Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Chapter 1: Evolution of the Foundation for Diagnostics and Pharmaceuticals Industries
1.1 Outline of Medical Discoveries between 1880 and 1980
1.2 The History of Infectious Diseases and Epidemiology in the late 19th and 20th Century
1.3 The Classification of Microbiota
1.4 Selected Contributions to Chemistry from 1880 to 1980
1.5 The Evolution of Clinical Chemistry in the 20th Century
1.6 Milestones in the Evolution of Diagnostics in the US HealthCare System: 1920s to Pre-Genomics
Chapter 2. The search for the evolution of function of proteins, enzymes and metal catalysts in life processes
2.1 The life and work of Allan Wilson
2.2 The evolution of myoglobin and hemoglobin
2.3 More complexity in proteins evolution
2.4 Life on earth is traced to oxygen binding
2.5 The colors of life function
2.6 The colors of respiration and electron transport
2.7 Highlights of a green evolution
Chapter 3. Evolution of New Relationships in Neuroendocrine States
3.1 Pituitary endocrine axis
3.2 Thyroid function
3.3 Sex hormones
3.4 Adrenal Cortex
3.5 Pancreatic Islets
3.6 Parathyroids
3.7 Gastointestinal hormones
3.8 Endocrine action on midbrain
3.9 Neural activity regulating endocrine response
3.10 Genomic Promise for Neurodegenerative Diseases, Dementias, Autism Spectrum, Schizophrenia, and Serious Depression
Chapter 4. Problems of the Circulation, Altitude, and Immunity
4.1 Innervation of Heart and Heart Rate
4.2 Action of hormones on the circulation
4.3 Allogeneic Transfusion Reactions
4.4 Graft-versus Host reaction
4.5 Unique problems of perinatal period
4.6. High altitude sickness
4.7 Deep water adaptation
4.8 Heart-Lung-and Kidney
4.9 Acute Lung Injury
4.10 Reconstruction of Life Processes requires both Genomics and Metabolomics to explain Phenotypes and Phylogenetics
Chapter 5. Problems of Diets and Lifestyle Changes
5.1 Anorexia nervosa
5.2 Voluntary and Involuntary S-insufficiency
5.3 Diarrheas – bacterial and nonbacterial
5.4 Gluten-free diets
5.5 Diet and cholesterol
5.6 Diet and Type 2 diabetes mellitus
5.7 Diet and exercise
5.8 Anxiety and quality of Life
5.9 Nutritional Supplements
Chapter 6. Advances in Genomics, Therapeutics and Pharmacogenomics
6.1 Natural Products Chemistry
6.2 The Challenge of Antimicrobial Resistance
6.3 Viruses, Vaccines and immunotherapy
6.4 Genomics and Metabolomics Advances in Cancer
6.5 Proteomics – Protein Interaction
6.6 Pharmacogenomics
6.7 Biomarker Guided Therapy
6.8 The Emergence of a Pharmaceutical Industry in the 20th Century: Diagnostics Industry and Drug Development in the Genomics Era: Mid 80s to Present
6.09 The Union of Biomarkers and Drug Development
6.10 Proteomics and Biomarker Discovery
6.11 Epigenomics and Companion Diagnostics
Chapter 7
Integration of Physiology, Genomics and Pharmacotherapy
7.1 Richard Lifton, MD, PhD of Yale University and Howard Hughes Medical Institute: Recipient of 2014 Breakthrough Prizes Awarded in Life Sciences for the Discovery of Genes and Biochemical Mechanisms that cause Hypertension
7.2 Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
7.3 Diagnostics and Biomarkers: Novel Genomics Industry Trends vs Present Market Conditions and Historical Scientific Leaders Memoirs
7.4 Synthetic Biology: On Advanced Genome Interpretation for Gene Variants and Pathways: What is the Genetic Base of Atherosclerosis and Loss of Arterial Elasticity with Aging
On its way for an IPO: mRNA platform, Moderna, Immune Oncology is recruiting 100 new Life Scientists in Cambridge, MA, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
On its way for an IPO: mRNA platform, Moderna, Immune Oncology is recruiting 100 new Life Scientists in Cambridge, MA
Curator: Aviva Lev-Ari, PhD, RN
Deals:
Moderna has now raised $1.9 billion from investors like AstraZeneca – 9% stack [AstraZeneca’s Pascal Soriot helped get that all started with a whopping $240 million upfront in its 2013 deal, which was tied to $180 million in milestones.], with another $230 million on the table from grants. In addition to the financing announcement this morning, Moderna is also unveiling a pact to develop a new Zika vaccine, with BARDA putting up $8 million to get the program started while offering an option on $117 million more to get through a successful development program.
Novel Strategy in Biotech:
in biotech. Instead of grabbing one or two new drugs and setting out to gather proof-of-concept data to help establish its scientific credibility, the company has harvested a huge windfall of cash and built a large organization before even entering the clinic. And it did that without turning to an IPO.
Pipeline include:
The deal with AstraZeneca covers new drugs for cardiovascular, metabolic and renal diseases as well as cancer.
partners filed a European application to start a Phase I study of AZD8601, an investigational mRNA-based therapy that encodes for vascular endothelial growth factor-A (VEGF-A)
Moderna CEO spelled out plans to get the first 6 new drugs in the clinic by the end of 2016.
The first human study was arranged for the infectious disease drug mRNA 1440, which began an early stage study in 2015.
Moderna built up a range of big preclinical partnerships.
CEO Bancel says the number of drugs in development has swelled to 11, with the first set of data slated to be released in 2017.
Moderna also plans to add about 10 drugs to the clinic by next summer,
SOURCES
UPDATED: Booming Moderna is raising $600M while ramping up manufacturing and clinical studies
First challenge to make use of the new NCI Cloud Pilots – Somatic Mutation Challenge – RNA: Best algorithms for detecting all of the abnormal RNA molecules in a cancer cell, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
First challenge to make use of the new NCI Cloud Pilots – Somatic Mutation Challenge – RNA: Best algorithms for detecting all of the abnormal RNA molecules in a cancer cell
Reporter: Aviva Lev-Ari, PhD, RN
Genomic rearrangements in cancer cells produce fusion transcripts, which may give rise to chimeric protein products not present in normal cells. In addition, cancer cells can express alternate forms of encoded messages that give rise to protein variants different from normal tissue. These chimeras and protein variants can serve as robust diagnostic markers or drug targets. Moreover, ongoing research efforts are beginning to unveil the potential clinical relevance of these variant RNA products. Increasing the “alterome” of tumors by fully characterizing their RNA landscapes will expand our understanding of cancer mechanisms, provide new biomarkers and reveal possible new RNA-based therapeutics, thus improving personalized patient treatment.
“Predicting RNA species in a cancer cell is a particularly challenging task,” says Josh Stuart, Professor at the UC Santa Cruz Genomics Institute and one of the challenge leaders. “RNA expression reflects much of the deranged complexity of the underlying cancer cell DNA and then adds another level of derangement on top of that.”
The goal of the SMC-RNA Challenge is to identify the best methods for detecting rearrangements in RNA sequencing (RNA-seq) data. Sub-challenges are focused on detecting and quantifying mRNA fusions and isoforms. Methods will be evaluated with both in silico and spiked-in data.
Two key questions that will be addressed are:
1) What is the best way to estimate the abundances of a set of known RNA isoforms? and
2) What is the best way to predict the presence of novel gene fusions?
Both of these questions will involve in silico generated and wet lab spiked-in RNA sequencing data.
We are launching the ICGC-TCGA DREAM Somatic Mutation Calling RNA Challenge (SMC-RNA), a community-based collaborative competition of researchers from across the world. We will rigorously assess the accuracy of methods to perform two key tasks in cancer RNA-Seq data analysis: the quantification of known isoforms and detecting novel fusion transcripts. We will generate RNA-Seq data for a synthetic-based challenge. Since synthetic data may not capture the complexity of real human tumours, we will also introduce a phase in which teams make predictions on real human-tumours. Challenge organizers will perform retrospective experimental validation on predictions to create a gold-standard. Validation will employ a combination of long-read sequencing and target-capture approaches. The SMC-RNA Challenge will analyze a couple of dozen samples created to have known alterations representing different tumor types, allowing confidence that the winning methods will be generalizable across the broad range of human cancers.
The ICGC-TCGA DREAM Somatic Mutation Calling – RNA Challenge (SMC-RNA) is an international effort to improve standard methods for identifying cancer-associated rearrangements in RNA sequencing (RNA-seq) data. Leaders of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA) cancer genomics projects are joining with Sage Bionetworks and IBM-DREAM to initiate this innovative open crowd-sourced Challenge [1-3].
Why is RNA biology important in cancer?
While only a small fraction of the genome encodes proteins, the majority is either transcribed or has putative regulatory functions, with the consequence that cellular functions are extensively regulated at the RNA level. The regulation of RNA, and its dramatic dysregulation in cancer cells, occurs in multiple ways. RNA abundances may be altered, and these have served as the basis for clinically-important prognostic biomarkers. Genomic rearrangements in cancer cells produce fusion transcripts which may give rise to chimeric protein products not present in normal cells. These can serve as robust diagnostic markers (e.g. TMPRSS2-ERG in prostate cancer) or drug targets (e.g. BCR-ABL in CML). Ongoing research efforts are beginning to unveil the potential clinical relevance of aberrant processing of RNA in cancer, such as defects in alternative-splicing. To fully document the molecular differences in transcription between tumor cells and their normal counterparts an assortment of computational methods are needed. Increasing the “alterome” of tumors by fully characterizing their RNA landscapes will expand our understanding of cancer mechanisms, provide new biomarkers and reveal possible new RNA-based therapeutics, improving personalized patient treatment.
What is RNA Sequencing?
RNA-seq is using next-generation sequencing techniques to sequence RNA. It allows the the transcriptome to be sequenced at high coverage, provides raw read counts that can be used to assess expression levels, and from it elucidate other biologically relevant information. RNA is reverse transcribed into cDNA and then sequenced at high depth by high-throughput technologies, such as Illumina HiSeq, Roche 454, and PacBio [4]. After sequencing, reads can be aligned de novo, to a reference genome, or to a reference transcriptome. Using RNA-seq has some advantages over microarrays, including no prior knowledge of the transcriptome needed and an unbiased expression analysis [5]. In comparison, microarrays, the current gold standard for RNA analysis require probe design and have been found to contain more bias in low-intensity genes [6]. Some key challenges in RNA-seq include biases occurring in RNA fragmentation, cDNA fragmentation, and library preparation, in addition to, potential PCR artifacts that skew expression levels, and possible alignment to multiple locations in a reference genome [5]. Due to these and other influences, methods for detecting and quantifying transcriptional isoforms, as well as fusion genes remains a challenging set of problems and competing methods for interpreting RNA-Seq results continues to be poor.
Fusion Prediction
Scientific Rationale: Gene fusions occur when two genes at the DNA level are joined and may be due to an oncogenic event. A fusion may also occur at the RNA level where a ligation between two transcripts occurs. Gene fusions have an important role in the initial steps of tumorigenesis. Specifically, gene fusions have been found to be the driver mutations in neoplasia and have been linked to various tumour subtypes. An increasing number of gene fusions are being recognized as important diagnostic and prognostic parameters in malignant haematological disorders and childhood sarcomas. Gene fusions occur in all malignancies and account for 20% of human cancer morbidity [7].
Isoform Prediction
Scientific Rationale: Isoforms are alternative expressions of a gene formed from splicing during post-transcriptional processing. Dysregulation of alternative splicing occurs in every category of Hanahan’s and Weinberg’s hallmarks of cancer. Modifications in splicing may occur due to mutations of cis-acting splicing elements, trans-acting regulators, and microRNAs. Moreover, cancer cell lines, regardless of their tissue of origin, can be effectively discriminated from non-cancer cell lines at isoform level, but not at gene level. Existence of an isoform signature, rather than a gene signature, could be used to distinguish cancer cells from normal cells [8].
Challenges
The goal of this Challenge is to use a crowd-based competition to identify the optimal methods for detecting (and quantifying) mRNA fusions and isoforms from RNA-seq data.
Sub-Challenge 1: Quantify Known Isoforms
Can algorithms estimate the levels of a set of provided isoforms?
1a: In silico simulated data challenge
1b: Spike-in data challenge on real data from long-reads and hybrid capture.
Sub-Challenge 2: Detect Gene Fusions
Can algorithms predict the presence of gene fusions?
2a: In silico simulated gene fusions.
2b: Spike-in long-read data.
Reward
All participants will be invited as consortium co-authors on Challenge marker papers
Winners will receive travel awards & speaking invitations at the next DREAM conference or Sage Congress
New methods will be considered for co-publication with the Challenge marker papers by our publishing partner
Other rewards will be announced as they are determined.
