
Novel Discoveries in Molecular Biology and Biomedical Science
Curator: Larry H. Bernstein, MD, FCAP
UPDATED on 6/1/2016
The following is a collection of current articles on noncoding DNA, synthetic genome engineering, protein regulation of apoptosis, drug design, and geometrics.
No longer ‘junk DNA’ — shedding light on the ‘dark matter’ of the genome
A new tool called “LIGR-Seq” enables scientists to explore in depth what non-coding RNAs actually do in human cells May 23, 2016
http://www.kurzweilai.net/no-longer-junk-dna-shedding-light-on-the-dark-matter-of-the-genome
http://www.kurzweilai.net/images/LIGR-seq-method.png
he LIGR-seq method for global-scale mapping of RNA-RNA interactions in vivo to reveal unexpected functions for uncharacterized RNAs that act via base-pairing interactions (credit: University of Toronto)
What used to be dismissed by many as “junk DNA” has now become vitally important, as accelerating genomic data points to the importance of non-coding RNAs (ncRNAs) — a genome’s messages that do not specifically code for proteins — in development and disease.
But our progress in understanding these molecules has been slow because of the lack of technologies that allow for systematic mapping of their functions.
Now, professor Benjamin Blencowe’s team at the University of Toronto’s Donnelly Centre has developed a method called “LIGR-seq” that enables scientists to explore in depth what ncRNAs do in human cells.
The study, described in Molecular Cell, was published on May 19, along with two other papers, in Molecular Cell and Cell, respectively, from Yue Wan’s group at the Genome Institute of Singapore and Howard Chang’s group at Stanford University in California, who developed similar methods to study RNAs in different organisms.
So what exactly do ncRNAs do?
http://www.kurzweilai.net/images/ncRNA.png
mRNAs vs. ncRNAs (credit: Thomas Shafee/CC)
Of the 3 billion letters in the human genome, only two per cent make up the protein-coding genes. The genes are copied, or transcribed, into messenger RNA (mRNA) molecules, which provide templates for building proteins that do most of the work in the cell. Much of the remaining 98 per cent of the genome was initially considered by some as lacking in functional importance. However, large swaths of the non-coding genome — between half and three quarters of it — are also copied into RNA.
So then what might the resulting ncRNAs do? That depends on whom you ask. Some researchers believe that most ncRNAs have no function, that they are just a by-product of the genome’s powerful transcription machinery that makes mRNA. However, it is emerging that many ncRNAs do have important roles in gene regulation — some ncRNAs act as carriages for shuttling the mRNAs around the cell, or provide a scaffold for other proteins and RNAs to attach to and do their jobs.
But the majority of available data has trickled in piecemeal or through serendipitous discovery. And with emerging evidence that ncRNAs could drive disease progression, such as cancer metastasis, there was a great need for a technology that would allow a systematic functional analysis of ncRNAs.
“Up until now, with existing methods, you had to know what you are looking for because they all require you to have some information about the RNA of interest. The power of our method is that you don’t need to preselect your candidates; you can see what’s occurring globally in cells, and use that information to look at interesting things we have not seen before and how they are affecting biology,” says Eesha Sharma, a PhD candidate in Blencowe’s group who, along with postdoctoral fellow Tim Sterne-Weiler, co-developed the method.
A new ncRNA identification tool
http://www.kurzweilai.net/images/rna-rna-interactions.jpg
The human RNA-RNA interactome, showing interactions detected by LIGR-seq (credit: University of Toronto)
The new ‘‘LIGation of interacting RNA and high-throughput sequencing’’ (LIGR-seq) tool captures interactions between different RNA molecules. When two RNA molecules have matching sequences — strings of letters copied from the DNA blueprint — they will stick together like Velcro. With LIGR-seq, the paired RNA structures are removed from cells and analyzed by state-of-the-art sequencing methods to precisely identify the RNAs that are stuck together.
“Most researchers in the life sciences agree that there’s an urgent need to understand what ncRNAs do. This technology will open the door to developing a new understanding of ncRNA function,” says Blencowe, who is also a professor in the Department of Molecular Genetics.
Not having to rely on pre-existing knowledge will boost the discovery of RNA pairs that have never been seen before. Scientists can also, for the first time, look at RNA interactions as they occur in living cells, in all their complexity, unlike in the juices of mashed up cells that they had to rely on before. This is a bit like moving on to explore marine biology from collecting shells on the beach to scuba-diving among the coral reefs, where the scope for discovery is so much bigger.
Actually, ncRNAs come in multiple flavors: there’s rRNA, tRNA, snRNA, snoRNA, piRNA, miRNA, and lncRNA, to name a few, where prefixes reflect the RNA’s place in the cell or some aspect of its function. But the truth is that no one really knows the extent to which these ncRNAs control what goes on in the cell, or how they do this.
Discoveries
Nonetheless, the new technology developed by Blencowe’s group has been able to pick up new interactions involving all classes of RNAs and has already revealed some unexpected findings.
The team discovered new roles for small nucleolar RNAs (snoRNAs), which normally guide chemical modifications of other ncRNAs. It turns out that some snoRNAs can also regulate stability of a set of protein-coding mRNAs. In this way, snoRNAs can also directly influence which proteins are made, as well as their abundance, adding a new level of control in cell biology.
And this is only the tip of the iceberg; the researchers plan to further develop and apply their technology to investigate the ncRNAs in different settings.
“We would like to understand how ncRNAs function during development. We are particularly interested in their role in the formation of neurons. But we will also use our method to discover and map changes in RNA-RNA interactions in the context of human diseases,” says Blencowe.
Abstract of Global Mapping of Human RNA-RNA Interactions
The majority of the human genome is transcribed into non-coding (nc)RNAs that lack known biological functions or else are only partially characterized. Numerous characterized ncRNAs function via base pairing with target RNA sequences to direct their biological activities, which include critical roles in RNA processing, modification, turnover, and translation. To define roles for ncRNAs, we have developed a method enabling the global-scale mapping of RNA-RNA duplexes crosslinked in vivo, “LIGation of interacting RNA followed by high-throughput sequencing” (LIGR-seq). Applying this method in human cells reveals a remarkable landscape of RNA-RNA interactions involving all major classes of ncRNA and mRNA. LIGR-seq data reveal unexpected interactions between small nucleolar (sno)RNAs and mRNAs, including those involving the orphan C/D box snoRNA, SNORD83B, that control steady-state levels of its target mRNAs. LIGR-seq thus represents a powerful approach for illuminating the functions of the myriad of uncharacterized RNAs that act via base-pairing interactions.
references:
Venter’s Research Team Creates an Artificial Cell and Reports That 32% of Genes Are Life-Essential but Contain Unknown Functions
http://www.radmailer.com/t/r-l-sttullk-ykogyktt-k/
May 27, 2016
Understanding the unknown functions of these genes may lead to the creation of new diagnostic tests for clinical laboratories and anatomic pathology groups
Once again, J. Craig Venter, PhD, is charting new ground in gene sequencing andgenomic science. This time his research team has built upon the first synthetic cell they created in 2010 to build a more sophisticated synthetic cell. Their findings from this work may give pathologists and medical laboratory scientists new tools to diagnose disease.
Recently the research team at the J. Craig Venter Institute (JCVI) and Synthetic Genomics, Inc. (SGI) published their latest findings. Among the things they learned is that science still does not understand the functions of about a third of the genes required for their synthetic cells to function.
JCVI-syn3.0 Could Radically Alter Understanding of Human Genome
Based in La Jolla, Calif., and Rockville, Md., JCVI is a not-for-profit research institute aiming to advance genomics. Building upon its first synthetic cell—Mycoplasma mycoides (M. mycoides) JCVI-syn1.0, which JCVI constructed in 2010—the same team of scientists created the first minimal synthetic bacterial cell, which they calledJCVI-syn3.0. This new artificial cell contains 531,560 base pairs and just 473 genes, which means it is the smallest genome of any organism that can be grown in laboratory media, according to a JCVI-SGI statement.
For pathologists and medical laboratory leaders, the creation of a synthetic life form is a milestone toward better understanding genome sequencing and how this new knowledge may help advance both diagnostics and therapeutics.
“What we’ve done is important because it is a step toward completely understanding how a living cell works,” Clyde Hutchison III, PhD, told New Scientist. “If we can really understand how the cell works, then we will be able to design cells efficiently for the production of pharmaceutical and other useful products.” Hutchison is Professor Emeritus of Microbiology and Immunology at the University of North Carolina at Chapel Hill, Distinguished Professor at the J. Craig Venter Institute, a member of the National Academy of Sciences, and a fellow of the American Academy of Arts and Sciences.
Clyde Hutchison, III, PhD (above), Professor Emeritus of Microbiology and Immunology at the University of North Carolina at Chapel Hill and Distinguished Professor at the J. Craig Venter Institute, stated that his team’s “goal is to have a cell for which the precise biological function of every gene is known.” (Photo credit: JCVI.)
Understanding a Gene’s True Purpose
According to the JCVI researchers, 149 genes have no known purpose. They are, however, necessary for life and health.
“We know about two-thirds of the essential biology, and we’re missing a third,” stated J. Craig Venter, PhD, Founder and CEO of JCVI, in a story published by MedPage Today.
This knowledge is based upon decades of research. JCVI seeks to create a minimal cell operating system to understand biology, while also providing what the JCVI statement called a “chassis for use in industrial applications.”
What Do these Genes Do Anyway?
The JCVI team found that among most genes’ biological functions:
- 41% are responsible for genome expressioninformation;
- 18% relate to cell membrane structure and function;
- 17% pertain to cytosolic metabolism;
- 7% suggest genome information.
“JCVI-syn3.0 is a working approximation of a minimal cellular genome—a compromise between a small genome size and a workable growth rate for an experimental organism. It retains almost all the genes that are involved in the synthesis and processing of macromolecules. Unexpectedly, it also contains 149 genes with unknown biological functions, suggesting the presence of undiscovered functions that are essential for life,” the researchers told the journal Science.
More research is needed, the scientists say, into the 149 genes that appear to lack specific biologic functions.
Unlocking Mystery of the 149 Genes Could Lead to Advances in Genomic Science
“Finding so many genes without a known function is unsettling, but it’s exciting because it’s left us with much still to learn. It’s like the ‘dark matter’ of biology,” said Alistair Elfick, PhD, Chair of Synthetic Biological Engineering, University of Edinburgh, UK, in the New Scientist article.
Studies such as JCVI’s research is key to broadening understanding and framing appropriate questions about scientific, ethical, and economic implications of synthetic biology.
The creation of a synthetic cell will have a profound and positive impact on understanding of biology and how life works, JCVI said.
Such research may inspire new whole genome synthesis tools and semi-automated processes that could dramatically affect clinical laboratory procedures. It also could lead to new techniques and tools for advanced vaccine and pharmaceuticals, JCVI pointed out.
—Donna Marie Pocius
Related Information:
CRISPR Versatility Inspires Molecular Biology Innovation
| GEN Tech Focus: CRISPR/Gene Editing |
|
||||||
| No single technique has set the molecular biology field ablaze with excitement and potential like the CRISPR-Cas9 genome editing system has following its introduction only a few short years ago. The following articles represent the flexibility of this technique to potentially treat a host of genetic disorders and possibly even prevent the onset of disease. | |||||||
| CRISPR Moves from Butchery to Surgery
Scientists recently convened at the CRISPR Precision Gene Editing Congress, held in Boston, to discuss the new technology. As with any new technique, scientists have discovered that CRISPR comes with its own set of challenges, and the Congress focused its discussion around improving specificity, efficiency, and delivery.
New CRISPR System Targets Both DNA and RNA With a staggering number of papers published in the past several years involving the characterization and use of the CRISPR/Cas9 gene editing system, it is surprising that researchers are still finding new features of the versatile molecular scissor enzyme.
High-Fidelity CRISPR-Cas9 Nucleases Virtually Free of Off-Target Noise If a Cas9 nuclease variant could be engineered that was less grabby, it might loosen its grip on DNA sequences throughout the genome—except those sequences representing on-target sites. That’s the assumption that guided a new investigation by researchers at Massachusetts General Hospital.
CRISPR Works Well but Needs Upgrades The gene-editing technology known as CRISPR-Cas9 is starting to raise expectations in the therapeutic realm. In fact, CRISPR-Cas9 and other CRISPR systems are moving so close to therapeutic uses that the technology’s ethical implications are starting to attract notice. |
A Guide to CRISPR Gene Activation
http://www.technologynetworks.com/rnai/news.aspx?ID=191776
Published: Tuesday, May 24, 2016
A comparison of synthetic gene-activating Cas9 proteins can help guide research and development of therapeutic approaches.
The CRISPR-Cas9 system has come to be known as the quintessential tool that allows researchers to edit the DNA sequences of many organisms and cell types. However, scientists are also increasingly recognizing that it can be used to activate the expression of genes. To that end, they have built a number of synthetic gene activating Cas9 proteins to study gene functions or to compensate for insufficient gene expression in potential therapeutic approaches.
“The possibility to selectively activate genes using various engineered variants of the CRISPR-Cas9 system left many researchers questioning which of the available synthetic activating Cas9 proteins to use for their purposes. The main challenge was that all had been uniquely designed and tested in different settings; there was no side-by-side comparison of their relative potentials,” said George Church, Ph.D., who is Core Faculty Member at the Wyss Institute for Biologically Inspired Engineering at Harvard University, leader of its Synthetic Biology Platform, and Professor of Genetics at Harvard Medical School. “We wanted to provide that side-by-side comparison to the biomedical research community.”
