Immune System in Perspective
Curator: Larry H. Bernstein, MD, FCAP
Updated 7/09/2023
Researchers find weaker immune response to viral infections in children with mitochondrial disorders
One of the first human studies on how mitochondrial function impacts immune cells to guide future treatments. Mitochondria convert food and oxygen into energy. Genomic variants in more than 350 genes have been linked to mitochondrial disorders.NHGRI In a new study, National Institutes of Health (NIH) researchers found that altered B cell function in children with mitochondrial disorders led to a weaker and less diverse antibody response to viral infections. The study, published in Frontiers in Immunology, was led by researchers at the National Human Genome Research Institute (NHGRI), who analyzed the gene activities of immune cells in children with mitochondrial disorders and found that B cells, which produce antibodies to fight viral infections, are less able to survive cellular stress.
“Our work is one of the first examples to study how B cells are affected in mitochondrial disease by looking at human patients,” said Eliza Gordon-Lipkin, M.D., assistant research physician in NHGRI’s Metabolism, Infection and Immunity Section and co-first author of the paper.
Mitochondria are important components of nearly every cell in the body because they convert food and oxygen into energy. Genomic variants in more than 350 genes have been linked to mitochondrial disorders with varied symptoms depending on which cells are affected.
“For children with mitochondrial disorders, infections can be life threatening or they can worsen the progression of their disorder,” said Peter McGuire, M.B.B.Ch., NHGRI investigator, head of the Metabolism, Infection and Immunity Section and senior author of the study. “We wanted to understand how immune cells differ in these patients and how that influences their response to infections.”
Around 1 in 5,000 people worldwide have a mitochondrial disorder. Examples of mitochondrial disorders are Leigh’s syndrome, which primarily affects the nervous system, and Kearns-Sayre syndrome, which primarily affects the eyes and heart. While mitochondrial disorders are known to affect organs such as the heart, liver, and brain, less is known how they affect the immune system. Using a genomic technique called single-cell RNA sequencing, which analyzes gene activity in different cell types, researchers studied immune cells found in blood. These cells include different types of white blood cells that help the body fight infections. During stressful conditions, these cells produce a microRNA called mir4485. MicroRNAs are small strings of RNA that help control when and where genes are turned on and off. mir4485 controls cellular pathways that help cells survive.
“We think that B cells in these patients undergo cellular stress when they turn into plasma cells and produce antibodies, and these B cells then try to survive by producing the microRNA to cope,” said Dr. McGuire. “But the B cells are too fragile due to their limited energy, so they are unable to survive the stressful conditions.”
Researchers used a technique called VirScan to look at all past viral infections, assess how well the immune system fought those infections and see the effects of B cells and plasma cells on antibody production. With a weaker antibody response, the immune systems in children with mitochondrial disorders are less able to recognize and neutralize invading viruses and clear infections. Researchers aim to use the results of this study to guide future treatment of patients with mitochondrial disorders, noting that more translational studies are needed in this research area.
Reference: Gordon-Lipkin et al. Primary oxidative phosphorylation defects lead to perturbations in the human B cell repertoire. Frontiers in Immunology. DOI: 10.3389/fimmu.2023.1142634. (2023)
5/29/2016
LPBI
How regulatory T cells work
Vignali DAA, Collison LW & Workman CJ
Nature Reviews Immunology 8, 523-532 (July 2008) | doi:10.1038/nri2343
http://www.nature.com/nri/journal/v8/n7/full/nri2343.html
Regulatory T (TReg) cells are essential for maintaining peripheral tolerance, preventing autoimmune diseases and limiting chronic inflammatory diseases. However, they also limit beneficial responses by suppressing sterilizing immunity and limiting antitumour immunity. Given that TReg cells can have both beneficial and deleterious effects, there is considerable interest in determining their mechanisms of action. In this Review, we describe the basic mechanisms used by TReg cells to mediate suppression and discuss whether one or many of these mechanisms are likely to be crucial for TReg-cell function. In addition, we propose the hypothesis that effector T cells may not be ‘innocent’ parties in this suppressive process and might in fact potentiate TReg-cell function.
How regulatory T cells work.
Nat Rev Immunol. 2008 Jul;8(7):523-32. http://dx.doi.org:/10.1038/nri2343.

Basic mechanisms used by Treg cells
This schematic depicts the various regulatory T (Treg)-cell mechanisms arranged into four groups centred around four basic modes of action. ‘Inhibitory cytokines’ include interleukin-10 (IL-10), interleukin-35 (IL-35) and transforming growth factor-β (TGF-β). ‘Cytolysis’ includes granzyme-A- and granzyme-B-dependent and perforin-dependent killing mechanisms. ‘Metabolic disruption’ includes high affinity IL-2 receptor α (CD25)-dependent cytokine-deprivation-mediated apoptosis, cyclic AMP (cAMP)-mediated inhibition, and CD39- and/or CD73-generated, adenosine–purinergic adenosine receptor (A2A)-mediated immunosuppression. ‘Targeting dendritic cells’ includes mechanisms that modulate DC maturation and/or function such as lymphocyte activation gene-3 (LAG3; also known as CD223)–MHC-class-II-mediated suppression of DC maturation, and cytotoxic T lymphocyte antigen-4 (CTLA4)–CD80/CD86-mediated induction of indoleamine 2,3-dioxygenase (IDO), which is an immunosuppressive molecule, by DCs.

Model for how effector T cells might boost Treg-cell function
This occurs in three stages. (a) Initial regulatory T (Treg)-cell activation induces production of regulatory factors such as interleukin-35 (IL-35). (b) Treg cells ‘sense’ the presence of recently activated effector T cells through a receptor–ligand interaction (cell surface or soluble). (c) This in turn boosts or potentiates Treg-cell function resulting in the enhanced production of regulatory mediators, such as IL-35, and perhaps the induction of new mediators.
Regulatory T (Treg) cells are essential for maintaining peripheral tolerance, preventing autoimmune diseases and limiting chronic inflammatory diseases. However, they also limit beneficial responses by suppressing sterilizing immunity and limiting anti-tumour immunity. Given that Treg cells can have both beneficial and deleterious effects, there is considerable interest in determining their mechanisms of action. In this Review, we discuss the basic mechanisms used by Treg cells to mediate suppression, and discuss whether one or many of these mechanisms are likely to be crucial for Tregcell function. In addition, we present the hypothesis that effector T cells may not be ‘innocent’ parties in this suppressive process and might in fact potentiate Treg-cell function.
Several sophisticated regulatory mechanisms are used to maintain immune homeostasis, prevent autoimmunity and moderate inflammation induced by pathogens and environmental insults. Chief amongst these are regulatory T (Treg) cells that are now widely regarded as the primary mediators of peripheral tolerance. Although Treg cells play a pivotal role in preventing autoimmune diseases, such as type 1 diabetes1,2, and limiting chronic inflammatory diseases, such as asthma and inflammatory bowel disease (IBD)3,4, they also block beneficial responses by preventing sterilizing immunity to certain pathogens5,6 and limiting anti-tumour immunity7. A seminal advance in the analysis of Treg cells came with the identification of a key transcription factor, forkhead box P3 (FOXP3), that is required for their development, maintenance and function8,9. Mice and patients that lack FOXP3 develop a profound autoimmune-like lymphoproliferative disease that graphically emphasizes the importance of Treg cells in maintaining peripheral tolerance10-12 (BOX 1). Although FOXP3 has been proposed as the master regulator of Treg cells that controls the expression of multiple genes that mediate their regulatory activity13,14, this has been recently challenged raising the possibility that other transcriptional events may operate upstream of and/or concurrently with FOXP3 to mediate Treg-cell development15.
While Foxp3 has proven to be an invaluable marker for murine Treg cells, its role in human Treg cells is less straightforward (see BOX 2 for a discussion of Treg-cell markers). Humans that lack FOXP3 develop immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), a severe autoimmune disease that presents early in infancy. Although FOXP3 appears to be required for human Treg-cell development and function, expression of FOXP3 alone is clearly not sufficient as a significant percentage of human activated T cells express FOXP3 and yet do not possess regulatory activity16-20. Furthermore, induction of FOXP3 in human T cells by transforming growth factor-β (TGFβ) does not confer a regulatory phenotype, in contrast to their murine counterparts20. Consequently, FOXP3 is not a good marker for human Treg cells (BOX 2). Whether this distinction is due to intrinsic differences between mouse and human FOXP3 and/or a requirement for an additional cofactor/ transcription factor is an important question that needs to be resolved.
Significant progress has been made over the last few years in delineating the molecules and mechanisms that Treg cells use to mediate suppression21,22. In this Review, we outline our current understanding of the mechanisms used by Treg cells to mediate suppression, and the challenges that lie ahead in defining their mode of action. We also discuss whether Treg cells are likely to depend on one, a few or many of these mechanisms. In addition, we propose that effector T cells may have a significant role in boosting and/or modulating Treg-cell function. Unless stated, we focus here primarily on the mechanisms that are used by thymus-derived natural CD4+CD25+ FOXP3+ Treg cells.
Basic mechanisms of Treg-cell function Defining the mechanisms of Treg-cell function is clearly of crucial importance. Not only would this provide insight into the control processes of peripheral tolerance but it would probably provide a number of potentially important therapeutic targets. Although this quest has been ongoing since interest in Treg cells was reignited in 199523, there has been significant progress in the last few years. From a functional perspective, the various potential suppression mechanisms of Treg cells can be grouped into four basic ‘modes of action’: suppression by inhibitory cytokines, suppression by cytolysis, suppression by metabolic disruption, and suppression by modulation of dendritic-cell (DC) maturation or function (FIG. 1).
Suppression by inhibitory cytokines Inhibitory cytokines, such as interleukin-10 (IL-10) and TGFβ, have been the focus of considerable attention as a mechanism of Treg-cell-mediated suppression. There has also been significant interest in their ability to generate induced (also known as adaptive) Treg-cell populations, either naturally in vivo or experimentally as a potential therapeutic modality (BOX 3). Although the general importance of IL-10 and TGFβ as suppressive mediators is undisputed, their contribution to the function of thymus-derived, natural Treg cells is still a matter of debate24. This is partly due to the general perception that Treg cells function in a contactdependent manner25,26. Indeed, in vitro studies using neutralizing antibodies or T cells that are unable to produce or respond to IL-10 and TGFβ suggested that these cytokines may not be essential for Treg-cell function25-28. However, this contrasts with data from in vivo studies29,30.
In allergy and asthma models, evidence suggests that both natural and antigen-specific Treg cells control disease in a manner that is, in part, dependent on IL-1029 and in some reports dependent on both IL-10 and TGFβ 31. Adoptive transfer of allergen-specific Treg cells induced significant IL-10 production by CD4+ effector T cells in the lung following allergen challenge and this Treg-cell-mediated control of disease was reversed by treatment with an IL-10- receptor-specific antibody32. However, suppression of allergic inflammation and airway hyper-reactivity, and increased production of IL-10 still occurred following transfer of IL-10- deficient Treg cells, suggesting that Treg cells can suppress the Th2-driven response to allergens in vivo through an IL-10-dependent mechanism, but that the production of IL-10 by Treg cells themselves is not required for the suppression observed. This contrasts with a recent study suggesting that the Treg-cell-specific ablation of IL-10 expression resulted in increased lung allergic inflammation and hyperreactivity33.
This scenario might occur in other disease models. For instance, the effects of IL-10 can only be partially attributed to Treg-cell-derived IL-10 in the immune response to hepatitis B virus34 and in the allograft tolerance response elicited by splenocytes exposed to non-inherited maternal antigens35. Recently, it was also shown that IL-10 is crucial for the control of various infections in which Treg cells have been reported to be involved including Mycobacterium tuberculosis36, Toxoplasma gondii37, Leishmania major38, and Trichinella spiralis39. However, Treg cells were not the source of IL-10 in all of these infection models.