Challenge Organizers / Scientific Advisory Board
Kyle Ellrott, Oregon Health Sciences University
Josh Stuart, University of California, Santa Cruz
Paul C. Boutros, Ontario Institute for Cancer Research
Wang, Z., Gerstein, M., & Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics, 10(1), 57-63.
Han, Y., Gao, S., Muegge, K., Zhang, W., & Zhou, B. (2015). Advanced Applications of RNA Sequencing and Challenges.Bioinformatics and biology insights, 9(Suppl 1), 29.
Robinson, D., Wang, J., & Storey, J. (2015). A nested parallel experiment demonstrates differences in intensity-dependence between RNA-seq and microarrays. Nucleic Acids Research. 43(20)
Mertens, F., Johansson, B., Fioretos, T., & Mitelman, F. (2015). The emerging complexity of gene fusions in cancer. Nature Reviews Cancer, 15(6), 371-381.
Liu S, Cheng C. Alternative RNA splicing and cancer. Wiley interdisciplinary reviews RNA. 2013,4(5),547-566.
The following are key points to remember from this update on the treatment of cardiac transthyretin amyloidosis:
Transthyretin (TTR) is a highly conserved protein involved in transportation of thyroxine (T4) and retinol-binding protein. TTR is synthesized mostly by the liver and is rich in beta strands with an intrinsic propensity to aggregate into insoluble amyloid fibers, which deposit within tissue leading to the development of TTR-related amyloidosis (ATTR). ATTR can follow the deposition of either variant TTR (ATTRv, previously known as mutant ATTR) or wild type TTR (ATTRwt).
Cardiac ATTR has a favorable survival rate compared to light chain (AL) amyloidosis, with a median survival of 75 versus 11 months. However, ATTR cardiomyopathy is a progressive disorder but newer therapeutic options include tafamidis (positive phase 3 clinical trial), and possibly patisiran and inotersen.
Inhibition of the Synthesis of Mutated Transthyretin
Liver transplantation removes the source of mutated TTR molecules and prolongs survival, with a 20-year survival of 55.3%. However, tissue accumulation of TTR can continue after liver transplantation because TTR amyloid fibers promote subsequent deposition of ATTRwt. Combined liver–heart transplantation is feasible in younger patients with ATTRv cardiomyopathy and a small series suggests better prognosis than cardiac transplantation.
Inhibition of TTR gene expression: Patisiran is a small interfering RNA blocking the expression of both variant and wt TTR. On the basis of the APOLLO trial, it was approved for therapy of adults with ATTRv-related polyneuropathy both in the United States and European Union. In this trial, patisiran promoted favorable myocardial remodeling based on echocardiographic and N-terminal B-type natriuretic peptide (NT-BNP) changes (this effect was not demonstrated for inotersen) and is still under investigation for tafamidis.
Antisense oligonucleotides inotersen inhibits the production of both variant and wt TTR. Based on the findings of the NEURO-TTR trial, the Food and Drug Administration (FDA) approved this agent for patients with ATTRv-related polyneuropathy. In the NEURO-TTR trial, cardiomyopathy was present in 63%, but the study was not powered to measure effects of inotersen on heart disease. Inotersen can cause thrombocytopenia and must be used cautiously with bleeding risk.
Tetramer Stabilization
Selective stabilizers include tafamidis and AG10. Tafamidis is a benzoxazole and a small molecule that inhibits the dissociation of TTR tetramers by binding the T4-binding sites. The phase ATTR-ACT study showed that when comparing the pooled tafamidis arms (80 and 20 mg) with the placebo arm, tafamidis was associated with lower all-cause mortality than placebo (78 of 264 [29.5%] vs. 76 of 177 [42.9%]; hazard ratio, 0.70; 95% confidence interval, 0.51-0.96) and a lower rate of cardiovascular hospitalizations. Based on the results of the ATTR-ACT trial, it has received Breakthrough Therapy designation from the FDA for treatment of ATTR cardiomyopathy.
Nonselective agents: Diflunisal, a nonsteroidal anti-inflammatory drug, is reported to stabilize TTR tetramers. More studies are needed to confirm its clinical efficacy.
Inhibition of Oligomer Aggregation and Oligomer Disruption
Epigallocatechin gallate is the most abundant catechin in green tea. One single-center open-label 12-month study did not show survival benefits or any change in echocardiographic parameters or NT-BNP compared to baseline.
Degradation and Reabsorption of Amyloid Fibers
Doxycycline-taurosodeoxycholic acid (TUDCA) has been evaluated in two small studies and the results appear to be modest. More data are needed to confirm its efficacy.
Antibodies targeting serum amyloid P protein or amyloid fibrils: Patient enrollment for miridesap followed by anti-SAP antibodies was suspended, and this approach is not being evaluated currently. However, a monoclonal antibody designed to specifically target TTR amyloid deposits (PRX004) has entered clinical evaluation, with an ongoing phase 1 study on ATTRv.
Supportive Treatment of Cardiac Involvement
Drug therapies: Although angiotensin-converting enzyme (ACE) inhibitors/angiotensin-receptor blockers (ARBs) and beta-blockers may have been poorly tolerated in the ATTR-ACT trial, 30% of the patients were on ACE inhibitors/ARBs. There are no data with digoxin in TTR amyloid, and non-dihydropyridine calcium channel blockers are contraindicated due to negative inotropy.
Implantable cardioverter-defibrillators (ICDs): In one study, which included 53 patients with amyloid, ICD shocks occurred exclusively in the AL amyloid group and none in the TTR amyloid patients. Higher defibrillation thresholds and complication rates are of concern.
Cardiac pacing: In a large series of ATTRv-related polyneuropathy (n = 262), a pacemaker was implanted in 110 patients with His ventricular interval >700 ms. The authors recommend that any conduction disturbance on 12-lead electrocardiogram (ECG) warrants further investigation with Holter monitoring to determine candidacy for a pacemaker.
Left ventricular assist device (LVAD): Although an LVAD is technically feasible, it is associated with high short-term mortality and worse outcomes than in dilated cardiomyopathy.
Cardiac transplantation: This is a valuable option for patients with end-stage heart failure when significant extracardiac disease is excluded. In one study with 10 patients, only episodes of amyloid recurrence occurred.
This is an outstanding overview of this topic and recommended reading for anyone who cares for patients with cardiac transthyretin amyloid.
First-Ever Evidence that Patisiran Reduces Pathogenic, Misfolded TTR Monomers and Oligomers in FAP Patients
We reported data from our ongoing Phase 2 open-label extension (OLE) study of patisiran, an investigational RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR amyloidosis) patients with familial amyloidotic polyneuropathy (FAP). Alnylam scientists and collaborators from The Scripps Research Institute and Misfolding Diagnostics, Inc. were able to measure the effects of patisiran on pathogenic, misfolded TTR monomers and oligomers in FAP patients. Results showed a rapid and sustained reduction in serum non-native conformations of TTR (NNTTR) of approximately 90%. Since NNTTR is pathogenic in ATTR amyloidosis and the level of NNTTR reduction correlated with total TTR knockdown, these results provide direct mechanistic evidence supporting the therapeutic hypothesis that TTR knockdown has the potential to result in clinical benefit. Furthermore, complete 12-month data from all 27 patients that enrolled in the patisiran Phase 2 OLE study showed sustained mean maximum reductions in total serum TTR of 91% for over 18 months and a mean 3.1-point decrease in mNIS+7 at 12 months, which compares favorably to an estimated increase in mNIS+7 of 13 to 18 points at 12 months based upon analysis of historical data sets in untreated FAP patients with similar baseline characteristics. Importantly, patisiran administration continues to be generally well tolerated out to 21 months of treatment.
We are encouraged by these new data that provide continued support for our hypothesis that patisiran has the potential to halt neuropathy progression in patients with FAP. If these results are replicated in a randomized, double-blind, placebo-controlled study, we believe that patisiran could emerge as an important treatment option for patients suffering from this debilitating, progressive and life-threatening disease.
Hereditary ATTR Amyloidosis with Polyneuropathy (hATTR-PN)
ATTR amyloidosis is a progressive, life-threatening disease caused by misfolded transthyretin (TTR) proteins that accumulate as amyloid fibrils in multiple organs, but primarily in the peripheral nerves and heart. ATTR amyloidosis can lead to significant morbidity, disability, and mortality. The TTR protein is produced primarily in the liver and is normally a carrier for retinol binding protein – one of the vehicles used to transport vitamin A around the body. Mutations in the TTR gene cause misfolding of the protein and the formation of amyloid fibrils that typically contain both mutant and wild-type TTR that deposit in tissues such as the peripheral nerves and heart, resulting in intractable peripheral sensory neuropathy, autonomic neuropathy, and/or cardiomyopathy.
ATTR represents a major unmet medical need with significant morbidity and mortality. There are over 100 reported TTR mutations; the particular TTR mutation and the site of amyloid deposition determine the clinical manifestations of the disease whether it is predominantly symptoms of neuropathy or cardiomyopathy.
Specifically, hereditary ATTR amyloidosis with polyneuropathy (hATTR-PN), also known as familial amyloidotic polyneuropathy (FAP), is an inherited, progressive disease leading to death within 5 to 15 years. It is due to a mutation in the transthyretin (TTR) gene, which causes misfolded TTR proteins to accumulate as amyloid fibrils predominantly in peripheral nerves and other organs. hATTR-PN can cause sensory, motor, and autonomic dysfunction, resulting in significant disability and death.
It is estimated that hATTR-PN, also known as FAP, affects approximately 10,000 people worldwide. Patients have a life expectancy of 5 to 15 years from symptom onset, and the only treatment options for early stage disease are liver transplantation and TTR stabilizers such as tafamidis (approved in Europe) and diflunisal. Unfortunately liver transplantation has limitations, including limited organ availability as well as substantial morbidity and mortality. Furthermore, transplantation eliminates the production of mutant TTR but does not affect wild-type TTR, which can further deposit after transplantation, leading to cardiomyopathy and worsening of neuropathy. There is a significant need for novel therapeutics to treat patients who have inherited mutations in the TTR gene.
Our ATTR program is the lead effort in our Genetic Medicine Strategic Therapeutic Area (STAr) product development and commercialization strategy, which is focused on advancing innovative RNAi therapeutics toward genetically defined targets for the treatment of rare diseases with high unmet medical need. We are developing patisiran (ALN-TTR02), an intravenously administered RNAi therapeutic, to treat the hATTR-PN form of the disease.
Patisiran for the Treatment hATTR-PN
APOLLO Phase 3 Trial
In 2012, Alnylam entered into an exclusive alliance with Genzyme, a Sanofi company, to develop and commercialize RNAi therapeutics, including patisiran and revusiran, for the treatment of ATTR amyloidosis in Japan and the broader Asian-Pacific region. In early 2014, this relationship was extended as a significantly broader alliance to advance RNAi therapeutics as genetic medicines. Under this new agreement, Alnylam will lead development and commercialization of patisiran in North America and Europe while Genzyme will develop and commercialize the product in the rest of world.
Hereditary ATTR Amyloidosis with Cardiomyopathy (hATTR-CM)
ATTR amyloidosis is a progressive, life-threatening disease caused by misfolded transthyretin (TTR) proteins that accumulate as amyloid fibrils in multiple organs, but primarily in the peripheral nerves and heart. ATTR amyloidosis can lead to significant morbidity, disability, and mortality. The TTR protein is produced primarily in the liver and is normally a carrier for retinol binding protein – one of the vehicles used to transport vitamin A around the body. Mutations in the TTR gene cause misfolding of the protein and the formation of amyloid fibrils that typically contain both mutant and wild-type TTR that deposit in tissues such as the peripheral nerves and heart, resulting in intractable peripheral sensory neuropathy, autonomic neuropathy, and/or cardiomyopathy.
ATTR represents a major unmet medical need with significant morbidity and mortality. There are over 100 reported TTR mutations; the particular TTR mutation and the site of amyloid deposition determine the clinical manifestations of the disease, whether it is predominantly symptoms of neuropathy or cardiomyopathy.
Specifically, hereditary ATTR amyloidosis with cardiomyopathy (hATTR-CM), also known as familial amyloidotic cardiomyopathy (FAC), is an inherited, progressive disease leading to death within 2 to 5 years. It is due to a mutation in the transthyretin (TTR) gene, which causes misfolded TTR proteins to accumulate as amyloid fibrils primarily in the heart. Hereditary ATTR amyloidosis with cardiomyopathy can result in heart failure and death.