In a study published on 23 May in Nature Methods, the Wyss Institute team reports how it rigorously compared and ranked the most commonly used artificial Cas9 activators in different cell types from organisms including humans, mice and flies. The findings provide a valuable guide to researchers, allowing them to streamline their endeavors.
The team also included Wyss Core Faculty Member James Collins, Ph.D., who also is the Termeer Professor of Medical Engineering & Science and Professor of Biological Engineering at the Massachusetts Institute of Technology (MIT)’s Department of Biological Engineering and Norbert Perrimon, Ph.D., a Professor of Genetics at Harvard Medical School.
Gene activating Cas9 proteins are fused to variable domains borrowed from proteins with well-known gene activation potentials and engineered so that the DNA editing ability is destroyed. In some cases, the second component of the CRISPR-Cas9 system, the guide RNA that targets the complex to specific DNA sequences, also has been engineered to bind gene-activating factors.
“We first surveyed seven advanced Cas9 activators, comparing them to each other and the original Cas9 activator that served to provide proof-of-concept for the gene activation potential of CRISPR-Cas9. Three of them, provided much higher gene activation than the other candidates while maintaining high specificities toward their target genes,” said Marcelle Tuttle, Research Fellow at the Wyss and a co-lead author of the study.
The team went on to show that the three top candidates were comparable in driving the highest level of gene expression in cells from humans, mice and fruit flies, irrespective of their tissue and developmental origins. The researchers also pinpointed ways to further maximize gene activation employing the three leading candidates.
“In some cases, maximum possible activation of a target gene is necessary to achieve a cellular or therapeutic effect. We managed to cooperatively enhance expression of specific genes when we targeted them with three copies of a top performing activator using three different guide RNAs,” said Alejandro Chavez, Ph.D., a Postdoctoral Fellow and the study’s co-first author.
“The ease of use of CRISPR-Cas9 offers enormous potential for development of genome therapeutics. This study provides valuable new design criteria that will help enable synthetic biologists and bioengineers to develop more effective targeted genome engineering technologies in the future,” said Wyss Institute Founding Director Donald Ingber, M.D., Ph.D., who is the Judah Folkman Professor of Vascular Biology at Harvard Medical School and the Vascular Biology Program at Boston Children’s Hospital, and also Professor of Bioengineering at the Harvard John A. Paulson School of Engineering and Applied Sciences.
Engineering T Cells to Functionally Cure HIV-1 Infection
and Molecular Therapy (21 April 2015) | http://dx.doi.org:/10.1038/mt.2015.70
Despite the ability of antiretroviral therapy to minimize human immunodeficiency virus type 1 (HIV-1) replication and increase the duration and quality of patients’ lives, the health consequences and financial burden associated with the lifelong treatment regimen render a permanent cure highly attractive. Although T cells play an important role in controlling virus replication, they are themselves targets of HIV-mediated destruction. Direct genetic manipulation of T cells for adoptive cellular therapies could facilitate a functional cure by generating HIV-1–resistant cells, redirecting HIV-1–specific immune responses, or a combination of the two strategies. In contrast to a vaccine approach, which relies on the production and priming of HIV-1–specific lymphocytes within a patient’s own body, adoptive T-cell therapy provides an opportunity to customize the therapeutic T cells prior to administration. However, at present, it is unclear how to best engineer T cells so that sustained control over HIV-1 replication can be achieved in the absence of antiretrovirals. This review focuses on T-cell gene-engineering and gene-editing strategies that have been performed in efforts to inhibit HIV-1 replication and highlights the requirements for a successful gene therapy–mediated functional cure.
Automated top-down design technique simplifies creation of DNA origami nanostructures
Nanoparticles for drug delivery and cell targeting, nanoscale robots, custom-tailored optical devices, and DNA as a storage medium are among the possible applications
May 27, 2016
The boldfaced line, known as a spanning tree, follows the desired geometric shape of the target DNA origami design method, touching each vertex just once. A spanning tree algorithm is used to map out the proper routing path for the DNA strand. (credit: Public Domain)
MIT, Baylor College of Medicine, and Arizona State University Biodesign Institute researchers have developed a radical new top-down DNA origami* design method based on a computer algorithm that allows for creating designs for DNA nanostructures by simply inputting a target shape.
DNA origami (using DNA to design and build geometric structures) has already proven wildly successful in creating myriad forms in 2- and 3- dimensions, which conveniently self-assemble when the designed DNA sequences are mixed together. The tricky part is preparing the proper DNA sequence and routing design for scaffolding and staple strands to achieve the desired target structure. Typically, this is painstaking work that must be carried out manually.
The new algorithm, which is reported together with a novel synthesis approach in the journal Science, promises to eliminate all that and expands the range of possible applications of DNA origami in biomolecular science and nanotechnology. Think nanoparticles for drug delivery and cell targeting, nanoscale robots in medicine and industry, custom-tailored optical devices, and most interesting: DNA as a storage medium, offering retention times in the millions of years.**
Shape-shifting, top-down software
Unlike traditional DNA origami, in which the structure is built up manually by hand, the team’s radical top-down autonomous design method begins with an outline of the desired form and works backward in stages to define the required DNA sequence that will properly fold to form the finished product.
“The Science paper turns the problem around from one in which an expert designs the DNA needed to synthesize the object, to one in which the object itself is the starting point, with the DNA sequences that are needed automatically defined by the algorithm,” said Mark Bathe, an associate professor of biological engineering at MIT, who led the research. “Our hope is that this automation significantly broadens participation of others in the use of this powerful molecular design paradigm.”
The algorithm, which is known as DAEDALUS (DNA Origami Sequence Design Algorithm for User-defined Structures) after the Greek craftsman and artist who designed labyrinths that resemble origami’s complex scaffold structures, can build any type of 3-D shape, provided it has a closed surface. This can include shapes with one or more holes, such as a torus.
A simplified version of the top-down procedure used to design scaffolded DNA origami nanostructures. It starts with a polygon corresponding to the target shape. Software translates a wireframe version of this structure into a plan for routing DNA scaffold and staple strands. That enables a 3D DNA-based atomic-level structural model that is then validated using 3D cryo-EM reconstruction. (credit: adapted from Biodesign Institute images)
With the new technique, the target geometric structure is first described in terms of a wire mesh made up of polyhedra, with a network of nodes and edges. A DNA scaffold using strands of custom length and sequence is generated, using a “spanning tree” algorithm — basically a map that will automatically guide the routing of the DNA scaffold strand through the entire origami structure, touching each vertex in the geometric form once. Complementary staple strands are then assigned and the final DNA structural model or nanoparticle self-assembles, and is then validated using 3D cryo-EM reconstruction.
The software allows for fabricating a variety of geometric DNA objects, including 35 polyhedral forms (Platonic, Archimedean, Johnson and Catalan solids), six asymmetric structures, and four polyhedra with nonspherical topology, using inverse design principles — no manual base-pair designs needed.
To test the method, simpler forms known as Platonic solids were first fabricated, followed by increasingly complex structures. These included objects with nonspherical topologies and unusual internal details, which had never been experimentally realized before. Further experiments confirmed that the DNA structures produced were potentially suitable for biological applications since they displayed long-term stability in serum and low-salt conditions.
Biological research uses
The research also paves the way for designing nanoscale systems mimicking the properties of viruses, photosynthetic organisms, and other sophisticated products of natural evolution. One such application is a scaffold for viral peptides and proteins for use as vaccines. The surface of the nanoparticles could be designed with any combination of peptides and proteins, located at any desired location on the structure, in order to mimic the way in which a virus appears to the body’s immune system.
The researchers demonstrated that the DNA nanoparticles are stable for more than six hours in serum, and are now attempting to increase their stability further.
The nanoparticles could also be used to encapsulate the CRISPR-Cas9 gene editing tool. The CRISPR-Cas9 tool has enormous potential in therapeutics, thanks to its ability to edit targeted genes. However, there is a significant need to develop techniques to package the tool and deliver it to specific cells within the body, Bathe says.
This is currently done using viruses, but these are limited in the size of package they can carry, restricting their use. The DNA nanoparticles, in contrast, are capable of carrying much larger gene packages and can easily be equipped with molecules that help target the right cells or tissue.
The most exciting aspect of the work, however, is that it should significantly broaden participation in the application of this technology, Bathe says, much like 3-D printing has done for complex 3-D geometric models at the macroscopic scale.
Hao Yan directs the Biodesign Center for Molecular Design and Biomimetics at Arizona State University and is the Milton D. Glick Distinguished Professor, College of Liberal Arts and Sciences, School of Molecular Sciences at ASU.
* DNA origami brings the ancient Japanese method of paper folding down to the molecular scale. The basics are simple: Take a length of single-stranded DNA and guide it into a desired shape, fastening the structure together using shorter “staple strands,” which bind in strategic places along the longer length of DNA. The method relies on the fact that DNA’s four nucleotide letters—A, T, C, & G stick together in a consistent manner — As always pairing with Ts and Cs with Gs.
The DNA molecule in its characteristic double stranded form is fairly stiff, compared with single-stranded DNA, which is flexible. For this reason, single stranded DNA makes for an ideal lace-like scaffold material. Further, its pairing properties are predictable and consistent (unlike RNA).
** A single gram of DNA can store about 700 terabytes of information — an amount equivalent to 14,000 50-gigabyte Blu-ray disks — and could potentially be operated with a fraction of the energy required for other information storage options.
Essential role of miRNAs in orchestrating the biology of the tumor microenvironment
Jamie N. Frediani and Muller Fabbri
Molecular Cancer (2016) 15:42 http://dx.doi.org:/10.1186/s12943-016-0525-3
MicroRNAs (miRNAs) are emerging as central players in shaping the biology of the Tumor Microenvironment (TME). They do so both by modulating their expression levels within the different cells of the TME and by being shuttled among different cell populations within exosomes and other extracellular vesicles. This review focuses on the state-of-the-art knowledge of the role of miRNAs in the complexity of the TME and highlights limitations and challenges in the field. A better understanding of the mechanisms of action of these fascinating micro molecules will lead to the development of new therapeutic weapons and most importantly, to an improvement in the clinical outcome of cancer patients. Keywords: Exosomes, microRNAs, Tumor microenvironment, Cancer
While cancer treatment and survival have improved worldwide, the need for further understanding of the underlying tumor biology remains. In recent years, there has been a significant shift in scientific focus towards the role of the tumor microenvironment (TME) on the development, growth, and metastatic spread of malignancies. The TME is defined as the surrounding cellular environment enmeshed around the tumor cells including endothelial cells, lymphocytes, macrophages, NK cells, other cells of the immune system, fibroblasts, mesenchymal stem cells (MSCs), and the extracellular matrix (ECM). Each of these components interacts with and influences the tumor cells, continually shifting the balance between pro- and anti-tumor phenotype. One of the predominant methods of communication between these cells is through extracellular vesicles and their microRNA (miRNA) cargo. Extracellular vesicles (EVs) are between 30 nm to a few microns in diameter, are surrounded by a phospholipid bilayer membrane, and are released from a variety of cell types into the local environment. There are three well characterized groups of EVs: 1) exosomes, typically 30–100 nm, 2) microvesicles (or ectosomes), typically 100–1000 nm, and 3) large oncosomes, typically 1–10 μm. Each of these categories has a distinctly unique biogenesis and purpose in cellcell communication despite the fact that current laboratory methods do not always allow precise differentiation. EVs are found to be enriched with membrane-bound proteins, lipid raft-associated and cytosolic proteins, lipids, DNA, mRNAs, and miRNAs, all of which can be transferred to the recipient cell upon fusion to allow cell-cell communications [1]. Of these, miRNAs have been of particular interest in cancer research, both as modifiers of transcription and translation as well as direct inhibitors or enhancers of key regulatory proteins. These miRNAs are a large family of small non-coding RNAs (19–24 nucleotides) and are known to be aberrantly expressed, both in terms of content as well as number, in both the tumor cells and the cells of the TME. Synthesis of these mature miRNA is a complex process, starting with the transcription of long, capped, and polyadenylated pri-miRNA by RNA polymerase II. These are cropped into a 60–100 nucleotide hairpinstructure pre-miRNA by the microprocessor, a heterodimer of Drosha (a ribonuclease III enzyme) and DGCR8 (DiGeorge syndrome critical region gene 8). The premiRNA is then exported to the cytoplasm by exportin 5, cleaved by Dicer, and separated into single strands by helicases. The now mature miRNA are incorporated into the RNA-induced silencing complex (RISC), a cytoplasmic effector machine of the miRNA pathway. The primary mechanism of action of the mature miRNA-RISC complex is through their binding to the 3’ untranslated region, or less commonly the 5’ untranslated region, of target mRNA, leading to protein downregulation either via translational repression or mRNA degradation. More recently, it has been shown that miRNAs can also upregulate the expression of target genes [2]. MiRNA genes are mostly intergenic and are transcribed by independent promoters [3] but can also be encoded by introns, sharing the same promoter of their host gene [4]. MiRNAs undergo the same regulatory mechanisms of any other protein coding gene (promoter methylation, histone modifications, etc.…) [5, 6]. Interestingly, each miRNA may have contradictory effects both within varying tumor cell lines and within different cells of the TME. In this review, we provide a state-of-the-art description of the key role that miRNAs have in the communication between tumor cells and the TME and their subsequent effects on the malignant phenotype. Finally, this review has made every effort to clarify, whenever possible, whether the reference is to the −3p or the -5p miRNA. Whenever such clarification has not been provided, this indicates that it was not possible to infer such information from the cited bibliography.
