Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Dr. Ahmad Masri and Dr. C Michael Gibson discuss outpatient worsening heart failure in transthyretin amyloid cardiomyopathy in the Attribute-CM trial, May 29, 2026
The following are key points to remember from this update on the treatment of cardiac transthyretin amyloidosis:
Transthyretin (TTR) is a highly conserved protein involved in transportation of thyroxine (T4) and retinol-binding protein. TTR is synthesized mostly by the liver and is rich in beta strands with an intrinsic propensity to aggregate into insoluble amyloid fibers, which deposit within tissue leading to the development of TTR-related amyloidosis (ATTR). ATTR can follow the deposition of either variant TTR (ATTRv, previously known as mutant ATTR) or wild type TTR (ATTRwt).
Cardiac ATTR has a favorable survival rate compared to light chain (AL) amyloidosis, with a median survival of 75 versus 11 months. However, ATTR cardiomyopathy is a progressive disorder but newer therapeutic options include tafamidis (positive phase 3 clinical trial), and possibly patisiran and inotersen.
Inhibition of the Synthesis of Mutated Transthyretin
Liver transplantation removes the source of mutated TTR molecules and prolongs survival, with a 20-year survival of 55.3%. However, tissue accumulation of TTR can continue after liver transplantation because TTR amyloid fibers promote subsequent deposition of ATTRwt. Combined liver–heart transplantation is feasible in younger patients with ATTRv cardiomyopathy and a small series suggests better prognosis than cardiac transplantation.
Inhibition of TTR gene expression: Patisiran is a small interfering RNA blocking the expression of both variant and wt TTR. On the basis of the APOLLO trial, it was approved for therapy of adults with ATTRv-related polyneuropathy both in the United States and European Union. In this trial, patisiran promoted favorable myocardial remodeling based on echocardiographic and N-terminal B-type natriuretic peptide (NT-BNP) changes (this effect was not demonstrated for inotersen) and is still under investigation for tafamidis.
Antisense oligonucleotides inotersen inhibits the production of both variant and wt TTR. Based on the findings of the NEURO-TTR trial, the Food and Drug Administration (FDA) approved this agent for patients with ATTRv-related polyneuropathy. In the NEURO-TTR trial, cardiomyopathy was present in 63%, but the study was not powered to measure effects of inotersen on heart disease. Inotersen can cause thrombocytopenia and must be used cautiously with bleeding risk.
Tetramer Stabilization
Selective stabilizers include tafamidis and AG10. Tafamidis is a benzoxazole and a small molecule that inhibits the dissociation of TTR tetramers by binding the T4-binding sites. The phase ATTR-ACT study showed that when comparing the pooled tafamidis arms (80 and 20 mg) with the placebo arm, tafamidis was associated with lower all-cause mortality than placebo (78 of 264 [29.5%] vs. 76 of 177 [42.9%]; hazard ratio, 0.70; 95% confidence interval, 0.51-0.96) and a lower rate of cardiovascular hospitalizations. Based on the results of the ATTR-ACT trial, it has received Breakthrough Therapy designation from the FDA for treatment of ATTR cardiomyopathy.
Nonselective agents: Diflunisal, a nonsteroidal anti-inflammatory drug, is reported to stabilize TTR tetramers. More studies are needed to confirm its clinical efficacy.
Inhibition of Oligomer Aggregation and Oligomer Disruption
Epigallocatechin gallate is the most abundant catechin in green tea. One single-center open-label 12-month study did not show survival benefits or any change in echocardiographic parameters or NT-BNP compared to baseline.
Degradation and Reabsorption of Amyloid Fibers
Doxycycline-taurosodeoxycholic acid (TUDCA) has been evaluated in two small studies and the results appear to be modest. More data are needed to confirm its efficacy.
Antibodies targeting serum amyloid P protein or amyloid fibrils: Patient enrollment for miridesap followed by anti-SAP antibodies was suspended, and this approach is not being evaluated currently. However, a monoclonal antibody designed to specifically target TTR amyloid deposits (PRX004) has entered clinical evaluation, with an ongoing phase 1 study on ATTRv.
Supportive Treatment of Cardiac Involvement
Drug therapies: Although angiotensin-converting enzyme (ACE) inhibitors/angiotensin-receptor blockers (ARBs) and beta-blockers may have been poorly tolerated in the ATTR-ACT trial, 30% of the patients were on ACE inhibitors/ARBs. There are no data with digoxin in TTR amyloid, and non-dihydropyridine calcium channel blockers are contraindicated due to negative inotropy.
Implantable cardioverter-defibrillators (ICDs): In one study, which included 53 patients with amyloid, ICD shocks occurred exclusively in the AL amyloid group and none in the TTR amyloid patients. Higher defibrillation thresholds and complication rates are of concern.
Cardiac pacing: In a large series of ATTRv-related polyneuropathy (n = 262), a pacemaker was implanted in 110 patients with His ventricular interval >700 ms. The authors recommend that any conduction disturbance on 12-lead electrocardiogram (ECG) warrants further investigation with Holter monitoring to determine candidacy for a pacemaker.
Left ventricular assist device (LVAD): Although an LVAD is technically feasible, it is associated with high short-term mortality and worse outcomes than in dilated cardiomyopathy.
Cardiac transplantation: This is a valuable option for patients with end-stage heart failure when significant extracardiac disease is excluded. In one study with 10 patients, only episodes of amyloid recurrence occurred.
This is an outstanding overview of this topic and recommended reading for anyone who cares for patients with cardiac transthyretin amyloid.
First-Ever Evidence that Patisiran Reduces Pathogenic, Misfolded TTR Monomers and Oligomers in FAP Patients
We reported data from our ongoing Phase 2 open-label extension (OLE) study of patisiran, an investigational RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR amyloidosis) patients with familial amyloidotic polyneuropathy (FAP). Alnylam scientists and collaborators from The Scripps Research Institute and Misfolding Diagnostics, Inc. were able to measure the effects of patisiran on pathogenic, misfolded TTR monomers and oligomers in FAP patients. Results showed a rapid and sustained reduction in serum non-native conformations of TTR (NNTTR) of approximately 90%. Since NNTTR is pathogenic in ATTR amyloidosis and the level of NNTTR reduction correlated with total TTR knockdown, these results provide direct mechanistic evidence supporting the therapeutic hypothesis that TTR knockdown has the potential to result in clinical benefit. Furthermore, complete 12-month data from all 27 patients that enrolled in the patisiran Phase 2 OLE study showed sustained mean maximum reductions in total serum TTR of 91% for over 18 months and a mean 3.1-point decrease in mNIS+7 at 12 months, which compares favorably to an estimated increase in mNIS+7 of 13 to 18 points at 12 months based upon analysis of historical data sets in untreated FAP patients with similar baseline characteristics. Importantly, patisiran administration continues to be generally well tolerated out to 21 months of treatment.
We are encouraged by these new data that provide continued support for our hypothesis that patisiran has the potential to halt neuropathy progression in patients with FAP. If these results are replicated in a randomized, double-blind, placebo-controlled study, we believe that patisiran could emerge as an important treatment option for patients suffering from this debilitating, progressive and life-threatening disease.
Hereditary ATTR Amyloidosis with Polyneuropathy (hATTR-PN)
ATTR amyloidosis is a progressive, life-threatening disease caused by misfolded transthyretin (TTR) proteins that accumulate as amyloid fibrils in multiple organs, but primarily in the peripheral nerves and heart. ATTR amyloidosis can lead to significant morbidity, disability, and mortality. The TTR protein is produced primarily in the liver and is normally a carrier for retinol binding protein – one of the vehicles used to transport vitamin A around the body. Mutations in the TTR gene cause misfolding of the protein and the formation of amyloid fibrils that typically contain both mutant and wild-type TTR that deposit in tissues such as the peripheral nerves and heart, resulting in intractable peripheral sensory neuropathy, autonomic neuropathy, and/or cardiomyopathy.
ATTR represents a major unmet medical need with significant morbidity and mortality. There are over 100 reported TTR mutations; the particular TTR mutation and the site of amyloid deposition determine the clinical manifestations of the disease whether it is predominantly symptoms of neuropathy or cardiomyopathy.
Specifically, hereditary ATTR amyloidosis with polyneuropathy (hATTR-PN), also known as familial amyloidotic polyneuropathy (FAP), is an inherited, progressive disease leading to death within 5 to 15 years. It is due to a mutation in the transthyretin (TTR) gene, which causes misfolded TTR proteins to accumulate as amyloid fibrils predominantly in peripheral nerves and other organs. hATTR-PN can cause sensory, motor, and autonomic dysfunction, resulting in significant disability and death.
It is estimated that hATTR-PN, also known as FAP, affects approximately 10,000 people worldwide. Patients have a life expectancy of 5 to 15 years from symptom onset, and the only treatment options for early stage disease are liver transplantation and TTR stabilizers such as tafamidis (approved in Europe) and diflunisal. Unfortunately liver transplantation has limitations, including limited organ availability as well as substantial morbidity and mortality. Furthermore, transplantation eliminates the production of mutant TTR but does not affect wild-type TTR, which can further deposit after transplantation, leading to cardiomyopathy and worsening of neuropathy. There is a significant need for novel therapeutics to treat patients who have inherited mutations in the TTR gene.
Our ATTR program is the lead effort in our Genetic Medicine Strategic Therapeutic Area (STAr) product development and commercialization strategy, which is focused on advancing innovative RNAi therapeutics toward genetically defined targets for the treatment of rare diseases with high unmet medical need. We are developing patisiran (ALN-TTR02), an intravenously administered RNAi therapeutic, to treat the hATTR-PN form of the disease.
Patisiran for the Treatment hATTR-PN
APOLLO Phase 3 Trial
In 2012, Alnylam entered into an exclusive alliance with Genzyme, a Sanofi company, to develop and commercialize RNAi therapeutics, including patisiran and revusiran, for the treatment of ATTR amyloidosis in Japan and the broader Asian-Pacific region. In early 2014, this relationship was extended as a significantly broader alliance to advance RNAi therapeutics as genetic medicines. Under this new agreement, Alnylam will lead development and commercialization of patisiran in North America and Europe while Genzyme will develop and commercialize the product in the rest of world.
Hereditary ATTR Amyloidosis with Cardiomyopathy (hATTR-CM)
ATTR amyloidosis is a progressive, life-threatening disease caused by misfolded transthyretin (TTR) proteins that accumulate as amyloid fibrils in multiple organs, but primarily in the peripheral nerves and heart. ATTR amyloidosis can lead to significant morbidity, disability, and mortality. The TTR protein is produced primarily in the liver and is normally a carrier for retinol binding protein – one of the vehicles used to transport vitamin A around the body. Mutations in the TTR gene cause misfolding of the protein and the formation of amyloid fibrils that typically contain both mutant and wild-type TTR that deposit in tissues such as the peripheral nerves and heart, resulting in intractable peripheral sensory neuropathy, autonomic neuropathy, and/or cardiomyopathy.
ATTR represents a major unmet medical need with significant morbidity and mortality. There are over 100 reported TTR mutations; the particular TTR mutation and the site of amyloid deposition determine the clinical manifestations of the disease, whether it is predominantly symptoms of neuropathy or cardiomyopathy.
Specifically, hereditary ATTR amyloidosis with cardiomyopathy (hATTR-CM), also known as familial amyloidotic cardiomyopathy (FAC), is an inherited, progressive disease leading to death within 2 to 5 years. It is due to a mutation in the transthyretin (TTR) gene, which causes misfolded TTR proteins to accumulate as amyloid fibrils primarily in the heart. Hereditary ATTR amyloidosis with cardiomyopathy can result in heart failure and death.
While the exact numbers are not known, it is estimated hATTR-CM, also known as FAC affects at least 40,000 people worldwide. hATTR-CM is fatal within 2 to 5 years of diagnosis and treatment is currently limited to supportive care. Wild-type ATTR amyloidosis (wtATTR amyloidosis), also known as senile systemic amyloidosis, is a nonhereditary, progressive disease leading to death within 2 to 5 years. It is caused by misfolded transthyretin (TTR) proteins that accumulate as amyloid fibrils in the heart. Wild-type ATTR amyloidosis can cause cardiomyopathy and result in heart failure and death. There are no approved therapies for the treatment of hATTR-CM or SSA; hence there is a significant unmet need for novel therapeutics to treat these patients.
Our ATTR program is the lead effort in our Genetic Medicine Strategic Therapeutic Area (STAr) product development and commercialization strategy, which is focused on advancing innovative RNAi therapeutics toward genetically defined targets for the treatment of rare diseases with high unmet medical need. We are developing revusiran (ALN-TTRsc), a subcutaneously administered RNAi therapeutic for the treatment of hATTR-CM.
Revusiran for the Treatment of hATTR-CM
ENDEAVOUR Phase 3 Trial
In 2012, Alnylam entered into an exclusive alliance with Genzyme, a Sanofi company, to develop and commercialize RNAi therapeutics, including patisiran and revusiran, for the treatment of ATTR amyloidosis in Japan and the broader Asian-Pacific region. In early 2014, this relationship was extended as a broader alliance to advance RNAi therapeutics as genetic medicines. Under this new agreement, Alnylam and Genzyme have agreed to co-develop and co-commercialize revusiran in North America and Europe, with Genzyme developing and commercializing the product in the rest of world. This broadened relationship on revusiran is aimed at expanding and accelerating the product’s global value.
We presented pre-clinical data with ALN-TTRsc02, an investigational RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR amyloidosis). In pre-clinical studies, including those in non-human primates (NHPs), ALN-TTRsc02 achieved potent and highly durable knockdown of serum TTR of up to 99% with multi-month durability achieved after just a single dose, supportive of a potentially once quarterly dose regimen. Results from studies comparing TTR knockdown activity of ALN-TTRsc02 to that of revusiran showed that ALN-TTRsc02 has a markedly superior TTR knockdown profile. Further, in initial rat toxicology studies, ALN-TTRsc02 was found to be generally well tolerated with no significant adverse events at doses as high as 100 mg/kg.
Heart failure with a preserved ejection fraction (HFPEF) is a clinical syndrome that has no pharmacologic therapies approved for this use to date. In light of failed medicines, cardiologists have refocused treatment strategies based on the theory that HFPEF is a heterogeneous clinical syndrome with different etiologies. Classification of HFPEF according to etiologic subtype may, therefore, identify cohorts with treatable pathophysiologic mechanisms and may ultimately pave the way forward for developing meaningful HFPEF therapies.1
A wealth of data now indicates that amyloid infiltration is an important mechanism underlying HFPEF. Inherited mutations in transthyretin cardiac amyloidosis (ATTRm) or the aging process in wild-type disease (ATTRwt) cause destabilization of the transthyretin (TTR) protein into monomers or oligomers, which aggregate into amyloid fibrils. These insoluble fibrils accumulate in the myocardium and result in diastolic dysfunction, restrictive cardiomyopathy, and eventual congestive heart failure (Figure 1). In an autopsy study of HFPEF patients, almost 20% without antemortem suspicion of amyloid had left ventricular (LV) TTR amyloid deposition.2 Even more resounding evidence for the contribution of TTR amyloid to HFPEF was a study in which 120 hospitalized HFPEF patients with LV wall thickness ≥12 mm underwent technetium-99m 3,3-diphosphono-1,2-propranodicarboxylic acid (99mTc-DPD) cardiac imaging,3,4 a bone isotope known to have high sensitivity and specificity for diagnosing TTR cardiac amyloidosis.5,6 Moderate-to-severe myocardial uptake indicative of TTR cardiac amyloid deposition was detected in 13.3% of HFPEF patients who did not have TTR gene mutations. Therefore, TTR cardiac amyloid deposition, especially in older adults, is not rare, can be easily identified, and may contribute to the underlying pathophysiology of HFPEF.
Figure 1
As no U.S. Food and Drug Administration-approved drugs are currently available for the treatment of HFPEF or TTR cardiac amyloidosis, the development of medications that attenuate or prevent TTR-mediated organ toxicity has emerged as an important therapeutic goal. Over the past decade, a host of therapies and therapeutic drug classes have emerged in clinical trials (Table 1), and these may herald a new direction for treating HFPEF secondary to TTR amyloid.
Table 1
TTR Silencers (siRNA and Antisense Oligonucleotides)
siRNA
Ribonucleic acid interference (RNAi) has surfaced as an endogenous cellular mechanism for controlling gene expression. Small interfering RNAs (siRNAs) delivered into cells can disrupt the production of target proteins.7,8 A formulation of lipid nanoparticle and triantennary N-acetylgalactosamine (GalNAc) conjugate that delivers siRNAs to hepatocytes is currently in clinical trials.9 Prior research demonstrated these GalNAc-siRNA conjugates result in robust and durable knockdown of a variety of hepatocyte targets across multiple species and appear to be well suited for suppression of TTR gene expression and subsequent TTR protein production.
The TTR siRNA conjugated to GalNAc, ALN-TTRSc, is now under active investigation as a subcutaneous injection in phase 3 clinical trials in patients with TTR cardiac amyloidosis.10 Prior phase 2 results demonstrated that ALN-TTRSc was generally well tolerated in patients with significant TTR disease burden and that it reduced both wild-type and mutant TTR gene expression by a mean of 87%. Harnessing RNAi technology appears to hold great promise for treating patients with TTR cardiac amyloidosis. The ability of ALN-TTRSc to lower both wild-type and mutant proteins may provide a major advantage over liver transplantation, which affects the production of only mutant protein and is further limited by donor shortage, cost, and need for immunosuppression.
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are under clinical investigation for their ability to inhibit hepatic expression of amyloidogenic TTR protein. Currently, the ASO compound, ISIS-TTRRx, is under investigation in a phase 3 multicenter, randomized, double-blind, placebo-controlled clinical trial in patients with familial amyloid polyneuropathy (FAP).11 The primary objective is to evaluate its efficacy as measured by change in neuropathy from baseline relative to placebo. Secondary measures will evaluate quality of life (QOL), modified body mass index (mBMI) by albumin, and pharmacodynamic effects on retinol binding protein. Exploratory objectives in a subset of patients with LV wall thickness ≥13 mm without a history of persistent hypertension will examine echocardiographic parameters, N-terminal pro–B-type natriuretic peptide (NT-proBNP), and polyneuropathy disability score relative to placebo. These data will facilitate analysis of the effect of antisense oligonucleotide-mediated TTR suppression on the TTR cardiac phenotype with a phase 3 trial anticipated to begin enrollment in 2016.
TTR Stabilizers (Diflunisal, Tafamidis)
Diflunisal
Several TTR-stabilizing agents are in various stages of clinical trials. Diflunisal, a traditionally used and generically available nonsteroidal anti-inflammatory drug (NSAID), binds and stabilizes familial TTR variants against acid-mediated fibril formation in vitro and is now in human clinical trials.12,13 The use of diflunisal in patients with TTR cardiac amyloidosis is controversial given complication of chronic inhibition of cyclooxygenase (COX) enzymes, including gastrointestinal bleeding, renal dysfunction, fluid retention, and hypertension that may precipitate or exacerbate heart failure in vulnerable individuals.14-17 In TTR cardiac amyloidosis, an open-label cohort study suggested that low-dose diflunisal with careful monitoring along with a prophylactic proton pump inhibitor could be safely administered to compensated patients.18 An association was observed, however, between chronic diflunisal use and adverse changes in renal function suggesting that advanced kidney disease may be prohibitive in diflunisal therapy.In FAP patients with peripheral or autonomic neuropathy randomized to diflunisal or placebo, diflunisal slowed progression of neurologic impairment and preserved QOL over two years of follow-up.19 Echocardiography demonstrated cardiac involvement in approximately 50% of patients.20 Longer-term safety and efficacy data over an average 38 ± 31 months in 40 Japanese patients with hereditary ATTR amyloidosis who were not candidates for liver transplantation showed that diflunisal was mostly well tolerated.12 The authors cautioned the need for attentive monitoring of renal function and blood cell counts. Larger multicenter collaborations are needed to determine diflunisal’s true efficacy in HFPEF patients with TTR cardiac amyloidosis.
Tafamidis
Tafamidis is under active investigation as a novel compound that binds to the thyroxine-binding sites of the TTR tetramer, inhibiting its dissociation into monomers and blocking the rate-limiting step in the TTR amyloidogenesis cascade.21 The TTR compound was shown in an 18-month double-blind, placebo-controlled trial to slow progression of neurologic symptoms in patients with early-stage ATTRm due to the V30M mutation.22 When focusing on cardiomyopathy in a phase 2, open-label trial, tafamidis also appeared to effectively stabilize TTR tetramers in non-V30M variants, wild-type and V122I, as well as biochemical and echocardiographic parameters.23,24 Preliminary data suggests that clinically stabilized patients had shorter disease duration, lower cardiac biomarkers, less myocardial thickening, and higher EF than those who were not stabilized, suggesting early institution of therapy may be beneficial. A phase 3 trial has completed enrollment and will evaluate the efficacy, safety, and tolerability of tafamidis 20 or 80 mg orally vs. placebo.25 This will contribute to long-term safety and efficacy data needed to determine the therapeutic effects of tafamidis among ATTRm variants.
Amyloid Degraders (Doxycycline/TUDCA and Anti-SAP Antibodies)
Doxycycline/TUDCA
While silencer and stabilizer drugs are aimed at lowering amyloidogenic precursor protein production, they cannot remove already deposited fibrils in an infiltrated heart. Removal of already deposited fibrils by amyloid degraders would be an important therapeutic strategy, particularly in older adults with heavily infiltrated hearts reflected by thick walls, HFPEF, systolic heart failure, and restrictive cardiomyopathy. Combined doxycycline and tauroursodeoxycholic acid (TUDCA) disrupt TTR amyloid fibrils and appeared to have an acceptable safety profile in a small phase 2 open-label study among 20 TTR patients. No serious adverse reactions or clinical progression of cardiac or neuropathic involvement was observed over one year.26 An active phase 2, single-center, open-label, 12-month study will assess primary outcome measures including mBMI, neurologic impairment score, and NT-proBNP.27 Another phase 2 study is examining the tolerability and efficacy of doxycycline/TUDCA over an 18-month period in patients with TTR amyloid cardiomyopathy.28 Additionally, a study in patients with TTR amyloidosis is ongoing to determine the effect of doxycycline alone on neurologic function, cardiac biomarkers, echocardiographic parameters, modified body mass index, and autonomic neuropathy.29
Anti-SAP Antibodies
In order to safely clear established amyloid deposits, the role of the normal, nonfibrillar plasma glycoprotein present in all human amyloid deposits, serum amyloid P component (SAP), needs to be more clearly understood.30 In mice with amyloid AA type deposits, administration of antihuman SAP antibody triggered a potent giant cell reaction that removed massive visceral amyloid deposits without adverse effects.31 In humans with TTR cardiac amyloidosis, anti-SAP antibody treatments could be feasible because the bis-D proline compound, CPHPC, is capable of clearing circulating human SAP, which allow anti-SAP antibodies to reach residual deposited SAP. In a small, open-label, single-dose-escalation, phase 1 trial involving 15 patients with systemic amyloidosis, none of whom had clinical evidence of cardiac amyloidosis, were treated with CPHPC followed by human monoclonal IgG1 anti-SAP antibody.32 No serious adverse events were reported and amyloid deposits were cleared from the liver, kidney, and lymph node. Anti-SAP antibodies hold promise as a potential amyloid therapy because of their potential to target all forms of amyloid deposits across multiple tissue types.
Mutant or wild-type TTR cardiac amyloidoses are increasingly recognized as a cause of HFPEF. Clinicians need to be aware of this important HFPEF etiology because the diverse array of emerging disease-modifying agents for TTR cardiac amyloidosis in human clinical trials has the potential to herald a new era for the treatment of HFPEF.
References
Maurer MS, Mancini D. HFpEF: is splitting into distinct phenotypes by comorbidities the pathway forward? J Am Coll Cardiol 2014;64:550-2.
Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2014;2:113-22.
González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015.
Castano A, Bokhari S, Maurer MS. Unveiling wild-type transthyretin cardiac amyloidosis as a significant and potentially modifiable cause of heart failure with preserved ejection fraction. Eur Heart J 2015 Jul 28. [Epub ahead of print]
Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009;120:1203-12.
Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013;6:195-201.
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-11.
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494-8.
Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nature Mater 2013;12:967-77.
U.S. National Institutes of Health. Phase 2 Study to Evaluate ALN-TTRSC in Patients With Transthyretin (TTR) Cardiac Amyloidosis (ClinicalTrials.gov website). 2014. Available at: https://www.clinicaltrials.gov/ct2/show/NCT01981837. Accessed 8/19/2015.
U.S. National Institutes of Health. Efficacy and Safety of ISIS-TTRRx in Familial Amyloid Polyneuropathy (Clinical Trials.gov Website. 2013. Available at: http://www.clinicaltrials.gov/ct2/show/NCT01737398. Accessed 8/19/2015.
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13:236-49.
Tojo K, Sekijima Y, Kelly JW, Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res 2006;56:441-9.
Epstein M. Non-steroidal anti-inflammatory drugs and the continuum of renal dysfunction. J Hypertens Suppl 2002;20:S17-23.
Wallace JL. Pathogenesis of NSAID-induced gastroduodenal mucosal injury. Best Pract Res Clin Gastroenterol 2001;15:691-703.
Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 2001;286:954-9.
Page J, Henry D. Consumption of NSAIDs and the development of congestive heart failure in elderly patients: an underrecognized public health problem. Arch Intern Med 2000;160:777-84.
Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 2012;18:315-9.
Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310:2658-67.
Quarta CCF, Solomon RH Suhr SD, et al. The prevalence of cardiac amyloidosis in familial amyloidotic polyneuropathy with predominant neuropathy: The Diflunisal Trial. International Symposium on Amyloidosis 2014:88-9.
Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A 2002;99 Suppl 4:16427-32.
Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79:785-92.
Merlini G, Plante-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res 2013;6:1011-20.
Maurer MS, Grogan DR, Judge DP, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015;8:519-26.
U.S. National Institutes of Health. Safety and Efficacy of Tafamidis in Patients With Transthyretin Cardiomyopathy (ATTR-ACT) (ClinicalTrials.gov website). 2014. Available at: http://www.clinicaltrials.gov/show/NCT01994889. Accessed 8/19/2015.
Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012;19 Suppl 1:34-6.
U.S. National Institutes of Health. Safety, Efficacy and Pharmacokinetics of Doxycycline Plus Tauroursodeoxycholic Acid in Transthyretin Amyloidosis (ClinicalTrials.gov website). 2011. Available at: http://www.clinicaltrials.gov/ct2/show/NCT01171859. Accessed 8/19/2015.
U.S. National Institutes of Health. Tolerability and Efficacy of a Combination of Doxycycline and TUDCA in Patients With Transthyretin Amyloid Cardiomyopathy (ClinicalTrials.gov website). 2013. Available at: http://www.clinicaltrials.gov/ct2/show/NCT01855360. Accessed 8/19/2015.
U.S. National Institutes of Health. Safety and Effect of Doxycycline in Patients With Amyloidosis (ClinicalTrials.gov website).2015. Available at: https://clinicaltrials.gov/ct2/show/NCT01677286. Accessed 8/19/2015.
Pepys MB, Dash AC. Isolation of amyloid P component (protein AP) from normal serum as a calcium-dependent binding protein. Lancet 1977;1:1029-31.
Bodin K, Ellmerich S, Kahan MC, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 2010;468:93-7.
Richards DB, Cookson LM, Berges AC, et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med 2015;373:1106-14.
…
The Acid-Mediated Denaturation Pathway of Transthyretin Yields a Conformational Intermediate That Can Self-Assemble into Amyloid†
Transthyretin (TTR) amyloid fibril formation is observed during partial acid denaturation and while refolding acid-denatured TTR, implying that amyloid fibril formation results from the self-assembly of a conformational intermediate. The acid denaturation pathway of TTR has been studied in detail herein employing a variety of biophysical methods to characterize the intermediate(s) capable of amyloid fibril formation. At physiological concentrations, tetrameric TTR remains associated from pH 7 to pH 5 and is incapable of amyloid fibril formation. Tetrameric TTR dissociates to a monomer in a process that is dependent on both pH and protein concentration below pH 5. The extent of amyloid fibril formation correlates with the concentration of the TTR monomer having an altered, but defined, tertiary structure over the pH range of 5.0−3.9. The inherent Trp fluorescence-monitored denaturation curve of TTR exhibits a plateau over the pH range where amyloid fibril formation is observed (albeit at a higher concentration), implying that a steady-state concentration of the amyloidogenic intermediate with an altered tertiary structure is being detected. Interestingly, 1-anilino-8-naphthalenesulfonate fluorescence is at a minimum at the pH associated with maximal amyloid fibril formation (pH 4.4), implying that the amyloidogenic intermediate does not have a high extent of hydrophobic surface area exposed, consistent with a defined tertiary structure. Transthyretin has two Trp residues in its primary structure, Trp-41 and Trp-79, which are conveniently located far apart in the tertiary structure of TTR. Replacement of each Trp with Phe affords two single Trp containing variants which were used to probe local pH-dependent tertiary structural changes proximal to these chromophores. The pH-dependent fluorescence behavior of the Trp-79-Phe mutant strongly suggests that Trp-41 is located near the site of the tertiary structural rearrangement that occurs in the formation of the monomeric amyloidogenic intermediate, likely involving the C-strand−loop−D-strand region. Upon further acidification of TTR (below pH 4.4), the structurally defined monomeric amyloidogenic intermediate begins to adopt alternative conformations that are not amyloidogenic, ultimately forming an A-state conformation below pH 3 which is also not amyloidogenic. In summary, analytical equilibrium ultracentrifugation, SDS−PAGE, far- and near-UV CD, fluorescence, and light scattering studies suggest that the amyloidogenic intermediate is a monomeric predominantly β-sheet structure having a well-defined tertiary structure.
Prevention of Transthyretin Amyloid Disease by Changing Protein Misfolding Energetics
Per Hammarström*, R. Luke Wiseman*, Evan T. Powers, Jeffery W. Kelly†+ Author Affiliations
Genetic evidence suggests that inhibition of amyloid fibril formation by small molecules should be effective against amyloid diseases. Known amyloid inhibitors appear to function by shifting the aggregation equilibrium away from the amyloid state. Here, we describe a series of transthyretin amyloidosis inhibitors that functioned by increasing the kinetic barrier associated with misfolding, preventing amyloidogenesis by stabilizing the native state. The trans-suppressor mutation, threonine 119 → methionine 119, which is known to ameliorate familial amyloid disease, also functioned through kinetic stabilization, implying that this small-molecule strategy should be effective in treating amyloid diseases.
Rational design of potent human transthyretin amyloid disease inhibitors
Thomas Klabunde1,2, H. Michael Petrassi3, Vibha B. Oza3, Prakash Raman3, Jeffery W. Kelly3 & James C. Sacchettini1
The human amyloid disorders, familial amyloid polyneuropathy, familial amyloid cardiomyopathy and senile systemic amyloidosis, are caused by insoluble transthyretin (TTR) fibrils, which deposit in the peripheral nerves and heart tissue. Several nonsteroidal anti-inflammatory drugs and structurally similar compounds have been found to strongly inhibit the formation of TTR amyloid fibrils in vitro. These include flufenamic acid, diclofenac, flurbiprofen, and resveratrol. Crystal structures of the protein–drug complexes have been determined to allow detailed analyses of the protein–drug interactions that stabilize the native tetrameric conformation of TTR and inhibit the formation of amyloidogenic TTR. Using a structure-based drug design approach ortho-trifluormethylphenyl anthranilic acid and N-(meta-trifluoromethylphenyl) phenoxazine 4,6-dicarboxylic acid have been discovered to be very potent and specific TTR fibril formation inhibitors. This research provides a rationale for a chemotherapeutic approach for the treatment of TTR-associated amyloid diseases.
First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy
Adams, Davida; Suhr, Ole B.b; Hund, Ernstc; Obici, Laurad; Tournev, Ivailoe,f; Campistol, Josep M.g; Slama, Michel S.h; Hazenberg, Bouke P.i; Coelho, Teresaj; from the European Network for TTR-FAP (ATTReuNET)
Purpose of review: Early and accurate diagnosis of transthyretin familial amyloid polyneuropathy (TTR-FAP) represents one of the major challenges faced by physicians when caring for patients with idiopathic progressive neuropathy. There is little consensus in diagnostic and management approaches across Europe.
Recent findings: The low prevalence of TTR-FAP across Europe and the high variation in both genotype and phenotypic expression of the disease means that recognizing symptoms can be difficult outside of a specialized diagnostic environment. The resulting delay in diagnosis and the possibility of misdiagnosis can misguide clinical decision-making and negatively impact subsequent treatment approaches and outcomes.
Summary: This review summarizes the findings from two meetings of the European Network for TTR-FAP (ATTReuNET). This is an emerging group comprising representatives from 10 European countries with expertise in the diagnosis and management of TTR-FAP, including nine National Reference Centres. The current review presents management strategies and a consensus on the gold standard for diagnosis of TTR-FAP as well as a structured approach to ongoing multidisciplinary care for the patient. Greater communication, not just between members of an individual patient’s treatment team, but also between regional and national centres of expertise, is the key to the effective management of TTR-FAP.
Transthyretin familial amyloid polyneuropathy (TTR-FAP) is a highly debilitating and irreversible neurological disorder presenting symptoms of progressive sensorimotor and autonomic neuropathy [1▪,2▪,3]. TTR-FAP is caused by misfolding of the transthyretin (TTR) protein leading to protein aggregation and the formation of amyloid fibrils and, ultimately, to amyloidosis (commonly in the peripheral and autonomic nervous system and the heart) [4,5]. TTR-FAP usually proves fatal within 7–12 years from the onset of symptoms, most often due to cardiac dysfunction, infection, or cachexia [6,7▪▪].
The prevalence and disease presentation of TTR-FAP vary widely within Europe. In endemic regions (northern Portugal, Sweden, Cyprus, and Majorca), patients tend to present with a distinct genotype in large concentrations, predominantly a Val30Met substitution in the TTR gene [8–10]. In other areas of Europe, the genetic footprint of TTR-FAP is more varied, with less typical phenotypic expression [6,11]. For these sporadic or scattered cases, a lack of awareness among physicians of variable clinical features and limited access to diagnostic tools (i.e., pathological studies and genetic screening) can contribute to high rates of misdiagnosis and poorer patient outcomes [1▪,11]. In general, early and late-onset variants of TTR-FAP, found within endemic and nonendemic regions, present several additional diagnostic challenges [11,12,13▪,14].
Delay in the time to diagnosis is a major obstacle to the optimal management of TTR-FAP. With the exception of those with a clearly diagnosed familial history of FAP, patients still invariably wait several years between the emergence of first clinical signs and accurate diagnosis [6,11,14]. The timely initiation of appropriate treatment is particularly pertinent, given the rapidity and irreversibility with which TTR-FAP can progress if left unchecked, as well as the limited effectiveness of available treatments during the later stages of the disease [14]. This review aims to consolidate the existing literature and present an update of the best practices in the management of TTR-FAP in Europe. A summary of the methods used to achieve a TTR-FAP diagnosis is presented, as well as a review of available treatments and recommendations for treatment according to disease status.
Patients with TTR-FAP can present with a range of symptoms [11], and care should be taken to acquire a thorough clinical history of the patient as well as a family history of genetic disease. Delay in diagnosis is most pronounced in areas where TTR-FAP is not endemic or when there is no positive family history [1▪]. TTR-FAP and TTR-familial amyloid cardiomyopathy (TTR-FAC) are the two prototypic clinical disease manifestations of a broader disease spectrum caused by an underlying hereditary ATTR amyloidosis [19]. In TTR-FAP, the disease manifestation of neuropathy is most prominent and definitive for diagnosis, whereas cardiomyopathy often suggests TTR-FAC. However, this distinction is often superficial because cardiomyopathy, autonomic neuropathy, vitreous opacities, kidney disease, and meningeal involvement all may be present with varying severity for each patient with TTR-FAP.
Among early onset TTR-FAP with usually positive family history, symptoms of polyneuropathy present early in the disease process and usually predominate throughout the progression of the disease, making neurological testing an important diagnostic aid [14]. Careful clinical examination (e.g., electromyography with nerve conduction studies and sympathetic skin response, quantitative sensation test, quantitative autonomic test) can be used to detect, characterize, and scale the severity of neuropathic abnormalities involving small and large nerve fibres [10]. Although a patient cannot be diagnosed definitively with TTR-FAP on the basis of clinical presentation alone, symptoms suggesting the early signs of peripheral neuropathy, autonomic dysfunction, and cardiac conduction disorders or infiltrative cardiomyopathy are all indicators that further TTR-FAP diagnostic investigation is warranted. Late-onset TTR-FAP often presents as sporadic cases with distinct clinical features (e.g., milder autonomic dysfunction) and can be more difficult to diagnose than early-onset TTR-FAP (Table 2) [1▪,11,12,13▪,14,20].
Genetic testing is carried out to allow detection of specific amyloidogenic TTR mutations (Table 1), using varied techniques depending on the expertise and facilities available in each country (Table S2, http://links.lww.com/CONR/A39). A targeted approach to detect a specific mutation can be used for cases belonging to families with previous diagnosis. In index cases of either endemic and nonendemic regions that do not have a family history of disease, are difficult to confirm, and have atypical symptoms, TTR gene sequencing is required for the detection of both predicted and new amyloidogenic mutations [26,27].
Following diagnosis, the neuropathy stage and systemic extension of the disease should be determined in order to guide the next course of treatment (Table 4) [3,30,31]. The three stages of TTR-FAP severity are graded according to a patient’s walking disability and degree of assistance required [30]. Systemic assessment, especially of the heart, eyes, and kidney, is also essential to ensure all aspects of potential impact of the disease can be detected [10].
The goals of cardiac investigations are to detect serious conduction disorders with the risk of sudden death and infiltrative cardiomyopathy. Electrocardiograms (ECG), Holter-ECG, and intracardiac electrophysiology study are helpful to detect conduction disorders. Echocardiograms, cardiac magnetic resonance imaging, scintigraphy with bone tracers, and biomarkers (e.g., brain natriuretic peptide, troponin) can all help to diagnose infiltrative cardiomyopathy[10]. An early detection of cardiac abnormalities has obvious benefits to the patient, given that the prophylactic implantation of pacemakers was found to prevent 25% of major cardiac events in TTR-FAP patients followed up over an average of 4 years [32▪▪]. Assessment of cardiac denervation with 123-iodine meta-iodobenzylguanidine is a powerful prognostic marker in patients diagnosed with FAP [33].
…..
Tafamidis
Tafamidis is a first-in-class therapy that slows the progression of TTR amyloidogenesis by stabilizing the mutant TTR tetramer, thereby preventing its dissociation into monomers and amyloidogenic and toxic intermediates [55,56]. Tafamidis is currently indicated in Europe for the treatment of TTR amyloidosis in adult patients with stage I symptomatic polyneuropathy to delay peripheral neurological impairment [57].
In an 18-month, double-blind, placebo-controlled study of patients with early-onset Val30Met TTR-FAP, tafamidis was associated with a 52% lower reduction in neurological deterioration (P = 0.027), a preservation of nerve function, and TTR stabilization versus placebo [58▪▪]. However, only numerical differences were found for the coprimary endpoints of neuropathy impairment [neuropathy impairment score in the lower limb (NIS-LL) responder rates of 45.3% tafamidis vs 29.5% placebo; P = 0.068] and quality of life scores [58▪▪]. A 12-month, open-label extension study showed that the reduced rates of neurological deterioration associated with tafamidis were sustained over 30 months, with earlier initiation of tafamidis linking to better patient outcomes (P = 0.0435) [59▪]. The disease-slowing effects of tafamidis may be dependent on the early initiation of treatment. In an open-label study with Val30Met TTR-FAP patients with late-onset and advanced disease (NIS-LL score >10, mean age 56.4 years), NIS-LL and disability scores showed disease progression despite 12 months of treatment with tafamidis, marked by a worsening of neuropathy stage in 20% and the onset of orthostatic hypotension in 22% of patients at follow-up [60▪].
Tafamidis is not only effective in patients exhibiting the Val30Met mutation; it also has proven efficacy, in terms of TTR stabilization, in non-Val30Met patients over 12 months [61]. Although tafamidis has demonstrated safe use in patients with TTR-FAP, care should be exercised when prescribing to those with existing digestive problems (e.g., diarrhoea, faecal incontinence) [60▪].
Diflunisal is a nonsteroidal anti-inflammatory drug (NSAID) that, similar to tafamidis, slows the rate of amyloidogenesis by preventing the dissociation, misfolding, and misassembly of the mutated TTR tetramer [62,63]. Off-label use has been reported for patients with stage I and II disease, although diflunisal is not currently licensed for the treatment of TTR-FAP.
Evidence for the clinical effectiveness of diflunisal in TTR-FAP derives from a placebo-controlled, double-blind, 24-month study in 130 patients with clinically detectable peripheral or autonomic neuropathy[64▪]. The deterioration in NIS scores was significantly more pronounced in patients receiving placebo compared with those taking diflunisal (P = 0.001), and physical quality of life measures showed significant improvement among diflunisal-treated patients (P = 0.001). Notable during this study was the high rate of attrition in the placebo group, with 50% more placebo-treated patients dropping out of this 2-year study as a result of disease progression, advanced stage of the disease, and varied mutations.
One retrospective analysis of off-label use of diflunisal in patients with TTR-FAP reported treatment discontinuation in 57% of patients because of adverse events that were largely gastrointestinal [65]. Conclusions on the safety of diflunisal in TTR-FAP will depend on further investigations on the impact of known cardiovascular and renal side-effects associated with the NSAID drug class [66,67].
Novel Discoveries in Molecular Biology and Biomedical Science, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Novel Discoveries in Molecular Biology and Biomedical Science
Curator: Larry H. Bernstein, MD, FCAP
UPDATED on 6/1/2016
The following is a collection of current articles on noncoding DNA, synthetic genome engineering, protein regulation of apoptosis, drug design, and geometrics.
No longer ‘junk DNA’ — shedding light on the ‘dark matter’ of the genome
A new tool called “LIGR-Seq” enables scientists to explore in depth what non-coding RNAs actually do in human cells May 23, 2016
he LIGR-seq method for global-scale mapping of RNA-RNA interactions in vivo to reveal unexpected functions for uncharacterized RNAs that act via base-pairing interactions (credit: University of Toronto)
What used to be dismissed by many as “junk DNA” has now become vitally important, as accelerating genomic data points to the importance of non-coding RNAs (ncRNAs) — a genome’s messages that do not specifically code for proteins — in development and disease.
But our progress in understanding these molecules has been slow because of the lack of technologies that allow for systematic mapping of their functions.
Now, professor Benjamin Blencowe’s team at the University of Toronto’s Donnelly Centre has developed a method called “LIGR-seq” that enables scientists to explore in depth what ncRNAs do in human cells.
The study, described in Molecular Cell, was published on May 19, along with two other papers, in Molecular Cell and Cell, respectively, from Yue Wan’s group at the Genome Institute of Singapore and Howard Chang’s group at Stanford University in California, who developed similar methods to study RNAs in different organisms.
Of the 3 billion letters in the human genome, only two per cent make up the protein-coding genes. The genes are copied, or transcribed, into messenger RNA (mRNA) molecules, which provide templates for building proteins that do most of the work in the cell. Much of the remaining 98 per cent of the genome was initially considered by some as lacking in functional importance. However, large swaths of the non-coding genome — between half and three quarters of it — are also copied into RNA.
So then what might the resulting ncRNAs do? That depends on whom you ask. Some researchers believe that most ncRNAs have no function, that they are just a by-product of the genome’s powerful transcription machinery that makes mRNA. However, it is emerging that many ncRNAs do have important roles in gene regulation — some ncRNAs act as carriages for shuttling the mRNAs around the cell, or provide a scaffold for other proteins and RNAs to attach to and do their jobs.
But the majority of available data has trickled in piecemeal or through serendipitous discovery. And with emerging evidence that ncRNAs could drive disease progression, such as cancer metastasis, there was a great need for a technology that would allow a systematic functional analysis of ncRNAs.
“Up until now, with existing methods, you had to know what you are looking for because they all require you to have some information about the RNA of interest. The power of our method is that you don’t need to preselect your candidates; you can see what’s occurring globally in cells, and use that information to look at interesting things we have not seen before and how they are affecting biology,” says Eesha Sharma, a PhD candidate in Blencowe’s group who, along with postdoctoral fellow Tim Sterne-Weiler, co-developed the method.
The human RNA-RNA interactome, showing interactions detected by LIGR-seq (credit: University of Toronto)
The new ‘‘LIGation of interacting RNA and high-throughput sequencing’’ (LIGR-seq) tool captures interactions between different RNA molecules. When two RNA molecules have matching sequences — strings of letters copied from the DNA blueprint — they will stick together like Velcro. With LIGR-seq, the paired RNA structures are removed from cells and analyzed by state-of-the-art sequencing methods to precisely identify the RNAs that are stuck together.
“Most researchers in the life sciences agree that there’s an urgent need to understand what ncRNAs do. This technology will open the door to developing a new understanding of ncRNA function,” says Blencowe, who is also a professor in the Department of Molecular Genetics.
Not having to rely on pre-existing knowledge will boost the discovery of RNA pairs that have never been seen before. Scientists can also, for the first time, look at RNA interactions as they occur in living cells, in all their complexity, unlike in the juices of mashed up cells that they had to rely on before. This is a bit like moving on to explore marine biology from collecting shells on the beach to scuba-diving among the coral reefs, where the scope for discovery is so much bigger.
Actually, ncRNAs come in multiple flavors: there’s rRNA, tRNA, snRNA, snoRNA, piRNA, miRNA, and lncRNA, to name a few, where prefixes reflect the RNA’s place in the cell or some aspect of its function. But the truth is that no one really knows the extent to which these ncRNAs control what goes on in the cell, or how they do this.
Discoveries
Nonetheless, the new technology developed by Blencowe’s group has been able to pick up new interactions involving all classes of RNAs and has already revealed some unexpected findings.
The team discovered new roles for small nucleolar RNAs (snoRNAs), which normally guide chemical modifications of other ncRNAs. It turns out that some snoRNAs can also regulate stability of a set of protein-coding mRNAs. In this way, snoRNAs can also directly influence which proteins are made, as well as their abundance, adding a new level of control in cell biology.
And this is only the tip of the iceberg; the researchers plan to further develop and apply their technology to investigate the ncRNAs in different settings.
“We would like to understand how ncRNAs function during development. We are particularly interested in their role in the formation of neurons. But we will also use our method to discover and map changes in RNA-RNA interactions in the context of human diseases,” says Blencowe.
Abstract of Global Mapping of Human RNA-RNA Interactions
The majority of the human genome is transcribed into non-coding (nc)RNAs that lack known biological functions or else are only partially characterized. Numerous characterized ncRNAs function via base pairing with target RNA sequences to direct their biological activities, which include critical roles in RNA processing, modification, turnover, and translation. To define roles for ncRNAs, we have developed a method enabling the global-scale mapping of RNA-RNA duplexes crosslinked in vivo, “LIGation of interacting RNA followed by high-throughput sequencing” (LIGR-seq). Applying this method in human cells reveals a remarkable landscape of RNA-RNA interactions involving all major classes of ncRNA and mRNA. LIGR-seq data reveal unexpected interactions between small nucleolar (sno)RNAs and mRNAs, including those involving the orphan C/D box snoRNA, SNORD83B, that control steady-state levels of its target mRNAs. LIGR-seq thus represents a powerful approach for illuminating the functions of the myriad of uncharacterized RNAs that act via base-pairing interactions.
Understanding the unknown functions of these genes may lead to the creation of new diagnostic tests for clinical laboratories and anatomic pathology groups
Once again, J. Craig Venter, PhD, is charting new ground in gene sequencing andgenomic science. This time his research team has built upon the first synthetic cell they created in 2010 to build a more sophisticated synthetic cell. Their findings from this work may give pathologists and medical laboratory scientists new tools to diagnose disease.
Recently the research team at the J. Craig Venter Institute (JCVI) and Synthetic Genomics, Inc. (SGI) published their latest findings. Among the things they learned is that science still does not understand the functions of about a third of the genes required for their synthetic cells to function.
JCVI-syn3.0 Could Radically Alter Understanding of Human Genome
Based in La Jolla, Calif., and Rockville, Md., JCVI is a not-for-profit research institute aiming to advance genomics. Building upon its first synthetic cell—Mycoplasma mycoides (M. mycoides) JCVI-syn1.0, which JCVI constructed in 2010—the same team of scientists created the first minimal synthetic bacterial cell, which they calledJCVI-syn3.0. This new artificial cell contains 531,560 base pairs and just 473 genes, which means it is the smallest genome of any organism that can be grown in laboratory media, according to a JCVI-SGI statement.
For pathologists and medical laboratory leaders, the creation of a synthetic life form is a milestone toward better understanding genome sequencing and how this new knowledge may help advance both diagnostics and therapeutics.
“What we’ve done is important because it is a step toward completely understanding how a living cell works,” Clyde Hutchison III, PhD, told New Scientist. “If we can really understand how the cell works, then we will be able to design cells efficiently for the production of pharmaceutical and other useful products.” Hutchison is Professor Emeritus of Microbiology and Immunology at the University of North Carolina at Chapel Hill, Distinguished Professor at the J. Craig Venter Institute, a member of the National Academy of Sciences, and a fellow of the American Academy of Arts and Sciences.
Clyde Hutchison, III, PhD (above), Professor Emeritus of Microbiology and Immunology at the University of North Carolina at Chapel Hill and Distinguished Professor at the J. Craig Venter Institute, stated that his team’s “goal is to have a cell for which the precise biological function of every gene is known.” (Photo credit: JCVI.)
Understanding a Gene’s True Purpose
According to the JCVI researchers, 149 genes have no known purpose. They are, however, necessary for life and health.
“We know about two-thirds of the essential biology, and we’re missing a third,” stated J. Craig Venter, PhD, Founder and CEO of JCVI, in a story published by MedPage Today.
This knowledge is based upon decades of research. JCVI seeks to create a minimal cell operating system to understand biology, while also providing what the JCVI statement called a “chassis for use in industrial applications.”
What Do these Genes Do Anyway?
The JCVI team found that among most genes’ biological functions:
“JCVI-syn3.0 is a working approximation of a minimal cellular genome—a compromise between a small genome size and a workable growth rate for an experimental organism. It retains almost all the genes that are involved in the synthesis and processing of macromolecules. Unexpectedly, it also contains 149 genes with unknown biological functions, suggesting the presence of undiscovered functions that are essential for life,” the researchers told the journal Science.
More research is needed, the scientists say, into the 149 genes that appear to lack specific biologic functions.
Unlocking Mystery of the 149 Genes Could Lead to Advances in Genomic Science
“Finding so many genes without a known function is unsettling, but it’s exciting because it’s left us with much still to learn. It’s like the ‘dark matter’ of biology,” said Alistair Elfick, PhD, Chair of Synthetic Biological Engineering, University of Edinburgh, UK, in the New Scientist article.
Studies such as JCVI’s research is key to broadening understanding and framing appropriate questions about scientific, ethical, and economic implications of synthetic biology.
The creation of a synthetic cell will have a profound and positive impact on understanding of biology and how life works, JCVI said.
Such research may inspire new whole genome synthesis tools and semi-automated processes that could dramatically affect clinical laboratory procedures. It also could lead to new techniques and tools for advanced vaccine and pharmaceuticals, JCVI pointed out.
No single technique has set the molecular biology field ablaze with excitement and potential like the CRISPR-Cas9 genome editing system has following its introduction only a few short years ago. The following articles represent the flexibility of this technique to potentially treat a host of genetic disorders and possibly even prevent the onset of disease.
Scientists recently convened at the CRISPR Precision Gene Editing Congress, held in Boston, to discuss the new technology. As with any new technique, scientists have discovered that CRISPR comes with its own set of challenges, and the Congress focused its discussion around improving specificity, efficiency, and delivery.
With a staggering number of papers published in the past several years involving the characterization and use of the CRISPR/Cas9 gene editing system, it is surprising that researchers are still finding new features of the versatile molecular scissor enzyme.
If a Cas9 nuclease variant could be engineered that was less grabby, it might loosen its grip on DNA sequences throughout the genome—except those sequences representing on-target sites. That’s the assumption that guided a new investigation by researchers at Massachusetts General Hospital.
The gene-editing technology known as CRISPR-Cas9 is starting to raise expectations in the therapeutic realm. In fact, CRISPR-Cas9 and other CRISPR systems are moving so close to therapeutic uses that the technology’s ethical implications are starting to attract notice.
Published: Tuesday, May 24, 2016 A comparison of synthetic gene-activating Cas9 proteins can help guide research and development of therapeutic approaches.
The CRISPR-Cas9 system has come to be known as the quintessential tool that allows researchers to edit the DNA sequences of many organisms and cell types. However, scientists are also increasingly recognizing that it can be used to activate the expression of genes. To that end, they have built a number of synthetic gene activating Cas9 proteins to study gene functions or to compensate for insufficient gene expression in potential therapeutic approaches.
“The possibility to selectively activate genes using various engineered variants of the CRISPR-Cas9 system left many researchers questioning which of the available synthetic activating Cas9 proteins to use for their purposes. The main challenge was that all had been uniquely designed and tested in different settings; there was no side-by-side comparison of their relative potentials,” said George Church, Ph.D., who is Core Faculty Member at the Wyss Institute for Biologically Inspired Engineering at Harvard University, leader of its Synthetic Biology Platform, and Professor of Genetics at Harvard Medical School. “We wanted to provide that side-by-side comparison to the biomedical research community.”
In a study published on 23 May in Nature Methods, the Wyss Institute team reports how it rigorously compared and ranked the most commonly used artificial Cas9 activators in different cell types from organisms including humans, mice and flies. The findings provide a valuable guide to researchers, allowing them to streamline their endeavors.
The team also included Wyss Core Faculty Member James Collins, Ph.D., who also is the Termeer Professor of Medical Engineering & Science and Professor of Biological Engineering at the Massachusetts Institute of Technology (MIT)’s Department of Biological Engineering and Norbert Perrimon, Ph.D., a Professor of Genetics at Harvard Medical School.
Gene activating Cas9 proteins are fused to variable domains borrowed from proteins with well-known gene activation potentials and engineered so that the DNA editing ability is destroyed. In some cases, the second component of the CRISPR-Cas9 system, the guide RNA that targets the complex to specific DNA sequences, also has been engineered to bind gene-activating factors.
“We first surveyed seven advanced Cas9 activators, comparing them to each other and the original Cas9 activator that served to provide proof-of-concept for the gene activation potential of CRISPR-Cas9. Three of them, provided much higher gene activation than the other candidates while maintaining high specificities toward their target genes,” said Marcelle Tuttle, Research Fellow at the Wyss and a co-lead author of the study.
The team went on to show that the three top candidates were comparable in driving the highest level of gene expression in cells from humans, mice and fruit flies, irrespective of their tissue and developmental origins. The researchers also pinpointed ways to further maximize gene activation employing the three leading candidates.
“In some cases, maximum possible activation of a target gene is necessary to achieve a cellular or therapeutic effect. We managed to cooperatively enhance expression of specific genes when we targeted them with three copies of a top performing activator using three different guide RNAs,” said Alejandro Chavez, Ph.D., a Postdoctoral Fellow and the study’s co-first author.
“The ease of use of CRISPR-Cas9 offers enormous potential for development of genome therapeutics. This study provides valuable new design criteria that will help enable synthetic biologists and bioengineers to develop more effective targeted genome engineering technologies in the future,” said Wyss Institute Founding Director Donald Ingber, M.D., Ph.D., who is the Judah Folkman Professor of Vascular Biology at Harvard Medical School and the Vascular Biology Program at Boston Children’s Hospital, and also Professor of Bioengineering at the Harvard John A. Paulson School of Engineering and Applied Sciences.
Engineering T Cells to Functionally Cure HIV-1 Infection
Despite the ability of antiretroviral therapy to minimize human immunodeficiency virus type 1 (HIV-1) replication and increase the duration and quality of patients’ lives, the health consequences and financial burden associated with the lifelong treatment regimen render a permanent cure highly attractive. Although T cells play an important role in controlling virus replication, they are themselves targets of HIV-mediated destruction. Direct genetic manipulation of T cells for adoptive cellular therapies could facilitate a functional cure by generating HIV-1–resistant cells, redirecting HIV-1–specific immune responses, or a combination of the two strategies. In contrast to a vaccine approach, which relies on the production and priming of HIV-1–specific lymphocytes within a patient’s own body, adoptive T-cell therapy provides an opportunity to customize the therapeutic T cells prior to administration. However, at present, it is unclear how to best engineer T cells so that sustained control over HIV-1 replication can be achieved in the absence of antiretrovirals. This review focuses on T-cell gene-engineering and gene-editing strategies that have been performed in efforts to inhibit HIV-1 replication and highlights the requirements for a successful gene therapy–mediated functional cure.
