proteins,” she said. “It is independent of gene mutations known from familial PD and when we introduced IFNβ-gene therapy, we could prevent neuronal death and disease development. Our hope is that this knowledge will enable development of more effective treatment of PD.”
Current treatments are effective at improving the early motor symptoms of the disease. However, as the disease progress, the treatment effect is lost. The next step for the research team will be to gain a better understanding of the molecular mechanisms by which IFNβ protects neurons and thereby prevents movement disorders and dementia. |
A review of heterogeneous data mining for brain disorder identification
- Bokai Cao , Xiangnan Kong, Philip S. Yu
Brain Informatics 30 Sept 2015, pp 1-12
http://dx.doi.org:/10.1007/s40708-015-0021-3
http://link.springer.com/article/10.1007/s40708-015-0021-3/fulltext.html
With rapid advances in neuroimaging techniques, the research on brain disorder identification has become an emerging area in the data mining community. Brain disorder data poses many unique challenges for data mining research. For example, the raw data generated by neuroimaging experiments is in tensor representations, with typical characteristics of high dimensionality, structural complexity, and nonlinear separability. Furthermore, brain connectivity networks can be constructed from the tensor data, embedding subtle interactions between brain regions. Other clinical measures are usually available reflecting the disease status from different perspectives. It is expected that integrating complementary information in the tensor data and the brain network data, and incorporating other clinical parameters will be potentially transformative for investigating disease mechanisms and for informing therapeutic interventions. Many research efforts have been devoted to this area. They have achieved great success in various applications, such as tensor-based modeling, subgraph pattern mining, and multi-view feature analysis. In this paper, we review some recent data mining methods that are used for analyzing brain disorders.
Many brain disorders are characterized by ongoing injury that is clinically silent for prolonged periods and irreversible by the time symptoms first present. New approaches for detection of early changes in subclinical periods will afford powerful tools for aiding clinical diagnosis, clarifying underlying mechanisms, and informing neuroprotective interventions to slow or reverse neural injury for a broad spectrum of brain disorders, including bipolar disorder, HIV infection on brain, Alzheimer’s disease, Parkinson’s disease, etc. Early diagnosis has the potential to greatly alleviate the burden of brain disorders and the ever increasing costs to families and society.
As the identification of brain disorders is extremely challenging, many different diagnosis tools and methods have been developed to obtain a large number of measurements from various examinations and laboratory tests. Especially, recent advances in the neuroimaging technology have provided an efficient and noninvasive way for studying the structural and functional connectivity of the human brain, either normal or in a diseased state [1]. This can be attributed in part to advances in magnetic resonance imaging (MRI) capabilities [2]. Techniques such as diffusion MRI, also referred to as diffusion tensor imaging (DTI), produce in vivo images of the diffusion process of water molecules in biological tissues. By leveraging the fact that the water molecule diffusion patterns reveal microscopic details about tissue architecture, DTI can be used to perform tractography within the white matter and construct structural connectivity networks [3–7]. Functional MRI (fMRI) is a functional neuroimaging procedure that identifies localized patterns of brain activation by detecting associated changes in the cerebral blood flow. The primary form of fMRI uses the blood-oxygenation-level-dependent (BOLD) response extracted from the gray matter [8–10]. Another neuroimaging technique is positron emission tomography (PET). Using different radioactive tracers (e.g., fluorodeoxyglucose), PET produces a three-dimensional image of various physiological, biochemical, and metabolic processes [11].
A variety of data representations can be derived from these neuroimaging experiments, which present many unique challenges for the data mining community. Conventional data mining algorithms are usually developed to tackle data in one specific representation, a majority of which are particularly for vector-based data. However, the raw neuroimaging data are in the form of tensors, from which we can further construct brain networks connecting regions of interest (ROIs). Both of them are highly structured considering correlations between adjacent voxels in the tensor data and that between connected brain regions in the brain network data. Moreover, it is critical to explore interactions between measurements computed from the neuroimaging and other clinical experiments which describe subjects in different vector spaces. In this paper, we review some recent data mining methods for (1) mining tensor imaging data; (2) mining brain networks; and (3) mining multi-view feature vectors.
Tensor imaging analysis
For brain disorder identification, the raw data generated by neuroimaging experiments are in tensor representations [11–13]. For example, in contrast to two-dimensional X-ray images, an fMRI sample corresponds to a four-dimensional array by recording the sequential changes of traceable signals in each voxel.1
Tensors are higher order arrays that generalize the concepts of vectors (first-order tensors) and matrices (second-order tensors), whose elements are indexed by more than two indices. Each index expresses amode of variation of the data and corresponds to a coordinate direction. In an fMRI sample, the first three modes usually encode the spatial information, while the fourth mode encodes the temporal information. The number of variables in each mode indicates the dimensionality of a mode. The order of a tensor is determined by the number of its modes. An mth-order tensor can be represented as X=(xi1,…,im)∈RI1×⋯×Im, where Ii is the dimension of X along the i-th mode.
Definition 1
(Tensor product) The tensor product of three vectors a∈RI1, b∈RI2, and c∈RI3, denoted by a⊗b⊗c, represents a third-order tensor with the elements (a⊗b⊗c)i1,i2,i3 = ai1bi2ci3.
Tensor product is also referred to as outer product in some literature [11, 12]. An mth-order tensor is a rank-one tensor if it can be defined as the tensor product of m vectors.
Definition 2
Given a third-order tensor X∈RI1×I2×I3 and an integer R, as illustrated in Fig. 1, a tensor factorization of X can be expressed as
X=X1+X2+⋯+XR=∑r=1Rar⊗br⊗cr
(1)
Fig. 1
Tensor factorization of a third-order tensor
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0021-3/MediaObjects/40708_2015_21_Fig1_HTML.gif
One of the major difficulties brought by the tensor data is the curse of dimensionality. The total number of voxels contained in a multi-mode tensor, say, X=(xi1,…,im)∈RI1×⋯×Im is I1×⋯×Im which is exponential to the number of modes. If we unfold the tensor into a vector, the number of features will be extremely high [14]. This makes traditional data mining methods prone to overfitting, especially with a small sample size. Both computational scalability and theoretical guarantee of the traditional models are compromised by such high dimensionality [13].
On the other hand, complex structural information is embedded in the tensor data. For example, in the neuroimaging data, values of adjacent voxels are usually correlated with each other [2]. Such spatial relationships among different voxels in a tensor image can be very important in neuroimaging applications. Conventional tensor-based approaches focus on reshaping the tensor data into matrices/vectors, and thus, the original spatial relationships are lost. The integration of structural information is expected to improve the accuracy and interpretability of tensor models.
2.1 Supervised learning
Suppose we have a set of tensor data D={(Xi,yi)}ni=1 for classification problem, where Xi∈RI1×⋯×Im is the neuroimaging data represented as an mth-order tensor and yi∈{−1,+1} is the corresponding binary class label of Xi. For example, if the i-th subject has Alzheimer’s disease, the subject is associated with a positive label, i.e., yi=+1. Otherwise, if the subject is in the control group, the subject is associated with a negative label, i.e., yi=−1.
Supervised tensor learning can be formulated as the optimization problem of support tensor machines (STMs) [15] which is a generalization of the standard support vector machines (SVMs) from vector data to tensor data. The objective of such learning algorithms is to learn a hyperplane by which the samples with different labels are divided as wide as possible. However, tensor data may not be linearly separable in the input space. To achieve a better performance on finding the most discriminative biomarkers or identifying infected subjects from the control group, in many neuroimaging applications, nonlinear transformation of the original tensor data should be considered. He et al. study the problem of supervised tensor learning with nonlinear kernels which can preserve the structure of tensor data [13]. The proposed kernel is an extension of kernels in the vector space to the tensor space which can take the multidimensional structure complexity into account. However, it cannot automatically consider the abundant and complicated information of the neuroimaging data in an integral manner. Han et al. apply a deep learning-based algorithm, the hierarchical convolutional sparse auto-encoder, to extract efficient and robust features and conserve abundant detail information for the neuroimaging classification [16].
Slightly different from classifying disease status (discrete label), another family of problems uses tensor neuroimages to predict cognitive outcome (continuous label). The problems can be formulated in a regression setup by treating clinical outcome as the real label, i.e., yi∈R, and treating tensor neuroimages as the input. However, most classical regression methods take vectors as input features. Simply reshaping a tensor into a vector is clearly an unsatisfactory solution.