While the exact numbers are not known, it is estimated hATTR-CM, also known as FAC affects at least 40,000 people worldwide. hATTR-CM is fatal within 2 to 5 years of diagnosis and treatment is currently limited to supportive care. Wild-type ATTR amyloidosis (wtATTR amyloidosis), also known as senile systemic amyloidosis, is a nonhereditary, progressive disease leading to death within 2 to 5 years. It is caused by misfolded transthyretin (TTR) proteins that accumulate as amyloid fibrils in the heart. Wild-type ATTR amyloidosis can cause cardiomyopathy and result in heart failure and death. There are no approved therapies for the treatment of hATTR-CM or SSA; hence there is a significant unmet need for novel therapeutics to treat these patients.
Our ATTR program is the lead effort in our Genetic Medicine Strategic Therapeutic Area (STAr) product development and commercialization strategy, which is focused on advancing innovative RNAi therapeutics toward genetically defined targets for the treatment of rare diseases with high unmet medical need. We are developing revusiran (ALN-TTRsc), a subcutaneously administered RNAi therapeutic for the treatment of hATTR-CM.
Revusiran for the Treatment of hATTR-CM
ENDEAVOUR Phase 3 Trial
In 2012, Alnylam entered into an exclusive alliance with Genzyme, a Sanofi company, to develop and commercialize RNAi therapeutics, including patisiran and revusiran, for the treatment of ATTR amyloidosis in Japan and the broader Asian-Pacific region. In early 2014, this relationship was extended as a broader alliance to advance RNAi therapeutics as genetic medicines. Under this new agreement, Alnylam and Genzyme have agreed to co-develop and co-commercialize revusiran in North America and Europe, with Genzyme developing and commercializing the product in the rest of world. This broadened relationship on revusiran is aimed at expanding and accelerating the product’s global value.
We presented pre-clinical data with ALN-TTRsc02, an investigational RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR amyloidosis). In pre-clinical studies, including those in non-human primates (NHPs), ALN-TTRsc02 achieved potent and highly durable knockdown of serum TTR of up to 99% with multi-month durability achieved after just a single dose, supportive of a potentially once quarterly dose regimen. Results from studies comparing TTR knockdown activity of ALN-TTRsc02 to that of revusiran showed that ALN-TTRsc02 has a markedly superior TTR knockdown profile. Further, in initial rat toxicology studies, ALN-TTRsc02 was found to be generally well tolerated with no significant adverse events at doses as high as 100 mg/kg.
Heart failure with a preserved ejection fraction (HFPEF) is a clinical syndrome that has no pharmacologic therapies approved for this use to date. In light of failed medicines, cardiologists have refocused treatment strategies based on the theory that HFPEF is a heterogeneous clinical syndrome with different etiologies. Classification of HFPEF according to etiologic subtype may, therefore, identify cohorts with treatable pathophysiologic mechanisms and may ultimately pave the way forward for developing meaningful HFPEF therapies.1
A wealth of data now indicates that amyloid infiltration is an important mechanism underlying HFPEF. Inherited mutations in transthyretin cardiac amyloidosis (ATTRm) or the aging process in wild-type disease (ATTRwt) cause destabilization of the transthyretin (TTR) protein into monomers or oligomers, which aggregate into amyloid fibrils. These insoluble fibrils accumulate in the myocardium and result in diastolic dysfunction, restrictive cardiomyopathy, and eventual congestive heart failure (Figure 1). In an autopsy study of HFPEF patients, almost 20% without antemortem suspicion of amyloid had left ventricular (LV) TTR amyloid deposition.2 Even more resounding evidence for the contribution of TTR amyloid to HFPEF was a study in which 120 hospitalized HFPEF patients with LV wall thickness ≥12 mm underwent technetium-99m 3,3-diphosphono-1,2-propranodicarboxylic acid (99mTc-DPD) cardiac imaging,3,4 a bone isotope known to have high sensitivity and specificity for diagnosing TTR cardiac amyloidosis.5,6 Moderate-to-severe myocardial uptake indicative of TTR cardiac amyloid deposition was detected in 13.3% of HFPEF patients who did not have TTR gene mutations. Therefore, TTR cardiac amyloid deposition, especially in older adults, is not rare, can be easily identified, and may contribute to the underlying pathophysiology of HFPEF.
Figure 1
As no U.S. Food and Drug Administration-approved drugs are currently available for the treatment of HFPEF or TTR cardiac amyloidosis, the development of medications that attenuate or prevent TTR-mediated organ toxicity has emerged as an important therapeutic goal. Over the past decade, a host of therapies and therapeutic drug classes have emerged in clinical trials (Table 1), and these may herald a new direction for treating HFPEF secondary to TTR amyloid.
Table 1
TTR Silencers (siRNA and Antisense Oligonucleotides)
siRNA
Ribonucleic acid interference (RNAi) has surfaced as an endogenous cellular mechanism for controlling gene expression. Small interfering RNAs (siRNAs) delivered into cells can disrupt the production of target proteins.7,8 A formulation of lipid nanoparticle and triantennary N-acetylgalactosamine (GalNAc) conjugate that delivers siRNAs to hepatocytes is currently in clinical trials.9 Prior research demonstrated these GalNAc-siRNA conjugates result in robust and durable knockdown of a variety of hepatocyte targets across multiple species and appear to be well suited for suppression of TTR gene expression and subsequent TTR protein production.
The TTR siRNA conjugated to GalNAc, ALN-TTRSc, is now under active investigation as a subcutaneous injection in phase 3 clinical trials in patients with TTR cardiac amyloidosis.10 Prior phase 2 results demonstrated that ALN-TTRSc was generally well tolerated in patients with significant TTR disease burden and that it reduced both wild-type and mutant TTR gene expression by a mean of 87%. Harnessing RNAi technology appears to hold great promise for treating patients with TTR cardiac amyloidosis. The ability of ALN-TTRSc to lower both wild-type and mutant proteins may provide a major advantage over liver transplantation, which affects the production of only mutant protein and is further limited by donor shortage, cost, and need for immunosuppression.
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are under clinical investigation for their ability to inhibit hepatic expression of amyloidogenic TTR protein. Currently, the ASO compound, ISIS-TTRRx, is under investigation in a phase 3 multicenter, randomized, double-blind, placebo-controlled clinical trial in patients with familial amyloid polyneuropathy (FAP).11 The primary objective is to evaluate its efficacy as measured by change in neuropathy from baseline relative to placebo. Secondary measures will evaluate quality of life (QOL), modified body mass index (mBMI) by albumin, and pharmacodynamic effects on retinol binding protein. Exploratory objectives in a subset of patients with LV wall thickness ≥13 mm without a history of persistent hypertension will examine echocardiographic parameters, N-terminal pro–B-type natriuretic peptide (NT-proBNP), and polyneuropathy disability score relative to placebo. These data will facilitate analysis of the effect of antisense oligonucleotide-mediated TTR suppression on the TTR cardiac phenotype with a phase 3 trial anticipated to begin enrollment in 2016.
TTR Stabilizers (Diflunisal, Tafamidis)
Diflunisal
Several TTR-stabilizing agents are in various stages of clinical trials. Diflunisal, a traditionally used and generically available nonsteroidal anti-inflammatory drug (NSAID), binds and stabilizes familial TTR variants against acid-mediated fibril formation in vitro and is now in human clinical trials.12,13 The use of diflunisal in patients with TTR cardiac amyloidosis is controversial given complication of chronic inhibition of cyclooxygenase (COX) enzymes, including gastrointestinal bleeding, renal dysfunction, fluid retention, and hypertension that may precipitate or exacerbate heart failure in vulnerable individuals.14-17 In TTR cardiac amyloidosis, an open-label cohort study suggested that low-dose diflunisal with careful monitoring along with a prophylactic proton pump inhibitor could be safely administered to compensated patients.18 An association was observed, however, between chronic diflunisal use and adverse changes in renal function suggesting that advanced kidney disease may be prohibitive in diflunisal therapy.In FAP patients with peripheral or autonomic neuropathy randomized to diflunisal or placebo, diflunisal slowed progression of neurologic impairment and preserved QOL over two years of follow-up.19 Echocardiography demonstrated cardiac involvement in approximately 50% of patients.20 Longer-term safety and efficacy data over an average 38 ± 31 months in 40 Japanese patients with hereditary ATTR amyloidosis who were not candidates for liver transplantation showed that diflunisal was mostly well tolerated.12 The authors cautioned the need for attentive monitoring of renal function and blood cell counts. Larger multicenter collaborations are needed to determine diflunisal’s true efficacy in HFPEF patients with TTR cardiac amyloidosis.
Tafamidis
Tafamidis is under active investigation as a novel compound that binds to the thyroxine-binding sites of the TTR tetramer, inhibiting its dissociation into monomers and blocking the rate-limiting step in the TTR amyloidogenesis cascade.21 The TTR compound was shown in an 18-month double-blind, placebo-controlled trial to slow progression of neurologic symptoms in patients with early-stage ATTRm due to the V30M mutation.22 When focusing on cardiomyopathy in a phase 2, open-label trial, tafamidis also appeared to effectively stabilize TTR tetramers in non-V30M variants, wild-type and V122I, as well as biochemical and echocardiographic parameters.23,24 Preliminary data suggests that clinically stabilized patients had shorter disease duration, lower cardiac biomarkers, less myocardial thickening, and higher EF than those who were not stabilized, suggesting early institution of therapy may be beneficial. A phase 3 trial has completed enrollment and will evaluate the efficacy, safety, and tolerability of tafamidis 20 or 80 mg orally vs. placebo.25 This will contribute to long-term safety and efficacy data needed to determine the therapeutic effects of tafamidis among ATTRm variants.
Amyloid Degraders (Doxycycline/TUDCA and Anti-SAP Antibodies)
Doxycycline/TUDCA
While silencer and stabilizer drugs are aimed at lowering amyloidogenic precursor protein production, they cannot remove already deposited fibrils in an infiltrated heart. Removal of already deposited fibrils by amyloid degraders would be an important therapeutic strategy, particularly in older adults with heavily infiltrated hearts reflected by thick walls, HFPEF, systolic heart failure, and restrictive cardiomyopathy. Combined doxycycline and tauroursodeoxycholic acid (TUDCA) disrupt TTR amyloid fibrils and appeared to have an acceptable safety profile in a small phase 2 open-label study among 20 TTR patients. No serious adverse reactions or clinical progression of cardiac or neuropathic involvement was observed over one year.26 An active phase 2, single-center, open-label, 12-month study will assess primary outcome measures including mBMI, neurologic impairment score, and NT-proBNP.27 Another phase 2 study is examining the tolerability and efficacy of doxycycline/TUDCA over an 18-month period in patients with TTR amyloid cardiomyopathy.28 Additionally, a study in patients with TTR amyloidosis is ongoing to determine the effect of doxycycline alone on neurologic function, cardiac biomarkers, echocardiographic parameters, modified body mass index, and autonomic neuropathy.29
Anti-SAP Antibodies
In order to safely clear established amyloid deposits, the role of the normal, nonfibrillar plasma glycoprotein present in all human amyloid deposits, serum amyloid P component (SAP), needs to be more clearly understood.30 In mice with amyloid AA type deposits, administration of antihuman SAP antibody triggered a potent giant cell reaction that removed massive visceral amyloid deposits without adverse effects.31 In humans with TTR cardiac amyloidosis, anti-SAP antibody treatments could be feasible because the bis-D proline compound, CPHPC, is capable of clearing circulating human SAP, which allow anti-SAP antibodies to reach residual deposited SAP. In a small, open-label, single-dose-escalation, phase 1 trial involving 15 patients with systemic amyloidosis, none of whom had clinical evidence of cardiac amyloidosis, were treated with CPHPC followed by human monoclonal IgG1 anti-SAP antibody.32 No serious adverse events were reported and amyloid deposits were cleared from the liver, kidney, and lymph node. Anti-SAP antibodies hold promise as a potential amyloid therapy because of their potential to target all forms of amyloid deposits across multiple tissue types.
Mutant or wild-type TTR cardiac amyloidoses are increasingly recognized as a cause of HFPEF. Clinicians need to be aware of this important HFPEF etiology because the diverse array of emerging disease-modifying agents for TTR cardiac amyloidosis in human clinical trials has the potential to herald a new era for the treatment of HFPEF.
References
Maurer MS, Mancini D. HFpEF: is splitting into distinct phenotypes by comorbidities the pathway forward? J Am Coll Cardiol 2014;64:550-2.
Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2014;2:113-22.
González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015.
Castano A, Bokhari S, Maurer MS. Unveiling wild-type transthyretin cardiac amyloidosis as a significant and potentially modifiable cause of heart failure with preserved ejection fraction. Eur Heart J 2015 Jul 28. [Epub ahead of print]
Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009;120:1203-12.
Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013;6:195-201.
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-11.
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494-8.
Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nature Mater 2013;12:967-77.
U.S. National Institutes of Health. Phase 2 Study to Evaluate ALN-TTRSC in Patients With Transthyretin (TTR) Cardiac Amyloidosis (ClinicalTrials.gov website). 2014. Available at: https://www.clinicaltrials.gov/ct2/show/NCT01981837. Accessed 8/19/2015.
U.S. National Institutes of Health. Efficacy and Safety of ISIS-TTRRx in Familial Amyloid Polyneuropathy (Clinical Trials.gov Website. 2013. Available at: http://www.clinicaltrials.gov/ct2/show/NCT01737398. Accessed 8/19/2015.
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13:236-49.
Tojo K, Sekijima Y, Kelly JW, Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res 2006;56:441-9.
Epstein M. Non-steroidal anti-inflammatory drugs and the continuum of renal dysfunction. J Hypertens Suppl 2002;20:S17-23.
Wallace JL. Pathogenesis of NSAID-induced gastroduodenal mucosal injury. Best Pract Res Clin Gastroenterol 2001;15:691-703.
Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 2001;286:954-9.
Page J, Henry D. Consumption of NSAIDs and the development of congestive heart failure in elderly patients: an underrecognized public health problem. Arch Intern Med 2000;160:777-84.
Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 2012;18:315-9.
Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310:2658-67.
Quarta CCF, Solomon RH Suhr SD, et al. The prevalence of cardiac amyloidosis in familial amyloidotic polyneuropathy with predominant neuropathy: The Diflunisal Trial. International Symposium on Amyloidosis 2014:88-9.
Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A 2002;99 Suppl 4:16427-32.
Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79:785-92.
Merlini G, Plante-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res 2013;6:1011-20.
Maurer MS, Grogan DR, Judge DP, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015;8:519-26.
U.S. National Institutes of Health. Safety and Efficacy of Tafamidis in Patients With Transthyretin Cardiomyopathy (ATTR-ACT) (ClinicalTrials.gov website). 2014. Available at: http://www.clinicaltrials.gov/show/NCT01994889. Accessed 8/19/2015.
Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012;19 Suppl 1:34-6.
U.S. National Institutes of Health. Safety, Efficacy and Pharmacokinetics of Doxycycline Plus Tauroursodeoxycholic Acid in Transthyretin Amyloidosis (ClinicalTrials.gov website). 2011. Available at: http://www.clinicaltrials.gov/ct2/show/NCT01171859. Accessed 8/19/2015.
U.S. National Institutes of Health. Tolerability and Efficacy of a Combination of Doxycycline and TUDCA in Patients With Transthyretin Amyloid Cardiomyopathy (ClinicalTrials.gov website). 2013. Available at: http://www.clinicaltrials.gov/ct2/show/NCT01855360. Accessed 8/19/2015.
U.S. National Institutes of Health. Safety and Effect of Doxycycline in Patients With Amyloidosis (ClinicalTrials.gov website).2015. Available at: https://clinicaltrials.gov/ct2/show/NCT01677286. Accessed 8/19/2015.
Pepys MB, Dash AC. Isolation of amyloid P component (protein AP) from normal serum as a calcium-dependent binding protein. Lancet 1977;1:1029-31.
Bodin K, Ellmerich S, Kahan MC, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 2010;468:93-7.
Richards DB, Cookson LM, Berges AC, et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med 2015;373:1106-14.
…
The Acid-Mediated Denaturation Pathway of Transthyretin Yields a Conformational Intermediate That Can Self-Assemble into Amyloid†
Transthyretin (TTR) amyloid fibril formation is observed during partial acid denaturation and while refolding acid-denatured TTR, implying that amyloid fibril formation results from the self-assembly of a conformational intermediate. The acid denaturation pathway of TTR has been studied in detail herein employing a variety of biophysical methods to characterize the intermediate(s) capable of amyloid fibril formation. At physiological concentrations, tetrameric TTR remains associated from pH 7 to pH 5 and is incapable of amyloid fibril formation. Tetrameric TTR dissociates to a monomer in a process that is dependent on both pH and protein concentration below pH 5. The extent of amyloid fibril formation correlates with the concentration of the TTR monomer having an altered, but defined, tertiary structure over the pH range of 5.0−3.9. The inherent Trp fluorescence-monitored denaturation curve of TTR exhibits a plateau over the pH range where amyloid fibril formation is observed (albeit at a higher concentration), implying that a steady-state concentration of the amyloidogenic intermediate with an altered tertiary structure is being detected. Interestingly, 1-anilino-8-naphthalenesulfonate fluorescence is at a minimum at the pH associated with maximal amyloid fibril formation (pH 4.4), implying that the amyloidogenic intermediate does not have a high extent of hydrophobic surface area exposed, consistent with a defined tertiary structure. Transthyretin has two Trp residues in its primary structure, Trp-41 and Trp-79, which are conveniently located far apart in the tertiary structure of TTR. Replacement of each Trp with Phe affords two single Trp containing variants which were used to probe local pH-dependent tertiary structural changes proximal to these chromophores. The pH-dependent fluorescence behavior of the Trp-79-Phe mutant strongly suggests that Trp-41 is located near the site of the tertiary structural rearrangement that occurs in the formation of the monomeric amyloidogenic intermediate, likely involving the C-strand−loop−D-strand region. Upon further acidification of TTR (below pH 4.4), the structurally defined monomeric amyloidogenic intermediate begins to adopt alternative conformations that are not amyloidogenic, ultimately forming an A-state conformation below pH 3 which is also not amyloidogenic. In summary, analytical equilibrium ultracentrifugation, SDS−PAGE, far- and near-UV CD, fluorescence, and light scattering studies suggest that the amyloidogenic intermediate is a monomeric predominantly β-sheet structure having a well-defined tertiary structure.
Prevention of Transthyretin Amyloid Disease by Changing Protein Misfolding Energetics
Per Hammarström*, R. Luke Wiseman*, Evan T. Powers, Jeffery W. Kelly†+ Author Affiliations
Genetic evidence suggests that inhibition of amyloid fibril formation by small molecules should be effective against amyloid diseases. Known amyloid inhibitors appear to function by shifting the aggregation equilibrium away from the amyloid state. Here, we describe a series of transthyretin amyloidosis inhibitors that functioned by increasing the kinetic barrier associated with misfolding, preventing amyloidogenesis by stabilizing the native state. The trans-suppressor mutation, threonine 119 → methionine 119, which is known to ameliorate familial amyloid disease, also functioned through kinetic stabilization, implying that this small-molecule strategy should be effective in treating amyloid diseases.
Rational design of potent human transthyretin amyloid disease inhibitors
Thomas Klabunde1,2, H. Michael Petrassi3, Vibha B. Oza3, Prakash Raman3, Jeffery W. Kelly3 & James C. Sacchettini1
The human amyloid disorders, familial amyloid polyneuropathy, familial amyloid cardiomyopathy and senile systemic amyloidosis, are caused by insoluble transthyretin (TTR) fibrils, which deposit in the peripheral nerves and heart tissue. Several nonsteroidal anti-inflammatory drugs and structurally similar compounds have been found to strongly inhibit the formation of TTR amyloid fibrils in vitro. These include flufenamic acid, diclofenac, flurbiprofen, and resveratrol. Crystal structures of the protein–drug complexes have been determined to allow detailed analyses of the protein–drug interactions that stabilize the native tetrameric conformation of TTR and inhibit the formation of amyloidogenic TTR. Using a structure-based drug design approach ortho-trifluormethylphenyl anthranilic acid and N-(meta-trifluoromethylphenyl) phenoxazine 4,6-dicarboxylic acid have been discovered to be very potent and specific TTR fibril formation inhibitors. This research provides a rationale for a chemotherapeutic approach for the treatment of TTR-associated amyloid diseases.
First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy
Adams, Davida; Suhr, Ole B.b; Hund, Ernstc; Obici, Laurad; Tournev, Ivailoe,f; Campistol, Josep M.g; Slama, Michel S.h; Hazenberg, Bouke P.i; Coelho, Teresaj; from the European Network for TTR-FAP (ATTReuNET)
Purpose of review: Early and accurate diagnosis of transthyretin familial amyloid polyneuropathy (TTR-FAP) represents one of the major challenges faced by physicians when caring for patients with idiopathic progressive neuropathy. There is little consensus in diagnostic and management approaches across Europe.
Recent findings: The low prevalence of TTR-FAP across Europe and the high variation in both genotype and phenotypic expression of the disease means that recognizing symptoms can be difficult outside of a specialized diagnostic environment. The resulting delay in diagnosis and the possibility of misdiagnosis can misguide clinical decision-making and negatively impact subsequent treatment approaches and outcomes.
Summary: This review summarizes the findings from two meetings of the European Network for TTR-FAP (ATTReuNET). This is an emerging group comprising representatives from 10 European countries with expertise in the diagnosis and management of TTR-FAP, including nine National Reference Centres. The current review presents management strategies and a consensus on the gold standard for diagnosis of TTR-FAP as well as a structured approach to ongoing multidisciplinary care for the patient. Greater communication, not just between members of an individual patient’s treatment team, but also between regional and national centres of expertise, is the key to the effective management of TTR-FAP.
Transthyretin familial amyloid polyneuropathy (TTR-FAP) is a highly debilitating and irreversible neurological disorder presenting symptoms of progressive sensorimotor and autonomic neuropathy [1▪,2▪,3]. TTR-FAP is caused by misfolding of the transthyretin (TTR) protein leading to protein aggregation and the formation of amyloid fibrils and, ultimately, to amyloidosis (commonly in the peripheral and autonomic nervous system and the heart) [4,5]. TTR-FAP usually proves fatal within 7–12 years from the onset of symptoms, most often due to cardiac dysfunction, infection, or cachexia [6,7▪▪].
The prevalence and disease presentation of TTR-FAP vary widely within Europe. In endemic regions (northern Portugal, Sweden, Cyprus, and Majorca), patients tend to present with a distinct genotype in large concentrations, predominantly a Val30Met substitution in the TTR gene [8–10]. In other areas of Europe, the genetic footprint of TTR-FAP is more varied, with less typical phenotypic expression [6,11]. For these sporadic or scattered cases, a lack of awareness among physicians of variable clinical features and limited access to diagnostic tools (i.e., pathological studies and genetic screening) can contribute to high rates of misdiagnosis and poorer patient outcomes [1▪,11]. In general, early and late-onset variants of TTR-FAP, found within endemic and nonendemic regions, present several additional diagnostic challenges [11,12,13▪,14].
Delay in the time to diagnosis is a major obstacle to the optimal management of TTR-FAP. With the exception of those with a clearly diagnosed familial history of FAP, patients still invariably wait several years between the emergence of first clinical signs and accurate diagnosis [6,11,14]. The timely initiation of appropriate treatment is particularly pertinent, given the rapidity and irreversibility with which TTR-FAP can progress if left unchecked, as well as the limited effectiveness of available treatments during the later stages of the disease [14]. This review aims to consolidate the existing literature and present an update of the best practices in the management of TTR-FAP in Europe. A summary of the methods used to achieve a TTR-FAP diagnosis is presented, as well as a review of available treatments and recommendations for treatment according to disease status.
Patients with TTR-FAP can present with a range of symptoms [11], and care should be taken to acquire a thorough clinical history of the patient as well as a family history of genetic disease. Delay in diagnosis is most pronounced in areas where TTR-FAP is not endemic or when there is no positive family history [1▪]. TTR-FAP and TTR-familial amyloid cardiomyopathy (TTR-FAC) are the two prototypic clinical disease manifestations of a broader disease spectrum caused by an underlying hereditary ATTR amyloidosis [19]. In TTR-FAP, the disease manifestation of neuropathy is most prominent and definitive for diagnosis, whereas cardiomyopathy often suggests TTR-FAC. However, this distinction is often superficial because cardiomyopathy, autonomic neuropathy, vitreous opacities, kidney disease, and meningeal involvement all may be present with varying severity for each patient with TTR-FAP.
Among early onset TTR-FAP with usually positive family history, symptoms of polyneuropathy present early in the disease process and usually predominate throughout the progression of the disease, making neurological testing an important diagnostic aid [14]. Careful clinical examination (e.g., electromyography with nerve conduction studies and sympathetic skin response, quantitative sensation test, quantitative autonomic test) can be used to detect, characterize, and scale the severity of neuropathic abnormalities involving small and large nerve fibres [10]. Although a patient cannot be diagnosed definitively with TTR-FAP on the basis of clinical presentation alone, symptoms suggesting the early signs of peripheral neuropathy, autonomic dysfunction, and cardiac conduction disorders or infiltrative cardiomyopathy are all indicators that further TTR-FAP diagnostic investigation is warranted. Late-onset TTR-FAP often presents as sporadic cases with distinct clinical features (e.g., milder autonomic dysfunction) and can be more difficult to diagnose than early-onset TTR-FAP (Table 2) [1▪,11,12,13▪,14,20].