Automated top-down design technique simplifies creation of DNA origami nanostructures
Nanoparticles for drug delivery and cell targeting, nanoscale robots, custom-tailored optical devices, and DNA as a storage medium are among the possible applications
May 27, 2016
The boldfaced line, known as a spanning tree, follows the desired geometric shape of the target DNA origami design method, touching each vertex just once. A spanning tree algorithm is used to map out the proper routing path for the DNA strand. (credit: Public Domain)
MIT, Baylor College of Medicine, and Arizona State University Biodesign Institute researchers have developed a radical new top-down DNA origami* design method based on a computer algorithm that allows for creating designs for DNA nanostructures by simply inputting a target shape.
DNA origami (using DNA to design and build geometric structures) has already proven wildly successful in creating myriad forms in 2- and 3- dimensions, which conveniently self-assemble when the designed DNA sequences are mixed together. The tricky part is preparing the proper DNA sequence and routing design for scaffolding and staple strands to achieve the desired target structure. Typically, this is painstaking work that must be carried out manually.
The new algorithm, which is reported together with a novel synthesis approach in the journal Science, promises to eliminate all that and expands the range of possible applications of DNA origami in biomolecular science and nanotechnology. Think nanoparticles for drug delivery and cell targeting, nanoscale robots in medicine and industry, custom-tailored optical devices, and most interesting: DNA as a storage medium, offering retention times in the millions of years.**
Shape-shifting, top-down software
Unlike traditional DNA origami, in which the structure is built up manually by hand, the team’s radical top-down autonomous design method begins with an outline of the desired form and works backward in stages to define the required DNA sequence that will properly fold to form the finished product.
“The Science paper turns the problem around from one in which an expert designs the DNA needed to synthesize the object, to one in which the object itself is the starting point, with the DNA sequences that are needed automatically defined by the algorithm,” said Mark Bathe, an associate professor of biological engineering at MIT, who led the research. “Our hope is that this automation significantly broadens participation of others in the use of this powerful molecular design paradigm.”
The algorithm, which is known as DAEDALUS (DNA Origami Sequence Design Algorithm for User-defined Structures) after the Greek craftsman and artist who designed labyrinths that resemble origami’s complex scaffold structures, can build any type of 3-D shape, provided it has a closed surface. This can include shapes with one or more holes, such as a torus.
A simplified version of the top-down procedure used to design scaffolded DNA origami nanostructures. It starts with a polygon corresponding to the target shape. Software translates a wireframe version of this structure into a plan for routing DNA scaffold and staple strands. That enables a 3D DNA-based atomic-level structural model that is then validated using 3D cryo-EM reconstruction. (credit: adapted from Biodesign Institute images)
With the new technique, the target geometric structure is first described in terms of a wire mesh made up of polyhedra, with a network of nodes and edges. A DNA scaffold using strands of custom length and sequence is generated, using a “spanning tree” algorithm — basically a map that will automatically guide the routing of the DNA scaffold strand through the entire origami structure, touching each vertex in the geometric form once. Complementary staple strands are then assigned and the final DNA structural model or nanoparticle self-assembles, and is then validated using 3D cryo-EM reconstruction.
The software allows for fabricating a variety of geometric DNA objects, including 35 polyhedral forms (Platonic, Archimedean, Johnson and Catalan solids), six asymmetric structures, and four polyhedra with nonspherical topology, using inverse design principles — no manual base-pair designs needed.
To test the method, simpler forms known as Platonic solids were first fabricated, followed by increasingly complex structures. These included objects with nonspherical topologies and unusual internal details, which had never been experimentally realized before. Further experiments confirmed that the DNA structures produced were potentially suitable for biological applications since they displayed long-term stability in serum and low-salt conditions.
Biological research uses
The research also paves the way for designing nanoscale systems mimicking the properties of viruses, photosynthetic organisms, and other sophisticated products of natural evolution. One such application is a scaffold for viral peptides and proteins for use as vaccines. The surface of the nanoparticles could be designed with any combination of peptides and proteins, located at any desired location on the structure, in order to mimic the way in which a virus appears to the body’s immune system.
The researchers demonstrated that the DNA nanoparticles are stable for more than six hours in serum, and are now attempting to increase their stability further.
The nanoparticles could also be used to encapsulate the CRISPR-Cas9 gene editing tool. The CRISPR-Cas9 tool has enormous potential in therapeutics, thanks to its ability to edit targeted genes. However, there is a significant need to develop techniques to package the tool and deliver it to specific cells within the body, Bathe says.
This is currently done using viruses, but these are limited in the size of package they can carry, restricting their use. The DNA nanoparticles, in contrast, are capable of carrying much larger gene packages and can easily be equipped with molecules that help target the right cells or tissue.
The most exciting aspect of the work, however, is that it should significantly broaden participation in the application of this technology, Bathe says, much like 3-D printing has done for complex 3-D geometric models at the macroscopic scale.
* DNA origami brings the ancient Japanese method of paper folding down to the molecular scale. The basics are simple: Take a length of single-stranded DNA and guide it into a desired shape, fastening the structure together using shorter “staple strands,” which bind in strategic places along the longer length of DNA. The method relies on the fact that DNA’s four nucleotide letters—A, T, C, & G stick together in a consistent manner — As always pairing with Ts and Cs with Gs.
The DNA molecule in its characteristic double stranded form is fairly stiff, compared with single-stranded DNA, which is flexible. For this reason, single stranded DNA makes for an ideal lace-like scaffold material. Further, its pairing properties are predictable and consistent (unlike RNA).
** A single gram of DNA can store about 700 terabytes of information — an amount equivalent to 14,000 50-gigabyte Blu-ray disks — and could potentially be operated with a fraction of the energy required for other information storage options.
Essential role of miRNAs in orchestrating the biology of the tumor microenvironment
MicroRNAs (miRNAs) are emerging as central players in shaping the biology of the Tumor Microenvironment (TME). They do so both by modulating their expression levels within the different cells of the TME and by being shuttled among different cell populations within exosomes and other extracellular vesicles. This review focuses on the state-of-the-art knowledge of the role of miRNAs in the complexity of the TME and highlights limitations and challenges in the field. A better understanding of the mechanisms of action of these fascinating micro molecules will lead to the development of new therapeutic weapons and most importantly, to an improvement in the clinical outcome of cancer patients. Keywords: Exosomes, microRNAs, Tumor microenvironment, Cancer
While cancer treatment and survival have improved worldwide, the need for further understanding of the underlying tumor biology remains. In recent years, there has been a significant shift in scientific focus towards the role of the tumor microenvironment (TME) on the development, growth, and metastatic spread of malignancies. The TME is defined as the surrounding cellular environment enmeshed around the tumor cells including endothelial cells, lymphocytes, macrophages, NK cells, other cells of the immune system, fibroblasts, mesenchymal stem cells (MSCs), and the extracellular matrix (ECM). Each of these components interacts with and influences the tumor cells, continually shifting the balance between pro- and anti-tumor phenotype. One of the predominant methods of communication between these cells is through extracellular vesicles and their microRNA (miRNA) cargo. Extracellular vesicles (EVs) are between 30 nm to a few microns in diameter, are surrounded by a phospholipid bilayer membrane, and are released from a variety of cell types into the local environment. There are three well characterized groups of EVs: 1) exosomes, typically 30–100 nm, 2) microvesicles (or ectosomes), typically 100–1000 nm, and 3) large oncosomes, typically 1–10 μm. Each of these categories has a distinctly unique biogenesis and purpose in cellcell communication despite the fact that current laboratory methods do not always allow precise differentiation. EVs are found to be enriched with membrane-bound proteins, lipid raft-associated and cytosolic proteins, lipids, DNA, mRNAs, and miRNAs, all of which can be transferred to the recipient cell upon fusion to allow cell-cell communications [1]. Of these, miRNAs have been of particular interest in cancer research, both as modifiers of transcription and translation as well as direct inhibitors or enhancers of key regulatory proteins. These miRNAs are a large family of small non-coding RNAs (19–24 nucleotides) and are known to be aberrantly expressed, both in terms of content as well as number, in both the tumor cells and the cells of the TME. Synthesis of these mature miRNA is a complex process, starting with the transcription of long, capped, and polyadenylated pri-miRNA by RNA polymerase II. These are cropped into a 60–100 nucleotide hairpinstructure pre-miRNA by the microprocessor, a heterodimer of Drosha (a ribonuclease III enzyme) and DGCR8 (DiGeorge syndrome critical region gene 8). The premiRNA is then exported to the cytoplasm by exportin 5, cleaved by Dicer, and separated into single strands by helicases. The now mature miRNA are incorporated into the RNA-induced silencing complex (RISC), a cytoplasmic effector machine of the miRNA pathway. The primary mechanism of action of the mature miRNA-RISC complex is through their binding to the 3’ untranslated region, or less commonly the 5’ untranslated region, of target mRNA, leading to protein downregulation either via translational repression or mRNA degradation. More recently, it has been shown that miRNAs can also upregulate the expression of target genes [2]. MiRNA genes are mostly intergenic and are transcribed by independent promoters [3] but can also be encoded by introns, sharing the same promoter of their host gene [4]. MiRNAs undergo the same regulatory mechanisms of any other protein coding gene (promoter methylation, histone modifications, etc.…) [5, 6]. Interestingly, each miRNA may have contradictory effects both within varying tumor cell lines and within different cells of the TME. In this review, we provide a state-of-the-art description of the key role that miRNAs have in the communication between tumor cells and the TME and their subsequent effects on the malignant phenotype. Finally, this review has made every effort to clarify, whenever possible, whether the reference is to the −3p or the -5p miRNA. Whenever such clarification has not been provided, this indicates that it was not possible to infer such information from the cited bibliography.
Angiogenesis and miRNAs Cellular plasticity, critical in the development of malignancy, includes the many diverse mechanisms elicited by cancer cells to increase their malignant potential and develop increasing treatment resistance. One such mechanism, angiogenesis, is critical to the development of metastatic disease, affecting both the growth of malignant cells locally and their survival at distant sites. In the last ten years, miRNAs, often packaged in tumor cell-derived exosomes, have emerged as important contributors to the complicated regulation and balance of pro- and anti-angiogenic factors.
Most commonly, miRNAs derived from cancer cells have oncogenic activity, promoting angiogenesis and tumor growth and survival. The most-well characterized of the pro-angiogenic miRNAs, the miR-17-92 cluster encoding six miRNAs (miR-17, −18a, −19a, −19b, −20a, and −92a), is found on chromosome 13, and is highly conserved among vertebrates [7]. The complex and multifaceted functions of the miR-17-92 cluster are summarized in Fig. 1. Amplification, both at the genetic and RNA level, of miR-17-92 was initially found in several lymphoma cell lines and has subsequently been observed in multiple mouse tumor models [7].
Central role of the miR-17-92 cluster in the biology of the TME. The miR-17-92 cluster encoding miR-17, −18a, −19b, −20a, and -92a is upregulated in multiple tumor types and interacts with various components of the TME to finely “tune” the TME through a complex combination of pro- and anti-tumoral effects
Most commonly, miRNAs derived from cancer cells have oncogenic activity, promoting angiogenesis and tumor growth and survival. The most-well characterized of the pro-angiogenic miRNAs, the miR-17-92 cluster encoding six miRNAs (miR-17, −18a, −19a, −19b, −20a, and −92a), is found on chromosome 13, and is highly conserved among vertebrates [7]. The complex and multifaceted functions of the miR-17-92 cluster are summarized in Fig. 1. Amplification, both at the genetic and RNA level, of miR-17-92 was initially found in several lymphoma cell lines and has subsequently been observed in multiple mouse tumor models [7]. Up-regulation of this particular locus has further been confirmed in miRnome analysis across multiple different tumor types, including lung, breast, stomach, prostate, colon, and pancreatic cancer [8]. The miR-17-92 cluster is directly activated by Myc and modulates a variety of downstream transcription factors important in cell cycle regulation and apoptosis including activation of E2F family and Cyclin-dependent kinase inhibitor (CDKN1A) and downregulation of BCL2L11/BIM and p21 [7]. In addition to promoting cell cycle progression and inhibiting apoptosis, the miR-17-92 cluster also downregulates thrombospondin-1 (Tsp1) and connective tissue growth factor (CTGF), important antiangiogenic proteins [7]. Similarly, microvesicles from colorectal cancer cells contain miR-1246 and TGF-β which are transferred to endothelial cells to silence promyelocytic leukemia protein (PML) and activate Smad 1/5/8 signaling promoting proliferation and migration [9]. Likewise, lung cancer cell line derived microvesicles contain miR-494, in response to hypoxia, which targets PTEN in the endothelial cells promoting angiogenesis through the Akt/eNOS pathway [10]. Lastly, exosomal miR-135b from multiple myeloma cells suppresses the HIF-1/FIH-1 pathway in endothelial cells, increasing angiogenesis [11]. A summary of the studies showing the functions of exosomal miRNAs in shaping the biology of the TME is provided in Table 1.
Table 1
Actions of exosomal miRNAs exchanged between cells of the TME
The most common target of anti-angiogenic therapy is VEGF, and not unsurprisingly, multiple miRNAs (including miR-9, miR-20b, miR-130, miR-150, and miR-497) promote angiogenesis through the induction of the VEGF pathway. The most studied of these is the up-regulation of miR-9 which has been linked to a poor prognosis in multiple tumor types, including breast cancer, non-small cell lung cancer, and melanoma [12]. The two oncogenes MYC and MYCN activate miR-9 and cause E-cadherin downregulation resulting in the upregulated transcription of VEGF [13]. In addition, miR-9 has been shown to upregulate the JAK-STAT pathway, supporting endothelial cell migration and tumor angiogenesis [13]. Both amplification of miR-20b and miR-130 as well as miR-497 suppression regulate VEGF through hypoxia inducible factor 1α (HIF-1α) supporting increased angiogenesis [14, 15, 16, 17]. …..
The pivotal discovery in 2012 by Mitra et al. laid the ground-work for our current knowledge on the interactions between tumor-derived miRNAs and fibroblasts. In combination, the down-regulation of miR-214 and miR-31 and the up-regulation of miR-155 trigger the reprogramming of quiescent fibroblasts to CAFs [32]. As expected, the reverse regulation of these miRNAs reduced the migration and invasion of co-cultured ovarian cancer cells [32]. While the pathway of miR-155’s involvement in CAF biology is still being elucidated, the pathways of miR-214 and miR-31 have been established. In endometrial cancer, miR-31 was found to target the homeobox gene SATB2, leading to enhanced tumor cell migration and invasion [33]. MiR-214 similarly has an inverse correlation with its chemokine target, C-C motif Ligand 5 (CCL5) [32]. CCL5 secretion has been associated with enhanced motility, invasion, and metastatic potential through NF-κB-mediated MMP9 activation and through generation and differentiation of myeloid-derived suppressor cells (MDSCs) [34, 35, 36]. Furthermore, miR-210 and miR-133b overexpression and miR-149 suppression have been subsequently found to independently trigger the conversion to CAFs, possibly through paracrine stimulation, and to additionally promote EMT in prostate and gastric cancer, respectively [37, 38,39]. MiR-210 additionally enlists monocytes and encourages angiogenesis [37]. …
Another function of CAFs is the destruction of the ECM and its remodeling with a tumor-supportive composition and structure which includes modulation of specific integrins and metalloproteinases as some of the most studied miRNA targets. The 23 matrix metalloproteinases (MMPs) are critical in the ECM degradation, disruption of the growth signal balance, resistance to apoptosis, establishment of a favorable metastatic niche, and promotion of angiogenesis [54]. As expected, miRNAs have been found to regulate the actions of MMPs, together working to promote cancer cell growth, invasiveness, and metastasis. In HCC, MMP2 and 9 expression is up-regulated by miR-21 via PTEN pathway downregulation. Similarly, in cholangiocarcinoma it was observed that reduced levels of miR-138 induced up-regulation of RhoC, leading to increased levels of the same two MMPs [55, 56]. ….
As has been shown throughout this review, miRNAs have an important and varied effect on human carcinogenesis by shaping the biology of the TME towards a more permissive pro-tumoral phenotype. The complex events leading to such an outcome are currently quite universally defined as the “educational” process of cancer cells on the surrounding TME. While the initial focus was on the direction from the cancer cell to the surrounding TME, increasingly interest is centered on the implications of a more dynamic bidirectional exchange of genetic information. MiRNAs represent only part of the cargo of the extracellular vesicles, but an increasing scientific literature points towards their pivotal role in creating the micro-environmental conditions for cancer cell growth and dissemination. The nearby future will have to address several questions still unanswered. First, it is absolutely necessary to clarify which miRNAs and to what extent they are involved in this process. The contradictory results of some studies can be explained by the differences in tumor-types and by different concentrations of miRNAs used for functional studies. Understanding whether different concentrations of the same miRNA elicit different target effects and therefore changes the biology of the TME, will represent a significant consideration in the development of this field. It is certainly very attractive (especially in an attempt to develop new and desperately needed better cancer biomarkers) to think that concentrations of miRNAs within the TME are reflected systemically in the circulating levels of that same miRNA, however this has not yet been irrefutably demonstrated. Moreover, the study of the paracrine interactions among different cell populations of the TME and their reciprocal effects has been limited to two, maximum three cell populations. This is still way too far from describing the complexity of the TME and only the development of new tridimensional models of the TME will be able to cast a more conclusive light on such complexity. Finally, the pharmacokinetics of miRNA-containing vesicles is in its infancy at best, and needs to be further developed if the goal is development of new therapies based on the use of exosomic miRNAs. Therefore, the future of miRNA research, particularly in its role in the TME, holds still a lot of questions that need answering. However, for these exact same reasons, this is an incredibly exciting time for research in this field. We can envision a not too far future in which these concerns will be satisfactorily addressed and our understanding of the role of miRNAs within the TME will allow us to use them as new therapeutic weapons to successfully improve the clinical outcome of cancer patients.
Researchers at the Walter and Eliza Hall Institute in Australia have discovered a new way to trigger cell death that could lead to drugs to treat cancer and autoimmune disease.
Programmed cell death (a.k.a. apoptosis) is a natural process that removes unwanted cells from the body. Failure of apoptosis can allow cancer cells to grow unchecked or immune cells to inappropriately attack the body.
The protein known as Bak is central to apoptosis. In healthy cells, Bak sits in an inert state but when a cell receives a signal to die, Bak transforms into a killer protein that destroys the cell.
Triggering the cancer-apoptosis trigger
Institute researchers Sweta Iyer, PhD, Ruth Kluck, PhD, and colleagues unexpectedly discovered that an antibody they had produced to study Bak actually bound to the Bak protein and triggered its activation. They hope to use this discovery to develop drugs that promote cell death.
The researchers used information about Bak’s three-dimensional structure to find out precisely how the antibody activated Bak. “It is well known that Bak can be activated by a class of proteins called ‘BH3-only proteins’ that bind to a groove on Bak. We were surprised to find that despite our antibody binding to a completely different site on Bak, it could still trigger activation,” Kluck said. “The advantage of our antibody is that it can’t be ‘mopped up’ and neutralized by pro-survival proteins in the cell, potentially reducing the chance of drug resistance occurring.”
Drugs that target this new activation site could be useful in combination with other therapies that promote cell death by mimicking the BH3-only proteins. The researchers are now working with collaborators to develop their antibody into a drug that can access Bak inside cells.
Their findings have just been published in the open-access journal Nature Communications. The research was supported by the National Health and Medical Research Council, the Australian Research Council, the Victorian State Government Operational Infrastructure Support Scheme, and the Victorian Life Science Computation Initiative.
Abstract of Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies
During apoptosis, Bak and Bax are activated by BH3-only proteins binding to the α2–α5 hydrophobic groove; Bax is also activated via a rear pocket. Here we report that antibodies can directly activate Bak and mitochondrial Bax by binding to the α1–α2 loop. A monoclonal antibody (clone 7D10) binds close to α1 in non-activated Bak to induce conformational change, oligomerization, and cytochrome c release. Anti-FLAG antibodies also activate Bak containing a FLAG epitope close to α1. An antibody (clone 3C10) to the Bax α1–α2 loop activates mitochondrial Bax, but blocks translocation of cytosolic Bax. Tethers within Bak show that 7D10 binding directly extricates α1; a structural model of the 7D10 Fab bound to Bak reveals the formation of a cavity under α1. Our identification of the α1–α2 loop as an activation site in Bak paves the way to develop intrabodies or small molecules that directly and selectively regulate these proteins.
“Cure” is a word that’s dominated the rhetoric in the war on cancer for decades. But it’s a word that medical professionals tend to avoid. While the American Cancer Society reports that cancer treatment has improved markedly over the decades and the five-year survival rate is impressively high for many cancers, oncologists still refrain from declaring their cancer-free patients cured. Why?
Patients are declared cancer-free (also called complete remission) when there are no more signs of detectable disease.
However, minuscule clusters of cancer cells below the detection level can remain in a patient’s body after treatment. Moreover, such small clusters of straggler cells may undergo metastasis, where they escape from the initial tumor into the bloodstream and ultimately settle in a distant site, often a vital organ such as the lungs, liver or brain.
When a colony of these metastatic cells reaches a detectable size, the patient is diagnosed with recurrent metastatic cancer. About one in three breast cancer patients diagnosed with early-stage cancer later develop metastatic disease, usually within five years of initial remission.
By the time metastatic cancer becomes evident, it is much more difficult to treat than when it was originally diagnosed.
What if these metastatic cells could be detected earlier, before they established a “foothold” in a vital organ? Better yet, could these metastatic cancer cells be intercepted, preventing them them from lodging in a vital organ in the first place?
The implant is a tiny porous polymer disc (basically a miniature sponge, no larger than a pencil eraser) that can be inserted just under a patient’s skin. Implantation triggers the immune system’s “foreign body response,” and the implant starts to soak up immune cells that travel to it. If the implant can catch mobile immune cells, then why not mobile metastatic cancer cells?
We gave implants to mice specially bred to model metastatic breast cancer. When the mice had palpable tumors but no evidence of metastatic disease, the implant was removed and analyzed.
Cancer cells were indeed present in the implant, while the other organs (potential destinations for metastatic cells) still appeared clean. This means that the implant can be used to spot previously undetectable metastatic cancer before it takes hold in an organ.
For patients with cancer in remission, an implant that can detect tumor cells as they move through the body would be a diagnostic breakthrough. But having to remove it to see if it has captured any cancer cells is not the most convenient or pleasant detection method for human patients.
Detecting cancer cells with noninvasive imaging
There could be a way around this, though: a special imaging method under development at Northwestern University called Inverse Spectroscopic Optical Coherence Tomography (ISOCT). ISOCT detects molecular-level differences in the way cells in the body scatter light. And when we scan our implant with ISOCT, the light scatter pattern looks different when it’s full of normal cells than when cancer cells are present. In fact, the difference is apparent when even as few as 15 out of the hundreds of thousands of cells in the implant are cancer cells.
There’s a catch – ISOCT cannot penetrate deep into tissue. That means it is not a suitable imaging technology for finding metastatic cells buried deep in internal organs. However, when the cancer cell detection implant is located just under the skin, it may be possible to detect cancer cells trapped in it using ISOCT. This could offer an early warning sign that metastatic cells are on the move.
This early warning could prompt doctors to monitor their patients more closely or perform additional tests. Conversely, if no cells are detected in the implant, a patient still in remission could be spared from unneeded tests.
The ISOCT results show that noninvasive imaging of the implant is feasible. But it’s a method still under development, and thus it’s not widely available. To make scanning easier and more accessible, we’re working to adapt more ubiquitous imaging technologies like ultrasound to detect tiny quantities of tumor cells in the implant.
Besides providing a way to detect tiny numbers of cancer cells before they can form new tumors in other parts of the body, our implant offers an even more intriguing possibility: diverting metastatic cells away from vital organs, and sequestering them where they cannot cause any damage.
In our mouse studies, we found that metastatic cells got caught in the implant before they were apparent in vital organs. When metastatic cells eventually made their way into the organs, the mice with implants still had significantly fewer tumor cells in their organs than implant-free controls. Thus, the implant appears to provide a therapeutic benefit, most likely by taking the metastatic cells it catches out of the circulation, preventing them from lodging anywhere vital.
Interestingly, we have not seen cancer cells leave the implant once trapped, or form a secondary tumor in the implant. Ongoing work aims to learn why this is. Whether the cells can stay safely immobilized in the implant or if it would need to be removed periodically will be important questions to answer before the implant could be used in human patients.
What the future may hold
For now, our work aims to make the implant more effective at drawing and detecting cancer cells. Since we tested the implant with metastatic breast cancer cells, we also want to see if it will work on other types of cancer. Additionally, we’re studying the cells the implant traps, and learning how the implant interacts with the body as a whole. This basic research should give us insight into the process of metastasis and how to treat it.
In the future (and it might still be far off), we envision a world where recovering cancer patients can receive a detector implant to stand guard for disease recurrence and prevent it from happening. Perhaps the patient could even scan their implant at home with a smartphone and get treatment early, when the disease burden is low and the available therapies may be more effective. Better yet, perhaps the implant could continually divert all the cancer cells away from vital organs on its own, like Iron Man’s electromagnet that deflects shrapnel from his heart.
This solution is still not a “cure.” But it would transform a formidable disease that one out of three cancer survivors would otherwise ultimately die from into a condition with which they could easily live.
New PSA Test Examines Protein Structures to Detect Prostate Cancers
5/16/2016 by Cleveland Clinic
A promising new test is detecting prostate cancer more precisely than current tests, by identifying molecular changes in the prostate specific antigen (PSA) protein, according to Cleveland Clinic research presented today at the American Urological Association annual meeting.
The study – part of an ongoing multicenter prospective clinical trial – found that the IsoPSATM test can also differentiate between high-risk and low-risk disease, as well as benign conditions.
Although widely used, the current PSA test relies on detection strategies that have poor specificity for cancer – just 25 percent of men who have a prostate biopsy due to an elevated PSA level actually have prostate cancer, according to the National Cancer Institute – and an inability to determine the aggressiveness of the disease.
The IsoPSA test, however, identifies prostate cancer in a new way. Developed by Cleveland Clinic, in collaboration with Cleveland Diagnostics, Inc., IsoPSA identifies the molecular structural changes in protein biomarkers. It is able to detect cancer by identifying these structural changes, as opposed to current tests that simply measure the protein’s concentration in a patient’s blood.
“While the PSA test has undoubtedly been one of the most successful biomarkers in history, its limitations are well known. Even currently available prostate cancer diagnostic tests rely on biomarkers that can be affected by physiological factors unrelated to cancer,” said Eric Klein, M.D., chair of Cleveland Clinic’s Glickman Urological & Kidney Institute. “These study results show that using structural changes in PSA protein to detect cancer is more effective and can help prevent unneeded biopsies in low-risk patients.”
The clinical trial involves six healthcare institutions and 132 patients, to date. It examined the ability of IsoPSA to distinguish patients with and without biopsy-confirmed evidence of cancer. It also evaluated the test’s precision in differentiating patients with high-grade (Gleason = 7) cancer from those with low-grade (Gleason = 6) disease and benign findings after standard ultrasound-guided biopsy of the prostate.
Substituting the IsoPSA structure-based composite index for the standard PSA resulted in improvement in diagnostic accuracy. Compared with serum PSA testing, IsoPSA performed better in both sensitivity and specificity.
“We took an ‘out of the box’ approach that has shown success in detecting prostate cancer but also has the potential to address other clinically important questions such as clinical surveillance of patients after treatment,” said Mark Stovsky, M.D., staff member, Cleveland Clinic Glickman Urological & Kidney Institute’s Department of Urology. Stovsky has a leadership position (Chief Medical Officer) and investment interest in Cleveland Diagnostics, Inc. “In general, the clinical utility of prostate cancer early detection and screening tests is often limited by the fact that biomarker concentrations may be affected by physiological processes unrelated to cancer, such as inflammation, as well as the relative lack of specificity of these biomarkers to the cancer phenotype. In contrast, clinical research data suggests that the IsoPSA assay can interrogate the entire PSA isoform distribution as a single stand-alone diagnostic tool which can reliably identify structural changes in the PSA protein that correlate with the presence or absence and aggressiveness of prostate cancer.”
Point of Care, Highly Accurate Cervical Cancer Screening
Fifty-five million times a year, American women go to their gynecologist for a Pap Smear. After waiting a few weeks for the results, more than 3.5 million of them are called back to the physician for a follow up visualization of the cervix. Beyond the stress related to possibly having cancer, the women are then subjected to a colposcopic exam, and all too often, a painful biopsy. Then more stressful waiting for a final diagnosis from the pathologist.
Cervical cancer develops slowly, allowing for successful treatment, when identified on time. Regions with high screening compliancy have low mortality rates from this cancer. In the US, for instance, where screening rates are close to 90%, only 4,200 women die from cervical cancer, annually, or 2.6 women per 100,000. However, the screening process in the developed world is long, complicated and not optimized.
In developing regions however, cervical cancer is a leading cause of women death. Over 85% of the total deaths from this cancer are in developing countries. Regions suffering from low screening rates include not only Africa, India and China, but many Eastern European countries as well. According to an OECD report from 2014, the cervical cancer screening rates in Romania and Hungary are as low as 14.6% and 36.7% respectively. The mortality rates in these countries are high, 16 in 100,000 women in Romania and 7.7 in 100,000 in Hungary.
The current screening process for cervical cancer detection is long, beginning with a Pap or HPV test. Cytology results take weeks to receive. A positive result requires follow-up testing by colposcopy and often biopsy. In countries where there is little access to medical care, or where screening compliancy is low, the chances of successful detection via this multi-step process are small. Developing regions and non-compliant countries require a point of care diagnostic method, which eliminates the need for return visits.
Additional limitations to cervical cancer screening are the low sensitivity and specificity rates of Pap tests and the high false positive rates of HPV test, leading to unnecessary colposcopies. Both cytology and colposcopy testing are highly dependent on operator proficiency for accurate diagnosis.
Biop has developed a new technology for the optimization of this process, into one, three minute, painless optical scan. The vaginal probe uses advanced optical, imaging and non-imaging technologies to identify and classify epithelium based cancers and pre-cancerous lesions. The probe is inserted into the vaginal canal, and scans the entire cervix. The resulting images and optical signatures created from the light, and captured by the sensors, are analyzed by the proprietary algorithm. The result is two pictures, on the physician’s screen; a high resolution photograph of the patient’s cervix, immediately next to a hot/cold map indicating a precise classification and location of any diseased lesions.
Deep learning applied to drug discovery and repurposing
Deep neural networks for drug discovery (credit: Insilico Medicine, Inc.)
Scientists from Insilico Medicine, Inc. have trained deep neural networks (DNNs) to predict the potential therapeutic uses of 678 drugs, using gene-expression data obtained from high-throughput experiments on human cell lines from Broad Institute’s LINCS databases and NIH MeSH databases.
The supervised deep-learning drug-discovery engine used the properties of small molecules, transcriptional data, and literature to predict efficacy, toxicity, tissue-specificity, and heterogeneity of response.
“We used LINCS data from Broad Institute to determine the effects on cell lines before and after incubation with compounds, co-author and research scientist Polina Mamoshina explained to KurzweilIAI.
“We used gene expression data of total mRNA from cell lines extracted and measured before incubation with compound X and after incubation with compound X to identify the response on a molecular level. The goal is to understand how gene expression (the transcriptome) will change after drug uptake. It is a differential value, so we need a reference (molecular state before incubation) to compare.”
The research is described in a paper in the upcoming issue of the journal Molecular Pharmaceutics.
Helping pharmas accelerate R&D
Alex Zhavoronkov, PhD, Insilico Medicine CEO, who coordinated the study, said the initial goal of their research was to help pharmaceutical companies significantly accelerate their R&D and increase the number of approved drugs. “In the process we came up with more than 800 strong hypotheses in oncology, cardiovascular, metabolic, and CNS spaces and started basic validation,” he said.
The team measured the “differential signaling pathway activation score for a large number of pathways to reduce the dimensionality of the data while retaining biological relevance.” They then used those scores to train the deep neural networks.*
“This study is a proof of concept that DNNs can be used to annotate drugs using transcriptional response signatures, but we took this concept to the next level,” said Alex Aliper, president of research, Insilico Medicine, Inc., lead author of the study.
Via Pharma.AI, a newly formed subsidiary of Insilico Medicine, “we developed a pipeline for in silico drug discovery — which has the potential to substantially accelerate the preclinical stage for almost any therapeutic — and came up with a broad list of predictions, with multiple in silico validation steps that, if validated in vitro and in vivo, can almost double the number of drugs in clinical practice.”
Despite the commercial orientation of the companies, the authors agreed not to file for intellectual property on these methods and to publish the proof of concept.
Deep-learning age biomarkers
According to Mamoshina, earlier this month, Insilico Medicine scientists published the first deep-learned biomarker of human age — aiming to predict the health status of the patient — in a paper titled “Deep biomarkers of human aging: Application of deep neural networks to biomarker development” by Putin et al, in Aging; and an overview of recent advances in deep learning in a paper titled “Applications of Deep Learning in Biomedicine” by Mamoshina et al., also in Molecular Pharmaceutics.
Insilico Medicine is located in the Emerging Technology Centers at Johns Hopkins University in Baltimore, Maryland, in collaboration with Datalytic Solutions and Mind Research Network.
* In this study, scientists used the perturbation samples of 678 drugs across A549, MCF-7 and PC-3 cell lines from the Library of Integrated Network-Based Cellular Signatures (LINCS) project developed by the National Institutes of Health (NIH) and linked those to 12 therapeutic use categories derived from MeSH (Medical Subject Headings) developed and maintained by the National Library of Medicine (NLM) of the NIH.
To train the DNN, scientists utilized both gene level transcriptomic data and transcriptomic data processed using a pathway activation scoring algorithm, for a pooled dataset of samples perturbed with different concentrations of the drug for 6 and 24 hours. Cross-validation experiments showed that DNNs achieve 54.6% accuracy in correctly predicting one out of 12 therapeutic classes for each drug.
One peculiar finding of this experiment was that a large number of drugs misclassified by the DNNs had dual use, suggesting possible application of DNN confusion matrices in drug repurposing. FutureTechnologies Media Group | Video presentation Insilico medicine
Abstract of Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data
Deep learning is rapidly advancing many areas of science and technology with multiple success stories in image, text, voice and video recognition, robotics and autonomous driving. In this paper we demonstrate how deep neural networks (DNN) trained on large transcriptional response data sets can classify various drugs to therapeutic categories solely based on their transcriptional profiles. We used the perturbation samples of 678 drugs across A549, MCF-7 and PC-3 cell lines from the LINCS project and linked those to 12 therapeutic use categories derived from MeSH. To train the DNN, we utilized both gene level transcriptomic data and transcriptomic data processed using a pathway activation scoring algorithm, for a pooled dataset of samples perturbed with different concentrations of the drug for 6 and 24 hours. When applied to normalized gene expression data for “landmark genes,” DNN showed cross-validation mean F1 scores of 0.397, 0.285 and 0.234 on 3-, 5- and 12-category classification problems, respectively. At the pathway level DNN performed best with cross-validation mean F1 scores of 0.701, 0.596 and 0.546 on the same tasks. In both gene and pathway level classification, DNN convincingly outperformed support vector machine (SVM) model on every multiclass classification problem. For the first time we demonstrate a deep learning neural net trained on transcriptomic data to recognize pharmacological properties of multiple drugs across different biological systems and conditions. We also propose using deep neural net confusion matrices for drug repositioning. This work is a proof of principle for applying deep learning to drug discovery and development.
A novel nanoscale organic transistor-based biosensor that can detect molecules associated with neurodegenerative diseases and some types of cancer has been developed by researchers at the National Nanotechnology Laboratory (LNNano) in Brazil.
The transistor, mounted on a glass slide, contains the reduced form of the peptide glutathione (GSH), which reacts in a specific way when it comes into contact with the enzyme glutathione S-transferase (GST), linked to Parkinson’s, Alzheimer’s and breast cancer, among other diseases.
Sensitive water-gated copper phthalocyanine (CuPc) thin-film transistor (credit: Rafael Furlan de Oliveira et al./Organic Electronics)
“The device can detect such molecules even when they’re present at very low levels in the examined material, thanks to its nanometric sensitivity,” explained Carlos Cesar Bof Bufon, Head of LNNano’s Functional Devices & Systems Lab (DSF).
Bufon said the system can be adapted to detect other substances by replacing the analytes (detection compounds). The team is working on paper-based biosensors to further lower the cost, improve portability, and facilitate fabrication and disposal.
The research is published in the journal Organic Electronics.
Abstract of Water-gated phthalocyanine transistors: Operation and transduction of the peptide–enzyme interaction
The use of aqueous solutions as the gate medium is an attractive strategy to obtain high charge carrier density (1012 cm−2) and low operational voltages (<1 V) in organic transistors. Additionally, it provides a simple and favorable architecture to couple both ionic and electronic domains in a single device, which is crucial for the development of novel technologies in bioelectronics. Here, we demonstrate the operation of transistors containing copper phthalocyanine (CuPc) thin-films gated with water and discuss the charge dynamics at the CuPc/water interface. Without the need for complex multilayer patterning, or the use of surface treatments, water-gated CuPc transistors exhibited low threshold (100 ± 20 mV) and working voltages (<1 V) compared to conventional CuPc transistors, along with similar charge carrier mobilities (1.2 ± 0.2) x 10−3 cm2 V−1 s−1. Several device characteristics such as moderate switching speeds and hysteresis, associated with high capacitances at low frequencies upon bias application (3.4–12 μF cm−2), indicate the occurrence of interfacial ion doping. Finally, water-gated CuPc OTFTs were employed in the transduction of the biospecific interaction between tripeptide reduced glutathione (GSH) and glutathione S-transferase (GST) enzyme, taking advantage of the device sensitivity and multiparametricity.
The study offers understanding of potential therapeutic targets.
Building on data from The Cancer Genome Atlas (TCGA) project, a multi-institutional team of scientists have completed the first large-scale “proteogenomic” study of breast cancer, linking DNA mutations to protein signaling and helping pinpoint the genes that drive cancer. Conducted by members of the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC), including Baylor College of Medicine, Broad Institute of MIT and Harvard, Fred Hutchinson Cancer Research Center, New York University Langone Medical Center, and Washington University School of Medicine, the study takes aim at proteins, the workhorses of the cell, and their modifications to better understand cancer.
Appearing in the Advance Online Publication of Nature, the study illustrates the power of integrating genomic and proteomic data to yield a more complete picture of cancer biology than either analysis could do alone. The effort produced a broad overview of the landscape of the proteome (all the proteins found in a cell) and the phosphoproteome (the sites at which proteins are tagged by phosphorylation, a chemical modification that drives communication in the cell) across a set of 77 breast cancer tumors that had been genomically characterized in the TCGA project. Although the TCGA produced an extensive catalog of somatic mutations found in cancer, the effects of many of those mutations on cellular functions or patients’ outcomes are unknown.
In addition, not all mutated genes are true “drivers” of cancer — some are merely “passenger” mutations that have little functional consequence. And some mutations are found within very large DNA regions that are deleted or present in extra copies, so winnowing the list of candidate genes by studying the activity of their protein products can help identify therapeutic targets. “We don’t fully understand how complex cancer genomes translate into the driving biology that causes relapse and mortality,” said Matthew Ellis, director of the Lester and Sue Smith Breast Center at Baylor College of Medicine and a senior author of the paper.
“These findings show that proteogenomic integration could one day prove to be a powerful clinical tool, allowing us to traverse the large knowledge gap between cancer genomics and clinical action.” In this study, the researchers at the Broad Institute analyzed breast tumors using accurate mass, high-resolution mass spectrometry, a technology that extends the coverage of the proteome far beyond the coverage that can be achieved by traditional antibody-based methods. This allowed them to scale their efforts and quantify more than 12,000 proteins and 33,000 phosphosites, an extremely deep level of coverage.
Scripps scientists have designed a drug candidate that decreases growth of breast cancer cells.
In a development that could lead to a new generation of drugs to precisely treat a range of diseases, scientists from the Florida campus of The Scripps Research Institute (TSRI) have for the first time designed a drug candidate that decreases the growth of tumor cells in animal models in one of the hardest to treat cancers—triple negative breast cancer.
“This is the first example of taking a genetic sequence and designing a drug candidate that works effectively in an animal model against triple negative breast cancer,” said TSRI Professor Matthew Disney. “The study represents a clear breakthrough in precision medicine, as this molecule only kills the cancer cells that express the cancer-causing gene—not healthy cells. These studies may transform the way the lead drugs are identified—by using the genetic makeup of a disease.”
The study, published by the journal Proceedings of the National Academy of Sciences, demonstrates that the Disney lab’s compound, known as Targaprimir-96, triggers breast cancer cells to kill themselves via programmed cell death by precisely targeting a specific RNA that ignites the cancer.
Short-Cut to Drug Candidates
While the goal of precision medicine is to identify drugs that selectively affect disease-causing biomolecules, the process has typically involved time-consuming and expensive high-throughput screens to test millions of potential drug candidates to identify those few that affect the target of interest. Disney’s approach eliminates these screens.
The new study uses the lab’s computational approach called Inforna, which focuses on developing designer compounds that bind to RNA folds, particularly microRNAs.
MicroRNAs are short molecules that work within all animal and plant cells, typically functioning as a “dimmer switch” for one or more genes, binding to the transcripts of those genes and preventing protein production. Some microRNAs have been associated with diseases. For example, microRNA-96, which was the target of the new study, promotes cancer by discouraging programmed cell death, which can rid the body of cells that grow out of control.
In the new study, the drug candidate was tested in animal models over a 21-day course of treatment. Results showed decreased production of microRNA-96 and increased programmed cell death, significantly reducing tumor growth. Since targaprimir-96 was highly selective in its targeting, healthy cells were unaffected.
In contrast, Disney noted, a typical cancer therapeutic targets and kills cells indiscriminately, often leading to side effects that can make these drugs difficult for patients to tolerate.
Benjamin Zealley and Aubrey D.N.J. de Grey Commentary on Some Recent Theses Relevant to Combating Aging: June 2015
Cancer Autoantibody Biomarker Discovery and Validation Using Nucleic Acid Programmable Protein Array Jie Wang, PhD, Arizona State University
Currently in the United States, many patients with cancer do not benefit from population-based screening due to challenges associated with the existing cancer screening scheme. Blood-based diagnostic assays have the potential to detect diseases in a non-invasive way. Proteins released from small early tumors may only be present intermittently and are diluted to tiny concentrations in the blood, making them difficult to use as biomarkers. However, they can induce autoantibody (AAb) responses, which can amplify the signal and persist in the blood even if the antigen is gone. Circulating autoantibodies are a promising class of molecules that have the potential to serve as early detection biomarkers for cancers. This PhD thesis aims to screen for autoantibody biomarkers for the early detection of two deadly cancers, basal-like breast cancer and lung adenocarcinoma. First, a method was developed to display proteins in both native and denatured conformations on a protein array. This method adopted a novel protein tag technology, called a HaloTag, to immobilize proteins covalently on the surface of a glass slide. The covalent attachment allowed these proteins to endure harsh treatment without becoming dissociated from the slide surface, which enabled the profiling of antibody responses against both conformational and linear epitopes. Next, a plasma screening protocol was optimized to increase significantly the signal-to-noise ratio of protein array–based AAb detection. Following this, the AAb responses in basal-like breast cancer were explored using nucleic acid programmable protein arrays (NAPPA) containing 10,000 full-length human proteins in 45 cases and 45 controls. After verification in a large sample set (145 basal-like breast cancer cases, 145 controls, 70 non-basal breast cancer) by enzyme-linked immunosorbent assay (ELISA), a 13-AAb classifier was developed to differentiate patients from controls with a sensitivity of 33% at 98% specificity. A similar approach was also applied to the lung cancer study to identify AAbs that distinguished lung cancer patients from computed tomography–positive benign pulmonary nodules (137 lung cancer cases, 127 smoker controls, 170 benign controls). In this study, two panels of AAbs were discovered that showed promising sensitivity and specificity. Six out of eight AAb targets were also found to have elevated mRNA levels in lung adenocarcinoma patients using TCGA data. These projects as a whole provide novel insights into the association between AAbs and cancer, as well as general B cell antigenicity against self-proteins.
Comment: There are two widely supported models for cancer development and progression—the clonal evolution (CE) model and the cancer stem cell (CSC) model. Briefly, the former claims that most or all cells in a tumor contribute to its maintenance; as newer and more aggressive clones develop by random mutation, they become responsible for driving growth. The range of different mutational profiles generated is assumed to be large enough to account for disease recurrence after therapy (due to rare resistant clones) and metastasis (clones arising with the ability to travel to distant sites). The CSC model instead asserts that a small number of mutated stem cells are the origin of the primary cell mass, drive metastasis through the intermittent release of undifferentiated, highly mobile progeny, and account for recurrence due to a generally quiescent metabolic profile conferring potent resistance to chemotherapy. In either case, the immunological visibility of an early tumor may be highly sporadic. Clones arising early in CE differ little in proteomic terms from healthy host cells; those that do trigger a response are unlikely to have acquired robust resistance to immune attack, so are destroyed quickly in favor of their stealthier brethren. Likewise, CSCs share some of the immune privilege of normal stem cells and, due to their inherent ability to produce differentiated progeny with distinct proteomic signatures, are partially protected from attacks on their descendants. Consequently, such well-hidden cells may remain in the body for years to decades. The autoantibody panel developed in this study for basal-like breast cancer exhibits exceptional specificity despite a comparatively small training set. Given its ease of application, this suggests great promise for a more exhaustively trained classifier as a populationlevel screening tool.
Condition-Specific Differential Sub-Network Analysis for Biological Systems Deepali Jhamb, PhD, Indiana University
Biological systems behave differently under different conditions. Advances in sequencing technology over the last decade have led to the generation of enormous amounts of condition-specific data. However, these measurements often fail to identify low-abundance genes and proteins that can be biologically crucial. In this work, a novel textmining system was first developed to extract condition-specific proteins from the biomedical literature. The literaturederived data was then combined with proteomics data to construct condition-specific protein interaction networks. Furthermore, an innovative condition-specific differential analysis approach was designed to identify key differences, in the form of sub-networks, between any two given biological systems. The framework developed here was implemented to understand the differences between limb regenerationcompetent Ambystoma mexicanum and regeneration-deficient Xenopus laevis. This study provides an exhaustive systems-level analysis to compare regeneration competent and deficient sub-networks to show how different molecular entities inter-connect with each other and are rewired during the formation of an accumulation blastema in regenerating axolotl limbs. This study also demonstrates the importance of literature-derived knowledge, specific to limb regeneration, to augment the systems biology analysis. Our findings show that although the proteins might be common between the two given biological conditions, they can have a high dissimilarity based on their biological and topological properties in the sub-network. The knowledge gained from the distinguishing features of limb regeneration in amphibians can be used in future to induce regeneration chemically in mammalian systems. The approach developed in this dissertation is scalable and adaptable to understanding differential sub-networks between any two biological systems. This methodology will not only facilitate the understanding of biological processes and molecular functions that govern a given system, but will also provide novel intuitions about the pathophysiology of diseases/conditions.
Comment: We have long advocated a principle of directly comparing young and old bodies as a means to identify the classes of physical damage that accumulate in the body during aging. This approach circumvents our ignorance of the full etiology of each particular disease manifestation, a phenomenally difficult question given the ethical issues of experimenting on human subjects, the lengthy ‘‘incubation time’’ of aging-related diseases, and the complex interconnections between their risk factors—innate and environmental. Repairing such damage has the potential to prevent pathology before symptoms appear, an approach now becoming increasingly mainstream.11 However, a naı¨ve comparison faces a number of difficulties, even given a sufficiently large sample set to compensate for inter-individual variation. Most importantly, the causal significance of a given species cannot be reliably determined from its simple prevalence.12 The catalytic nature of cell biology means that those entities whose abundance changes the most profoundly in absolute terms are quite unlikely to be the drivers of that change and may even spontaneously revert to baseline levels in the absence of on-going stimulation. Meanwhile, functionality is often heavily influenced independently of abundance by post-translational modifications that may escape direct detection. Sub-network analysis uses computational means to identify groups of genes and/or proteins that vary in a synchronized way with some parameter, indicating functional connectivity. The application of methods such as those developed here to the comparison of a wide range of younger and older conditions will facilitate the identification of processes—not merely individual factors—that are impaired with age, and thus will help greatly in identifying the optimal points for intervention.
Development of a Light Actuated Drug Delivery-on-Demand System Chase Linsley, PhD, University of California, Los Angeles
The need for temporal–spatial control over the release of biologically active molecules has motivated efforts to engineer novel drug delivery-on-demand strategies actuated via light irradiation. Many systems, however, have been limited to in vitro proof-of-concept due to biocompatibility issues with the photo-responsive moieties or the light wavelength, intensity, and duration. To overcome these limitations, the objective of this dissertation was to design a light-actuated drug delivery-on-demand strategy that uses biocompatible chromophores and safe wavelengths of light, thereby advancing the clinical prospects of light-actuated drug delivery-on-demand systems. This was achieved by: (1) Characterizing the photothermal response of biocompatible visible light and near-infrared-responsive chromophores and demonstrating the feasibility and functionality of the light actuated on-demand drug delivery system in vitro; and (2) designing a modular drug delivery-on-demand system that could control the release of biologically active molecules over an extended period of time. Three biocompatible chromophores—Cardiogreen, Methylene Blue, and riboflavin—were identified and demonstrated significant photothermal response upon exposure to near-infrared and visible light, and the amount of temperature change was dependent upon light intensity, wavelength, as well as chromophore concentration. As a proof-of-concept, pulsatile release of a model protein from a thermally responsive delivery vehicle fabricated from poly(N-isopropylacrylamide) was achieved over 4 days by loading the delivery vehicle with Cardiogreen and irradiating with near-infrared light. To extend the useful lifetime of the light-actuated drug delivery-on-demand system, a modular, reservoir-valve system was designed. Using poly(ethylene glycol) as a reservoir for model small molecule drugs combined with a poly(N-isopropylacrylamide) valve spiked with chromophore-loaded liposomes, pulsatile release was achieved over 7 days upon light irradiation. Ultimately, this drug delivery strategy has potential for clinical applications that require explicit control over the presentation of biologically active molecules. Further research into the design and fabrication of novel biocompatible thermally responsive delivery vehicles will aid in the advancement of the light-actuated drug delivery-on-demand strategy described here. Comment: Our combined comments on this thesis and the next one appear after the next abstract.
Light-Inducible Gene Regulation in Mammalian Cells Lauren Toth, PhD, Duke University
The growing complexity of scientific research demands further development of advanced gene regulation systems. For instance, the ultimate goal of tissue engineering is to develop constructs that functionally and morphologically resemble the native tissue they are expected to replace. This requires patterning of gene expression and control of cellular phenotype within the tissue-engineered construct. In the field of synthetic biology, gene circuits are engineered to elucidate mechanisms of gene regulation and predict the behavior of more complex systems. Such systems require robust gene switches that can quickly turn gene expression on or off. Similarly, basic science requires precise genetic control to perturb genetic pathways or understand gene function. Additionally, gene therapy strives to replace or repair genes that are responsible for disease. The safety and efficacy of such therapies require control of when and where the delivered gene is expressed in vivo.
Unfortunately, these fields are limited by the lack of gene regulation systems that enable both robust and flexible cellular control. Most current gene regulation systems do not allow for the manipulation of gene expression that is spatially defined, temporally controlled, reversible, and repeatable. Rather, they provide incomplete control that forces the user to choose to control gene expression in either space or time, and whether the system will be reversible or irreversible. The recent emergence of the field of optogenetics—the ability to control gene expression using light—has made it possible to regulate gene expression with spatial, temporal, and dynamic control. Light-inducible systems provide the tools necessary to overcome the limitations of other gene regulation systems, which can be slow, imprecise, or cumbersome to work with. However, emerging light-inducible systems require further optimization to increase their efficiency, reliability, and ease of use.
Initially, we engineered a light-inducible gene regulation system that combines zinc finger protein technology and the light-inducible interaction between Arabidopsis thaliana plant proteins GIGANTEA (GI) and the light oxygen voltage (LOV) domain of FKF1. Zinc finger proteins (ZFPs) can be engineered to target almost any DNA sequence through tandem assembly of individual zinc finger domains that recognize a specific 3-bp DNA sequence. Fusion of three different ZFPs to GI (GI-ZFP) successfully targeted the fusion protein to the specific DNA target sequence of the ZFP. Due to the interaction between GI and LOV, co-expression of GI-ZFP with a fusion protein consisting of LOV fused to three copies of the VP16 transactivation domain (LOV-VP16) enabled blue-light dependent recruitment of LOV-VP16 to the ZFP target sequence. We showed that placement of three to nine copies of a ZFP target sequence upstream of a luciferase or enhanced green fluorescent protein (eGFP) transgene enabled expression of the transgene in response to blue light. Gene activation was both reversible and tunable on the basis of duration of light exposure, illumination intensity, and the number of ZFP binding sites upstream of the transgene. Gene expression could also be patterned spatially by illuminating the cell culture through photomasks containing various patterns.
Although this system was useful for controlling the expression of a transgene, for many applications it is useful to control the expression of a gene in its natural chromosomal position. Therefore, we capitalized on recent advances in programmed gene activation to engineer an optogenetic tool that could easily be targeted to new, endogenous DNA sequences without re-engineering the light inducible proteins. This approach took advantage of CRISPR/Cas9 technology, which uses a gene-specific guide RNA (gRNA) to facilitate Cas9 targeting and binding to a desired sequence, and the light-inducible heterodimerizers CRY2 and CIB1 from Arabidopsis thaliana to engineer a lightactivated CRISPR/Cas9 effector (LACE) system. We fused the full-length (FL) CRY2 to the transcriptional activator VP64 (CRY2FL-VP64) and the amino-terminal fragment of CIB1 to the amino, carboxyl, or amino and carboxyl terminus of a catalytically inactive Cas9. When CRY2-VP64 and one of the CIBN/dCas9 fusion proteins are expressed with a gRNA, the CIBN/dCas9 fusion protein localizes to the gRNA target. In the presence of blue light, CRY2FL binds to CIBN, which translocates CRY2FL-VP64 to the gene target and activates transcription. Unlike other optogenetic systems, the LACE system can be targeted to new endogenous loci by solely manipulating the specificity of the gRNA without having to re-engineer the light-inducible proteins. We achieved light-dependent activation of the IL1RN, HBG1/2, or ASCL1 genes by delivery of the LACE system and four gene-specific gRNAs per promoter region. For some gene targets, we achieved equivalent activation levels to cells that were transfected with the same gRNAs and the synthetic transcription factor dCas9-VP64. Gene activation was also shown to be reversible and repeatable through modulation of the duration of blue light exposure, and spatial patterning of gene expression was achieved using an eGFP reporter and a photomask.
Finally, we engineered a light-activated genetic ‘‘on’’ switch (LAGOS) that provides permanent gene expression in response to an initial dose of blue light illumination. LAGOS is a lentiviral vector that expresses a transgene only upon Cre recombinase–mediated DNA recombination. We showed that this vector, when used in conjunction with a light-inducible Cre recombinase system, could be used to express MyoD or the synthetic transcription factor VP64- MyoD in response to light in multiple mammalian cell lines, including primary mouse embryonic fibroblasts. We achieved light-mediated up-regulation of downstream myogenic markers myogenin, desmin, troponin T, and myosin heavy chains I and II as well as fusion of C3H10T1/2 cells into myotubes that resembled a skeletal muscle cell phenotype. We also demonstrated LAGOS functionality in vivo by engineering the vector to express human VEGF165 and human ANG1 in response to light. HEK 293T cells stably expressing the LAGOS vector and transiently expressing the light-inducible Cre recombinase proteins were implanted into mouse dorsal window chambers. Mice that were illuminated with blue light had increased micro-vessel density compared to mice that were not illuminated. Analysis of human vascular endothelial growth factor (VEGF) and human ANG1 levels by enzyme-linked immunosorbent assay (ELISA) revealed statistically higher levels of VEGF and ANG1 in illuminated mice compared to non-illuminated mice.
In summary, the objective of this work was to engineer robust light-inducible gene regulation systems that can control genes and cellular fate in a spatial and temporal manner. These studies combine the rapid advances in gene targeting and activation technology with natural light-inducible plant protein interactions. Collectively, this thesis presents several optogenetic systems that are expected to facilitate the development of multicellular cell and tissue constructs for use in tissue engineering, synthetic biology, gene therapy, and basic science both in vitro and in vivo.