Zhou et al. exploit the tensor structure in imaging data and integrate tensor decomposition within a statistical regression paradigm to model multidimensional arrays [14]. By imposing a low-rank approximation to the extremely high-dimensional complex imaging data, the curse of dimensionality is greatly alleviated, thereby allowing development of a fast estimation algorithm and regularization. Numerical analysis demonstrates its potential applications in identifying ROI in brains that are relevant to a particular clinical response. In scenarios where the objective is to predict a set of dependent variables, Cichocki et al. introduce a generalized multilinear regression model, higher order partial least squares, which projects the electrocorticogram data into a latent space and performs regression on the corresponding latent variables [17, 18].
2.2 Unsupervised learning
Modern imaging techniques have allowed us to study the human brain as a complex system by modeling it as a network [19]. For example, the fMRI scans consist of activations of thousands of voxels over time embedding a complex interaction of signals and noise [20], which naturally presents the problem of eliciting the underlying network from brain activities in the spatio-temporal tensor data. A brain connectivity network, also called a connectome [21], consists of nodes (gray matter regions) and edges (white matter tracts in structural networks or correlations between two BOLD time series in functional networks).
Although the anatomical atlases in the brain have been extensively studied for decades, task/subject specific networks have still not been completely explored with consideration of functional or structural connectivity information. An anatomically parcellated region may contain subregions that are characterized by dramatically different functional or structural connectivity patterns, thereby significantly limiting the utility of the constructed networks. There are usually trade-offs between reducing noise and preserving utility in brain parcellation [2]. Thus, investigating how to directly construct brain networks from tensor imaging data and understanding how they develop, deteriorate, and vary across individuals will benefit disease diagnosis [12].
Davidson et al. pose the problem of network discovery from fMRI data which involves simplifying spatio-temporal data into regions of the brain (nodes) and relationships between those regions (edges) [12]. Here the nodes represent collections of voxels that are known to behave cohesively over time; the edges can indicate a number of properties between nodes such as facilitation/inhibition (increases/decreases activity) or probabilistic (synchronized activity) relationships; and the weight associated with each edge encodes the strength of the relationship.
A tensor can be decomposed into several factors. However, unconstrained tensor decomposition results of the fMRI data may not be good for node discovery because each factor is typically not a spatially contiguous region nor does it necessarily match an anatomical region. That is to say, many spatially adjacent voxels in the same structure are not active in the same factor which is anatomically impossible. Therefore, to achieve the purpose of discovering nodes while preserving anatomical adjacency, known anatomical regions in the brain are used as masks and constraints are added to enforce that the discovered factors should closely match these masks [12].
Yang et al. investigate the inference of mouse brain networks and propose a hierarchical graphical model framework with tree-structural regularization [22]. In the hierarchical structure, voxels serve as the leaf nodes of the tree, and a node in the intermediate layer represents a region formed by voxels in the subtree rooted at that node. For edge discovery problem, Papalexakis et al. leverage control theory to model the dynamics of neuron interactions and infer the functional connectivity [23]. It is assumed that in addition to the linear influence of the input stimulus, there are hidden neuron regions of the brain, which interact with each other, causing the voxel activities. Veeriah et al. propose a deep learning algorithm for predicting if the two brain neurons are causally connected given their activation time-series data [24]. It reveals that the exploitation of the deep architecture is critical, which jointly extracts sequences of salient patterns of activation and aligns them to predict neural connections.
Overall, current research on tensor imaging analysis presents two directions: (1) supervised: for a particular brain disorder, a classifier can be trained by modeling the relationship between a set of neuroimages and their associated labels (disease status or clinical response); (2) unsupervised: regardless of brain disorders, a brain network can be discovered from a given neuroimage.
3 Brain network analysis
We have briefly introduced that brain networks can be constructed from neuroimaging data where nodes correspond to brain regions, e.g., insula, hippocampus, thalamus, and links correspond to the functional/structural connectivity between brain regions. The linkage structure in brain networks can encode tremendous information about the mental health of human subjects. For example, in brain networks derived from fMRI, functional connections can encode the correlations between the functional activities of brain regions. While structural links in DTI brain networks can capture the number of neural fibers connecting different brain regions. The complex structures and the lack of vector representations for the brain network data raise major challenges for data mining.
Next, we will discuss different approaches on how to conduct further analysis for constructed brain networks, which are also referred to as graphs hereafter.
Definition 3
(Binary graph) A binary graph is represented as G=(V,E), where V={v1,…,vnv} is the set of vertices, and E⊆V×V is the set of deterministic edges.
3.1 Kernel learning on graphs
In the setting of supervised learning on graphs, the target is to train a classifier using a given set of graph data D={(Gi,yi)}ni=1, so that we can predict the label y^ for a test graph G. With applications to brain networks, it is desirable to identify the disease status for a subject based on his/her uncovered brain network. Recent development of brain network analysis has made characterization of brain disorders at a whole-brain connectivity level possible, thus providing a new direction for brain disease classification.
Due to the complex structures and the lack of vector representations, graph data cannot be directly used as the input for most data mining algorithms. A straightforward solution that has been extensively explored is to first derive features from brain networks and then construct a kernel on the feature vectors.
Wee et al. use brain connectivity networks for disease diagnosis on mild cognitive impairment (MCI), which is an early phase of Alzheimer’s disease (AD) and usually regarded as a good target for early diagnosis and therapeutic interventions [25–27]. In the step of feature extraction, weighted local clustering coefficients of each ROI in relation to the remaining ROIs are extracted from all the constructed brain networks to quantify the prevalence of clustered connectivity around the ROIs. To select the most discriminative features for classification, statistical t test is performed and features with p values smaller than a predefined threshold are selected to construct a kernel matrix. Through the employment of the multi-kernel SVM, Wee et al. integrate information from DTI and fMRI and achieve accurate early detection of brain abnormalities [27].
However, such strategy simply treats a graph as a collection of nodes/links, and then extracts local measures (e.g., clustering coefficient) for each node or performs statistical analysis on each link, thereby blinding the connectivity structures of brain networks. Motivated by the fact that some data in real-world applications are naturally represented by means of graphs, while compressing and converting them to vectorial representations would definitely lose structural information, kernel methods for graphs have been extensively studied for a decade [28].
A graph kernel maps the graph data from the original graph space to the feature space and further measures the similarity between two graphs by comparing their topological structures [29]. For example, product graph kernel is based on the idea of counting the number of walks in product graphs [30]; marginalized graph kernel works by comparing the label sequences generated by synchronized random walks of labeled graphs [31]; and cyclic pattern kernels for graphs count pairs of matching cyclic/tree patterns in two graphs [32].
To identify individuals with AD/MCI from healthy controls, instead of using only a single property of brain networks, Jie et al. integrate multiple properties of fMRI brain networks to improve the disease diagnosis performance [33]. Two different yet complementary network properties, i.e., local connectivity and global topological properties are quantified by computing two different types of kernels, i.e., a vector-based kernel and a graph kernel. As a local network property, weighted clustering coefficients are extracted to compute a vector-based kernel. As a topology-based graph kernel, Weisfeiler-Lehman subtree kernel [29] is used to measure the topological similarity between paired fMRI brain networks. It is shown that this type of graph kernel can effectively capture the topological information from fMRI brain networks. The multi-kernel SVM is employed to fuse these two heterogeneous kernels for distinguishing individuals with MCI from healthy controls.
3.2 Subgraph pattern mining
In brain network analysis, the ideal patterns we want to mine from the data should take care of both local and global graph topological information. Graph kernel methods seem promising, which, however, are not interpretable. Subgraph patterns are more suitable for brain networks, which can simultaneously model the network connectivity patterns around the nodes and capture the changes in local area [2].
Definition 4
(Subgraph) Let G′=(V′,E′) and G=(V,E) be two binary graphs. G′ is a subgraph of G (denoted as G′⊆G) iff V′⊆V and E′⊆E. If G′ is a subgraph of G, then G is supergraph of G′.