By contrast, several studies have shown that IL-10 production by Treg cells is essential for the prevention of colitis in mouse models of IBD40. Moreover, it appears that the tumour microenvironment promotes the generation of FOXP3+ Treg cells that mediate IL-10- dependent, cell-contact independent, suppression41. Similarly, in UV-radiation-induced carcinogenesis, IL-10 production by Treg cells appears to be important for blocking anti-tumour immunity42. IL-10 produced by Treg cells also appears to be crucial for IL-10-mediated tolerance in a model of hepatitis induced by concanavalin A43 and tolerance to bacterial and viral superantigens44. In addition, recent papers suggest new roles for Treg-cell-derived IL-10 in the induction of feto-maternal tolerance45 and B-cell-enhanced recovery from experimental autoimmune encephalomyelitis46. Collectively, the picture that appears to be emerging is that the relative importance of Treg-cell-derived IL-10 is very dependent on the target organism or disease and on the experimental system. Furthermore, the Treg-cell-specific deletion of IL-10 did not result in the development of spontaneous systemic autoimmunity, but did result in enhanced pathology in the colon of older mice and in the lungs of mice with induced airway hypersensitivity, suggesting that the function of Treg-cell-derived IL-10 may be restricted to controlling inflammatory responses induced by pathogens or environmental insults33.
While some early in vitro studies using neutralizing antibodies to TGFβ or Treg cells lacking TGFβ 25,47 indicated that TGFβ was not required for natural Treg-cell function, other studies, both in vitro and in vivo suggested a critical role for Treg-cell surface bound TGFβ 48,49. Therefore, the importance of TGFβ for natural Treg-cell function has also been a controversial topic. Indeed, there has been considerably more focus recently on the importance of TGFβ in the development of induced Treg cells and perhaps in Treg-cell maintenance in general (BOX 3). However, there are studies that suggest that TGFβ produced by Treg cells may directly participate in effector T-cell suppression. For instance, effector T cells that are resistant to TGFβ-mediated suppression cannot be controlled by Treg cells in an IBD model50. In addition, TGFβ produced by Treg cells has been found to be important in the control of the host immune response to M. tuberculosis36, suppression of allergic responses31 and prevention of colitis in an IBD model51. Interestingly, TGFβ produced by Treg cells has also been implicated in limiting anti-tumour immunity in head and neck squamous-cell carcinoma52 and in follicular lymphoma53 by rendering T cells unresponsive to the tumour. TGFβ also appears to limit the anti-tumour activity of cytokine-induced killer cells54.
Membrane-tethered TGFβ can also mediate suppression by Treg cells in a cell-cell contactdependent manner48. Treg cells can control islet infiltration of CD8+ T cells and delay the progress of diabetes through membrane-tethered TGFβ 49. However, experiments using mice deficient in TGFβ-receptor (TGFβR) signalling in effector T cells or using TGFβ or TGFβR blocking reagents failed to show that membrane-tethered TGFβ is required for natural Treg cell development or function47. More recently, however, interest in membrane-tethered TGFβ has re-surfaced with the description of a previously unappreciated role for it in the tumour microenvironment. TGFβ associated with tumour exosome membranes appears to enhance the suppressive function of Treg cells and skew T cells away from their effector functions and towards regulatory functions55. Furthermore, ovalbumin-induced airway inflammation can be attenuated by heme oxygenase-1 through membrane-tethered TGFβ and IL-10 secretion by Treg cells56, a process that activates the Notch1–HES1 (hairy and enhancer of split 1) axis in target cells57. Thus, in light of the most current data, it now appears that soluble and/or membrane-tethered TGFβ may have a previously unappreciated role in natural Treg-cell function.
Recently, a new inhibitory cytokine, IL-35, has been described that is preferentially expressed by Treg cells and is required for their maximal suppressive activity58. IL-35 is a novel member of the IL-12 heterodimeric cytokine family and is formed by the pairing of Epstein–Barr virus induced gene 3 (Ebi3), which normally pairs with p28 to form IL-27, and p35 (also known as Il12a), which normally pairs with p40 to form IL-12. Both Ebi3 and Il12a are preferentially expressed by murine Foxp3+ Treg cells58,59, but not resting or active effector T cells, and are significantly upregulated in actively suppressing Treg cells58. As predicted for a heterodimeric cytokine, both Ebi3−/− and Il12a−/− Treg cells had significantly reduced regulatory activity in vitro and failed to control homeostatic proliferation and cure IBD in vivo. This precise phenocopy suggested that IL-35 is required for the maximal suppressive activity of Treg cells. Importantly IL-35 was not only required but sufficient, as ectopic expression of IL-35 conferred regulatory activity on naive T cells and recombinant IL-35 suppressed T cell proliferation in vitro58. Although IL-35 is an exciting addition to the Treg-cell portfolio, there is clearly much that remains to be defined about this cytokine and its contribution to Treg-cell function. For instance, it remains to be determined if IL-35 suppresses the development and/or function of other cell types such as DCs and macrophages.
It is now clear that three inhibitory cytokines, IL-10, IL-35 and TGFβ, are key mediators of Treg-cell function. Although they are all inhibitory, the extent to which they are utilized in distinct pathogenic/homeostatic settings differs suggesting a non-overlapping function, which needs further refinement.
……….
How many mechanisms do Treg cells need? Although efforts to define the suppressive mechanisms used by Treg cells continue, an important question looms large. Is it likely that all these molecules and mechanisms will be crucial for Treg-cell function? There are three broad possibilities.
One, a single, overriding suppressive mechanism is required by all Treg cells Until the entire mechanistic panoply of Treg cells is defined, one cannot completely rule out this possibility. However, this possibility would seem unlikely as none of the molecules and/ or mechanisms that have been defined to date, when blocked or deleted, result in the complete absence of regulatory activity — a consequence that one might predict would result in a ‘Scurfy-like’ phenotype (BOX 1). So, although Treg cells that lack a single molecule, for instance IL-10, IL-35 or granzyme B, exhibit significantly reduced suppressor function, a scurfy phenotype does not ensue. Given that none of the current Treg-cell mechanisms can exclusively claim this distinction, it seems unlikely that any ‘unknown’ molecules or mechanisms could do so either.
Two, multiple, non-redundant mechanisms are required for maximal Treg-cell function In the studies conducted to date, Treg cells that lack various suppressive molecules have been shown to be functionally defective. This favours a scenario where there are multiple mechanisms that can be used by Treg cells but they are non-redundant, with each molecule contributing to the mechanistic whole. At present, this possibility would seem plausible. Indeed, this is supported by the recent analysis of mice possessing a Treg-cell-specific ablation of IL-10 expression, in which enhanced pathology was observed following environmental insult33. One would predict that at some point we should be able to generate knockout mice that lack a particular set of genes which results in a complete loss of Treg-cell activity. For this to be truly non-redundant, this list would probably be restricted and small (2–4 genes).
Three, multiple, redundant mechanisms are required for maximal Treg-cell function With the plethora of regulatory mechanisms described to date and the possibility of more yet to be identified, it is conceivable that there are multiple mechanisms that function redundantly. Such a redundant system would help to mitigate against effector T-cell escape from regulatory control. Also, given the very small size of the Treg-cell population, a sizable arsenal may be required at the height of an effector T-cell attack. Of course, it is possible that a semi-redundant scenario exists.
These possibilities have been discussed from the perspective of there being a single homogeneous Treg-cell population. However, as for helper T cell subsets it remains possible that a few or even many different Treg-cell subsets exist24. Each of these may rely on one or multiple regulatory mechanisms. Several recent studies have provided support for both phenotypic and functional heterogeneity amongst Treg cells. For instance, it has recently been shown that a small sub-population of Treg cells express the chemokine receptor CCR6, which is associated with T cells possessing an effector-memory phenotype102. CCR6+ Treg cells appeared to accumulate in the central nervous systems of mice with experimental autoimmune encephalomyelitis (EAE) suggesting that they may have a prevalent role in controlling responses in inflamed tissues. Heterogeneous expression of HLA-DR has also been suggested to mark different subpopulations of functionally distinct human Treg cells103. Indeed, HLADR positive Treg cells were found to be more suppressive than their DR negative counterparts. One might speculate that their enhanced inhibitory activity is due to DR-mediated ligation of the inhibitory molecule LAG3 expressed by activated effector T cells95,96.
So, if multiple suppressor mechanisms exist, how might these be integrated and used productively by Treg cells in vivo? We would propose the following possible models21. First, a ‘hierarchical’ model in which Treg cells possess many mechanisms that could be used but only one or two that are really crucial and consistently important in a variety of regulatory settings. Second, a ‘contextual’ model where different mechanisms become more or less important depending on the background or context in which the Treg cells reside and the type of target cell that they have to repress. For example, some cell types may be inhibited primarily by cytokines, whereas others are most effectively suppressed through lysis by Treg cells. Alternatively, different mechanisms may be more effective in different tissue compartments or in different disease settings. This notion is supported by the recent analysis of mice in which IL-10 expression was specifically ablated in Treg cells33. Whereas Treg-cell-derived IL-10 was not required for the systemic control of autoimmunity, it did seem to be required from the control of inflammatory events at mucosal interfaces such as the lungs and colon. As a clear picture of the available Treg-cell weaponry emerges, an important challenge will be to determine their relative importance and contribution to Treg-cell function in different disease models.
A hypothesis: effector T cells potentiate Treg-cell function? Most cellular interactions within the immune system are bidirectional, with molecular signals moving in both directions even though the interaction has broader unidirectional intentions (for example, CD4+ T-cell help). However, to date the general perception is that Treg cells suppress and effector T cells capitulate. We hypothesize that this is in fact an incomplete picture and that effector T cells have a very active role in their own functional demise. Three recent observations support this view. First, we have recently examined the molecular signature of activated Treg cells in the presence and absence of effector T cells and were surprised to find that it was strikingly different, with hundreds of genes differentially modulated as a consequence of the presence of effector T cells (C.J.W. and D.A.A.V., unpublished observations). Second, we have shown that Ebi3 and Il12a mRNA are markedly upregulated in Treg cells that were co-cultured with effector T cells, supporting the idea that effector T cells may provide signals which boost IL-35 production in trans58. Third, we found that Treg cells were able to mediate suppression of effector T cells across a permeable membrane when placed in direct contact with effector T cells in the upper chamber of a Transwell™ plate (L.W.C. and D.A.A.V., unpublished observations). Interestingly, this suppression was IL-35 dependent, as Ebi3−/− Treg cells were unable to mediate this ‘long-distance’ suppression. Collectively, these data suggest that it is the ‘induction’, rather than the ‘function’, of Treg-cell suppression that is contact-dependent and that effector T cells have an active role in potentiating Treg-cellmediated suppression. Therefore, we hypothesize that receptor–ligand interactions between the co-cultured CD4+ effector T cells and Treg cells initiate a signalling pathway that leads to enhanced IL-35 secretion and regulatory activity (FIG. 2). While the molecule that mediates this enhanced Treg-cell suppression is unknown, it is possible that IL-2 may serve this function104. Given the contrasting genetic profiles of activated Treg cells in the presence and absence of effector T cells, it seems possible that this interaction may boost the expression of other regulatory proteins. It may well be that effector T cells unwittingly perform the ultimate act of altruism.
Concluding remarks Although significant progress has been made over the last few years in defining the mechanisms that Treg cells use to mediate their suppressive function, there is clearly much that remains to be elucidated and many questions persist. First, are there more undiscovered mechanisms and/ or molecules that mediate Treg-cell suppression? What is clear is that the transcriptional landscape of Treg cells is very different from naive or activated effector T cells. There are literally thousands of genes that are upregulated (or downregulated) in Treg cells compared with effector T cells. Although it seems unlikely that all or many of these will be crucial for Treg-cell function, it is quite possible that a few undiscovered genes might be important. It should be noted that although we are discussing mechanisms here, it is clear that some of these molecules may perform key Treg-cell functions, such as Treg-cell homing and homeostasis, which are likely to indirectly influence their suppressive capacity in vivo but don’t directly contribute to their inhibitory activity. It is also possible that some of these unknown molecules may represent more specific markers for the characterization and isolation of Treg cells, a particularly important issue for the analysis and use of human Treg cells (BOX 2).
Second, which mechanisms are most important? An important but potentially complex challenge will be to determine if a few mechanisms are important in many Treg-cell settings or whether different mechanisms are required in different cellular scenarios. At present it is difficult to assess this objectively as these mechanisms have predominantly been elucidated in different labs using distinct experimental systems and thus none have really been compared in side-by-side experiments. Furthermore, only recently have conditional mutant mice been examined that have a regulatory component specifically deleted in Treg cells33.