Comment: Although it is easy to characterize technological progress as following in the wake of scientific discoveries, the reverse is almost equally true; advances in technique open the door to types of experiment previously intractable or impossible. Such is currently the case for the field of optically controlled biotechnology, which has exploded into prominence, particularly over the last half-decade. Light of an appropriate wavelength can penetrate mammalian tissues to a depth of up to a couple of centimeters, rendering much of the living body accessible to optical study and control—still more if the detector/source is integrated into an endoscopic or fiber optic probe. Techniques borrowed from the semiconductor industry allow patterns of illumination to be controlled down to the nanometer scale, ideal for addressing individual cells. The highly controlled time course of such experiments, as compared to traditional means of gene activation, such as the addition of a chemical agent to the medium, eliminates confounding variables, and simplifies data analysis. Furthermore, this level of immediate control opens the door to closed-loop systems where the activity of entities under optical control can be continuously tuned in relation to some parameter(s). In the first of these two illuminating theses, a vehicle is developed that permits light-driven release of a small molecule. Such a system could be employed to target a systemically administered antibiotic or anti-neoplastic agent to a site of infection or cancer while sparing other bodily tissues from toxicity. Because most modern drugs cannot be produced in the body, even given arbitrarily good control of cellular biochemistry, this technique will have lasting value in numerous clinical contexts. In the second thesis, the level of precision achieved is even more profound; the CRISPR/Cas9 system has received much recent attention13 in its own right for its capacity to target arbitrary genetic sequences without an arduous protein-engineering step. The LACE system described stands to permit genetic manipulation with almost arbitrarily good spatial, temporal, and genomic site-specific control, using only means available to a typical university laboratory.
Targeting T Cells for the Immune-Modulation of Human Diseases Regina Lin, PhD, Duke University
Dysregulated inflammation underlies the pathogenesis of a myriad of human diseases ranging from cancer to autoimmunity. As coordinators, executers, and sentinels of host immunity, T cells represent a compelling target population for immune-modulation. In fact, the antigen-specificity, cytotoxicity, and promise of long-lived of immune-protection make T cells ideal vehicles for cancer immunotherapy. Interventions for autoimmune disorders, on the other hand, aim to dampen T cell–mediated inflammation and promote their regulatory functions. Although significant strides have been made in targeting T cells for immune modulation, current approaches remain less than ideal and leave room for improvement. In this dissertation, I seek to improve on current T cell-targeted immunotherapies, by identifying and pre-clinically characterizing their mechanisms of action and in vivo therapeutic efficacy.
CD8+ cytotoxic T cells have potent anti-tumor activity and therefore are leading candidates for use in cancer immunotherapy. The application of CD8+ T cells for clinical use has been limited by the susceptibility of ex vivo– expanded CD8+ T cells to become dysfunctional in response to immunosuppressive microenvironments. To enhance the efficacy of adoptive cell transfer therapy (ACT), we established a novel microRNA (miRNA)-targeting approach that augments CTL cytotoxicity and preserves immunocompetence. Specifically, we screened for miRNAs that modulate cytotoxicity and identified miR-23a as a strong functional repressor of the transcription factor Blimp-1, which promotes CTL cytotoxicity and effector cell differentiation. In a cohort of advanced lung cancer patients, miR- 23a was up-regulated in tumor-infiltrating CD8+ T cells, and its expression correlated with impaired anti-tumor potential of patient CD8+ T cells. We determined that tumor-derived transforming growth factor-b (TGF-b) directly suppresses CD8+ T cell immune function by elevating miR-23a and down-regulating Blimp-1. Functional blockade of miR-23a in human CD8+ T cells enhanced granzyme B expression; and in mice with established tumors, immunotherapy with just a small number of tumor-specific CD8+ T cells in which miR-23a was inhibited robustly hindered tumor progression. Together, our findings provide a miRNA-based strategy that subverts the immunosuppression of CD8+ T cells that is often observed during adoptive cell transfer tumor immunotherapy and identify a TGF-bmediated tumor immune-evasion pathway
Having established that miR-23a-inhibition can enhance the quality and functional resilience of anti-tumor CD8+ T cells, especially within the immune-suppressive tumor microenvironment, we went on to interrogate the translational applicability of this strategy in the context of chimeric antigen receptor (CAR)-modified CD8+ T cells. Although CAR T cells hold immense promise for ACT, CAR T cells are not completely curative due to their in vivo functional suppression by immune barriers—such as TGF-b—within the tumor microenvironment. Because TGF-b poses a substantial immune barrier in the tumor microenvironment, we sought to investigate whether inhibiting miR-23a in CAR T cells can confer immune competence to afford enhanced tumor clearance. To this end, we retrovirally transduced wild-type and miR-23a–deficient CD8+ T cells with the EGFRvIII-CAR, which targets the PepvIII tumorspecific epitope expressed by glioblastomas (GBM). Our in vitro studies demonstrated that while wild-type EGFRvIIICAR T cells were vulnerable to functional suppression by TGF-b, miR-23a abrogation rendered EGFRvIII-CAR T cells immune-resistant to TGF-b. Rigorous preclinical studies are currently underway to evaluate the efficacy of miR-23adeficient EGFRvIII-CAR T cells for GBM immunotherapy.
Last, we explored novel immune-suppressive therapies by the biological characterization of pharmacological agents that could target T cells. Although immune-suppressive drugs are classical therapies for a wide range of autoimmune diseases, they are accompanied by severe adverse effects. This motivated our search for novel immunesuppressive agents that are efficacious and lack undesirable side effects. To this end, we explored the potential utility of subglutinol A, a natural product isolated from the endophytic fungus Fusarium subglutinans. We showed that subglutinol A exerts multimodal immune-suppressive effects on activated T cells in vitro. Subglutinol A effectively blocked T cell proliferation and survival, while profoundly inhibiting pro-inflammatory interferon-c (IFN-c) and interleukin-17 (IL-17) production by fully differentiated effector Th1 and Th17 cells. Our data further revealed that subglutinol A might exert its anti-inflammatory effects by exacerbating mitochondrial damage in T cells, but not in innate immune cells or fibroblasts. Additionally, we demonstrated that subglutinol A significantly reduced lymphocytic infiltration into the footpad and ameliorated footpad swelling in the mouse model of Th1-driven delayed-type hypersensitivity. These results suggest the potential of subglutinol A as a novel therapeutic for inflammatory diseases.
Comment: Immunotherapy is among the most promising approaches to cancer treatment, having the specificity and scope to selectively target transformed cells wherever they may reside within the body and the potential to install a permanent defense against disease recurrence. By the time a typical cancer is clinically diagnosed, however, it has already found means to survive a prolonged period of potential immune attack. The mechanisms by which tumors evade immune surveillance are beginning to be elucidated,15,16 and include both direct suppression of effector cells and progressive editing of the host’s immune repertoire to disfavor future attack. It is inherently difficult to interfere with these defenses directly, due to the selection pressures in genetically heterogeneous neoplastic tissue. Much effort is thus being focused on methods for rendering therapeutically delivered immune cells resistant to their effects. The cytokine TGF-b is paradoxically known to function as both a tumor suppressor in healthy tissue and as a tumorderived species associated with multiple cancer-promoting activities, including enhanced immune evasion. This work identifies the pathway by which TGF-b compromises cytotoxic T cell function in the tumor microenvironment, and demonstrates an effective method for blocking this signal. In many clinical cases, however, editing of the patient’s immune repertoire has already removed or rendered anergic those immune cells able to recognize their cancer. Thus, the finding that blocking TGF-b signaling also appears to enhance the effectiveness of CAR-modified T cells— engineered with an antibody fragment targeting them with high affinity to a particular tumor-associated epitope—is a welcome addition to these already promising results.
Novel Fibonacci and non-Fibonacci structure in the sunflower: results of a citizen science experiment
Jonathan Swinton, Erinma Ochu, The MSI Turing’s Sunflower Consortium
This citizen science study evaluates the occurrence of Fibonacci structure in the spirals of sunflower (Helianthus annuus) seedheads. This phenomenon has competing biomathematical explanations, and our core premise is that observation of both Fibonacci and non-Fibonacci structure is informative for challenging such models. We collected data on 657 sunflowers. In our most reliable data subset, we evaluated 768 clockwise or anticlockwise parastichy numbers of which 565 were Fibonacci numbers, and a further 67 had Fibonacci structure of a predefined type. We also found more complex Fibonacci structures not previously reported in sunflowers. This is the third, and largest, study in the literature, although the first with explicit and independently checkable inclusion and analysis criteria and fully accessible data. This study systematically reports for the first time, to the best of our knowledge, seedheads without Fibonacci structure. Some of these are approximately Fibonacci, and we found in particular that parastichy numbers equal to one less than a Fibonacci number were present significantly more often than those one more than a Fibonacci number. An unexpected further result of this study was the existence of quasi-regular heads, in which no parastichy number could be definitively assigned.
Introduction
Fibonacci structure can be found in hundreds of different species of plants [1]. This has led to a variety of competing conceptual and mathematical models that have been developed to explain this phenomenon. It is not the purpose of this paper to survey these: reviews can be found in [1–4], with more recent work including [5–10]. Instead, we focus on providing empirical data useful for differentiating them.
These models are in some ways now very mathematically satisfying in that they can explain high Fibonacci numbers based on a small number of plausible assumptions, though they are not so satisfying to experimental scientists [11]. Despite an increasingly detailed molecular and biophysical understanding of plant organ positioning [12–14], the very parsimony and generality of the mathematical explanations make the generation and testing of experimental hypotheses difficult. There remains debate about the appropriate choice of mathematical models, and whether they need to be central to our understanding of the molecular developmental biology of the plant. While sunflowers provide easily the largest Fibonacci numbers in phyllotaxis, and thus, one might expect, some of the stronger constraints on any theory, there is a surprising lack of systematic data to support the debate. There have been only two large empirical studies of spirals in the capitulum, or head, of the sunflower: Weisse [15] and Schoute [16], which together counted 459 heads; Schoute found numbers from the main Fibonacci sequence 82% of the time and Weise 95%. The original motivation of this study was to add a third replication to these two historical studies of a widely discussed phenomenon. Much more recently, a study of a smaller sample of 21 seedheads was carried out by Couder [17], who specifically searched for non-Fibonacci examples, whereas Ryan et al. [18] studied the arrangement of seeds more closely in a small sample of Helianthus annuus and a sample of 33 of the related perennial H. tuberosus.
Neither the occurrence of Fibonacci structure nor the developmental biology leading to it are at all unique to sunflowers. As common in other species, the previous sunflower studies found not only Fibonacci numbers, but also the occasional occurrence of the double Fibonacci numbers, Lucas numbers and F4 numbers defined below [1]. It is worth pointing out the warning of Cooke [19] that numbers from these sequences make up all but three of the first 17 integers. This means that it is particularly valuable to look at specimens with large parastichy numbers, such as the sunflowers, where the prevalence of Fibonacci structure is at its most striking.
Neither Schoute nor Weisse reported their precise technique for assigning parastichy numbers to their samples, and it is noteworthy that neither author reported any observation of non-Fibonacci structure. One of the objectives of this study was to rigorously define Fibonacci structure in advance and to ensure that the assignment method, though inevitably subjective, was carefully documented.
This paper concentrates on the patterning of seeds towards the outer rim of sunflower seedheads. The number of ray florets (the ‘petals’, typically bright yellow) or the green bracts behind them tends to have a looser distribution around a Fibonacci number. In the only mass survey of these, Majumder & Chakravarti [20] counted ray florets on 1002 sunflower heads and found a distribution centred on 21.
This citizen science study evaluates the occurrence of Fibonacci structure in the spirals of sunflower (Helianthus annuus) seedheads. This phenomenon has competing biomathematical explanations, and our core premise is that observation of both Fibonacci and non-Fibonacci structure is informative for challenging such models. We collected data on 657 sunflowers. In our most reliable data subset, we evaluated 768 clockwise or anticlockwise parastichy numbers of which 565 were Fibonacci numbers, and a further 67 had Fibonacci structure of a predefined type. We also found more complex Fibonacci structures not previously reported in sunflowers. This is the third, and largest, study in the literature, although the first with explicit and independently checkable inclusion and analysis criteria and fully accessible data. This study systematically reports for the first time, to the best of our knowledge, seedheads without Fibonacci structure. Some of these are approximately Fibonacci, and we found in particular that parastichy numbers equal to one less than a Fibonacci number were present significantly more often than those one more than a Fibonacci number. An unexpected further result of this study was the existence of quasi-regular heads, in which no parastichy number could be definitively assigned.
Incorporation of irregularity into the mathematical models of phyllotaxis is relatively recent: [17] gave an example of a disordered pattern arising directly from the deterministic model while more recently the authors have begun to consider the effects of stochasticity [10,21]. Differentiating between these models will require data that go beyond capturing the relative prevalence of different types of Fibonacci structure, so this study was also designed to yield the first large-scale sample of disorder in the head of the sunflower.
The Fibonacci sequence is the sequence of integers 1,2,3,5,8,13,21,34,55,89,144… in which each member after the second is the sum of the two preceding. The Lucas sequence is the sequence of integers 1,3,4,7,11,18,29,47,76,123… obeying the same rule but with a different starting condition; the F4 sequence is similarly 1,4,5,9,14,23,37,60,97,…. The double Fibonacci sequence 2,4,6,10,16,26,42,68,110,… is double the Fibonacci sequence. We say that a parastichy number which is any of these numbers has Fibonacci structure. The sequencesF5=1,5,6,11,17,28,45,73,… and F8=1,8,9,17,26,43,69,112… also arise from the same rule, but as they had not been previously observed in sunflowers we did not include these in the pre-planned definition of Fibonacci structure for parsimony. One example of adjacent pairs from each of these sequences was, in fact, observed but both examples are classified as non-Fibonacci below. A parastichy number which is any of 12,20,33,54,88,143 is also not classed as having Fibonacci structure but is distinguished as a Fibonacci number minus one in some of the analyses, and similarly 14,22,35,56,90,145 as Fibonacci plus one.
When looking at a seedhead such as in figure 1 the eye naturally picks out at least one family of parastichies or spirals: in this case, there is a clockwise family highlighted in blue in the image on the right-hand side.
Figure 5 plots the individual pairs observed. On the reference line, the ratio of the numbers is equal to the golden ratio so departures from the line mark departures from Fibonacci structure, which are less evident in the more reliable photoreviewed dataset. It can be seen from table 3 that Fibonacci pairings dominate the dataset.
Observed pairings of Fibonacci types of clockwise and anticlockwise parastichy numbers. Other means any parastichy number which neither has Fibonacci structure nor is Fibonacci ±1. Of all the Fibonacci ±1/Fibonacci pairs, only sample 191, a (21,20) pair, was not close to an adjacent Fibonacci pair.
One typical example of a Fibonacci pair is shown in figure 6, with a double Fibonacci case infigure 1 and a Lucas one in figure 7. There was no photoreviewed example of an F4 pairing. The sole photoreviewed assignment of a parastichy number to the F4 sequence was the anticlockwise parastichy number 37 in sample 570, which was relatively disordered. The clockwise parastichy number was 55, lending support to the idea this may have been a perturbation of a (34,55) pattern. We also found adjacent members of higher-order Fibonacci series. Figures 8 and 9 each show well-ordered examples with parastichy counts found adjacent in the F5 and F8 series, respectively: neither of these have been previously reported in the sunflower.
Sunflower 095. An (89,55) example with 89 clockwise parastichies and 55 anticlockwise ones, extending right to the rim of the head. Because these are clear and unambiguous, the other parastichy families which are visible towards the centre are not counted here.
Sunflower 667. Anticlockwise parastichies only, showing competing parastichy families which are distinct but in some places overlapping.
Our core results are twofold. First, and unsurprisingly, Fibonacci numbers, and Fibonacci structure more generally, are commonly found in the patterns in the seedheads of sunflowers. Given the extent to which Fibonacci patterns have attracted pseudo-scientific attention [33], this substantial replication of limited previous studies needs no apology. We have also published, for the first time, examples of seedheads related to the F5 and F8 sequences but by themselves they do not add much to the evidence base. Our second core result, though, is a systematic survey of cases where Fibonacci structure, defined strictly or loosely, did not appear. Although not common, such cases do exist and should shed light on the underlying developmental mechanisms. This paper does not attempt to shed that light, but we highlight the observations that any convincing model should explain. First, the prevalence of Lucas numbers is higher than those of double Fibonacci numbers in all three large datasets in the literature, including ours, and there are sporadic appearances of F4, F5 and F8 sequences. Second, counts near to but not exactly equal to Fibonacci structure are also observable: we saw a parastichy count of 54 more often than the most common Lucas count of 47. Sometimes, ambiguity arises in the counting process as to whether an exact Fibonacci-structured number might be obtained instead, but there are sufficiently many unambiguous cases to be confident this is a genuine phenomenon. Third, among these approximately Fibonacci counts, those which are a Fibonacci number minus one are significantly more likely to be seen than a Fibonacci number plus one. Fourth, it is not uncommon for the parastichy families in a seedhead to have strong departures from rotational symmetry: this can have the effect of yielding parastichy numbers which have large departures from Fibonacci structure or which are completely uncountable. This is related to the appearance of competing parastichy families. Fifth, it is common for the parastichy count in one direction to be more orderly and less ambiguous than that in the other. Sixth, seedheads sometimes possess completely disordered regions which make the assignment of parastichy numbers impossible. Some of these observations are unsurprising, some can be challenged by different counting protocols, and some are likely to be easily explained by the mathematical properties of deformed lattices, but taken together they pose a challenge for further research.
It is in the nature of this crowd-sourced experiment with multiple data sources that it is much easier to show variability than it is to find correlates of that variability. We tried a number of cofactor analyses that found no significant effect of geography, growing conditions or seed type but if they do influence Fibonacci structure, they are likely to be much easier to detect in a single-experimenter setting.
We have been forced by our results to extend classifications of seedhead patterns beyond structured Fibonacci to approximate Fibonacci ones. Clearly, the more loose the definition of approximate Fibonacci, the easier it is to explain away departures from model predictions. Couder [17] found one case of a (54,87) pair that he interpreted as a triple Lucas pair 3×(18,29). While mathematically true, in the light of our data, it might be more compellingly be thought of as close to a (55,89) ideal than an exact triple Lucas one. Taken together, this need to accommodate non-exact patterns, the dominance of one less over one more than Fibonacci numbers, and the observation of overlapping parastichy families suggest that models that explicitly represent noisy developmental processes may be both necessary and testable for a full understanding of this fascinating phenomenon. In conclusion, this paper provides a testbed against which a new generation of mathematical models can and should be built.
Researchers find weaker immune response to viral infections in children with mitochondrial disorders
One of the first human studies on how mitochondrial function impacts immune cells to guide future treatments. Mitochondria convert food and oxygen into energy. Genomic variants in more than 350 genes have been linked to mitochondrial disorders.NHGRI In a new study, National Institutes of Health (NIH) researchers found that altered B cell function in children with mitochondrial disorders led to a weaker and less diverse antibody response to viral infections. The study, published in Frontiers in Immunology, was led by researchers at the National Human Genome Research Institute (NHGRI), who analyzed the gene activities of immune cells in children with mitochondrial disorders and found that B cells, which produce antibodies to fight viral infections, are less able to survive cellular stress.
“Our work is one of the first examples to study how B cells are affected in mitochondrial disease by looking at human patients,” said Eliza Gordon-Lipkin, M.D., assistant research physician in NHGRI’s Metabolism, Infection and Immunity Section and co-first author of the paper.
Mitochondria are important components of nearly every cell in the body because they convert food and oxygen into energy. Genomic variants in more than 350 genes have been linked to mitochondrial disorders with varied symptoms depending on which cells are affected.
“For children with mitochondrial disorders, infections can be life threatening or they can worsen the progression of their disorder,” said Peter McGuire, M.B.B.Ch., NHGRI investigator, head of the Metabolism, Infection and Immunity Section and senior author of the study. “We wanted to understand how immune cells differ in these patients and how that influences their response to infections.”
Around 1 in 5,000 people worldwide have a mitochondrial disorder. Examples of mitochondrial disorders are Leigh’s syndrome, which primarily affects the nervous system, and Kearns-Sayre syndrome, which primarily affects the eyes and heart. While mitochondrial disorders are known to affect organs such as the heart, liver, and brain, less is known how they affect the immune system. Using a genomic technique called single-cell RNA sequencing, which analyzes gene activity in different cell types, researchers studied immune cells found in blood. These cells include different types of white blood cells that help the body fight infections. During stressful conditions, these cells produce a microRNA called mir4485. MicroRNAs are small strings of RNA that help control when and where genes are turned on and off. mir4485 controls cellular pathways that help cells survive.
“We think that B cells in these patients undergo cellular stress when they turn into plasma cells and produce antibodies, and these B cells then try to survive by producing the microRNA to cope,” said Dr. McGuire. “But the B cells are too fragile due to their limited energy, so they are unable to survive the stressful conditions.”
Researchers used a technique called VirScan to look at all past viral infections, assess how well the immune system fought those infections and see the effects of B cells and plasma cells on antibody production. With a weaker antibody response, the immune systems in children with mitochondrial disorders are less able to recognize and neutralize invading viruses and clear infections. Researchers aim to use the results of this study to guide future treatment of patients with mitochondrial disorders, noting that more translational studies are needed in this research area.
Reference: Gordon-Lipkin et al. Primary oxidative phosphorylation defects lead to perturbations in the human B cell repertoire. Frontiers in Immunology. DOI: 10.3389/fimmu.2023.1142634. (2023)
Regulatory T (TReg) cells are essential for maintaining peripheral tolerance, preventing autoimmune diseases and limiting chronic inflammatory diseases. However, they also limit beneficial responses by suppressing sterilizing immunity and limiting antitumour immunity. Given that TReg cells can have both beneficial and deleterious effects, there is considerable interest in determining their mechanisms of action. In this Review, we describe the basic mechanisms used by TReg cells to mediate suppression and discuss whether one or many of these mechanisms are likely to be crucial for TReg-cell function. In addition, we propose the hypothesis that effector T cells may not be ‘innocent’ parties in this suppressive process and might in fact potentiate TReg-cell function.
This schematic depicts the various regulatory T (Treg)-cell mechanisms arranged into four groups centred around four basic modes of action. ‘Inhibitory cytokines’ include interleukin-10 (IL-10), interleukin-35 (IL-35) and transforming growth factor-β (TGF-β). ‘Cytolysis’ includes granzyme-A- and granzyme-B-dependent and perforin-dependent killing mechanisms. ‘Metabolic disruption’ includes high affinity IL-2 receptor α (CD25)-dependent cytokine-deprivation-mediated apoptosis, cyclic AMP (cAMP)-mediated inhibition, and CD39- and/or CD73-generated, adenosine–purinergic adenosine receptor (A2A)-mediated immunosuppression. ‘Targeting dendritic cells’ includes mechanisms that modulate DC maturation and/or function such as lymphocyte activation gene-3 (LAG3; also known as CD223)–MHC-class-II-mediated suppression of DC maturation, and cytotoxic T lymphocyte antigen-4 (CTLA4)–CD80/CD86-mediated induction of indoleamine 2,3-dioxygenase (IDO), which is an immunosuppressive molecule, by DCs.
Model for how effector T cells might boost Treg-cell function
This occurs in three stages. (a) Initial regulatory T (Treg)-cell activation induces production of regulatory factors such as interleukin-35 (IL-35). (b) Treg cells ‘sense’ the presence of recently activated effector T cells through a receptor–ligand interaction (cell surface or soluble). (c) This in turn boosts or potentiates Treg-cell function resulting in the enhanced production of regulatory mediators, such as IL-35, and perhaps the induction of new mediators.
Regulatory T (Treg) cells are essential for maintaining peripheral tolerance, preventing autoimmune diseases and limiting chronic inflammatory diseases. However, they also limit beneficial responses by suppressing sterilizing immunity and limiting anti-tumour immunity. Given that Treg cells can have both beneficial and deleterious effects, there is considerable interest in determining their mechanisms of action. In this Review, we discuss the basic mechanisms used by Treg cells to mediate suppression, and discuss whether one or many of these mechanisms are likely to be crucial for Tregcell function. In addition, we present the hypothesis that effector T cells may not be ‘innocent’ parties in this suppressive process and might in fact potentiate Treg-cell function.
Several sophisticated regulatory mechanisms are used to maintain immune homeostasis, prevent autoimmunity and moderate inflammation induced by pathogens and environmental insults. Chief amongst these are regulatory T (Treg) cells that are now widely regarded as the primary mediators of peripheral tolerance. Although Treg cells play a pivotal role in preventing autoimmune diseases, such as type 1 diabetes1,2, and limiting chronic inflammatory diseases, such as asthma and inflammatory bowel disease (IBD)3,4, they also block beneficial responses by preventing sterilizing immunity to certain pathogens5,6 and limiting anti-tumour immunity7. A seminal advance in the analysis of Treg cells came with the identification of a key transcription factor, forkhead box P3 (FOXP3), that is required for their development, maintenance and function8,9. Mice and patients that lack FOXP3 develop a profound autoimmune-like lymphoproliferative disease that graphically emphasizes the importance of Treg cells in maintaining peripheral tolerance10-12 (BOX 1). Although FOXP3 has been proposed as the master regulator of Treg cells that controls the expression of multiple genes that mediate their regulatory activity13,14, this has been recently challenged raising the possibility that other transcriptional events may operate upstream of and/or concurrently with FOXP3 to mediate Treg-cell development15.
While Foxp3 has proven to be an invaluable marker for murine Treg cells, its role in human Treg cells is less straightforward (see BOX 2 for a discussion of Treg-cell markers). Humans that lack FOXP3 develop immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), a severe autoimmune disease that presents early in infancy. Although FOXP3 appears to be required for human Treg-cell development and function, expression of FOXP3 alone is clearly not sufficient as a significant percentage of human activated T cells express FOXP3 and yet do not possess regulatory activity16-20. Furthermore, induction of FOXP3 in human T cells by transforming growth factor-β (TGFβ) does not confer a regulatory phenotype, in contrast to their murine counterparts20. Consequently, FOXP3 is not a good marker for human Treg cells (BOX 2). Whether this distinction is due to intrinsic differences between mouse and human FOXP3 and/or a requirement for an additional cofactor/ transcription factor is an important question that needs to be resolved.
Significant progress has been made over the last few years in delineating the molecules and mechanisms that Treg cells use to mediate suppression21,22. In this Review, we outline our current understanding of the mechanisms used by Treg cells to mediate suppression, and the challenges that lie ahead in defining their mode of action. We also discuss whether Treg cells are likely to depend on one, a few or many of these mechanisms. In addition, we propose that effector T cells may have a significant role in boosting and/or modulating Treg-cell function. Unless stated, we focus here primarily on the mechanisms that are used by thymus-derived natural CD4+CD25+ FOXP3+ Treg cells.
Basic mechanisms of Treg-cell function Defining the mechanisms of Treg-cell function is clearly of crucial importance. Not only would this provide insight into the control processes of peripheral tolerance but it would probably provide a number of potentially important therapeutic targets. Although this quest has been ongoing since interest in Treg cells was reignited in 199523, there has been significant progress in the last few years. From a functional perspective, the various potential suppression mechanisms of Treg cells can be grouped into four basic ‘modes of action’: suppression by inhibitory cytokines, suppression by cytolysis, suppression by metabolic disruption, and suppression by modulation of dendritic-cell (DC) maturation or function (FIG. 1).
Suppression by inhibitory cytokines Inhibitory cytokines, such as interleukin-10 (IL-10) and TGFβ, have been the focus of considerable attention as a mechanism of Treg-cell-mediated suppression. There has also been significant interest in their ability to generate induced (also known as adaptive) Treg-cell populations, either naturally in vivo or experimentally as a potential therapeutic modality (BOX 3). Although the general importance of IL-10 and TGFβ as suppressive mediators is undisputed, their contribution to the function of thymus-derived, natural Treg cells is still a matter of debate24. This is partly due to the general perception that Treg cells function in a contactdependent manner25,26. Indeed, in vitro studies using neutralizing antibodies or T cells that are unable to produce or respond to IL-10 and TGFβ suggested that these cytokines may not be essential for Treg-cell function25-28. However, this contrasts with data from in vivo studies29,30.
In allergy and asthma models, evidence suggests that both natural and antigen-specific Treg cells control disease in a manner that is, in part, dependent on IL-1029 and in some reports dependent on both IL-10 and TGFβ 31. Adoptive transfer of allergen-specific Treg cells induced significant IL-10 production by CD4+ effector T cells in the lung following allergen challenge and this Treg-cell-mediated control of disease was reversed by treatment with an IL-10- receptor-specific antibody32. However, suppression of allergic inflammation and airway hyper-reactivity, and increased production of IL-10 still occurred following transfer of IL-10- deficient Treg cells, suggesting that Treg cells can suppress the Th2-driven response to allergens in vivo through an IL-10-dependent mechanism, but that the production of IL-10 by Treg cells themselves is not required for the suppression observed. This contrasts with a recent study suggesting that the Treg-cell-specific ablation of IL-10 expression resulted in increased lung allergic inflammation and hyperreactivity33.
This scenario might occur in other disease models. For instance, the effects of IL-10 can only be partially attributed to Treg-cell-derived IL-10 in the immune response to hepatitis B virus34 and in the allograft tolerance response elicited by splenocytes exposed to non-inherited maternal antigens35. Recently, it was also shown that IL-10 is crucial for the control of various infections in which Treg cells have been reported to be involved including Mycobacterium tuberculosis36, Toxoplasma gondii37, Leishmania major38, and Trichinella spiralis39. However, Treg cells were not the source of IL-10 in all of these infection models.
By contrast, several studies have shown that IL-10 production by Treg cells is essential for the prevention of colitis in mouse models of IBD40. Moreover, it appears that the tumour microenvironment promotes the generation of FOXP3+ Treg cells that mediate IL-10- dependent, cell-contact independent, suppression41. Similarly, in UV-radiation-induced carcinogenesis, IL-10 production by Treg cells appears to be important for blocking anti-tumour immunity42. IL-10 produced by Treg cells also appears to be crucial for IL-10-mediated tolerance in a model of hepatitis induced by concanavalin A43 and tolerance to bacterial and viral superantigens44. In addition, recent papers suggest new roles for Treg-cell-derived IL-10 in the induction of feto-maternal tolerance45 and B-cell-enhanced recovery from experimental autoimmune encephalomyelitis46. Collectively, the picture that appears to be emerging is that the relative importance of Treg-cell-derived IL-10 is very dependent on the target organism or disease and on the experimental system. Furthermore, the Treg-cell-specific deletion of IL-10 did not result in the development of spontaneous systemic autoimmunity, but did result in enhanced pathology in the colon of older mice and in the lungs of mice with induced airway hypersensitivity, suggesting that the function of Treg-cell-derived IL-10 may be restricted to controlling inflammatory responses induced by pathogens or environmental insults33.
While some early in vitro studies using neutralizing antibodies to TGFβ or Treg cells lacking TGFβ 25,47 indicated that TGFβ was not required for natural Treg-cell function, other studies, both in vitro and in vivo suggested a critical role for Treg-cell surface bound TGFβ 48,49. Therefore, the importance of TGFβ for natural Treg-cell function has also been a controversial topic. Indeed, there has been considerably more focus recently on the importance of TGFβ in the development of induced Treg cells and perhaps in Treg-cell maintenance in general (BOX 3). However, there are studies that suggest that TGFβ produced by Treg cells may directly participate in effector T-cell suppression. For instance, effector T cells that are resistant to TGFβ-mediated suppression cannot be controlled by Treg cells in an IBD model50. In addition, TGFβ produced by Treg cells has been found to be important in the control of the host immune response to M. tuberculosis36, suppression of allergic responses31 and prevention of colitis in an IBD model51. Interestingly, TGFβ produced by Treg cells has also been implicated in limiting anti-tumour immunity in head and neck squamous-cell carcinoma52 and in follicular lymphoma53 by rendering T cells unresponsive to the tumour. TGFβ also appears to limit the anti-tumour activity of cytokine-induced killer cells54.
Membrane-tethered TGFβ can also mediate suppression by Treg cells in a cell-cell contactdependent manner48. Treg cells can control islet infiltration of CD8+ T cells and delay the progress of diabetes through membrane-tethered TGFβ 49. However, experiments using mice deficient in TGFβ-receptor (TGFβR) signalling in effector T cells or using TGFβ or TGFβR blocking reagents failed to show that membrane-tethered TGFβ is required for natural Treg cell development or function47. More recently, however, interest in membrane-tethered TGFβ has re-surfaced with the description of a previously unappreciated role for it in the tumour microenvironment. TGFβ associated with tumour exosome membranes appears to enhance the suppressive function of Treg cells and skew T cells away from their effector functions and towards regulatory functions55. Furthermore, ovalbumin-induced airway inflammation can be attenuated by heme oxygenase-1 through membrane-tethered TGFβ and IL-10 secretion by Treg cells56, a process that activates the Notch1–HES1 (hairy and enhancer of split 1) axis in target cells57. Thus, in light of the most current data, it now appears that soluble and/or membrane-tethered TGFβ may have a previously unappreciated role in natural Treg-cell function.
Recently, a new inhibitory cytokine, IL-35, has been described that is preferentially expressed by Treg cells and is required for their maximal suppressive activity58. IL-35 is a novel member of the IL-12 heterodimeric cytokine family and is formed by the pairing of Epstein–Barr virus induced gene 3 (Ebi3), which normally pairs with p28 to form IL-27, and p35 (also known as Il12a), which normally pairs with p40 to form IL-12. Both Ebi3 and Il12a are preferentially expressed by murine Foxp3+ Treg cells58,59, but not resting or active effector T cells, and are significantly upregulated in actively suppressing Treg cells58. As predicted for a heterodimeric cytokine, both Ebi3−/− and Il12a−/− Treg cells had significantly reduced regulatory activity in vitro and failed to control homeostatic proliferation and cure IBD in vivo. This precise phenocopy suggested that IL-35 is required for the maximal suppressive activity of Treg cells. Importantly IL-35 was not only required but sufficient, as ectopic expression of IL-35 conferred regulatory activity on naive T cells and recombinant IL-35 suppressed T cell proliferation in vitro58. Although IL-35 is an exciting addition to the Treg-cell portfolio, there is clearly much that remains to be defined about this cytokine and its contribution to Treg-cell function. For instance, it remains to be determined if IL-35 suppresses the development and/or function of other cell types such as DCs and macrophages.
It is now clear that three inhibitory cytokines, IL-10, IL-35 and TGFβ, are key mediators of Treg-cell function. Although they are all inhibitory, the extent to which they are utilized in distinct pathogenic/homeostatic settings differs suggesting a non-overlapping function, which needs further refinement.
……….
How many mechanisms do Treg cells need? Although efforts to define the suppressive mechanisms used by Treg cells continue, an important question looms large. Is it likely that all these molecules and mechanisms will be crucial for Treg-cell function? There are three broad possibilities.
One, a single, overriding suppressive mechanism is required by all Treg cells Until the entire mechanistic panoply of Treg cells is defined, one cannot completely rule out this possibility. However, this possibility would seem unlikely as none of the molecules and/ or mechanisms that have been defined to date, when blocked or deleted, result in the complete absence of regulatory activity — a consequence that one might predict would result in a ‘Scurfy-like’ phenotype (BOX 1). So, although Treg cells that lack a single molecule, for instance IL-10, IL-35 or granzyme B, exhibit significantly reduced suppressor function, a scurfy phenotype does not ensue. Given that none of the current Treg-cell mechanisms can exclusively claim this distinction, it seems unlikely that any ‘unknown’ molecules or mechanisms could do so either.
Two, multiple, non-redundant mechanisms are required for maximal Treg-cell function In the studies conducted to date, Treg cells that lack various suppressive molecules have been shown to be functionally defective. This favours a scenario where there are multiple mechanisms that can be used by Treg cells but they are non-redundant, with each molecule contributing to the mechanistic whole. At present, this possibility would seem plausible. Indeed, this is supported by the recent analysis of mice possessing a Treg-cell-specific ablation of IL-10 expression, in which enhanced pathology was observed following environmental insult33. One would predict that at some point we should be able to generate knockout mice that lack a particular set of genes which results in a complete loss of Treg-cell activity. For this to be truly non-redundant, this list would probably be restricted and small (2–4 genes).
Three, multiple, redundant mechanisms are required for maximal Treg-cell function With the plethora of regulatory mechanisms described to date and the possibility of more yet to be identified, it is conceivable that there are multiple mechanisms that function redundantly. Such a redundant system would help to mitigate against effector T-cell escape from regulatory control. Also, given the very small size of the Treg-cell population, a sizable arsenal may be required at the height of an effector T-cell attack. Of course, it is possible that a semi-redundant scenario exists.
These possibilities have been discussed from the perspective of there being a single homogeneous Treg-cell population. However, as for helper T cell subsets it remains possible that a few or even many different Treg-cell subsets exist24. Each of these may rely on one or multiple regulatory mechanisms. Several recent studies have provided support for both phenotypic and functional heterogeneity amongst Treg cells. For instance, it has recently been shown that a small sub-population of Treg cells express the chemokine receptor CCR6, which is associated with T cells possessing an effector-memory phenotype102. CCR6+ Treg cells appeared to accumulate in the central nervous systems of mice with experimental autoimmune encephalomyelitis (EAE) suggesting that they may have a prevalent role in controlling responses in inflamed tissues. Heterogeneous expression of HLA-DR has also been suggested to mark different subpopulations of functionally distinct human Treg cells103. Indeed, HLADR positive Treg cells were found to be more suppressive than their DR negative counterparts. One might speculate that their enhanced inhibitory activity is due to DR-mediated ligation of the inhibitory molecule LAG3 expressed by activated effector T cells95,96.
So, if multiple suppressor mechanisms exist, how might these be integrated and used productively by Treg cells in vivo? We would propose the following possible models21. First, a ‘hierarchical’ model in which Treg cells possess many mechanisms that could be used but only one or two that are really crucial and consistently important in a variety of regulatory settings. Second, a ‘contextual’ model where different mechanisms become more or less important depending on the background or context in which the Treg cells reside and the type of target cell that they have to repress. For example, some cell types may be inhibited primarily by cytokines, whereas others are most effectively suppressed through lysis by Treg cells. Alternatively, different mechanisms may be more effective in different tissue compartments or in different disease settings. This notion is supported by the recent analysis of mice in which IL-10 expression was specifically ablated in Treg cells33. Whereas Treg-cell-derived IL-10 was not required for the systemic control of autoimmunity, it did seem to be required from the control of inflammatory events at mucosal interfaces such as the lungs and colon. As a clear picture of the available Treg-cell weaponry emerges, an important challenge will be to determine their relative importance and contribution to Treg-cell function in different disease models.
A hypothesis: effector T cells potentiate Treg-cell function? Most cellular interactions within the immune system are bidirectional, with molecular signals moving in both directions even though the interaction has broader unidirectional intentions (for example, CD4+ T-cell help). However, to date the general perception is that Treg cells suppress and effector T cells capitulate. We hypothesize that this is in fact an incomplete picture and that effector T cells have a very active role in their own functional demise. Three recent observations support this view. First, we have recently examined the molecular signature of activated Treg cells in the presence and absence of effector T cells and were surprised to find that it was strikingly different, with hundreds of genes differentially modulated as a consequence of the presence of effector T cells (C.J.W. and D.A.A.V., unpublished observations). Second, we have shown that Ebi3 and Il12a mRNA are markedly upregulated in Treg cells that were co-cultured with effector T cells, supporting the idea that effector T cells may provide signals which boost IL-35 production in trans58. Third, we found that Treg cells were able to mediate suppression of effector T cells across a permeable membrane when placed in direct contact with effector T cells in the upper chamber of a Transwell™ plate (L.W.C. and D.A.A.V., unpublished observations). Interestingly, this suppression was IL-35 dependent, as Ebi3−/− Treg cells were unable to mediate this ‘long-distance’ suppression. Collectively, these data suggest that it is the ‘induction’, rather than the ‘function’, of Treg-cell suppression that is contact-dependent and that effector T cells have an active role in potentiating Treg-cellmediated suppression. Therefore, we hypothesize that receptor–ligand interactions between the co-cultured CD4+ effector T cells and Treg cells initiate a signalling pathway that leads to enhanced IL-35 secretion and regulatory activity (FIG. 2). While the molecule that mediates this enhanced Treg-cell suppression is unknown, it is possible that IL-2 may serve this function104. Given the contrasting genetic profiles of activated Treg cells in the presence and absence of effector T cells, it seems possible that this interaction may boost the expression of other regulatory proteins. It may well be that effector T cells unwittingly perform the ultimate act of altruism.
Concluding remarks Although significant progress has been made over the last few years in defining the mechanisms that Treg cells use to mediate their suppressive function, there is clearly much that remains to be elucidated and many questions persist. First, are there more undiscovered mechanisms and/ or molecules that mediate Treg-cell suppression? What is clear is that the transcriptional landscape of Treg cells is very different from naive or activated effector T cells. There are literally thousands of genes that are upregulated (or downregulated) in Treg cells compared with effector T cells. Although it seems unlikely that all or many of these will be crucial for Treg-cell function, it is quite possible that a few undiscovered genes might be important. It should be noted that although we are discussing mechanisms here, it is clear that some of these molecules may perform key Treg-cell functions, such as Treg-cell homing and homeostasis, which are likely to indirectly influence their suppressive capacity in vivo but don’t directly contribute to their inhibitory activity. It is also possible that some of these unknown molecules may represent more specific markers for the characterization and isolation of Treg cells, a particularly important issue for the analysis and use of human Treg cells (BOX 2).
Second, which mechanisms are most important? An important but potentially complex challenge will be to determine if a few mechanisms are important in many Treg-cell settings or whether different mechanisms are required in different cellular scenarios. At present it is difficult to assess this objectively as these mechanisms have predominantly been elucidated in different labs using distinct experimental systems and thus none have really been compared in side-by-side experiments. Furthermore, only recently have conditional mutant mice been examined that have a regulatory component specifically deleted in Treg cells33.
It almost goes without saying that although defining the Treg-cell mode of action is of great academic importance, it is also essential in order to develop effective approaches for the clinical manipulation of Treg cells. Given the capacity of Treg cells to control inflammation and autoimmunity, and their implication in blocking effective anti-tumour immunity and preventing sterilizing immunity, it seems probable that a clear understanding of how Treg cells work will present definitive opportunities for therapeutic intervention.
Mice that carry a spontaneous loss-of-function mutation (known as Scurfy mice) or a deletion of Foxp3 develop a fatal autoimmune-like disease with hyperresponsive CD4+ T cells9,12. More recently Foxp3:diptheria toxin receptor (DTR) knockin mice have allowed for the selective depletion of Treg cells following DT treatment105. These mice have been invaluable for dissecting the role of Foxp3 in Treg-cell function. Given the profound phenotype in these mice, there is a general expectation that genetic disruption of any key Treg-cell inhibitory molecule or mechanism would probably result in a Scurfy-like phenotype. Of course, it is also possible that deletion of a key Treg-cell gene may be more synonymous with DT-mediated Treg-cell depletion where Foxp3 may still serve to prevent expression of proinflammatory cytokines105. Nonetheless, this has lead to the notion that if mutant mice don’t have a Scurfy-like or a Treg-cell-depleted phenotype, then the disrupted gene probably isn’t important for Treg-cell function. This may not necessarily be correct. Indeed, it is possible that no mouse lacking a Treg-cell inhibitory effector molecule will ever be generated that develops a profound, spontaneous autoimmune disease21. It should be noted that mutant mice that are Helicobacter spp. and/or Citrobacter rodentium positive may have an exacerbated phenotype, as several studies have shown that opportunistic enteric bacteria can significant exacerbate gut pathology4. Ultimately, the occurrence of disease in knockout mice will depend on whether Treg cells rely on a single or multiple suppressive mechanisms. Given the number of genes induced or modulated by FOXP3, it is probable that a programme of intrinsic and extrinsic regulation is induced that involves multiple proteins9,13. Therefore, it would not be surprising if deletion of a single molecule does not provoke the profound Scurfy-like phenotype seen in mice that lack Foxp3.
Box 2. Treg-cell markers
Identifying discriminatory cell surface markers for the characterization and isolation of Treg cells has always been a critical goal. Although excellent markers exist for murine Treg cells, this goal has remained elusive for human Treg cells. Traditionally, murine and human Treg cells have been characterized as CD4+CD25+ (also known as interleukin-2 receptor α (IL-2Rα)). Indeed, murine Treg cells can be effectively isolated based on staining for CD4+CD25+CD45RBlow expression. However, the purity of isolated human Treg cells has always been an issue because T cells up-regulate CD25 upon activation106. Indeed, during the influenza or allergy season a substantial proportion of human CD4+ T cells can express CD25. Although the identification of forkhead box P3 (Foxp3) as a key regulator of Treg-cell development and function has facilitated their identification in the mouse8, many activated (non-regulatory) human T cells express FOXP3, precluding it as a useful marker for human Treg cells16-20. Consequently, the search for Treg-cell-specific cellsurface markers, particularly in humans, has continued in earnest with a growing number of candidates proposed (reviewed by Zhao and colleagues107). For instance, it was shown that the expression of CD127 (also known as IL-7R) is down-regulated on Treg cells and that this could be used to increase the purity of human Treg-cell isolation. Indeed, there is a 90% correlation between CD4+CD25+CD127low T cells and FOXP3 expression108, 109. In addition, it was recently found that Treg cells expressed a higher level of folate receptor 4 (FR4) compared with activated effector T cells110. It is also important to recognize that Treg cells, like their T helper cell counterparts, may be heterogeneous and thus a collection of cell surface markers could facilitate their isolation and functional characterization. Indeed, such heterogeneity has recently been described based on differential expression of HLA-DR or CCR6102,103. However, the general use of both markers remains to be fully established so it is quite probable that the search for better Treg-cell markers will continue for some time.
Box 3 Induced or adaptive Treg cells: development and mode of action
Naturally occurring FOXP3+CD4+CD25+ Treg cells develop in the thymus and display a diverse T-cell receptor (TCR) repertoire that is specific for self-antigens111,112. However, Treg cells can also be ‘induced’, ‘adapted’ or ‘converted’ from effector T cells during inflammatory processes in peripheral tissues, or experimentally generated as a possible therapeutic29,113,114. For instance, T regulatory 1 cells (Tr1) and T helper 3 cells (Th3) can be generated experimentally by, and mediate their suppressive activity through interleukin-10 (IL-10) and transforming growth factor-β (TGFβ), respectively114,115. Typically, these regulatory populations do not express FOXP3. In vivo, it has recently been suggested that stimulation of mouse effector T cells by CD103+ dendritic cells (DCs) in the presence of TGFβ and retinoic acid induces the generation of Foxp3+ T cells in the gutassociated lymphoid tissue (GALT)116-121. Furthermore, Treg cells can be preferentially induced in the periphery by exposure to αVβ8-integrin-expressing DCs122 or suppressor of cytokine signalling 3 (Socs3) −/− DCs123. Interestingly, independent of its role in generating induced Treg cells, TGFβ may also have an important role in helping to maintain Foxp3 expression in natural Treg cells124, a process that can be blocked by IL-4 or interferon-γ (IFNγ) 125. In contrast to mouse T cells, FOXP3 induction by TCR stimulation in the presence of TGFβ in human T cells does not confer a regulatory phenotype20. The mechanism of action of adaptive Treg cells may not necessarily be restricted to suppressive cytokines. Indeed, human adaptive Treg cells (CD4+CD45RA+ T cells stimulated with CD3- and CD46-specific antibodies) have also been shown to express granzyme B and killing target cells in a perforin-dependent manner126. Treg cells often have a restricted specificity for particular cell types, tumours or foreign antigens127. Therefore, induced Treg cells may be ideally suited to respond to infectious agents. This may also be of particular importance in the GALT and in the tumour microenvironment where TGFβ drives the conversion of induced Treg cells118,128. A significant challenge in deciphering data from in vivo experiments is to assess the contribution of natural Treg cells versus induced Treg cells, and to determine whether inhibitory molecules, such as IL-10 or TGFβ, were derived from the former or the latter (or elsewhere).
Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers.
Successful treatment of many patients with advanced cancer using antibodies against programmed cell death 1 (PD-1; also known as PDCD1) and its ligand (PD-L1; also known as CD274) has highlighted the critical importance of PD-1/PD-L1-mediated immune escape in cancer development1, 2, 3, 4, 5, 6. However, the genetic basis for the immune escape has not been fully elucidated, with the exception of elevated PD-L1 expression by gene amplification and utilization of an ectopic promoter by translocation, as reported in Hodgkin and other B-cell lymphomas, as well as stomach adenocarcinoma6, 7, 8, 9, 10. Here we show a unique genetic mechanism of immune escape caused by structural variations (SVs) commonly disrupting the 3′ region of the PD-L1 gene. Widely affecting multiple common human cancer types, including adult T-cell leukaemia/lymphoma (27%), diffuse large B-cell lymphoma (8%), and stomach adenocarcinoma (2%), these SVs invariably lead to a marked elevation of aberrant PD-L1 transcripts that are stabilized by truncation of the 3′-untranslated region (UTR). Disruption of the Pd-l1 3′-UTR in mice enables immune evasion of EG7-OVA tumour cells with elevated Pd-l1 expression in vivo, which is effectively inhibited by Pd-1/Pd-l1 blockade, supporting the role of relevant SVs in clonal selection through immune evasion. Our findings not only unmask a novel regulatory mechanism of PD-L1 expression, but also suggest that PD-L1 3′-UTR disruption could serve as a genetic marker to identify cancers that actively evade anti-tumour immunity through PD-L1 overexpression.
Viruses are a dominant driver of protein adaptation in mammals.
Viruses interact with hundreds to thousands of proteins in mammals, yet adaptation against viruses has only been studied in a few proteins specialized in antiviral defense. Whether adaptation to viruses typically involves only specialized antiviral proteins or affects a broad array of virus-interacting proteins is unknown. Here, we analyze adaptation in ~1300 virus-interacting proteins manually curated from a set of 9900 proteins conserved in all sequenced mammalian genomes. We show that viruses (i) use the more evolutionarily constrained proteins within the cellular functions they interact with and that (ii) despite this high constraint, virus-interacting proteins account for a high proportion of all protein adaptation in humans and other mammals. Adaptation is elevated in virus-interacting proteins across all functional categories, including both immune and non-immune functions. We conservatively estimate that viruses have driven close to 30% of all adaptive amino acid changes in the part of the human proteome conserved within mammals. Our results suggest that viruses are one of the most dominant drivers of evolutionary change across mammalian and human proteomes.
Purdue scientists use adaptors to advance CAR-T therapy
Chimeric antigen receptor (CAR) T cells, developed in the 1990s, are a genetically engineered type of T cell that can target a specific cancer. Now, scientists at Purdue University say they’ve made improvements in this strategy–overcoming the several limitations of traditional CAR-T therapy.
Purdue professor of chemistry Philip Low and his team presented their findings at the American Association for Cancer Research meeting in New Orleans last month.
T cells are a type of immune cell that recognizes and clears the body of invading cells or pathogens, like cancer. They are fine-tuned by the immune system in order to specifically target and kill these foreign invaders–but cancer cells may respond by jumping these safety barriers.
CAR-T therapy was therefore proposed and has been recently used for cancer treatment. It has been hailed for its promising remission rates after early stage clinical trials for acute lymphoblastic leukemia.
“The problem is that the traditional engineered T-cell treatment can be too effective, sometimes killing tumor cells too fast and triggering a toxic reaction in a patient, and sometimes not stopping once the tumor has been destroyed and continuing to seek out and destroy healthy cells important to bodily functions,” Low said in a university news release. “We have found a potential way to control the engineered immune cells to overcome the limitations posed by CAR T-cell therapy.”
They did this by teaming up with Endocyte ($ECYT) scientist Haiyan Chu and designing CAR T cells that require activation by a small molecule adaptor before proceeding. In this way, they can carefully control the amount of active CAR T cells in the circulation.
So far, they have only tried the novel therapy in animal models, but when they tested it in mice they observed antitumor activity only when both the CAR T cells and the correct adaptor molecules were present.
Low believes it will allow clinicians to target multiple cancer subtypes at once. “Most tumors are heterogeneous and contain cancer cells that express different characteristics, including having different tumor-specific proteins on their surface,” he said in the release. “The cancer-targeting molecule on the adaptor we designed can be swapped out to target different molecules on other unrelated cancer cell surfaces. The idea is that a mixture of these adaptors can be given to a patient so that a single CAR T cell clone can be targeted to all of the relevant cancer subtypes in a patient.”
“In the past a new CAR T cell had to be designed for each desired cancer target,” Low said. “This system uses the same blind CAR T cell for all treatments. The adaptor molecule is what needs to be changed, and it is far easier to manipulate and swap pieces in and out of it than the T cells.”
A graphic depicting the activation and inactivation of CAR T cells through a small molecule adaptor is shown. Philip S. Low, Purdue’s Ralph C. Corley Distinguished Professor of Chemistry and director of the Purdue Center for Drug Discovery, and graduate student Yong Gu Lee led a team that designed new engineered CAR T cells that must be activated and targeted by a small molecule adaptor before they can kill cancer cells. The system has the potential to control the engineered cells to overcome existing limitations in CAR T-cell therapy. CREDIT Purdue University image courtesy of Yong Gu Lee
Purdue University researchers may have figured out a way to call off a cancer cell assassin that sometimes goes rogue and assign it a larger tumor-specific “hit list.”
T cells are the immune system’s natural defense against cancer and other harmful entities in the human body. However, the cells must be activated and taught by the immune system to recognize cancer cells in order to seek out and destroy them. Unfortunately, many types of cancer manage to thwart this process.
In the 1990s scientists found a way to genetically engineer T cells to recognize a specific cancer. These engineered T cells, called chimeric antigen receptor, or CAR, T cells, have been recently used as treatment for cancer, said Philip S. Low, Purdue’s Ralph C. Corley Distinguished Professor of Chemistry and director of the Purdue Center for Drug Discovery who led the work.
“The problem is that the traditional engineered T-cell treatment can be too effective, sometimes killing tumor cells too fast and triggering a toxic reaction in a patient, and sometimes not stopping once the tumor has been destroyed and continuing to seek out and destroy healthy cells important to bodily functions,” Low said. “We have found a potential way to control the engineered immune cells to overcome the limitations posed by CAR T-cell therapy.”
Low and Purdue graduate student Yong Gu Lee collaborated with Endocyte Inc. scientist Haiyan Chu to design genetically engineered CAR T cells that must be activated and targeted by a small molecule adaptor before they can kill cancer cells. The technology has been tested in animal models but no human trials have been performed. A poster presentation describing the work was presented Tuesday (April 19, 2016) at the American Association for Cancer Research annual meeting in New Orleans.
“While the traditional CAR T cells could remain and replicate in the human body for many years, the adaptors we have created are expected to be excreted fairly quickly,” Lee said. “By controlling the level of adaptors in the system, we can control the numbers and potencies of active CAR T cells. Those that aren’t stimulated by an adaptor molecule are blind and do not recognize or target any cells. Eventually, if they remain inactive for a while, they should die and be eliminated from the body.”
A study in mice showed the anti-tumor activity was induced only when both the engineered CAR T cell and the correct adaptor molecules were present.
The system also offers the potential to treat multiple cancer subtypes at once, Low said.
“Most tumors are heterogeneous and contain cancer cells that express different characteristics, including having different tumor-specific proteins on their surface,” he said. “The cancer-targeting molecule on the adaptor we designed can be swapped out to target different molecules on other unrelated cancer cell surfaces. The idea is that a mixture of these adaptors can be given to a patient so that a single CAR T cell clone can be targeted to all of the relevant cancer subtypes in a patient.”
The adaptor molecule serves as a bridge between the CAR T-cell and the cancer cell. It is made with a yellow dye called fluorescein isothiocyanate on one end, to which the engineered CAR T cells have been designed to bind, and a cancer-targeting molecule on the other.
Low’s research has focused on the design and synthesis of technologies for targeted delivery of therapeutic and imaging agents to treat cancer, inflammatory and autoimmune diseases, and infectious diseases.
He has developed molecules that target folate-receptors and prostate-specific membrane antigen on the surfaces of cancer cells. Approximately 85 percent of ovarian cancers; 80 percent of endometrial and lung cancers; and 50 percent of breast, kidney and colon cancers express folate receptors on their cellular surfaces. Prostate-specific membrane antigen receptors are found on nearly 90 percent of all prostate cancers. Other tumor-specific ligands developed by Low’s lab can target each of the other major human cancers, he said.
Each CAR T cell has thousands of receptors on its surface to which an adaptor molecule can bind. One CAR T cell could have a variety of adaptor molecules bound to its surface and the cancer cell it targets will depend on which of those adaptors first encounters a targeted cancer cell. Once the CAR T cell binds to a cancer cell, it begins the process of destroying it. When that process is complete, the CAR T cell is released and can bind to a new cancer cell, he said.
“In the past a new CAR T cell had to be designed for each desired cancer target,” Low said. “This system uses the same blind CAR T cell for all treatments. The adaptor molecule is what needs to be changed, and it is far easier to manipulate and swap pieces in and out of it than the T cells.”
In addition to Low, Chu and Lee, members of the research group include Purdue postdoctoral research associates at the time of the study Srinivasarao Tenneti and Ananda Kumar Kanduluru.