A subgraph pattern, in a brain network, represents a collection of brain regions and their connections. For example, as shown in Fig. 2, three brain regions should work collaboratively for normal people and the absence of any connection between them can result in Alzheimer’s disease in different degrees. Therefore, it is valuable to understand which connections collectively play a significant role in disease mechanism by finding discriminative subgraph patterns in brain networks.
Mining subgraph patterns from graph data has been extensively studied by many researchers [34–37]. In general, a variety of filtering criteria are proposed. A typical evaluation criterion is frequency, which aims at searching for frequently appearing subgraph features in a graph dataset satisfying a prespecified threshold. Most of the frequent subgraph mining approaches are unsupervised. For example, Yan and Han develop a depth-first search algorithm: gSpan [38]. This algorithm builds a lexicographic order among graphs, and maps each graph to a unique minimum DFS code as its canonical label. Based on this lexicographic order, gSpan adopts the depth-first search strategy to mine frequent connected subgraphs efficiently. Many other approaches for frequent subgraph mining have also been proposed, e.g., AGM [39], FSG [40], MoFa [41], FFSM [42], and Gaston [43].
Fig. 2
An example of discriminative subgraph patterns in brain networks
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0021-3/MediaObjects/40708_2015_21_Fig2_HTML.gif
Moreover, the problem of supervised subgraph mining has been studied in recent work which examines how to improve the efficiency of searching the discriminative subgraph patterns for graph classification. Yan et al. introduce two concepts structural leap search and frequency-descending mining, and propose LEAP [37] which is one of the first work in discriminative subgraph mining. Thoma et al. propose CORK which can yield a near-optimal solution using greedy feature selection [36]. Ranu and Singh propose a scalable approach, called GraphSig, that is capable of mining discriminative subgraphs with a low-frequency threshold [44]. Jin et al. propose COM which takes into account the co-occurrences of subgraph patterns, thereby facilitating the mining process [45]. Jin et al. further propose an evolutionary computation method, called GAIA, to mine discriminative subgraph patterns using a randomized searching strategy [34]. Zhu et al. design a diversified discrimination score based on the log ratio which can reduce the overlap between selected features by considering the embedding overlaps in the graphs [46].
Conventional graph mining approaches are best suited for binary edges, where the structure of graph objects is deterministic, and the binary edges represent the presence of linkages between the nodes [2]. In fMRI brain network data, however, there are inherently weighted edges in the graph linkage structure, as shown in Fig. 3 (left). A straightforward solution is to threshold weighted networks to yield binary networks. However, such simplification will result in great loss of information. Ideal data mining methods for brain network analysis should be able to overcome these methodological problems by generalizing the network edges to positive and negative weighted cases, e.g., probabilistic weights in fMRI brain networks and integral weights in DTI brain networks.
Fig. 3
An example of fMRI brain networks (left) and all possible instantiations of linkage structures between red nodes (right) [47]. (Color figure online)
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0021-3/MediaObjects/40708_2015_21_Fig3_HTML.gif
Definition 5
A weighted graph is represented as G˜=(V,E,p), where V={v1,…,vnv} is the set of vertices, and E⊆V×V is the set of nondeterministic edges. p:E→(0,1] is a function that assigns a probability of existence to each edge in E.
fMRI brain networks can be modeled as weighted graphs where each edge e∈E is associated with a probability p(e) indicating the likelihood of whether this edge should exist or not [47, 48]. It is assumed thatp(e) of different edges in a weighted graph are independent from each other. Therefore, by enumerating the possible existence of all edges in a weighted graph, we can obtain a set of binary graphs. For example, in Fig. 3 (right), consider the three red nodes and links between them as a weighted graph. There are 23=8binary graphs that can be implied with different probabilities. For a weighted graph G˜, the probability of G˜containing a subgraph feature G′ is defined as the probability that a binary graph G implied by G˜ contains subgraph G′. Kong et al. propose a discriminative subgraph feature selection method based on dynamic programming to compute the probability distribution of the discrimination scores for each subgraph pattern within a set of weighted graphs [48].
For brain network analysis, usually we only have a small number of graph instances [48]. In these applications, the graph view alone is not sufficient for mining important subgraphs. Fortunately, the side information is available along with the graph data for brain disorder identification. For example, in neurological studies, hundreds of clinical, immunologic, serologic, and cognitive measures may be available for each subject, apart from brain networks. These measures compose multiple side views which contain a tremendous amount of supplemental information for diagnostic purposes. It is desirable to extract valuable information from a plurality of side views to guide the process of subgraph mining in brain networks.
Fig. 4
Two strategies of leveraging side views in feature selection process for graph classification: late fusion and early fusion
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0021-3/MediaObjects/40708_2015_21_Fig4_HTML.gif
Figure 4 illustrates two strategies of leveraging side views in the process of selecting subgraph patterns. Conventional graph classification approaches treat side views and subgraph patterns separately and may only combine them at the final stage of training a classifier. Obviously, the valuable information embedded in side views is not fully leveraged in the feature selection process. In order to fuse heterogeneous data sources at an early stage thereby exploring their correlations, Cao et al. introduce an effective algorithm for discriminative subgraph selection using multiple side views as guidance [49]. Side information consistency is first validated via statistical hypothesis testing which suggests that the similarity of side view features between instances with the same label should have higher probability to be larger than that with different labels. Based on such observations, it is assumed that the similarity/distance between instances in the space of subgraph features should be consistent with that in the space of a side view. That is to say, if two instances are similar in the space of a side view, they should also be close to each other in the space of subgraph features. Therefore the target is to minimize the distance between subgraph features of each pair of similar instances in each side view [49]. In contrast to existing subgraph mining approaches that focus on the graph view alone, the proposed method can explore multiple vector-based side views to find an optimal set of subgraph features for graph classification.
For graph classification, brain network analysis approaches can generally be put into three groups: (1) extracting some local measures (e.g., clustering coefficient) to train a standard vector-based classifier; (2) directly adopting graph kernels for classification; and (3) finding discriminative subgraph patterns. Different types of methods model the connectivity embedded in brain networks in different ways.
4 Multi-view feature analysis
Medical science witnesses everyday measurements from a series of medical examinations documented for each subject, including clinical, imaging, immunologic, serologic, and cognitive measures [50], as shown in Fig. 5. Each group of measures characterizes the health state of a subject from different aspects. This type of data is named as multi-view data, and each group of measures form a distinct view quantifying subjects in one specific feature space. Therefore, it is critical to combine them to improve the learning performance, while simply concatenating features from all views and transforming a multi-view data into a single-view data, as the method (a) shown in Fig. 6, would fail to leverage the underlying correlations between different views.
4.1 Multi-view learning and feature selection
Suppose we have a multi-view classification task with n labeled instances represented from m different views: D={(x(1)i,x(2)i,…,x(m)i,yi)}ni=1, where x(v)i∈RIv, Iv is the dimensionality of the v-th view, and yi∈{−1,+1} is the class label of the i-th instance.
Fig. 5
An example of multi-view learning in medical studies [51]
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0021-3/MediaObjects/40708_2015_21_Fig5_HTML.gif
Representative methods for multi-view learning can be categorized into three groups: co-training, multiple kernel learning, and subspace learning [52]. Generally, the co-training style algorithm is a classic approach for semi-supervised learning, which trains in alternation to maximize the mutual agreement on different views. Multiple kernel learning algorithms combine kernels that naturally correspond to different views, either linearly [53] or nonlinearly [54, 55] to improve learning performance. Subspace learning algorithms learn a latent subspace, from which multiple views are generated. Multiple kernel learning and subspace learning are generalized as co-regularization style algorithms [56], where the disagreement between the functions of different views is taken as a part of the objective function to be minimized. Overall, by exploring the consistency and complementary properties of different views, multi-view learning is more effective than single-view learning.
In the multi-view setting for brain disorders, or for medical studies in general, a critical problem is that there may be limited subjects available (i.e., a small n) yet introducing a large number of measurements (i.e., a large ∑mi=1Ii). Within the multi-view data, not all features in different views are relevant to the learning task, and some irrelevant features may introduce unexpected noise. The irrelevant information can even be exaggerated after view combinations thereby degrading performance. Therefore, it is necessary to take care of feature selection in the learning process. Feature selection results can also be used by researchers to find biomarkers for brain diseases. Such biomarkers are clinically imperative for detecting injury to the brain in the earliest stage before it is irreversible. Valid biomarkers can be used to aid diagnosis, monitor disease progression, and evaluate effects of intervention [48].