It almost goes without saying that although defining the Treg-cell mode of action is of great academic importance, it is also essential in order to develop effective approaches for the clinical manipulation of Treg cells. Given the capacity of Treg cells to control inflammation and autoimmunity, and their implication in blocking effective anti-tumour immunity and preventing sterilizing immunity, it seems probable that a clear understanding of how Treg cells work will present definitive opportunities for therapeutic intervention.
Box 1 Scurfy mice: misplaced mechanistic expectations?
Mice that carry a spontaneous loss-of-function mutation (known as Scurfy mice) or a deletion of Foxp3 develop a fatal autoimmune-like disease with hyperresponsive CD4+ T cells9,12. More recently Foxp3:diptheria toxin receptor (DTR) knockin mice have allowed for the selective depletion of Treg cells following DT treatment105. These mice have been invaluable for dissecting the role of Foxp3 in Treg-cell function. Given the profound phenotype in these mice, there is a general expectation that genetic disruption of any key Treg-cell inhibitory molecule or mechanism would probably result in a Scurfy-like phenotype. Of course, it is also possible that deletion of a key Treg-cell gene may be more synonymous with DT-mediated Treg-cell depletion where Foxp3 may still serve to prevent expression of proinflammatory cytokines105. Nonetheless, this has lead to the notion that if mutant mice don’t have a Scurfy-like or a Treg-cell-depleted phenotype, then the disrupted gene probably isn’t important for Treg-cell function. This may not necessarily be correct. Indeed, it is possible that no mouse lacking a Treg-cell inhibitory effector molecule will ever be generated that develops a profound, spontaneous autoimmune disease21. It should be noted that mutant mice that are Helicobacter spp. and/or Citrobacter rodentium positive may have an exacerbated phenotype, as several studies have shown that opportunistic enteric bacteria can significant exacerbate gut pathology4. Ultimately, the occurrence of disease in knockout mice will depend on whether Treg cells rely on a single or multiple suppressive mechanisms. Given the number of genes induced or modulated by FOXP3, it is probable that a programme of intrinsic and extrinsic regulation is induced that involves multiple proteins9,13. Therefore, it would not be surprising if deletion of a single molecule does not provoke the profound Scurfy-like phenotype seen in mice that lack Foxp3.
Box 2. Treg-cell markers
Identifying discriminatory cell surface markers for the characterization and isolation of Treg cells has always been a critical goal. Although excellent markers exist for murine Treg cells, this goal has remained elusive for human Treg cells. Traditionally, murine and human Treg cells have been characterized as CD4+CD25+ (also known as interleukin-2 receptor α (IL-2Rα)). Indeed, murine Treg cells can be effectively isolated based on staining for CD4+CD25+CD45RBlow expression. However, the purity of isolated human Treg cells has always been an issue because T cells up-regulate CD25 upon activation106. Indeed, during the influenza or allergy season a substantial proportion of human CD4+ T cells can express CD25. Although the identification of forkhead box P3 (Foxp3) as a key regulator of Treg-cell development and function has facilitated their identification in the mouse8, many activated (non-regulatory) human T cells express FOXP3, precluding it as a useful marker for human Treg cells16-20. Consequently, the search for Treg-cell-specific cellsurface markers, particularly in humans, has continued in earnest with a growing number of candidates proposed (reviewed by Zhao and colleagues107). For instance, it was shown that the expression of CD127 (also known as IL-7R) is down-regulated on Treg cells and that this could be used to increase the purity of human Treg-cell isolation. Indeed, there is a 90% correlation between CD4+CD25+CD127low T cells and FOXP3 expression108, 109. In addition, it was recently found that Treg cells expressed a higher level of folate receptor 4 (FR4) compared with activated effector T cells110. It is also important to recognize that Treg cells, like their T helper cell counterparts, may be heterogeneous and thus a collection of cell surface markers could facilitate their isolation and functional characterization. Indeed, such heterogeneity has recently been described based on differential expression of HLA-DR or CCR6102,103. However, the general use of both markers remains to be fully established so it is quite probable that the search for better Treg-cell markers will continue for some time.
Box 3 Induced or adaptive Treg cells: development and mode of action
Naturally occurring FOXP3+CD4+CD25+ Treg cells develop in the thymus and display a diverse T-cell receptor (TCR) repertoire that is specific for self-antigens111,112. However, Treg cells can also be ‘induced’, ‘adapted’ or ‘converted’ from effector T cells during inflammatory processes in peripheral tissues, or experimentally generated as a possible therapeutic29,113,114. For instance, T regulatory 1 cells (Tr1) and T helper 3 cells (Th3) can be generated experimentally by, and mediate their suppressive activity through interleukin-10 (IL-10) and transforming growth factor-β (TGFβ), respectively114,115. Typically, these regulatory populations do not express FOXP3. In vivo, it has recently been suggested that stimulation of mouse effector T cells by CD103+ dendritic cells (DCs) in the presence of TGFβ and retinoic acid induces the generation of Foxp3+ T cells in the gutassociated lymphoid tissue (GALT)116-121. Furthermore, Treg cells can be preferentially induced in the periphery by exposure to αVβ8-integrin-expressing DCs122 or suppressor of cytokine signalling 3 (Socs3) −/− DCs123. Interestingly, independent of its role in generating induced Treg cells, TGFβ may also have an important role in helping to maintain Foxp3 expression in natural Treg cells124, a process that can be blocked by IL-4 or interferon-γ (IFNγ) 125. In contrast to mouse T cells, FOXP3 induction by TCR stimulation in the presence of TGFβ in human T cells does not confer a regulatory phenotype20. The mechanism of action of adaptive Treg cells may not necessarily be restricted to suppressive cytokines. Indeed, human adaptive Treg cells (CD4+CD45RA+ T cells stimulated with CD3- and CD46-specific antibodies) have also been shown to express granzyme B and killing target cells in a perforin-dependent manner126. Treg cells often have a restricted specificity for particular cell types, tumours or foreign antigens127. Therefore, induced Treg cells may be ideally suited to respond to infectious agents. This may also be of particular importance in the GALT and in the tumour microenvironment where TGFβ drives the conversion of induced Treg cells118,128. A significant challenge in deciphering data from in vivo experiments is to assess the contribution of natural Treg cells versus induced Treg cells, and to determine whether inhibitory molecules, such as IL-10 or TGFβ, were derived from the former or the latter (or elsewhere).
Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers.
Keisuke Kataoka, Yuichi Shiraishi, Yohei Takeda, Seiji Sakata, et al.
Nature may 23,2016; http://dx.doi.org:/10.1038/nature18294
Successful treatment of many patients with advanced cancer using antibodies against programmed cell death 1 (PD-1; also known as PDCD1) and its ligand (PD-L1; also known as CD274) has highlighted the critical importance of PD-1/PD-L1-mediated immune escape in cancer development1, 2, 3, 4, 5, 6. However, the genetic basis for the immune escape has not been fully elucidated, with the exception of elevated PD-L1 expression by gene amplification and utilization of an ectopic promoter by translocation, as reported in Hodgkin and other B-cell lymphomas, as well as stomach adenocarcinoma6, 7, 8, 9, 10. Here we show a unique genetic mechanism of immune escape caused by structural variations (SVs) commonly disrupting the 3′ region of the PD-L1 gene. Widely affecting multiple common human cancer types, including adult T-cell leukaemia/lymphoma (27%), diffuse large B-cell lymphoma (8%), and stomach adenocarcinoma (2%), these SVs invariably lead to a marked elevation of aberrant PD-L1 transcripts that are stabilized by truncation of the 3′-untranslated region (UTR). Disruption of the Pd-l1 3′-UTR in mice enables immune evasion of EG7-OVA tumour cells with elevated Pd-l1 expression in vivo, which is effectively inhibited by Pd-1/Pd-l1 blockade, supporting the role of relevant SVs in clonal selection through immune evasion. Our findings not only unmask a novel regulatory mechanism of PD-L1 expression, but also suggest that PD-L1 3′-UTR disruption could serve as a genetic marker to identify cancers that actively evade anti-tumour immunity through PD-L1 overexpression.
Viruses are a dominant driver of protein adaptation in mammals.
David Enard, Le Cai, Carina Gwennap and Dmitri A Petrov.
eLife May 16, 2016; 5:e12469. http://dx.doi.org/10.7554/eLife.12469
Viruses interact with hundreds to thousands of proteins in mammals, yet adaptation against viruses has only been studied in a few proteins specialized in antiviral defense. Whether adaptation to viruses typically involves only specialized antiviral proteins or affects a broad array of virus-interacting proteins is unknown. Here, we analyze adaptation in ~1300 virus-interacting proteins manually curated from a set of 9900 proteins conserved in all sequenced mammalian genomes. We show that viruses (i) use the more evolutionarily constrained proteins within the cellular functions they interact with and that (ii) despite this high constraint, virus-interacting proteins account for a high proportion of all protein adaptation in humans and other mammals. Adaptation is elevated in virus-interacting proteins across all functional categories, including both immune and non-immune functions. We conservatively estimate that viruses have driven close to 30% of all adaptive amino acid changes in the part of the human proteome conserved within mammals. Our results suggest that viruses are one of the most dominant drivers of evolutionary change across mammalian and human proteomes.
Purdue scientists use adaptors to advance CAR-T therapy
by Oliver Worsley | May 25, 2016
http://www.fiercebiotech.com/research/purdue-scientists-use-adaptors-to-advance-car-t-therapy
Chimeric antigen receptor (CAR) T cells, developed in the 1990s, are a genetically engineered type of T cell that can target a specific cancer. Now, scientists at Purdue University say they’ve made improvements in this strategy–overcoming the several limitations of traditional CAR-T therapy.
Purdue professor of chemistry Philip Low and his team presented their findings at the American Association for Cancer Research meeting in New Orleans last month.
T cells are a type of immune cell that recognizes and clears the body of invading cells or pathogens, like cancer. They are fine-tuned by the immune system in order to specifically target and kill these foreign invaders–but cancer cells may respond by jumping these safety barriers.
CAR-T therapy was therefore proposed and has been recently used for cancer treatment. It has been hailed for its promising remission rates after early stage clinical trials for acute lymphoblastic leukemia.
“The problem is that the traditional engineered T-cell treatment can be too effective, sometimes killing tumor cells too fast and triggering a toxic reaction in a patient, and sometimes not stopping once the tumor has been destroyed and continuing to seek out and destroy healthy cells important to bodily functions,” Low said in a university news release. “We have found a potential way to control the engineered immune cells to overcome the limitations posed by CAR T-cell therapy.”
They did this by teaming up with Endocyte ($ECYT) scientist Haiyan Chu and designing CAR T cells that require activation by a small molecule adaptor before proceeding. In this way, they can carefully control the amount of active CAR T cells in the circulation.
So far, they have only tried the novel therapy in animal models, but when they tested it in mice they observed antitumor activity only when both the CAR T cells and the correct adaptor molecules were present.
Low believes it will allow clinicians to target multiple cancer subtypes at once. “Most tumors are heterogeneous and contain cancer cells that express different characteristics, including having different tumor-specific proteins on their surface,” he said in the release. “The cancer-targeting molecule on the adaptor we designed can be swapped out to target different molecules on other unrelated cancer cell surfaces. The idea is that a mixture of these adaptors can be given to a patient so that a single CAR T cell clone can be targeted to all of the relevant cancer subtypes in a patient.”
“In the past a new CAR T cell had to be designed for each desired cancer target,” Low said. “This system uses the same blind CAR T cell for all treatments. The adaptor molecule is what needs to be changed, and it is far easier to manipulate and swap pieces in and out of it than the T cells.”