Drug discovery is one of the priorities within Purdue Moves, an initiative designed to broaden the university’s global impact and enhance educational opportunities for its students. All of the moves fall into four broad categories: science, technology, engineering and math (STEM) leadership; world-changing research; transformative education; and affordability and accessibility.
The Purdue University Center for Drug Discovery supports more than 100 faculty in six colleges with research focused on several major disease categories: cancer; diabetes, obesity and cardiovascular; immune and infectious disease; and neurological disorders and trauma.
The center and drug discovery initiative builds upon Purdue’s strengths along all points of the drug discovery pipeline, including 14 core units to provide shared resources for analysis, screening, synthesis and testing of potential therapeutic compounds.
With more than 44 Purdue-developed compounds at various stages of preclinical development, and 16 in human clinical trials, Purdue is among the most productive universities in the world of drug discovery.
The center also is aligned with the university’s recently announced $250 million investment in the life sciences.
Endocyte Inc., a Purdue Research Park-based company that develops receptor-targeted therapeutics for the treatment of cancer and autoimmune diseases, funded the study, holds exclusive rights to the technology and assisted Purdue researchers in the development of the technology. Low is a founder and chief science officer of Endocyte Inc. and serves on the Endocyte board of directors.
Yong Gu Lee, Haiyan Chu, Srinivasarao Tenneti, Ananda Kumar Kanduluru, Philip S. Low
Chimeric antigen receptor (CAR) T cells show significant potential for treating cancer due to their tumor-specific activation and ability to focus their killing activity on cells that express a tumor antigen. Unfortunately, this promising therapeutic technology is still limited by: (1) an inability to control the rate of cytokine release and tumor lysis; (2) the absence of an “off switch” that can terminate cytotoxic activity when tumor eradication is complete; (3) a failure to eliminate tumor cells that do not express the targeted antigen; and (4) a requirement to generate a different CAR T cell for each unique tumor antigen. In order to address these limitations, we have exploited a low molecular weight bi-specific adaptor molecule that must bridge between the CAR T cell and its targeted tumor cell by simultaneously binding to the chimeric antigen receptor on the CAR T cell and the unique antigen on the tumor. Using this bispecific adaptor, one can control CAR T cell cytotoxicity by adjusting the concentration and rate of administration of the adaptor. Because the half life of the adapter is <20 minutes in vivo, termination of CAR T cell killing can be accomplished by cessation of adapter administration. Moreover, when heterogeneous tumors containing cells that express orthogonal antigens must be treated, the same CAR T cell can be targeted to multiple antigens by attachment of the same CAR ligand to the appropriate selection of tumor-specific ligands. Finally, when the targeted tumor antigen is also expressed at low levels on normal cells, tumor specificity can be achieved by adjusting the affinity of the tumor-specific ligand to enable CAR T cell engagement only when a highly multivalent interaction is possible. To experimentally demonstrate the aforementioned benefits of using low molecular weight bispecific adaptors, CAR T cells were constructed by fusing an anti-fluorescein isothiocyanate (FITC) scFv to a CD3 zeta chain containing the intracellular domain of CD137 (i.e. CAR4-1BBZ T cells). Then, to enable their tumor-specific cytotoxicity, a bispecific adaptor molecule comprised of fluorescein linked to a small organic ligand with high affinity and specificity for a tumor-specific antigen (FITC-SMC) was synthesized. For these studies, the tumor-specific ligands were: i) folate for recognition of the folate receptor that is over-expressed on ~1/3 of human cancers, ii) DUPA for binding to prostate specific membrane antigen that is over-expressed on prostate cancers, and iii) NK-1R ligand that is over-expressed on neuroendocrine tumors. The ability of the same clone of CAR4-1BBZ T cells to eliminate tumors expressing each of the above antigens was then demonstrated by administration of the desired FITC-SMC to mice injected with the CAR4-1BBZ T cells. Our data show that anti-tumor activity: i) is only induced when both CAR4-1BBZ T cells and the correct antigen-specific FITC-SMC are present, ii) anti-tumor activity and toxicity can be sensitively controlled by adjusting the dosing of FITC-SMC, and iii) treatment of antigenically heterogeneous tumors can be achieved by administration of a mixture of the desired FITC-SMCs. Taken together, these data show that many of the limitations of CAR T cell technology can be addressed by use of a bi-specific adaptor molecule to mediate tumor cell recognition and killing.
CTLA-4 found in dendritic cells suggests New cancer treatment possibilities
Both dendritic cells and T cells are important in triggering the immune response, whereas antigen presenting dendritic cells act as the “general” leading T cells “soldiers” to chase and eliminate enemies in the battle against cancer. The well-known immune checkpoint break, CTLA-4, is believed to be present only in T cells (and cells of the same lineage). However, a new study published in Stem Cells and Development suggests that CTLA-4 also presents in dendritic cells. It further explores the mechanism on how turning off the dendritic cells in the immune response against tumors.
Dendritic Cell-Secreted Cytotoxic T-Lymphocyte-Associated Protein-4 Regulates the T-cell Response by Downmodulating Bystander Surface B7.
The remarkable functional plasticity of professional antigen-presenting cells (APCs) allows the adaptive immune system to respond specifically to an incredibly diverse array of potential pathogenic insults; nonetheless, the specific molecular effectors and mechanisms that underpin this plasticity remain poorly characterized. Cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), the target of the blockbuster cancer immunotherapeutic ipilimumab, is one of the most well-known and well-studied members of the B7 superfamily and negatively regulates T cell responses by a variety of known mechanisms. Although CTLA-4 is thought to be expressed almost exclusively among lymphoid lineage hematopoietic cells, a few reports have indicated that nonlymphoid APCs can also express the CTLA-4 mRNA transcript and that transcript levels can be regulated by external stimuli. In this study, we substantially build upon these critical observations, definitively demonstrating that mature myeloid lineage dendritic cells (DC) express significant levels of intracellular CTLA-4 that they constitutively secrete in microvesicular structures. CTLA-4(+) microvesicles can competitively bind B7 costimulatory molecules on bystander DC, resulting in downregulation of B7 surface expression with significant functional consequences for downstream CD8(+) T-cell responses. Hence, the data indicate a previously unknown role for DC-derived CTLA-4 in immune cell functional plasticity and have significant implication for the design and implementation of immunomodulatory strategies intended to treat cancer and infectious disease.
Non-invasive strategy to guide personalized cancer immunotherapy
Cancer immunotherapy is the rising hope to offer ultimate solutions for cancer. Neoantigens, derived from products of mutated genes in tumor cells, are found to be closely related to the efficacy of cancer immunotherapies. A non-invasive approach to identify unique, patient-specific neoantigens has been advanced by Dr. Steven Rosenberg’s group. A recent article published in Nature Medicine reported that a small population of circulating CD8+PD-1+ tumor-reactive T lymphocytes can be used to identify neoantigens, in addition to tumor-infiltrating T cells. The study paves the way for designing personalized cancer immunotherapy with a novel non-invasive approach.
Detection of lymphocytes that target tumor-specific mutant neoantigens-derived from products encoded by mutated genes in the tumor-is mostly limited to tumor-resident lymphocytes, but whether these lymphocytes often occur in the circulation is unclear. We recently reported that intratumoral expression of the programmed cell death 1 (PD-1) receptor can guide the identification of the patient-specific repertoire of tumor-reactive CD8(+) lymphocytes that reside in the tumor. In view of these findings, we investigated whether PD-1 expression on peripheral blood lymphocytes could be used as a biomarker to detect T cells that target neoantigens. By using a high-throughput personalized screening approach, we identified neoantigen-specific lymphocytes in the peripheral blood of three of four melanoma patients. Despite their low frequency in the circulation, we found that CD8(+)PD-1(+), but not CD8(+)PD-1(-), cell populations had lymphocytes that targeted 3, 3 and 1 unique, patient-specific neoantigens, respectively. We show that neoantigen-specific T cells and gene-engineered lymphocytes expressing neoantigen-specific T cell receptors (TCRs) isolated from peripheral blood recognized autologous tumors. Notably, the tumor-antigen specificities and TCR repertoires of the circulating and tumor-infiltrating CD8(+)PD-1(+) cells appeared similar, implying that the circulating CD8(+)PD-1(+) lymphocytes could provide a window into the tumor-resident antitumor lymphocytes. Thus, expression of PD-1 identifies a diverse and patient-specific antitumor T cell response in peripheral blood, providing a novel noninvasive strategy to develop personalized therapies using neoantigen-reactive lymphocytes or TCRs to treat cancer.
PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors
Adoptive transfer of tumor-infiltrating lymphocytes (TILs) can mediate regression of metastatic melanoma; however, TILs are a heterogeneous population, and there are no effective markers to specifically identify and select the repertoire of tumor-reactive and mutation-specific CD8+ lymphocytes. The lack of biomarkers limits the ability to study these cells and develop strategies to enhance clinical efficacy and extend this therapy to other malignancies. Here, we evaluated unique phenotypic traits of CD8+ TILs and TCR β chain (TCRβ) clonotypic frequency in melanoma tumors to identify patient-specific repertoires of tumor-reactive CD8+lymphocytes. In all 6 tumors studied, expression of the inhibitory receptors programmed cell death 1 (PD-1; also known as CD279), lymphocyte-activation gene 3 (LAG-3; also known as CD223), and T cell immunoglobulin and mucin domain 3 (TIM-3) on CD8+ TILs identified the autologous tumor-reactive repertoire, including mutated neoantigen-specific CD8+ lymphocytes, whereas only a fraction of the tumor-reactive population expressed the costimulatory receptor 4-1BB (also known as CD137). TCRβ deep sequencing revealed oligoclonal expansion of specific TCRβ clonotypes in CD8+PD-1+ compared with CD8+PD-1– TIL populations. Furthermore, the most highly expanded TCRβ clonotypes in the CD8+ and the CD8+PD-1+ populations recognized the autologous tumor and included clonotypes targeting mutated antigens. Thus, in addition to the well-documented negative regulatory role of PD-1 in T cells, our findings demonstrate that PD-1 expression on CD8+ TILs also accurately identifies the repertoire of clonally expanded tumor-reactive cells and reveal a dual importance of PD-1 expression in the tumor microenvironment.
Cancer immunotherapy has experienced major progress in the last decade. Adoptive transfer of ex vivo–expanded tumor-infiltrating lymphocytes (TILs) can cause substantial regression of metastatic melanoma (1, 2). Blockade of the interaction of cytotoxic T lymphocyte antigen 4 (CTLA-4; also known as CD152) or programmed cell death 1 receptor (PD-1; also known as CD279) with their ligands using blocking antibodies alone or in combination have been shown to unleash an otherwise-ineffective immune response against melanoma (3–7), renal cell carcinoma (3), and non–small cell lung cancer (3). The antitumor responses observed in these clinical trials support the presence of naturally occurring tumor-reactive CD8+ T cells and their immunotherapeutic potential. In the particular case of TIL therapy, persistence of transferred tumor-specific T cell clones is associated with tumor regression (8). Moreover, retrospective clinical studies have shown an association of autologous tumor recognition by TILs and clinical response (9, 10), which suggests that enrichment of tumor-reactive cells could enhance clinical efficacy. However, the identification of the diverse repertoire of tumor-reactive cells limits the ability to study these cells, enhance clinical efficacy, and extend this therapy to other malignancies.
Melanoma TILs represent a heterogeneous population that can target a variety of antigens, including melanocyte differentiation antigens, cancer germline antigens, self-antigens overexpressed by the tumor, and mutated tumor neoantigens (11). The latter appear to be of critical importance for the antitumor responses observed after transfer of TILs, given the substantial regression of metastatic melanoma in up to 72% of patients in phase 2 clinical trials, in the absence of any autoimmune side effects in the great majority of patients (2). This contrasts with the modest antitumor activity but high prevalence of severe autoimmune manifestations observed after transfer of peripheral blood gene-engineered T cells expressing TCRs targeting shared melanocyte differentiation antigens MART1 and gp100 (12,13). Furthermore, T cells targeting mutated neoepitopes are not subject to negative selection in the thymus and may constitute the predominant naturally occurring tumor-reactive population in cancer patients. In support of this notion, a recent study reported the frequent detection and dominance of T cell populations targeting mutated epitopes in melanoma-derived TILs (14). Conversely, T cells targeting shared melanocyte differentiation antigens and cancer germline antigens in bulk melanoma TILs were represented at a strikingly low frequency (15). These findings have shifted our interest from the more accessible and commonly studied T cells targeting melanocyte differentiation antigens to T cells targeting unique patient-specific mutations. However, the often rare availability of autologous tumor cell lines necessary to study these reactivities, and the hurdles associated with the identification of the unique mutations targeted, have thus far hindered immunobiological studies of these T cell populations in the tumor.
Naturally occurring tumor-reactive cells are exposed to their antigen at the tumor site. Thus, the immunobiological characterization of T cells infiltrating tumors represents a unique opportunity to study their function and to identify the patient-specific repertoire of tumor-reactive cells. TCR stimulation triggers simultaneous upregulation of both costimulatory and coinhibitory receptors, which can either promote or inhibit T cell activation and function. Expression of the inhibitory receptors PD-1, CTLA-4, lymphocyte-activation gene 3 (LAG-3; also known as CD223), and T cell immunoglobulin and mucin domain 3 (TIM-3) is regulated in response to activation and throughout differentiation (16, 17). Chronic antigen stimulation has been shown to induce coexpression of inhibitory receptors and is associated with T cell hyporesponsiveness, termed exhaustion (18). Exhaustion in response to persistent exposure to antigen was first delineated in a murine model of chronic lymphocytic choriomeningitis virus (19), but has been observed in multiple human chronic viral infections (20–22) as well as in tumor-reactive MART1-specific TILs (23, 24), and has provided the rationale for restoring immune function using immune checkpoint blockade. Conversely, 4-1BB (also known as CD137) is a costimulatory member of the TNF receptor family that has emerged as an important mediator of survival and proliferation, particularly in CD8+ T cells (25–27). 4-1BB is transiently expressed upon TCR stimulation, and its expression has been used to enrich for antigen-specific T cells in response to acute antigen stimulation (28). However, expression of this marker has not been extensively explored in CD8+ lymphocytes infiltrating human tumors. In addition to changes in the expression of cosignaling receptors on the surface of T cells, antigen-specific stimulation typically results in clonal expansion. TCR sequence immunoprofiling can be used to monitor T cell responses to a given immune challenge even without a priori knowledge of the specific epitope targeted, through determination of the abundance of specific clonotypes (29, 30). However, there is limited knowledge regarding the TCR repertoire and the frequency of tumor-reactive clonotypes infiltrating human tumors.
We hypothesized that the assessment of unique phenotypic traits expressed by CD8+ TILs and TCR β chain (TCRβ; encoded by TRB) clonotypic immunoprofiling of lymphocytes infiltrating the tumor could provide a powerful platform to study antitumor T cell responses and evaluated their usefulness in identifying the diverse repertoire of tumor-reactive cells. Despite the accepted negative regulatory role of PD-1 in T cells, our findings establish that expression of PD-1 on CD8+ melanoma TILs accurately identifies the repertoire of clonally expanded tumor-reactive, mutation-specific lymphocytes and suggest that cells derived from this population play a critical role in tumor regression after TIL administration.
PD-1 was initially described to be expressed on a T cell hybridoma undergoing cell death (37). Its negative effect on T cell responses was first delineated in PD-1 knockout mice (38, 39). Since then, PD-1 expression and coexpression of other inhibitory receptors such as CTLA-4, TIM-3, BTLA, CD160, LAG-3, and 2B4 have become a hallmark of chronically stimulated T cells during chronic infection or in the tumor microenvironment. This altered phenotype, and the interaction of these receptors with their corresponding ligands on target cells, is associated with impaired proliferation and effector function frequently referred to as exhaustion (18, 24, 40). Expression of PD-1 in patients with chronic viral infections correlates with disease progression (22, 41). Additionally, CD8+ lymphocytes targeting melanoma differentiation antigens in the tumor express PD-1, CTLA-4, TIM-3, and LAG-3 and exhibit impaired IFN-γ and IL-2 secretion (23, 24), supporting a negative regulatory role of PD-1 and inhibitory receptors in naturally occurring T cell responses to cancer and providing a rationale for the treatment of cancer with immune checkpoint inhibitors.
In the present study, we found that expression of PD-1 on CD8+ melanoma TILs captured the diverse repertoire of clonally expanded tumor-reactive lymphocytes. TCRβ sequencing revealed that tumor-reactive and mutation-specific clonotypes were highly expanded in the CD8+ population and preferentially expanded in the PD-1+ population. This is consistent with the TCR stimulation-driven expression of this receptor on T cells (42). The inhibitory receptors TIM-3 and LAG-3 and the costimulatory receptor 4-1BB were also expressed on CD8+PD-1+ TILs and could also be used to enrich for tumor-reactive cells. PD-1 was consistently expressed at a higher frequency and was found to be more comprehensive at identifying the diverse repertoire of tumor-reactive cells infiltrating melanoma tumors, although the less frequent PD-1–/TIM-3+ and PD-1–/LAG-3+ subpopulations could also represent tumor-reactive cells (Supplemental Figure 4 and Supplemental Table 6). Additionally, previous studies from our laboratory showing coexpression of PD-1 and CTLA-4 (23), and our preliminary data supporting coexpression of PD-1 and ICOS (Supplemental Figure 5), suggest that other receptors may also be used to distinguish tumor-reactive cells. Our present results further support immunotherapeutic intervention using immune checkpoint blockade using PD-1, TIM-3, and LAG-3 blocking antibodies or 4-1BB agonistic antibody to restore the function of tumor-reactive lymphocytes, which is currently being actively pursued in the clinic (3, 4, 6, 7, 43). The potential cooperative mechanisms of inhibition of these receptors when engaged with their ligands (44, 45) suggests that the combined targeting of different inhibitory receptors can further enhance antitumor efficacy, as already shown with the combination of anti–PD-1 and anti–CTLA-4 (5). Our present results demonstrate that PD-1 identifies the clonally expanded CD8+ tumor-reactive population and suggest that expression of PD-1 on CD8+TILs could function as a potential predictive biomarker of antitumor efficacy using immune checkpoint inhibitors.
Naturally occurring tumor-reactive cells play a pivotal role in mediating antitumor responses after TIL transfer. Currently, expansion of TILs for patient treatment involves nonspecific growth of TILs from tumor fragments in IL-2, and the diversity and frequency of antitumor T cells present in the final T cell product used for treatment remains largely uncharacterized. Prospective clinical studies have reported that in vitro recognition of autologous tumor by TILs is associated with a higher probability of clinical response (9, 10), which suggests that enrichment of tumor-reactive cells could enhance clinical efficacy. This is consistent with the idea that both tumor-reactive and non–tumor-reactive cells may compete for cytokines in vivo, especially in the absence of vaccination. However, the isolation of the patient-specific repertoire of tumor-reactive cells is not possible with current technologies (14, 28, 46–50). Our findings established that expression of PD-1, TIM-3, LAG-3, and 4-1BB in CD8+ TILs can be used to enrich for tumor-reactive cells, regardless of the specific antigen targeted. One potential concern with isolating T cells expressing inhibitory receptors for therapy is that these cells may be exhausted or functionally impaired (23, 24, 44, 51, 52). However, we found that PD-1+, TIM-3+, and LAG-3+ CD8+ cells expanded in IL-2 were capable of secreting IFN-γ and lyse tumor in vitro. This supports the notion that immune dysfunction associated with coexpression of inhibitory receptors on CD8+ TILs can be reversed (21, 41, 51, 53), and may enable the reproducible enrichment of tumor-reactive cells for patient treatment. Notably, in a preliminary experiment (n= 8 nonresponders; 14 responders), there was no association between the frequency of expression of any of the markers studied in the CD8+ TILs in the fresh tumor and the clinical response to TILs derived from these tumor samples. However, the fresh tumors included in this study belonged to patients treated in several TIL protocols over the course of 10 years, and TILs were generated from these tumors using different methods, which makes these data difficult to interpret. In addition, the frequency of cells initially expressing PD-1 in the tumor may not reflect the frequency of the PD-1 derived cells in the infusion bag. For example, a low frequency of PD-1+ cells may be highly enriched during the process of TIL culture as a result of the presence of tumor cells. Although in vivo antitumor activity of tumor-isolated TILs based on PD-1 expression requires testing in a clinical trial, the observation that the overwhelming majority of tumor-reactive cells were derived from cells expressing PD-1 suggests that cells expressing PD-1 and inhibitory receptors in the tumor play a critical role in tumor regression after TIL administration.
The functional implications of selecting PD-1–, LAG-3–, TIM-3–, or 4-1BB–expressing T cells to enrich for tumor-reactive cells for patient treatment remain unclear. Although previous studies have reported differential expression of PD-1, LAG-3, and TIM-3 throughout differentiation (17), or preferential expression of TIM-3 in IFN-γ–secreting cells (54), our preliminary results have failed to show consistent phenotypic or functional differences between PD-1+, LAG-3+, TIM-3+, and 4-1BB+ selected TILs, including cytokine secretion, proliferation, and susceptibility to apoptosis (data not shown). We found that PD-1 expression was almost completely lost in the PD-1+ derived populations upon in vitro culture in IL-2. Conversely, TIM-3 and LAG-3 expression increased in the TIM-3– and LAG-3– populations after expansion. Overall, there were no differences in the expression of PD-1, TIM-3, or LAG-3 between any the populations after expansion. Thus, in agreement with previous reports (55, 56), we conclude that expansion in IL-2 alters the expression of these markers and compromises the potential use of inhibitory receptors to select for tumor-reactive cells after in vitro expansion. Recent work in animal models suggests that chronic antigen stimulation (57–59) or a tolerizing microenvironment (60) may lead to permanent epigenetic changes in T cells, raising the possibility that the restoration of function observed in previously exhausted or tolerized cells in presence of cytokines may only be transient. These results have not yet been corroborated in human tumor-specific cells. However, given that the overwhelming majority of tumor-reactive cells appear to derive from cells expressing PD-1 in the tumor, studying permanent versus transient reversion of exhaustion may have important implications for adoptive cell transfer of TILs.
Tumor-reactive cells can also be found infiltrating other tumor malignancies, such as renal cell carcinoma (61) or ovarian (62), cervical (63), or gastrointestinal tract cancers (64), albeit at lower frequencies. Our findings provide alternatives to enrich and study tumor-reactive CD8+ TILs through selection of cells expressing the cell surface receptors PD-1, LAG-3, TIM-3, and 4-1BB, a hypothesis that we are actively investigating. Additionally, our present findings showed that the frequency of a specific clonotype in the CD8+ and PD-1+ populations can be used to predict its ability to recognize tumor and isolate tumor-specific TCRs, thus providing means to overcome potential irreversible functional impairments of TILs (52).
2 reports with opposing results have generated controversy regarding which may be the optimal marker for the identification of the tumor-reactive repertoire, PD-1 or 4-1BB. In one report studying PD-1 expression in the tumor, the authors showed promising although inconsistent ability to enrich for shared melanoma-reactive cells (55). In a more recent article studying the role of 4-1BB in fresh ovarian TILs, Ye et al. concluded that expression of 4-1BB, but not PD-1, on lymphocytes defines the population of tumor-reactive cells in the tumor (65). The results of Ye et al. appear to contradict our present findings, showing that expression of PD-1 rather than 4-1BB more comprehensively identifies the repertoire of tumor-reactive cells in the tumor. However, these inconsistencies can be explained by different experimental approaches undertaken to study the immunobiology of TILs. First, Ye et al. found that expression of 4-1BB in fresh ovarian TILs and tumor-associated lymphocytes was low, and thus exposed the tumor to IL-7 and IL-15 (65). In the 1 patient sample in which the authors enriched for tumor-reactive cells from fresh ovarian TILs or tumor-associated lymphocytes exposed to IL-7 and IL-15, expression of 4-1BB was dependent on in vitro activation, but no longer represented the natural expression of 4-1BB in the fresh tumor. Second, with the exception of the 1 experiment described above, the enrichment experiments reported were carried out with melanoma or ovarian TIL lines expanded in IL-2 and cocultured with tumor cell lines in vitro. It is well known that IL-2 can change the activation status and also the expression of inhibitory receptors on T cells (data not shown and ref. 56). Thus, the experiment comparing expression of PD-1 and 4-1BB performed by Ye et al. (65) addressed the significance of these receptors after in vitro coculture of a highly activated melanoma TIL line with a tumor cell line, rather than the role of PD-1 and 4-1BB expression in CD8+ lymphocytes in the fresh tumor. Finally, both Inozume et al. and Ye et al. used matched HLA-A2 cell lines to assess tumor reactivity (55, 65). However, the use of HLA-matched tumor cell lines does not enable the assessment of reactivities against unique mutations that are present only in the autologous tumor cell line. In our current study, we used fresh melanoma tumors for all our experiments, and these were rested in the absence of cytokines to preserve the phenotype of TILs. Moreover, we used autologous tumor cell lines to assess tumor recognition. We believe that our experimental approach overcomes the limitations described above, enabling us to conclude that tumor-reactive cells can be detected in both the PD-1+/4-1BB+ and PD-1+/4-1BB– CD8+ TIL populations.
In summary, expression of PD-1 in CD8+ TILs in the fresh tumor identified and selected for the diverse patient-specific repertoire of tumor-reactive cells, including mutation-specific cells. In addition, analysis of the CD8+ TIL TCRβ repertoire in 2 melanomas showed that the frequency of a specific TCRβ clonotype in the CD8+ and PD-1+ populations could be used to predict its ability to recognize the autologous tumor. The use of inhibitory receptors and the frequency of individual TCRs to prospectively identify and select the diverse repertoire of tumor-reactive cells holds promise for the personalized treatment of cancer with T cell therapies, but may also facilitate the dissection and understanding of the immune response in human cancer patients.
Anti-PD-1 is poised to be a blockbuster, which other immune-checkpoint targeting drugs are on the horizon?
Clinical studies of anti-immune-checkpoint protein therapeutics have shown not only an improved overall survival, but also a long-term durable response, compared to chemotherapy and genomically-targeted therapy. To expand the success of immune-checkpoint therapeutics into more tumor types and improving efficacy in difficult-to-treat tumors, additional targets involved in checkpoint-blockade need to be explored, as well as testing the synergy between combining approaches.
Currently, CTLA-4 and PD-1/PD-L1 are furthest along in development, and have shown very promising results in metastatic melanoma patients. This is just a fraction of targets involved in the checkpoint-blockade pathway. Several notable targets include:
LAG-3 – Furthest along in clinical development with both a fusion protein and antibody approach, antibody apporach being tested in combination with anti-PD-1
TIM-3 – Also in clinical development. Pre-clinical studies indicate that it co-expresses with PD-1 on tumor-infiltrating lymphocytes. Combination with anti-PD-improves anti-tumor response
VISTA – Antibody targeting VISTA was shown to improve anti-tumor immune response in mice
In addition, there are also co-stimulatory factors that are also being explored as viable therapeutic targets
OX40 – Both OX40 and 4-1BB are part of the TNF-receptor superfamily. Phase I data shows acceptable safety profile, and evidence of anti-tumor response in some patients
4-1BB – Phase I/II data on an antibody therapeutic targeting OX40 shows promising clinical response for melanoma, renal cell carcinoma and ovarian cancer.
Inducible co-stimulator (ICOS) – Member of the CD28/B7 family. Its expression was found to increase upon T-cell activation. Anti-CTLA-4 therapy increases ICOS-positive effector T-cells, indicating that it may work in synergy with anti-CTLA-4. Clinical trials of anti-ICOS antibody are planned for 2015.
Targeting single immune-checkpoint proteins has proven to be clinically effective at treating specific tumor types; can targeting two different proteins synergize effects?
Despite the success of targeting immune-checkpoint proteins, such as CTLA-4, PD-1, LAG-3, TIM-3 among others, percentages of patient response vary and rarely exceed 50%. It is highly tempting to speculate a strategy of dual-targeting of these checkpoint proteins. A recent presentation at the Keystone Symposium for Tumor Immunology: Multidisciplinary Science Driving Combination Therapy detailed findings of dual-targeting two immune-checkpoint proteins in mouse tumor models. Their key findings are summarized below:
Dual-targeting PD-1 and LAG-3 demonstrates superior efficacy over blocking either target alone
In addition to previous reported data on superior dual-targeting efficacy against fibrosarcoma (Sa1N) and colorectal adenosarcoma (MC38) tumor types1, anti-tumor activity against myeloma (SC J558L) and B-cell lymphoma (A20) hematological tumor types were also reported to be effacious.2
These exciting pre-clinical findings may result in further exploration of dual-targeting antibodies in the clinic, either as combination of existing antibody therapies, or as a new bi-specific antibody therapeutic.
Camelid single domain antibodies are a novel bi-specific antibody platform that may be used to develop a new generation of dual-targeting antibodies against multiple immune-checkpoint proteins.
New insight behind the success of fighting cancer by targeting immune checkpoint proteins
Immune checkpoint blockade has proven to be highly successful in the clinic at treating aggressive and difficult-to-treat forms of cancer. The mechanism of the blockade, targeting CTLA-4 and PD-1 receptors which act as on/off switches in T cell-mediated tumor rejection, is well understood. However, little is known about the tumor antigen recognition profile of these affected T-cells, once the checkpoint blockade is initiated.
In a recent published study, the authors used genomics and bioinformatics approaches to identify critical epitopes on 3-methylcholanthrene induced sarcoma cell lines, d42m1-T3 and F244. CD8+ T cells in anti-PD-1 treated tumor bearing mice were isolated and fluorescently labeled with tetramers loaded with predicted mutant epitopes. Out of 66 predicted mutants, mLama4 and mAlg8 were among the highest in tetramer-positive infiltrating T-cells. To determine whether targeting these epitopes alone would yield similar results as anti-PD-1 treatment, vaccines against these two epitopes were developed and tested in mice. Prophylactic administration of the combined vaccine against mLama4 and mAlg8 yielded an 88% survival in tumor bearing mice, thus demonstrating that these two epitopes are the major antigenic targets from checkpoint-blockade and therapies against these two targets are similarly efficacious.
In addition to understanding the mechanism, identification of these tumor-specific mutant antigens is the first step in discovering the next wave of cancer immunotherapies via vaccines or antibody therapeutics. Choosing the right antibody platform can speed the discovery of a new therapeutics against these new targets. Single domain antibodies have the advantage of expedited optimization, flexibility of incorporating multiple specificity and functions, superior stability, and low COG over standard antibody approaches.
Myeloid-derived-suppressor cells as regulators of the immune system
Dmitry I. Gabrilovich and Srinivas Nagaraj Nat Rev Immunol. 2009 March ; 9(3): 162–174. http://dx.doi.org:/10.1038/nri2506
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells that expands during cancer, inflammation and infection, and that has a remarkable ability to suppress T-cell responses. These cells constitute a unique component of the immune system that regulates immune responses in healthy individuals and in the context of various diseases. In this Review, we discuss the origin, mechanisms of expansion and suppressive functions of MDSCs, as well as the potential to target these cells for therapeutic benefit.
The first observations of suppressive myeloid cells were described more than 20 years ago in patients with cancer1-3. However, the functional importance of these cells in the immune system has only recently been appreciated due to accumulating evidence that has demonstrated their contribution to the negative regulation of immune responses during cancer and other diseases. It is now becoming increasingly clear that this activity is contained within a population known as myeloid-derived suppressor cells (MDSCs). Features common to all MDSCs are their myeloid origin, immature state and a remarkable ability to suppress T-cell responses (Box 1). In addition to their suppressive effects on adaptive immune responses, MDSCs have also been reported to regulate innate immune responses by modulating the cytokine production of macrophages4. Non-immunological functions of MDSC have also been described, such as the promotion of tumour angiogenesis, tumour-cell invasion and metastasis. However, as a discussion of these aspects of MDSC biology is beyond the scope of this article, the reader is referred to another recent Review on this topic5.
MDSCs represent an intrinsic part of the myeloid-cell lineage and are a heterogeneous population that is comprised of myeloid-cell progenitors and precursors of myeloid cells. In healthy individuals, immature myeloid cells (IMCs) generated in bone marrow quickly differentiate into mature granulocytes, macrophages or dendritic cells (DCs). In pathological conditions such as cancer, various infectious diseases, sepsis, trauma, bone marrow transplantation or some autoimmune disorders, a partial block in the differentiation of IMCs into mature myeloid cells results in an expansion of this population. Importantly, the activation of these cells in a pathological context results in the upregulated expression of immune suppressive factors such as arginase (encoded by ARG1) and inducible nitric oxide synthase (iNOS; also known as NOS2) and an increase in the production of NO (nitric oxide) and reactive oxygen species (ROS). Together, this results in the expansion of an IMC population that has immune suppressive activity; these cells are now collectively known as MDSCs. In this
Origin and subsets of MDSCs It is important to note that MDSCs that are expanded in pathological conditions (see later) are not a defined subset of myeloid cells but rather a heterogeneous population of activated IMCs that have been prevented from fully differentiating into mature cells. MDSCs lack the expression of cell-surface markers that are specific for monocytes, macrophages or DCs and are comprised of a mixture of myeloid cells with granulocytic and monocytic morphology6. Early studies showed that 1–5% of MDSCs are able to form myeloid-cell colonies7-9 and that about one third of this population can differentiate into mature macrophages and DCs in the presence of appropriate cytokines in vitro and in vivo7-9. In mice, MDSCs are characterized by the co-expression of the myeloid lineage differentiation antigen Gr1 (also known as Ly6G) and CD11b (also known as αM-integrin)10. Normal bone marrow contains 20–30% of cells with this phenotype, but these cells make up only a small proportion (2–4%) of spleen cells and are absent from the lymph nodes in mice (Fig. 1). In humans, MDSCs are most commonly defined as CD14-CD11b+ cells or, more narrowly, as cells that express the common myeloid marker CD33 but lack the expression of markers of mature myeloid and lymphoid cells and the MHC-class-II molecule HLA-DR11, 12. MDSCs have also been identified within a CD15+ population in human peripheral blood13. In healthy individuals, immature myeloid cells with described above phenotype comprise ∼0.5% of peripheral blood mononuclear cells.
Recently, the morphological heterogeneity of these cells has been defined more precisely in part based on their expression of Gr1. Notably, Gr1-specific antibodies bind to both Ly6G and Ly6C, which are encoded by separate genes. However, these epitopes are recognized by different antibodies specific for each individual epitopes: anti-Ly6C and anti-Ly6G. Granulocytic MDSCs have a CD11b+Ly6G+Ly6Clow phenotype, whereas MDSCs with monocytic morphology are CD11b+Ly6G-Ly6Chigh 6,14. Importantly, evidence indicates that these two subpopulations may have different functions in cancer and infectious and autoimmune diseases15-17. During the analysis of ten different experimental tumour models, we found that both of these subsets of MDSCs were expanded. In most cases, however, the expansion of the granulocytic MDSC population was much greater than that of the monocytic subset6 and, interestingly, the two subpopulations used different mechanisms to suppress Tcell function (see later). In addition, the ability to differentiate into mature DCs and macrophages in vitro has been shown to be restricted to monocytic MDSCs6.
In recent years, several other surface molecules have been used to identify additional subsets of suppressive MDSCs, including CD80 (also known as B7.1)18, CD115 (the macrophage colony-stimulating factor receptor)19, 20 and CD124 (the IL-4 receptor α-chain)20. In our own studies, we observed that many MDSCs in tumour-bearing mice co-express CD115 and CD1246; however, direct comparison of MDSCs from tumour-bearing mice and Gr1+CD11b+ cells from naive mice showed that they expressed similar levels of CD115 and CD124. In addition, sorted CD115+ or CD124+ MDSCs from EL-4 tumour-bearing mice had the same ability to suppress T-cell proliferation on a per cell basis as did CD115- or CD124-MDSCs. This suggests that, although these molecules are associated with MDSCs, they might not be involved in the immunosuppressive function of these cells in all tumour models.
Overall, current data suggest that MDSCs are not a defined subset of cells but rather a group of phenotypically heterogeneous myeloid cells that have common biological activity.
MDSCs in pathological conditions MDSCs were first characterized in tumour-bearing mice or in patients with cancer. Inoculation of mice with transplantable tumour cells, or the spontaneous development of tumours in transgenic mice with tissue-restricted oncogene expression, results in a marked systemic expansion of these cells (Fig. 1 and Table 1). In addition, up to a tenfold increase in MDSC numbers was detected in the blood of patients with different types of cancer11, 12, 21, 22. In many mouse tumour models, as many as 20–40% of nucleated splenocytes are represented by MDSCs (in contrast to the 2-4% seen in normal mice). In addition, these cells are found in tumour tissues and in the lymph nodes of tumour-bearing mice.
Although initial observations and most of the current information regarding the role of MDSCs in immune responses has come from studies in the cancer field, accumulating evidence has shown that MDSCs also regulate immune responses in bacterial and parasitic infections, acute and chronic inflammation, traumatic stress, surgical sepsis and transplantation. A systemic expansion of both the granulocytic and monocytic subset of MDSCs was observed in mice primed with Mycobacterium tuberculosis as part of complete Freund’s adjuvant (CFA). Acute Trypanosoma cruzi infection, which induces T-cell activation and increased production of interferon-γ (IFNγ), also leads to the expansion of MDSCs23, 24. A similar expansion of MDSCs has been reported during acute toxoplasmosis25, polymicrobial sepsis26, acute infection with Listeria monocytogenes or chronic infection with Leishmania major27 and infection with helminths28,29, 30, Candida albicans31 or Porphyromonas gingivalis32.
MDSC expansion is also associated with autoimmunity and inflammation. In experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, an increase in CD11b+Ly6ChiLy6G− MDSCs was observed in the spleen and blood and these cells were found to enter the central nervous system during the inflammatory phase of the disease16. A significant increase in the number of MDSCs was also detected in experimental autoimmune uveoretinitis, an animal model of human intraocular inflammatory disease33, in the skin and spleens of mice that were repeatedly treated with a contact sensitizer to induce an inflammatory response34 and in inflammatory bowel diseases35. MDSCs were also found to infiltrate the spleen and suppress T-cell function in a model of traumatic stress36. Finally, a significant transient increase in MDSC numbers was also demonstrated in normal mice following immunization with different antigens such as ovalbumin or peptide together with CFA, a recombinant vaccinia virus expressing interleukin-2 (IL-2) or staphylococcal enterotoxin A 8, 37, 38. Therefore, current information clearly indicates that the expansion of an immunosuppressive MDSC population is frequently observed in many pathological conditions.
Expansion and activation of MDSCs Studies have demonstrated that the MDSC population is influenced by several different factors (Table 1), which can be divided into two main groups. The first group includes factors that are produced mainly by tumour cells and promote the expansion of MDSC through stimulation of myelopoiesis and inhibiting of the differentiation of mature myeloid cells. The second group of factors is produced mainly by activated T cells and tumour stroma, and is involved in directly activating MDSCs. Mechanisms of MDSC expansion—Factors that induce MDSC expansion can include cyclooxygenase-2 (COX2), prostaglandins 39-41, stem-cell factor (SCF)39, macrophage colony-stimulating factor (M-CSF), IL-642, granulocyte/macrophage colony-stimulating factor (GM-CSF)41 and vascular endothelial growth factor (VEGF) 43 (Table 1). The signalling pathways in MDSCs that are triggered by most of these factors converge on Janus kinase (JAK) protein family members and signal transducer and activator of transcription 3 (STAT3) (Fig. 2), which are signalling molecules that are involved in cell survival, proliferation, differentiation and apoptosis44. STAT3 is arguably the main transcription factor that regulates the expansion of MDSCs. MDSCs from tumour-bearing mice have markedly increased levels of phosphorylated STAT3 compared with IMCs from naive mice45. Exposure of haematopoietic progenitor cells to tumour-cell-conditioned medium resulted in the activation of JAK2 and STAT3 and was associated with an expansion of MDSCs in vitro, whereas inhibition of STAT3 expression in haematopoietic progenitor cells abrogated the effect of tumour-derived factors on MDSC expansion46. Ablation of STAT3 expression in conditional knockout mice or selective STAT3 inhibitors markedly reduced the expansion of MDSCs and increased T-cell responses in tumour-bearing mice45, 47. STAT3 activation is associated with increased survival and proliferation of myeloid progenitor cells, probably through upregulated expression of STAT3 target genes including B-cell lymphoma XL, (BCL-XL), cyclin D1, MYC and survivin. So, abnormal and persistent activation of STAT3 in myeloid progenitors prevents their differentiation into mature myeloid cells and thereby promotes MDSC expansion.
Recent findings suggest that STAT3 also regulates MDSC expansion through inducing the expression of S100A8 and S100A9 proteins. In addition, it has been shown that MDSCs also express receptors for these proteins on their cell surface. S100A8 and S100A9 belong to the family of S100 calcium-binding proteins that have been reported to have an important role in inflammation48. STAT3-dependent upregulation of S100A8 and S100A9 expression by myeloid progenitor cells prevented their differentiation and resulted in the expansion of MDSCs in the spleens of tumor-bearing and naive S100A9-transgenic mice. By contrast, MDSCs did not expand in the peripheral blood and spleens of mice deficient for S100A9 following challenge with tumour cells or CFA49. In a different study, S100A8 and S100A9 proteins were shown to promote MDSC migration to the tumour site through binding to carboxylated N-glycan receptors expressed on the surface of these cells 50. Blocking the binding of S100A8 and S100A9 to their receptors on MDSCs in vivo with a carboxylated glycan-specific antibody reduced MDSC levels in the blood and secondary lymphoid organs of tumour-bearing mice50. In human colon tumour tissue, and in a mouse model of colon cancer, myeloid progenitor cells expressing S100A8 and S100A9 have been shown to infiltrate regions of dysplasia and adenoma. Furthermore, administration of a carboxylated glycan-specific monoclonal antibody (mAbGB3.1) was found to markedly reduced chronic inflammation and tumorigenesis51. Although the mechanisms involved require further study, these studies suggest that S100A9 and/or S100A8 proteins have a crucial role in regulating MDSC expansion, and may provide a link between inflammation and immune suppression in cancer.
Mechanisms of MDSC activation—Recently, it has become clear that the suppressive activity of MDSCs requires not only factors that promote their expansion but those that induce their activation. The expression of these factors, which are produced mainly by activated T cells and tumour stromal cells, is induced by different bacterial or viral products or as a result of tumour cell death 26. These factors, which include IFNγ, ligands for Toll-like receptors (TLRs), IL-13, IL-4 and transforming growth factor-β (TGFβ), activate several different signalling pathways in MDSCs that involve STAT6, STAT1, and nuclear factor-κB (NF-κB) (Fig. 2).
Blockade of IFNγ, which is produced by activated T cells, abolishes MDSC-mediated T-cell suppression17, 52. STAT1 is the major transcription factor activated by IFNγ-mediated signalling and, in the tumour microenvironment, the upregulation of ARG1 and iNOS expression in MDSCs involved a STAT1-dependent mechanism. Indeed, MDSCs from Stat1-/- mice failed to up regulate ARG1 and iNOS expression and therefore did not inhibit Tcell responses53. Consistent with other findings, IFNγ produced by activated T cells and by MDSCs triggered iNOS expression and synergized with IL-4Rα and ARG1 pathways that have been implicated in the suppressive function of MDSCs20.
An important role for the signalling pathway that involves IL-4 receptor α-chain (IL-4Rα) and STAT6 (which is activated by the binding of either IL-4 or IL-13 to IL-4Rα) in MDSC activation has been demonstrated in several studies. It has been shown that ARG1 expression is induced by culturing freshly isolated MDSCs or cloned MDSC lines with IL-454. In addition, IL-4 and IL-13 upregulate arginase activity, which increases the suppressive function of MDSCs55. In line with these observations, other experiments have shown that STAT6 deficiency prevents signalling downstream of the IL-4Rα and thereby blocks the production of ARG1 by MDSCs56. In addition, the IL-4Rα–STAT6 pathway was also found to be involved in IL-13-induced TGFβ1 production by MDSCs in mice with sarcoma, which resulted in decreased tumour immunosurveillance57. This could be regulated by neutralizing both TGFβ and IL-1357. However, in breast tumor model IL-4Rα knockout mice retain high levels of MDSC after surgery56. In a different study that evaluated the separate role of TGFβ (not involving study of IL-4Rα) TGFβ-specific blocking antibody failed to reverse T-cell anergy in B-cell lymphoma in vitro58. It is possible that, the IL4Rα–STAT6 pathway might not be involved in promoting tumour immunosuppression in all tumour models.
TLRs have a central role in the activation of innate immune responses. Polymicrobial sepsis induced by the ligation and puncture of the caecum, which releases microbial products into the peritoneum and systemic circulation, was shown to result in an expansion of the MDSC population in the spleen that was dependent on the TLR adaptor molecule myeloid differentiation primary-response gene 88 (MyD88)26. However, wild-type mice and mice lacking a functional TLR4 protein had comparable expansion of the MDSC during polymicrobial sepsis, which suggests that signalling through TLR4 is not required for MDSC expansion and that MyD88-dependent signalling pathways that are triggered by other TLRs probably contribute to the expansion of MDSCs in sepsis26. This indicates that the activation of MDSCs is a fundamental outcome of the host innate immune response to pathogens that express TLR ligands.
It is important to note that an increase in the production and/or recruitment of IMCs in the context of acute infectious diseases or following vaccination does not necessarily represent an expansion of an immunosuppressive MDSC population. It is likely that under pathological conditions, the expansion of a suppressive MDSC population is regulated by two different groups of factors that have partially overlapping activity: those that induce MDSC expansion and those that induce their activation (which leads to increased levels of ROS, arginase, and/ or NO). This two-tiered system may allow for flexibility in the regulation of these cells under physiological and pathological conditions.
Mechanisms of MDSC suppressive activity Most studies have shown that the immunosuppressive functions of MDSCs require direct cell– cell contact, which suggests that they act either through cell-surface receptors and/or through the release of short-lived soluble mediators. The following sections describe the several mechanisms that have been implicated in MDSC-mediated suppression of T-cell function.
Arginase and iNOS—Historically, the suppressive activity of MDSCs has been associated with the metabolism of L-arginine. L-arginine serves as a substrate for two enzymes: iNOS, which generates NO, and arginase, which converts L-arginine into urea and L-ornithine. MDSCs express high levels of both arginase and iNOS, and a direct role for both of these enzymes in the inhibition of T-cell function is well established; this has been reviewed recently59, 60. Recent data suggest that there is a close correlation between the availability of arginine and the regulation of T-cell proliferation11, 61. The increased activity of arginase in MDSCs leads to enhanced L-arginine catabolism, which depletes this non-essential amino acid from the microenvironment. The shortage of L-arginine inhibits T-cell proliferation through several different mechanisms, including decreasing their CD3ζ expression62 and preventing their upregulation of the expression of the cell cycle regulators cyclin D3 and cyclin-dependent kinase 4 (CDK4)63. NO suppresses T-cell function through a variety of different mechanisms that involve the inhibition of JAK3 and STAT5 in T cells64, the inhibition of MHC class II expression 65 and the induction of T-cell apoptosis66.
ROS—Another important factor that contributes to the suppressive activity of MDSCs is ROS. Increased production of ROS has emerged as one of the main characteristics of MDSCs in both tumour-bearing mice and patients with cancer6, 10, 13, 53, 67-70. Inhibition of ROS production by MDSCs isolated from mice and patients with cancer completely abrogated the suppressive effect of these cells in vitro10, 13, 67. Interestingly, ligation of integrins expressed on the surface of MDSCs was shown to contribute to increased ROS production following the interaction of MDSCs with T cells10. In addition, several known tumour-derived factors, such as TGFβ, IL-10, IL-6, IL-3, platelet-derived growth factor (PDGF) and GM-CSF, can induce the production of ROS by MDSCs (for review see Ref 71).
The involvement of ROS and NO in mechanisms of MDSC suppression are not restricted to neoplastic conditions, as inflammation and microbial products are also known to induce the development of a MDSC population that produces ROS and NO following interactions with activated T cells15. Similar findings were observed in models of EAE16 and acute Toxoplasmosis infection 16. In addition, it has been observed that MDSCs mediated their suppressive function through IFNγ-dependent NO production in an experimental model of Trypanosoma cruzi infection23.
Peroxynitrite—More recently, it has emerged that peroxynitrite (ONOO-) is a crucial mediator of MDSC-mediated suppression of T-cell function. Peroxynitrite is a product of a chemical reaction between NO and superoxide anoion (O2-) and is one of the most powerful oxidants produced in the body. It induces the nitration and nitrosylation of the amino acids cystine, methionine, tryptophan and tyrosine72. Increased levels of peroxynitrite are present at sites of MDSC and inflammatory-cell accumulation, including sites of ongoing immune reactions. In addition, high levels of peroxynitrite are associated with tumour progression in many types of cancer72, 73,74-78, which has been linked with T-cell unresponsiveness. Bronte and colleagues reported that human prostate adenocarcinomas were infiltrated by terminallydifferentiated CD8+ T cells that were in an unresponsive state. High levels of nitrotyrosine were present in the T cells, which suggested the production of peroxynitrites in the tumour environment. Inhibiting the activity of arginase and iNOS, which are expressed in malignant but not in normal prostate tissue and are key enzymes of L-arginine metabolism,, led to decreased tyrosine nitration and restoration of T-cell responsiveness to tumour antigens79. In addition, we have demonstrated that peroxynitrite production by MDSCs during direct contact with T cells results in nitration of the T-cell receptor (TCR) and CD8 molecules, which alters the specific peptide binding of the T cells and renders them unresponsive to antigen-specific stimulation. However, the T cells maintained their responsiveness to nonspecific stimuli80. This phenomenon of MDSC induced antigen-specific T-cell unresponsiveness was also observed in vivo in tumour-bearing mice53.
Subset-specific suppressive mechanisms?—Recent findings indicate that different subsets of MDSC might use different mechanisms by which to suppress T-cell proliferation. As described earlier, two main subsets of MDSCs have been identified: a granulocytic subset and a monocytic subset. The granulocytic subset of MDSC was found to express high levels of ROS and low levels of NO, whereas the monocytic subset expressed low levels of ROS and high levels of NO and both subsets expressed ARG16 (Fig.3). Interestingly, both populations suppressed antigen-specific T-cell proliferation to an equal extent, despite their different mechanisms of action. Consistent with these observations, Movahedi et al. also reported two distinct MDSC subsets in tumour-bearing mice, one that consisted of mononuclear cells that resembled inflammatory monocytes and a second that consisted of polymorphonuclear cells that were similar to immature granulocytes. Again, both populations were found to suppress antigen-specific T-cell responses, although by using distinct effector molecules and signalling pathways. The suppressive activity of the granulocytic subset was ARG1-dependent, in contrast to the STAT1- and iNOS-dependent mechanism of the monocyte fraction17. Finally, the same trend was observed in Trypanosoma cruzii infection. In this case, monocytic MDSCs produced NO and strongly inhibited T-cell proliferation, and granulocytic MDSCs produced low levels of NO and did not inhibit T-cell proliferation, although they did produce superoxide15. The biological significance of such functional dichotomy of these two MDSC subsets remains to be elucidated. Induction of TReg cells—Recently, the ability of MDSCs to promote the de novo development of FOXP3+ regulatory T (TReg) cells in vivo has been described18, 19. The induction of TReg cells by MDSCs was found to require the activation of tumour-specific Tcells and the presence of IFNγ and IL-10 but was independent of NO19. In mice bearing 1D8 ovarian tumours, the induction of TReg cells by MDSCs required the expression of cytotoxic lymphocyte antigen 4 (CTLA-4; also known as CD152) by MDSCs18. In a mouse model of lymphoma, MDSCs were shown to induce TReg-cell expansion through a mechanism that required arginase and the capture, processing and presentation of tumour-associated antigens by MDSCs, but not TGFβ58. By contrast, Movahedi et al. found that the percentage of TReg cells was invariably high throughout tumour growth and did not relate to the kinetics of expansion of the MDSC population, suggesting that MDSCs were not involved in TReg-cell expansion17. Furthermore, in a rat model of kidney allograft tolerance that was induced with a CD28-specific antibody, MDSCs that were co-expressing CD80 and CD86 were found to have a limited effect on the expansion of the TReg-cell population81. Although further work is required to resolve these discrepancies and to determine the physiological relevance of these studies, it seems possible that MDSCs are involved in TReg-cell differentiation through the production of cytokines or direct cell–cell interactions. Furthermore, MDSCs and TReg cells might be linked in a common immunoregulatory network (see later).
Tissue-specific effects on MDSCs A major unresolved question in this field is whether MDSCs mediate antigen-specific or nonspecific suppression of T-cell responses. Provided that MDSCs and T cells are in close proximity, the factors that mediate MDSC suppressive function (ROS, arginase and NO) can inhibit T-cell proliferation regardless of the antigen specificity of the T cells. Indeed, numerous in vitro studies have demonstrated the antigen nonspecific nature of MDSC-mediated suppression of T cells82 83. However, whether the situation is the same in vivo is not clear, and evidence suggests that MDSC-mediated immunosuppression in peripheral lymphoid organs is mainly antigen-specific. The idea that MDSC-mediated T-cell suppression occurs in an antigen-specific manner is based on findings that antigen-specific interactions between antigen-presenting cells and T cells result in much more stable and more prolonged cell–cell contact than nonspecific interactions82, 84, 85. Such stable contacts are necessary for MDSCderived ROS and peroxynitrite to mediate effects on the molecules on the surface of T cells that render the T cells unresponsive to specific antigen. It should be noted that such modification of cell-surface molecules does not lead to T-cell death nor prevent nonspecific T-cell activation. Other evidence that supports the idea that MDSCs mediate antigen-specific suppression is the finding that that MDSCs can take up soluble antigens, including tumourassociated antigens, and process and present them to T cells17 80; blockade of MDSC–T-cell interactions with a MHC-class-I-specific antibody abrogated MDSC-mediated inhibition of T cell responses in vitro86. The MHC-class-I-restricted nature of MDSC-mediated CD8+ T-cell suppression has also been demonstrated in vivo in tumor models53 and in the model of inflammatory bowel disease 35. This is consistent with the recent observation that large numbers of tumour-induced MDSCs did not inhibit CD8+ T-cell responses specific for unrelated antigens in a model of sporadic cancer87. Notably, it is currently unclear whether similar antigen-specific mechanisms of MDSC-mediated suppression operate on CD4+ T cells, as published studies have only assessed the effects of MDSCs on CD8+ T cells. Addressing this question is complicated by the fact that only a small proportion of MDSCs in many tumour models expresses MHC class II molecules.
The theory that MDSCs suppress T-cell responses in an antigen-specific manner helps to explain the finding that T cells in the peripheral lymphoid organs of tumour-bearing mice and in the peripheral blood of cancer patients can still respond to stimuli other than tumourassociated antigens, including viruses, lectins, co-stimulatory molecules, IL-2 and CD3- and CD28-specific antibodies21, 80, 88-90. Furthermore, even patients with advanced stage cancer do not have systemic immunodeficiency except in cases in which the patient has received high doses of chemotherapy or is at a terminal stage of the disease.
Evidence suggets that the nature of MDSC-mediated suppression at the tumour site is quite different to that which occurs in the periphery. MDSCs actively migrate into the tumour site10, where they upregulate the expression of ARG1 and iNOS, downregulate the production of ROS and/or rapidly differentiate into tumour-associated macrophages (TAMs) 52. The levels of NO and arginase produced by tumour-associated MDSCs and TAMs are much higher than those of MDSCs found in peripheral lymphoid organs of the same animals. In addition, TAMs produce several cytokines (reviewed in REFs91, 92) that suppress T-cell responses in a nonspecific manner (Fig. 4). The mechanisms by which MDSC functions are regulated within the tumour microenvironment, and how they differ from those that operate at peripheral sites, remain unclear. It is possible that tumour stroma, hypoxia and/or the acidophilic environment have a role.
Therapeutic targeting of MDSCs The recognition that immune suppression has a crucial role in promoting tumour progression and contributes to the frequent failure of cancer vaccines to elicit an immune response has resulted in a paradigm shift with respect to approaches for cancer immunotherapy. Indeed, it has become increasingly clear that successful cancer immunotherapy will be possible only with a strategy that involves the elimination of suppressive factors from the body. As MDSCs are one of the main immunosuppressive factors in cancer and other pathological conditions, several different therapeutic strategies that target these cells are currently being explored (Table 2). Although the studies described below were carried out in tumor-bearing hosts, it is likely that the same strategies will be useful in other pathological conditions in which inhibition or elimination of MDSCs is a therapeutic aim.