Conventional feature selection approaches can be divided into three main directions: filter, wrapper, and embedded methods [57]. Filter methods compute a discrimination score of each feature independently of the other features based on the correlation between the feature and the label, e.g., information gain, Gini index, Relief [58, 59]. Wrapper methods measure the usefulness of feature subsets according to their predictive power, optimizing the subsequent induction procedure that uses the respective subset for classification [51,60–63]. Embedded methods perform feature selection in the process of model training based on sparsity regularization [64–67]. For example, Miranda et al. add a regularization term that penalizes the size of the selected feature subset to the standard cost function of SVM, thereby optimizing the new objective function to conduct feature selection [68]. Essentially, the process of feature selection and learning algorithm interact in embedded methods which means the learning part and the feature selection part cannot be separated, while wrapper methods utilize the learning algorithm as a black box.
However, directly applying these feature selection approaches to each separate view would fail to leverage multi-view correlations. By taking into account the latent interactions among views and the redundancy triggered by multiple views, it is desirable to combine multi-view data in a principled manner and perform feature selection to obtain consensus and discriminative low-dimensional feature representations.
4.2 Modeling view correlations
Recent years have witnessed many research efforts devoted to the integration of feature selection and multi-view learning. Tang et al. study multi-view feature selection in the unsupervised setting by constraining that similar data instances from each view should have similar pseudo-class labels [69]. Considering brain disorder identification, different neuroimaging features may capture different but complementary characteristics of the data. For example, the voxel-based tensor features convey the global information, while the ROI-based automated anatomical labeling (AAL) [70] features summarize the local information from multiple representative brain regions. Incorporating these data and additional nonimaging data sources can potentially improve the prediction. For Alzheimer’s disease (AD) classification, Ye et al. propose a kernel-based method for integrating heterogeneous data, including tensor and AAL features from MRI images, demographic information, and genetic information [11]. The kernel framework is further extended for selecting features (biomarkers) from heterogeneous data sources that play more significant roles than others in AD diagnosis.
Huang et al. propose a sparse composite linear discriminant analysis model for identification of disease-related brain regions of AD from multiple data sources [71]. Two sets of parameters are learned: one represents the common information shared by all the data sources about a feature, and the other represents the specific information only captured by a particular data source about the feature. Experiments are conducted on the PET and MRI data which measure structural and functional aspects, respectively, of the same AD pathology. However, the proposed approach requires the input as the same set of variables from multiple data sources. Xiang et al. investigate multi-source incomplete data for AD and introduce a unified feature learning model to handle block-wise missing data which achieves simultaneous feature-level and source-level selection [72].
For modeling view correlations, in general, a coefficient is assigned for each view, either at the view-level or feature-level. For example, in multiple kernel learning, a kernel is constructed from each view and a set of kernel coefficients are learned to obtain an optimal combined kernel matrix. These approaches, however, fail to explicitly consider correlations between features.
4.3 Modeling feature correlations
One of the key issues for multi-view classification is to choose an appropriate tool to model features and their correlations hidden in multiple views, since this directly determines how information will be used. In contrast to modeling on views, another direction for modeling multi-view data is to directly consider the correlations between features from multiple views. Since taking the tensor product of their respective feature spaces corresponds to the interaction of features from multiple views, the concept of tensor serves as a backbone for incorporating multi-view features into a consensus representation by means of tensor product, where the complex multiple relationships among views are embedded within the tensor structures. By mining structural information contained in the tensor, knowledge of multi-view features can be extracted and used to establish a predictive model.
Smalter et al. formulate the problem of feature selection in the tensor product space as an integer quadratic programming problem [73]. However, this method is computationally intractable on many views, since it directly selects features in the tensor product space resulting in the curse of dimensionality, as the method (b) shown in Fig. 6. Cao et al. propose to use a tensor-based approach to model features and their correlations hidden in the original multi-view data [51]. The operation of tensor product can be used to bringm-view feature vectors of each instance together, leading to a tensorial representation for common structure across multiple views, and allowing us to adequately diffuse relationships and encode information among multi-view features. In this manner, the multi-view classification task is essentially transformed from an independent domain of each view to a consensus domain as a tensor classification problem.
By using Xi to denote ∏mv=1⊗x(v)i, the dataset of labeled multi-view instances can be represented as D={(Xi,yi)}ni=1. Note that each multi-view instance Xi is an mth-order tensor that lies in the tensor product space RI1×⋯×Im. Based on the definitions of inner product and tensor norm, multi-view classification can be formulated as a global convex optimization problem in the framework of supervised tensor learning [15]. This model is named as multi-view SVM [51], and it can be solved with the use of optimization techniques developed for SVM.
Fig. 6
Schematic view of the key differences among three strategies of multi-view feature selection [51]
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0021-3/MediaObjects/40708_2015_21_Fig6_HTML.gif
Furthermore, a dual method for multi-view feature selection is proposed in [51] that leverages the relationship between original multi-view features and reconstructed tensor product features to facilitate the implementation of feature selection, as the method (c) in Fig. 6. It is a wrapper model which selects useful features in conjunction with the classifier and simultaneously exploits the correlations among multiple views. Following the idea of SVM-based recursive feature elimination [60], multi-view feature selection is consistently formulated and implemented in the framework of multi-view SVM. This idea can extend to include lower order feature interactions and to employ a variety of loss functions for classification or regression [74].
5 Future work
The human brain is one of the most complicated biological structures in the known universe. While it is very challenging to understand how it works, especially when disorders and diseases occur, dozens of leading technology firms, academic institutions, scientists, and other key contributors to the field of neuroscience have devoted themselves to this area and made significant improvements in various dimensions.2 Data mining on brain disorder identification has become an emerging area and a promising research direction.
This paper provides an overview of data mining approaches with applications to brain disorder identification, which have attracted increasing attention in both data mining and neuroscience communities in recent years. A taxonomy is built based upon data representations, i.e., tensor imaging data, brain network data, and multi-view data, following which the relationships between different data mining algorithms and different neuroimaging applications are summarized. We briefly present some potential topics of interest in the future.
5.1 Bridging heterogeneous data representations
As introduced in this paper, we can usually derive data from neuroimaging experiments in three representations, including raw tensor imaging data, brain network data, and multi-view vector-based data. It is critical to study how to train a model on a mixture of data representations, although it is very challenging to combine data that are represented in tensor space, vector space, and graph space, respectively. There is a straightforward idea of defining different kernels on different feature spaces and combing them through multi-kernel algorithms. However, it is usually hard to interpret the results. The concept of side view has been introduced to facilitate the process of mining brain networks, which may also be used to guide supervised tensor learning. It is even more interesting if we can learn on tensors and graphs simultaneously.
5.2 Integrating multiple neuroimaging modalities
There are a variety of neuroimaging techniques available characterizing subjects from different perspectives and providing complementary information. For example, DTI contains local microstructural characteristics of water diffusion; structural MRI can be used to delineate brain atrophy; fMRI records BOLD response related to neural activity; and PET measures metabolic patterns [27]. Based on such multimodality representation, it is desirable to find useful patterns with rich semantics. For example, it is important to know which connectivity between brain regions is significant in the sense of both structure and functionality. On the other hand, by leveraging the complementary information embedded in the multimodality representation, better performance on disease diagnosis can be expected.
Fig. 7
A bioinformatics heterogeneous information network schema
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0021-3/MediaObjects/40708_2015_21_Fig7_HTML.gif
5.3 Mining bioinformatics information networks
Bioinformatics network is a rich source of heterogeneous information involving disease mechanisms, as shown in Fig. 7. The problems of gene-disease association and drug-target binding prediction have been studied in the setting of heterogeneous information networks [75, 76]. For example, in gene-disease association prediction, different gene sequences can lead to certain diseases. Researchers would like to predict the association relationships between genes and diseases. Understanding the correlations between brain disorders and other diseases and the causality between certain genes and brain diseases can be transformative for yielding new insights concerning risk and protective relationships, for clarifying disease mechanisms, for aiding diagnostics and clinical monitoring, for biomarker discovery, for identification of new treatment targets, and for evaluating effects of intervention.