Angiogenesis and miRNAs Cellular plasticity, critical in the development of malignancy, includes the many diverse mechanisms elicited by cancer cells to increase their malignant potential and develop increasing treatment resistance. One such mechanism, angiogenesis, is critical to the development of metastatic disease, affecting both the growth of malignant cells locally and their survival at distant sites. In the last ten years, miRNAs, often packaged in tumor cell-derived exosomes, have emerged as important contributors to the complicated regulation and balance of pro- and anti-angiogenic factors.
Most commonly, miRNAs derived from cancer cells have oncogenic activity, promoting angiogenesis and tumor growth and survival. The most-well characterized of the pro-angiogenic miRNAs, the miR-17-92 cluster encoding six miRNAs (miR-17, −18a, −19a, −19b, −20a, and −92a), is found on chromosome 13, and is highly conserved among vertebrates [7]. The complex and multifaceted functions of the miR-17-92 cluster are summarized in Fig. 1. Amplification, both at the genetic and RNA level, of miR-17-92 was initially found in several lymphoma cell lines and has subsequently been observed in multiple mouse tumor models [7].
Central role of the miR-17-92 cluster in the biology of the TME. The miR-17-92 cluster encoding miR-17, −18a, −19b, −20a, and -92a is upregulated in multiple tumor types and interacts with various components of the TME to finely “tune” the TME through a complex combination of pro- and anti-tumoral effects
Most commonly, miRNAs derived from cancer cells have oncogenic activity, promoting angiogenesis and tumor growth and survival. The most-well characterized of the pro-angiogenic miRNAs, the miR-17-92 cluster encoding six miRNAs (miR-17, −18a, −19a, −19b, −20a, and −92a), is found on chromosome 13, and is highly conserved among vertebrates [7]. The complex and multifaceted functions of the miR-17-92 cluster are summarized in Fig. 1. Amplification, both at the genetic and RNA level, of miR-17-92 was initially found in several lymphoma cell lines and has subsequently been observed in multiple mouse tumor models [7]. Up-regulation of this particular locus has further been confirmed in miRnome analysis across multiple different tumor types, including lung, breast, stomach, prostate, colon, and pancreatic cancer [8]. The miR-17-92 cluster is directly activated by Myc and modulates a variety of downstream transcription factors important in cell cycle regulation and apoptosis including activation of E2F family and Cyclin-dependent kinase inhibitor (CDKN1A) and downregulation of BCL2L11/BIM and p21 [7]. In addition to promoting cell cycle progression and inhibiting apoptosis, the miR-17-92 cluster also downregulates thrombospondin-1 (Tsp1) and connective tissue growth factor (CTGF), important antiangiogenic proteins [7]. Similarly, microvesicles from colorectal cancer cells contain miR-1246 and TGF-β which are transferred to endothelial cells to silence promyelocytic leukemia protein (PML) and activate Smad 1/5/8 signaling promoting proliferation and migration [9]. Likewise, lung cancer cell line derived microvesicles contain miR-494, in response to hypoxia, which targets PTEN in the endothelial cells promoting angiogenesis through the Akt/eNOS pathway [10]. Lastly, exosomal miR-135b from multiple myeloma cells suppresses the HIF-1/FIH-1 pathway in endothelial cells, increasing angiogenesis [11]. A summary of the studies showing the functions of exosomal miRNAs in shaping the biology of the TME is provided in Table 1.
Table 1
Actions of exosomal miRNAs exchanged between cells of the TME
|
Angiogenesis: |
|||||
|
miRNA |
Cell of origin |
Accepting cell |
Pathway/target |
Effect on TME |
Ref. |
|
miR-135b |
Multiple myeloma |
Endothelial cells |
HIF-1/FIH-1 |
↑angiogenesis |
[11] |
|
miR-494 |
Lung cancer |
Endothelial cells |
PTEN/AKT/eNOS |
↑angiogenesis |
[10] |
|
miR-503 |
Endothelial cells |
Breast cancer |
Cyclin D2 and D3 |
↓Tumor growth and invasion |
[22] |
|
miR-1246 |
Colorectal cancer |
Endothelial Cells |
PML/Smad 1/5/8 |
↑ Growth & migration |
[9] |
|
Stromal compartment: |
|||||
|
miR-105 |
Breast cancer |
Endothelial cells |
ZO-1 |
↓Tight junctions ↑Metastatic progression |
[68] |
|
miR-202-3p |
CLL |
Stromal cells |
c-fos/ATM |
↑Tumor growth |
[53] |
|
Immune system: |
|||||
|
miR-29a |
NSCLC |
TAM |
TLR8/NF-κB |
↑Growth & metastasis |
[75] |
|
miR-21 |
NSCLC |
TAM |
TLR8/NF-κB |
↑Growth & metastasis |
[75] |
|
NBL |
TAM |
TLR8/NF-κB |
↑miR-155 |
[76] |
|
|
miR-155 |
TAM |
NBL |
TERF1 |
↑ Drug resistance |
[76] |
|
miR-23a |
Hypoxic tumor derived |
NK cells |
CD107a |
↓ NK cell response |
[95] |
|
miR-210 |
|||||
|
miR-214 |
Tumor cells (various) |
Regulatory T cells |
PTEN |
↑Immunosuppression |
[96] |
|
miR-223 |
TAM |
Breast cancer |
Mef2c/β-catenin |
↑ Invasion |
[82] |
The most common target of anti-angiogenic therapy is VEGF, and not unsurprisingly, multiple miRNAs (including miR-9, miR-20b, miR-130, miR-150, and miR-497) promote angiogenesis through the induction of the VEGF pathway. The most studied of these is the up-regulation of miR-9 which has been linked to a poor prognosis in multiple tumor types, including breast cancer, non-small cell lung cancer, and melanoma [12]. The two oncogenes MYC and MYCN activate miR-9 and cause E-cadherin downregulation resulting in the upregulated transcription of VEGF [13]. In addition, miR-9 has been shown to upregulate the JAK-STAT pathway, supporting endothelial cell migration and tumor angiogenesis [13]. Both amplification of miR-20b and miR-130 as well as miR-497 suppression regulate VEGF through hypoxia inducible factor 1α (HIF-1α) supporting increased angiogenesis [14, 15, 16, 17]. …..
The pivotal discovery in 2012 by Mitra et al. laid the ground-work for our current knowledge on the interactions between tumor-derived miRNAs and fibroblasts. In combination, the down-regulation of miR-214 and miR-31 and the up-regulation of miR-155 trigger the reprogramming of quiescent fibroblasts to CAFs [32]. As expected, the reverse regulation of these miRNAs reduced the migration and invasion of co-cultured ovarian cancer cells [32]. While the pathway of miR-155’s involvement in CAF biology is still being elucidated, the pathways of miR-214 and miR-31 have been established. In endometrial cancer, miR-31 was found to target the homeobox gene SATB2, leading to enhanced tumor cell migration and invasion [33]. MiR-214 similarly has an inverse correlation with its chemokine target, C-C motif Ligand 5 (CCL5) [32]. CCL5 secretion has been associated with enhanced motility, invasion, and metastatic potential through NF-κB-mediated MMP9 activation and through generation and differentiation of myeloid-derived suppressor cells (MDSCs) [34, 35, 36]. Furthermore, miR-210 and miR-133b overexpression and miR-149 suppression have been subsequently found to independently trigger the conversion to CAFs, possibly through paracrine stimulation, and to additionally promote EMT in prostate and gastric cancer, respectively [37, 38,39]. MiR-210 additionally enlists monocytes and encourages angiogenesis [37]. …
Another function of CAFs is the destruction of the ECM and its remodeling with a tumor-supportive composition and structure which includes modulation of specific integrins and metalloproteinases as some of the most studied miRNA targets. The 23 matrix metalloproteinases (MMPs) are critical in the ECM degradation, disruption of the growth signal balance, resistance to apoptosis, establishment of a favorable metastatic niche, and promotion of angiogenesis [54]. As expected, miRNAs have been found to regulate the actions of MMPs, together working to promote cancer cell growth, invasiveness, and metastasis. In HCC, MMP2 and 9 expression is up-regulated by miR-21 via PTEN pathway downregulation. Similarly, in cholangiocarcinoma it was observed that reduced levels of miR-138 induced up-regulation of RhoC, leading to increased levels of the same two MMPs [55, 56]. ….
As has been shown throughout this review, miRNAs have an important and varied effect on human carcinogenesis by shaping the biology of the TME towards a more permissive pro-tumoral phenotype. The complex events leading to such an outcome are currently quite universally defined as the “educational” process of cancer cells on the surrounding TME. While the initial focus was on the direction from the cancer cell to the surrounding TME, increasingly interest is centered on the implications of a more dynamic bidirectional exchange of genetic information. MiRNAs represent only part of the cargo of the extracellular vesicles, but an increasing scientific literature points towards their pivotal role in creating the micro-environmental conditions for cancer cell growth and dissemination. The nearby future will have to address several questions still unanswered. First, it is absolutely necessary to clarify which miRNAs and to what extent they are involved in this process. The contradictory results of some studies can be explained by the differences in tumor-types and by different concentrations of miRNAs used for functional studies. Understanding whether different concentrations of the same miRNA elicit different target effects and therefore changes the biology of the TME, will represent a significant consideration in the development of this field. It is certainly very attractive (especially in an attempt to develop new and desperately needed better cancer biomarkers) to think that concentrations of miRNAs within the TME are reflected systemically in the circulating levels of that same miRNA, however this has not yet been irrefutably demonstrated. Moreover, the study of the paracrine interactions among different cell populations of the TME and their reciprocal effects has been limited to two, maximum three cell populations. This is still way too far from describing the complexity of the TME and only the development of new tridimensional models of the TME will be able to cast a more conclusive light on such complexity. Finally, the pharmacokinetics of miRNA-containing vesicles is in its infancy at best, and needs to be further developed if the goal is development of new therapies based on the use of exosomic miRNAs. Therefore, the future of miRNA research, particularly in its role in the TME, holds still a lot of questions that need answering. However, for these exact same reasons, this is an incredibly exciting time for research in this field. We can envision a not too far future in which these concerns will be satisfactorily addressed and our understanding of the role of miRNAs within the TME will allow us to use them as new therapeutic weapons to successfully improve the clinical outcome of cancer patients.
Triggering the protein that programs cancer cells to kill themselves
http://www.kurzweilai.net/triggering-the-protein-that-programs-cancer-cells-to-kill-themselves
May 24, 2016
https://youtu.be/DR80Huxp4y8
WEHI | Apoptosis
Researchers at the Walter and Eliza Hall Institute in Australia have discovered a new way to trigger cell death that could lead to drugs to treat cancer and autoimmune disease.
Programmed cell death (a.k.a. apoptosis) is a natural process that removes unwanted cells from the body. Failure of apoptosis can allow cancer cells to grow unchecked or immune cells to inappropriately attack the body.
The protein known as Bak is central to apoptosis. In healthy cells, Bak sits in an inert state but when a cell receives a signal to die, Bak transforms into a killer protein that destroys the cell.
Triggering the cancer-apoptosis trigger
Institute researchers Sweta Iyer, PhD, Ruth Kluck, PhD, and colleagues unexpectedly discovered that an antibody they had produced to study Bak actually bound to the Bak protein and triggered its activation. They hope to use this discovery to develop drugs that promote cell death.
The researchers used information about Bak’s three-dimensional structure to find out precisely how the antibody activated Bak. “It is well known that Bak can be activated by a class of proteins called ‘BH3-only proteins’ that bind to a groove on Bak. We were surprised to find that despite our antibody binding to a completely different site on Bak, it could still trigger activation,” Kluck said. “The advantage of our antibody is that it can’t be ‘mopped up’ and neutralized by pro-survival proteins in the cell, potentially reducing the chance of drug resistance occurring.”
Drugs that target this new activation site could be useful in combination with other therapies that promote cell death by mimicking the BH3-only proteins. The researchers are now working with collaborators to develop their antibody into a drug that can access Bak inside cells.
Their findings have just been published in the open-access journal Nature Communications. The research was supported by the National Health and Medical Research Council, the Australian Research Council, the Victorian State Government Operational Infrastructure Support Scheme, and the Victorian Life Science Computation Initiative.
Abstract of Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies
During apoptosis, Bak and Bax are activated by BH3-only proteins binding to the α2–α5 hydrophobic groove; Bax is also activated via a rear pocket. Here we report that antibodies can directly activate Bak and mitochondrial Bax by binding to the α1–α2 loop. A monoclonal antibody (clone 7D10) binds close to α1 in non-activated Bak to induce conformational change, oligomerization, and cytochrome c release. Anti-FLAG antibodies also activate Bak containing a FLAG epitope close to α1. An antibody (clone 3C10) to the Bax α1–α2 loop activates mitochondrial Bax, but blocks translocation of cytosolic Bax. Tethers within Bak show that 7D10 binding directly extricates α1; a structural model of the 7D10 Fab bound to Bak reveals the formation of a cavity under α1. Our identification of the α1–α2 loop as an activation site in Bak paves the way to develop intrabodies or small molecules that directly and selectively regulate these proteins.
references:
Catching metastatic cancer cells before they grow into tumors: a new implant shows promise
“Cure” is a word that’s dominated the rhetoric in the war on cancer for decades. But it’s a word that medical professionals tend to avoid. While the American Cancer Society reports that cancer treatment has improved markedly over the decades and the five-year survival rate is impressively high for many cancers, oncologists still refrain from declaring their cancer-free patients cured. Why?