Promoting myeloid-cell differentiation—One of the most promising approaches by which to target MDSCs for therapy is to promote their differentiation into mature myeloid cells that do not have suppressive abilities. Vitamin A has been identified as a compound that can mediate this effect: vitamin A metabolites such as retinoic acid have been found to stimulate the differentiation of myeloid progenitors into DCs and macrophages 86, 93. Mice that are deficient in vitamin A94 or that have been treated with a pan-retinoic-acid-receptor antagonist95, show an expansion of MDSCs in the bone marrow and spleen. Conversely, therapeutic concentrations of all-trans retinoic acid (ATRA) results in substantial decrease in the presence of MDSCs in cancer patients and tumour-bearing mice. ATRA induced MDSCs to differentiate into DCs and macrophages in vitro and in vivo 12, 86, 96. It is probable that ATRA preferentially induces the differentiation of the monocytic subset of MDSCs, whereas it causes apoptosis of the granulocytic subset. The main mechanism of ATRA-mediated differentiation involved an upregulation of glutathione synthesis and a reduction in ROS levels in MDSCs 97. Decreasing the number of MDSCs in tumour-bearing mice resulted in increased tumour-specific T-cell responses, and the combination of ATRA and two different types of cancer vaccine prolonged the anti-tumour effect of the vaccine treatment in two different tumour models 96. Moreover, administration of ATRA to patients with metastatic renal cell carcinoma resulted in a substantial decrease in the number of MDSCs in the peripheral blood and improved antigen-specific response of T cells 21. Further studies will lead to identification of other agents that have a similar effect. So far, evidence suggests that Vitamin D3 may be another agent with the potential to decrease MDSC numbers in patients with cancer, as it is also known to promote myeloid-cell differentiation98.
Inhibition of MDSC expansion—Because MDSC expansion is known to be regulated by tumour-derived factors (Table 1), several studies have focused on neutralizing the effects of these factors. Recently, SCF has been implicated in causing MDSC expansion in tumourbearing mice39. Inhibition of SCF-mediated signalling by blocking its interaction with its receptor, c-kit, decreased MDSC expansion and tumor angiogenesis39. VEGF, another tumourderived factor that is involved in promoting MDSC expansion, might also be a useful target by which to manipulate MDSC. However, in a clinical trial of 15 patients with refractory solid tumours, treatment with VEGF–trap (a fusion protein that binds all forms of VEGF-A and placental growth factor) showed no effect on MDSC numbers and did not result in increased T-cell responses99. By contrast, treatment of patients with metastatic renal cell cancer with a VEGF-specific blocking antibody (known as avastin) resulted in a decrease in the size of a CD11b+VEGFR1+ population of MDSCs in the peripheral blood 100. However, whether avastatin treatment resulted in an improvement in antitumour responses in these patients has not been determined. Finally, inhibition of matrix metalloproteinase 9 function in tumorbearing mice decreased the number of MDSCs in the spleen and tumour tissues and resulted in a significant delay in the growth of spontaneous NeuT tumours in transgenic BALB/c mice101. However, the mechanism responsible for this outcome remains to be elucidated.
Inhibition of MDSC function—Another approach by which to inhibit MDSCs is to block the signalling pathways that regulate the production of suppressive factors by these cells. One potential target by which this might be achieved is COX2. COX2 is required for the production of prostaglandin E2, which in 3LL tumour cells61 and mammary carcinoma40 has been shown to induce the upregulation of ARG1 expression by MDSCs, thereby inducing their suppressive function. Accordingly, COX2 inhibitors were found to downregulate the expression of ARG1 by MDSCs, which improved antitumour T-cell responses and enhanced the therapeutic efficacy of immunotherapy102, 103. Similarly, phosphodiesterase-5 inhibitors such as sildenafil were found to downregulate the expression of arginase and iNOS expression by MDSCs, thereby inhibiting their suppressive function in growing tumours104. This resulted in the induction of a measurable anti-tumour immune response and a marked delay of tumour progression in several mouse models 104.
ROS inhibitors have also been shown to be effective for decreasing MDSC-mediated immune suppression in tumour-bearing mice. The coupling of a NO-releasing moiety to a conventional non-steroidal anti-inflammatory drug has proven to be an efficient means by which to inhibit the production of ROS. One such drug, nitroaspirin, was found to limit the activity of ARG1 and iNOS in spleen MDSCs105. In combination with vaccination with endogenous retroviral gp70 antigen, nitroaspirin inhibited MDSCs function and increased the number and function of tumour-antigen-specific T cells105.
Elimination of MDSCs—MDSCs can be directly eliminated in pathological settings by using some chemotherapeutic drugs. Administration of one such drug, gemcitabine, to mice that were bearing large tumours resulted in a dramatic reduction in the number of MDSCs in the spleen and resulted in a marked improvement in the anti-tumour response induced by immunotherapy106, 107. This effect was specific to MDSCs, as a significant decrease in the number of T or B cells was not observed in these animals. Furthermore, in a study of 17 patients with early-stage breast cancer that were treated with doxorubicin–cyclophosphamide chemotherapy, a decrease in the level of MDSCs in the peripheral blood was observed22.
Evidence suggests that there is a broad range of methods that will be effective for targeting of the number and/or function of MDSCs in vivo. These strategies will undoubtedly help to further investigate the biology of these cells as well as expedite clinical applications to treat cancer and other pathological conditions.
MDSCs as regulatory myeloid cells? The wealth of information that has accumulated in recent years regarding the biology of MDSCs suggests that these cells might have evolved as a regulatory component of the immune system. These cells are absent under physiological conditions, as IMCs in naive mice are an intrinsic part of normal haematopoiesis that are not immunosuppressive in an unactivated state. In conditions of acute stress, infection or immunization, there is a transient expansion of this IMC population, which then quickly differentiates into mature myeloid cells. This transient IMC population can mediate the suppressive functions that are characteristic of MDSCs but, because the acute conditions are short-lived, the suppressive functions of this transient population have a minimal impact on the overall immune response. However, these cells probably function as important ‘gatekeepers’ that prevent pathological immune-mediated damage.
The role of the MDSC population in settings of chronic infections and cancer is very different. In these pathological conditions, the prolonged and marked expansion of IMCs and their subsequent activation leads to the expansion of a large population of MDSCs with immunosuppressive abilities. MDSCs accumulate in peripheral lymphoid organs and migrate to tumour sites, where they contribute to immunosuppression. Furthermore, some evidence suggests that MDSCs can also induce expansion of regulatory T cells. Future studies will reveal whether MDSCs can be considered part of a natural immune regulatory network.
Concluding remarks The field of MDSC research has more outstanding questions than answers. The roles of specific MDSC subsets in mediating T-cell suppression, and the molecular mechanisms responsible for inhibition of myeloid-cell differentiation, need to be elucidated. The issue of whether Tcell suppression occurs in an antigen-specific manner remains to be clarified, as do the mechanisms that cause MDSC migration to peripheral lymphoid organs. Some of the main priorities in this field should include a better characterization of human MDSCs and a clear understanding of whether targeting these cells in patients with various pathological conditions will be of clinical significance. Conversely, adoptive cellular therapy with MDSCs may be an attractive opportunity by which to inhibit immune responses in the setting of autoimmune disease or transplantation. The challenge for these approaches will be to devise methods by which to generate these cells ex vivo in clinical-grade conditions such that they are suitable for administration to patients. If the past 5–6 years are an indication of the potential for progress in this area, it is safe to estimate that there will soon be significantly more discoveries that further our understanding about the biology and clinical utility of MDSCs.
Box 1. Definition of myeloid-derived suppressor cells (MDSCs)
• a heterogeneous population of cells of myeloid origin that consist of myeloid progenitors and immature macrophages, immature granulocytes and immature dendritic cells
• present in activated state that is characterized by the increased production of reactive oxygen and nitrogen species, and of arginase
• potent suppressors of various T-cell functions • in mice, their phenotype is CD11b+Gr1+, although functionally distinct subsets within this population have been identified (see main text)
• in humans, their phenotype is Lin-HLA-DR-CD33+ or CD11b+CD14-CD33+.
Human cells do not express a marker homologous to mouse Gr1. MDSC have also been identified within a CD15+ population in human peripheral blood.
• in the steady state, immature myeloid cells lack suppressive activity and are present in the bone marrow, but not in secondary lymphoid organs
• accumulation of MDSCs in lymphoid organs and in tumours in response to various growth factors and cytokines is associated with various pathological conditions (most notably cancer)
• in tumour tissues, MDSCs can be differentiated from tumour-associated macrophages (TAMs) by their high expression of Gr1 (not expressed by TAMs) by their low expression of F4/80 (expressed by TAMs), by the fact that a large proportion of MDSCs have a granulocytic morphology and based the upregulated expression of both arginase and inducible nitric oxide synthase by MDSCs but not TAMs.
References
1. Young MRI, Newby M, Wepsic TH. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res 1987;47:100–106. [PubMed: 2947676]
2. Buessow SC, Paul RD, Lopez DM. Influence of mammary tumor progression on phenotype and function of spleen and in situ lymphocytes in mice. J Natl Cancer Inst 1984;73:249–255. [PubMed: 6610791]
3. Seung L, Rowley D, Dubeym P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci U S A 1995;92:6254–6258. [PubMed: 7603979]
4. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Crosstalk between myeloidderived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 2007;179:977–983. [PubMed: 17617589]
5. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008;8:618–631. [PubMed: 18633355]
6. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumorbearing mice. J Immunol 2008;181:5791–5802. [PubMed: 18832739] Together with reference # 17 this paper described functional differences between subsets of MDSC.
7. Bronte V, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood 2000;96:3838. [PubMed: 11090068]
8. Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J Leukoc Biol 2003;74:186–196. [PubMed: 12885935]
9. Li Q, Pan PY, Gu P, Xu D, Chen SH. Role of immature myeloid Gr-1+ cells in the development of antitumor immunity. Cancer Res 2004;64:1130–1139. [PubMed: 14871848] …..
“The proto-oncogenic transcription factor Myc is known to promote transcription of genes for the cell cycle as well as aerobic glycolysis and glutamine metabolism. Recently, Myc has been shown to play an essential role to induce the expression of glycolytic and glutamine metabolism genes in the initial hours of T cell activation. In a similar fashion, the transcription factor HIF1a can up-regulate glycolytic genes to allow cancer cells to survive under hypoxic conditions. “
Outsource a part of the T cell’s immune value chain, propose cancer immunotherapy researchers, from patient T cells to donor T cells. The novel allogeneic approach could rely on T-cell receptor gene transfer to generate broad and tumor-specific T-cell immune responses. [NIAID]
A new cancer immunotherapy approach could essentially outsource a crucial T-cell function. This function, T-cell reactivity to specific cancer antigens, is sometimes lacking in cancer patients. Yet, according to a new proof-of-principle study, these patients could benefit from T cells provided by healthy donors. Specifically, the healthy donors’ T cells could be used to broaden the T-cell receptor repertoires of the cancer patients’ T cells.
Ultimately, this approach relies on a cancer immunotherapy technique called T-cell receptor (TCR) transfer, or the genetic transfer of TCR chains. TCR transfer can be used to outsource the T cell’s learning function, the process by which a T cell acquires the ability to recognize foreign antigens—in this case, the sort of proteins that can be expressed on the surface of cancer cells. Because cancer cells harbor faulty proteins, they can also display foreign protein fragments, also known as neoantigens, on their surface, much in the way virus-infected cells express fragments of viral proteins.
The approach was detailed in a paper that appeared May 19 in the journal Science, in an article entitled, “Targeting of Cancer Neoantigens with Donor-Derived T Cell Receptor Repertoires.” This article, by scientists based at the Netherlands Cancer Institute and the University of Oslo, describes a novel strategy to broaden neoantigen-specific T-cell responses. Such a strategy would be useful in overcoming a common limitation seen in the immune response to cancer: Neoantigen-specific T-cell reactivity is generally limited to just a few mutant epitopes, even though the number of predicted epitopes is large.
“We demonstrate that T cell repertoires from healthy donors provide a rich source of T cells that specifically recognize neoantigens present on human tumors,” the study’s authors wrote. “Responses to 11 epitopes were observed, and for the majority of evaluated epitopes, potent and specific recognition of tumor cells endogenously presenting the neoantigens was detected.”
First, the researchers mapped all possible neoantigens on the surface of melanoma cells from three different patients. In all three patients, the cancer cells seemed to display a large number of different neoantigens. But when the researchers tried to match these to the T cells derived from within the patient’s tumors, most of these aberrant protein fragments on the tumor cells went unnoticed.
Next, the researchers tested whether the same neoantigens could be seen by T cells derived from healthy volunteers. Strikingly, these donor-derived T cells could detect a significant number of neoantigens that had not been seen by the patients’ T cells.
“Many of the T cell reactivities [among donor T cells] involved epitopes that in vivo were neglected by patient autologous tumor-infiltrating lymphocytes,” the authors of the Science article continued. “T cells re-directed with T cell receptors identified from donor-derived T cells efficiently recognized patient-derived melanoma cells harboring the relevant mutations, providing a rationale for the use of such ‘outsourced’ immune responses in cancer immunotherapy.”
“In a way, our findings show that the immune response in cancer patients can be strengthened; there is more on the cancer cells that makes them foreign that we can exploit. One way we consider doing this is finding the right donor T cells to match these neoantigens,” said Ton Schumacher, Ph.D., a principal investigator at the Netherlands Cancer Institute. “The receptor that is used by these donor T cells can then be used to genetically modify the patient’s own T cells so these will be able to detect the cancer cells.”
“Our study shows that the principle of outsourcing cancer immunity to a donor is sound,” added Johanna Olweus, M.D., Ph.D., who heads a research group at the University of Oslo. “However, more work needs to be done before patients can benefit from this discovery. Thus, we need to find ways to enhance the throughput.”
“We are currently exploring high-throughput methods to identify the neoantigens that the T cells can ‘see’ on the cancer and isolate the responding cells. But the results showing that we can obtain cancer-specific immunity from the blood of healthy individuals are already very promising.”
Targeting of cancer neoantigens with donor-derived T cell receptor repertoires
Accumulating evidence suggests that clinically efficacious cancer immunotherapies are driven by T cell reactivity against DNA mutation-derived neoantigens. However, among the large number of predicted neoantigens, only a minority is recognized by autologous patient T cells, and strategies to broaden neoantigen specific T cell responses are therefore attractive. Here, we demonstrate that naïve T cell repertoires of healthy blood donors provide a source of neoantigen-specific T cells, responding to 11/57 predicted HLA-A2-binding epitopes from three patients. Many of the T cell reactivities involved epitopes that in vivo were neglected by patient autologous tumor-infiltrating lymphocytes. Finally, T cells re-directed with T cell receptors identified from donor-derived T cells efficiently recognized patient-derived melanoma cells harboring the relevant mutations, providing a rationale for the use of such “outsourced” immune responses in cancer immunotherapy.
Metabolic maintenance of cell asymmetry following division in activated T lymphocytes.
Asymmetric cell division, the partitioning of cellular components in response to polarizing cues during mitosis, has roles in differentiation and development. It is important for the self-renewal of fertilized zygotes in Caenorhabditis elegans and neuroblasts in Drosophila, and in the development of mammalian nervous and digestive systems. T lymphocytes, upon activation by antigen-presenting cells (APCs), can undergo asymmetric cell division, wherein the daughter cell proximal to the APC is more likely to differentiate into an effector-like T cell and the distal daughter is more likely to differentiate into a memory-like T cell. Upon activation and before cell division, expression of the transcription factor c-Myc drives metabolic reprogramming, necessary for the subsequent proliferative burst. Here we find that during the first division of an activated T cell in mice, c-Myc can sort asymmetrically. Asymmetric distribution of amino acid transporters, amino acid content, and activity of mammalian target of rapamycin complex 1 (mTORC1) is correlated with c-Myc expression, and both amino acids and mTORC1 activity sustain the differences in c-Myc expression in one daughter cell compared to the other. Asymmetric c-Myc levels in daughter T cells affect proliferation, metabolism, and differentiation, and these effects are altered by experimental manipulation of mTORC1 activity or c-Myc expression. Therefore, metabolic signalling pathways cooperate with transcription programs to maintain differential cell fates following asymmetric T-cell division.
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive malignancy associated with Notch pathway mutations. While both normal activated and leukemic T cells can utilize aerobic glycolysis to support proliferation, it is unclear to what extent these cell populations are metabolically similar and if differences reveal T-ALL vulnerabilities. Here we show that aerobic glycolysis is surprisingly less active in T-ALL cells than proliferating normal T cells and that T-ALL cells are metabolically distinct. Oncogenic Notch promoted glycolysis but also induced metabolic stress that activated 5′ AMP-activated kinase (AMPK). Unlike stimulated T cells, AMPK actively restrained aerobic glycolysis in T-ALL cells through inhibition of mTORC1 while promoting oxidative metabolism and mitochondrial Complex I activity. Importantly, AMPK deficiency or inhibition of Complex I led to T-ALL cell death and reduced disease burden. Thus, AMPK simultaneously inhibits anabolic growth signaling and is essential to promote mitochondrial pathways that mitigate metabolic stress and apoptosis in T-ALL.
Obesity and diabetes are associated with excessive inflammation and impaired wound healing. Increasing evidence suggests that macrophage dysfunction is responsible for these inflammatory defects. In the setting of excess nutrients, particularly dietary saturated fatty acids (SFAs), activated macrophages develop lysosome dysfunction, which triggers activation of the NLRP3 inflammasome and cell death. The molecular pathways that connect lipid stress to lysosome pathology are not well understood, but may represent a viable target for therapy. Glutamine uptake is increased in activated macrophages leading us to hypothesize that in the context of excess lipids glutamine metabolism could overwhelm the mitochondria and promote the accumulation of toxic metabolites. To investigate this question we assessed macrophage lipotoxicity in the absence of glutamine using LPS-activated peritoneal macrophages exposed to the SFA palmitate. We found that glutamine deficiency reduced lipid induced lysosome dysfunction, inflammasome activation, and cell death. Under glutamine deficient conditions mTOR activation was decreased and autophagy was enhanced; however, autophagy was dispensable for the rescue phenotype. Rather, glutamine deficiency prevented the suppressive effect of the SFA palmitate on mitochondrial respiration and this phenotype was associated with protection from macrophage cell death. Together, these findings reveal that crosstalk between activation-induced metabolic reprogramming and the nutrient microenvironment can dramatically alter macrophage responses to inflammatory stimuli.
Immunoregulatory Protein B7-H3 Reprograms Glucose Metabolism in Cancer Cells by ROS-Mediated Stabilization of HIF1α
CD8(+) T cells can respond to unrelated infections in an Ag-independent manner. This rapid innate-like immune response allows Ag-experienced T cells to alert other immune cell types to pathogenic intruders. In this study, we show that murine CD8(+) T cells can sense TLR2 and TLR7 ligands, resulting in rapid production of IFN-γ but not of TNF-α and IL-2. Importantly, Ag-experienced T cells activated by TLR ligands produce sufficient IFN-γ to augment the activation of macrophages. In contrast to Ag-specific reactivation, TLR-dependent production of IFN-γ by CD8(+) T cells relies exclusively on newly synthesized transcripts without inducing mRNA stability. Furthermore, transcription of IFN-γ upon TLR triggering depends on the activation of PI3K and serine-threonine kinase Akt, and protein synthesis relies on the activation of the mechanistic target of rapamycin. We next investigated which energy source drives the TLR-induced production of IFN-γ. Although Ag-specific cytokine production requires a glycolytic switch for optimal cytokine release, glucose availability does not alter the rate of IFN-γ production upon TLR-mediated activation. Rather, mitochondrial respiration provides sufficient energy for TLR-induced IFN-γ production. To our knowledge, this is the first report describing that TLR-mediated bystander activation elicits a helper phenotype of CD8(+) T cells. It induces a short boost of IFN-γ production that leads to a significant but limited activation of Ag-experienced CD8(+) T cells. This activation suffices to prime macrophages but keeps T cell responses limited to unrelated infections.
The bidirectional interaction between the immune system and whole-body metabolism has been well recognized for many years. Via effects on adipocytes and hepatocytes, immune cells can modulate whole-body metabolism (in metabolic syndromes such as type 2 diabetes and obesity) and, reciprocally, host nutrition and commensal-microbiota-derived metabolites modulate immunological homeostasis. Studies demonstrating the metabolic similarities of proliferating immune cells and cancer cells have helped give birth to the new field of immunometabolism, which focuses on how the cell-intrinsic metabolic properties of lymphocytes and macrophages can themselves dictate the fate and function of the cells and eventually shape an immune response. We focus on this aspect here, particularly as it relates to regulatory T cells.
Figure 1: Proposed model for the metabolic signatures of various Treg cell subsets.
(a) Activated CD4+ T cells that differentiate into the Teff cell lineage (green) (TH1 or TH17 cells) are dependent mainly on carbon substrates such as glucose and glutamine for their anabolic metabolism. In contrast to that, pTreg cells…
T-bet is a key modulator of IL-23-driven pathogenic CD4+ T cell responses in the intestine
IL-23 is a key driver of pathogenic Th17 cell responses. It has been suggested that the transcription factor T-bet is required to facilitate IL-23-driven pathogenic effector functions; however, the precise role of T-bet in intestinal T cell responses remains elusive. Here, we show that T-bet expression by T cells is not required for the induction of colitis or the differentiation of pathogenic Th17 cells but modifies qualitative features of the IL-23-driven colitogenic response by negatively regulating IL-23R expression. Consequently, absence of T-bet leads to unrestrained Th17 cell differentiation and activation characterized by high amounts of IL-17A and IL-22. The combined increase in IL-17A/IL-22 results in enhanced epithelial cell activation and inhibition of either IL-17A or IL-22 leads to disease amelioration. Our study identifies T-bet as a key modulator of IL-23-driven colitogenic responses in the intestine and has important implications for understanding of heterogeneity among inflammatory bowel disease patients.
Th17 cells are enriched at mucosal sites, produce high amounts of IL-17A, IL-17F and IL-22, and have an essential role in mediating host protective immunity against a variety of extracellular pathogens1. However, on the dark side, Th17 cells have also been implicated in a variety of autoimmune and chronic inflammatory conditions, including inflammatory bowel disease (IBD)2. Despite intense interest, the cellular and molecular cues that drive Th17 cells into a pathogenic state in distinct tissue settings remain poorly defined.
The Th17 cell programme is driven by the transcription factor retinoid-related orphan receptor gamma-t (RORγt) (ref. 3), which is also required for the induction and maintenance of the receptor for IL-23 (refs 4, 5). The pro-inflammatory cytokine IL-23, composed of IL-23p19 and IL-12p40 (ref. 6), has been shown to be a key driver of pathology in various murine models of autoimmune and chronic inflammatory disease such as experimental autoimmune encephalomyelitis (EAE)7, collagen induced arthritis8 and intestinal inflammation9, 10, 11, 12. Several lines of evidence, predominantly derived from EAE, suggest that IL-23 promotes the transition of Th17 cells to pathogenic effector cells9, 10, 11, 12. Elegant fate mapping experiments of IL-17A-producing cells during EAE have shown that the majority of IL-17A+IFN-γ+ and IL-17A−IFN-γ+ effector cells arise from Th17 cell progeny13. This transition of Th17 cells into IFN-γ-producing ‘ex’ Th17 cells required IL-23 and correlated with increased expression of T-bet. The T-box transcription factor T-bet drives the Th1 cell differentiation programme14 and directly transactivates the Ifng gene by binding to its promoter as well as multiple enhancer elements15. Indeed, epigenetic analyses have revealed that the loci for T-bet and IFN-γ are associated with permissive histone modifications in Th17 cells suggesting that Th17 cells are poised to express T-bet which could subsequently drive IFN-γ production16, 17.
A similar picture is emerging in the intestine where IL-23 drives T-cell-mediated intestinal pathology which is thought to be dependent on expression of T-bet18 and RORγt (ref. 19) by T cells. In support of this we have recently shown that IL-23 signalling in T cells drives the emergence of IFN-γ producing Th17 cells in the intestine during chronic inflammation20. Collectively these studies suggest a model whereby RORγt drives differentiation of Th17 cells expressing high amounts of IL-23R, and subsequently, induction of T-bet downstream of IL-23 signalling generates IL-17A+IFN-γ+ T cells that are highly pathogenic. Indeed, acquisition of IFN-γ production by Th17 cells has been linked to their pathogenicity in several models of chronic disease13, 21, 22, 23, 24 and a population of T cells capable of producing both IL-17A and IFN-γ has also been described in intestinal biopsies of IBD patients25, 26.
However, in the context of intestinal inflammation, it remains poorly defined whether the requirement for RORγt and T-bet reflects a contribution of Th17 and Th1 cells to disease progression or whether Th17 cells require T-bet co-expression to exert their pathogenic effector functions. Here, we use two distinct models of chronic intestinal inflammation and make the unexpected finding that T-bet is dispensable for IL-23-driven colitis. Rather the presence of T-bet serves to modify the colitogenic response restraining IL-17 and IL-22 driven pathology. These data identify T-bet as a key modulator of IL–23-driven colitogenic effector responses in the intestine and have important implications for understanding of heterogeneous immune pathogenic mechanisms in IBD patients.
Figure 1: IL-23 signalling is required for bacteria-driven T-cell-dependent colitis and the emergence of IL-17A+IFN-γ+ T cells.
C57BL/6 WT and Il23r−/− mice were infected orally with Hh and received weekly i.p. injections of IL-10R blocking antibody. Mice were killed at 4 weeks post infection and assessed for intestinal inflammation. (a) Colitis scores. (b) Typhlitis sores. (c) Representative photomicrographs of colon and caecum (× 10 magnification; scale bars, 200μM). (d) Representative flow cytometry plots of colonic lamina propria gated on viable CD4+ T cells. (e) Frequencies of IL-17A+ and/or IFN-γ+ CD4+ T cells present in the colon. Data represent pooled results from two independent experiments (n=12 for WT, n=10 for Il23r−/−). Bars are the mean and each symbol represents an individual mouse. *P<0.05, ***P<0.001 as calculated by Mann–Whitney U test.
C57BL/6 Rag1−/− mice were injected i.p. with 4 × 105 CD4+CD25−CD45RBhi T cells from C57BL/6 WT,Rorc−/− or Tbx21−/− donors. Mice were killed when recipients of Tbx21−/− T cells developed clinical signs of disease (4–6 weeks) and assessed for intestinal inflammation. (a) Colitis scores. (b) Representative photomicrographs of proximal colon sections (× 10 magnification; scale bars, 200μM). (c) Concentration of cytokines released from colon explants into the medium after overnight culture. Data represent pooled results from two independent experiments (n=14 for WT, n=11 for Rorc−/−, n=14 forTbx21−/−). Bars are the mean and each symbol represents an individual mouse. Bars are the mean and error bars represent s.e.m. *P<0.05, **P<0.01, ***P<0.001 as calculated by Kruskal–Wallis one-way ANOVA with Dunn’s post-test.
C57BL/6 Rag1−/− mice were injected i.p. with 4×105 CD4+CD25−CD45RBhi T cells from C57BL/6 WT,Rorc−/− or Tbx21−/− donors. Mice were killed when recipients of Tbx21−/−T cells developed clinical signs of disease (4–6 weeks). (a) Representative plots of IL-17A and IFN-γ expression in colonic CD4+ T cells. (b) Frequencies of IL-17A+ and/or IFN-γ+ cells among colonic CD4+ T cells. (c) Total numbers of IL-17A+and/or IFN-γ+ CD4+ T cells present in the colon. Data represent pooled results from three independent experiments (n=20 for WT, n=18 for Tbx21−/−, n=12 for Rorc−/−). Bars are the mean and each symbol represents an individual mouse. *P<0.05, **P<0.01, ***P<0.001 as calculated by Kruskal–Wallis one-way ANOVA with Dunn’s post-test.
T-bet deficiency promotes an exacerbated Th17-type response
Our transfer of Tbx21−/− T cells revealed a striking increase in the frequency of IL-17A+IFN-γ−cells (Fig. 3) and we reasoned that T-bet-deficiency could impact on Th17 cell cytokine production. Therefore, we transferred WT or Tbx21−/− CD4+ T cells into Rag1−/− recipients and measured the expression of RORγt, IL-17A, IL-17F and IL-22 by CD4+ T cells isolated from the colon. In agreement with our earlier findings, Tbx21−/− T cells gave rise to significantly increased frequencies of RORγt-expressing T cells capable of producing IL-17A (Fig. 4a). Furthermore, T-bet deficiency also led to a dramatic expansion of IL-17F and IL-22-expressing cells, which constituted only a minor fraction in WT T cells (Fig. 4a,b). By contrast, the frequency of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IFN-γ producing cells was significantly reduced in T-bet-deficient T cells as compared with WT T cells. When analysed in more detail we noted that the production of IL-17A, IL-17F and IL-22 increased specifically in T-bet-deficient IL-17A+IFN-γ+ T cells as compared with WT T cells whereas IFN-γ production decreased overall in the absence of T-bet as expected (Supplementary Fig. 4A). Similarly, GM-CSF production was also generally reduced in Tbx21−/− CD4+ T cells further suggesting a shift in the qualitative nature of the T cell response.
Figure 4: T-bet-deficient CD4+ T cells promote an exacerbated Th17-type inflammatory response.
C57BL/6 Rag1−/− mice were injected i.p. with 4×105 CD4+CD25−CD45RBhi T cells from C57BL/6 WT orTbx21−/− donors. Mice were killed when recipients of Tbx21−/−T cells developed clinical signs of disease (4–6 weeks). (a) Representative plots of cytokines and transcription factors in WT or Tbx21−/− colonic CD4+ T cells. (b) Frequency of IL-17A+, IL-17F+, IL-22+, GM-CSF+ or IFN-γ+ colonic T cells in WT orTbx21−/−. (c) quantitative reverse transcription PCR (qRT-PCR) analysis of mRNA levels of indicated genes in colon tissue homogenates. (d) Total number of neutrophils (CD11b+ Gr1high) in the colon. (e) Primary epithelial cells were isolated from the colon of steady state C57BL/6 Rag1−/− mice and stimulated with 10ngml−1 cytokines for 4h after which cells were harvested and analysed by qRT-PCR for the indicated genes. Data in b–d represent pooled results from two independent experiments (n=14 for WT, n=11 for Tbx21−/−). Bars are the mean and error bars represent s.e.m. Data in e are pooled results from four independent experiments, bars are the mean and error bars represent s.e.m. *P<0.05, **P<0.01,***P<0.001 as calculated by Mann–Whitney U test.
T-bet-deficient colitis depends on IL-23, IL-17A and IL-22
In the present study we show that bacteria-driven colitis is associated with the IL-23-dependent emergence of IFN-γ-producing Th17 cells co-expressing RORγt and T-bet. Strikingly, while RORγt is required for the differentiation of IFN-γ-producing Th17 cells and induction of colitis, T-bet is dispensable for the emergence of IL-17A+IFN-γ+ T cells and intestinal pathology. Our results show that instead of a mandatory role in the colitogenic response, the presence of T-bet modulates the qualitative nature of the IL-23-driven intestinal inflammatory response. In the presence of T-bet, IL-23-driven colitis is multifunctional in nature and not functionally dependent on either IL-17A or IL-22. By contrast, in the absence of T-bet a highly polarized colitogenic Th17 cell response ensues which is functionally dependent on both IL-17A and IL-22. T-bet-deficient T cells are hyper-responsive to IL-23 resulting in enhanced STAT3 activation and downstream cytokine secretion providing a mechanistic basis for the functional changes. These data newly identify T-bet as a key modulator of IL-23-driven colitogenic CD4+ T cell responses.
Contrary to our expectations T-bet expression by CD4 T cells was not required for their pathogenicity. In keeping with the negative effect of T-bet on Th17 differentiation40, 41, 42, we observed highly polarized Th17 responses in T-bet-deficient intestinal T cells. Early studies demonstrated that IFN-γ could suppress the differentiation of Th17 cells40 and thus the reduced IFN-γ production by Tbx21−/−T cells could facilitate Th17 cell generation. However, our co-transfer studies revealed unrestrained Th17 differentiation of Tbx21−/− T cells even in the presence of WT T cells, suggesting a cell autonomous role for T-bet-mediated suppression of the Th17 programme. Indeed, the role of T-bet as a transcriptional repressor of the Th17 cell fate has been described recently. For example, T-bet physically interacts with and sequesters Runx1, thereby preventing Runx1-mediated induction of RORγt and Th17 cell differentiation43. In addition, T-bet binds directly to and negatively regulates expression of many Th17-related genes15, 34 and we identified IL23r to be repressed in a T-bet-dependent manner. In line with this we show here that T-bet-deficient intestinal T cells express higher amounts of Il23r as well as Rorc. This resulted in enhanced IL-23-mediated STAT3 activation and increased production of IL-17A and IL-22. It has also been suggested that T-bet activation downstream of IL-23R signalling is required for pathogenic IL-23-driven T cell responses43, 44. However, we did not find a role for IL-23 in the induction and/or maintenance of T-bet expression and colitis induced by T-bet-deficient T cells was IL-23 dependent. Collectively, these findings demonstrate that T-bet deficiency leads to unrestrained expansion of colitogenic Th17 cells, which is likely mediated through enhanced activation of the IL-23R-STAT3 pathway.
The observation that T-bet-deficient T cells retain their colitogenic potential is in stark contrast to earlier studies. Neurath et al.18 convincingly showed that adoptive transfer of Tbx21−/− CD4+ T cells into severe combined immunodeficiency (SCID) recipients failed to induce colitis and this correlated with reduced IFN-γ and increased IL-4 production. Another study revealed that IL-4 plays a functional role in inhibiting the colitogenic potential of Tbx21−/− T cells, as recipients ofStat6−/−Tbx21−/− T cells developed severe colitis37. Importantly, the intestinal inflammation that developed in recipients of Stat6−/−Tbx21−/− T cells could be blocked by administration of IL-17A neutralizing antibody, suggesting that the potent inhibitory effect of IL-4/STAT6 signals on Th17 differentiation normally prevent colitis induced by Tbx21−/− T cells37. Various explanations could account for the discrepancy between our study and those earlier findings. First, in contrast to the published reports, we used naïve Tbx21−/− CD4+ T cells from C57BL/6 mice instead of BALB/c mice. An important difference between Tbx21−/− CD4+ T cells from these genetic backgrounds appears to be their differential susceptibility to suppression by IL-4/STAT6 signals. We found that transfer of Tbx21−/− T cells induced IL-17A-dependent colitis despite increased frequencies of IL-4-expressing cells in the intestine. This discrepancy may be due to higher amounts of IL-4 produced by activated CD4+ T cells from BALB/c versus C57BL/6 mice45, leading to the well-described Th2-bias of the BALB/c strain45. Second, differences in the composition of the intestinal microbiota between animal facilities can have a substantial effect on skewing CD4+ T cells responses. In particular, the Clostridium-related segmented filamentous bacteria (SFB) have been shown to drive the emergence of IL-17 and IL-22 producing CD4+ T cells in the intestine46. Importantly, the ability of naïve CD4+ T cells to induce colitis is dependent on the presence of intestinal bacteria, as germ-free mice do not develop pathology upon T cell transfer47. In line with this, we previously described that colonization of germ-free mice with intestinal microbiota containing SFB was necessary to restore the development of colitis47. Since our Rag1−/− colony is SFB+ and the presence of SFB was not reported in the previous studies, it is possible that differences in SFB colonization status contributed to the observed differences in pathogenicity ofTbx21−/− T cells.
It is important to note that T-bet-deficient T cells did not induce more severe colitis than WT T cells but rather promoted a distinct mucosal inflammatory response. Colitis induced by WT T cells is characterized by a multifunctional response with high amounts of IFN-γ and GM-CSF and a lower IL-17A and IL-22 response. Consistent with this, we have shown that blockade of GM-CSF abrogates T cell transfer colitis48 as well as bacteria-driven intestinal inflammation49 in T-bet sufficiency whereas blockade of IL-17A or IL-22 fails to do so. By contrast T-bet deficiency leads to production of high amounts of IL-17A and IL-22 in the colon and neutralization of either was sufficient to reduce intestinal pathology. Our in vitro experiments suggest that IL-17A and IL-22 synergise to promote intestinal epithelial cell responses, which may in part explain the efficacy of blocking IL-17A or IL-22 in colitis induced by T-bet-deficient T cells. A similar synergistic interplay has been described in the lung where IL-22 served a tissue protective function in homeostasis but induced airway inflammation in the presence of IL-17A (ref. 50). This highlights the complexity of the system in health and disease, and the need for a controlled production of both cytokines. We describe here only one mechanism of how IL-17A/IL-22 induce a context-specific epithelial cell response that potentially impacts on the order or composition of immune cell infiltration. Overall, these results provide a new perspective on T-bet, revealing its role in shaping the qualitative nature of the IL-23-driven colitogenic T cell response.
We also describe here the unexpected finding that a substantial proportion of T-bet-deficient intestinal T cells retain the ability to express IFN-γ. To investigate the potential mechanisms responsible for T-bet-independent IFN-γ production by intestinal CD4+ T cells we focused on two transcription factors, Runx3 and Eomes. Runx3 has been shown to promote IFN-γ expression directly through binding to the Ifng promoter38 and Eomes is known to compensate for IFN-γproduction in T-bet-deficient Th1 cells37. We found IL-23-mediated induction of Runx3 protein in WT and Tbx21−/− T cells isolated from the intestine, thus identifying Runx3 downstream of IL-23R signalling. By contrast, we could only detect Eomes protein and its induction by IL-23 in T-bet-deficient but not WT T cells. Thus, Runx3 and Eomes are activated in response to IL-23 in T-bet-deficient cells and are likely to be drivers of T-bet-independent IFN-γ production. In support of this we found that the majority of T-bet-deficient IL-17A−IFN-γ+ T cells expressed Eomes. However, only a minor population of IL-17A+IFN-γ+ T cells stained positive for Eomes, suggesting the existence of alternative pathways for IFN-γ production by Th17 cells. Intriguingly, a recent study identified Runx3 and Runx1 as the transcriptional regulators critical for the differentiation of IFN-γ-producing Th17 cells51. The author’s demonstrated that ectopic expression of Runx transcription factors was sufficient to induce IFN-γ production by Th17 cells even in the absence of T-bet. These findings, combined with our data on Runx3 activation downstream of IL-23R signalling strongly suggest that Runx3 rather than Eomes is driving IFN-γ expression by intestinal Th17 cells.
We have not formally addressed the role of IFN-γ in colitis driven by T-bet-deficient T cells. A recent report by Zimmermann et al.52 found that antibody-mediated blockade of IFN-γ ameliorates colitis induced by WT or T-bet-deficient T cells suggesting IFN-γ also contributes to the colitogneic response mediated by T-bet-deficient T cells as originally described for WT T cells53, 54. By contrast with our results the Zimmerman study found that IL-17A blockade exacerbated colitis following transfer of Tbx21−/− T cells. The reason for the differential role of IL-17A in the two studies is not clear but it is notable that the Zimmerman study was performed in the presence of co-infection with SFB and Hh, and this strong inflammatory drive may alter the pathophysiological role of particular cytokines. Together the data indicate that T-bet deficiency in T cells does not impede their colitogenic activity but that the downstream effector cytokines of the response are context dependent.
In conclusion, our data further underline the essential role for IL-23 in intestinal inflammation and demonstrate that T-bet is an important modulator of the IL–23-driven effector T cell response. The colitogenic T cell response in a T-bet sufficient environment is multifunctional with a dominant GM-CSF and IFN-γ response. By contrast T-bet-deficient colitogenic responses are dominated by IL-17A and IL-22-mediated immune pathology. These results may have significant bearing on human IBD where it is now recognized that differential responsiveness to treatment may reflect considerable disease heterogeneity. As such, identification of suitable biomarkers such as immunological parameters, that allow stratification of patient groups, is becoming increasingly important55. Genome-wide association studies have identified polymorphisms in loci related to innate and adaptive immune arms that confer increased susceptibility to IBD. Among these are Th1 (STAT4, IFNG and STAT1) as well as Th17-related genes (RORC, IL23R and STAT3) (refs56, 57). Thus, detailed profiling of the T cell response in IBD patients may help identify appropriate patient groups that are most likely to benefit from therapeutic blockade of certain effector cytokines. Finally, our studies highlight the importance of IL-23 in the intestinal inflammatory hierarchy and suggest that IL-23 could be an effective therapeutic target across a variety of patient groups.
Yale study: How antibodies access neurons to fight infection
Yale scientists have solved a puzzle of the immune system: how antibodies enter the nervous system to control viral infections. Their finding may have implications for the prevention and treatment of a range of conditions, including herpes and Guillain-Barre syndrome, which has been linked to the Zika virus.
Many viruses — such as West Nile, Zika, and the herpes simplex virus — enter the nervous system, where they were thought to be beyond the reach of antibodies. Yale immunobiologists Akiko Iwasaki and Norifumi Iijima used mice models to investigate how antibodies could gain access to nerve tissue in order to control infection.
In mice infected with herpes, they observed a previously under-recognized role of CD4 T cells, a type of white blood cell that guards against infection by sending signals to activate the immune system. In response to herpes infection, CD4 T cells entered the nerve tissue, secreted signaling proteins, and allowed antibody access to infected sites. Combined, CD4 T cells and antibodies limited viral spread.
“This is a very elegant design of the immune system to allow antibodies to go to the sites of infection,” said Iwasaki. “The CD4 T cells will only go to the site where there is a virus. It’s a targeted delivery system for antibodies.”
Access of protective antiviral antibody to neuronal tissues requires CD4 T-cell help
Circulating antibodies can access most tissues to mediate surveillance and elimination of invading pathogens. Immunoprivileged tissues such as the brain and the peripheral nervous system are shielded from plasma proteins by the blood–brain barrier1 and blood–nerve barrier2, respectively. Yet, circulating antibodies must somehow gain access to these tissues to mediate their antimicrobial functions. Here we examine the mechanism by which antibodies gain access to neuronal tissues to control infection. Using a mouse model of genital herpes infection, we demonstrate that both antibodies and CD4 T cells are required to protect the host after immunization at a distal site. We show that memory CD4 T cells migrate to the dorsal root ganglia and spinal cord in response to infection with herpes simplex virus type 2. Once inside these neuronal tissues, CD4 T cells secrete interferon-γ and mediate local increase in vascular permeability, enabling antibody access for viral control. A similar requirement for CD4 T cells for antibody access to the brain is observed after intranasal challenge with vesicular stomatitis virus. Our results reveal a previously unappreciated role of CD4 T cells in mobilizing antibodies to the peripheral sites of infection where they help to limit viral spread.
T Cells Help Reverse Ovarian Cancer Drug Resistance
T cells (red) attack ovarian cancer cells (green). [University of Michigan Health System]
Researchers at the University of Michigan have recently published the results from a new study that they believe underscores why so many ovarian tumors develop resistance to chemotherapy. The tumor microenvironment is made up of an array of cell types, yet effector T cells and fibroblasts constitute the bulk of the tissue. The investigators believe that understanding the interplay between these two cell types holds the key to how ovarian cancer cells develop resistance.
The new study suggests that the fibroblasts surrounding the tumor work to block chemotherapy, which is why nearly every woman with ovarian cancer becomes resistant to treatment. Conversely, the scientists published evidence that T cells in the microenvironment can reverse the resistance phenotype—suggesting a whole different way of thinking about chemotherapy resistance and the potential to harness immunotherapy drugs to treat ovarian cancer.
“Ovarian cancer is often diagnosed at late stages, so chemotherapy is a key part of treatment,” explained co-senior study author J. Rebecca Liu, M.D., associate professor of obstetrics and gynecology at the University of Michigan. “Most patients will respond to it at first, but everybody develops chemoresistance. And that’s when ovarian cancer becomes deadly.”
Dr. Liu continued, stating that “in the past, we’ve thought the resistance was caused by genetic changes in tumor cells. But we found that’s not the whole story.”
The University of Michigan team looked at tissue samples from ovarian cancer patients and separated the cells by type to study the tumor microenvironment in vitro and in mice. More importantly, the scientists linked their findings back to actual patient outcomes.
The results of this study were published recently in Cell through an article entitled “Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer.”
Ovarian cancer is typically treated with cisplatin, a platinum-based chemotherapy. The researchers found that fibroblasts blocked platinum. These cells prevented platinum from accumulating in the tumor and protected tumor cells from being killed off by cisplatin.
Diagram depicting how T cells can reverse chemotherapeutic resistance. [Cell, Volume 165, Issue 5, May 19, 2016]
“We show that fibroblasts diminish the nuclear accumulation of platinum in ovarian cancer cells, resulting in resistance to platinum-based chemotherapy,” the authors wrote. “We demonstrate that glutathione and cysteine released by fibroblasts contribute to this resistance.”
T cells, on the other hand, overruled the protection of the fibroblasts. When researchers added the T cells to the fibroblast population, the tumor cells began to die off.
“CD8+ T cells abolish the resistance by altering glutathione and cystine metabolism in fibroblasts,” the authors explained. “CD8+ T-cell-derived interferon (IFN)γ controls fibroblast glutathione and cysteine through upregulation of gamma-glutamyltransferases and transcriptional repression of system xc−cystine and glutamate antiporter via the JAK/STAT1 pathway.”
By boosting the effector T cell numbers, the researchers were able to overcome the chemotherapy resistance in mouse models. Moreover, the team used interferon, an immune cell-secreted cytokine, to manipulate the pathways involved in cisplatin.
“T cells are the soldiers of the immune system,” noted co-senior study author Weiping Zou, M.D., Ph.D., professor of surgery, immunology, and biology at the University of Michigan. “We already know that if you have a lot of T cells in a tumor, you have better outcomes. Now we see that the immune system can also impact chemotherapy resistance.”
The researchers suggest that combining chemotherapy with immunotherapy may be effective against ovarian cancer. Programmed death ligand 1 (PD-L1) and PD-1 pathway blockers are currently FDA-approved treatments for some cancers, although not ovarian cancer.
“We can imagine re-educating the fibroblasts and tumor cells with immune T cells after chemoresistance develops,” Dr. Zou remarked.
“Then we could potentially go back to the same chemotherapy drug that we thought the patient was resistant to. Only now we have reversed that, and it’s effective again,” Dr. Liu concluded.
Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer
•Fibroblasts diminish platinum content in cancer cells, resulting in drug resistance
•GSH and cysteine released by fibroblasts contribute to platinum resistance
•T cells alter fibroblast GSH and cystine metabolism and abolish the resistance
•Fibroblasts and CD8+ T cells associate with patient chemotherapy response
Summary
Effector T cells and fibroblasts are major components in the tumor microenvironment. The means through which these cellular interactions affect chemoresistance is unclear. Here, we show that fibroblasts diminish nuclear accumulation of platinum in ovarian cancer cells, resulting in resistance to platinum-based chemotherapy. We demonstrate that glutathione and cysteine released by fibroblasts contribute to this resistance. CD8+ T cells abolish the resistance by altering glutathione and cystine metabolism in fibroblasts. CD8+ T-cell-derived interferon (IFN)γ controls fibroblast glutathione and cysteine through upregulation of gamma-glutamyltransferases and transcriptional repression of system xc− cystine and glutamate antiporter via the JAK/STAT1 pathway. The presence of stromal fibroblasts and CD8+ T cells is negatively and positively associated with ovarian cancer patient survival, respectively. Thus, our work uncovers a mode of action for effector T cells: they abrogate stromal-mediated chemoresistance. Capitalizing upon the interplay between chemotherapy and immunotherapy holds high potential for cancer treatment.
Activation of effect or T cells leads to increased glucose uptake, glycolysis, and lipid synthesis to support growth and proliferation. Activated T cells were identified with CD7, CD5, CD3, CD2, CD4, CD8 and CD45RO. Simultaneously, the expression of CD95 and its ligand causes apoptotic cells death by paracrine or autocrine mechanism, and during inflammation, IL1-β and interferon-1α.. The receptor glucose, Glut 1, is expressed at a low level in naive T cells, and rapidly induced by Myc following T cell receptor (TCR) activation. Glut1 trafficking is also highly regulated, with Glut1 protein remaining in intracellular vesicles until T cell activation. CD28 co-stimulation further activates the PI3K/Akt/mTOR pathway in particular, and provides a signal for Glut1 expression and cell surface localization. Mechanisms that control T cell metabolic reprogramming are now coming to light, and many of the same oncogenes importance in cancer metabolism are also crucial to drive T cell metabolic transformations, most notably Myc, hypoxia inducible factor (HIF)1a, estrogen-related receptor (ERR) a, and the mTOR pathway. The proto-oncogenic transcription factor, Myc, is known to promote transcription of genes for the cell cycle, as well as aerobic glycolysis and glutamine metabolism. Recently, Myc has been shown to play an essential role in inducing the expression of glycolytic and glutamine metabolism genes in the initial hours of T cell activation. In a similar fashion, the transcription factor (HIF)1a can up-regulate glycolytic genes to allow cancer cells to survive under hypoxic conditions
UPDATE 6/11/2021
Bispecific Antibodies Emerging as Effective Cancer Therapeutics
Science 28 May 2021: Vol. 372, Issue 6545, pp. 916-917 DOI: 10.1126/science.abg1209
Bispecific antibodies (bsAbs) bind two different epitopes on the same or different antigens. Through this dual specificity for soluble or cell-surface antigens, bsAbs exert activities beyond those of natural antibodies, offering numerous opportunities for therapeutic applications. Although initially developed for retargeting T cells to tumors, with a first bsAb approved in 2009 (catumaxomab, withdrawn in 2017), exploring new modes of action opened the door to many additional applications beyond those of simply combining the activity of two different antibodies within one molecule. Examples include agonistic “assembly activities” that mimic the activity of natural ligands and cofactors (for example, factor VIII replacement in hemophilia A), inactivation of receptors or ligands, and delivery of payloads to cells or tissues or across biological barriers. Over the past years, the bsAb field transformed from early research to clinical applications and drugs. New developments offer a glimpse into the future promise of this exciting and rapidly progressing field.
Monoclonal antibodies (mAbs) comprise antigen-binding sites formed by the variable domains of the heavy and light chain and an Fc region that mediates immune responses. BsAbs, produced through genetic engineering, combine the antigen-binding sites of two different antibodies within one molecule, with a plethora of formats available (1). Conceptually, one can discriminate between bsAbs with combinatorial modes of action where the antigen-binding sites act independently from each other, and bsAbs with obligate modes of action where activity needs binding of both, either in a sequential (temporal) way or dependent on the physical (spatial) linkage of both (see the figure) (2). BsAbs approved as drugs are so far in the obligate dual-binding category: A T cell recruiter (blinatumomab) against cancer and a factor VIIIa mimetic to treat hemophilia A (emicizumab). Most but not all of the more than 100 bsAbs in clinical development address cancers. Some are in late stage (such as amivantamab, epcoritamab, faricimab, and KNO46), but most are still in early stages (2). Most of these entities enable effector cell retargeting to induce target cell destruction.
An increasing number of programs also explore alternative modes of action. This includes bsAbs that target pathways involved in tumor proliferation (such as amivantamab), invasion, ocular angiogenesis (such as faricimab), or immune regulation by blocking receptors and/or ligands, mainly in a combinatorial manner. Challenges for all of these entities are potential adverse effects, toxicity in normal tissues, and overshooting and systemic immune responses, especially with T cell retargeting or immune-modulating or activating entities. Such issues need to be carefully addressed.
Most of the bispecific T cell engagers comprise a binding site for a tumor-associated antigen and CD3 [a component of the T cell receptor (TCR) activation complex] as trigger molecule on T cells. To prevent or ameliorate “on-target, off-tumor” effects of T cell recruiters, approaches currently investigated include the modulation of target affinities and mechanisms to allow conditional activation upon target cell binding. Thus, a reduced affinity for CD3 increased tolerability by reducing peripheral cytokine concentrations that are associated with nonspecific or overshooting immune reactions (3). Similarly, reduced affinity for the target antigen was shown to ameliorate cytokine release and damage of target-expressing tissues (4). Tumor selectivity can be further increased by implementing avidity effects—for example, by using 2+1 bsAb formats with two low-affinity binding sites for target antigens and monovalent binding to CD3 (4).
In further approaches, binders to CD3 were identified that efficiently trigger target cell destruction without inducing undesired release of cytokines, demonstrating the importance of epitope specificity to potentially uncouple efficacy from cytokine release (5). Complementing these T cell–recruiting principles, the nonclassical T cell subset of γ9d2 T cells with strong cytotoxic activity emerged as potent effectors, which can be retargeted with bsAbs binding to the γ9d2 TCR. Thereby, global activation of all T cells, including inhibitory regulatory T cells (Treg cells), through CD3 binding, may be avoided (6). However, even these approaches might result in a narrow therapeutic window to treat solid tumors because of T cell activation in normal tissues.
Consequently, there are several approaches to conditionally activate T cells within tumors, including a local liberation of the CD3-binding sites or triggering local assembly of CD3-binding sites from two half-molecules. For example, CD3-binding sites have been masked by fusing antigen binding or blocking moieties—such as peptides, aptamers, or anti-idiotypic antibody fragments—to one or both variable domains. These moieties are released within the tumor by tumor-associated proteases, or through biochemical responses to hypoxia or low pH (7). This approach can also be applied to confer specific binding of antibody therapeutics, including bsAbs, to antigens on tumor cells (8).
An on-target restoration of CD3-binding sites requires application of two target-binding entities, each comprising parts of the CD3-binding site, which assemble into functional binding sites upon close binding of both half-antibodies. The feasibility of this approach was recently shown, for example, for a split T cell–engaging antibody derivative (Hemibody) that targets a cell surface antigen (9). Such approaches can also be applied to half-antibodies that recognize two different targets expressed on the same cell, further increasing tumor selectivity.
Regarding T cell engagers, increasing efforts are made to target not only cell-surface antigens expressed on tumor cells but also human leukocyte antigen (HLA)–presented tumor-specific peptides. This expands the target space of bsAbs toward tumor-specific intracellular antigens and can be achieved by using either recombinant TCRs or antibodies with TCR-like specificities combined with, for example, CD3-binding arms to engage T cell responses. A first TCR–anti-CD3 bispecific molecule is in phase I and II trials to treat metastatic melanoma (10). A challenge of this approach is the identification of TCRs or TCR-like antibodies that bind the peptide in the context of HLA with high affinity and specificity, without cross-reacting with related peptides to reduce or avoid off-target activities. Comprehensive screening tools and implementation of computational approaches are being developed to achieve this task.
A rapidly growing area of bsAbs in cancer therapy is their use to foster antitumor immune responses. Here, they are especially applied for dual inhibition of checkpoints that prevent immune responses—for example, programmed cell death protein 1 (PD-1) and its ligand (PD-L1), cytotoxic T lymphocyte–associated antigen 4 (CTLA-4), or lymphocyte activation gene 3 (LAG-3; for example, KNO46). Tumor-targeted bsAbs can also target costimulatory factors such as CD28 or 4-1BB ligand (4-1BBL) to enhance T cell responses when combined with PD-1 blockade or to provide an activity-enhancing costimulatory signal in combination with CD3-based bsAbs (11). Furthermore, bsAbs are being developed for local effects by targeting one arm to antigens that are expressed by tumor cells or cells of the tumor microenvironment (2).
Clinical application of bsAbs now expands to other therapeutic areas, including chronic inflammatory, autoimmune, and neurodegenerative diseases; vascular, ocular, and hematologic disorders; and infections. In contrast to mAbs, bsAbs can inactivate the signaling of different cytokines with one molecule to treat inflammatory diseases (12). Simultaneous dual-target binding is not essential to elicit activity for bsAbs against combinations of proinflammatory cytokines, such as tumor necrosis factor (TNF), interleukin-1α (IL-1α), IL-1β, IL-4, IL-13, IL-17, inducible T cell costimulator ligand (ICOSL), or B cell–activating factor (BAFF). This presumably also applies to blockade of immune cell receptors, although dual targeting might confer increased efficacy due to avidity effects and increased selectivity through simultaneous binding of two different receptors.
A further application of combinatorial dual targeting is in ophthalmology. Loss of vision in wet age-related macular degeneration (AMD) results from abnormal proliferation and leakiness of blood vessels in the macula. This can be treated with antibodies that bind and inactivate factors that stimulate their proliferation (13). In contrast to mAbs or fragments that recognize individual factors, bsAbs bind two such factors. For example, faricimab that binds vascular endothelial growth factor A (VEGF-A) and angiopoietin-2 (ANG2), demonstrated dual efficacy in preclinical studies, and is currently in phase 3 trials.
BsAbs with obligate modes of action that mandate simultaneous dual-target binding are “assemblers” that replace the function of factors necessary to form functional protein complexes. One of these bsAbs with an assembly role (emicizumab, approved in 2018) replaces factor VIIIa in the clotting cascade. Deficiency of factor VIII causes hemophilia A, which can be overcome by substitution with recombinant factor VIII. However, a proportion of patients develop factor VIII–neutralizing immune responses and no longer respond to therapy. To overcome this, a bsAb was developed with binding sites that recognize and physically connect factors IXa and X, a process normally mediated by factor VIIIa. Extensive screening of a large set of bsAbs was required to identify those that combine suitable epitopes with optimized affinities and geometry to serve as functional factor VIIIa mimetics (14). This exemplifies the complexity of identifying the best bsAb for therapeutic applications.