Footnotes
1 A voxel is the smallest three-dimensional point volume referenced in a neuroimaging of the brain.
2 http://www.whitehouse.gov/BRAIN
References
- 1.
Rubinov M, Sporns O (2010) Complex network measures of brain connectivity: uses and interpretations. Neuroimage 52(3):1059–1069CrossRef
- 2.
Kong X, Yu PS (2014) Brain network analysis: a data mining perspective. ACM SIGKDD Explor Newsl 15(2):30–38MathSciNetCrossRef
- 3.
Basser PJ, Pierpaoli C (1996) Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson Ser B 111(3):209–219CrossRef
- 4.
Le Bihan D, Breton E, Lallemand D, Grenier P, Cabanis E, Laval-Jeantet M (1986) MR imaging of intravoxel incoherent motions: application to diffusion and perfusion in neurologic disorders. Radiology 161(2):401–407CrossRef
- 5.
Chenevert TL, Brunberg JA, Pipe J (1990) Anisotropic diffusion in human white matter: demonstration with mr techniques in vivo. Radiology 177(2):401–405CrossRef
- 6.
McKeown MJ, Makeig S, Brown GG, Jung T-P, Kindermann SS, Bell AJ, Sejnowski TJ (1998) Analysis of fMRI data by blind separation into independent spatial components. Hum Brain Mapp 6:160–188CrossRef
- 7.
Moseley ME, Cohen Y, Kucharczyk J, Mintorovitch J, Asgari H, Wendland M, Tsuruda J, Norman D (1990) Diffusion-weighted MR imaging of anisotropic water diffusion in cat central nervous system. Radiology 176(2):439–445CrossRef
- 8.
Biswal B, Yetkin FZ, Haughton VM, Hyde JS (1995) Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn Reson Med 34(4):537–541CrossRef
- 9.
Ogawa S, Lee T, Kay A, Tank D (1990) Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc Natl Acad Sci 87(24):9868–9872CrossRef
- 10.
Ogawa S, Lee T-M, Nayak AS, Glynn P (1990) Oxygenation-sensitive contrast in magnetic resonance image of rodent brain at high magnetic fields. Magn Reson Med 14(1):68–78CrossRef
- 11.
Ye J, Chen K, Wu T, Li J, Zhao Z, Patel R, Bae M, Janardan R, Liu H, Alexander G et al (2008) Heterogeneous data fusion for Alzheimer’s disease study. In: KDD. ACM, pp 1025–1033
- 12.
Davidson I, Gilpin S, Carmichael O, Walker P (2013) Network discovery via constrained tensor analysis of fMRI data. In: KDD. ACM, pp 194–202
- 13.
He L, Kong X, Yu PS, Ragin AB, Hao Z, Yang X (2014) Dusk: a dual structure-preserving kernel for supervised tensor learning with applications to neuroimages. In: SDM. SIAM
- 14.
Zhou H, Li L, Zhu H (2013) Tensor regression with applications in neuroimaging data analysis. J Am Stat Assoc 108(502):540–552MATHMathSciNetCrossRef
- 15.
Tao D, Li X, Wu X, Hu W, Maybank SJ (2007) Supervised tensor learning. Knowl Inf Syst 13(1):1–42CrossRef
- 16.
Han X, Zhong Y, He L, Philip SY, Zhang L (2015) The unsupervised hierarchical convolutional sparse auto-encoder for neuroimaging data classification. In: Brain informatics and health. Springer, pp 156–166
- 17.
Cichocki A, Mandic D, De Lathauwer L, Zhou G, Zhao Q, Caiafa C, Phan HA (2015) Tensor decompositions for signal processing applications: from two-way to multiway component analysis. Signal Process Mag 32(2):145–163CrossRef
- 18.
Zhao Q, Caiafa CF, Mandic DP, Chao ZC, Nagasaka Y, Fujii N, Zhang L, Cichocki A (2013) Higher order partial least squares (HOPLS): a generalized multilinear regression method. Pattern Anal Mach Intell 35(7):1660–1673CrossRef
- 19.
Ajilore O, Zhan L, GadElkarim J, Zhang A, Feusner JD, Yang S, Thompson PM, Kumar A, Leow A (2013) Constructing the resting state structural connectome. Front Neuroinform 7:30CrossRef
- 20.
Genovese CR, Lazar NA, Nichols T (2002) Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage 15(4):870–878CrossRef
… more…
Multimodal neuroimaging computing: a review of the applications in neuropsychiatric disorders
Sidong Liu , Weidong Cai, Siqi Liu, Fan Zhang, Michael Fulham, Dagan Feng, Sonia Pujol, Ron Kikinis
Brain Informatics Sept 2015; 2(3): 167-180
http://dx.doi.org:/10.1007/s40708-015-0019-x
Multimodal neuroimaging is increasingly used in neuroscience research, as it overcomes the limitations of individual modalities. One of the most important applications of multimodal neuroimaging is the provision of vital diagnostic data for neuropsychiatric disorders. Multimodal neuroimaging computing enables the visualization and quantitative analysis of the alterations in brain structure and function, and has reshaped how neuroscience research is carried out. Research in this area is growing exponentially, and so it is an appropriate time to review the current and future development of this emerging area. Hence, in this paper, we review the recent advances in multimodal neuroimaging (MRI, PET) and electrophysiological (EEG, MEG) technologies, and their applications to the neuropsychiatric disorders. We also outline some future directions for multimodal neuroimaging where researchers will design more advanced methods and models for neuropsychiatric research.
Neuroimaging has advanced rapidly in the past two decades. The advanced non-invasive neuroimaging techniques, e.g., magnetic resonance imaging (MRI), positron emission tomography (PET), electroencephalography (EEG), and magnetoencephalography (MEG), have enabled the visualization and analysis of the brain function and structure in unprecedented detail and transformed the way we study the nervous system under normal and pathological conditions [1], particularly neuropsychiatric disorders including neurological and psychiatric disorders that affect the nervous system [2–4].
In the US, President Obama’s announcement of the ‘Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative’ on his state of the union address on April 2013 fueled resurgent interest in the neuroscience with a bold commitment to better understand the brain over the forthcoming decade [4]. Similar projects have been undertaken in the European Union [5] and Asia [6].
Multimodal neuroimaging, which we declare as the summation of information from different neuroimaging modalities, has become one of the major drivers in neuroimaging research due to the recognition of the clinical benefits of multimodal data [7, 8], and the better access to hybrid devices, e.g., PET/CT [9, 10], PET/MRI [11], and PET/MRI/EEG [12]. Multimodal neuroimaging data can either be obtained from simultaneous imaging measurement (EEG/fMRI [13], PET/CT[14]), or integration of separate measurements (PET and sMRI [15], sMRI and dMRI [16], fMRI and dMRI [17]).
Multimodal neuroimaging advances neuroscience research, i.e., neurology, psychiatry, neurophysiology, and neurosurgery, by overcoming the limitation of individual modalities and by allowing a more comprehensive picture of the brain. For instance, we can jointly analyze the structure and function using the data provided by PET/CT and PET/MRI; EEG combined with functional MRI (fMRI) improves the spatiotemporal resolution that cannot be achieved by the single modality alone. Multimodal neuroimaging can also cross-validate findings from different sources and identify associations and patterns, e.g., causality of brain activity can be deduced by linking dynamics in different imaging readings. It can provide access, in an experimental setting, to determine the roles of different brain areas from multiple perspectives.
The growth of neuroimaging has spurred a parallel development of multimodal neuroimaging computing, which focuses on computational analysis of multimodal neuroimaging data, including pre-processing, feature extraction, image fusion, machine learning, visualization, and post-processing. These computational advances help to address the variations in spatiotemporal resolution and merge the biophysical/biochemical information in images [18].
Fig. 1
The explosive growth of multimodal neuroimaging studies over the past two decades. (Color figure online)
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0019-x/MediaObjects/40708_2015_19_Fig1_HTML.gif
We conducted a search on PubMed using the keywords ‘multimodal AND neuroimaging’ up to ‘31 Dec 2014.’ There were 1461 relevant publications retrieved from the database. Figure 1 illustrates how multimodal neuroimaging in neuroscience research has rapidly expanded over the past 10 years. In 2004, there were 30 publications, and in 2014, there were close to 300 (indicated by the green area). There is a wide range of applications of multimodal neuroimaging, clinical and non-clinical, including building a brain machine interface (BMI) [19], tracing neural activities and information pathways [20], mapping mind and behavior to brain regions [21–23], evaluating the effects of pharmacological treatments [24, 25], and image-guided therapy (IGT) [26–28].