Patients are declared cancer-free (also called complete remission) when there are no more signs of detectable disease.
However, minuscule clusters of cancer cells below the detection level can remain in a patient’s body after treatment. Moreover, such small clusters of straggler cells may undergo metastasis, where they escape from the initial tumor into the bloodstream and ultimately settle in a distant site, often a vital organ such as the lungs, liver or brain.
Cancer cells can move throughout the body, like these metastatic melanoma cells. NIH Image Gallery/Flickr, CC BY
When a colony of these metastatic cells reaches a detectable size, the patient is diagnosed with recurrent metastatic cancer. About one in three breast cancer patients diagnosed with early-stage cancer later develop metastatic disease, usually within five years of initial remission.
By the time metastatic cancer becomes evident, it is much more difficult to treat than when it was originally diagnosed.
What if these metastatic cells could be detected earlier, before they established a “foothold” in a vital organ? Better yet, could these metastatic cancer cells be intercepted, preventing them them from lodging in a vital organ in the first place?
To catch a cancer cell
With these goals in mind, our biomaterials lab joined forces with surgical oncologist Jacqueline Jeruss to create an implantable medical device that acts as a metastatic cancer cell trap.
The implant is a tiny porous polymer disc (basically a miniature sponge, no larger than a pencil eraser) that can be inserted just under a patient’s skin. Implantation triggers the immune system’s “foreign body response,” and the implant starts to soak up immune cells that travel to it. If the implant can catch mobile immune cells, then why not mobile metastatic cancer cells?
The disc can detect cancer cells in mice. Lab mouse via www.shutterstock.com.
We gave implants to mice specially bred to model metastatic breast cancer. When the mice had palpable tumors but no evidence of metastatic disease, the implant was removed and analyzed.
Cancer cells were indeed present in the implant, while the other organs (potential destinations for metastatic cells) still appeared clean. This means that the implant can be used to spot previously undetectable metastatic cancer before it takes hold in an organ.
For patients with cancer in remission, an implant that can detect tumor cells as they move through the body would be a diagnostic breakthrough. But having to remove it to see if it has captured any cancer cells is not the most convenient or pleasant detection method for human patients.
Detecting cancer cells with noninvasive imaging
There could be a way around this, though: a special imaging method under development at Northwestern University called Inverse Spectroscopic Optical Coherence Tomography (ISOCT). ISOCT detects molecular-level differences in the way cells in the body scatter light. And when we scan our implant with ISOCT, the light scatter pattern looks different when it’s full of normal cells than when cancer cells are present. In fact, the difference is apparent when even as few as 15 out of the hundreds of thousands of cells in the implant are cancer cells.
There’s a catch – ISOCT cannot penetrate deep into tissue. That means it is not a suitable imaging technology for finding metastatic cells buried deep in internal organs. However, when the cancer cell detection implant is located just under the skin, it may be possible to detect cancer cells trapped in it using ISOCT. This could offer an early warning sign that metastatic cells are on the move.
This early warning could prompt doctors to monitor their patients more closely or perform additional tests. Conversely, if no cells are detected in the implant, a patient still in remission could be spared from unneeded tests.
The ISOCT results show that noninvasive imaging of the implant is feasible. But it’s a method still under development, and thus it’s not widely available. To make scanning easier and more accessible, we’re working to adapt more ubiquitous imaging technologies like ultrasound to detect tiny quantities of tumor cells in the implant.
Detect and capture. Joseph Xu, Michigan Engineering, CC BY-NC-ND
Not just detecting, but quarantining cancer
Besides providing a way to detect tiny numbers of cancer cells before they can form new tumors in other parts of the body, our implant offers an even more intriguing possibility: diverting metastatic cells away from vital organs, and sequestering them where they cannot cause any damage.
In our mouse studies, we found that metastatic cells got caught in the implant before they were apparent in vital organs. When metastatic cells eventually made their way into the organs, the mice with implants still had significantly fewer tumor cells in their organs than implant-free controls. Thus, the implant appears to provide a therapeutic benefit, most likely by taking the metastatic cells it catches out of the circulation, preventing them from lodging anywhere vital.
Interestingly, we have not seen cancer cells leave the implant once trapped, or form a secondary tumor in the implant. Ongoing work aims to learn why this is. Whether the cells can stay safely immobilized in the implant or if it would need to be removed periodically will be important questions to answer before the implant could be used in human patients.
What the future may hold
For now, our work aims to make the implant more effective at drawing and detecting cancer cells. Since we tested the implant with metastatic breast cancer cells, we also want to see if it will work on other types of cancer. Additionally, we’re studying the cells the implant traps, and learning how the implant interacts with the body as a whole. This basic research should give us insight into the process of metastasis and how to treat it.
In the future (and it might still be far off), we envision a world where recovering cancer patients can receive a detector implant to stand guard for disease recurrence and prevent it from happening. Perhaps the patient could even scan their implant at home with a smartphone and get treatment early, when the disease burden is low and the available therapies may be more effective. Better yet, perhaps the implant could continually divert all the cancer cells away from vital organs on its own, like Iron Man’s electromagnet that deflects shrapnel from his heart.
This solution is still not a “cure.” But it would transform a formidable disease that one out of three cancer survivors would otherwise ultimately die from into a condition with which they could easily live.
New PSA Test Examines Protein Structures to Detect Prostate Cancers
5/16/2016 by Cleveland Clinic
A promising new test is detecting prostate cancer more precisely than current tests, by identifying molecular changes in the prostate specific antigen (PSA) protein, according to Cleveland Clinic research presented today at the American Urological Association annual meeting.
The study – part of an ongoing multicenter prospective clinical trial – found that the IsoPSATM test can also differentiate between high-risk and low-risk disease, as well as benign conditions.
Although widely used, the current PSA test relies on detection strategies that have poor specificity for cancer – just 25 percent of men who have a prostate biopsy due to an elevated PSA level actually have prostate cancer, according to the National Cancer Institute – and an inability to determine the aggressiveness of the disease.
The IsoPSA test, however, identifies prostate cancer in a new way. Developed by Cleveland Clinic, in collaboration with Cleveland Diagnostics, Inc., IsoPSA identifies the molecular structural changes in protein biomarkers. It is able to detect cancer by identifying these structural changes, as opposed to current tests that simply measure the protein’s concentration in a patient’s blood.
“While the PSA test has undoubtedly been one of the most successful biomarkers in history, its limitations are well known. Even currently available prostate cancer diagnostic tests rely on biomarkers that can be affected by physiological factors unrelated to cancer,” said Eric Klein, M.D., chair of Cleveland Clinic’s Glickman Urological & Kidney Institute. “These study results show that using structural changes in PSA protein to detect cancer is more effective and can help prevent unneeded biopsies in low-risk patients.”
The clinical trial involves six healthcare institutions and 132 patients, to date. It examined the ability of IsoPSA to distinguish patients with and without biopsy-confirmed evidence of cancer. It also evaluated the test’s precision in differentiating patients with high-grade (Gleason = 7) cancer from those with low-grade (Gleason = 6) disease and benign findings after standard ultrasound-guided biopsy of the prostate.
Substituting the IsoPSA structure-based composite index for the standard PSA resulted in improvement in diagnostic accuracy. Compared with serum PSA testing, IsoPSA performed better in both sensitivity and specificity.
“We took an ‘out of the box’ approach that has shown success in detecting prostate cancer but also has the potential to address other clinically important questions such as clinical surveillance of patients after treatment,” said Mark Stovsky, M.D., staff member, Cleveland Clinic Glickman Urological & Kidney Institute’s Department of Urology. Stovsky has a leadership position (Chief Medical Officer) and investment interest in Cleveland Diagnostics, Inc. “In general, the clinical utility of prostate cancer early detection and screening tests is often limited by the fact that biomarker concentrations may be affected by physiological processes unrelated to cancer, such as inflammation, as well as the relative lack of specificity of these biomarkers to the cancer phenotype. In contrast, clinical research data suggests that the IsoPSA assay can interrogate the entire PSA isoform distribution as a single stand-alone diagnostic tool which can reliably identify structural changes in the PSA protein that correlate with the presence or absence and aggressiveness of prostate cancer.”
Point of Care, Highly Accurate Cervical Cancer Screening
5/20/2016 by Avi Rosenzweig, VP of Business Development, Biop Medical
http://www.mdtmag.com/article/2016/05/point-care-highly-accurate-cervical-cancer-screening
Fifty-five million times a year, American women go to their gynecologist for a Pap Smear. After waiting a few weeks for the results, more than 3.5 million of them are called back to the physician for a follow up visualization of the cervix. Beyond the stress related to possibly having cancer, the women are then subjected to a colposcopic exam, and all too often, a painful biopsy. Then more stressful waiting for a final diagnosis from the pathologist.
Cervical cancer develops slowly, allowing for successful treatment, when identified on time. Regions with high screening compliancy have low mortality rates from this cancer. In the US, for instance, where screening rates are close to 90%, only 4,200 women die from cervical cancer, annually, or 2.6 women per 100,000. However, the screening process in the developed world is long, complicated and not optimized.
In developing regions however, cervical cancer is a leading cause of women death. Over 85% of the total deaths from this cancer are in developing countries. Regions suffering from low screening rates include not only Africa, India and China, but many Eastern European countries as well. According to an OECD report from 2014, the cervical cancer screening rates in Romania and Hungary are as low as 14.6% and 36.7% respectively. The mortality rates in these countries are high, 16 in 100,000 women in Romania and 7.7 in 100,000 in Hungary.
The current screening process for cervical cancer detection is long, beginning with a Pap or HPV test. Cytology results take weeks to receive. A positive result requires follow-up testing by colposcopy and often biopsy. In countries where there is little access to medical care, or where screening compliancy is low, the chances of successful detection via this multi-step process are small. Developing regions and non-compliant countries require a point of care diagnostic method, which eliminates the need for return visits.
Additional limitations to cervical cancer screening are the low sensitivity and specificity rates of Pap tests and the high false positive rates of HPV test, leading to unnecessary colposcopies. Both cytology and colposcopy testing are highly dependent on operator proficiency for accurate diagnosis.
Biop has developed a new technology for the optimization of this process, into one, three minute, painless optical scan. The vaginal probe uses advanced optical, imaging and non-imaging technologies to identify and classify epithelium based cancers and pre-cancerous lesions. The probe is inserted into the vaginal canal, and scans the entire cervix. The resulting images and optical signatures created from the light, and captured by the sensors, are analyzed by the proprietary algorithm. The result is two pictures, on the physician’s screen; a high resolution photograph of the patient’s cervix, immediately next to a hot/cold map indicating a precise classification and location of any diseased lesions.
Deep learning applied to drug discovery and repurposing
May 27, 2016 http://www.kurzweilai.net/deep-learning-applied-to-drug-discovery-and-repurposing
Deep neural networks for drug discovery (credit: Insilico Medicine, Inc.)
Scientists from Insilico Medicine, Inc. have trained deep neural networks (DNNs) to predict the potential therapeutic uses of 678 drugs, using gene-expression data obtained from high-throughput experiments on human cell lines from Broad Institute’s LINCS databases and NIH MeSH databases.
The supervised deep-learning drug-discovery engine used the properties of small molecules, transcriptional data, and literature to predict efficacy, toxicity, tissue-specificity, and heterogeneity of response.
“We used LINCS data from Broad Institute to determine the effects on cell lines before and after incubation with compounds, co-author and research scientist Polina Mamoshina explained to KurzweilIAI.
“We used gene expression data of total mRNA from cell lines extracted and measured before incubation with compound X and after incubation with compound X to identify the response on a molecular level. The goal is to understand how gene expression (the transcriptome) will change after drug uptake. It is a differential value, so we need a reference (molecular state before incubation) to compare.”
The research is described in a paper in the upcoming issue of the journal Molecular Pharmaceutics.
Helping pharmas accelerate R&D
Alex Zhavoronkov, PhD, Insilico Medicine CEO, who coordinated the study, said the initial goal of their research was to help pharmaceutical companies significantly accelerate their R&D and increase the number of approved drugs. “In the process we came up with more than 800 strong hypotheses in oncology, cardiovascular, metabolic, and CNS spaces and started basic validation,” he said.
The team measured the “differential signaling pathway activation score for a large number of pathways to reduce the dimensionality of the data while retaining biological relevance.” They then used those scores to train the deep neural networks.*
“This study is a proof of concept that DNNs can be used to annotate drugs using transcriptional response signatures, but we took this concept to the next level,” said Alex Aliper, president of research, Insilico Medicine, Inc., lead author of the study.
Via Pharma.AI, a newly formed subsidiary of Insilico Medicine, “we developed a pipeline for in silico drug discovery — which has the potential to substantially accelerate the preclinical stage for almost any therapeutic — and came up with a broad list of predictions, with multiple in silico validation steps that, if validated in vitro and in vivo, can almost double the number of drugs in clinical practice.”
Despite the commercial orientation of the companies, the authors agreed not to file for intellectual property on these methods and to publish the proof of concept.