Genetic testing is carried out to allow detection of specific amyloidogenic TTR mutations (Table 1), using varied techniques depending on the expertise and facilities available in each country (Table S2, http://links.lww.com/CONR/A39). A targeted approach to detect a specific mutation can be used for cases belonging to families with previous diagnosis. In index cases of either endemic and nonendemic regions that do not have a family history of disease, are difficult to confirm, and have atypical symptoms, TTR gene sequencing is required for the detection of both predicted and new amyloidogenic mutations [26,27].
Following diagnosis, the neuropathy stage and systemic extension of the disease should be determined in order to guide the next course of treatment (Table 4) [3,30,31]. The three stages of TTR-FAP severity are graded according to a patient’s walking disability and degree of assistance required [30]. Systemic assessment, especially of the heart, eyes, and kidney, is also essential to ensure all aspects of potential impact of the disease can be detected [10].
The goals of cardiac investigations are to detect serious conduction disorders with the risk of sudden death and infiltrative cardiomyopathy. Electrocardiograms (ECG), Holter-ECG, and intracardiac electrophysiology study are helpful to detect conduction disorders. Echocardiograms, cardiac magnetic resonance imaging, scintigraphy with bone tracers, and biomarkers (e.g., brain natriuretic peptide, troponin) can all help to diagnose infiltrative cardiomyopathy[10]. An early detection of cardiac abnormalities has obvious benefits to the patient, given that the prophylactic implantation of pacemakers was found to prevent 25% of major cardiac events in TTR-FAP patients followed up over an average of 4 years [32▪▪]. Assessment of cardiac denervation with 123-iodine meta-iodobenzylguanidine is a powerful prognostic marker in patients diagnosed with FAP [33].
…..
Tafamidis
Tafamidis is a first-in-class therapy that slows the progression of TTR amyloidogenesis by stabilizing the mutant TTR tetramer, thereby preventing its dissociation into monomers and amyloidogenic and toxic intermediates [55,56]. Tafamidis is currently indicated in Europe for the treatment of TTR amyloidosis in adult patients with stage I symptomatic polyneuropathy to delay peripheral neurological impairment [57].
In an 18-month, double-blind, placebo-controlled study of patients with early-onset Val30Met TTR-FAP, tafamidis was associated with a 52% lower reduction in neurological deterioration (P = 0.027), a preservation of nerve function, and TTR stabilization versus placebo [58▪▪]. However, only numerical differences were found for the coprimary endpoints of neuropathy impairment [neuropathy impairment score in the lower limb (NIS-LL) responder rates of 45.3% tafamidis vs 29.5% placebo; P = 0.068] and quality of life scores [58▪▪]. A 12-month, open-label extension study showed that the reduced rates of neurological deterioration associated with tafamidis were sustained over 30 months, with earlier initiation of tafamidis linking to better patient outcomes (P = 0.0435) [59▪]. The disease-slowing effects of tafamidis may be dependent on the early initiation of treatment. In an open-label study with Val30Met TTR-FAP patients with late-onset and advanced disease (NIS-LL score >10, mean age 56.4 years), NIS-LL and disability scores showed disease progression despite 12 months of treatment with tafamidis, marked by a worsening of neuropathy stage in 20% and the onset of orthostatic hypotension in 22% of patients at follow-up [60▪].
Tafamidis is not only effective in patients exhibiting the Val30Met mutation; it also has proven efficacy, in terms of TTR stabilization, in non-Val30Met patients over 12 months [61]. Although tafamidis has demonstrated safe use in patients with TTR-FAP, care should be exercised when prescribing to those with existing digestive problems (e.g., diarrhoea, faecal incontinence) [60▪].
Diflunisal is a nonsteroidal anti-inflammatory drug (NSAID) that, similar to tafamidis, slows the rate of amyloidogenesis by preventing the dissociation, misfolding, and misassembly of the mutated TTR tetramer [62,63]. Off-label use has been reported for patients with stage I and II disease, although diflunisal is not currently licensed for the treatment of TTR-FAP.
Evidence for the clinical effectiveness of diflunisal in TTR-FAP derives from a placebo-controlled, double-blind, 24-month study in 130 patients with clinically detectable peripheral or autonomic neuropathy[64▪]. The deterioration in NIS scores was significantly more pronounced in patients receiving placebo compared with those taking diflunisal (P = 0.001), and physical quality of life measures showed significant improvement among diflunisal-treated patients (P = 0.001). Notable during this study was the high rate of attrition in the placebo group, with 50% more placebo-treated patients dropping out of this 2-year study as a result of disease progression, advanced stage of the disease, and varied mutations.
One retrospective analysis of off-label use of diflunisal in patients with TTR-FAP reported treatment discontinuation in 57% of patients because of adverse events that were largely gastrointestinal [65]. Conclusions on the safety of diflunisal in TTR-FAP will depend on further investigations on the impact of known cardiovascular and renal side-effects associated with the NSAID drug class [66,67].
Compared to other mammals, humans have the largest cerebral cortex. A sheet of brain cells that folds in on itself multiple times in order to fit inside the skull, the cortex is the seat of higher functions. It is what enables us to process everything we see and hear and think.
The expansion of the cerebral cortex sets humans apart from the rest of their fellow primates. Yet scientists have long wondered what mechanisms are responsible for this evolutionary development.
New research from the Kosik Molecular and Cellular Neurobiology Lab at UC Santa Barbara has pinpointed a specific long nocoding ribonucleic acid (lncRNA) that regulates neural development (ND). The findings appear in the journal Neuron.
“This lncND, as we’ve called it, can be found only in the branch of primates that leads to humans. It is a stretch of nucleotides that does not code a protein,” said senior author Kenneth S. Kosik, the Harriman Professor of Neuroscience Research in UCSB’s Department of Molecular, Cellular, and Developmental Biology. “We demonstrate that lncND is turned on during development and turned off when the cell matures.”
Lead author Neha Rani, a postdoctoral scholar in the Kosik Lab, idenfitied several binding sites on lncND for another type of RNA called a microRNA. One of them, called microRNA-143, binds to lncND.
“We found that lncND could sequester this microRNA and in doing so regulate the expression of Notch proteins,” Rani said. “Notch proteins are very important regulators during neuronal development. They are involved in cell differentiation and cell fate and are critical in the neural development pathway.”
Kosik describes lncND as a platform that binds these microRNAs like a sponge. “This allows Notch to do what it’s supposed to do during development,” he explained. “Then as the brain matures, levels of lncND go down and when they do, those microRNAs come flying off the platform and glom onto Notch to bring its levels down. You want Notch levels to be high while the brain is developing but not once maturation occurs. This lncND is an elegant way to change Notch levels quickly.”
To replicate these cell culture results, Rani used human stem cells to grow neurons into what is called a mini brain. In this pea-sized gob of brain tissue, she identified a subpopulation — radial glial cells (neuronal stem cells) and other neural progenitors — responsible for making lncND.
But the researchers wanted to see the radial glial cells in actual human brain tissue, so they turned to colleagues in the Developmental & Stem Cell Biology Graduate Program at the UC San Francisco School of Medicine. Using in situ hybridization, UCSF scientists found lncND in neural precursor cells but not in mature neurons.
“It was right where we thought it would be in brain tissue,” said Kosik, who is also the co-director of UCSB’s Neuroscience Research Institute. “But we still had one more thing we had to do because people would still not be satisfied that we had done everything possible to show that lncND was really doing something functionally.”
So the UCSF team introduced lncND into the fetal brain of a gestating mouse. Green fluorescent protein labeling allowed them to see the early development pattern and show that lncND, which ordinarily is not present in mice — lncND is present only in some primates including humans — had a functional effect on development.
“When we overexpressed lncND in the mouse fetus, we actually affected development in the predicted manner,” Kosik said. “The early developmental pattern was shifted toward more precursor cells, even though the mouse does not make lncND at all.”
According to Kosik, this work not only identifies a very critical gene for human brain development but also offers a clue about a component that likely contributed to brain expansion in humans. “We have shown that lncND might be an important player in human brain expansion, which is exciting in itself,” Rani said. “Another interesting aspect of this work is that lncND appears to help regulate the key developmental pathway of Notch signaling.”
Recent studies highlighted long noncoding RNAs (lncRNAs) to play an important role in cardiac development. However, understanding of lncRNAs in cardiac diseases is still limited. Global lncRNA expression profiling indicated that several lncRNA transcripts are deregulated during pressure overload-induced cardiac hypertrophy in mice. Using stringent selection criteria, we identified Chast (cardiac hypertrophy-associated transcript) as a potential lncRNA candidate that influences cardiomyocyte hypertrophy. Cell fractionation experiments indicated that Chast is specifically up-regulated in cardiomyocytes in vivo in transverse aortic constriction (TAC)-operated mice. In accordance, CHAST homolog in humans was significantly up-regulated in hypertrophic heart tissue from aortic stenosis patients and in human embryonic stem cell-derived cardiomyocytes upon hypertrophic stimuli. Viral-based overexpression of Chast was sufficient to induce cardiomyocyte hypertrophy in vitro and in vivo. GapmeR-mediated silencing of Chast both prevented and attenuated TAC-induced pathological cardiac remodeling with no early signs on toxicological side effects. Mechanistically, Chast negatively regulated Pleckstrin homology domain-containing protein family M member 1 (opposite strand of Chast), impeding cardiomyocyte autophagy and driving hypertrophy. These results indicate that Chast can be a potential target to prevent cardiac remodeling and highlight a general role of lncRNAs in heart diseases.
Diagnosing complex autoimmune diseases and related conditions is a challenging endeavor, as these multifaceted conditions often present with non-specific symptoms across several organ systems. Historically, physicians have diagnosed autoimmune disease through detailed physical examination, collection of medical and family history, multiple laboratory tests and monitoring symptoms over time. Current diagnostic algorithms can lead to a protracted and costly diagnostic process of elimination.
However, emerging RNA-based diagnostic technologies can dramatically reduce the time to diagnosis of complex diseases, including autoimmune disease. With the completion of the Human Genome Project and basic research in the field of RNA biology over the past two decades, it is now accepted that the majority of the human genome is transcribed into unique patterns of information that exhibit cell type specificity and are consistent in a variety of chronic conditions, including autoimmune disease. With the invention of new technologies, including next-generation RNA sequencing, we are able to assess global changes in gene expression.
As such, RNA-based tools are now poised to provide physicians with actionable information early in the diagnostic process. While RNA-based testing is a relatively new innovation in the clinical setting and widespread adoption efforts are still in their infancy, the science behind these techniques makes them a reliable method for helping physicians make fast, accurate decisions.
The science behind RNA
Analysis of RNA expression paints a molecular portrait of what’s going on in an individual’s cells at a given point in time. This can provide a picture of disease manifestation. At a high level, current RNA testing involves collecting a patient’s blood sample via standard venipuncture, isolating the RNA and then detecting RNA expression patterns in real time using polymerase chain reaction (PCR). The turning on or off of these expression patterns can serve as an indicator of the presence or absence of disease.
In contrast, DNA tests examine changes in nucleotide sequence to establish a patient’s risk of developing a particular condition. These genotype associations do not always reveal information about disease manifestation. A DNA test, for example, might indicate that a patient is at high risk for an autoimmune disorder, such as rheumatoid arthritis (RA) or multiple sclerosis (MS), but the patient may never develop the condition. Just because the patient has a particular DNA risk marker does not mean he or she actually has—or will have—the disease. Thus, a major limitation with DNA methodology is its inability to reliably forecast active disease. RNA tests are more specific in this regard.
Other types of testing, such as pharmacologic and serologic testing, have also been used to detect autoimmune disorders. However, due to the complexity of these diseases, a single test can only put a physician one small step closer to diagnosis and additional tests are often required. For instance, to accurately identify RA, a clinician may need to perform approximately 10 to 15 different molecular tests over what is often a lengthy time period. If the doctor concludes that a portion of these tests point to the presence of RA, then he or she may be able to make a diagnosis of RA. In some cases, it can take years for the patient to receive a definitive diagnosis. In comparison, new RNA-based diagnostic testing for the same condition offers an accuracy of greater than 90 percent, enabling a higher degree of certainty for establishing the presence or absence of the disease.