A mode of action requiring sequential binding of two targets is the transport of bsAbs across the blood-brain barrier (BBB). This is a tight barrier of brain capillary endothelial cells that controls the transport of substances between the blood and the cerebrospinal fluid—the brain parenchyma. Passage of large molecules, including antibodies, across the BBB is thereby restricted. Some proteins, such as transferrin or insulin, pass through the BBB by way of transporters on endothelial cells. Antibodies that bind these shuttle molecules, such as the transferrin receptor (TfR), can hitchhike across the BBB. BsAbs that recognize brain targets (such as β-amyloid for Alzheimer’s disease) and TfR with optimized affinities, epitopes, and formats can thereby enter the brain. Such bsAbs are currently in clinical evaluation to treat neurodegenerative diseases (15).
In the past years, there has been a transition from a technology-driven phase, solving hurdles to generate bsAbs with defined composition, toward exploring and extending the modes of action for new therapeutic options. The challenge of generating bsAbs is not only to identify suitable antigen pairs to be targeted in a combined manner. It is now recognized that the molecular composition has a profound impact on bsAb functionality (13). That more than 30 different bsAb formats are in clinical trials proves that development is now driven by a “fit for purpose” or “format defines function” rationale. Many candidates differ in their composition, affecting valency, geometry, flexibility, size, and half-life (1). Not all members of this “zoo of bsAb formats” qualify to become drugs. Strong emphasis is therefore on identifying candidates that exhibit drug-like properties and fulfill safety, developability, and manufacturability criteria. There is likely to be an exciting new wave of bsAb therapeutics available in the coming years.
UC Berkeley researchers have found a long-elusive Achilles’ heel within “triple-negative” breast tumors, a common type of breast cancer that is difficult to treat. The scientists then used a drug-like molecule to successfully target this vulnerability, killing cancer cells in the lab and shrinking tumors in mice.
“We were looking for targets that drive cancer metabolism in triple-negative breast cancer, and we found one that was very specific to this type of cancer,” said Daniel K. Nomura, an associate professor of chemistry and of nutritional sciences and toxicology at UC Berkeley and senior author for the study, which is published online ahead of print in Cell Chemical Biology.
Triple-negative breast cancers account for about one in five breast cancers, and they are deadlier than other forms of breast cancer, in part because no drugs have been developed to specifically target these tumors.
Triple-negative breast cancers do not rely on the hormones estrogen and progesterone for growth, nor on human epidermal growth factor receptor 2 (HER2). Because they do not depend on these three targets, they are not vulnerable to modern hormonal therapies or to the HER2-targeted drug Herceptin (trastuzumab).
Instead, oncologists treat triple-negative breast cancer with older chemotherapies that target all dividing cells. If triple-negative breast cancer spreads beyond the breast to distant sites within the body, an event called metastasis, there are few treatment options.
Tumor cells develop abnormal metabolism, which they rely on to get the energy boost they need to fuel their rapid growth. In their new study, the research team used an innovative approach to search for active enzymes that triple-negative breast cancers use differently for metabolism in comparison to other cells and even other tumors.
Inhibiting cancer metabolism
They discovered that cells from triple-negative breast cancer cells rely on vigorous activity by an enzyme called glutathione-S-transferase Pi1 (GSTP1). They showed that in cancer cells, GSTP1 regulates a type of metabolism called glycolysis, and that inhibition of GSTP1 impairs glycolytic metabolism in triple-negative cancer cells, starving them of energy, nutrients and signaling capability. Normal cells do not rely as much on this particular metabolic pathway to obtain usable chemical energy, but cells within many tumors heavily favor glycolysis.
Co-author Eranthie Weerapana, an associate professor of chemistry at Boston College, developed a molecule named LAS17 that tightly and irreversibly attaches to the target site on the GSTP1 molecule. By binding tightly to GSTP1, LAS17 inhibits activity of the enzyme. The researchers found that LAS17 was highly specific for GSTP1, and did not attach to other proteins in cells.
According to Nomura, LAS17 did not appear to have toxic side effects in mice, where it shrank tumors grown to an invasive stage from surgically transplanted, human, triple-negative breast cancer cells that had long been maintained in lab cultures.
The research team intends to continue studying LAS17, Nomura said, with the next step being to study tumor tissue resected from human triple-negative breast cancers and transplanted directly into mice.
“Inhibiting GSTP1 impairs glycolytic metabolism,” Nomura said. “More broadly, this inhibition starves triple-negative breast cancer cells, preventing them from making the macromolecules they need, including the lipids they need to make membranes and the nucleic acids they need to make DNA. It also prevents these cells from making enough ATP, the molecule that is the basic energy fuel for cells.”
Beyond the metabolic role they first sought to track down, GSTP1 also appears to aid signaling within triple-negative breast cancer cells, helping to spur tumor growth, the researchers found.
Technique identifies Achilles’ heels
Nomura said it was surprising that a single, unique target emerged from the research team’s search.
The method used by the researchers, called “reactivity-based chemoproteomics,” can quickly lead to specific targetable sites — the Achilles’ heels — on proteins of interest, and eventually to drug development strategies, Nomura said.
The approach is to search for protein targets that are actively functioning within cells, instead of first using the well-trod path of surveying all genes to identify the specific genes that have taken the first step toward protein production. With that more conventional strategy, the switching on, or “expression,” of genes is evidenced by the easily quantified molecule called messenger RNA, made by the cell from a gene’s DNA template.
Nomura’s team instead first used chemical probes that can react with certain configurations of two of the amino acid building blocks of protein — cysteine and lysine — known to be involved in several kinds of important structural and functional transitions that active proteins can undergo.
“A lot can happen after the first step in protein production, and we believe our method for identifying fully formed, active proteins is more useful for tracking down relevant differences in cellular physiology,” Nomura said.
The researchers analyzed and compared cells from five distinct triple-negative breast cancers that had been grown in cell cultures for generations, along with cells from four distinct breast cancers that were not triple negative.
The scientists used a chemical identification technique known as mass spectrometry to narrow down the set of proteins that had active lysines and cysteines to just those that were metabolic enzymes. Only then did they use the more conventional approach of measuring gene expression in the different cancer cell types.
GSTP1 was the only metabolically active enzyme that was specifically expressed only in triple-negative breast cancer cells compared to other breast cancer cell types, the researchers found. Separate analysis of databases of human breast cancer by UC San Francisco co-authors confirmed that GSTP1 is overexpressed in patients with triple-negative breast cancers in comparison to patients with other breast cancers.
In addition to Nomura and Weerapana, study authors included Sharon Louie, Elizabeth Grossman, Lucky Ding, Tucker Huffman and David Miyamoto, from UC Berkeley; Roman Camarda and Andrei Goga, from UC San Francisco, and Lisa Crawford, from Boston College. Study funders included the National Institutes of Health, the American Cancer Society, the U.S. Department of Defense, and the Searle Scholar Foundation.
Breast cancer metastasized to the liver (Courtesy of National Cancer Institute)
UC Berkeley researchers have found a long-elusive Achilles’ heel within “triple-negative” breast tumors, a common type of breast cancer that is difficult to treat. The scientists then used a drug-like molecule to successfully target this vulnerability, killing cancer cells in the lab and shrinking tumors in mice.
“We were looking for targets that drive cancer metabolism in triple-negative breast cancer, and we found one that was very specific to this type of cancer,” said Daniel K. Nomura, an associate professor of chemistry and of nutritional sciences and toxicology at UC Berkeley and senior author for the study, which is published online ahead of print on May 12 in Cell Chemical Biology.
Triple-negative breast cancers account for about one in five breast cancers, and they are deadlier than other forms of breast cancer, in part because no drugs have been developed to specifically target these tumors.
Triple-negative breast cancers do not rely on the hormones estrogen and progesterone for growth, nor on human epidermal growth factor receptor 2 (HER2). Because they do not depend on these three targets, they are not vulnerable to modern hormonal therapies or to the HER2-targeted drug Herceptin (trastuzumab).
Instead, oncologists treat triple-negative breast cancer with older chemotherapies that target all dividing cells. If triple-negative breast cancer spreads beyond the breast to distant sites within the body, an event called metastasis, there are few treatment options.
Tumor cells develop abnormal metabolism, which they rely on to get the energy boost they need to fuel their rapid growth. In their new study, the research team used an innovative approach to search for active enzymes that triple-negative breast cancers use differently for metabolism in comparison to other cells and even other tumors.
Inhibiting cancer metabolism
They discovered that cells from triple-negative breast cancer cells rely on vigorous activity by an enzyme called glutathione-S-transferase Pi1 (GSTP1). They showed that in cancer cells, GSTP1 regulates a type of metabolism called glycolysis, and that inhibition of GSTP1 impairs glycolytic metabolism in triple-negative cancer cells, starving them of energy, nutrients and signaling capability. Normal cells do not rely as much on this particular metabolic pathway to obtain usable chemical energy, but cells within many tumors heavily favor glycolysis.
•We used chemoproteomics to profile metabolic drivers of breast cancer
•GSTP1 is a novel triple-negative breast cancer-specific target
•GSTP1 inhibition impairs triple-negative breast cancer pathogenicity
•GSTP1 inhibition impairs GAPDH activity to affect metabolism and signaling
Breast cancers possess fundamentally altered metabolism that fuels their pathogenicity. While many metabolic drivers of breast cancers have been identified, the metabolic pathways that mediate breast cancer malignancy and poor prognosis are less well understood. Here, we used a reactivity-based chemoproteomic platform to profile metabolic enzymes that are enriched in breast cancer cell types linked to poor prognosis, including triple-negative breast cancer (TNBC) cells and breast cancer cells that have undergone an epithelial-mesenchymal transition-like state of heightened malignancy. We identified glutathione S-transferase Pi 1 (GSTP1) as a novel TNBC target that controls cancer pathogenicity by regulating glycolytic and lipid metabolism, energetics, and oncogenic signaling pathways through a protein interaction that activates glyceraldehyde-3-phosphate dehydrogenase activity. We show that genetic or pharmacological inactivation of GSTP1 impairs cell survival and tumorigenesis in TNBC cells. We put forth GSTP1 inhibitors as a novel therapeutic strategy for combatting TNBCs through impairing key cancer metabolism and signaling pathways.
Disease related changes in proteomics, protein folding, protein-protein interaction, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Disease related changes in proteomics, protein folding, protein-protein interaction
Curator: Larry H. Bernstein, MD, FCAP
LPBI
Frankenstein Proteins Stitched Together by Scientists
The Frankenstein monster, stitched together from disparate body parts, proved to be an abomination, but stitched together proteins may fare better. They may, for example, serve specific purposes in medicine, research, and industry. At least, that’s the ambition of scientists based at the University of North Carolina. They have developed a computational protocol called SEWING that builds new proteins from connected or disconnected pieces of existing structures. [Wikipedia]
Unlike Victor Frankenstein, who betrayed Promethean ambition when he sewed together his infamous creature, today’s biochemists are relatively modest. Rather than defy nature, they emulate it. For example, at the University of North Carolina (UNC), researchers have taken inspiration from natural evolutionary mechanisms to develop a technique called SEWING—Structure Extension With Native-substructure Graphs. SEWING is a computational protocol that describes how to stitch together new proteins from connected or disconnected pieces of existing structures.
“We can now begin to think about engineering proteins to do things that nothing else is capable of doing,” said UNC’s Brian Kuhlman, Ph.D. “The structure of a protein determines its function, so if we are going to learn how to design new functions, we have to learn how to design new structures. Our study is a critical step in that direction and provides tools for creating proteins that haven’t been seen before in nature.”
Traditionally, researchers have used computational protein design to recreate in the laboratory what already exists in the natural world. In recent years, their focus has shifted toward inventing novel proteins with new functionality. These design projects all start with a specific structural “blueprint” in mind, and as a result are limited. Dr. Kuhlman and his colleagues, however, believe that by removing the limitations of a predetermined blueprint and taking cues from evolution they can more easily create functional proteins.
Dr. Kuhlman’s UNC team developed a protein design approach that emulates natural mechanisms for shuffling tertiary structures such as pleats, coils, and furrows. Putting the approach into action, the UNC team mapped 50,000 stitched together proteins on the computer, and then it produced 21 promising structures in the laboratory. Details of this work appeared May 6 in the journal Science, in an article entitled, “Design of Structurally Distinct Proteins Using Strategies Inspired by Evolution.”
“Helical proteins designed with SEWING contain structural features absent from other de novo designed proteins and, in some cases, remain folded at more than 100°C,” wrote the authors. “High-resolution structures of the designed proteins CA01 and DA05R1 were solved by x-ray crystallography (2.2 angstrom resolution) and nuclear magnetic resonance, respectively, and there was excellent agreement with the design models.”
Essentially, the UNC scientists confirmed that the proteins they had synthesized contained the unique structural varieties that had been designed on the computer. The UNC scientists also determined that the structures they had created had new surface and pocket features. Such features, they noted, provide potential binding sites for ligands or macromolecules.
“We were excited that some had clefts or grooves on the surface, regions that naturally occurring proteins use for binding other proteins,” said the Science article’s first author, Tim M. Jacobs, Ph.D., a former graduate student in Dr. Kuhlman’s laboratory. “That’s important because if we wanted to create a protein that can act as a biosensor to detect a certain metabolite in the body, either for diagnostic or research purposes, it would need to have these grooves. Likewise, if we wanted to develop novel therapeutics, they would also need to attach to specific proteins.”
Currently, the UNC researchers are using SEWING to create proteins that can bind to several other proteins at a time. Many of the most important proteins are such multitaskers, including the blood protein hemoglobin.
Histone Mutation Deranges DNA Methylation to Cause Cancer
In some cancers, including chondroblastoma and a rare form of childhood sarcoma, a mutation in histone H3 reduces global levels of methylation (dark areas) in tumor cells but not in normal cells (arrowhead). The mutation locks the cells in a proliferative state to promote tumor development. [Laboratory of Chromatin Biology and Epigenetics at The Rockefeller University]
They have been called oncohistones, the mutated histones that are known to accompany certain pediatric cancers. Despite their suggestive moniker, oncohistones have kept their oncogenic secrets. For example, it has been unclear whether oncohistones are able to cause cancer on their own, or whether they need to act in concert with additional DNA mutations, that is, mutations other than those affecting histone structures.
While oncohistone mechanisms remain poorly understood, this particular question—the oncogenicity of lone oncohistones—has been resolved, at least in part. According to researchers based at The Rockefeller University, a change to the structure of a histone can trigger a tumor on its own.
This finding appeared May 13 in the journal Science, in an article entitled, “Histone H3K36 Mutations Promote Sarcomagenesis Through Altered Histone Methylation Landscape.” The article describes the Rockefeller team’s study of a histone protein called H3, which has been found in about 95% of samples of chondoblastoma, a benign tumor that arises in cartilage, typically during adolescence.
The Rockefeller scientists found that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo.
After the scientists inserted the H3 histone mutation into mouse mesenchymal progenitor cells (MPCs)—which generate cartilage, bone, and fat—they watched these cells lose the ability to differentiate in the lab. Next, the scientists injected the mutant cells into living mice, and the animals developed the tumors rich in MPCs, known as an undifferentiated sarcoma. Finally, the researchers tried to understand how the mutation causes the tumors to develop.
The scientists determined that H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases.
“Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation,” the authors of the Science study wrote. “After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation.”
Essentially, when the H3K36M mutation occurs, the cell becomes locked in a proliferative state—meaning it divides constantly, leading to tumors. Specifically, the mutation inhibits enzymes that normally tag the histone with chemical groups known as methyls, allowing genes to be expressed normally.
In response to this lack of modification, another part of the histone becomes overmodified, or tagged with too many methyl groups. “This leads to an overall resetting of the landscape of chromatin, the complex of DNA and its associated factors, including histones,” explained co-author Peter Lewis, Ph.D., a professor at the University of Wisconsin-Madison and a former postdoctoral fellow in laboratory of C. David Allis, Ph.D., a professor at Rockefeller.
The finding—that a “resetting” of the chromatin landscape can lock the cell into a proliferative state—suggests that researchers should be on the hunt for more mutations in histones that might be driving tumors. For their part, the Rockefeller researchers are trying to learn more about how this specific mutation in histone H3 causes tumors to develop.
“We want to know which pathways cause the mesenchymal progenitor cells that carry the mutation to continue to divide, and not differentiate into the bone, fat, and cartilage cells they are destined to become,” said co-author Chao Lu, Ph.D., a postdoctoral fellow in the Allis lab.
Once researchers understand more about these pathways, added Dr. Lewis, they can consider ways of blocking them with drugs, particularly in tumors such as MPC-rich sarcomas—which, unlike chondroblastoma, can be deadly. In fact, drugs that block these pathways may already exist and may even be in use for other types of cancers.
“One long-term goal of our collaborative team is to better understand fundamental mechanisms that drive these processes, with the hope of providing new therapeutic approaches,” concluded Dr. Allis.
Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape
Missense mutations (that change one amino acid for another) in histone H3 can produce a so-called oncohistone and are found in a number of pediatric cancers. For example, the lysine-36–to-methionine (K36M) mutation is seen in almost all chondroblastomas. Lu et al. show that K36M mutant histones are oncogenic, and they inhibit the normal methylation of this same residue in wild-type H3 histones. The mutant histones also interfere with the normal development of bone-related cells and the deposition of inhibitory chromatin marks.
Several types of pediatric cancers reportedly contain high-frequency missense mutations in histone H3, yet the underlying oncogenic mechanism remains poorly characterized. Here we report that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo. H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases. Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation. After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation. Our findings are mirrored in human undifferentiated sarcomas in which novel K36M/I mutations in H3.1 are identified.
Mitochondria? We Don’t Need No Stinking Mitochondria!
Diagram comparing typical eukaryotic cell to the newly discovered mitochondria-free organism. [Karnkowska et al., 2016, Current Biology 26, 1–11]
The organelle that produces a significant portion of energy for eukaryotic cells would seemingly be indispensable, yet over the years, a number of organisms have been discovered that challenge that biological pretense. However, these so-called amitochondrial species may lack a defined organelle, but they still retain some residual functions of their mitochondria-containing brethren. Even the intestinal eukaryotic parasite Giardia intestinalis, which was for many years considered to be mitochondria-free, was proven recently to contain a considerably shriveled version of the organelle.
Now, an international group of scientists has released results from a new study that challenges the notion that mitochondria are essential for eukaryotes—discovering an organism that resides in the gut of chinchillas that contains absolutely no trace of mitochondria at all.
“In low-oxygen environments, eukaryotes often possess a reduced form of the mitochondrion, but it was believed that some of the mitochondrial functions are so essential that these organelles are indispensable for their life,” explained lead study author Anna Karnkowska, Ph.D., visiting scientist at the University of British Columbia in Vancouver. “We have characterized a eukaryotic microbe which indeed possesses no mitochondrion at all.”
Mysterious Eukaryote Missing Mitochondria
Researchers uncover the first example of a eukaryotic organism that lacks the organelles.
Monocercomonoides sp. PA203VLADIMIR HAMPL, CHARLES UNIVERSITY, PRAGUE, CZECH REPUBLIC
Scientists have long thought that mitochondria—organelles responsible for energy generation—are an essential and defining feature of a eukaryotic cell. Now, researchers from Charles University in Prague and their colleagues are challenging this notion with their discovery of a eukaryotic organism,Monocercomonoides species PA203, which lacks mitochondria. The team’s phylogenetic analysis, published today (May 12) in Current Biology,suggests that Monocercomonoides—which belong to the Oxymonadida group of protozoa and live in low-oxygen environments—did have mitochondria at one point, but eventually lost the organelles.
“This is quite a groundbreaking discovery,” said Thijs Ettema, who studies microbial genome evolution at Uppsala University in Sweden and was not involved in the work.
“This study shows that mitochondria are not so central for all lineages of living eukaryotes,” Toni Gabaldonof the Center for Genomic Regulation in Barcelona, Spain, who also was not involved in the work, wrote in an email to The Scientist. “Yet, this mitochondrial-devoid, single-cell eukaryote is as complex as other eukaryotic cells in almost any other aspect of cellular complexity.”
Charles University’s Vladimir Hampl studies the evolution of protists. Along with Anna Karnkowska and colleagues, Hampl decided to sequence the genome of Monocercomonoides, a little-studied protist that lives in the digestive tracts of vertebrates. The 75-megabase genome—the first of an oxymonad—did not contain any conserved genes found on mitochondrial genomes of other eukaryotes, the researchers found. It also did not contain any nuclear genes associated with mitochondrial functions.
“It was surprising and for a long time, we didn’t believe that the [mitochondria-associated genes were really not there]. We thought we were missing something,” Hampl told The Scientist. “But when the data kept accumulating, we switched to the hypothesis that this organism really didn’t have mitochondria.”
Because researchers have previously not found examples of eukaryotes without some form of mitochondria, the current theory of the origin of eukaryotes poses that the appearance of mitochondria was crucial to the identity of these organisms.
“We now view these mitochondria-like organelles as a continuum from full mitochondria to very small . Some anaerobic protists, for example, have only pared down versions of mitochondria, such as hydrogenosomes and mitosomes, which lack a mitochondrial genome. But these mitochondrion-like organelles perform essential functions of the iron-sulfur cluster assembly pathway, which is known to be conserved in virtually all eukaryotic organisms studied to date.
Yet, in their analysis, the researchers found no evidence of the presence of any components of this mitochondrial pathway.
Like the scaling down of mitochondria into mitosomes in some organisms, the ancestors of modernMonocercomonoides once had mitochondria. “Because this organism is phylogenetically nested among relatives that had conventional mitochondria, this is most likely a secondary adaptation,” said Michael Gray, a biochemist who studies mitochondria at Dalhousie University in Nova Scotia and was not involved in the study. According to Gray, the finding of a mitochondria-deficient eukaryote does not mean that the organelles did not play a major role in the evolution of eukaryotic cells.
To be sure they were not missing mitochondrial proteins, Hampl’s team also searched for potential mitochondrial protein homologs of other anaerobic species, and for signature sequences of a range of known mitochondrial proteins. While similar searches with other species uncovered a few mitochondrial proteins, the team’s analysis of Monocercomonoides came up empty.
“The data is very complete,” said Ettema. “It is difficult to prove the absence of something but [these authors] do a convincing job.”
To form the essential iron-sulfur clusters, the team discovered that Monocercomonoides use a sulfur mobilization system found in the cytosol, and that an ancestor of the organism acquired this system by lateral gene transfer from bacteria. This cytosolic, compensating system allowed Monocercomonoides to lose the otherwise essential iron-sulfur cluster-forming pathway in the mitochondrion, the team proposed.
“This work shows the great evolutionary plasticity of the eukaryotic cell,” said Karnkowska, who participated in the study while she was a postdoc at Charles University. Karnkowska, who is now a visiting researcher at the University of British Columbia in Canada, added: “This is a striking example of how far the evolution of a eukaryotic cell can go that was beyond our expectations.”
“The results highlight how many surprises may await us in the poorly studied eukaryotic phyla that live in under-explored environments,” Gabaldon said.
Ettema agreed. “Now that we’ve found one, we need to look at the bigger picture and see if there are other examples of eukaryotes that have lost their mitochondria, to understand how adaptable eukaryotes are.”
Karnkowska et al., “A eukaryote without a mitochondrial organelle,” Current Biology,doi:10.1016/j.cub.2016.03.053, 2016.
•Monocercomonoides sp. is a eukaryotic microorganism with no mitochondria
•The complete absence of mitochondria is a secondary loss, not an ancestral feature
•The essential mitochondrial ISC pathway was replaced by a bacterial SUF system
The presence of mitochondria and related organelles in every studied eukaryote supports the view that mitochondria are essential cellular components. Here, we report the genome sequence of a microbial eukaryote, the oxymonad Monocercomonoides sp., which revealed that this organism lacks all hallmark mitochondrial proteins. Crucially, the mitochondrial iron-sulfur cluster assembly pathway, thought to be conserved in virtually all eukaryotic cells, has been replaced by a cytosolic sulfur mobilization system (SUF) acquired by lateral gene transfer from bacteria. In the context of eukaryotic phylogeny, our data suggest that Monocercomonoides is not primitively amitochondrial but has lost the mitochondrion secondarily. This is the first example of a eukaryote lacking any form of a mitochondrion, demonstrating that this organelle is not absolutely essential for the viability of a eukaryotic cell.
This method catches a bait protein together with its associated protein partners in virus-like particles that are budded from human cells. Like this, cell lysis is not needed and protein complexes are preserved during purification.
With his feet in both a proteomics lab and an interactomics lab, VIB/UGent professor Sven Eyckerman is well aware of the shortcomings of conventional approaches to analyze protein complexes. The lysis conditions required in mass spectrometry–based strategies to break open cell membranes often affect protein-protein interactions. “The first step in a classical study on protein complexes essentially turns the highly organized cellular structure into a big messy soup”, Eyckerman explains.
Inspired by virus biology, Eyckerman came up with a creative solution. “We used the natural process of HIV particle formation to our benefit by hacking a completely safe form of the virus to abduct intact protein machines from the cell.” It is well known that the HIV virus captures a number of host proteins during its particle formation. By fusing a bait protein to the HIV-1 GAG protein, interaction partners become trapped within virus-like particles that bud from mammalian cells. Standard proteomic approaches are used next to reveal the content of these particles. Fittingly, the team named the method ‘Virotrap’.
The Virotrap approach is exceptional as protein networks can be characterized under natural conditions. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. The researchers showed the method was suitable for detection of known binary interactions as well as mass spectrometry-based identification of novel protein partners.
Virotrap is a textbook example of bringing research teams with complementary expertise together. Cross-pollination with the labs of Jan Tavernier (VIB/UGent) and Kris Gevaert (VIB/UGent) enabled the development of this platform.
Jan Tavernier: “Virotrap represents a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. It is complementary to the arsenal of existing interactomics methods, but also holds potential for other fields, like drug target characterization. We also developed a small molecule-variant of Virotrap that could successfully trap protein partners for small molecule baits.”
Kris Gevaert: “Virotrap can also impact our understanding of disease pathways. We were actually surprised to see that this virus-based system could be used to study antiviral pathways, like Toll-like receptor signaling. Understanding these protein machines in their natural environment is essential if we want to modulate their activity in pathology.“
Trapping mammalian protein complexes in viral particles
Cell lysis is an inevitable step in classical mass spectrometry–based strategies to analyse protein complexes. Complementary lysis conditions, in situ cross-linking strategies and proximal labelling techniques are currently used to reduce lysis effects on the protein complex. We have developed Virotrap, a viral particle sorting approach that obviates the need for cell homogenization and preserves the protein complexes during purification. By fusing a bait protein to the HIV-1 GAG protein, we show that interaction partners become trapped within virus-like particles (VLPs) that bud from mammalian cells. Using an efficient VLP enrichment protocol, Virotrap allows the detection of known binary interactions and MS-based identification of novel protein partners as well. In addition, we show the identification of stimulus-dependent interactions and demonstrate trapping of protein partners for small molecules. Virotrap constitutes an elegant complementary approach to the arsenal of methods to study protein complexes.
Proteins mostly exert their function within supramolecular complexes. Strategies for detecting protein–protein interactions (PPIs) can be roughly divided into genetic systems1 and co-purification strategies combined with mass spectrometry (MS) analysis (for example, AP–MS)2. The latter approaches typically require cell or tissue homogenization using detergents, followed by capture of the protein complex using affinity tags3 or specific antibodies4. The protein complexes extracted from this ‘soup’ of constituents are then subjected to several washing steps before actual analysis by trypsin digestion and liquid chromatography–MS/MS analysis. Such lysis and purification protocols are typically empirical and have mostly been optimized using model interactions in single labs. In fact, lysis conditions can profoundly affect the number of both specific and nonspecific proteins that are identified in a typical AP–MS set-up. Indeed, recent studies using the nuclear pore complex as a model protein complex describe optimization of purifications for the different proteins in the complex by examining 96 different conditions5. Nevertheless, for new purifications, it remains hard to correctly estimate the loss of factors in a standard AP–MS experiment due to washing and dilution effects during treatments (that is, false negatives). These considerations have pushed the concept of stabilizing PPIs before the actual homogenization step. A classical approach involves cross-linking with simple reagents (for example, formaldehyde) or with more advanced isotope-labelled cross-linkers (reviewed in ref. 2). However, experimental challenges such as cell permeability and reactivity still preclude the widespread use of cross-linking agents. Moreover, MS-generated spectra of cross-linked peptides are notoriously difficult to identify correctly. A recent lysis-independent solution involves the expression of a bait protein fused to a promiscuous biotin ligase, which results in labelling of proteins proximal to the activity of the enzyme-tagged bait protein6. When compared with AP–MS, this BioID approach delivers a complementary set of candidate proteins, including novel interaction partners7, 8. Such particular studies clearly underscore the need for complementary approaches in the co-complex strategies.
The evolutionary stress on viruses promoted highly condensed coding of information and maximal functionality for small genomes. Accordingly, for HIV-1 it is sufficient to express a single protein, the p55 GAG protein, for efficient production of virus-like particles (VLPs) from cells9, 10. This protein is highly mobile before its accumulation in cholesterol-rich regions of the membrane, where multimerization initiates the budding process11. A total of 4,000–5,000 GAG molecules is required to form a single particle of about 145 nm (ref. 12). Both VLPs and mature viruses contain a number of host proteins that are recruited by binding to viral proteins. These proteins can either contribute to the infectivity (for example, Cyclophilin/FKBPA13) or act as antiviral proteins preventing the spreading of the virus (for example, APOBEC proteins14).
We here describe the development and application of Virotrap, an elegant co-purification strategy based on the trapping of a bait protein together with its associated protein partners in VLPs that are budded from the cell. After enrichment, these particles can be analysed by targeted (for example, western blotting) or unbiased approaches (MS-based proteomics). Virotrap allows detection of known binary PPIs, analysis of protein complexes and their dynamics, and readily detects protein binders for small molecules.
Concept of the Virotrap system
Classical AP–MS approaches rely on cell homogenization to access protein complexes, a step that can vary significantly with the lysis conditions (detergents, salt concentrations, pH conditions and so on)5. To eliminate the homogenization step in AP–MS, we reasoned that incorporation of a protein complex inside a secreted VLP traps the interaction partners under native conditions and protects them during further purification. We thus explored the possibility of protein complex packaging by the expression of GAG-bait protein chimeras (Fig. 1) as expression of GAG results in the release of VLPs from the cells9, 10. As a first PPI pair to evaluate this concept, we selected the HRAS protein as a bait combined with the RAF1 prey protein. We were able to specifically detect the HRAS–RAF1 interaction following enrichment of VLPs via ultracentrifugation (Supplementary Fig. 1a). To prevent tedious ultracentrifugation steps, we designed a novel single-step protocol wherein we co-express the vesicular stomatitis virus glycoprotein (VSV-G) together with a tagged version of this glycoprotein in addition to the GAG bait and prey. Both tagged and untagged VSV-G proteins are probably presented as trimers on the surface of the VLPs, allowing efficient antibody-based recovery from large volumes. The HRAS–RAF1 interaction was confirmed using this single-step protocol (Supplementary Fig. 1b). No associations with unrelated bait or prey proteins were observed for both protocols.
Figure 1: Schematic representation of the Virotrap strategy.
Expression of a GAG-bait fusion protein (1) results in submembrane multimerization (2) and subsequent budding of VLPs from cells (3). Interaction partners of the bait protein are also trapped within these VLPs and can be identified after purification by western blotting or MS analysis (4).
Virotrap for the detection of binary interactions
We next explored the reciprocal detection of a set of PPI pairs, which were selected based on published evidence and cytosolic localization15. After single-step purification and western blot analysis, we could readily detect reciprocal interactions between CDK2 and CKS1B, LCP2 and GRAP2, and S100A1 and S100B (Fig. 2a). Only for the LCP2 prey we observed nonspecific association with an irrelevant bait construct. However, the particle levels of the GRAP2 bait were substantially lower as compared with those of the GAG control construct (GAG protein levels in VLPs; Fig. 2a, second panel of the LCP2 prey). After quantification of the intensities of bait and prey proteins and normalization of prey levels using bait levels, we observed a strong enrichment for the GAG-GRAP2 bait (Supplementary Fig. 2).
…..
Virotrap for unbiased discovery of novel interactions
For the detection of novel interaction partners, we scaled up VLP production and purification protocols (Supplementary Fig. 5 and Supplementary Note 1 for an overview of the protocol) and investigated protein partners trapped using the following bait proteins: Fas-associated via death domain (FADD), A20 (TNFAIP3), nuclear factor-κB (NF-κB) essential modifier (IKBKG), TRAF family member-associated NF-κB activator (TANK), MYD88 and ring finger protein 41 (RNF41). To obtain specific interactors from the lists of identified proteins, we challenged the data with a combined protein list of 19 unrelated Virotrap experiments (Supplementary Table 1 for an overview). Figure 3 shows the design and the list of candidate interactors obtained after removal of all proteins that were found in the 19 control samples (including removal of proteins from the control list identified with a single peptide). The remaining list of confident protein identifications (identified with at least two peptides in at least two biological repeats) reveals both known and novel candidate interaction partners. All candidate interactors including single peptide protein identifications are given in Supplementary Data 2 and also include recurrent protein identifications of known interactors based on a single peptide; for example, CASP8 for FADD and TANK for NEMO. Using alternative methods, we confirmed the interaction between A20 and FADD, and the associations with transmembrane proteins (insulin receptor and insulin-like growth factor receptor 1) that were captured using RNF41 as a bait (Supplementary Fig. 6). To address the use of Virotrap for the detection of dynamic interactions, we activated the NF-κB pathway via the tumour necrosis factor (TNF) receptor (TNFRSF1A) using TNFα (TNF) and performed Virotrap analysis using A20 as bait (Fig. 3). This resulted in the additional enrichment of receptor-interacting kinase (RIPK1), TNFR1-associated via death domain (TRADD), TNFRSF1A and TNF itself, confirming the expected activated complex20.
Figure 3: Use of Virotrap for unbiased interactome analysis
Lysis conditions used in AP–MS strategies are critical for the preservation of protein complexes. A multitude of lysis conditions have been described, culminating in a recent report where protein complex stability was assessed under 96 lysis/purification protocols5. Moreover, the authors suggest to optimize the conditions for every complex, implying an important workload for researchers embarking on protein complex analysis using classical AP–MS. As lysis results in a profound change of the subcellular context and significantly alters the concentration of proteins, loss of complex integrity during a classical AP–MS protocol can be expected. A clear evolution towards ‘lysis-independent’ approaches in the co-complex analysis field is evident with the introduction of BioID6 and APEX25 where proximal proteins, including proteins residing in the complex, are labelled with biotin by an enzymatic activity fused to a bait protein. A side-by-side comparison between classical AP–MS and BioID showed overlapping and unique candidate binding proteins for both approaches7, 8, supporting the notion that complementary methods are needed to provide a comprehensive view on protein complexes. This has also been clearly demonstrated for binary approaches15 and is a logical consequence of the heterogenic nature underlying PPIs (binding mechanism, requirement for posttranslational modifications, location, affinity and so on).
In this report, we explore an alternative, yet complementary method to isolate protein complexes without interfering with cellular integrity. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. This constitutes a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. A comparison of our Virotrap approach with AP–MS shows complementary data, with specific false positives and false negatives for both methods (Supplementary Fig. 7).
The current implementation of the Virotrap platform implies the use of a GAG-bait construct resulting in considerable expression of the bait protein. Different strategies are currently pursued to reduce bait expression including co-expression of a native GAG protein together with the GAG-bait protein, not only reducing bait expression but also creating more ‘space’ in the particles potentially accommodating larger bait protein complexes. Nevertheless, the presence of the bait on the forming GAG scaffold creates an intracellular affinity matrix (comparable to the early in vitro affinity columns for purification of interaction partners from lysates26) that has the potential to compete with endogenous complexes by avidity effects. This avidity effect is a powerful mechanism that aids in the recruitment of cyclophilin to GAG27, a well-known weak interaction (Kd=16 μM (ref. 28)) detectable as a background association in the Virotrap system. Although background binding may be increased by elevated bait expression, weaker associations are readily detectable (for example, MAL—MYD88-binding study; Fig. 2c).
The size of Virotrap particles (around 145 nm) suggests limitations in the size of the protein complex that can be accommodated in the particles. Further experimentation is required to define the maximum size of proteins or the number of protein complexes that can be trapped inside the particles.
….
In conclusion, Virotrap captures significant parts of known interactomes and reveals new interactions. This cell lysis-free approach purifies protein complexes under native conditions and thus provides a powerful method to complement AP–MS or other PPI data. Future improvements of the system include strategies to reduce bait expression to more physiological levels and application of advanced data analysis options to filter out background. These developments can further aid in the deployment of Virotrap as a powerful extension of the current co-complex technology arsenal.
New Autism Blood Biomarker Identified
Researchers at UT Southwestern Medical Center have identified a blood biomarker that may aid in earlier diagnosis of children with autism spectrum disorder, or ASD
In a recent edition of Scientific Reports, UT Southwestern researchers reported on the identification of a blood biomarker that could distinguish the majority of ASD study participants versus a control group of similar age range. In addition, the biomarker was significantly correlated with the level of communication impairment, suggesting that the blood test may give insight into ASD severity.
“Numerous investigators have long sought a biomarker for ASD,” said Dr. Dwight German, study senior author and Professor of Psychiatry at UT Southwestern. “The blood biomarker reported here along with others we are testing can represent a useful test with over 80 percent accuracy in identifying ASD.”
ASD1 – was 66 percent accurate in diagnosing ASD. When combined with thyroid stimulating hormone level measurements, the ASD1-binding biomarker was 73 percent accurate at diagnosis
A Search for Blood Biomarkers for Autism: Peptoids
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impairments in social interaction and communication, and restricted, repetitive patterns of behavior. In order to identify individuals with ASD and initiate interventions at the earliest possible age, biomarkers for the disorder are desirable. Research findings have identified widespread changes in the immune system in children with autism, at both systemic and cellular levels. In an attempt to find candidate antibody biomarkers for ASD, highly complex libraries of peptoids (oligo-N-substituted glycines) were screened for compounds that preferentially bind IgG from boys with ASD over typically developing (TD) boys. Unexpectedly, many peptoids were identified that preferentially bound IgG from TD boys. One of these peptoids was studied further and found to bind significantly higher levels (>2-fold) of the IgG1 subtype in serum from TD boys (n = 60) compared to ASD boys (n = 74), as well as compared to older adult males (n = 53). Together these data suggest that ASD boys have reduced levels (>50%) of an IgG1 antibody, which resembles the level found normally with advanced age. In this discovery study, the ASD1 peptoid was 66% accurate in predicting ASD.
….
Peptoid libraries have been used previously to search for autoantibodies for neurodegenerative diseases19 and for systemic lupus erythematosus (SLE)21. In the case of SLE, peptoids were identified that could identify subjects with the disease and related syndromes with moderate sensitivity (70%) and excellent specificity (97.5%). Peptoids were used to measure IgG levels from both healthy subjects and SLE patients. Binding to the SLE-peptoid was significantly higher in SLE patients vs. healthy controls. The IgG bound to the SLE-peptoid was found to react with several autoantigens, suggesting that the peptoids are capable of interacting with multiple, structurally similar molecules. These data indicate that IgG binding to peptoids can identify subjects with high levels of pathogenic autoantibodies vs. a single antibody.
In the present study, the ASD1 peptoid binds significantly lower levels of IgG1 in ASD males vs. TD males. This finding suggests that the ASD1 peptoid recognizes antibody(-ies) of an IgG1 subtype that is (are) significantly lower in abundance in the ASD males vs. TD males. Although a previous study14 has demonstrated lower levels of plasma IgG in ASD vs. TD children, here, we additionally quantified serum IgG levels in our individuals and found no difference in IgG between the two groups (data not shown). Furthermore, our IgG levels did not correlate with ASD1 binding levels, indicating that ASD1 does not bind IgG generically, and that the peptoid’s ability to differentiate between ASD and TD males is related to a specific antibody(-ies).
ASD subjects underwent a diagnostic evaluation using the ADOS and ADI-R, and application of the DSM-IV criteria prior to study inclusion. Only those subjects with a diagnosis of Autistic Disorder were included in the study. The ADOS is a semi-structured observation of a child’s behavior that allows examiners to observe the three core domains of ASD symptoms: reciprocal social interaction, communication, and restricted and repetitive behaviors1. When ADOS subdomain scores were compared with peptoid binding, the only significant relationship was with Social Interaction. However, the positive correlation would suggest that lower peptoid binding is associated with better social interaction, not poorer social interaction as anticipated.
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
It is interesting that in serum samples from older men, the ASD1 binding is similar to that in the ASD boys. This is consistent with the observation that with aging there is a reduction in the strength of the immune system, and the changes are gender-specific25. Recent studies using parabiosis26, in which blood from young mice reverse age-related impairments in cognitive function and synaptic plasticity in old mice, reveal that blood constituents from young subjects may contain important substances for maintaining neuronal functions. Work is in progress to identify the antibody/antibodies that are differentially binding to the ASD1 peptoid, which appear as a single band on the electrophoresis gel (Fig. 4).
……..
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
Titration of IgG binding to ASD1 using serum pooled from 10 TD males and 10 ASD males demonstrates ASD1’s ability to differentiate between the two groups. (B)Detecting IgG1 subclass instead of total IgG amplifies this differentiation. (C) IgG1 binding of individual ASD (n=74) and TD (n=60) male serum samples (1:100 dilution) to ASD1 significantly differs with TD>ASD. In addition, IgG1 binding of older adult male (AM) serum samples (n=53) to ASD1 is significantly lower than TD males, and not different from ASD males. The three groups were compared with a Kruskal-Wallis ANOVA, H = 10.1781, p<0.006. **p<0.005. Error bars show SEM. (D) Receiver-operating characteristic curve for ASD1’s ability to discriminate between ASD and TD males.
Association between peptoid binding and ADOS and ADI-R subdomains
Higher scores in any domain on the ADOS and ADI-R are indicative of more abnormal behaviors and/or symptoms. Among ADOS subdomains, there was no significant relationship between Communication and peptoid binding (z = 0.04, p = 0.966), Communication + Social interaction (z = 1.53, p = 0.127), or Stereotyped Behaviors and Restrictive Interests (SBRI) (z = 0.46, p = 0.647). Higher scores on the Social Interaction domain were significantly associated with higher peptoid binding (z = 2.04, p = 0.041).
Among ADI-R subdomains, higher scores on the Communication domain were associated with lower levels of peptoid binding (z = −2.28, p = 0.023). There was not a significant relationship between Social Interaction (z = 0.07, p = 0.941) or Restrictive/Repetitive Stereotyped Behaviors (z = −1.40, p = 0.162) and peptoid binding.
Computational Model Finds New Protein-Protein Interactions
Researchers at University of Pittsburgh have discovered 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia.
Using a computational model they developed, researchers at the University of Pittsburgh School of Medicine have discovered more than 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia. The findings, published online in npj Schizophrenia, a Nature Publishing Group journal, could lead to greater understanding of the biological underpinnings of this mental illness, as well as point the way to treatments.
There have been many genome-wide association studies (GWAS) that have identified gene variants associated with an increased risk for schizophrenia, but in most cases there is little known about the proteins that these genes make, what they do and how they interact, said senior investigator Madhavi Ganapathiraju, Ph.D., assistant professor of biomedical informatics, Pitt School of Medicine.
“GWAS studies and other research efforts have shown us what genes might be relevant in schizophrenia,” she said. “What we have done is the next step. We are trying to understand how these genes relate to each other, which could show us the biological pathways that are important in the disease.”
Each gene makes proteins and proteins typically interact with each other in a biological process. Information about interacting partners can shed light on the role of a gene that has not been studied, revealing pathways and biological processes associated with the disease and also its relation to other complex diseases.
Dr. Ganapathiraju’s team developed a computational model called High-Precision Protein Interaction Prediction (HiPPIP) and applied it to discover PPIs of schizophrenia-linked genes identified through GWAS, as well as historically known risk genes. They found 504 never-before known PPIs, and noted also that while schizophrenia-linked genes identified historically and through GWAS had little overlap, the model showed they shared more than 100 common interactors.
“We can infer what the protein might do by checking out the company it keeps,” Dr. Ganapathiraju explained. “For example, if I know you have many friends who play hockey, it could mean that you are involved in hockey, too. Similarly, if we see that an unknown protein interacts with multiple proteins involved in neural signaling, for example, there is a high likelihood that the unknown entity also is involved in the same.”
Dr. Ganapathiraju and colleagues have drawn such inferences on protein function based on the PPIs of proteins, and made their findings available on a website Schizo-Pi. This information can be used by biologists to explore the schizophrenia interactome with the aim of understanding more about the disease or developing new treatment drugs.
Schizophrenia interactome with 504 novel protein–protein interactions
(GWAS) have revealed the role of rare and common genetic variants, but the functional effects of the risk variants remain to be understood. Protein interactome-based studies can facilitate the study of molecular mechanisms by which the risk genes relate to schizophrenia (SZ) genesis, but protein–protein interactions (PPIs) are unknown for many of the liability genes. We developed a computational model to discover PPIs, which is found to be highly accurate according to computational evaluations and experimental validations of selected PPIs. We present here, 365 novel PPIs of liability genes identified by the SZ Working Group of the Psychiatric Genomics Consortium (PGC). Seventeen genes that had no previously known interactions have 57 novel interactions by our method. Among the new interactors are 19 drug targets that are targeted by 130 drugs. In addition, we computed 147 novel PPIs of 25 candidate genes investigated in the pre-GWAS era. While there is little overlap between the GWAS genes and the pre-GWAS genes, the interactomes reveal that they largely belong to the same pathways, thus reconciling the apparent disparities between the GWAS and prior gene association studies. The interactome including 504 novel PPIs overall, could motivate other systems biology studies and trials with repurposed drugs. The PPIs are made available on a webserver, called Schizo-Pi at http://severus.dbmi.pitt.edu/schizo-pi with advanced search capabilities.
Schizophrenia (SZ) is a common, potentially severe psychiatric disorder that afflicts all populations.1 Gene mapping studies suggest that SZ is a complex disorder, with a cumulative impact of variable genetic effects coupled with environmental factors.2 As many as 38 genome-wide association studies (GWAS) have been reported on SZ out of a total of 1,750 GWAS publications on 1,087 traits or diseases reported in the GWAS catalog maintained by the National Human Genome Research Institute of USA3 (as of April 2015), revealing the common variants associated with SZ.4 The SZ Working Group of the Psychiatric Genomics Consortium (PGC) identified 108 genetic loci that likely confer risk for SZ.5 While the role of genetics has been clearly validated by this study, the functional impact of the risk variants is not well-understood.6,7 Several of the genes implicated by the GWAS have unknown functions and could participate in possibly hitherto unknown pathways.8 Further, there is little or no overlap between the genes identified through GWAS and ‘candidate genes’ proposed in the pre-GWAS era.9
Interactome-based studies can be useful in discovering the functional associations of genes. For example,disrupted in schizophrenia 1 (DISC1), an SZ related candidate gene originally had no known homolog in humans. Although it had well-characterized protein domains such as coiled-coil domains and leucine-zipper domains, its function was unknown.10,11 Once its protein–protein interactions (PPIs) were determined using yeast 2-hybrid technology,12 investigators successfully linked DISC1 to cAMP signaling, axon elongation, and neuronal migration, and accelerated the research pertaining to SZ in general, and DISC1 in particular.13 Typically such studies are carried out on known protein–protein interaction (PPI) networks, or as in the case of DISC1, when there is a specific gene of interest, its PPIs are determined by methods such as yeast 2-hybrid technology.
Knowledge of human PPI networks is thus valuable for accelerating discovery of protein function, and indeed, biomedical research in general. However, of the hundreds of thousands of biophysical PPIs thought to exist in the human interactome,14,15 <100,000 are known today (Human Protein Reference Database, HPRD16 and BioGRID17 databases). Gold standard experimental methods for the determination of all the PPIs in human interactome are time-consuming, expensive and may not even be feasible, as about 250 million pairs of proteins would need to be tested overall; high-throughput methods such as yeast 2-hybrid have important limitations for whole interactome determination as they have a low recall of 23% (i.e., remaining 77% of true interactions need to be determined by other means), and a low precision (i.e., the screens have to be repeated multiple times to achieve high selectivity).18,19Computational methods are therefore necessary to complete the interactome expeditiously. Algorithms have begun emerging to predict PPIs using statistical machine learning on the characteristics of the proteins, but these algorithms are employed predominantly to study yeast. Two significant computational predictions have been reported for human interactome; although they have had high false positive rates, these methods have laid the foundation for computational prediction of human PPIs.20,21
We have created a new PPI prediction model called High-Confidence Protein–Protein Interaction Prediction (HiPPIP) model. Novel interactions predicted with this model are making translational impact. For example, we discovered a PPI between OASL and DDX58, which on validation showed that an increased expression of OASL could boost innate immunity to combat influenza by activating the RIG-I pathway.22 Also, the interactome of the genes associated with congenital heart disease showed that the disease morphogenesis has a close connection with the structure and function of cilia.23Here, we describe the HiPPIP model and its application to SZ genes to construct the SZ interactome. After computational evaluations and experimental validations of selected novel PPIs, we present here 504 highly confident novel PPIs in the SZ interactome, shedding new light onto several uncharacterized genes that are associated with SZ.
We developed a computational model called HiPPIP to predict PPIs (see Methods and Supplementary File 1). The model has been evaluated by computational methods and experimental validations and is found to be highly accurate. Evaluations on a held-out test data showed a precision of 97.5% and a recall of 5%. 5% recall out of 150,000 to 600,000 estimated number of interactions in the human interactome corresponds to 7,500–30,000 novel PPIs in the whole interactome. Note that, it is likely that the real precision would be higher than 97.5% because in this test data, randomly paired proteins are treated as non-interacting protein pairs, whereas some of them may actually be interacting pairs with a small probability; thus, some of the pairs that are treated as false positives in test set are likely to be true but hitherto unknown interactions. In Figure 1a, we show the precision versus recall of our method on ‘hub proteins’ where we considered all pairs that received a score >0.5 by HiPPIP to be novel interactions. In Figure 1b, we show the number of true positives versus false positives observed in hub proteins. Both these figures also show our method to be superior in comparison to the prediction of membrane-receptor interactome by Qi et al’s.24 True positives versus false positives are also shown for individual hub proteins by our method in Figure 1cand by Qi et al’s.23 in Figure 1d. These evaluations showed that our predictions contain mostly true positives. Unlike in other domains where ranked lists are commonly used such as information retrieval, in PPI prediction the ‘false positives’ may actually be unlabeled instances that are indeed true interactions that are not yet discovered. In fact, such unlabeled pairs predicted as interactors of the hub gene HMGB1 (namely, the pairs HMGB1-KL and HMGB1-FLT1) were validated by experimental methods and found to be true PPIs (See the Figures e–g inSupplementary File 3). Thus, we concluded that the protein pairs that received a score of ⩾0.5 are highly confident to be true interactions. The pairs that receive a score less than but close to 0.5 (i.e., in the range of 0.4–0.5) may also contain several true PPIs; however, we cannot confidently say that all in this range are true PPIs. Only the PPIs predicted with a score >0.5 are included in the interactome.
Computational evaluation of predicted protein–protein interactions on hub proteins: (a) precision recall curve. (b) True positive versus false positives in ranked lists of hub type membrane receptors for our method and that by Qi et al. True positives versus false positives are shown for individual membrane receptors by our method in (c) and by Qi et al. in (d). Thick line is the average, which is also the same as shown in (b). Note:x-axis is recall in (a), whereas it is number of false positives in (b–d). The range of y-axis is observed by varying the threshold from 1.0–0 in (a), and to 0.5 in (b–d).
SZ interactome
By applying HiPPIP to the GWAS genes and Historic (pre-GWAS) genes, we predicted over 500 high confidence new PPIs adding to about 1400 previously known PPIs.
Schizophrenia interactome: network view of the schizophrenia interactome is shown as a graph, where genes are shown as nodes and PPIs as edges connecting the nodes. Schizophrenia-associated genes are shown as dark blue nodes, novel interactors as red color nodes and known interactors as blue color nodes. The source of the schizophrenia genes is indicated by its label font, where Historic genes are shown italicized, GWAS genes are shown in bold, and the one gene that is common to both is shown in italicized and bold. For clarity, the source is also indicated by the shape of the node (triangular for GWAS and square for Historic and hexagonal for both). Symbols are shown only for the schizophrenia-associated genes; actual interactions may be accessed on the web. Red edges are the novel interactions, whereas blue edges are known interactions. GWAS, genome-wide association studies of schizophrenia; PPI, protein–protein interaction.
We have made the known and novel interactions of all SZ-associated genes available on a webserver called Schizo-Pi, at the addresshttp://severus.dbmi.pitt.edu/schizo-pi. This webserver is similar to Wiki-Pi33 which presents comprehensive annotations of both participating proteins of a PPI side-by-side. The difference between Wiki-Pi which we developed earlier, and Schizo-Pi, is the inclusion of novel predicted interactions of the SZ genes into the latter.
Despite the many advances in biomedical research, identifying the molecular mechanisms underlying the disease is still challenging. Studies based on protein interactions were proven to be valuable in identifying novel gene associations that could shed new light on disease pathology.35 The interactome including more than 500 novel PPIs will help to identify pathways and biological processes associated with the disease and also its relation to other complex diseases. It also helps identify potential drugs that could be repurposed to use for SZ treatment.
Functional and pathway enrichment in SZ interactome
When a gene of interest has little known information, functions of its interacting partners serve as a starting point to hypothesize its own function. We computed statistically significant enrichment of GO biological process terms among the interacting partners of each of the genes using BinGO36 (see online at http://severus.dbmi.pitt.edu/schizo-pi).
Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution
Massimo Stefani · Christopher M. Dobson
Abstract The deposition of proteins in the form of amyloid fibrils and plaques is the characteristic feature of more than 20 degenerative conditions affecting either the central nervous system or a variety of peripheral tissues. As these conditions include Alzheimer’s, Parkinson’s and the prion diseases, several forms of fatal systemic amyloidosis, and at least one condition associated with medical intervention (haemodialysis), they are of enormous importance in the context of present-day human health and welfare. Much remains to be learned about the mechanism by which the proteins associated with these diseases aggregate and form amyloid structures, and how the latter affect the functions of the organs with which they are associated. A great deal of information concerning these diseases has emerged, however, during the past 5 years, much of it causing a number of fundamental assumptions about the amyloid diseases to be reexamined. For example, it is now apparent that the ability to form amyloid structures is not an unusual feature of the small number of proteins associated with these diseases but is instead a general property of polypeptide chains. It has also been found recently that aggregates of proteins not associated with amyloid diseases can impair the ability of cells to function to a similar extent as aggregates of proteins linked with specific neurodegenerative conditions. Moreover, the mature amyloid fibrils or plaques appear to be substantially less toxic than the prefibrillar aggregates that are their precursors. The toxicity of these early aggregates appears to result from an intrinsic ability to impair fundamental cellular processes by interacting with cellular membranes, causing oxidative stress and increases in free Ca2+ that eventually lead to apoptotic or necrotic cell death. The ‘new view’ of these diseases also suggests that other degenerative conditions could have similar underlying origins to those of the amyloidoses. In addition, cellular protection mechanisms, such as molecular chaperones and the protein degradation machinery, appear to be crucial in the prevention of disease in normally functioning living organisms. It also suggests some intriguing new factors that could be of great significance in the evolution of biological molecules and the mechanisms that regulate their behaviour.
The genetic information within a cell encodes not only the specific structures and functions of proteins but also the way these structures are attained through the process known as protein folding. In recent years many of the underlying features of the fundamental mechanism of this complex process and the manner in which it is regulated in living systems have emerged from a combination of experimental and theoretical studies [1]. The knowledge gained from these studies has also raised a host of interesting issues. It has become apparent, for example, that the folding and unfolding of proteins is associated with a whole range of cellular processes from the trafficking of molecules to specific organelles to the regulation of the cell cycle and the immune response. Such observations led to the inevitable conclusion that the failure to fold correctly, or to remain correctly folded, gives rise to many different types of biological malfunctions and hence to many different forms of disease [2]. In addition, it has been recognised recently that a large number of eukaryotic genes code for proteins that appear to be ‘natively unfolded’, and that proteins can adopt, under certain circumstances, highly organised multi-molecular assemblies whose structures are not specifically encoded in the amino acid sequence. Both these observations have raised challenging questions about one of the most fundamental principles of biology: the close relationship between the sequence, structure and function of proteins, as we discuss below [3].