Deep-learning age biomarkers
According to Mamoshina, earlier this month, Insilico Medicine scientists published the first deep-learned biomarker of human age — aiming to predict the health status of the patient — in a paper titled “Deep biomarkers of human aging: Application of deep neural networks to biomarker development” by Putin et al, in Aging; and an overview of recent advances in deep learning in a paper titled “Applications of Deep Learning in Biomedicine” by Mamoshina et al., also in Molecular Pharmaceutics.
Insilico Medicine is located in the Emerging Technology Centers at Johns Hopkins University in Baltimore, Maryland, in collaboration with Datalytic Solutions and Mind Research Network.
* In this study, scientists used the perturbation samples of 678 drugs across A549, MCF-7 and PC-3 cell lines from the Library of Integrated Network-Based Cellular Signatures (LINCS) project developed by the National Institutes of Health (NIH) and linked those to 12 therapeutic use categories derived from MeSH (Medical Subject Headings) developed and maintained by the National Library of Medicine (NLM) of the NIH.
To train the DNN, scientists utilized both gene level transcriptomic data and transcriptomic data processed using a pathway activation scoring algorithm, for a pooled dataset of samples perturbed with different concentrations of the drug for 6 and 24 hours. Cross-validation experiments showed that DNNs achieve 54.6% accuracy in correctly predicting one out of 12 therapeutic classes for each drug.
One peculiar finding of this experiment was that a large number of drugs misclassified by the DNNs had dual use, suggesting possible application of DNN confusion matrices in drug repurposing.
FutureTechnologies Media Group | Video presentation Insilico medicine
Abstract of Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data
Deep learning is rapidly advancing many areas of science and technology with multiple success stories in image, text, voice and video recognition, robotics and autonomous driving. In this paper we demonstrate how deep neural networks (DNN) trained on large transcriptional response data sets can classify various drugs to therapeutic categories solely based on their transcriptional profiles. We used the perturbation samples of 678 drugs across A549, MCF-7 and PC-3 cell lines from the LINCS project and linked those to 12 therapeutic use categories derived from MeSH. To train the DNN, we utilized both gene level transcriptomic data and transcriptomic data processed using a pathway activation scoring algorithm, for a pooled dataset of samples perturbed with different concentrations of the drug for 6 and 24 hours. When applied to normalized gene expression data for “landmark genes,” DNN showed cross-validation mean F1 scores of 0.397, 0.285 and 0.234 on 3-, 5- and 12-category classification problems, respectively. At the pathway level DNN performed best with cross-validation mean F1 scores of 0.701, 0.596 and 0.546 on the same tasks. In both gene and pathway level classification, DNN convincingly outperformed support vector machine (SVM) model on every multiclass classification problem. For the first time we demonstrate a deep learning neural net trained on transcriptomic data to recognize pharmacological properties of multiple drugs across different biological systems and conditions. We also propose using deep neural net confusion matrices for drug repositioning. This work is a proof of principle for applying deep learning to drug discovery and development.
references:
Transistor-based biosensor detects molecules linked to cancer, Alzheimer’s, and Parkinson’s
May 23, 2016 http://www.kurzweilai.net/transistor-based-biosensor-detects-molecules-linked-to-cancer-alzheimers-and-parkinsons
An inexpensive portable biosensor developed by researchers at Brazil’s National Nanotechnology Laboratory (credit: LNNano) http://www.kurzweilai.net/images/Biosensor-LNNano.jpg
A novel nanoscale organic transistor-based biosensor that can detect molecules associated with neurodegenerative diseases and some types of cancer has been developed by researchers at the National Nanotechnology Laboratory (LNNano) in Brazil.
The transistor, mounted on a glass slide, contains the reduced form of the peptide glutathione (GSH), which reacts in a specific way when it comes into contact with the enzyme glutathione S-transferase (GST), linked to Parkinson’s, Alzheimer’s and breast cancer, among other diseases.
http://www.kurzweilai.net/images/CuPc-transistor.png
Sensitive water-gated copper phthalocyanine (CuPc) thin-film transistor (credit: Rafael Furlan de Oliveira et al./Organic Electronics)
“The device can detect such molecules even when they’re present at very low levels in the examined material, thanks to its nanometric sensitivity,” explained Carlos Cesar Bof Bufon, Head of LNNano’s Functional Devices & Systems Lab (DSF).
Bufon said the system can be adapted to detect other substances by replacing the analytes (detection compounds). The team is working on paper-based biosensors to further lower the cost, improve portability, and facilitate fabrication and disposal.
The research is published in the journal Organic Electronics.
Abstract of Water-gated phthalocyanine transistors: Operation and transduction of the peptide–enzyme interaction
The use of aqueous solutions as the gate medium is an attractive strategy to obtain high charge carrier density (1012 cm−2) and low operational voltages (<1 V) in organic transistors. Additionally, it provides a simple and favorable architecture to couple both ionic and electronic domains in a single device, which is crucial for the development of novel technologies in bioelectronics. Here, we demonstrate the operation of transistors containing copper phthalocyanine (CuPc) thin-films gated with water and discuss the charge dynamics at the CuPc/water interface. Without the need for complex multilayer patterning, or the use of surface treatments, water-gated CuPc transistors exhibited low threshold (100 ± 20 mV) and working voltages (<1 V) compared to conventional CuPc transistors, along with similar charge carrier mobilities (1.2 ± 0.2) x 10−3 cm2 V−1 s−1. Several device characteristics such as moderate switching speeds and hysteresis, associated with high capacitances at low frequencies upon bias application (3.4–12 μF cm−2), indicate the occurrence of interfacial ion doping. Finally, water-gated CuPc OTFTs were employed in the transduction of the biospecific interaction between tripeptide reduced glutathione (GSH) and glutathione S-transferase (GST) enzyme, taking advantage of the device sensitivity and multiparametricity.
references:
First Large-Scale Proteogenomic Study of Breast Cancer
Tues, May 31, 2016 http://www.technologynetworks.com/rnai/news.aspx?ID=191934
The study offers understanding of potential therapeutic targets.
Building on data from The Cancer Genome Atlas (TCGA) project, a multi-institutional team of scientists have completed the first large-scale “proteogenomic” study of breast cancer, linking DNA mutations to protein signaling and helping pinpoint the genes that drive cancer. Conducted by members of the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC), including Baylor College of Medicine, Broad Institute of MIT and Harvard, Fred Hutchinson Cancer Research Center, New York University Langone Medical Center, and Washington University School of Medicine, the study takes aim at proteins, the workhorses of the cell, and their modifications to better understand cancer.
Appearing in the Advance Online Publication of Nature, the study illustrates the power of integrating genomic and proteomic data to yield a more complete picture of cancer biology than either analysis could do alone. The effort produced a broad overview of the landscape of the proteome (all the proteins found in a cell) and the phosphoproteome (the sites at which proteins are tagged by phosphorylation, a chemical modification that drives communication in the cell) across a set of 77 breast cancer tumors that had been genomically characterized in the TCGA project. Although the TCGA produced an extensive catalog of somatic mutations found in cancer, the effects of many of those mutations on cellular functions or patients’ outcomes are unknown.
In addition, not all mutated genes are true “drivers” of cancer — some are merely “passenger” mutations that have little functional consequence. And some mutations are found within very large DNA regions that are deleted or present in extra copies, so winnowing the list of candidate genes by studying the activity of their protein products can help identify therapeutic targets. “We don’t fully understand how complex cancer genomes translate into the driving biology that causes relapse and mortality,” said Matthew Ellis, director of the Lester and Sue Smith Breast Center at Baylor College of Medicine and a senior author of the paper.
“These findings show that proteogenomic integration could one day prove to be a powerful clinical tool, allowing us to traverse the large knowledge gap between cancer genomics and clinical action.” In this study, the researchers at the Broad Institute analyzed breast tumors using accurate mass, high-resolution mass spectrometry, a technology that extends the coverage of the proteome far beyond the coverage that can be achieved by traditional antibody-based methods. This allowed them to scale their efforts and quantify more than 12,000 proteins and 33,000 phosphosites, an extremely deep level of coverage.
Breakthrough Approach to Breast Cancer Treatment
May 24, 2016 http://www.technologynetworks.com/rnai/news.aspx?ID=191771
Scripps scientists have designed a drug candidate that decreases growth of breast cancer cells.
In a development that could lead to a new generation of drugs to precisely treat a range of diseases, scientists from the Florida campus of The Scripps Research Institute (TSRI) have for the first time designed a drug candidate that decreases the growth of tumor cells in animal models in one of the hardest to treat cancers—triple negative breast cancer.
“This is the first example of taking a genetic sequence and designing a drug candidate that works effectively in an animal model against triple negative breast cancer,” said TSRI Professor Matthew Disney. “The study represents a clear breakthrough in precision medicine, as this molecule only kills the cancer cells that express the cancer-causing gene—not healthy cells. These studies may transform the way the lead drugs are identified—by using the genetic makeup of a disease.”
The study, published by the journal Proceedings of the National Academy of Sciences, demonstrates that the Disney lab’s compound, known as Targaprimir-96, triggers breast cancer cells to kill themselves via programmed cell death by precisely targeting a specific RNA that ignites the cancer.
Short-Cut to Drug Candidates
While the goal of precision medicine is to identify drugs that selectively affect disease-causing biomolecules, the process has typically involved time-consuming and expensive high-throughput screens to test millions of potential drug candidates to identify those few that affect the target of interest. Disney’s approach eliminates these screens.
The new study uses the lab’s computational approach called Inforna, which focuses on developing designer compounds that bind to RNA folds, particularly microRNAs.
MicroRNAs are short molecules that work within all animal and plant cells, typically functioning as a “dimmer switch” for one or more genes, binding to the transcripts of those genes and preventing protein production. Some microRNAs have been associated with diseases. For example, microRNA-96, which was the target of the new study, promotes cancer by discouraging programmed cell death, which can rid the body of cells that grow out of control.
In the new study, the drug candidate was tested in animal models over a 21-day course of treatment. Results showed decreased production of microRNA-96 and increased programmed cell death, significantly reducing tumor growth. Since targaprimir-96 was highly selective in its targeting, healthy cells were unaffected.
In contrast, Disney noted, a typical cancer therapeutic targets and kills cells indiscriminately, often leading to side effects that can make these drugs difficult for patients to tolerate.
Benjamin Zealley and Aubrey D.N.J. de Grey
Commentary on Some Recent Theses Relevant to Combating Aging: June 2015
REJUVENATION RESEARCH 2015; 18(3), 282 – 287 http://dx.doi.org:/10.1089/rej.2015.1728
Cancer Autoantibody Biomarker Discovery and Validation Using Nucleic Acid Programmable Protein Array
Jie Wang, PhD, Arizona State University
Currently in the United States, many patients with cancer do not benefit from population-based screening due to challenges associated with the existing cancer screening scheme. Blood-based diagnostic assays have the potential to detect diseases in a non-invasive way. Proteins released from small early tumors may only be present intermittently and are diluted to tiny concentrations in the blood, making them difficult to use as biomarkers. However, they can induce autoantibody (AAb) responses, which can amplify the signal and persist in the blood even if the antigen is gone. Circulating autoantibodies are a promising class of molecules that have the potential to serve as early detection biomarkers for cancers. This PhD thesis aims to screen for autoantibody biomarkers for the early detection of two deadly cancers, basal-like breast cancer and lung adenocarcinoma. First, a method was developed to display proteins in both native and denatured conformations on a protein array. This method adopted a novel protein tag technology, called a HaloTag, to immobilize proteins covalently on the surface of a glass slide. The covalent attachment allowed these proteins to endure harsh treatment without becoming dissociated from the slide surface, which enabled the profiling of antibody responses against both conformational and linear epitopes. Next, a plasma screening protocol was optimized to increase significantly the signal-to-noise ratio of protein array–based AAb detection. Following this, the AAb responses in basal-like breast cancer were explored using nucleic acid programmable protein arrays (NAPPA) containing 10,000 full-length human proteins in 45 cases and 45 controls. After verification in a large sample set (145 basal-like breast cancer cases, 145 controls, 70 non-basal breast cancer) by enzyme-linked immunosorbent assay (ELISA), a 13-AAb classifier was developed to differentiate patients from controls with a sensitivity of 33% at 98% specificity. A similar approach was also applied to the lung cancer study to identify AAbs that distinguished lung cancer patients from computed tomography–positive benign pulmonary nodules (137 lung cancer cases, 127 smoker controls, 170 benign controls). In this study, two panels of AAbs were discovered that showed promising sensitivity and specificity. Six out of eight AAb targets were also found to have elevated mRNA levels in lung adenocarcinoma patients using TCGA data. These projects as a whole provide novel insights into the association between AAbs and cancer, as well as general B cell antigenicity against self-proteins.