The impact on patients, providers and the scientific community
As RNA testing becomes more widespread, its impact on patients, providers and other stakeholders promises to be significant. Once widely adopted, this kind of testing could cut down on the number of tests and amount of the time needed to confirm diagnosis. This, in turn, would enhance the patient experience, as individuals would no longer have to undergo repeated or invasive testing to determine if they have a disease.
Current autoimmune therapies are reasonably effective at slowing disease advancement. Across many autoimmune diseases, the best outcomes are achieved when therapies are initiated early in the disease process. Providing physicians with actionable diagnostic information enables the provider to place patients on the appropriate treatments faster, limiting disease progression and supporting better long-term health outcomes.
RNA testing can improve outcomes for providers by helping to get patients on the path to suitable treatment earlier. This not only facilitates optimal medication performance, but also lessens the cost of diagnosis and management of disease. In turn, this ensures a better quality of life for patients.
A little adversity builds character, or so the saying goes. True or not, the saying does seem an apt description of a developmental phenomenon that shapes gene expression. While it knows nothing of character, the gene expression apparatus appears to respond well to short-term mitochondrial stress that occurs early in development. In fact, transient stress seems to result in lasting benefits. These benefits, which include improved metabolic function and increased longevity, have been observed in both worms and mice, and may even occur—or be made to occur—in humans.
Gene expression is known to be subject to reprogramming by epigenetic modifiers, but such modifiers generally affect metabolism or lifespan, not both. A new set of epigenetic modifiers, however, has been found to trigger changes that do just that—both improve metabolism and extend lifespan.
Scientists based at the University of California, Berkeley, and the École Polytechnique Fédérale de Lausanne (EPFL) have discovered enzymes that are ramped up after mild stress during early development and continue to affect the expression of genes throughout the animal’s life. When the scientists looked at strains of inbred mice that have radically different lifespans, those with the longest lifespans had significantly higher expression of these enzymes than did the short-lived mice.
“Two of the enzymes we discovered are highly, highly correlated with lifespan; it is the biggest genetic correlation that has ever been found for lifespan in mice, and they’re both naturally occurring variants,” said Andrew Dillin, a UC Berkeley professor of molecular and cell biology. “Based on what we see in worms, boosting these enzymes could reprogram your metabolism to create better health, with a possible side effect of altering lifespan.”
Details of the work, which appeared online April 29 in the journal Cell, are presented in a pair of papers. One paper (“Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity”) resulted from an effort led by Dillin and the EPFL’s Johan Auwerx. The other paper (“Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPRmt”) resulted from an effort led by Dillin and his UC Berkeley colleague Barbara Meyer.
According to these papers, mitochondrial stress activates enzymes in the brain that affect DNA folding, exposing a segment of DNA that contains the 1500 genes involved in the work of the mitochondria. A second set of enzymes then tags these genes, affecting their activation for much or all of the lifetime of the animal and causing permanent changes in how the mitochondria generates energy.
The first set of enzymes—methylases, in particular LIN-65—add methyl groups to the DNA, which can silence promoters and thus suppress gene expression. By also opening up the mitochondrial genes, these methylases set the stage for the second set of enzymes—demethylases, in this case jmjd-1.2 and jmjd-3.1—to ramp up transcription of the mitochondrial genes. When the researchers artificially increased production of the demethylases in worms, all the worms lived longer, a result identical to what is observed after mitochondrial stress.
“By changing the epigenetic state, these enzymes are able to switch genes on and off,” Dillin noted. This happens only in the brain of the worm, however, in areas that sense hunger or satiety. “These genes are expressed in neurons that are sensing the nutritional status of the animal, and these signals emanate out to the periphery to change peripheral metabolism,” he continued.
When the scientists profiled enzymes in short- and long-lived mice, they found upregulation of these genes in the brains of long-lived mice, but not in other tissues or in the brains of short-lived mice. “These genes are expressed in the hypothalamus, exactly where, when you eat, the signals are generated that tell you that you are full. And when you are hungry, signals in that region tell you to go and eat,” Dillin explained said. “These genes are all involved in peripheral feedback.”
Among the mitochondrial genes activated by these enzymes are those involved in the body’s response to proteins that unfold, which is a sign of stress. Increased activity of the proteins that refold other proteins is another hallmark of longer life.
These observations suggest that the reversal of aging by epigenetic enzymes could also take place in humans.
“It seems that, while extreme metabolic stress can lead to problems later in life, mild stress early in development says to the body, ‘Whoa, things are a little bit off-kilter here, let’s try to repair this and make it better.’ These epigenetic switches keep this up for the rest of the animal’s life,” Dillin stated.
Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity
Carsten Merkwirth6, Virginija Jovaisaite6, Jenni Durieux,…., Reuben J. Shaw, Johan Auwerx, Andrew Dillin
•H3K27 demethylases jmjd-1.2 and jmjd-3.1 are required for ETC-mediated longevity
•jmjd-1.2 and jmjd-3.1 extend lifespan and are sufficient for UPRmt activation
•UPRmt is required for increased lifespan due to jmjd-1.2 or jmjd-3.1 overexpression
•JMJD expression is correlated with UPRmt and murine lifespan in inbred BXD lines
Across eukaryotic species, mild mitochondrial stress can have beneficial effects on the lifespan of organisms. Mitochondrial dysfunction activates an unfolded protein response (UPRmt), a stress signaling mechanism designed to ensure mitochondrial homeostasis. Perturbation of mitochondria during larval development in C. elegans not only delays aging but also maintains UPRmt signaling, suggesting an epigenetic mechanism that modulates both longevity and mitochondrial proteostasis throughout life. We identify the conserved histone lysine demethylases jmjd-1.2/PHF8 and jmjd-3.1/JMJD3 as positive regulators of lifespan in response to mitochondrial dysfunction across species. Reduction of function of the demethylases potently suppresses longevity and UPRmt induction, while gain of function is sufficient to extend lifespan in a UPRmt-dependent manner. A systems genetics approach in the BXD mouse reference population further indicates conserved roles of the mammalian orthologs in longevity and UPRmt signaling. These findings illustrate an evolutionary conserved epigenetic mechanism that determines the rate of aging downstream of mitochondrial perturbations.
Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPRmt
Ye Tian, Gilberto Garcia, Qian Bian, Kristan K. Steffen, Larry Joe, Suzanne Wolff, Barbara J. Meyer, Andrew Dillin
•LIN-65 accumulates in the nucleus in response to mitochondrial stress
•Mitochondrial stress-induced chromatin changes depend on MET-2 and LIN-65
•LIN-65 and DVE-1 exhibit interdependence in nuclear accumulation
•met-2 and atfs-1 act in parallel to affect mitochondrial stress-induced longevity
Organisms respond to mitochondrial stress through the upregulation of an array of protective genes, often perpetuating an early response to metabolic dysfunction across a lifetime. We find that mitochondrial stress causes widespread changes in chromatin structure through histone H3K9 di-methylation marks traditionally associated with gene silencing. Mitochondrial stress response activation requires the di-methylation of histone H3K9 through the activity of the histone methyltransferase met-2 and the nuclear co-factor lin-65. While globally the chromatin becomes silenced by these marks, remaining portions of the chromatin open up, at which point the binding of canonical stress responsive factors such as DVE-1 occurs. Thus, a metabolic stress response is established and propagated into adulthood of animals through specific epigenetic modifications that allow for selective gene expression and lifespan extension
Siddharta Mukherjee’s Writing Career Just Got Dealt a Sucker Punch
Siddharha Mukherjee won the 2011 Pulitzer Prize in non-fiction for his book, The Emperor of All Maladies. The book has received widespread acclaim among lay audience, physicians, and scientists alike. Last year the book was turned into a special PBS series. But, according to a slew of scientists, we should all be skeptical of his next book scheduled to hit book shelves this month, The Gene, An Intimate History.
Publishing an article on epigenetics in the New Yorker this week–perhaps a selection from his new book–Mukherjee has waltzed into one of the most active scientific debates in all of biology: that of gene regulation, or epigenetics.
Jerry Coyne, the evolutionary biologist known for keeping journalists honest, has published a two part critique of Mukherjee’s New Yorker piece. The first part–wildly tweeted yesterday–is a list of quotes from Coyne’s colleagues and those who have written in to the New Yorker, including two Nobel prize winners, Wally Gilbert and Sidney Altman, offering some very unfriendly sentences.
Wally Gilbert: “The New Yorker article is so wildly wrong that it defies rational analysis.”
Sidney Altman: “I am not aware that there is such a thing as an epigenetic code. It is unfortunate to inflict this article, without proper scientific review, on the audience of the New Yorker.”
The second part is a thorough scientific rebuttal of the Mukherjee piece. It all serves as a great drama about one of the most contested ideas in biology and also as a cautionary tale to journalists, even experienced writers such as Mukherjee, about the dangers of wading into scientific arguments. Readers may remember that a few years ago, science writer, David Dobbs, similarly skated into the same topic with his piece, Die, Selfish Gene, Die, and which raised a similar shitstorm, much of it from Coyne.
Mukherjee’s mistake is in giving credence to only one side of a very fierce debate–that the environment causes changes in the genome which can be passed on; another kind of evolution–as though it were settled science. Either Mukherjee, a physicisan coming off from a successful book and PBS miniseries on cancer, is setting himself up as a scientist, or he has been a truly naive science reporter. If he got this chapter so wrong, what does it mean about an entire book on the gene?
Coyne quotes one of his colleagues who raised some questions about the New Yorker’s science reporting, one particular question we’ve been asking here at Mendelspod. How do we know what we know? Does science now have an edge on any other discipline for being able to create knowledge?
Coyne’s colleague is troubled by science coverage in the New Yorker, and goes so far as to write that the New Yorker has been waging a “war on behalf of cultural critics and literary intellectuals against scientists and technologists.”
From my experience, it’s not quite that tidy. First of all, the New Yorker is the best writing I read each week. Period. Second, I haven’t found their science writing to have the slant claimed in the quote above. For example, most other mainstream outlets–including the New York Times with the Amy Harmon pieces–have given the anti-GMO crowd an equal say in the mistaken search for a “balance” on whether GMOs are harmful. (Remember John Stewart’s criticism of Fox News? That they give a false equivalent between two sides even when there is no equivalent on the other side?)
But the New Yorker has not fallen into this trap on GMOs and most of their pieces on the topic–mainly by Michael Specter–have been decidedly pro science and therefore decided pro GMO.
So what led Mukherjee to play scientist as well as journalist? There’s no question about whether I enjoy his prose. His writing beautifully whisks me away so that I don’t feel that I’m really working to understand. There is a poetic complexity that constantly brings different threads effortlessly together, weaving them into the same light. At one point he uses the metaphor of a web for the genome, with the epigenome being the stuff that sticks to the web. He borrows the metaphor from the Hindu notion of “being”, or jaal.
“Genes form the threads of the web; the detritus that adheres to it transforms every web into a singular being.”
There have been a few writers on Twitter defending Mukherjee’s piece. Tech Review’s Antonio Regalado called Coyne and his colleagues “tedious literalists” who have an “issue with epigenetic poetry.”
At his best, Mukherjee can take us down the sweet alleys of his metaphors and family stories with a new curiosity for the scientific truth. He can hold a mirror up to scientists, or put the spotlight on their work. At their worst, Coyne and his scientific colleagues can reek of a fear of language and therefore metaphor. The always outspoken scientist and author, Richard Dawkins, who made his name by personifying the gene, was quick to personify epigentics in a tweet: “It’s high time the 15 minutes of underserved fame for “epigenetics” came to an overdue end.” Dawkins is that rare scientist who has consistently been as comfortable with rhetoric and language as he is with data.
Hats off to Coyne who reminds us that a metaphor–however lovely–does not some science make. If Mukherjee wants to play scientist, let him create and gather data. If it’s the role of science journalist he wants, let him collect all the science he can before he begins to pour it into his poetry.
Same but Different
How epigenetics can blur the line between nature and nurture.
The author’s mother (right) and her twin are a study in difference and identity. CREDIT: PHOTOGRAPH BY DAYANITA SINGH FOR THE NEW YORKER
October 6, 1942, my mother was born twice in Delhi. Bulu, her identical twin, came first, placid and beautiful. My mother, Tulu, emerged several minutes later, squirming and squalling. The midwife must have known enough about infants to recognize that the beautiful are often the damned: the quiet twin, on the edge of listlessness, was severely undernourished and had to be swaddled in blankets and revived.
The first few days of my aunt’s life were the most tenuous. She could not suckle at the breast, the story runs, and there were no infant bottles to be found in Delhi in the forties, so she was fed through a cotton wick dipped in milk, and then from a cowrie shell shaped like a spoon. When the breast milk began to run dry, at seven months, my mother was quickly weaned so that her sister could have the last remnants.