– here’s the release
Related Articles:
Novartis-allied scientist at Penn creates a new CAR-T model for solid tumors
Baxalta dives into CAR-T with a $1.6B bet
Kite’s CAR-T gets a ‘breakthrough’ tag with pivotal data on the way
Purdue research may expand engineered T-cell cancer treatment
PURDUE UNIVERSITY
http://www.eurekalert.org/multimedia/pub/116141.php
A graphic depicting the activation and inactivation of CAR T cells through a small molecule adaptor is shown. Philip S. Low, Purdue’s Ralph C. Corley Distinguished Professor of Chemistry and director of the Purdue Center for Drug Discovery, and graduate student Yong Gu Lee led a team that designed new engineered CAR T cells that must be activated and targeted by a small molecule adaptor before they can kill cancer cells. The system has the potential to control the engineered cells to overcome existing limitations in CAR T-cell therapy. CREDIT Purdue University image courtesy of Yong Gu Lee
Purdue University researchers may have figured out a way to call off a cancer cell assassin that sometimes goes rogue and assign it a larger tumor-specific “hit list.”
T cells are the immune system’s natural defense against cancer and other harmful entities in the human body. However, the cells must be activated and taught by the immune system to recognize cancer cells in order to seek out and destroy them. Unfortunately, many types of cancer manage to thwart this process.
In the 1990s scientists found a way to genetically engineer T cells to recognize a specific cancer. These engineered T cells, called chimeric antigen receptor, or CAR, T cells, have been recently used as treatment for cancer, said Philip S. Low, Purdue’s Ralph C. Corley Distinguished Professor of Chemistry and director of the Purdue Center for Drug Discovery who led the work.
“The problem is that the traditional engineered T-cell treatment can be too effective, sometimes killing tumor cells too fast and triggering a toxic reaction in a patient, and sometimes not stopping once the tumor has been destroyed and continuing to seek out and destroy healthy cells important to bodily functions,” Low said. “We have found a potential way to control the engineered immune cells to overcome the limitations posed by CAR T-cell therapy.”
Low and Purdue graduate student Yong Gu Lee collaborated with Endocyte Inc. scientist Haiyan Chu to design genetically engineered CAR T cells that must be activated and targeted by a small molecule adaptor before they can kill cancer cells. The technology has been tested in animal models but no human trials have been performed. A poster presentation describing the work was presented Tuesday (April 19, 2016) at the American Association for Cancer Research annual meeting in New Orleans.
“While the traditional CAR T cells could remain and replicate in the human body for many years, the adaptors we have created are expected to be excreted fairly quickly,” Lee said. “By controlling the level of adaptors in the system, we can control the numbers and potencies of active CAR T cells. Those that aren’t stimulated by an adaptor molecule are blind and do not recognize or target any cells. Eventually, if they remain inactive for a while, they should die and be eliminated from the body.”
A study in mice showed the anti-tumor activity was induced only when both the engineered CAR T cell and the correct adaptor molecules were present.
The system also offers the potential to treat multiple cancer subtypes at once, Low said.
“Most tumors are heterogeneous and contain cancer cells that express different characteristics, including having different tumor-specific proteins on their surface,” he said. “The cancer-targeting molecule on the adaptor we designed can be swapped out to target different molecules on other unrelated cancer cell surfaces. The idea is that a mixture of these adaptors can be given to a patient so that a single CAR T cell clone can be targeted to all of the relevant cancer subtypes in a patient.”
The adaptor molecule serves as a bridge between the CAR T-cell and the cancer cell. It is made with a yellow dye called fluorescein isothiocyanate on one end, to which the engineered CAR T cells have been designed to bind, and a cancer-targeting molecule on the other.
Low’s research has focused on the design and synthesis of technologies for targeted delivery of therapeutic and imaging agents to treat cancer, inflammatory and autoimmune diseases, and infectious diseases.
He has developed molecules that target folate-receptors and prostate-specific membrane antigen on the surfaces of cancer cells. Approximately 85 percent of ovarian cancers; 80 percent of endometrial and lung cancers; and 50 percent of breast, kidney and colon cancers express folate receptors on their cellular surfaces. Prostate-specific membrane antigen receptors are found on nearly 90 percent of all prostate cancers. Other tumor-specific ligands developed by Low’s lab can target each of the other major human cancers, he said.
Each CAR T cell has thousands of receptors on its surface to which an adaptor molecule can bind. One CAR T cell could have a variety of adaptor molecules bound to its surface and the cancer cell it targets will depend on which of those adaptors first encounters a targeted cancer cell. Once the CAR T cell binds to a cancer cell, it begins the process of destroying it. When that process is complete, the CAR T cell is released and can bind to a new cancer cell, he said.
“In the past a new CAR T cell had to be designed for each desired cancer target,” Low said. “This system uses the same blind CAR T cell for all treatments. The adaptor molecule is what needs to be changed, and it is far easier to manipulate and swap pieces in and out of it than the T cells.”
In addition to Low, Chu and Lee, members of the research group include Purdue postdoctoral research associates at the time of the study Srinivasarao Tenneti and Ananda Kumar Kanduluru.
Drug discovery is one of the priorities within Purdue Moves, an initiative designed to broaden the university’s global impact and enhance educational opportunities for its students. All of the moves fall into four broad categories: science, technology, engineering and math (STEM) leadership; world-changing research; transformative education; and affordability and accessibility.
The Purdue University Center for Drug Discovery supports more than 100 faculty in six colleges with research focused on several major disease categories: cancer; diabetes, obesity and cardiovascular; immune and infectious disease; and neurological disorders and trauma.
The center and drug discovery initiative builds upon Purdue’s strengths along all points of the drug discovery pipeline, including 14 core units to provide shared resources for analysis, screening, synthesis and testing of potential therapeutic compounds.
With more than 44 Purdue-developed compounds at various stages of preclinical development, and 16 in human clinical trials, Purdue is among the most productive universities in the world of drug discovery.
The center also is aligned with the university’s recently announced $250 million investment in the life sciences.
Endocyte Inc., a Purdue Research Park-based company that develops receptor-targeted therapeutics for the treatment of cancer and autoimmune diseases, funded the study, holds exclusive rights to the technology and assisted Purdue researchers in the development of the technology. Low is a founder and chief science officer of Endocyte Inc. and serves on the Endocyte board of directors.
AACR press release: http://www.aacr.org/Newsroom/Pages/News-Release-Detail.aspx?ItemID=874#.VxZFs2N8V0c
Endocyte press release: http://investor.endocyte.com/releasedetail.cfm?ReleaseID=965753
ABSTRACT
A Universal Remedy for CAR T Cell Limitations
Yong Gu Lee, Haiyan Chu, Srinivasarao Tenneti, Ananda Kumar Kanduluru, Philip S. Low
Chimeric antigen receptor (CAR) T cells show significant potential for treating cancer due to their tumor-specific activation and ability to focus their killing activity on cells that express a tumor antigen. Unfortunately, this promising therapeutic technology is still limited by: (1) an inability to control the rate of cytokine release and tumor lysis; (2) the absence of an “off switch” that can terminate cytotoxic activity when tumor eradication is complete; (3) a failure to eliminate tumor cells that do not express the targeted antigen; and (4) a requirement to generate a different CAR T cell for each unique tumor antigen. In order to address these limitations, we have exploited a low molecular weight bi-specific adaptor molecule that must bridge between the CAR T cell and its targeted tumor cell by simultaneously binding to the chimeric antigen receptor on the CAR T cell and the unique antigen on the tumor. Using this bispecific adaptor, one can control CAR T cell cytotoxicity by adjusting the concentration and rate of administration of the adaptor. Because the half life of the adapter is <20 minutes in vivo, termination of CAR T cell killing can be accomplished by cessation of adapter administration. Moreover, when heterogeneous tumors containing cells that express orthogonal antigens must be treated, the same CAR T cell can be targeted to multiple antigens by attachment of the same CAR ligand to the appropriate selection of tumor-specific ligands. Finally, when the targeted tumor antigen is also expressed at low levels on normal cells, tumor specificity can be achieved by adjusting the affinity of the tumor-specific ligand to enable CAR T cell engagement only when a highly multivalent interaction is possible. To experimentally demonstrate the aforementioned benefits of using low molecular weight bispecific adaptors, CAR T cells were constructed by fusing an anti-fluorescein isothiocyanate (FITC) scFv to a CD3 zeta chain containing the intracellular domain of CD137 (i.e. CAR4-1BBZ T cells). Then, to enable their tumor-specific cytotoxicity, a bispecific adaptor molecule comprised of fluorescein linked to a small organic ligand with high affinity and specificity for a tumor-specific antigen (FITC-SMC) was synthesized. For these studies, the tumor-specific ligands were: i) folate for recognition of the folate receptor that is over-expressed on ~1/3 of human cancers, ii) DUPA for binding to prostate specific membrane antigen that is over-expressed on prostate cancers, and iii) NK-1R ligand that is over-expressed on neuroendocrine tumors. The ability of the same clone of CAR4-1BBZ T cells to eliminate tumors expressing each of the above antigens was then demonstrated by administration of the desired FITC-SMC to mice injected with the CAR4-1BBZ T cells. Our data show that anti-tumor activity: i) is only induced when both CAR4-1BBZ T cells and the correct antigen-specific FITC-SMC are present, ii) anti-tumor activity and toxicity can be sensitively controlled by adjusting the dosing of FITC-SMC, and iii) treatment of antigenically heterogeneous tumors can be achieved by administration of a mixture of the desired FITC-SMCs. Taken together, these data show that many of the limitations of CAR T cell technology can be addressed by use of a bi-specific adaptor molecule to mediate tumor cell recognition and killing.
CTLA-4 found in dendritic cells suggests New cancer treatment possibilities
Matthew Halpert, et al. Dendritic Cell Secreted CTLA-4 Regulates the T-cell Response by Downmodulating Bystander Surface B7.
Stem Cells and Development, 2016; http://dx.doi.org:/10.1089/scd.2016.0009
Both dendritic cells and T cells are important in triggering the immune response, whereas antigen presenting dendritic cells act as the “general” leading T cells “soldiers” to chase and eliminate enemies in the battle against cancer. The well-known immune checkpoint break, CTLA-4, is believed to be present only in T cells (and cells of the same lineage). However, a new study published in Stem Cells and Development suggests that CTLA-4 also presents in dendritic cells. It further explores the mechanism on how turning off the dendritic cells in the immune response against tumors.
The remarkable functional plasticity of professional antigen-presenting cells (APCs) allows the adaptive immune system to respond specifically to an incredibly diverse array of potential pathogenic insults; nonetheless, the specific molecular effectors and mechanisms that underpin this plasticity remain poorly characterized. Cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), the target of the blockbuster cancer immunotherapeutic ipilimumab, is one of the most well-known and well-studied members of the B7 superfamily and negatively regulates T cell responses by a variety of known mechanisms. Although CTLA-4 is thought to be expressed almost exclusively among lymphoid lineage hematopoietic cells, a few reports have indicated that nonlymphoid APCs can also express the CTLA-4 mRNA transcript and that transcript levels can be regulated by external stimuli. In this study, we substantially build upon these critical observations, definitively demonstrating that mature myeloid lineage dendritic cells (DC) express significant levels of intracellular CTLA-4 that they constitutively secrete in microvesicular structures. CTLA-4(+) microvesicles can competitively bind B7 costimulatory molecules on bystander DC, resulting in downregulation of B7 surface expression with significant functional consequences for downstream CD8(+) T-cell responses. Hence, the data indicate a previously unknown role for DC-derived CTLA-4 in immune cell functional plasticity and have significant implication for the design and implementation of immunomodulatory strategies intended to treat cancer and infectious disease.
Non-invasive strategy to guide personalized cancer immunotherapy
Cancer immunotherapy is the rising hope to offer ultimate solutions for cancer. Neoantigens, derived from products of mutated genes in tumor cells, are found to be closely related to the efficacy of cancer immunotherapies. A non-invasive approach to identify unique, patient-specific neoantigens has been advanced by Dr. Steven Rosenberg’s group. A recent article published in Nature Medicine reported that a small population of circulating CD8+PD-1+ tumor-reactive T lymphocytes can be used to identify neoantigens, in addition to tumor-infiltrating T cells. The study paves the way for designing personalized cancer immunotherapy with a novel non-invasive approach.
Gros, A. et al.
Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients.