Comment: There are two widely supported models for cancer development and progression—the clonal evolution (CE) model and the cancer stem cell (CSC) model. Briefly, the former claims that most or all cells in a tumor contribute to its maintenance; as newer and more aggressive clones develop by random mutation, they become responsible for driving growth. The range of different mutational profiles generated is assumed to be large enough to account for disease recurrence after therapy (due to rare resistant clones) and metastasis (clones arising with the ability to travel to distant sites). The CSC model instead asserts that a small number of mutated stem cells are the origin of the primary cell mass, drive metastasis through the intermittent release of undifferentiated, highly mobile progeny, and account for recurrence due to a generally quiescent metabolic profile conferring potent resistance to chemotherapy. In either case, the immunological visibility of an early tumor may be highly sporadic. Clones arising early in CE differ little in proteomic terms from healthy host cells; those that do trigger a response are unlikely to have acquired robust resistance to immune attack, so are destroyed quickly in favor of their stealthier brethren. Likewise, CSCs share some of the immune privilege of normal stem cells and, due to their inherent ability to produce differentiated progeny with distinct proteomic signatures, are partially protected from attacks on their descendants. Consequently, such well-hidden cells may remain in the body for years to decades. The autoantibody panel developed in this study for basal-like breast cancer exhibits exceptional specificity despite a comparatively small training set. Given its ease of application, this suggests great promise for a more exhaustively trained classifier as a populationlevel screening tool.
Condition-Specific Differential Sub-Network Analysis for Biological Systems
Deepali Jhamb, PhD, Indiana University
Biological systems behave differently under different conditions. Advances in sequencing technology over the last decade have led to the generation of enormous amounts of condition-specific data. However, these measurements often fail to identify low-abundance genes and proteins that can be biologically crucial. In this work, a novel textmining system was first developed to extract condition-specific proteins from the biomedical literature. The literaturederived data was then combined with proteomics data to construct condition-specific protein interaction networks. Furthermore, an innovative condition-specific differential analysis approach was designed to identify key differences, in the form of sub-networks, between any two given biological systems. The framework developed here was implemented to understand the differences between limb regenerationcompetent Ambystoma mexicanum and regeneration-deficient Xenopus laevis. This study provides an exhaustive systems-level analysis to compare regeneration competent and deficient sub-networks to show how different molecular entities inter-connect with each other and are rewired during the formation of an accumulation blastema in regenerating axolotl limbs. This study also demonstrates the importance of literature-derived knowledge, specific to limb regeneration, to augment the systems biology analysis. Our findings show that although the proteins might be common between the two given biological conditions, they can have a high dissimilarity based on their biological and topological properties in the sub-network. The knowledge gained from the distinguishing features of limb regeneration in amphibians can be used in future to induce regeneration chemically in mammalian systems. The approach developed in this dissertation is scalable and adaptable to understanding differential sub-networks between any two biological systems. This methodology will not only facilitate the understanding of biological processes and molecular functions that govern a given system, but will also provide novel intuitions about the pathophysiology of diseases/conditions.
Comment: We have long advocated a principle of directly comparing young and old bodies as a means to identify the classes of physical damage that accumulate in the body during aging. This approach circumvents our ignorance of the full etiology of each particular disease manifestation, a phenomenally difficult question given the ethical issues of experimenting on human subjects, the lengthy ‘‘incubation time’’ of aging-related diseases, and the complex interconnections between their risk factors—innate and environmental. Repairing such damage has the potential to prevent pathology before symptoms appear, an approach now becoming increasingly mainstream.11 However, a naı¨ve comparison faces a number of difficulties, even given a sufficiently large sample set to compensate for inter-individual variation. Most importantly, the causal significance of a given species cannot be reliably determined from its simple prevalence.12 The catalytic nature of cell biology means that those entities whose abundance changes the most profoundly in absolute terms are quite unlikely to be the drivers of that change and may even spontaneously revert to baseline levels in the absence of on-going stimulation. Meanwhile, functionality is often heavily influenced independently of abundance by post-translational modifications that may escape direct detection. Sub-network analysis uses computational means to identify groups of genes and/or proteins that vary in a synchronized way with some parameter, indicating functional connectivity. The application of methods such as those developed here to the comparison of a wide range of younger and older conditions will facilitate the identification of processes—not merely individual factors—that are impaired with age, and thus will help greatly in identifying the optimal points for intervention.
Development of a Light Actuated Drug Delivery-on-Demand System
Chase Linsley, PhD, University of California, Los Angeles
The need for temporal–spatial control over the release of biologically active molecules has motivated efforts to engineer novel drug delivery-on-demand strategies actuated via light irradiation. Many systems, however, have been limited to in vitro proof-of-concept due to biocompatibility issues with the photo-responsive moieties or the light wavelength, intensity, and duration. To overcome these limitations, the objective of this dissertation was to design a light-actuated drug delivery-on-demand strategy that uses biocompatible chromophores and safe wavelengths of light, thereby advancing the clinical prospects of light-actuated drug delivery-on-demand systems. This was achieved by: (1) Characterizing the photothermal response of biocompatible visible light and near-infrared-responsive chromophores and demonstrating the feasibility and functionality of the light actuated on-demand drug delivery system in vitro; and (2) designing a modular drug delivery-on-demand system that could control the release of biologically active molecules over an extended period of time. Three biocompatible chromophores—Cardiogreen, Methylene Blue, and riboflavin—were identified and demonstrated significant photothermal response upon exposure to near-infrared and visible light, and the amount of temperature change was dependent upon light intensity, wavelength, as well as chromophore concentration. As a proof-of-concept, pulsatile release of a model protein from a thermally responsive delivery vehicle fabricated from poly(N-isopropylacrylamide) was achieved over 4 days by loading the delivery vehicle with Cardiogreen and irradiating with near-infrared light. To extend the useful lifetime of the light-actuated drug delivery-on-demand system, a modular, reservoir-valve system was designed. Using poly(ethylene glycol) as a reservoir for model small molecule drugs combined with a poly(N-isopropylacrylamide) valve spiked with chromophore-loaded liposomes, pulsatile release was achieved over 7 days upon light irradiation. Ultimately, this drug delivery strategy has potential for clinical applications that require explicit control over the presentation of biologically active molecules. Further research into the design and fabrication of novel biocompatible thermally responsive delivery vehicles will aid in the advancement of the light-actuated drug delivery-on-demand strategy described here. Comment: Our combined comments on this thesis and the next one appear after the next abstract.
Light-Inducible Gene Regulation in Mammalian Cells
Lauren Toth, PhD, Duke University
The growing complexity of scientific research demands further development of advanced gene regulation systems. For instance, the ultimate goal of tissue engineering is to develop constructs that functionally and morphologically resemble the native tissue they are expected to replace. This requires patterning of gene expression and control of cellular phenotype within the tissue-engineered construct. In the field of synthetic biology, gene circuits are engineered to elucidate mechanisms of gene regulation and predict the behavior of more complex systems. Such systems require robust gene switches that can quickly turn gene expression on or off. Similarly, basic science requires precise genetic control to perturb genetic pathways or understand gene function. Additionally, gene therapy strives to replace or repair genes that are responsible for disease. The safety and efficacy of such therapies require control of when and where the delivered gene is expressed in vivo.
Unfortunately, these fields are limited by the lack of gene regulation systems that enable both robust and flexible cellular control. Most current gene regulation systems do not allow for the manipulation of gene expression that is spatially defined, temporally controlled, reversible, and repeatable. Rather, they provide incomplete control that forces the user to choose to control gene expression in either space or time, and whether the system will be reversible or irreversible. The recent emergence of the field of optogenetics—the ability to control gene expression using light—has made it possible to regulate gene expression with spatial, temporal, and dynamic control. Light-inducible systems provide the tools necessary to overcome the limitations of other gene regulation systems, which can be slow, imprecise, or cumbersome to work with. However, emerging light-inducible systems require further optimization to increase their efficiency, reliability, and ease of use.
Initially, we engineered a light-inducible gene regulation system that combines zinc finger protein technology and the light-inducible interaction between Arabidopsis thaliana plant proteins GIGANTEA (GI) and the light oxygen voltage (LOV) domain of FKF1. Zinc finger proteins (ZFPs) can be engineered to target almost any DNA sequence through tandem assembly of individual zinc finger domains that recognize a specific 3-bp DNA sequence. Fusion of three different ZFPs to GI (GI-ZFP) successfully targeted the fusion protein to the specific DNA target sequence of the ZFP. Due to the interaction between GI and LOV, co-expression of GI-ZFP with a fusion protein consisting of LOV fused to three copies of the VP16 transactivation domain (LOV-VP16) enabled blue-light dependent recruitment of LOV-VP16 to the ZFP target sequence. We showed that placement of three to nine copies of a ZFP target sequence upstream of a luciferase or enhanced green fluorescent protein (eGFP) transgene enabled expression of the transgene in response to blue light. Gene activation was both reversible and tunable on the basis of duration of light exposure, illumination intensity, and the number of ZFP binding sites upstream of the transgene. Gene expression could also be patterned spatially by illuminating the cell culture through photomasks containing various patterns.
Although this system was useful for controlling the expression of a transgene, for many applications it is useful to control the expression of a gene in its natural chromosomal position. Therefore, we capitalized on recent advances in programmed gene activation to engineer an optogenetic tool that could easily be targeted to new, endogenous DNA sequences without re-engineering the light inducible proteins. This approach took advantage of CRISPR/Cas9 technology, which uses a gene-specific guide RNA (gRNA) to facilitate Cas9 targeting and binding to a desired sequence, and the light-inducible heterodimerizers CRY2 and CIB1 from Arabidopsis thaliana to engineer a lightactivated CRISPR/Cas9 effector (LACE) system. We fused the full-length (FL) CRY2 to the transcriptional activator VP64 (CRY2FL-VP64) and the amino-terminal fragment of CIB1 to the amino, carboxyl, or amino and carboxyl terminus of a catalytically inactive Cas9. When CRY2-VP64 and one of the CIBN/dCas9 fusion proteins are expressed with a gRNA, the CIBN/dCas9 fusion protein localizes to the gRNA target. In the presence of blue light, CRY2FL binds to CIBN, which translocates CRY2FL-VP64 to the gene target and activates transcription. Unlike other optogenetic systems, the LACE system can be targeted to new endogenous loci by solely manipulating the specificity of the gRNA without having to re-engineer the light-inducible proteins. We achieved light-dependent activation of the IL1RN, HBG1/2, or ASCL1 genes by delivery of the LACE system and four gene-specific gRNAs per promoter region. For some gene targets, we achieved equivalent activation levels to cells that were transfected with the same gRNAs and the synthetic transcription factor dCas9-VP64. Gene activation was also shown to be reversible and repeatable through modulation of the duration of blue light exposure, and spatial patterning of gene expression was achieved using an eGFP reporter and a photomask.
Finally, we engineered a light-activated genetic ‘‘on’’ switch (LAGOS) that provides permanent gene expression in response to an initial dose of blue light illumination. LAGOS is a lentiviral vector that expresses a transgene only upon Cre recombinase–mediated DNA recombination. We showed that this vector, when used in conjunction with a light-inducible Cre recombinase system, could be used to express MyoD or the synthetic transcription factor VP64- MyoD in response to light in multiple mammalian cell lines, including primary mouse embryonic fibroblasts. We achieved light-mediated up-regulation of downstream myogenic markers myogenin, desmin, troponin T, and myosin heavy chains I and II as well as fusion of C3H10T1/2 cells into myotubes that resembled a skeletal muscle cell phenotype. We also demonstrated LAGOS functionality in vivo by engineering the vector to express human VEGF165 and human ANG1 in response to light. HEK 293T cells stably expressing the LAGOS vector and transiently expressing the light-inducible Cre recombinase proteins were implanted into mouse dorsal window chambers. Mice that were illuminated with blue light had increased micro-vessel density compared to mice that were not illuminated. Analysis of human vascular endothelial growth factor (VEGF) and human ANG1 levels by enzyme-linked immunosorbent assay (ELISA) revealed statistically higher levels of VEGF and ANG1 in illuminated mice compared to non-illuminated mice.
In summary, the objective of this work was to engineer robust light-inducible gene regulation systems that can control genes and cellular fate in a spatial and temporal manner. These studies combine the rapid advances in gene targeting and activation technology with natural light-inducible plant protein interactions. Collectively, this thesis presents several optogenetic systems that are expected to facilitate the development of multicellular cell and tissue constructs for use in tissue engineering, synthetic biology, gene therapy, and basic science both in vitro and in vivo.
Comment: Although it is easy to characterize technological progress as following in the wake of scientific discoveries, the reverse is almost equally true; advances in technique open the door to types of experiment previously intractable or impossible. Such is currently the case for the field of optically controlled biotechnology, which has exploded into prominence, particularly over the last half-decade. Light of an appropriate wavelength can penetrate mammalian tissues to a depth of up to a couple of centimeters, rendering much of the living body accessible to optical study and control—still more if the detector/source is integrated into an endoscopic or fiber optic probe. Techniques borrowed from the semiconductor industry allow patterns of illumination to be controlled down to the nanometer scale, ideal for addressing individual cells. The highly controlled time course of such experiments, as compared to traditional means of gene activation, such as the addition of a chemical agent to the medium, eliminates confounding variables, and simplifies data analysis. Furthermore, this level of immediate control opens the door to closed-loop systems where the activity of entities under optical control can be continuously tuned in relation to some parameter(s). In the first of these two illuminating theses, a vehicle is developed that permits light-driven release of a small molecule. Such a system could be employed to target a systemically administered antibiotic or anti-neoplastic agent to a site of infection or cancer while sparing other bodily tissues from toxicity. Because most modern drugs cannot be produced in the body, even given arbitrarily good control of cellular biochemistry, this technique will have lasting value in numerous clinical contexts. In the second thesis, the level of precision achieved is even more profound; the CRISPR/Cas9 system has received much recent attention13 in its own right for its capacity to target arbitrary genetic sequences without an arduous protein-engineering step. The LACE system described stands to permit genetic manipulation with almost arbitrarily good spatial, temporal, and genomic site-specific control, using only means available to a typical university laboratory.