An important clinical application is the provision of functional and anatomical data for diagnosis of neuropsychiatric disorders [3, 4]. In another PubMed search on these 1461 publications, using the keywords ‘(multimodal AND neuroimaging) AND (neuropsychiatric OR neurological OR psychiatric),’ a substantial proportion (over 30%) of the relevant results focused on the neuropsychiatric disorders (see blue area in Fig. 1). The number of publications dramatically increased each year from 10 to 121 in the period 2004–2014.
Previous reviews mainly focused on a single neuropsychiatric disorder, and summarize the image-based findings of them. For Alzheimer’s disease (AD), for example, Perrin briefly reviewed the multimodal techniques, including PET, fMRI, structural MRI (sMRI), and biochemical examination of cerebrospinal fluid (CSF), to detect AD pathology [29]. Ewers et al. integrated the findings on changes in cortical gray matter volume, white matter fiber tracts, and brain metabolism of patients [30], and discussed the sequential changes in neuroimaging biomarkers during different disease stages [31], similar to the review of Lin et al. [32]. In a more recent review, Nasrallah et al. extended a review to other forms of neurodegenerative dementia [33]. More in-depth reviews on other neuropsychiatric disorders can be found in Sect. 3.
The goal of this review differs from those above in that our interest is to review the recent advances in multimodal neuroimaging and evaluate its applications in neuropsychiatric disorders. Such a review will provide a clearer picture of the current status and offer insights and inspiration to researchers as they design better models/methods for future research.
An extensive review of the image-based findings in neuropsychiatric disorders is beyond the scope of this paper, and we instead review recent studies with a focus on the applications of multimodal neuroimaging, and refer the readers to other reviews for the detailed findings. In Sect. 2, we provide an overview of the common multimodal neuroimaging techniques, and analyze the spatial/temporal resolution, functional/structural connectivity, sensitivity/specificity to brain changes, risks/benefits for clinical applications, computing workflows, and future potential. In Sect. 3, we discuss how these neuroimaging techniques can complement each other, and how they are applied in neuropsychiatric disorders. In Sect. 4, we outline future directions for multimodal neuroimaging in neuropsychiatric research.
An overview of neuroimaging techniques
The different neuroimaging techniques have different biophysical/biochemical mechanisms, and vary in imaging capabilities. Current neuroimaging techniques could be broadly classified into functional and structural neuroimaging. For example, sMRI reveals the detailed anatomy of the brain, and diffusion MRI (dMRI) provides information about fiber tracts. Functional modalities, including fMRI, PET, and EEG/MEG, provide data in brain metabolism and neural activity.
In the following paragraphs, we briefly summarize these neuroimaging techniques with respect to
- spatial resolution; exploring the brain anatomy and detecting morphological changes
- temporal resolution; monitoring neural activities and interactions, tracing information pathways
- structural connectivity; tracing the major brain white matter pathways
- functional connectivity; recording the neural co-activation, in the resting state
- molecular imaging; detecting the molecular activity using agents to target specific functions
- safety and risks
- clinical availability, accessibility, and ease of use
- future developments
Fig. 2
The overview of the properties of sMRI (blue), dMRI (green), fMRI (orange), PET (red), EEG (violet), and multimodal neuroimaging (gray), as indicated by the polar diagrams. Each axis in the diagram represents an attribute, and greater distance from the origin means better performance. Note the indexes in the diagrams are merely indicative and should not be interpreted in a quantitative way. (Color figure online)
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0019-x/MediaObjects/40708_2015_19_Fig2_HTML.gif
……
Applications to neuropsychiatric disorders
Neuropsychiatric disorders represent the most disabling and costly category, based on the systematic analysis of descriptive epidemiology of 291 diseases and injuries from 1990 to 2010 for 187 countries [58]. As shown in Fig. 3, neuropsychiatric disorders caused the largest number of years lost due to illness, disability, and early death measured by disability-adjusted life years (DALYs) in US, and the socioeconomic burden of neuropsychiatric disorders will be aggravated as people live longer.
Fig. 3
The disability-adjusted life years (DALYs) of 291 diseases and injuries based on the systematic analysis of descriptive epidemiology from 1990 to 2010 in US [58]. (Color figure online)
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0019-x/MediaObjects/40708_2015_19_Fig3_HTML.gif
Neuroimaging techniques have expanded beyond a traditional diagnostic role to have a fundamental role in patient management from diagnosis, to selection and assessment of treatment and to prognosis stratification. There is a rising trend of using the multimodal neuroimaging approaches in neuropsychiatric disorders, as shown in Fig. 1. In this section, we summarize how these neuroimaging techniques can be integrated using the multimodal computing methods, and further demonstrate their applications in neuropsychiatric disorders as well as in stroke, traumatic brain injury (TBI), brain tumors, and the brain connectome (Fig. 4).
Fig. 4
The applications of the multimodal neuroimaging approaches in a variety of neuropsychiatric disorders, as well as in stroke, brain injury, brain tumor, and connectome. The color of circle indicates various neuroimaging techniques, same as in Fig. 2. The size of the circle indicates the prevalence of use the technique in specific applications. Note the sizes are only indicative and should not be interpreted in a quantitative way. (Color figure online)
http://static-content.springer.com/image/art%3A10.1007%2Fs40708-015-0019-x/MediaObjects/40708_2015_19_Fig4_HTML.gif
These multimodal approaches can be separated into categories that include a structural–structural combination, a functional–functional combination, and a structural–functional combination. Each category has different applications, and requires different computing workflows. In brief, a structural–structural combination, e.g., sMRI-dMRI, is used to extract and fuse various morphological features and is applied to disorders that affect both gray matter and white matter, such as TBI and stroke. The functional–functional combination can be used to explore brain activation/metabolism patterns and is mainly applied to cognition and consciousness-related disorders, e.g., epilepsy and obsessive-compulsive disorder (OCD). The structural–functional combination is virtually applicable to all disorders, but more frequently used for identifying the structure–function associations in neurodegenerative disorders, neurodevelopmental disorders, multiple sclerosis, schizophrenia, bipolar disorder, brain tumors, and the brain connectome.
Structural–structural combination
sMRI-dMRI methods dominate the structural–structural category, as they take clinical benefits of sMRI and dMRI by integrating the gray matter and white matter morphometry. It has become a useful tool to detect lesions and evaluate treatments for various neuropsychiatric disorders that cause brain morphological changes. Here, we list a few examples of clinical uses of sMRI-dMRI.
Traumatic brain injury (TBI) has very high incidence, resulting in 6.8 million TBI cases every year in the US, and causes impairment of memory, information processing, attention, and executive function [59]. Multimodal structural neuroimaging can assist neurosurgeons, intensive care specialists, neurologists, and rehabilitation specialists in the management of TBI [60]. Conventional brain CT usually fails to detect the subtle structural abnormalities in mild TBI, and sMRI and dMRI are the methods of choice to evaluate and predict outcome in TBI. The sMRI sequences (T1, T2, FLAIR, susceptibility-weighted imaging (SWI) and gradient-recalled echo (GRE)) provide highly accurate depiction of pathological lesions, and dMRI detects the effects of TBI on brain connectivity and non-hemorrhagic diffuse axonal injury (DAI), which are not detected by CT. The sMRI-dMRI methods are widely used in TBI [61, 62]. There are also some studies that have used dMRI and fMRI to validate the connectivity information in TBI patients in the recovery phase [63,64].
The sMRI-dMRI methods have been routinely used in the assessment and treatment planning for stroke. Stroke is a leading cause of death worldwide. There are different types of stroke, and each requires a different diagnostic approach and treatment. T2*-weighted sMRI, e.g., SWI and GRE, is primarily used to detect hemorrhagic stroke, and has equal sensitivity to standard CT methods. However, dMRI is 4-5 times more sensitive in detecting acute ischemic stroke than CT. Other structural imaging techniques, such as perfusion CT (PCT), CT angiography (CTA), digital subtraction angiography (DSA), perfusion-weighted imaging (PWI), and MR angiography (MRA), can also be used to evaluate suspected vascular occlusion, edema, and cerebral infarction. Tong et al. [65] recently published a comprehensive comparison of these methods in the evaluation and management of stroke. Another review on multimodal neuroimaging in stroke is given by Copen et al. [66].
sMRI-dMRI methods have also been used to analyze the gray and white matter alterations in schizophrenia [67] and Autism spectrum disorders (ASDs) [16, 68], neurodegeneration simulation [69], classification of AD and frontotemporal dementia (FTD) [70], and Parkinson’s Disease (PD) staging [71].