Nat. Med. (2016) http://dx. doi.org:/10.1038/nm.4501
Detection of lymphocytes that target tumor-specific mutant neoantigens-derived from products encoded by mutated genes in the tumor-is mostly limited to tumor-resident lymphocytes, but whether these lymphocytes often occur in the circulation is unclear. We recently reported that intratumoral expression of the programmed cell death 1 (PD-1) receptor can guide the identification of the patient-specific repertoire of tumor-reactive CD8(+) lymphocytes that reside in the tumor. In view of these findings, we investigated whether PD-1 expression on peripheral blood lymphocytes could be used as a biomarker to detect T cells that target neoantigens. By using a high-throughput personalized screening approach, we identified neoantigen-specific lymphocytes in the peripheral blood of three of four melanoma patients. Despite their low frequency in the circulation, we found that CD8(+)PD-1(+), but not CD8(+)PD-1(-), cell populations had lymphocytes that targeted 3, 3 and 1 unique, patient-specific neoantigens, respectively. We show that neoantigen-specific T cells and gene-engineered lymphocytes expressing neoantigen-specific T cell receptors (TCRs) isolated from peripheral blood recognized autologous tumors. Notably, the tumor-antigen specificities and TCR repertoires of the circulating and tumor-infiltrating CD8(+)PD-1(+) cells appeared similar, implying that the circulating CD8(+)PD-1(+) lymphocytes could provide a window into the tumor-resident antitumor lymphocytes. Thus, expression of PD-1 identifies a diverse and patient-specific antitumor T cell response in peripheral blood, providing a novel noninvasive strategy to develop personalized therapies using neoantigen-reactive lymphocytes or TCRs to treat cancer.
PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors
Anti-PD-1 is poised to be a blockbuster, which other immune-checkpoint targeting drugs are on the horizon?
Clinical studies of anti-immune-checkpoint protein therapeutics have shown not only an improved overall survival, but also a long-term durable response, compared to chemotherapy and genomically-targeted therapy. To expand the success of immune-checkpoint therapeutics into more tumor types and improving efficacy in difficult-to-treat tumors, additional targets involved in checkpoint-blockade need to be explored, as well as testing the synergy between combining approaches.
Currently, CTLA-4 and PD-1/PD-L1 are furthest along in development, and have shown very promising results in metastatic melanoma patients. This is just a fraction of targets involved in the checkpoint-blockade pathway. Several notable targets include:
- LAG-3 – Furthest along in clinical development with both a fusion protein and antibody approach, antibody apporach being tested in combination with anti-PD-1
- TIM-3 – Also in clinical development. Pre-clinical studies indicate that it co-expresses with PD-1 on tumor-infiltrating lymphocytes. Combination with anti-PD-improves anti-tumor response
- VISTA – Antibody targeting VISTA was shown to improve anti-tumor immune response in mice
In addition, there are also co-stimulatory factors that are also being explored as viable therapeutic targets
- OX40 – Both OX40 and 4-1BB are part of the TNF-receptor superfamily. Phase I data shows acceptable safety profile, and evidence of anti-tumor response in some patients
- 4-1BB – Phase I/II data on an antibody therapeutic targeting OX40 shows promising clinical response for melanoma, renal cell carcinoma and ovarian cancer.
- Inducible co-stimulator (ICOS) – Member of the CD28/B7 family. Its expression was found to increase upon T-cell activation. Anti-CTLA-4 therapy increases ICOS-positive effector T-cells, indicating that it may work in synergy with anti-CTLA-4. Clinical trials of anti-ICOS antibody are planned for 2015.
Targeting single immune-checkpoint proteins has proven to be clinically effective at treating specific tumor types; can targeting two different proteins synergize effects?
Despite the success of targeting immune-checkpoint proteins, such as CTLA-4, PD-1, LAG-3, TIM-3 among others, percentages of patient response vary and rarely exceed 50%. It is highly tempting to speculate a strategy of dual-targeting of these checkpoint proteins. A recent presentation at the Keystone Symposium for Tumor Immunology: Multidisciplinary Science Driving Combination Therapy detailed findings of dual-targeting two immune-checkpoint proteins in mouse tumor models. Their key findings are summarized below:
- Dual-targeting PD-1 and LAG-3 demonstrates superior efficacy over blocking either target alone
- In addition to previous reported data on superior dual-targeting efficacy against fibrosarcoma (Sa1N) and colorectal adenosarcoma (MC38) tumor types1, anti-tumor activity against myeloma (SC J558L) and B-cell lymphoma (A20) hematological tumor types were also reported to be effacious.2
These exciting pre-clinical findings may result in further exploration of dual-targeting antibodies in the clinic, either as combination of existing antibody therapies, or as a new bi-specific antibody therapeutic.
Camelid single domain antibodies are a novel bi-specific antibody platform that may be used to develop a new generation of dual-targeting antibodies against multiple immune-checkpoint proteins.
New insight behind the success of fighting cancer by targeting immune checkpoint proteins
Immune checkpoint blockade has proven to be highly successful in the clinic at treating aggressive and difficult-to-treat forms of cancer. The mechanism of the blockade, targeting CTLA-4 and PD-1 receptors which act as on/off switches in T cell-mediated tumor rejection, is well understood. However, little is known about the tumor antigen recognition profile of these affected T-cells, once the checkpoint blockade is initiated.
In a recent published study, the authors used genomics and bioinformatics approaches to identify critical epitopes on 3-methylcholanthrene induced sarcoma cell lines, d42m1-T3 and F244. CD8+ T cells in anti-PD-1 treated tumor bearing mice were isolated and fluorescently labeled with tetramers loaded with predicted mutant epitopes. Out of 66 predicted mutants, mLama4 and mAlg8 were among the highest in tetramer-positive infiltrating T-cells. To determine whether targeting these epitopes alone would yield similar results as anti-PD-1 treatment, vaccines against these two epitopes were developed and tested in mice. Prophylactic administration of the combined vaccine against mLama4 and mAlg8 yielded an 88% survival in tumor bearing mice, thus demonstrating that these two epitopes are the major antigenic targets from checkpoint-blockade and therapies against these two targets are similarly efficacious.
In addition to understanding the mechanism, identification of these tumor-specific mutant antigens is the first step in discovering the next wave of cancer immunotherapies via vaccines or antibody therapeutics. Choosing the right antibody platform can speed the discovery of a new therapeutics against these new targets. Single domain antibodies have the advantage of expedited optimization, flexibility of incorporating multiple specificity and functions, superior stability, and low COG over standard antibody approaches.
Myeloid-derived-suppressor cells as regulators of the immune system
Dmitry I. Gabrilovich and Srinivas Nagaraj Nat Rev Immunol. 2009 March ; 9(3): 162–174. http://dx.doi.org:/10.1038/nri2506
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells that expands during cancer, inflammation and infection, and that has a remarkable ability to suppress T-cell responses. These cells constitute a unique component of the immune system that regulates immune responses in healthy individuals and in the context of various diseases. In this Review, we discuss the origin, mechanisms of expansion and suppressive functions of MDSCs, as well as the potential to target these cells for therapeutic benefit.
The first observations of suppressive myeloid cells were described more than 20 years ago in patients with cancer1-3. However, the functional importance of these cells in the immune system has only recently been appreciated due to accumulating evidence that has demonstrated their contribution to the negative regulation of immune responses during cancer and other diseases. It is now becoming increasingly clear that this activity is contained within a population known as myeloid-derived suppressor cells (MDSCs). Features common to all MDSCs are their myeloid origin, immature state and a remarkable ability to suppress T-cell responses (Box 1). In addition to their suppressive effects on adaptive immune responses, MDSCs have also been reported to regulate innate immune responses by modulating the cytokine production of macrophages4. Non-immunological functions of MDSC have also been described, such as the promotion of tumour angiogenesis, tumour-cell invasion and metastasis. However, as a discussion of these aspects of MDSC biology is beyond the scope of this article, the reader is referred to another recent Review on this topic5.
MDSCs represent an intrinsic part of the myeloid-cell lineage and are a heterogeneous population that is comprised of myeloid-cell progenitors and precursors of myeloid cells. In healthy individuals, immature myeloid cells (IMCs) generated in bone marrow quickly differentiate into mature granulocytes, macrophages or dendritic cells (DCs). In pathological conditions such as cancer, various infectious diseases, sepsis, trauma, bone marrow transplantation or some autoimmune disorders, a partial block in the differentiation of IMCs into mature myeloid cells results in an expansion of this population. Importantly, the activation of these cells in a pathological context results in the upregulated expression of immune suppressive factors such as arginase (encoded by ARG1) and inducible nitric oxide synthase (iNOS; also known as NOS2) and an increase in the production of NO (nitric oxide) and reactive oxygen species (ROS). Together, this results in the expansion of an IMC population that has immune suppressive activity; these cells are now collectively known as MDSCs. In this
Origin and subsets of MDSCs It is important to note that MDSCs that are expanded in pathological conditions (see later) are not a defined subset of myeloid cells but rather a heterogeneous population of activated IMCs that have been prevented from fully differentiating into mature cells. MDSCs lack the expression of cell-surface markers that are specific for monocytes, macrophages or DCs and are comprised of a mixture of myeloid cells with granulocytic and monocytic morphology6. Early studies showed that 1–5% of MDSCs are able to form myeloid-cell colonies7-9 and that about one third of this population can differentiate into mature macrophages and DCs in the presence of appropriate cytokines in vitro and in vivo7-9. In mice, MDSCs are characterized by the co-expression of the myeloid lineage differentiation antigen Gr1 (also known as Ly6G) and CD11b (also known as αM-integrin)10. Normal bone marrow contains 20–30% of cells with this phenotype, but these cells make up only a small proportion (2–4%) of spleen cells and are absent from the lymph nodes in mice (Fig. 1). In humans, MDSCs are most commonly defined as CD14-CD11b+ cells or, more narrowly, as cells that express the common myeloid marker CD33 but lack the expression of markers of mature myeloid and lymphoid cells and the MHC-class-II molecule HLA-DR11, 12. MDSCs have also been identified within a CD15+ population in human peripheral blood13. In healthy individuals, immature myeloid cells with described above phenotype comprise ∼0.5% of peripheral blood mononuclear cells.
Recently, the morphological heterogeneity of these cells has been defined more precisely in part based on their expression of Gr1. Notably, Gr1-specific antibodies bind to both Ly6G and Ly6C, which are encoded by separate genes. However, these epitopes are recognized by different antibodies specific for each individual epitopes: anti-Ly6C and anti-Ly6G. Granulocytic MDSCs have a CD11b+Ly6G+Ly6Clow phenotype, whereas MDSCs with monocytic morphology are CD11b+Ly6G-Ly6Chigh 6,14. Importantly, evidence indicates that these two subpopulations may have different functions in cancer and infectious and autoimmune diseases15-17. During the analysis of ten different experimental tumour models, we found that both of these subsets of MDSCs were expanded. In most cases, however, the expansion of the granulocytic MDSC population was much greater than that of the monocytic subset6 and, interestingly, the two subpopulations used different mechanisms to suppress Tcell function (see later). In addition, the ability to differentiate into mature DCs and macrophages in vitro has been shown to be restricted to monocytic MDSCs6.
In recent years, several other surface molecules have been used to identify additional subsets of suppressive MDSCs, including CD80 (also known as B7.1)18, CD115 (the macrophage colony-stimulating factor receptor)19, 20 and CD124 (the IL-4 receptor α-chain)20. In our own studies, we observed that many MDSCs in tumour-bearing mice co-express CD115 and CD1246; however, direct comparison of MDSCs from tumour-bearing mice and Gr1+CD11b+ cells from naive mice showed that they expressed similar levels of CD115 and CD124. In addition, sorted CD115+ or CD124+ MDSCs from EL-4 tumour-bearing mice had the same ability to suppress T-cell proliferation on a per cell basis as did CD115- or CD124-MDSCs. This suggests that, although these molecules are associated with MDSCs, they might not be involved in the immunosuppressive function of these cells in all tumour models.
Overall, current data suggest that MDSCs are not a defined subset of cells but rather a group of phenotypically heterogeneous myeloid cells that have common biological activity.
MDSCs in pathological conditions MDSCs were first characterized in tumour-bearing mice or in patients with cancer. Inoculation of mice with transplantable tumour cells, or the spontaneous development of tumours in transgenic mice with tissue-restricted oncogene expression, results in a marked systemic expansion of these cells (Fig. 1 and Table 1). In addition, up to a tenfold increase in MDSC numbers was detected in the blood of patients with different types of cancer11, 12, 21, 22. In many mouse tumour models, as many as 20–40% of nucleated splenocytes are represented by MDSCs (in contrast to the 2-4% seen in normal mice). In addition, these cells are found in tumour tissues and in the lymph nodes of tumour-bearing mice.