It is well established that proteins that are ‘misfolded’, i.e. that are not in their functionally relevant conformation, are devoid of normal biological activity. In addition, they often aggregate and/or interact inappropriately with other cellular components leading to impairment of cell viability and eventually to cell death. Many diseases, often known as misfolding or conformational diseases, ultimately result from the presence in a living system of protein molecules with structures that are ‘incorrect’, i.e. that differ from those in normally functioning organisms [4]. Such diseases include conditions in which a specific protein, or protein complex, fails to fold correctly (e.g. cystic fibrosis, Marfan syndrome, amyotonic lateral sclerosis) or is not sufficiently stable to perform its normal function (e.g. many forms of cancer). They also include conditions in which aberrant folding behaviour results in the failure of a protein to be correctly trafficked (e.g. familial hypercholesterolaemia, α1-antitrypsin deficiency, and some forms of retinitis pigmentosa) [4]. The tendency of proteins to aggregate, often to give species extremely intractable to dissolution and refolding, is of course also well known in other circumstances. Examples include the formation of inclusion bodies during overexpression of heterologous proteins in bacteria and the precipitation of proteins during laboratory purification procedures. Indeed, protein aggregation is well established as one of the major difficulties associated with the production and handling of proteins in the biotechnology and pharmaceutical industries [5].
Considerable attention is presently focused on a group of protein folding diseases known as amyloidoses. In these diseases specific peptides or proteins fail to fold or to remain correctly folded and then aggregate (often with other components) so as to give rise to ‘amyloid’ deposits in tissue. Amyloid structures can be recognised because they possess a series of specific tinctorial and biophysical characteristics that reflect a common core structure based on the presence of highly organised βsheets [6]. The deposits in strictly defined amyloidoses are extracellular and can often be observed as thread-like fibrillar structures, sometimes assembled further into larger aggregates or plaques. These diseases include a range of sporadic, familial or transmissible degenerative diseases, some of which affect the brain and the central nervous system (e.g. Alzheimer’s and Creutzfeldt-Jakob diseases), while others involve peripheral tissues and organs such as the liver, heart and spleen (e.g. systemic amyloidoses and type II diabetes) [7, 8]. In other forms of amyloidosis, such as primary or secondary systemic amyloidoses, proteinaceous deposits are found in skeletal tissue and joints (e.g. haemodialysis-related amyloidosis) as well as in several organs (e.g. heart and kidney). Yet other components such as collagen, glycosaminoglycans and proteins (e.g. serum amyloid protein) are often present in the deposits protecting them against degradation [9, 10, 11]. Similar deposits to those in the amyloidoses are, however, found intracellularly in other diseases; these can be localised either in the cytoplasm, in the form of specialised aggregates known as aggresomes or as Lewy or Russell bodies or in the nucleus (see below).
The presence in tissue of proteinaceous deposits is a hallmark of all these diseases, suggesting a causative link between aggregate formation and pathological symptoms (often known as the amyloid hypothesis) [7, 8, 12]. At the present time the link between amyloid formation and disease is widely accepted on the basis of a large number of biochemical and genetic studies. The specific nature of the pathogenic species, and the molecular basis of their ability to damage cells, are however, the subject of intense debate [13, 14, 15, 16, 17, 18, 19, 20]. In neurodegenerative disorders it is very likely that the impairment of cellular function follows directly from the interactions of the aggregated proteins with cellular components [21, 22]. In the systemic non-neurological diseases, however, it is widely believed that the accumulation in vital organs of large amounts of amyloid deposits can by itself cause at least some of the clinical symptoms [23]. It is quite possible, however, that there are other more specific effects of aggregates on biochemical processes even in these diseases. The presence of extracellular or intracellular aggregates of a specific polypeptide molecule is a characteristic of all the 20 or so recognised amyloid diseases. The polypeptides involved include full length proteins (e.g. lysozyme or immunoglobulin light chains), biological peptides (amylin, atrial natriuretic factor) and fragments of larger proteins produced as a result of specific processing (e.g. the Alzheimer βpeptide) or of more general degradation [e.g. poly(Q) stretches cleaved from proteins with poly(Q) extensions such as huntingtin, ataxins and the androgen receptor]. The peptides and proteins associated with known amyloid diseases are listed in Table 1. In some cases the proteins involved have wild type sequences, as in sporadic forms of the diseases, but in other cases these are variants resulting from genetic mutations associated with familial forms of the diseases. In some cases both sporadic and familial diseases are associated with a given protein; in this case the mutational variants are usually associated with early-onset forms of the disease. In the case of the neurodegenerative diseases associated with the prion protein some forms of the diseases are transmissible. The existence of familial forms of a number of amyloid diseases has provided significant clues to the origins of the pathologies. For example, there are increasingly strong links between the age at onset of familial forms of disease and the effects of the mutations involved on the propensity of the affected proteins to aggregate in vitro. Such findings also support the link between the process of aggregation and the clinical manifestations of disease [24, 25].
The presence in cells of misfolded or aggregated proteins triggers a complex biological response. In the cytosol, this is referred to as the ‘heat shock response’ and in the endoplasmic reticulum (ER) it is known as the ‘unfolded protein response’. These responses lead to the expression, among others, of the genes for heat shock proteins (Hsp, or molecular chaperone proteins) and proteins involved in the ubiquitin-proteasome pathway [26]. The evolution of such complex biochemical machinery testifies to the fact that it is necessary for cells to isolate and clear rapidly and efficiently any unfolded or incorrectly folded protein as soon as it appears. In itself this fact suggests that these species could have a generally adverse effect on cellular components and cell viability. Indeed, it was a major step forward in understanding many aspects of cell biology when it was recognised that proteins previously associated only with stress, such as heat shock, are in fact crucial in the normal functioning of living systems. This advance, for example, led to the discovery of the role of molecular chaperones in protein folding and in the normal ‘housekeeping’ processes that are inherent in healthy cells [27, 28]. More recently a number of degenerative diseases, both neurological and systemic, have been linked to, or shown to be affected by, impairment of the ubiquitin-proteasome pathway (Table 2). The diseases are primarily associated with a reduction in either the expression or the biological activity of Hsps, ubiquitin, ubiquitinating or deubiquitinating enzymes and the proteasome itself, as we show below [29, 30, 31, 32], or even to the failure of the quality control mechanisms that ensure proper maturation of proteins in the ER. The latter normally leads to degradation of a significant proportion of polypeptide chains before they have attained their native conformations through retrograde translocation to the cytosol [33, 34].
….
It is now well established that the molecular basis of protein aggregation into amyloid structures involves the existence of ‘misfolded’ forms of proteins, i.e. proteins that are not in the structures in which they normally function in vivo or of fragments of proteins resulting from degradation processes that are inherently unable to fold [4, 7, 8, 36]. Aggregation is one of the common consequences of a polypeptide chain failing to reach or maintain its functional three-dimensional structure. Such events can be associated with specific mutations, misprocessing phenomena, aberrant interactions with metal ions, changes in environmental conditions, such as pH or temperature, or chemical modification (oxidation, proteolysis). Perturbations in the conformational properties of the polypeptide chain resulting from such phenomena may affect equilibrium 1 in Fig. 1 increasing the population of partially unfolded, or misfolded, species that are much more aggregation-prone than the native state.
Fig. 1 Overview of the possible fates of a newly synthesised polypeptide chain. The equilibrium ① between the partially folded molecules and the natively folded ones is usually strongly in favour of the latter except as a result of specific mutations, chemical modifications or partially destabilising solution conditions. The increased equilibrium populations of molecules in the partially or completely unfolded ensemble of structures are usually degraded by the proteasome; when this clearance mechanism is impaired, such species often form disordered aggregates or shift equilibrium ② towards the nucleation of pre-fibrillar assemblies that eventually grow into mature fibrils (equilibrium ③). DANGER! indicates that pre-fibrillar aggregates in most cases display much higher toxicity than mature fibrils. Heat shock proteins (Hsp) can suppress the appearance of pre-fibrillar assemblies by minimising the population of the partially folded molecules by assisting in the correct folding of the nascent chain and the unfolded protein response target incorrectly folded proteins for degradation.
……
Little is known at present about the detailed arrangement of the polypeptide chains themselves within amyloid fibrils, either those parts involved in the core βstrands or in regions that connect the various β-strands. Recent data suggest that the sheets are relatively untwisted and may in some cases at least exist in quite specific supersecondary structure motifs such as β-helices [6, 40] or the recently proposed µ-helix [41]. It seems possible that there may be significant differences in the way the strands are assembled depending on characteristics of the polypeptide chain involved [6, 42]. Factors including length, sequence (and in some cases the presence of disulphide bonds or post-translational modifications such as glycosylation) may be important in determining details of the structures. Several recent papers report structural models for amyloid fibrils containing different polypeptide chains, including the Aβ40 peptide, insulin and fragments of the prion protein, based on data from such techniques as cryo-electron microscopy and solid-state magnetic resonance spectroscopy [43, 44]. These models have much in common and do indeed appear to reflect the fact that the structures of different fibrils are likely to be variations on a common theme [40]. It is also emerging that there may be some common and highly organised assemblies of amyloid protofilaments that are not simply extended threads or ribbons. It is clear, for example, that in some cases large closed loops can be formed [45, 46, 47], and there may be specific types of relatively small spherical or ‘doughnut’ shaped structures that can result in at least some circumstances (see below).
…..
The similarity of some early amyloid aggregates with the pores resulting from oligomerisation of bacterial toxins and pore-forming eukaryotic proteins (see below) also suggest that the basic mechanism of protein aggregation into amyloid structures may not only be associated with diseases but in some cases could result in species with functional significance. Recent evidence indicates that a variety of micro-organisms may exploit the controlled aggregation of specific proteins (or their precursors) to generate functional structures. Examples include bacterial curli [52] and proteins of the interior fibre cells of mammalian ocular lenses, whose β-sheet arrays seem to be organised in an amyloid-like supramolecular order [53]. In this case the inherent stability of amyloid-like protein structure may contribute to the long-term structural integrity and transparency of the lens. Recently it has been hypothesised that amyloid-like aggregates of serum amyloid A found in secondary amyloidoses following chronic inflammatory diseases protect the host against bacterial infections by inducing lysis of bacterial cells [54]. One particularly interesting example is a ‘misfolded’ form of the milk protein α-lactalbumin that is formed at low pH and trapped by the presence of specific lipid molecules [55]. This form of the protein has been reported to trigger apoptosis selectively in tumour cells providing evidence for its importance in protecting infants from certain types of cancer [55]. ….
Amyloid formation is a generic property of polypeptide chains ….
It is clear that the presence of different side chains can influence the details of amyloid structures, particularly the assembly of protofibrils, and that they give rise to the variations on the common structural theme discussed above. More fundamentally, the composition and sequence of a peptide or protein affects profoundly its propensity to form amyloid structures under given conditions (see below).
Because the formation of stable protein aggregates of amyloid type does not normally occur in vivo under physiological conditions, it is likely that the proteins encoded in the genomes of living organisms are endowed with structural adaptations that mitigate against aggregation under these conditions. A recent survey involving a large number of structures of β-proteins highlights several strategies through which natural proteins avoid intermolecular association of β-strands in their native states [65]. Other surveys of protein databases indicate that nature disfavours sequences of alternating polar and nonpolar residues, as well as clusters of several consecutive hydrophobic residues, both of which enhance the tendency of a protein to aggregate prior to becoming completely folded [66, 67].
……
Precursors of amyloid fibrils can be toxic to cells
It was generally assumed until recently that the proteinaceous aggregates most toxic to cells are likely to be mature amyloid fibrils, the form of aggregates that have been commonly detected in pathological deposits. It therefore appeared probable that the pathogenic features underlying amyloid diseases are a consequence of the interaction with cells of extracellular deposits of aggregated material. As well as forming the basis for understanding the fundamental causes of these diseases, this scenario stimulated the exploration of therapeutic approaches to amyloidoses that focused mainly on the search for molecules able to impair the growth and deposition of fibrillar forms of aggregated proteins. ….
Structural basis and molecular features of amyloid toxicity
The presence of toxic aggregates inside or outside cells can impair a number of cell functions that ultimately lead to cell death by an apoptotic mechanism [95, 96]. Recent research suggests, however, that in most cases initial perturbations to fundamental cellular processes underlie the impairment of cell function induced by aggregates of disease-associated polypeptides. Many pieces of data point to a central role of modifications to the intracellular redox status and free Ca2+ levels in cells exposed to toxic aggregates [45, 89, 97, 98, 99, 100, 101]. A modification of the intracellular redox status in such cells is associated with a sharp increase in the quantity of reactive oxygen species (ROS) that is reminiscent of the oxidative burst by which leukocytes destroy invading foreign cells after phagocytosis. In addition, changes have been observed in reactive nitrogen species, lipid peroxidation, deregulation of NO metabolism [97], protein nitrosylation [102] and upregulation of heme oxygenase-1, a specific marker of oxidative stress [103]. ….
Results have recently been reported concerning the toxicity towards cultured cells of aggregates of poly(Q) peptides which argues against a disease mechanism based on specific toxic features of the aggregates. These results indicate that there is a close relationship between the toxicity of proteins with poly(Q) extensions and their nuclear localisation. In addition they support the hypotheses that the toxicity of poly(Q) aggregates can be a consequence of altered interactions with nuclear coactivator or corepressor molecules including p53, CBP, Sp1 and TAF130 or of the interaction with transcription factors and nuclear coactivators, such as CBP, endowed with short poly(Q) stretches ([95] and references therein)…..
Concluding remarks
The data reported in the past few years strongly suggest that the conversion of normally soluble proteins into amyloid fibrils and the toxicity of small aggregates appearing during the early stages of the formation of the latter are common or generic features of polypeptide chains. Moreover, the molecular basis of this toxicity also appears to display common features between the different systems that have so far been studied. The ability of many, perhaps all, natural polypeptides to ‘misfold’ and convert into toxic aggregates under suitable conditions suggests that one of the most important driving forces in the evolution of proteins must have been the negative selection against sequence changes that increase the tendency of a polypeptide chain to aggregate. Nevertheless, as protein folding is a stochastic process, and no such process can be completely infallible, misfolded proteins or protein folding intermediates in equilibrium with the natively folded molecules must continuously form within cells. Thus mechanisms to deal with such species must have co-evolved with proteins. Indeed, it is clear that misfolding, and the associated tendency to aggregate, is kept under control by molecular chaperones, which render the resulting species harmless assisting in their refolding, or triggering their degradation by the cellular clearance machinery [166, 167, 168, 169, 170, 171, 172, 173, 175, 177, 178].
Misfolded and aggregated species are likely to owe their toxicity to the exposure on their surfaces of regions of proteins that are buried in the interior of the structures of the correctly folded native states. The exposure of large patches of hydrophobic groups is likely to be particularly significant as such patches favour the interaction of the misfolded species with cell membranes [44, 83, 89, 90, 91, 93]. Interactions of this type are likely to lead to the impairment of the function and integrity of the membranes involved, giving rise to a loss of regulation of the intracellular ion balance and redox status and eventually to cell death. In addition, misfolded proteins undoubtedly interact inappropriately with other cellular components, potentially giving rise to the impairment of a range of other biological processes. Under some conditions the intracellular content of aggregated species may increase directly, due to an enhanced propensity of incompletely folded or misfolded species to aggregate within the cell itself. This could occur as the result of the expression of mutational variants of proteins with decreased stability or cooperativity or with an intrinsically higher propensity to aggregate. It could also occur as a result of the overproduction of some types of protein, for example, because of other genetic factors or other disease conditions, or because of perturbations to the cellular environment that generate conditions favouring aggregation, such as heat shock or oxidative stress. Finally, the accumulation of misfolded or aggregated proteins could arise from the chaperone and clearance mechanisms becoming overwhelmed as a result of specific mutant phenotypes or of the general effects of ageing [173, 174].
The topics discussed in this review not only provide a great deal of evidence for the ‘new view’ that proteins have an intrinsic capability of misfolding and forming structures such as amyloid fibrils but also suggest that the role of molecular chaperones is even more important than was thought in the past. The role of these ubiquitous proteins in enhancing the efficiency of protein folding is well established [185]. It could well be that they are at least as important in controlling the harmful effects of misfolded or aggregated proteins as in enhancing the yield of functional molecules.
Nutritional Status is Associated with Faster Cognitive Decline and Worse Functional Impairment in the Progression of Dementia: The Cache County Dementia Progression Study1
Nutritional status may be a modifiable factor in the progression of dementia. We examined the association of nutritional status and rate of cognitive and functional decline in a U.S. population-based sample. Study design was an observational longitudinal study with annual follow-ups up to 6 years of 292 persons with dementia (72% Alzheimer’s disease, 56% female) in Cache County, UT using the Mini-Mental State Exam (MMSE), Clinical Dementia Rating Sum of Boxes (CDR-sb), and modified Mini Nutritional Assessment (mMNA). mMNA scores declined by approximately 0.50 points/year, suggesting increasing risk for malnutrition. Lower mMNA score predicted faster rate of decline on the MMSE at earlier follow-up times, but slower decline at later follow-up times, whereas higher mMNA scores had the opposite pattern (mMNA by time β= 0.22, p = 0.017; mMNA by time2 β= –0.04, p = 0.04). Lower mMNA score was associated with greater impairment on the CDR-sb over the course of dementia (β= 0.35, p < 0.001). Assessment of malnutrition may be useful in predicting rates of progression in dementia and may provide a target for clinical intervention.
Shared Genetic Risk Factors for Late-Life Depression and Alzheimer’s Disease
Background: Considerable evidence has been reported for the comorbidity between late-life depression (LLD) and Alzheimer’s disease (AD), both of which are very common in the general elderly population and represent a large burden on the health of the elderly. The pathophysiological mechanisms underlying the link between LLD and AD are poorly understood. Because both LLD and AD can be heritable and are influenced by multiple risk genes, shared genetic risk factors between LLD and AD may exist. Objective: The objective is to review the existing evidence for genetic risk factors that are common to LLD and AD and to outline the biological substrates proposed to mediate this association. Methods: A literature review was performed. Results: Genetic polymorphisms of brain-derived neurotrophic factor, apolipoprotein E, interleukin 1-beta, and methylenetetrahydrofolate reductase have been demonstrated to confer increased risk to both LLD and AD by studies examining either LLD or AD patients. These results contribute to the understanding of pathophysiological mechanisms that are common to both of these disorders, including deficits in nerve growth factors, inflammatory changes, and dysregulation mechanisms involving lipoprotein and folate. Other conflicting results have also been reviewed, and few studies have investigated the effects of the described polymorphisms on both LLD and AD. Conclusion: The findings suggest that common genetic pathways may underlie LLD and AD comorbidity. Studies to evaluate the genetic relationship between LLD and AD may provide insights into the molecular mechanisms that trigger disease progression as the population ages.
Association of Vitamin B12, Folate, and Sulfur Amino Acids With Brain Magnetic Resonance Imaging Measures in Older Adults: A Longitudinal Population-Based Study
Importance Vitamin B12, folate, and sulfur amino acids may be modifiable risk factors for structural brain changes that precede clinical dementia.
Objective To investigate the association of circulating levels of vitamin B12, red blood cell folate, and sulfur amino acids with the rate of total brain volume loss and the change in white matter hyperintensity volume as measured by fluid-attenuated inversion recovery in older adults.
Design, Setting, and Participants The magnetic resonance imaging subsample of the Swedish National Study on Aging and Care in Kungsholmen, a population-based longitudinal study in Stockholm, Sweden, was conducted in 501 participants aged 60 years or older who were free of dementia at baseline. A total of 299 participants underwent repeated structural brain magnetic resonance imaging scans from September 17, 2001, to December 17, 2009.
Main Outcomes and Measures The rate of brain tissue volume loss and the progression of total white matter hyperintensity volume.
Results In the multi-adjusted linear mixed models, among 501 participants (300 women [59.9%]; mean [SD] age, 70.9 [9.1] years), higher baseline vitamin B12 and holotranscobalamin levels were associated with a decreased rate of total brain volume loss during the study period: for each increase of 1 SD, β (SE) was 0.048 (0.013) for vitamin B12 (P < .001) and 0.040 (0.013) for holotranscobalamin (P = .002). Increased total homocysteine levels were associated with faster rates of total brain volume loss in the whole sample (β [SE] per 1-SD increase, –0.035 [0.015]; P = .02) and with the progression of white matter hyperintensity among participants with systolic blood pressure greater than 140 mm Hg (β [SE] per 1-SD increase, 0.000019 [0.00001]; P = .047). No longitudinal associations were found for red blood cell folate and other sulfur amino acids.
Conclusions and Relevance This study suggests that both vitamin B12 and total homocysteine concentrations may be related to accelerated aging of the brain. Randomized clinical trials are needed to determine the importance of vitamin B12supplementation on slowing brain aging in older adults.
Notes from Kurzweill
This vitamin stops the aging process in organs, say Swiss researchers
A potential breakthrough for regenerative medicine, pending further studies
Improved muscle stem cell numbers and muscle function in NR-treated aged mice: Newly regenerated muscle fibers 7 days after muscle damage in aged mice (left: control group; right: fed NR). (Scale bar = 50 μm). (credit: Hongbo Zhang et al./Science) http://www.kurzweilai.net/images/improved-muscle-fibers.png
EPFL researchers have restored the ability of mice organs to regenerate and extend life by simply administering nicotinamide riboside (NR) to them.
NR has been shown in previous studies to be effective in boosting metabolism and treating a number of degenerative diseases. Now, an article by PhD student Hongbo Zhang published in Science also describes the restorative effects of NR on the functioning of stem cells for regenerating organs.
As in all mammals, as mice age, the regenerative capacity of certain organs (such as the liver and kidneys) and muscles (including the heart) diminishes. Their ability to repair them following an injury is also affected. This leads to many of the disorders typical of aging.
Mitochondria —> stem cells —> organs
To understand how the regeneration process deteriorates with age, Zhang teamed up with colleagues from ETH Zurich, the University of Zurich, and universities in Canada and Brazil. By using several biomarkers, they were able to identify the molecular chain that regulates how mitochondria — the “powerhouse” of the cell — function and how they change with age. “We were able to show for the first time that their ability to function properly was important for stem cells,” said Auwerx.
Under normal conditions, these stem cells, reacting to signals sent by the body, regenerate damaged organs by producing new specific cells. At least in young bodies. “We demonstrated that fatigue in stem cells was one of the main causes of poor regeneration or even degeneration in certain tissues or organs,” said Zhang.
How to revitalize stem cells
Which is why the researchers wanted to “revitalize” stem cells in the muscles of elderly mice. And they did so by precisely targeting the molecules that help the mitochondria to function properly. “We gave nicotinamide riboside to 2-year-old mice, which is an advanced age for them,” said Zhang.
“This substance, which is close to vitamin B3, is a precursor of NAD+, a molecule that plays a key role in mitochondrial activity. And our results are extremely promising: muscular regeneration is much better in mice that received NR, and they lived longer than the mice that didn’t get it.”
Parallel studies have revealed a comparable effect on stem cells of the brain and skin. “This work could have very important implications in the field of regenerative medicine,” said Auwerx. This work on the aging process also has potential for treating diseases that can affect — and be fatal — in young people, like muscular dystrophy (myopathy).
So far, no negative side effects have been observed following the use of NR, even at high doses. But while it appears to boost the functioning of all cells, it could include pathological ones, so further in-depth studies are required.
Abstract of NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice
Adult stem cells (SCs) are essential for tissue maintenance and regeneration yet are susceptible to senescence during aging. We demonstrate the importance of the amount of the oxidized form of cellular nicotinamide adenine dinucleotide (NAD+) and its impact on mitochondrial activity as a pivotal switch to modulate muscle SC (MuSC) senescence. Treatment with the NAD+ precursor nicotinamide riboside (NR) induced the mitochondrial unfolded protein response (UPRmt) and synthesis of prohibitin proteins, and this rejuvenated MuSCs in aged mice. NR also prevented MuSC senescence in the Mdx mouse model of muscular dystrophy. We furthermore demonstrate that NR delays senescence of neural SCs (NSCs) and melanocyte SCs (McSCs), and increased mouse lifespan. Strategies that conserve cellular NAD+ may reprogram dysfunctional SCs and improve lifespan in mammals.
Discriminating the gene target of a distal regulatory element from other nearby transcribed genes is a challenging problem with the potential to illuminate the causal underpinnings of complex diseases. We present TargetFinder, a computational method that reconstructs regulatory landscapes from diverse features along the genome. The resulting models accurately predict individual enhancer–promoter interactions across multiple cell lines with a false discovery rate up to 15 times smaller than that obtained using the closest gene. By evaluating the genomic features driving this accuracy, we uncover interactions between structural proteins, transcription factors, epigenetic modifications, and transcription that together distinguish interacting from non-interacting enhancer–promoter pairs. Most of this signature is not proximal to the enhancers and promoters but instead decorates the looping DNA. We conclude that complex but consistent combinations of marks on the one-dimensional genome encode the three-dimensional structure of fine-scale regulatory interactions.
Cyclic Dinucleotides and Histone deacetylase inhibitors
Curators: Larry H. Bernsten, MD, FCAP and Aviva Lev-Ari, PhD, RN
LPBI
New Class of Immune System Stimulants: Cyclic Di-Nucleotides (CDN): Shrink Tumors and bolster Vaccines, re-arm the Immune System’s Natural Killer Cells, which attack Cancer Cells and Virus-infected Cells
Reporter: Aviva Lev-Ari, PhD, RN
The Immunotherapeutics and Vaccine Research Initiative (IVRI), a UC Berkeley effort funded by Aduro Biotech, Inc.
A new class of immune system stimulants called cyclic di-nucleotides have shown promise in shrinking tumors and bolstering vaccines against tuberculosis, and research that could help re-arm the immune system’s natural killer cells, which normally attack cancer cells and virus-infected cells, to better fight tumors.
Much of the excitement around combining these two areas — the immunology of cancer and the immunology of infectious disease — comes from the amazing success of immunotherapy against cancer, which started with the work of James Allison when he was a professor of immunology at UC Berkeley and director of the Cancer Research Laboratory from 1985 to 2004. Allison, now at the University of Texas MD Anderson Cancer Center, discovered a way to release a brake on the body’s immune response to cancer that has proved highly successful at unleashing the immune system to attack melanoma and is being tried against other types of cancer. Allison’s technique uses an antibody that blocks an immune suppressor called CTLA4, antibodies that block another immune suppressor, PD1. This has been successful in treating melanoma, renal cancer and a type of lung cancer. Both CTLA4 and PD1 antibodies are now FDA-approved as cancer therapies.
Another promising avenue involves a protein in cells that responds to foreign DNA to launch an innate immune response — the first response of the body’s immune system. The protein, dubbed STING, is triggered by small molecules called cyclic di-nucleotides (CDN), and makes immune cells release interferon and other cytokines that activate disease-fighting T cells and stimulate the production of antibodies that together kill invading pathogens and destroy cancer cells. Listeria bacteria, for example, secrete a CDN directly into infected cells that activates STING.
Russell Vance, a UC Berkeley professor of molecular and cell biology and current head of the Cancer Research Laboratory, discovered several years ago that the chemical structure of these di-nucleotides is critical to their ability to work in humans. Aduro has since developed a CDN designed to work in humans and found that injecting it directly into a tumor in mice caused the tumor to shrink.
Sarah Stanley, an assistant professor of public health, has found evidence that CDNs can help improve the imperfect vaccines we have today against tuberculosis.
Researchers at UC Berkeley will have access to Aduro’s novel technology platforms for research use, including its STING pathway activators, proprietary monoclonal antibodies and the engineered listeria bacteria, referred to as LADD (listeria attenuated double-deleted). David Raulet, professor of molecular and cell biology and director of IVRI has contributed to making these cells a new focus of cancer research. As tumors advance, NK cells inside the tumors appear to become desensitized, he said. Recent research shows that some cytokines and other immune mediators Raulet discovered are able to “wake them up” and get them to resume their elimination of cancer cells.
Histone deacetylase inhibitors: molecular mechanisms of action
This review focuses on the mechanisms of action of histone deacetylase (HDAC) inhibitors (HDACi), a group of recently discovered ‘targeted’ anticancer agents. There are 18 HDACs, which are generally divided into four classes, based on sequence homology to yeast counterparts. Classical HDACi such as the hydroxamic acid-based vorinostat (also known as SAHA and Zolinza) inhibits classes I, II and IV, but not the NAD+-dependent class III enzymes. In clinical trials, vorinostat has activity against hematologic and solid cancers at doses well tolerated by patients. In addition to histones, HDACs have many other protein substrates involved in regulation of gene expression, cell proliferation and cell death. Inhibition of HDACs causes accumulation of acetylated forms of these proteins, altering their function. Thus, HDACs are more properly called ‘lysine deacetylases.’ HDACi induces different phenotypes in various transformed cells, including growth arrest, activation of the extrinsic and/or intrinsic apoptotic pathways, autophagic cell death, reactive oxygen species (ROS)-induced cell death, mitotic cell death and senescence. In comparison, normal cells are relatively more resistant to HDACi-induced cell death. The plurality of mechanisms of HDACi-induced cell death reflects both the multiple substrates of HDACs and the heterogeneous patterns of molecular alterations present in different cancer cells.
Acetylation and deacetylation of histones play an important role in transcription regulation of eukaryotic cells (Lehrmann et al., 2002;Mai et al., 2005). The acetylation status of histones and non-histone proteins is determined by histone deacetylases (HDACs) and histone acetyl-transferases (HATs). HATs add acetyl groups to lysine residues, while HDACs remove the acetyl groups. In general, acetylation of histone promotes a more relaxed chromatin structure, allowing transcriptional activation. HDACs can act as transcription repressors, due to histone deacetylation, and consequently promote chromatin condensation. HDAC inhibitors (HDACi) selectively alter gene transcription, in part, by chromatin remodeling and by changes in the structure of proteins in transcription factor complexes (Gui et al., 2004). Further, the HDACs have many non-histone proteins substrates such as hormone receptors, chaperone proteins and cytoskeleton proteins, which regulate cell proliferation and cell death (Table 1). Thus, HDACi-induced transformed cell death involves transcription-dependent and transcription-independent mechanisms (Marks and Dokmanovic, 2005; Rosato and Grant, 2005; Bolden et al., 2006;Minucci and Pelicci, 2006).
In humans, 18 HDAC enzymes have been identified and classified, based on homology to yeast HDACs (Blander and Guarente, 2004;Bhalla, 2005; Marks and Dokmanovic, 2005). Class I HDACs include HDAC1, 2, 3 and 8, which are related to yeast RPD3 deacetylase and have high homology in their catalytic sites. Recent phylogenetic analyses suggest that this class can be divided into classes Ia (HDAC1 and -2), Ib (HDAC3) and Ic (HDAC8) (Gregoretti et al., 2004). Class II HDACs are related to yeast Hda1 (histone deacetylase 1) and include HDAC4, -5, -6, -7, -9 and -10 (Bhalla, 2005; Marks and Dokmanovic, 2005). This class is divided into class IIa, consisting of HDAC4, -5, -7 and -9, and class IIb, consisting of HDAC6 and -10, which contain two catalytic sites. All class I and II HDACs are zinc-dependent enzymes. Members of a third class, sirtuins, require NAD+ for their enzymatic activity (Blander and Guarente, 2004) (see review by E Verdin, in this issue). Among them, SIRT1 is orthologous to yeast silent information regulator 2. The enzymatic activity of class III HDACs is not inhibited by compounds such as vorinostat or trichostatin A (TSA), that inhibit class I and II HDACs. Class IV HDAC is represented by HDAC11, which, like yeast Hda 1 similar 3, has conserved residues in the catalytic core region shared by both class I and II enzymes (Gao et al., 2002).
HDACs are not redundant in function (Marks and Dokmanovic, 2005; Rosato and Grant, 2005; Bolden et al., 2006). Class I HDACs are primarily nuclear in localization and ubiquitously expressed, while class II HDACs can be primarily cytoplasmic and/or migrate between the cytoplasm and nucleus and are tissue-restricted in expression.
The structural details of the HDAC–HDACi interaction has been elucidated in studies of a histone deacetylase-like protein from an anerobic bacterium with TSA and vorinostat (Finnin et al., 1999). More recently, the crystal structure of HDAC8–hydroxamate interaction has been solved (Somoza et al., 2004; Vannini et al., 2004). These studies provide an insight into the mechanism of deacetylation of acetylated substrates. The hydroxamic acid moiety of the inhibitor directly interacts with the zinc ion at the base of the catalytic pocket.
This review focuses on the molecular mechanisms triggered by inhibitors of zinc-dependent HDACs in tumor cells that explain in part: (I) the effects of these compounds in inducing transformed cell death and (II) the relative resistance of normal and certain cancer cells to HDACi induced cell death. HDACi, for example, the hydroxamic acid-based vorinostat (SAHA, Zolinza), are promising drugs for cancer treatment (Richon et al., 1998; Marks and Breslow, 2007). Several HDACi are in phase I and II clinical trials, being tested in different tumor types, such as cutaneous T-cell lymphoma, acute myeloid leukemia, cervical cancer, etc (Bug et al., 2005; Chavez-Blanco et al., 2005; Kelly and Marks, 2005;Duvic and Zhang, 2006) (Table 2). Although considerable progress has been made in elucidating the role of HDACs and the effects of HDACi, these areas are still in early stages of discovery.
Recent phylogenetic analyses of bacterial HDACs suggest that all four HDAC classes preceded the evolution of histone proteins (Gregoretti et al., 2004). This suggests that the primary activity of HDACs may be directed against non-histone substrates. At least 50 non-histone proteins of known biological function have been identified, which may be acetylated and substrates of HDACs (Table 1) (Glozak et al., 2005; Marks and Dokmanovic, 2005;Rosato and Grant, 2005; Bolden et al., 2006; Minucci and Pelicci, 2006; Zhao et al., 2006). In addition, two recent proteomic studies identified many lysine-acetylated substrates (Iwabata et al., 2005; Kim et al., 2006). In view of all these findings, HDACs may be better called ‘N-epsilon-lysine deacetylase’. This designation would also distinguish them from the enzymes that catalyse other types of deacetylation in biological reactions, such as acylases that catalyse the deacetylation of a range of N-acetyl amino acids (Anders and Dekant, 1994).
Non-histone protein targets of HDACs include transcription factors, transcription regulators, signal transduction mediators, DNA repair enzymes, nuclear import regulators, chaperone proteins, structural proteins, inflammation mediators and viral proteins (Table 1). Acetylation can either increase or decrease the function or stability of the proteins, or protein–protein interaction (Glozak et al., 2005). These HDAC substrates are directly or indirectly involved in many biological processes, such as gene expression and regulation of pathways of proliferation, differentiation and cell death. These data suggest that HDACi could have multiple mechanisms of inducing cell growth arrest and cell death (Figure 1).
HDACi have been discovered with different structural characteristics, including hydroximates, cyclic peptides, aliphatic acids and benzamides (Table 2) (Miller et al., 2003; Yoshida et al., 2003; Marks and Breslow, 2007). Certain HDACi may selectively inhibit different HDACs. For example, MS-275 preferentially inhibits HDAC1 with IC50, at 0.3 m, compared to HDAC3 with an IC50 of about 8 m, and has little or no inhibitory effect against HDAC6 and HDAC8 (Hu et al., 2003). Two novel synthetic compounds, SK7041 and SK7068, preferentially target HDAC1 and 2 and exhibit growth inhibitory effects in human cancer cell lines and tumor xenograft models (Kim et al., 2003a). A small molecule, tubacin, selectively inhibits HDAC6 activity and causes an accumulation of acetylated -tubulin, but does not affect acetylation of histones, and does not inhibit cell cycle progression (Haggarty et al., 2003). No other HDACi for a specific HDAC has been reported.
HDACi selectively alters gene expression
HDACi-induced antitumor pathways
HDACi induces cell cycle arrest
HDACi activates the extrinsic apoptotic pathways
HDACi activates the intrinsic apoptotic pathways
HDACi induces mitotic cell death
HDACi induces autophagic cell death and senescence
ROS, thioredoxin and Trx binding protein 2 in HDACi-induced cell death
Antitumor effects of HDAC6 inhibition
Activation of protein phosphatase 1
Disruption of the function of chaperonin HSP90
Disruption of the aggresome pathway
HDACi inhibits angiogenesis
HDACi can block tumor angiogenesis by inhibition of hypoxia inducible factors (HIF) (Liang et al., 2006). HIF-1 and HIF-2 are transcription factors for angiogenic genes (Brown and Wilson, 2004). The oxygen level can control HIF activity through two mechanisms. First, under normoxic conditions, HIF-1 binds to von Hippel–Lindau protein (pVHL) and is degraded by the ubiquitination–proteasome system. Second, HIF activity depends on its transactivation potential (TAP), which is affected by the interaction with the coactivator p300/CBP among others. This complex can be disrupted by Factor Inhibiting HIF (FIH). Hypoxic conditions activate HIF through repression of the hydroxylases responsible for HIF degradation and loss of function.
Combination of HDACi with other antitumor agents
The HDACi have shown synergistic or additive antitumor effects with a wide range of antitumor reagents, including chemotherapeutic drugs, new targeted therapeutic reagents and radiation, by various mechanisms, some unique for particular combinations (Rosato and Grant, 2004; Bhalla, 2005; Marks and Dokmanovic, 2005; Bolden et al., 2006).
Clinical development of HDACi
At least 14 different HDACi are in some phase of clinical trials as monotherapy or in combination with retinoids, taxols, gemcitabine, radiation, etc, in patients with hematologic and solid tumors, including cancer of lung, breast, pancreas, renal and bladder, melanoma, glioblastoma, leukemias, lymphomas, multiple myeloma (see National Cancer Institute website for CTEP clinical trials, ctep.cancer.gov or clinicaltrials.gov, and website of companies developing HDACi; Table 2).
The resistance to HDACi
Conclusions and perspectives
HDACs have multiple substrates involved in many biological processes, including proliferation, differentiation, apoptosis and other forms of cell death. Indeed, the fact that HDACs have histone and multiple nonhistone protein substrates suggests these enzymes should be referred to as ‘lysine deacetylases’. HDACi can cause transformed cells to undergo growth arrest, differentiation and/or cell death. Normal cells are relatively resistant to HDACi. HDACi are selective in altering gene expression, which may reflect, in part, the proteins composing the transcription factor complex to which HDACs are recruited. Both altered gene expression and changes in non-histone proteins caused by HDACi-induced acetylation play a role in the antitumor activity of HDACi. This is reflected in the different inducer-activated antitumor pathways in transformed cells (Figure 1). The functions of HDACs are not redundant. Thus, a pan-HDAC inhibitor such as vorinostat may activate more antitumor pathways and have therapeutic advantages compared to HDAC isotype-specific inhibitors.
Almost all cancers have multiple defects in the expression and/or structure of proteins that regulate cell proliferation and death. Compared to other antitumor reagents, the plurality of action of HDACi potentially confers efficacy in a wide spectrum of cancers, which have heterogeneity and multiple defects, both among different types of cancer and within different individual tumors of the same type. The multiple defects in a cancer cell may be the reason for transformed cells being more sensitive than normal cells to HDACi. Thus, given the relatively rapid reversibility of vorinostat inhibition of HDACs, normal cells may be able to compensate for HDACi-induced changes more effectively than cancer cells.
HDACi have synergistic or additive antitumor effects with many other antitumor reagents – suggesting that combination of HDACi and other anticancer agents may be very attractive therapeutic strategies for using these agents. Complete understanding of the mechanisms underlying the resistance and sensitivity to HDACi has obvious therapeutic importance. Targeting resistant factors will enhance the antitumor efficacy of HDACi. Identifying markers that can predict response to HDACi is a high priority for expanding the efficacy of these novel anticancer agents.
The complexity of cancer has been known for almost a century, in large part from the seminal work of Otto Warburg in the 1920s using manometry, and following the work of Louis Pasteur 60 years earlier with fungi.
The volume of work and our unlocking of mitotic activity, apoptosis, mitochondria, and the cytoskeleton has taken us further into the cell interior, cell function, metabolic regulation, and pathophysiology. Despite the enormous contributions to our knowledge of genomics, there is a large body of work in pathways of cell function that resides in no small part in activity of protein catalysts and enzymes.
The work that has been described covers only cyclic dinucleotides and HDACi’s. Some of the activities described have relevance to microorganisms as well as cancer. As we have seen, blocking HDACs boosts the activity of regulatory T-cells. There are many specific functional alterations elucidated above.
The first presentation is of an antibody that blocks an immune suppressor called CTLA4, antibodies that block another immune suppressor, PD1. This also involves a protein in cells that responds to foreign DNA to launch an innate immune response — the first response of the body’s immune system. The protein, dubbed STING, is triggered by small molecules called cyclic di-nucleotides (CDN), and makes immune cells release interferon and other cytokines that activate disease-fighting T cells and stimulate the production of antibodies that together kill invading pathogens and destroy cancer cells. Listeria bacteria, for example, secrete a CDN directly into infected cells that activates STING.
The second is resident in acetylation status of histones and non-histone proteins is determined by histone deacetylases (HDACs) and histone acetyl-transferases (HATs). HATs add acetyl groups to lysine residues, while HDACs remove the acetyl groups. In general, acetylation of histone promotes a more relaxed chromatin structure, allowing transcriptional activation. HDACs can act as transcription repressors, due to histone deacetylation, and consequently promote chromatin condensation. HDAC inhibitors (HDACi) selectively alter gene transcription, in part, by chromatin remodeling and by changes in the structure of proteins in transcription factor complexes (Gui et al., 2004). The description focuses on the molecular mechanisms triggered by inhibitors of zinc-dependent HDACs in tumor cells that explain in part: (I) the effects of these compounds in inducing transformed cell death and (II) the relative resistance of normal and certain cancer cells to HDACi induced cell death.
HDACs have multiple substrates involved in many biological processes, including proliferation, differentiation, apoptosis and other forms of cell death. Indeed, the fact that HDACs have histone and multiple nonhistone protein substrates suggests these enzymes should be referred to as ‘lysine deacetylases’. HDACi can cause transformed cells to undergo growth arrest, differentiation and/or cell death. Normal cells are relatively resistant to HDACi. HDACi are selective in altering gene expression, which may reflect, in part, the proteins composing the transcription factor complex to which HDACs are recruited. Both altered gene expression and changes in non-histone proteins caused by HDACi-induced acetylation play a role in the antitumor activity of HDACi.
Below are a curation of reports highlighting both clinical trial failures and serious adverse events reported from clinical trials of various antibody drug or antibody radioligand conjugates. As see below, there have been mutliple failures of these types of biological entitites in oncology clinical trials, either displaying issues related to efficacy and/or safety.
ASCO: J&J’s radioligand spurs responses, but 4 deaths mar early results
By Annalee Armstrong May 24, 2024
Johnson & Johnson’s radiopharmaceutical spurred “profound and durable” responses, however, four patient deaths in the early-stage trial marred the results.
JNJ-6420 is an anti-hK2 antibody-based targeted radioligand therapy that’s designed to deliver a high-energy, short-range alpha-particle emitter to prostate cancer cells. The first-in-human study tested the radiopharmaceutical in patients with metastatic castration-resistant prostate cancer who have previously received at least one prior androgen receptor pathway inhibitor. The results are to be presented next week at the American Society of Clinical Oncology conference. The goal of the phase 1 dose-escalation study was to demonstrate the drug’s safety and to find a dose to move into phase 2. In one trial group, 37 patients were on a fixed dosing starting schedule, receiving between 50 μCi and up to 300 μCi. Twenty-nine patients in the other trial group were capped at a cumulative 500-μCi dose. As of the Jan. 5 data cutoff, 64 patients had received at least one dose of JNJ-6420, with safety data being recorded for 57 patients who had received 150 μCi. Of these patients, 35, or 61%, experienced grade 3 or higher treatment-emergent adverse events (TEAEs), and 21, or 37%, had a serious TEAE. Almost all patients experienced some sort of TEAE. There were four deaths due to TEAEs, which were associated with repeated dosing of JNJ-6420. The full data set linked two of the deaths to interstitial lung disease (ILD), one to respiratory failure related to COVID-19 and one to decreased appetite/hypotension. ILD is a common adverse event in oncology treatment, particularly for antibody-drug conjugates. The condition causes progressive scarring of lung tissue. The deaths related to ILD occurred in patients who had received cumulative doses greater than or equal to 750 μCi. To address the risk of ILD and thrombocytopenia, the study investigators are recommending a cumulative dose cap and an adaptive dose schedule. Evaluation of adaptive dosing is ongoing. Other common TEAEs in the study included anemia and two conditions related to low white blood cells, lymphopenia and leukopenia. Nine patients discontinued treatment. As for the responses, the data showed a reported PSA50 rate of 45.6%. This is a measure of prostatic-specific antigen, which is a key biomarker in prostate cancer. A PSA response is associated with prolonged overall survival.
ADC puts Zynlonta study on hold after 7 patient deaths, 5 other severe adverse events (2023)
DC Therapeutics has slammed the brakes on enrollment in a phase 2 combination trial for Zynlonta as it investigates seven patient deaths and five other severe respiratory events among patients who received the drug.
For the study in unfit or frail patients with previously untreated diffuse large B-cell lymphoma (DLBCL), investigators had enrolled 40 participants. After receiving the ADC drug, 12 of them experienced respiratory-related, treatment-emergent adverse events, ADC said in a Tuesday release.
The investigators concluded that 11 of the events, including six of the deaths, were “unrelated” to the Zynlonta treatment or unlikely to be related to the drug, ADC said. All of the patients who died suffered from at least one “significant” comorbidity, including obstructive pulmonary disease, pulmonary edema, chronic bronchiectasis, idiopathic pulmonary fibrosis or recent COVID-19 infection. All of the patients who passed away were at least 80 years of age, according to the company. ADC said it put a “voluntary pause” on the trial to gain more time to “evaluate data … and determine next steps.” The study was testing ADC’s medicine in combination with Roche’s Rituxan. “Our top priority is the safety of every patient who participates in our clinical trials,” CEO Ameet Mallik said in the company’s statement. “This trial includes a very difficult-to-treat patient population with limited treatment options, and we will provide an update on next steps when available.” ADC has notified the FDA and the European Medicines Agency (EMA) and doesn’t expect to report any additional trial data by the end of the year.
ZYNLONTA®14Q 2023 net sales expected to be ~$16.5 million, a double-digit percentage increase as compared to 3Q 2023
LOTIS-7: Study of ZYNLONTA in combination with bispecifics cleared first dosing cohort with no DLT and with early signs of efficacy
ADCT-601 (targeting AXL): Reached MTD and currently in dose optimization; Early signs of antitumor activity in both monotherapy and in combination
Multiple data catalysts expected in 2024 and with a cash runway now expected into 4Q 2025
LAUSANNE, Switzerland, Jan. 04, 2024 (GLOBE NEWSWIRE) — ADC Therapeutics SA (NYSE: ADCT) today provided business updates.
“During 2023, we took a number of decisive actions to help position the Company for success in 2024 and beyond. We prioritized our pipeline, strengthened our organization and implemented a disciplined capital allocation model to generate cost efficiencies,” said Ameet Mallik, Chief Executive Officer of ADC Therapeutics. “We believe we are starting to see signs of the commercial turnaround. We are also encouraged to see positive initial signals in the LOTIS-7 trial of ZYNLONTA in combination with bispecifics as well as early signs of antitumor activity in the Phase 1b trial of ADCT-601. We now expect our cash runway to extend into the fourth quarter of 2025 and believe we are on a path to unlock the substantial value in the Company.”
Antibody drug conjugates (ADCs) are synthesized by conjugating a cytotoxic drug or “payload” to a monoclonal antibody. The payloads are conjugated using amino or sulfhydryl specific linkers that react with lysines or cysteines on the antibody surface. A typical antibody contains over 60 lysines and up to 12 cysteines as potential conjugation sites. The desired DAR (drugs/antibody ratio) depends on a number of different factors and ranges from two to eight drugs/antibody. The discrepancy between the number of potential conjugation sites and the desired DAR, combined with use of conventional conjugation methods that are not site-specific, results in heterogeneous ADCs that vary in both DAR and conjugation sites. Heterogeneous ADCs contain significant fractions with suboptimal DARs that are known to possess undesired pharmacological properties. As a result, new methods for synthesizing homogeneous ADCs have been developed in order to increase their potential as therapeutic agents. This article will review recently reported processes for preparing ADCs with improved homogeneity. The advantages and potential limitations of each process are discussed, with emphasis on efficiency, quality, and in vivo efficacy relative to similar heterogeneous ADCs.
Antibody drug conjugates (ADCs) are a rapidly growing class of targeted therapeutic agents for treatment of cancer.(1-8) Although the number of ADCs in clinical trials has steadily increased since 2005, many have failed to reach the later stages of clinical development; one has been withdrawn from the market (Mylotarg in 2002), and only two (Adcetris and Kadcyla) are currently approved by the FDA for cancer indications (Figure 1A).(9-11) Thus, far, the approval rate for ADCs has not met early expectations and is lagging behind other antibody-based therapeutics. Based on the number of approved ADCs versus those that have failed to progress into later stage clinical trials, the success rate is reminiscent of that for small molecule drugs. The reasons for the clinical failures of ADCs are often not known or they are still under investigation. More commonly, when the reasons for clinical failure are clear, the information is not made available to the public domain. Emerging preclinical data suggests that heterogeneity, a property shared by most ADCs currently in clinical development (Table 1), may ultimately limit their potential as therapeutic agents.(12, 13)
Table 1. Examples of Heterogeneous ADCs Currently in Clinical Trials for Cancer Indicationsa
Figure 1. (A) Number of ADCs in different stages of clinical development from 2006 to 2014. (B) Structure of a typical IgG antibody showing lysines (red), cysteines (yellow), and glycans (green) as potential conjugation sites.(16)
ADCs are composed of a cytotoxic drug or “payload” conjugated to a tumor selective monoclonal antibody. The heterogeneity of conventional ADCs arises from the synthetic processes currently used for conjugation.(14) Payloads are typically conjugated to the antibody using amino or thiol specific linkers that react with lysines or cysteines on the antibody surface.(15) A typical antibody contains more than 50 lysines and up to 12 cysteines as potential conjugation sites (Figure 1B).(16) The optimal DAR (drugs/antibody ratio) for most ADCs, however, ranges from 2 to 8 drugs/antibody and is dependent upon a variety of different factors. The discrepancy between the number of potential conjugation sites and the desired DAR, combined with the use of conjugation methods that are not site-specific, result in heterogeneous ADCs that vary in both DAR and conjugation sites. Consequently, conventional heterogeneous ADCs often contain significant amounts of unconjugated antibody in addition to fractions with suboptimal DARs. Unconjugated antibodies can compete for antigen binding and inhibit ADC activity, while fractions with suboptimal DARs are frequently prone to aggregation, poor solubility, and/or instability that ultimately result in a poor therapeutic window.(17, 18)
The relative degree of ADC heterogeneity depends on the methods used for conjugation. For example, Kadcyla, an ADC approved in 2013 for breast cancer, is synthesized using a two-step process in which the linker and payload are conjugated in separate steps (Scheme 1A).(19-21)The linker contains an amino-specific NHS ester that reacts with antibody lysines in the first step and a thiol-specific maleimide group that reacts with a maytansinoid payload in the second step. The process affords a highly heterogeneous mixture of ADC molecules containing from 0 to 10 payloads/antibody with an average DAR of 3.5 drugs/antibody.(22, 23) Additional heterogeneity arises due to distribution of the payloads across dozens of potential conjugation sites. As a result, Kadcyla contains hundreds of different ADC molecules, each with its own unique pharmacological properties.(24)
Scheme 1. (A) General Process for Synthesizing ADCs such as Kadcyla via Lysine Conjugation; (B) General Process for Synthesizing ADCs, such as Adcetris, via Cysteine Conjugation
Conjugation of payloads to antibodies through interchain cysteines reduces ADC heterogeneity relative to lysine conjugation because there are fewer potential conjugation sites. Adcetris, an ADC approved in 2011 for treatment of Hodgkin’s lymphoma, is an example of a cysteine conjugated ADC.(25-27) The process for cysteine conjugation involves partial reduction of four antibody interchain disulfide bonds to generate up to eight reactive thiol groups. The partially reduced antibody is subsequently conjugated to a payload containing a thiol-specific maleimide linker. The payload used for Adcetris is monomethyl auristatin E (MMAE) and contains a protease cleavable maleimide linker (Scheme 1B). Although Adcetris is less heterogeneous than Kadcyla, it is composed of dozens of different ADC molecules containing 0 to 8 payloads with an average DAR of 3.6 drugs/antibody.(28) Like most cysteine conjugated ADCs, Adcetris has a reduced half-life in vivo compared to the parent antibody, cAC10. The diminished half-life has been attributed to rapid clearance of high DAR species (>4 drugs/antibody) and to partial loss of interchain disulfide bonds during the conjugation process.(29, 30)
Although different processes for lysine and cysteine conjugation are used to synthesize Adcetris and Kadcyla, both ADCs contain thio-succinimide bonds between the payload and the antibody, which originate from the use of maleimide linkers in the conjugation processes. Kadcyla contains a thio-succinimide between the linker and the payload (Scheme 1A), while Adcetris contains a thio-succinimide bond between the linker and the antibody (Scheme 1B). Thio-succinimide groups are known to undergo undesired side reactions such as elimination or thiol exchange that can result in premature release of the payloads from the ADC and lead to reduced potency and/or increased systemic toxicity.(31, 32)
Despite the known limitations of conventional heterogeneous ADCs, most ADCs currently in clinical development utilize similar conjugation methods to those described in Scheme 1. As a result, they are likely to possess similar pharmacological properties to Adcetris and Kadcyla, in addition to other less successful ADCs that may have performed poorly in clinical trials. In order to improve the pharmacological properties of current and future ADCs, new site-specific conjugation processes for synthesizing homogeneous ADCs are now being developed.(33-36)
Site-specific conjugation processes for constructing homogeneous ADCs can be divided into three different categories. Two are focused on antibody modification (engineered amino acids and enzyme mediated), while the third category is focused on linker modification. The categories can be subdivided further based on the specific processes that are used (Table 2). Examples from each process were selected based on availability of sufficient preclinical data to enable comparison with similar conventional heterogeneous ADCs. Homogeneous ADCs derived from these processes have only just begun to enter clinical trials. Whether they will outperform their heterogeneous counterparts in clinical trials remains uncertain, but preclinical data suggest that homogeneous ADCs are likely to dominate future clinical trials and will lead to improved clinical results.
Table 2. Summary of Different Processes for Constructing Homogeneous ADCs
…….
All of the processes reviewed here were successfully used to construct ADCs with improved homogeneity over ADCs synthesized using conventional methods. A majority of approaches utilize recombinant antibody engineering to introduce unique functional groups for site-specific conjugation. The unique functional groups were introduced either as point mutations for cysteine and non-natural amino acids or as enzyme recognition tags. These recombinant engineering approaches offer several potential advantages over nonrecombinant approaches. For example, engineered cysteines can be incorporated into dozens of different sites with minimal impact on the functional properties of the antibody. This enables ADCs to be optimized for conjugation efficiency, linker stability, and potency. Engineered non-natural amino acids offer additional advantages due to the diverse array of different functional groups that can be introduced. Furthermore, non-natural amino acids enable a variety of new linker chemistries to be investigated that are not possible with conventional conjugation processes.
The flexibility offered by recombinant processes may also represent their greatest challenge. The importance of the conjugation site for ADC activity is well-established, but additional factors should be considered before selecting a development candidate. Potential effects on antibody expression, conjugation efficiency, linker stability, aggregation, and other factors need to be considered before selecting a specific conjugation site. These factors can ultimately determine the success or failure of an ADC development program. Since antibodies share many of the same properties, it seems likely that optimal conjugation sites will be identified that are broadly effective when used with different antibodies. Other potential challenges for processes involving antibody engineering include increased development time and costs, immunogenicity of engineered sequence tags, scalability, and use of novel linkers and payloads that are not yet clinically validated.
In addition to homogeneity, improvements in other ADC properties such as potency, stability and half-life were observed. In fact, many of the homogeneous ADCs derived from these processes out-performed conventional heterogeneous ADCs in efficacy and safety studies. Much of their success has been attributed to elimination of high DAR species present in conventional ADCs. In general, experimental results are consistent with this conclusion, and many would agree that substantial progress has resulted from these efforts to improve ADC homogeneity. Ironically, the relative contribution of homogeneity to the improved properties of the engineered ADCs could not be determined from most studies because other factors known to effect ADC activity could not be ruled out.
For instance, recombinant approaches for making homogeneous ADCs were designed to introduce conjugation sites in different locations from those used in conventional methods. Since it is now well-established that “location matters”, the observed differences in activity between TDCs (or NDCs) and the conventional ADC controls could result from different conjugation sites, rather than from elimination of high DAR species. Enzyme mediated approaches face similar challenges when comparing homogeneous and heterogeneous ADCs because the conjugation sites are different. Other variables such as linker type (cleavable or noncleavable) and payload (maytansine or PBD) need to be carefully controlled before reaching conclusions about the benefits of homogeneity.