Functional–functional combination
EEG-fMRI is valued in functional brain research due to the complementary nature of EEG and fMRI. EEG-fMRI can provide simultaneous cortical and subcortical recording of brain activity with high spatiotemporal resolution.
Epilepsy is one of the most prevalent neurological disorders worldwide. EEG-fMRI is increasingly used to provide clinical support for the diagnosis of epilepsy, in addition to the routinely used sMRI [72] and PET [14, 73]. Researches have used EEG-fMRI to identify a set of brain functional regions that collectively form ‘consciousness,’ including contributions from the DMN, ascending arousal systems, and the thalamus, as summarized by Bagshaw et al. [74]. The activation of these regions and the connection of the networks are important in the evaluation of epilepsy, and together may provide a more fundamental understanding of the alterations of consciousness experienced in epilepsy. Abela et al. [75] focused on altered network compositions in epilepsy, and identified the specific connectivity pathways that characterize the underlying epilepsy syndromes, such as mesial temporal lobe epilepsy (MTLE), lateral temporal lobe epilepsy (LTLE), frontal lobe epilepsy (FLE), idiopathic generalized epilepsy (IGE), and absence epilepsy (AE). A substantial proportion of patients have refractory epilepsy and surgery offers the potential to reduce seizure frequency. Successful surgical treatments, however, require accurate localization of the seizure onset zones and an understanding of surrounding functional cortex to avoid iatrogenic disability. PET, MRI, and intracranial EEG (iEEG) are all needed for optimal surgical planning and treatment evaluation of refractory epilepsy [76, 77].
Another important application of EEG-fMRI is to evaluate patients with obsessive-compulsive disorder (OCD). OCD is a chronic and relatively common neuropsychiatric disorder that characterized by stereotyped and repetitive behaviors. Patients with OCD feel intense need to carry out these behaviors, and have impaired ability to recognize an error and to adjust future responses. OCD may result in social disability. Two neuroimaging biomarkers of error commission, the error-related negativity (ERN) and the dorsal anterior cingulate cortex activation, have been identified using EEG and fMRI, respectively [78]. However, Agam et al. [79] recently suggested that these biomarkers have different neural and genetic mediation. dMRI is also increasingly being used to examine the microstructural integrity of white matter in OCD patients, since white matter abnormalities have long been suspected in OCD, but the findings are inconsistent. For example, one recent study indicated that patients with OCD had decreased fractional anisotropy in the anterior cingulum bundle [80], but in another recent study, the OCD patients showed increased fractional anisotropy of the cingulum bundle [81]. Further investigation on large datasets is needed to confirm these findings.
Structural–functional combination
sMRI-dMRI-fMRI has been ubiquitously used in neuropsychiatric research largely because of high clinical availability, and partially due to its capability to link brain function, structure, and connectivity. It has been increasingly used in research in attention-deficit hyperactivity disorder (ADHD), Autism spectrum disorder (ASD), bipolar disorder, schizophrenia, and clinically in multiple Sclerosis (MS).
ADHD is one of the most commonly diagnosed childhood behavioral disorders. It is characterized by persistent inattention (ADHD-I), hyperactivity-impulsivity (ADHD-H), or a combination of both (ADHD-C). ADHD affects at least 5–11% of school-age children, and symptoms may persist into adulthood [82]. Previous studies using sMRI have reported various findings, such as decreased total brain volume and abnormalities in specific brain regions. The task-evoked and resting-state fMRI approaches were also used in ADHD studies to detect the abnormal brain activation. The use of sMRI and fMRI was reported recently in ADHD [83, 84]. It is only quite recently that dMRI has been applied to ADHD to characterize the disrupted interconnected structural networks in the brain. Shenton et al. provided a brief summary of the latest studies [85]. For example, Hong et al. used dMRI and whole-brain tractography to investigate the altered white matter connectivity in 71 children with ADHD, and identified a single network (comprising 23 brain regions and 25 links) that differentiates the ADHD group from the normal control group [86].
ASDs are neurodevelopmental disorders characterized by deficits in social reciprocity, impaired communication, and restricted interests and repetitive behaviors. Previous studies using sMRI have shown that infants with ASD might have excessive brain growth followed by abnormally slow or even arrested growth as compared to normal developing control infants in early childhood [87]. Subsequent research indicated ASD affects both gray and white matter volumes. Therefore, dMRI has been exploited to describe the microstructural integrity and orientation of white matter. fMRI has enhanced the understanding of the neural circuity of ASDs by demonstrating the convergent structural and functional changes [88, 89]. For example, Mueller et al. used sMRI-dMRI-fMRI approach and identified three brain areas with strong correlations between the structural and functional abnormalities: right temporoparietal junction and the left frontal lobe, bilateral superior temporal gyri, and the right temporoparietal region [90].
MS is a demyelinating disease commonly seen in young people. The cause of MS is unknown. Symptoms and signs vary across patients and can include cognitive impairment, fatigue, vertigo, diplopia, ataxia, hemiparesis, and paraparesis in severe MS patients. Histopathologic and neuroimaging examinations suggest that both white matter and gray matter are affected. In particular, the thalamus can be affected frequently in MS [91], which can lead to impaired cognition. sMRI can detect the thalamic atrophy; dMRI can be used to demonstrate the altered thalamocortical white matter pathways, and fMRI can be used to show the association between the resting-state thalamocortical functional connectivity and cognitive impairment. Recently, sMRI-dMRI-fMRI was jointly used in several studies [92, 93].
Bipolar disorder is a psychotic disorder that characterized states of depression and mania, and sometimes with symptoms common to schizophrenia. It is therefore difficult to conceptualize bipolar disorder and its subtypes, and differentiate it from other psychiatric disorders. The multimodal MRI methods have been applied to bipolar disorder and clearly demonstrate abnormalities in brain networks associated with emotion processing, emotion regulation, and reward processing. In a recent study, Sui et al. proposed a joint analysis model for fMRI and DTI for discriminating bipolar disorder from schizophrenia [94]. Common abnormalities were seen in dorsolateral prefrontal cortex, thalamus, and uncinate fasciculus, whereas differences were found in medial frontal and visual cortex, as well as occipitofrontal white matter tracts. Phillips and Swartz recently published an extensive review of these neuroimaging findings and further pointed out the future directions of neuroimaging research in bipolar disorder [95].
Schizophrenia is a major psychosis that is characterized by altered perception, thought processes, and behaviors. It can be highly heritable disorder [96], and can be triggered by a combination of genetic factors and environmental interactions [97]. Disconnection in white matter pathways and alteration of cortex are assumed to underlie the cognitive abnormalities in schizophrenia, although this is a hypothesis and as yet there is no direct proof. The approaches used for characterizing schizophrenia are very similar to those for bipolar disorder, primarily using sMRI-dMRI-fMRI. Various findings in schizophrenia studies have been reported, based on the investigation on microstructure of white matter [98] or gray matter [97], or the connectivity between different brain regions [67, 99].