Although initial observations and most of the current information regarding the role of MDSCs in immune responses has come from studies in the cancer field, accumulating evidence has shown that MDSCs also regulate immune responses in bacterial and parasitic infections, acute and chronic inflammation, traumatic stress, surgical sepsis and transplantation. A systemic expansion of both the granulocytic and monocytic subset of MDSCs was observed in mice primed with Mycobacterium tuberculosis as part of complete Freund’s adjuvant (CFA). Acute Trypanosoma cruzi infection, which induces T-cell activation and increased production of interferon-γ (IFNγ), also leads to the expansion of MDSCs23, 24. A similar expansion of MDSCs has been reported during acute toxoplasmosis25, polymicrobial sepsis26, acute infection with Listeria monocytogenes or chronic infection with Leishmania major27 and infection with helminths28,29, 30, Candida albicans31 or Porphyromonas gingivalis32.
MDSC expansion is also associated with autoimmunity and inflammation. In experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, an increase in CD11b+Ly6ChiLy6G− MDSCs was observed in the spleen and blood and these cells were found to enter the central nervous system during the inflammatory phase of the disease16. A significant increase in the number of MDSCs was also detected in experimental autoimmune uveoretinitis, an animal model of human intraocular inflammatory disease33, in the skin and spleens of mice that were repeatedly treated with a contact sensitizer to induce an inflammatory response34 and in inflammatory bowel diseases35. MDSCs were also found to infiltrate the spleen and suppress T-cell function in a model of traumatic stress36. Finally, a significant transient increase in MDSC numbers was also demonstrated in normal mice following immunization with different antigens such as ovalbumin or peptide together with CFA, a recombinant vaccinia virus expressing interleukin-2 (IL-2) or staphylococcal enterotoxin A 8, 37, 38. Therefore, current information clearly indicates that the expansion of an immunosuppressive MDSC population is frequently observed in many pathological conditions.
Expansion and activation of MDSCs Studies have demonstrated that the MDSC population is influenced by several different factors (Table 1), which can be divided into two main groups. The first group includes factors that are produced mainly by tumour cells and promote the expansion of MDSC through stimulation of myelopoiesis and inhibiting of the differentiation of mature myeloid cells. The second group of factors is produced mainly by activated T cells and tumour stroma, and is involved in directly activating MDSCs.
Mechanisms of MDSC expansion—Factors that induce MDSC expansion can include cyclooxygenase-2 (COX2), prostaglandins 39-41, stem-cell factor (SCF)39, macrophage colony-stimulating factor (M-CSF), IL-642, granulocyte/macrophage colony-stimulating factor (GM-CSF)41 and vascular endothelial growth factor (VEGF) 43 (Table 1). The signalling pathways in MDSCs that are triggered by most of these factors converge on Janus kinase (JAK) protein family members and signal transducer and activator of transcription 3 (STAT3) (Fig. 2), which are signalling molecules that are involved in cell survival, proliferation, differentiation and apoptosis44. STAT3 is arguably the main transcription factor that regulates the expansion of MDSCs. MDSCs from tumour-bearing mice have markedly increased levels of phosphorylated STAT3 compared with IMCs from naive mice45. Exposure of haematopoietic progenitor cells to tumour-cell-conditioned medium resulted in the activation of JAK2 and STAT3 and was associated with an expansion of MDSCs in vitro, whereas inhibition of STAT3 expression in haematopoietic progenitor cells abrogated the effect of tumour-derived factors on MDSC expansion46. Ablation of STAT3 expression in conditional knockout mice or selective STAT3 inhibitors markedly reduced the expansion of MDSCs and increased T-cell responses in tumour-bearing mice45, 47. STAT3 activation is associated with increased survival and proliferation of myeloid progenitor cells, probably through upregulated expression of STAT3 target genes including B-cell lymphoma XL, (BCL-XL), cyclin D1, MYC and survivin. So, abnormal and persistent activation of STAT3 in myeloid progenitors prevents their differentiation into mature myeloid cells and thereby promotes MDSC expansion.
Recent findings suggest that STAT3 also regulates MDSC expansion through inducing the expression of S100A8 and S100A9 proteins. In addition, it has been shown that MDSCs also express receptors for these proteins on their cell surface. S100A8 and S100A9 belong to the family of S100 calcium-binding proteins that have been reported to have an important role in inflammation48. STAT3-dependent upregulation of S100A8 and S100A9 expression by myeloid progenitor cells prevented their differentiation and resulted in the expansion of MDSCs in the spleens of tumor-bearing and naive S100A9-transgenic mice. By contrast, MDSCs did not expand in the peripheral blood and spleens of mice deficient for S100A9 following challenge with tumour cells or CFA49. In a different study, S100A8 and S100A9 proteins were shown to promote MDSC migration to the tumour site through binding to carboxylated N-glycan receptors expressed on the surface of these cells 50. Blocking the binding of S100A8 and S100A9 to their receptors on MDSCs in vivo with a carboxylated glycan-specific antibody reduced MDSC levels in the blood and secondary lymphoid organs of tumour-bearing mice50. In human colon tumour tissue, and in a mouse model of colon cancer, myeloid progenitor cells expressing S100A8 and S100A9 have been shown to infiltrate regions of dysplasia and adenoma. Furthermore, administration of a carboxylated glycan-specific monoclonal antibody (mAbGB3.1) was found to markedly reduced chronic inflammation and tumorigenesis51. Although the mechanisms involved require further study, these studies suggest that S100A9 and/or S100A8 proteins have a crucial role in regulating MDSC expansion, and may provide a link between inflammation and immune suppression in cancer.
Mechanisms of MDSC activation—Recently, it has become clear that the suppressive activity of MDSCs requires not only factors that promote their expansion but those that induce their activation. The expression of these factors, which are produced mainly by activated T cells and tumour stromal cells, is induced by different bacterial or viral products or as a result of tumour cell death 26. These factors, which include IFNγ, ligands for Toll-like receptors (TLRs), IL-13, IL-4 and transforming growth factor-β (TGFβ), activate several different signalling pathways in MDSCs that involve STAT6, STAT1, and nuclear factor-κB (NF-κB) (Fig. 2).
Blockade of IFNγ, which is produced by activated T cells, abolishes MDSC-mediated T-cell suppression17, 52. STAT1 is the major transcription factor activated by IFNγ-mediated signalling and, in the tumour microenvironment, the upregulation of ARG1 and iNOS expression in MDSCs involved a STAT1-dependent mechanism. Indeed, MDSCs from Stat1-/- mice failed to up regulate ARG1 and iNOS expression and therefore did not inhibit Tcell responses53. Consistent with other findings, IFNγ produced by activated T cells and by MDSCs triggered iNOS expression and synergized with IL-4Rα and ARG1 pathways that have been implicated in the suppressive function of MDSCs20.
An important role for the signalling pathway that involves IL-4 receptor α-chain (IL-4Rα) and STAT6 (which is activated by the binding of either IL-4 or IL-13 to IL-4Rα) in MDSC activation has been demonstrated in several studies. It has been shown that ARG1 expression is induced by culturing freshly isolated MDSCs or cloned MDSC lines with IL-454. In addition, IL-4 and IL-13 upregulate arginase activity, which increases the suppressive function of MDSCs55. In line with these observations, other experiments have shown that STAT6 deficiency prevents signalling downstream of the IL-4Rα and thereby blocks the production of ARG1 by MDSCs56. In addition, the IL-4Rα–STAT6 pathway was also found to be involved in IL-13-induced TGFβ1 production by MDSCs in mice with sarcoma, which resulted in decreased tumour immunosurveillance57. This could be regulated by neutralizing both TGFβ and IL-1357. However, in breast tumor model IL-4Rα knockout mice retain high levels of MDSC after surgery56. In a different study that evaluated the separate role of TGFβ (not involving study of IL-4Rα) TGFβ-specific blocking antibody failed to reverse T-cell anergy in B-cell lymphoma in vitro58. It is possible that, the IL4Rα–STAT6 pathway might not be involved in promoting tumour immunosuppression in all tumour models.
TLRs have a central role in the activation of innate immune responses. Polymicrobial sepsis induced by the ligation and puncture of the caecum, which releases microbial products into the peritoneum and systemic circulation, was shown to result in an expansion of the MDSC population in the spleen that was dependent on the TLR adaptor molecule myeloid differentiation primary-response gene 88 (MyD88)26. However, wild-type mice and mice lacking a functional TLR4 protein had comparable expansion of the MDSC during polymicrobial sepsis, which suggests that signalling through TLR4 is not required for MDSC expansion and that MyD88-dependent signalling pathways that are triggered by other TLRs probably contribute to the expansion of MDSCs in sepsis26. This indicates that the activation of MDSCs is a fundamental outcome of the host innate immune response to pathogens that express TLR ligands.
It is important to note that an increase in the production and/or recruitment of IMCs in the context of acute infectious diseases or following vaccination does not necessarily represent an expansion of an immunosuppressive MDSC population. It is likely that under pathological conditions, the expansion of a suppressive MDSC population is regulated by two different groups of factors that have partially overlapping activity: those that induce MDSC expansion and those that induce their activation (which leads to increased levels of ROS, arginase, and/ or NO). This two-tiered system may allow for flexibility in the regulation of these cells under physiological and pathological conditions.
Mechanisms of MDSC suppressive activity Most studies have shown that the immunosuppressive functions of MDSCs require direct cell– cell contact, which suggests that they act either through cell-surface receptors and/or through the release of short-lived soluble mediators. The following sections describe the several mechanisms that have been implicated in MDSC-mediated suppression of T-cell function.
Arginase and iNOS—Historically, the suppressive activity of MDSCs has been associated with the metabolism of L-arginine. L-arginine serves as a substrate for two enzymes: iNOS, which generates NO, and arginase, which converts L-arginine into urea and L-ornithine. MDSCs express high levels of both arginase and iNOS, and a direct role for both of these enzymes in the inhibition of T-cell function is well established; this has been reviewed recently59, 60. Recent data suggest that there is a close correlation between the availability of arginine and the regulation of T-cell proliferation11, 61. The increased activity of arginase in MDSCs leads to enhanced L-arginine catabolism, which depletes this non-essential amino acid from the microenvironment. The shortage of L-arginine inhibits T-cell proliferation through several different mechanisms, including decreasing their CD3ζ expression62 and preventing their upregulation of the expression of the cell cycle regulators cyclin D3 and cyclin-dependent kinase 4 (CDK4)63. NO suppresses T-cell function through a variety of different mechanisms that involve the inhibition of JAK3 and STAT5 in T cells64, the inhibition of MHC class II expression 65 and the induction of T-cell apoptosis66.
ROS—Another important factor that contributes to the suppressive activity of MDSCs is ROS. Increased production of ROS has emerged as one of the main characteristics of MDSCs in both tumour-bearing mice and patients with cancer6, 10, 13, 53, 67-70. Inhibition of ROS production by MDSCs isolated from mice and patients with cancer completely abrogated the suppressive effect of these cells in vitro10, 13, 67. Interestingly, ligation of integrins expressed on the surface of MDSCs was shown to contribute to increased ROS production following the interaction of MDSCs with T cells10. In addition, several known tumour-derived factors, such as TGFβ, IL-10, IL-6, IL-3, platelet-derived growth factor (PDGF) and GM-CSF, can induce the production of ROS by MDSCs (for review see Ref 71).
The involvement of ROS and NO in mechanisms of MDSC suppression are not restricted to neoplastic conditions, as inflammation and microbial products are also known to induce the development of a MDSC population that produces ROS and NO following interactions with activated T cells15. Similar findings were observed in models of EAE16 and acute Toxoplasmosis infection 16. In addition, it has been observed that MDSCs mediated their suppressive function through IFNγ-dependent NO production in an experimental model of Trypanosoma cruzi infection23.