Targeting T Cells for the Immune-Modulation of Human Diseases
Regina Lin, PhD, Duke University
Dysregulated inflammation underlies the pathogenesis of a myriad of human diseases ranging from cancer to autoimmunity. As coordinators, executers, and sentinels of host immunity, T cells represent a compelling target population for immune-modulation. In fact, the antigen-specificity, cytotoxicity, and promise of long-lived of immune-protection make T cells ideal vehicles for cancer immunotherapy. Interventions for autoimmune disorders, on the other hand, aim to dampen T cell–mediated inflammation and promote their regulatory functions. Although significant strides have been made in targeting T cells for immune modulation, current approaches remain less than ideal and leave room for improvement. In this dissertation, I seek to improve on current T cell-targeted immunotherapies, by identifying and pre-clinically characterizing their mechanisms of action and in vivo therapeutic efficacy.
CD8+ cytotoxic T cells have potent anti-tumor activity and therefore are leading candidates for use in cancer immunotherapy. The application of CD8+ T cells for clinical use has been limited by the susceptibility of ex vivo– expanded CD8+ T cells to become dysfunctional in response to immunosuppressive microenvironments. To enhance the efficacy of adoptive cell transfer therapy (ACT), we established a novel microRNA (miRNA)-targeting approach that augments CTL cytotoxicity and preserves immunocompetence. Specifically, we screened for miRNAs that modulate cytotoxicity and identified miR-23a as a strong functional repressor of the transcription factor Blimp-1, which promotes CTL cytotoxicity and effector cell differentiation. In a cohort of advanced lung cancer patients, miR- 23a was up-regulated in tumor-infiltrating CD8+ T cells, and its expression correlated with impaired anti-tumor potential of patient CD8+ T cells. We determined that tumor-derived transforming growth factor-b (TGF-b) directly suppresses CD8+ T cell immune function by elevating miR-23a and down-regulating Blimp-1. Functional blockade of miR-23a in human CD8+ T cells enhanced granzyme B expression; and in mice with established tumors, immunotherapy with just a small number of tumor-specific CD8+ T cells in which miR-23a was inhibited robustly hindered tumor progression. Together, our findings provide a miRNA-based strategy that subverts the immunosuppression of CD8+ T cells that is often observed during adoptive cell transfer tumor immunotherapy and identify a TGF-bmediated tumor immune-evasion pathway
Having established that miR-23a-inhibition can enhance the quality and functional resilience of anti-tumor CD8+ T cells, especially within the immune-suppressive tumor microenvironment, we went on to interrogate the translational applicability of this strategy in the context of chimeric antigen receptor (CAR)-modified CD8+ T cells. Although CAR T cells hold immense promise for ACT, CAR T cells are not completely curative due to their in vivo functional suppression by immune barriers—such as TGF-b—within the tumor microenvironment. Because TGF-b poses a substantial immune barrier in the tumor microenvironment, we sought to investigate whether inhibiting miR-23a in CAR T cells can confer immune competence to afford enhanced tumor clearance. To this end, we retrovirally transduced wild-type and miR-23a–deficient CD8+ T cells with the EGFRvIII-CAR, which targets the PepvIII tumorspecific epitope expressed by glioblastomas (GBM). Our in vitro studies demonstrated that while wild-type EGFRvIIICAR T cells were vulnerable to functional suppression by TGF-b, miR-23a abrogation rendered EGFRvIII-CAR T cells immune-resistant to TGF-b. Rigorous preclinical studies are currently underway to evaluate the efficacy of miR-23adeficient EGFRvIII-CAR T cells for GBM immunotherapy.
Last, we explored novel immune-suppressive therapies by the biological characterization of pharmacological agents that could target T cells. Although immune-suppressive drugs are classical therapies for a wide range of autoimmune diseases, they are accompanied by severe adverse effects. This motivated our search for novel immunesuppressive agents that are efficacious and lack undesirable side effects. To this end, we explored the potential utility of subglutinol A, a natural product isolated from the endophytic fungus Fusarium subglutinans. We showed that subglutinol A exerts multimodal immune-suppressive effects on activated T cells in vitro. Subglutinol A effectively blocked T cell proliferation and survival, while profoundly inhibiting pro-inflammatory interferon-c (IFN-c) and interleukin-17 (IL-17) production by fully differentiated effector Th1 and Th17 cells. Our data further revealed that subglutinol A might exert its anti-inflammatory effects by exacerbating mitochondrial damage in T cells, but not in innate immune cells or fibroblasts. Additionally, we demonstrated that subglutinol A significantly reduced lymphocytic infiltration into the footpad and ameliorated footpad swelling in the mouse model of Th1-driven delayed-type hypersensitivity. These results suggest the potential of subglutinol A as a novel therapeutic for inflammatory diseases.
Comment: Immunotherapy is among the most promising approaches to cancer treatment, having the specificity and scope to selectively target transformed cells wherever they may reside within the body and the potential to install a permanent defense against disease recurrence. By the time a typical cancer is clinically diagnosed, however, it has already found means to survive a prolonged period of potential immune attack. The mechanisms by which tumors evade immune surveillance are beginning to be elucidated,15,16 and include both direct suppression of effector cells and progressive editing of the host’s immune repertoire to disfavor future attack. It is inherently difficult to interfere with these defenses directly, due to the selection pressures in genetically heterogeneous neoplastic tissue. Much effort is thus being focused on methods for rendering therapeutically delivered immune cells resistant to their effects. The cytokine TGF-b is paradoxically known to function as both a tumor suppressor in healthy tissue and as a tumorderived species associated with multiple cancer-promoting activities, including enhanced immune evasion. This work identifies the pathway by which TGF-b compromises cytotoxic T cell function in the tumor microenvironment, and demonstrates an effective method for blocking this signal. In many clinical cases, however, editing of the patient’s immune repertoire has already removed or rendered anergic those immune cells able to recognize their cancer. Thus, the finding that blocking TGF-b signaling also appears to enhance the effectiveness of CAR-modified T cells— engineered with an antibody fragment targeting them with high affinity to a particular tumor-associated epitope—is a welcome addition to these already promising results.
Novel Fibonacci and non-Fibonacci structure in the sunflower: results of a citizen science experiment
Jonathan Swinton, Erinma Ochu, The MSI Turing’s Sunflower Consortium
Published 18 May 2016. DOI http://dx.doi.org:/10.1098/rsos.160091
This citizen science study evaluates the occurrence of Fibonacci structure in the spirals of sunflower (Helianthus annuus) seedheads. This phenomenon has competing biomathematical explanations, and our core premise is that observation of both Fibonacci and non-Fibonacci structure is informative for challenging such models. We collected data on 657 sunflowers. In our most reliable data subset, we evaluated 768 clockwise or anticlockwise parastichy numbers of which 565 were Fibonacci numbers, and a further 67 had Fibonacci structure of a predefined type. We also found more complex Fibonacci structures not previously reported in sunflowers. This is the third, and largest, study in the literature, although the first with explicit and independently checkable inclusion and analysis criteria and fully accessible data. This study systematically reports for the first time, to the best of our knowledge, seedheads without Fibonacci structure. Some of these are approximately Fibonacci, and we found in particular that parastichy numbers equal to one less than a Fibonacci number were present significantly more often than those one more than a Fibonacci number. An unexpected further result of this study was the existence of quasi-regular heads, in which no parastichy number could be definitively assigned.
- Introduction
Fibonacci structure can be found in hundreds of different species of plants [1]. This has led to a variety of competing conceptual and mathematical models that have been developed to explain this phenomenon. It is not the purpose of this paper to survey these: reviews can be found in [1–4], with more recent work including [5–10]. Instead, we focus on providing empirical data useful for differentiating them.
These models are in some ways now very mathematically satisfying in that they can explain high Fibonacci numbers based on a small number of plausible assumptions, though they are not so satisfying to experimental scientists [11]. Despite an increasingly detailed molecular and biophysical understanding of plant organ positioning [12–14], the very parsimony and generality of the mathematical explanations make the generation and testing of experimental hypotheses difficult. There remains debate about the appropriate choice of mathematical models, and whether they need to be central to our understanding of the molecular developmental biology of the plant. While sunflowers provide easily the largest Fibonacci numbers in phyllotaxis, and thus, one might expect, some of the stronger constraints on any theory, there is a surprising lack of systematic data to support the debate. There have been only two large empirical studies of spirals in the capitulum, or head, of the sunflower: Weisse [15] and Schoute [16], which together counted 459 heads; Schoute found numbers from the main Fibonacci sequence 82% of the time and Weise 95%. The original motivation of this study was to add a third replication to these two historical studies of a widely discussed phenomenon. Much more recently, a study of a smaller sample of 21 seedheads was carried out by Couder [17], who specifically searched for non-Fibonacci examples, whereas Ryan et al. [18] studied the arrangement of seeds more closely in a small sample of Helianthus annuus and a sample of 33 of the related perennial H. tuberosus.
Neither the occurrence of Fibonacci structure nor the developmental biology leading to it are at all unique to sunflowers. As common in other species, the previous sunflower studies found not only Fibonacci numbers, but also the occasional occurrence of the double Fibonacci numbers, Lucas numbers and F4 numbers defined below [1]. It is worth pointing out the warning of Cooke [19] that numbers from these sequences make up all but three of the first 17 integers. This means that it is particularly valuable to look at specimens with large parastichy numbers, such as the sunflowers, where the prevalence of Fibonacci structure is at its most striking.
Neither Schoute nor Weisse reported their precise technique for assigning parastichy numbers to their samples, and it is noteworthy that neither author reported any observation of non-Fibonacci structure. One of the objectives of this study was to rigorously define Fibonacci structure in advance and to ensure that the assignment method, though inevitably subjective, was carefully documented.
This paper concentrates on the patterning of seeds towards the outer rim of sunflower seedheads. The number of ray florets (the ‘petals’, typically bright yellow) or the green bracts behind them tends to have a looser distribution around a Fibonacci number. In the only mass survey of these, Majumder & Chakravarti [20] counted ray florets on 1002 sunflower heads and found a distribution centred on 21.
This citizen science study evaluates the occurrence of Fibonacci structure in the spirals of sunflower (Helianthus annuus) seedheads. This phenomenon has competing biomathematical explanations, and our core premise is that observation of both Fibonacci and non-Fibonacci structure is informative for challenging such models. We collected data on 657 sunflowers. In our most reliable data subset, we evaluated 768 clockwise or anticlockwise parastichy numbers of which 565 were Fibonacci numbers, and a further 67 had Fibonacci structure of a predefined type. We also found more complex Fibonacci structures not previously reported in sunflowers. This is the third, and largest, study in the literature, although the first with explicit and independently checkable inclusion and analysis criteria and fully accessible data. This study systematically reports for the first time, to the best of our knowledge, seedheads without Fibonacci structure. Some of these are approximately Fibonacci, and we found in particular that parastichy numbers equal to one less than a Fibonacci number were present significantly more often than those one more than a Fibonacci number. An unexpected further result of this study was the existence of quasi-regular heads, in which no parastichy number could be definitively assigned.
Incorporation of irregularity into the mathematical models of phyllotaxis is relatively recent: [17] gave an example of a disordered pattern arising directly from the deterministic model while more recently the authors have begun to consider the effects of stochasticity [10,21]. Differentiating between these models will require data that go beyond capturing the relative prevalence of different types of Fibonacci structure, so this study was also designed to yield the first large-scale sample of disorder in the head of the sunflower.
The Fibonacci sequence is the sequence of integers 1,2,3,5,8,13,21,34,55,89,144… in which each member after the second is the sum of the two preceding. The Lucas sequence is the sequence of integers 1,3,4,7,11,18,29,47,76,123… obeying the same rule but with a different starting condition; the F4 sequence is similarly 1,4,5,9,14,23,37,60,97,…. The double Fibonacci sequence 2,4,6,10,16,26,42,68,110,… is double the Fibonacci sequence. We say that a parastichy number which is any of these numbers has Fibonacci structure. The sequencesF5=1,5,6,11,17,28,45,73,… and F8=1,8,9,17,26,43,69,112… also arise from the same rule, but as they had not been previously observed in sunflowers we did not include these in the pre-planned definition of Fibonacci structure for parsimony. One example of adjacent pairs from each of these sequences was, in fact, observed but both examples are classified as non-Fibonacci below. A parastichy number which is any of 12,20,33,54,88,143 is also not classed as having Fibonacci structure but is distinguished as a Fibonacci number minus one in some of the analyses, and similarly 14,22,35,56,90,145 as Fibonacci plus one.
When looking at a seedhead such as in figure 1 the eye naturally picks out at least one family of parastichies or spirals: in this case, there is a clockwise family highlighted in blue in the image on the right-hand side.
http://d3hu9binmobce5.cloudfront.net/content/royopensci/3/5/160091/F1.medium.gif
Distribution and type of parastichy pairs
Figure 5 plots the individual pairs observed. On the reference line, the ratio of the numbers is equal to the golden ratio so departures from the line mark departures from Fibonacci structure, which are less evident in the more reliable photoreviewed dataset. It can be seen from table 3 that Fibonacci pairings dominate the dataset.
http://d3hu9binmobce5.cloudfront.net/content/royopensci/3/5/160091/F5.medium.gif
- Table 3. View inline View popup
Table 3.