The study of brain networks, the connectome, is the focus of intense current neuroscience research [100]. Exploration on the neural systems and brain connections is critical to advance our understanding of normal brain reaction and is one of the greatest challenges of the twenty first century. The Human Connectome Project1 is directed at tackling this challenge using the highest quality imaging data available today, predominantly MRI data, complemented by EEG and MEG. The information about brain anatomy, structural connectivity, and functional connectivity is being obtained using dMRI and resting-state fMRI. Additional information about brain function is being obtained using task-evoked fMRI, EEG, and MEG to record the brain activity.
sMRI-PET is a new structural–functional combination that is being applied to neurodegenerative diseases and brain tumors to improve the localization and targeting of diseased tissue with high accuracy and sensitivity. AD is the most common neurodegenerative disorder among aging people, and it accounts for close to 70% of all dementia cases. In AD, activities of daily living deteriorate over a number of years, ultimately leading to death. There is no cure [101]. AD neuroimaging biomarkers can detect the changes in brain structure (e.g., atrophy on sMRI) and function (e.g., hypometabolism, amyloid plaque, and NFT formation on PET) before there is cognitive impairment. As a result, sMRI and PET with 18F-FDG and amyloid tracers are being increasingly used in the evaluation of patients with early dementia in the research setting [8, 102–106]. These studies also demonstrated clear benefits of multimodal neuroimaging over any single technique alone. Recently, dMRI [107, 108] and fMRI [109] have also been used in the evaluation of dementia as there is evidence that suggests the functional connection between networks is disrupted [110–112]. There are many extensive reviews which summarized these imaging techniques and the image-based findings [29–31, 33].
Over 200,000 individuals are diagnosed with primary or metastatic brain tumors in the US each year [28]. The primary use of sMRI-PET in brain tumors is to accurately localize and label the lesion, e.g., tumor and edema. PET has the potential to more accurately detect the peripheral tumor boundary than using sMRI alone [11, 113]. For brain tumor surgery, dMRI is usually combined with sMRI and PET for pre-operative surgical planning and intra-operative surgical navigation. For example, Durst et al. used dMRI to predict tumor infiltration in patients with gliomas [114]. Tempany et al. used sMRI and dMRI tractography to display a complete brain map for surgical planning [28]. They further demonstrated how to optimize the separation between tumor and normal brain in intrinsic brain tumors with sMRI, and how to avoid inadequate resection of the tumor.
Future directions
Multimodal neuroimaging approaches have been increasingly used in detection, diagnosis, prognosis, and treatment planning of neuropsychiatric disorders. In this paper, we have briefly summarized the recent advances in neuroimaging techniques, and reviewed their applications to neuropsychiatric disorders to provide an overview of the current status. We have also outlined some future directions for multimodal neuroimaging research.
Improved neuroimaging capabilities Neuroimaging techniques will continue to advance rapidly, with higher spatial/temporal/angular resolutions, shorter scan time, and better image contrast. In particular, hybrid scanners, e.g., PET/CT and PET/MRI, will become more clinically accessible. These technologies will enable more discoveries in the neuropsychiatric disorders. The improved imaging capabilities will offer better neuroimaging biomarkers to evaluate neuropsychiatric disorders, and various subtypes or different stages of the same disorder with higher statistical power. These biomarkers will be standardized so they can be widely used clinically and evaluated in large-scale sample sets. In addition, once the biomarkers reach a satisfactory level or the treatment, appropriate clinical guidelines must be developed to support and encourage widespread clinical testing.
Enhanced neuroimaging computing models and methods The continued growth in the complexity and dimensionality of the neuroimaging data will spur the parallel advances of computation models and methods to analyze such complex data. Future neuroimaging analysis models will integrate the longitudinal information to track the long-term changes in the biomarkers [115]. This is essential for us to understand the pathology of the disorders and its degeneration trajectory. With sufficiently large longitudinal datasets, we may be able to identify the causes and detect the early signs, as well as predict the course of the disorders. Future studies will also focus on subject-centered therapy. However, no matter how large the datasets are, they cannot include the entire population, and there will always be inter-subject variations. Personalized/patient-centered care is highly demanded and is the ultimate goal of neuroimaging studies [116]. Neuroimaging computing models and methods also need to keep increasing the degree of automation, accuracy, reproducibility, and robustness, and eventually need to be integrated into the clinical workflow to facilitate clinical testing of the new neuroimaging biomarkers.
Converged neurotechnologies Another future direction will be to combine imaging with non-imaging studies. The multidisciplinary nature of neuroimaging computing will keep bringing together clinicians, biologists, computer scientists, engineers, physicists, and other researchers. Imaging genetics is a very promising area for the future, where the aim is to identify the genetic basis of anatomical and functional abnormalities of the human brain and show how this is connected with neuropsychiatric disorders. There is a trend to use imaging findings in brain disorders to reveal the endophenotypes for various gene mutations. By converting the endophenotype data to novel genetic biomarkers, it may be possible to identify individuals at greater risk of developing brain disorders, and in the near future provide treatment options before the symptoms appear.
Footnotes
1 http://www.neuroscienceblueprint.nih.gov/connectome
References
- 1.
Kikinis R, Pieper SD, Vosburgh K (2014) 3D Slicer: a platform for subject-specific image analysis, visualization, and clinical support. Intraoper Imaging Image-Guided Ther 3(19):277–289
- 2.
Alzheimer’s Association (2015) Changing the trajectory of Alzheimer’s disease: how a treatment by 2025 saves lives and dollars. http://www.alz.org/alzheimers_disease_trajectory.asp
- 3.
Brookmeyer B, Johnson E, Ziegler-Graham K, Arrighi H (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement 3(3):186–191
- 4.
Insel TR, Landis SC, Collins FS (2013) The NIH BRAIN initiative. Science 340(6133):687–688
- 5.
Amunts K, Linder A, Zilles K (2014) The human brain project: neuroscience perspectives and German contributions. e-Neuroforum 5(2):43–50
- 6.
Jiang T (2013) Brainnetome: a new-ome to understand the brain and its disorders. NeuroImage 80:263–272
- 7.
Hinrichs C, Singh V, Xu G, Johnson S (2011) Predictive markers for AD in a multi-modality framework: an analysis of MCI progression in the ADNI population. NeuroImage 55:574–589
- 8.
Zhang D, Wang Y, Zhou L, Yuan H, Shen D (2011) Multimodal classification of Alzheimer’s disease and mild cognitive impairment. NeuroImage 55(3):856–867
- 9.
Beyer T, Townsend DW, Brun T, Kinahan PE, Charron M, Robby R et al (2000) A combined PET/CT scanner for clinical oncology. J Nucl Med 41(8):1369–1379
- 10.
Townsend DW (2001) A combined PET/CT scanner: the choices. J Nucl Med 42(3):533–534
- 11.
Bisdas S, Nagele T, Schlemmer P, Boss A, Claussen C, Pichler B, Ernemann U (2010) Switching on the lights for real-time multimodality tumor neuroimaging: the integrated positron-emission tomography/MR imaging system. Am J Neuroradiol 31(4):610–614
- 12.
Shah NJ, Oros-Peusquens AM, Arrbula J, Zhang K, Warbrick T et al (2013) Advances in multimodal neuroimaging: hybrid MR-PET and MR-PET-EEG at 3 T and 9.4 T. J Magn Reson 229:101–115
- 13.
He B, Liu Z (2008) Multimodal functional neuroimaging: integrating functional MRI and EEG/MEG. IEEE Rev Biomed Eng 1:23–40
- 14.
Knopman AA, Wong CH, Stevenson RJ et al (2015) The relationship between neuropsychological functioning and FDG-PET hypometabolism in intractable mesial temporal lobe epilepsy. Epilepsy Behav 44:136–142
- 15.
Liu S, Zhang L, Cai W, Song Y, Wang Z, Wen L, Feng D (2013b) A supervised multiview spectral embedding method for neuroimaging classification. In: The 20th IEEE international conference on image processing (ICIP), IEEE, pp 601–605
…more…
Read Full Post »







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
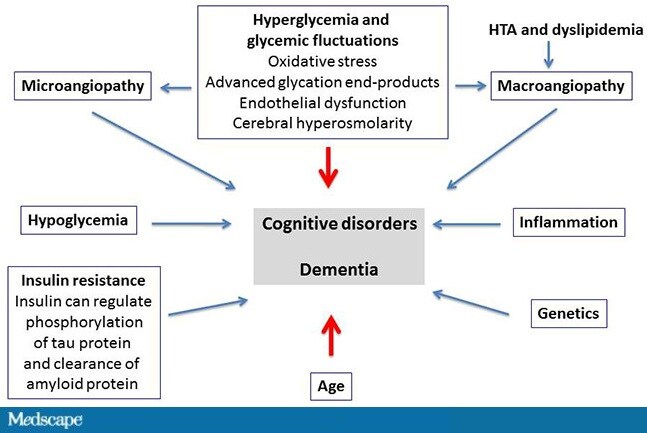{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}