Peroxynitrite—More recently, it has emerged that peroxynitrite (ONOO-) is a crucial mediator of MDSC-mediated suppression of T-cell function. Peroxynitrite is a product of a chemical reaction between NO and superoxide anoion (O2-) and is one of the most powerful oxidants produced in the body. It induces the nitration and nitrosylation of the amino acids cystine, methionine, tryptophan and tyrosine72. Increased levels of peroxynitrite are present at sites of MDSC and inflammatory-cell accumulation, including sites of ongoing immune reactions. In addition, high levels of peroxynitrite are associated with tumour progression in many types of cancer72, 73,74-78, which has been linked with T-cell unresponsiveness. Bronte and colleagues reported that human prostate adenocarcinomas were infiltrated by terminallydifferentiated CD8+ T cells that were in an unresponsive state. High levels of nitrotyrosine were present in the T cells, which suggested the production of peroxynitrites in the tumour environment. Inhibiting the activity of arginase and iNOS, which are expressed in malignant but not in normal prostate tissue and are key enzymes of L-arginine metabolism,, led to decreased tyrosine nitration and restoration of T-cell responsiveness to tumour antigens79. In addition, we have demonstrated that peroxynitrite production by MDSCs during direct contact with T cells results in nitration of the T-cell receptor (TCR) and CD8 molecules, which alters the specific peptide binding of the T cells and renders them unresponsive to antigen-specific stimulation. However, the T cells maintained their responsiveness to nonspecific stimuli80. This phenomenon of MDSC induced antigen-specific T-cell unresponsiveness was also observed in vivo in tumour-bearing mice53.
Subset-specific suppressive mechanisms?—Recent findings indicate that different subsets of MDSC might use different mechanisms by which to suppress T-cell proliferation. As described earlier, two main subsets of MDSCs have been identified: a granulocytic subset and a monocytic subset. The granulocytic subset of MDSC was found to express high levels of ROS and low levels of NO, whereas the monocytic subset expressed low levels of ROS and high levels of NO and both subsets expressed ARG16 (Fig.3). Interestingly, both populations suppressed antigen-specific T-cell proliferation to an equal extent, despite their different mechanisms of action. Consistent with these observations, Movahedi et al. also reported two distinct MDSC subsets in tumour-bearing mice, one that consisted of mononuclear cells that resembled inflammatory monocytes and a second that consisted of polymorphonuclear cells that were similar to immature granulocytes. Again, both populations were found to suppress antigen-specific T-cell responses, although by using distinct effector molecules and signalling pathways. The suppressive activity of the granulocytic subset was ARG1-dependent, in contrast to the STAT1- and iNOS-dependent mechanism of the monocyte fraction17. Finally, the same trend was observed in Trypanosoma cruzii infection. In this case, monocytic MDSCs produced NO and strongly inhibited T-cell proliferation, and granulocytic MDSCs produced low levels of NO and did not inhibit T-cell proliferation, although they did produce superoxide15. The biological significance of such functional dichotomy of these two MDSC subsets remains to be elucidated.
Induction of TReg cells—Recently, the ability of MDSCs to promote the de novo development of FOXP3+ regulatory T (TReg) cells in vivo has been described18, 19. The induction of TReg cells by MDSCs was found to require the activation of tumour-specific Tcells and the presence of IFNγ and IL-10 but was independent of NO19. In mice bearing 1D8 ovarian tumours, the induction of TReg cells by MDSCs required the expression of cytotoxic lymphocyte antigen 4 (CTLA-4; also known as CD152) by MDSCs18. In a mouse model of lymphoma, MDSCs were shown to induce TReg-cell expansion through a mechanism that required arginase and the capture, processing and presentation of tumour-associated antigens by MDSCs, but not TGFβ58. By contrast, Movahedi et al. found that the percentage of TReg cells was invariably high throughout tumour growth and did not relate to the kinetics of expansion of the MDSC population, suggesting that MDSCs were not involved in TReg-cell expansion17. Furthermore, in a rat model of kidney allograft tolerance that was induced with a CD28-specific antibody, MDSCs that were co-expressing CD80 and CD86 were found to have a limited effect on the expansion of the TReg-cell population81. Although further work is required to resolve these discrepancies and to determine the physiological relevance of these studies, it seems possible that MDSCs are involved in TReg-cell differentiation through the production of cytokines or direct cell–cell interactions. Furthermore, MDSCs and TReg cells might be linked in a common immunoregulatory network (see later).
Tissue-specific effects on MDSCs A major unresolved question in this field is whether MDSCs mediate antigen-specific or nonspecific suppression of T-cell responses. Provided that MDSCs and T cells are in close proximity, the factors that mediate MDSC suppressive function (ROS, arginase and NO) can inhibit T-cell proliferation regardless of the antigen specificity of the T cells. Indeed, numerous in vitro studies have demonstrated the antigen nonspecific nature of MDSC-mediated suppression of T cells82 83. However, whether the situation is the same in vivo is not clear, and evidence suggests that MDSC-mediated immunosuppression in peripheral lymphoid organs is mainly antigen-specific. The idea that MDSC-mediated T-cell suppression occurs in an antigen-specific manner is based on findings that antigen-specific interactions between antigen-presenting cells and T cells result in much more stable and more prolonged cell–cell contact than nonspecific interactions82, 84, 85. Such stable contacts are necessary for MDSCderived ROS and peroxynitrite to mediate effects on the molecules on the surface of T cells that render the T cells unresponsive to specific antigen. It should be noted that such modification of cell-surface molecules does not lead to T-cell death nor prevent nonspecific T-cell activation. Other evidence that supports the idea that MDSCs mediate antigen-specific suppression is the finding that that MDSCs can take up soluble antigens, including tumourassociated antigens, and process and present them to T cells17 80; blockade of MDSC–T-cell interactions with a MHC-class-I-specific antibody abrogated MDSC-mediated inhibition of T cell responses in vitro86. The MHC-class-I-restricted nature of MDSC-mediated CD8+ T-cell suppression has also been demonstrated in vivo in tumor models53 and in the model of inflammatory bowel disease 35. This is consistent with the recent observation that large numbers of tumour-induced MDSCs did not inhibit CD8+ T-cell responses specific for unrelated antigens in a model of sporadic cancer87. Notably, it is currently unclear whether similar antigen-specific mechanisms of MDSC-mediated suppression operate on CD4+ T cells, as published studies have only assessed the effects of MDSCs on CD8+ T cells. Addressing this question is complicated by the fact that only a small proportion of MDSCs in many tumour models expresses MHC class II molecules.
The theory that MDSCs suppress T-cell responses in an antigen-specific manner helps to explain the finding that T cells in the peripheral lymphoid organs of tumour-bearing mice and in the peripheral blood of cancer patients can still respond to stimuli other than tumourassociated antigens, including viruses, lectins, co-stimulatory molecules, IL-2 and CD3- and CD28-specific antibodies21, 80, 88-90. Furthermore, even patients with advanced stage cancer do not have systemic immunodeficiency except in cases in which the patient has received high doses of chemotherapy or is at a terminal stage of the disease.
Evidence suggets that the nature of MDSC-mediated suppression at the tumour site is quite different to that which occurs in the periphery. MDSCs actively migrate into the tumour site10, where they upregulate the expression of ARG1 and iNOS, downregulate the production of ROS and/or rapidly differentiate into tumour-associated macrophages (TAMs) 52. The levels of NO and arginase produced by tumour-associated MDSCs and TAMs are much higher than those of MDSCs found in peripheral lymphoid organs of the same animals. In addition, TAMs produce several cytokines (reviewed in REFs91, 92) that suppress T-cell responses in a nonspecific manner (Fig. 4). The mechanisms by which MDSC functions are regulated within the tumour microenvironment, and how they differ from those that operate at peripheral sites, remain unclear. It is possible that tumour stroma, hypoxia and/or the acidophilic environment have a role.
Therapeutic targeting of MDSCs The recognition that immune suppression has a crucial role in promoting tumour progression and contributes to the frequent failure of cancer vaccines to elicit an immune response has resulted in a paradigm shift with respect to approaches for cancer immunotherapy. Indeed, it has become increasingly clear that successful cancer immunotherapy will be possible only with a strategy that involves the elimination of suppressive factors from the body. As MDSCs are one of the main immunosuppressive factors in cancer and other pathological conditions, several different therapeutic strategies that target these cells are currently being explored (Table 2). Although the studies described below were carried out in tumor-bearing hosts, it is likely that the same strategies will be useful in other pathological conditions in which inhibition or elimination of MDSCs is a therapeutic aim.
Promoting myeloid-cell differentiation—One of the most promising approaches by which to target MDSCs for therapy is to promote their differentiation into mature myeloid cells that do not have suppressive abilities. Vitamin A has been identified as a compound that can mediate this effect: vitamin A metabolites such as retinoic acid have been found to stimulate the differentiation of myeloid progenitors into DCs and macrophages 86, 93. Mice that are deficient in vitamin A94 or that have been treated with a pan-retinoic-acid-receptor antagonist95, show an expansion of MDSCs in the bone marrow and spleen. Conversely, therapeutic concentrations of all-trans retinoic acid (ATRA) results in substantial decrease in the presence of MDSCs in cancer patients and tumour-bearing mice. ATRA induced MDSCs to differentiate into DCs and macrophages in vitro and in vivo 12, 86, 96. It is probable that ATRA preferentially induces the differentiation of the monocytic subset of MDSCs, whereas it causes apoptosis of the granulocytic subset. The main mechanism of ATRA-mediated differentiation involved an upregulation of glutathione synthesis and a reduction in ROS levels in MDSCs 97. Decreasing the number of MDSCs in tumour-bearing mice resulted in increased tumour-specific T-cell responses, and the combination of ATRA and two different types of cancer vaccine prolonged the anti-tumour effect of the vaccine treatment in two different tumour models 96. Moreover, administration of ATRA to patients with metastatic renal cell carcinoma resulted in a substantial decrease in the number of MDSCs in the peripheral blood and improved antigen-specific response of T cells 21. Further studies will lead to identification of other agents that have a similar effect. So far, evidence suggests that Vitamin D3 may be another agent with the potential to decrease MDSC numbers in patients with cancer, as it is also known to promote myeloid-cell differentiation98.
Inhibition of MDSC expansion—Because MDSC expansion is known to be regulated by tumour-derived factors (Table 1), several studies have focused on neutralizing the effects of these factors. Recently, SCF has been implicated in causing MDSC expansion in tumourbearing mice39. Inhibition of SCF-mediated signalling by blocking its interaction with its receptor, c-kit, decreased MDSC expansion and tumor angiogenesis39. VEGF, another tumourderived factor that is involved in promoting MDSC expansion, might also be a useful target by which to manipulate MDSC. However, in a clinical trial of 15 patients with refractory solid tumours, treatment with VEGF–trap (a fusion protein that binds all forms of VEGF-A and placental growth factor) showed no effect on MDSC numbers and did not result in increased T-cell responses99. By contrast, treatment of patients with metastatic renal cell cancer with a VEGF-specific blocking antibody (known as avastin) resulted in a decrease in the size of a CD11b+VEGFR1+ population of MDSCs in the peripheral blood 100. However, whether avastatin treatment resulted in an improvement in antitumour responses in these patients has not been determined. Finally, inhibition of matrix metalloproteinase 9 function in tumorbearing mice decreased the number of MDSCs in the spleen and tumour tissues and resulted in a significant delay in the growth of spontaneous NeuT tumours in transgenic BALB/c mice101. However, the mechanism responsible for this outcome remains to be elucidated.
Inhibition of MDSC function—Another approach by which to inhibit MDSCs is to block the signalling pathways that regulate the production of suppressive factors by these cells. One potential target by which this might be achieved is COX2. COX2 is required for the production of prostaglandin E2, which in 3LL tumour cells61 and mammary carcinoma40 has been shown to induce the upregulation of ARG1 expression by MDSCs, thereby inducing their suppressive function. Accordingly, COX2 inhibitors were found to downregulate the expression of ARG1 by MDSCs, which improved antitumour T-cell responses and enhanced the therapeutic efficacy of immunotherapy102, 103. Similarly, phosphodiesterase-5 inhibitors such as sildenafil were found to downregulate the expression of arginase and iNOS expression by MDSCs, thereby inhibiting their suppressive function in growing tumours104. This resulted in the induction of a measurable anti-tumour immune response and a marked delay of tumour progression in several mouse models 104.