Observed pairings of Fibonacci types of clockwise and anticlockwise parastichy numbers. Other means any parastichy number which neither has Fibonacci structure nor is Fibonacci ±1. Of all the Fibonacci ±1/Fibonacci pairs, only sample 191, a (21,20) pair, was not close to an adjacent Fibonacci pair.
One typical example of a Fibonacci pair is shown in figure 6, with a double Fibonacci case infigure 1 and a Lucas one in figure 7. There was no photoreviewed example of an F4 pairing. The sole photoreviewed assignment of a parastichy number to the F4 sequence was the anticlockwise parastichy number 37 in sample 570, which was relatively disordered. The clockwise parastichy number was 55, lending support to the idea this may have been a perturbation of a (34,55) pattern. We also found adjacent members of higher-order Fibonacci series. Figures 8 and 9 each show well-ordered examples with parastichy counts found adjacent in the F5 and F8 series, respectively: neither of these have been previously reported in the sunflower.
Figure 6.
http://d3hu9binmobce5.cloudfront.net/content/royopensci/3/5/160091/F6.medium.gif
Sunflower 095. An (89,55) example with 89 clockwise parastichies and 55 anticlockwise ones, extending right to the rim of the head. Because these are clear and unambiguous, the other parastichy families which are visible towards the centre are not counted here.
Figure 7. Sunflower 171. A Lucas series (76,47) example.
- Figure 12. Download figure Open in new tab
Sunflower 667. Anticlockwise parastichies only, showing competing parastichy families which are distinct but in some places overlapping.
Our core results are twofold. First, and unsurprisingly, Fibonacci numbers, and Fibonacci structure more generally, are commonly found in the patterns in the seedheads of sunflowers. Given the extent to which Fibonacci patterns have attracted pseudo-scientific attention [33], this substantial replication of limited previous studies needs no apology. We have also published, for the first time, examples of seedheads related to the F5 and F8 sequences but by themselves they do not add much to the evidence base. Our second core result, though, is a systematic survey of cases where Fibonacci structure, defined strictly or loosely, did not appear. Although not common, such cases do exist and should shed light on the underlying developmental mechanisms. This paper does not attempt to shed that light, but we highlight the observations that any convincing model should explain. First, the prevalence of Lucas numbers is higher than those of double Fibonacci numbers in all three large datasets in the literature, including ours, and there are sporadic appearances of F4, F5 and F8 sequences. Second, counts near to but not exactly equal to Fibonacci structure are also observable: we saw a parastichy count of 54 more often than the most common Lucas count of 47. Sometimes, ambiguity arises in the counting process as to whether an exact Fibonacci-structured number might be obtained instead, but there are sufficiently many unambiguous cases to be confident this is a genuine phenomenon. Third, among these approximately Fibonacci counts, those which are a Fibonacci number minus one are significantly more likely to be seen than a Fibonacci number plus one. Fourth, it is not uncommon for the parastichy families in a seedhead to have strong departures from rotational symmetry: this can have the effect of yielding parastichy numbers which have large departures from Fibonacci structure or which are completely uncountable. This is related to the appearance of competing parastichy families. Fifth, it is common for the parastichy count in one direction to be more orderly and less ambiguous than that in the other. Sixth, seedheads sometimes possess completely disordered regions which make the assignment of parastichy numbers impossible. Some of these observations are unsurprising, some can be challenged by different counting protocols, and some are likely to be easily explained by the mathematical properties of deformed lattices, but taken together they pose a challenge for further research.
It is in the nature of this crowd-sourced experiment with multiple data sources that it is much easier to show variability than it is to find correlates of that variability. We tried a number of cofactor analyses that found no significant effect of geography, growing conditions or seed type but if they do influence Fibonacci structure, they are likely to be much easier to detect in a single-experimenter setting.
We have been forced by our results to extend classifications of seedhead patterns beyond structured Fibonacci to approximate Fibonacci ones. Clearly, the more loose the definition of approximate Fibonacci, the easier it is to explain away departures from model predictions. Couder [17] found one case of a (54,87) pair that he interpreted as a triple Lucas pair 3×(18,29). While mathematically true, in the light of our data, it might be more compellingly be thought of as close to a (55,89) ideal than an exact triple Lucas one. Taken together, this need to accommodate non-exact patterns, the dominance of one less over one more than Fibonacci numbers, and the observation of overlapping parastichy families suggest that models that explicitly represent noisy developmental processes may be both necessary and testable for a full understanding of this fascinating phenomenon. In conclusion, this paper provides a testbed against which a new generation of mathematical models can and should be built.





































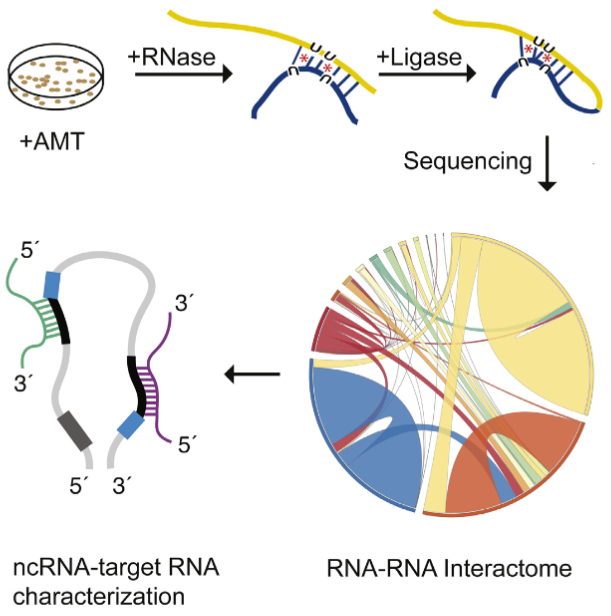{kind=link}
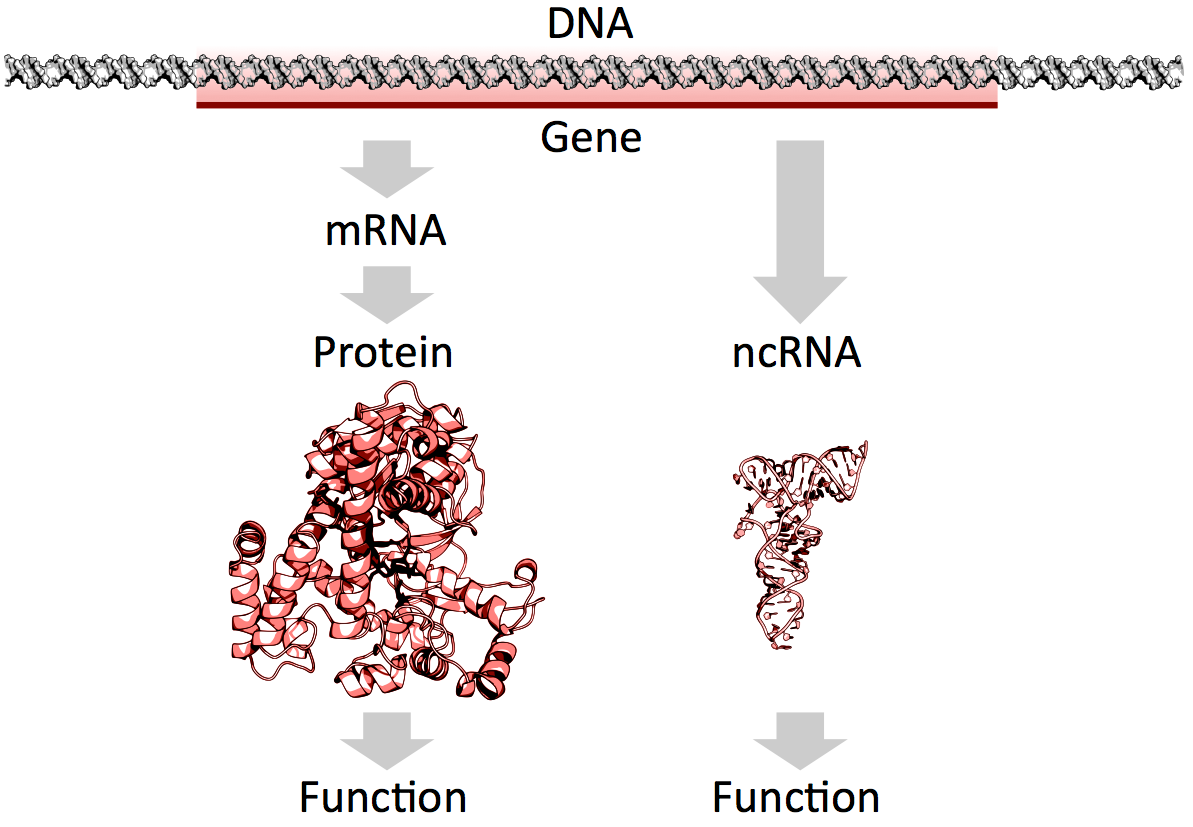{kind=link}
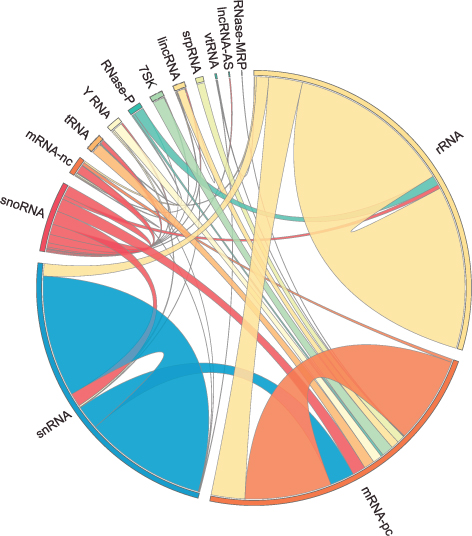{kind=link}
{kind=link}
{kind=link}
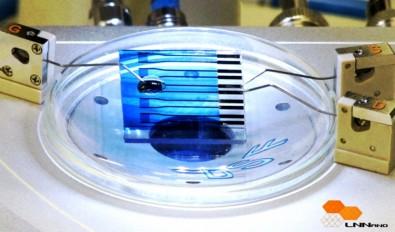{kind=link}
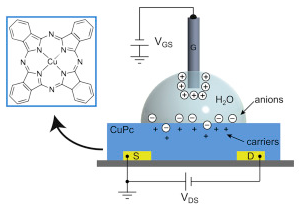{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
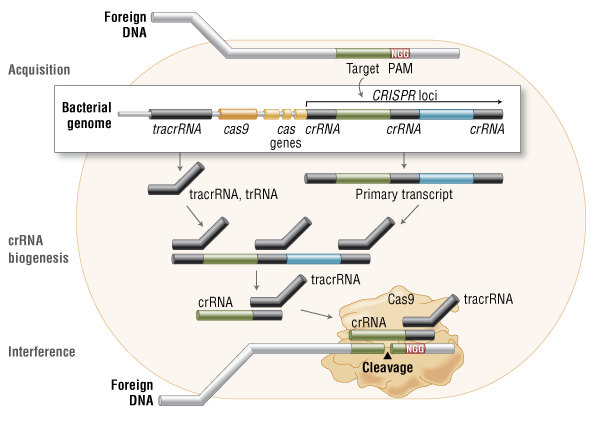{kind=link}
{kind=link}
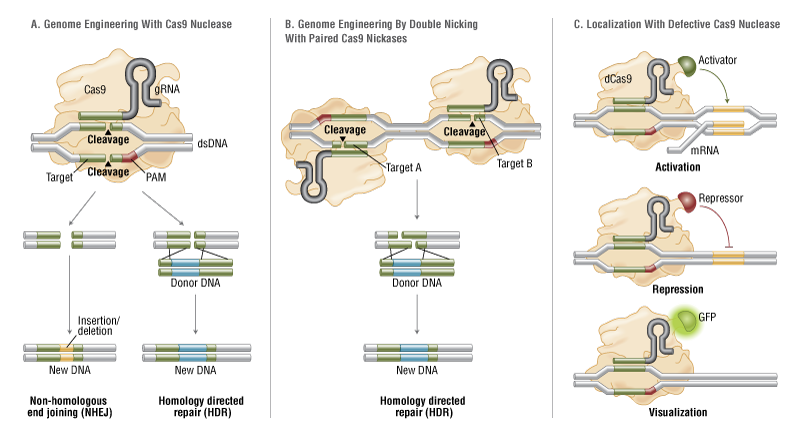{kind=link}
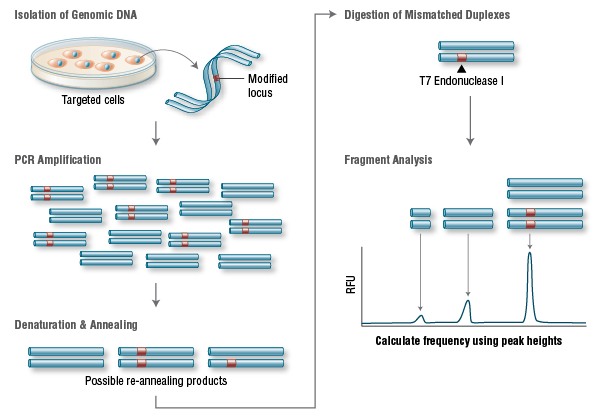{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}