Tulu and Bulu grew up looking strikingly similar: they had the same freckled skin, almond-shaped face, and high cheekbones, unusual among Bengalis, and a slight downward tilt of the outer edge of the eye, something that Italian painters used to make Madonnas exude a mysterious empathy. They shared an inner language, as so often happens with twins; they had jokes that only the other twin understood. They even smelled the same: when I was four or five and Bulu came to visit us, my mother, in a bait-and-switch trick that amused her endlessly, would send her sister to put me to bed; eventually, searching in the half-light for identity and difference—for the precise map of freckles on her face—I would realize that I had been fooled.
But the differences were striking, too. My mother was boisterous. She had a mercurial temper that rose fast and died suddenly, like a gust of wind in a tunnel. Bulu was physically timid yet intellectually more adventurous. Her mind was more agile, her tongue sharper, her wit more lancing. Tulu was gregarious. She made friends easily. She was impervious to insults. Bulu was reserved, quieter, and more brittle. Tulu liked theatre and dancing. Bulu was a poet, a writer, a dreamer.
….. more
Why are identical twins alike? In the late nineteen-seventies, a team of scientists in Minnesota set out to determine how much these similarities arose from genes, rather than environments—from “nature,” rather than “nurture.” Scouring thousands of adoption records and news clips, the researchers gleaned a rare cohort of fifty-six identical twins who had been separated at birth. Reared in different families and different cities, often in vastly dissimilar circumstances, these twins shared only their genomes. Yet on tests designed to measure personality, attitudes, temperaments, and anxieties, they converged astonishingly. Social and political attitudes were powerfully correlated: liberals clustered with liberals, and orthodoxy was twinned with orthodoxy. The same went for religiosity (or its absence), even for the ability to be transported by an aesthetic experience. Two brothers, separated by geographic and economic continents, might be brought to tears by the same Chopin nocturne, as if responding to some subtle, common chord struck by their genomes.
One pair of twins both suffered crippling migraines, owned dogs that they had named Toy, married women named Linda, and had sons named James Allan (although one spelled the middle name with a single “l”). Another pair—one brought up Jewish, in Trinidad, and the other Catholic, in Nazi Germany, where he joined the Hitler Youth—wore blue shirts with epaulets and four pockets, and shared peculiar obsessive behaviors, such as flushing the toilet before using it. Both had invented fake sneezes to diffuse tense moments. Two sisters—separated long before the development of language—had invented the same word to describe the way they scrunched up their noses: “squidging.” Another pair confessed that they had been haunted by nightmares of being suffocated by various metallic objects—doorknobs, fishhooks, and the like.
The Minnesota twin study raised questions about the depth and pervasiveness of qualities specified by genes: Where in the genome, exactly, might one find the locus of recurrent nightmares or of fake sneezes? Yet it provoked an equally puzzling converse question: Why are identical twins different? Because, you might answer, fate impinges differently on their bodies. One twin falls down the crumbling stairs of her Calcutta house and breaks her ankle; the other scalds her thigh on a tipped cup of coffee in a European station. Each acquires the wounds, calluses, and memories of chance and fate. But how are these changes recorded, so that they persist over the years? We know that the genome can manufacture identity; the trickier question is how it gives rise to difference.
….. more
But what turns those genes on and off, and keeps them turned on or off? Why doesn’t a liver cell wake up one morning and find itself transformed into a neuron? Allis unpacked the problem further: suppose he could find an organism with two distinct sets of genes—an active set and an inactive set—between which it regularly toggled. If he could identify the molecular switches that maintain one state, or toggle between the two states, he might be able to identify the mechanism responsible for cellular memory. “What I really needed, then, was a cell with these properties,” he recalled when we spoke at his office a few weeks ago. “Two sets of genes, turned ‘on’ or ‘off’ by some signal.”
more…
“Histones had been known as part of the inner scaffold for DNA for decades,” Allis went on. “But most biologists thought of these proteins merely as packaging, or stuffing, for genes.” When Allis gave scientific seminars in the early nineties, he recalled, skeptics asked him why he was so obsessed with the packing material, the stuff in between the DNA. …. A skein of silk tangled into a ball has very different properties from that same skein extended; might the coiling or uncoiling of DNA change the activity of genes?
In 1996, Allis and his research group deepened this theory with a seminal discovery. “We became interested in the process of histone modification,” he said. “What is the signal that changes the structure of the histone so that DNA can be packed into such radically different states? We finally found a protein that makes a specific chemical change in the histone, possibly forcing the DNA coil to open. And when we studied the properties of this protein it became quite clear that it was also changing the activity of genes.” The coils of DNA seemed to open and close in response to histone modifications—inhaling, exhaling, inhaling, like life.
Allis walked me to his lab, a fluorescent-lit space overlooking the East River, divided by wide, polished-stone benches. A mechanical stirrer, whirring in a corner, clinked on the edge of a glass beaker. “Two features of histone modifications are notable,” Allis said. “First, changing histones can change the activity of a gene without affecting the sequence of the DNA.” It is, in short, formally epi-genetic, just as Waddington had imagined. “And, second, the histone modifications are passed from a parent cell to its daughter cells when cells divide. A cell can thus record ‘memory,’ and not just for itself but for all its daughter cells.”
…..
The New Yorker screws up big time with science: researchers criticize the Mukherjee piece on epigenetics
Abstract: This is a two part-post about a science piece on gene regulation that just appeared in the New Yorker. Today I give quotes from scientists criticizing that piece; tomorrow I’ll present a semi-formal critique of the piece by two experts in the field.
esterday I gave readers an assignment: read the new New Yorkerpiece by Siddhartha Mukherjee about epigenetics. The piece, called “Same but different” (subtitle: “How epigenetics can blur the line between nature and nurture”) was brought to my attention by two readers, both of whom praised it. Mukherjee, a physician, is well known for writing the Pulitzer-Prize-winning book (2011) The Emperor of All Maladies: A Biography of Cancer. (I haven’t read it yet, but it’s on my list.) Mukherjee has a new book that will be published in May: The Gene: An Intimate History. As I haven’t seen it, the New Yorker piece may be an excerpt from this book.
Everyone I know who has read The Emperor of All Maladies gives it high praise. I wish I could say the same for Mukherjee’s New Yorker piece. When I read it at the behest of the two readers, I found his analysis of gene regulation incomplete and superficial. Although I’m not an expert in that area, I knew that there was a lot of evidence that regulatory proteins called “transcription factors”, and not “epigenetic markers” (see discussion of this term tomorrow) or modified histones—the factors emphasized by Mukherjee—played hugely important roles in gene regulation. The speculations at the end of the piece about “Lamarckian evolution” via environmentally induced epigenetic changes in the genome were also unfounded, for we have no evidence for that kind of adaptive evolution. Mukherjee does, however, mention that lack of evidence, though I wish he’d done so more strongly given that environmental modification of DNA bases is constantly touted as an important and neglected factor in evolution.
Unbeknownst to me, there was a bit of a kerfuffle going on in the community of scientists who study gene regulation, with many of them finding serious mistakes and omissions in Mukherjee’s piece. There appears to have been some back-and-forth emailing among them, and several wrote letters to the New Yorker, urging them to correct the misconceptions, omissions, and scientific errors in “Same but different.” As I understand it, both Mukherjee and the New Yorker simply batted these criticisms away, and, as far as I know, will not publish any corrections. So today and tomorrow I’ll present the criticisms here, just so they’ll be on the record.
Because Mukherjee writes very well, and because even educated laypeople won’t know the story of gene regulation revealed over the last few decades, they may not see the big lacunae in his piece. It is, then, important to set matters straight, for at least we should know what science has told us about how genes are turned on and off. The criticism of Mukherjee’s piece, coming from scientists who really are experts in gene regulation, shows a lack of care on the part of Mukherjee and theNew Yorker: both a superficial and misleading treatment of the state of the science, and a failure of the magazine to properly vet this piece (I have no idea whether they had it “refereed” not just by editors but by scientists not mentioned in the piece).
Let me add one thing about science and the New Yorker. I believe I’ve said this before, but the way the New Yorker treats science is symptomatic of the “two cultures” problem. This is summarized in an email sent me a while back by a colleague, which I quote with permission:
The New Yorker is fine with science that either serves a literary purpose (doctors’ portraits of interesting patients) or a political purpose (environmental writing with its implicit critique of modern technology and capitalism). But the subtext of most of its coverage (there are exceptions) is that scientists are just a self-interested tribe with their own narrative and no claim to finding the truth, and that science must concede the supremacy of literary culture when it comes to anything human, and never try to submit human affairs to quantification or consilience with biology. Because the magazine is undoubtedly sophisticated in its writing and editing they don’t flaunt their postmodernism or their literary-intellectual proprietariness, but once you notice it you can make sense of a lot of their material.
. . . Obviously there are exceptions – Atul Gawande is consistently superb – but as soon as you notice it, their guild war on behalf of cultural critics and literary intellectuals against scientists, technologists, and analytic scholars becomes apparent.
…. more
Researchers criticize the Mukherjee piece on epigenetics: Part 2
Trigger warning: Long science post!
Yesterday I provided a bunch of scientists’ reactions—and these were big names in the field of gene regulation—to Siddhartha Mukherjee’s ill-informed piece in The New Yorker, “Same but different” (subtitle: “How epigenetics can blur the line between nature and nurture”). Today, in part 2, I provide a sentence-by-sentence analysis and reaction by two renowned researchers in that area. We’ll start with a set of definitions (provided by the authors) that we need to understand the debate, and then proceed to the critique.
Let me add one thing to avoid confusion: everything below the line, including the definition (except for my one comment at the end) was written by Ptashne and Greally.
by Mark Ptashne and John Greally
Introduction
Ptashne is The Ludwig Professor of Molecular Biology at the Memorial Sloan Kettering Cancer Center in New York. He wrote A Genetic Switch, now in its third edition, which describes the principles of gene regulation and the workings of a ‘switch’; and, with Alex Gann, Genes and Signals, which extends these principles and ideas to higher organisms and to other cellular processes as well. John Greally is the Director of the Center for Epigenomics at the Albert Einstein College of Medicine in New York.
The New Yorker (May 2, 2016) published an article entitled “Same But Different” written by Siddhartha Mukherjee. As readers will have gathered from the letters posted yesterday, there is a concern that the article is misleading, especially for a non-scientific audience. The issue concerns our current understanding of “gene regulation” and how that understanding has been arrived at.
First some definitions/concepts:
Gene regulation refers to the “turning on and off of genes”. The primary event in turning a gene “on” is to transcribe (copy) it into messenger RNA (mRNA). That mRNA is then decoded, usually, into a specific protein. Genes are transcribed by the enzyme called RNA polymerase.
Development: the process in which a fertilized egg (e.g., a human egg) divides many times and eventually forms an organism. During this process, many of the roughly 23,000 genes of a human are turned “on” or “off” in different combinations, at different times and places in the developing organism. The process produces many different cell types in different organs (e.g. liver and brain), but all retain the original set of genes.
Transcription factors: proteins that bind to specific DNA sequences near specific genes and turn transcription of those genes on and off. A transcriptional ‘activator’, for example, bears two surfaces: one binds a specific sequence in DNA, and the other binds to, and thereby recruits to the gene, protein complexes that include RNA polymerase. It is widely acknowledged that the identity of a cell in the body depends on the array of transcription factors present in the cell, and the cell’s history. RNA molecules can also recognize specific genomic sequences, and they too sometimes work as regulators. Neither transcription factors nor these kinds of RNA molecules – the fundamental regulators of gene expression and development – are mentioned in the New Yorker article.
Signals: these come in many forms (small molecules like estrogen, larger molecules (often proteins such as cytokines) that determine the ability of transcription factors to work. For example, estrogen binds directly to a transcription factor (the estrogen receptor) and, by changing its shape, permits it to bind DNA and activate transcription.
“Memory”: a dividing cell can (often does) produce daughters that are identical, and that express identical genes as does the mother cell. This occurs because the transcription factors present in the mother cell are passively transmitted to the daughters as the cell divides, and they go to work in their new contexts as before. To make two different daughters, the cell must distribute its transcription factors asymmetrically.
PositiveFeedback: An activator can maintain its own expression by positive feedback. This requires, simply, that a copy of the DNA sequence to which the activator binds is present near its own gene. Expression of the activator then becomes self-perpetuating. The activator (of which there now are many copies in the cell) activates other target genes as it maintains its own expression. This kind of ‘memory circuit’, first described in bacteria, is found in higher organisms as well. Positive feedback can explain how a fully differentiated cell (that is, a cell that has reached its developmental endpoint) maintains its identity.