ROS inhibitors have also been shown to be effective for decreasing MDSC-mediated immune suppression in tumour-bearing mice. The coupling of a NO-releasing moiety to a conventional non-steroidal anti-inflammatory drug has proven to be an efficient means by which to inhibit the production of ROS. One such drug, nitroaspirin, was found to limit the activity of ARG1 and iNOS in spleen MDSCs105. In combination with vaccination with endogenous retroviral gp70 antigen, nitroaspirin inhibited MDSCs function and increased the number and function of tumour-antigen-specific T cells105.
Elimination of MDSCs—MDSCs can be directly eliminated in pathological settings by using some chemotherapeutic drugs. Administration of one such drug, gemcitabine, to mice that were bearing large tumours resulted in a dramatic reduction in the number of MDSCs in the spleen and resulted in a marked improvement in the anti-tumour response induced by immunotherapy106, 107. This effect was specific to MDSCs, as a significant decrease in the number of T or B cells was not observed in these animals. Furthermore, in a study of 17 patients with early-stage breast cancer that were treated with doxorubicin–cyclophosphamide chemotherapy, a decrease in the level of MDSCs in the peripheral blood was observed22.
Evidence suggests that there is a broad range of methods that will be effective for targeting of the number and/or function of MDSCs in vivo. These strategies will undoubtedly help to further investigate the biology of these cells as well as expedite clinical applications to treat cancer and other pathological conditions.
MDSCs as regulatory myeloid cells? The wealth of information that has accumulated in recent years regarding the biology of MDSCs suggests that these cells might have evolved as a regulatory component of the immune system. These cells are absent under physiological conditions, as IMCs in naive mice are an intrinsic part of normal haematopoiesis that are not immunosuppressive in an unactivated state. In conditions of acute stress, infection or immunization, there is a transient expansion of this IMC population, which then quickly differentiates into mature myeloid cells. This transient IMC population can mediate the suppressive functions that are characteristic of MDSCs but, because the acute conditions are short-lived, the suppressive functions of this transient population have a minimal impact on the overall immune response. However, these cells probably function as important ‘gatekeepers’ that prevent pathological immune-mediated damage.
The role of the MDSC population in settings of chronic infections and cancer is very different. In these pathological conditions, the prolonged and marked expansion of IMCs and their subsequent activation leads to the expansion of a large population of MDSCs with immunosuppressive abilities. MDSCs accumulate in peripheral lymphoid organs and migrate to tumour sites, where they contribute to immunosuppression. Furthermore, some evidence suggests that MDSCs can also induce expansion of regulatory T cells. Future studies will reveal whether MDSCs can be considered part of a natural immune regulatory network.
Concluding remarks The field of MDSC research has more outstanding questions than answers. The roles of specific MDSC subsets in mediating T-cell suppression, and the molecular mechanisms responsible for inhibition of myeloid-cell differentiation, need to be elucidated. The issue of whether Tcell suppression occurs in an antigen-specific manner remains to be clarified, as do the mechanisms that cause MDSC migration to peripheral lymphoid organs. Some of the main priorities in this field should include a better characterization of human MDSCs and a clear understanding of whether targeting these cells in patients with various pathological conditions will be of clinical significance. Conversely, adoptive cellular therapy with MDSCs may be an attractive opportunity by which to inhibit immune responses in the setting of autoimmune disease or transplantation. The challenge for these approaches will be to devise methods by which to generate these cells ex vivo in clinical-grade conditions such that they are suitable for administration to patients. If the past 5–6 years are an indication of the potential for progress in this area, it is safe to estimate that there will soon be significantly more discoveries that further our understanding about the biology and clinical utility of MDSCs.
Box 1. Definition of myeloid-derived suppressor cells (MDSCs)
• a heterogeneous population of cells of myeloid origin that consist of myeloid progenitors and immature macrophages, immature granulocytes and immature dendritic cells
• present in activated state that is characterized by the increased production of reactive oxygen and nitrogen species, and of arginase
• potent suppressors of various T-cell functions • in mice, their phenotype is CD11b+Gr1+, although functionally distinct subsets within this population have been identified (see main text)
• in humans, their phenotype is Lin-HLA-DR-CD33+ or CD11b+CD14-CD33+.
Human cells do not express a marker homologous to mouse Gr1. MDSC have also been identified within a CD15+ population in human peripheral blood.
• in the steady state, immature myeloid cells lack suppressive activity and are present in the bone marrow, but not in secondary lymphoid organs
• accumulation of MDSCs in lymphoid organs and in tumours in response to various growth factors and cytokines is associated with various pathological conditions (most notably cancer)
• in tumour tissues, MDSCs can be differentiated from tumour-associated macrophages (TAMs) by their high expression of Gr1 (not expressed by TAMs) by their low expression of F4/80 (expressed by TAMs), by the fact that a large proportion of MDSCs have a granulocytic morphology and based the upregulated expression of both arginase and inducible nitric oxide synthase by MDSCs but not TAMs.
References
1. Young MRI, Newby M, Wepsic TH. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res 1987;47:100–106. [PubMed: 2947676]
2. Buessow SC, Paul RD, Lopez DM. Influence of mammary tumor progression on phenotype and function of spleen and in situ lymphocytes in mice. J Natl Cancer Inst 1984;73:249–255. [PubMed: 6610791]
3. Seung L, Rowley D, Dubeym P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci U S A 1995;92:6254–6258. [PubMed: 7603979]
4. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Crosstalk between myeloidderived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 2007;179:977–983. [PubMed: 17617589]
5. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008;8:618–631. [PubMed: 18633355]
6. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumorbearing mice. J Immunol 2008;181:5791–5802. [PubMed: 18832739] Together with reference # 17 this paper described functional differences between subsets of MDSC.
7. Bronte V, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood 2000;96:3838. [PubMed: 11090068]
8. Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J Leukoc Biol 2003;74:186–196. [PubMed: 12885935]
9. Li Q, Pan PY, Gu P, Xu D, Chen SH. Role of immature myeloid Gr-1+ cells in the development of antitumor immunity. Cancer Res 2004;64:1130–1139. [PubMed: 14871848] …..
 |
Aurelian Udristioiu commented on your update | |
| “The proto-oncogenic transcription factor Myc is known to promote transcription of genes for the cell cycle as well as aerobic glycolysis and glutamine metabolism. Recently, Myc has been shown to play an essential role to induce the expression of glycolytic and glutamine metabolism genes in the initial hours of T cell activation. In a similar fashion, the transcription factor HIF1a can up-regulate glycolytic genes to allow cancer cells to survive under hypoxic conditions. “ | ||















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
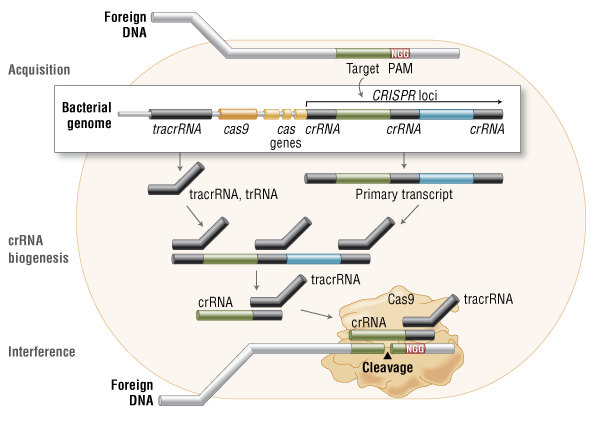{kind=link}
{kind=link}
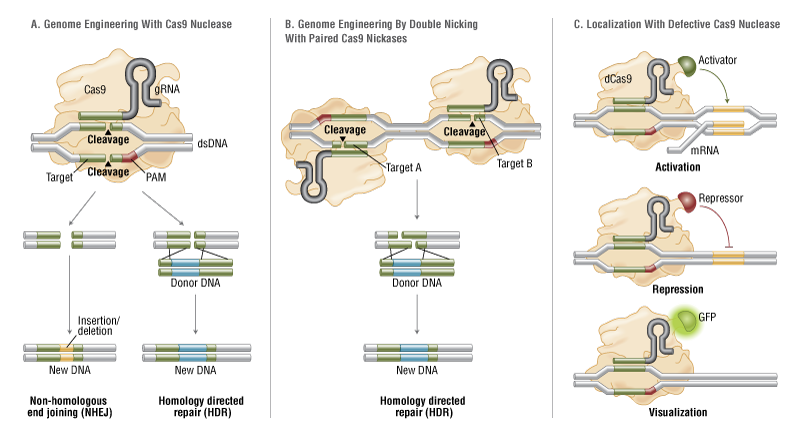{kind=link}
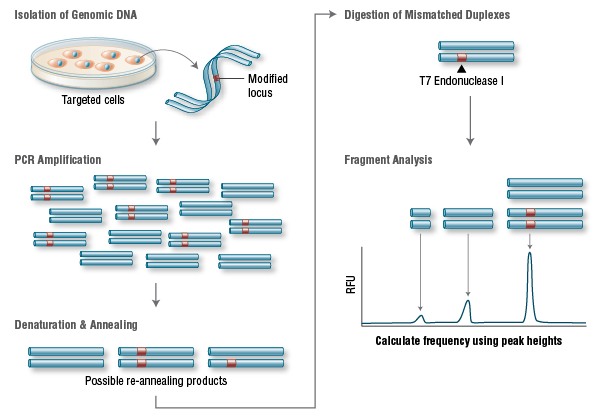{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This is a lovely method and should find wide applicability in many settings, especially for microorganisms and cell lines. However, it is not clear that this approach will be, as implied by the discussion, an efficient mapping method for all multicellular organisms. I have performed similar experiments in Drosophila, focused on meiotic recombination, on a much smaller scale, and found that CRISPR-Cas9 can indeed generate targeted recombination at gRNA target sites. In every case I tested, I found that the recombination event was associated with a deletion at the gRNA site, which is probably unimportant for most mapping efforts, but may be a concern in some specific cases, for example for clinical applications. It would be interesting to know how often mutations occurred at the targeted gRNA site in this study.
The wider issue, however, is whether CRISPR-mediated recombination will be more efficient than other methods of mapping. After careful consideration of all the costs and the time involved in each of the steps for Drosophila, we have decided that targeted meiotic recombination using flanking visible markers will be, in most cases, considerably more efficient than CRISPR-mediated recombination. This is mainly due to the large expense of injecting embryos and the extensive effort and time required to screen injected animals for appropriate events. It is both cheaper and faster to generate markers (with CRISPR) and then perform a large meiotic recombination mapping experiment than it would be to generate the lines required for CRISPR-mediated recombination mapping. It is possible to dramatically reduce costs by, for example, mapping sequentially at finer resolution. But this approach would require much more time than marker-assisted mapping. If someone develops a rapid and cheap method of reliably introducing DNA into Drosophila embryos, then this calculus might change.
However, it is possible to imagine situations where CRISPR-mediated mapping would be preferable, even for Drosophila. For example, some genomic regions display extremely low or highly non-uniform recombination rates. It is possible that CRISPR-mediated mapping could provide a reasonable approach to fine mapping genes in these regions.
The authors also propose the exciting possibility that CRISPR-mediated loss of heterozygosity could be used to map traits in sterile species hybrids. It is not entirely obvious to me how this experiment would proceed and I hope the authors can illuminate me. If we imagine driving a recombination event in the early embryo (with maternal Cas9 from one parent and gRNA from a second parent), then at best we would end up with chimeric individuals carrying mitotic clones. I don’t think one could generate diploid animals where all cells carried the same loss of heterozygosity event. Even if we could, this experiment would require construction of a substantial number of stable transgenic lines expressing gRNAs. Mapping an ~20Mbp chromosome arm to ~10kb would require on the order of two-thousand transgenic lines. Not an undertaking to be taken lightly. It is already possible to perform similar tests (hemizygosity tests) using D. melanogaster deficiency lines in crosses with D. simulans, so perhaps CRISPR-mediated LOH could complement these deficiency screens for fine mapping efforts. But, at the moment, it is not clear to me how to do the experiment.