Linker based processes are more suitable for comparing homogeneous ADCs with conventional heterogeneous ADCs because they utilize the same conjugation sites. Once other variables that might impact ADC activity were carefully controlled, the relative benefits of homogeneity were revealed for the first time and the results confirmed that efforts to improve ADC homogeneity have been a worthwhile endeavor.
Most of the processes reviewed here are still in early phases of clinical development. All of the methods have advantages and limitations that will ultimately decide which approach will become the preferred process for manufacturing homogeneous ADCs. It is not yet clear which process will rise above the others as a preferred method, but all of these approaches have contributed valuable information to our knowledge base and resulted in ADCs with improved pharmacological properties over conventional heterogeneous ADCs. Our future challenge will be to apply this knowledge to develop ADCs that will be more effective as therapeutic agents. Our ability to synthesize homogeneous ADCs provides another reason to be optimistic about the future of ADCs.
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
Gene Editing with CRISPR gets Crisper, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Gene Editing with CRISPR gets Crisper
Curators: Larry H. Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
CRISPR Moves from Butchery to Surgery
More Genomes Are Going Under the CRISPR Knife, So Surgical Standards Are Rising
The Dharmacon subsidary of GE Healthcare provides the Edit-R Lentiviral Gene Engineering platform. It is based on the natural S. pyrogenes system, but unlike that system, which uses a single guide RNA (sgRNA), the platform uses two component RNAs, a gene-specific CRISPR RNA (crRNA) and a universal trans-activating crRNA (tracrRNA). Once hybridized to the universal tracrRNA (blue), the crRNA (green) directs the Cas9 nuclease to a specific genomic region to induce a double- strand break.
Scientists recently convened at the CRISPR Precision Gene Editing Congress, held in Boston, to discuss the new technology. As with any new technique, scientists have discovered that CRISPR comes with its own set of challenges, and the Congress focused its discussion around improving specificity, efficiency, and delivery.
In the naturally occurring system, CRISPR-Cas9 works like a self-vaccination in the bacterial immune system by targeting and cleaving viral DNA sequences stored from previous encounters with invading phages. The endogenous system uses two RNA elements, CRISPR RNA (crRNA) and trans-activating RNA (tracrRNA), which come together and guide the Cas9 nuclease to the target DNA.
Early publications that demonstrated CRISPR gene editing in mammalian cells combined the crRNA and tracrRNA sequences to form one long transcript called asingle-guide RNA (sgRNA). However, an alternative approach is being explored by scientists at the Dharmacon subsidiary of GE Healthcare. These scientists have a system that mimics the endogenous system through a synthetic two-component approach thatpreserves individual crRNA and tracrRNA. The tracrRNA is universal to any gene target or species; the crRNA contains the information needed to target the gene of interest.
Predesigned Guide RNAs
In contrast to sgRNAs, which are generated through either in vitro transcription of a DNA template or a plasmid-based expression system, synthetic crRNA and tracrRNA eliminate the need for additional cloning and purification steps. The efficacy of guide RNA (gRNA), whether delivered as a sgRNA or individual crRNA and tracrRNA, depends not only on DNA binding, but also on the generation of an indel that will deliver the coup de grâce to gene function.
“Almost all of the gRNAs were able to create a break in genomic DNA,” said Louise Baskin, senior product manager at Dharmacon. “But there was a very wide range in efficiency and in creating functional protein knock-outs.”
To remove the guesswork from gRNA design, Dharmacon developed an algorithm to predict gene knockout efficiency using wet-lab data. They also incorporated specificity as a component of their algorithm, using a much more comprehensive alignment tool to predict potential off-target effects caused by mismatches and bulges often missed by other alignment tools. Customers can enter their target gene to access predesigned gRNAs as either two-component RNAs or lentiviral sgRNA vectors for multiple applications.
“We put time and effort into our algorithm to ensure that our guide RNAs are not only functional but also highly specific,” asserts Baskin. “As a result, customers don’t have to do any design work.”
MilliporeSigma’s CRISPR Epigenetic Activator is based on fusion of a nuclease-deficient Cas9 (dCas9) to the catalytic histone acetyltransferase (HAT) core domain of the human E1A-associated protein p300. This technology allows researchers to target specific DNA regions or gene sequences. Researchers can localize epigenetic changes to their target of interest and see the effects of those changes in gene expression.
Knockout experiments are a powerful tool for analyzing gene function. However, for researchers who want to introduce DNA into the genome, guide design, donor DNA selection, and Cas9 activity are paramount to successful DNA integration.MilliporeSigma offers two formats for donor DNA: double-stranded DNA (dsDNA) plasmids and single-stranded DNA (ssDNA) oligonucleotides. The most appropriate format depends on cell type and length of the donor DNA. “There are some cell types that have immune responses to dsDNA,” said Gregory Davis, Ph.D., R&D manager, MilliporeSigma.
The ssDNA format can save researchers time and money, but it has a limited carrying capacity of approximately 120 base pairs.In addition to selecting an appropriate donor DNA format, controlling where, how, and when the Cas9 enzyme cuts can affect gene-editing efficiency. Scientists are playing tug-of-war, trying to pull cells toward the preferred homology-directed repair (HDR) and away from the less favored nonhomologous end joining (NHEJ) repair mechanism.One method to achieve this modifies the Cas9 enzyme to generate a nickase that cuts only one DNA strand instead of creating a double-strand break. Accordingly, MilliporeSigma has created a Cas9 paired-nickase system that promotes HDR, while also limiting off-target effects and increasing the number of sequences available for site-dependent gene modifications, such as disease-associated single nucleotide polymorphisms (SNPs).“The best thing you can do is to cut as close to the SNP as possible,” advised Dr. Davis. “As you move the double-stranded break away from the site of mutation you get an exponential drop in the frequency of recombination.”
Ribonucleo-protein Complexes
Another strategy to improve gene-editing efficiency, developed by Thermo Fisher, involves combining purified Cas9 protein with gRNA to generate a stable ribonucleoprotein (RNP) complex. In contrast to plasmid- or mRNA-based formats, which require transcription and/or translation, the Cas9 RNP complex cuts DNA immediately after entering the cell. Rapid clearance of the complex from the cell helps to minimize off-target effects, and, unlike a viral vector, the transient complex does not introduce foreign DNA sequences into the genome.
To deliver their Cas9 RNP complex to cells, Thermo Fisher has developed a lipofectamine transfection reagent called CRISPRMAX. “We went back to the drawing board with our delivery, screened a bunch of components, and got a brand-new, fully optimized lipid nanoparticle formulation,” explained Jon Chesnut, Ph.D., the company’s senior director of synthetic biology R&D. “The formulation is specifically designed for delivering the RNP to cells more efficiently.”
Besides the reagent and the formulation, Thermo Fisher has also developed a range of gene-editing tools. For example, it has introduced the Neon® transfection system for delivering DNA, RNA, or protein into cells via electroporation. Dr. Chesnut emphasized the company’s focus on simplifying complex workflows by optimizing protocols and pairing everything with the appropriate up- and downstream reagents.
From Mammalian Cells to Microbes
One of the first sources of CRISPR technology was the Feng Zhang laboratory at the Broad Institute, which counted among its first licensees a company called GenScript. This company offers a gene-editing service called GenCRISPR™ to establish mammalian cell lines with CRISPR-derived gene knockouts.
“There are a lot of challenges with mammalian cells, and each cell line has its own set of issues,” said Laura Geuss, a marketing specialist at GenScript. “We try to offer a variety of packages that can help customers who have difficult-to-work-with cells.” These packages include both viral-based and transient transfection techniques.
However, the most distinctive service offered by GenScript is its microbial genome-editing service for bacteria (Escherichia coli) and yeast (Saccharomyces cerevisiae). The company’s strategy for gene editing in bacteria can enable seamless knockins, knockouts, or gene replacements by combining CRISPR with lambda red recombineering. Traditionally one of the most effective methods for gene editing in microbes, recombineering allows editing without restriction enzymes through in vivo homologous recombination mediated by a phage-based recombination system such as lambda red.
On its own, lambda red technology cannot target multiple genes, but when paired with CRISPR, it allows the editing of multiple genes with greater efficiency than is possible with CRISPR alone, as the lambda red proteins help repair double-strand breaks in E. coli. The ability to knockout different gene combinations makes Genscript’s microbial editing service particularly well suited for the optimization of metabolic pathways.
Pooled and Arrayed Library Strategies
Scientists are using CRISPR technology for applications such as metabolic engineering and drug development. Yet another application area benefitting from CRISPR technology is cancer research. Here, the use of pooled CRISPR libraries is becoming commonplace. Pooled CRISPR libraries can help detect mutations that affect drug resistance, and they can aid in patient stratification and clinical trial design.
Pooled screening uses proliferation or viability as a phenotype to assess how genetic alterations, resulting from the application of a pooled CRISPR library, affect cell growth and death in the presence of a therapeutic compound. The enrichment or depletion of different gRNA populations is quantified using deep sequencing to identify the genomic edits that result in changes to cell viability.
MilliporeSigma provides pooled CRISPR libraries ranging from the whole human genome to smaller custom pools for these gene-function experiments. For pharmaceutical and biotech companies, Horizon Discovery offers a pooled screening service, ResponderSCREEN, which provides a whole-genome pooled screen to identify genes that confer sensitivity or resistance to a compound. This service is comprehensive, taking clients from experimental design all the way through to suggestions for follow-up studies.
Horizon Discovery maintains a Research Biotech business unit that is focused on target discovery and enabling translational medicine in oncology. “Our internal backbone gives us the ability to provide expert advice demonstrated by results,” said Jon Moore, Ph.D., the company’s CSO.
In contrast to a pooled screen, where thousands of gRNA are combined in one tube, an arrayed screen applies one gRNA per well, removing the need for deep sequencing and broadening the options for different endpoint assays. To establish and distribute a whole-genome arrayed lentiviral CRISPR library, MilliporeSigma partnered with the Wellcome Trust Sanger Institute. “This is the first and only arrayed CRISPR library in the world,” declared Shawn Shafer, Ph.D., functional genomics market segment manager, MilliporeSigma. “We were really proud to partner with Sanger on this.”
Pooled and arrayed screens are powerful tools for studying gene function. The appropriate platform for an experiment, however, will be determined by the desired endpoint assay.
The QX200 Droplet Digital PCR System from Bio-Rad Laboratories can provide researchers with an absolute measure of target DNA molecules for EvaGreen or probe-based digital PCR applications. The system, which can provide rapid, low-cost, ultra-sensitive quantification of both NHEJ- and HDR-editing events, consists of two instruments, the QX200 Droplet Generator and the QX200 Droplet Reader, and their associated consumables.
Finally, one last challenge for CRISPR lies in the detection and quantification of changes made to the genome post-editing. Conventional methods for detecting these alterations include gel methods and next-generation sequencing. While gel methods lack sensitivity and scalability, next-generation sequencing is costly and requires intensive bioinformatics.
To address this gap, Bio-Rad Laboratories developed a set of assay strategies to enable sensitive and precise edit detection with its Droplet Digital PCR (ddPCR) technology. The platform is designed to enable absolute quantification of nucleic acids with high sensitivity, high precision, and short turnaround time through massive droplet partitioning of samples.
Using a validated assay, a typical ddPCR experiment takes about five to six hours to complete. The ddPCR platform enables detection of rare mutations, and publications have reported detection of precise edits at a frequency of <0.05%, and of NHEJ-derived indels at a frequency as low as 0.1%. In addition to quantifying precise edits, indels, and computationally predicted off-target mutations, ddPCR can also be used to characterize the consequences of edits at the RNA level.
According to a recently published Science paper, the laboratory of Charles A. Gersbach, Ph.D., at Duke University used ddPCR in a study of muscle function in a mouse model of Duchenne muscular dystrophy. Specifically, ddPCR was used to assess the efficiency of CRISPR-Cas9 in removing the mutated exon 23 from the dystrophin gene. (Exon 23 deletion by CRISPR-Cas9 resulted in expression of the modified dystrophin gene and significant enhancement of muscle force.)
Quantitative ddPCR showed that exon 23 was deleted in ~2% of all alleles from the whole-muscle lysate. Further ddPCR studies found that 59% of mRNA transcripts reflected the deletion.
“There’s an overarching idea that the genome-editing field is moving extremely quickly, and for good reason,” asserted Jennifer Berman, Ph.D., staff scientist, Bio-Rad Laboratories. “There’s a lot of exciting work to be done, but detection and quantification of edits can be a bottleneck for researchers.”
The gene-editing field is moving quickly, and new innovations are finding their way into the laboratory as researchers lay the foundation for precise, well-controlled gene editing with CRISPR.
Researchers utilized a systems biology approach to develop new methods to assess drug sensitivity in cells. [The Institute for Systems Biology]
Understanding how cells respond and proliferate in the presence of anticancer compounds has been the foundation of drug discovery ideology for decades. Now, a new study from scientists at Vanderbilt University casts significant suspicion on the primary method used to test compounds for anticancer activity in cells—instilling doubt on methods employed by the entire scientific enterprise and pharmaceutical industry to discover new cancer drugs.
“More than 90% of candidate cancer drugs fail in late-stage clinical trials, costing hundreds of millions of dollars,” explained co-senior author Vito Quaranta, M.D., director of the Quantitative Systems Biology Center at Vanderbilt. “The flawed in vitro drug discovery metric may not be the only responsible factor, but it may be worth pursuing an estimate of its impact.”
The Vanderbilt investigators have developed what they believe to be a new metric for evaluating a compound’s effect on cell proliferation—called the DIP (drug-induced proliferation) rate—that overcomes the flawed bias in the traditional method.
The findings from this study were published recently in Nature Methods in an article entitled “An Unbiased Metric of Antiproliferative Drug Effect In Vitro.”
For more than three decades, researchers have evaluated the ability of a compound to kill cells by adding the compound in vitro and counting how many cells are alive after 72 hours. Yet, proliferation assays that measure cell number at a single time point don’t take into account the bias introduced by exponential cell proliferation, even in the presence of the drug.
“Cells are not uniform, they all proliferate exponentially, but at different rates,” Dr. Quaranta noted. “At 72 hours, some cells will have doubled three times and others will not have doubled at all.”
Dr. Quaranta added that drugs don’t all behave the same way on every cell line—for example, a drug might have an immediate effect on one cell line and a delayed effect on another.
The research team decided to take a systems biology approach, a mixture of experimentation and mathematical modeling, to demonstrate the time-dependent bias in static proliferation assays and to develop the time-independent DIP rate metric.
“Systems biology is what really makes the difference here,” Dr. Quaranta remarked. “It’s about understanding cells—and life—as dynamic systems.”This new study is of particular importance in light of recent international efforts to generate data sets that include the responses of thousands of cell lines to hundreds of compounds. Using the
Cancer Cell Line Encyclopedia (CCLE) and
Genomics of Drug Sensitivity in Cancer (GDSC) databases
will allow drug discovery scientists to include drug response data along with genomic and proteomic data that detail each cell line’s molecular makeup.
“The idea is to look for statistical correlations—these particular cell lines with this particular makeup are sensitive to these types of compounds—to use these large databases as discovery tools for new therapeutic targets in cancer,” Dr. Quaranta stated. “If the metric by which you’ve evaluated the drug sensitivity of the cells is wrong, your statistical correlations are basically no good.”
The Vanderbilt team evaluated the responses from four different melanoma cell lines to the drug vemurafenib, currently used to treat melanoma, with the standard metric—used for the CCLE and GDSC databases—and with the DIP rate. In one cell line, they found a glaring disagreement between the two metrics.
“The static metric says that the cell line is very sensitive to vemurafenib. However, our analysis shows this is not the case,” said co-lead study author Leonard Harris, Ph.D., a systems biology postdoctoral fellow at Vanderbilt. “A brief period of drug sensitivity, quickly followed by rebound, fools the static metric, but not the DIP rate.”
Dr. Quaranta added that the findings “suggest we should expect melanoma tumors treated with this drug to come back, and that’s what has happened, puzzling investigators. DIP rate analyses may help solve this conundrum, leading to better treatment strategies.”
The researchers noted that using the DIP rate is possible because of advances in automation, robotics, microscopy, and image processing. Moreover, the DIP rate metric offers another advantage—it can reveal which drugs are truly cytotoxic (cell killing), rather than merely cytostatic (cell growth inhibiting). Although cytostatic drugs may initially have promising therapeutic effects, they may leave tumor cells alive that then have the potential to cause the cancer to recur.
The Vanderbilt team is currently in the process of identifying commercial entities that can further refine the software and make it widely available to the research community to inform drug discovery.
An unbiased metric of antiproliferative drug effect in vitro
In vitro cell proliferation assays are widely used in pharmacology, molecular biology, and drug discovery. Using theoretical modeling and experimentation, we show that current metrics of antiproliferative small molecule effect suffer from time-dependent bias, leading to inaccurate assessments of parameters such as drug potency and efficacy. We propose the drug-induced proliferation (DIP) rate, the slope of the line on a plot of cell population doublings versus time, as an alternative, time-independent metric.
Researchers develop a technique to direct chromosome recombination with CRISPR/Cas9, allowing high-resolution genetic mapping of phenotypic traits in yeast.
Researchers used CRISPR/Cas9 to make a targeted double-strand break (DSB) in one arm of a yeast chromosome labeled with a green fluorescent protein (GFP) gene. A within-cell mechanism called homologous repair (HR) mends the broken arm using its homolog, resulting in a recombined region from the site of the break to the chromosome tip. When this cell divides by mitosis, each daughter cell will contain a homozygous section in an outcome known as “loss of heterozygosity” (LOH). One of the daughter cells is detectable because, due to complete loss of the GFP gene, it will no longer be fluorescent.REPRINTED WITH PERMISSION FROM M.J. SADHU ET AL., SCIENCE
When mapping phenotypic traits to specific loci, scientists typically rely on the natural recombination of chromosomes during meiotic cell division in order to infer the positions of responsible genes. But recombination events vary with species and chromosome region, giving researchers little control over which areas of the genome are shuffled. Now, a team at the University of California, Los Angeles (UCLA), has found a way around these problems by using CRISPR/Cas9 to direct targeted recombination events during mitotic cell division in yeast. The team described its technique today (May 5) in Science.
“Current methods rely on events that happen naturally during meiosis,” explained study coauthor Leonid Kruglyak of UCLA. “Whatever rate those events occur at, you’re kind of stuck with. Our idea was that using CRISPR, we can generate those events at will, exactly where we want them, in large numbers, and in a way that’s easy for us to pull out the cells in which they happened.”
Generally, researchers use coinheritance of a trait of interest with specific genetic markers—whose positions are known—to figure out what part of the genome is responsible for a given phenotype. But the procedure often requires impractically large numbers of progeny or generations to observe the few cases in which coinheritance happens to be disrupted informatively. What’s more, the resolution of mapping is limited by the length of the smallest sequence shuffled by recombination—and that sequence could include several genes or gene variants.
“Once you get down to that minimal region, you’re done,” said Kruglyak. “You need to switch to other methods to test every gene and every variant in that region, and that can be anywhere from challenging to impossible.”
But programmable, DNA-cutting champion CRISPR/Cas9 offered an alternative. During mitotic—rather than meiotic—cell division, rare, double-strand breaks in one arm of a chromosome preparing to split are sometimes repaired by a mechanism called homologous recombination. This mechanism uses the other chromosome in the homologous pair to replace the sequence from the break down to the end of the broken arm. Normally, such mitotic recombination happens so rarely as to be impractical for mapping purposes. With CRISPR/Cas9, however, the researchers found that they could direct double-strand breaks to any locus along a chromosome of interest (provided it was heterozygous—to ensure that only one of the chromosomes would be cut), thus controlling the sites of recombination.
Combining this technique with a signal of recombination success, such as a green fluorescent protein (GFP) gene at the tip of one chromosome in the pair, allowed the researchers to pick out cells in which recombination had occurred: if the technique failed, both daughter cells produced by mitotic division would be heterozygous, with one copy of the signal gene each. But if it succeeded, one cell would end up with two copies, and the other cell with none—an outcome called loss of heterozygosity.
“If we get loss of heterozygosity . . . half the cells derived after that loss of heterozygosity event won’t have GFP anymore,” study coauthor Meru Sadhu of UCLA explained. “We search for these cells that don’t have GFP out of the general population of cells.” If these non-fluorescent cells with loss of heterozygosity have the same phenotype as the parent for a trait of interest, then CRISPR/Cas9-targeted recombination missed the responsible gene. If the phenotype is affected, however, then the trait must be linked to a locus in the recombined, now-homozygous region, somewhere between the cut site and the GFP gene.
By systematically making cuts using CRISPR/Cas9 along chromosomes in a hybrid, diploid strain ofSaccharomyces cerevisiae yeast, picking out non-fluorescent cells, and then observing the phenotype, the UCLA team demonstrated that it could rapidly identify the phenotypic contribution of specific gene variants. “We can simply walk along the chromosome and at every [variant] position we can ask, does it matter for the trait we’re studying?” explained Kruglyak.
For example, the team showed that manganese sensitivity—a well-defined phenotypic trait in lab yeast—could be pinpointed using this method to a single nucleotide polymorphism (SNP) in a gene encoding the Pmr1 protein (a manganese transporter).
Jason Moffat, a molecular geneticist at the University of Toronto who was not involved in the work, toldThe Scientist that researchers had “dreamed about” exploiting these sorts of mechanisms for mapping purposes, but without CRISPR, such techniques were previously out of reach. Until now, “it hasn’t been so easy to actually make double-stranded breaks on one copy of a pair of chromosomes, and then follow loss of heterozygosity in mitosis,” he said, adding that he hopes to see the approach translated into human cell lines.
Applying the technique beyond yeast will be important, agreed cell and developmental biologist Ethan Bier of the University of California, San Diego, because chromosomal repair varies among organisms. “In yeast, they absolutely demonstrate the power of [this method],” he said. “We’ll just have to see how the technology develops in other systems that are going to be far less suited to the technology than yeast. . . . I would like to see it implemented in another system to show that they can get the same oomph out of it in, say, mammalian somatic cells.”
Kruglyak told The Scientist that work in higher organisms, though planned, is still in early stages; currently, his team is working to apply the technique to map loci responsible for trait differences between—rather than within—yeast species.
“We have a much poorer understanding of the differences across species,” Sadhu explained. “Except for a few specific examples, we’re pretty much in the dark there.”
Linkage and association studies have mapped thousands of genomic regions that contribute to phenotypic variation, but narrowing these regions to the underlying causal genes and variants has proven much more challenging. Resolution of genetic mapping is limited by the recombination rate. We developed a method that uses CRISPR to build mapping panels with targeted recombination events. We tested the method by generating a panel with recombination events spaced along a yeast chromosome arm, mapping trait variation, and then targeting a high density of recombination events to the region of interest. Using this approach, we fine-mapped manganese sensitivity to a single polymorphism in the transporter Pmr1. Targeting recombination events to regions of interest allows us to rapidly and systematically identify causal variants underlying trait differences.
Thank you, David, for the kind words and comments. We agree that the most immediate applications of the CRISPR-based recombination mapping will be in unicellular organisms and cell culture. We also think the method holds a lot of promise for research in multicellular organisms, although we did not mean to imply that it “will be an efficient mapping method for all multicellular organisms”. Every organism will have its own set of constraints as well as experimental tools that will be relevant when adapting a new technique. To best help experts working on these organisms, here are our thoughts on your questions.
You asked about mutagenesis during recombination. We Sanger sequenced 72 of our LOH lines at the recombination site and did not observe any mutations, as described in the supplementary materials. We expect the absence of mutagenesis is because we targeted heterozygous sites where the untargeted allele did not have a usable PAM site; thus, following LOH, the targeted site is no longer present and cutting stops. In your experiments you targeted sites that were homozygous; thus, following recombination, the CRISPR target site persisted, and continued cutting ultimately led to repair by NHEJ and mutagenesis.
As to the more general question of the optimal mapping strategies in different organisms, they will depend on the ease of generating and screening for editing events, the cost and logistics of maintaining and typing many lines, and generation time, among other factors. It sounds like in Drosophila today, your related approach of generating markers with CRISPR, and then enriching for natural recombination events that separate them, is preferable. In yeast, we’ve found the opposite to be the case. As you note, even in Drosophila, our approach may be preferable for regions with low or highly non-uniform recombination rates.
Finally, mapping in sterile interspecies hybrids should be straightforward for unicellular hybrids (of which there are many examples) and for cells cultured from hybrid animals or plants. For studies in hybrid multicellular organisms, we agree that driving mitotic recombination in the early embryo may be the most promising approach. Chimeric individuals with mitotic clones will be sufficient for many traits. Depending on the system, it may in fact be possible to generate diploid individuals with uniform LOH genotype, but this is certainly beyond the scope of our paper. The calculation of the number of lines assumes that the mapping is done in a single step; as you note in your earlier comment, mapping sequentially can reduce this number dramatically.
This is a lovely method and should find wide applicability in many settings, especially for microorganisms and cell lines. However, it is not clear that this approach will be, as implied by the discussion, an efficient mapping method for all multicellular organisms. I have performed similar experiments in Drosophila, focused on meiotic recombination, on a much smaller scale, and found that CRISPR-Cas9 can indeed generate targeted recombination at gRNA target sites. In every case I tested, I found that the recombination event was associated with a deletion at the gRNA site, which is probably unimportant for most mapping efforts, but may be a concern in some specific cases, for example for clinical applications. It would be interesting to know how often mutations occurred at the targeted gRNA site in this study.
The wider issue, however, is whether CRISPR-mediated recombination will be more efficient than other methods of mapping. After careful consideration of all the costs and the time involved in each of the steps for Drosophila, we have decided that targeted meiotic recombination using flanking visible markers will be, in most cases, considerably more efficient than CRISPR-mediated recombination. This is mainly due to the large expense of injecting embryos and the extensive effort and time required to screen injected animals for appropriate events. It is both cheaper and faster to generate markers (with CRISPR) and then perform a large meiotic recombination mapping experiment than it would be to generate the lines required for CRISPR-mediated recombination mapping. It is possible to dramatically reduce costs by, for example, mapping sequentially at finer resolution. But this approach would require much more time than marker-assisted mapping. If someone develops a rapid and cheap method of reliably introducing DNA into Drosophila embryos, then this calculus might change.
However, it is possible to imagine situations where CRISPR-mediated mapping would be preferable, even for Drosophila. For example, some genomic regions display extremely low or highly non-uniform recombination rates. It is possible that CRISPR-mediated mapping could provide a reasonable approach to fine mapping genes in these regions.
The authors also propose the exciting possibility that CRISPR-mediated loss of heterozygosity could be used to map traits in sterile species hybrids. It is not entirely obvious to me how this experiment would proceed and I hope the authors can illuminate me. If we imagine driving a recombination event in the early embryo (with maternal Cas9 from one parent and gRNA from a second parent), then at best we would end up with chimeric individuals carrying mitotic clones. I don’t think one could generate diploid animals where all cells carried the same loss of heterozygosity event. Even if we could, this experiment would require construction of a substantial number of stable transgenic lines expressing gRNAs. Mapping an ~20Mbp chromosome arm to ~10kb would require on the order of two-thousand transgenic lines. Not an undertaking to be taken lightly. It is already possible to perform similar tests (hemizygosity tests) using D. melanogaster deficiency lines in crosses with D. simulans, so perhaps CRISPR-mediated LOH could complement these deficiency screens for fine mapping efforts. But, at the moment, it is not clear to me how to do the experiment.
For the first time, treatment with genetically engineered T-cells has used CD4 T-cells instead of the CD8 T-cells, which are used in the chimeric antigen receptor (CAR) T-cell approach. Early data suggest that this CD4 T-cell approach has activity against solid tumors, whereas the CAR T-cell approach so far has achieved dramatic success in hematologic malignancies.
In the new approach, CD4 T-cells were genetically engineered to target MAGE-A3, a protein found on many tumor cells. The treatment was found to be safe in patients with metastatic cancers, according to data from a phase 1 clinical study presented here at the American Association for Cancer Research (AACR) 2016 Annual Meeting.
“This is the first trial testing an immunotherapy using genetically engineered CD4 T-cells,” senior author Steven A. Rosenberg, MD, PhD, chief of the Surgery Branch at the National Cancer Institute (NCI), told Medscape Medical News.
Most approaches use CD8 T-cells. Although CD8 T-cells are known be cytotoxic and CD4 T-cells are normally considered helper cells, CD4 T-cells can induce tumor regression, he said.
Louis M. Weiner, MD, director of the Lombardi Comprehensive Cancer Center at Georgetown University, in Washington, DC, indicated that in contrast with CAR T-cells, these CD4 T-cells target proteins on solid tumors. “CAR T-cells are not tumor specific and do not target solid tumors,” he said.
Engineering CD4 Cells
Immunotherapy with engineered CD4 T-cells was personalized for each patient whose tumors had not responded to or had recurred following treatment with least one standard therapy. The immunotherapy was specific for patients in whom a specific human leukocyte antigen (HLA) — HLA-DPB1*0401 — was found to be expressed on their cells and whose tumors expressed MAGE-A3.
MAGE-A3 belongs to a class of proteins expressed during fetal development. The expression is lost in normal adult tissue but is reexpressed on tumor cells, explained presenter Yong-Chen William Lu, PhD, a research fellow in the Surgery Branch of the NCI.
Targeting MAGE-A3 is relevant, because it is frequently expressed in a variety of cancers, such as melanoma and urothelial, esophageal, and cervical cancers, he pointed out.
Researchers purified CD4 T-cells from the peripheral blood of patients. Next, the CD4 T-cells were genetically engineered with a retrovirus carrying the T-cell receptor (TCR) gene that recognizes MAGE-A3. The modified cells were grown ex vivo and were transferred back into the patient.
Clinical Results
Dr Lu presented data for 14 patients enrolled into the study: eight patients received cell doses from 10 million to 30 billion cells, and six patients received up to 100 billion cells.
This was similar to a phase 1 dose-finding study, except the researchers were seeking to determine the maximum number of genetically engineered CD4 T-cells that a patient could safely receive.
One patient with metastatic cervical cancer, another with metastatic esophageal cancer, and a third with metastatic urothelial cancer experienced partial objective responses. At 15 months, the response is ongoing in the patient with cervical cancer; after 7 months of treatment, the response was durable in the patient with urothelial cancer; and a response lasting 4 months was reported for the patient with esophageal cancer.
Dr Lu said that a phase 2 trial has been initiated to study the clinical responses of this T-cell receptor therapy in different types of metastatic cancers.
In his discussion of the paper, Michel Sadelain, MD, of the Memorial Sloan Kettering Cancer Center, New York City, said, “Although therapy with CD4 cells has been evaluated using endogenous receptor, this is the first study using genetically engineered CD4 T-cells.”
Although the study showed that therapy with genetically engineered T-cells is safe and efficacious at least in three patients, the mechanism of cytotoxicity remains unclear, Dr Sadelain indicated.
Comparison With CAR T-cells
CAR T-cells act in much the same way. CARs are chimeric antigen receptors that have an antigen-recognition domain of an antibody (the V region) and a “business end,” which activates T-cells. In this case, CD8 T-cells from the patients are used to genetically engineer T-cells ex vivo. In the majority of cases, dramatic responses have been seen in hematologic malignancies.
CARs, directed against self-proteins, result in on-target, off-tumor effects, Gregory L. Beatty, MD, PhD, assistant professor of medicine at the University of Pennsylvania, in Philadelphia, indicated when he reported the first success story of CAR T-cells in a solid pancreatic cancer tumor.
Side effects of therapy with CD4 T-cells targeting MAGE-A3 were different and similar to side effects of chemotherapy, because patients received a lymphodepleting regimen of cyclophosphamide and fludabarine. Toxicities included high fever, which was experienced by the majority of patients (12/14). The fever lasted 1 to 2 weeks and was easily manageable.
High levels of the cytokine interleukin-6 (IL-6) were detected in the serum of all patients after treatment. However, the elevation in IL-6 levels was not considered to be a cytokine release syndrome, because no side effects occurred that correlated with the syndrome, Dr Liu indicated.
He also indicated that future studies are planned that will employ genetically engineered CD4 T-cells in combination with programmed cell death protein 1–blocking antibodies.
This study was funded by Intramural Research Program of the National Institutes of Health. The NCI’s research and development of T-cell receptor therapy targeting MAGE-A3 are supported in part under a cooperative research and development agreement between the NCI and Kite Pharma, Inc. Kite has an exclusive, worldwide license with the NIH for intellectual property relating to retrovirally transduced HLA-DPB1*0401 and HLA A1 T-cell receptor therapy targeting MAGE-A3 antigen. Dr Lu and Dr Rosenberg have disclosed no relevant financial relationships.
American Association for Cancer Research (AACR) 2016 Annual Meeting: Abstract CT003, presented April 17, 2016.
Searches Related to immunotherapy using genetically engineered CD4 T-cells
To be effective for the treatment of cancer and infectious diseases, T cell adoptive immunotherapy requires large numbers of cells with abundant proliferative reserves and intact effector functions. We are achieving these goals using a gene therapy strategy wherein the desired characteristics are introduced into a starting cell population, primarily by high efficiency lentiviral vector-mediated transduction. Modified cells are then expanded using ex vivo expansion protocols designed to minimally alter the desired cellular phenotype. In this article, we focus on strategies to (1) dissect the signals controlling T cell proliferation; (2) render CD4 T cells resistant to HIV-1 infection; and (3) redirect CD8 T cell antigen specificity.
Adoptive T cell therapy is a form of transfusion therapy involving the infusion of large numbers of T cells with the aim of eliminating, or at least controlling, malignancies or infectious diseases. Successful applications of this technique include the infusion of CMV-or EBVspecific CTLs to protect immunosuppressed patients from these transplantation-associated diseases [1,2]. Furthermore, donor lymphocyte infusions of ex vivo-expanded allogeneic T cells have been used to successfully treat hematological malignancies in patients with relapsed disease following allogeneic hematopoietic stem cell transplant [3]. However, in many other malignancies and chronic viral infections such as HIV-1, adoptive T cell therapy has achieved inconsistent and/or marginal successes. Nevertheless, there are compelling reasons for optimism on this strategy. For example, the existence of HIV-positive elite non-progressors [4], as well as the correlation between the presence of intratumoral T cells and a favorable prognosis in malignancies such as ovarian [5,6] and colon carcinoma [7,8], provides in vivo evidence for the critical role of the immune system in controlling both HIV and cancer.
The key to successful adoptive immunotherapy strategies appears to consist of (1) using the “right” T cell type(s) and (2) obtaining therapeutically effective numbers of these cells without compromising their effector functions or their ability to engraft within the host. This article is focused on strategies employed in our laboratory to generate the “right” cell through genetic engineering approaches, with an emphasis on redirecting the antigen specificity of CD8 T cells, and rendering CD4 T cells resistant to HIV-1 infection. The article by Paulos et al. describes the evolving process of how to best obtain therapeutically effective numbers of the “right” cells by optimizing ex vivo cell expansion strategies.
Our laboratory’s overall strategy and flow plan for development and evaluation of engineered T cells is depicted in Fig. 1. We work almost exclusively with primary human T cells; little or no work is performed with conventional established cell lines. Thus, we benefit substantially from our close association with the UPenn Human Immunology Core. The Core performs leukaphereses on healthy donors 2–3 times a week, and provides purified peripheral blood mononuclear cell subsets, ensuring a constant influx of fresh human T cells into our laboratory. We have extensive experience in developing both bead- and cell-based artificial antigen presenting cells (aAPCs), as described in detail in the article by Paulos et al. The ability to genetically modify T cells at high efficiency is critical for virtually every project within the laboratory. We have adapted the lentiviral vector system described by Dull [15] for most, but not all, of the engineering applications in our laboratory.
CD4 T cells are the primary target of HIV-1, and decreasing CD4 T cell numbers is a hallmark of advancing HIV-1 disease [34]. Thus, strategies that protect CD4 T cells from HIV-1 infection in vivo would conceivably provide sufficient immunological help to control HIV-1 infection. Our early observations that CD3/CD28 costimulation resulted in improved ex vivo expansion of CD4 T cells from both healthy and HIV-infected donors, as well as enhanced resistance to HIV-1 infection [35,36], ultimately led to the first-in-human trial of lentiviral vector-modified CD4 T cells [37]. In this trial, CD4 T cells from HIV-positive subjects who had failed antiretroviral therapy were transduced with a lentiviral vector encoding an antisense RNA that targeted a 937 bp region in the HIV-1 envelope gene. Preclinical studies demonstrated that this antisense region, directed against the HIV-1NL4-3 envelope, provided robust protection from a broad range of both R5-and X4-tropic HIV-1 isolates [38]. One year after administration of a single dose of the gene-modified cells, four of the five enrolled patients had increased peripheral blood CD4 T cell counts, and in one subject, a 1.7 log decrease in viral load was observed. Finally, in two of the five patients, persistence of the gene-modified cells was detected one year post-infusion.
Since its identification as the primary co-receptor involved in HIV transmission, CCR5 has attracted considerable attention as a target for HIV therapy [42,43]. Indeed, “experiments of nature” have shown that individuals with a homozygous CCR5 Δ32 deletion are highly resistant to HIV-1 infection. Thus, we hypothesized that knocking out the CCR5 locus would generate CD4 T cells permanently resistant to infection by R5 isolates of HIV-1. To test this hypothesis we took advantage of zinc-finger nuclease (ZFN) technology [44]. ZFNs introduce sequencespecific double-strand DNA breakage, which is imperfectly repaired by non-homologous endjoining. This results in the permanent disruption of the genomic target, a process termed genome editing (Fig. 3).
Genetic modification of T cells to redirect antigen specificity is an attractive strategy compared to the lengthy process of growing T cell lines or CTL clones for adoptive transfer. Genetically modified, adoptively transferred T cells are capable of long-term persistence in humans [37, 46,47], demonstrating the feasibility of this approach. When compared to the months it can take to generate an infusion dose of antigen-specific CTL lines or clones from a patient, a homogeneous population of redirected antigen-specific cells can be expanded to therapeutically relevant numbers in about two weeks [3]. Several strategies are being explored to bypass the need to expand antigen-specific T cells for adoptive T cell therapy. The approaches currently studied in our laboratory involve the genetic transfer of chimeric antigen receptors and supraphysiologic T cell receptors.
Chimeric antigen receptors (CARs or T-bodies) are artificial T cell receptors that combine the extracellular single-chain variable fragment (scFv) of an antibody with intracellular signaling domains, such as CD3ζ or Fc(ε)RIγ [48–50]. When expressed on T cells, the receptor bypasses the need for antigen presentation on MHC since the scFv binds directly to cell surface antigens. This is an important feature, since many tumors and virus-infected cells downregulate MHCI, rendering them invisible to the adaptive immune system. The high-affinity nature of the scFv domain makes these engineered T cells highly sensitive to low antigen densities. In addition, new chimeric antigen receptors are relatively easy to produce from hybridomas. The key to this approach is the identification of antigens with high surface expression on tumor cells, but reduced or absent expression on normal tissues. Since one can redirect both CD4 and CD8 T cells, the T-body approach to immunotherapy represents a near universal “off the shelf” method to generate large numbers of antigen-specific helper and cytotoxic T cells.
Many T-bodies targeting diverse tumors have been developed [51], and four have been evaluated clinically [52–55]. Three of the four studies were characterized by poor transgene expression and limited T-body engraftment. However, in a study of metastatic renal cell carcinoma using a T-body directed against carbonic anhydrase IX [55], T-body-expressing cells were detectable in the peripheral blood for nearly 2 months post-administration.
The major goals in the T-body field currently are to optimize their engraftment and maximize their effector functions. Our laboratory is addressing both problems simultaneously through an in-depth study of the requirements for T-body activation. We hypothesize that their limited persistence is due to incomplete cell activation due to the lack of costimulation. While naïve T cells depend on costimulation through CD28 ligation to avoid anergy and undergo full activation in response to antigen, it is recognized that effector cells also require costimulation to properly proliferate and produce cytokines [56]. Previous studies have shown that providing CD28 costimulation is crucial for the antitumoral function of adoptively transferred T cells and T-bodies [57–59]. Unlike conventional T cell activation, which requires two discrete signals, T-bodies can be engineered to provide both costimulation and CD3 signaling through one binding event.
A different approach for redirecting specificity to T cells for adoptive immunotherapy involves the genetic transfer of full-length TCR genes. A T cell’s specificity for its cognate antigen is solely determined by its TCR. Genes encoding the α and β chains of a T cell receptor (TCR) can be isolated from a T cell specific for the antigen of interest and restricted to a defined HLA allele, inserted into a vector, and then introduced into large numbers of T cells of individual patients that share the restricting HLA allele as well as the targeted antigen. In 1999, Clay and colleagues from Rosenberg’s group at the National Cancer Institute were the first to report the transfer of TCR genes via a retroviral vector into human lymphocytes and to show that T cells gained stable reactivity to MART-1 [67]. To date, many others have shown that the same approach can be used to transfer specificity for multiple viral and tumor associated antigens in mice and human systems. These T cells gain effector functions against the transferred TCR’s cognate antigen, as defined by proliferation, cytokine production, lysis of targets presenting the antigen, trafficking to tumor sites in vivo, and clearance of tumors and viral infection.
In 2006, Rosenberg’s group redirected patients’ PBLs with the naturally occurring, MART-1- specific TCR reported in 1999 by Clay. In the first clinical trial to test TCR-transfer immunotherapy, these modified T cells were infused into melanoma patients [68]. While the transduced T cells persisted in vivo, only two of the 17 patients had an objective response to this therapy. One issue revealed by the study was the poor expression of the transgenic TCRs by the transferred T cells. Nonetheless, the results from this trial showed the potential of TCR transfer immunotherapy as a safe form of therapy for cancer and highlighted the need to optimize such therapy to attain maximum potency.
The adoptive immunotherapy field is advancing by a tried-and-true method: learning from disappointments and moving forward. Our ability to fully realize the therapeutic potential of adoptive T cell therapy is tied to a more complete understanding of how human T cells receive signals, kill targets, and modulate effective immune responses. Our goal is to perform labbased experiments that provide insight into how primary T cells function in a manner that will facilitate and enable adoptive T cell therapy clinical trials. Our ability to efficiently modify (and expand) T cells ex vivo provides the opportunity to deliver sufficient immune firepower where it has heretofore been lacking. Sustained transgene expression, coupled with enhanced in vivo engraftment capability, will move adoptive immunotherapy into a realm where longterm therapeutic benefits are the norm rather than the exception.
Genetic Modification of T Lymphocytes for Adoptive Immunotherapy
Adoptive transfer of T lymphocytes is a promising therapy for malignancies—particularly of the hemopoietic system—and for otherwise intractable viral diseases. Efforts to broaden the approach have been limited by the physiology of the T cells themselves and by a range of immune evasion mechanisms developed by tumor cells. In this review we show how genetic modification of T cells is being used preclinically and in patients to overcome these limitations, by incorporation of novel receptors, resistance mechanisms, and control genes. We also discuss how the increasing safety and effectiveness of gene transfer technologies will lead to an increase in the use of gene-modified T cells for the treatment of a wider range of disorders.
That gene transfer could be used to improve the effectiveness of T lymphocytes was apparent from the beginning of clinical studies in the field. T cells were the very first targets for genetic modification in human gene transfer experiments. Rosenberg’s group marked tumor-infiltrating lymphocytes ex vivo with a Moloney retroviral vector encoding neomycin phosphotransferase before reinfusing them and attempting to demonstrate selective accumulation at tumor sites. Shortly thereafter, Blaese and Anderson led a group that infused corrected T cells into two children with severe combined immunodeficiency due to ADA deficiency. While neither study was completely successful in terms of outcome, both showed the feasibility of ex vivo gene transfer into human cells and set the stage for many of the studies that followed. More recently, a second wave of interest in adoptive T cell therapies has developed, based on their success in the prevention and treatment of viral infections such as EBV and cytomegalovirus (CMV) and on their apparent ability to eradicate hematologic and perhaps solid malignancies1,2,3,4,5,6. There has been a corresponding increase in studies directed toward enhancing the antineoplastic and antiviral properties of the T cells. In this article we will review how gene transfer may be used to produce the desired improvements focusing on vectors and genes that have had clinical application.
Currently available viral and nonviral vector systems lack a pattern of biodistribution that would favor T cell transduction in vivo—as occurs, for example, with adenovectors and the liver or liposomal vectors and the lung. This lack of favorable biodistribution cannot yet be compensated for by the introduction of specific T-cell-targeting ligands into vectors. Hence, all T cell gene transfer studies conducted to date have used ex vivo transduction followed by adoptive transfer of gene-modified cells. This approach is inherently less attractive for commercial development than directin vivo gene transfer and has probably restricted interest in developing clinical applications using these cells. On the other hand, ex vivo transduction may be more readily controlled, characterized, and standardized than in vivo efforts and may ultimately produce a better defined final product (the transduced cell).
The gene products of suicide and coexpressed resistance genes are highly immunogenic and may induce immune-mediated rejection of the transduced cells. In one study, the persistence of adoptively transferred autologous CD8+ HIV-specific CTL clones modified to express the hygromycin phosphotransferase (Hy) gene and the herpesvirus thymidine kinase gene as a fusion gene was limited by the induction of a potent CD8+ class I MHC-restricted CTL response specific for epitopes derived from the Hy-tk protein126. Less immunogenic suicide and selection marker genes, preferably of human origin, may reduce the immunological inactivation of genetically modified donor lymphocytes. Human-derived prodrug-activating systems include the human folylpolyglutamate synthetase/methotrexate127, the deoxycytidine/cytosine arabinoside128, or the carboxylesterase/irinotecan129 systems. These systems do not activate nontoxic prodrugs but are based on enhancement of already potent chemotherapeutic agents. The administration of methotrexate to treat severe GVHD may not only kill transduced donor lymphocytes but may also have additional inhibitory activity on nontransduced but activated T cells.
Finally, endogenous proapoptotic molecules have been proposed as nonimmunogenic suicide genes. A chimeric protein that contains the FK506-binding protein FKBP12 linked to the intracellular domain of human Fas130 was recently introduced. Addition of the dimerizing prodrug induces Fas crosslinking with subsequent triggering of an apoptotic death signal.
Genetic engineering of T lymphocytes should help deliver on the promise of immunotherapies for cancer, infection, and autoimmune disease. Improvements in transduction, selection, and expansion techniques and the development of new viral vectors incapable of insertional mutagenesis will reduce the risks and further enhance the integration of T cell and gene therapies. Nonetheless, successful application of the proposed modifications to the clinical setting still requires many iterative studies to allow investigators to optimize the individual components of the approach.
Genetically modified T cells in cancer therapy: opportunities and challenges
Tumours use many strategies to evade the host immune response, including downregulation or weak immunogenicity of target antigens and creation of an immune-suppressive tumour environment. T cells play a key role in cell-mediated immunity and, recently, strategies to genetically modify T cells either through altering the specificity of the T cell receptor (TCR) or through introducing antibody-like recognition in chimeric antigen receptors (CARs) have made substantial advances. The potential of these approaches has been demonstrated in particular by the successful use of genetically modified T cells to treat B cell haematological malignancies in clinical trials. This clinical success is reflected in the growing number of strategic partnerships in this area that have attracted a high level of investment and involve large pharmaceutical organisations. Although our understanding of the factors that influence the safety and efficacy of these therapies has increased, challenges for bringing genetically modified T-cell immunotherapy to many patients with different tumour types remain. These challenges range from the selection of antigen targets and dealing with regulatory and safety issues to successfully navigating the routes to commercial development. However, the encouraging clinical data, the progress in the scientific understanding of tumour immunology and the improvements in the manufacture of cell products are all advancing the clinical translation of these important cellular immunotherapies.
The immune system is split into two arms: the innate and adaptive immune systems (Fig. 1). Through immune surveillance, any molecules that are identified as non-self are eliminated. Targets include not only virally infected cells but also transformed (tumour) cells, which can acquire antigenicity (see Box 1for a glossary of terms) and hence immunogenicity through the expression of neo-antigens that can be recognised as non-self. However, cancer cells have developed strategies to escape and suppress the immune system (Pinzon-Charry et al., 2005; Blankenstein et al., 2012), which results in a failure to initiate and maintain adequate antitumour immunity, and consequently facilitates tumour survival and progression. Strategies include tumour antigens being only weakly immunogenic. Alternatively, the tumour might downregulate or modulate the expression of antigens, thus evading immune-cell detection. In addition, tumours can suppress the immune response through the synthesis of various immune suppressants that have roles in maintaining self-tolerance, or that inhibit effector immune cell function. Tumour immune suppression affects all branches of the immune system and can result in tumour escape from the immune system.
Fig. 1. Cells of the innate and adaptive immune systems. The innate immune system provides an immediate response to foreign targets, with responses typically within minutes to hours. It consists of a number of soluble factors and proteins as well as a diverse set of cells, including granulocytes, macrophages, dendritic cells and natural killer cells. The second branch of the immune system is the adaptive or acquired immune system, which provides specific, long-lasting immune responses. The adaptive and innate immune systems are linked; for example dendritic cells are important adaptive immune system cell activators. The adaptive immune system consists of antibodies, B cells, and CD4+ and CD8+ T cells, and these enable a highly specific response against a particular target. Natural killer T cells and γδ T cells are cytotoxic lymphocytes that overlap both innate and adaptive immunity. Cells from both arms of the immune system are in development as potential cellular immunotherapies.
The concept of transferring T cells to patients (adoptive T-cell transfer) to treat disease has been established over many years through the ex vivo manipulation, expansion and reinfusion of T cells that target specific viruses, for example to treat viral infections, such as cytomegalovirus or Epstein Barr virus infections following haematopoietic stem cell transplantation (Walter et al., 1995; Heslop et al., 2010; Rooney and Leen, 2012). As described above, rare populations of tumour-antigen-specific T cells do exist and specifically can be isolated at the site of the tumour, and these are known as tumour infiltrating lymphocytes (TILs) (Kawakami et al., 1994; Robbins et al., 2013). TILs can be isolated from excised tumour tissue, cultivated, activated and expanded ex vivo, and, on reinfusion, have shown promising efficacy in the clinic, particularly in the treatment of melanoma (Rosenberg et al., 1988,Besser et al., 2013; Kvistborg et al., 2012; Dudley et al., 2013), supporting the therapeutic potential of tumour-specific T cells.
An alternative option to these approaches that is now starting to generate compelling clinical data is based on the premise that the antigen specificity of T cells can be manipulated by genetic modification and redirected to successfully target antigens that are expressed by tumours. In particular, T cells can be engineered to express modified TCRs (so-called TCR therapies) or protein-fusion-derived chimeric antigen receptors (CARs) that have enhanced antigen specificity (Fig. 3). These approaches could overcome the fundamental limitations associated with central and peripheral tolerance, and generate T cells that will be more efficient at targeting tumours without the requirement for de novo T-cell activation in the patient.
Fig. 3. Genetically modified TCRs for cancer immunotherapy. (A) T-cell response can be manipulated and redirected against cancer, with improved specificity and affinity for tumour antigens, via genetic engineering of the endogenous TCR. (B) Genetically modified TCR: gene sequences are transferred to the T cell to encode new TCR α and β chains with different peptide specificity. In addition, there can also be transmembrane changes (red bars). To minimise interchain mispairing with the endogenous TCR, modifications such as the addition of a disulphide bridge (ss) are made. (C) Alternatively, a fusion receptor can be generated, a chimeric antigen receptor (CAR). Typically, these consist of three parts: a recognition sequence [represented here by an antibody-derived single-chain variable fragment (scFv)], a transmembrane element and an intracellular bespoke signalling domain (CD3ζ), which also contains co-stimulatory molecules, such as CD28 and tumour necrosis factor receptors (TNFr) such as OX-40.
The feasibility of T-cell adoptive transfer was first reported nearly 20 years ago (Walter et al., 1995) and the field of T-cell therapies is now poised for significant clinical advances. Recent clinical trial successes have been achieved through multiple small advances, improved understanding of immunology and emerging technologies. As the key challenges of T-cell avidity, persistence and ability to exert the desired anti-tumour effects as well as the identification of new target antigens are addressed, a broader clinical application of these therapies could be achieved. As the clinical data emerges, the challenge of making these therapies available to patients shifts to implementing robust, scalable and cost-effective manufacture and to the further evolution of the regulatory requirements to ensure an appropriate but proportionate system that is adapted to the characteristics of these innovative new medicines.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
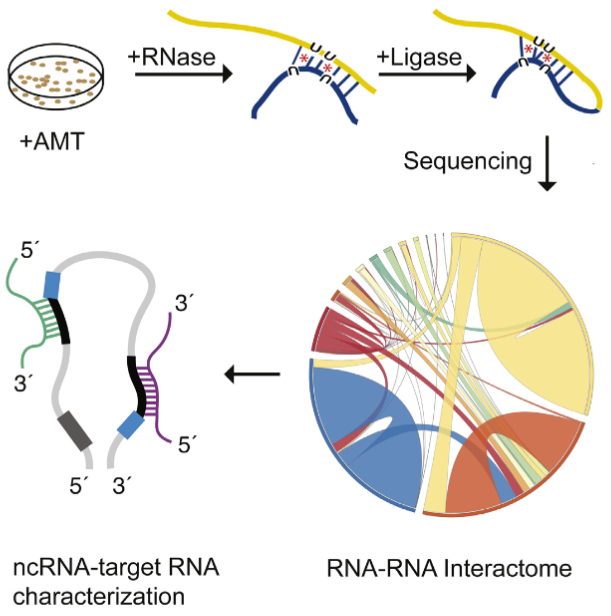{kind=link}
{kind=link}
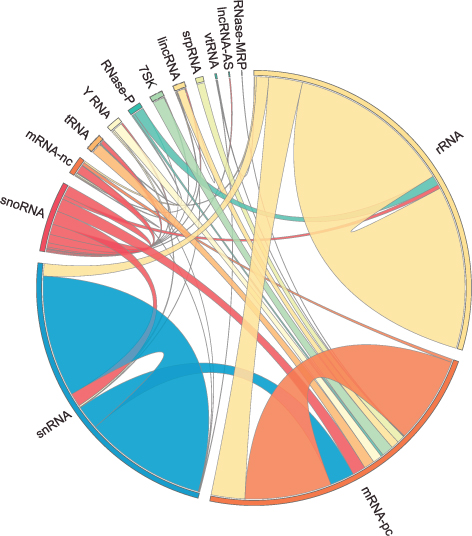{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This is a lovely method and should find wide applicability in many settings, especially for microorganisms and cell lines. However, it is not clear that this approach will be, as implied by the discussion, an efficient mapping method for all multicellular organisms. I have performed similar experiments in Drosophila, focused on meiotic recombination, on a much smaller scale, and found that CRISPR-Cas9 can indeed generate targeted recombination at gRNA target sites. In every case I tested, I found that the recombination event was associated with a deletion at the gRNA site, which is probably unimportant for most mapping efforts, but may be a concern in some specific cases, for example for clinical applications. It would be interesting to know how often mutations occurred at the targeted gRNA site in this study.
The wider issue, however, is whether CRISPR-mediated recombination will be more efficient than other methods of mapping. After careful consideration of all the costs and the time involved in each of the steps for Drosophila, we have decided that targeted meiotic recombination using flanking visible markers will be, in most cases, considerably more efficient than CRISPR-mediated recombination. This is mainly due to the large expense of injecting embryos and the extensive effort and time required to screen injected animals for appropriate events. It is both cheaper and faster to generate markers (with CRISPR) and then perform a large meiotic recombination mapping experiment than it would be to generate the lines required for CRISPR-mediated recombination mapping. It is possible to dramatically reduce costs by, for example, mapping sequentially at finer resolution. But this approach would require much more time than marker-assisted mapping. If someone develops a rapid and cheap method of reliably introducing DNA into Drosophila embryos, then this calculus might change.
However, it is possible to imagine situations where CRISPR-mediated mapping would be preferable, even for Drosophila. For example, some genomic regions display extremely low or highly non-uniform recombination rates. It is possible that CRISPR-mediated mapping could provide a reasonable approach to fine mapping genes in these regions.
The authors also propose the exciting possibility that CRISPR-mediated loss of heterozygosity could be used to map traits in sterile species hybrids. It is not entirely obvious to me how this experiment would proceed and I hope the authors can illuminate me. If we imagine driving a recombination event in the early embryo (with maternal Cas9 from one parent and gRNA from a second parent), then at best we would end up with chimeric individuals carrying mitotic clones. I don’t think one could generate diploid animals where all cells carried the same loss of heterozygosity event. Even if we could, this experiment would require construction of a substantial number of stable transgenic lines expressing gRNAs. Mapping an ~20Mbp chromosome arm to ~10kb would require on the order of two-thousand transgenic lines. Not an undertaking to be taken lightly. It is already possible to perform similar tests (hemizygosity tests) using D. melanogaster deficiency lines in crosses with D. simulans, so perhaps CRISPR-mediated LOH could complement these deficiency screens for fine mapping efforts. But, at the moment, it is not clear to me how to do the experiment.