Nucleosomes: DNA in higher organisms (eukaryotes) is wrapped, like beads on a string, around certain proteins (called histones), to form nucleosomes. The histones are subject to enzymatic modifications: e.g., acetyl, methyl, phosphate, etc. groups can be added to these structures. In bacteria there are no nucleosomes, and the DNA is more or less ‘naked’.
“Epigenetic modifications”: please don’t worry about the word ”epigenetic”; it is misused in any case. What Mukherjee refers to by this term are the histone modifications mentioned above, and a modification to DNA itself: the addition of methyl groups. Keep in mind that the organisms that have taught us the most about development – flies (Drosophila) and worms (C. elegans)—do not have the enzymes required for DNA methylation. That does not mean that DNA methylation cannot do interesting things in humans, for example, but it is obviously not at the heart of gene regulation.
Specificity Development requires the highly specific sequential turning on and off of sets of genes. Transcription factors and RNA supply this specificity, but enzymes that impart modifications to histones cannot: every nucleosome (and hence every gene) appears the same to the enzyme. Thus such enzymes cannot pick out particular nucleosomes associated with particular genes to modify. Histone modifications might be imagined to convey ‘memory’ as cells divide – but there are no convincing indications that this happens, nor are there molecular models that might explain why they would have the imputed effects.
Analysis and critique of Mukherjee’s article
The picture we have just sketched has taken the combined efforts of many scientists over 50 years to develop. So what, then, is the problem with the New Yorker article?
There are two: first, the picture we have just sketched, emphasizing the primary role of transcription factors and RNA, is absent. Second, that picture is replaced by highly dubious speculations, some of which don’t make sense, and none of which has been shown to work as imagined in the article.
(Quotes from the Mukherjee article are indented and in plain text; they are followed by comments, flush left and in bold, by Ptashne and Greally.)
In 1978, having obtained a Ph.D. in biology at Indiana University, Allis began to tackle a problem that had long troubled geneticists and cell biologists: if all the cells in the body have the same genome, how does one become a nerve cell, say, and another a blood cell, which looks and functions very differently?
The problems referred to were recognized long before 1978. In fact, these were exactly the problems that the great French scientists François Jacob and Jacques Monod took on in the 1950s-60s. In a series of brilliant experiments, Jacob and Monod showed that in bacteria, certain genes encode products that regulate (turn on and off) specific other genes. Those regulatory molecules turned out to be proteins, some of which respond to signals from the environment. Much of the story of modern biology has been figuring out how these proteins – in bacteria and in higher organisms – bind to and regulate specific genes. Of note is that in higher organisms, the regulatory proteins look and act like those in bacteria, despite the fact that eukaryotic DNA is wrapped in nucleosomes whereas bacterial DNA is not. We have also learned that certain RNA molecules can play a regulatory role, a phenomenon made possible by the fact that RNA molecules, like regulatory proteins, can recognize specific genomic sequences.
In the nineteen-forties, Conrad Waddington, an English embryologist, had proposed an ingenious answer: cells acquired their identities just as humans do—by letting nurture (environmental signals) modify nature (genes). For that to happen, Waddington concluded, an additional layer of information must exist within a cell—a layer that hovered, ghostlike, above the genome. This layer would carry the “memory” of the cell, recording its past and establishing its future, marking its identity and its destiny but permitting that identity to be changed, if needed. He termed the phenomenon “epigenetics”—“above genetics.”
This description greatly misrepresents the original concept. Waddington argued that development proceeds not by the loss (or gain) of genes, which would be a “genetic” process, but rather that some genes would be selectively expressed in specific and complex cellular patterns as development proceeds. He referred to this intersection of embryology (then called “epigenesis”) and genetics as “epigenetic”.We now understand that regulatory proteins work in combinations to turn on and off genes, including their own genes, and that sometimes the regulatory proteins respond to signals sent by other cells. It should be emphasized that Waddington never proposed any “ghost-like” layer of additional information hovering above the gene. This is a later misinterpretation of a literal translation of the term epigenetics, with “epi-“ meaning “above/upon” the genetic information encoded in DNA sequence. Unfortunately, this new and pervasive definition encompasses all of transcriptional regulation and is of no practical value.
…..more
By 2000, Allis and his colleagues around the world had identified a gamut of proteins that could modify histones, and so modulate the activity of genes. Other systems, too, that could scratch different kinds of code on the genome were identified (some of these discoveries predating the identification of histone modifications). One involved the addition of a chemical side chain, called a methyl group, to DNA. The methyl groups hang off the DNA string like Christmas ornaments, and specific proteins add and remove the ornaments, in effect “decorating” the genome. The most heavily methylated parts of the genome tend to be dampened in their activity.
It is true that enzymes that modify histones have been found—lots of them. A striking problem is that, after all this time, it is not at all clear what the vast majority of these modifications do. When these enzymatic activities are eliminated by mutation of their active sites (a task substantially easier to accomplish in yeast than in higher organisms) they mostly have little or no effect on transcription. It is not even clear that histones are the biologically relevant substrates of most of these enzymes.
In the ensuing decade, Allis wrote enormous, magisterial papers in which a rich cast of histone-modifying proteins appear and reappear through various roles, mapping out a hatchwork of complexity. . . These protein systems, overlaying information on the genome, interacted with one another, reinforcing or attenuating their signals. Together, they generated the bewildering intricacy necessary for a cell to build a constellation of other cells out of the same genes, and for the cells to add “memories” to their genomes and transmit these memories to their progeny. “There’s an epigenetic code, just like there’s a genetic code,” Allis said. “There are codes to make parts of the genome more active, and codes to make them inactive.”
By ‘epigenetic code’ the author seems to mean specific arrays of nucleosome modifications, imparted over time and cell divisions, marking genes for expression. This idea has been tested in many experiments and has been found not to hold.
….. and more
Larry H. Bernstein, MD, FCAP
I hope that this piece brings greater clarity to the discussion. I have heard the use of the term “epigenetics” for over a decade. The term was never so clear. I think that the New Yorker article was a reasonable article for the intended audience. It was not intended to clarify debates about a mechanism for epigenetic based changes in evolutionary science. I think it actually punctures the “classic model” of the cell depending only on double stranded DNA and transcription, which deflates our concept of the living cell. The concept of epigenetics was never really formulated as far as I have seen, and I have done serious work in enzymology and proteins at a time that we did not have the technology that exists today. I have considered with the critics that protein folding, protein misfolding, protein interactions with proximity of polar and nonpolar groups, and the regulatory role of microRNAs that are not involved in translation, and the evolving concept of what is “dark (noncoding) DNA” lend credence to the complexity of this discussion. Even more interesting is the fact that enzymes (and isoforms of enzymes) have a huge role in cellular metabolic differences and in the function of metabolic pathways. What is less understood is the extremely fast reactions involved in these cellular reactions. These reactions are in my view critical drivers. This is brought out by Erwin Schroedinger in the book What is Life? which infers that there can be no mathematical expression of life processes.
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….
Scientists publish the first RNA interactome of the human nucleus
Reporter: Aviva Lev-Ari, PhD, RN
Studying sequence and function of DNA has been in the focus of life sciences for decades, but now the interest of many researchers has turned to the RNA. Today, many scientists believe that RNA molecules, together with a variety of different proteins, play a regulatory or structural role in virtually all cellular processes. However, the mechanisms underlying these RNA-protein interactions are still largely unknown. A team of scientists from the Max Planck Institute for Molecular Genetics in Berlin has now successfully identified hundreds of proteins that interact with RNA molecules in the nucleus of human cells. The researchers present the first RNA interactome of a human nucleus and describe how they have identified the bulk of RNA-binding proteins in the nucleus of human cells, using their newly developed method of “serial RNA interactome capturing”.
For decades, proteins have been regarded as the main functional components in living cells. However, in recent years their paramount importance for cellular processes has been rivaled by the growing knowledge about the involvement of RNA molecules. RNA in the shape of messenger RNA (mRNA) and transfer RNA (tRNA) has been believed to act as a mere mediator between the DNA, carrying the genetic information, and the proteins, being the building blocks of the cell. But for a few years it has been shown now, that in addition to being a messenger for the genomic information, the RNA mediates several other functions. These non-coding functions of RNA include tasks in the regulation of gene transcription and protein production as well as the determination of the positions of other molecules within the cell.
Elucidating the interplay between proteins and RNA has therefore become crucial for understanding the molecular mechanisms of the development of organisms and the emergence of diseases.
A research group headed by Ulf Andersson Ørom at the Max Planck Institute for Molecular Genetics in Berlin has now for the first time created an overview of the numerous interactions between RNA molecules and proteins in a human nucleus. For this, the scientists had to modify the “RNA interactome capture technique” for analyzing RNA-protein interactions at first, so that they could use it to identify the RNA-protein interactions inside specific compartments of the cell, e.g., the nucleus. With the modified method, the researchers investigated the nuclei of a total of one billion cells in order to capture and catalogue all possible RNA-protein interactions in the nucleus.
“It has been particularly interesting for us that many of the discovered RNA-binding proteins do not only control the activity of genes and the fate of the resulting RNA molecules, but are also involved in the detection and repair of damaged DNA”, Ørom explains. DNA damage such as false or missing bases or breakage of one or both strands of the DNA double helix can occur as a consequence of reactive oxygen species, UV-radiation or other external or internal stimuli. DNA-damage occurs thousands of times each day within every cell in the body. The cell responds with a complex repair process involving numerous proteins and RNA molecules that specifically repair damaged DNA, thus maintaining the functionality of the cell.
“A role for RNA in the repair of damaged DNA has been suspected for some time, but how RNA can impact on this process has remained unknown”, says Ørom. “By identifying the protein factors that link RNA to the DNA damage response, this study contributes to a better understanding of these mechanisms.” The scientists hope that their findings will contribute to a better understanding of the emergence of human diseases and the development of new therapies against cancer.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
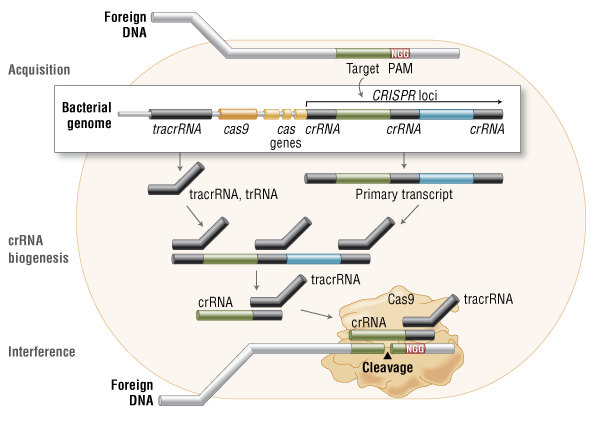{kind=link}
{kind=link}
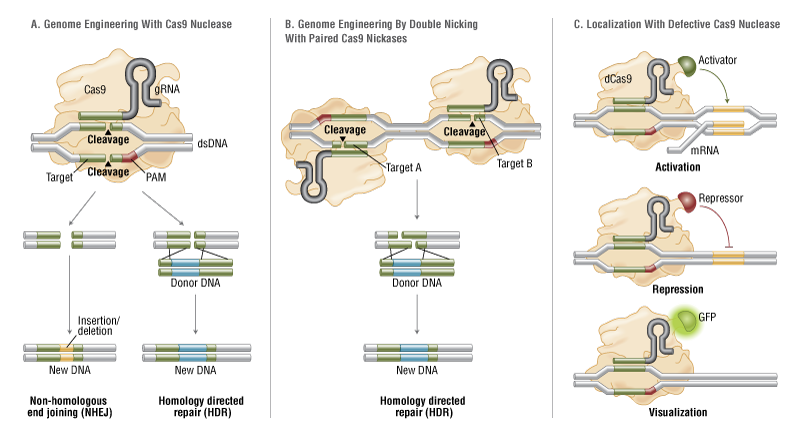{kind=link}
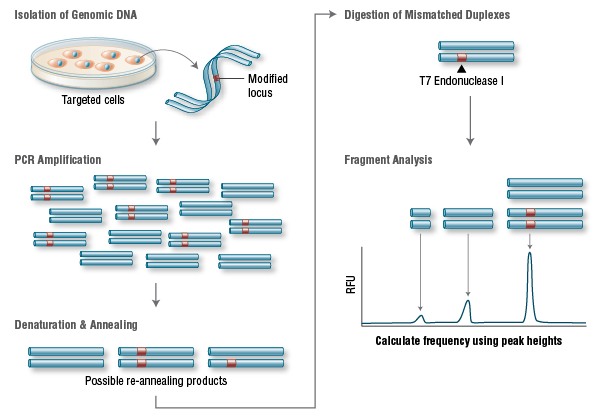{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}